Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02506238 2005-05-03
CHROMAN DERIVED COMPOUNDS & FORMULATIONS niEREOF FOR USE
IN THERAPY
Field of the Invention
The present invention relates to certain novel isoflavan derivatives,
compositions
containing same, methods for their preparation and uses thereof as therapeutic
agents
particularly as anti-cancer and chemotherapeutic selective agents.
Background of the Invention
Over 700 different naturally occurring isoflavones are known some of which
have
biological properties with potential therapeutic benefit.
US 5,726,202 generically discloses certain isoflavan compounds, particularly
3,4-
diarylchroman and centchroman for the treatment of benign prostatic
hypertrophy.
WO 01/17986 also discloses certain isoflavan compounds.
Summary of the Invention
Surprisingly, the present inventors have found a novel group of compounds of
the general
formula (I) which exhibit important therapeutic activities including strong
anti-cancer
activity, chemotherapeutic selectivity and radiosensitisation of cancers.
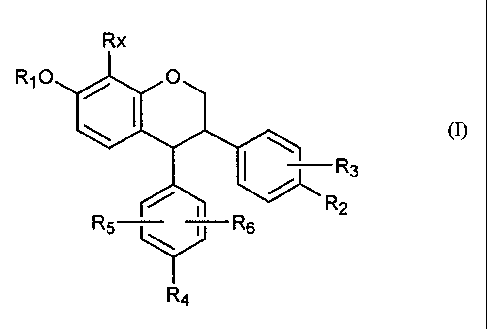
Thus according to an aspect of the present invention there is provided a
compound of the
general formula (I):
Rx
Ri0 0
(I)
I ¨R3
R2
R6 -7- R6
R4
CA 02506238 2012-05-18
71884-135
- 2 -
wherein
R1 is hydrogen, alkyl, cycloalkyl or C(0)R7,
R2 and R3 are independently hydrogen, hydroxy, alkoxy, alkyl, cycloalkyl, halo
or
OC(0)R7, with the exception that R2 and R3 are not both hydrogen,
R4, R5 and R6 are independently hydrogen, hydroxy, alkoxy, alkyl, cycloalkyl,
acyl,
NH2, NHC1-C4 alkyl or N(C1-a4 alky1)2, OC(0)R7 or OR8,
R7 is hydrogen, alkyl, cycloalkyl, aryl, arylalkyl or amino,
R8 is aryl such as phenyl or arylalkyl such as benzyl, and
Rx is hydrogen, hydroxy, alkoxy, alkyl, cycloalkyl or halo
or a pharmaceutically acceptable salt or derivative thereof.
According to another aspect of the invention, there is provided a compound of
a
general formula (I):
Rx
R10 Op 0
-R3
R2
R5yR6
R4 (I)
wherein
R1 is hydrogen,
R2 and R3 are independently hydrogen, hydroxy, alkoxy, alkyl, cycloalkyl, halo
or
OC(0)R7, with the exception that R2 and R3 are not both hydrogen,
CA 02506238 2012-05-18
71884-135
- 2a -
R4, R5 and R6 are independently hydrogen, hydroxy, alkoxy, alkyl, cycloalkyl,
NH2,
NHC1-C4 alkyl or N(C1-C4alky1)2, OC(0)R7 or OR8,
R7 is hydrogen, alkyl, cycloalkyl, aryl, arylalkyl or amino,
R8 is phenyl or benzyl, and
Rx is hydrogen,
or a pharmaceutically acceptable salt thereof.
According to another aspect of the invention, there is provided a compound
having a
structure of:
HO si 0
40 10 OH
OH =
According to another aspect of the present invention there is provided a
process for
the preparation of a compound of formula (I) comprising the step of reacting
the keto
group of a compound of the formula (II):
R10 0
0
R2
or the analogue thereof including a substituent which corresponds to Rx in
compounds of formula (I)
wherein
CA 02506238 2012-05-18
71884-135
- 2b -
R1, is alkyl or a protecting group such as Si(RA)3,
R2 and R3 are independently hydrogen, alkoxy or OSi(RA)3, with the exception
that R2
and R3 are not both hydrogen, and
RA is independently alkyl or aryl,
with an arylating agent IN-M+,
wherein
1./V- is optionally substituted aryl radical, and
M+ is one or more counter ions, preferably [MgBr],
to form the intermediate tertiary alcohol of formula (Ill):
CA 02506238 2005-05-03
- 3 -
R10 0
(HI)
HO I ¨ R 3
R2
or protected derivative thereof or a salt thereof (or an analogue thereof
including a
substituent which corresponds to Rx in compounds of formula (I)) and which is
dehydrated
to form a compound of formula (IV):
R10 0
(IV)
====
I ¨R3
(or an analogue thereof including a substituent which corresponds to Rx in
compounds of
formula (I)) the double bond of which is subsequently reduced, for example, by
hydrogenation and optionally deprotected to form a compound of formula (I).
According to another aspect of the present invention there is provided a
compound of the
general formula (III), compositions containing same and uses thereof.
In another aspect, there is provided a compound of the general formula (IV),
compositions
containing same and uses thereof.
Thus, according to another aspect of the present invention there is provided
the use of a
compound of formula (I) in therapy, particularly chemotherapy and/or as
radiosensitising
or chemosensitising agents.
According to another aspect of the present invention there is provided a
method for the
treatment, prevention or amelioration of a disease or disorder, which
comprises
administering to a subject one or more compounds of the formula (I) or a
pharmaceutically
acceptable salt or derivative thereof optionally in association with a carrier
and/or
CA 02506238 2005-05-03
- 4 -
excipient.
According to another aspect of the present invention there is provided the use
of one or
more compounds of formula (I) or a pharmaceutically acceptable salt or
derivative thereof
in the manufacture of a medicament for the treatment of a disease or disorder.
According to another aspect of the present invention there is provided an
agent for the
treatment, prophylaxis or amelioration of a disease or disorder which agent
comprises one
or more compounds of formula (I) or a pharmaceutically acceptable salt or
derivative
thereof.
According to another aspect of the present invention there is provided a
pharmaceutical
composition which comprises one or more compounds of formula (I) or a
pharmaceutically
acceptable salt or derivative thereof in association with one or more
pharmaceutical
carriers, excipients, auxiliaries and/or diluents.
According to another aspect of the present invention there is provided a drink
or food-stuff,
which contains one or more compounds of formula (I) or a pharmaceutically
acceptable
salt or derivative thereof.
These and other aspects of the invention will become evident from the
description and
claims which follow, together with the accompanying drawings.
Brief Description of the Figures
Fig. 1 represents a comparison of dehydroequol (DHE graph A), 3-(4-
hydroxypheny1)-4-
(4-methoxyphenyl)chroman-7-ol (HMC compound 1 according to the invention graph
B)
and cisplatin (graph C) toxicity in neonatal foreskin fibroblasts.
Fig. 2 represents HMC efficacy in melanoma cells in comparison with cisplatin.
Fig. 3 represents a phannacokinetic profile of free and total forms of HMC (A)
and DHE
(B) after p.o (pen i oral) administration to BALB/c mice (50 mg/kg).
CA 02506238 2005-05-03
- 5 -
Fig. 4 represents a comparison of the pharmacokinetic profile the HMC
concentration in
serum after i.v (intravenously) and i.p (intraperitoneally) administration of
}WC
formulated in 20% hydroxypropyl-beta-cyclodextrin at a dose of 50 mg/kg.
Fig. 5 represents comparative mean tumour volume data taken from nude mice
bearing
HPAC pancreatic cancer tumours treated with either i.p dosed 20% HPBCD
(vehicle
control, qdx15) or HMC (100 mg/kg, qdx15). Data represented as mean SEM *,
student's T-test,p <0.01.
Fig. 6 represents comparative mean terminal tumour mass data taken from nude
mice
bearing HPAC pancreatic cancer tumours treated with either i.p dosed 20% HPBCD
(vehicle control, qdx15) or HMC (100 mg/kg, qdx15). Data represented as mean
SEM
*, student's T-test,p <0.01.
Fig. 7 represents comparative mean terminal tumour mass data taken from nude
mice
bearing HPAC pancreatic cancer tumours treated with either i.p dosed 20% HPBCD
(vehicle control, qdx15) or HMC (100 mg/kg, qdx15). Data represented as mean
SEM.
*, student's T-test, p <0.01.
Fig. 8 represents a summary of apoptosis incidence in DHE and HMC treated
melanoma
cells over a 24 and 48 hour period.
Fig. 9 represents selective initiation of programmed cell death in HMC and DHE
treated
malignant melanoma cells (Mel-RM and Me4405). The same concentration of DHE
and
HMC and exposure times do not induce apoptosis in normal fibroblasts (MRC-5).
Fig. 10 represents a 3D analysis of HMC-cisplatin synergy cytotoxicity data in
the MIV1200
melanoma cell line. HMC-cisplatin combinations were assessed using a 5-day
combination protocol (Fig 10 A), or a 24 hr anti-cancer
sequence (Fig 10 B). For
each combination experiment HMC was assessed at 10, 5, 2 and 1 [M. See Table 8
for raw
data.
Fig. 11 represents the percentage inhibition of TNFa in murine macrophages by
compounds 6 and 7 of the invention.
Fig. 12 represents the chromatograph of 3-(4-hydroxypheny1)-4-(4-
hydroxyphenyl)chroman-7-ol when subjected to preparative HPLC.
CA 02506238 2005-05-03
- 6 -
Detailed Description of the Invention
The present inventors have found that a class of isoflavan derivatives of the
general
formula (I) show surprising and unexpected biological and pharmaceutical
properties.
The compounds of formula (I) of the invention are believed to have favourable
toxicity
profiles with normal cells and good bioavailability. Surprisingly the
compounds of the
invention exhibit anti-cancer activity, significantly better than or at least
comparable to
known cancer treatments.
The compounds of formula (I) are cytostatic and cytotoxic against a broad
range of cancer
cells of human and animal origin. By cancer cells, it is meant cells that
display malignant
characteristics and which are distinguished from non-cancer cells by
unregulated growth
and behaviour which usually ultimately is life-threatening unless successfully
treated.
The cancer cells that have been found to be responsive to compounds of formula
(I) are of
epithelial origin (for example, prostate, ovarian, cervical, breast, gall-
bladder, pancreatic,
colorectal, renal, and non-small lung cancer cells), of mesenchymal origin
(for example,
melanoma, mesothelioma and sarcoma cancer cells), and of neural origin (for
example
glioma cancer cells). It is highly unusual and surprising to find a related
group of
compounds that display such potent cytotoxicity against cancer cells, but with
low toxicity
against non-cancer cells such as keratinocytes derived from human foreskin.
Such cancer
cell selectivity is highly unusual and unexpected.
Advantageously the compounds of formula (I) show cytotoxicity against cancer
cells that
are well recognized for being poorly sensitive to standard anti-cancer drugs.
It is highly
unusual and unexpected to find such potent activity against cancers, for
example,
cholangiocarcinoma, pancreatic adenocarcinoma and melanoma.
Advantageously the compounds of formula (I) also unexpectedly display an
ability to
radio-sensitise cancer cells, by which it is meant that these compounds either
lower the
amount of gamma-irradiation that is required to kill the cells, or they
convert cancer cells
from a state of radio-resistance to a radio-sensitive state.
CA 02506238 2005-05-03
- 7 -
Additionally the compounds of formula (I) are thought to possess chemo-
sensitising
activity, that is they increase the cytotoxicity of chemotherapeutic agents,
especially to
cancer cells, and/or convert cancerous cells from a state of chemo-resistance
to a chemo-
sensitive state.
Compounds of the invention may also provide chemo and/or radio-protective
properties for
non-cancerous cells. This has significant therapeutic implications because the
traumatic
side-effects of chemotherapy and radiotherapy are caused by the toxicity of
the traditional
treatments to non-cancerous cells.
The properties described above offer significant clinical advantages.
The radio and/or chemo-protective properties of the compounds of the invention
may be
employed to protect healthly individuals from the effects of radiation and/or
chemical
toxins, or lessen the effects of the same.
