Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
FROPARGYLAMINO INDAN DERIVATIVES AND PROPARGYLAMINO
TETRALIN DERIVATIVES AS BRAIN-SELECTIVE MAO INHIBITORS
This .application claims the benefit of U.S, Serial No.
10/085,674, filed February 27, 2002, the contents of which are
hereby incorporated by reference.
Throughout this application, various references are referenced
by short citations within parenthesis. Full citations for these
references may be found at the end of the specification,
immediately preceding the claims. These references, in their
entireties, are hereby incorporated by reference to more fully
describe the state of the art to which this invention pertains.
Field of the Invention
The subject of this invention provides for derivatives of
propargylaminoindans and propargylaminotetralins that are
irreversible inhibitors of the enzyme monoamine oxidase A and/or
B and also for prodrugs far the administration of these
compounds. Such compounds may be useful in the treatment of
Parkinson's disease, Alzheimer's disease, depression and other
neurological disorders.
Background of the Invention
The enzyme monoamine oxidase (MAO) plays an essential role in
the metabolic degradation of important amine neurotransmitters
including dopamine, serotonin and noradrenaline. Thus, agents
that inhibit MAO are of potential therapeutic benefit for a
variety of neurological disease indications, including
Parkinson's disease, Alzheimer's~disease, depression, epilepsy,
narcolepsy, amyotrophic lateral sclerosis (ALS), etc. (Szelnyi,
I.; Bentue-Ferrer et al.; .Loscher et al.; White et al.; U.S.
Patent No. 5,744,500). Other diseases and conditions which have
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
_7_
been associated with toxic levels of monoamine oxidase-B are
memory disorders (The interaction of L-deprenyl and scopolamine
on spatial learning/memory in rats), panic, post-traumatic
stress disorder (PTSD), sexual dysfunction, attention deficit
and hyperactivity syndrome (ADHD)(Potential applications for
monoamine oxidase B inhibitors), attention deficit disorder
(ICleywegt), and Tourette's syndrome (Treatment of Tourette's:
Overview).
Many inhibitors of MAO are chiral molecules (U~.S. Patent No.
5,744,500). Although one enantiomer often shows some
stereoselectivity in relative potency towards MAO-A and -B, a
given enantiomeric configuration is not always more selective
than its isomer in discriminating between MAO-A and -B
(Hazelhoff et al., Naunyn-Schmeideberg's Arch. Pharmacol.).
MAO inhibitors can also be classified as reversible inhibitors
which inhibit the enzyme by a competitive mechanism or as
irreversible inhibitors which are generally mechanism based
(suicide inhibitors) (Dostert). For example, moclobemide is a
reversible MAO-A-specific inhibitor (Fitton et al.) developed as
an anti-depressant. Likewise, rasagiline (U.S. Patent No.
5,744,500) and selegiline,(Chrisp et al.) are MAO-B-selective
irreversible inhibitors.
30
Irreversible ,inhibitors have the advantage of lower, less
frequent dosing since their MAO inhibition is not based directly
on the drugs' pharmacokinetic behavior, but rather on the de
novo regeneration of the MAO enzyme.
MAO also plays an essential role in the oxidative deamination of
biogenic and food-derived amines, both in the central nervous
system and in peripheral tissues.. MAO is found in two
functional isoenzyme forms, MAO-A and MAO-B, each of which shows
preferential affinity for substrates and specificity toward
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-3-
inhibitors. Thus, MAO-A preferentially oxidizes serotonin,
noradrenaline and adrenaline, whereas MAO-B preferentially
metabolizes phenylethylamine. Dopamine is a substrate fpr both
forms of the enzyme(Szelenyi, I.).
N-Propargyl-(1R)-aminoindan is known to be a potent B-selective
inhibitor of MAO (U. S. Patent No. 5,457,133). Various
derivatives of this compound have been prepared and shown to
have varying degrees of potency and selectivity for the
inhibition of MAO-A andlor -B. There is no currently accepted
theory explaining the effect of structure on the activity (SAR)
of the various substituted propargylaminoindans.
The dopamine agonistic activity and MAO inhibitory properties of
7-(methyl-prop-2-ynylamino)-tetralin-2-of and 7-(methyl-prop-2-
ynylamino)-tetralin-2,3-diol have been reported (Hazelhoff et
al., Eur. J. Pharmacol.). The details of the synthesis of these
compounds have not been published, however.
?0 6,7-di-0-benzoyl-2-aminotetralin has been reported as a prodrug
of the dopaminergic agonist 6,7-di-hydroxy-2-aminotetralin (Horn
et al.). However, no N-propargyl derivatives were reported and
the compounds were not shown to have MAO inhibitory or
neuroprotective activities.
7-(propyl-prop-2-ynylamino)-tetralin-2-of has been reported as
an intermediate in the preparation .of 7-[(3-iodoallyl)-
propylamino]-tetralin-2-ol. Only the latter has been
pharmacologically characterized as D3-dopamine receptor ligand
(Churnpradit et al.). No other N-alkyl substituents were
described.
Florvall et al. report the preparation of amino acid-based
prodrugs of amiflamine analogues. Amiflamine is a reversible
MAO-A inhibitor. ,
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
PCT International Application No. PCT/US97/24155 concerns
carbamate aminoindan derivatives, including propargylamines, as
inhibitors of MAO-A and M.AO-B for the treatment of Alzheimer's
disease and other neurological conditions. However, the
compounds of PCT/US97/24155 are not selective for MAO over
acetylcholinesterase ("AChE"). Thus, the compounds generally
inhibit acetylcholinesterase along with MAO.
Acetylcholinesterase inhibition is a route implicated in certain
neurological disorders, but is a different route from the route
of MAO inhibition.
U.S. Patent No. 6,303,650 discloses derivatives of 1-aminoindan
as selective MAO B inhibitors that additionally inhibit
acetylcholinesterase. The reference teaches that its compounds
can be used to treat depression, Attention Deficit Disorder
(ADC), Attention Deficit and Hyperactivity Disorder (ADHD),
Tourette's Syndrome, Alzheimer's Disease and other dementias
such as senile dementia, dementia of the Parkinson's type.,
vascular~dementia and Lewy body dementia.
Many irreversible MAO inhibitors contain the propargyl amine
functionality. This pharmacophore is responsible for the MAO
inhibitory activity of such compounds. Some propargylamines
have been shown to have neuroprotective/neurorescue properties
independent of their MAO inhibition activity (U.S. Patent No.
4,844,033; Krageten et al.).
PCT International Application No. PCTlIL96/00115 relates to
pharmaceutical compositions comprising racemic, (S), and (R)-N-
propargyl-1-aminoindan. (R)-N-propargyl-1-aminoindan selectively
inhibits MAO-B in the treatment of Parkinson's disease and other
neurological disorders(PCT/IL96/00115).
Derivatives of 1-aminoindan, including propargyl aminoindan, and
their salts are described in many U. S . patents (U. S . . Patents No.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-5-
5,639,913, 5,877,221, 5,880,159, 5,877,218, 5,914,349,
5,994,408) and a PCT International Application (PCT/US95100245).
These references disclose racemic, R and S enantiomer~ of 1-
aminoindan derivatives for the treatment of Parkinson's disease
S and other neurological conditions (U. S. Patents No. 5,639,913,
5,87.7,221, 5,880,159, 5,877,218, 5,914,349, 5,994,408,
PCT/US95/00245).
pCT International Application No. PCT/US97/24155 concerns
10. aminoindan derivatives, including propargyl aminoindan, as
inhibitors of MAO-A and MAO-B for the treatment of Parkinson's
disease and other neurological conditions. The publication
reveals that the disclosed compounds exhibit a greater
selectivity for MAO-A and MAO-B in the brain than in the liver
15 or intestine.
U.S. Patent No. 6,316,504 discloses that the R(+) enantiomer
of N-propargyl-1-aminoindan is a selective irreversible
inhibitor of MAQ-B. The patent indicates that (R)-
20 N-propargyl-1-aminoindan is useful for the treatment of
Parkinson's disease, a memory disorder, dementia, depression,
hyperactive syndrome, an affective illness, a neurodegenerative
disease, a neurotoxic injury, stroke, again ischemia, a head
trauma injury, a spinal traumG injury, neurotrauma,
25 schizophrenia, an attention deficit disorder, , multiple
sclerosis, and withdrawal symptoms.
European Patent No. 436492 discloses the R enantiomer of N-
propargyl-1-aminoindan as a selective irreversible inhibitor of
30 MAO-B in the treatment of Parkinson's disease and other
neurological conditions. Numerous U.S. patents also relate to
the MAO B inhibition of (R) -N-propargyl-1-aminoindan and its use
for treating patients suffering from Parkinson's Disease and
other neurological disorders (U. S. Patents No. 5,387,612,
35 5,453,446, 5,457,13.3, 5,519,061, 5,532,415, 5,576,353,
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-6-
5,668,181, 5,744,500, 5,786,390 and 5,891,923).
PCT International Application No. PCT/IL97/00205 disclpses 5-
(-)-N-propargyl-1-aminoindan or a pharmaceutically acceptable
salt thereof for the treatment of a neurological disorder of
neurotrauma or for improving memory. The compounds were found
to be neuroprotective, but not inhibitory of MAO-A or MAO-B
(PCT/IL97/00205).
U.S. Patent No. 5,486,541 provides N-propargyl-1-amonoindan
monofluorinated in the phenyl ring as selective inhibitors. of
MAO-B. These compounds are presented as useful in the.treatment
of Parkinson's disease, memory disorders, dementia of the
Alzheimer's type, depression and the hyperactive .syndrome in
children.
Among.the many derivatives of propargylaminoindan mentioned in
the prior art are hydroxy-propargylaminoindans. U.S. Patent No.
3,513,244 lists some racemic N-propargylamino indanols and
tetralinols for use as antihypertensives. These compounds are
not exemplified chemically and are not pharmacologically,
characterized (U.S.'Patent No. 3,513,244).
N-propargylamino indanol also appears in E.P. 267024 as a
hydrofluorene derivative, i.e., 3-amino-4-indanol (7-OH
fluorene). The hydrofluorene derivatives and salts in E.P.
267024 are employed as cerebral activators in.the treatment of
anoxemia and hypoxemia. In addition, such derivatives help
prevent arrhythmia and heart failure caused by lack of oxygen
(E.P. 267024). The derivatives also act as antioxidants and
cholinergic nerve system activating agents (E. P. 267024).
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
_'7_
Summary of the Invention
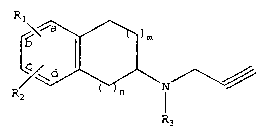
The subject invention provides a compound having the structure:
R1
~~ ) m
Wd.
( n
R2
R;
wherein R1 is OC(0)R4 and Rz is H,
wherein R9 is branched or unbranched C, to C6 alkyl,
aryl, or aralkyl, or
R1 i s OC ( 0 ) R4 and RZ i s OC ( O ) RQ ,
wherein R9 is branched or unbranched C1 to, Cs alkyl,
aryl, aralkyl or NRSR6,
wherein RS and RE are each independently H, C1 to
CB alkyl, CE to C1z aryl, C6 to C,z aralkyl or C6 to
Cia cycloalkyl, each optionally substituted;
wherein R3 is H or C1 to C6 alkyl;
wherein n is 0 or 1; and
wherein m is 1 or 2,
or a pharmaceutically acceptable salt thereof.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
_g_
The subject invention also provides a compound having the
structure:
R1
~m
\~'
( n N
R2
R3
wherein Rl is OH;
wherein R2 is H or OC(0)R9 when R1 is attached to the "a"
carbon or the "d" carbon, or
R~ is OC (0) Rq when R1 is attached to the "b" carbon or
the "c" carbon;
wherein R9 is C1 to C6 branched or unbranched alkyl'
aryl , aralkyl or NRSR6,
wherein RS and R6 are each independently H, C1 to
C8 alkyl, C6 to C12 aryl, C6 to C12 aralkyl or C6 to
C1z cycloalkyl, each optionally substituted;
wherein n is 0 or 1, and m is 1 or 2; and
wherein R3 is H or Me when n is 1 and m is 1, or R3 is H or
C1 to C6 alkyl when n is 0 or m is 2 '
or a pharmaceutically acceptable salt thereof.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-9-
In addition, the subject invention provides a compound having
the structure:
R1
s. ~ia )
m
~~d ( n N
R2
R.,
. wherein the compound is an optically pure enantiomer;
wherein R1 is OH;
wherein R2 is H;
wherein R3 is H or C1 to C6 alkyl;
wherein n is 0 or 1; and
wherein m is 1 or 2,
or a pharmaceutically acceptable salt thereof.
