Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02519568 2005-09-19
WO 2004/094444 PCT/US2003/008530
6"-Amino-6 --deoxygalactosylceramides
TECHNICAL FIELD
This invention relates to galactosylceramide compounds.
BACI' GROUND
Peptide antigen presentation via major histacompatability complexes has long
been recognized as a central element in adaptive immune responses. Recently, a
parallel pathway that can elicit potent immune responses has begun to be
elucidated.
This pathway involves the presentation of glycolipids by CD1 proteins and is
believed
to be responsible for a portion of the innate immunity of mammals to bacteria.
The CD1 locus of mammals includes five distinct isotypes, CD1a, CD lb,
CD 1 c, CD 1 d, and CD 1 e. These nonpolymorphic, membrane-bound proteins are
characterized by their ability to present classes of glycolipids to T cells.
The CD 1 d
member of the gene family has been characterized by its ability to bind and
present c-
galactosylceramides to natural killer T cells (NIT cells).
Complex formation between glycolipid-loaded CDId proteins and T cell
receptors can subsequently lead to the stimulation of T cells. T cell
stimulation
initiates, inter alia, the cellular production of certain key immunoresponsive
biochemicals.
SUMMARY
This invention relates to 6"-amino-6"-deoxygalactosylcerainide compounds
and their methods of use.
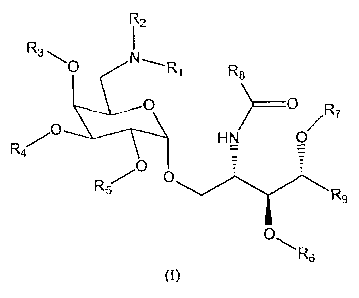
One aspect of this invention features compounds of Formula (I):
R
i2
R3,0 N-.R1 R8
0 HN ~,R7 (1)
R4 -
R5 00
R9
0,R
6
CA 02519568 2005-09-19
WO 2004/094444 PCT/US2003/008530
wherein,
R1 is:
(i) hydrogen; or
(ii) -S02RIo,
wherein R10 is:
halo; hydroxy; OR11; OR12; amino; NHRI1; N(RII)2; NHR12; N(R12)2;
aralkylamino; or
C1-C12 alkyl optionally substituted with halo, hydroxy, oxo, nitro,
OR,,, OR12, acyloxy, amino, NHR11, N(R11)2, NHR12, N(R12)2, aralkylamino,
mercapto, thioalkoxy, S(O)R11, S(O)R12, S02R11, S02R12, NHSO2R11,
NHS02R12, sulfate, phosphate, cyano, carboxyl, C(O)R11, C(O)R12,
C(O)OR11, C(O)NH2, C(O)NHR11, C(O)N(R11)2, C3-C10 cycloalkyl containing
0-3 R13, C3-Clo heterocyclyl containing 0-3 R13, C2-C6 alkenyl, C2-C6 alkynyl,
C5-C10 cycloalkenyl, C5-Clo heterocycloallcenyl, C6-C20 aryl containing 0-3
R14, or heteroaryl containing 0-3 R14i or
C3-C10 cycloalkyl, C3-Clo heterocyclyl, C5-C10 cycloalkenyl, or C5-C10
heterocycloalkenyl optionally substituted with one or more halo, hydroxy,
oxo, OR,,, OR12, acyloxy, nitro, amino, NHR11, N(R11)2, NHR12, N(R12)2,
aralkylamino, mercapto, thioalkoxy, S(O)R11, S(O)R12, S02R11, SO2R12,
NHSO2R11, NHSO2R12, sulfate, phosphate, cyano, carboxyl, C(O)R11,
C(O)R12, C(O)OR11, C(O)NH2, C(O)NHR11, C(O)N(R11)2, alkyl, haloallcyl,
C3-Clo cycloalkyl containing 0-3 R13, C3-C10 heterocyclyl containing 0-3 R13,
C2-C6 alkenyl, C2-C6 alkenyl, C5-C10 cycloalkenyl, C5-Clo heterocycloalkenyl,
C6-C20 aryl heteroaryl containing 0-3 R14, or C6-C20 heteroaryl containing 0-3
R14; or
C2-C6 alkenyl, C2-C6 alkynyl, aryl, or heteroaryl optionally substituted
with one or more halo, hydroxy, OR,,, OR12, acyloxy, nitro, amino, NHR11,
N(R11)2, NHR12, N(R12)2, aralkylamino, mercapto, thioalkoxy, S(O)R11,
S(O)R12, SO2R11a S02R12, NHSO2R11, NHSO2R12, sulfate, phosphate, cyano,
carboxyl, C(O)R11, C(O)R12, C(O)OR1 1, C(O)NH2, C(O)NHR11, C(O)N(R11)2,
2
CA 02519568 2005-09-19
WO 2004/094444 PCT/US2003/008530
alkyl, haloalkyl, C3-C10 cycloalkyl containing 0-3 R13, C3-C10 heterocyclyl
containing 0-3 R13, C2-C6 alkenyl, C2-C6 alkynyl, C5-C10 cycloalkenyl, C5-C10
heterocycloalkenyl, C6-C20 aryl containing 0-3 R14, or C6-C20 heteroaryl
containing 0-3 R14; or
(iii) -C(O)R1o, wherein Rio is defined as above; or
(iv) -C(R10)2(R15), wherein Rio is defined as above; R15 is hydrogen,
R10, or R15 and R2 taken together forms a double bond between the carbon and
nitrogen atoms to which they are attached; or
(v) R1 and R2 taken together forms a heterocyclyl of 3-10 ring atoms
optionally substituted with Rio;
R2 is hydrogen, or R2 and R15 taken together forms a double bond between the
carbon and nitrogen atoms to which they are attached, or R2 and Ri taken
together
forms a heterocyclyl of 3-10 ring atoms optionally substituted with Rio;
R3, R4, R5, R6, and R7 are each independently hydrogen, C1-C6 alkyl, C6-C12
aralkyl, or Ci-C6 acyl;
R8 is -(CH2),tCR3;
R9 is a linear or branched C3-C100 alkyl;
R11 is Cl-C20 alkyl optionally substituted with halo, hydroxy, alkoxy, amino,
alkylamino, dialkylamino, sulfate, or phosphate;
R12 is aryl optionally substituted with halo, haloalkyl, hydroxy, alkoxy,
nitro,
amino, alkylamino, dialkylamino, sulfate, or phosphate;
Each R13 is independently halo, haloalkyl, hydroxy, alkoxy, oxo, amino,
alkylamino, dialkylamino, sulfate, or phosphate;
Each R14 is independently halo, haloalkyl, hydroxy, alkoxy, nitro, amino,
alkylamino, dialkylamino, sulfate, or phosphate; and
xis 1-100.
Referring to Formula (I) above, a subset of compounds described above are
those in which x is 24 and R9 is n-tetradecyl.
In some embodiments, R1 is SO2R10 and R10 can be aryl substituted with
N(R11)2, e.g.:
3
CA 02519568 2005-09-19
WO 2004/094444 PCT/US2003/008530
H3C\ /CH3
N
I
ti
In other embodiments, RI is C(O)R10 and R10 can be C1-C6 alkyl substituted
with halo, hydroxy, oxo, nitro, OR, I, OR12, acyloxy, amino, NHRII, N(R11)2,
NHR12,
N(R12)2, aralkylamino, mercapto, thioalkoxy, S(O)R11, S(O)R12, S02R11, S02R12,
NHSO2R11, NHS02R12, sulfate, phosphate, cyano, carboxyl, C(O)R11, C(O)R12,
C(O)OR11, C(O)NH2, C(O)NHR11, C(O)N(R1i)2, C3-C10 cycloalkyl containing 0-3
R13, C3-C10 heterocyclyl containing 0-3 R13, C2-C6 alkenyl, C2-C6 alkenyl, C5-
Clo
cycloalkenyl, C5-C10 heterocycloalkenyl, C6-C20 aryl containing 0-3 R14, or C6-
C20
heteroaryl containing 0-3 R14.
In certain embodiments, R10 can be C1-C6 alkyl substituted with NHS02R12, in
which R12 is e.g.:
H3C\ /CH3
N
~LfULfL
4
CA 02519568 2005-09-19
WO 2004/094444 PCT/US2003/008530
In certain embodiments, R10 can be alkyl substituted with C(O)R12, in which
e.g. R12 is:
H3C` /CH3
N
In certain embodiments, R10 can be alkyl substituted with C5-C10 heterocyclyl
containing 0-3 R13 in which the heterocyclyl is e.g.:
15
O
HN NH
ds
5
CA 02519568 2005-09-19
WO 2004/094444 PCT/US2003/008530
In another aspect, this invention relates to a probe for observing glycolipid
association with CDId and NKT cell receptors during NKT cell stimulation
having a
compound of Formula (II):
H
OH
HO O C(O)C25H51
HN OH
OHO
C14H29
OH
(II)
wherein:
X is -S02-,-C(O)-, or absent;
Y is a linker group; and
Z is a reporter group.
In a further aspect, this invention relates to a method of quantifying
glycolipid
association with CD 1 d and NKT cell receptors during NKT cell stimulation
including: (i) contacting a compound of Formula (II) with a CD 1 d protein;
(ii)
allowing the compound to associate with the CDId protein; (iii) measuring
fluorescence emitted by the compound during steps (i) and (ii) to provide one
or more
pre-NKT cell contact fluorescence measurements; (iv) contacting the compound
and
CD1d protein with an NKT cell line; (v) measuring fluorescence emitted by the
compound during step (iv) to provide one or more NKT cell contact fluorescence
measurements.
Embodiments can include one or more of the following features.
Step (v) can be repeated over time.
6
CA 02519568 2005-09-19
WO 2004/094444 PCT/US2003/008530
The method can further include the step of comparing the fluorescence
measurements in steps (iii) and (v).
In one aspect, this invention features a method of stimulating NKT cells,
which includes contacting an NKT cell with a compound of Formula (I) and a CD1
protein.
The protein can be a CDld protein.
In another aspect, this invention features a method of stimulating the immune
system of a subject (e.g., mammal, human, dog, horse, cat) in need of such
stimulation, the method includes administering a compound of Formula (I) to
the
subject.
In a further aspect, this invention features a method of treating an
autoimmune
disease in a subject (e.g., mammal, human, dog, horse, cat) in need of such
treatment,
the method includes administering an effective amount of a compound of Formula
(I).
The subject can be a mammal, preferably a human. Identifying a subject in
need of such treatment can be in the judgement of a subject or a health care
professional and cap be subjective (e.g., opinion) or objective (e.g.,
measurable by a
test or diagnostic method).