Thus, the invention also provides the use of compounds of formula (I) to treat
patients with
cancer by either reducing the rate of growth of such tumors or by reducing the
size of such
tumors through therapy with said compounds alone, and/or in combination with
each other,
and/or in combination with other anti-cancer agents, and/or in combination
with
radiotherapy.
The use of compounds of the present invention either alone or in combination
therapy as
described above may reduce the adverse side-effects often experienced by
patients when
treated with standard anti-cancer treatments. The use of compounds of the
invention may
mean that lower doses can be employed in such therapy which represents an
important
advance for cancer sufferers.
Preferably R3 in compounds of formula (I) is in the 3-position.
In one aspect of the invention Rx is not hydrogen. However, preferably in
compounds of
formula (I) Rx is hydrogen or CI-Ca alkyl such as methyl, especially hydrogen.
CA 02506238 2005-05-03
- 8 -
References to compounds of formula (I) in this specification are intended to
include other
compounds/formulas of the invention unless otherwise indicated.
Compounds of the invention include those of formula (I-a):
Ri el 0
toR3 (I-a)
R5-- R6 R2
R4
wherein
R1 is hydrogen, alkyl, cycloalkyl or C(0)R7,
R2 and R3 are independently hydrogen, hydroxy, alkoxy, halo or OC(0)R7,
with the
exception that R2 and R3 are not both hydrogen,
R4, R5 and R6 are independently hydrogen, hydroxy, alkoxy, alkyl,
cycloalkyl, acyl,
OC(0)R7, and
R7 is hydrogen, alkyl, cycloalkyl, aryl or arylalkyl.
Preferably in compounds of formula (I-a):
R1 is hydrogen, CI-Ca alkyl or C(0)R7,
R2 and R3 are independently hydrogen, hydroxy, CI-Ca alkoxy, halo or
OC(0)R7,
provided that R2 and R3 are not both hydrogen,
R4, R5 and R6 are independently hydrogen, hydroxy, alkoxy, alkyl,
cycloalkyl, acyl,
OC(0)R7, and
R7 is C1-C4 alkyl, phenyl or benzyl,
or a pharmaceutically acceptable salt or derivative thereof.
More preferably in compounds of formula (I-a):
R1 is hydrogen, methyl, ethyl, propyl, isopropyl or acetyl,
R2 and R3 are independently hydrogen, hydroxy, methoxy, ethoxy, propoxy,
isopropoxy, bromo, chloro, fluoro or acetyloxy, with the exception that R2 and
R3
CA 02506238 2005-05-03
- 9 -
are not both hydrogen,
R4 is hydrogen, hydroxy, methoxy, ethoxy, propoxy, isopropoxy or acetyloxy,
and
R5 and R6 are independently hydrogen, hydroxy, methoxy, ethoxy, propoxy,
isopropoxy, acetyl, or acetyloxy,
or a pharmaceutically acceptable salt or derivative thereof.
Particular preferred compounds of formula (I-a) have the following
substituents where:
R1 is hydrogen, methyl or acetyl,
R2 and R3 are independently hydrogen, hydroxy, methoxy, bromo or acetyloxy,
with
the exception that R2 and R3 are not both hydrogen,
R4 and R6 are independently hydrogen, hydroxy, methoxy or acetyloxy, and
R5 is hydrogen,
or a pharmaceutically acceptable salt or derivative thereof.
The invention also extends to compounds of formula (I-b)
R10 0
R3
(I-b)
R2
R5
R4
wherein:
R1 represents hydrogen or C1-C6 alkyl, more preferably hydrogen or methyl,
especially
hydrogen.
R2 represents hydrogen, hydroxy or C1-C6 alkoxy such as methoxy, ethoxy,
propoxy,
more preferably hydroxy or methoxy, especially hydroxy.
R3 represents hydrogen, hydroxy C1-C6 alkoxy such as methoxy, ethoxy,
propoxy,
more preferably hydrogen or methoxy, especially hydrogen,
with the proviso that R2 and R3 do not both represent hydrogen,
represents hydrogen, hydroxy C1-C6 alkoxy such as methoxy, ethoxy, propoxy, CI-
CA 02506238 2005-05-03
- 1 0 -
C6 alkyl such as methyl, ethyl, propyl, isopropyl, especially hydrogen,
hydroxy,
methoxy or methyl particularly methoxy or hydroxy,
R5 represents hydrogen, C1-C6 alkoxy, C1-C6 alkyl, especially hydrogen,
methoxy,
hydroxy, particularly hydrogen,
or a pharmaceutically acceptable salt or derivative thereof.
Preferred compounds of the invention include those of the general formula (I-
c):
R10 40 0
0-0
411 OR2a
OR3a
wherein:
R1 is hydrogen or CI-C6 alkyl such as methyl, ethyl, propyl, isopropyl,
butyl, isobutyl,
secbutyl, tertiary butyl,
R2a is hydrogen or CI-C6 alkyl such as methyl, ethyl, propyl, isopropyl,
butyl, isobutyl,
secbutyl, tertiary butyl, and
R3a is hydrogen or C1-C6 alkyl such as methyl, ethyl, propyl, isopropyl,
butyl, isobutyl,
secbutyl, tertiary butyl,
or a pharmaceutically acceptable salt or a derivative thereof.
More preferably in compounds of formula (I-c) R1 is hydrogen or methyl,
especially
hydrogen.
More preferably in compounds of formula (I-c) R2a is hydrogen or methyl,
especially
hydrogen.
More preferably in compounds of formula (I-c) R3a is hydrogen or methyl,
especially
methyl.
CA 02506238 2005-05-03
- 11 -
Especially preferred compounds of formula (I) include:
3-(4-hydroxypheny1)-4-(4-methoxyphenyl)chroman-7-ol (HMC) (Cpd. 1);
3-(4-hydroxypheny1)-4-pheny1chroman-7-ol (Cpd. 2);
3-(4-hydroxypheny1)-4-(3-methoxyphenyl)chroman-7-ol (Cpd. 3);
3-(3,4-dimethoxypheny1)-4-(4-methoxyphenyl)chroman-7-01(Cpd. 4);
3-(4-hydroxypheny1)-4-(4-methylphenyl)chroman-7-ol (Cpd. 5);
3-(4-methoxypheny1)-4-(4-methoxypheny1)-7-methoxychroman (Cpd. 6);
3-(4-hydroxypheny1)-4-(2,6-dimethoxy-4-hydroxyphenyl)chroman-7-ol (Cpd. 7);
3-(4-hydroxypheny1)-4-(2-hydroxyphenyl)chroman-7-ol (Cpd. 8);
3-(4-hydroxypheny1)-4-(3-acy1-2-hydroxy-4-methoxyphenyl)chroman-7-ol (Cpd. 9);
3-(3-hydroxypheny1)-4-(3-methoxyphenyl)chroman-7-01(Cpd. 10);
3-(4-hydroxypheny1)-4-(4-hydroxyphenyl)chroman-7-ol (Cpd. 11);
3-(4-bromopheny1)-4-(4-methoxyphenyl)chroman-7-ol (Cpd. 12);
3-(4-hydroxypheny1)-4-(3-methoxyphenyl)chroman-7-ol (Cpd. 13);
3-(4-hydroxypheny1)-4-(3-aminophenyl)chroman-7-ol (Cpd. 14);
3-(4-hydroxypheny1)-4-(4-phenoxyphenyl)chroman-7-ol (Cpd 15);
3-(3,4-dimethoxypheny1)-4-(4-methoxypheny1)-8-methylchroman-7-ol (Cpd 16).
HO is 0 HO si 0
(1) 411 (2)
411 OH
1411:1 OH
OMe
HO arith 0 HO 0
IN(3) OMe (4)
1.1 OH
0111 OMe
OMe
OMe
CA 02506238 2005-05-03
-12-
HO, 0 Me0 op 0
11011 (5)
Si (8)
Si OH
lei OMe
Me OMe
HO 0 0 HO 0 0
(7) (8)
Me0 11101
1410 OMe OH HO 0 Oil
OH
OH
HO 0 0 HO 40 0
(9) la OH (10)
HO 0 0
OH
411
Me
OMe
0 OMe
HO 0 0 HO I. 0
1101 (11)
101 (12)
0 OH
el Br
OH OMe
HO 0 0 HO 40 0
(010 (13)
1.1 (14)
el OH
'N
OH
OMe NH2
CA 02506238 2005-05-03
- 13 -
HO 0
(15) HO 0
4111 OH OMe
OMe
0
(16)
OMe
or a pharmaceutically acceptable salt thereof.
The compounds of formula (I) according to the invention include two chiral
centres. The
present invention includes all the enantiomers and diastereoisomers as well as
mixtures
thereof in any proportions. The invention also extends to isolated enantiomers
or pairs of
enantiomers. Methods of separating enantiomers and diastereoisomers are well
known to
person skilled in the art.
It will be clear to persons skilled in the art that the in compounds of
formula (I) the aryl
substituents on the heterocyclic ring can be cis or trans relative to each
other. Preferably
in the compounds of formula (I) these substituents will be cis.
A particularly preferred compound of the present invention is the cis-isomer
of HMC:
HO 0
Olt OH
OMe
or a pharmaceutically acceptable salt thereof.
Likewise, particularly preferred compounds are compound Nos. (2) to (16) in
the cis-
conformation.
CA 02506238 2005-05-03
- 14 -
In an alternative aspect the invention provides compounds of formula (I-d):
R10el 0
R2b
4111 (I-d)
OR3,,
wherein:
R1 is as defined above for compounds of formula (I),
R2b represents hydroxy, C1-C6 alkyl or C1-C6 alkoxy, especially hydroxy or
methoxy,
and
R3a is as defined above for compounds of formula (I-c)
In a further alternative aspect the invention provides compounds of formula (1-
e):
R10 oil 0
I R3
R2 (I-e)
111
wherein
R2 and R3 are as defmed above for compouds of formula (I).
CA 02506238 2005-05-03
- 15 -
Preferably in compounds of formula (I-e) R1 represents hydrogen or methyl,
especially
hydrogen.
Preferably in compounds of formula (I-e) R2 represents hydroxy or CI-C6 alkoxy
such as
methoxy.
In compounds of formula (I-e) preferably R3 represents hydrogen, hydroxy or
methoxy,
especially hydrogen.
In a further alternative aspect the invention provides compounds of formula (I-
f):
R10 oil 0
I R3
R2 (1-0
R4a
wherein
RI, R2 and R3 are as defined above for compouds of formula (I), and
Raa represents hydroxy, alkoxy, alkyl, cycloallcyl, acyl, NH2, NHCI-C4 alkyl
or N(C1-C4
alky1)2, OC(0)R7 or OR8, and
R8 is defined above for compounds of formula (I).
Preferably in compounds of formula (I-f) R1 represents hydrogen or methyl,
especially
hydrogen.
Preferably in compounds of formula (I-f) R2 represents hydroxy or C1-C6 alkoxy
such as
methoxy, especially hydroxy.
Preferably in compounds of formula (I-f) R3 represents hydrogen or C1-C6
alkoxy such as
methoxy, especially hydrogen.
CA 02506238 2005-05-03
- 16 -
Preferably in compounds of formula (I-f) R3 is in the 3-position.
Preferably in compounds of formula (I-f) R4a represents NH2, NHC1-C4 alkyl or
N(C1-C4
alIcy1)2, especially NH2.
The compounds of formulae (III) and (IV) are intermediates as set out herein.
Each
corresponding isoflavan-4-ol and isoflavan-3-ene intermediate of compound Nos.
(1) to
(12) are also preferred compounds of the present invention.
W in compounds of formula (III) and (IV) may, for example, represent the
following
radicals:
R6 leo
(V-1)
R5 R5 R6 (V-2)
R4 R4
=IVVIA.
R5 411 R6
(V-3) 40 R6 (V-4)
R5
R4 R4
or a protected derivative thereof wherein R4, R5 and R6 are as defined above
for
compounds of formula (I).
