Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
METHODS AND COMPOSITIONS FOR TREATING T CELL MEDIATED
INFLAMMATORY/AUTOIMMUNE DISEASES AND DISORDERS IN
SUBJECTS HAVING A GLUCOCORTICOID REGULATION
DEFICIENCY
BACKGROUND OF THE INVENTION
(1 ) Field of the Invention:
(0001 ] The present invention relates to methods and compositions for
limiting morbidity and mortality arising from T cell activation in subjects
having a glucocorticoid regulation deficiency, and more particularly to
methods and compositions for preventing and treating T cell mediated
inflammatory/autoimmune diseases and disorders in subjects having a
glucocorticoid regulation deficiency.
(2) Description of the Related Art:
[0002] T cells are lymphocytes that play a key role in the immune
system. During normal immune system function in a vertebrate, the
activation of T cells triggers the production of a number of inflammatory
active molecules, including various cytokines and eicosanoids, including
the prostaglandins. The presence of the cytokines in the hypothalamus
and pituitary is known to cause the production of adrenocorticotropic
hormone (ACTH, corticotropin), which acts on the adrenal gland to cause
the production of glucocorticoids. The increased level of glucocortic,oids,
in turn, down-regulates the expression of the inflammatory cytokines in the
T cells, thereby modulating the inflammatory immune response in the
subject. Dysfunction of the feedback regulatory effect by glucocorticoids
can cause morbidity and death. See, e.g., Webster, J. I. et al., Annu. Rev.
Immunol., 20:125 - 163 (2002).
[0003] The direct or indirect administration of glucocorticoids (GCs) is
a mainstay therapy for treatment of inflammatory conditions, autoimmune
diseases, and lymphomas. Webster, J. I. et al., Annu. Rev. Immunol.,
20:125 - 163 (2002). Consistent with these immunosuppressive effects of
steroids, in vivo and in vitro studies have shown that pharmacologic levels
of GCs, acting through the glucocorticoid receptor (GR), modulate cytokine
1
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
synthesis and affect T cell and macrophage function. Almawi, W. Y. et al.,
J. Leukoc. Biol, 60:563 - 572 (1996). Additionally, physiologic GCs, which
are released at high levels into the blood stream via cytokine activation of
the hypothalamic-pituitary-adrenal (HPA) axis, have also been shown to
be critical in maintaining homeostasis. Removal of systemic
glucocorticoids via adrenalectomy in animal models or adrenal
insufficiency in humans (as with Addison's disease) has demonstrated the
requirement for endogenous GC production for regulation of physiologic
immune responses. Bertini, R. et al., J. Exp. Med., 1671708 - 1712
(1988). However, critical cellular and molecular targets of endogenous GR
action in modulating physiological inflammatory responses remain unclear.
[0004] One important function of GR in maintaining normal
homeostasis is its participation in modulation of the immune response of
the HPA axis. In this negative-feedback loop, an adrenal gland-derived
glucocorticoid (corticosterone in rodents, cortisol in humans) acts via the
hypothalamus and pituitary to regulate its own production. Additionally,
the HPA axis can be regulated by cytokines, neuropeptides and the
sympathetic nervous system. Da Silva, J. A., Ann N Y. Acad. Sci.,
876:102 - 117 (1999).
[0005] Glucocorticoids have also been shown to regulate expression of
pro-inflammatory mediators in addition to cytokines. Of note,
cyclooxygenase-2 (Cox-2) was discovered as a GC-modulated enzyme
that was induced in monocytes after lipopolysaccharide (LPS)
administration, and subsequently has been shown to be induced in vitro in
T cells after activation. See, e.g., Iniguez, M. A. et al., J. Immunol,
163:111 - 119 (1999); Masferrer, J. L. et al., Proc. Natl. Acad. Sci. USA,
89:3917 - 3921 (1992).
[0006] In subjects wherein the glucocorticoid/glucocorticoid receptor
regulatory mechanism does not function normally, immune system
challenge results in the hyperproduction of cytokines and eicosanoids.
The overproduction of these compounds can have significant
cardiovascular effects, such as changes in blood vessel tone and blood
2
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
vessel permeability. Chronic or acute dysfunction of the normal regulation
can result in morbidity and even mortality of the subject. However, it is
unclear which component, or components, of the array of eicosanoids,
cytokines, associated enzymes, and other associated compounds, directly
or indirectly results in morbidity or mortality of the host.
(0007] Dysfunction of the normal glucocorticoid/glucocorticoid receptor
regulatory system may be due to glucocorticoid insufficiency, ,
glucocorticoid resistance, or to the occurrence of a T cell-activating
stimulus that simply overwhelms the subject's T cell mediated immune
response regulatory capacity. It is known that in subjects showing
glucocorticoid insufficiency, about 70% of primary or chronic
adrenocortical insufficiency (Addison's disease) is due to idiopathic
atrophy of the adrenal cortex. The rest is probably caused by autoimmune
processes. See, e.g., The Merck Manual, 17 Ed., M. H. Beers and R.
Berkow, Eds., pp. 101 - 105, Merck Research Laboratories, Whitehouse
Station, NJ (1999)). Subjects can show a glucocorticoid resistance for any
of several reasons. One explanation is an abnormal GRa/GR~i ratio. See,
e.g., Bantel, H. et al., Gastroenterology, 114(4):1178 (2000). Another is
resistance developed in response to either chronic inflammatory stimuli or
chronic GC treatment. Glucocorticoid resistance can also be iatrogenic,
as with the withdrawal of GC administration from a subject for whom GC
use has become chronic. Furthermore, there are cases where subjects
having a normally functioning T cell-mediated immune response are
challenged with a T cell activating stimulus, such as toxic shock, bacterial
or viral sepsis, or a graft vs, host response, that is sufficiently strong
that it
overwhelms the GC regulatory system.
(0008] Despite the success of therapy involving the administration of
glucocorticoids to modulate the immune response in cases involving T cell
activation, there continue to be instances, particularly in subjects having a
glucocorticoid regulation deficiency, where such therapy is insufficient. In
these cases, it would be useful to provide compositions and methods that
could be used to prevent and to treat the morbidity and mortality that
3
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
results from such conditions. It would be particularly useful if the
compositions and methods supplemented or enhanced therapies that are
known.
SUMMARY OF THE INVENTION
[0009] Briefly, therefore, the present invention is directed to a novel
method of preventing or treating a T cell mediated
inflammatory/autoimmune disease or disorder in a subject having a
glucocorticoid regulation deficiency, where the subject is in need of such
treatment, the method comprising administering to the subject an effective
amount of a cyclooxygenase-2 inhibitor or prodrug thereof. In some
embodiments, the cyclooxygenase-2 inhibitor can be a cyclooxygenase-2
selective inhibitor.
[00010] The present invention is also directed to a novel method of
preventing or treating morbidity and mortality associated with T cell
activation in a subject having a glucocorticoid regulation deficiency, the
method comprising administering to the subject an effective amount of a
cyclooxygenase-2 inhibitor.
[00011] The present invention is also directed to a novel method of
limiting morbidity and mortality in a subject having a glucocorticoid
regulation deficiency, the method comprising administering to the subject
an effective amount of a cyclooxygenase-2 inhibitor prior to, during, or
after the subject has undergone a T cell activating process.
[00012] The present invention is also directed to a novel method of
treating a subject for a T cell mediated inflammatory/autoimmune disease
or disorder, the method comprising administering an effective amount of a
cyclooxygenase-2 inhibitor to a subject having a glucocorticoid regulation
deficiency after the subject has undergone a T cell activation process.
[00013] The present invention is also directed to a novel method of
treating a subject for a T cell mediated disease or disorder, wherein
method comprises treating the subject with a cyclooxygenase-2 inhibitor in
combination with a glucocorticoid.
4
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
[00014] The present invention is also directed to a novel composition for
the prevention andlor treatment of T cell mediated
inflammatory/autoimmune diseases and disorders in a subject having a
glucocorticoid regulation deficiency, the composition comprising a
combination of a cyclooxygenase-2 inhibitor and a glucocorticoid.
[00015] The present invention is also directed to a novel pharmaceutical
composition for the prevention and/or treatment of T cell mediated
inflammatory/autoimmune diseases and disorders in a subject having a
glucocorticoid regulation deficiency, the pharmaceutical composition
comprising a pharmaceutically acceptable excipient and a combination of
a cyclooxygenase-2 inhibitor and a glucocorticoid.
[00016] The present invention is also directed to a novel kit for the
prevention and/or treatment of T cell mediated inflammatory/autoimmune
disease or disorder in a subject having a glucocorticoid regulation
deficiency, the kit comprising one dosage form comprising a
cyclooxygenase-2 inhibitor and a second dosage form comprising a
glucocorticoid, wherein the cyclooxygenase-2 inhibitor and a glucocorticoid
are present each in an amount sufficient that the kit provides an effective
amount of the combination.
[00017] Among the several advantages found to be achieved by the
'present invention, therefore, may be noted the provision of compositions
and methods that could be used to prevent and to treat the morbidity and
mortality that results from T cell mediated inflammatory/autoimmune
diseases and disorders, particularly in subjects having a glucocorticoid
regulation deficiency. It would be particularly useful if the compositions
and methods supplemented or enhanced therapies that are known.
BRIEF DESCRIPTION OF THE DRAWINGS
[00018] Figure 1 shows: (A) a schematic illustration of the targeted
deletion of GR exon two, wherein a targeting vector was designed in which
exon two was flanked by IoxP sites (triangles); (B) a Western blot test for
total protein that was extracted from whole thymus or CD4+ thymocytes
purified by flow cytometry and probed for expression of GR and where
5
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
blots were re-probed for expression of actin as a loading control; and (C) a
bar chart showing plasma corticosterone levels in T cell glucocorticoid
receptor knock out (TGRko) and control mice in the morning, evening, two
or eight hours after injection of 100 ~.g anti-CD3s antibody (CD3s is the
epsilon component of the T cell receptor complex); which, taken together,
illustrate that the deletion of T cell glucocorticoid receptor does not alter
HPA axis regulation;
[00019] Figure 2 shows: (A) Kaplan-Meyer plots of survival (O control, n
= 10; ~ TGRko, n = 4; O control + dexamethasone (DEX), n = 10;
TGRko + DEX, n = 7); (B) bar charts indicating plasma cytokine levels in
TGRko and control mice two and eight hours after injection of anti-CD3E
antibody (100 ~.g) ~ dexamethasone administration (200 p.g 1 hour before
and 8 hours after anti-CD3E antibody administration) analyzed by enzyme
linked immunosorbent assay (ELISA); and (C) a quantitated
phosphorimager display of a ribonuclease protection assay (RPA) analysis
of splenic RNA (2 ~.g) from TGRko and control mice at baseline or eight
hours after injection of anti-CD3e antibody ((+) denotes positive control
RNA provided by the manufacturer), where expression was normalized to
glyceraldehyde phosphate dehydrogenase (GAPDH), which, taken
together, illustrate that T cell GR is required for prevention of lethality
and
downregulation of multiple cytokines after activation; and
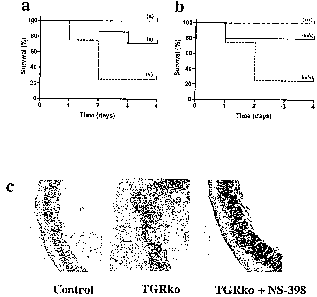
[00020] Figure 3 shows plots of survival vs. time for: (A) TGRko mice
which had been treated with a cyclooxygenase-2 selective inhibitor (SC-
236) (solid line (s), n = 7), NS-398 (alternately dashed line (n)), or vehicle
(dashed line (v), n = 3) one hour before anti-CD3s antibody administration,
and twice a day for two days thereafter; and (B) control mice which had
been treated with mifepristone + SC-236 (m/s, solid line, n = 10),
mifepristone (RU-486, a GR antagonist) + vehicle (m/v, dashed line, n =
8), or vehicle + vehicle (v/v, mixed line, n = 3), and shows that the
administration of a cyclooxygenase-2 selective inhibitor protects against
mortality induced by polyclonal T cell activation in GR-deficient mice; and
6
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
histological examination of the cecum in mice treated with anti-CD3~
antibody (C) demonstrates marked edema, inflammation, and mucosal
disruption in the TGRko mice and rescue with Cox-2 inhibition with NS-
398.
7
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENTS
[00021 ] In accordance with the present invention, it has been
discovered that morbidity and mortality associated with T cell-mediated
inflammatory/autoimmune diseases and disorders in subjects that have a
glucocorticoid regulation deficiency, and that are in need of such
treatment, can be prevented and/or treated by the administration of a
cyclooxygenase-2 inhibitor to the subject. In preferred embodiments, the
cyclooxygenase-2 inhibitor can be a cyclooxygenase-2 selective inhibitor.
[00022] The inventors have shown that glucocorticoid receptor function
within the T cell is essential in order to maintain the survival of a subject
in
the setting of polyclonal T cell activation. While T cell glucocorticoid
receptor deficiency results in dysregulation of several cytokines, redundant
mechanisms for down-regulation of some of these molecules, interleukin-2
(IL-2) and tumor necrosis factor-alpha (TNFa), for example, have evolved.
Interferon gamma (IFN~) regulation is relatively unique in its requirement
for glucocorticoid receptor inhibition, but the inventors have discovered
that IFNy immunoneutrali~ation does not attenuate mortality.
[00023] The inventors have now found that, surprisingly, a critical role
for glucocorticoid receptor action in the activated T cell is the modulation
of
Cox-2 expression and eicosanoid production, and that the administration
of a cyclooxygenase-2 (Cox-2) inhibitor is therapeutically useful for limiting
morbidity and mortality in patients with a glucocorticoid regulation
deficiency during or after a T cell activating process.
[00024] Furthermore, many patients with systemic lupus erythematosis,
graft versus host disease, and other chronic T cell-dependent autoimmune
and inflammatory diseases requiring chronic glucocorticoid treatment
experience life-threatening flares in disease severity despite continued
glucocorticoid administration. Often, these glucocorticoid-resistant states
are treated with extremely high dose glucocorticoid pulses that cause
diabetes mellitus, osteoporosis, and a host of other side effects. It has
been discovered, however, that institution of Cox-2 inhibitor therapy can
limit disease symptoms in these glucocorticoid regulation-deficient
8
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
circumstances, and prevent morbidity incurred with institution of high-dose
glucocorticoid therapy. Thus, one feature of the invention is the
administration of a Cox-2 inhibitor in the same instances as a treating
physician would presently administer a glucocorticoid -- either in place of,
or in .combination with, a glucocorticoid -- with the advantageous result
being that the Cox-2 inhibitor acts more quickly to limit the morbidity
associated with the T cell activating stimulus -- even reducing the danger
of death -- and also provides a complementary mechanism to provide
additive or synergistic therapeutic efficacy.
(00025] As used herein, the term "morbidity" should be understood to
mean the state of being not sound and healthful; induced by a diseased or
abnormal condition; or diseased. By way of example, morbidity should be
interpreted to include the major clinical symptoms surrounding the OKT-3
treatment syndrome: fever, headache, chills, diarrhoea, vomiting,
meningismus, respiratory distress, hypotension, intestinal hypomotility,
and (in mice) piloerection.
[00026] In the course of the present invention, the inventors generated
T cell-specific, GR knockout mice to aid in the determination of the role of
GR in lymphocyte development and regulation. It was shown that these
animals die following polyclonal T cell activation, whereas normal mice
uniformly survive. This mortality is associated with dysregulation of
cytokine and Cox-2 synthesis, and can be very effectively blocked with
Cox-2 inhibitors, but not by cytokine neutralization. These data
demonstrated that Cox-2 in T cells is a critical target for glucocorticoid
effects to maintain survival. Together with data in human and animal
systems, these findings strongly implicate the utility of Cox-2 inhibition in
settings of human glucocorticoid insufficiency (e.g., iatrogenic adrenal
suppression in GC-treated patients, Addison's disease, etc.) with infection
or inflammation, or in the context of glucocorticoid resistant autoimmune
and inflammatory diseases as an adjunct therapy to limit morbidity (e.g.,
Lupus exacerbations, rejection or graft versus host disease in transplant
patients).
9
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
[00027] The present invention includes a method of preventing or
treating T cell mediated inflammatory diseases and disorders, and is
particularly useful for treating such maladies in a subject having a
glucocorticoid regulation deficiency. The method comprises administering
to the subject an effective amount of a cyclooxygenase-2 inhibitor. The
Cox-2 inhibitor can be administered to the subject alone, or in combination
with a glucocorticoid.
[00028] The method can also be used for prophylactic purposes, such
as by administering an effective amount of a cyclooxygenase-2 inhibitor,
with or without glucocorticoids, to the subject prior to the subject's
undergoing a T cell activating process.
[00029] The cyclooxygenase-2 inhibitor of the present invention can be
any compound that inhibits the activity or production of the
cyclooxygenase-2 enzyme. Included within the meaning of the terms
"cyclooxygenase-2 inhibitor", as used herein, are Cox-2 inhibiting
compounds such as acetaminophin and nonsteroidal anti-inflammatory
drugs (NSAIDs), which can be non-selective, or selective (such as are
described below); nitric oxide (NO) NSAIDs (i.e., NSAIDs or NSAID
analogs containing a nitrite and/or nitrite esters) which upon release can
be GI-sparing); misoprostol/NSAID combinations (e.g., ArthrotecTM); Cox-2
transcription inhibitors; and Cox-2 mRNA translation inhibitors. Cox-2
inhibitors can be synthetic or natural, and natural Cox-2 inhibitors can be
plant-derived, animal-derived, or microbe derived.
[00030] Examples of Cox-2 inhibitors that are useful in the present
invention include, without limitation, indoles, such as etodolac,
indomethacin, sulindac and tolmetin; naphthylalkanones, such as
nabumetone; oxicams, such as piroxicam; para-aminophenol derivatives,
such as acetaminophen; propionic acids, such as fenoprofen, flurbiprofen,
ibuprofen, ketoprofen, naproxen, naproxen sodium, and oxaprozin;
salicylates, such as aspirin, choline magnesium trisalicylate and diflunisal;
fenamates, such as meclofenamic acid and mefenamic acid; and
pyrazoles, such as phenylbutazone.
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
[00031 ] In preferred embodiments, the Cox-2 inhibitor can be a
cyclooxygenase-2 selective inhibitor. The terms "cyclooxygenase-2
selective inhibitor", or "Cox-2 selective inhibitor", which can be used
interchangeably herein, embrace compounds which selectively inhibit
cyclooxygenase-2 over cyclooxygenase-1, and also include
pharmaceutically acceptable salts of those compounds.
[00032] In practice, the selectivity of a Cox-2 inhibitor varies depending
upon the condition under which the test is performed and on the inhibitors
being tested. However, for the purposes of this specification, the
selectivity of a Cox-2 inhibitor can be measured as a ratio of the in vitro or
in vivo IC5o value for inhibition of Cox-1, divided by the IC5o value for
inhibition of Cox-2 (Cox-1 ICS~/Cox-2 IC5o). A Cox-2 selective inhibitor is
any inhibitor for which the ratio of Cox-1 IC5o to Cox-2 IC5o is greater than
1. In preferred embodiments, this ratio is greater than 2, more preferably
greater than 5, yet more preferably greater than 10, still more preferably
greater than 50, and more preferably still greater than 100.
[00033] As used herein, the term "IC5o" refers to the concentration of a
compound that is required to produce 50% inhibition of cyclooxygenase
activity. Preferred cyclooxygenase-2 selective inhibitors of the present
invention have a cyclooxygenase-2 IC5o of less than about 1 p,M, more
preferred of less than about 0.5 p.M, and even more preferred of less than
about 0.2 ~,M.
[00034] Preferred cycloxoygenase-2 selective inhibitors have a
cyclooxygenase-1 IC5o of greater than about 1 p,M, and more preferably of
greater than 20 p.M. Such preferred selectivity may indicate an ability to
reduce the incidence of common NSAID-induced side effects.
[00035] Also included within the scope of the present invention are
compounds that act as prodrugs of cyclooxygenase-2-selective inhibitors.
As used herein in reference to Cox-2 selective inhibitors, the term
"prodrug" refers to a chemical compound that can be converted into an
active Cox-2 selective inhibitor by metabolic or simple chemical processes
11
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
within the body of the subject. One example of a prodrug for a Cox-2
selective inhibitor is parecoxib, which is a therapeutically effective prodrug
of the tricyclic cyclooxygenase-2 selective inhibitor valdecoxib. An
example of a preferred Cox-2 selective inhibitor prodrug is parecoxib
sodium. A class of prodrugs of Cox-2 inhibitors is described in U.S. Patent
No. 5,932,598.
[00036] The cyclooxygenase-2 selective inhibitor of the present
invention can be, for example, the Cox-2 selective inhibitor meloxicam,
Formula B-1 (CAS registry number 71125-38-7), or a pharmaceutically
acceptable salt or prodrug thereof.
OH O
/
N S CH3 B-1
H
N
S O \CHs
[00037] In another embodiment of the invention the cyclooxygenase-2
selective inhibitor can be the Cox-2 selective inhibitor RS 57067, 6-[[5-(4-
chlorobenzoyl)-1,4-dimethyl-1 H-pyrrol-2-yl]methyl]-3(2H)-pyridazinone,
Formula B-2 (CAS registry number 179382-91-3), or a pharmaceutically
acceptable salt or prodrug thereof.
~ H3 ll
HN~N\ N
~ ~ / B_2
O CH3 Cl
[00038] In a another embodiment of the invention the cyclooxygenase-2
selective inhibitor is of the chromene/chroman structural class that is a
substituted benzopyran or a substituted benzopyran analog, and even more
preferably selected from the group consisting of substituted
12
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
benzothiopyrans, dihydroquinolines, or dihydronaphthalenes having the
structure of any one of the compounds having a structure shown by general
Formulas I, II, III, IV, V, and VI, shown below, and possessing, by way of
example and; not limitation, the structures disclosed in Table 1, including
the diastereomers, enantiomers, racemates, tautomers, salts, esters,
amides and prodrugs thereof.
[00039] Benzopyrans that can serve as a cyclooxygenase-2 selective
inhibitor of the present invention include substituted benzopyran
derivatives that are described in U.S. Patent No. 6,271,253. One such
class of compounds is defined by the general formula shown below in
formulas I:
;1
A
A2
R4 I A
A3
~A'- x- R3
wherein Xi is selected from O, S, CR° R'~ and NRa ;
wherein Ra is selected from hydrido, C1 -C3 -alkyl, (optionally
substituted phenyl)-C1 -C3 -alkyl, acyl and carboxy-C1 -C6 -alkyl;
wherein each of Rb and R~ is independently selected from hydrido,
C1 -C3 -alkyl, phenyl-C1 -C3 -alkyl, C1 -C3 -perfluoroalkyl, chloro, Ci -C6 -
alkylthio, Ci -C6 -alkoxy, nitro, cyano and cyano-C1 -C3 -alkyl; or wherein
CRS R° forms a 3-6 membered cycloalkyl ring;
wherein R1 is selected from carboxyl, aminocarbonyl, C1 -C6 -
alkylsulfonylaminocarbonyl and Ci -C6 -alkoxycarbonyl;
wherein R2 is selected from hydrido, phenyl, thienyl, Ci -C6 -alkyl and C2 -
C6 -alkenyl;
wherein R3 is selected from C1 -C3 -perfluoroalkyl, chloro, C1 -C6 -
alkylthio, C1 -C6 -alkoxy, nitro, cyano and cyano-C1 -C3 -alkyl;
13
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
wherein R4 is one or more radicals independently selected from
hydrido, halo, C1 -C6 -alkyl, C2 -C6 -alkenyl, C2 -C6 -alkynyl, halo-C2 -C6 -
alkynyl, aryl-C1 -Cs -alkyl, aryl-C2 -C6 -alkynyl, aryl-C2 -C6 -alkenyl, C1 -
C6
-alkoxy, methylenedioxy, Ci -C6 -alkylthio, C1 -C6 -alkylsulfinyl, aryloxy,
arylthio, arylsulfinyl, heteroaryloxy, C1 -C6 -alkoxy-C1 -C6 -alkyl, aryl-C1 -
C6 -alkyloxy, heteroaryl-C1 -C6 -alkyloxy, aryl-C1 -C6 -alkoxy-Ci -C6 -alkyl,
C1 -C6 -haloalkyl, C1 -C6 -haloalkoxy, C1 -C6 -haloalkylthio, C1 -C6 -
haloalkylsulfinyl, C1 -C6 -haloalkylsulfonyl, C1 -C3 -(haloalkyl-1 -C3 -
hydroxyalkyl, C1 -C6 -hydroxyalkyl, hydroxyimino-C1 -C6 -alkyl, C1 -C6 -
alkylamino, arylamino, aryl-C1 -C6 -alkylamino, heteroarylamino,
heteroaryl-C1 -C6 -alkylamino, vitro, cyano, amino, aminosulfonyl, Ci -C6 -
alkylaminosulfonyl, arylaminosulfonyl, heteroarylaminosulfonyl, aryl-C1 -C6
-alkylaminosulfonyl, heteroaryl-C1 -C6 -alkylaminosulfonyl,
heterocyclylsulfonyl, C1 -C6 -alkylsulfonyl, aryl-C1 -C6 -alkylsulfonyl,
optionally substituted aryl, optionally substituted heteroaryl, aryl-C1 -C6 -
alkylcarbonyl, heteroaryl-C1 -C6 -alkylcarbonyl, heteroarylcarbonyl,
arylcarbonyl, aminocarbonyl, C1 -C1 -alkoxycarbonyl, formyl, Ci -C6 -
haloalkylcarbonyl and C1 -C6 -alkylcarbonyl; and
wherein the A ring atoms A1, A2, A3 and A4 are independently
selected from carbon and nitrogen with the proviso that at least two of Ai,
A2, A3 and A4 are carbon;
or wherein R~ together with ring A forms a radical selected from
naphthyl, quinolyl, isoquinolyl, quinolizinyl, quinoxalinyl and dibenzofuryl;
or an isomer or pharmaceutically acceptable salt thereof.