The subject invention further provides a compound having the
structure:
R7 ~
R7
NR3R8
wherein R, is H, Cl to C6 alkyl, aryl, aralkyl or C(O)R9,
wherein R4 is branched or unbranched C1 to C6 alkyl,
aryl, aralkyl or NRSR6,
wherein RS and R6are each independently H, C1 to
C8 alkyl, C6 to Clz aryl, C6 to C12 aralkyl or C6 to
C12 cycloalkyl, each optionally substituted;
wherein R3 is H or C1 to C6 alkyl;
wherein R8 is H or t-butoxycarbonyl (Boc).
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-10-
The subject invention also provides a method of treating a~
subject afflicted with a neurological disease comprising
administering to the subject a compound having the structure:
m
~d
R ( n N
2
R3
wherein R1 is~ OH or OC (O) R9, and wherein Ro is branched or
unbranched C1 to C6 alkyl, aryl, or aralkyl;
RZ is H or OC (O) R4, or both R1 and R~ are OC (O) R4,
wherein R4 is branched or unbranched C1 to C6 alkyl,
aryl, aralkyl or NR;R6,
wherein RS and R6 are each independently H, Cl to
Ce alkyl, C6 to Clz aryl, C6 to C12 aralkyl or C6 to
C12 cycloalkyl, each optionally substituted;
wherein R3 is H or C1 to C6 alkyl;
wherein n is 0 or l; and
wherein m is 1 or 2,
or a pharmaceutically acceptable salt thereof, or a prodrug
which becomes the compound in the subject, so as to thereby
treat the neurological disease in the subject.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-11-
Furthermore, the subject invention provides a method of treating
a subject afflicted with a neurological disease comprising
administering to the subject a compound having the structure:
y
~/a
~ ~ .
~a
( n N
R3
wherein R1 is OH or OC(0)R9;
wherein Ra is H or OC (O) R9,
wherein R9 is branched or unbranched C1 to C6 alkyl,
aryl, aralkyl or NRSR6,
wherein RS and R6 are each independently H, C1 to'
Ca alkyl, C6 to C12 aryl, C6 to C12 aralkyl or C6 to
C12 cycloalkyl, each optionally substituted;
wherein R3 is H or C1 to C6 alkyl;
wherein n is 0 or 1; and
wherein m is 1 or 2,
or a pharmaceutically acceptable salt thereof , or a prodrug
which becomes the compound in the subject, so as to thereby
treat the neurological disease in the subject.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-12-
The subject invention additionally provides a process for
preparing a compound having the structure:
)m
0
( n N
R9
wherein n is 0 or 1, and m is 1 or 2;
wherein R~ is H or C1 to C6 alkyl; and
wherein R~ is branched or unbranched C1 to C6 alkyl,
aryl, or aralkyl;
comprising the step of reacting
~ )m
HO' ~ ( n
boc
or
)m
Ho/
R3
with
R9 \Cl
in the presence of.an acid or 4-dimethylaminopyridine (DMAP) to
form the compound.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-13-
The subject invention also provides a process for preparing a
compound having the structure:
0 R4
O
0
HN
0 R"
wherein R~ is branched or unbranched C1 to C6 alkyl, aryl,
aralkyl or NR~R6,
wherein R~ and R6 are each independently H, Cl to Ce
alkyl, C6 to C1z aryl, C6 to C12 aralkyl or C6 to Cla
cycloalkyl, each optionally substituted;
which process comprises:
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-14-
(a) reacting a compound having the structure:
M e~
Me
O
l0
with A1C1, or BBr3 in the presence of toluene to produce a
compound having the structure:
HO
HO
O
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-15
(b) reacting the product formed in step (a) with benzyl
chloride and KZC03 in the presence of dimethyl formamide
(DMF) to produce a compound having the structure: ,
Phy
Phi
to
O
(c) reacting the product formed in step (b) with MeNH2~HCl,
NaCNBH3 in tetrahydrofuran (THF) /MeOH to produce a compound
having the structure:
Phi O
Phi 0
HN
(d) reacting the product formed in step (c) with.H2, Pd/C and
MeOH to produce a compound having the structure:
HN'
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-16-
(e) reacting the product formed in step (d) with BoczO,
dioxane/Hz0 and NaHC03 to produce a compound having the
structure:
HO
HO
boc
(f) reacting the product formed in step (e) with R4COC1, Et3N
in CH2Clz in the presence of Q-dimethylaminopyridine (DMAP)
to produce a compound having the structure:
O R4
O
0
0 R4
boc
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-17-
(g) reacting the product formed in step (f) with HC1/dioxane to
produce a compound having the structure:
0 R4
0
O
io
o R4
(h) reacting the product formed in step (g) with propargyl
bromide, K~CO, in CH3CN and then with HC1/ether and MeOH
to produce a compound having the structure:
O R4
O
O
HN
0 Re
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-18-
The subject invention also provides the use of a compound or a
prodrug of a compound which becomes the compound ,having the
structure:
R
wherein Rl is OH or OC (O) R4;
'wherein R2 is H, OH or OC (O) R9,
wherein R4 is branched or unbranched C1 to C6 alkyl,
aryl, aralkyl. or NRSR6,
wherein RS and R6 are each independently H, C1 to
CB alkyl , C~ to C1~ aryl , C6 to C12 aralkyl or C6 to
ClZ cycloalkyl, each optionally substituted;
wherein R3 is H or C1 to C6 alkyl;
wherein n is 0 or 1; and
wherein m is 1. or 2,
or a pharmaceutically acceptable salt thereof,
for the manufacture of a medicament for treating a subject
afflicted with a neurological disease, wherein the compound
is to be periodically administered to the subject in a
therapeutically effective dose.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-19-
Additionally, the subject invention provides the use of a
compound or a prodrug of a compound which becomes the compound
having the structure:
~~~d
N
R2
R3
wherein R1 is OH or OC (O) R9, and
wherein R9 is branched or unbranched C1 to C6 alkyl,
aryl, or aralkyl;
R2 is H or OC(0)Rg, or both R1 and R2 are OC(O)R4,
wherein RQ is C1 to C6 alkyl, aryl, aralkyl or NR5R6,
wherein RS and R6 are each independently H, C1 to
C8 alkyl, C6 to C12 aryl, C6 to ClZ aralkyl or C6 to
. C12 cycloalkyl, each optionally substituted;
wherein R3 is H or C1 to C6 alkyl;
wherein n is 0 or 1; and
wherein m is 1 or 2,
or a pharmaceutically acceptable salt thereof, for the
manufacture of a medicament for treating neurological
disease in a subject, wherein the compound is to be
periodically administered to the ,subject in a
therapeutically effective dose.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-20-
Descrit~tion of the Drawings .
Figure 1 presents routes for the manufacture of compounds with
the following structures:
5. ~ ~ )m
HO ~ n N
R-
. and
0
~4 ~ ~ ( n N
° I
R3
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-21-
Figure 2 displays~routes for the manufacture of a compound with
the following structure:
0 R4
O
O
N
O R4 R3
In Figure 2, the letters a) - i) are used to designat a the
following: a) A1C13, toluene; b) BnCl, KzC03, DMF; c) R3-NHz, HC1,
NaCNBH3, THF/MeOH; d) H2, Pd/C, MeOH; e) Boc20, dioxane/H20,
NaHC03; f) RS-COCl, Et3N, DMAP, CH2C12; g) HC1/dioxane; h)
propargyl bromide, KaC03, CH3CN; and i) HC1/ether, MeOH.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-22-
Figure 3 depicts routes for the manufacture of compounds with
the structures:
O Rq
O
O ~ N
O Rq
and
20
In Figure 3, the letters g) - 1) are used to designate the
following: g.) NaCNBHj, NH90Ac; h) propargyl bromide, ACN, KzC03;
i ) NaCNBH3 , paraf ormaldehyde ; j ) N-methylpropargylamine, NaCNBH3 ;
k ) BBr3 ; and 1 ) RqCOCl , TFA or DMAP .
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-23-
Detailed Description of the Invention
The subject invention provides a compound having the structure:
)
m
~d
C n N
R2
R~
wherein R1 is OC (0) Ro and Ra is H,
wherein Rq is branched or unbranched C1 to C6 alkyl,
aryl, or aralkyl, or
R1 i s OC ( 0 ) R4 and Rz i s OC ( O ) R4 ,
wherein R4 is branched or unbranched C1 to C6 alkyl,
aryl, aralkyl or NRSR6,
wherein R~ and R6 are each independently H, C1 to
' Ce alkyl , CE to C12 aryl , CE to C12 aralkyl or C6 to
Cl~ cycloalkyl, each optionally substituted;
wherein R3 is H or C1 to C6 alkyl;
wherein n is 0 or 1; and
wherein m is 1 or 2,
or a pharmaceutically acceptable salt thereof.
In one embodiment, the pharmaceutically acceptable salt is the
acetate salt, mesylate salt, esylate, tartarate salt, hydrogen
tartarate salt, benzoate salt, phenylbutyrate salt, phosphate
salt, citrate salt, ascorbate salt, mandelate salt, adipate
salt, octanoate salt, the myristate salt, the succinate salt, or
fumarate salt.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-24-
In another embodiment, the compound has the structure:
m
2
R3
In a further embodiment, the compound has the structure:
R1
R-,
In yet another embodiment, the compound has the structure:
Rg
R3
In one embodiment, n is 1.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-25-
In a further embodiment, the compound has the structure:
O
Ph \0
In an added embodiment, n is U.
In yet another embodiment, the compound has the structure:
0
R o 0 .~i/N
~3
In still another embodiment, the compound has the structure:
0
_5
Rg O ~ N
R3
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-26-
In one embodiment, R9 is Me and R3 is H.
In another embodiment, R9 is tBu and R3 is H. ,
In a further embodiment, R9 is nBu and R3 is H.
In yet another embodiment, R~ is CHaPh and R, is H.
In an additional embodiment, R9 is Ph and R3 is H.
In still another embodiment, wherein R9 is Me and R3 is Me..
In a further embodiment, R9 is nBu and R3 is Me
In one embodiment, R9 is Ph and R3 is Me.
In an added embodiment, R9 is tBu and R3 is Me.
In another embodiment, R9 is Ph(Me) and R3 is Me.
In still another embodiment, R9 is Ph(OMe)2 and R3 is Me.
In a further embodiment , R4 i s Ph ( OMe ) Z and R3 i s H .
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
_'7~_
In one embodiment, the compound has the structure:
0 R;
0
R~
In.an additional embodiment, R3 is Me and R9 is Me.
In a further embodiment, R3 is Me and R9 is Ph.
In another embodiment , R3 i s Me and R9 i s Ph ( OMe ) z .
In yet another embodiment, the compound has the structure:
Ro
25
In an added embodiment, R; is Me and R9 is Me.
In still another embodiment, R3 is H and R9 is Ph.