The term "isoflavone" as used herein is to be taken broadly to include as
isoflavones,
isoflavenes, isoflavans, isoflavanones, isoflavanols and the like.
The term "alkyl" is taken to include straight chain and branched chain
saturated alkyl
groups of 1 to 6 carbon atoms, such as methyl, ethyl, propyl, isopropyl,
butyl, isobutyl,
secbutyl, tertiary butyl, pentyl and the like. The alkyl group more preferably
contains
CA 02506238 2005-05-03
- 17 -
preferably from 1 to 4 carbon atoms, especially methyl, ethyl, propyl or
isopropyl.
Cycloalkyl includes C3-6 cycloalkyl such as cylopropyl, cyclobutyl,
cyclopentyl and
cyclohexyl.
The alkyl group or cycloalkyl group may optionally be substituted by one or
more of
fluorine, chlorine, bromine, iodine, carboxyl, Ci-C4-alkoxycarbonyl, Ci-C4-
alkylamino-
carbonyl, di-(CI-C4-alkyl)-amino-carbonyl, hydroxyl, Ci-C4-alkoxy, formyloxy,
CI-Cr
alkyl-carbonyloxy, CI-C4-alkylthio, C3-C6-cycloalkyl or phenyl.
Preferably the alkyl group does not bear any substituents.
The term "aryl" is taken to include phenyl, benzyl, biphenyl and naphthyl and
may be
optionally substituted by one or more CI-C4-alkyl, hydroxy, Ci-C4-alkoxy,
carbonyl, Cr
C4-alkoxycarbonyl, C1-C4-alkylcarbonyloxy, nitro or halo.
The term "halo" is taken to include fluoro, chloro, bromo and iodo, preferably
fluoro and
chloro, more preferably fluoro. Reference to for example "haloallcyl" will
include
monohalogenated, dihalogenated and up to perhalogenated alkyl groups.
Preferred
haloalkyl groups are trifluoromethyl and pentafluoroethyl.
The compounds of the invention include all salts, such as acid addition salts,
anionic salts
and zwitterionic salts, and in particular include pharmaceutically acceptable
salts as would
be known to those skilled in the art. The term "pharmaceutically acceptable
salt" refers to
an organic or inorganic moiety that carries a charge and that can be
administered in
association with a pharmaceutical agent, for example, as a counter-cation or
counter-anion
in a salt. Pharmaceutically acceptable cations are known to those of skilled
in the art, and
include but are not limited to sodium, potassium, calcium, zinc and quaternary
amine.
Pharmaceutically acceptable anions are known to those of skill in the art, and
include but
are not limited to chloride, acetate, tosylate, citrate, bicarbonate and
carbonate.
Pharmaceutically acceptable salts include those formed from: acetic, ascorbic,
aspartic,
CA 02506238 2005-05-03
- 18 -
benzoic, benzenesulphonic, citric, cinnamic, ethanesulphonic, fumaric,
glutamic, glutaric,
gluconic, hydrochloric, hydrobromic, lactic, maleic, malic, methanesulphonic,
naphthoic,
hydroxynaphthoic, naphthalenesulphonic, naphthalenedisulphonic,
naphthaleneacrylic,
oleic, oxalic, oxaloacetic, phosphoric, pyruvic, p-toluenesulphonic, tartaric,
trifluoroacetic,
triphenylacetic, tricarballylic, salicylic, sulphuric, sulphamic, sulphanilic
and succinic
acid.
The term "pharmaceutically acceptable derivative" or "prodrug" refers to a
derivative of
the active compound that upon administration to the recipient is capable of
providing
directly or indirectly, the parent compound or metabolite, or that exhibits
activity itself
and includes for example phosphate derivatives and sulphonate derivatives.
Thus,
derivatives include solvates, pharmaceutically active esters, prodrugs or the
like. This also
includes derivatives with physiologically cleavable leaving groups that can be
cleaved in
vivo to provide the compounds of the invention or their active moiety. The
leaving groups
may include acyl, phosphate, sulfate, sulfonate, and preferably are mono-, di-
and per-acyl
oxy-substituted compounds, where one or more of the pendant hydroxy groups are
protected by an acyl group, preferably an acetyl group. Typically acyloxy
substituted
compounds of the invention are readily cleavable to the corresponding hydroxy
substituted
compounds.
Chemical functional group protection, deprotection, synthons and other
techniques known
to those skilled in the art may be used where appropriate to aid in the
synthesis of the
compounds of the present invention, and their starting materials.
The protection of functional groups on the compounds and derivatives of the
present
invention can be carried out by well established methods in the art, for
example as
described in T. W. Greene, Protective Groups in Organic Synthesis, John Wiley
& Sons,
New York, 1981.
Hydroxyl protecting groups include but are not limited to carboxylic acid
esters, eg acetate
esters, aryl esters such as benzoate, acetals/ketals such as acetonide and
benzylidene, ethers
such as o-benzyl and p-methoxy benzyl ether, tetrahydropyranyl ether and silyl
ethers such
CA 02506238 2005-05-03
- 19 -
as t-butyldimethyl silyl ether.
Protecting groups can be removed by, for example, acid or base catalysed
hydrolysis or
reduction, for example, hydrogenation. Silyl ethers may require hydrogen
fluoride or
tetrabutylammonium fluoride to be cleaved.
It will be clear to persons skilled in the art of medicinal chemistry that
compounds of
formula (I) may be converted into other compounds of formula (I), for example,
where a
compound of formula (I) bears one or more hydroxyl substituents then one or
more of
these substituents can be converted in to a halo substituent such as bromo,
chloro or iodo
by treating the alcohol with a halogenating agent. Halogenating agents include
compounds
like NBS, hydrobromic acid, chlorine gas etc. It may be necessary during
processes such
as halogenation to use protecting groups to protect other functionality in the
molecule.
Phenolic type hydroxyls may not be readily convertible to the corresponding
halogen
compound by treatment with a halogenating agent. However, the desired halogen
compound may be prepared by, for example, treating an appropriate aryl amine
starting
material with NaNO2 in the presence of HCI under reduced temperature
conditions such as
0 C, to form the corresponding azide salt. Subsequent treatment with CuCI,
CuBr, KI or
IMF,' may be used to convert the azide into the required halo-compound.
A general process for preparing compounds of formula (I) comprises the step of
treating a
compound of formula (IV):
R10 0
(IV)
I ¨R3
R2
wherein RI, R2, R3 and W are as defined above in relation to compounds of
formula (II)
with a reducing agent to provide a compounds of formula (I) or a protected
derivative
CA 02506238 2005-05-03
- 20 -
thereof.
Reducing agents are well known to persons skilled in the art and can include
hydride
sources like borohydrides and alkali metal borohydrides, but would include
hydrogen in
catalytic hydrogenation where a suitable catalyst such as palladium on carbon
may be used.
Other suitable hydride sources include sodium triacetoxyborohydride tetrabutyl
ammonium
triacetoxyborohydride and sodium cyanoborohydride.
Preferably the double bond in compounds of formula (IV) is reduced by
hydrogenation.
Compounds of formula (IV) are prepared by dehydrating a compound of formula
(III):
R10 0
(III)
HO I ¨R3
R2
wherein RI, R2, R3 and W are as defined above, in relation to compounds of
formula (II) or
a protected derivative thereof.
Dehydration can, for example, be catalysed by acid, by base or facilitated by
conversion of
the tertiary alcohol into a better leaving group as would be known to those
skilled in the
art.
Preferably compounds of formula (III) are dehydrated, for example, by
treatment with
para-toluene sulphonic acid.
Compounds of formula (III) may be prepared by treating compounds of formula
(II):
CA 02506238 2005-05-03
- 21 -
R10 0
(II)
I ¨R3
0
R2
wherein RI, R2, R3 are as defmed above for compounds of formula (II) or a
protected
derivative thereof with an arylating agent, for example, a compound of formula
WM+
wherein W is an optionally substituted aryl radical and NI+ is one ore more
counter ions,
preferably [MgBr].
The arylating agent \VW+ may be prepared by Grignard chemistry where the
haloaly1
compound (V):
X
R5¨ R6 (V)
R4
or a protected derivative thereof wherein
R4, R5 and R6 are independently hydrogen, alkoxy, alkyl, acyl, OC(0)R7 or a
protected
hydroxy such as OSKRA)3, and
RA is independently alkyl or aryl, and
X is halo, preferably bromo,
is reacted with a metal such as magnesium to form the arylating agent.
Preferably the haloaryl compound (V) is selected from:
X X X
R6 40
(V R5
A) 41101 D N R5 R6B) NC)
R5
R4 R4 R4
CA 02506238 2005-05-03
- 22 -
wherein R4, R5, R6 and X are as defined above for compounds of formula (V).
Reaction of the arylating agent with the ketone of formula (II) provides
access to the
corresponding isoflavan-4-ols (III), isoflav-3-enes (IV) and isoflavans (I) of
the present
invention.
Alternatively compounds of formula (III) may be prepared by reacting compounds
of
formula (II) with a compound analogous to compounds of formula (V) wherein X
represents any appropriate leaving group L which is lost in the formation of
the product by
nucleophilic addition of the aryl moiety to a ketone by reactions well known
by those
skilled in the art.
Preferably any free alcohols, esters or other such reactive groups in the keto
compounds of
formula (II) will be protected, for example, as t-butyldimethylsilyl ethers
during the
nucleophilic addition reaction.
Compounds of formula (H) can be prepared by reducing the eneone double bond in
compounds of formula (VI):
Ri 0
(VI), _R3
0
R2
or a protected derivative thereof, wherein RI, R2 and R3 are as defined above,
for
compounds of formula (II).
Suitable reducing agents are described above. Preferably reduction of the
carbon-carbon
double bond can be effected, for example, by hydrogenation.
Access to compounds of general formula (VI) is available by general synthetic
methods as
set out in Scheme 1 below and as described in published International
application No.
CA 02506238 2005-05-03
- 23 -
W001/17986, the disclosure of which is incorporated herein by reference.
Ri0 OH
SI + HO 0 BF3/Et20
I )---R3 70 C
R2
R10 = OH R10 0 (VI)
MeS02C1
I ¨R3 DMF
0 0
R2
r%2
Scheme 1
Access to the variatious 3-phenyl substituted chromans is available by varying
the
substitution pattern on the phenylacetic acid derived group.
Access to the 4-phenyl substituted chromans is available by varying the
substitution pattern
of the arylating agent (V).
Analogues of compounds employed in the processes may be used which include a
substituent which corresponds to Rx as defined for compounds of formula (I).
As used herein, the terms "treatment", "prophylaxis" or "prevention",
"amelioration" and
the like are to be considered in their broadest context. In particular, the
term "treatment"
does not necessarily imply that an animal is treated until total recovery.
Accordingly,
"treatment" includes amelioration of the symptoms or severity of a particular
condition or
preventing or otherwise reducing the risk of developing a particular
condition.
The amount of one or more compounds of formula (I) which is required in a
therapeutic
treatment according to the invention will depend upon a number of factors,
which include
the specific application, the nature of the particular compound used, the
condition being
treated, the mode of administration and the condition of the patient.
CA 02506238 2005-05-03
- 24 -
Compounds of formula (I) may be administered in a manner and amount as is
conventionally practised. See, for example, Goodman and Gilman, "The
pharmacological
basis of therapeutics", 7th Edition, (1985). The specific dosage utilised will
depend upon
the condition being treated, the state of the subject, the route of
administration and other
well known factors as indicated above. In general, a daily dose per patient
may be in the
range of 0.1 mg to 5 g; typically from 0.5 mg to 1 g; preferably from 50 mg to
200 mg.
The length of dosing may range from a single dose given once every day or two,
to twice
or thrice daily doses given over the course of from a week to many months to
many years
as required, depending on the severity of the condition to be treated or
alleviated.
It will be further understood that for any particular subject, specific dosage
regimens
should be adjust over time according to the individual need and the
professional judgment
of the person administering or supervising the administration of the
compositions.