[00040] Another class of benzopyran derivatives that can serve as the
Cox-2 selective inhibitor of the present invention includes a compound
having the structure of formula II:
14
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
R6
R$ ~ 6 D ~~ ~I II
~pa~X2~R~
wherein X2 is selected from O, S, CRS Rb and NRa ;
wherein Ra is selected from hydrido, C1 -C3 -alkyl, (optionally
substituted phenyl)-Ci -C3 -alkyl, alkylsulfonyl, phenylsulfonyl,
benzylsulfonyl, acyl and carboxy-C1 -C~ -alkyl;
wherein each of R'~ and R° is independently selected from hydrido,
Ci -C3 -alkyl, phenyl-C1 -C~ -alkyl, C1 -C3 -perfluoroalkyl, chloro, C1 -C6 -
alkylthio, C1 -Cs -alkoxy, vitro, cyano and cyano-C1 -C3 -alkyl;
or wherein CRS R'~ form a cyclopropyl ring;
wherein R5 is selected from carboxyl, aminocarbonyl, C1 -C6 -
alkylsulfonylaminocarbonyl and C1 -C6 -alkoxycarbonyl;
wherein R6 is selected from hydrido, phenyl, thienyl, C2 -C6 -alkynyl
and C2 -C6 -alkenyl;
wherein R' is selected from C1 -C3 -perfluoroalkyl, chloro, C1 -C6 -
alkylthio, Ci -C6 -alkoxy, vitro, cyano and cyano-Ci -C3 -alkyl;
wherein Rs is one or more radicals independently selected from hydrido,
halo, C1 -C6 -alkyl, C2 -C6 -alkenyl, C2 -C6 -alkynyl, halo-C2 -C6 -alkynyl,
aryl-C1 -C3 -alkyl, aryl-C2 -C6 -alkynyl, aryl-C2 -C6 -alkenyl, C1 -C6 -
alkoxy,
methylenedioxy, C1 -C6 -alkylthio, C1 -C6 -alkylsulfinyl, -O(CF2)2 O-,
aryloxy, arylthio, arylsulfinyl, heteroaryloxy, C1 -C6 -alkoxy-C1 -C6 -alkyl,
aryl-C1 -C6 -alkyloxy, heteroaryl-C1 -C6 -alkyloxy, aryl-Ci -C6 -alkoxy-Ci -C6
-alkyl, C1 -C6 -haloalkyl, C1 -C6 -haloalkoxy, C1 -C6 -haloalkylthio, C1 -C6 -
haloalkylsulfinyl, C1 -C6 -haloalkylsulfonyl, Ci -C3 -(haloalkyl-C1 -C3 -
hydroxyalkyl), C1 -C6 -hydroxyalkyl, hydroxyimino-C1 -C6 -alkyl, Ci -C6 -
alkylamino, arylamino, aryl-C1 -C6 -alkylamino, heteroarylamino,
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
heteroaryl-C1 -C6 -alkylamino, nitro, cyano, amino, aminosulfonyl, C1 -C6 -
alkylaminosulfonyl, arylaminosulfonyl, heteroarylaminosulfonyl, aryl-C1 -Cs
-alkylaminosulfonyl, heteroaryl-C1 -C6 -alkylaminosulfonyl,
heterocyclylsulfonyl, C1 -C6 -alkylsulfonyl, aryl-C1 -C6 -alkylsulfonyl,
optionally substituted aryl, optionally substituted heteroaryl, aryl-C1 -C6 -
alkylcarbonyl, heteroaryl-C1 -C6 -alkylcarbonyl, heteroarylcarbonyl,
arylcarbonyl, aminocarbonyl, C1 -C6 -alkoxycarbonyl, formyl, C1 -C6 -
haloalkylcarbonyl and C1 -C6 -alkylcarbonyl; and
wherein the D ring atoms D1, D2, D3 and D4 are independently
selected from carbon and nitrogen with the proviso that at least two of D1,
D2, D3 and D4 are carbon; or
wherein R8 together with ring D forms a radical selected from
naphthyl, quinolyl, isoquinolyl, quinolizinyl, quinoxalinyl and dibenzofuryl;
or an isomer or pharmaceutically acceptable salt thereof.
(00041] Other benzopyran Cox-2 selective inhibitors useful in the
practice of the present invention are described in U.S. Patent Nos.
6,034,256 and 6,077,850. The general formula for these compounds is
shown in formula III:
[00042] Formula III is:
R9
R1o
R12 E III
X3 R11
wherein X3 is selected from the group consisting of O or S or NR~;
wherein Ra is alkyl;
wherein R9 is selected from the group consisting of H and aryl;
wherein R1° is selected from the group consisting of carboxyl,
aminocarbonyl, alkylsulfonylaminocarbonyl and alkoxycarbonyl;
16
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
wherein Rii is selected from the group consisting of haloalkyl, alkyl,
aralkyl, cycloalkyl and aryl optionally substituted with one or more radicals
selected from alkylthio, nitro and alkylsulfonyl; and
wherein R12 is selected from the group consisting of one or more
radicals selected from H, halo, alkyl, aralkyl, alkoxy, aryloxy,
heteroaryloxy, aralkyloxy, heteroaralkyloxy, haloalkyl, haloalkoxy,
alkylamino, arylamino, aralkylamino, heteroarylamino,
heteroarylalkylamino, nitro, amino, aminosulfonyl, alkylaminosulfonyl,
arylaminosulfonyl, heteroarylaminosulfonyl, aralkylaminosulfonyl,
heteroaralkylaminosulfonyl, heterocyclosulfonyl, alkylsulfonyl,
hydroxyarylcarbonyl, nitroaryl, optionally substituted aryl, optionally
substituted heteroaryl, aralkylcarbonyl, heteroarylcarbonyl, arylcarbonyl,
aminocarbonyl, and alkylcarbonyl; or
wherein R12 together with ring E forms a naphthyl radical; or an
isomer or pharmaceutically acceptable salt thereof; and
including the diastereomers, enantiomers, racemates, tautomers, salts,
esters, amides and prodrugs thereof.
[00043] A related class of compounds useful as cyclooxygenase-2
selective inhibitors in the present invention is described by Formulas IV
and V:
Ris
R15 G ~V
X4 R14
wherein X4 is selected from O or S or NRa ;
wherein Ra is alkyl;
wherein R13 is selected from carboxyl, aminocarbonyl,
alkylsulfonylaminocarbonyl and alkoxycarbonyl;
17
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
wherein R14 is selected from haloalkyl, alkyl, aralkyl, cycloalkyl and
aryl optionally substituted with one or more radicals selected from
alkylthio, nitro and alkylsulfonyl; and
wherein R15 is one or more radicals selected from hydrido, halo,
alkyl, aralkyl, alkoxy, aryloxy, heteroaryloxy, aralkyloxy, heteroaralkyloxy,
haloalkyl, haloalkoxy, alkylamino, arylamino, aralkylamino,
heteroarylamino, heteroarylalkylamino, vitro, amino, aminosulfonyl,
alkylaminosulfonyl, arylaminosulfonyl, heteroarylaminosulfonyl,
aralkylaminosulfonyl, heteroaralkylaminosulfonyl, heterocyclosulfonyl,
alkylsulfonyl, optionally substituted aryl, optionally substituted heteroaryl,
aralkylcarbonyl, heteroarylcarbonyl, arylcarbonyl, aminocarbonyl, and
alkylcarbonyl;
or wherein R15 together with ring G forms a naphthyl radical;
or an isomer or pharmaceutically acceptable salt thereof.
18
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
[00044] Formula V is:
R1s
R1a AI V
X5 R17
wherein:
X5 is selected from the group consisting of Q or S or NRb;
Rb is alkyl;
R16 is selected from the group consisting of carboxyl,
aminocarbonyl, alkylsulfonylaminocarbonyl and alkoxycarbonyl;
R17 is selected from the group consisting of haloalkyl, alkyl, aralkyl,
cycloalkyl and aryl, wherein haloalkyl, alkyl, aralkyl, cycloalkyl, and aryl
each is independently optionally substituted with one or more radicals
selected from the group consisting of alkylthio, nitro and alkylsulfonyl; and
R1$ is one or more radicals selected from the group consisting of
hydrido, halo, alkyl, aralkyl, alkoxy, aryloxy, heteroaryloxy, aralkyloxy,
heteroaralkyloxy, haloalkyl, haloalkoxy, alkylamino, arylamino,
aralkylamino, heteroarylamino, heteroarylalkylamino, nitro, amino,
aminosulfonyl, alkylaminosulfonyl, arylaminosulfonyl,
heteroarylaminosulfonyl, aralkylaminosulfonyl, heteroaralkylaminosulfonyl,
heterocyclosulfonyl, alkylsulfonyl, optionally substituted aryl, optionally
substituted heteroaryl, aralkylcarbonyl, heteroarylcarbonyl, arylcarbonyl,
aminocarbonyl, and alkylcarbonyl; or wherein R1$ together with ring A
forms a naphthyl radical;
or an isomer or pharmaceutically acceptable salt thereof.
(00045] The cyclooxygenase-2 selective inhibitor may also be a
compound of Formula V, wherein:
X5 is selected from the group consisting of oxygen and sulfur;
19
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
R16 is selected from the group consisting of carboxyl, lower alkyl,
lower aralkyl and lower alkoxycarbonyl;
Ri' is selected from the group consisting of lower haloalkyl, lower
cycloalkyl and phenyl; and
R'8 is one or more radicals selected from the group of consisting of
hydrido, halo, lower alkyl, lower alkoxy, lower haloalkyl, lower haloalkoxy,
lower alkylamino, nitro, amino, aminosulfonyl, lower alkylaminosulfonyl, 5-
membered heteroarylalkylaminosulfonyl, 6-membered
heteroarylalkylaminosulfonyl, lower aralkylaminosulfonyl, 5-membered
nitrogen-containing heterocyclosulfonyl, 6-membered-nitrogen containing
heterocyclosulfonyl, lower alkylsulfonyl, optionally substituted phenyl,
lower aralkylcarbonyl, and lower alkylcarbonyl; or
wherein Ri$ together with ring A forms a naphthyl radical;
or an isomer or pharmaceutically acceptable salt thereof.
[00046] The cyclooxygenase-2 selective inhibitor may also be a
compound of Formula V, wherein:
X5 is selected from the group consisting of oxygen and sulfur;
R16 is carboxyl;
Ri' is lower haloalkyl; and
R'$ is one or more radicals selected from the group consisting of
hydrido, halo, lower alkyl, lower haloalkyl, lower haloalkoxy, lower
alkylamino, amino, aminosulfonyl, lower alkylaminosulfonyl, 5-membered
heteroarylalkylaminosulfonyl, 6-membered heteroarylalkylaminosulfonyl,
lower aralkylaminosulfonyl, lower alkylsulfonyl, 6-membered nitrogen-
containing heterocyclosulfonyl, optionally substituted phenyl, lower
aralkylcarbonyl, and lower alkylcarbonyl; or wherein R'$ together with ring
A forms a naphthyl radical;
or an isomer or pharmaceutically acceptable salt thereof.
(00047] The cyclooxygenase-2 selective inhibitor may also be a
compound of Formula V, wherein:
X5 is selected from the group consisting of oxygen and sulfur;
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
R'6 is selected from the group consisting of carboxyl, lower alkyl,
lower aralkyl and lower alkoxycarbonyl;
R1' is selected from the group consisting of fluoromethyl,
chloromethyl, dichloromethyl, trichloromethyl, pentafluoroethyl,
heptafluoropropyl, difluoroethyl, difluoropropyl, dichloroethyl,
dichloropropyl, difluoromethyl, and trifluoromethyl; and
Ri$ is one or more radicals selected from the group consisting of
hydrido, chloro, fluoro, bromo, iodo, methyl, ethyl, isopropyl, tent butyl,
butyl, isobutyl, pentyl, hexyl, methoxy, ethoxy, isopropyloxy, tertbutyloxy,
trifluoromethyl, difluoromethyl, trifluoromethoxy, amino, N,N-
dimethylamino, N,N-diethylamino, N-phenylmethylaminosulfonyl, N-
phenylethylaminosulfonyl, N-(2-furylmethyl)aminosulfonyl, vitro, N,N-
dimethylaminosulfonyl, aminosulfonyl, N-methylaminosulfonyl, N-
ethylsulfonyl, 2,2-dimethylethylaminosulfonyl, N,N-dimethylaminosulfonyl,
N-(2-methylpropyl)aminosulfonyl, N-morpholinosulfonyl, methylsulfonyl,
benzylcarbonyl, 2,2-dimethylpropylcarbonyl, phenylacetyl and phenyl; or
wherein R2 together with ring A forms a naphthyl radical;
or an isomer or pharmaceutically acceptable salt thereof.
[00048] The cyclooxygenase-2 selective inhibitor may also be a
compound of Formula V, wherein:
X5 is selected from the group consisting of oxygen and sulfur;
R16 is selected from the group consisting of carboxyl, lower alkyl,
lower aralkyl and lower alkoxycarbonyl;
Ri' is selected from the group consisting trifluoromethyl and
pentafluoroethyl; and
R1$ is one or more radicals selected from the group consisting of
hydrido, chloro, fluoro, bromo, iodo, methyl, ethyl, isopropyl, tent butyl,
methoxy, trifluoromethyl, trifluoromethoxy, N-phenylmethylaminosulfonyl,
N-phenylethylaminosulfonyl, N-(2-furylmethyl)aminosulfonyl, N,N-
dimethylaminosulfonyl, N-methylaminosulfonyl, N-(2,2-
dimethylethyl)aminosulfonyl, dimethylaminosulfonyl, 2-
methylpropylaminosulfonyl, N-morpholinosulfonyl, methylsulfonyl,
21
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
benzylcarbonyl, and phenyl; or wherein R1$ together with ring A forms a
naphthyl radical;
or an isomer or prodrug thereof.
[00049] The cyclooxygenase-2 selective inhibitor of the present
invention can also be a compound having the structure of Formula VI:
R2o
R21
VI
R2a
wherein:
X6 is selected from the group consisting of O and S;
R19 is lower haloalkyl;
R2° is selected from the group consisting of hydrido, and halo;
R21 is selected from the group consisting of hydrido, halo, lower
alkyl, lower haloalkoxy, lower alkoxy, lower aralkylcarbonyl, lower
dialkylaminosulfonyl, lower alkylaminosulfonyl, lower aralkylaminosulfonyl,
lower heteroaralkylaminosulfonyl, 5-membered nitrogen-containing
heterocyclosulfonyl, and 6- membered nitrogen-containing
heterocyclosulfonyl;
R22 is selected from the group consisting of hydrido, lower alkyl,
halo, lower alkoxy, and aryl; and
R23 is selected from the group consisting of the group consisting of
hydrido, halo, lower alkyl, lower alkoxy, and aryl;
or an isomer or prodrug thereof.
[00050] The cyclooxygenase-2 selective inhibitor can also be a
compound of having the structure of Formula VI, wherein:
X6 is selected from the group consisting of O and S;
22
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
Ri9 is selected from the group consisting of trifluoromethyl and
pentafluoroethyl;
R2° is selected from the group consisting of hydrido, chloro, and
fluoro;
R2' is selected from the group consisting of hydrido, chloro, bromo,
fluoro, iodo, methyl, tert-butyl, trifluoromethoxy, methoxy, benzylcarbonyl,
dimethylaminosulfonyl, isopropylaminosulfonyl, methylaminosulfonyl,
benzylaminosulfonyl, phenylethylaminosulfonyl,
methylpropylaminosulfonyl, methylsulfonyl, and morpholinosulfonyl;
R22 is selected from the group consisting of hydrido, methyl, ethyl,
isopropyl, tert-butyl, chloro, methoxy, diethylamino, and phenyl; and
R23 is selected from the group consisting of hydrido, chloro, bromo,
fluoro, methyl, ethyl, tert-butyl, methoxy, and phenyl;
or an isomer or prodrug thereof.
23
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
Table 1. Examples of Chromene Cox-2 Selective Inhibitors
Compound Structural Formula
Number
B_3 0
o~N
OH
0 CF3
6-Nitro-2-trifluoromethyl-2H-1
-benzopyran-3-carboxylic acid
B_4 0
cl
~OH
/ 0 CF3
CH3
6-Chloro-8-methyl-2-trifluoromethyl
-2H-1-benzopyran-3-carboxylic acid
B_5 0
cl
~OH
O CF3
((S)-6-Chloro-7-(1,1-dimethylethyl)-2-(trifluo
romethyl-2H-1-benzopyran-3-carboxylic acid
24
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
Compound Structural Formula
Number
B-6
\ \ \ ~oH
O CF3
2-Trifluoromethyl-2H-naphtho[2,3-b]
pyran-3-carboxylic acid
B_7 O
OZN ~ \ Cl ~ \ \
OOH
/ /
O O CF3
6-Chloro-7-(4-nitrophenoxy)-2-(trifluoromethyl)-2H-1-
ben~opyran-3-carboxylic acid
B_$ 0
C1 \ \
OH
O CF3
Cl
((S)-6,8-Dichloro-2-(trifluoromethyl)-
2H-1-benzopyran-3-carboxylic acid
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
Compound Structural Formula
Number
B-9
i
0
cl
OH
0 "CF3
6-Chloro-2-(trifluoromethyl)-4-phenyl-2H
1-benzopyran-3-carboxylic acid
B-1 O O O
\OH
HO / / O CF3
6-(4-Hydroxybenzoyl)-2-(trifluoromethyl)
-2H-1-benzopyran-3-carboxylic acid
B-11 °
s
F3C~ ~ ~ ~ \OH
S CF3
2-(Trifluoromethyl)-6-[(trifluoromethyl)thio]
-2H-1-benzothiopyran-3-carboxylic acid
26
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
Compound Structural Formula
Number
B-12 °
cl \ \ ,
~OH
S CF3
C1
6,8-Dichloro-2-trifluoromethyl-2H-1
benzothiopyran-3-carboxylic acid
B-13 °
\ \ ~oH
s cF3
6-(l,1-Dimethylethyl)-2-(trifluoromethyl)
-2H-1-benzothiopyran-3-carboxylic acid
B-14 °
F \ \
-OH
F ~ H CF3
6,7-Difluoro-1,2-dihydro-2-(trifluoro
methyl)-3-quinolinecarboxylic acid
27
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
Compound Structural Formula
Number
B-15 °
cl
off
° i cF3
CH3
6-Chloro-1,2-dihydro-1-methyl-2-(trifluoro
methyl)-3-quinolinecarboxylic acid
B-16 °
cl
off
N H CF3
6-Chloro-2-(trifluoromethyl)-1,2-dihydro
[1,8]naphthyridine-3-carboxylic acid
B-17 °
C1
OH
° H CF3
((S)-6-Chloro-1,2-dihydro-2-(t'rifluoro
methyl)-3-quinolinecarboxylic acid
[00051 ] Examples of specific compounds that are useful for the
cyclooxygenase-2 selective inhibitor include (without limitation):
a1 ) 8-acetyl-3-(4-fluorophenyl)-2-(4-methylsulfonyl)phenyl-imidazo(1,2-
a)pyridine;
a2) 5,5-dimethyl-4-(4-methylsulfonyl)phenyl-3-phenyl-2-(5H)-furanone;
28
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
a3) 5-(4-fluorophenyl)-1-[4-(methylsulfonyl)phenyl]-3-
(trifluoromethyl)pyrazole;
a4) 4-(4-fluorophenyl)-5-[4-(methylsulfonyl)phenyl]-1-phenyl-3-
(trifluoromethyl)pyrazole;
a5) 4-(5-(4-chlorophenyl)-3-(4-methoxyphenyl)-1 H-pyrazol-1-
yl)benzenesulfonamide
a6) 4-(3,5-bis(4-methylphenyl)-iH-pyrazol-1-yl)benzenesulfonamide;
a7) 4-(5-(4-chlorophenyl)-3-phenyl-1 H-pyrazol-1-
yl)benzenesulfonamide;
a8) 4-(3,5-bis(4-methoxyphenyl)-1 H-pyrazol-1-yl)benzenesulfonamide;
a9) 4-(5-(4-chlorophenyl)-3-(4-methylphenyl)-1 H-pyrazol-1-
yl)benzenesulfonamide;
a10) 4-(5-(4-chlorophenyl)-3-(4-nitrophenyl)-1H-pyrazol-1-
yl)benzenesulfonamide;
b1) 4-(5-(4-chlorophenyl)-3-(5-chloro-2-thienyl)-1H-pyrazol-1-
yl)benzenesulfonamide;
b2) 4-(4-chloro-3,5-diphenyl-1 H-pyrazol-1-yl)benzenesulfonamide
b3) 4-[5-(4-chlorophenyl)-3-(trifluoromethyl)-1 H-pyrazol-1-
yl]benzenesulfonamide;
b4) 4-[5-phenyl-3-(trifluoromethyl)-1 H-pyrazol-1-yl]benzenesulfonamide;
b5) 4-[5-(4-fluorophenyl)-3-(trifluoromethyl)-1 H-pyrazol-1-
yl]benzenesulfonamide;
b6) 4-[5-(4-methoxyphenyl)-3-(trifluoromethyl)-1 H-pyrazol-1-
yl]benzenesulfonamide;
b7) 4-[5-(4-chlorophenyl)-3-(difluoromethyl)-1 H-pyrazol-1-
yl]benzenesulfonamide;
b8) 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1 H-pyrazol-1-
yl]benzenesulfonamide;
b9) 4-[4-chloro-5-(4-chlorophenyl)-3-(trifluoromethyl)-1 H-pyrazol-1-
yl]benzenesulfonamide;
b10) 4-[3-(difluoromethyl)-5-(4-methylphenyl)-1H-pyrazol-1-
yl]benzenesulfonamide;
29
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
c1 ) 4-[3-(difluoromethyl)-5-phenyl-1 H-pyrazol-1-yl]benzenesulfonamide;
c2) 4-[3-(difluoromethyl)-5-(4-methoxyphenyl)-1 H-pyrazol-1-
yl]benzenesulfonamide;
c3) 4-[3-cyano-5-(4-fluorophenyl)-1 H-pyrazol-1-yl]benzenesulfonamide;
c4) 4-[3-(difluoromethyl)-5-(3-fluoro-4-methoxyphenyl)-1 H-pyrazol-1-
yl]benzenesulfonamide;
c5) 4-[5-(3-fluoro-4-methoxyphenyl)-3-(trifluoromethyl)-1 H-pyrazol-1-
yl]benzenesulfonamide;
c6) 4-[4-chloro-5-phenyl-1 H-pyrazol-1-yl]benzenesulfonamide;
c7) 4-[5-(4-chlorophenyl)-3-(hydroxymethyl)-1 H-pyrazol-1-
yl]benzenesulfonamide;
c8) 4-[5-(4-(N,N-dimethylamino)phenyl)-3-(trifluoromethyl)-1 H-pyrazol-
1-yl]benzenesulfonamide;
c9) 5-(4-fluorophenyl)-6-[4-(methylsulfonyl)phenyl]spiro[2.4]hept-5-ene;
c10) 4-[6-(4-fluorophenyl)spiro[2.4]hept-5-en-5-yl]benzenesulfonamide;
d1 ) 6-(4-fluorophenyl)-7-[4-(methylsulfonyl)phenyl]spiro[3.4]oct-6-ene;
d2) 5-(3-chloro-4-methoxyphenyl)-6-[4-
(methylsulfonyl)phenyl]spiro[2.4]hept-5-ene;
d3) 4-[6-(3-chloro-4-methoxyphenyl)spiro[2.4]hept-5-en-5-
yl]benzenesulfonamide;
d4) 5-(3,5-dichloro-4-methoxyphenyl)-6-[4-
(methylsulfonyl)phenyl]spiro[2.4]hept-5-ene;
d5) 5-(3-chloro-4-fluorophenyl)-6-[4-
(methylsulfonyl)phenyl]spiro[2.4]hept-5-ene;
d6) 4-[6-(3,4-dichlorophenyl)spiro[2.4]hept-5-en-5-
yl]benzenesulfonamide;
d7) 2-(3-chloro-4-fluorophenyl)-4-(4-fluorophenyl)-5-(4-
methylsulfonylphenyl)thiazole;
