Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
OPTICALLY ACTIVE, HETEROAROMATIC BETA-HYDROXY ESTERS, PROCESSES FOR THEIR
PREPARATION FROM BETA-KETO ESTERS AND PROCESSES FOR THE PREPARATION OF THOSE
BETA-KETO ESTERS.
The invention relates to the subject matter characterized in the claims, i.e.
novel
optically active, heteroaromatic (3-hydroxy esters and processes for the
preparation
as well as to their use as intermediate products in the total synthesis of
epothilones
and epothilone derivatives. The process for the production of the intermediate
products yields products in high chemical purity, optical purity, in very good
yields
and allows an industrial-scale production.
Hofle et al. describes the cytotoxic effect of the natural substances
epothilone A (R =
hydrogen) and epothilone B (R = methyl)
)H
Epothilone A (R = H), Epothilone B (R=CH3)
e.g. in Angew. Chem. 1996, 108, 1671-1673. Epothilones are representatives of
a
class of promising anti-tumor agents that were tested and found to be
efFective
against a number of cancer lines. An overview of the syntheses for these
compounds
has been described by J. Mulzer in Monatsh. Chem. 2000, 131, 205-238. These
agents have the same biological mode of action as paclitaxel and other taxanes
(See
for paclitaxel, D.G.I. Kingston, Chem Commun. 2001, 867-880), however,
epothilones have also been shown to be active against a number of resistant
cell
lines (See S. J. Stachel et al., CurrPharmaceut. Design 2001, 7, 1277-1290; K.-
H.
Altmann, Curr. Opin. Chem. Biol. 2001, 5, 424-431).
Due to their in vitro selectivity to breast and intestinal cell lines and
their activity
against P-glycoprotein forming, multi-resistant tumor lines, which is
distinctly higher
than that of Taxol, as well as their improved physical properties with respect
to Taxol,
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
2
e.g. a water solubility that is higher by the factor of 30, this new class of
compounds
is of special interest for the development of drugs for the treatment of
malignant
tumors.
In addition to the natural epothilones, the literature describes a large
number of
synthetically modified epothilone derivatives, including, inter alia,
derivatives which
contain an aromatic and/or heteroaromatic grouping in the 1-position instead
of the
methyl thiazole methyl vinyl side chains.
Epothilone derivatives with anellated aromatic heterocycles in the 1-position
are also
known in patent literature (Schering AG, WO 00/66589 and Novartis US
6,387,927).
Since these compounds are very potent antitumor agents, the development of an
economic and efficient synthesis for producing them was of great interest. The
intermediates of formulae 11 and III, representing some of the key compounds
for the
synthesis of this structural class, have already been described in the patent
literature.
The goal of the present invention is to provide a novel process for the
production of
novel intermediate compounds of general formula I for use in the synthesis of
these
epothilone derivatives:
A
~'COZR ~ 1 )
OH
wherein A stands for a bicyclic heteroaromatic residue of the formula:
Hetero-
aromatic ~ / ,
-.
wherein "heteroaromatic" stands for a 5- or 6-membered heteroaromatic ring
having
up to 2 heteroatoms selected from oxygen, nitrogen or sulphur, which is
optionally
substituted with one or two substituents selected from alkyl, such as for
example,
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
3
methyl or ethyl; optionally protected hydroxyalkyl such as for example, TBDMS-
OCH2-; halo-alkyl such as for example, F-CH2-; halogen such as CI, F, or Br;
or CN;
and,
wherein R stands for a straight-chain or branched, optionally saturated alkyl
chain,
which optionally contains 1-3 oxygen atoms, such as for example, methyl,
ethyl,
propyl, 2-propyl, n-butyl, tert-butyl, -CH2CH=CH2, -CH2CH20CH3, or
-CH2CH20CH2CH20CH3 group; or stands for a phenyl, cyclohexyl or benzyl
radical.
A "bicyclic heteroaromatic residue" can, for example, stand for one of the
following
groups:
I \ \ N \ \ v \
N / / .~~ I / / .., I / / .~~
~.~ S \ ~ S \ ..