In one embodiment,. R3 is H and R9 is Ph (OMe) 2 .
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
O Ph
-28-
In mother embodiment, the compound has the structure:
O R4 ,
O
O ~ C n \N
O~ \R R
9
In a further embodiment, n is 0.
In yet another embodiment, R4 is Ph and R3 is Me.
In one embodiment, n is 1.
25
In still another embodiment, R3 is Me.
In an added embodiment, the compound has the structure:
O
O
0 Ph
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-29-
The subject invention also provides a compound having the
structure:
R1
R2
R3
wherein Rl is OH;
wherein R~ is H or OC (O) R~ when ' Rl is attached to the "a"
carbon or the "d" carbon, or
RZ is OC (0) R4 when R1 is attached to the "b" carbon or
the "c" carbon;
wherein R4 is C1 to C6 branched or unbranched alkyl,
aryl, aralkyl or NRSR6,
wherein RS and RE, are each independently H, C1 to
C~ alkyl , C6 to Clz aryl , C6 to C12 aralkyl or C6 to
C1~ cycloalkyl, each optionally substituted;
wherein.n is 0 or 1, and m is 1 or 2; and
wherein R; is H or Me when n is 1 and m is 1, or R3 is H or
C1 to C6 alkyl when n is 0 or m is 2,
or a pharmaceutically acceptable salt thereof.
In one embodiment, the pharmaceutically acceptable salt is the
acetate salt, mesylate salt, esylate, tartarate salt, hydrogen
tartarate salt, benzoate salt, phenylbutyrate salt, phosphate
salt, citrate salt, ascorbate salt, mandelate salt, adipate
salt, octanoate salt, the myristate salt, the succinate salt, or'
fumarate salt.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-30-
In another embodiment, the compound has the structure:
N
.
OH R3
In an additional embodiment, R,.is H.
In a'further embodiment, R3 is Me.
In yet another embodiment, the compound has the structure:
20
R3
In still another embodiment, R3 is H.
In one embodiment, R3 is Me.
In a further embodiment, n is 0.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-31-
Additionally, the subject invention provides a compound having
the structure:
R1
~~a
m
~d
R/
R,
wherein the compound is an optically pure enantiomer;
wherein R1 is OH;
wherein Rz is H;
wherein R3 is H or Cs to C6 alkyl;
wherein n is 0 or 1; and
wherein m is 1 or 2,
or a pharmaceutically acceptable salt thereof.
In one embodiment, the pharmaceutically acceptable salt is the
acetate salt, mesylate salt, esylate, tartarate salt, hydrogen
tartarate salt, benzoate salt, phenylbutyrate salt, phosphate
salt, citrate salt, ascorbate salt, mandelate salt, adipate
salt, octanoate salt, the myristate salt, the succinate salt, or
fumarate salt.
In a further embodiment, the compound has the structure:
HO
3
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-32-
In another embodiment, the compound has the structure:
HO
R,
In an added embodiment, R3 is H.
In yet another embodiment, R3 is Me.
In a further embodiment, the compound has the structure:
HO ~ .~~''i~~,
.
In one embodiment, R3 is H.
In another embodiment, R3 is Me.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-33-
The subject invention further provides a compound raving the
structure:
H71
R7
NR3Rs
wherein R~ is H, C1 to C6 alkyl, aryl, aralkyl or C (O) R4,
wherein R~ is branched or unbranched C1 to C6 alkyl,
aryl, aralkyl or NR~R6,
wherein RS and RE are each independently H, C1 to
Ce alkyl , C6 to C12 aryl , C6 to C12 aralkyl or C6 to
C12 cycloalkyl, each optionally substituted;
wherein R~ is H or C1 to C6 alkyl;'
wherein Re is H or t-butoxycarbonyl (Boc).
In one errJ~odiment, the compound has the structure:
HO
HO
/ N \
Boc
30
In another embodiment, the compound has the structure:
_/
HN
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-34-
In still another 'embodiment, the compound has the structure:
Phi O
Phi 0
In an added embodiment,, the compound has the structure:
l0 ~ R4
0',~ R
4 BOC
In yet another embodiment, Ft9 is Ph.
In one embodiment, the compound has the structure:
R4
30 i
d ~4
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-35-
In a further embodiment, R4 is Ph.
The subject invention additionally provides a pharmaceutical
composition comprising a compound having the structure:
R1
)
\G
R~ (
2
R3
wherein R1 is OC (O) Ro and R2 is H,
wherein R4 is branched or unbranched C1 to C6 alkyl,
aryl, or aralkyl, or
Rl i s OC ( O ) R4 and Rz i s OC ( 0 ) R4 ,
wherein R4 is branched or unbranched C1 to C6 alkyl,
aryl, aralkyl or NRSR6,
wherein RS and R6 are each independently H, C1 to
C8 alkyl , C6 to C12 aryl , C6 to C1z aralkyl or C6 to
Cla cycloalkyl, each optionally substituted;
wherein R3 is H or C1 ~to C6 alkyl;
wherein n is 0 or 1; and
wherein m is 1 or 2,
or a pharmaceutically acceptable salt thereof.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-36-
The subject invention further provides a pharmaceutical
composition comprising a compound having the structure:
R1
)_
m
~d
R ~~ ( n N
R3
wherein R1 is OH; .
wherein R2 is H or OC(O)R4 when Rl is attached to the "a"
carbon or the "d" carbon, or
RZ is OC (0) R9 when R1 is attached to the "b" carbon or
the "c" carbon;
wherein R4 is C1 to C6 branched or unbranched alkyl,
aryl, aralkyl or NRSR6,
wherein R5 and R6 are each independently H, Cl to
Ce alkyl, CE to C1z aryl, C6 to C12 aralkyl or C6 to
C12 cycloalkyl, each optionally substituted;
wherein n is 0 or 1, and m is 1 or 2; and
wherein R3 is H or Me when n is 1 and m is 1, or R3 is H or
C1 to C6 alkyl when n is 0 or m is 2,
or a pharmaceutically.acceptable salt thereof.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-37-
The subject invention also provides a pharmaceutical composition
comprising a compound having the structure:
R1
m
~d
N
R3
wherein the compound is an optically pure enantiomer;
wherein R1 i s OH;
wherein Rz is H;
wherein R3 is H or C1 to C6 alkyl;
wherein n is 0 or 1; and
wherein m is 1 or 2,
or a pharmaceutically acceptable salt thereof.
The subject invention also provides a method of treating a
subject afflicted with a neurological disease comprising
administering to the subject a compound having the structure:
R,
2$
N .
R3
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-38-
wherein Rl is OH or OC (0) R4;
wherein R~ is H, OH o~r OC(0)R4,
wherein R4 is branched or unbranched C1 to C6 , alkyl ,
aryl, aralkyl or NRSRE,
wherein RS and R6 are each independently H, C1 to
Ce alkyl, C6 to Cla aryl, C6 to C12 aralkyl or C6 to
C12 cycloalkyl, each optionally substituted;
wherein R3 is H or C1 to CE alkyl;
wherein n is 0 or 1; and
wherein m is 1 or 2,
or a pharmaceutically acceptable salt thereof , or a prodrug
which becomes the compound in the subject, so as to thereby
treat the neurological disease in the subject.
Additionally, the subject invention' provides a method of
treating a subject afflicted with a neurological disease,
. comprising administering to the subject a compound having the
structure:
R1
~ia
m
N
R , ( n
2
R3
when ein R1 is OH or OC (0) R9, and Rz is H or OC (0) R9, or both
Rl and R2 are OC ( 0 ) R9 , .
wherein R9 is branched or unbranched C1 to C6 alkyl,
aryl, or aralkyl; .
wherein R4 is branched or unbranched C1 to C6 alkyl,
aryl, aralkyl or NRSR6,
wherein R~ and Rbare each independently H, C1 to
Ce alkyl, C6 to C~2 aryl, C6 to Cla aralkyl or C6 to
C12 cycloalkyl, each optionally substituted;
wherein R3 is H or C1 to C6 alkyl;
wherein n is 0 or 1; and
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-39-
wherein m is 1 or 2,
or a pharmaceutically acceptable salt thereof, or. a prodrug
which becomes the compound in the subject, so as to thereby
treat the neurological disease in. the subject.
In one embodiment of the method, the compound has the structure:
Rl~/a )
m
N
R.,
J
wherein Rl is OC (0) Ra and RZ is H,
wherein R9 is branched or unbranched C1 to C6 alkyl,
aryl, or aralkyl, or
R1 i s OC ( 0 ) R9 and R2 i s OC ( O ) R4 ,
wherein R~ is branched or unbranched C1 to C6 alkyl,
aryl, aralkyl or NRSR6, .
wherein RS and R6 are each independently H, C1 to
Ca alkyl, C6 to Clz aryl, C6 to Cla aralkyl or C6 to
C12 cycloalkyl, each optionally substituted;
wherein R3 i s H or C1 to C6 alkyl ;
wherein n is 0 or 1; and
wherein m is 1 or 2.
In another embodiment of the method, the compound has the
structure:
~~i a )
~d
N
R2
R3
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-40-
wherein Rl is OH;
wherein R2 is H or OC(0)RQ when Rl is, attached to the "a"
carbon or the "d" carbon, or .
RZ is OC (0) R4 when R1 is attached to the "b" carbon or
the "c" carbon;
wherein R4 is C1 to C6 branched or unbranched alkyl,
aryl , aralkyl or NRSR6,
wherein RS and Rsare each independently H, C1 to
Ce alkyl, C6 to C12 aryl, CE to C12 aralkyl or C6 to
Clz cycloalkyl, each optionally substituted;
wherein R3 is H or C1 to C6 alkyl;
wherein n is 0 or 1; and
"wherein m is 1 or 2.
In a further embodiment of the method, the compound has the
structure:
R1
. ~ia )
m
RG ( n N
R3
wherein the compound is an optically pure enantiomer;
wherein R1 is OH;
wherein R2 is H; .
wherein R; is H or C1 to C6 alkyl;
wherein n is 0 or 1; and
wherein m is 1 or 2.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-41-
In one embodiment, the subject is human.
In a further embodiment, the aaministration comprises oral,
parenteral, intravenous, transdermal, or rectal administration.
In one embodiment, the effective amount is from about 0.01 mg
per day to about 100.0 mg per day.
In yet another embodiment, the effective amount is from about
0.01 mg per day to about 50.0 mg per day.
In still another embodiment, the effective amount is from about
0.1 mg per day to about 100.0 mg per day.
In an added embodiment, the effective amount is from about 0.1
mg per day to about 10.0 mg per day.
In yet another embodiment, the effective amount is from about
0.01 mg to about 100.0 mg.
In one embodiment, the effective amount is from about 0.01 mg to
about 50.0 mg.
In a further embodiment, the effective amount is from about 0.1
mg to about 100.0 mg.
In another embodiment, the ef f active amount i s, from about 0 .1 mg
to about 10.0 mg.
In an additional embodiment, the neurological disease is
Parkinson's disease, Alzheimer's disease, depression,. epilepsy,
narcolepsy, amyotrophic lateral sclerosis (ALS), memory
disorders, panic, post-traumatic stress disorder (PTSD), sexual
dysfunction, attention deficit and hyperactivity syndrome
(ADHD), attention deficit disorder, or Tourette's syndrome. The
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-42-
disease may also be neuropathy, hyperactive syndrome,
neurotrauma, stroke, Parkinson's disease, Huntington's disease,
and other dementia such as senile dementia, dementia ,of the
vascular dementia or Lewy body dementia.
In still another embodiment, the ,neurological disease is
depression.
