Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02474687 2004-07-27
WO 03106599 1 PCT/EP03/00538
79950pct.210
N-allyloxyethyl-1,2,3,4,5,6-hexahydro-2,6-methano-3-benzazocines and
the use thereof as medicaments
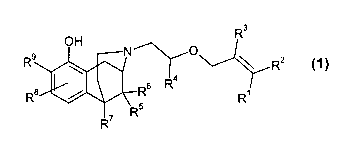
The planned patent application relates to substituted N-allyloxyethyl-
1,2,3,4,5,6-hexahydro-2,6-methano-3-benzazocin-10-ols of general formula 1
R3
OH
R9 N O \ R2
4
R$ / Rs R R~
R7 Ft5
1
wherein
R1, R2 and R3 which may be identical or different, may denote
hydrogen, methyl or ethyl;
R4 may denote hydrogen, methyl or ethyl;
R5, R6 and R7 which may be identical or different, may denote hydrogen,
methyl or ethyl;
R$ and R9 which may be identical or different, may denote hydrogen,
fluorine, chlorine, bromine, methyl, ethyl, hydroxy or
methoxy,
optionally in the form of the racemates, the enantiomers, the diastereomers
and the mixtures thereof, as well as optionally the pharmacologically
acceptable acid addition salts thereof.
Preferred compounds of general formula 1 are those wherein
R1, R2 and R3 which may be identical or different, may denote hydrogen
or methyl;
R4 may denote hydrogen or methyl;
R5, R6 and R7 which may be identical or different, may denote hydrogen
or methyl, preferably methyl;
R$ may denote hydrogen, methyl, hydroxy or methoxy,
preferably hydrogen or methyl,
R9 may denote hydrogen or methyl,
CA 02474687 2004-07-27
WO 03/066599 2 PCT/EP03100538
optionally in the form of the racemates, the enantiomers, the diastereomers
and the mixtures thereof, as well as optionally the pharmacologically
acceptable acid addition salts thereof.
Particularly preferred are the compounds of general formula 1 wherein
R1, R2 and R3 which may be identical or different, may denote hydrogen
or methyl;
R4 may denote hydrogen or methyl;
R5, R6 and R7 may denote methyl;
R$ may denote hydrogen or methyl, preferably hydrogen;
Rg may denote hydrogen or methyl,
optionally in the form of the racemates, the enantiomers, the diastereomers
and the mixtures thereof, as well as optionally the pharmacologically
acceptable acid addition salts thereof.
Of exceptional importance according to the invention are those compounds of
formula 1 which are in the 1 R configuration and, if R4 does not denote
hydrogen, in the 2"S configuration. These stereoisomers which are preferred
according to the invention may also be represented by general formula 1'
R3
OH
R9 ~ N ~ O ~ R2
4
R$ / Rs R R~
R7 Rs
wherein the groups R1, R2, R3, R4,R5, R6, R7, R$ and R9 may have the
meanings given above.
Of particular interest are the following compounds of general formula 1:
- (2R)-N-allyloxyethyl-1,2,3,4,5,6-hexahydro-6,11,11-trimethyl-2,6-
methano-3-benzazocin-10-ol-hydrochloride;
- (2R,2"S)-N-(2-allyloxy-propyl)-1,2,3,4,5,6-hexahydro-6,11,11-trimethyl-
2,6-methano-3-benzazocin-10-oi-hydrochloride.
If desired, the compounds of general formula (1 ) may be converted into the
salts thereof, particularly, for pharmaceutical use, into the
pharmacologically
CA 02474687 2004-07-27
WO 03/066599 3 PCTBP03/00538
acceptable acid addition salts thereof with an inorganic or organic acid.
Examples of acids for this purpose include for example succinic acid,
hydrobromic acid, acetic acid, fumaric acid, malefic acid, methanesulphonic
acid, lactic acid, phosphoric acid, hydrochloric acid, sulphuric acid,
tartaric
acid or citric acid. Moreover, mixtures of these acids may also be used.