I /~. ~~ I /
N N
\ p \ ,. S
---y I / ~~ --~N I / --~N / ~~.
N a
S \ .. ~ \ .. ~ \
'~N I / ~N I / ~CN I / ..~
\ \ \ \ \ \
I ... I ~..~ I / / .~~
N
S \ S \
OTBS- N I / ~'- F' N ( / ~'.
The compounds of general formula I are valuable intermediate compounds for the
preparation of the intermediate compounds of general formulae II and III:
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
4
A A
~COZR ECHO
OSg OSg
(II) (III)
wherein Sg stands for an alcohol protecting group, such as for example TBDMS,
THP, MEM, Mom, TROC, -CHZ-C6H4-OCH3, or benzyl, with TBDMS being preferred.
The compounds of general formula I are, therefore, also valuable in the total
synthesis of epothilone derivatives.
Compounds of general formula II are prepared according to processes for the
protection of secondary alcohols known to a person skilled in the art (See for
example T.W. Greene, "Protecting groups in organic synthesis", John Wiley and
Sons, Inc., edition 1999; P.J. Kociensky, "Protecting Groups", Georg Thieme
Verlag
Stuttgart, 1994).
Scheme 1
A A
~'COzR ~COZR
OH OSg
Compounds of the general formula III can either be prepared directly from the
ester
of formula II by means of a reduction with DIBAH (See Tetrahedron Lett. 1977,
3195
- 3198; Liebigs Ann. Chemie 1992, 145 - 158 ; JACS, 107, 1985, 3640 - 3645 ;
Tetrahedron Lett. 31, 10, 1990, 1443 - 1446; Tetrahedron Lett. 31, 16, 1990,
2235
- 2238; Chem. Communications, 1999, 2049 - 2050; Bull. Chem. Soc. Jp. 66, 2,
1993, 523) or, in 2 stages, by first carrying out a reduction to alcohol and,
subsequently, oxidizing to the aldehyde (See for reduction: Tetrahedron Lett.
58, 1,
2002, 61; JAGS, 123, 34, 2001, 8420; Chem. Europ. J., 7, 24, 2001, 5286;
Tetrahedron Asym. 12, 20, 2001, 2835; Org. Lett. 3, 20, 2001, 3149; JACS, 123,
13,
2001, 2946; Chem. Europ. J. 6, 18, 2000, 3313; and for oxidation: JACS, 123,
38,
2001, 9313; Org. Lett. 4, 11, 2002, 1879; JACS, 123, 44, 2001, 10942; JOC, 66,
24,
2001, 8037; JOC, 66, 25, 2001, 8370; Tetrahedron Asym. 12, 20, 2001, 2835;
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
Angewandte Chemie, 131, 2001, 3324; Org. Lett. 3, 22, 2001, 3549; Chem.
Commun. 15, 2001, 1392).
Scheme 2
A A
~COZR ECHO
OSg -----~ pSg
(11)
(III)
A
~CHZOH
OSg
(11a)
The methods presently existing in the literature for the preparation of the
compounds
of the general formulae II and III involve long synthesis sequences and poor
total
yields. In some cases, these processes also involve the use of technically
expensive
and complex processes such as cryo temperature reactions, irradiation, and
very
expensive raw materials and reagents.
The following syntheses for the synthesis of compounds of formula II are, for
example, found in the literature:
1. Schering AG (WO 00166589)
Number of steps : 5
Number of chromatography steps: 4
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
6
S a , b S / c
~N ~ ' CI ~ ~N ~ I / O~ ~ ~N ~ ~ , O
i
O
d
~S / I O a
_ -
TBDMSO
S
-'.'~~N \ I O
TBDMSO O
a ) 1. Nal, NiBr2, DMF, 150°C; 2. ethyl acrylate, NEt3,
Tris(dibenzylidene
acetone)-dipalladium, Tris(o-tolyl)phosphin, 150°C.