d8) 2-(2-chlorophenyl)-4-(4-fluorophenyl)-5-(4-
methylsulfonylphenyl)thiazole;
d9) 5-(4-fluorophenyl)-4-(4-methylsulfonylphenyl)-2-methylthiazole;
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
d10) 4-(4-fluorophenyl)-5-(4-methylsulfonylphenyl)-2-
trifluoromethylthiazole;
e1 ) 4-(4-fluorophenyl)-5-(4-methylsulfonylphenyl)-2-(2-thienyl)thiazole;
e2) 4-(4-fluorophenyl)-5-(4-methylsulfonylphenyl)-2-
benzylaminothiazole;
e3) 4-(4-fluorophenyl)-5-(4-methylsulfonylphenyl)-2-(1-
propylamino)thiazole;
e4) 2-[(3,5-dichlorophenoxy)methyl)-4-(4-fluorophenyl)-5-[4-
(methylsulfonyl)phenyl]thiazole;
e5) 5-(4-fluorophenyl)-4-(4-methylsulfonylphenyl)-2-
trifluoromethylthiazole;
e6) 1-methylsulfonyl-4-[1,1-dimethyl-4-(4-fluorophenyl)cyclopenta-2,4-
dien-3-yl]benzene;
e7) 4-[4-(4-fluorophenyl)-1,1-dimethylcyclopenta-2,4-dien-3-
yl]benzenesulfonamide;
e8) 5-(4-fluorophenyl)-6-[4-(methylsulfonyl)phenyl]spiro[2.4]hepta-4,6-
diene;
e9) 4-[6-(4-fluorophenyl)spiro[2.4]hepta-4,6-dien-5-
yl]benzenesulfonamide;
e10) 6-(4-fluorophenyl)-2-methoxy-5-[4-(methylsulfonyl)phenyl]-pyridine-
3-carbonitrile;
f1 ) 2-bromo-6-(4-fluorophenyl)-5-[4-(methylsulfonyl)phenyl]-pyridine-3-
carbonitrile;
f2) 6-(4-fluorophenyl)-5-[4-(methylsulfonyl)phenyl]-2-phenyl-pyridine-3-
carbonitrile;
f3) 4-[2-(4-methylpyridin-2-yl)-4-(trifluoromethyl)-1 H-imidazol-1-
yl]benzenesulfonamide;
f4) 4-[2-(5-methylpyridin-3-yl)-4-(trifluoromethyl)-1 H-imidazol-1-
yl]benzenesulfonamide;
f5) 4-[2-(2-methylpyridin-3-yl)-4-(trifluoromethyl)-1 H-imidazol-1-
yl]benzenesulfonamide;
31
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
f6) 3-[1-[4-(methylsulfonyl)phenyl]-4-(trifluoromethyl)-1 H-imidazol-2-
yl]pyridine;
f7) 2-[1-[4-(methylsulfonyl)phenyl-4-(trifluoromethyl)-1H-imidazol-2-
yl]pyridine;
f8) 2-methyl-4-[1-[4-(methylsulfonyl)phenyl-4-(trifluoromethyl)-1 H-
imidazol-2-yl]pyridine;
f9) 2-methyl-6-[1-[4-(methylsulfonyl)phenyl-4-(trifluoromethyl)-1 H-
imidazol-2-yl]pyridine;
f10) 4-[2-(6-methylpyridin-3-yl)-4-(trifluoromethyl)-1 H-imidazol-1-
yl]benzenesulfonamide;
g1) 2-(3,4-difluorophenyl)-1-[4-(methylsulfonyl)phenyl]-4-
(trifluoromethyl)-1 H-imidazole;
g2) 4-[2-(4-methylphenyl)-4-(trifluoromethyl)-1 H-imidazol-1-
yl]benzenesulfonamide;
g3) 2-(4-chlorophenyl)-1-[4-(methylsulfonyl)phenyl]-4-methyl-iH-
imidazole;
g4) 2-(4-chlorophenyl)-1-[4-(methylsulfonyl)phenyl]-4-phenyl-1 H-
imidazole;
g5) 2-(4-chlorophenyl)-4-(4-fluorophenyl)-1-[4-(methylsulfonyl)phenyl]-
1 H-imidazole;
g6) 2-(3-fluoro-4-methoxyphenyl)-1-[4-(methylsulfonyl)phenyl-4-
(trifluoromethyl)-1 H-imidazole;
g7) 1-[4-(methylsulfonyl)phenyl]-2-phenyl-4-trifluoromethyl-1 H-
imidazole;
g8) 2-(4-methylphenyl)-1-[4-(methylsulfonyl)phenyl]-4-trifluoromethyl-
1 H-imidazole;
g9) 4-[2-(3-chloro-4-methylphenyl)-4-(trifluoromethyl)-1 H-imidazol-1-
yl]benzenesulfonamide;
g10) 2-(3-fluoro-5-methylphenyl)-1-[4-(methylsulfonyl)phenyl]-4-
(trifluoromethyl)-1 H-imidazole;
h1 ) 4-[2-(3-fluoro-5-methylphenyl)-4-(trifluoromethyl)-1 H-imidazol-1-
yl]benzenesulfonamide;
32
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
h2) 2-(3-methylphenyl)-1-[4-(methylsulfonyl)phenyl]-4-trifluoromethyl-
1 H-imidazole;
h3) 4-[2-(3-methylphenyl)-4-trifluoromethyl-1 H-imidazol-1-
yl]benzenesulfonamide;
h4) 1-[4-(methylsulfonyl)phenyl]-2-(3-chlorophenyl)-4-trifluoromethyl-
1 H-imidazole;
h5) 4-[2-(3-chlorophenyl)-4-trifluoromethyl-1 H-imidazol-1-
yl]benzenesulfonamide;
h6) 4-[2-phenyl-4-trifluoromethyl-1 H-imidazol-1-yl]benzenesulfonamide;
h7) 4-[2-(4-methoxy-3-chlorophenyl)-4-trifluoromethyl-1H-imidazol-1-
yl]benzenesulfonamide;
h8) 1-allyl-4-(4-fluorophenyl)-3-[4-(methylsulfonyl)phenyl]-5-
(trifluoromethyl)-1 H-pyrazole;
h10) 4-[1-ethyl-4-(4-fluorophenyl)-5-(trifluoromethyl)-1 H-pyrazol-3-
yl]benzenesulfonamide;
i1) N-phenyl-[4-(4-luorophenyl)-3-[4-(methylsulfonyl)phenyl]-5-
(trifluoromethyl)-1 H-pyrazol-1-yl]acetamide;
i2) ethyl [4-(4-fluorophenyl)-3-[4-(methylsulfonyl)phenyl]-5-
(trifluoromethyl)-1 H-pyrazol-1-yl]acetate;
i3) 4-(4-fluorophenyl)-3-[4-(methylsulfonyl)phenyl]-1-(2-phenylethyl)-
1 H-pyrazole; _
i4) 4-(4-fluorophenyl)-3-[4-(methylsulfonyl)phenyl]-1-(2-phenylethyl)-5-
(trifluoromethyl)pyrazole;
i5) 1-ethyl-4-(4-fluorophenyl)-3-[4-(methylsulfonyl)phenyl]-5-
(trifluoromethyl)-1 H-pyrazole;
i6) 5-(4-fluorophenyl)-4-(4-methylsulfonylphenyl)-2-trifluoromethyl-1 H-
imidazole;
i7) 4-[4-(methylsulfonyl)phenyl]-5-(2-thiophenyl)-2-(trifluoromethyl)-1 H-
imidazole;
i8) 5-(4-fluorophenyl)-2-methoxy-4-[4-(methylsulfonyl)phenyl]-6-
(trifluoromethyl)pyridine;
33
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
i9) 2-ethoxy-5-(4-fluorophenyl)-4-[4-(methylsulfonyl)phenyl]-6-
(trifluoromethyl)pyridine;
i10) 5-(4-fluorophenyl)-4-[4-(methylsulfonyl)phenyl]-2-(2-propynyloxy)-6-
(trifluoromethyl)pyridine;
j1 ) 2-bromo-5-(4-fluorophenyl)-4-[4-(methylsulfonyl)phenyl]-6-
(trifluoromethyl)pyridine;
j2) 4-[2-(3-chloro-4-methoxyphenyl)-4,5-
difluorophenyl]benzenesulfonamide;
j3) 1-(4-fluorophenyl)-2-[4-(methylsulfonyl)phenyl]benzene;
r
j4) 5-difluoromethyl-4-(4-methylsulfonylphenyl)-3-phenylisoxazole;
j5) 4-[3-ethyl-5-phenylisoxazol-4-yl]benzenesulfonamide;
j6) 4-[5-difluoromethyl-3-phenylisoxazol-4-yl]benzenesulfonamide;
j7) 4-[5-hydroxymethyl-3-phenylisoxazol-4-yl]benzenesulfonamide;
j8) 4-[5-methyl-3-phenyl-isoxazol-4-yl]benzenesulfonamide;
j9) 1-[2-(4-fluorophenyl)cyclopenten-1-yl]-4-(methylsulfonyl)benzene;
j t 0) 1-[2-(4-fluoro-2-methylphenyl)cyclopenten-1-yl]-4-
(methylsulfonyl)benzene;
k1 ) 1-[2-(4-chlorophenyl)cyclopenten-1-yl]-4-(methylsulfonyl)benzene;
k2) 1-(2-(2,4-dichlorophenyl)cyclopenten-1-yl]-4-
(methylsulfonyl)benzene;
k3) 1-[2-(4-trifluoromethylphenyl)cyclopenten-1-yl]-4-
(methylsulfonyl)benzene;
k4) 1-[2-(4-methylthiophenyl)cyclopenten-1-yl]-4-
(methylsulfonyl)benzene;
k5) 1-[2-(4-fluorophenyl)-4,4-dimethylcyclopenten-1-yl]-4-
(methylsulfonyl)benzene;
k6) 4-[2-(4-fluorophenyl)-4,4-dimethylcyclopenten-1-
yl]benzenesulfonamide;
k7) 1-[2-(4-chlorophenyl)-4,4-dimethylcyclopenten-1-yl]-4-
(methylsulfonyl)benzene;
k8) 4-[2-(4-chlorophenyl)-4,4-dimethylcyclopenten-1-
yl]benzenesulfonamide;
34
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
k9) 4-[2-(4-fluorophenyl)cyclopenten-1-yl]benzenesulfonamide;
k10) 4-[2-(4-chlorophenyl)cyclopenten-1-yl]benzenesulfonamide;
11 ) 1-[2-(4-methoxyphenyl)cyclopenten-1-yl]-4-
(methylsulfonyl)benzene;
12) 1-[2-(2,3-difluorophenyl)cyclopenten-1-yl]-4-
(methylsulfonyl)benzene;
13) 4-[2-(3-fluoro-4-methoxyphenyl)cyclopenten-1-
yl]benzenesulfonamide;
14) 1-[2-(3-chloro-4-methoxyphenyl)cyclopenten-1-yl]-4-
(methylsulfonyl)benzene;
15) 4-[2-(3-chloro-4-fluorophenyl)cyclopenten-1-yl]benzenesulfonamide;
16) 4-[2-(2-methylpyridin-5-yl)cyclopenten-1-yl]benzenesulfonamide;
17) ethyl 2-[4-(4-fluorophenyl)-5-[4-(methylsulfonyl) phenyl]oxazol-2-yl]-
2-benzyl-acetate;
18) 2-[4-(4-fluorophenyl)-5-[4-(methylsulfonyl)phenyl]oxazol-2-yl]acetic
acid;
19) 2-(tert butyl)-4-(4-fluorophenyl)-5-[4-(methylsulfonyl)phenyl]oxazole;
110) 4-(4-fluorophenyl)-5-[4-(methylsulfonyl)phenyl]-2-phenyloxazole;
m1) 4-(4-fluorophenyl)-2-methyl-5-[4-(methylsulfonyl)phenyl]oxazole;
and
m2) 4-[5-(3-fluoro-4-methoxyphenyl)-2-trifluoromethyl-4-
oxazolyl]benzenesulfonamide.
m3) 6-chloro-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic acid;
m4) 6-chloro-7-methyl-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic
acid;
m5) 8-(1-methylethyl)-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic
acid;
m6) 6-chloro-7-(1,1-dimethylethyl)-2-trifluoromethyl-2H-1-benzopyran-3-
carboxylic acid; ,
m7) 6-chloro-8-(1-methylethyl)-2-trifluoromethyl-2H-1-benzopyran-3-
carboxylic acid;
m8) 2-trifluoromethyl-3H-naphthopyran-3-carboxylic acid ;
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
m9) 7-(1,1-dimethylethyl)-2-trifluoromethyl-2H-1-benzopyran-3-
carboxylic acid;
m10) 6-bromo-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic acid;
n1) 8-chloro-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic acid;
n2) 6-trifluoromethoxy-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic
acid;
n3) 5,7-dichloro-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic acid;
n4) 8-phenyl-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic
acid;
n5) 7,8-dimethyl-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic
acid;
n6) 6,8-bis(dimethylethyl)-2-trifluoromethyl-2H-1-benzopyran-3-
carboxylic
acid;
n7) 7-(1-methylethyl)-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic
acid;
n8) 7-phenyl-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic
acid;
n9) 6-chloro-7-ethyl-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic
acid;
n10) 6-chloro-8-ethyl-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic
acid;
01 ) 6-chloro-7-phenyl-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic
acid;
02) 6,7-dichloro-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic
acid;
03) 6,8-dichloro-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic
acid;
04) 2-trifluoromethyl-3H-naptho[2,1-b]pyran-3-carboxylic
acid;
05) 6-chloro-8-methyl-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic
acid;
06) 8-chloro-6-methyl-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic
acid;
07) 8-chloro-6-methoxy-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic
acid;
08) 6-bromo-8-chloro-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic
acid;
36
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
09) 8-bromo-6-fluoro-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic
acid;
010) 8-bromo-6-methyl-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic
acid;
p1) 8-bromo-5-fluoro-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic
acid;
p2) 6-chloro-8-fluoro-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic
acid;
p3) 6-bromo-8-methoxy-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic
acid;
p4) 6-[[(phenylmethyl)amino]sulfonyl]-2-trifluoromethyl-2H-1-
benzopyran-3-carboxylic acid;
p5) 6-[(dimethylamino)sulfonyl]-2-trifluoromethyl-2H-1-benzopyran-3-
carboxylic acid;
p6) 6-[(methylamino)sulfonyl]-2-trifluoromethyl-2H-1-benzopyran-3-
carboxylic acid;
p7) 6-[(4-morpholino)sulfonyl]-2-trifluoromethyl-2H-1-benzopyran-3-
carboxylic acid;
p8) 6-[(1,1-dimethylethyl)aminosulfonyl]-2-trifluoromethyl-2H-1-
benzopyran-3-carboxylic acid;
p9) 6-[(2-methylpropyl)aminosulfonyl]-2-trifluoromethyl-2H-1-
benzopyran-3-carboxylic acid;
p10) 6-methylsulfonyl-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic
acid;
q1) 8-chloro-6-[[(phenylmethyl)amino]sulfonyl]-2-trifluoromethyl-2H-1-
benzopyran-3-carboxylic acid;
q2) 6-phenylacetyl-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic acid;
q3) 6,8-dibromo-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic acid;
q4) 8-chloro-5,6-dimethyl-2-trifluoromethyl-2H-1-benzopyran-3-
carboxylic acid;
q5) 6,8-dichloro-(S)-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic
acid;
37
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
q6) 6-benzylsulfonyl-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic
acid;
q7) 6-[[N-(2-furylmethyl)amino]sulfonyl]-2-trifluoromethyl-2H-1-
benzopyran-3-carboxylic acid;
q8) 6-[[N-(2-phenylethyl)amino]sulfonyl] -2-trifluoromethyl-2H-1-
benzopyran-3-carboxylic acid;
q9) 6-iodo-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic acid;
q10) 7-(1,1-dimethylethyl)-2-pentafluoroethyl-2H-1-benzopyran-3-
carboxylic acid;
r1 ) 5,5-dimethyl-3-(3-fluorophenyl)-4-(4-methyl-sulphonyl-2(5H)-
fluranone;
r2) 6-chloro-2-trifluoromethyl-2H-1-benzothiopyran-3-carboxylic acid;
r3) 4-[5-(4-chlorophenyl)-3-(trifluoromethyl)-1 H-pyrazol-1-
yl]benzenesulfonamide;
r4) 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1 H-pyrazol-1-
yl]benzenesulfonamide;
r5) 4-[5-(3-fluoro-4-methoxyphenyl)-3-(difluoromethyl)-1 H-pyrazol-1-
yl]benzenesulfonamide;
r6) 3-[1-[4-(methylsulfonyl)phenyl]-4-trifluoromethyl-1H-imidazol-2-
yl]pyridine;
r7) 2-methyl-5-[1-[4-(methylsulfonyl)phenyl]-4-trifluoromethyl-1H-
imidazol-2-yl]pyridine;
r8) 4-[2-(5-methylpyridin-3-yl)-4-(trifluoromethyl)-1H-imidazol-1-
yl]benzenesulfonamide;
r9) 4-[5-methyl-3-phenylisoxazol-4-yl]benzenesulfonamide;
r10) 4-[5-hydroxymethyl-3-phenylisoxazol-4-yl]benzenesulfonamide;
si ) [2-trifluoromethyl-5-(3,4-difluorophenyl)-4-
oxazolyl]benzenesulfonamide;
s2) 4-[2-methyl-4-phenyl-5-oxazolyl]benzenesulfonamide; or
s3) 4-[5-(3-fluoro-4-methoxyphenyl-2-trifluoromethyl)-4-
oxazolyl]benzenesulfonamide;
or a pharmaceutically acceptable salt or prodrug thereof.
38
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
[00052] In a further preferred embodiment of the invention the
cyclooxygenase inhibitor can be selected from the class of tricyclic
cyclooxygenase-2 selective inhibitors represented by the general structure
of formula VII:
O R24
O\ ~~ Z1~ V~~
\ ~ \
R 26
wherein:
Zi is selected from the group consisting of partially unsaturated or
unsaturated heterocyclyl and partially unsaturated or unsaturated
carbocyclic rings;
R24 is selected from the group consisting of heterocyclyl, cycloalkyl,
cycloalkenyl and aryl, wherein R24 is optionally substituted at a
substitutable position with one or more radicals selected from alkyl,
haloalkyl, cyano, carboxyl, alkoxycarbonyl, hydroxyl, hydroxyalkyl,
haloalkoxy, amino, alkylamino, arylamino, nitro, alkoxyalkyl, alkylsulfinyl,
halo, alkoxy and alkylthio;
R25 is selected from the group consisting of methyl or amino; and
R26 is selected from the group consisting of a radical selected from
H, halo, alkyl, alkenyl, alkynyl, oxo, cyano, carboxyl, cyanoalkyl,
heterocyclyloxy, alkyloxy, alkylthio, alkylcarbonyl, cycloalkyl, aryl,
haloalkyl, heterocyclyl, cycloalkenyl, aralkyl, heterocyclylalkyl, acyl,
alkylthioalkyl, hydroxyalkyl, alkoxycarbonyl, arylcarbonyl, aralkylcarbonyl,
aralkenyl, alkoxyalkyl, arylthioalkyl, aryloxyalkyl, aralkylthioalkyl,
aralkoxyalkyl, alkoxyaralkoxyalkyl, alkoxycarbonylalkyl, aminocarbonyl,
aminocarbonylalkyl, alkylaminocarbonyl, N- arylaminocarbonyl, N-alkyl-N-
arylaminocarbonyl, alkylaminocarbonylalkyl, carboxyalkyl, alkylamino, N-
arylamino, N-aralkylamino, N-alkyl-N-aralkylamino, N-alkyl-N-arylamino,
aminoalkyl, alkylaminoalkyl, N-arylaminoalkyl, N-aralkylaminoalkyl, N-alkyl-
39
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
N-aralkylaminoalkyl, N-alkyl-N-arylaminoalkyl, aryloxy, aralkoxy, arylthio,
aralkylthio, alkylsulfinyl, alkylsulfonyl, aminosulfonyl, alkylaminosulfonyl,
N-
arylaminosulfonyl, arylsulfonyl, N-alkyl-N-arylaminosulfonyl;
or a prodrug thereof.
(00053] In a preferred embodiment of the invention the cyclooxygenase-
2 selective inhibitor represented by the above Formula VII is selected from
the group of compounds, illustrated in Table 2, which includes celecoxib
(B-18), valdecoxib (B-19), deracoxib (B-20), rofecoxib (B-21 ), etoricoxib
(MK-663; B-22), JTE-522 (B-23), or a prodrug thereof.
[00054] Additional information about selected examples of the Cox-2
selective inhibitors discussed above can be found as follows: celecoxib
(CAS RN 169590-42-5, C-2779, SC-58653, and in U.S. Patent No.
5,466,823); deracoxib (CAS RN 169590-41-4); rofecoxib (CAS RN
162011-90-7); compound B-24 (U.S. Patent No. 5,840,924); compound B-
26 (WO 00/25779); and etoricoxib (CAS RN 202409-33-4, MIC-663, SC-
86218, and in WO 98/03484).
Table 2. Examples of Tricyclic COX-2 Selective Inhibitors
Compound Structural
Formula
Number
B-18 ~\S%
CH
~ ~ / 3
H2N ~
N
N~
CF3
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
Compound Structural Formula
Number
o~s,o
B-19
H~N~ \
\N
H3C O
B-2~ F
H N s/O OCH3
N
N~
CHFZ
B-21
oOs~
H3C~ \
0
B-22 O\S O CH
H3~~ \ ~~ 3
\ N
\N
C1~
41
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
Compound Structural Formula
Number
B-23
oOsi
HzN~ \
/
p' / N
YICH3
[00055] In a more preferred embodiment of the invention, the Cox-2
selective inhibitor is selected from the group consisting of celecoxib,
rofecoxib and etoricoxib.
[00056] In a preferred embodiment of the invention, parecoxib (See,
e.g. U.S. Patent No. 5,932,598), having the structure shown in B-24, which
is a therapeutically effective prodrug of the tricyclic cyclooxygenase-2
selective inhibitor valdecoxib, B-19, (See, e.g., U.S. Patent No. 5,633,272),
may be advantageously employed as a source of a cyclooxygenase
inhibitor.
~S o
HN~ I \ /
o / \ B-24
~N
H3C 0
[00057] A preferred form of parecoxib is sodium parecoxib.
[00058] In another embodiment of the invention, the compound ABT-
963 having the formula B-25 that has been previously described in
International Publication number WO 00/24719, is another tricyclic
42
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
cyclooxygenase-2 selective inhibitor which may be advantageously
employed.
O / F
OH
O
N \ F
~N
~S
//
0
B-25
[00059] In a further embodiment of the invention, the cyclooxygenase
inhibitor can be selected from the class of phenylacetic acid derivative
cyclooxygenase-2 selective inhibitors represented by the general structure
of Formula VIII:
wherein:
R2' is methyl, ethyl, or propyl;
R2$ is chloro or fluoro;
R29 is hydrogen, fluoro, or methyl;
R2~ O
OH
NH
R32
R31
VIII
43
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
R3° is hydrogen, fluoro, chloro, methyl, ethyl, methoxy, ethoxy or
v hyd roxy;
R31 IS hydrogen, fluoro, or methyl; and
R32 is chloro, fluoro, trifluoromethyl, methyl, or ethyl,
provided that R28, R29, R3° and R31 are not all fluoro when R2' is
ethyl and
R3° is H.
[00060] A phenylacetic acid derivative cyclooxygenase-2 selective
inhibitor that is described in WO 99/11605 is a compound that has the
structure shown in Formula VIII,
wherein:
R2' is ethyl;
R28 and R3° are chloro;
R29 and R31 are hydrogen; and
R32 is methyl.
[00061 ] Another phenylacetic acid derivative cyclooxygenase-2
selective inhibitor is a compound that has the structure shown in Formula
VIII,
wherein:
R2' is propyl;
R28 and R3° are chloro;
R29 and R31 are methyl; and
R32 is ethyl.
[00062] Another phenylacetic acid derivative cyclooxygenase-2
selective inhibitor that is described in WO 02/20090 is a compound that is
referred to as COX-189 (also termed lumiracoxib), having CAS Reg. No.
220991-20-8, and having the structure shown in Formula VIII,
wherein:
R2' is methyl;
R28 is fluoro;
R32 is chloro; and
R29, R3°, and R31 are hydrogen.
44
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
(00063] Compounds that have a structure similar to that shown in
Formula VIII, which can serve as the Cox-2 selective inhibitor of the
present invention, are described in U.S. Patent Nos. 6,310,099, 6,291,523,
and 5,958,978.