In still another embodiment, the compound has the structure:
HO
HO ~ or
H/
H
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-43-
The subject invention further provides a process for p.r,eparing
a compound having the structure:
) ,
0
( n N
Rs
Rz
wherein n is 0 or 1, and m is 1 or 2;
wherein R3 i s H or C1 to CE alkyl ; and
wherein RG is branched or unbranched C1 to C6 alkyl,
aryl, or aralkyl;
comprising the step of reacting
)m
HO' ~ ~ n _ H
boc
or
?5 . H
R3
with
0
R9 \C 1
in the presence of an acid or 4-dimethylaminopyridine (DMAP) to
form the compound.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-44-
The subject invention also provides a process for preparing a
compound having the structure:
~m
0
0 ~ n
Ro
wherein R5 is branched or unbranched C1 to C6 alkyl, aryl,
or aralkyl;
which process comprises:
(a) reacting a compound having the structure:
~ ) n'
H0~ n NH 2
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-45-
with a compound having the structure:
X
5~ wherein X is a leaving group,
to produce a compound having the structure:
)m
H
( n
(b) reacting the compound formed in step (a) with a compound
having the structure:
O
R9 \C 1
in the presence of trifluoroacetic acid (TFA) and an
aprotic solvent to produce a compound having the structure:
)m
O
( . n NH
R9 .
In one embodiment, the leaving group in step (a) is selected
from the group consisting of a halogen and benzene sulfonate and
the aprotic solvent in step (b) is CHC13.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-46-
The subject invention further provides a process for preparing
a compound having the structure:
S
r /
Ph O ( n
which comprises:
(a) reacting a compound having the structure:
t )m
IS H0~ n NH 2
with a compound having the structure:
z~ x
wherein X is a leaving group,
to produce a compound. having the structure:
25 / ) m
HO~ ( n
30 (b) N-protecting the compound formed in step (a) with tert-
butoxycarbonyl _(Boc) to produce a compound having the
structure:
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-47-
~m
//
HO t n
boc
(c) reac ing the compound formed in step (b) with a compound
having the structure:
O
Ph C1
in the presence of 4-dimethylaminopyridine (DMAP) to
produce a compound having the structure:
. o
Ph O
boc
(d) deprotecting the compound formed in step (c) with HC1 to
produce a compound having the structure:
\ )
O ~ ~ ( n NH
Ph
In one embodiment, the leaving group in step (a) is selected
from the group consisting of a halogen and benzene sulfonate and
the aprotic solvent in step (b) is CHC13.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-48-
The subject invention additionally provides a process for
preparing a compound having the structure:
0
( n IM
Rs I
wherein Ra is branched or unbranched C1 to C6 alkyl, aryl,
or aralkyl;
which process comprises:
(a) reacting a compound having the structure:
( )
w
H0~ n NH a
?0 with a compound having the structure:
x
?5 wherein X is a leaving group,
to produce a compound having the structure:
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-49-
)m
HO ~ ( n NH
(b) reacting the compound formed in step (a) with NaCNBH3 and
paraformaldehyde to produce a compound having the
structure:
l0 \ )m
~/ N
Ho
(c) reacting the compound formed in step (b) with a compound
having the structure:
0
R9 \C1
in the presence of trifluoroacetic acid (TFA) and an
aprotic solvent to form a compound having the structure:
O ~ ) m
.-
R OA ~ ( n N
9
In one embodiment, the leaving group in step (a) is selected
from the group consisting of a halogen and benzene sulfonate and
the aprotic solvent in step (c) is CHC13.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-50-
The subject invention provides another process for preparing a
compound having the structure: .
R
.9
wherein R9 is branched or unbranched C1 to C6 alkyl, aryl,
or aralkyl; .
which process comprises:
(a) reacting a compound having the structure:
w
HO~ n NH
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-51-
with ethyl formate to produce a compound having the
structure:
~m
s
H ~\ ( NH H
n
(b) reacting the compound formed in step (a) with lithium
aluminum hydride to produce a compound having the
structure:
. ~m
( n NH
1S
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-52-
(c) reacting the compound formed in step (b) with a compound
having the structure:
wherein X is a leaving group,
to form a compound having the structure:
H(
(d) reacting the compound formed in step (c) with a compound
having the structure:
O
0 R~ ~C1
in the presence of trifluoroacetic acid (TFA) and an
aprotic solvent to form a compound having the structure:
O
Rg C
In one embodiment, the aprotic solvent in step (c) is CHC13.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-53-
The subject invention provides yet another process for preparing
a compound having the structure:
0
R9 C
wherein R9 is branched or unbranched C; to Cs alkyl, aryl,
or aralkyl;
which process comprises:
(a) reacting a compound having the structure:
is / c ) m
H 0~ j, NH 2
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-54-
with NaCNBH3/paraformaldehyde to produce a compound having
the structure:
)m
H0~ ~ ( n NH
(b) reacting the compound formed in step (a) with a compound
having the structure:
x~~
wherein X is a leaving group,
IS to form a compound having the structure:
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-55-
)m
I
(c) reacting the compound formed in step (b) with a compound
having the structure:
0
Rg -C 1
in the presence of trifluoroacetic acid (TFA) and an
aprotic solvent to form a compound having the structure:
o ~ ) m.
Ra ~ ~ ( n N
-
In one embodiment, the aprotic solvent in step (d) is CHC13.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-56-
Additionally, the subject invention provides a process for
preparing a compound having the structure:
o ~ )m
Ph D~ ~ ( n
which comprises:
(a) reacting a compound having the structure:
c )m '
HO~ n NI-:2
with a compound having the structure:
X
wherein X is a leaving group,
to produce a compound having the structure:
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-57-
)m
.
H ~ ( n NH
(b) reacting the compound formed in step (a) with NaCNBH3 and
paraformaldehyde to produce G compound having the
structure:
)m
H ~ ~ , ( n N
(c) reacting the compound formed in step (b) with a compound
having the structure:
0
Ph \C1
in the presence of 4-dimethylaminopyridine (DMAP) and an
aprotic solvent to form a compound having the structure:
)m
0
( n N
Ph 0
In one embodiment, the leaving group in step (a) is selected
from the group consisting of a halogen and benzene sulfonate and
the aprotic solvent in step (c) is CHC13.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-5 8-
The subject .invention provides another process for preparing a
compound having the structure:
)m
O
( n . N
Ph
which comprises:
(a) reacting a compound having the structure:
( )
NH ~
H O
with ethyl formate to produce a compound having the
structure:
. ~ )m
H ~\ ( NH~CHO
n
(b) reacting the compound formed in step (a) with lithium
aluminum hydride to produce a compound having the
structure:
)m
NH
H ~ .( n.
.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-59-
(c) reacting the compound formed in step (b) with a compound
having the structure:
x~
wherein X is a leaving group,
to form a compound having the structure:
)m
HO~ ~ (
(d) reacting the compound formed in step (c) with a compound
having the structure:
0
?0 ph C1
in the presence of 4-dimethylaminopyridine (DMAP) and an
aprotic solvent to form a compound having the structure:
2s ~ ) m
o
n N
Ph ~ I
In one embodiment, the aprotic solvent in step (c) is CHC13.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-60-
The subject invention provides yet another process for preparing
a compound having the structure:
o ~ )m
Ph ( n
which comprises:
(a) reacting a compound having the structure:
()
H 0~ " NH
1$
with NaCNBH3/paraformaldehyde to produce a compound having
the structure:
) n,
NH
HO/ ( n
2$ (b) reacting the compound formed in step (a) with a compound
having the structure:
X
wherein X is a leaving group,
to form a compound having the structure:
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-61-
)m
HO/ ~ ( n N
(c) reacting the compound formed in step (b)~with a compound
having the structure:
O
Ph C 1
in the presence of 4-dimethylaminopyridine (DMAP) and an
aprotic solvent to form a compound having the structure:
?0
)m
O
Ph ~ ~ (
In one embodiment, the aprotic solvent in step (d) is CHC13.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-62-
The subject invention further provides a process for preparing
a compound having the structure:
~ ~4
0
O~~R4 ~HN
which comprises:
(a) reacting a compound having the structure:
M~
M
0
with A1C13 or BBr3 in the presence of toluene to produce a
compound having the structure: .
HO
HO
O
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-63
(b) reacting the product formed in step (a) with benzyl
chloride and KzC03 in the presence of dimethyl formamide
(DMF) to produce a compound having the structure: ,
Phi 0
Ph~O
O
(c) reacting the product formed in step (b) with MeNH2~HC1,
NaCNBH; in tetrahydrofuran (THF)/MeOH to produce a compound
having the structure:
Phi 0
Ph~O
HN'
(d) reacting the product formed in step (c) with Ha, Pd/C and
MeOH to produce a compound having the structure:
HO
HO
HN' .
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-64-
(e) reacting the product formed in step (d) with Boc20,
dioxane/H~0 and NaHCO; to produce a compound having the
structure:
HO
HO
~N~
bo \c
15
(f) reacting the product formed in step (e) with RQCOCl, Et3N
in CH2Clz in the presence of 4-dimethylaminopyridine (DMAP)
to produce a compound having the structure:
n n
25
0'~R4 ~N~
bo \c
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-65-
(g) reacting the product formed in step (f) with HClldioxane to
produce a compound having the structure:
O R4
0
0
NH
O ~R4
(h) reacting the product formed in step (g) with propargyl
bromide, K~C03 in CH3CN and then with HCllether and MeOH
to produce a compound having the structure:
,
R4
0
0
/ HN
0 Rd
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-66-
Also, the subject invention provides a process for preparing a
compound having the structure:
0 Ph
O
0
O Ph
which comprises:
(a) reacting a compound having the structure:
M
M
O
with A1C13 or BBr3 in the presence of toluene to produce a
compound having the structure:
0
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-67
(b) reacting the product formed in step (a) with benzyl
chloride and K~C03 in the presence of dimethyl formamide
(DMF) to produce a compound having the structure: ,
Ph~O
Ph~O
O
(c) reacting the product formed in step (b) with MeNH2~HC1,
NaCNBH3 in tetrahydrofuran (THF)/MeOH to produce a compound
having the structure:
Ph~O
Phi 0
HN
25
(d) reacting the product formed in step (c) with Hz, Pd/C and
MeOH to produce a compound having the structure:
HO
HO
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-68-
(e) reacting the product formed in step (d) with Boc20,
dioxane/Hz0 and NaHC03 to produce a compound having the
structure: .
HO
HO
~N~
bo \c
(f) reacting the product formed in step (e) with PhCOCl, Et3N
in CH~Clz in the presence of 4-dimethylaminopyridine (DMAP)
to produce a compound having the structure:
, O Ph
boc
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-69-
(g) reacting the product formed in step (f) with HClldioxane to
produce a compound having the structure:
O Ph
O
O
O Ph
20
(h) 'reacting the product formed in step (g) with propargyl
bromide, KzC03 in CH3CN and then with HC1/ether and,lMeOH
to produce a compound having the structure:
O Ph
0
0
/ HN
0 Ph
'
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-70-
The subject invention further provides the use of a compound or
a prodrug of a compound which becomes the compound having the
structure: ,
R1
~~a ~
m
__
~d
( n N
R~
wherein R1 is OH or OC (O) Rs;
wherein R2 is H, OH or OC (O) R9,
wherein R4 is branched or unbranched C1 to C6 alkyl,
aryl, aralkyl or NRSR6,
wherein RS and R6 are each independently H, C1 to
Ce alkyl, C6 to C12 aryl, C6 to C12 aralkyl or C6 to
C1~ cycloalkyl, each optionally substituted;
wherein R3 is H or C1 to C6 alkyl;
wherein n is 0 or 1; and
whexein m is 1 or 2, ,
or a pharmaceutically acceptable salt thereof,
for the manufacture of a medicament for treating a subject
afflicted with a neurological disease, wherein the compound
is to be periodically aaministered to the subject in a
therapeutically effective dose.