25
In one aspect, this invention relates to a method of making a compound
described herein. In some embodiments, the method can be a method of making a
compound of Formula (I) including: (i) converting a compound of Formula (III)
to a
compound of Formula (IV):
7
CA 02519568 2005-09-19
WO 2004/094444 PCT/US2003/008530
R3\0 N3 R8
>=:::o R7
HN 0
R4.
R5 R
9
(III) ' R6
R30 NH2
R8
O O R7
H N O
R4 -0 O
R5 R9
(IV) R6
and (ii) contacting a compound of Formula (N) with R1-LG to afford a
compound of Formula (I), wherein:
R1 is:
(i) -S02R1o,
wherein R10 is:
halo; hydroxy; OR, I; OR12; amino; NHRI I; N(Ri i)2; NHR12i N(R12)2;
aralkylamino; or
C1-C12 alkyl optionally substituted with halo, hydroxy, oxo, nitro,
OR,,, OR12, acyloxy, amino, NHR11, N(R11)2, N 12, N(R12)2, aralkylamino,
mercapto, thioalkoxy, S(O)R11, S(O)R12, S02R11, S02RI2, NHSO2R11,
8
CA 02519568 2005-09-19
WO 2004/094444 PCT/US2003/008530
NHSO2R12, sulfate, phosphate, cyano, carboxyl, C(O)R11, C(O)RI2,
C(O)ORII, C(O)NH2, C(O)NHR11, C(O)N(Rll)2, C3-C10 cycloalkyl containing
0-3 R13, C3-Clo heterocyclyl containing 0-3 R13, C2-C6 alkenyl, C2-C6 alkenyl,
C5-C10 cycloalkenyl, C5-C10 heterocycloalkenyl, C6-C20 aryl containing 0-3
R14, or C6-C20 heteroaryl containing 0-3 R14; or
C3-Clo cycloalkyl, C3-CIO heterocyclyl, C5-C10 cycloalkenyl, or C5-CI0
heterocycloalkenyl optionally substituted with one or more halo, hydroxy,
oxo, OR,,, OR12, acyloxy, nitro, amino, NHR11, N(R11)2, NHR12, N(R12)2,
aralkylamino, mercapto, thioalkoxy, S(O)R11, S(O)R12, S02R11, S02R12,
NHS02R11, NHS02R12, sulfate, phosphate, cyano, carboxyl, C(O)R11,
C(O)R12, C(O)OR11, C(O)NH2, C(O)NHRII, C(O)N(R11)2, alkyl, haloalkyl,
C3-C10 cycloalkyl containing 0-3 R13, C3-C10 heterocyclyl containing 0-3 R13,
C2-C6 alkenyl, C2-C6 alkynyl, C5-C10 cycloalkenyl, C5-C10 heterocycloalkenyl,
C6-C20 aryl containing 0-3 R14, or C6-C20 heteroaryl containing 0-3 R14; or
C2-C6 alkenyl, C2-C6 alkynyl, aryl, or heteroaryl optionally substituted
with one or more halo, hydroxy, OR,,,. OR12, acyloxy, nitro, amino, NHR11,
N(RI1)2, NHR12, N(R12)2, aralkylamino, mercapto, thioalkoxy, S(O)R11,
S(O)R12, S02R11, SO2RI2, NHS02RII, NHSO2R12, sulfate, phosphate, cyano,
carboxyl, C(O)R11, C(O)R12, C(O)ORI1, C(O)NH2, C(O)NHR1I, C(O)N(R11)2,
alkyl, haloalkyl, C3-C10 cycloalkyl containing 0-3 R13, C3-C10 heterocyclyl
containing 0-3 R13, C2-C6 alkenyl, C2-C6 alkynyl, C5-C10 cycloalkenyl, C5-CI0
heterocycloalkenyl, C6-C20 aryl containing 0-3 R14, or C6-C20 heteroaryl
containing 0-3 R14; or
(ii) -C(O)Rlo, wherein RIO is defined as above; or
(iii) -C(RIO)2(RI5), wherein RIO is defined as above; R15 is hydrogen,
RIO, or R15 and R2 taken together forms a double bond between the carbon and
nitrogen atoms to which they are attached; or
R3, R4, R5, R6, and R7 are each independently hydrogen, CI-C6 alkyl, C6-C12
aralkyl, or C1-C6 acyl;
Rs is -(CH2),,CH3;
R9 is a linear or branched C3-C100 alkyl;
9
CA 02519568 2005-09-19
WO 2004/094444 PCT/US2003/008530
R11 is C1-C20 alkyl optionally substituted with halo, hydroxy, alkoxy, amino,
alkylamino, dialkylamino, sulfate, or phosphate;
R12 is aryl optionally substituted with halo, haloalkyl, hydroxy, alkoxy,
nitro,
amino, alkylamino, dialkylamino, sulfate, or phosphate;
Each R13 is independently halo, haloalkyl, hydroxy, alkoxy, oxo, amino,
alkylamino, dialkylamino, sulfate, or phosphate;
Each R14 is independently halo, haloalkyl, hydroxy, alkoxy, nitro, amino,
alkylamino, dialkylamino, sulfate, or phosphate;
x is 1-100;
LG is halo, -OS02R16, B(OH)2, or
O
N-0
O
and
R16 is alkyl, haloalkyl or aryl optionally substituted with alkyl, halo or
nitro.
In another aspect, this invention features a pharmaceutical composition
including a compound of Formula (I) and a pharmaceutically acceptable carrier.
Also within the scope of this invention is a composition containing one or
more of the compounds described herein for use in treating cancer or
autoimmune
disorders, diseases, or disease symptoms, including any of those delineated
herein,
and the use of such a composition for the manufacture of a medicament for the
just-
mentioned use.
Also within the scope of this invention is a packaged product. The packaged
product includes a container, one of the aforementioned compounds in the
container,
and a legend (e.g., a label or insert) associated with the container and
indicating
CA 02519568 2010-05-21
62396-1079
administration of the compound for treating cancer or autoimmune disorders,
diseases, or disease symptoms, including any of those delineated herein.
Embodiments of the invention may have one or more of the following
advantages. For example, the replacement of the parent sugar's C6'-hydroxyl by
the more reactive amino group allows for the efficient synthesis of a more
expansive range of C6"-amino-C6"-deoxygalactosylceramides. Further, having the
derivitization handle situated at Cs' allows a-g a lactosylce ram ides to be
modified
without significantly altering their binding to the proteins and receptors
involved in
T cell stimulation because the C6'-amino substituents are sufficiently
distanced
from the lipid portion of the molecule, which is known to engage in relatively
strong
interactions with the deep hydrophobic pockets of the CD1d proteins.
The details of one or more embodiments of the invention are set
forth in the accompanying drawings and the description below. Other features,
objects, and advantages of the invention will be apparent from the description
and
drawings, and from the claims.
According to one aspect of the present invention, there is provided a
compound of Formula (I):
R2
a R8
O O R7
HN 0
l0
R5 C I
R6
(t)
11
CA 02519568 2011-02-24
62396-1079
wherein,
R1 is -C(O)R10, wherein R10 is C1-C12 alkyl optionally substituted with
carboxyl;
R2, R3, R4, R5, R6 and R7 are each hydrogen;
R8 is -(CH2)XCH3;
R9 is a linear or branched C3-C100 alkyl;
and
x is 1-100.
According to another aspect of the present invention, there is provided
use of the compound described herein for stimulating the immune system of a
subject, for treating an autoimmune disease in a subject or, in combination
with a
CD1d protein for stimulating NKT cells.
According to yet another aspect of the present invention, there is
provided a method of making a compound of formula (I) as described herein
comprising: (i) converting a compound of Formula (III):
R3,, N3
O Rx
O 0
HN 0
/ R
7
R4 O
O 0 RS R
O~
R6
(III)
lla
CA 02519568 2011-02-24
62396-1079
to a compound of Formula (IV)
R3,,,~' NI-12 O Rs
O O O R
7
HN O/
R4
O O
R5 R9
R6
(IV)
and (ii) contacting the compound of Formula (IV) with R1-LG to afford a
compound of Formula (I), wherein:
R1 is -C(O)R10, wherein R10 is C1-C12 alkyl optionally substituted with
carboxyl;
R2, R3, R4, R5, R6, and R7 are each hydrogen;
R8 is -(CH2)xCH3;
R9 is a linear or branched C3-C100 alkyl;
xis 1-100; and
LG is halo, -0S02R16, B(OH)2, or
0
O
lib
CA 02519568 2010-05-21
62396-1079
wherein
R16 is alkyl, halo alkyl or aryl optionally substituted with alkyl,
halo or nitro.
DESCRIPTION OF DRAWINGS
FIG. 1 shows NKT cell stimulatory activity of fluorophore-appended
C6"-amino-C6"-deoxygalactosylceramides 8, 10, 12.
FIG. 2 shows NKT cell stimulatory activity of biotin-appended
C6"-amino-C6"-deoxygalactosylceramide 13.
DETAILED DESCRIPTION
As used herein, the term "halo" or "halogen" refers to any radical of
fluorine, chlorine, bromine or iodine.
The term "alkyl" refers to a hydrocarbon chain that may be a straight
chain or branched chain, containing the indicated number of carbon atoms. For
example, C1-C12 alkyl indicates that the group may have from 1 to 12
(inclusive)
carbon atoms in it. The terms "arylalkyl" or "aralkyl" refer to an alkyl
moiety in
which an alkyl hydrogen atom is replaced by an aryl group. Examples of
"arylalkyl" or "aralkyl" include benzyl and 9-fluorenyl groups.
11c
CA 02519568 2005-09-19
WO 2004/094444 PCT/US2003/008530
The terms "alkylamino" and "dialkylamino" refer to -NH(alkyl) and -
NH(alkyl)2 radicals respectively. The term "aralkylamino" refers to a -
NH(aralkyl)
radical. The term "alkoxy" refers to an - -alkyl radical. The term "mercapto"
refers
to an Sal radical. The term "thioalkoxy" refers to an -S-alkyl radical.
The term "aryl" refers to an aromatic monocyclic, bicyclic, or tricyclic
hydrocarbon ring system, wherein any ring atom capable of substitution can be
substituted by a substituent. Examples of aryl moieties include, but are not
limited to,
phenyl, naphthyl, and anthracenyl.
The term "cycloalkyl" as employed herein includes saturated cyclic, bicyclic,
1 o tricyclic,or polycyclic hydrocarbon groups having 3 to 12 carbons, wherein
any ring
atom capable of substitution can be substituted by a substituent. Examples of
cycloalkyl moieties include, but are not limited to, cyclohexyl and adamantyl.
