Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02516426 2005-08-18
Boehringer Ingelheim Pharma GmbH & Co. KG Case 1/1463
D-55216 INGELHEIM foreign filing text
82982fft
Bicyclic heterocycles, pharmaceutical compositions containing these
compounds, their use and processes for preparing them
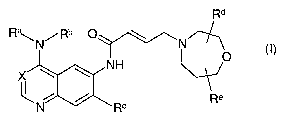
The present invention relates to bicyclic heterocycles of general formula
Rd
Ra\N/Rb ~ \ N
NH
X ~
/ Re
N R~ (I),
the tautomers, the stereoisomers, the mixtures thereof and the salts thereof,
particularly the physiologically acceptable salts thereof with inorganic or
organic acids, which have valuable pharmacological properties, particularly an
inhibitory effect on signal transduction mediated by tyrosine kinases, the use
thereof for treating diseases, particularly tumoral diseases and benign
prostatic hyperplasia (BPH), diseases of the lungs and respiratory tract, and
the preparation thereof.
In the above general formula I
Ra denotes a hydrogen atom or a C»-alkyl group,
Rb denotes a phenyl, benzyl or 1-phenylethyl group, wherein the phenyl
nucleus in each case is substituted by the groups R' to R3, while
R' and R2, which may be identical or different, in each case denote a
hydrogen, fluorine, chlorine, bromine or iodine atom,
a C~.~-alkyl, hydroxy, C»-alkoxy, CZ_3-alkenyl or C2_3-alkynyl group,
an aryl, aryloxy, arylmethyl or arylmethoxy group,
CA 02516426 2005-08-18
2
a heteroaryl, heteroaryloxy, heteroarylmethyl or heteroarylmethoxy
group,
a methyl or methoxy group substituted by 1 to 3 fluorine atoms or
a cyano, nitro or amino group, and
R3 denotes a hydrogen, fluorine, chlorine or bromine atom,
a methyl or trifluoromethyl group,
R° denotes a hydrogen atom or a fluorine, chlorine or bromine
atom,
a hydroxy or C~.~-alkyloxy group,
a methoxy group substituted by 1 to 3 fluorine atoms,
an ethyloxy group substituted by 1 to 5 fluorine atoms,
a C2~-alkyloxy group which is substituted by a group R4, while
R4 denotes a hydroxy, C~_3-alkyloxy, C3_6-cycloalkyloxy, C3_6-cycloalkyl-
C,_3-alkyloxy, amino, C~_3-alkylamino, di-(C~_3-alkyl)amino, bis-(2-C~_3-
alkyloxy-ethyl)-amino, bis-(3-C~_3-alkyloxy-propyl)-amino, pyrrolidin-1-yl,
piperidin-1-yl, homopiperidin-1-yl, morpholin-4-yl, homomorpholin-4-yl,
piperazin-1-yl, 4-(C~_3-alkyl)-piperazin-1-yl, homopiperazin-1-yl or 4-
(C~_3-alkyl)-homopiperazin-1-yl group,
a C3_~-cycloalkyloxy or C3_,-cycloalkyl-C,~-alkyloxy group,
a tetrahydrofuran-3-yloxy, tetrahydropyran-3-yloxy or tetrahydropyran-4-yloxy
group,
a tetrahydrofuranyl-C~_4-alkyloxy or tetrahydropyranyl-C~~-alkyloxy group,
CA 02516426 2005-08-18
3
a pyrrolidin-3-yloxy, piperidin-3-yloxy or piperidin-4-yloxy group,
a 1-(C~_3-alkyl)-pyrrolidin-3-yloxy, 1-(C~_3-alkyl)-piperidin-3-yloxy or 1-
(C~_3-alkyl)-piperidin-4-yloxy group,
a C~~-alkoxy group which is substituted by a pyrrolidinyl, piperidinyl or
homopiperidinyl group substituted in the 1 position by the group R5, where
R5 denotes a hydrogen atom or a C,_3-alkyl group,
or a C»-alkoxy group which is substituted by a morpholinyl or
homomorpholinyl group substituted in the 4 position by the group R5, where
R5 is as hereinbefore defined,
Re and Rd, which may be identical or different, in each case denote a
hydrogen atom or a C~_3-alkyl group
and
X denotes a methyne group substituted by a cyano group or a nitrogen atom,
while by the aryl groups mentioned in the definition of the above groups is
meant in each case a phenyl group which is mono- or disubstituted by R6,
while the substituents may be identical or different and
Rs denotes a hydrogen atom, a fluorine, chlorine, bromine or iodine
atom or a C~_3-alkyl, hydroxy, C~_3-alkyloxy, difluoromethyl,
trifluoromethyl, difluoromethoxy, trifluoromethoxy or cyano group,
by the heteroaryl groups mentioned in the definition of the above groups is
meant a pyridinyl, pyridazinyl, pyrimidinyl or pyrazinyl group, while the
above-
mentioned heteroaryl groups are mono- or disubstituted in each case by the
CA 02516426 2005-08-18
4
group R6, while the substituents may be identical or different and R6 is as
hereinbefore defined, and
unless otherwise stated, the above-mentioned alkyl groups may be straight-
chain or branched.