Relatively short-term treatments with the active compounds can be used to
cause
stabilisation or shrinkage or remission of cancers. Longer-term treatments can
be
employed to prevent the development of cancers in high-risk patients.
The production of pharmaceutical compositions for the treatment of the
therapeutic
indications herein described are typically prepared by admixture of the
compounds of the
invention (for convenience hereafter referred to as the "active compounds")
with one or
more pharmaceutically or veterinary acceptable carriers and/or excipients as
are well
known in the art.
The carrier must, of course, be acceptable in the sense of being compatible
with any other
ingredients in the formulation and must not be deleterious to the subject. The
carrier or
excipient may be a solid or a liquid, or both, and is preferably formulated
with the
compound as a unit-dose, for example, a tablet, which may contain up to 100%
by weight
of the active compound, preferably from 0.5% to 59% by weight of the active
compound.
One or more active compounds may be incorporated in the formulations of the
invention,
which may be prepared by any of the well known techniques of pharmacy
consisting
CA 02506238 2005-05-03
- 25 -
essentially of admixing the components, optionally including one or more
accessory
ingredients. The preferred concentration of active compound in the drug
composition will
depend on absorption, distribution, inactivation, and excretion rates of the
drug as well as
other factors known to those of skill in the art.
The formulations of the invention include those suitable for oral, rectal,
ocular, buccal (for
example, sublingual), parenteral (for example, subcutaneous, intramuscular,
intradermal, or
intravenous), transdermal administration including mucosal administration via
the nose,
mouth, vagina or rectum, and as inhalants, although the most suitable route in
any given
case will depend on the nature and severity of the condition being treated and
on the nature
of the particular active compound which is being used.
Formulation suitable for oral administration may be presented in discrete
units, such as
capsules, sachets, lozenges, or tablets, each containing a predetermined
amount of the
active compound; as a powder or granules; as a solution or a suspension in an
aqueous or
non-aqueous liquid; or as an oil-in-water or water-in-oil emulsion. Such
formulations may
be prepared by any suitable method of pharmacy which includes the step of
bringing into
association the active compound and a suitable carrier (which may contain one
or more
accessory ingredients as noted above).
In general, the formulations of the invention are prepared by uniformly and
intimately
admixing the active compound with a liquid or finely divided solid carrier, or
both, and
then, if necessary, shaping the resulting mixture such as to form a unit
dosage. For
example, a tablet may be prepared by compressing or moulding a powder or
granules
containing the active compound, optionally with one or more other ingredients.
Compressed tablets may be prepared by compressing, in a suitable machine, the
compound
of the free-flowing, such as a powder or granules optionally mixed with a
binder, lubricant,
inert diluent, and/or surface active/dispersing agent(s). Moulded tablets may
be made by
moulding, in a suitable machine, the powdered compound moistened with an inert
liquid
binder.
CA 02506238 2005-05-03
- 26 -
Formulations suitable for buccal (sublingual) administration include lozenges
comprising
the active compound in a flavoured base, usually sucrose and acacia or
tragacanth; and
pastilles comprising the compound in an inert base such as gelatine and
glycerin or sucrose
and acacia.
Formulations suitable for ocular administration include liquids, gels and
creams
comprising the active compound in an ocularly acceptable carrier or diluent.
Compositions of the present invention suitable for parenteml administration
conveniently
comprise sterile aqueous preparations of the active compounds, which
preparations are
preferably isotonic with the blood of the intended recipient. These
preparations are
preferably administered intravenously, although administration may also be
effected by
means of subcutaneous, intramuscular, or intradermal injection. Such
preparations may
conveniently be prepared by admixing the compound with water or a glycine
buffer and
rendering the resulting solution sterile and isotonic with the blood.
Injectable formulations
according to the invention generally contain from 0.1% to 60% w/v of active
compound
and can be administered at a rate of 0.1 ml/minute/kg.
Formulations for infusion, for example, may be prepared employing saline as
the carrier
and a solubilising agent such as a cyclodextrin or derivative thereof.
Suitable
cyclodextrins include a-cyclodextrin, P-cyclodextrin, y-cyclodextrin, dimethyl-
P-
cyclodextrin, 2-hydroxyethyl-3-cyclodextrin, 2-
hydroxypropyl-cyclodextrin, 3-
hydroxypropyl-P-cyclodextrin and tri-methyl-P-cyclodextrin. More
preferably the
cyclodextrin is hydroxypropyl-P-cyclodextrin. Suitable derivatives of
cyclodextrins
include Captisol a sulfobutyl ether derivative of cyclodextrin and analogues
thereof as
described in US 5,134,127.
Formulations suitable for rectal administration are preferably presented as
unit dose
suppositories. Formulations suitable for vaginal administration are preferably
presented as
unit dose pessaries. These may be prepared by admixing the active compound
with one or
more conventional solid carriers, for example, cocoa butter, and then shaping
the resulting
mixture.
CA 02506238 2005-05-03
- 27 -
Formulations or compositions suitable for topical administration to the skin
preferably take
the form of an ointment, cream, lotion, paste, gel, spray, aerosol, or oil.
Carriers which
may be used include Vasoline, lanoline, polyethylene glycols, alcohols, and
combination
of two or more thereof. The active compound is generally present at a
concentration of
from 0.1% to 5% w/w, more particularly from 0.5% to 2% w/w. Examples of such
compositions include cosmetic skin creams.
Formulations suitable for transdermal administration may be presented as
discrete patches
adapted to remain in intimate contact with the epidermis of the recipient for
a prolonged
period of time. Such patches suitably contain the active compound as an
optionally
buffered aqueous solution of, for example, 0.1 M to 0.2 M concentration with
respect to
the said active compound. See for example Brown, L., et al. (1998).
Formulations suitable for transdermal administration may also be delivered by
iontophoresis (see, for example, Panchagnula R, etal., 2000) and typically
take the form of
an optionally buffered aqueous solution of the active compound. Suitable
formulations
comprise citrate or Bis/Tris buffer (pH 6) or ethanol/water and contain from
0.1 M to 0.2
M active ingredient.
Formulations suitable for inhalation may be delivered as a spray composition
in the form
of a solution, suspension or emulsion. The inhalation spray composition may
further
comprise a pharmaceutically acceptable propellant such as carbon dioxide or
nitrous oxide
or a hydrogen containing fluorocarbon such as 1,1,1,2-tetrafluoroethane,
1,1,1,2,3,3,3-
heptafluoro-n-propane or mixtures thereof.
The active compounds may be provided in the form of food stuffs, such as being
added to,
admixed into, coated, combined or otherwise added to a food stuff. The term
food stuff is
used in its widest possible sense and includes liquid formulations such as
drinks including
dairy products and other foods, such as health bars, desserts, etc. Food
formulations
containing compounds of the invention can be readily prepared according to
standard
practices.
CA 02506238 2005-05-03
- 28 -
Therapeutic methods, uses and compositions may be for administration to humans
or other
animals, including mammals such as companion and domestic animals (such as
dogs and
cats) and livestock animals (such as cattle, sheep, pigs and goats), birds
(such as chickens,
turkeys, ducks), marine animals including those in the aquaculture setting
(such as fish,
crustaceans and shell fish) and the like.
The active compound or pharmaceutically acceptable derivatives prodrugs or
salts thereof
can also be co-administered with other active materials that do not impair the
desired
action, or with materials that supplement the desired action, such as
antibiotics,
antifungals, antiinflammatories, or antiviral compounds. The active agent can
comprise
two or more isoflavones or derivatives thereof in combination or synergistic
mixture. The
active compounds can also be administered with lipid lowering agents such as
probucol
and nicotinic acid; platelet aggregation inhibitors such as aspirin;
antithrombotic agents
such as coumadin; calcium channel blockers such as verapamil, diltiazem, and
nifedipine;
angiotensin converting enzyme (ACE) inhibitors such as captopril and
enalapril, and 13-
blockers such as propanolol, terbutalol, and labetalol. The compounds can also
be
administered in combination with nonsteriodal antiinflammatories such as
ibuprofen,
indomethacin, aspirin, fenoprofen, mefenamic acid, flufenamic acid and
sulindac. The
compounds can also be administered with corticosteroids or an anti-emetic such
as
zofran .
Compounds of formula (I) seem to be particularly suitable for co-
administration with one
or more anti-cancer drugs such as cisplatin, dehydroequol (DHE), taxol
(paclitaxel),
gemcitabine, doxorubicin, topotecan and/or camptothecin, especially cisplatin,
dehydroequol (DHE), taxol. This may result in improved effects in the
treatment, for
example in the form of synergistic effects, in comparison to when only one of
the
medicaments is employed. Particularly the compounds of the presently claimed
invention,
especially HMC (ie compound 1) seem to be chemosensitisers and increase the
cytotoxicity of the one or more anticancer drug co-administered therewith.
This seems to
be the case even though said anticancer drugs work through a variety of
different
mechanisms, for example cisplatin is thought to work by interacting with
nuclear DNA,
taxol is thought to work by blocking cells in the G2/M phase of the cell cycle
and prevent
CA 02506238 2012-05-18
71884-135
- 29 -
them forming normal mitotic apparatus, gemcitabine is thought to work by
incorporating
itself into the DNA of the cell, ultimately preventing mitosis, doxorubicin is
though to be a
topoisomerase 11 inhibitor thereby preventing DNA replication and
transcription and
topotecan is thought to be a topoisomerase I inhibitor.
Interestingly, in some situations this increased cytotoxicity to cancerous
cells is not
associated with a corresponding increase in toxicity to non-cancerous cells.
Whilst this observation has important implications for the treatment of many
cancers, it is
especially important to the treatment of cancers such as melanoma, which are
extremely
difficult to treat.
The co-administration may be simultaneous or sequential. Simultaneous
administration
may be effected by the compounds being in the same unit dose, or in individual
and
discrete unit doses administered at the same or similar time. Sequential
administration
may be in any order as required and typically will require an ongoing
physiological effect
of the first or initial active agent to be current when the second or later
active agent is
administered, especially where a cumulative or synergistic effect is desired.
The invention also extends to a pack comprising the combination therapy.
Compounds for use in the preferred synthetic methods of the present invention
may be
derived from any number of sources readily identifiable to a person skilled in
the art. For
example, daidzein is readily available or can be synthesised by standard
methods known in
the art. Suitable methods may be found in, for example, published
international patent
applications WO 98/08503 and WO 00/49009, and references cited therein
Compounds of the general formulae (II), (III) and (IV) described above are
intermediates
in the production of the active isoflavan compounds of formula (1). These
intermediates
also represent fiwther aspects of the present invention.
CA 02506238 2005-05-03
- 30 -
Whilst not wishing to be bound by theory the compounds of the present
invention are
thought to regulate a wide variety of signal transduction processes within
animal cells and
that these signal transduction processes are involved in a wide range of
functions that are
vital to the survival and function of all animal cells. Therefore, these
compounds have
broad-ranging and important health benefits in animals including humans, and
in particular
have the potential to prevent and treat important and common human diseases,
disorders
and functions, which represents a substantial unexpected benefit.
Thus it seems that the compounds of the present invention have activity as
TNFa
inhibitors. It Is hypothesised that TNFft is part of a tightly regulated
cytokine network,
activating multiple signal transduction pathways and inducing or suppressing a
wide
variety of genes. TNFa can provide a survival signal for cancer cells and
hence it has been
referred to as a tumour promoting factor. As a central mediator of
inflammation, TNFa
provides a molecular link between chronic inflammatory stimuli and the
subsequent
development of malignant disease. Consequently its inhibition by the compounds
of the
invention may provide one mechanism by which they exert anti-cancer and/or
anti-
inflammatory activity. Alternatively, these compounds may be used as
chemopreventative
agents.
The particular benefits of this invention lie in (a) the large range of signal
transduction
processes targeted by the compounds, (b) the fact that regulation of these
various processes
includes both up-regulation of some processes and down-regulation of others,
and (c) that
such a broad and varied effect on signal transduction processes also is
accompanied by an
independent effect on a range of important enzymes that are fundamental to
metabolism
and steroidogenesis.