The term "heterocyclyl" refers to a nonaromatic 3-10 membered monocyclic,
8-12 membered bicyclic, or 11-14 membered tricyclic ring system having 1-3
heteroatoms if monocyclic, 1-6 heteroatoms if bicyclic, or 1-9 heteroatoms if
tricyclic,
said heteroatoms selected from 0, N, or S (e.g., carbon atoms and 1-3, 1-6, or
1-9
heteroatoms of N, 0, or S if monocyclic, bicyclic, or tricyclic,
respectively), wherein
any ring atom capable of substitution can be substituted by a substituent.
The term "cycloalkenyl" as employed herein includes partially unsaturated,
nonaromatic, cyclic, bicyclic, tricyclic,or polycyclic hydrocarbon groups
having 5 to
12 carbons, preferably 5 to 8 carbons, wherein any ring atom capable of
substitution
can be substituted by a substituent. Examples of cycloalkyl moieties include,
but are
not limited to cyclohexenyl, cyclohexadienyl, or norbornenyl.
The term "heterocycloalkenyl" refers to a partially saturated, nonaromatic 5-
10 membered monocyclic, 8-12 membered bicyclic, or 11-14 membered tricyclic
ring
system having 1-3 heteroatoms if monocyclic, 1-6 heteroatoms if bicyclic, or 1-
9
heteroatoms if tricyclic, said heteroatoms selected from 0, N, or S (e.g.,
carbon atoms
and 1-3, 1-6, or 1-9 heteroatoms of N, 0, or S if monocyclic, bicyclic, or
tricyclic,
respectively), wherein any ring atom capable of substitution can be
substituted by a
substituent.
The term "heteroaryl" refers to an aromatic 5-8 membered monocyclic, 8-12
membered bicyclic, or 11-14 membered tricyclic ring system having 1-3
heteroatoms
12
CA 02519568 2005-09-19
WO 2004/094444 PCT/US2003/008530
if monocyclic, 1-6 heteroatoms if bicyclic, or 1-9 heteroatoms if tricyclic,
said
heteroatoms selected from 0, N, or S (e.g., carbon atoms and 1-3, 1-6, or 1-9
heteroatoms of N, 0, or S if monocyclic, bicyclic, or tricyclic,
respectively), wherein
any ring atom capable of substitution can be substituted by a substituent.
The term "oxo" refers to an oxygen atom, which forms a carbonyl when
attached to carbon, an N-oxide when attached to nitrogen, and a sulfoxide or
sulfone
when attached to sulfur.
The term "acyl" refers to an alkylcarbonyl, cycloalkylcarbonyl, arylcarbonyl,
heterocyclylcarbonyl, or heteroarylcarbonyl substituent, any of which may be
further
lo substituted by substituents.
The term "substituents" refers to a group "substituted" on an alkyl,
cycloalkyl,
alkenyl, alkynyl, heterocyclyl, heterocycloalkenyl, cycloalkenyl, aryl, or
heteroaryl
group at any atom of that group. Suitable substituents include, without
limitation,
alkyl, alkenyl, alkynyl, alkoxy, halo, hydroxy, cyano, nitro, amino, SO3H,
sulfate,
phosphate, perfluoroalkyl, perfluoroalkoxy, methylenedioxy, ethylenedioxy,
carboxyl,
oxo, thioxo, imino (alkyl, aryl, aralkyl), S(O)õalkyl (where n is 0-2), S(O)õ
aryl
(where n is 0-2), S(O)õ heteroaryl (where n is 0-2), S(O)õ heterocyclyl (where
n is 0-
2), amine (mono-, di-, alkyl, cycloalkyl, aralkyl, heteroaralkyl, and
combinations
thereof), ester (alkyl, aralkyl, heteroaralkyl), amide (mono-, di-, alkyl,
aralkyl,
heteroaralkyl, and combinations thereof), sulfonamide (mono-, di-, alkyl,
aralkyl,
heteroaralkyl, and combinations thereof), unsubstituted aryl, unsubstituted
heteroaryl,
unsubstituted heterocyclyl, and unsubstituted cycloalkyl. In one aspect, the
substituents on a group are independently any one single, or any subset of the
aforementioned substituents.
The term "treating" or "treated" refers to administering a compound described
herein to a subject with the purpose to cure, heal, alleviate, relieve, alter,
remedy,
ameliorate, improve, or affect a disease, the symptoms of the disease or the
predisposition toward the disease.
"An effective amount" refers to an amount of a compound that confers a
therapeutic effect on the treated subject. The therapeutic effect may be
objective (i.e.,
measurable by some test or marker) or subjective (i.e., subject gives an
indication of
or feels an effect). An effective amount of the compound described above may
range
13
CA 02519568 2005-09-19
WO 2004/094444 PCT/US2003/008530
from about 0.1 mg/Kg to about 500 mg/Kg, alternatively from about 1 to about
50
mg/Kg. Effective doses will also vary depending on route of administration, as
well
as the possibility of co-usage with other agents.
The term "mammal" includes organisms, which include mice, rats, cows,
sheep, pigs, goats, and horses, monkeys, dogs, cats, and preferably humans.
Structure of 6' -amino-6 "-deoxygalactosylceramide compounds
In general, the 6' '-amino-6' '-deoxygalactosylceramide compounds include a
"glyco" portion and a "lipid" portion as indicated in Formula (V). The two
portions
1o are appended to one another via an a-glycosidic bond between the anomeric
carbon,
Ca, of the "glyco" portion and the oxygen (bolded) bound to C1 of the "lipid"
portion.
R2
R3 I
O C6/ `R1 R$
` /R7
O >=::::o
R4 Ca H N O
O i C C4
/ 4
C1 C3 R9
1
"glyco" portion O"1-, R6
"lipid" portion
(V)
The lipid portion is a chain of carbon atoms having functionalized and
unfuctionalized segments. The functionalized segment includes carbons C1-C4.
The
functionalized segment terminus C1 is the carbon through which the lipid
portion is
appended to the glyco portion of the molecule. The remaining members of this
14
CA 02519568 2005-09-19
WO 2004/094444 PCT/US2003/008530
segment, C2, C3, and C4, each contain a heteroatom substituent. The C2
nitrogen is
acylated with C(O)R8, in which R8 is a hydrocarbon chain represented by the
formula
(CH2)xCH3. The hydrocarbon chain R8 can have 1 to 100 methylene (CH2) groups
(e.g., 1 to 75 CH2 groups, 1 to 50 CH2 groups, 1 to 25 CH2 groups, 1 to 20 CH2
groups, 1 to 15 CH2 groups, 1 to 10 CH2 groups, or 1 to 5 CH2 groups). In
certain
embodiments, R8 contains 24 CH2 groups. The oxygens on C3, and C4, may be
substituted with hydrogen, alkyl, aralkyl, trisubstituted silyl, or acyl
groups. In some
embodiments, R5 and R6 are hydrogen, and in other embodiments, they are tert-
butyldimethylsilyl (TBS).
The unfunctionalized segment is represented by R9, which can include any
branched or unbranched alkyl group containing 3-100 carbons atoms (e.g, 3-75
carbons atoms, 3-50 carbons atoms, 3-25 carbons atoms, 3-20 carbons atoms, 3-
15
carbons atoms, or 3-10 carbons atoms). In certain embodiments, R9 contains an
unbranched alkyl group composed of 14 carbon atoms.
The glyco portion is a derivative of cc-D- galactose. Each of R3, R4, and R5
may be hydrogen, alkyl, aralkyl or acyl groups. When R3-R5 are substituted
with a
group other than hydrogen, the group is preferably one that is readily removed
using
carbohydrate deprotection chemistries that are well known in the art. In
certain
embodiments, these groups include methyl, benzyl or acetyl.
The C6"-hydroxyl group of the parent sugar is replaced by a substituted (R1
and/or R2 are substituents other than hydrogen) or unsubstituted (R1 and R2
are both
hydrogen) amino group.
In certain embodiments, the C6"-amino group is monosubstituted (R1 =
substitutent and R2 = H). For example, the C6"-nitrogen can form part of a
sulfonamide (R1 = -SO2R) or amide (R1 = C(O)R group. R may include e.g.,
substituted or unsubstituted alkyl, cycloalkyl, aryl, heteroaryl, etc. These
groups can
be formed upon the reaction of the unsubstituted amino group with e.g., the
corresponding sulfonyl halide or activated acyl derivative. Alternatively, the
nitrogen
may form part of a secondary alkyl-alkyl or alkyl-aryl amino group (e.g., R1=
substituted or unsubstituted alkyl or aryl). Introduction of an alkyl group at
R1 can be
carried out e.g., by first exposing the unsubstituted amino group to a
carbonyl
compound and then performing a reductive alkylation on the resulting,
intermediary
CA 02519568 2005-09-19
WO 2004/094444 PCT/US2003/008530
aldimine or ketenimine. Introduction of an aryl group at Rl can be
accomplished e.g.,
by transition metal mediated coupling between the unsubstituted amino group
and an
aryl halide or triflate.
In other embodiments, the C6"-amino group is disubstituted (Ri and R2 =
substituent). In some embodiments, Rt and R2 may form a cyclic structure in
which
one or more of the ring atoms may be a heteroatom (e.g., N, 0, or S). Further,
any
one of the ring atoms may be substituted with e.g., halo, hydroxy, alkyl,
haloalkyl,
aryl, herteroaryl, etc.
In some embodiments, the C6 '-nitrogen can form part of an imino group, i.e.,
1 o C=N. The imino group can be an aldimine, which may be obtained in a
condensation
reaction between the C6"-amino group and a substituted or unsubstituted alkyl
or aryl
aldehyde. Similarly, the imino group can be a ketenimine, which may be
obtained in
a condensation reaction between the C6-amino group and a substituted or
unsubstituted dialkyl ketone, a diarylketone, aryl-alkyl ketone, etc.
In certain embodiments, it can be advantageous for a reporter group to be
linked either directly or indirectly to the C6"-amino group. While not wishing
to be
bound by theory, it believed that labelling galactosylceramides with
fluorophores or
other small molecules (e.g., biotin) would allow observation of the compounds
at low
concentrations and/or provide a means of quantifying association with CD 1 d
and
NKT cell receptors. Thus compounds containing reporter groups could be useful
as
probes for determining e.g., specific structural requirements for glycolipid
binding by
CD 1 d and T cell receptors.