Preferred compounds of the above general formula I are those wherein
Ra denotes a hydrogen atom,
Rb denotes a phenyl group substituted by the groups R~ to R3, while
R' denotes a hydrogen, fluorine, chlorine or bromine atom,
a methyl, trifluoromethyl or ethynyl group,
a phenyloxy or phenylmethoxy group, while the phenyl moiety of the
above-mentioned groups is optionally substituted by a fluorine or
chlorine atom, or
a pyridinyloxy or pyridinylmethoxy group, while the pyridinyl moiety of
the above-mentioned groups is optionally substituted by a methyl or
trifluoromethyl group,
R2 denotes a hydrogen, fluorine or chlorine atom and
R3 denotes a hydrogen atom,
R' denotes a hydrogen atom,
a C~_3-alkyloxy group,
a C4_6-cycloalkyloxy or C3~-cycloalkyl-C,_2-alkyloxy group,
CA 02516426 2005-08-18
a tetrahydrofuran-3-yloxy, tetrahydropyran-3-yloxy, tetrahydropyran-4-yloxy,
tetrahydrofuranyl-C~_2-alkyloxy or tetrahydropyranyl-C~_Z-alkyloxy group,
an ethyloxy group which is substituted in the 2 position by a group R4, where
R4 denotes a hydroxy, C~_3-alkyloxy, amino, C~_3-alkylamino, di-
(C~_3-alkyl)amino, bis-(2-methoxyethyl)-amino, pyrrolidin-1-yl, piperidin-
1-yl, homopiperidin-1-yl, morpholin-4-yl, homomorpholin-4-yl, piperazin-
1-yl, 4-(C~_3-alkyl)-piperazin-1-yl, homopiperazin-1-yl or 4-(C~_3-alkyl)-
homopiperazin-1-yl group,
a propyloxy group which is substituted by a group R4 in the 3 position, while
R4 is as hereinbefore defined, or
a butyloxy group which is substituted by a group R4 in the 4 position, while
R4
is as hereinbefore defined,
Re and Rd, which may be identical or different, in each case denote a
hydrogen atom or a methyl group
and
X denotes a nitrogen atom,
while, unless otherwise stated, the above-mentioned alkyl groups may be
straight-chain or branched,
the tautomers, their stereoisomers, the mixtures thereof and the salts
thereof.
Particularly preferred compounds of the above general formula I are those
wherein
Ra denotes a hydrogen atom,
CA 02516426 2005-08-18
6
Rb denotes a 3-ethynylphenyl, 3-bromophenyl, 3,4-difluorophenyl or 3-chloro-
4-fluoro-phenyl group,
R~ denotes a hydrogen atom,
a methoxy, ethyloxy, 2-(methoxy)ethyloxy, 3-(morpholin-4-yl)propyloxy, cyclo-
butyloxy, cyclopentyloxy, cyclohexyloxy, cyclopropylmethoxy,
cyclopentylmethoxy, tetrahydrofuran-3-yloxy, tetrahydropyran-3-yloxy,
tetrahydropyran-4-yloxy, tetrahydrofuran-2-ylmethoxy, tetrahydrofuran-3-
ylmethoxy or tetrahydropyran-4-ylmethoxy group,
Re and Rd in each case denote a hydrogen atom
and
X denotes a nitrogen atom,
the tautomers, their stereoisomers, the mixtures thereof and the salts
thereof.
Most particularly preferred compounds of general formula I are those wherein
Ra denotes a hydrogen atom,
Rb denotes a 3-chloro-4-fluoro-phenyl group,
R° denotes a tetrahydrofuran-3-yloxy group,
Re and Rd in each case denote a hydrogen atom
and
X denotes a nitrogen atom,
the tautomers, their stereoisomers, the mixtures thereof and the salts
thereof.
CA 02516426 2005-08-18
7
The following particularly preferred compound of general formula I is
mentioned by way of example:
4-[(3-chloro-4-fluoro-phenyl)amino]-6-{[4-(homomorpholin-4-yl)-1-oxo-2-buten-
1-yl]amino}-7-[(S)-(tetrahydrofuran-3-yl)oxy]-quinazoline
and the salts thereof.