(00064] Other cyclooxygenase-2 selective inhibitors that can be used in
the present invention have the general structure shown in formula IX,
where the J group is a carbocycle or a heterocycle. Preferred
embodiments have the structure:
R33
J
34
R3s
wherein:
IX
X is O; J is 1-phenyl; R33 is 2-NHS02CH3; R34 is 4-N02; and there is
no R35 group, (nimesulide), and
X is O; J is 1-oxo-inden-5-yl; R33 is 2-F; R34 is 4-F; and R35 is 6-
NHS02CH3, (flosulide); and
X is O; J is cyclohexyl; R33 is 2-NHS02CH3; R34 is 5-N02; and there
is no R35 group, (NS-398); and
X is S; J is 1-oxo-inden-5-yl; R33 is 2-F; R34 is 4-F; and R35 is 6-N-
S02CH3 ~ Na+, (L-745337); and
X is S; J is thiophen-2-yl; R33 is 4-F; there is no R34 group; and R3s
is 5-NHS02CH3, (RWJ-63556); and
X is O; J is 2-oxo-5(R)-methyl-5-(2,2,2-trifluoroethyl)furan-(5H)-3-yl;
R33 is 3-F; R34 is 4-F; and R35 is 4-(p-S02CH3)C6H4, (L-784512).
(00065] Further information on the applications of the Cox-2 selective
inhibitor N-(2-cyclohexyloxynitrophenyl) methane sulfonamide (NS-398,
CAS RN 123653-11-2), having a structure as shown in formula B-26, have
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
been described by, for example, Yoshimi, N. et al., in Japanese J. Cancer
Res., 90(4):406 - 412 (1999); Falgueyret, J.-P. et al., in Science Spectra,
available at: http://www.gbhap.com/Science_Spectra/20-1-article.htm
(06/06/2001 ); and Iwata, K. et al., in Jpn. J. Pharmacol., 75(2):191 - 194
(1997).
B-26
[00066] An evaluation of the anti-inflammatory activity of the
cyclooxygenase-2 selective inhibitor, RWJ 63556, in a canine model of
inflammation, was described by Kirchner et al., in J Pharmacol Exp Ther
282, 1094-1101 (1997).
[00067] Materials that can serve as the cyclooxygenase-2 selective
inhibitor of the present invention include diarylmethylidenefuran derivatives
that are described in U.S. Patent No. 6,180,651. Such
diarylmethylidenefuran derivatives have the general formula shown below
in formula X:
i
39
Rss
X
L2
46
H~ ~S02CH3
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
wherein:
the rings T and M independently are:
a phenyl radical,
a naphthyl radical,
a radical derived from a heterocycle comprising 5 to 6 members
and possessing from 1 to 4 heteroatoms, or
a radical derived from a saturated hydrocarbon ring having from 3
to 7 carbon atoms;
at least one of the substituents Qi, Q2, L~ or L2 is:
an -S(O)" -R group, in which n is an integer equal to 0, 1 or 2 and R is:
a lower alkyl radical having 1 to 6 carbon atoms or
a lower haloalkyl radical having 1 to 6 carbon atoms, or
an -S02NH2 group;
and is located in the para position,
the others independently being:
a hydrogen atom,
a halogen atom,
a lower alkyl radical having 1 to 6 carbon atoms,
a trifluoromethyl radical, or
a lower O-alkyl radical having 1 to 6 carbon atoms, or
Q' and Q2 or L1 and L2 are a methylenedioxy group; and
R36, R3', R3$ and R39 independently are:
a hydrogen atom,
a halogen atom,
a lower alkyl radical having 1 to 6 carbon atoms,
a lower haloalkyl radical having 1 to 6 carbon atoms, or
an aromatic radical selected from the group consisting of phenyl,
naphthyl, thienyl, furyl and pyridyl; or,
R36, R3' or R38, R39 are an oxygen atom, or
47
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
R36, R3' or R38, R39, together with the carbon atom to which they are
attached, form a saturated hydrocarbon ring having from 3 to 7 carbon
atoms;
or an isomer or prodrug thereof.
[00068] Particular materials that are included in this family of
compounds, and which can serve as the cyclooxygenase-2 selective
inhibitor in the present invention, include N-(2- .
cyclohexyloxynitrophenyl)methane sulfonamide, and (E)-4-[(4-
methylphenyl)(tetrahydro-2-oxo-3-furanylidene)
methyl]benzenesulfonamide.
[00069] Cyclooxygenase-2 selective inhibitors that are useful in the
present invention include darbufelone (Pfizer), CS-502 (Sankyo), LAS
34475 (Almirall Profesfarma), LAS 34555 (Almirall Profesfarma), S-33516
(Servier), SD 8381 (Pharmacia, described in U.S. Patent No. 6,034,256),
BMS-347070 (Bristol Myers Squibb, described in U.S. Patent No.
6,180,651), MK-966 (Merck), L-783003 (Merck), T-614 (Toyarna), D-1367
(Chiroscience), L-748731 (Merck), CT3 (Atlantic Pharmaceutical), CGP-
28238 (Novartis), BF-389 (Biofor/Scherer), GR-253035 (Glaxo Wellcome),
6-dioxo-9H-purin-8-yl-cinnamic acid (Glaxo Wellcome), and S-2474
(Shionogi).
[00070] Information about S-33516, mentioned above, can be found in
Current Drugs Headline News, at http://www.current-
drugs.com/NEWS/Inflaml .htm, 10/04/2001, where it was reported that S-
33516 is a tetrahydroisoinde derivative which has IC5o values of 0.1 and
0.001 mM against cyclooxygenase-1 and cyclooxygenase-2, respectively.
In human whole blood, S-33516 was reported to have an EDSO = 0.39
mg/kg.
[00071] Compounds that may act as cyclooxygenase-2 selective
inhibitors include multibinding compounds containing from 2 to 10 ~ligands
covanlently attached to one or more linkers, as described in U.S. Patent
No. 6,395,724.
48
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
[00072] Compounds that may act as cyclooxygenase-2 inhibitors
include conjugated linoleic acid that is described in U.S. Patent No.
6,077,868.
[00073] Materials that can serve as a cyclooxygenase-2 selective
inhibitor of the present invention include heterocyclic aromatic oxazole
compounds that are described in U.S. Patents 5,994,381 and 6,362,209.
Such heterocyclic aromatic oxazole compounds have the formula shown
below in formula XI:
R4o
N
XI
R42
~2
wherein:
Z2 is an oxygen atom;
one of R4° and R41 is a group of the formula
R44
R43 ~2~ ;47
t
wherein:
R43 is lower alkyl, amino or lower alkylamino; and
R44~ R45~ R4s and R47 are the same or different and each is
hydrogen atom, halogen atom, lower alkyl, lower alkoxy, trifluoromethyl,
49
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
hydroxy or amino, provided that at least one of R44, R45, R4s and R4' is not
hydrogen atom, and the other is an optionally substituted cycloalkyl, an
optionally substituted heterocyclic group or an optionally substituted aryl;
and
R3° is a lower alkyl or a halogenated lower alkyl, and a
pharmaceutically acceptable salt thereof.
[00074] Cox-2 selective inhibitors that are useful in the subject method
and compositions can include compounds that are described in U.S.
Patent Nos. 6,080,876 and 6,133,292, and described by formula XII:
O
XII
Rso
R48o2s
wherein:
Z3 is selected from the group consisting of:
(a) linear or branched C1_6 alkyl,
(b) linear or branched C1_6 alkoxy,
(c) unsubstituted, mono-, di- or tri-substituted phenyl or naphthyl
wherein the substituents are selected from the group consisting of:
(1 ) hydrogen,
(2) halo,
(3) C1_3 alkoxy,
(4) CN,
(5) Cy_3 fluoroalkyl
(6) C1_3 alkyl,
(7) -C02 H;
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
R48 is selected from the group consisting of NH2 and CH3,
R49 is selected from the group consisting of:
Cy_6 alkyl unsubstituted or substituted with C3_6 cycloalkyl, and
C3_6 cycloalkyl;
R5° is selected from the group consisting of:
Ci_6 alkyl unsubstituted or substituted with one, two or three fluoro
atoms; and
C3_g cycloalkyl;
with the proviso that R49 and R5° are not the same.
[00075] Materials that can serve as cyclooxygenase-2 selective
inhibitors include pyridines that are described in U.S. Patent Nos.
6,369,275, 6,127,545, 6,130,334, 6,204,387, 6,071,936, 6,001,843 and
6,040,450, and which have the general formula described by formula XIII:
R51
R52
wherein:
R51 is selected from the group consisting of:
(a) CH3,
(b) NH2,
(c) NHC(O)CF3,
(d) NHCH3 ;
Z4 is a mono-, di-, or trisubstituted phenyl or pyridinyl (or the N-
oxide thereof),
wherein the substituents are chosen from the group consisting of:
(a) hydrogen,
51
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
(b) halo,
(c) C1_6 alkoxy,
(d) C1_6 alkylthio,
(e) CN,
(f) C1_6 alkyl,
(g) e1-6 fluoroalkyl,
(h) Ns~
(i) -CO2R53,
(j) hydroxy,
(k) -C(Rs4)(Rss)-OH,
(I) -C1_6alkyl-CO2-R56,
(m) C~_6fluoroalkoxy;
R52 is chosen from the group consisting of:
(a) halo,
(b) C1_6alkoxy,
(c) C1_6 alkylthio,
(d) CN,
(e) C1_6 alkyl,
(f) C1_g fluoroalkyl,
(g) Ns,
(h) -C02R5',
(i) hydroxy,
(l) -C(R5$)(Rss)-OH~
(k) -C1_6alkyl-CO2-R6°, .
(I) C1_6fluoroalkoxy,
(m) N02,
(n) N R6' R62, and
(o) NHCOR6s;
R53, R54, R55, R56, R5', R58, R59, R60, R61, R62, R63, are each
independently chosen from the group consisting of:
(a) hydrogen, and
(b) C1_6alkyl;
52
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
or R54 and R55, R5$ and R59 or R61 and R62 together with the atom to which
they are attached form a saturated monocyclic ring of 3, 4, 5, 6, or 7
atoms.
[00076] Materials that can serve as the cyclooxygenase-2 selective
inhibitor of the present invention include diarylbenzopyran derivatives that
are described in U.S. Patent No. 6,340,694. Such diarylbenzopyran
derivatives have the general formula shown below in formula XIV:
x8
XIV
wherein:
X$ is an oxygen atom or a sulfur atom;
R64 and R65, identical to or different from each other, are
independently a hydrogen atom, a halogen atom, a C1 -C6 lower alkyl
group, a trifluoromethyl group, an alkoxy group, a hydroxy group, a vitro
group, a nitrite group, or a carboxyl group;
R66 is a group of a formula: S(O)"R6$ wherein n is an integer of 0~2,
R6$ is a hydrogen atom, a C1 -C6 lower alkyl group, or a group of a
formula: NR69 R'° wherein R69 and R'°, identical to or different
from each
other, are independently a hydrogen atom, or a C1 -C6 lower alkyl group;
and
R6' is oxazolyl, benzo[b]thienyl, furanyl, thienyl, naphthyl, thiazolyl,
indolyl, pyrolyl, benzofuranyl, pyrazolyl, pyrazolyl substituted with a C1 -C6
lower alkyl group, indanyl, pyrazinyl, or a substituted group represented by
the following structures:
53
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
R'1
N
R'°
R~s
N~ N
R~s
R~s
wherein:
R" through R'5, identical to or different from one another, are
independently a hydrogen atom, a halogen atom, a C1 -C6 lower alkyl
group, a trifluoromethyl group, an alkoxy group, a hydroxy group, a
hydroxyalkyl group, a nitro group, a group of a formula: S(O)"R68, a group
of a formula: NR69 R'°, a trifluoromethoxy group, a nitrite group a
carboxyl
group, an acetyl group, or a formyl group,
wherein n, R68, R69 and R'° have the same meaning as defined by
R66 above; and
R'6 is a hydrogen atom, a halogen atom, a C1 -C6 lower alkyl group,
a trifluoromethyl group, an alkoxy group, a hydroxy group, a
trifluoromethoxy group, a carboxyl group, or an acetyl group.
[00077] Materials that can serve as the cyclooxygenase-2 selective
inhibitor of the present invention include 1-(4-sulfamylaryl)-3-substituted-5-
aryl-2-pyrazolines that are described in U.S. Patent No. 6,376,519. Such
54
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
1-(4-sulfamylaryl)-3-substituted-5-aryl-2-pyrazolines have the formula
shown below in formula XV:
xs
Z5 XV
S02NH2
wherein:
Xs is selected from the group consisting of Cy -C6 trihalomethyl,
preferably trifluoromethyl; Cy -C6 alkyl; and an optionally substituted or di-
substituted phenyl group of formula XVI:
R~~
_ ' XVI
R~$
wherein:
R" and R'$ are independently selected from the group consisting of
hydrogen, halogen, preferably chlorine, fluorine and bromine; hydroxyl;
nitro; C1 -C6 alkyl, preferably C1 -C3 alkyl; C1 -C6 alkoxy, preferably C1 -C3
alkoxy; carboxy; C1 -C6 trihaloalkyl, preferably trihalomethyl, most
preferably trifluoromethyl; and cyano;
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
Z5 is selected from the group consisting of substituted and
unsubstituted aryl.
[00078] Materials that can serve as the cyclooxygenase-2 selective
inhibitor of the present invention include heterocycles that are described in
U.S. Patent No. 6,153,787. Suoh heterocycles have the general formulas
shown below in formulas XVII and XVII1:
R'9
O
R$OS~~)2
XVII
wherein:
R'9 is a mono-, di-, or tri-substituted C1_12 alkyl, or a mono-, or an
unsubstituted or mono-, di- or tri-substituted linear or branched C2_1o
alkenyl, or an unsubstituted or mono-, di- or tri-substituted linear or
branched C2_io alkynyl, or an unsubstituted or mono-, di- or tri-substituted
C3-12 cycloalkenyl, or an unsubstituted or mono-, di- or tri-substituted C5_12
cycloalkynyl, wherein the substituents are chosen from the group
consisting of:
(a) halo, selected from F, CI, Br, and I,
(b) OH,
(c) C F3,
(d) C3_6 cycloalkyl,
(e) =O,
(f) dioxolane,
(g) CN; and
R$° is selected from the group consisting of:
(a) CH3,
56
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
(b) NH2,
(c) NHC(O)CF3,
(d) NHCH3 ;
R81 and R82 are independently chosen from the group consisting of:
(a) hydrogen,
(b) C1-io alkyl;
or R81 and R82 together with the carbon to which they are attached
form a saturated monocyclic carbon ring of 3, 4, 5, 6 or 7 atoms.
[00079] Formula XVIII is:
X10
O
~~~2SH3C
X1° is fluoro or chloro.
XVIII
[00080] Materials that can serve as the cyclooxygenase-2 selective
inhibitor of the present invention include 2,3,5-trisubstituted pyridines that
are described in U.S. Patent No. 6,046,217. Such pyridines have the
general formula shown below in formula XIX:
57
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
Rs4
S02Rss
XIX
Rss Ray
Rs9
X11- ~ - 0891
~~~n
R9o
Rss Rss
or a pharmaceutically acceptable salt thereof,
wherein:
X11 is selected from the group consisting of:
(a) O,
(b) S,
(c) bond;
nis0orl;
R83 is selected from the group consisting of:
(a) CH3,
(b) NH2,
(c) NHC(O)CF3;
R84 is chosen from the group consisting of:
(a) halo,
(b) C1_6 alkoxy,
(c) C1_6 alkylthio,
(d) CN,
(e) C1_6 alkyl,
(f) C1_g fluoroalkyl,
(g) Ns~
(h) -CO2 R92,
(i) hydroxy,
(i) -~(R93)(R94)-oH,
(k) -C1_6 alkyl-C02 -R95,
58
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
(I) C1_6 fluoroalkoxy,
(m) N02,
(n) NR96 R9',
(o) NHCOR98;
R85 to R9$ are independantly chosen from the group consisting of
(a) hydrogen,
(b) C1_6 alkyl;
or R85 and R89, or R89 and R9° together with the atoms to which they
are attached form a carbocyclic ring of 3, 4, 5, 6 or 7 atoms, or R85 and R$'
are joined to form a bond.
[00081] One preferred embodiment of the Cox-2 selective inhibitor of
formula XIX is that wherein X is a bond.
[00082] Another preferred embodiment of the Cox-2 selective inhibitor of
formula XIX is that wherein X is O.
[00083] Another preferred embodiment of the Cox-2 selective inhibitor of
formula XIX is that wherein X is S.
[00084] Another preferred embodiment of the Cox-2 selective inhibitor of
formula XIX is that wherein R83 is CH3.
[00085] Another preferred embodiment of the Cox-2 selective inhibitor
of formula XIX is that wherein R84 is halo or C1_g fluoroalkyl.
[00086] Materials that can serve as the cyclooxygenase-2 selective
inhibitor of the present invention include diaryl bicyclic heterocycles that
are described in U.S. Patent No. 6,329,421. Such diaryl bicyclic
heterocycles have the general formula shown below in formula XX:
R99
8101 A6 =~ H~ XX
v
Rioo
Riot ~
59
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
and pharmaceutically acceptable salts thereof wherein:
-A5=A6-A~=A8- is selected from the group consisting of:
(a) -CH=CH-CH=CH-,
(b) -CH2 -CH2 -CH2 -C(O)-, -CH2 -CH2 -C(O)-CH2 -,
-CH2 -C(O)-CH2 -CH2, -C(O)-CH2 -CH2 -CH2,
(c) -CH2 -CH2 -C(O)-, -CH2 -C(O)-CH2 -, -C(O)-CH2
-CH2 -
(d) -CH2 -CH2 -O-C(O)-, CH2 -O-C(O)-CH2 -, -O-
C(O)-CH2 -CH2 -,
(e) -CH2 -CH2 -C(O)-O-, -CH2 -C(O)-OCH2 -, -C(O)-
O-CH2 -CH2 -,
(f) -C(R~os)2 -O-C(O)-, -C(O)-O-~(R105)2 -, -O-C(O)-
C(R105)2 -, -C(R105)2 -C(O)-O-,
(g) -N=CH-CH=CH-,
(h) -CH=N-CH=CH-,
(i) -CH=CH-N=CH-,
(j) -CH=CH-CH=N-,
(k) -N=CH-CH=N-,
(I) -N=CH-N=CH-,
(m) -CH=N-CH=N-,
(n) -S-CH=N-,
(o) -S-N=CH-,
(p) -N=N-NH-,
(q) -CH=N-S-, and
(r) -N=CH-S-;
R99 is selected from the group consisting of:
(a) S(O)2 CH3,
(b) S(O)2 NH2,
(c) S(O)2 NHCOCF3,
(d) S(O)(NH)CH3,
(e) S(O)(NH)NH2,
(f) S(O)(NH)NHCOCF3,
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
(g) P(O)(CH3)OH, and
(h) P(O)(CH3)NH2;
R1°° is selected from the group consisting of:
(a) C1_6 alkyl,
(b) C3_~, cycloalkyl,
(c) mono- or di-substituted phenyl or naphthyl wherein the
substituent is selected from the group consisting of:
(1 ) hydrogen,
(2) halo, including F, CI, Br, I,
(3) C1_6 alkoxy,
(4) C1_6 alkylthio,
(5) CN,
(5) CFs
(7) C1_6 alkyl,
(8) N3,
(9) -C02 H,
(10) -C02 -Cy_4 alkyl,
(11 ) -C~R103)(R104)-OH'
(12) -C(Rlos)(Rloa)-O-C~_4 alkyl, and
(13) -C1_6 alkyl-C02 -8106;
(d) mono- or di-substituted heteroaryl wherein the heteroaryl is a
monocyclic aromatic ring of 5 atoms, said ring having one hetero atom
which is S, O, or N, and optionally 1, 2, or 3 additional N atoms; or the
heteroaryl is a monocyclic ring of 6 atoms, said ring having one hetero
atom which is N, and optionally 1, 2, 3, or 4 additional N atoms; said
substituents are selected from the group consisting of:
(1 ) hydrogen,
(2) halo, including fluoro, chloro, bromo and iodo,
(3) C1_6 alkyl,
(4) C1_6 alkoxy,
(5) C1_6 alkylthio,
(6) CN,
61
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
(~) CFs
(8) Ns~
(9) -C(R103)(R104)-OH, and
(1 0) -C(R103)(R104)-O-C1-4 alkyl;
(e) benzoheteroaryl which includes the benzo fused analogs of (d);
R~°' and R1°~ are the substituents residing on any position
of -A5=A6-
A'=A$- and are selected independently from the group consisting of:
(a) hydrogen,
(b) CF3,
(c) CN,
(d) Ci_6 alkyl,
(e) -Q3 wherein Q3 is Q4, C02 H, C(Rlos)(R7o4)OH,
(f) -O-Qa.
(g) -S-Q4, and
(h) optionally substituted:
(1 ) -C1_5 alkyl-Q3,
(2) -O-C1_5 alkyl-Q3,
(3) -S-C1_5 alkyl-Q3,
(4) -C1_3 alkyl-O-Ci_3 alkyl-Q3,
(5) -Cy_3 alkyl-S-C1_3 alkyl-Q3,
(6) -C1_5 alkyl-O-Q4,
(7) -C1_5 alkyl-S-Q4,
wherein the substituent resides on the alkyl chain and the
substituent is C1_3 alkyl, and Q3 is Q4, C02 H, C(Rio3)(R~o~)OH Q4 is C02
-C1_4 alkyl, tetrazolyl-5-yl, or C(Rlos)(Rloa.)O-C1_~ alkyl;
8103' 8104 and R'°5 are each independently selected from the group
consisting of
(a) hydrogen,
(b) C1_g alkyl; or
R~°3 and Rlo4 together with the carbon to which they are attached
form a saturated monocyclic carbon ring of 3, 4, 5, 6 or 7 atoms, or two
62
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
8105 groups on the same carbon form a saturated monocyclic carbon ring
of 3, 4, 5, 6 or 7 atoms;
8106 is hydrogen or C1_g alkyl;
R1°' is hydrogen, C1_6 alkyl or aryl;
X' is O, S, NRio', CO, C~R10')2, C(Rlo~)(OH)~ -C(Rio~)=C(Rlo~)-; -
C(Rlo~)-N-; -N=C(Rio~)-.
[00087] Compounds that may act as cyclooxygenase-2 inhibitors
include salts of 5-amino or a substituted amino 1,2,3-triazole compound
that are described in U.S. Patent No. 6,239,137. The salts are of a class
of compounds of formula XXI:
8110
N
xxl
Rlos N
108
wherein:
RioB is:
~(R112)
n
X13
-(CH2)p
\(R111)
m
wherein:
p is 0 to 2; m is 0 to 4; and n is 0 to 5; X13 is O, S, SO, S02, CO,
CHCN, CH2 or C=NR113 where 8113 IS hydrogen, loweralkyl, hydroxy,
loweralkoxy, amino, loweralkylamino, diloweralkylamino or cyano; and,
R' 11 and 8112 are independently halogen, cyano, trifluoromethyl,
63
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
loweralkanoyl, nitro, loweralkyl, loweralkoxy, carboxy, lowercarbalkoxy,
trifuloromethoxy, acetamido, loweralkylthio, loweralkylsulfinyl,
loweralkylsulfonyl, trichlorovinyl, trifluoromethylthio,
trifluoromethylsulfinyl,
or trifluoromethylsulfonyl; R1°9 is amino, mono or diloweralkyl amino,
acetamido, acetimido, ureido, formamido, formamido or guanidino; and
8110 is carbamoyl, cyano, carbazoyl, amidino or N-hydroxycarbamoyl;
wherein the loweralkyl, loweralkyl containing, loweralkoxy and
loweralkanoyl groups contain from 1 to 3 carbon atoms.
(00088] Materials that can serve as a cyclooxygenase-2 selective
inhibitor of the present invention include pyrazole derivatives that are
described in U.S. Patent 6,136,831. Such pyrazole derivatives have the
formula shown below in formula XXII:
8114
~i N
8115
8117
x14 ~ N XXII
116
R N
Z6
wherein:
8114 IS hydrogen or halogen, 8115 and 8116 are each independently
hydrogen, halogen, lower alkyl, lower alkoxy, hydroxy or lower
alkanoyloxy;
8117 is lower haloalkyl or lower alkyl;
X14 is sulfur, oxygen or NH; and
Z6 is lower alkylthio, lower alkylsulfonyl or sulfamoyl;
or a pharmaceutically acceptable salt thereof.