In some embodiments, probes are compounds in which the reporter group may
be directly attached to the C6"-amino group in either a covalent or
noncovalent
manner. In other embodiments, the reporter group may be indirectly attached to
the
C6-amino group via covalent or noncovalent linkages. For example, when the
reporter group is indirectly attached, the C6"-amino group can be attached to
a moiety
-X-Y-Z, wherein X is -S02-, -C(0)-, or absent; Y is a linker group, and Z is a
reporter
group.
The linker group Y can be any carbon-containing chain or ring. For example,
the linker can be -(CII2)t-, in which the chain optionally contains one or
more terminal
heteroatoms (e.g., N, 0, S), and/or one or more heteroatoms, rings, double
bonds,
16
CA 02519568 2010-05-21
62396-1079
triple bonds that are inserted into the chain. The value of"t" can be 1-100.
The linker
may also be one ring, or a series-of two or more rings.
The reporter groups may be selected as desired. Selection of the reporter
groups is within skill of the art. Examples of reporter groups include
labelling
reagents, e.g., radiolabelled moieties, functional small molecules, e.g.,
biotin, or
fluorophores e.g., acridines, Cy5.5Tm, Dabcyl, Dansyl, Fluorescien, Oregon
GreenM
488, Prodan, Tamra, etc. Representative reporter groups may be selected and
obtained from e.g., Molecular Probes, Inc.
Combinations of substituents and variables envisioned by this invention are
only those that result in the formation of stable compounds. The term
"stable", as used
herein, refers to compounds which possess stability sufficient to allow
manufacture
and which maintains the integrity of the compound for a sufficient period of
time to
be useful for the purposes detailed herein (e.g., therapeutic or prophylactic
administration to a subject).
Synthesis of 6"-amino-6"-deoxygaiactosylceramide compounds
The synthesis of 6""-amino-6"-deoxygalactosylceramides may be carried out
using conventional methods including those described herein for exemplary
compound 1 (R3-R7 = H; R$ = (CH2)24CH3; and R9 = C14H29). In general, compound
1 may be obtained from the reaction between compound 2 and a desired
electrophile,
e.g., RI-leaving group, as shown below.
17
CA 02519568 2005-09-19
WO 2004/094444 PCT/US2003/008530
R1
I
OH NH C25H51
O O
HO H= OH
OH
C14H29
OH
OH NH2 C25H51
O O
HO HN OH
OHO
C14H29
OH
2
The synthesis of compound 2 can be carried out as follows. The amine
functionality was incorporated early onto the carbohydrate as the azide using
the
procedure of Corey et al., J. Airs. Chem. Soc. 1984, 106, 3682. (3, Scheme 1).
Following the procedure of Singh, P. P., et al., Carbohyd. Res. 1970, 12, 261,
the
acetonides were hydrolyzed with concomitant methylgalactoside formation and
benzyl ethers at C2, C3 and C4 were formed giving 4. The methoxy group was
then
18
CA 02519568 2005-09-19
WO 2004/094444 PCT/US2003/008530
replaced by an acetoxy group, followed by conversion to the anomeric fluoride
(5)
according to the methods of Davis, N. J., et al., J Chem. Soc. Perkin Trans. 1
1994,
359 and Hayashi, M., et al., Chem. Lett. 1984, 1747 respectively. Glycosyl
bond
formation with compound 6 gave 7 via Hashimoto, et al., Tetrahedron Lett.
1984, 25,
1379. Compound 6, (2S, 3S, 4R)-3,4-bis-t-butyldimethylsisyloxy-2-
hexaosanoylamino-4-octadecanol, was prepared by coupling of phytosphingosine
(Avanti Polar Lipids) with hexacosanoic acid using 2-dimethylaminoisopropyl
chloride hydrochloride (DIC) and 1-hydroxybenzotriazole (HOBT), followed by
the
protection/deprotection scheme reported by Takikawa, et al., Tetrahedron 1998,
54,
3141. The silyl protecting groups were removed (Takikawa, et al.) followed by
reduction of the azide (Vaultier, M. et al., Tetrahedron Lett. 1983, 24, 763)
and
removal of the benzyl groups giving 2 (Sakai, T. et al., J. Med. Chem. 1998,
41, 650).
SCHEME 1
Bn Bit
B,O Okb e,d> Bn
X Bn0 BnOF
4 5
3 Bn ~-
5 + H OTBS e Bn HN OTBS
BnO
H0.~~C1 aH2s ~~~C1 aHzs
6 OTBS 7 OTBS
H H2 O
C25H51
f q H HO HN OH
\C1 4H29
2 OH
Reagents (yields in parentheses): a) AcCl, MeOH (86% yield). b) BnBr, 18-crown-
6, NaH, THE (95% yield). c)
AcOH, H2SO4 (84% yield). d) HF=pyridine, pyridine (78% yield). e) MS 4A,
AgC1O4, SnC12, THE (44% yield).
f) TBAF, THE (81% yield). g) PPh3/H20, THE (quant. yield). h) NH3/Na, -78 C
(53% yield).
Reaction of 2 with acid chlorides and N-hydroxysuccinimidyl (NHS) esters
can provide reasonable yields of the corresponding amides. For example,
compounds
8 and 10 were prepared from 2 and dansyl chloride and 9 respectively (Scheme
2).
The latter compound is a dansyl amide tethered to an N-hydroxysuccinimidyl
ester
19
CA 02519568 2005-09-19
WO 2004/094444 PCT/US2003/008530
(Wang, F.; Schwabacher, A. W. J. Org. Chem. 1999, 64, 8922). NHS ester 11 can
be
prepared from 4-(6-methoxy-[2]naphthyl)-4-oxo-butyric acid (Khan, M. et al.,
Indian
J Chem. Sect. B 2000, 39, 614) by nucleophilic displacement of the methoxy
group
with lithium dimethylamide (Weber, G., et al., Biocheinistiy 1979, 18, 3075)
followed
by reaction with dicyclohexylcarbodiimide (DCC) and N- hydroxysuccinimide.
Reaction of NHS ester 11 with 2 gave 12 in 46% yield. Similarly, reaction of 2
with
the N-hydroxysuccinimidyl ester of biotin gave 13 in 52% yield (Scheme 3).
Glycosylceramides can exhibit relatively limited solubility in many organic
solvents,
and this relatively high insolubility may result in loss of yield, e.g.,
during
purification.
SCHEME 2
0.Q NMe2
dansyl chloride i-V NH
2 -C25H51
Et3N HN OH
IC14H29
OH
8 (60% yield)
H ';6-Me2
NH S
Mee 0
5p`O
O g HO HN OH 1
2
pyridine c149
OH
10 (53% yield)
SCHEME 3
O / ^ /NMe2
O 11 O
1-. H
Me2 HH 'H
H H NN H
H9H 51-151 ~HN--PFH51
HC~_ ` 1
HO HO
2~^C14H29 C14Hz9
1
OH 13 OH
CA 02519568 2005-09-19
WO 2004/094444 PCT/US2003/008530
The synthesized 6''-amino-6 "-deoxygalactosylceramide compounds can be
separated from a reaction mixture and further purified by a method such as
column
chromatography, high pressure liquid chromatography, or recrystallization. As
can be
appreciated by the skilled artisan, further methods of synthesizing the
compounds of
the formulae herein will be evident to those of ordinary skill in the art.
Additionally,
the various synthetic steps may be performed in an alternate sequence or order
to give
the desired compounds. Synthetic chemistry transformations and protecting
group
methodologies (protection and deprotection) useful in synthesizing the
compounds
described herein are known in the art and include, for example, those such as
1o described in R. Larock, Comprehensive Organic Transformations, VCH
Publishers
(1989); T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis,
2d.
Ed., John Wiley and Sons (1991); L. Fieser and M. Fieser, Fieser and Fieser's
Reagents for Organic Synthesis, John Wiley and Sons (1994); and L. Paquette,
ed.,
Encyclopedia of Reagents for Organic Synthesis, John Wiley and Sons (1995),
and
subsequent editions thereof.
The 6 "-amino-6 "-deoxygalactosylceramide compounds of this invention may
contain one or more asymmetric centers and thus occur as racemates and racemic
mixtures, single enantiomers, individual diastereomers and diastereomeric
mixtures.
All such isomeric forms of these compounds are expressly included in the
present
invention. The compounds of this invention may also be represented in multiple
tautomeric forms, in such instances, the invention expressly includes all
tautomeric
forms of the compounds described herein (e.g., alkylation of a ring system may
result
in alkylation at multiple sites, the invention expressly includes all such
reaction
products). All such isomeric forms of such compounds are expressly included in
the
present invention. All crystal forms of the compounds described herein are
expressly
included in the present invention.
The 6"-amino-6"-deoxygalactosylceramide compounds of this invention
include the compounds themselves, as well as their salts and their prodrugs,
if
applicable. A salt, for example, can be formed between an anion and a
positively
charged substituent (e.g., amino) on a 6"-amino-6"-deoxygalactosylceramide
compound. Suitable anions include chloride, bromide, iodide, sulfate, nitrate,
phosphate, citrate, methanesulfonate, trifluoroacetate, and acetate. Likewise,
a salt
21
CA 02519568 2005-09-19
WO 2004/094444 PCT/US2003/008530
can also be formed between a cation and a negatively charged substituent
(e.g.,
carboxylate) on a 6"-amino-6"-deoxygalactosylceramide compound. Suitable
cations include sodium ion, potassium ion, magnesium ion, calcium ion, and an
ammonium cation such as tetramethylammonium ion. Examples of prodrugs include
esters and other pharmaceutically acceptable derivatives, which, upon
administration
to a subject, are capable of providing active 6''-amino-6''-
deoxygalactosylceramide
compounds.
The compounds of this invention may be modified by appending appropriate
functionalities to enhance selective biological properties. Such modifications
are
lo known in the art and include those which increase biological penetration
into a given
biological compartment (e.g., blood, lymphatic system, central nervous
system),
increase oral availability, increase solubility to allow administration by
injection, alter
metabolism and alter rate of excretion.
Methods of Using 6 "-amino-6"-deoxy alg actosylceramide compounds
The effect of a particular C6"-substitution on a 6''-amino-6 "-
deoxygalactosylceramide compound's ability to stimulate NKT cells can be
evaluated
e.g., by measuring interleukin (IL)-2 production using an immobilized CD1d
assay
(Benlagha, K.; Weiss, A.; Beavis, A.; Teyton, L.; Bendalac, A. J. Exp. Med.
2000,
191, 1895). The assay includes loading soluble, biotinylated CD 1 d onto
precoated
avidin plates, pulsing the plates with incrementally varied concentrations of
glycolipids, washing the plates, treating the plates with a CD 1 d-restricted
Va24 NKT
cell hybridoma, and measuring IL-2 release using ELISA (enzyme-linked
immunosorbent assay).