The compounds of general formula I may be prepared by the following
methods, for example:
a) reacting a compound of general formula
R~
R ~ , Rb O ~ ,O
N ~P ~R8
NH O
X
~N / R
(II)
wherein
Ra, Rb, R' and X are as hereinbefore defined and R' and R8, which may be
identical or different, denote C~~-alkyl groups, with a compound of general
formula
Rd
O
~N
H O
Re , (III)
wherein
Rd and Re are as hereinbefore defined.
CA 02516426 2005-08-18
8
The reaction is expediently carried out in a solvent or mixture of solvents
such
as tetrahydrofuran, tetrahydrofuran/water, acetonitrile, acetonitrile/water,
dioxane, ethyleneglycol dimethyl ether, isopropanol, methylene chloride,
dimethylformamide or sulpholane, optionally in the presence of an inorganic
or organic base, e.g. sodium carbonate, potassium hydroxide or 1,8-
diazabicyclo[5.4.0]undec-7-ene and optionally in the presence of a lithium
salt
such as lithium chloride at temperatures between -50 and 150°C, but
preferably at temperatures between -20 and 80°C. The reaction may also
be
carried out with a reactive derivative of the compound of general formula III,
for example the hydrate or a hemiacetal.
b) reacting a compound of general formula
Ra~N~Rb O ~ Z~
NH
X
~N / R
(IV)
wherein
Ra, Rb, R° and X are as hereinbefore defined and Z' denotes a
leaving group
such as a halogen atom or a substituted sulphonyloxy group such as a
chlorine or bromine atom, a methanesulphonyloxy or p-toluenesulphonyloxy
group, with a compound of general formula
Rd
H
N
O
Re , ~V)
wherein Rd and Re are as hereinbefore defined.
CA 02516426 2005-08-18
9
The reaction is expediently carried out in a solvent such as isopropanol,
butanol, tetrahydrofuran, dioxane, acetonitrile, dimethylformamide,
sulpholane, toluene or methylene chloride or mixtures thereof, optionally in
the presence of an inorganic or organic base, e.g. sodium carbonate,
potassium carbonate, potassium hydroxide, triethylamine or N-ethyl-
diisopropylamine and optionally in the presence of a reaction accelerator such
as an alkali metal iodide at temperatures between -20 and 150°C, but
preferably at temperatures between 0 and 100°C. The reaction may
however
also be carried out without a solvent or in an excess of the compound of
general formula V used.
In the reactions described hereinbefore, any reactive groups present such as
hydroxy, amino, alkylamino or imino groups may be protected during the
reaction by conventional protecting groups which are cleaved again after the
reaction.
For example, a protecting group for a hydroxy group may be a trimethylsilyl,
acetyl, trityl, benzyl or tetrahydropyranyl group.
Protecting groups for an amino, alkylamino or imino group may be a formyl,
acetyl, trifluoroacetyl, ethoxycarbonyl, tert.butoxycarbonyl,
benzyloxycarbonyl,
benzyl, methoxybenzyl or 2,4-dimethoxybenzyl group.
Any protecting group used is optionally subsequently cleaved for example by
hydrolysis in an aqueous solvent, e.g. in water, isopropanol/water, acetic
acid/water, tetrahydrofuran/water or dioxane/water, in the presence of an acid
such as trifluoroacetic acid, hydrochloric acid or sulphuric acid or in the
presence of an alkali metal base such as sodium hydroxide or potassium
hydroxide or aprotically, e.g. in the presence of iodotrimethylsilane, at
temperatures between 0 and 120°C, preferably at temperatures between 10
and 100°C.
However, a benzyl, methoxybenzyl or benzyloxycarbonyl group is cleaved, for
example, hydrogenolytically, e.g. with hydrogen in the presence of a catalyst
CA 02516426 2005-08-18
such as palladium/charcoal in a suitable solvent such as methanol, ethanol,
ethyl acetate or glacial acetic acid, optionally with the addition of an acid
such
as hydrochloric acid at temperatures between 0 and 100°C, but
preferably at
temperatures between 20 and 60°C, and at a hydrogen pressure of 1 to 7
bar,
but preferably 3 to 5 bar. A 2,4-dimethoxybenzyl group, however, is preferably
cleaved in trifluoroacetic acid in the presence of anisol.
A tert.butyl or tert.butyloxycarbonyl group is preferably cleaved by treating
with
an acid such as trifluoroacetic acid or hydrochloric acid or by treating with
iodotrimethylsilane, optionally using a solvent such as methylene chloride,
dioxane, methanol or diethylether.
A trifluoroacetyl group is preferably cleaved by treating with an acid such as
hydrochloric acid, optionally in the presence of a solvent such as acetic acid
at
temperatures between 50 and 120°C or by treating with sodium hydroxide
solution optionally in the presence of a solvent such as tetrahydrofuran at
temperatures between 0 and 50°C.