64
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
[00089] Materials that can serve as a cyclooxygenase-2 selective
inhibitor of the present invention include substituted derivatives of
benzosulphonamides that are described in U.S. Patent 6,297,282. Such
benzosulphonamide derivatives have the formula shown below in formula
XXIII:
8118
S O 8119
~m
0 0 ~ ~N~ XXIII
' 120
R
8123 NH
r,124
wherein:
X15 denotes oxygen, sulphur or NH;
10 Rii$ is an optionally unsaturated alkyl or alkyloxyalkyl group,
optionally mono- or polysubstituted or mixed substituted by halogen,
alkoxy, oxo or cyano, a cycloalkyl, aryl or heteroaryl group optionally
mono- or polysubstituted or mixed substituted by halogen, alkyl, CF3,
cyano or alkoxy;
15 8119 and Rl2o, independently from one another, denote hydrogen,
an optionally polyfluorised alkyl group, an aralkyl, aryl or heteroaryl group
or a group (CH2)~ -X16; or
8119 and R12°, together with the N- atom, denote a 3 to 7-
membered, saturated, partially or completely unsaturated heterocycle with
one or more heteroatoms N, O or S, which can optionally be substituted by
oxo, an alkyl, alkylaryl or aryl group, or a group (CH2)n X16;
X16 denotes halogen, NO2, -OR121, -COR121, -C02 8121, -OCO2 8121'
-CN, -CONR121 OR122' -CONR121 8122' -SR121~ -S(O)R121~ -S(~)2
R121~ -NR121 R122~ -NHC(O)R121, -NHS(O)2 8121
n denotes a whole number from 0 to 6;
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
8123 denotes a straight-chained or branched alkyl group with 1-10
C- atoms, a cycloalkyl group, an alkylcarboxyl group, an aryl group, aralkyl
group, a heteroaryl or heteroaralkyl group which can optionally be mono-
or polysubstituted or mixed substituted by halogen or alkoxy;
8124 denotes halogen, hydroxy, a straight-chained or branched
alkyl, alkoxy, acyloxy or alkyloxycarbonyl group with 1-6 C- atoms, which
can optionally be mono- or polysubstituted by halogen, N02, -OR121~ -
COR121' -CO2 8121' -OCO2 R121~ -CND -CONR121 OR122' -CONR121
R122~ -SR121' -S(O)R121' -S(O)2 8121' -NR121 R122~ -NHC(O)R121~ -
NHS(O)2 8121, or a polyfluoroalkyl group;
8121 and 8122, independently from one another, denote hydrogen,
alkyl, aralkyl or aryl; and
m denotes a whole number from 0 to 2;
and the pharmaceutically-acceptable salts thereof.
[00090] Materials that can serve as a cyclooxygenase-2 selective
inhibitor of the present invention include 3-phenyl-4-
(4(methylsulfonyl)phenyl)-2-(5H)-furanones that are described in U.S.
Patent 6,239,173. Such 3-phenyl-4-(4(methylsulfonyl)phenyl)-2-(5H)-
furanones have the formula shown below in formula XXIV:
8125
XXIV
8126 p ~e_ . ~~~7
0
a
a'
' 1
~i~_.-Y
66
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
or pharmaceutically acceptable salts thereof wherein:
X1'-Y1-Z'-is selected from the group consisting of:
(a) -CH2 CH2 CH2 -,
(b) -C(O)CH2 CH2 -,
(c) -CH2 CH2 C(O)-,
(d) -CR129 (R129')-O-C(O)-
(e) -G,(~)-O-G,R129 (R129')-a
(f) -CH2 -NR12' -CH2 -,
(g) -CR129 (R129')-NR127 -C(O)-
(h) -CR128=CR'28~ -S-,
(I) -S-CR128=~rR128' -~
(j) -S-N=CH-,
(k) -CH=N-S-,
(I) -N=CR128 -O-,
(m) -O-CR4=N- ,
(n) -N=CR128 -NH-,
(o) -N=CR12$ -S-, and
(p) -S-CRl2s=N-
(a) -C(~)-NR127 -CR129 (R129')-s
(r) -R12' N-CH=CH- provided 8122 is not -S(O)2CH3,
(s) -CH=CH-NR12' - provided 8125 is not -S(O)2CH3,
when side b is a double bond, and sides a and c are single bonds;
and
X1'-Y1-Z'-is selected from the group consisting of:
(a) =CH-O-CH=, and
(b) =CH-NR12' -CH=,
(c) =N-S-CH=,
(d) =CH-S-N=,
(e) =N-O-CH=,
(f) =CH-O-N=,
(g) =N-S-N=,
(h) =N-O-N=,
67
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
when sides a and c are double bonds and side b is a single bond;
R'25 is selected from the group consisting of:
(a) S(O)2 CH3,
(b) S(O)2 NH2,
(c) S(O)2 NHC(O)CF3,
(d) S(O)(NH)CH3,
(e) S(O)(NH)NH2,
(f) S(O)(NH)NHC(O)CF3,
(g) P(O)(CH3)OH, and
(h) P(O)(CH3)NH2;
R'26 is selected from the group consisting of
(a) C1_g alkyl,
(b) C3, C4, C5, C6, and C~, cycloalkyl,
(c) mono-, di- or tri-substituted phenyl or naphthyl,
wherein the substituent is selected from the group consisting of:
(1 ) hydrogen,
(2) halo,
(3) C1_~ alkoxy,
(4) C1_6 alkylthio,
(5) CN,
(6) CF3,
(7) C1_6 alkyl,
(8) Ns~
(9) -C02 H,
(10) -C02 -C1_4 alkyl,
(11 ) -C(R129)(R130)-OH,
(12) -C(R'29)(Riso)-O-C1_4 alkyl, and
(13) -C1_6 alkyl-C02 -R'29 ;
(d) mono-, di- or tri-substituted heteroaryl wherein the heteroaryl is
a monocyclic aromatic ring of 5 atoms, said ring having one hetero atom
which is S, O, or N, and optionally 1, 2, or 3 additionally N atoms; or the
heteroaryl is a monocyclic ring of 6 atoms, said ring having one hetero
68
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
atom which is N, and optionally 1, 2, 3, or 4 additional N atoms; said
substituents are selected from the group consisting of:
(1 ) hydrogen,
(2) halo, including fluoro, chloro, bromo and iodo,
(3) C1_6 alkyl,
(4) C1_6 alkoxy,
(5) C1_6 alkylthio,
(6) CN,
(~) CFs
(8) N3,
(9) -C(Rl2s)(Rlso)-~H, and
(1 O) -C(R129)(R130)-O-C1_4 alkyl;
(e) benzoheteroaryl which includes the benzo fused analogs of (d);
R'2' is selected from the group consisting of:
(a) hydrogen,
(b) CF3,
(c) CN,
(d) C1_6 alkyl,
(e) hydroxyCl_6 alkyl,
(f) -C(O)-C1_6 alkyl,
(g) optionally substituted:
(1 ) -C1_5 alkyl-Q5,
2) -C1_3 alkyl-O-C1_3 alkyl-Q5,
(3) -Cy_3 alkyl-S-Cy_3 alkyl-Q5,
(4) -C1_5 alkyl-Q-Q5, or
(5) -Cy_5 alkyl-S-Q5,
wherein the substituent resides on the alkyl and the substituent is
C1_3 alkyl;
(h) -Q5~
R~2$ and R128~ are each independently selected from the group
consisting of:
69
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
(a) hydrogen,
(b) CF3,
(c) CN,
(d) C1_6 alkyl,
(e) _Q5,
(f) -O-Q5
(g) -S-Q5, and
(h) optionally substituted:
(1 ) -C1_5 alkyl-Q5,
(2) -O-C1_5 alkyl-Q5,
(3) -S-C1_5 alkyl-Q5,
(4) -C,_3 alkyl-O-C1_3
alkyl-Q5,
(5) -C1_3 alkyl-S-C1_3
alkyl-Q5,
(6) -C1_5 alkyl-O-Q5,
(7) -C1_5 alkyl-S-Q5,
wherein the substituent resides on the alkyl and the substituent is
C1_3 alkyl, and
8129' R129'~ 8130' 8131 and 8132 are each independently selected
from the group consisting of:
(a) hydrogen,
(b) C1_g alkyl;
or 8129 and R13° or 8131 and 8132 together with the carbon to which
they are attached form a saturated monocyclic carbon ring of 3, 4, 5, 6 or 7
atoms;
Q5 is C02 H, C02 -C1_4 alkyl, tetrazolyl-5-yl, C(Rls1)(Rls2)(OH), or
~(R131)(R132)(O-C1_4 alkyl);
provided that when X-Y-Z is -S-CR12$=CR128~, then R12$ and
R128~ are other than CF3.
[00091 ] Materials that can serve as a cyclooxygenase-2 selective
inhibitor of the present invention include bicycliccarbonyl indole
compounds that are described in U.S. Patent No. 6,303,628. Such
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
bicycliccarbonyl indole compounds have the formula shown below in
formula XXV:
~s
9
(X19)n-~ I ~~ XXV
~(CH2)q
// \Z10
( ~ H2)
(C~"~2)r_Y2~ m
or the pharmaceutically acceptable salts thereof wherein
A9 is C1_6 alkylene or-NR133-;
Z$ IS C(=L3)R134~ Or SO2 8135 ;
Z9 is CH or N;
Zi° and Y2 are independently selected from -CH2 -, O, S and -
N-8133 ;
misl,2or3;
q and r are independently 0, 1 or 2;
Xi$ is independently selected from halogen, C1_4 alkyl, halo-
substituted Ci_4 alkyl, hydroxy, C1_4 alkoxy, halo-substituted C1_4 alkoxy, C1-
4
alkylthio, nitro, amino, mono- or di-(C1_4 alkyl)amino and cyano;
n is 0, 1, 2, 3 or 4;
L3 is oxygen or sulfur;
8133 IS hydrogen or Ci_4 alkyl;
8134 iS hydroxy, C1_6 alkyl, halo-substituted C1_6 alkyl, C1_6 alkoxy,
halo-substituted C1_6 alkoxy, C3_~ cycloalkoxy, C1_4 alkyl(C3_~ cycloalkoxy),
-NR136 R137~ C1_4 alkylphenyl-O- or phenyl-O-, said phenyl being
optionally substituted with one to five substituents independently selected
71
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
from halogen, C1_4 alkyl, hydroxy, C1_4 alkoxy and nitro;
8135 is Ci_6 alkyl or halo-substituted Ci_6 alkyl; and
8136 and R13' are independently selected from hydrogen, C1_6 alkyl
and halo-substituted C1_6 alkyl.
[00092] Materials that can serve as a cyclooxygenase-2 selective
inhibitor of the present invention include benzimidazole compounds that
are described in U.S. Patent No. 6,310,079. Such benzimidazole
compounds have the formula shown below in formula XXVI:
N
(X21)n ~ \ CR140 CR139 8138
N
1 o-(x2o)
m
or a pharmaceutically acceptable salt thereof, wherein:
Ai° is heteroaryl selected from a 5-membered monocyclic aromatic
ring having one hetero atom selected from O, S and N and optionally
containing one to three N atoms) in addition to said hetero atom, or
a 6-membered monocyclic aromatic ring having one N atom and optionally
containing one to four N atoms) in addition to said N atom; and
said heteroaryl being connected to the nitrogen atom on the benzimidazole
through a carbon atom on the heteroaryl ring;
X2° is independently selected from halo, C1 -C4 alkyl, hydroxy, C1
-
C4 alkoxy, halo-substituted C1 -C4 alkyl, hydroxy-substituted C1 -C4 alkyl,
(Ci -C4 alkoxy)C1 -C4 alkyl, halo-substituted Ci -C4 alkoxy, amino, N-(C1 -
C4 alkyl)amino, N, N-di(C1 -C4 alkyl)amino, [N-(C1 -C4 alkyl)amino]C1 -C4
alkyl, [N, N-di(C1 -C4 alkyl)amino]Ci -C4 alkyl, N-(Ci -C4 alkanoyl)amonio,
N-(C1 -C4 alkyl)(Ci -C4 alkanoyl)amino, N-[(C1 -C4 alkyl)sulfonyl]amino, N-
[(halo-substituted Ci -C4 alkyl)sulfonyl]amino, C1 -C4 alkanoyl, carboxy, (Ci
-C4 alkoxy)carbonyl, carbamoyl, [N-(C1 -C4 alkyl)amino]carbonyl, [N, N-
di(C1 -C4 alkyl)amino]carbonyl, cyano, nitro, mercapto, (Ci -C4 alkyl)thio,
72
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
(C1 -C4 alkyl)sulfinyl, (C1 -C4 alkyl)sulfonyl, aminosulfonyl, [N-(C, -C4
alkyl)amino]sulfonyl and [N, N-di(Ci -C4 alkyl)amino]sulfonyl;
X2' is independently selected from halo, C1 -C4 alkyl, hydroxy, C1 -C4
alkoxy, halo-substituted C1 -C4 alkyl, hydroxy-substituted C1 -C4 alkyl, (C1 -
C4 alkoxy)C1 -C4 alkyl, halo-substituted C1 -C4 alkoxy, amino, N-(C1 -C4
alkyl)amino, N, N-di(C1 -C4 alkyl)amino, [N-(C1 -C4 alkyl)amino]C1 -C4
alkyl, [N, N-di(C1 -C4 alkyl)amino]Cy -C4 alkyl, N-(C1 -C4 alkanoyl)amino, N-
(C1 -C4 alkyl)-N-(C1 -C4 alkanoyl) amino, N-[(Ci -C4 alkyl)sulfonyl]amino,
N-[(halo-substituted C1 -C4 alkyl)sulfonyl]amino, C1 -C4 alkanoyl, carboxy,
(C1 -C4 alkoxy)cabonyl, cabamoyl, [N-(C1 -C4 alkyl) amino]carbonyl, [N, N-
di(C1 -C4 alkyl)amino]carbonyl, N-carbomoylamino, cyano, nitro, mercapto,
(C1 -C4 alkyl)thio, (C1 -C4 alkyl)sulfinyl, (C1 -C4 alkyl)sulfonyl,
aminosulfonyl, [N-(C1 -C4 alkyl)amino]sulfonyl and [N, N-di(Cy -C4
alkyl)amino]sulfonyl;
8138 is selected from hydrogen,
straight or branched C1 -C4 alkyl optionally substituted with one to
three substituent(s) wherein said substituents are independently selected
from halo hydroxy, Ci -C4 alkoxy, amino, N-(C1 -C4 alkyl)amino and N, N-
di(C1 -C4 alkyl)amino,
C3 -C8 cycloalkyl optionally substituted with one to three
substituent(s) wherein said substituents are indepently selected from halo,
C1 -C4 alkyl, hydroxy, C1 -C4 alkoxy, amino, N-(C1 -C4 alkyl)amino and N,
N-di(C1 -C4 alkyl)amino,
C4 -C8 cycloalkenyl optionally substituted with one to three
substituent(s) wherein said substituents are independently selected from
halo, C1 -C4 alkyl, hydroxy, C1 -C4 alkoxy, amino, N-(C1 -C4 alkyl)amino
and N, N-di(C1 -C4 alkyl)amino,
phenyl optionally substituted with one to three substituent(s)
wherein said substituents are independently selected from halo, C1 -C4
alkyl, hydroxy, C1 -C4 alkoxy, halo-substituted C1 -C4 alkyl, hydroxy-
substituted C1 -C4 alkyl, (C1 -C4 alkoxy)C1 -C4 alkyl, halo-substituted C1 -C4
alkoxy, amino, N-(C1 -C4 alkyl)amino, N, N-di(Ci -C4 alkyl)amino, [N-(C1 -
73
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
C4 alkyl)amino]C1 -C4 alkyl, [N, N-di(C1 -C4 alkyl)amino]C1 -C4 alkyl, N-(Ci
-C4 alkanoyl)amino, N-[C1 -C4 alkyl)(C1 -C4 alkanoyl)]amino, N-[(C1 -C4
alkyl)sulfony]amino, N-[(halo-substituted C1 -C4 alkyl)sulfonyl]amino, Cy -
C4 alkanoyl, carboxy, (Ci -C4 alkoxy)carbonyl, carbomoyl, [N-(Ci -C4
alky)amino]carbonyl, [N, N-di(C1 -C4 alkyl)amino]carbonyl, cyano, nitro,
mercapto, (Cy -C4 alkyl)thio, (Cy -C4 alkyl)sulfinyl, (Ci -C4 alkyl)sulfonyl,
aminosulfonyl, [N-(C1 -C4 alkyl)amino]sulfonyl and [N, N-di(C1 -C4
alkyl)amino]sulfonyl; and
heteroaryl selected from:
a 5-membered monocyclic aromatic ring having one hetero atom
selected from O, S and N and optionally containing one to three N atoms)
in addition to said hetero atom; or a 6-membered monocyclic aromatic ring
having one N atom and optionally containing one to four N atoms) in
addition to said N atom; and
said heteroaryl being optionally substituted with one to three
substituent(s) selected from X2o ;
R'39 and R'~° are independently selected from:
hydrogen,
halo,
C1 -C4 alkyl,
phenyl optionally substituted with one to three substituent(s)
wherein said substituents are independently selected from halo, C1 -C4
alkyl, hydroxy, C1 -C4 alkoxy, amino, N-(C1 -C4 alkyl)amino and N, N-di(C1
-C~ alkyl)amino,
or 8738 and 8139 can form, together with the carbon atom to which
they are attached, a C3 -C~ cycloalkyl ring;
m is 0, 1, 2, 3, 4 or 5; and
n is 0, 1, 2, 3 or 4.
[00093] Materials that can serve as a cyclooxygenase-2 selective
inhibitor of the present invention include indole compounds that are
described in U.S. Patent No. 6,300,363. Such indole compounds have the
formula shown below in formula XXVI1:
74
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
8141
8142
~4 XXVII
(X22) n
3-Q6
N
H
and the pharmaceutically acceptable salts thereof,
wherein:
L4 is oxygen or sulfur;
Y3 is a direct bond or C1_4 alkylidene;
Q6 is:
(a) C1_6 alkyl or halosubstituted Ci_6 alkyl, said alkyl being optionally
substituted with up to three substituents independently selected from
hydroxy, C1_4 alkoxy, amino and mono- or di-(C1_4 alkyl)amino,
(b) C3_~ cycloalkyl optionally substituted with up to three substituents
independently selected from hydroxy, C1_4 alkyl and C1_4 alkoxy,
(c) phenyl or naphthyl, said phenyl or naphthyl being optionally
substituted with up to four substituents independently selected from:
(c-1 ) halo, C1_4 alkyl, halosubstituted Ci_4 alkyl, hydroxy, C1_4 alkoxy,
halosubstituted C1_4 alkoxy, S(O)m 8143, SO2 NH2, S02 N(C1_4 alkyl)2,
amino, mono- or di-(C1_4 alkyl)amino, NHS02 8143, NHC(O)R143, CN, C02
H, C02 (C1_4 alkyl), C1_4 alkyl-OH, Ci_4 alkyl-OR143, CONH2, CONH(C1_4
alkyl), CON(C1_4 alkyl)2 and -O-Y-phenyl, said phenyl being optionally
substituted with one or two substituents independently selected from halo,
C1_4 alkyl, CF3, hydroxy, OR143, S(O)mR143, amino, mono- or dn(C1-4.
alkyl)amino and CN;
(d) a monocyclic aromatic group of 5 atoms, said aromatic group
having one heteroatom selected from O, S and N and optionally containing
up to three N atoms in addition to said heteroatom, and said aromatic
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
group being substituted with up to three substitutents independently
selected from:
(d-1 ) halo, Cy_4 alkyl, halosubstituted C1_4 alkyl, hydroxy, Ci_4 alkoxy,
halosubstituted C1_4 alkoxy, C1_4 alkyl-OH, S(O)m 8143, S02 NH2, S02 N(C1_
4 alkyl)2, amino, mono- or di-(C1_4 alkyl)amino, NHS02 8143, NHC(O)R143,
CN, C02 H, C02 (C1_4 alkyl), Ci_4 alkyl-OR143, CONH2, CONH(C1_4 alkyl),
CON(Ci_4 alkyl)2, phenyl, and mono-, di- or tri-substituted phenyl wherein
the substituent is independently selected from halo, CF3, C~_4 alkyl,
hydroxy, C1_4 alkoxy, OCF3, SR143, SO2 CH3, S02 NH2, amino, C1_~.
alkylamino and NHS02 8143;
(e) a monocyclic aromatic group of 6 atoms, said aromatic group
having one heteroatom which is N and optionally containing up to three
atoms in addition to said heteroatom, and said aromatic group being
substituted with up to three substituents independently selected from the
above group (d-1 );
R1~1 IS hydrogen or C1_g alkyl optionally substituted with a
substituent selected independently from hydroxy, 011as, vitro, amino,
mono- or dl-(C1_4 alkyl)amino, C02 H, C02 (C1_4 alkyl), CONH2, CONH(Cy_4
alkyl) and CON(C1_4 alkyl)2 ;
R~42 is:
(a) hydrogen,
(b) C1_4 alkyl,
(c) C(O)Rl4.s,
wherein 1145 is selected from:
(c-1 ) C1-22 alkyl or C2_22 alkenyl, said alkyl or alkenyl being optionally
substituted with up to four substituents independently selected from:
(c-1-1) halo, hydroxy, 01143' S(O)m R143~ vitro, amino, mono- or dl-(C1-4
alkyl)amino, NHS02 1143, C02 H, C02 (C1_4 alkyl), CONH2, CONH(C1_4
alkyl), CON(C1_4 alkyl)2, OC(O)R'43, thienyl, naphthyl and groups of the
following formulae:
76
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
(X22)n
NHS02 ~ NHS02
a
(X22)n ~ (X22)n
\ / a a
0
O
N/(CH2)p N/(CH2)p
, a
O
(CH2)q
(CH2)a
11
N/ ~Zii , and N Z
(c-2) C1_22 alkyl or C2_22 alkenyl, said alkyl or alkenyl being optionally
substituted with five to forty-five halogen atoms,
(c-3) -Y5-Cg_7 cycloalkyl or -Y5-C3_~ cycloalkenyl, said cycloalkyl
or cycloalkenyl being optionally substituted with up to three substituent
independently selected from:
(c-3-1 ) C1_4 alkyl, hydroxy, OR143' S(a)m R143~ amino, mono- or di-
(C1_4 alkyl)amino, CONH2, CONH(C1_4 alkyl) and CON(C1_4 alkyl)2,
(c-4) phenyl or naphthyl, said phenyl or naphthyl being optionally
substituted with up to seven (preferably up to seven) substituents
independently selected from:
77
~X22~ n
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
(c-4-1 ) halo, Ci_8 alkyl, C1_4 alkyl-OH, hydroxy, Ci_8 alkoxy,
halosubstituted Ci_$ alkyl, halosubstituted C1_$ alkoxy, CN, nitro, S(O)m
8143' SO2 NH2, SO2 NH(C1_4 alkyl), SO2 N(C1_4 alkyl)2, amino, C1-4
alkylamino, di-(C1_4 alkyl)amino, CONH2, CONH(C1_4 alkyl), CON(C1_4
alkyl)2, OC(O)R143, and phenyl optionally substituted with up to three
substituents independently selected from halo, C1_4 alkyl, hydroxy, OCH3,
CF3, OCF3, CN, nitro, amino, mono- or dl-(Ci_4 alkyl)amino, C02 H, C02
(C1_4 alkyl) and CONH2,
(c-5) a monocyclic aromatic group as defined in (d) and (e) above,
said aromatic group being optionally substituted with up to three
substituents independently selected from:
(c-5-1) halo, C1_8 alkyl, C1_4 alkyl-OH, hydroxy, C1_8 alkoxy, CF3,
OCF3, CN, nitro, S(O)m 8143, amino, mono- or di-(Ci_4 alkyl)amino, CONH2,
CONH(Ci_4 alkyl), CON(C1_4 alkyl)2, CO2 H and C02 (Ci_4 alkyl), and -Y-
phenyl, said phenyl being optionally substituted with up to three
substituents independently selected halogen, C1_4 alkyl, hydroxy, C1_4
alkoxy, CF3, OCF3, CN, nitro, S(O)m 8143, amino, mono- or di-(C1_4
alkyl)amino, C02 H, C02 (C1_4 alkyl), CONH2, CONH(C1_4 alkyl) and
CON(C1_4 alkyl)2,
(c-6) a group of the following formula:
(CH2)q
'Zi i
s
(CH2)n
X22 is halo, C1_4 alkyl, hydroxy, C1_4 alkoxy, halosubstitutued Ci_4
alkoxy, S(O)m 8143, amino, mono- or di-(C1_4 alkyl)amino, NHS02 8143,
nitro, halosubstitutued C1_4 alkyl, CN, C02 H, C02 (C1_4 alkyl), C1_4 alkyl-
OH, C1_ø alkylOR1431 CONH2, CONH(C1_4 alkyl) or CON(C1_4 alkyl)2 ;
8143 is C1-4 alkyl or halosubstituted C1_4 alkyl;
78
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
m is 0, 1 or 2; n is 0, 1, 2 or 3; p is 1, 2, 3, 4 or 5; p is 2 or 3;
Z~1 is oxygen, sulfur or NR144 ; and
8144 iS hydrogen, C1_6 alkyl, halosubstitutued C1_4 alkyl or-Y5-
phenyl, said phenyl being optionally substituted with up to two substituents
independently selected from halo, C1_4 alkyl, hydroxy, C1_4 alkoxy, S(O)m
R143~ amino, mono- or di-(C1_4 alkyl)amino, CF3, OCF3, CN and nitro;
with the proviso that a group of formula -Y5-Q is not methyl or
ethyl when X22 is hydrogen;
L4 is oxygen;
R'4~ is hydrogen; and
R~42 is acetyl.
[00094] Materials that can serve as a cyclooxygenase-2 selective
inhibitor of the present invention include aryl phenylhydrazides that are
described in U.S. Patent No. 6,077,869. Such aryl phenylhydrazides have
the formula shown below in formula XXVIII:
0
H
~N
XXVIII
i
Xz3 Ys
wherein:
X23 and Y6 are selected from hydrogen, halogen, alkyl, nitro, amino or
other oxygen and sulfur containing functional groups such as hydroxy,
methoxy and methylsulfonyl.