The compounds claimed are blockers of the voltage-dependent sodium
channel. These are compounds which displace batrachotoxin (BTX) with a
high affinity (Ki < 500 nM) competitively or non-competitively from the
binding
site on the sodium channel. Such substances exhibit "use-dependency" while
the sodium channels are blocked, i.e. in order to bind the substances at the
sodium channel, the sodium channels first have to be activated. Maximum
blockage of the sodium channels is only achieved after repeated stimulation
of the sodium channels. Consequently, the substances bind preferentially to
sodium channels which are activated a number of times. As a result, the
substances are in a position to become effective particularly in those parts
of
the body which are pathologicaNy overstimulated. The compounds of general
formula 1 according to the invention can thus be used to treat diseases which
are caused by a functional disorder resulting from overstimulation. These
include diseases such as arrhythmias, spasms, cardiac and cerebral
ischaemias, pain and neurodegenerative diseases of various origins. These
include, for example: epilepsy, hypoglycaemia, hypoxia, anoxia, brain trauma,
brain oedema, cerebral stroke, perinatal asphyxia, degeneration of the
cerebellum, amyotropic lateral sclerosis, Huntington's disease, Alzheimer's
disease, Parkinson's disease, cyclophrenia, hypotonia, cardiac infarction,
heart rhythm disorders, angina pectoris, chronic pain, neuropathic pain and
local anaesthesia.
Consequently, in another aspect, the invention relates to the use of
compounds of general formula 1 as pharmaceutical compositions, particularly
as pharmaceutical compositions wherein the blocking of the voltage-
dependent sodium channel may have a therapeutic benefit.
Preferably, the compounds of general formula 1 are used according to the
invention to prepare a pharmaceutical composition for the prevention or
CA 02474687 2004-07-27
WO 03/066599 4 PCT/EP03/00538
treatment of arrhythmias, spasms, cardiac and cerebral ischaemias, pain and
neurodegenerative diseases.
Most preferably, the compounds of general formula 1 are used as described
above according to the invention to prepare a pharmaceutical composition for
the prevention or treatment of epilepsy, hypoglycaemia, hypoxia, anoxia, brain
trauma, brain oedema, cerebral stroke, perinatal asphyxia, degeneration of
the cerebellum, amyotropic lateral sclerosis, Huntington's disease,
Alzheimer's disease, Parkinson's disease, cyclophrenia, hypotonia, cardiac
infarction, heart rhythm disorders, angina pectoris, chronic pain, neuropathic
pain and local anaesthesia.
The blocking action on the sodium channel may be demonstrated by the test
system which tests the BTX binding to the sodium channel [S.W. Postma &
W.A. Catterall, Mol. Pharmacol 25, 219-227 (1984)] as well as by patch-clamp
experiments which show that the compounds according to the invention block
the electrically stimulated sodium channel in a "use-dependent" manner [W.A.
Catterall, Trends Pharmacol. Sci., 8, 57-65 (1987)]. By a suitable choice of
cell system (e.g. neuronal, cardiac, DRG cells) it is possible to test the
effect
of the substances on different subtypes of sodium channel.
The sodium channel blocking property of the compounds according to the
invention can be demonstrated by the blocking of the veratridine-induced
release of glutamate [S. Villanueva, P. Frenz, Y. Dragnic, F. Orrego, Brain
Res. 461, 377-380 (1988)]. Veratridine is a toxin which opens the sodium
channel permanently. This leads to an increased influx of sodium ions into the
cell. This sodium influx leads to an increased release of glutamate in the
neuronal tissue. The compounds according to the invention antagonise this
release of glutamate.
Neuroprotective properties were demonstrated by a protective effect in a rat
MCAO model [U. Pschorn & A. J. Carter, J. Stroke Cerebrovascular Diseases,
6, 93-99 (1996)] and a malonate-induced lesion model [M.F. Beal, Annals of
Neurology, 38, 357-366 (1995) and J.B. Schulz, R.T. Matthews, D.R.
Henshaw and M.F. Beal, Neuroscience, 71, 1043-1048 (1996)].
CA 02474687 2004-07-27
WO 03/06699 5 PCT/EP03/00538
Analgesic effects can be investigated in models of diabetic neuropathy and in
a ligature model [C. Courteix, M. Bardin, C. Chantelauze, J. Lavarenne, A.
Eschalier, Pain 57, 153-160 (1994); C. Courteix, A. Eschalier, J. Lavarenne,
Pain 53, 81-88 (1993); G. J. Bennett and Y.-K. Xie, Pain 33, 87-107 (1988)].
It has also been reported that sodium channel blockers can be used to treat
cyclophrenia (manic depressive disorder) [J. R. Calabrese, C. Bowden, M.J.
Woyshville; in: Psychopharmacology: The Fourth Generation of Progress
(Eds.: D. E. Bloom and D. J. Kupfer) 1099-1111. New York: Raven Press
Ltd. ].
The compounds 1 according to the invention may be prepared analogously to
methods of synthesis known per se. One possible method of synthesis is
shown in Diagram 1. The 1,2,3,4,5,6-hexahydro-2,6-methano-3-benzazocin-
10-ols (2) shown as starting compounds in Diagram 1 may be obtained by
methods of synthesis known in the art. In this respect reference is hereby
made to European Patent Application EP-A-521422 and to International
Patent Applications WO 97/06146 and WO 99/14199.