Moreover, the compounds of general formula I obtained may be resolved into
their enantiomers and/or diastereomers, as mentioned hereinbefore. Thus, for
example, cis/trans mixtures may be resolved into their cis and trans isomers,
and compounds with at least one optically active carbon atom may be
separated into their enantiomers.
Thus, for example, the cisltrans mixtures may be resolved by chromatography
into the cis and trans isomers thereof, the compounds of general formula I
obtained which occur as racemates may be separated by methods known per
se (cf. Allinger N. L. and Eliel E. L. in "Topics in Stereochemistry", Vol. 6,
Wiley
Interscience, 1971) into their optical antipodes and compounds of general
formula I with at least 2 asymmetric carbon atoms may be resolved into their
diastereomers on the basis of their physical-chemical differences using
methods known per se, e.g. by chromatography and/or fractional
crystallisation, and, if these compounds are obtained in racemic form, they
may subsequently be resolved into the enantiomers as mentioned above.
CA 02516426 2005-08-18
11
The enantiomers are preferably separated by column separation on chiraf
phases or by recrystallisation from an optically active solvent or by reacting
with an optically active substance which forms salts or derivatives such as
e.g.
esters or amides with the racemic compound, particularly acids and the
activated derivatives or alcohols thereof, and separating the diastereomeric
mixture of salts or derivatives thus obtained, e.g. on the basis of their
differences in solubility, whilst the free antipodes may be released from the
pure diastereomeric salts or derivatives by the action of suitable agents.
Optically active acids in common use are e.g. the D- and L-forms of tartaric
acid or dibenzoyltartaric acid, di-O-p-tolyltartaric acid, malic acid,
mandelic
acid, camphorsulphonic acid, glutamic acid, aspartic acid or quinic acid. An
optically active alcohol may be for example (+) or (-)-menthol and an
optically
active acyl group in amides, for example, may be a (+)-or
(-)-menthyloxycarbonyl.
Furthermore, the compounds of formula I obtained may be converted into the
salts thereof, particularly for pharmaceutical use into the physiologically
acceptable salts with inorganic or organic acids. Acids which may be used for
this purpose include for example hydrochloric acid, hydrobromic acid,
sulphuric acid, methanesulphonic acid, phosphoric acid, fumaric acid, succinic
acid, lactic acid, citric acid, tartaric acid or malefic acid
As already mentioned hereinbefore, the compounds of general formula I
according to the invention and the physiologically acceptable salts thereof
have valuable pharmacological properties, particularly an inhibiting effect on
signal transduction mediated by the Epidermal Growth Factor receptor
(EGF-R), whilst this may be achieved for example by inhibiting ligand
bonding, receptor dimerisation or tyrosinekinase itself. It is also possible
to
block the transmission of signals to components located further downstream.
The biological properties of the new compounds were investigated as follows:
The inhibition of human EGF-receptor kinase was determined using the
cytoplasmatic tyrosine kinase domain (methionine 664 to alanine 1186, based
CA 02516426 2005-08-18
12
on the sequence published in Nature 309 (1984), 418). To do this, the protein
was expressed in Sf9 insect cells as a GST fusion protein using the
Baculovirus expression system.
The enzyme activity was measured in the presence or absence of the test
compounds in serial dilutions. The polymer pEY (4:1) produced by SIGMA
was used as the substrate. Biotinylated pEY (bio-pEY) was added as the
tracer substrate. Every 100 NI of reaction solution contained 10 NI of the
inhibitor in 50% DMSO, 20 NI of the substrate solution (200 mM HEPES pH
7.4, 50 mM magnesium acetate, 2.5 mglml of poly(EY), 5 Ng/ml of bio-pEY)
and 20 NI of enzyme preparation. The enzyme reaction was started by the
addition of 50N1 of a 100NM ATP solution in 10 mM magnesium chloride. The
dilution of the enzyme preparation was adjusted so that the incorporation of
phosphate into the bio-pEY was linear in terms of time and quantity of
enzyme. The enzyme preparation was diluted in 20 mM HEPES pH 7.4, 1 mM
EDTA, 130 mM common salt, 0.05% Triton X-100, 1 mM DTT and 10%
glycerol.