b ) Os04, Na104, THF/H20, room temperature
c ) 3-Acetyl-(4R,5S)-4-methyl-5-phenyl-2-oxazolidinone, LiHMDS, THF or
3-bromoacetyl-(4R,5S)-4-methyl-5-phenyl-2-oxazolidinone, CrCl2, THF,
40°C
d ) TBDMS-OTf, NEt3, 0°C
a ) Ti(OEt)4, EtOH, reflux
This 5-stage synthesis starts from the very expensive chlorobenzotriazole
compound
and requires from the beginning the use of heavy metals such as nickel,
palladium.
The scaling-up of this reaction is additionally rendered more difficult by the
high
reaction temperature. In the second stage, the breaking of the double bond
with
osmium tetraoxide is carried out. Due to the high toxicity of this reagent, a
transfer to
a pilot plant scale is not feasible. The optical activity is achieved by means
of an
Evans aldol reaction, whereby the Evans auxiliary agent is used in excess (it
must be
produced in a two-stage sequence). The transfer of this process to an
industrial
scale is difficult, since expensive and partly very toxic raw materials are
used. In
addition, several chromatographic purifications are carried out.
2. NOVARTIS (US 6,387,927 and PCT/EP99/10129)
a)
Number of steps: 4
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
7
Number of chromatography steps: 4
Total yield: Cannot be ascertained, since no individual yields are indicated.
s ~ 1. Nss/irradiation s
CCh ~N i ~ CHO
2. aqu. HOAc I Urotropine/110°C
s. Flash chromatography
1) Sultam
CF,SOxOH 1 BEt~
s~
Htinig's baSa N
2) -78°C / AldehydP. SOx N
3. Flash chromatography O OH
1.TBSCI/DMF/Imidazole / S
_ i ~/J~''--
2. Flash chromatography SO--'N ~ N
x
p OTBS
1.DIBALHIMethylenechl0tida ~ S
'78 C
H ~ N
2. Flash chromatography p OTBS
The first synthesis step is conducted by means of an irradiation (which is, in
general
unsuited for the implementation on an industrial scale) in the solvent carbon
tetrachloride, a solvent which is no longer acceptable from an environmental
protection perspective. Subsequently, the non-isolated brominated product is
hydrolysed to aldehyde with a mixture of aqueous acetic acid and urotropine
(110°C/80 min.). Purification is effected by means of flash
chromatography on silica
gel. The very expensive Oppolzer sultam (Tetrahedron Lett. 33, 2439, 1992) is
stoichiometrically used for the asymmetric aldol reaction in the subsequent
reaction
and reacted in a process that takes place in a relatively complex fashion (via
boron
enolate). Purification is once again done using flash chromatography. A
reaction to
silyl ether is subsequently carried out under standard conditions using TBDMS-
CI
and another purification with chromatography is effected. The splitting off of
the
sultam residue is successful with DIBAH in dichloromethane at -78°C.
The
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
8
purification of the product takes place by means of chromatography on silica
gel, This
process involves several chromatography steps which result in additional costs
and
makes it difficult to transfer to an industrial scale. The use of the
expensive sultam
auxiliary agent is also a hurdle since it may be quite difficult to obtain
this reagent in
bulk amounts (> 50 kg).
b)
Number of stages: 6
Number of chromatographies: 5
Total yield: Cannot be ascertained, since no individual yields are indicated.
N \ MeOHlH2S04 \N °
DVBAH THFI -98 C
~ ~~
N COOH N COZMe plash chromatography
(COCI)z IDMSO
\N \ NEta N \
'N I ~ -78 C N I ~ CHO
CH20H
F~ash chromatography
1) Sultam
CF,SOZOH I BEt3
Hiinlg's base
2) '78°C I Aldehyde
3. Flash chromatography
1.TBSGIIDMFIImidazole
2. Flash chromatography
1. DIBALHIMethylenechloride ~ N
.78°C
H ~ N
2. Flash chromatography p OTBS
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
9
The key intermediate was synthesized starting from the methyl benzimidazole
acid in
a relatively long sequence. Upscaling is also rendered difficult by the fact
that two
low-temperature reactions are additionally used (DIBAH reduction and Swern
oxidation).