The isoflavan compounds of the present invention exhibit good in vitro
toxicity profiles
against normal cells. The isoflavans have broad activity, markedly better than
or at least
comparable with dehydroequol. The isoflavans are highly active against cancer
cells
representative of leukaemia, glioma, prostate, ovarian, breast and lung
cancer. The
isoflavan compounds show potent activity against melanoma and
cholangiocarcinoma
(gall bladder cancer) cell lines. Good activity was observed against
colorectal cancer
CA 02506238 2005-05-03
-31 -
cells.
Radio-sensitisation in vivo may be tested for example employing human
epidermoid vulval
carcinoma A431 tumours established on the upper leg and subjected to several
doses of
local radiation (to the tumour bearing leg only). A radiation treatment
regimen of
2.5Gy/day for 4 days will delay tumour growth, and the effect of the radiation
dose in
combination with the test compound could be assessed by monitoring tumour
growth
delay. Tumour growth delay of ¨6 days can be expected using radiation alone.
Tumour
growth delay using orally dosed test compound can be determined separately.
Evidence of
test compound mediated radio-sensitisation of A431 tumours is then determined
by
measuring tumour growth delay using a regimen of orally dosed test compound
pre-treated
animals followed by the standard radiation therapy regimen described above. A
mean
growth delay of up to 30 days using the combination treatment compared to up
to 10 days
using either radiation or test compound monotherapy regimens is evidence of
the radio-
sensitisation properties of the compounds of the invention.
Radio-sensitisation in vitro may be tested, for example, employing clonogenic
assays using
human the human epidermoid vulva carcinoma A431 cell line to measure response
to
radiation alone or in combination with test compounds. A drug dose causing 10%
toxicity
to the cells may be used in combination with graded doses of radiation. The
appropriate
dose of compound would be determined by clonogenic assay. Evidence of test
compound
mediated radio-sensitisation is shown by, for example, a >20% toxicity to
cells using
chemoradiation therapy compared to 10% toxicity using the corresponding
monotherapy
regimens.
The invention is further illustrated by the following non-limiting Examples
and
accompanying drawings.
Examples
In the Examples and accompanying drawings which follow the abbreviation "DHE"
is used
for dehydroequol and the abbreviation "HMC" is used herein for the compound, 3-
(4-
hydroxypheny1)-4-(4-methoxypheny1)-chroman-7-ol.
CA 02506238 2005-05-03
-32-
1Ø Synthesis
Example 1: 4',7-Diacetoxydaidzein
HO 0 ,Ir0 0
Acetic Anhydnde
OH Pyndine
0 ojC(
0 101
A mixture of daidzein (2.0 g), acetic anhydride (10 ml) and pyridine (2 ml)
was heated at
105-110 C for lh. After cooling the mixture to room temperature, it was
stirred for a
further 30 min during which time the diacetate crystallised from solution. The
product was
filtered, washed thoroughly with water and recrystallised from methanol to
yield 4',7-
diacetoxydaidzein as colourless prisms (2.4 g, 90%).
Example 2: 7-Acetoxy-3-(4-acetoxyphenyl)chroman-4-one
õ...õ00 0
H2, Catalyst
II 1110
0
0 IN ,
0 )1'N''
01
Palladium-on-charcoal (5%, 0.02g) was added to a solution of 4',7-
diacetoxydaidzein
(0.50g, 1.5 mmol) in ethyl acetate (80 ml) and the mixture was stirred at room
temperature
under a hydrogen atmosphere for 72h. The catalyst was removed by filtration
through
Celite and the resulting filtrate was evaporated in vacuo. The residue was
recrystallised
from ethanol to yield 7-acetoxy-3-(4-acetoxyphenyl)chroman-4-one (0.40g, 80%)
as
colourless plates.
Example 3: 7-Hydroxy-3-(4-hydroxyphenyl)chroman-4-one
CA 02506238 2005-05-03
-33-
0 Imidazole HO 0
0
IR
0 0
OH
Imidazole (0.63g) was added to a suspension of 4',7-diacetoxydihydrodaidzein
(0.26g, 0.08
mmol) in absolute ethanol (5.0 ml) and the mixture was refluxed for 45 min
under argon.
The solution was concentrated under reduced pressure and distilled water (10
ml) was
added to the residue. The mixture was left overnight under refrigeration and
the resulting
precipitate was filtered. The crude product was recrystallised from ethyl
acetate/dichloromethane to yield 7-hydroxy-3-(4-hydroxyphenyl)chroman-4-one
(0.14g,
71%) as a white powder.
Example 4: 7-(tert-Butyldimethlysilyloxy)-3-(4-(tert-butyldimethlysilyloxy)
phenyl)ehroman-4-one
HO 0 __Fdi_0 0
tBMSCI
1111
Imidazole
0
OH 0 04 I
I I
7-Hydroxy-3-(4-hydroxyphenyl)chroman-4-one 42g, imidazole 130g, tert-
butyldimethylsily1 chloride 127g, and N,N-dimethylformamide (500m1) were
combined in
a 2L round bottom flask and stirred under nitrogen at room temperature for 16
hours. The
reaction was quenched with the addition of chilled H20 (200m1) with the
reaction mix
cooled in an ice bath. The resultant white solid was filtered and rinsed with
water.
Recrystallisation from ethanol afforded the product as white fluffy crystals
(35.7g).
Example 5: 7-(tert-Butyldimethylsilyloxy)-3-(4-(tert-
butyldimethylsilyloxy)phenyI)-4-
(4-methoxyphenyl)chroman-4-ol
CA 02506238 2005-05-03
- 34 -
MgBr
I 0
OMe I Si¨.
101
CI 0- s ____________________ HO
41 I. 0-SI+
OMe
7-(tert-Butyldimethylsilyloxy)-3-(4-(tert-butyldimethylsilyloxy)pheny1)-4-(4-
methoxypenyl)chroman-4-ol 25g was weighed in a 2-neck round bottom flask, and
flushed
under nitrogen. Anhydrous THF 80m1 was added to the reaction vessel to give a
clear
slightly yellow solution. A condenser was attached and the reaction vessel
placed in an ice
bath. Commercial 4-methoxyphenylmagnesium bromide (0.5M solution in THF) 225m1
was added to the reaction mix dropwise over 10 minutes. The reaction was
quenched by
the dropwise addition of wet ether (50:50 H20: diethyl ether) while still
under nitrogen,
with a white precipitate forming as increasing amounts of H20 was added. A
further
amount of H20 was added to the reaction mix before extraction with diethyl
ether.
The organic layers were combined and washed with water, brine, dried over
anhydrous
MgSO4 and solvent removed on rotovap to give clear yellow oil which solidified
overnight
to give an off-white solid. The crude product was used in the next step
without
purification.
Example 6: 3-(4-Hydroxypheny1)-4-(4-methoxypheny1)-2H-chromen-7-ol
0 HO 0
40 pTs0H
HOtairi IP I __
OH
OMe OMe
7-(tert-Butyldimethylsilyloxy)-3-(4-(tert-butyldimethylsilyloxy)pheny1)-4-(4-
methoxyphenyl)chroman-4-ol (42g) , pTs0H (435g), boiling chips and 2.5L of
ethanol
CA 02506238 2005-05-03
- 35 -
were combined in a 2-neck 5L round bottom flask with condenser attached. The
reaction
was heated at reflux for 3 hours. The solvent was concentrated in vacuo to
¨100m1 before
being poured into chilled, stirred water (700m1). The mixture was then
extracted with
ethyl acetate, the combined organic layers washed with water (3 x 2L), brine
(1 x 500m1),
dried over anhydrous magnesium sulphate and filtered and solvent removed in
vacuo to
give red/ brown oil. The oil was dissolved in methanol (-100m1) and put in
freezer
overnight.
A white precipitate had formed overnight, which was filtered off and rinsed
with methanol.
The filtrate was concentrated in vacuo to give a brown oil.
Example 7: 3-(4-11ydroxypheny1)-4-(4-methoxypheny1)-chroman-7-ol
HO 401 0 HO 0
H2 /Pd
OH ethanol
4111 OH
OMe OMe
3-(4-hydroxypheny1)-4-(4-methoxypheny1)-2H-chromen-7-ol 25,5g (70mmoles), 10%
Pd/A1203 3.95g and 200m1 of ethanol were combined in a 2-neck 500m1round
bottom
flask. The reaction was hydrogenated at low pressure using standard conditions
for 3
hours. The reaction was filtered through Celite to remove the catalyst, rinsed
through with
ethanol (300m1). The filtrate was concentrated to ¨50m1 before being poured
into chilled,
stirred water (1.4L). A pale orange precipitate formed which then formed a
brown oil.
The mixture was then extracted with diethyl ether, the combined organic layers
washed
with water (3 x 1L), brine (1 x 500m1), dried over anhydrous magnesium
sulphate and
filtered. The solvent was removed in vacuo to give red/ brown oil. The product
was
recrystallised from diethyl ether (-100m1), to give brown solid which was
rinsed with
chilled diethyl ether to give off-white crystals 11.3g. The 1H NMR spectrum
and
numbering scheme being shown below.
CA 02506238 2005-05-03
-36-
8
HO 7 8a 0 2
3= 2'
6
4- 3
4
OH
5 41/ 2" 5'
4" 3"
9/0
C2equatorial 4.14 Dd 10.98 1
C2axial 4.35 Dd , 1 Dd is overlapping
C3 3.47 Ddd 1
C4 4.20 Dd 5.12 1
C5 6.71 D 8.05 1
C6 6.36 Dd 2.56, 8.42 1 ,
C8 6.41 D 2.20 1
C9 3.71 3
C2', C6' 6.61 2 Doublets overlapping for C2',C3',C2"
C3"
C3', C5' 6.61 2 Total integration is 8
C2", C6" 6.61 2
C3", C5" 6.61 2
In the above general methods, the structures may be optionally substituted or
protected
with appropriate substituents, or synthons or derivatives thereof The
compounds may be
present as, for example, their salts, acetates, benzyl or silyloxy derivatives
as can be
determined by a skilled synthetic chemist. Hydroxy groups can be readily
alkylated
(Mel/base), acylated (Ac20/Py) or silylated (C1-SiR3/base) and likewise
deprotected by
standard methods known in the art.
Example 8: 3-(4-Hydroxypheny1)-4-(4-hydroxyphenyI)-chroman-7-ol
3-(4-hydroxypheny1)-4-(4-methoxyphenyl)chroman-7-ol (3.17g) was transferred to
a round
bottom flask and the flask was purged with nitrogen. 33 wt.% Hydrogen bromide
in acetic
acid (13m1) was added drop-wise to the flask and the contents were heated to
reflux at
130 C for 7 hours. The reaction mixture was placed in an ice bath and adjusted
to pH 6
using sodium hydroxide solution (40%w/v). The mixture was extracted with ethyl
acetate
and the ethyl acetate layer was further washed with water and brine prior to
drying over
CA 02506238 2005-05-03
- 37 -
magnesium sulphate. The mixture was filtered and the solvent was removed in
vacuo to
yield a brown solid (2.89g). The solid was dissolved in minimal ethyl acetate
and purified
by column chromatography (Silica 60H, 200-400mesh using ethyl
acetate:chloroform
(40:60) eluant). 3-(4-hydroxypheny1)-4-(4-hydroxyphenyl)chroman-7-ol was
obtained in
¨80% purity and was further purified by semi-preparative high performance
liquid
chromatography (HPLC) see Fig. 12.