The subject invention also provides the use of a compound or a
prodrug of a compound which becomes the compound having the
structure:
~~ia
m
~,d
R2 ( n N
R3
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-71-
wherein Rl is OH or OC (O) R9, and
wherein Ro is branched or unbranched C1 to C6 alkyl,
aryl, or aralkyl;
R2 is H or OC (O) RQ, or both R1 and RZ are OC (0) R9,
wherein Rg is branched or unbranched Cl to CE alkyl,
aryl, aralkyl or NR~R6,
wherein RS and R6are each independently H, C1 to
Ce alkyl, C6 to Cla aryl, CE to C1~ aralkyl or C6 to
C12 cycloalkyl, each optionally substituted;
wherein R3 ~ s H or C1 to C6 alkyl ;
wherein n is 0 or 1; and
wherein m is 1 or 2,
or a pharmaceutically acceptable salt thereof, for the
manufacture of a medicament for treating neurological
disease in a Subject, wherein the compound is to be
periodically aaministered to the subject in a
therapeutically effective dose.
In one embodiment of the use, the compound has the structure:
R
~N
n
?5 . Rs
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-72-
wherein Rl is OC (0) R9 and RZ is H,
wherein R9 is branched or unbranched C1 to. CE alkyl,
aryl, or aralkyl, or
R1 is OC (0) RQ and Ra ~is OC (O) R9,
wherein R9 is branched or unbranched Cl to C6 alkyl,
$ aryl , arahkyl or NR5R6,
wherein RS and RE are each independently H, Cl to
CE alkyl, C6 to C1z aryl, C6 to C12 aralkyl or C6 to
Clz cycloalkyl, each optionally substituted;
wherein. R, is H or C1 .to C6 alkyl;
wherein n is 0 or 1; and
wherein m is 1 or 2.
In another embodiment of the use, the compound has the
structure:
1$
R1
~~a )
m
R~~d ' (
R3
wherein R1 is OH; .
wherein Rz is H or OC(0)R9 when R~ is attached to the "a"
carbon or the "d" carbon, or
RZ is OC (O) R4 when R1 is attached to the "b" carbon or
the "c" carbon;
wherein RQ is C1 to C6 branched or unbrariched alkyl,
aryl, aralkyl or NRSRs.
wherein RS and R6 are each independently H, C1 to
CE alkyl, C6 to C1~ aryl, C6 to Cla aralkyl or C6 to
C12 cycloalkyl, each optionally substituted;
wherein R3 is H or C1 to C6 alkyl;
wherein n is 0 or 1; and
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-73-
wherein m is 1 or 2.
In an additional embodiment of the use, the compound,has the
structure:
R1
\~a )
m
~d
R~ ~ n N
2
R3
'wherein the compound is an optically pure enantiomer;
wherein R1 is OH;
wherein Rz is H;
wherein R, is H or C1 to C6 alkyl;
wherein n is 0 or 1; and
wherein m is 1 or 2.
In a further embodiment of the use; the subject is human.
In yet another embodiment of the use, the medicament is
formulated for oral, parenteral, intravenous, transdermal, or
rectal administration.
25~ In an embodiment of the use, the therapeutically effective
amount is from about 0.01 mg per day to about 50.0 mg per day.
In an added embodiment of the use, the therapeutically effective
amount is from about O.l mg per day to about 100.0 mg per day.
Iw still another embodiment of the use, the therapeutically
effective amount is from about 0.1 mg per day to about 10.0 mg
per day.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-74-
In an embodiment of the use, the neurological disease is
Parkinson's disease, Alzheimer's disease, depression, epilepsy,
narcolepsy, amyotrophic lateral sclerosis (ALS), , memory
disorders, panic, post-traurr~atic stress disorder (PTSD), sexual
dysfunction, attention deficit and hyperactivity syndrome
(P.DHD), attention deficit disorder, or Tourette's syndrome.
In a further embodiment of the use, the neurological disease is
depression. In one embodiment, the compound has the structure:
\ \
HO ~ ~ HO
o r ~N
The subject invention thus discloses various derivatives and
isomers of hydroxylated propargylamino indan and tetralin which
have surprisingly varied potency and selectivity for MAO
inhibition. The subject invention also provides modifications
of the hydroxy compounds which have surprisingly varied MAO
inhibitory properties depending upon the substitution pattern,
however, the hydroxy compound is always a m~re potent inhibitor
than the modified version. Thus, the modified version may be
considered a prodrug of the more active hydroxy compound into
which it will be metabolized in vivo.
In one embodiment of the invention, the prodrug compound is a
carboxylic acid ester of the hydroxy compound. In another
embodiment, the parent is a carbamate derivative of the hydroxy
compound.
As discussed above, carbamate propargylamino indans and
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-75-
tetralins have been reported in PCT International Application
No. PCT/US97/24155 as both MAO inhibitors and AchE inhibitors.
However, it is a further embodiment of this invention that such
a prodrug compound will not be a potent inhibitor of AchE (ICS,
>500 micromolar), and the ICSO for MAO-A inhibition of the
corresponding hydroxy metabolite be at least 100 times more
potent than the prodrug.
In one embodiment, the compounds are dihydroxy derivatives of
propargylamino indan .or tetralin. These derivatives are
expected to be antioxidants, as well as MAO inhibitors. In
another embodiment, the subject invention provides ester
prodrugs.
Thus, the subject invention provides esters or carbamat~es of
propargylamino indanols, propargylamino indandiols,
propargylamino tetralinols or propargylamino tetralindiols, and
may be prepared by methods of esterification or carbamoylation
of hydr~oxy compounds. Ester derivatives (Figur.e 1) when Ra
equals hydrogen were prepared by reacting the propargylamino
indanols with acyl chlorides in the presence of a strong organic
acid such as trifluoroacetic acid or an acylation catalyst such
as 4-dimethylaminopyridine (DMAP), with or without an inert
orcranic solvent such as chloroform. Compounds when R3 equals
hydrogen were prepared either by direct acylation as described
above, or by first N-protecting the amine moiety, e.g. , by a
tert-butoxycarbonyl (Boc) group, followed by acylation as above,
and finally removing the protecting group. The preparation of
compounds of the subject invention which are carbamates is
described in PCT/US97/24155.
Propargylamino indanols may :be prepared by reacting amino
indanols with propargyl bromide in a polar organic solvent such
as N,N-dimethylacetamide or acetonitrile in the presence of a.
base such as potassium carbonate. N-Methyl, N-pr.oparaylamino
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-76-
indanols may be prepared by reductive alkylation of
propargylamino indanols by methods known to those skilled in the
art, e.g., with NaCNBH, and paraformaldehyde. Alternatively, N-
methyl,N-propargylamino indanols were prepared by first
methylating amino indanols either by NaCNBH3/paraformaldehyde or
by ethyl formate followed by LiAlH~ reduction, and then reacting
the N-methylamino indanols thus obtained with propargyl bromide
as described above.
The N-propargyl derivatives of, inter alia, 3-amino-indan-4-ol,
1-amino-indan-4-ol, 3-amino-indan-5-of and 7-amino-5,6,7,8-
tetrahydro-naphthalen-2-of were prepared.
Compounds of the subject invention with both R1 and Rz equal to
OCOR4 (see Figure 2, compound numbered 9) were prepared by
propargylation of 5,6-di-0-benzoyl-1-methylamino-1-indan (Figure
2, compound numbered 8), as described above. 5,6-Di-O-benzoyl-
1-methylamino-1-indan (Figure 2, compound numbered 8) was
prepared from 5;6-bis-benzyloxy-1-indanone 3 as follows:
1) reductive amination of the compound numbered 3 in Figure
2 as described above gave 5,6-bis-benzyloxy-1
indanyl)methylamine (Figure 2, compound numbered 4);
2) the compound numbered 4 in Figure 2 was debenzylated by
catalytic hydrogenation and protected by the Boc group to
give N-Boc-1-methylamino-indan-5,6-diol (Figure 2, compound
numbered 6); and
3) Compound 6 in Figure 2 was esterified as described above
and the protecting group removed as previously described to
give 5,6-di-0-benzoyl-1-methylamino-1-indan (Figure 2,
compound numbered 8).
The diester tetralin derivative numbered 12 (Figure 3) was
prepared by esterification of the dihydr.oxy tetralin numbered 11
(Figure 3).
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
_77_
Table 1. Chemical Data
cmpd ster Rz R, R, R3 n m mp . formula yield
#i pos
100* S H OH 6 H 0 1 175-7 C,3H"NO,S45
,
101 R H OH I 6 H 0 1 173-5 C,3H"N04S42
*
102 S H OCOMe 6 H 0 1 13S-40 C,aH,6C1N0z46
103 R H OCOMe 6 H 0 1 156-8 C,4H,6C1N0277
104 S H OCOtBu 6 H 0 1 126-8 C,~H::CINOZ67
I ~
105 R H OCOtBu 6 H 0 1 128-30 C,~H_,C1N0246
106 S H OCOnBu 6 H 0 1 149-50 C,~HZ:CINOZ37
107 R H OCOnBu 6 H 0 1 155-7 C,~H2,CINOi85
. I
108 S H OCOCH:Ph 6 H 0 1 144-5 C~oHzoClNOz22
I
109 R H OCOCH:Ph 6 H 0 1 145-7 C~oH~oCINO252
110 S H . OCOPh 6 H 0 1 202-4 C,9H,eC1N0z~ 18
111 R H OCOPh 6 H 0 1 210-I1 C,9H,HClN0261
I
112 rac H OH 6 Me 0 1 210-11 C,;H,~CINO70
I
113 S H OH 6 Me 0 1 82-4 C,3H,6C1N072
114 R H OH 6 Me 0 1 71-2 C,3H,6C1N078
115 S H OCOMe 6 Me 0 1 168-70 C,sH,gCINOz95
116 R H OCOMe 6 Me 0 1 168-70 C,SH,eCIN0293
117 rac H OH 4 Me 0 1 160-62 C,3H,6NC1089
118 rac H OH 7 Me 0 1 83-5 C,3H,6NCI053
119 rac H OCOA9e 4 Me 0 1 148-50 C,SH,pC1N02.72
120 rac H OCOPh 4 Me 0 1 176-8 C,a1-l2oCIN0259
_
121 rac H OCOPh(OMe)24 Me 0 1 183-5 Cz,HZ,C1N0439
_
122 rac H OCOPh 7 Me 0 1.185-7 C:oH2oC1N0245
123 rac H OH 7 Me 1 1 220-1 C,4H,8NC1066
124 rac H OCOPh 7 Me 1 1 104-6 CZ,HZ:C1N0271
125 S H OCOnBu 6 Me 0 1 78-80 C,$H2,C1N0273
126 R H OCOnBu 6 Me 0 1 96-8 C,aHZ4CIN0272
~
127 S H OCOPh 6 Me 0 I 73-5 CZOHzoC1N0252
128 R H OCOPh 6 Me 0 1 82-4 C_oHzoC1N0256
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
_78_
Table 1. Chemical Data cont.
129 S H . OCOtBu 6 Me 0 1 153-5 C,BHZ,ClN0273
130 R H OCOtBu 6 Me 0 I 155-7 C,eH2aCIN0=78
..