Due to the complexity of these stages and the difficulties in scaling-up,
there was a
need for shorter and less expensive alternatives which ofiFer the possibility
of
producing such intermediates on a scale of 100 kg.
These problems are avoided by the use of the processes described in the
present
invention, which allow us to obtain compounds of general formula I in a high
total
yield and high optical purity (>98% e.e.) from a starting material known in
the
literature in a very short sequence (2 steps). The conversion of compounds of
general formula i to compounds of general formulae II and III is then easily
carried-
out as previously described in Schemes 1 and 2, i.e. using 1 or 2 further
steps.
The preparation of compounds of general formula I is carried out by means of a
chemical, microbiological or enzymatic reduction of a f3-ketoester of general
formula
IV:
A
(IV)
O
wherein A and R, have the same meaning as indicated above under general
formula
The reduction takes place in accordance with the process of f3-ketoester
reduction
that is known to a person skilled in the art. The processes described in the
experimental part serve as examples only.
a) Chemical Reduction
Examples of chemical reductions in the literature
1. Asymmetric hydrogenations and transfer hydrogenations
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
M. Beller, C. Bolm "Transition Metals for Organic Synthesis", vol. 2, pages.
25 et
seq., Wiley - VCH, 1998 ;
R. Noyori , Angew. Chemie 2001, 113, 4.0-75 (and the literature quoted in this
article);
R. Noyori, Acc. Res. 1997, 30, 97-102; and
K. Murata, JOC 1999, 64, 2186-2187.
The catalyst used is also indicated in each case:
Ru(R-Xyl-P-Phos)(C6H6)C12
Wu, Jing; Chen, Hua; Kwok, Wai Him; Lam, Kim Hung; Zhou, Zhong Yuan; Yeung,
Chi Hung; Chan, Albert S. C.; Tetrahedron Lett., 43, 8, 2002, 1539 - 1544.
[Ru(cod)(C~.H~)Z]/HBr ferrocenyl ligand
lreland, Tania; Grossheimann, Gabriele; Wiener-Jeunesse, Catherine; Knochel,
Paul;
Angew. Chem. Int. Ed., 38, 21, 1999, 3212 - 3215; Angew. Chem., 111, 1999,
3397 -
3400.
(R)-Me0-BIPHEP-RuBr2
Ratovelomanana-Vidal, Virginia; Genet, Jean-Pierre; JORCAI; J. Organomet.
Chem.
567, 1-2, 1998, 163-172 ;
Genet, J. P.; Ratovelomanana-Vidal, V.; Cano de Andrade, M. C.; Pfister, X.;
Guerreiro, P.; Lenoir, J. Y., Tetrahedron Lett., 36, 27, 1995, 4801-4804.
(R)-Diamo-BINAPRuBr2
Guerreiro, Patricio; Ratovelomanana-Vidal, Virginia; Genet, Jean-Pierre;
Dellis,
Philippe, Tetrahedron Lett., 42, 20, 2001, 3423 - 3426.
(+)-[(4,4'-PPh2-2,2',5,5'-Me-3,3'-bithiophene)RuCi(C6H6)]CI HBF4
Benincori, Tiziana; Cesarotti, Edoardo; Piccolo, Oreste; Sannicolo, Franco; J.
Org.
Chem., 65, 7, 2000, 2043 - 2047.
(-)-2,2'-bis(diphenylphosphino)-4,4',6,6'-tetramethyl-3,3'-bibenzo<b>thiophene-
RuCl2
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
11
Benincori, Tiziana; Brenna, Elisabetta; Sannicolo, Franco; Trimarco, Licia;
Antognazza, Patrizia; Cesarotti, Edoardo; J. Chem. Soc. Chem. Commun., 6,
1995,
685-686.