Diagram 1:
0
R3
X O
.s -~- ~ R2
' Ra
R'
2 3
OH Rs
Rs N~O
R2
Ra Rs Ra R~
Rs
R
1
The synthesis component 3 contains a leaving group X, which is preferably
chlorine, bromine, hydroxy or a methoxy or ethoxy group. For a detailed
CA 02474687 2004-07-27
WO 03/066599 ( PCT/EP03f00538
explanation of the synthesis of the compounds of formula 1 according to the
invention reference is made to the experimental procedures described below.
One possible method of obtaining compounds of formula 1 wherein R9
denotes methyl is shown in Diagram 2.
Diagram 2:
R3
OH
H N~O \~ Z 1. CH20
R6 R4 YR 2. SOCK
'R
R$ RS 3. LiAIH4
R'
R3
(with R9= H) OH N O
Me ~ ~ ~ Rz
Rs R4
R
R$ RS
R'
1 (with R9= Methyl)
For a detailed explanation of the synthesis of the compounds of formula 1
according to the invention as shown in Diagram 2 reference is again made to
the experimental procedures described below.
The Examples that follow serve to illustrate the invention more fully without
restricting it to the compounds and processes disclosed by way of example.
CA 02474687 2004-07-27
WO 03/066599 '7 PCTlEP03/00538
Example 1: (2R)-N-allyloxyethyl-1,2,3,4,5,6-hexahydro-6,11 11-trimethyl-2 6-
methano-3-benzazocin-10-ol-hydrochloride
)H
N~O
/~\ ~Me
Me
Me
1.8 g of allyloxyacetic acid are placed in 15 mL dichloromethane, combined
with 4.8 g of TBTU and 7.5 mL of ethyldiisopropylamine and stirred for 15 min.
at RT. Then the mixture is cooled to -5°C and 3.1 g of 1,2,3,4,5,6-
hexahydro-
6,11,11-trimethyl-2,6-methano-3-benzazocin-10-0l are added. The mixture is
stirred for 30 min at 0°C, and for 1 h at RT. Then it is washed once
with 100
ml of 2N HCL and 100m1 of 10% potassium carbonate solution, dried and
evaporated down in vacuo. The residue is taken up in 50 mL of THF and
added dropwise under nitrogen to a suspension of 1.0 g of lithium aluminium
hydride in 50 ml of THF (temp. increases to 35°C). Then the mixture is
heated
to 50°C, stirred for 1 h, cooled and 1 ml of water is added dropwise at
0 -
10°C, the mixture is stirred for 30 min, 3ml of NaOH are added and the
mixture is stirred for another 30 min. The precipitate is suction filtered,
the
mother liquor is evaporated down in vacuo and the residue is filtered through
a short column (about 75 ml of silica gel; dichloromethane 70, ethyl acetate
20, methanol 10). The appropriate fractions are evaporated down in vacuo,
and crystallised from acetone + eth. HCI.
Yield 2.8 g (77%), melting point: 236 °C; [a]p~°= -78,3
° (c = 1; methanol).
Example 2: (2R,2"S)-N-(2-allyloxy-propyl)-1,2,3,4,5,6-hexahydro-6 11 11-
trimethyl-2,6-methano-3-benzazocin-10-ol-hydrochloride
)H
N O ~~
/ ' Me Me
Me
Me
This is prepared analogously to the method according to Example 1.
Yield 56%, melting point: 239 °C; [a]p °_ -33.9 ° (c
= 1; methanol).
CA 02474687 2004-07-27
WO 03!066599 g PCT/EP03/00538
Example 3: (2R,2"S)-N-(2-but-2-epoxy-propyl)-1,2,34 5,6-hexahydro-
6,11,11-trimethyl-2,6-methano-3-benzazocin-10-ol-hydrochloride
)H
N \, O
I\
Me Me
Me
Me
This is prepared analogously to the method according to Example 1.
Yield 47%, melting point: 205 °C
Example 4: (2R,2"S)-N-I2-(2-metal-propenoxy~-proQyll-1,2,3,4,5,6-
hexahydro-6,11,11-trimethyl-2,6-methano-3-benzazocin-10-ol-hydrochloride
OH
N O
Me Me
Me
This is prepared analogously to the method according to Example 1.
Yield 12%, melting point: 240 °C; [a,]o2°- -29.6 °
(c = 1; methanol).