The enzyme assays were carried out at ambient temperature over a period of
30 minutes and were ended by the addition of 50 NI of a stopping solution
(250 mM EDTA in 20 mM HEPES pH 7.4). 100 NI were placed on a
streptavidin-coated microtitre plate and incubated for 60 minutes at ambient
temperature. Then the plate was washed with 200 NI of a washing solution (50
mM Tris, 0.05% Tween 20). After the addition of 100 NI of a HRPO-labelled
anti-PY antibody (PY20H Anti-PTyr:HRP produced by Transduction
Laboratories, 250 ng/ml) it was incubated for 60 minutes. Then the microtitre
plate was washed three times with 200 NI of washing solution. The samples
were then combined with 100 NI of a TMB-peroxidase solution (A:B = 1:1,
Kirkegaard Perry Laboratories). After 10 minutes the reaction was stopped.
The extinction was measured at OD450nm with an ELISA reader. All data points
were measured three times.
The data were matched using an iterative calculation using an analytical
programme for sigmoidal curves (Graph Pad Prism Version 3.0; sigmoid
CA 02516426 2005-08-18
13
curves, variable pitch). All the iteration data released showed a correlation
coefficient of more than 0.9. The maxima and minima of the curves showed a
spread of at least a factor of 5. The ICSO (concentration of active substance
which inhibits the activity of EGF-receptor kinase by 50%) was determined
from the curves.
The following results were obtained:
Inhibition of EGF-
Compound
receptor kinase
(Example No.)
IC50 [nM]
1 1.5
The compounds of general formula I according to the invention thus inhibit
signal transduction by tyrosine kinases, as demonstrated by the example of
the human EGF receptor, and are therefore useful for treating
pathophysiological processes caused by hyperfunction of tyrosine kinases.
These are e.g. benign or malignant tumours, particularly tumours of epithelial
and neuroepithelial origin, metastasisation and the abnormal proliferation of
vascular endothelial cells (neoangiogenesis).
The compounds according to the invention are also useful for preventing and
treating diseases of the airways and lungs which are accompanied by
increased or altered production of mucus caused by stimulation by tyrosine
kinases, e.g. in inflammatory diseases of the airways such as chronic
bronchitis, chronic obstructive bronchitis, asthma, bronchiectasis, allergic
or
non-allergic rhinitis or sinusitis, cystic fibrosis, a1-antitrypsin
deficiency, or
coughs, pulmonary emphysema, pulmonary fibrosis and hyperreactive
airways.
The compounds are also suitable for treating diseases of the gastrointestinal
tract and bile duct and gall bladder which are associated with disrupted
activity of the tyrosine kinases, such as may be found e.g. in chronic
CA 02516426 2005-08-18
14
inflammatory changes such as cholecystitis, Crohn's disease, ulcerative
colitis, and ulcers in the gastrointestinal tract or such as may occur in
diseases of the gastrointestinal tract which are associated with increased
secretions, such as Menetrier's disease, secreting adenomas and protein loss
syndrome
In addition, the compounds of general formula I and the physiologically
acceptable salts thereof may be used to treat other diseases caused by
abnormal function of tyrosine kinases, such as e.g. epidermal
hyperproliferation (psoriasis), benign prostatic hyperplasia (BPH),
inflammatory processes, diseases of the immune system, hyperproliferation of
haematopoietic cells, treatment of nasal polyps, etc.
By reason of their biological properties the compounds according to the
invention may be used on their own or in conjunction with other
pharmacologically active compounds, for example in tumour therapy, in
monotherapy or in conjunction with other anti-tumour therapeutic agents, for
example in combination with topoisomerase inhibitors (e.g. etoposide), mitosis
inhibitors (e.g. vinblastine), compounds which interact with nucleic acids
(e.g.
cis-platin, cyclophosphamide, adriamycin), hormone antagonists (e.g.
tamoxifen), inhibitors of metabolic processes (e.g. 5-FU etc.), cytokines
(e.g.
interferons), antibodies, etc. For treating respiratory tract diseases, these
compounds may be used on their own or in conjunction with other therapeutic
agents for the airways, such as substances with a secretolytic (e.g. ambroxol,
N-acetylcysteine), broncholytic (e.g. tiotropium or ipratropium or fenoterol,
salmeterol, salbutamol) and/or anti-inflammatory activity (e.g. theophylline
or
glucocorticoids). For treating diseases in the region of the gastrointestinal
tract, these compounds may also be administered on their own or in
conjunction with substances having an effect on motility or secretion. These
combinations may be administered either simultaneously or sequentially.
These compounds may be administered either on their own or in conjunction
with other active substances by intravenous, subcutaneous, intramuscular,
CA 02516426 2005-08-18
' 15
intraperitoneal or intranasal route, by inhalation or transdermally or orally,
whilst aerosol formulations are particularly suitable for inhalation.