Each compound's NKT cell stimulating ability is compared against that of the
reference compound, KRN7000,14 (Morita, M., et al., I Med. Chem. 1995, 38,
2176.).
OH OH
HO O C(O)C25H51
HN OH
OHO
22
C14H29
CA 02519568 2005-09-19
WO 2004/094444 PCT/US2003/008530
Compounds 8, 10, 12 and 13 were determined to possess relatively high NKT
cell stimulating abilities according to the above assay. The results in
Figures 1 and 2
indicate that there is a dose-dependent response to the glycolipids (i.e.,
cytokine
release) and that this response is comparable to that of the reference
compound.
Although 8 and 12 appear slightly less efficient in the results from the
experiment
shown in Figure 1, no significant differences among the compounds were found
in
repeated experiments (at least three experiments for each compound). In a
separate
1o series of experiments (e.g., Figure 2), compound 13 was slightly, but
reproducibly
more efficient in stimulating NKT cells than 1. Similar results were observed
using
CD 1 d transfected rat basophilic leukemia cells for antigen presentation to
NKT cell
hybridomas.
The attachment of a dansyl group directly at C6" (as in 8) or through a five
carbon tether (as in 10) did not cause a significant loss of stimulating
properties.
Similarly, alteration of the appended group (i.e., dansyl vs, prodan vs.
biotin) -did not
greatly affect the abilities of these glycolipids to stimulate NKT cells.
Binding of the glycolipids with CD1d and NKT cell receptors can be
visualized by fluorescence modulation studies, fluorescence and surface
plasmon
resonance studies, which employ 6 "-amino-6''-deoxygalactosylceramide probe
compounds that contain one or more reporter groups attached directly or
indirectly to
the 6"-amino group (e.g., probe compounds described herein). In certain
embodiments, probes that contain fluorophores as the reporting group can be
used to
quantify the association between the glycolipids and CD1d and the NKT cell
receptors during NKT cell stimulation. The selection of experimental protocols
to
observe association are within the art and are described in Kasten, F.H.,
"Introduction
to Fluorescent Probes: Properties, History and Applications" in Fluorescent
and
Luminescent Probes for Biological Activit, W.T. Mason, Ed., Academic Press
(1993)
pp. 12-33 and Lakowicz, J.R., Ed., Topics in Fluorescence Spectroscopy: Probe
Design and Chemical Sensing (Volume 4), Plenum Publishing (1994).
Pharmaceutically acceptable salts of the compounds of this invention include
those derived from pharmaceutically acceptable inorganic and organic acids and
23
CA 02519568 2005-09-19
WO 2004/094444 PCT/US2003/008530
bases. Examples of suitable acid salts include acetate, adipate, alginate,
aspartate,
benzoate, benzenesulfonate, bisulfate, butyrate, citrate, camphorate,
camphorsulfonate, digluconate, dodecylsulfate, ethanesulfonate, formate,
fumarate,
glucoheptanoate, glycolate, hemisulfate, heptanoate, hexanoate, hydrochloride,
hydrobromide, hydroiodide, 2-hydroxyethanesulfonate, lactate, maleate,
malonate,
methanesulfonate, 2-naphthalenesulfonate, nicotinate, nitrate, palmoate,
pectinate,
persulfate, 3-phenylpropionate, phosphate, picrate, pivalate, propionate,
salicylate,
succinate, sulfate, tartrate, thiocyanate, tosylate and undecanoate. Other
acids, such
as oxalic, while not in themselves pharmaceutically acceptable, may be
employed in
1o the preparation of salts useful as intermediates in obtaining the compounds
of the
invention and their pharmaceutically acceptable' acid addition salts. Salts
derived
from appropriate bases include alkali metal (e.g., sodium), alkaline earth
metal (e.g.,
magnesium), ammonium and N-(alkyl)4+ salts. This invention also envisions the
quaternization of any basic nitrogen-containing groups of the compounds
disclosed
herein. Water or oil-soluble or dispersible products may be obtained by such
quaternization. Salt forms of the compounds of any 'of the formulae herein can
be
amino acid salts of carboxy groups (e.g. L-arginine, -lysine, -histidine
salts).
The compounds of the formulae described herein can, for example, be
administered by injection, intravenously, intraarterially, subdermally,
intraperitoneally, intramuscularly, or subcutaneously; or orally, buccally,
nasally,
transmucosally, topically, in an ophthalmic preparation, or by inhalation,
with a
dosage ranging from about 0.5 to about 100 mg/kg of body weight, alternatively
dosages' between 1 mg and 1000 mg/dose, every 4 to 120 hours, or according to
the
requirements of the particular drug. The methods herein contemplate
administration
of an effective amount of compound or compound composition to achieve the
desired
or stated effect. Typically, the pharmaceutical compositions of this invention
will be
administered from about 1 to about 6 times per day or alternatively, as a
continuous
infusion. Such administration can be used as a chronic or acute therapy. The
amount
of active ingredient that may be combined with the carrier materials to
produce a
single dosage form will vary depending upon the host treated and the
particular mode
of administration.. typical preparation will contain from about 5% to about
95%
24
_~^e CA 02519568 2010-05-21
62396-1079
active compound (w/w). Alternatively, such preparations contain from about 20%
to
about 80% active compound.
Lower or higher doses than those recited above may be required. Specific
dosage and treatment regimens for any particular patient will depend upon a
variety of
factors, including the activity of the specific compound employed, the age,
body
weight, general health status, sex, diet, time of administration, rate of
excretion, drug
combination, the severity and course of the disease, condition or symptoms,
the
patient's disposition to the disease, condition or symptoms, and the judgment
of the
treating physician-
Upon improvement of a patient's condition, a maintenance dose of a
compound, composition or combination of this invention maybe administered, if
necessary. Subsequently, the dosage or frequency of administration, or both,
may be
reduced, as a function of the symptoms, to a level at which- the improved
condition is
retained when the symptoms have been alleviated to the, desired level.
Patients may,
however, require intermittent treatment on a long-term basis upon any
recurrence of
disease symptoms.
The compositions delineated herein include the compounds of the formulae
delineated herein, as well as additional therapeutic agents if present, in
amounts
effective for achieving a modulation of disease or disease symptoms, including
those
described herein.
The term "pharmaceutically acceptable carrier or adjuvant" refers to a carrier
or adjuvant that may be administered to a patient, together with a compound of
this
invention, and which does not destroy the pharmacological activity thereof and
is
nontoxic when administered in doses sufficient to deliver a therapeutic amount
of the
compound.
Pharmaceutically acceptable carriers, adjuvants and vehicles that may be used
in the pharmaceutical compositions of this invention include, but are not
limited to,
ion exchangers, alumina, aluminum stearate, lecithin, self-emulsifying drug
delivery
systems (SEDDS) such as d-a-tocopherol polyethyleneglycol 1000 succinate,
TM
surfactants used in pharmaceutical dosage forms such as Tweens or other
similar
polymeric delivery matrices, serum proteins, such as human serum albumin,
buffer
substances such as phosphates, glycine, sorbic acid, potassium sorbate,
partial
CA 02519568 2005-09-19
WO 2004/094444 PCT/US2003/008530
glyceride mixtures of saturated vegetable fatty acids, water, salts or
electrolytes, such
as protamine sulfate, disodium hydrogen phosphate, potassium hydrogen
phosphate,
sodium chloride, zinc salts, colloidal silica, magnesium trisilicate,
polyvinyl
pyrrolidone, cellulose-based substances, polyethylene glycol, sodium
carboxymethylcellulose, polyacrylates, waxes, polyethylene-polyoxypropylene-
block
polymers, polyethylene glycol and wool fat. Cyclodextrins such as a-, J3-, and
y-
cyclodextrin, or chemically modified derivatives such as
hydroxyalkylcyclodextrins,
including 2- and 3-hydroxypropyl-(3-cyclodextrins, or other solubilized
derivatives
may also be advantageously used to enhance delivery of compounds of the
formulae
1o described herein.
The pharmaceutical compositions of this invention may be administered
orally, parenterally, by inhalation spray, topically, rectally, nasally,
buccally,
vaginally or via an implanted reservoir, preferably by oral administration or
administration by injection. The pharmaceutical compositions of this invention
may
contain any conventional non-toxic pharmaceutically-acceptable carriers,
adjuvants or
vehicles. In some cases, the pH of the formulation may be adjusted with
pharmaceutically acceptable acids, bases or buffers to enhance the stability
of the
formulated compound or its delivery form. The term parenteral as used herein
includes subcutaneous, intracutaneous, intravenous, intramuscular,
intraarticular,
intraarterial, intrasynovial, intrasternal, intrathecal, intralesional and
intracranial
injection or infusion techniques.
The pharmaceutical compositions may be in the form of a sterile injectable
preparation, for example, as a sterile injectable aqueous or oleaginous
suspension.
This suspension may be formulated according to techniques known in the art
using
suitable dispersing or wetting agents (such as, for example, Tween 80) and
suspending agents. The sterile injectable preparation may also be a sterile
injectable
solution or suspension in a non-toxic parenterally acceptable diluent or
solvent, for
example, as a solution in 1,3-butanediol. Among the acceptable vehicles and
solvents
that may be employed are mannitol, water, Ringer's solution and isotonic
sodium
chloride solution. In addition, sterile, fixed oils are conventionally
employed as a
solvent or suspending medium. For this purpose, any bland fixed oil may be
employed including synthetic mono- or diglycerides. Fatty acids, such as oleic
acid
26
CA 02519568 2010-05-21
62396-1079
and its glyceride derivatives are useful in the preparation of injectables, as
are natural
pharmaceutically-acceptable oils, such as olive oil or castor oil, especially
in their
polyoxyethylated versions. These oil solutions or suspensions may also contain
a
long-chain alcohol diluent or dispersant, or carboxymethyl cellulose or
similar
dispersing agents which are commonly used in the formulation of
pharmaceutically
acceptable dosage forms such as emulsions and or suspensions. Other commonly
used
TM TM
surfactants such as Tweens or Spans and/or other similar emulsifying agents or
bioavailability enhancers which are commonly used in the manufacture of
pharmaceutically acceptable solid, liquid, or other dosage forms may also be
used for
lo the purposes of formulation.
The pharmaceutical compositions of this invention may be orally administered
in, any orally acceptable dosage form including, but not limited to, capsules,
tablets,
emulsions and aqueous suspensions, dispersions and solutions. In the case of
tablets
for oral use, carriers which are commonly used include lactose and corn
starch.