Example 5: (2R)-N-f2-allyloxyethy)-1,2L3,4,5,6-hexahydro-6,9,11,11-
tetramethyl-2,6-methano-3-benzazocin-10-ol-hydrochloride
~O~
Ma
Me
9e
Me
1.9 g of (2R)-N-allyloxyethyl-1,2,3,4,5,6-hexahydro-6,11,11-trimethyl-2,6-
methano-3-benzazocin-10-ol-hydrochloride (Example 1) are dissolved in 40
mL methanol and combined with 3 g of 30% formalin solution and 3 mL of 4 N
NaOH. The mixture is heated to 50 °C for 12 hours, the solvent is
eliminated
in vacuo, the residue is combined with 100 mL of water and extracted twice
with 200 ml of ether. The organic phase is washed with water, dried and
evaporated down in vacuo. The residue is dissolved in 20 mL of
CA 02474687 2004-07-27
WO 03/066599 9 PCT/EP03/00538
dichloromethane and 1.5 g of SOC12 are added dropwise at RT. After 30 min.
the mixture is evaporated down in vacuo, the residue is taken up in 20 ml of
THF and added dropwise under nitrogen to a suspension of 0.5 g of lithium
aluminium hydride in 20 ml of tetrahydrofuran. Then it is heated to
50°C for 2
h, cooled, 1.5mL of 4N NaOH are added dropwise and the resulting mixture is
stirred for 30 min. The precipitate is suction filtered and the mother liquor
evaporated down in vacuo. The residue is filtered through a short silica gel
column (about 30 mL of silica gel, about 250 mL of ethyl acetate). The
appropriate fractions are evaporated down in vacuo and crystallised from
acetone + eth. HCI.
Yield 1.1 g (56%), melting point: 212 °C, [a]~2°= - 71.6
° (c = 1; methanol).
Example 6: (2R,2"S)-N-(2-allyloxy-eropyll-1,2,3,4,5,6-hexahydro-6,9,11,11-
tetrameth»I-2,6-methano-3-benzazocin-10-ol-hydrochloride
O'
Me
Me Me
1e
Me
This is prepared analogously to the method according to Example 5, starting
from Example 2.
Yield 60°I°, melting point: 215 °C; [oc]o2°= -
29.3° (c = 1; methanol).
The compounds according to the invention may be administered orally,
transdermally, by inhalation or parenterally. The compounds according to the
invention occur as active ingredients in conventional preparations, for
example in compositions which consist essentially of an inert pharmaceutical
carrier and an effective dose of the active substance, such as for example
tablets, coated tablets, capsules, lozenges, powders, solutions, suspensions,
emulsions, syrups, suppositories, transdermal systems etc. An effective dose
of the compounds according to the invention is between 1 and 1000,
preferably between 1 and 500, most preferably between 5-300 mgJdose for
oral administration, and between 0.001 and 50, preferably between 0.1 and
mgldose for intravenous, subcutaneous or intramuscular administration.
For inhalation, according to the invention, solutions containing 0.01 to 1.0,
CA 02474687 2004-07-27
WO 03/066'599 10 PCTBP03/00538
preferably 0.1 to 0.5 % active substance are suitable. For administration by
inhalation the use of powders is preferred. By virtue of their particular
physico-
chemical properties it is also possible to use the compounds according to the
invention as a solution for infusion, preferably in a physiological saline or
nutrient saline solution. In an infusion, 10-100 mg/h, preferably 25-60 mg/h
might be administered. This latter method of administration is of exceptional
importance according to the invention.
The compounds according to the invention may be used on their own or in
conjunction with other active substances according to the invention,
optionally
also in conjunction with other pharmacologically active substances. Suitable
preparations include for example tablets, capsules, suppositories, solutions,
elixirs, emulsions or dispersible powders. Suitable tablets may be obtained,
for example, by mixing the active substances) with known excipients, for
example inert diluents such as calcium carbonate, calcium phosphate or
lactose, disintegrants such as corn starch or alginic acid, binders such as
starch or gelatine, lubricants such as magnesium stearate or talc and/or
agents for delaying release, such as carboxymethyl cellulose, cellulose
acetate phthalate, or polyvinyl acetate. The tablets may also comprise several
layers.
Coated tablets may be prepared accordingly by coating cores produced
analogously to the tablets with substances normally used for tablet coatings,
for example collidone or shellac, gum arabic, talc, titanium dioxide or sugar.
To achieve delayed release or prevent incompatibilities the core may also
consist of a number of layers. Similarly the tablet coating may consist of a
number of layers to achieve delayed release, possibly using the excipients
mentioned above for the tablets.