(+)-3-[(2-Ph2P-5-Me0)C6H3]-2-(Ph2P)naphtho[2,1-b]thiophene
Sannicolo, Franco; Benincori, Tiziana; Rizzo, Simona; Gladiali, Serafino;
Pulacchini,
Sonia; Zotti, Gianni; Synthesis, 15, 2001, 2327 - 2336.
(+)-3-[2-(Ph2P)-C6H4]-2-(Ph2P)naphtho[2,1-b]thiophene Ru(II)
Benincori, Tiziana; Gladiali, Serafino; Rizzo, Simona; Sannicolo, Franco; J.
Org.
Chem., 66, 17, 2001, 5940 - 5942.
(R,R)-1,3-dicyclohexyl-1,3-propanediol cyclic sulfate j(C6H6)RuCl2]z
Marinetti, Angela; Jus, Sebastien; Genet, Jean-Pierre; Ricard, Louis; J.
Organomet.
Chem., 624, 1-2, 2001, 162 - 166.
RuCl3+(S)-Me0-BIPHEP
Madec, J.; Pfister, X.; Phansavath, P.; Ratovelomanana-Vidal, V.; Genet, J. -
P.;
Tetrahedron, 57, 13, 2001, 2563 - 2568.
(-)-(6,6'-O(CH2)~O-biphenyl-2,2'-diyl)bis(diphenylphosphine) [Ru(C6H6)CI2]a
Zhang, Zhaoguo; Qian, Hu; Longmire, James; Zhang, Xumu; J. Org. Chem., 65, 19,
2000, 6223 - 6226.
[Ru(cod)(C4H~)2]/HBr ferrocenyl ligand
Ireland, Tania; Grossheimann, Gabriele; Wieser-Jeunesse, Catherine; Knochel,
Paul;
Angew. Chem. Int. Ed., 38, 21, 1999, 3212 - 3215; Angew. Chem., 111, 1999,
3397
- 3400.
[RuCl2(p-cymene)]2 (1 S,2R)-ephedrine i-PrOK
Everaere, Kathelyne; Carpentier, Jean-Francois; Mortreux, Andre; Bulliard,
Michel;
Tetrahedron: Asymmetry, 10, 24, 1999, 4663 - 4666.
(-)-[4,4'-PPh2-2,2',5,5'-Me-3,3'-bithiophene] RuCl2
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
12
Marinetti, Angela; Genet, Jean-Pierre; Jus, Sebastien; Bianc, Delphine;
Ratovelomanana-Vidal, Virginie; Chem. Europ. J., 5, 4, 1999, 1160 - 1165.
<Ru(p-cymene)CIZ>2, (1 S,2R)-(+)-ephedrine, i-PrOK
Everaere, Kathelyne; Carpentier, Jean-Francois; Mortreux, Andre; Bulliard,
Michel;
Tetrahedron: Asymmetry, 9, 17, 1998, 2971 - 2974.
1,2-bis(t-butylmethyfphosphino)ethane, hydrogen RuBr2
Yamano, Toru; Taya, Naohiro; Kawada, Mitsuru; Huang, Taisheng; Imamoto,
Tsuneo; Tetrahedron Lett., 40, 13, 1999, 2577 - 2580.
<(-)-2,2'-bis(diphenylphosphino)-4,4',6,6'-tetramethyl-3,3'-
ibenzo<b>thiophene>RuCl2
Benincori, Tiziana; Brenna, Elisabetta; Sannicolo, Franco; Trimarco, Licia;
Antognazza, Patrizia; et al.; J. Org. Chem., 61, 18, 1996, 6244-6251.
RuBr2<(R)-binap>
Noyori, R.; Ohkuma, T.; Kitamura, M.; Takaya, H.; Sayo, N.; et al.; J. Amer.
Chem.
Soc.,109, 19, 1987, 5856-5858.