For pharmaceutical use the compounds according to the invention are
generally used for warm-blooded vertebrates, particularly humans, in doses of
0.01-100 mg/kg of body weight, preferably 0.1-15 mg/kg. For administration
they are formulated with one or more conventional inert carriers and/or
diluents, e.g. with corn starch, lactose, glucose, microcrystalline cellulose,
magnesium stearate, polyvinylpyrrolidone, citric acid, tartaric acid, water,
water/ethanol, water/glycerol, water/sorbitol, water/polyethylene glycol,
propylene glycol, stearyl alcohol, carboxymethylcellulose or fatty substances
such as hard fat or suitable mixtures thereof in conventional galenic
preparations such as plain or coated tablets, capsules, powders, suspensions,
solutions, sprays or suppositories.
The following Examples are intended to illustrate the present invention
without
restricting it:
CA 02516426 2005-08-18
16
Preparation of the starting compounds:
Example I
4-f (3-chloro-4-fluoro-phenyl)aminol-6-f (diethoxy-phosphoryl)-acetylaminol-7-
((S)-(tetrahyd rofura n-3-yl)oxyl-a ui nazol ine
60.07 g of diethoxyphosphorylacetic acid are placed in 750 ml of N,N-
dimethylformamide and at ambient temperature combined with 48.67 g of
N,N'-carbonyldiimidazole. After the development of gas has ceased 90.00 g of
4-[(3-chloro-4-fluoro-phenyl)amino]-6-amino-[(S)-(tetrahydrofuran-3-yl)oxy]-
quinazoline are added and the reaction mixture is stirred for about 4-5 hours
at ambient temperature until the reaction is complete. The reaction mixture is
then heated gently in the water bath and 750 ml of water are added twice. The
thick suspension is stirred overnight and the next morning another 350 ml of
water are added. The suspension is cooled in the ice bath, stirred for one
hour
and suction filtered. The filter cake is washed again with 240 ml of N,N-
dimethylformamide/water (1:2) and 240 ml of diisopropylether and dried at
40°C in the circulating air dryer.
Yield: 117.30 g of (88 % of theory)
Rf value: 0.37 (silica gel, methylene chloride/methanol = 9:1 )
Mass spectrum (ESI+): m/z = 553, 555 [M+H]+
The following compounds are obtained analogously to Example I:
(1) 4-[(3-chloro-4-fluoro-phenyl)amino]-6-[(diethoxy-phosphoryl)-acetylamino]-
7-[(R)-(tetra hyd rofuran-3-yl)oxy]-q ui nazoli ne
Mass spectrum (ESI+): m/z = 553, 555 [M+H]+
(2) 4-[(3-chloro-4-fluoro-phenyl)amino]-6-[(diethoxy-phosphoryl)-acetylamino]-
7-cyclopropylmethoxy-quinazoline
melting point: 185-187°C
(3) 4-[(3-bromophenyl)amino]-6-[(diethoxy-phosphoryl)-acetylamino]-
quinazoline
Mass spectrum (ESI-): m/z = 491, 493 [M-H]-
CA 02516426 2005-08-18
' 17
(4) 4-[(3-chloro-4-fluoro-phenyl)amino]-6-[(diethoxy-phosphoryl)-acetylamino)-
7-cyclopentyloxy-quinazoline
Rf value: 0.54 (silica gel, methylene chloridelethanol = 20:1 )
Example II
Homomorpholin-4,i1-acetaldehyde-hydrochloride
Prepared by stirring (2.5 hours) 4-(2,2-dimethoxy-ethyl)-homomorpholine with
semiconcentrated hydrochloric acid at 80°C. The solution obtained is
further
reacted directly as in Example 1.
Example III
4-(2,2-dimethoxy-ethyl)-homomorpholine
Prepared by stirring (5 hours) homomorpholine-hydrochloride with
bromoacetaldehyde-dimethylacetal in the presence of potassium carbonate in
N-methylpyrrolidinone at 80°C.
Rf value: 0.2 (silica gel, ethyl acetate/methanol/conc. aqueous ammonia =
90:10:1)
CA 02516426 2005-08-18
18
Preparation of the final compounds:
Example 1
4-f (3-chloro-4-fluoro-phenyl)aminol-6-~f4-(homomorpholin-4-yl)-1-oxo-2-buten-
1-yllamino}-7-f(S)-(tetrahydrofuran-3-yl)oxyl-auinazoline
CI ~N~
NH
O
O
A solution of 3.9 g of 4-[(3-chloro-4-fluoro-phenyl)amino]-6-[2-(diethoxy-
phosphoryl)-acetylamino]-7-[(S)-(tetrahydrofuran-3-yl)oxy]-quinazoline in 20
ml of tetrahydrofuran is added to a solution of 300 mg of lithium chloride in
20
ml of water at ambient temperature. Then 2.35 g of potassium hydroxide
flakes are added and the reaction mixture is cooled to -3°C in an
ice/acetone
cooling bath. The solution of the homomorpholin-4-yl-acetaldehyde hydro-
chloride obtained in Example II is then added dropwise within 5 min at a
temperature of 0°C. After the addition has ended the reaction mixture
is
stirred for another 10 min at 0°C and for a further hour at ambient
temperature. For working up 100 ml of ethyl acetate are added and the
aqueous phase is separated off. The organic phase is washed with saturated
sodium chloride solution, dried over magnesium sulphate and evaporated
down. The crude product is purified by chromatography over a silica gel
column using ethyl acetate/methanol/conc. methanolic ammonia as eluant.