Syrups or elixirs containing the active substances or combinations thereof
according to the invention may additionally contain a sweetener such as
saccharine, cyclamate, glycerol or sugar and a flavour enhancer, e.g. a
flavouring such as vanillin or orange extract. They may also contain
suspension adjuvants or thickeners such as sodium carboxymethyl cellulose,
wetting agents such as, for example, condensation products of fatty alcohols
with ethylene oxide, or preservatives such as p-hydroxybenzoates.
CA 02474687 2004-07-27
WO 03/066599 11 PCTBP03/00538
Solutions for injection are prepared in the usual way, e.g. with the addition
of
preservatives such as p-hydroxybenzoates, or stabilisers such as alkali metal
salts of ethylenediamine tetraacetic acid, and transferred into injection
vials
or ampoules.
Capsules containing one or more active substances or combinations of active
substances may for example be prepared by mixing the active substances
with inert carriers such as lactose or sorbitol and packing them into gelatine
capsules.
Suitable suppositories may be made for example by mixing with carriers
provided for this purpose, such as neutral fats or polyethyleneglycol or the
derivatives thereof.
A therapeutically effective daily dose is between 1 and 2500 mg, preferably 10
to 1000 mg per adult.
CA 02474687 2004-07-27
WO 03/066'599 12 PCT/EP03/00538
The Examples which follow illustrate the present invention without restricting
its scope:
Examples of pharmaceutical formulations
A) Tablets per tablet
active substance 100 mg
lactose 140 mg
corn starch 240 mg
polyvinylpyrrolidone 15 mg
magnesium stearate 5 mg
500 m g
The finely ground active substance, lactose and some of the corn starch are
mixed together. The mixture is screened, then moistened with a solution of
polyvinylpyrrolidone in water, kneaded, wet-granulated and dried. The
granules, the remaining corn starch and the magnesium stearate are
screened and mixed together. The mixture is compressed to produce tablets
of suitable shape and size.
B) Tablets per tablet
active substance 80 mg
corn starch 190 mg
lactose 55 mg
microcrystalline cellulose 35 mg
polyvinylpyrrolidone 15 mg
sodium-carboxymethyl starch 23 mg
magnesium stearate 2 mg
400 m g
The finely ground active substance, some of the corn starch, lactose,
microcrystalline cellulose and polyvinylpyrrolidone are mixed together, the
mixture is screened and worked with the remaining corn starch and water to
form a granulate which is dried and screened. The sodium-carboxymethyl
CA 02474687 2004-07-27
WO 03/066599 j 3 PCT/EP03/00538
starch and the magnesium stearate are added and mixed in and the mixture is
compressed to form tablets of a suitable size.
C) Coated tablets per coated tablet
Active substance 5 mg
Corn starch 41.5 mg
Lactose 30 mg
Polyvinylpyrrolidone 3 mg
Magnesium stearate 0.5 mg
80 mg
The active substance, com starch, lactose and polyvinylpyrrolidone are
thoroughly mixed and moistened with water. The moist mass is pushed
through a screen with a 1 mm mesh size, dried at about 45°C and the
granules are then passed through the same screen. After the magnesium
stearate has been mixed in, convex tablet cores with a diameter of 6 mm are
compressed in a tablet-making machine . The tablet cores thus produced are
coated in known manner with a covering consisting essentially of sugar and
talc. The finished coated tablets are polished with wax.
D) Capsules per capsule
Active substance 50 mg
Corn starch 268.5 mg
Magnesium stearate 1.5 mg
320 mg
The substance and com starch are mixed and moistened with water. The
moist mass is screened and dried. The dry granules are screened and mixed
with magnesium stearate. The finished mixture is packed into size 1 hard
gelatine capsules.
E) Ampoule solution
active substance 50 mg
sodium chloride 50 mg
water for inj. 5 ml
CA 02474687 2004-07-27
WO 03/066599 14 PCT/EP03/00538
The active substance is dissolved in water at its own pH or optionally at pH
5.5 to 6.5 and sodium chloride is added to make it isotonic. The solution
obtained is filtered free from pyrogens and the filtrate is transferred under
aseptic conditions into ampoules which are then sterilised and sealed by
fusion. The ampoules contain 5 mg, 25 mg and 50 mg of active substance.
F) Suppositories
Active substance 50 mg
Solid fat 1650 mg
1700 mg
The hard fat is melted. At 40°C the ground active substance is
homogeneously dispersed. It is cooled to 38°C and poured into slightly
chilled
suppository moulds.