131 S H OCOPh(Me 6 Me 0 1 Cz,Hz:CINO,51
)
132 R H OCOPh(Me) 6 Me 0 1 82-4 C2,H22C1NOz46
133 S H OCOPh(OMe)6 Me .01 118-20C~:H"CINO=58
2
134 R H OCOPh(OMe)6 Me 0 1 73-5 ~C2:H2,C1N0,68
Z
135 rac H OH 7 H 0 1 166-8 C,.H,QC1N035
136 ra H OH 4 H I 1 196-8 ~ C,,H"C1N066
c 0
137 rac OCOPh OCOPh ~ Me 0 1 114-5 C:~HZ,C1N0459
(5-pos) 6
138 rac OCOPh OCOPh 7 Me 1 1 180-2 CZaH=6C1N0458
( 6-pos)
stet = stereochemistry
pos = position
mesylate salts
,»
wide range, hygroscopic
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-79-
Table 2. 1H-iv~lR Data (Rl = R3 = H) (300 142-iz, dimethyl sulfoxide
,,".~rc~W _.a ~
~ ._
L. ,
~.
,
Cmpd Ph indan Pg R4 ~:
C3-H C2-H C1-H CH2 CH
7.52(d)
$ 102 7.35(d)4.79(m)2.43(m)2.83(m)3.88(m)3.71 2.27 10.2
(m) (Me,s) (br
s)
103 7.10(dd) 2.28(m)3.12(m)
7.48(a)
104 7.36(d)4.79(m)2.45(m)2.85(m)3.90(m)3.72(m)1.30 10.15
(tBu,s)
105 7.07(dd) 2.27(m)3.12(m) (br s)
l~ 7.48(d)
108 7.36(d)4.80(m)2.45(m)2.85(m)3.91(m)3.72(m)7.38(m,IN)10.2
109 ~7.07(dd) 2.28(m)3.13(m) 7.33(m,4H)(br s)
3.99(CH2,s)
7.48(d) 2.57(t,2H)
106 7.35(d)4.79(m)2.45(m)2.85(m)3.90(m)3.71(m)1.61(m,2H)10.1
15 107 7.08(dd) 2.26(m)3.11(m) 1.38(m,2H)(br s)
0.91
(t,3H)
,
7.67(d)
110 7.42(d)4.83(m)2.46(m)2.86(m)3.93(m)3.72(m)8.13(d,2H)10.15
11I 7.28(dd)
2.30(m)3.16(m) 7.76(t,lH)(d)
7.61(t,2H)
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-80-
Table 3. 1H-NMR Data (R1 = H, R3 = Me) (300 Mhiz,DzO)
Cmpd Ph Indan Propargyl Ra N-Me
C3-H C2-H C1-H CH, CH
7.50(d)
116 7.35(d) 5.22(m)2.46(m)3.07(m)4.05 3.15(m)2.37(Me,s)2.83(s)
(m)
115 7:25(dd) 2.60(m)3.17(m)
7.50(d) 2.69(t,2H)
126 7.33(d) 5.23(m)2.49(m)3.07(m)4.05(m)3.17(m)1.73(m 2.82(s)
~ ~H)
125 7.22(dd) 2.62(m)3.17(m) 1.44(m,2H)
0.97(t,3H)
7.50(d) 8.11 (dd,2H)
128 7.40(d) 5.17(m)2.57(m)3.06(m)4.00(m)3.15(m)7.74(dt,lH)2.81(s)
127 7.29(dd) 2.47(m)3.17(m) 7.57(t,2H)
7.49(d) .
129 7.28(d) 5.20(m)2.60(m)3.05(m)4.03(m)3.17(m)1.37(s,9H)2.81(s)
130 7.21(dd) 2.45(m)3.16(m)
7.90(d,lH) For Ar
H's,
131 7.44(t.lH)5.02 2.50(m)3.08(m)3.93(m)3.14(m)see 2.72(s)
(m)
132 7.36(m.2H) 2.40(m)2.95(m) under.Ph.
7.21(m.2H) 2.43(s.Me)
,
7.OS
(dd.l '
H)
For Ar
H's,
133 7.5-7.1 5.05 2.50(m)3.08(m)3.92(m)3.16(m)see 2.73(s)
134 (m,4H) (br 2.41(m)2.95(m) under
d) Ph.
6.74(dd,2H) 3.84 (s,6H,
OMe)
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-81-
Table 3. 'H-NMR Data (R1 = H, R3 = Me) (300 I~3z,Dz0) cont.
7.62(t,lH)5.19(m)2.46(m)2.98(m)4.09(m)3.84(s)8.13(d,2H)
~
120 7.44(t,lH) 2.80(m) 7.77(t,2H)
*
7.35(d,lH) 7.62(t,lH)
119 7.50(m,2H)5.29(dd)2.60(m)2.97(m)4.05(m)3.15(m)2.39(s,3H)2.81(s)
7.26(d,lH) 2.47(m)
121 7.46(t,2H)5.15 2.46(m)2.93(m)3.97(m)3.16(m)7.42(t,lH)2.73(s)
7.23(dd, (br 6.75(d,2H)
d)
1H) 3.83(s,6H,
OMe)
7.50(t,lH)5.18 2.95(m)~3.50(m)4.40- 3.80(m)8.20(dd,2H)2.68(s)
122 7.35(d,lH)(br 2.36(m) 3.90(m) 7.78(t,lH)2.56(s)
* s)
' 7:25(d,lH)4.96 7.61(t.2H)
'
(br
s)
* DM80-d6
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
82
V
. ,
ct1 '-r
(~1 'r
.a
_ N n
C. .~ cy1 ~ O
i~l ~r 1T'~
~ O n _ ' O ~ fV
0
_ ,-. N ~ c~l
~_ O
D _
~i 01
~ Z ~ ~: ~ O ~ Ov 'fl
ct ~ (, '~ '-~ ~ -p ,~"D
p
O
~'i
v
z ~
'v -."°v ~ vv t'1v
._
Q
c~ Cr N g op O ~ 00
r~ ~ rl 00 00'
r''1 O~ ~
U ~ ~ n
u_. .
V
Z
v.
~r G) Z Z
.
o p
O ~--~ , u1 'D
O O
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
83
!p .v ~1
f~ N SV
,'Z' r., j ' ,
. !'~,..~ O ,
i ~1 ~ ~ 1~!
~_ t~l a frl wr . t'rl .
d,
O ~ ~ n O ~ n
~ ~ '~ ~ ~i ~ - '~ ~r
v ~ (~ ~1 ~ ~1 n 'n ~ O ~ Y1 n
1 0 ~ o; ~ ~o E ...,
I cri ~ N '~ '~' ~. cri '. N ...
O
n
r- ~ .
n ~-~ .-~ ~ ~ o ~ O n Ov n
G~ j .~n ~ .~ ~ E
c_z O . ~ N ~_. N w. N c-~W N ~ N '..
_~ ~
N
T
C~ . ~ p~
0o U vyp v'i
z .~
...
.o ~_ ~ w
1 ~p ~ " v ~ 'r '.s 'r
F- . O ~ ~-~ c~1
cTl (~ ~ O oho oT1 ~ 00
f~ ~ 'p I~ ~O ~O
V ~U
.x
'. z
z
z
/ \ ~ ~ / \
/ \ o
U
x
y c~ ~ r~ ~ 00
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
84
p C~1 N
O O.
_° p N O O O. N ,
C~ o c~ 0 0
.>
= o ~ '--,
O O O
'n O O
_L
.'
C
~G' ~
L.
=' _a ~ \~ \
V
z
Z.
C/7 .
u_
O
r,..~ O N ~' cYl
rd
p p ~.
.
U
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
r-~
00
O
r~
. C~ ~ . 00 .
oN. W O O
(~ : p ~ O
0
.>
r_- cn
.-. °o C~ ~ u1
Q O O
i O O O ~- ,
O
v
n
._
C
.>
C
O
v
- = Z
v = \\
= ~ v \~
z
E.-.
z
z ~ / \ _ / \
/ \ / \ o
0
i z
00 w crl
b
--a ~ N ~
U
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
86
v N
p ~ ~ p
0
O vD
,_..,,
p ~ .-~ ~ N
'" O
. ....
~ , oo N o0
Q O O ' ' ~'1
O c~1
O O
.r
C
C
V
CC ~t.
W
c
_L
0 ~ \\ _ ~
z
z
U ~ / \
a
/ \
V7 z o ~ o = o i
0
o .o
o
b ~ ~. 00 t~ O
N N N M
w
U
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
87
v-~ ~ O
O
. . , .
O O O O .
o~ ~ O_
. U . m .-, ov . ~ N
0
.>
~= . ~ . r-, N .~i'
N
v
ea
p
c
.,- w F
a / \ ~. \
~ ~ ci~ ~ ~ ~ o / \
0 0 ~
v
t
a a
. .
N
N c'r~ cYl eY1
w
U
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
88
~ O
o r-, ~--~ N
U W os '-- cn .--.
0 0
0
. >.
~ , ..
c c~1 .-- c-
.,.i ~ .,--~
...
c -
L
..r
.>
C
L I
_47
U z
c~
E"' 4~" i \~ i \~ ~ v ~ ~ Z
O o O
O
p ~ ~ p / \
n'~.
. b . O ~ . ~ .--~
N N
U
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
89
~ ~
C~ C~
O O
0
Gp ' ~ N O
U:
0
'>
N
. , c~'1
c '~ : r--I V' 'O
v ~,
e~
,.
L
y_
.'
U
x
v~ 3~ 2 - Z
a~ L,
E~ v
\ /
C%7 / ' o z zW=
0
o \ ./ ~ z
v
'n. a
M ~~ O ,
M
U
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
O
O O ~ ,
O
~. .
N M
0
_, .
.....
c
......, . N ~O .--
v:. Q O ~ .
..
c
a
c-.
r
c
_L r
O ~L Z ~~ z
Z
a
(/7 ~ .~ o ~ o
0'-1 _ Z
Z _ ~ 2
U
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
91
C~
V
V
0 0
O O
. , 0 0
.-, .-,
n n
0
L '
. ..r
Q C~~ O
Cw M
.r
c
c
v
ea I
C
C
.7 . I =
Z z C7
C ~ Z= Z
v
~.. co
E= .r O o ~ o
o ~ o
O
.z z _ z
~ ~ .
b
~ ~ .
U
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
92
N
~ O
O .
O
O O
U ~ ,~
Q
-, O .
' Q O N
oc
w
c
a
.r
m
C
_L
C
r~
__
Z=
W ... Z
~ .~ \ v \ i
_ /
0
o
. o o~
z-
00 . d1
U
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
93
~r1 l~ ~ ..~ N I~
W Q ~D I~ ~ O~ c~
+J
. ,..,
.--» 'C
O ~1 t~ 1~ v1 04
Q t~ v~ oo O~ O~ N ~D
O O ~ ~ ~ O O
ap
ea O . 0,..X0 00
C
.>
c_
O
4 ~ ~
N
a
z z_z.
a
2- x
o x o x.
v~
.-~ oO M
p", p N O
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
94
. ..
. ,..,
,.D
. ..,
0
Q
N ~?
U ~ U
.~ cC c0 W
c
_ _
V
C O~ O O O O
o --
.>
c_
O
a
N
s ~ c~ . c~
w
E~
P
a ~ j
~ ~ 2
z z
~ w
U
sv
~ ~ / ~ x
4.. . ,~
o ~ o
0 0
0
U o
a,
/\ o /
j ~ . . N ~t
M M M
~> >
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
N ~~ i~ O cr1 ~ v~ ~ ~
~n r o0
O
..r
+~
. ...
\,
.... v1 ~1 O
Q ~ ~ ~ ~ FWD ~ N o0
~ v~ I~ ~1 ~1 ~1 ~1 ~1 v1
N
C
C
V
p cn ~ O $ ~ N O O ~y O O
o O O 'n .--~ .-, ~ ~ .-~ v1
.a
e_
O
a
N tr
~ cr . ~ .~-;
o cn ~o o c~
m
'z-x i
~\
0
O =( S-° ° ~ o
(> z_ ~ z -
z ._
.