2Ø Materials and Methods
2.1. Tissue culture
The human pancreatic cancer cell line, HPAC (CRL-2119) was routinely cultured
in 1:1
mixture DMEM (Dulbecco's Modified Eagle Medium Sigma) plus Ham's F12 (Sigma)
medium containing HEPES (15 mM), insulin (0.002 mg/ml), transferrin (0.005
mg/ml),
hydrocortisone, (40 ng/ml), epidermal growth factor (10 ng/ml). The ovarian
cancer cell
lines; CP70 was obtained as a gift from Dr. Gil Mor (Yale University) and
cultured in a 1:1
mixture DMEM plus Ham's F12 medium, and SKOV-3 (ovarian cancer cell line) was
purchased from ATCC and cultured in McCoys 5a medium. The breast cancer cell
line
MDA-MB-468 cultured in Leibovitz's L-15 medium. The melanoma cell line MM200
was obtained as a gift from Peter Hersey (University of Newcastle) and A2058
was
obtained as a gift from Dr Peter Parsons (QIMR). Both were cultured in DMEM
medium.
All cultures were supplemented with 10% FCS (fetal calf serum CSL, Australia),
penicillin
(100U/m1), streptomycin (100mg/m1), L-glutamine (2mM) and sodium bicarbonate
(1.2
g/L), and cultured at 37 C in a humidified atmosphere of 5% CO2. All cell
lines were
purchased from ATCC (Maryland, USA) except where noted.
The normal cell line NFF (neonatal foreskin fibroblasts) was a gift from Dr.
Peter Parsons
(Queensland Institute of Medical Research). RK (rabbit kidney) cells were
obtained from
Miller Whalley (Macquarie University). Both cell lines were cultured in RPMI
supplemented with 10% FCS (CSL, Australia), penicillin (100U/m1), streptomycin
(100mg/m1), L-glutamine (2mM) and sodium bicarbonate (1.2 g/L), and cultured
at 37 C
in a humidified atmosphere of 5% CO2.
CA 02506238 2005-05-03
-38-
2.2. Proliferation assays
1050 values were determined for each cell line. Cells were seeded in 96-well
plates at an
appropriate cell density as determined from growth kinetics analysis and
cultured for 5
days in the absence and presence of the test compounds. Cell proliferation was
assessed
after the addition of 20 ul of 3-4,5 dimethylthiazol-2,5-diphenyl tetrazolium
bromide
(MTT, 2.5mg/m1 in PBS, Sigma) for 3-4hrs at 37 C according to manufacturer's
instructions. 1050 values were calculated from semi-log plots of % of control
proliferation
on the y-axis against log dose on the x-axis.
2.3. DHE and HMC Pharmacokinetics - Oral
HMC and DHE were prepared as homogenous suspensions in 1% CMC (m:v, water).
Both
formulations were delivered orally by gavage to female BALB/c mice at a dosage
of 50
mg/kg. Three animals were allocated to each timepoint (15 min, 30 min, lhr, 4
hr and 24
hr). At each respective timepoint, animals were euthanased by cervical
dislocation and
blood collected. Free HMC was analysed by mass spectroscopy.
2.4. AMC Pharmacokinetics ¨ i.v. and i.p.
HMC was prepared as a solution in 20% hydroxypropyl-beta-cyclodextrin (m:v,
water).
The formulation was delivered either orally by gavage or by i.p. injection to
female
BALB/c mice at a dosage of 50 mg/kg. Three animals were allocated to each
timepoint (15
min, 30 min, 1 lir, 4 hr and 24 hr). At each respective timepoint, animals
were euthanased
by cervical dislocation and blood collected. Urine was also collected and
analysed for
HMC. Free 11/v1C was analysed by mass spectroscopy.
2.5 Pilot in vivo efficacy study ¨ HPAC tumour bearing mice
Subconfluent (80%) flasks of HPAC cells were trypsinised, washed in Hanks
balanced salt
solution (Sigma), resuspended in dubellco's minimal essential medium (Sigma)
and an
equal volume of matrige1174 (Becton Dickson) at a density of 3.7 x 107cells
per ml.
Athymic nu/nu BALB/c mice were s.c. inoculated with 3.7x106 HPAC cells bi-
laterally
midway along the dorsal surface. For the HMC (n=3 per dosage regimen) and
control
groups (n=2), treatment commenced five days post inoculation to allow tumour
formation.
HMC was formulated 20% HPBCD and delivered i.p. daily for 15 days. The control
group
CA 02506238 2005-05-03
- 39 -
received equivalent (weight:weight) i.p. doses of 20% HPBCD. Tumour
measurements
commenced on day 5 post inoculation (10 x10 mm2) and were measured in 2
dimensions,
length (a) and width (b), using calipers. Tumor weight (W) was calculated by
the formula
W=ab2 /2, where a, is the longer of the 2 measurements (Odwyer et al., 1994).
Tumour
proliferation curves were analyzed with respect to maximal tumour inhibition
(treated/control, TIC). On sacrifice, liver, kidney, femur, stomach and colon
tissue were
fixed in buffered formalin, embedded in paraffin, sections cut and stained
with H&E.
Stained sections were then submitted to Rothwell consulting for histopathology
analysis.
Serum biochemistry was conducted on bloods taken from control, vehicle control
and
HMC treatment groups. Serum analysis was conducted by Veterinary Clinical
pathology
(U. Syd).
2.6 Three-D model analysis of synergy
3-D model analysis of the cytotoxic interaction between drug A and drug B
enables the
representation of predicted inhibitory effect of two drugs in combination in 3
dimensions
to reveal actual regions of synergy or antagonism. The 3D synergy plots are
based on a
theory of "Theretical Additivity" (TA) as outlined by Kanzawa et al (Int. J.
Cancer 71,
311-319 (1997)). Theoretical Additivity was calculated from the cytotoxicities
of drug A
and drug B as monotherapies using the following formula which assumes the
drugs are
mutually exclusive inhibitors:
(fa)A (fal3 2(f0A(fa)B
TA(1)
1 - (1a)A(T'a)13
Where: (f.)A = fraction of cells affected by drug A
(fa)B = fraction of cells affected by drug B
The TA is calculated for each combination of drug concentrations and
subtracted from the
observed experimental effect for each combination to give a measurement of
synergistic
action. A positive difference indicates that more cells are affected by the
drug combination
than would be expected in theory if the two drugs were administered together ¨
hence
synergism. A negative difference indicates that less cells were affected than
theoretically
expected ¨ hence antagonism.
CA 02506238 2005-05-03
- 40 -
3Ø Results
3.1. Normal cell toxicity
Dehydroequol (DHE) was less toxic to both NFF and rabbit kidney cells with
IC50 values
above 150 1.1.M when compared to HMC (86 and 611.1M respectively) (Table 1 and
Figure
1). When compared to cisplatin a benchmark chemotherapeutic agent, the degree
of
toxicity exhibited by HMC is mild (9.9 M vs 86 M).
Table 1. Relative toxicity of DHE, HMC and cisplatin against Neonatal foreskin
fibroblasts (NFF) and rabbit kidney cells.
Antineoplastic
Tissue/cell Type Designation Analogue (IC50 uM) (IC50 uM)
DHE HMC Cisplatin
Neonatal Foreskin Fibroblasts
Fibroblast >150 86.12 7.6 9.85 5
(Human, NFF)
Kidney Rabbit Kidney >150 61 4.3 Not tested
3.2. In vitro efficacy against cancer cells
When compared to DBE IC50 values, HMC demonstrated markedly superior activity
(-5-
fold greater) against the multi-drug resistant, p53 mt ovarian cancer cell
line (SKOV-3),
the AR negative, p53 Mt prostate cancer cell line (PC3), both ER positive (p53
wt) and
negative (p53 mt) breast cancer cell lines (MCF-7 and MDA-MB-468
respectively), p53
Mt Glioma (HTB-138), p53 Mt pancreatic cancer (HPAC) and p53 Mt large cell
lung
cancer (Table 2). HMC exhibited anti-cancer activity comparable to that of DHE
against
all other cell lines tested (Table 1). Particular efficacy of HMC was noted
against
melanoma cells. (Table 2 and Fig 2). This represents a substantial advantage
over the
prior art.
HMC was differentially active against 2 separate colorectal cell lines, with
marked activity
observed against HT-29 cells and somewhat less activity against HCT-15. It is
noted that
HT-29 and HCT-15 are COX-2 positive and deficient respectively. When examined
microscopically and compared to cells treated with vehicle only, HMC treated
SKOV-3
cells exhibited morphological changes consistent with cells undergoing
apoptosis (cell
enlargement, granular appearance in cytosol and blebbling of plasma membrane).
In
CA 02506238 2005-05-03
- 41 -
contrast SKOV-3 cells exposed to 100 1.1M Dehydroequol after 18 hr retained a
relatively
normal morphology, comparable with that of vehicle only treated cells.
Table 2. Comparison of Dehydroequol and HMC cytoxicity against cell lines
representative of different malignancies.
Antineoplastic
Analogue (IC50 uM)
Indication Designation (IC50 uM)
DHE HMC Cisplatin
A2780 1.7 0.61 1.58 0.59 2.10
Ovarian CP70 1.69 0.62 1.21 0.29 10.30
SKOV-3 21.83 4.65 2.26 5.40
PC3 9.09 t 8.12 2.9 0.92 2.11
Prostate LNCaP 4.8 3.8 4.52 >10
DU145 5.95 1.5 , 3.78 2.07
MCF-7 21.5 13.2 7.15 7 3.69
Breast
MDA-MB-468 7.9 3.5 1.1 0.35 0.58
Glioma HTB-138 7.35 0.89 1.9 0.27 , 42.30
Pancreatic CRL-2119 56.62 16.8 14.1 1.16 9.36
RPMI-8226 3.72 0.08 NT NT
Leukemic
CCRF-CEM 1.7 0.68 1.90 NT
NCI-H23 8.75 7.2 3.75 2.5 , NT
Lung
NCI-H460 10.6 3.8 2.23 0.15 22.29
HT-29 50.45 21.9 3.7 1.4 22.7 35
Colorectal
HCT-15 24.4 12.57 37.8 33 129.9 39
MM200 2.90 0.7 .03 8.3 0.7
Melanoma A2058 NT 1.2 0.65 5.73 2.3
IGR-3 NT 0.53 0.02 NT
3.3. HNC Pharmaeokinetics - oral
When compared with the phannacokinetic profile of orally dosed DHE, HMC
administered via the same route and dosage (50 mg/kg), HMC exhibited a Cmax of
141pM (achieved after 1 hr) compared to 511 M for DHE (achieved after 15 min)
(Table
3 and Figure 3). Like DHE, HMC is also subject to conjugation with low plasma
concentrations of the free form of the molecule observed (1.3 p.M after 30
min) (Table 3
and Figure 3). This is less than half the maximum concentration of free
dehydroequol
achieved using the same dosage regimen (3.3 ;AM after 15 min) (Figure 3). The
ratio of
free:total is greater for HMC when compared to DHE (0.92 vs 0.64
respectively).
Table 3. Comparison of free and total plasma concentrations achieved in mice
dosed with
50 mg/kg of either HMC or DHE p.o.
CA 02506238 2005-05-03
- 42 -
HMC (uM) DHE uM)
Time
Total Free Total Free
0.25 72 4.4 0.38 0.04 511.5 99 3.3 0.13
0.5 122 18.4 1.3 0.2 357 82 2.9 0.05
1 141 45.8 0.95 0.4 387 22.8 1.5 0.11
4 33.9 12.7 0.19 0.08 117.6 42 1.3 0.07
24 0 0 0.13 0.1 0.15 0.04
3.4. HMC pharmacokineties - i.v. and i.p.
When formulated in HPBCD and delivered i.v., extremely high levels of HNC were
observed in the blood equating to 1 mM of drug, 15 min post administration
(Figure 4).
Elimination kinetics of i.v. delivered HMC were biphasic with HMC being
rapidly
excreted from blood at a rate of -1000 uM/hr in the first hr post
administration. Assuming
linear excretion, this rate slowed to 0.97 uM/hr in hours 1-4 hr post
administration. When
the same formulation was administered i.p., approximately 1 log less HMC was
observed
in plasma (131 1.L114 by i.p. administration vs 1069 AM by i.v.
administration) up to 1 hour
post administration (Fig 4). Elimination kinetics by i.p. administration
however, was much
slower during this period (112 p,M/hr) thus resulting in a serum concentration
some 4.5
fold higher at 1 hr post administration (18.7 by i.p. vs 3.98 by i.v.).