The product obtained is stirred with a little diisopropyl ether, suction
filtered
and dried.
Yield: 2.40 g of (63 % of theory)
Rf value: 0.09 (silica gel, ethyl acetate/methanol/conc. aqueous ammonia =
90:10:1 )
Mass spectrum (ESI+): m/z = 542, 544 [M+H]+
CA 02516426 2005-08-18
19
The following compounds may also be prepared analogously to the foregoing
Examples and other methods known from the literature:
Example No. Structure
F /
CI \ NH O~~'~N
N H ~O
N ~~~~
N
(2) F /
CI \ NH O~~N
NH
N ~%
_N / O
~3) F /
CI \ NH O~~~N
INH
N' ~~~~
_N / O
(4) F /
w I o w
CI NH ~~~N~
N H ~O
N \~
~N / O
O~
CA 02516426 2005-08-18
20
Example No. Structure
(5) F /
CI \ NH O~~~N
NH
N' \~
_N / O
N
~O
~6) F /
CI \ NH O~~'~N
NH
N ~~~~
~N / O
(7) F /
0
CI NH ~~'~N~
INH ~O
N \~
~N / O
~8) F /
CI \ NH O~~~N
INH
N' \
_N / O
CA 02516426 2005-08-18
21
Example No. Structure
(9) F i
o w ~
CI NH ~~N~
NH
N ~%
~N / 0
(10) F i
CI \ NH O~~~N
IN H ~O
N ~%
~N / O
(11)
CI \ NH O~~~N
INH
N
~N
O
(12)
CI \ NH O~~~N
NH
N ~
~N / O
O
CA 02516426 2005-08-18
22
Example No. Structure
(13) F i
0
CI NH ~~N~
N H ~O
N ~%
~N ~ O
OJ
(14) F i
0
CI NH ~~N~
NH
N ~~~~
~N ~ O
O
(15) F i
CI \ NH O~~'~N
NH
N ~i
~N / O
(16) F i
0
CI NH ~~N~
N H ~O
N \~
~N / O
O
CA 02516426 2005-08-18
23
Example No. Structure
F /
CI \ NH O~~~N
NH
N \~
~N / O
O
NH O~~~N
NH
N
~N / O
O
(19) F /
o w
F NH ~~N~
NH
N' ~~~~
_N / O
X20) /
CI \ NH O~~~N
INH
N'
_N / O
O
CA 02516426 2005-08-18
24
Example No. Structure
(21)
0
Br NH ~~N~
NH
N ~%
~N / O
O
Example 2
Coated tablets containing 75 ma of active substance
1 tablet core contains:
active substance 75.0 mg
calcium phosphate 93.0 mg
corn starch 35.5 mg
polyvinylpyrrolidone 10.0 mg
hydroxypropylmethylcellulose 15.0 mg
magnesium stearate 1.5 ma
230.0 mg
Preparation:
The active substance is mixed with calcium phosphate, corn starch, polyvin-
ylpyrrolidone, hydroxypropylmethylcellulose and half the specified amount of
magnesium stearate. Blanks 13 mm in diameter are produced in a tablet-
making machine and these are then rubbed through a screen with a mesh size
of 1.5 mm using a suitable machine and mixed with the rest of the magnesium
stearate. This granulate is compressed in a tablet-making machine to form
tablets of the desired shape.
Weight of core: 230 mg
die: 9 mm, convex
CA 02516426 2005-08-18
The tablet cores thus produced are coated with a film consisting essentially
of
hydroxypropylmethylcellulose. The finished film-coated tablets are polished
with beeswax.
Weight of coated tablet: 245 mg.
Example 3
Tablets containing 100 m4 of active substance
Composition:
1 tablet contains:
active substance 100.0 mg
lactose 80.0 mg
corn starch 34.0 mg
polyvinylpyrrolidone 4.0 mg
magnesium stearate 2.0 mg
220.0 mg
Method of Preparation:
The active substance, lactose and starch are mixed together and uniformly
moistened with an aqueous solution of the polyvinylpyrrolidone. After the
moist
composition has been screened (2.0 mm mesh size) and dried in a rack-type
drier at 50°C it is screened again (1.5 mm mesh size) and the lubricant
is
added. The finished mixture is compressed to form tablets.