,..d . . ~ Q O
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-96-
~'xperimental Details
EXAMPLE 1: GENERAL PROCEDURE FOR PROPYN-2-YLAMINO
(PROPARGYLP.MINO) INDANOLS (R3-H)H)
A mixture of amino indanol ( 3 5 mmol ) , propargyl bromide ( 3 5 mmol )
and potassium carbonate (35 mmol) in DMA (100 ml) was stirred at
room temperature (RT) for 24 hours. The reaction mixture was
filtered, diluted with water (200 ml) and extracted with toluene
(4 x 100 ml). The organic extracts were combined, dried and
evaporated to dryness under reduced pressure. The residue was
then subjected to flash column chromatography (hexane . EtOAc,
1:1).~ The free base was optionally converted to an acid
addition salt.
Alternatively, the' propargylation reaction was run in
acetonitrile at elevated temperature, e.g., 60°C ~or 4 hours.
The reaction mixture was then filtered, and the cake washed with
acetonitrile. The combined layers were evaporated. to dryness,
and the residue (brown oil) subjected to flash column
chromatography (hexane : EtOAc, 2:1). The product (white solid)
was thus obtained in 40 - 55 ~ yield.
Thus were prepared: (R)-3-prop-2-ynylamino-5-indanol mesylate,
(S)-3-prop-2-ynylamino-5-indanol mesylate, 1-prop-2-ynylamino-4-
indanol HC1, and 3-prop-2-ynylamino-4-indanol HC1.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-97-
EXAMPLE 2: GENERAL PROCEDURES FOR N-METHYL-PROP-2-YNYLAMINO
INDPNOLS (R;=Me) EXEMPLIFIED BY f-(METHYL-PROP-2-YNYL AMINO)-5-
INDANOL
Experiment 2A
A mixture of (S)-3-prop-2-ynylamino-5-indanol (5.0 g, 26.7
mmol) , paraformaldehyde (3 .6 g, 30 mmol) and NaCNBH3 (1.96 g,
31.2 mmol) in abs MeOH (90 ml) was refluxed under argon for 4
hours. The crude product obtained after evaporation .of the
solvent was purified by flash chromatography (hexane: EtOAc,
70:30) and was converted to its HC1 salt (etheral HC1: 4.2g(17.6
mmol, 66~)). 1H NMR (DMSO-d6):11.7(br d,NH), 9.62(br s, OH),
6.8-7.3(3H), 4.98(m,lH), 3.98(ABq,2H), 3.0(m;lH), 2.90(m,lH),
2.77(s,Me), 2.48(m1H), 2.40(m,lH)ppm.
1H NMR(D20):7.29(d,lH),6,.95-7.02(2H),5.09(m,lH),4.0(AB q, 2H),
3.0(m,lH),2.90(m,lH),2.77(s,Me),2.48(m,lH),2.40(m,lH)ppm.
Thus were prepared (R)3-(methyl-prop-2-ynylamino)-5-indanol and
1-(methyl-prop-2-ynylamino)-4-indanol.
Example 2B: 3-(methyl-prop-2-ynylamino)-4-indanol
Experiment 2B1
3-amino-4-indanol (3.70 g, 24.8 mmol) in ethylformate (200 ml)
was refluxed for 18 hr. The solvent was then removed under
reduced pressure, and the residue was purifed by flash
chromatography to give 4 .10 g ( 93 0 ) of N- ( 7-hydroxy-indan-1-yl )
formamide as a yellow solid.
Experiment 2B2
Lithium aluminium hydride (4.-5 g) was added portionwise to
stirred and cooled dry T.HF (100 ml) at 0°C. A solution of N-(7-
hydroxy-indan-1-yl)-f.ormamide (4.1 g) in dry THF (70 ml) was
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-98-
added while maintaining the temperature at 5-'! 0°C. The reaction
mixture was stirred at ambient temperature for 9 hr, cooled and
treated with water (100 ml). The pH was adjusted to,8-9, water
(200 ml) was added, and the mixture was extracted with ether (6
X 300 ml). The etheral extract was evaporated to dryness to
give 3.2 g (94~).
Experiment 2B3
3-Methylamino-4-indanol was reacted with propargyl bromide in
acetonitrile as described in Example 1.
Example 2C
7-lmethyl-prop-2-ynylamino)-2-tetralinol and 6-(methyl-prop-2-
ynylamino)-2,3-tetralindiol were prepared according to
Chumpradit et al. and Horn et al.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-99-
EXAMPLE 3: GENERAL PROCEDURE FOR ESTERIFICATION OF PROP-2-YNYL
AMINO II~1DANOLS P.ND TERALINOLS EXEMPLIFIED BY PENTANOIC ACID
(R)-3-PROP-2-1'NYLP.MINO-INDAN-5-YL ESTER HCL (Cmnd # 107)
To a solution of (R)3-prop-2-ynylamino-5-indanol (2.5 g, 13.4
mmol) in CHC13(30m1) and TFA (5 ml), was added valeryl chloride
(2.03 g, 2.0 ml, 16.7 mmo1) . The solution was heated at 60° for
8 hours and cooled to RT. Water (250 ml) was added, and the pH
adjusted to 7 by means of concentrated aqueous ammonia.
Extracted with methylene chloride (4x100 ml), dried and
evaporated to dryness under reduced pressure. The residue
(brown oil, 3.65 g) was purified by flash chromatography (Si02,
CHZCIz:MeOH 99:1) . The free base thus obtained (3.25 g) was
dissolved in dry ether (80 ml), and 20~ etheral HC1 was added.
The resulting suspension was stirred for 2 hours at RT, the
solid product was collected by filtration and washed with ether
(20 ml) and dried at 60° to give 3.45 g (11.2 mmol, 85~) of the
ester HC1.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-100-
Ex.Pr?PLE 4: ALTERNATIVE PROCEDURE EXEMPLIFIED BY BENZOIC ACID
(R)-3-PROP-2-YNYLAMINO-INDAN-5-YL ESTER (Cm~d # 111)
(R) 3-prop-2-ynylamino-5-indanol (3.0 g, 16 mmol) was dissolved
in dry THF (75 ml), and triethylamine~ (3.15 ml, 22.6 mmol)
followed by BoczO(4.5 g, 20.6 mmol) was added. The solution was
stirred at RT for 24 hours and evaporated to dryness. The
residue was taken up in water (200 ml) and extracted with
CHzCl2(4x100 ml). The organic layers were combined, dried and
evaporated to dryness. The crude product was purified by flash
column chromatography (hexane: EtOAc 3:1) to give 3.75 g (81.50
of a white solid.
1H h'I~lR ( DMSO-d6 ) ( a 1 : 1 mixture of 2 rotamers ) : 9 . 17 ( s , OH) , 7
. 0
(d,lH), 6.62 (dd, 1H), 6.5(br s, 1H), 5.51 & 5.22 (brs, 1H),
4.05, 3.72, 3.60, 3.38(m,2H), 3.06(br s, 1H), 2.83 (m,lH),
2. 64 (m, 1H) , 2.30 (br s, 1H, 2 .10 (br s, 1H) , 1 .4 & 1.27 (2s, 9H)ppm.
(R) N-Boc 3-prop-2-ynylamino-5-indanol(2.65 g, 9.23 mmol) was
dissolved in dry methylene chloride (20 ml), and triethylamine
(2.65 ml, 18.5, mmol), DMAP (0.11 g, 0.9 mmol) and benzoyl
chloride (1.7 ml, 18.5 mmol) was added. The solution was
stirred at RT for 3 hours, water (100 ml) was added and
acidified to pH 4 (aq HC1). The organic layer was separated and
washed with 10~ HC1. The aqueous layer was washed with
methylene chloride (100 ml), and the combined organic phases
were dried and evaporated to dryness in vacuo. The crude
product ~(5.2 g brown oil) was purified ,by flash column
chromatography (hexane: EtOAc 3:1) to give 4.1 g (90~) of a
white solid.
(R) N-Boc-3-prop-2-ynylamino-5-benzoyloxy indan (2.55 g, 6.5
mmol) was dissolved in dioxan~~(25 ml), and HC1/dioxan (25 ml)
was added. The mixture was stirred at RT for 4 hours and the
solvent was evaporated to dryness in vacuo. Ether (50 ml) was
added, the suspension was then stirred at RT for 2 hours. The
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-101-
solid was~collected by filtration, washed with ether and dried
(1.5 g). The crude product was crystallized from iPrOH (90 ml)
to give 1.3 g (3.96 mmol, 61~), mp 210-2°C. .
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-102-
EXAMPLE 5- PREPARATION OF BENZOIC ACID iS)-3-(METHYL-PROP-2-
YNYL-AMINO)-INDAN-5-YL ESTER HCL Cmpd # 127
(S)3-(methyl-prop-2-ynylamino)-5-indanol (1.5 g, 7.46 mmol) was
dissolved in dry methylene chloride (15 ml), and triethylamine
5~ (2.15 ml, 15.5 mmol), DMAP (0.08 g, 0.66 mmol) and benzoyl
chloride .(2.1 ml, 18.1 mmol) was added. The solution was
stirred at RT for 2 hours, water (100 ml) was added and
acidified to pH 4 (aq HC1). The organic layer was separated,
washed with 10~ HC1. The ac_ueous layer was washed with
methylene chloride (4x100 ml), and the combined organic phases
were dried evaporated to dryness in vacuo. The crude product
(3.78 g brown oil) was purified by flash column chromatography
(hexane: EtOAc 4:1) to give 1.6 g (5.3 mmol, 71~) of a yellow
oil. The free base was converted to the HC1 salt (etheral HCl,
IS 2 hours, RT), 1.39g (4.07 mmol, 77~, 55~ from the hydroxy
compound).
By the same procedure was prepared 7-O-benzoyl-2-(methyl-prop-2-
ynylamino)-tetralin Hcl, 1HNMR (D~0): 7.20, 6.98, 6.95 (3H,
ArOCO); 8.05, 7.71, 7.53 (5H, PhC00), 4.15 (m, 2H, CH2CCH), 3.80
(m, 1H, C~-H) , 3 .15 (t, 1H, CHaCCH) , 3 .14, 3 .01 (m, 2H, C8-H) ,
2.8-3.0 (m, 2H, C5-H), 2.31, 1.87 (m, 2H, C6-H), 3.0 (S, 3H, Me)
ppm.
The same procedure was also used to prepare 6,7-di-O-benzoyl-2-
(methyl-prop-2-ynylamino)-tetralin HC1, 1HNMR (DM80-d6): 7.91
(dd, 4H) , 7 .65 (t,2H) , 7.46 (t, 4H) , 7.28 (s, 2H) , 4.24 (br s,
2H), 3.87 (br s, 1H), 3.74 (m, 1H), 3.35-2.90 (m, 4H), 2.87 (s,
3H), 2.39 (m, 1H), 1.90 (m, 1H) ppm.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-103-
EXAMPLE 6' PREPARATION OF 5 6-DI-O-3ENZOYL-1-(METHYL-PROP-2-
YNYL-P.MINO)-INDAN HCL Cmpd ~ 137):
Experiment 6A: (5,6-Bis-benzylcxy-1-indan-1-yl)-methylamiae HCl
(Figure 2, Compound 4)
5.
A mixture of 5,6-dibenzyloxy-1-indanone (10.0 g, 29 mmol), 8M
ethanolic methylamine (~30 ml, 240 mmol), methylamine HC1 (7.15
g, 106 mmol), and NaCNBH3 (2.95 g, 47 mmol) in dry THF (750 ml)
and methanol (250 ml) was refluxed under nitrogen for 4 hours.
The reaction mixture was cooled to 5°C, acidified with
concentrated HC1 to pH 1.5, and evaporated to dryness. The
solid residue was treated with a mixture of methylene chloride
(600 ml) and water (400 ml). The aqueous layer was separated,
extracted with rnethylene chloride (4x100 ml), and the combined
organic layers were evaporated to dryness. The crude product
thus obtained was slurried in EtOAc (80 ml) for 30 min at RT,
filtered and purified by flash column chromatography (CHZC12 .