[00095] Materials that can serve as a cyclooxygenase-2 selective
inhibitor of the present invention include 2-aryloxy, 4-aryl furan-2-ones that
are described in U.S. Patent No. 6,140,515. Such 2-aryloxy, 4-aryl furan-
2-ones have the formula shown below in formula XXIX:
79
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
8146
R148y
XX~X
or a pharmaceutical salt thereof,
wherein:
8146 is selected from the group consisting of SCH3, -S(O)2 CHI
and -S(O)2 NH2 ;
R14' is selected from the group consisting of ORl5o, mono or di-
substituted phenyl or pyridyl wherein the substituents are selected from
the group consisting of methyl, chloro and F;
R15° is unsubstituted or mono or di-substituted phenyl or pyridyl
wherein the substituents are selected from the group consisting of methyl,
chloro and F;
8148 is H, C1_4 alkyl optionally substituted with 1 to 3 groups of F, CI
or Br; and
8149 is H, C1_4 alkyl optionally substituted with 1 to 3 groups of F, CI
or Br, with the proviso that 8148 and 8149 are not the same.
[00096] Materials that can serve as a cyclooxygenase-2 selective
inhibitor of the present invention include bisaryl compounds that are
described in U.S. Patent No. 5,994,379. Such bisaryl compounds have
the formula shown below in formula XXX:
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
(R151 ~0_i
113 8152
Y~ /
A
R 153
R 154
XXX
or a pharmaceutically acceptable salt, ester or tautomer thereof,
wherein:
Z13 is C or N;
when Z13 is N, 8151 represents H or is absent, or is taken in
conjunction with 8152 as described below:
when Z13 is C, 8151 represents H and 8152 is a moiety which has the
following characteristics:
(a) it is a linear chain of 3-4 atoms containing 0-2 double bonds,
which can adopt an energetically stable transoid configuration and if a
double bond is present, the bond is in the trans configuration,
(b) it is lipophilic except for the atom bonded directly to ring A,
which is either lipophilic or non-lipophilic, and
(c) there exists an energetically stable configuration planar with ring
A to within about 15 degrees;
or 8151 and 8152 are taken in combination and represent a 5- or 6-
membered aromatic or non-aromatic ring D fused to ring A, said ring D
containing 0-3 heteroatoms selected from O, S and N;
81
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
said ring D being lipophilic except for the atoms attached directly to
ring A, which are lipophilic or non-lipophilic, and said ring D having
available an energetically stable configuration planar with ring A to within
about 15 degrees;
said ring D further being substituted with 1 Ra group selected from
the group consisting of: C1_2 alkyl, -OC1_2 alkyl, -NHC1_2 alkyl, -N(C1_2
alkyl)2, -C(O)C1_2 alkyl, -S-C1_2 alkyl and -C(S)Ci_2 alkyl;
Y' represents N, CH or C-OC1_3 alkyl, and when Z13 is N, Y' can
also represent a carbonyl group;
8153 represents H, Br, CI or F; and
8154 represents H or CH3.
[00097] Materials that can serve as a cyclooxygenase-2 selective
inhibitor of the present invention include 1,5-diarylpyrazoles that are
described in U.S. Patent No. 6,028,202. Such 1,5-diarylpyrazoles have
the formula shown below in formula XXXI:
R 158
8160
R157~'
N N O Risi
XXXI
N
8156-
8159
wherein:
8155' 8156' R15~~ and R15$ are independently selected from the
groups consisting of hydrogen, C1_5 alkyl, C1_5 alkoxy, phenyl, halo,
82
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
hydroxy, Ci_5 alkylsulfonyl, C1_5 alkylthio, trihaloCl_5 alkyl, amino, vitro
and
2-quinolinylmethoxy;
R~59 IS hydrogen, C1_5 alkyl, trihaloCi_5 alkyl, phenyl, substituted
phenyl where the phenyl substitutents are halogen, C1_5 alkoxy, trihaloCl-s
alkyl or vitro or R'59 IS heteroaryl of 5-7 ring members where at least one of
the ring members is nitrogen, sulfur or oxygen;
8160 jS hydrogen, C1_5 alkyl, phenyl C1_5 alkyl, substituted phenyl C1_
5 alkyl where the phenyl substitutents are halogen, C1_5 alkoxy, trihaloCl-5
alkyl or vitro, or Rlso is C1_5 alkoxycarbonyl, phenoxycarbonyl, substituted
phenoxycarbonyl where the phenyl substitutents are halogen, Cy_5 alkoxy,
trihaloCl_5 alkyl or: vitro;
8161 is 01_10 alkyl, substituted C1-1o alkyl where the substituents are
halogen, trihaloCl_5 alkyl, C1_5 alkoxy, carboxy, C1_5 alkoxycarbonyl, amino,
C1_5 alkylamino, diCy_5 alkylamino, dIC1_5 alkylaminoC7_5 alkylamino, C1-s
alkylaminoCl_5 alkylamino or a heterocycle containing 4-8 ring atoms where
one more of the ring atoms is nitrogen, oxygen or sulfur, where said
heterocycle may be optionally substituted with C1_5 alkyl; or 8161 IS phenyl,
substituted phenyl (where the phenyl substitutents are one or more of C1-5
alkyl, halogen, Cy_5 alkoxy, trihaloCl_5 alkyl or vitro), or 8161 Is
heteroaryl
having 5-7 ring atoms where one or more atoms are nitrogen, oxygen or
sulfur, fused heteroaryl where one or more 5-7 membered aromatic rings
are fused to the heteroaryl; or
8161 jS NR163 8764 where 8163 and ~R164 are independently selected
from hydrogen and C1_5 alkyl or 8163 and 8164 may be taken together with
the depicted nitrogen to form a heteroaryl ring of 5-7 ring members where
one or more of the ring members is nitrogen, sulfur or oxygen where said
heteroaryl ring may be optionally substituted with C1_5 alkyl;
8162 is hydrogen, C1_5 alkyl, vitro, amino, and halogen;
and pharmaceutically acceptable salts thereof.
[00098] Materials that can serve as a cyclooxygenase-2 selective
inhibitor of the present invention include 2-substituted imidazoles that are
83
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
described in U.S. Patent No. 6,040,320. Such 2-substituted imidazoles
have the formula shown below in formula XXXII:
8166
8165
N
Rise XXXII
8164 N
wherein:
8164 is phenyl, heteroaryl wherein the heteroaryl contains 5 to 6 ring
atoms, or
substituted phenyl;
wherein the substituents are independently selected from one or
members of the group consisting of C1_5 alkyl, halogen, nitro,
trifluoromethyl and nitrite;
8165 is phenyl, heteroaryl wherein the heteroaryl contains 5 to 6 ring
atoms,
substituted heteroaryl;
wherein the substituents are independently selected from one or
more members of the group consisting Of C1_5 alkyl and halogen, or
substituted phenyl,
wherein the substituents are independently selected from one or
members of the group consisting of Ci_5 alkyl, halogen, nitro,
trifluoromethyl and nitrite;
8166 is hydrogen, SEM, C1_5 alkoxycarbonyl, aryloxycarbonyl,
arylCi_5 alkyloxycarbonyl, arylCi_5 alkyl, phthalimidoCi_5 alkyl, aminoCi-5
alkyl, diaminoCi_5 alkyl, succinimidoCi_5 alkyl, C1_5 alkylcarbonyl,
arylcarbonyl, C1_5 alkylcarbonylCi_5 alkyl, aryloxycarbonylCi_5 alkyl,
heteroarylCi_5 alkyl where the heteroaryl contains 5 to 6 ring atoms, or
substituted arylCi_5 alkyl,
84
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
wherein the aryl substituents are independently selected from one
or more members of the group consisting Of C1_5 alkyl, C1_5 alkoxy,
halogen, amino, C1_5 alkylamino, and diC1_5 alkylamino;
R'167 iS ~All~n -~CH165~~-X24 wherein:
A'~ is sulfur or carbonyl;
nis0orl;
q is 0-9;
X24 is selected from the group consisting of hydrogen, hydroxy,
halogen, vinyl, ethynyl, Cy_5 alkyl, C3_~ cycloalkyl, C1_5 alkoxy, phenoxy,
phenyl, arylCl_5 alkyl, amino, Cy_5 alkylamino, nitrite, phthalimido, amido,
phenylcarbonyl, C1_5 alkylaminocarbonyl, phenylaminocarbonyl, arylCl-s
alkylaminocarbonyl, C1_5 alkylthio, C1_5 alkylsulfonyl, phenylsulfonyl,
substituted sulfonamido,
wherein the sulfonyl substituent is selected from the group
consisting Of C1_5 alkyl, phenyl, araC1_5 alkyl, thienyl, furanyl, and
naphthyl;
substituted vinyl,
wherein the substituents are independently selected from one or
members of the group consisting of fluorine, bromine, chlorine and iodine,
substituted ethynyl,
wherein the substituents are independently selected from one or
more members of the group consisting of fluorine, bromine chlorine and
iodine,
substituted C1_5 alkyl,
wherein the substituents are selected from the group consisting of
one or more C1_5 alkoxy, trihaloalkyl, phthalimido and amino,
substituted phenyl,
wherein the phenyl substituents are independently selected from
one or more members of the group consisting Of C1_5 alkyl, halogen and
Cy_5 alkoxy,
substituted phenoxy,
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
wherein the phenyl substituents are independently selected from
one or more members of the group consisting of C1_5 alkyl, halogen and
Ci_5 alkoxy,
substituted C1_5 alkoxy,
wherein the alkyl substituent is selected from the group consisting of
phthalimido and amino,
substituted arylCl_5 alkyl,
wherein the alkyl substituent is hydroxyl,
substituted arylCi_5 alkyl,
wherein the phenyl substituents are independently selected from
one or more members of the group consisting Of C1_5 alkyl, halogen and
C1_5 alkoxy,
substituted amido,
wherein the carbonyl substituent is selected from the group
consisting Of C1_5 alkyl, phenyl, arylCl_5 alkyl, thienyl, furanyl, and
naphthyl,
substituted phenylcarbonyl,
wherein the phenyl substituents are independently selected from
one or members of the group consisting of C1_5 alkyl, halogen and C1_5
alkoxy,
substituted C1_5 alkylthio,
wherein the alkyl substituent is selected from the group consisting
of hydroxy and phthalimido,
substituted C1_5 alkylsulfonyl,
wherein the alkyl substituent is selected from the group consisting
of hydroxy and phthalimido,
substituted phenylsulfonyl,
wherein the phenyl substituents are independently selected from
one or members of the group consisting of bromine, fluorine, chlorine, C1_5
alkoxy and trifluoromethyl,
with the proviso:
86
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
if A11 is sulfur and X24 is other than hydrogen, Ci-s
alkylaminocarbonyl, phenylaminocarbonyl, arylCi_5 alkylaminocarbonyl, C1_
alkylsulfonyl or phenylsulfonyl, then q must be equal to or greater than 1;
if Aii is sulfur and q is 1, then X24 cannot be C1_2 alkyl;
5 if Aii is carbonyl and q is 0, then X24 cannot be vinyl, ethynyl, C1_5
alkylaminocarbonyl, phenylaminocarbonyl, arylCi_5 alkylaminocarbonyl,Ci-s
alkylsulfonyl or phenylsulfonyl;
if Aii is carbonyl, q is 0 and X24 is H, then 8166 is not SEM (2-
(trimethylsilyl)ethoxymethyl);
if n is 0 and q is 0, then X24 cannot be hydrogen;
and pharmaceutically acceptable salts thereof.
[00099] Materials that can serve as a cyclooxygenase-2 selective
inhibitor of the present invention include 1,3- and 2,3-diarylcycloalkano
and cycloalkeno pyrazoles that are described in U.S. Patent No.
6,083,969. Such 1,3- and 2,3-diarylpyrazole compounds have the general
formulas shown below in formulas XXXIII and XXXIV:
R 169
XXXIII
8168
87
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
8169
8168
I
wherein:
R16$ and 8169 are independently selected from the group consisting
of hydrogen, halogen, (Ci -C6)alkyl, (C1 -C6)alkoxy, nitro, amino, hydroxy,
trifluoro, -S(C1 -C6)alkyl, -SO(C1 -C6)alkyl and -S02 (C1 -C6)alkyl; and
the fused moiety M is a group selected from the group consisting of an
optionally substituted cyclohexyl and cycloheptyl group having the
formulae:
;173
8172
,or
172
wherein:
R17° is selected from the group consisting of hydrogen, halogen,
hydroxy and carbonyl;
88
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
or Ri'° and Ri" taken together form a moiety selected from the
group consisting of -OCOCH2 -, -ONH(CH3)COCH2 -, -
OCOCH= and -O-;
Ri" and R"2 are independently selected from the group consisting
of hydrogen, halogen, hydroxy, carbonyl, amino, (C1 -C6)alkyl, (C1 -
C6)alkoxy, =NOH, -NR"4 R175~ -~CH3, -OCH2 CH3, -OS02 NHC02
CH3, =CHC02 CH2 CH3, -CH2 C02 H, -CH2 C02 CH3, -CH2 C02 CH2
CH3, -CH2 CON(CH3)2, -CH2 CO2 NHCH3, -CHCHC02 CH2 CH3, -
OCON(CH3)OH, -C(COCH3)2, di(Ci -C6)alkyl and di(C1 -C6)alkoxy;
R"3 is selected from the group consisting of hydrogen, halogen,
hydroxy, carbonyl, amino, (C1 -C6)alkyl, (C1 -C6)alkoxy and optionally
substituted carboxyphenyl, wherein substituents on the carboxyphenyl
group are selected from the group consisting of halogen, hydroxy, amino,
(C1 -C6)alkyl and (C1 -C6)alkoxy;
or R~'2 and R"3 taken together form a moiety selected from the
group consisting of -O-and
F
R1'4 is selected from the group consisting of hydrogen, OH, -
OCOCH3, -COCH3 and (C1 -C6)alkyl; and
R1'5 is selected from the group consisting of hydrogen, OH, -
OCOCH3, -COCH3, (C1 -C6)alkyl, -CONH2 and -S02 CH3 ;
with the proviso that
if M is a cyclohexyl group, then R1'° through R1'3 may not all be
hydrogen; and
89
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
pharmaceutically acceptable salts, esters and pro-drug forms
thereof.
(000100] Materials that can serve as a cyclooxygenase-2 selective
inhibitor of the present invention include esters derived from indolealkanols
and novel amides derived from indolealkylamides that are described in
U.S. Patent No. 6,306,890. Such compounds have the general formula
shown below in formula XXXV:
0
8176
-X25
8177
XXXV
Rl7a
179
wherein:
8176 is C1 to C6 alkyl, C1 to C6 branched alkyl, C4 to C$ cycloalkyl,
C1 to C6 hydroxyalkyl, branched C1 to C6 hydroxyalkyl, hydroxy substituted
Ca. to C$ aryl, primary, secondary or tertiary C1 to C6 alkylamino, primary,
secondary or tertiary branched C1 to C6 alkylamino, primary, secondary or
tertiary C4 to C8 arylamino, C1 to C6 alkylcarboxylic acid, branched C1 to C6
alkylcarboxylic acid, C1 to C6 alkylester, branched C1 to C6 alkylester, C4 to
C$ aryl, C~ to C$ arylcarboxylic acid, C4 to C$ arylester, C4 to C$ aryl
substituted C1 to C6 alkyl, C4 to C$ heterocyclic alkyl or aryl with O, N or S
in the ring, alkyl-substituted or aryl-substituted C4 to C8 heterocyclic alkyl
or aryl with O, N or S in the ring, or halo-substituted versions thereof,
where halo is chloro, bromo, fluoro or iodo;
R1'7 is C1 to C6 alkyl, C1 to C6 branched alkyl, C4 to C$ cycloalkyl,
C4 to C$ aryl, C4 to C$ aryl-substituted C1 to C6 alkyl, C1 to C6 alkoxy, C1
to
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
C6 branched alkoxy, C4 to C8 aryloxy, or halo-substituted versions thereof
or R1" is halo where halo is chloro, fluoro, bromo, or iodo;
R1'$ is hydrogen, C1 to C6 alkyl or C1 to C6 branched alkyl;
Ri'9 is Ci to C6 alkyl, C4 to C$ aroyl, C4 to C8 aryl, C4 to Cs
heterocyclic alkyl or aryl with O, N or S in the ring, C4 to C$ aryl-
substituted
C1 to C6 alkyl, alkyl-substituted or aryl-substituted C4 to C8 heterocyclic
alkyl or aryl with O, N or S in the ring, alkyl-substituted C4 to C$ aroyl, or
alkyl-substituted C4 to Cs aryl, or halo-substituted versions thereof where
halo is chloro, bromo, or iodo;
n is 1, 2, 3, or 4; and
X25 is O, NH, or N-Ri$°, where Ri$° is C1 to C6 alkyl or C1
to C6
branched alkyl.
[000101 ] Materials that can serve as a cyclooxygenase-2 selective
inhibitor of the present invention include pyridazinone compounds that are
described in U.S. Patent No. 6,307,047. Such pyridazinone compounds
have the formula shown below in formula XXXVI:
8184 N 8181
~N~
XXXVI
8183
or a pharmaceutically acceptable salt, ester, or prodrug thereof,
wherein:
X26 is selected from the group consisting of O, S, -NR185, -NORa,
and -NNRb R~ ;
8185 is selected from the group consisting of alkenyl, alkyl, aryl,
arylalkyl, cycloalkenyl, cycloalkenylalkyl, cycloalkyl, cycloalkylalkyl,
heterocyclic, and heterocyclic alkyl;
Ra, Rb, and R° are independently selected from the group
consisting
of alkyl, aryl, arylalkyl, cycloalkyl, and cycloalkylalkyl;
91
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
R~81 is selected from the group consisting of alkenyl, alkoxy,
alkoxyalkyl, alkoxyiminoalkoxy, alkyl, alkylcarbonylalkyl, alkylsulfonylalkyl,
alkynyl, aryl, arylalkenyl, arylalkoxy, arylalkyl, arylalkynyl, arylhaloalkyl,
arylhydroxyalkyl, aryloxy, aryloxyhaloalkyl, aryloxyhydroxyalkyl,
arylcarbonylalkyl, carboxyalkyl, cyanoalkyl, cycloalkenyl, cycloalkenylalkyl,
cycloalkyl, cycloalkylalkyl, cycloalkylidenealkyl, haloalkenyl,
haloalkoxyhydroxyalkyl, haloalkyl, haloalkynyl, heterocyclic, heterocyclic
alkoxy, heterocyclic alkyl, heterocyclic oxy, hydroxyalkyl,
hydroxyiminoalkoxy, -(CH2)" C(O)Rls6, -(CH2)n CH(OH)R's6, -(CH2)"
C(NORd)Rla6, -(CH2)" CH(NORd)R7a6, -(CH2)n CH(NRd Re)Ris6, -R1s'
Rl8s' -(CH2)" C=CR~as~ -(CH2)r, [CH(CX26~3)]m (CI-la)p Riss~ -(CH2)"
(C~(26~2)m (CH2)p Rias~ and -(CH2)" (CHX26~)m (CH2)m Risa ;
Riss is selected from the group consisting of hydrogen, alkenyl,
alkyl, alkynyl, aryl, arylalkyl, cycloalkenyl, cycloalkyl, haloalkenyl,
haloalkyl,
haloalkynyl, heterocyclic, and heterocyclic alkyl;
R's' is selected from the group consisting of alkenylene, alkylene,
halo-substituted alkenylene, and halo-substituted alkylene;
Rlss is selected from the group consisting of hydrogen, alkenyl,
alkyl, alkynyl, aryl, arylalkyl, cycloalkyl, cycloalkenyl, haloalkyl,
heterocyclic, and heterocyclic alkyl;
Rd and Re are independently selected from the group consisting of
hydrogen, alkenyl, alkyl, alkynyl, aryl, arylalkyl, cycloalkenyl, cycloalkyl,
haloalkyl, heterocyclic, and heterocyclic alkyl;
X26 is halogen;
m is an integer from 0-5;
n is an integer from 0-10; and
p is an integer from 0-10; and
Rise, R~s3, and Rlsa. are independently selected from the group
consisting of hydrogen, alkenyl, alkoxyalkyl, alkoxyiminoalkoxy,
alkoxyiminoalkyl, alkyl, alkynyl, alkylcarbonylalkoxy, alkylcarbonylamino,
alkylcarbonylaminoalkyl, aminoalkoxy, aminoalkylcarbonyloxyalkoxy
aminocarbonylalkyl, aryl, arylalkenyl, arylalkyl, arylalkynyl,
92
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
carboxyalkylcarbonyloxyalkoxy, cyano, cycloalkenyl, cycloalkyl,
cycloalkylidenealkyl, haloalkenyloxy, haloalkoxy, haloalkyl, halogen,
heterocyclic, hydroxyalkoxy, hydroxyiminoalkoxy, hydroxyiminoalkyl,
mercaptoalkoxy, nitro, phosphonatoalkoxy, Y8, and Z14;
provided that one of Ri82, R183~ or Ri84 must be Z14, and further
provided that only one of Rls2, Ri83, or Ri84 Is Z14;
Z14 is selected from the group consisting of:
X28
X28
X27 8190 and
X27 8190
S
2~ is selected from the group consisting of S(O)2, S(O)(NRi91), S(O),
Se(O)2, P(O)(OR192), and P(O)(NRi93 8194);
X2$ is selected from the group consisting of hydrogen, alkenyl, alkyl,
alkynyl and halogen;
Ri9° is selected from the group consisting of alkenyl, alkoxy,
alkyl,
alkylamino, alkylcarbonylamino, alkynyl, amino, cycloalkenyl, cycloalkyl,
dialkylamino, -NHNH2, and -NCHN(Ri91)Ri92 ;
8191' 8192' R193~ and Ri94 are independently selected from the group
consisting of hydrogen, alkyl, and cycloalkyl, or Ri93 and 8194 can be taken
together, with the nitrogen to which they are attached, to form a 3-6
membered ring containing 1 or 2 heteroatoms selected from the group
consisting of O, S, and NRi$$ ;
Y8 is selected from the group consisting of -ORi95, -SRi95, -
e(R197)(R198)R195' -C(O)R195' -C(O)OR195' -N(R197)C(O)R195~ -
NC(R19')R195~ and -N(R197)R195 ;
Ri95 is selected from the group consisting of hydrogen, alkenyl,
alkoxyalkyl, alkyl, alkylthioalkyl, alkynyl, cycloalkenyl, cycloalkenylalkyl,
93
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
cycloalkyl, cycloalkylalkyl, aryl, arylalkyl, heterocyclic, heterocyclic
alkyl,
hydroxyalkyl, and NR199 R2oo ; and
Ri9', 8198, 8199, and R2°° are independently selected from
the group
consisting of hydrogen, alkenyl, alkoxy, alkyl, cycloalkenyl, cycloalkyl,
aryl,
arylalkyl, heterocyclic, and heterocyclic alkyl.
(000102] Materials that can serve as a cyclooxygenase-2 selective
inhibitor of the present invention include benzosulphonamide derivatives
that are described in U.S. Patent No. 6,004,948. Such
benzosulphonamide derivatives have the formula shown below in formula
XXXVII:
8201
1 12
\ / 5 XXXVII
\S/ D
8206
H
herein:
A12 denotes oxygen, sulphur or NH;
R2°1 denotes a cycloalkyl, aryl or heteroaryl group optionally
mono-
or polysubstituted by halogen, alkyl, CF3 or alkoxy;
D5 denotes a group of formula XXXVIII or XXXIX:
S (\m 8202
XXXVIII
8203 ,
or
S(o)m
R202~ XXXIX
Z15
94
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
R2°2 and R2os independently of each other denote hydrogen, an
optionally polyfluorinated alkyl radical, an aralkyl, aryl or heteroaryl
radical
or a radical (CH2)n -X29; or
8202 and R2°3 together with the N-atom denote a three- to seven-
membered, saturated, partially or totally unsaturated heterocycle with one
or more heteroatoms N, O, or S, which may optionally be substituted by
oxo, an alkyl, alkylaryl or aryl group or a group (CH2)" -X29, R2°2'
denotes
hydrogen, an optionally polyfluorinated alkyl group, an aralkyl, aryl or
heteroaryl group or a group (CH2)" -X29
wherein:
X29 denotes halogen, N02, -OR2°4, -COR2°4, -C02 R2o4~ -
OCO2 8204'-CN,-CONR2o4 OR2o5, -CONR204 8205' -SR204'
-
S(O)R204'S(O)2R204' -NR2o4 -NHC(O)R2o4, -NHS(O)2
- R25~ 8204;
Z15 denotes-CH2-, -CH2 -CH2 -CH2 -CH2 -CH2 -, -CH2
-, -
CH=CH-, -CH=CH-CH2 -, -CH2 -CO-, -CO-CH2 -, -
NHCO-, -CONH-, -NHCH2 -, -CH2 NH-, -N=CH-, -NHCH-,
-CH2-CH2-NH-, -CH=CH-, >N-R2o3, >C=O, >S(O)rr,;
8204 and R2°5 independently of each other denote hydrogen, alkyl,
aralkyl or aryl;
n is an integer from 0 to 6;
R2os is a straight-chained or branched C1_4 -alkyl group which may
optionally be mono- or polysubstituted by halogen or alkoxy, or R2°s
denotes CF3; and
m denotes an integer from 0 to 2;
with the proviso that A12 does not represent O if R2°s denotes CF3;
and the pharmaceutically acceptable salts thereof.