Lubricating agents, such as magnesium stearate, are also typically added. For
oral
administration in a capsule form, useful diluents include lactose and dried
corn starch.
When aqueous suspensions and/or emulsions are administered orally, the active
ingredient may be suspended or dissolved in an oily phase is combined with
emulsifying and/or suspending agents. If desired, certain sweetening and/or
flavoring
and/or.. coloring agents maybe added.
The pharmaceutical compositions, of this invention may also be administered
in the form of suppositories for rectal administration. These compositions can
be
prepared by mixing a compound of this invention with a suitable non irritating
excipient which is solid at room temperature but liquid at the rectal
temperature and
therefore will melt in the rectum to release the active components. Such
materials
include, but are not limited to, cocoa butter, beeswax and polyethylene
glycols.
Topical administration of the pharmaceutical compositions of this invention is
useful when the desired treatment involves areas or organs readily accessible
by
topical application. For application topically to the skin, the pharmaceutical
composition should be formulated with a suitable ointment containing the
active
components suspended or dissolved in a carrier. Carriers for topical
administration of
the compounds of this invention include, but are not limited to, mineral oil,
liquid
27
CA 02519568 2005-09-19
WO 2004/094444 PCT/US2003/008530
petroleum, white petroleum, propylene glycol, polyoxyethylene polyoxypropylene
compound, emulsifying wax and water. Alternatively, the pharmaceutical
composition can be formulated with a suitable lotion or cream containing the
active
compound suspended or dissolved in a carrier with suitable emulsifying agents.
Suitable carriers include, but are not limited to, mineral oil, sorbitan
monostearate,
polysorbate 60, cetyl esters wax, cetearyl alcohol, 2-octyldodecanol, benzyl
alcohol
and water. The pharmaceutical compositions of this invention may also be
topically
applied to the lower intestinal tract by rectal suppository formulation or in
a suitable
enema formulation. Topically-transdermal patches are also included in this
invention.
The pharmaceutical compositions of this invention may be administered by
nasal aerosol or inhalation. Such compositions are prepared according to
techniques
well-known in the art of pharmaceutical formulation and may be prepared as
solutions
in saline, employing benzyl alcohol or other suitable preservatives,
absorption
promoters to enhance bioavailability, fluorocarbons, and/or other solubilizing
or
dispersing agents known in the art.
A composition having the compound of the formulae herein and an additional
agent (e.g., a therapeutic agent) can be administered using an implantable
device.
Implantable devices and related technology are known in the art and are useful
as
delivery systems where a continuous, or timed-release delivery of compounds or
compositions delineated herein is desired. Additionally, the implantable
device
delivery system is useful for targeting specific points of compound or
composition
delivery (e.g., localized sites, organs). Negrin et al., Biomaterials,
22(6):563 (2001).
Timed-release technology involving alternate delivery methods can also be used
in
this invention. For example, timed-release formulations based on polymer
technologies, sustained-release techniques and encapsulation techniques (e.g.,
polymeric, liposomal) can also be used for delivery of the compounds and
compositions delineated herein.
Also within the invention is a patch to deliver active chemotherapeutic
combinations herein. A patch includes a material layer (e.g., polymeric,
cloth, gauze,
bandage) and the compound of the formulae herein as delineated herein. One
side of
the material layer can have a protective layer adhered to it to resist passage
of the
compounds or compositions. The patch can additionally include an adhesive to
hold
28
CA 02519568 2005-09-19
WO 2004/094444 PCT/US2003/008530
the patch in place on a subject. An adhesive is a composition, including those
of either
natural or synthetic origin, that when contacted with the skin of a subject,
temporarily
adheres to the skin. It can be water resistant. The adhesive can be placed on
the patch
to hold it in contact with the skin of the subject for an extended period of
time. The
adhesive can be made of a tackiness, or adhesive strength, such that it holds
the
device in place subject to incidental contact, however, upon an affinnative
act (e.g.,
ripping, peeling, or other intentional removal) the adhesive gives way to the
external
pressure placed on the device or the adhesive itself, and allows for breaking
of the
adhesion contact. The adhesive can be pressure sensitive, that is, it can
allow for
1o positioning of the adhesive (and the device to be adhered to the skin)
against the skin
by the application of pressure (e.g., pushing, rubbing,) on the adhesive or
device.
When the compositions of this invention comprise a combination of a
compound of the formulae described herein and one or more additional
therapeutic or
prophylactic agents, both the compound and the additional agent should be
present at
dosage levels of between about 1 to 100%, and more preferably between about 5
to
95% of the dosage normally administered in a monotherapy regimen. The
additional
agents maybe administered separately, as part'of a multiple dose regimen, from
the
compounds of this invention. Alternatively, those agents may be part of a
single
dosage form, mixed together with the compounds of this invention in a single
composition.
The compounds of the invention can be used in the treatment of cancer. The
cancer can be, but is not limited to: a human leukemia, sarcoma, osteosarcoma,
lymphoma, melanoma, ovarian, skin, testicular, gastric, pancreatic, renal,
breast,
prostate colorectal, head and neck, brain, esophageal, bladder, adrenal
cortical, lung,
bronchus, endometrial, cervical or hepatic cancer, or cancer of unknown
primary site.
The compounds of the invention can also be used in the treatment of an
autoimmune diseases. The autoimmune disease can be, but is not limited to: (1)
a
rheumatic disease such as rheumatoid arthritis, systemic lupus erythematosus,
Sjogren's syndrome, scleroderma, mixed connective tissue disease,
dermatomyositis,
polymyositis, Reiter's syndrome or Behcet's disease (2) type I or type II
diabetes
(3) an autoimmune disease of the thyroid, such as Hashimoto's thyroiditis or
Graves'
Disease (4) an autoimmune disease of the central nervous system, such as
multiple
29
CA 02519568 2005-09-19
WO 2004/094444 PCT/US2003/008530
sclerosis, myasthenia gravis, or encephalomyelitis (5) a variety of
phemphigus, such
as phemphigus vulgaris, phemphigus vegetans, phemphigus foliaceus, Senear-
Usher
syndrome, or Brazilian phemphigus, (6) diseases of the skin such as psoriasis
or
neurodermitis, and (7) inflammatory bowel disease (e.g., ulcerative colitis or
Crohn's
Disease).
The invention will be further described in the following examples. It should
be
understood that these examples are for illustrative purposes only and are not
to be
construed as limiting this invention in any manner.
Example 1
Preparation of 6-azido-2,3,4-tetra-O-benzyl-6-deox meths alg actopyranoside
(4).
Compound 3 (1.86 g, 6.52 nunol) was dissolved in MeOH (20 mL), cooled to
0 C, and acetyl chloride (4.35 mL) was added. The mixture was allowed warm to
room temperature and stirred for 12 h. The solvent was removed in vacuo, and
the
residue was chromatographed (Si02, 10% MeOH in CH2C12) to afford 6-azido-6-
deoxymethylgalactopyranoside (mixture of anomers) as a white solid (1.23 g,
86%
yield). 1H NMR (10% CD3OD in CDC13) S 4.79 (d, J= 2.5 Hz, 1 H), 4.46 (br, 1
H),
3.92 (dd, J= 8.5, 4.0 Hz, 1 H), 3.84 - 3.75 (m, 3 H), 3.63 (dd, J= 12.5, 8.5
Hz, 1 H),
3.46 (s, 3 H), 3.31 (dd, J=13.0, 4.5 Hz, 1 H); 13C NMR (10% CD3OD in CDC13) 6
99.80, 69.84, 69.66, 69.54, 68.55, 55.08, 51.21; HRFAB-MS (thioglycerol + H+
matrix) m/e ([M + H]+) 220.0951(3.1%), calcd 220.0933.
To a mixture of 6-azido-6-deoxymethylgalactopyranoside (482 mg, 2.2 mmol)
in THE (30 mL) was added benzyl bromide (1.57 mL, 13.2 minol), K2C03 (2.4 g,
17.6 mmol) and 18-crown-6 (120 mg). The suspension was stirred for 15 min, and
NaH (0.396 g, 60% in mineral oil, 16.5 mmol) was added. After 12 h, brine (30
mL)
was added and the product was extracted with 10% EtOAc in hexane (3 x 20 mL).
The combined extracts were dried over Na2S04 and concentrated in vacuo. The
CA 02519568 2005-09-19
WO 2004/094444 PCT/US2003/008530
desired product 4 (1.02 g, 95% yield) was obtained as a clear oil after
chromatography
(Si02, EtOAc:hexanes 1:2). NMR (1H, CDC13) 6 7.40 - 7.25 (m, 15 H), 5.02 -
4.62
(m, 7 H), 4.14 - 3.76 (m, 4 H), 3.57 - 3.48 (in, 1 H), 3.39 (s, 3 H), 2.94
(dd, J=12.4,
4.4 Hz, 1 H); NMR (13C, CDC13) S 138.65, 138.58, 138.34, 128.68, 128.61,
128.32,
128.12, 128.01, 127.87, 127.78, 99.01, 79.16, 76.48, 75.45, 74.81, 73.89,
69.98,
55.71, 51.64; HRFAB-MS (thioglycerol + H+ matrix) in/e ([M + H]+)
490.2347(3.6%), calcd 490.2342.
Example 2
Preparation of 6-azido-2 3 4-tetra-O-benzyl-6-deoxy-a-galactosyl fluoride (5)
Acetic anhydride (0.45 mL) was added to a solution of 4 (398 mg, 0.81 mmol)
in acetic acid (0.33 mL). The mixture was cooled to 0 C, and conccentrated
H2SO4
(6.8 l) was added. The mixture was stirred at 0 C for 8 h, and H2O (5 mL)
was
. added. The product was extracted with CH2Cl2 (3 x 5 mL), and the combined
extracts were dried over Na2SO4 and concentrated in vacuo. After
chromatography
(Si02, EtOAc:hexanes 1:2), 6-azido-2,3,4-tetra-O-benzyl-6-deoxy-a-galactosyl 1-
acetate (354 mg, 84% yield) was obtained as a clear oil. NMR (1H, CDC13) 6
7.39 -
7.28 (m, 15 H), 6.38 (d, J= 3.5 Hz, 1 H), 5.02 4.58 (m, 6 H), 4.17 (dd, J=
11.0, 4.0
Hz,' 1 H), 3.91- 3.88 (m, 3 H), 3.47 (dd, J= 12.5, 7.0 Hz, 1 H), 3.15 (dd,
J=12.5, 7.0
Hz, 1 H), 2.12 (s, 3 H); NMR (13C, CDC13) S 169.55, 138.59, 138.13, 138.00,
128.68,
128.62, 128.57, 128.56, 128.53, 128.51, 128.18', 128.13, 128.10, 128.03,
127.98,
127.85, 127.79, 127.60, 90.65, 78.67, 75.45, 75.31, 74.95, 74.69, 74.60,
74.42, 73.57,
73.53, 71.89, 50.85, 21.28; HRFAB-MS (thioglycerol + Nat matrix) m/e ([M +
Na]+)
540.2112(100%), calcd 540.2111.