MeOH, 80:20), to give 6.3 g (54.8 ~), mp: 180-182°C.
Experiment 6B: 1- Methylamino -1-indan- 5,6-diol HC1 (Figure 2,.
Compound 5)
A solution of (5,6-bis-ben~zyloxy-1-indan-1-yl)-methylamiize HC1
(3.15 g, 7.96 mmol) in MeOH (250 ml) was hydrogenated (44 psi)
over 10~ Pd/C (1.05 g) at RT for 3 hours. The mixture was
filtered (Filteraid), and the filtrate evaporated to dryness.
The residue was treated with charcoal in boiling MeOH, filtered
and evaporated to dryness, to give 1.6 g of a light grey solid,
mp.. 153-5°C.
1H NMR (DM80-d6): 9.3-8.8 (3H, br m, OH, NH2), 7.02 (s, 1H, Ar),
~6 . 08 (s, 1H, Ar) , 4 .7 (dd, 1H, C3-H) , 2 . 92 (m, 1H, C1-H) . 2. 66
(m, 1H, Cl-H), 2.44 (s, 3H, Me), 2.33 (m, 1H, CZ-H), 2.11 (m, 1H,
C2-H' ) PPm.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-104-
Experiment 6C: N-Boc-1- methylamino -1-indan- 5,6-diol (Figure
2, Compound 6)
To a solution of 1-methylamino-1-indan-5,6-diol HC1 (0.5 g, 2.32
mmol) in water (30 ml) was added dioxane (30 ml), NaHC03 (0.6 g)
and BocaO (0.6 g). The reaction mixture was stirred at RT for
4 hours under nitrogen, evaporated to dryness, and the solid
residue taken up in a mixture of water (100 ml)~and methylene
chloride (100 ml). The aqueous layer was separated and
extracted with methylene chloride (5x50 ml). The latter was
filtered, washed with water, dried and evaporated to dryness to
give a viscous oil which was purified by flash column
chromatography (CHZC12 . MeOH, 95:5) to give 0.35 g (54 ~) of a
viscous oil which soon solidified.
Experiment 6D: N-Boc=(5,6-di-O-benzoyl-1-indan-1-yl)-methylamine
(Figure 2, Compound 7)
To a solution of N-Boc-1-methylamino-1-indan-5,6-diol (0.34 g,
1.22 mmol) in methylene chloride (15 ml) was added triethylamine
(0.49 g, 4.88 mmol), DMAP (0.03 g, 0.244 mmol) and benzoyl
chloride (0.69 g, 4.88 mmol), and the solution was stirred at RT
for 4.5 hours. V~later (100 ml) was added, acidified to pH 4 with
dilute HC1. The organic layer was separated and washed with 10$
HC1. The aqueous layer was extracted with methylene chloride
(2x75 ml.) , and the latter was washed with 10~ HC1. , The combined
organic layers were dried, evaporated to dryness, and the
residue purified by flash column chromatography (hexane : EtOAc,
50:50) to give 0.50 g (40~) of a yellow oil.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-105-
Experiment 6E: (5,6-di-O-Benzoyl-1-indan-1-yl)-methylamine HC1
(Figure 2, Compound 8)
To a solution of N-Boc-(5,6-di-0-benzoyl-1-indan-1-yl)-
methylamine (0.29 g, 0.59 mmol) in dioxane (5 ml) was added 20~
HCl,in dioxane (5 ml), and the mixture stirred at RT for 4 hours
under nitrogen. The solvent was removed and ether (40 ml) was
added to the residue, and the suspension stirred at RT for 1
hour. The solvent was removed'to give 0.11 g (89 ~) of a white
solid, mp . 192-3°C.
1H NMR (CDC13) : 8.1-7.2 (12H, Ar) , 4.79 (br s, 1H, C3-H) , 3.40
(m, 1H, C1-H), 3.01 (m,lH,C1-H), 2.60 (s, 3H, Me), 2.50 (m, 1H,
C2-H) , 1 . 83 (m, 1H, C2-H' ) ppm.
Experiment 6F: 5,6-di-O-Benzoyl-1-(methyl-prop-2-ynyl-amino)-
indan HC1 (Figure 2, Compound 9 (Cmpd # 137))
To a solution of (5,6-di-O-benzoyl-1-indan-1-yl)-methylamine
HC1 (~0.2 g, 0.48 mmol) in acetonitrile (100 ml) was added KzC03
(130 mg, 0.96 mmol), followed after 15 min by a solution of
propargyh bromide (56 mg, 0.48 mmol) in acetonitrile (10 ml).
The reaction mixture was stirred under nitrogen at RT for 20
hours, filtered and evaporated to dryness. The crude product
was purified by flash column chromatography (hexane . EtOAc,
50:50I) to give 0.15 g (0.35 mmol, 75 0) of a viscous light tan
oil.
The free base. was dissolved in MeOH (30 ml), and saturated
etheral HC1 (4 ml) was added. The solution was stirred at RT
for 30 min and evaporated to dryness. The oily residue was
triturated three times in ether, to give 120 mg (0.26 mmol, 74~)
.of a light tan solid.
NMR (CDC13): 8.1 - 7.2 (m, 12H, Ar), 5.1 (br d, 1H, C1-H), 3.91
(br s, 2H, CHZCCH) , 3 .6 - 2.5 (m, 8H, indan CHZ's, Me, CHZCCH) .
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-106-
EXAMPLE 7: INHIBITION OF MAO ACTIVITY IN VITRO
EXPERIMENTAL PROTOCOL
The MAO enzyme source was a homogenate of rat brain in 0.3 M
sucrose 1:20 w/v. The homogenate was pre-incubated with serial
dilutions of the test compounds (Table 5) for 60 minutes at
37°C. 19C-labeled substrates (2-phenylethylamine, hereinafter
PEA; 5-hydroxytryptamine, hereinafter 5-HT) were then added, and
the incubation continued for a further 20 minutes (PEA?, or 30-
45 minutes (5-HT) . In the case of PEA, the enzyme concentration
was chosen so that not more than 10~ of the substrate was
metabolized during the course of the reaction. The reaction was
then stopped by addition of citric acid. Radioactivity
indicates the production of 5-HT and~PER metabolites formed as
a result of MAO activity. Activity of MAO in the sample was
expressed as a percentage of control activity in the absence of
test compounds after subtraction of appropriate blank values.
The activity determined using PEA as substrate is referred to as
MAO-B, and that determined using 5-HT as MAO-A.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-107-
EX.AS'lPLE 8: INHIBITION OF MAO ACTIVITY IN VIVO: CHRONIC TREATMENT
EXPERIMENTAL PROTOCOL
Rats were treated with the test compounds (Table 5) at several
dose levels by oral administration, one dose daily for 7-21
5' days, and decapitated 2 hours after the last dose. The
activities of MAO-A and MAO-B were determined in the brain,
liver and intestine as described in the previous example.
Inhibition'I of MAO activity was calculated by dividing MAO
activity in the treated rats by MAO activity in. the control rats
(saline treated, MAO activity in these rats was taken as 1000 .
A mixture of toluene: ethyl acetate (1:1) was added to the
reaction and mixed for 10 minutes, followed by 5 minutes of
centrifugation at 1760 g. The upper phase was taken for
radioactive determination by liquid scintillation spectrometry.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-108-
References
U.S. Patent No. 3,513,244, Gittos et al.~, issued May 19., 1970.
U.S. Patent No. 4,844,033, hbinet.t et al., issued July 4, 1989.
U. S. Patent No. ~5, 387, 612, Youdim et al. , issued FebruGry 7,
1995.
U.S. Patent No. 5,453,446, Youdim et al., issued September 26,
1995.
U:S. Patent No. 5,457,133, Youdim et al., issued October 10,
1995'.
U.S. Patent No.5,519,061,Youdim et al.,issued May 21, 1996.
U.S. Patent No.5,532,415;Youdim et al.,issued July 2, 1996.
U.S.' Patent No.5,576,353,Youdim et al. , issued November 19,
1996.
U.S. Patent No.5,649,913,Cohen, issued July 22, 1997.
U.S. Patent No.5,668,181,Youdim et al.,issued September 16,
1997.
U.S. Patent No.5,744,500,Youdim issued April 28, 1998.
et al.,
U.S. Patent No.5,786,390,Youdim et al.,issued July 28, 1998.
U.S. Patent No.5,844,003,Tatton,
issued
December
1, 1998.
U.S. Patent No.5.,877,221,Cohen,et issued March 2, 1999..
al.,
U.S. Patent No.5,877,218,Herzig et al.,issued March 2, 1999.
U.S. Patent No.5,880,159,Herzig et al.,issued March 9, 1999.
U.S. Patent No.5,891,923,~Youdim et al.,issued April 6, 1999.
U.S. Patent No.5,914,349,Cohen et al.,issued June 22, 1999.
U. Patent No.5, 994, , Cohen et al , issued November 30,
S. 408 .
1999.
U.S. Patent No.6,303,650 , Chorev ., issued October 16,
et al
2001.
U.S. Patent No. 6,316,504, Youdim et al., issued November 13,
2001.
.PCT International Application N~o . PCT/ IL9 6 / 00115 , Berger et al . ,
published April 10, 1997.
PCT International Application No.~PCTlUS95/00245, Cohen et al.,
published July 13, 1995.
CA 02477218 2004-08-25
WO 03/072055 PCT/US03/05871
-109-
PCT International Application No. PCT/US97/24155, Chorev et al.,
published June 25, 1998. .
European Patent No. 0 436 492, Youdim et al. , issued ,7une 8,
1994.
Bentue-Ferrer et al., CNS Drugs 6, 217, 1996.
3 Chrisp,et al., Drugs & Aging, 1, 228, 1991.
Chumpradit, S. et al., J. Med. Chem., 36, 4308, 1993.
Dostert, J... Neurol. Transm. ~Suppl 41, 269, 1994.
Fitton et al., Drugs, 43, 561, 1992.
Florvall et al., Eur. J. Med. Chem. 34, 137, 1999.
Kleywegt, Monoamine oxidase, 1999 (http://alpha2.bmc.uu.se/ .
gerard/mao.html).
Krageten et al., J. Biol. Chem., 273, 5821, 1998.
Hazelhoff et al., Naunyn-Schmeideberg's Arch. Pharmacol.,
1985, 330, 50.
Hazelhoff, B. et al, Eur. J. Pharmacol., 109, 229, 1985.
Horn, A.S. et al., J. Med. Chem., 25, 993, 1982.
Lidor et al., 1997, Organic Preparations and Procedures
International (OPPI) 29,701.
Loscher et al., J. Pharmacol and Expt. Therap. 288, 984, 1999.
Palfreyman et al., J. Neurochemistry, 45, 1850, 1985.
Szelenyi, I., Inhibitors of Monoamine Oxidase B: Pharmacology
and Clinical Use in Neurodegenerative Disorders, 1992,
Bierkhauser Berlag, Switzerland.
White et al., Drug Development kesearch, 25, 191, 1992.
Youdim et al., Handbook of Experimental Pharmacology,
Trendelenburg and Weiner, eds. Springer-Verlag, 1988, 90, ch.3.
The interaction of L-deprenyl and scopolamine on. spatial
~learning/memory in rats, J. Neural Trans. Parkinson's Dis.
Dementia Sect., 1993, 6(3): 189-197.
Potential applications for monoamine oxidase B inhibitors,
Dementia, 1990, 1(6): 323-348.
Treatment of Tourette's: An Overview, (http://www.haverford.
edu/psych/biopsych217b/tourettes/TSwebtreat.caf.html).~