[000103] Cox-2 selective inhibitors that are useful in the subject method
and compositions can include the compounds that are described in U.S.
Patent Nos. 6,169,188, 6,020,343, 5,981,576 ((methylsulfonyl)phenyl
furanones); U.S. Patent No. 6,222,048 (diaryl-2-(5H)-furanones); U.S.
Patent No. 6,057,319 (3,4-diaryl-2-hydroxy-2,5-dihydrofurans); U.S. Patent
No. 6,046,236 (carbocyclic sulfonamides); U.S. Patent Nos. 6,002,014 and
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
5,945,539 (oxazole derivatives); and U.S. Patent No. 6,359,182 (C-nitroso
compounds).
[000104] Cyclooxygenase-2 selective inhibitors that are useful in the
present invention can be supplied by any source as long as the
cyclooxygenase-2-selective inhibitor is pharmaceutically acceptable.
Cyclooxygenase-2-selective inhibitors can be isolated and purified from
natural sources or can be synthesized. Cyclooxygenase-2-selective
inhibitors should be of a quality and purity that is conventional in the trade
for use in pharmaceutical products.
[000105] In an embodiment of the present method, the Cox-2 inhibitor is
administered in combination with a glucocorticoid. Glucocorticoids that are
useful in the present invention are steroid hormones that are produced by
the adrenal cortex that help the body of a subject respond to stress and
fatigue by increasing metabolism and inhibiting the inflammatory response.
Examples of useful glucocorticoids include mometasone, fluticasone-17-
proprionate, budesonide, beclomethasone, betamethasone, methyl-
prednisolone, dexamethasone, prednisolone, hydrocortisone (cortisol),
triamcinolone, cortisone, corticosterone and prednisone. Each of these
glucocorticoids can be supplied in the form of a salt, or prodrug, if
desirable. Also included in the meaning of glucocorticoids in the present
invention, are non-steroidal GC mimics that are not dissociated, and
steroidal and non-steroidal GC analogs and mimics, respectively, that are
dissociated. When the term "dissociated" is used herein to describe
glucocorticoid analogs and mimics, what is meant are steroidal or non-
steroidal glucocorticoid analogs or mimics, respectively, that retain anti-
inflammatory/immunosuppressive efficacy, but manifest the reduction of
one or multiple side effects.
[000106] Hydrocortisone, also known as cortisol, is a steroid with
glucocorticoid activity and some mineralocorticoid effects. In addition to all
conventional uses for a glucocorticoid, it is indicated for septic shock,
adrenal insufficiency, congenital adrenal hyperplasia and allergic reaction,
and is available as hydrocortisone, hydrocortisone acetate, hydrocortisone
96
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
butyrate, hydrocortisone cypionate, hydrocortisone probutate,
hydrocortisone sodium phosphate, hydrocortisone sodium succinate, and
hydrocortisone valerate. Dose, depending on disease, is 20 - 240 mg/day.
[000107] Beclomethasone is available as beclomethasone dipropionate.
In addition to all conventional uses for a glucocorticoid, it is used for
rhinitis, to prevent recurrence of nasal polyps following surgical removal,
and for bronchial asthma. Dosage for adults and children over 12 years
old, administered by inhalation, is from 84 micrograms/day to 840
micrograms/day.
[000108] Cortisone is available as cortisone acetate. In addition to all
conventional uses for a glucocorticoid, it is used in replacement therapy in
chronic cortical insufficiency, and on a short-term for inflammatory or
allergic disorders. Dosage for initial treatment, or during crisis, is from 25
to 300 mg/day; as an inflammatory is 25 - 150 mg/day; and for acute
rheumatic fever is 200 mg/day. Maintenance dose is 0.5 to 0.75
mg/kg/day.
[000109] Dexamethasone is available as dexamethasone,
dexamethasone sodium phosphate, and dexamethasone acetate. In
addition to all conventional uses for a glucocorticoid, it is used for acute
allergic disorders, to test for adrenal cortical hyperfunction, cerebral
edema due to brain tumor, craniotomy, or head injury. Dosage is initially
0.75 - 9 mg/day, gradually reduced to a maintenance dose of 0.5 - 3
mg/day.
[000110] Methylprednisolone is available as methylprednisolone,
' 25 methylprednisolone acetate, and methylprednisolone sodium succinate. In
addition to all conventional uses for a glucocorticoid, it is used for
rheumatoid arthritis, severe hepatitis due to alcoholism, within 8 hr of
severe spinal cord injury (to improve neurologic function), and for septic
shock. Dosage for adults for rheumatoid arthritis is 6 - 16 mg/day,
decreased gradually; for acute indications is 20 - 96 mg/day, decreasing to
a maintenance dosage of 8 - 20 mg/day.
97
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
[000111 ] Betamethasone is available as betamethasone, betamethasone
dipropionate, betamethasone sodium phosphate, betamethasone acetate,
and betamethasone valerate. In addition to all conventional uses for a
glucocorticoid, it is used for prevention of respiratory distress syndrome is
premature infants. Dosage is from 0.5 to 9 mg/day, with dosages adjusted
downward to maintenance level.
[000112] Glucocorticoids that are useful in the present invention can be
of any purity or grade, as long as the preparation is of a quality suitable
for
pharmaceutical use. The glucocorticoid can be provided in pure form, or it
can be accompanied with impurities or commonly associated compounds
that do not affect its physiological activity or safety. The glucocorticoid
can
be supplied as a pure compound, or in the form of a pharmaceutically
active salt. The glucocorticoid can be supplied in the form of a prodrug, an
isomer, a racemic mixture, or in any other chemical form or combination
that, under physiological conditions, provides the glucocorticoid.
[000113] The term "subject", for purposes of treatment, includes any
vertebrate. The subject is typically a mammal. "Mammal", as that term is
used herein, refers to any animal classified as a mammal, including
humans, domestic and farm animals, and zoo, sports, or pet animals, such
as dogs, horses, cats, cattle, etc. Preferably, the mammal is a human.
[000114] Diseases and disorders that~are amenable to prevention or
treatment by the present methods and compositions are "T cell mediated
inflammatory/autoimmune diseases and disorders". As those-terms are
used herein, they should be understood to mean those diseases or
disorders that are associated with T cell-mediated inflammatory processes
or T cell mediated autoimmune processes. In particular, the present
invention is useful for diseases or disorders that are mediated by activated
circulatory T cells that are present in the blood, the spleen and the lymph
nodes. The benefits of the present invention are particularly useful in
those subjects having a deficiency of glucocorticoid regulation of immune
response.
98
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
[000115] T cells can be activated by contact with a T cell activating
agent. Such agents include exogenous or endogenous T cell activating
antigens (attached to suitable presenter cells), or can be T cell-specific
antibodies, such as a CD3s antibody.
[000116] T cell-mediated inflammatory/autoimmune diseases and
disorders have been discussed above, and examples include, without
limitation, graft vs. host disease, toxic shock syndrome, bacterial sepsis,
viral sepsis, food poisoning (superantigen mediated), transplant rejection,
immunosuppression using anti-CD3 antibodies or other compounds '
(OKT-3, etc), multiple sclerosis, systemic lupus erythematosus,
rheumatoid arthritis, and inflammatory bowel disease.
[000117] As used herein, the terms "subject in need of such treatment"
refer to a subject having some type of glucocorticoid regulation deficiency,
where the subject is suffering from, or at risk of suffering from, symptoms
associated with a T cell mediated inflammatory/autoimmune disease
and/or disorder. In some embodiments of the invention, the subject in
need of such treatment is one that is already receiving treatment with
glucocorticoids.
[000118] When it is said that the subject has a "glucocorticoid regulation
deficiency", it is meant that the subject has a glucocorticoid resistance, a
glucocorticoid insufficiency, or has experienced a T cell activating stimulus
of a strength sufficient to overwhelm the subject's endogenous
glucocorticoid regulation of the production of inflammation and immune-
related compounds, such as eicosanoids, cytokines, and associated
enzymes and other compounds.
[000119] Clinical syndromes of glucocorticoid resistance can be familial
or acquired, and can be generalized or tissue-specific. Examples of
generalized glucocorticoid resistance include generalized inherited
glucocorticoid resistance (GIGR), and acquired generalized glucocorticoid
resistance, which can occur in a subgroup of patients with acquired
immunodeficiency syndrome (AIDS). Subjects could show a
glucocorticoid resistance on account of an abnormal GRa/GRa ratio, or
99
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
due to resistance developed in response to either chronic inflammatory
stimuli or chronic GC treatment. Glucocorticoid resistance can be
iatrogenic.
[000120] Subjects can show glucocorticoid insufficiency on account of
primary or chronic adrenocortical insufficiency (Addison's disease), or
autoimmune processes.
[000121] Glucocorticoid regulation deficiency also includes cases where
subjects having an otherwise normally functioning T cell-mediated immune
response are challenged with a T cell activating stimulus, such as are
present in toxic shock, a graft vs. host response, immune response
triggered by trauma, or infectious disease, that is sufficiently strong that
it
overwhelms the GC/GR regulatory system and causes hyper-production of
Cox-2.
[000122] In an embodiment of the present method, a subject having a
glucocorticoid regulation deficiency can be prevented from experiencing,
or treated for the symptoms of, T cell mediated inflammatory/autoimmune
diseases and disorders. The method comprises administering to a subject
in need of such prevention or treatment an effective amount of a
cyclooxygenase-2 inhibitor or prodrug thereof. The Cox-2 inhibitor can be
administered to the subject alone, or in combination with a glucocorticoid.
In preferred embodiments, the effective amount constitutes a
therapeutically effective amount. For methods of prevention, the subject is
any human or animal subject, and preferably is a subject that is in need of
prevention and/or treatment of a T cell mediated inflammatory/autoimmune
disease or disorder. The subject may be at risk due to genetic
predisposition, sedentary lifestyle, diet, exposure to disorder-causing
agents, exposure to traumatic event, exposure to pathogenic agents and
the like.
[000123] In another embodiment'of the present method, the subject is
treated with a cyclooxygenase-2 inhibitor or prodrug thereof and a
glucocorticoid. In one embodiment, the subject is treated with an amount
of a Cox-2 inhibitor and an amount of a glucocorticoid, where the amount
100
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
of the Cox-2 inhibitor and the amount of the glucocorticoid together
provide a dosage or amount of the combination that is sufficient to
constitute an effective amount of the combination. In preferred
embodiments, the effective amount is a therapeutically effective amount.
[000124] As used herein, an "effective amount" means the dose or
effective amount to be administered to a patient and the frequency of
administration to the subject which is readily determined by one of ordinary
skill in the art, by the use of known techniques and by observing results
obtained under analogous circumstances. The dose or effective amount
to be administered to a patient and the frequency of administration to the
subject can be readily determined by one of ordinary skill in the art by the
use of known techniques and by observing results obtained under
analogous circumstances. In determining the effective amount or dose, a
number of factors are considered by the attending diagnostician, including,
but not limited to, the potency and duration of action of the compounds
used, the nature and severity of the illness to be treated, as well as on the
sex, age, weight, general health and individual responsiveness of the
patient to be treated, and other relevant circumstances.
[000125] The phrase "therapeutically-effective" indicates the capability of
an agent to prevent, or mitigate the severity of, the disorder, while avoiding
adverse side effects typically associated with alternative therapies. The
phrase "therapeutically-effective" is intended to qualify the amount of one
or more agents for use in the therapy which will achieve the goal of
improvement in the severity of symptoms associated with T cell mediated
inflammatory/autoimmune diseases or disorders, while limiting adverse
side effects typically associated with alternative therapies.
[000126] Those skilled in the art will appreciate that dosages may also be
determined with guidance from Goodman & Goldman's The
Pharmacological Basis of Therapeutics, Ninth Edition (1996), Appendix II,
pp.1707-1711.
[000127] The amount of Cox-2 inhibitor that is used in the subject
method may be an amount that is sufficient to constitute an effective
101
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
amount. Preferably, such amount would be a therapeutically effective
amount. The therapeutically effective amount can also be considered to
be a maximally saturating amount, or, alternatively, as the maximum
amount that can be administered while avoiding 'the incidence of
gastrointestinal ulcers caused by crossover Cox-1 inhibition.
[000128] In an embodiment of the present method where the Cox-2
inhibitor is a Cox-2 selective inhibitor, the amount of Cox-2 selective
inhibitor that is used in the novel method of treatment preferably ranges
from about 0.01 to about 100 milligrams per day per kilogram of body
weight of the subject (mg/day~kg), more preferably from about 0.1 to about
50 mg/day~kg, even more preferably from about 1 to about 20 mg/day~kg.
[000129] When the Cox-2 selective inhibitor comprises rofecoxib, it is
preferred that the amount used is within a range of from about 0.15 to
about 1.0 mglday~kg, and even more preferably from about 0.18 to about
0.4 mg/day~kg.
[000130] When the Cox-2 selective inhibitor comprises etoricoxib, it is
preferred that the amount used is within a range of from about 0.5 to about
5 mglday~kg, and even more preferably from about 0.8 to about 4
mg/day~kg.
[000131 ] When the Cox-2 selective inhibitor comprises celecoxib, it is
preferred that the amount used is within a range of from about 1 to about
10 mg/day~kg, even more preferably from about 1.4 to about 8.6
mg/day~kg, and yet more preferably from about 2 to about 3 mg/day~kg.
[000132] When the Cox-2 selective inhibitor comprises valdecoxib or
parecoxib sodium, it is preferred that the amount used is within a range of
from about 0.1 to about 3 mg/day~kg, and even more preferably from about
0.3 to about 1 mglday~kg.
[000133] In those embodiments of the present invention where a
glucocorticoid is administered in combination with the cyclooxygenase-2
inhibitor, the amount of the glucocorticoid that is administered is an
effective amount. The amount of a glucocorticoid that constitutes an
102
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
effective amount depends upon the type of glucocorticoid that is used and
the route of administration. The effective amount for commercially
available glucocorticoid preparations is provided in the prescribing
information that is available from the manufacturers and suppliers of the
particular glucocorticoid of interest. By way of example, equivalent
dosages (expressed in milligrams) have been determined for
betamethasone (0.6 - 0.75 mg), dexamethasone (0.75 mg), hydrocortisone
(20), methylprednisolone (4), prednisolone (5), and prednisone (5). See
Am. Soc, of Health System Pharmacists, Dexamethasone Sodium
Phosphate for Injection, at
http://www.ashp.org/shortage/dexamethasome.html, on 05/08/2002.
[000134] The frequency of dose will depend upon the half-life of the
cyclooxygenase-2 inhibitor. If the Cox-2 inhibitor has a short half-life (e.g.
from about 2 to 10 hours) it may be necessary to give one or more doses
per day. Alternatively, if the Cox-2 inhibitor has a long half-life (e.g. from
about 2 to about 15 days) it may only be necessary to give a dosage once
per day, per week, or even once every 1 or 2 months. A preferred dosage
rate is to administer the dosage amounts described above to a subject
once per day.
[000135] For the purposes of calculating and expressing a dosage rate,
all dosages that are expressed herein are calculated on an average
amount-per-day basis irrespective of the dosage rate. For example, one
100 mg dosage of an ingredient taken once every two days would be
expressed as a dosage rate of 50 mg/day. Similarly, the dosage rate of an
ingredient where 50 mg is taken twice per day would be expressed as a
dosage rate of 100 mg/day.
[000136] For the purposes of calculation of a dosage rate for the present
method, the weight of an adult human is assumed to be 70 kg.
[000137] In the present method, and in the subject compositions, the
Cox-2 inhibitor may be administered alone, or in combination with a
glucocorticoid. When the Cox-2 inhibitor is a Cox-2 selective inhibitor, it is
preferred that the weight ratio of the amount of the amount of Cox-2
103
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
selective inhibitor to the amount of the glucocorticoid that is administered
to the subject is within a range of from about 0.03:1 to about 35,000:1,
more preferred is a range of from about 0.3:1 to about 14,000:1, even
more preferred is a range of from about 0.5:1 to about 100:1.
[000138] The combination of a Cox-2 inhibitor and a glucocorticoid can
be supplied in the form of a novel therapeutic composition that is believed
to be within the scope of the present invention. The relative amounts of
each component in the therapeutic composition may be varied and may be
as described just above. The Cox-2 inhibitor and the glucocorticoid that
are described above can be provided in the therapeutic composition so
that the preferred amounts of each of the components are supplied by a
single dosage, a single injection or a single capsule for example, or, by up
to four, or more, single dosage forms.
[000139] When the novel combination is supplied along with a
pharmaceutically acceptable carrier, a pharmaceutical composition is
formed. A pharmaceutical composition of the present invention is directed
to a composition suitable for the prevention or treatment of T cell mediated
inflammatory/autoimmune diseases and disorders.
[000140] The pharmaceutical composition comprises a pharmaceutically
acceptable carrier, a cyclooxygenase-2 inhibitor and a glucocorticoid.
[000141 ] Pharmaceutically acceptable carriers include, but are not limited
to, physiological saline, Ringer's solution, phosphate solution or buffer,
buffered saline and other carriers known in the art. Pharmaceutical
compositions may also include stabilizers, anti-oxidants, colorants, and
diluents. Pharmaceutically acceptable carriers and additives are chosen
such that side effects from the pharmaceutical compounds) are minimized
and the performance of the compounds) is not canceled or inhibited to
such an extent that treatment is ineffective.
[000142] The term "pharmacologically effective amount" shall mean that
amount of a drug or pharmaceutical agent that will elicit the biological or
medical response of a tissue, system, animal or human that is being
104
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
sought by a researcher or clinician. This amount can be a therapeutically
effective amount.
[000143] The term "pharmaceutically acceptable" is used herein to mean
that the modified noun is appropriate for use in a pharmaceutical product.
Pharmaceutically acceptable cations include metallic ions and organic
ions. More preferred metallic ions include, but are not limited to,
appropriate alkali metal salts, alkaline earth metal salts and other
physiological acceptable metal ions. Exemplary ions include aluminum,
calcium, lithium, magnesium, potassium, sodium and zinc in their usual
valences. Preferred organic ions include protonated tertiary amines and
quaternary ammonium cations, including in part, trimethylamine,
diethylamine, N,N'-dibenzylethylenediamine, chloroprocaine, choline,
diethanolamine, ethylenediamine, meglumine (N-methylglucamine) and
procaine. Exemplary pharmaceutically acceptable acids include, without
limitation, hydrochloric acid, hydroiodic acid, hydrobromic acid, phosphoric
acid, sulfuric acid, methanesulfonic acid, acetic acid, formic acid, tartaric
acid, malefic acid, malic acid, citric acid, isocitric acid, succinic acid,
lactic
acid, gluconic acid, glucuronic acid, pyruvic acid oxaloacetic acid, fumaric
acid, propionic acid, aspartic acid, glutamic acid, benzoic acid, and the
like.
[000144] Also included in the combination of the invention are the
isomeric forms and tautomers and the pharmaceutically acceptable salts
of cyclooxygenase-2 inhibitors and glucocorticoids. Illustrative
pharmaceutically acceptable salts are prepared from formic, acetic,
propionic, succinic, glycolic, gluconic, lactic, malic, tartaric, citric,
ascorbic,
glucuronic, malefic, fumaric, pyruvic, aspartic, glutamic, benzoic,
anthranilic, mesylic, stearic, salicylic, p-hydroxybenzoic, phenylacetic,
mandelic, embonic (pamoic), methanesulfonic, ethanesulfonic,
benzenesulfonic, pantothenic, toluenesulfonic, 2-hydroxyethanesulfonic,
sulfanilic, cyclohexylaminosulfonic, algenic, (i-hydroxybutyric, galactaric
and galacturonic acids.
105
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
[000145] Suitable pharmaceutically acceptable base addition salts of
compounds of the present invention include metallic ion salts and organic
ion salts. More preferred metallic ion salts include, but are not limited to,
appropriate alkali metal (group la) salts, alkaline earth metal (group Ila)
salts and other physiological acceptable metal ions. Such salts can be
made from the ions of aluminum, calcium, lithium, magnesium, potassium,
sodium and zinc. Preferred organic salts can be made from tertiary amines
and quaternary ammonium salts, including in part, trimethylamine,
diethylamine, N,N'-dibenzylethylenediamine, chloroprocaine, choline,
diethanolamine, ethylenediamine, meglumine (N-methylglucamine) and
procaine. All of the above salts can be prepared by those skilled in the art
by conventional means from the corresponding compound of the present
invention.
[000146] The terms "treating" or "to treat," mean to alleviate signs and
symptoms, eliminate the causation either on a temporary or permanent
basis, prevent or slow the appearance of symptoms, or to retard or halt
disease progression. The term "treatment" includes alleviation, elimination
of causation of, or prevention of symptoms associated with, but not limited
to, any of the diseases or disorders described herein, and also retarding or
halting of disease progression for these diseases or disorders.
[000147] The subject pharmaceutical compositions may be administered
enterally and parenterally. Parenteral administration includes
subcutaneous, intramuscular, intradermal, intramammary, intravenous,
and other administrative methods known in the art. Enteral administration
includes solution, tablets, sustained release capsules, enteric coated
capsules, and syrups. When administered, the pharmaceutical
composition may be at or near body temperature.
[000148] The phrases "combination therapy", "co-administration",
"administration with", or "co-therapy", in defining the use of a
cyclooxygenase-2 inhibitor agent and a glucocorticoid agent, is intended to
embrace administration of each agent in a sequential manner in a regimen
that will provide beneficial effects of the drug combination, and is intended
106
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
as well to embrace co-administration of these agents in a substantially
simultaneous manner, such as in a single capsule or dosage device
having a fixed ratio of these active agents or in multiple, separate capsules
or dosage devices for each agent, where the separate capsules or dosage
devices can be taken together contemporaneously, or taken within a
period of time sufficient to receive a beneficial effect from both of the
constituent agents of the combination.
[000149] Although the combination of the present invention may include
administration of a cyclooxygenase-2 inhibitor component and a
glucocorticoid component within an effective time of each respective
component, it is preferable to administer both respective components
contemporaneously, and more preferable to administer both respective
components in a single delivery dose.
[000150] In particular, the compositions and pharmaceutical
compositions of the present invention can be administered orally, for
example, as tablets, coated tablets, dragees, troches, lozenges, aqueous
or oily suspensions, dispersible powders or granules, emulsions, hard or
soft capsules, or syrups or elixirs. Compositions intended for oral use may
be prepared according to any method known in the art for the manufacture
of pharmaceutical compositions and such compositions may contain one
or more agents selected from the group consisting of sweetening agents,
flavoring agents, coloring agents and preserving agents in order to provide
pharmaceutically elegant and palatable preparations. Tablets contain the
active ingredient in admixture with non-toxic pharmaceutically acceptable
excipients, which are suitable for the manufacture of tablets. These
excipients may be, for example, inert diluents, such as calcium carbonate,
sodium carbonate, lactose, calcium phosphate or sodium phosphate;
granulating and disintegrating agents, for example, maize starch, or alginic
acid; binding agents, for example starch, gelatin or acacia, and lubricating
agents, for example magnesium stearate, stearic acid or talc. The tablets
may be uncoated or they may be coated by known techniques to delay
disintegration and adsorption in the gastrointestinal tract and thereby
107
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
provide a sustained action over a longer period. For example, a time delay
material such as glyceryl monostearate or glyceryl distearate may be
employed.
[000151] Formulations for oral use may also be presented as hard gelatin
capsules wherein the active ingredients are mixed with an inert solid
diluent, for example, calcium carbonate, calcium phosphate or kaolin, or
as soft gelatin capsules wherein the active ingredients are present as
such, or mixed with water or an oil medium, for example, peanut oil, liquid
paraffin, or olive oil.
[000152] Aqueous suspensions can be produced that contain the active
materials in admixture with excipients suitable for the manufacture of
aqueous suspensions. Such excipients are suspending agents, for
example, sodium carboxymethylcellulose, methylcellulose,
hydroxypropylmethyl-cellulose, sodium alginate, polyvinylpyrrolidone gum
tragacanth and gum acacia; dispersing or wetting agents may be naturally-
occurring phosphatides, for example lecithin, or condensation products of
an alkylene oxide with fatty acids, for example polyoxyethylene stearate,
or condensation products of ethylene oxide with long chain aliphatic
alcohols, for example heptadecaethyleneoxycetanol, or condensation
products of ethylene oxide with partial esters derived from fatty acids and
a hexitol such as polyoxyethylene sorbitol monooleate, or condensation
products of ethylene oxide with partial esters derived from fatty acids and
hexitol anhydrides, for example polyoxyethylene sorbitan monooleate.
[000153] The aqueous suspensions may also contain one or more
preservatives, for example, ethyl or n-propyl p-hydroxybenzoate, one or
more coloring agents, one or more flavoring agents, or one or more
sweetening agents, such as sucrose or saccharin.