Anhydrous pyridine (0.6 mL) and 70% hydrogen fluoride-pyridine (1.5 mL)
were placed in a 50-ml polyethylene vessel. To this mixture cooled to -20 C
was
added a solution of 6-azido-2,3,4-tetra-O-benzyl-6-deoxy-a-galactosyl 1-
acetate (401
mg, 0.77 mmol) in toluene (0.3 mL). The mixture was allowed to warm to 0 C
and
stir for 6 h then poured into a mixture of ether (10 mL) and saturated aqueous
31
CA 02519568 2010-05-21
62396-1079
potassium fluoride (30 n1L). The product was extracted with a 3:1 ether -
hexane
solution (2 x 50 mL), and the combined extracts were washed with saturated
aqueous
potassium fluoride (30 n1L) and brine (30 mL). The organics were dried over
Na2SO4
and the solvent was removed in vacuo. The residue was purified
chromatographically
(SiO2, EtOAc:hexanes 1:2) to give 6-azido-2,3,4-tetra-O-benzyl-6-deoxy-a-
galactosyl fluoride as a clear oil (230 mg, 78% yield). NMR (1H, CDC13) S 7.40
-
7.25 (m, 15 H), 5.63 (dd, J= 54.0, 2.5 Hz, 1 H), 5.00 (d, J= 11.5 Hz, I H),
4.88 -
4.72 (m, 4 H), 4.61 (d, J= 11.0 Hz, 1 H), 4.01- 3.88 (m, 4 H), 3.51 (dd,
J=12.5, 7.5
Hz, 1 H), 3.13 (dd, J= 12. 0, 6.0 Hz, I H); NMR (13C ,CDC13) S 138.38, 138.09,
138.07, 128.74, 128.71, 128.66, 128.56, 128.23, 128.20, 128.16, 128.04,
127.80,
107.12, 105.32, 78.45, 75.85, 75.67, 74.99, 74.48, 73.99, 73.67, 72.14, 72.11,
50.96;
HRFAB-MS (thioglycerol + Nat matrix) mle ([M + Na]}) 500.1956(100%), calcd
500.1962.
Example 3
Preparation of Compound 7
To a solution of (2S, 3S, 4R)-3,4, bis-t-butyldimethylsilyloxy-2-
hexacosanoylamino-4-octadecanol (6) (266 mg, 0.28 mmol) in THE (10 mL), SnC12
(163.7 mg, 0.86 mmol), AgCIO4 (179 mg, 0.86 mmol) and powdered 4A molecular
sieves (1.34 g) were added. A solution of 5 (214 mg, 0.45 mmol) in THE (2 mL)
was
then added at -10 C. The reaction mixture was allowed to warm gradually to
room
temperature with stirring over the course of 2 h. The mixture was filtered
through
TM
Celite, and the filter cake was washed with Et2O. The combined filtrate was
concentrated under reduced pressure. The residue was purified
chromatographically
(SiO2, EtOAc:hexanes 1:2) to give compound 7 (175 mg, 44% yield) as a clear
oil.
NMR (1H, CDC13) S 7.40 - 7.31 (m, 15 H), 5.92 (d, J= 8.0 Hz,1 H), 5.02 (d, J=
11.0 Hz, 1 H), 4.85 - 4.59 (m, 6 H), 4.21 (m,1 H), 4.06 - 3.99 (m, 2 H), 3.91
(dd, J=
13.0, 3.0 Hz, I H), 3.86 - 3.83 (m, 3 H), 3.79 (m, 1 H), 3.50 (dd, J=12.0, 7.5
Hz, 1
H), 3.17 (dd, J=12.0, 7.5 Hz, 1 H), 2.03 (t, J= 7.5 Hz, 2 H), 1.58 -1.24 (m,
73 H),
32
CA 02519568 2005-09-19
WO 2004/094444 PCT/US2003/008530
0.92 (s, 9 H), 0.91 (s, 9 H), 0.89 (m, 6 H), 0.09 (s, 3 H), 0.08 (s, 3 H),
0.06 (s, 3 H),
0.05 (s, 3 H); NMR (13C ,CDC13) S 173.24, 138.69, 138.61, 138.35, 128.64,
128.58,
128.57, 128.06, 128.03, 127.94, 127.85, 127.64, 100.18, 79.16, 76.59, 76.20,
75.99,
75.04, 74.87, 73.71, 73.49, 70.11, 69.56, 51.78, 51.38, 37.05, 33.59, 32.14,
32.13,
30.10, 29.93, 29.91, 29.89, 29.87, 29.81, 29.77, 29.68, 29.66, 29.58, 26.31,
26.24,
25.86, 22.90, 18.52, 18.37, 14.34, -3.471, -3.756, -4.442, -4.705; HRFAB-MS
(thioglycerol + H+ matrix) nile ([M + H]+) 1382.0592(81.3%), calcd 1382.0601.
Example 4
Preparation of Compound 2
To a solution of 7 (175 mg, 0.12 mmol) in THE (4,mL), TBAF (1.0 M in THF,
0.5 mL, 0.5 mmol) was added dropwise at room temperature. After stirring for
1.5 h,
the mixture was diluted with water and extracted with Et20. The extract was
washed
with water and brine, dried (MgSO4), and concentrated under reduced pressure.
The
residue was purified by column chromatography (Si02, EtOAc:hexanes 1:2) to
give
the corresponding diol (118 mg, 81% yield) as a clear glass. NMR (1H, CDC13) S
7.39 - 7.25 (m, 15 H), 6.25 (d, J= 8.0 Hz, 1 H), 4.99 (d, J=11.0 Hz, 1 H),
4.88 -
4.57 (m, 6 H), 4.27 (m, 1 H), 4.05 (dd, J= 9.5, 3.0 Hz, 1 H), 3.93 (dd, J =
10.0, 3.0
Hz, 1 H), 3.87 - 3.80 (m, 2 H), 3.72 (m, 1 H), 3.51- 3.45 (m, 3 H), 3.03 (dd,
J= 13.0,
6.0 Hz, 1 H), 2.25 (d, J= 5.5 Hz, 1 H), 2.15 (t, ,J = 7.0 Hz, 2 H), 2.13 -
1.25 (m, 76
H), 0.88 (t, J= 7.5 Hz, 6 H); NMR (13C, CDC13) 6 173.07, 138.30, 138.12,
137.84,
128.75, 128.74, 128.64, 128.58, 128.32, 128.29, 128.20, 128.02, 127.72, 99.08,
79.38,
76.41, 75.93, 74.84, 74.60, 74.49, 73.54, 73.27, 70.42, 69.99, 51.22, 49.28,
37.03,
33.58, 32.13, 29.94, 29.91, 29.88, 29.75, 29.63, 29.57, 29.52, 26.12, 25.98,
22.90,
14.33; HRFAB-MS (thioglycerol + H+ matrix) mle ([M + H]+) 1151.9072(89.4%),
calcd 1151.9079.
To a solution of the diol (118 mg, 0.1 mmol) in THF/H20 (1.5 mL/0.3 mL)
was added triphenylphosphine (40.3 mg). The reaction mixture was stirred at
room
temperature for 12 h. The resulting amine (114 mg, 99% yield) was obtained as
a
clear glass after chromatography (Si02, CHC13:MeOH:NH3-H20 1:0.4:0.02). NMR
33
CA 02519568 2005-09-19
WO 2004/094444 PCT/US2003/008530
(1H, CDC13) 6 7.59 - 7.18 (m, 15 H), 6.49 (d, J= 8.5 Hz, 1 H), 4.87 (d, J=
11.5 Hz, 1
H), 4.78 - 4.52 (m, 6 H), 4.18 (m, 1H), 3.97 (dd, J=10.0, 3.5 Hz, 1 H), 3.81
(dd, J=
10.0, 4.5 Hz, 1 H), 3.77 (dd, J= 10.0, 2.5 Hz, 1 H), 3.72 (m, 2 H), 3.67 (dd,
J= 10.0,
4.0 Hz, 1 H), 3.52 (dd, J = 8.0, 5.0 Hz, 1 H), 3.41 (m, 2 H), 2.82 (dd, J
=13.0, 8.0 Hz,
1 H), 2.42 (dd, J= 13.0, 5.0 Hz, 1 H), 2.37 (m, 1 H), 2.04 (t, J= 8.0 Hz, 2
H), 1.49 -
0.82 (m, 74 H), 0.79 (t, J= 7.0 Hz, 6 H); ); NMR (13C,CDC13) 6 173.54, 138.60,
138.33, 138.12, 132.96, 132.31, 132.23, 132.19, 132.17, 132.14, 128.76,
128.74,
128.66, 128.59, 128.24, 128.11, 128.09, 127.86, 127.69, 98.86, 79.81, 76.50,
76.35,
74.79, 74.62, 74.08, 73.37, 73.03, 72.58, 68.51, 53.93, 50.42, 42.43, 36.97,
33.88,
32.11, 29.96, 29.90, 29.84, 29.74, 29.61, 29.55, 29.52, 26.16, 25.97, 25.86,
22.88,
20.92, 14.32; HRFAB-MS (thioglycerol + Nat matrix) m/e ([M + Na]+)
1149.8790(100%), calcd 1149.8786.
To liquid NH3 (ca. 8 mL) under N2 at -78 C was added Na (20 mg), and the
mixture was stirred for 2 min. To the blue solution was added the amine (18
mg,
0.016 mmol) in THE (1 mL), and the mixture was stirred for 40 min at -78 C.