Conversely, in hours 1-
4 post administration, elimination kinetics was faster after i.p.
administration when
compared to i.v. (4.6 AM/hr by i.p. vs 0.97 ilM/hr by i.v.). These data
confirm that HMC
is highly bioavailable in its free state when administered by i.v. or i.p.
routes. In
conjunction with oral PK data, these data also suggest that HMC is susceptible
to rapid
removal by GI detoxification enzymes. Large concentrations of free HMC were
observed
in urine over 0.5, 1 and 4hr where collected (3.3 mM, 3.9 mM and 0.093 mM).
Table 4. Comparison of the pharmacokinetic profile of IIMC in serum after i.v
and i.p
administration of HMC formulated in 20% hydroxypropyl beta cyclodextrin at a
dose of 50
mg/kg). Inset shows HMC concentrations in serum.
CA 02506238 2005-05-03
- 43 -
Serum HMC (uM)
Time (hr)
IV ip
0.25 1069.75 131.37
0.50 198.66 31.78
1 3.98 18.74
4 0.07 0.15
24 0.05 0.17
3.5. Pilot in vivo efficacy study ¨ HPAC tumour bearing mice
HMC when dosed daily, i.p. at 100 mg/kg significantly retarded the
proliferation of HPAC
tumours over the treatment period when compared to vehicle control (Figure5).
When the
mean terminal tumour mass was assessed a significant reduction in final tumour
burden
(%T/C = 62) was also noted (Figure 6). Importantly, no signs of toxicity were
noted in
animals dosed with HMC at 100 mg/kg daily for 15 days as determined by weight
loss.
Indeed animals treated with HMC appeared to thrive when compared to control
(Figure 7).
Organs (liver, kidney, spleen, femur, stomach and colon) were collected and
submitted for
histopathological assessment by Rothwell consulting. A limited serum
biochemistry
analysis was also conducted. These data confirm that HMC demonstrates
antitumorigenic
activity against HPAC tumours in vivo.
3.5.1. Histpathological examination of HMC treated groups
Histopathological examination of haematoxylin and eosin-stained sections cut
from
formalin-fixed tissues from two series of experimental mice was made. The
liver, kidney,
stomach and colon were examined for evidence of toxic damage, the spleen and
bone
marrow for evidence of myelosuppression and the tumour for degree of necrosis.
A score
of 0-5 was allocated to each tumour specimen for the degree of necrosis
present, a 0 score
representing, no necrosis and a score of 5, total necrosis. The sections were
scored 'blind'
on two separate occasions and the final score given in the results is the mean
of these two
scores.
CA 02506238 2005-05-03
- 44 -
Table 5. HMC Toxicology
Sample Description Necrosis Score 0-5
4/04 & 1/8 Vehicle control 0.5
4/04 & 2/8 2
4/04 & 4/8 HMC 2
4/04 & 5/8 2
4/04 & 1/11 No treatment control 0.5
3.5.1.1. Overview of results
No evidence of toxicity or myelosuppression was detected in sections cut from
the tissues
of the drug-treated mice. However, in all the drug-treated mice there were
patchy
mild/moderately severe chronic inflammatory changes affecting the serosa and
attached
mesentery, as well as reactive changes of the mesothelial cells, in some of
the tissues
examined. These changes are consistent with the intra-peritoneal injection of
a mildly
irritant material.
Significant necrosis of tumour tissue was not detected in control specimens
1/8 and 1/11.
However, there was considerable necrosis in the tumour sections from the drug-
treated
mice.
3.5.1.2. Serum biochemistry of HIVIC treated mice in comparison to control
Alakalines phosphatase (ALP), alanine transferase (ALT) and creatine (Cre)
were assessed
in HMC treated vs control animals. ALP and Cre levels were similar to control
and fell
within normal ranges (for rat) however, ALT levels in vehicle control and HMC
treated
groups were much lower than no treatment control levels.
Table 6
Clinical marker (mice)
Sample Group ALP ALT Cr. Urea
U/L U/L uM mM
Control 1/11 116 713 7 6.92
Vechicle Control 1/8 152 441 26 9.81
HMC treated 2/8 74 505 17 7.27
100
4/8 111 482 8 8.11
mg/kg)
(
5/8 100 494 8 7.79
Normal ranges) 86- 246 84 - 143 1.5 - 6 6.3- 8*
= for rat
ALP: Alakaline phosphatase
ALT: Alanine aminotransferase
Cre: Creatinine
. CA 02506238 2005-05-03
- 45 -
3.6. HMC induced apoptosis in melanoma cells and normal fibroblasts
3.6.1. Melanoma
HMC induced apoptosis in all TRAIL-sensitive and ¨resistant melanoma cells at
concentrations down to 2pM (-7-10% apoptosis) over 24 and 48 hrs of exposure
(Table 7
and Figure 8A). At the clinically significant drug concentration of 4 ILM, the
incidence of
apoptotic cells after 24 hr exposure to HMC rose to 25% and 39% in TRAIL
sensitive
(MEL-RM) and TRAIL negative (IGR3) cell lines respectively (Table 7 and Figure
8A).
The incidence of HMC induced apoptosis at 4 j.tM after 24 hr exposure in the
other cell
lines was ¨9%. In comparison, the incidence apoptosis in DBE- treated cells at
a
concentratoin of 4 p.M after 24 hr exposure was 0-1%. Over 48 hr at the same
concentration of HMC (41.1M), the incidence of apoptosis rose to 21-42 % in
all cell lines
examined (Table 7 and Figure 8B). DHE was the only other agent to induce
moderate
levels of apoptosis after 48 hr exposure at a concentration of 4p,M, but only
in ME4405
(14%) and Mel-AT (15%) cell lines (Table 7 and Figure 8B).
Table 7. Summary of apoptosis incidence in DHE and HMC treated melanoma cells
over
24 and 48 hr.
Drug Percent Apoptosis
Concentration TRAIL sensitive Trail-resistant .. Trail
+ve /
(1M) Caspase 8 -
ye
MEL-RM ME4405 IGR3 Mel-AT
DHE HMC DHE HMC , DHE . HMC DHE HMC
Control 0 0 0 0 0 0 0 0
co 1 0 0 0 0 0 8 o o
1" g 2 0 8 2 8 0 11 3 6
7, g. 4 0 25 2 9 1 _ 39 4 9
01
ell 8 2 39 6 11 3 41 8 23
20 5 38 11 19 4 32 10 24 _
Control 0 0 0 0 0 0 0 0
a) 1 1 1.5 1 1.5 1 1 1.5 1
1..
1 g 2 1.5 8 3 8 1 9 5 7
4 3 22 14 42 2 21 15 25
m
8 10 38 40 20 8 38 43 59
20 12 30 35 18 8 38 40 35
CA 02506238 2005-05-03
- 46 -
3.6.2. Normal fibroblasts
Studies were conducted on normal fibroblasts (MRC-5) and TRAIL-sensitive
melanoma
cells (ME4405 and MEL-RM) using 81.tM of either DHE or HMC over 24 and 48 hr
of
exposure (Figure 9). These data demonstrate that HMC, and to a lesser extent
DHE,
induced marked levels of apoptosis in both melanoma cell lines over 24 and
48hr.
Importantly, while promoting programmed cell death in malignant cells, normal
fibroblasts
were shown to both HMC and DHE induced apoptosis at 81AM drug over 24 and 48
hr of
exposure. These data confirm that HMC is selectively cytotoxic to cancer
cells.
The isoflavan compounds of the invention exhibit a superior efficacy profile
against all
cancers tested when compared to DHE. While HMC is marginally more toxic than
DBE in
NFF and RK cells, HMC is markedly less toxic than cisplatin. HMC delivered
orally in
mice is less bioavailable when compared to DBE but the ratio of free:total is
greater.
HPBCD-formulated HMC was markedly bioavailable in its free form when delivered
i.v
and i.p. Significant serum concentrations of free HMC post delivery i.p. were
some 18 fold
above that of orally delivered HMC. It has been demonstrated that HMC,
formulated in
20% HPBCD and delivered i.p., exerts a moderate antitumorigenic activity
against HPAC
tumours in vivo. HMC when delivered at 100 mg/kg to mice is not toxic to major
organs as
determined by histopathology however, in all the drug-treated mice there were
patchy
mild/moderately severe chronic inflammatory changes affecting the serosa and
attached
mesentery, as well as reactive changes of the mesothelial cells which are
consistent with
the intra-peritoneal injection of a mildly irritant material.
EIMC induced moderate-strong levels of apoptosis in TRAIL-resistant and TRAIL-
sensitive melanoma cells after both 24 and 48 hrs of exposure. Normal
fibroblast cells
were resistant to apoptosis after 48 hrs exposure DHE induces mild-moderate
levels of
apoptosis in TRAIL-resistant and TRAIL-sensitive melanoma cells after 48 hrs
of
exposure. Normal fibroblast cells were resistant to apoptosis after 48 hr
exposure. The
ability of both HMC and DHE to induce apoptosis in caspase negative cells
suggest that an
operational extrinsic programmed cell death pathway is not essential for HMC
and DBE
mediated apoptosis.
CA 02506238 2005-05-03
- 47 -
3.7 In vitro HMC synergistic toxicity in cancer cells when combined with
cisplatin,
paclitaxel and gemcitabine, camptothecin, topotecan and doxorubicin
3.7.1. AMC combination with cisplatin against the MA/200 melanoma cell line.
HMC synergy with cisplatin was assessed either in combination over 5 days
exposure or in
sequence (HMC ¨> cisplatin) against the MM200 melanoma cell line. It was
difficult to
assess for synergisitic toxicity using a change in IC50 as a measure of
synergy due to HMC
toxicity as monotherapy (Table 8). 3D analysis of the data reveals that only
additive
toxicity was apparent using the 5-day combination protocol (Fig. 11). Evidence
of synergy
using the HMC-cisplatin combination was further assessed using the HMC-->
cisplatin
sequence (24hr exposure to each compound in sequence) against the melanoma
cell line
MM200. Using change in IC50 to assess for synergy it was noted that HMC at
concentrations of 2 M markedly chemosensitised the MM200 cells to cisplatin
by >1000
fold (Table 8). HMC induced chemosensitisation of M1v1200 cells to cisplatin
was
confirmed using 3D analysis of the data (Fig. 11B). These data demonstrate
that HMC is
able to chemosensitise cancer cells, in this case melanoma, to cisplatin.
Table 8. Comparative assessment of synergy between HMC and the cisplatin
against the
Mel-RM melanoma cell line. Average IC50 data for each agent when assessed as a
monotherapy or in combination are shown
TREATMENT (1050 uM)
DRUGChange HMC Change
Combined 24 hr
Factor first Factor
cisplatin 8.82 6.32
HMC 0.72 20.64
--
HMC 10uM 1.00E-06 HMC effect 1.28E-05 >10000
11
HMC 5uM 1.00E-06 2.71E-05 >10000
11
HMC 2uM 1.00E-06 1.00E-06 >10000
HMC luM 0.45 8.29 -1.31
3.7.2 n:mc combination with gemcitabine against the Mel-RM melanoma cells.
HMC synergy with gemcitabine was assessed either in combination over 5 days
exposure
or in sequence (HMC --> gemcitabine) against the Mel-RM melanoma cell line. It
was
CA 02506238 2005-05-03
- 48 -
difficult to assess for synergisitic toxicity using a change in IC50 as a
measure of synergy
due to HMC toxicity as monotherapy (Table 9). 3D analysis of the data reveals
that 5-day
combination protocol did not elicit synergisitic toxicity against the Mel-RM
cell line.