[000154] Oily suspensions may be formulated by suspending the active
ingredients in an omega-3 fatty acid, a vegetable oil, for example, arachis
oil, olive oil, sesame oil or coconut oil, or in a mineral oil such as liquid
paraffin. The oily suspensions may contain a thickening agent, for
example, beeswax, hard paraffin or cetyl alcohol.
108
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
[000155] Sweetening agents, such as those set forth above, and
flavoring agents may be added to provide a palatable oral preparation.
These compositions may be preserved by the addition of an antioxidant
such as ascorbic acid.
[000156] Dispersible powders and granules suitable for preparation of an
aqueous suspension by the addition of water provide the active ingredient
in admixture with a dispersing or wetting agent, a suspending agent and
one or more preservatives. Suitable dispersing or wetting agents and
suspending agents are exemplified by those already mentioned above.
Additional excipients, for example sweetening, flavoring and coloring
agents, may also be present.
[000157] Syrups and elixirs containing the novel combination may be
formulated with sweetening agents, for example glycerol, sorbitol or
sucrose. Such formulations may also contain a demulcent, a preservative
and flavoring and coloring agents.
[000158] The subject combinations can also be administered
parenterally, either subcutaneously, or intravenously, or intramuscularly, or
intrasternally, or by infusion techniques, in the form of sterile injectable
aqueous or olagenous suspensions. Such suspensions may be formulated
according to the known art using those suitable dispersing of wetting
agents and suspending agents which have been mentioned above, or
other acceptable agents. The sterile injectable preparation may also be a
sterile injectable solution or suspension in a non-toxic parenterally
acceptable diluent or solvent, for example as a solution in 1,3-butanediol.
Among the acceptable vehicles and solvents that may be employed are
water, Ringer's solution and isotonic sodium chloride solution. In addition,
sterile, fixed oils are conventionally employed as a solvent or suspending
medium. For this purpose, any bland fixed oil may be employed including
synthetic mono- or diglycerides. In addition, n-3 polyunsaturated fatty
acids may find use in the preparation of injectables.
[000159] The subject combination can also be administered by
inhalation, in the form of aerosols or solutions for nebulizers, or rectally,
in
109
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
the form of suppositories prepared by mixing the drug with a suitable non-
irritating excipient which is solid at ordinary temperature but liquid at the
rectal temperature and will therefore melt in the rectum to release the
drug. Such materials are cocoa butter and polyethylene glycols.
[000160] The novel compositions can also be administered topically, in
the form of creams, ointments, jellies, collyriums, solutions or suspensions.
[000161 ] Daily dosages can vary within wide limits and will be adjusted to
the individual requirements in each particular case. In general, for
administration to adults, an appropriate daily dosage has been described
above, although the limits that were identified as being preferred may be
exceeded if expedient. The daily dosage can be administered as a single
dosage or in divided dosages.
[000162] The present invention further comprises kits that are suitable for
use in performing the methods of treatment, prevention or inhibition
described above. In one embodiment, the kit contains a first dosage form
comprising a glucocorticoid in one or more of the forms identified above
and a second dosage form comprising one or more of the
cyclooxygenase-2 inhibitors or prodrugs thereof identified above, in
quantities sufficient to carry out the methods of the present invention.
Preferably, the first dosage form and the second dosage form together
comprise a therapeutically effective amount of the compounds for the
prevention or treatment of T cell mediated inflammatory/autoimmune
diseases and disorders.
[000163] The following examples describe embodiments of the invention.
~25 Other embodiments within the scope of the claims herein will be apparent
to one skilled in the art from consideration of the specification or practice
of the invention as disclosed herein. It is intended that the specification,
together with the examples, be considered to be exemplary only, with the
scope and spirit of the invention being indicated by the claims which follow
the examples. In the examples, all percentages are given on a weight
basis unless otherwise indicated.
110
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
EXAMPLES: GENERAL INFORMATION
Materials and Methods:
[000164] Animal husbandry and plasma sampling: Mice were housed on
a 12 h/12 h light/dark cycle with ad libitum access to rodent chow. Blood
for measurement of corticosterone and cytokines was obtained by rapid
retroorbital phlebotomy into heparinized capillary tubes with a total time
from first handling the animal to completion of bleeding not exceeding 30
s. Plasma was separated by centrifugation and stored at -80° C until
assay. Unless otherwise noted, all mice used were 6-10 weeks old and of
a C57BL/6 X 129/Sv genetic background.
[000165] For a detailed description of the experimental methods and data
provided herein, see Brewer, J. A., The role of glucocorticoids in immune
system development and regulation, Dissertation presented to Washington
University, St. Louis, MO (2002).
[000166] Generation of TGRko mice: To build a TGRko targeting vector
(pGRIoxPneo), a IoxP site was inserted into the unique Sac I.site in the
G R gene region upstream of exon 2 between exons 1 B and 1 C. A
PGKneo (gene encoding for resistance to neomycin) cassette was then
subcloned containing flanking IoxP sites into intron 2 of the GR gene using
oligonucleotide linkers. To obtain ES clones having replaced one copy of
the endogenous murine GR locus with the GRIoxPneo allele, TC1 ES cells
were electroporated in the presence of linearized pGRIoxPneo as has
previously been described (See, e.g., Muglia, L. J. et al., J. Clin. Invest.,
93:2066 - 2072 (1994)). DNA from 87 6418 resistant embryonic stem cell
(ES) clones was analyzed by Southern blot, employing a probe external to
the flanking regions within our targeting vector. Five clones demonstrated
homologous recombination of the targeting vector into the endogenous GR
locus as evidenced by the appearance of a 4 kb restriction fragment length
polymorphism. A clone heterozygous for the floxed exon 2 allele was
injected into C57BL/6 blastocysts. After germline transmission,
heterozygous mice harboring the floxed exon 2 allele were mated to Lck-
Cre transgenic mice. Mice were then mated to homozygosity for the
111
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
floxed exon 2 allele. Matings were set up between Lck-Cre(+) floxed exon
2 homozygotes and Lck-Cre(-) floxed exon 2 homozygotes to generate
Lck-Cre(+) GRf~°'~fn" (referred to as TGRko) and GRfn'~fn"
(referred to as
control). Unless otherwise noted, all experiments were performed using
age and sex-matched wild-type (WT) and TGRko littermates. (Lck-Cre,
refers to the Cre recombinase under control of the Lck proximal promoter).
[000167] Antibody detection of GR protein: Total protein was harvested
from whole thymus or CD4+ thymocytes that had been sorted by flow
cytometry (MoFlo~, Cytomation Inc.). Fifteen p.g of the protein was
resolved on a 4-12% bis-tris polyacrilamide gel, probed with anti-GR anti-
sera (M-20, Santa Cruz) at a 1:200 dilution, anti-actin anti-sera (Sigma) at
a 1:1000 dilution, and developed using ECL detection reagents
(Amersham). Membranes were then stained with Ponceau S solution
(Sigma) to ensure equal loading of protein.
[000168] Cytokine Measurements: Plasma cytokines were measured
according to manufacturer's instructions (Pharmingen).
[000169] Ribonuclease Protection Assay and Microarray Analysis: Total
splenic RNA was isolated from WT and TGRko mice eight hours after anti-
CD3s antibody challenge using a RNEasy kit (Qiagen). An RNAse
protection assay was then performed on 2 p,g of total RNA according to
manufacturer's instructions (Pharmingen). Microarray experiments were
performed on pooled splenic RNA from the above samples according to
manufacturer's instructions (Affymetrix). Data were analyzed using
Microarray Suite Version 5.0 (Affymetrix).
[000170] Corticosterone Assay: Plasma concentration of corticosterone
was determined by RIA (ICN) from blood collected by retroorbital
phlebotomy at indicated the timepoints in singly housed adult male mice
as previously described. See, e.g., Bethin (2000) ibid.
[000171 ] Flow Cytometry: Thymocytes were dispersed through nylon
mesh into PBS, washed, counted on a hemocytometer using trypan blue
to exclude non-viable cells, stained for cell surface markers (PE-anti-
CD25, PerCP-anti-CDB, APC-anti-CD4, FITC-anti-CD69, PE-anti-TCR~
112
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
from PharMingen), washed, resuspended in PBS and analyzed on a
FACSCaliber~ (Becton Dickinson). Unless otherwise indicated, non-
viable cells were excluded from analysis based on forward and side
scatter profiles.
[000172] Adrenalectomy: Mice were adrenalectomized as previously
described and rested one week before being subjected to experimentation.
See, e.g., Muglia, L. J. et al, J. Clin. Invest., 105:1269-1277 (2000).
[000173] Pharmacologic and antibody treatment: Mice were injected
intraperitoneally 100 p,g anti-CD3s antibody (145-2C11 ) diluted in 250 p.l
PBS. Dexamethasone-treated mice were injected intraperitoneally with
200 p,g dexamethasone phosphate just prior to and eight hours after anti-
CD3s antibody challenge. Neutralizing anti-IFNy antibody (H22, 50 p,g)
was injected intraperitoneally one day before anti-CD3s antibody
administration as described previously. See, e.g., Ferran, C. et al., Eur. J.
Immunol, 21:2349 - 2353 (1991 ). Mifepristone (RU 486, Sigma) was
dissolved in 100% ethanol (50mg/ml). Mifepristone-treated mice were
given 0.5 mg (diluted in sesame oil) s.c. the night before and one hour
prior to inflammatory challenge. Cox-2 selective inhibitor-treated mice
were given 300 p.g of either SC-236 or NS-398 suspended in PBS/1
Tween-80 or vehicle twice a day for two days by oral gavage as previously
described by Gross, G. et al, Am J. Physiol. Regul. Integr. Comp. Physiol.,
278:81415 - 81423 (2000).
[000174] Statistical Methods: All results are expressed as mean ~ SEM
unless otherwise stated. Statistical analysis was done by ANOVA with P <
0.05 considered significant.
EXAMPLE 1
[000175] This example describes the generation of T cell specific
glucocorticoid receptor knockout (TGRko) mice.
[000176] To define specific roles that GR may play in thymocyte
development and peripheral T cell activation, specific T cell glucocorticoid
receptor (GR) knock out mice were generated by the use of Lck promoter-
113
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
driven, Cre recombinase-mediated excision of exon 2 of the GR gene
(shown in Fig. 1 a). While global inactivation of the GR gene results in
perinatal lethality due to abnormal lung maturation (See. e.g., Cole, T. J. et
al., Genes & Development, 9:1608 - 1621 (1995)), mice homozygous for
the floxed GR gene segment and expression of the Lck-Cre transgene
(TGRko mice) were born alive and appeared as healthy as their Lck-Cre (-
), but otherwise genetically identical littermates (control mice). Very little
GR was noted in whole thymus, and no detectable protein in purified CD4+
thymocytes (encompassing both CD4/8+ double positive (DP) and CD4+
single positive (SP) subpopulations, Fig. 1 b). These results indicate that
GR is efficiently deleted early in thymocyte development in TGRko mice.
[000177] One important function of GR in maintaining normal
homeostasis is feedback inhibition of the HPA axis. In this negative
feedback loop, adrenally-derived corticosterone acts via the hypothalamus
and pituitary to regulate its own production. Additionally, the HPA axis can
be regulated by cytokines, neuropeptides and the sympathetic nervous
system. See, e.g., Da Silva, J. A., Ann. N YAcad. Sci. 876:102 - 117;
discussion 117 - 118 (1999). To determine whether deletion of T cell GR
modulates the HPA axis basally, plasma corticosterone levels were
analyzed at circadian nadir (morning) and peak (evening). No differences
were noted between TGRko and control mice (Fig. 1 c). Additionally,
TGRko mice mounted a corticosterone response equal to their littermate
controls, when challenged with a polyclonal T cell activation stimulus (anti-
CD3E antibody, Fig. 1 c). Taken together, these data showed that deletion
of GR in the T cell did not alter basal HPA axis function, and indicated that
GR signaling in T cells did not alter activation of this axis during an
inflammatory response.
[000178] In summary, the data that are shown in Figure 1 indicate that
the deletion of T cell glucocorticoid receptor does not alter HPA axis
regulation. Fig. 1 (A) shows a schematic for targeted deletion of GR exon
two. A targeting vector was designed in which exon two was flanked by
IoxP sites (triangles). Deletion of GR exon two in mice homozygous for
114
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
the floxed GR allele was mediated by transgenic expression of Cre
recombinase controlled by the T cell-specific Lck promoter (TGRko).
Littermates homozygous for the floxed GR allele, but not expressing Cre
recombinase, served as controls (control). Figure 1 (B) shows total protein,
which was extracted from whole thymus or CD4+ thymocytes purified by
flow cytometry and probed for expression of GR by Western blot analysis.
Blots were re-probed for expression of actin as a loading control. Figure
1 (C) shows plasma corticosterone levels, which were measured in TGRko
and control mice in the morning, evening, two and eight hours after
injection of 100 p.g anti-CD3s antibody (145-2C11, n = 4/group).
EXAMPLE 2
[000179] This example illustrated that T cell glucocorticoid receptor is not
required for thymocyte development or peripheral distribution of T
lymphocytes.
[000180] Previous studies using pharmacologic blockade of steroid
biosynthesis in fetal thymic organ culture (FTOC), GR anti-sense
transgenic mice, and GR hypomorph alleles have yielded conflicting data
on the role of GR in thymocyte development. See, e.g., King, L. B. et al.,
Immunity, 3:647 - 656 (1995); Purton, J. F. et al., Immunity, 13:179 - 186
(2000). To determine whether GR signaling affects thymocyte
development in mice with T cell-specific GR deletion, thymocytes were
analyzed from 8-10 week old sex-matched TGRko and control mice. No
significant difference was noted in total thymus cellularity or subset
distribution between genotypes (Total cells (x106): TGRko = 85.8 ~ 10.5, n
= 13; control = 102.5 ~ 11.6, n = 14). Additionally TCR(3 and CD25
surface expression did not differ between TGRko and control thymocytes
(data not shown). In concert with these findings, there was a normal
'distribution of T cells in spleen and lymph nodes of TGRko mice. These
data indicated that GR is not required for T cell development.
115
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
EXAMPLE 3
[000181] This example~shows that T cell glucocorticoid receptor is
required for prevention of lethality and downregulation of multiple
cytokines after T cell activation.
[000182] To evaluate regulation of cytokines and other pro-inflammatory
molecules by T cell GR, anti-CD3s antibodies were administered to TGRko
and control mice. In the spleen, this polyclonal T cell activation stimulus
has been shown to induce rapid, but transient transcription of IL-2, IL-3, IL-
4, IL-6, IFN~y and TNFa, leading to measurable, but ephemeral plasma
levels of these cytokines between one and eight hours after administration.
(See, e.g., Scott, D. E. et al., J. Immunol., 145:2183 - 2188 (1990). In
mice and humans, this leads to an acute, but self-limited clinical syndrome
characterized by hypotension, hypomotility, fever and hypoglycemia, which
can be modulated by pharmacologic GC administration. See, Charpentier,
B. et al., Transplantation, 54:997 - 1001 (1992); Ferran, C. et al.,
Transplantation, 50:642 - 648 (1990). In contrast to uniform survival in
control mice, high mortality was noted in TGRko mice after anti-CD3s
antibody administration, which could not be rescued by pre-treatment with
the synthetic GC dexamethasone (DEX, Fig. 2a). These results
suggested that the T cell is a critical target for GC down-regulation of
immune responses.
[000183] To determine whether TGRko mice were dying from altered
cytokine regulation, plasma cytokine levels were measured after polyclonal
T cell activation. Significant increases in TNFa, IFNy, and IL-6, but not IL-
2, were noted in TGRko mice after T cell activation (Fig. 2b). Interestingly,
DEX administration reduced plasma levels of TNFa in both TGRko and
control mice, but had no significant effect on plasma IL-2 in TGRko mice.
Plasma IFN~ywas not affected by DEX administration in either genotype
(Fig. 2b). These data suggest that in contrast to other cytokines, IFNy
regulation is specifically controlled by T cell GR signaling.
116
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
[000184] To determine the role of endogenous T cell GR signaling in
transcriptional regulation of these and other inflammatory genes, gene
expression in spleens of TGRko and control mice were compared eight
hours after T cell activation by microarray analysis and ribonuclease
protection assay (RPA). Of 21 known genes that were induced 2.5 fold or
greater in microarray analysis of TGRko compared to control splenocytes,
have documented immune function. For example, a listing of genes
(with fold induction values in parenthesis) shows: T cell and activation
regulated chemokine (7.0), small inducible cytokine B subfamily, member
10 5 (6.1), IL-6 (4.3), COX-2 (3.5), Src-suppressed C kinase substrate (3.5),
MMP-1 (3.2), Eotaxin precursor (3.2), IFNy (2.8), SOCS-3 (2.6), protein
kinase inhibitor (2.6). Providing validation of these data, expression of 9l9
cytokines from the same samples, as well as from pooled lymph nodes
(data not shown), analyzed by RPA showed the same degree of induction
(or lack thereof) as that shown by microarray (Fig. 2c).
[000185] Interestingly, IFNy, but not TNFa or IL-2 was elevated in TGRko
microarray and RPA samples (IL-6 was induced in one of two RPA
samples). The low levels of IL-2 and TNFa RNA correlated with reduced
plasma cytokine measurements eight hours after stimulation (IL-2 and
TNFa were not statistically different in TGRko and control mice, data not
shown). These data suggest that endogenous GCs acting through T cell
GR are required for IFNytranscriptional suppression (possibly mediated
through inhibition of Stat4 phosphorylation (See, e.g., Franchimont, D. et
al., J. Immunol., 164:1768 - 1774 (2000)), but not for TNFa or IL-2.
[000186] In summary, the data shown in Figure 2 indicate that T cell GR
is required for prevention of lethality and downregulation of multiple
cytokines after activation. (A) Survival presented as a ICaplan-Meyer plot
(D control, n = 10; ~ TGRko, n = 4; O control + DEX, n = 10; ~ TGRko +
DEX, n = 7). No further mortality was noted after four days. P < 0.01
between TGRko and control mice in both plots (B) Plasma cytokine levels
were measured in TGRko and control mice two and eight hours after
117
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
injection of anti-CD3s antibody (100 ~.g) ~ dexamethasone administration
(200 p.g 1 hour before and 8 hours after anti-CD3E antibody) by ELISA (n =
6-9/group for TGRko, 8-13/group for control mice). (C) Splenic RNA (2 p.g)
from TGRko and control mice was analyzed by RPA at baseline or eight
hours after injection of anti-CD3s antibody ((+) denotes positive control
RNA provided by the manufacturer). Expression was normalized to
GAPDH and quantitated using a phosphorimager. The same samples
were pooled and analyzed by microarray. *, P < 0.05, **, P < 0.01
between TGRko and control.
EXAMPLE 4
[000187] This example shows that overproduction of IFNywas not the
cause of mortality in T cell activated TGRko mice.
[000188] To determine if unchecked IFNY production was inducing a high
degree of mortality in TGRko mice, neutralizing anti-IFNyantibodies were
administered to the mice before in vivo T cell stimulation. Neutralizing
anti-IFN~y antibodies decreased the level of plasma IFN~y below the limit of
detection (624 pg/ml), however, there was no reduction in mortality (3/3
mice died at 1.67 ~ 0.44 days). These results suggested that although GR
signaling in T cells is important in the regulation of IFNy production, this
dysregulation does not directly result in the high mortality seen in TGRko
mice after T cell activation.
EXAMPLE 5
[000189] This example shows that Cox-2 dysregulation is directly
involved in the mortality of TGRko mice and indicates the efficacy of the
administration of a Cox-2 selective inhibitor to reduce the mortality.
[000190] Glucocorticoids have been shown to regulate expression of pro-
inflammatory mediators in addition to cytokines. Of note, cyclooxygenase
2 (Cox-2) was discovered as a GC-modulated enzyme that was induced in
monocytes after LPS administration, and subsequently has been shown to
be induced in vitro in T cells after activation. See, e.g., Iniguez, M. A. et
al,
J. Immunol., 163:111 - 119 (1999); Masferrer, J. L. et al., Proc. Natl. Acad.
118
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
Sei. USA, 89:3917 - 3921 (1992). In this work, it was found that Cox-2
mRNA levels were 3.5 fold higher in TGRko spleenocytes compared to
controls eight hours after anti-TCR~ antibody administration by microarray
analysis. To determine whether Cox-2 dysregulation was directly involved
in induction of mortality by polyclonal T cell activation in TGRko mice, mice
were treated with each of two selective Cox-2 inhibitors (SC-236, which
was provided by the Pharmacia Company, and NS-398, available from
Cayman Chemical, Ann Arbor, MI) before and after anti-CD3s antibody
administration. Protection from lethality was noted in TGRko mice treated
with either SC-236 (s) or NS-398 (n) compared to vehicle-treated mice (v)
(Fig. 3a).
(000191] To bring these observations into the more physiologically and
clinically relevant context of global GC deficiency/resistance, these
experiments were repeated in control mice pre-treated with mifepristone
(RU-486), a GR antagonist. Highlighting the importance of GR signaling in
this system, mice treated with mifepristone + vehicle (m/v) were
significantly more susceptible to the lethal affects of T cell activation
compared to vehicle controls (v/v) (Fig. 3b). As with TGRko mice treated
with SC-236, control mice treated with mifepristone + SC-236 (m/s) (were
significantly protected compared to their vehicle-treated counterparts (Fig.
3b). Additionally, SC-236 treatment of adrenalectomized control mice
showed essentially the same degree of rescue, though on a shorter
timescale (0/3 Sham operated + vehicle, 2/6 ADX + SC-236, and 4/5 ADX
+ vehicle mice died within eight hours of anti-CD3s antibody treatment).
Taken together, these data directly show that endogenous T cell GR
modulation of Cox-2 expression is required to prevent polyclonal T cell
activation from becoming lethal. Histological analysis in the cecum in mice
treated with anti-CD3s antibody demonstrated marked edema,
inflammation, and mucosal disruption in the TGRko mice and rescue with
Cox-2 inhibition with NS-398. See, Figure 3C. These sections are
representative of 3 - 5 mice per group analyzed.
119
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
[000192] In summary, the data shown in Figure 3 show that
cyclooxygenase 2 inhibition protects against mortality induced by
polyclonal T cell activation in GR-deficient mice. (A) TGRko were treated
with SC-236, (solid line (s), n = 7), NS-398 (alternately dashed line (n)), or
vehicle (dashed line (v), n = 3) one hour before anti-CD3s antibody
administration, and twice a day for two days thereafter. (B) Control mice
were treated with mifepristone + SC-236 (m/s, solid line, n = 10),
mifepristone + vehicle (m/v, dashed line, n = 8), or vehicle + vehicle (v/v,
mixed line, n = 3) as described in materials and methods. P < 0.05
between SC-236 and vehicle treated mice.
EXAMPLE 6
[000193] This illustrates the production of a composition containing
parecoxib sodium and dexamethasone sodium phosphate, and of a
pharmaceutical composition containing the combination.
[000194] Parecoxib sodium can be produced according to the
procedures described in U.S. Patent No. 5,932,598. Dexamethasone
sodium phosphate for injection can be obtained from Merck, Wyeth-Ayerst,
and other suppliers, under the trade name "Decadron".
[000195] A therapeutic composition of the present invention can be
formed by intermixing parecoxib sodium (40 g) into 187.5 ml of
dexamethasone sodium phosphate sterile injection solution (containing
0.75 g of dexamethasone phosphate, 1.5 g of creatinine, 1.875 g sodium
citrate, sodium hydroxide to adjust pH, and water for injection p.s., with
187.5 mg sodium bisulfite, 281 mg methylparaben, and 37.5 mg
propylparaben as preservatives; available as Decadron Phosphate
injection, 4 mg/ml, from Merck & Co., Inc., Whitehouse Station, NJ).
Additional water may be added if necessary for the complete dissolution of
all solid components.
[000196] After mixing, the combination of parecoxib and dexamethasone
form a therapeutic composition that is sufficient for the production of about
1000 human single dose units. Each single dose unit contains about 40
mg of parecoxib sodium and about 0.75 mg of dexamethasone phosphate.
120
CA 02484989 2004-10-29
WO 03/096970 PCT/US03/13548
[000197] Therapeutic and pharmaceutical compositions comprising a
combination of any of the cyclooxygenase-2 inhibitors and any of the
glucocorticoids that are described above can be formed by similar
methods.
[000198] All references cited in this specification, including without
limitation, all papers, publications, patents, patent applications,
presentations, texts, reports, manuscripts, brochures, books, Internet
postings, journal articles, periodicals, and the like, are hereby incorporated
by reference into this specification in their entireties. The discussion of
the
references herein is intended merely to summarize the assertions made by
their authors and no admission is made that any reference constitutes
prior art. Applicants reserve the right to challenge the accuracy and
pertinency of the cited references.
[000199] In view of the above, it will be seen that the several advantages
of the invention are achieved and other advantageous results obtained.
[000200] As various changes could be made in the above methods and
compositions without departing from the scope of the invention, it is
intended that all matter contained in the above description shall be
interpreted as illustrative and not in a limiting sense.
121