Ru(R-Tol-P-Phos)(C6H6)Cl2
Wu, Jing; Chen, Hua; Zhou, Zhong-Yuan; Yeung, Chi Hung; Chan, Albert S. C.;
Syn.
Lett., 2001, 1050 - S1054.
RuCl2[(-)-N,N'-Me2-3,3'-bis(Ph2P)-2,2'-biindoleJ
Benincori, Tiziana; Piccolo, Oreste; Rizzo, Simona; Sannicolo, Franco; J. Org.
Chem., 65, 24, 2000, 8340 - 8347.
(R)-Me-Duphos-RuBr2
Genet, J. P.; Ratovelomanana-Vidaf, V.; Cano de Andrade, M. C.; Pfister, X.;
Guerreiro, P.; Lenoir, J. Y.; Tetrahedron Lett., 36, 27, 1995, 4801-4804.
[NH2Mez][~RuCI[(R)-segphos]}2(~.-CI)s]
Saito, Takao; Yokozawa, Tohru; Ishizaki, Takero; Moroi, Takashi; Sayo, Noboru;
Miura, Takashi; Kumobayashi, Hidenori; Adv. Synth. Catal., 343, 3, 2001, 264 -
268.
CA 02546981 2006-05-23
WO 2005/064006 13 PCT/EP2004/014753
The asymmetric hydrogenation is carried out in solvents such as methanol,
ethanol,
trifluoroethanol, THF, 2-methyl-THF, dichloromethane and mixtures of these
solvents.
The reaction temperature is from 0 to 100°C and the reaction times are
from 3 to 72
hours. The catalyst is added with 0.01 to 5 mole % (based on the substrate).
It
proved to be useful in some cases to add 0.1 to 30% of water in the case of
solvents
miscible with water. Sometimes it is advantageous to add 0.01 to 5 mole eq.
(based
on the substrate) of an inorganic or organic acid, such as HCI, H3P04, H2S04,
acetic
acid, methane sulfonic acid, p-TsOH, phenyl sulfonic acid, camphor sulfonic
acid.
Hydrogenation takes place at temperatures of 0°C to 100°C and
hydrogen pressures
of 1 to 270 bar.
2. Asymmetric reduction with complex hydrides
LiBH4, (R,R')-N,N'-dibenzoylcystine, t-BuOH
Soai, Kenso; Yamanoi, Takashi; Hikima, Hitoshi; Oyamada, Hidekazu; J. Chem.
Soc.
Chem. Commun., 3, 1985, 138-139.
Tartaric acid, NaBH4
J. Chem. Soc. Perkin Trans. 1, 1990, 1826.
b) Microbiological reduction
In general, the use of microorganisms of the following species:
baker's yeast, Bretfanomyces bruxellensis, Candida albicans, Candida boidinii,
Candida gropengiesseri, Candida guilliermondii, Candida kefyr, Candida pini,
Candida rugosa, Candida solani, Candida tropicalis, Candida utilis, Candida
valida,
Clostridium beijerinckii, Clostridium pasteurianum, Cryptococcus laurentii,
Cryptococcus macerans, Debaryomyces hansenii, Debaryomyces kloeckeri,
Debaryomyces nicotianae, Debaryomyces vini, Endomycopsis fibuliger,
Hanseniaspora guilliermondii, Hanseniaspora osmophila, Hanseniaspora uvarum,
Hansenula capsuiata, Hansenula holstii, Hansenula polymorpha, Hansenula
saturnus, Hansenula silvicola, Issatchenkia orientalis, Kloeckera apiculata,
Kloeckera
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
14
corticis, Kloeckera javanica, Kloeckera sp., Kluyveromyces lactis,
Kluyveromyces
marxianus Kluveromyces sphaerica, Lactobacillus kefir, Nadsonia fulvescens,
Octosporomyces octosporus, Pichia anomala, Pichia cactophila, Pichia farinosa,
Pichia fermentans, Pichia holstii, Pichia jadinii, Pichia membranaefaciens,
Pichia
pijperi, Pichia silvicola, Pichia subpelliculosa, Pichia wickerhamii,
Rhodoforula flava,
Rhodotorula glutinis, Rhodotorula minuta var. minuta, Saccharomyces
acidificans,
Saccharomyces bailiff, Saccharomyces bayanus, Saccharomyces carlsb. strain
Herrliberg, Saccharomyces carlsbergensis, Saccharomyces cerevisiae,
Saccharomyces chevaliers, Saccharomyces exiguus, Saccharomycopsis fibuligera,
Saccharomyces paradoxus, Saccharomyces pastorianus, Saccharomyces
pastorianus formerly Saccharomycopsis capsularis, Saccharomyces sp.,
Schizosaccharomyces pombe, Schizosaccharomyces octosporus, Sporobolomyces
coralliformis, Sporobolomyces salmonicolor, Torulopsis pinus, Trigonopsis
varabilis,
Tremella fuciformis, Waltomyces lipofer, Zygosaccharomyces fermentati or
Zygosaccharomyces rouxii, are used for the microbiological reduction.