Evidence of synergy using the HMC-gemcitabine combination was further assessed
using
the HMC¨* gemcitabine sequence (24hr exposure to each compound in sequence)
against
the melanoma cell line Mel-RM. Using change in IC50 to assess for synergy it
was noted
that HMC at concentrations of 2 and 11.1M markedly chemosensitised the Mel-RM
cells to
gemcitabine by >1000 fold. HMC-induced chemosensitisation of Mel-RM cells to
gemcitabine was confirmed using 3D analysis of the data. These data
demonstrate that
HMC is able to chemosesnsitise cancer cells to gemcitabine.
Table 9. Comparative assessment of synergy between HMC and gemcitabine against
the
Mel-RM melanoma cell line. Average IC50 data for each agent when assessed as a
monotherapy or in combination are shown.
TREATMENT (IC50 uM)
DRUGChange HMC Change
Combined 24 hr
Factor first Factor
HMC 0.51 27.79
HMC 1 uM 1.00E-06 ?HMC effect 1.76E-03 >1000
HMC 2 uM 1.00E-06 ?HMC effect 1.00E-06 >1000
Gemcitabine 3.88E-03 4.67E-03
HMC 0.51 27.79
HMC 1 uM 1.00E-06 ?HMC effect 1.00E-06 >1000
HMC 2 uM 1.00E-06 ?HMC effect 3.89E-05 >100
3.73. HMC combination with paclitaxel against the 4405 melanoma cell line.
HMC synergy with paclitaxel was assessed either in combination over 5 days
exposure or
in sequence (HMC ¨> paclitaxel) against the 4405 melanoma cell line. It was
difficult to
assess for synergisitic toxicity using a change in IC50 as a measure of
synergy due to HMC
toxicity as monotherapy (Table 10). A 30 fold reduction in IC50 was noted in
the
combination experiment when compared to the paclitaxel monotherapy. However,
3D
analysis of the data revealed that the 5-day combination protocol did not
elicit synergisitic
toxicity against the 4405 cell line. Evidence of synergy using the HMC-
paclitaxel
combination was further assessed using the HMC¨* paclitaxel sequence (24hr
exposure to
= CA 02506238 2005-05-03
- 49 -
each compound in sequence) against the melanoma cell line 4405. Using change
in IC50
to assess for synergy it was noted that filvIC at concentrations of 21.1M
markedly
chemosensitised the 4405 cells to paclitaxel by >1000 fold (Table 10). HMC-
induced
chemosensitisation of MM200 cells to paclitaxel was confirmed using 3D
analysis of the
data. These data demonstrate that HMC is able to chemosesnsitise cancer cells
to
pacltaxel.
Table 10. Comparative assessment of synergy between AMC and paclitaxel against
the
4405 melanoma cell line. Average IC50 data for each agent when assessed as a
monotherapy or in combination are shown.
TREATMENT (IC50 uM)
DRUGChange HMC Change
Combined 24 hr
Factor first Factor
HMC 1 uM 0.006 1.00 0.03 -
4.44
HMC 2 uM 1.00E-06 5964.400 0.01 -
1.035
Paclitaxel 1.25E-07 5.84E-04
HMC 1.26 55.50
HMC 1 uM 4.12E-09 30.36 5.31E-05
11.00
HMC 2 uM 3.91E-10 ?HMC effect 5.48E-09
106633.75
3.7.4. KWIC combination with topotecan against the MM200 melanoma cell line.
HMC synergy with topotecan was assessed either in combination over 5 days
exposure or
in sequence (HMC topotecan) against the MM200 melanoma cell line. It was
difficult
to assess for synergisitic toxicity using a change in IC50 as a measure of
synergy due to
HMC toxicity as monotherapy (Table 8). 3D analysis of the data confirmed that
the 5-day
combination protocol did not elicit synergisitic toxicity against the MM200
cell line.
Evidence of synergy using the HMC-gemcitabine combination was further assessed
using
the HMC¨) topotecan sequence (24hr exposure to each compound in sequence)
against the
melanoma cell line M1v1200. Using change in IC50 to assess for synergy it was
noted that
HMC at a concentration of 21.1M markedly chemosensitised the MM200 cells to
topotecan
by >1000 fold (Table 11). HMC-induced chemosensitisation of MM200 cells to
topotecan
was confirmed using 3D analysis of the data. These data demonstrate that HMC
is able to
chemosesnsitise cancer cells to topotecan. From the 3D analysis the optimum
combination
CA 02506238 2005-05-03
- 50 -
of HMC and topotecan against the MM200 melanoma cell line would appear to be 2
M
HMC and between 1 and 0.1 M topotecan.
Table 11. Comparative assessment of synergy between HMC and topotecan against
the
MM200 melanoma cell line. Average IC50 data for for each agent when assessed
as a
monotherapy or in combination are shown.
TREATMENT (1050 uM)
DRUGChange HMC Change
Combined 24 hr
Factor first Factor
HMC 1 uM 0.115 -14.145 0.009 9.304
IBC 2 uM 1.00E-06 ?HMC effect 7.85E-05 >1000
Topotecan 0.095 2.216
HMC 0.702 17.952
HMC 1 uM 0.000 ?HMC effect 0.044 50.656
HMC 2 uM 1.00E-06 ?HCM effect -- 1.00E-06 >10000
3.7.5. HMC combination with camptothecin against the Mel-RM melanoma cell
line.
HMC synergy with doxorubicin was assessed either in combination over 5 days
exposure
or in sequence (HMC --> doxorubicin) against the Mel-RM melanoma cell line. It
was
difficult to assess for synergisitic toxicity using a change in IC50 as a
measure of synergy
due to HMC toxicity as monotherapy (Table 12). 3D analysis of the data
confirmed that
the 5-day combination protocol did not elicit synergisitic toxicity against
the Mel-RM cell
line, indeed evidence of antagonism was noted. Evidence of synergy using the
HMC-
doxorubicin combination was further assessed using the HMC--> doxorubicin
sequence
protocol (24hr exposure to each compound in sequence) against the melanoma
cell line
Mel-RM. Using change in IC50 to assess for synergy it was noted that HMC at a
concentration of 2 M chemosensitised the Mel-RM cells to camptothecin by ¨12
fold
(Table 12). 3D analysis of the data, however, reveal a marked degree of
synergy between
HMC and doxorubicin against Mel-RM cells. These data demonstrate that HMC is
able to
chemosesnsitise cancer cells to pacltaxel. From the 3D analysis the optimum
combination
of HMC and camptothecin against the MM200 melanoma cell line would appear to
be 2
M HMC and between 1 and 0.1 M doxorubicin.
CA 02506238 2005-05-03
- 51 -
Table 12. Comparative assessment of synergy between HMC and doxorubicin
against the
Mel-RM melanoma cell line. Average 1050 data for each agent when assessed as a
monotherapy or in combination are shown.
TREATMENT (IC50 uM)
DRUGChange HMC Change
Combined 24 hr
Factor first Factor
Doxorubic in 0.19 0.100
1EVIC 0.54 50.515
HMC 1 uM 0.40 -2.06 0.062 1.62
HMC 2 uM 0.18 ?HMC effect -- 8.16E-03 12.22
3.8 Inhibition of TNFa in murine macrophages (RAW 264.7) by compounds 4, 6 & 7
The mouse macrophage cell line RAW 264.7 was cultured in DMEM supplemented
with
FCS, 2mM glutamine and 50U/m1 penicillin/streptomycin. Subconfluent cells were
detached from the flask by gentle scraping and 24-well plates seeded at 5 x
105 cells per
well and allowed to adhere for lhr. Cells were treated with either test agent
(in 0.025%
DMSO) or vehicle alone, 1hr prior to the addition of 5Ong/m1 LPS. After
incubation for 16
hrs, culture media was collected and stored at -80 C for TNFa measurement
using an
enzyme immunometric assay (Becton Dickinson).
Compound 6 and compound 7 of the present invention were tested and the results
are
shown in Figure 12, which indicated that the compounds tested inhibit TNFa in
murine
macrophages in a dose dependent manner over the concentration ranges tested.
The raw data for compounds 6, 7 and additionally compound 4 is shown in Table
13
below.
Table 13
Concentration Compound 6 Compound 7 Compound 4
pM
-48.7 -26.2 -6.1
1 -13.0 -25.4 -11.9
0.1 -10.4 -1.4 -6.3
0.001 1.0 19.2 -14.3
0.01 -11.3
CA 02506238 2012-05-18
71884-135
- 52 -
The invention has been described herein, with reference to certain preferred
embodiments,
in order to enable the reader to practice the invention without undue
experimentation.
However, a person having ordinary skill in the art will readily recognise that
many of the
components and parameters may be varied or modified to a certain extent
without
departing from the scope of the invention. Furthermore, titles, headings, or
the like are
provided to enhance the reader's comprehension of this document, and should
not be read
as limiting the scope of the present invention.
Throughout this specification and the claims which follow, unless the context
requires
otherwise, the word "comprise", and variations such as "comprises" or
"comprising", will
be understood to imply the inclusion of a stated integer or step or group of
integers or steps
but not the exclusion of any other integer or step or group of integers or
steps.
Those skilled in the art will appreciate that the invention described herein
is susceptible to
variations and modifications other than those specifically described. It is to
be understood
that the invention includes all such variations and modifications. The
invention also
includes all of the steps, features, compositions and compounds referred to or
indicated in
this specification individually or collectively, and any and all combinations
of any two or
more of said steps or features.
The reference to any prior art in this specification is not, and should not be
taken as, an
acknowledgment or any form of suggestion that that prior art forms part of the
common
general knowledge in the field of endeavour.
Selected Reference Articles
Constantinou AI, Mehta R, Husband A. 2003 Phenoxodiol, a novel isoflavone
derivative,
inhibits dimethylbenz[a]anthracene (DMBA)-induced mammary carcinogenesis in
female
Sprague-Dawley rats. Eur J Cancer. 39, 1012-8.
Constantinou Al, Husband A. 2002 Phenoxodiol (2H-1-benzopyran-7-0,1,3-(4-
CA 02506238 2005-05-03
- 53 -
hydroxypheny1)), a novel isoflavone derivative, inhibits DNA topoisomerase II
by
stabilizing the cleavable complex. Anticancer Res. 22, 2581-5.
Gamble, JR., Xia, P., Hahn, C., Drew, J., Drogemuller, C., Carter, C., Walker,
C., Brown,
DM., Vadas, MA. 2003 Phenoxodiol, a derivative of plant flavanoids, shows
potent anti-
tumour and anti-angiogenic properties. Nature Medicine. Submitted.
Hersey, P and Zhang, X. D. 2001 How melanoma cells evade Trail-induced
apoptosis.
Nature reviews, Cancer, 1, 142-150.
Kamsteeg, M., Rutherford, T., Sapi, E., Hanczaruk, B., Shahabi, S., Flick, M.,
Brown, D.M
and Mor, G. 2003 Phenoxodiol-an isoflvone analogue- induces apoptosis in chemo-
resistant ovarian cancer cells. Oncogene, ;22, 2611-20.
O'Dwyer PJ, Moyer JD, Suffness M, Harrison SD Jr, Cysyk R, Hamilton TC,
Plowman J.
1994 Antitumor activity and biochemical effects of aphidicolin glycinate (NSC
303812)
alone and in combination with cisplatin in vivo. Cancer Res. 54, 724-9
Todorov PT, Field WN, Tisdale MJ 1999 Role of a proteolysis-inducing factor
(PIF) in
cachexia induced by a human melanoma (G361). Br J Cancer. 80,1734-7.
Bellisarii, F. L., S. Gallina, et al. (2001). "Tumor necrosis factor-alpha and
cardiovascular
diseases." Italian Heart Journal: Official Journal of the Italian Federation
of Cardiology.
2(6): 408-17.
Szlosarek, P. W. and F. R. Balkwill (2003). "Tumour necrosis factor alpha: a
potential
target for the therapy of solid tumours." Lancet Oncol 4(9): 565-73.
Nakata E, Hunter N, Mason K, Fan Z, Ang KX, Milas L. 2004 C225 antiepidermal
growth
factor receptor antibody enhances the efficacy of docetaxel chemoradiotherapy.
Int J
Radiat Oncol Biol Phys. 59(4):1163-73.