The
reaction was quenched by addition of MeOH (4 mL). Ammonia was removed with a
stream of N2, and the solution was diluted with MeOH to 8 mL. The solution was
concentrated under reduced pressure, and the residue was purified by column
(Si02,
CHC13:MeOH:NH3-H20 1:0.4:0.02) to give 2 (7.3 mg, 53%) as a white solid. NMR
(1H, 5% CD3OD in CDC13) 6 4.91 (d, J= 4.0 Hz, 1 H), 4.21 (m, 1 H), 3.88 (m, 2
H),
3.80 (dd, J= 10.0, 3.5 Hz, 1 H), 3.75 (m, 1 H), 3.70 (dd, J= 10.0, 3.5 Hz, 1
H), 3.62 -
3.51 (m, 10 H), 3.06 (dd, J = 13.0, 7.5 Hz, 1 H), 2.90 (dd, J =13.0, 4.0 Hz, 1
H), 2.19
(t, J= 8.0 Hz, 2 H), 1.68 - 1.25 (m, 73 H). 0.88 (t, J= 7.0 Hz, 6 H); NMR
(13C, 5%
CD3OD in CDC13) 8 174.36, 99.75, 75.17, 72.06, 70.84, 70.37, 70.22, 68.92,
67.31,
50.36, 42.40, 36.62, 33.02, 31.97, 29.83, 29.77, 29.74, 29.71, 29.70, 29.61,
29.46,
29.42, 25.90, 25.87, 22.73, 14.11; HRFAB-MS (thioglycerol + Nat matrix) m/e
([M +
Na]+) 879.7384(100%), calcd 879.7377.
34
CA 02519568 2005-09-19
WO 2004/094444 PCT/US2003/008530
Example 5
Preparation of Compound 8
Dansyl chloride (1.5 mg, 0.0055 mmol) was added to a solution of 2 (4.5 mg,
0.005 mmol) in pyridine (1 mL), and the mixture was stirred for 5 h. The
pyridine
was removed in vacuo, and the product was purified chromatographically (Si02,
10%
MeOH in CH2C12) giving a light yellow glass (3.4 mg, 60% yield). NMR (1H, 5%
CD30D in CDC13) 8 8.55 (d, J= 9.0 Hz, 1 H), 8.26 (d, J= 8.5 Hz, 1 H), 8.19
(dd, J
7.0, 1.5 Hz, 1 H), 7.58 - 7.50 (m, 2 H), 7.20 (d, J= 7.5 Hz, 1 H), 6.93 (d, J=
8.5 Hz,
1 H), 4.83 (d, J= 2.5 Hz, 1 H), 4.22 (m, 1 H), 3.91 (m, 3 H), 3.72 (m, 2 H),
3.64 -
3.55 (m, 4 H), 3.07 (m, 2 H), 2.88 (s, 6 H), 2.22 (br, 7 H), 1.62 -1.25 (m, 72
H), 0.87
(t, J= 7.0 Hz, 6 H); NMR (13C, 5% CD3OD in CDC13) 8 174.45, 152.09, 134.62,
130.69,130.10,129.71,129.36,123.31,118:91,115.43, 99.68, 75.36, 72.41, 70.18,
69.55, 69.13, 69.03, 67.96, 50.42, 45.56, 43.28, 36.78, 33.14, 32.07, 29.93,
29.88,
29.85, 29.82, 29.80, 29.72, 29.57, 29.52, 25.96, 25.94, 22.84, 14.25; HRFAB-MS
(thioglycerol + Nat matrix) na/e ([M + Na]+) 1112.7867(100%), calcd 1112.7887.
Example 6
Preparation of Compound 10
Ester 9 (9.4 mg, 0.02 mmol) was added to a solution of 2 (5.1 mg, 0.0059
mmol) in pyridine (1 mL), and the mixture was stirred for 12 h. The pyridine
was
removed in vacuo, and the product was purified by column chromatography (Si02,
10% MeOH in CH2C12) giving a light yellow glass (3.7 mg, 53% yield). NMR (1H,
5% CD3OD in CDC13) 8 8.53 (d, J= 9.0 Hz, 1 H), 8.28 (d, J= 8.5 Hz, 1 H), 8.20
(dd,
J= 7.5, 1.2 Hz, 1 H), 7.59 - 7.50 (m, 2 H), 7.20 (d, J= 7.5 Hz, 1 H), 7.04 (d,
J= 8.5
Hz, 1 H), 4.91 (d, J= 3.0 Hz, 1 H), 4.17 (m, 1 H), 3.90 (dd, J=10.5, 4.5 Hz, 1
H),
3.84 (m, 4 H), 3.75 (dd, J= 11.0, 4.5 Hz, 1 H), 3.68 - 3.64 (m, 2 H), 3.57 -
3.54 (m, 2
H), 3.20 (dd, J= 13.5, 5.5 Hz, 1 H), 2.89 (s, 6 H), 2.80 (m, 1 H), 2.46 - 2.03
(m, 11
3o H), 1.62 -1.25 (m, 78 H), 0.87 (t, J= 7.5 Hz, 6 H); NMR (13C, 5% CD3OD in
CA 02519568 2005-09-19
WO 2004/094444 PCT/US2003/008530
CDC13) 8 175.38, 174.50, 152.04, 134.69, 130.52, 130.01, 129.72, 129.61,
128.46,
123.31, 118.98, 115.40, 99.71, 74.97, 72.45, 69.94, 69.01, 68.95, 68.03,
50.39, 45.54,
42.76, 42.61, 39.45, 36.57, 35.87, 32.79, 32.06, 29.85, 29.68, 29.50, 29.41,
28.97,
26.85, 26.00, 25.92, 25.49, 25.03, 24.77, 24.37, 22.82, 14.23; HRFA.B-MS
(thioglycerol + Nat matrix) m/e ([M + Na]+) 1225.8741(100%), calcd 1225.8728.
Example 7
Preparation of Compound 12
Ester 11 (6.6 mg, 0.018 mmol) was added to a solution of 2 (5.0 mg, 0.0058
mmol) in pyridine (1 mL), and the mixture was stirred for 12 h. The pyridine
was
removed in vacuo, and the product was purified by column chromatography (Si02,
10% MeOH in CH2C12) giving compound 12 as a clear glass (3.0 mg, 46% yield).
NMR (1H, 5% CD3OD in CDC13) 6 8.36 (m, 1 H), 7.89 (dd, J= 8.0, 1.5 Hz, 1 H),
7.81 (d, J = 8.5 Hz, 1 H), 7.64 (d, J= 8.5 Hz, 1 H), 7.18 (dd, J = 8.5, 2.5
Hz, 1 'H),
7.01 (d, J= 8.0 Hz, 1 H), 6.86 (d, J= 2.5 Hz, 1 H), 4.89 (d, J= 4.0 Hz, 1 H),
4.19 (m,
1 H), 3.92 (dd, J= 10.0, 4.5 Hz, 1 H), 3.82 - 3.38 (m, 9 H), 3.22 (dd, J=
13.5, 6.0 Hz,
1 H), 3.19 (s, 6 H), 2.65 (t, J= 7.0 Hz, 2 H), 2.34 (br, 5 H), 2.18 (m, 2H),
2.05 - 2.08
(m, 2 H), 1.65 - 1.23 (m, 72 H), 0.87 (t, J= 7.0 Hz, 6 H); NMR (13C, 5% CD3OD
in
CDC13) 8 199.01, 174.46, 150.56, 138.06, 130.98, 130.42, 129.86, 126.43,
125.10,
124.35, 121.72, 116.49, 110.59, 105.36, 99.72, 84.07, 75.29, 72.47, 70.11,
69.23,
68.99, 68.69, 67.80, 60.06, 50.56, 40.54, 36.76, 33.62, 33.11, 32.07, 30.23,
29.86,
29.85, 29.81, 29.80, 29.71, 29.57, 29.51, 25.99, 22.83, 14.25; HRFAB-MS
(thioglycerol + Nat matrix) m/e ([M + Na]+) 1132.8484(100%), calcd 1132.8480.
Example 8
Preparation of Compound 13
N-hydroxysuccinimidobiotin (5.9 mg, 0.017 mmol) and Et3N (30 ul) were
added to a solution of 2 (5.0 mg, 0.0058 mmol) in DMF(1.5 mL). The mixture was
36
CA 02519568 2010-05-21
62396-1079
stirred for 12 h, and applied directly to an SiO2 column. Elution with 10%
MeOH in
CH2C12 gave the product 13 as a clear glass (3.2 mg, 52% yield). NMR (1H,
Pyridine-
d5) 5 8.86 (m, 1 H), 8.61 (d, J= 9.0 Hz, 1 H), 7.54 - 7.40 (m, 4 H), 5.54 (d,
J= 4.0
Hz, 1 H), 5.28 (br, OH), 4.66 - 4.61 (m, 2 H), 4.56 - 4.50 (m, 3 H), 4.41-
4.31 (m, 6
H), 4.23 (m, 1 H), 3.92 (m, l H), 3.27 (m, 1 H), 3.16 (m, 1 H), 3.0 - 2.85 (m,
10 H),
2.55 - 2.44 (m, 6 H), 2.32 (m,1 H), 1.96 -1.50 (m, 20 H),1.32 -1.26 (m, 44 H),
0.87 (t, J= 7.0 Hz, 611); NMR (13C, pyridine-d5) S
170.36,169.46,164.28,101.41,
76.88, 72.57, 71.30, 71.21, 70.74, 70.10, 68.62, 62.41, 62.36, 60.57,
56.30,56.13,
51.24,41.22,41.15,36.86,36.27,34.52, 32.17, 30.89, 30.46, 30.23, 30.11, 29.98,
29.89, 29.85, 29.67, 29.12, 28.87, 28.53, 26.56, 26.50, 26.27, 26.16, 26.10,
24.85,
22.98, 14.33; HRFAB-MS (thioglycerol + Nat matrix) m/e ([M + Na]+)
1105.8143(100%), calcd 1105.8153.
Example 9
In Vivo Antitumor Assay
Experiment is performed with groups consisting of 6 female BDF1 mice, 6
weeks old, B16 mouse melanoma cells (1 x 10)are inoculated subcutaneously in
the
rear part of mice (day 0). On 1, 5, and 9 days after inoculation, a sample in
a level of
0.1 mg/kg is administered to the tail vein in a dose of 0.2 ml/20g/mouse. The
volume,
of tumor in the subcutaneous rear partis measured on 8, 12, 16, and 20 days to
determine the tumor growth inhibiting rate of each sample.
Example 10
In Vivo Antiautoimmune Activity Assay
Representative compounds of the formulae herein are screened for
antiautoimmune activity in a nonobese diabetic (NOD) mouse assay essentially
as
described in Wang, B.; Geng, Y.-B.; Wang, CAL J. Exp. Med. 2001,194, 313-319.
37
CA 02519568 2010-05-21
62396-1079
Example 11
In Vivo Antiautoimmune Activity Assay
Representative compounds of the formulae herein are screened for
antiautoimmune activity in an experimental autoimmune encephalomyelitis (EAE)
assay essentially as described in Pal, E.; Tabira, T.; Kawano, T.; Taniguchi,
M.;
Miyake, S.; Yamamura, T. J..bnmunol. 2001, 166, 662-668.
Other embodiments are in the claims.
38