Weight of tablet: 220 mg
Diameter: 10 mm, biplanar, facetted on both sides and notched on
one side.
CA 02516426 2005-08-18
~ 26
Example 4
Tablets containing 150 mg of active substance
Composition:
1 tablet contains:
active substance 150.0
mg
powdered lactose 89.0 mg
corn starch 40.0 mg
colloidal silica 10.0 mg
polyvinylpyrrolidone10.0 mg
magnesium stearate1.0 mg
300.0
mg
Preparation:
The active substance mixed with lactose, corn starch and silica is moistened
with a 20% aqueous polyvinylpyrrolidone solution and passed through a
screen with a mesh size of 1.5 mm. The granules, dried at 45°C, are
passed
through the same screen again and mixed with the specified amount of
magnesium stearate. Tablets are pressed from the mixture.
Weight of tablet: 300 mg
die: 10 mm, flat
Example 5
Hard gelatine capsules containing 150 ma of active substance
1 capsule contains:
active substance150.0 mg
corn starch (dried)approx. 180.0
mg
lactose (powdered)approx. 87.0
mg
magnesium stearate3.0 mp
approx. 420.0
mg
CA 02516426 2005-08-18
27
Preparation:
The active substance is mixed with the excipients, passed through a screen
with a mesh size of 0.75 mm and homogeneously mixed using a suitable
apparatus. The finished mixture is packed into size 1 hard gelatine capsules.
Capsule filling: approx. 320 mg
Capsule shell: size 1 hard gelatine capsule.
Example 6
Suppositories containing 150 mg of active substance
1 suppository contains:
active substance 150.0 mg
polyethyleneglycol 1500 550.0 mg
polyethyleneglycol 6000 460.0 mg
polyoxyethylene sorbitan monostearate 840.0 mg
2,000.0 mg
Preparation:
After the suppository mass has been melted the active substance is
homogeneously distributed therein and the melt is poured into chilled moulds.
CA 02516426 2005-08-18
. 28
Example 7
Suspension containing 50 mg of active substance
100 ml of suspension contain:
active substance 1.00
g
carboxymethylcellulose-Na-salt0.10
g
methyl p-hydroxybenzoate 0.05
g
propyl p-hydroxybenzoate 0.01
g
glucose 10.00
g
glycerol 5.00
g
70% sorbitol solution 20.00
g
flavouring 0.30
g
dist. water ad 100
ml
Preparation:
The distilled water is heated to 70°C. The methyl and propyl
p-hydroxybenzoates together with the glycerol and sodium salt of
carboxymethylcellulose are dissolved therein with stirring. The solution is
cooled to ambient temperature and the active substance is added and
homogeneously dispersed therein with stirring. After the sugar, the sorbitol
solution and the flavouring have been added and dissolved, the suspension is
evacuated with stirring to eliminate air.
ml of suspension contain 50 mg of active substance.
Example 8
Ampoules containing 10 m4 active substance
Composition:
active substance 10.0 mg
0.01 N hydrochloric acid q.s.
double-distilled water ad 2.0 ml
CA 02516426 2005-08-18
' ' 29
Preparation:
The active substance is dissolved in the requisite amount of 0.01 N HCI, made
isotonic with common salt, filtered sterile and transferred into 2 ml
ampoules.
Example 9
Ampoules containin~gi of active substance
Composition:
active substance 50.0 mg
0.01 N hydrochloric acid q.s.
double-distilled water ad 10.0 ml
Preparation:
The active substance is dissolved in the necessary amount of 0.01 N HCI,
made isotonic with common salt, filtered sterile and transferred into 10 ml
ampoules.
Example 10
Capsules for powder inhalation containing 5 mg of active substance
1 capsule contains:
active substance 5.0 mg
lactose for inhalation 15.0 mg
20.0 mg
CA 02516426 2005-08-18
Preparation:
The active substance is mixed with lactose for inhalation. The mixture is
packed into capsules in a capsule-making machine (weight of the empty
capsule approx. 50 mg).
weight of capsule: 70.0 mg
size of capsule 3
Example 11
Solution for inhalation for hand-held nebulisers containing 2.5 mg active
substance
1 spray contains:
active substance 2.500 mg
benzalkonium chloride 0.001 mg
1 N hydrochloric acid q.s.
ethanol/water (50/50) ad 15.000 mg
Preparation:
The active substance and benzalkonium chloride are dissolved in
ethanol/water (50/50). The pH of the solution is adjusted with 1 N
hydrochloric
acid. The resulting solution is filtered and transferred into suitable
containers
for use in hand-held nebulisers (cartridges).
Contents of the container: 4.5 g