Preferred is
the Pichia wickerhamii microorganism.
Literature:
Bardot, Valerie; Besse, Pascale; Gelas-Miahle, Yvonne; Remuson, Roland;
Veschambre, Henri; Tetrahedron: Asymmetry, 7, 4, 1996, 1077-1088.
Bhalerao, U.T.; Chandraprakash, Y.; Babu, R. Luke; Fadnavis, N.W.; Synth.
Commun., 23, 9, 1993, 1201-1208.
Chenevert, Robert; Fortier, Genevieve; Rhlid, Rachid Bel; Tetrahedron, 48, 33,
1992,
6769-6776.
Mochiziki, Naoki; Sugai, Takeshi; Ohta, Hiromichi; Biosci. Biotechnol.
Biochem., 58,
9, 1994, 1666-1670.
Kumar, Ashok; Ner, Dilip H.; Dike, Suneel Y.; Tetrahedron Lett., 32, 16, 1991,
1901-
1904.
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
Mochiziki, Naoki; Sugai, Takeshi; Ohta, Hiromichi; Biosci. Biotechnol.
Biochem., 58,
9, 1994, 1666-1670.
Manzocchi, Ada; Casati, Rosangela; Fiecchi, Alberto; Santaniello, Enzo; J.
Chem.
Soc. Perkin Trans.1, 1987, 2753-2758.
c) Enzymatic reduction
Literature:
Deof, B.S. et al.; Aust. J. Chem., 29, 1976, 2459-2467.
Ema, Tadashi; Moriya, Hiroyuki; Kofukuda, Toru; lshida, Tomomasa; Maehara,
Kentaro; Utaka, Masanori; Sakai, Takashi; J. Org. Chem., 66, 25, 2001, 8682 -
8684.
However, the use of microbiological reductions with yeasts andlor modified
yeasts
and the processes for asymmetric hydrogenation and transfer hydrogenation
relying
on Noyori are preferred.
The methods described above also allow for the preparation of the antipode of
compounds of general formula I, namely la:
A
( la )
,OH
The f3-ketoesters of the general formula IV
A
(IV)
O
wherein A and R have the same meaning as indicated above under general formula
I, can be prepared using known methods by reacting activated acid derivatives
of the
general formula V with malonic acid ester derivatives of the general formula
Vl:
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
16
R
O O
A X
(V) O~Y (VI)
O i
O
Wherein X stands fior a chlorine, bromine, 4-nitrophenol or the imidazoyl
residue,
R has the same meaning as indicated above under general formula I, and
Y stands for hydrogen, Li, Na, K or Mg/2 or a silyl protective group such as
trimethylsilyl.
Examples of 13-ketoester syntheses are described in the literature:
Synthesis 1993(3), 290/292; Med. Chem. 1985, 28(12), 1864; Tetrahedron Lett.
1984, 25, 5681; J. Heterocycl. Chem. 1996, 33(4), 1407; Org.' Prep. Proceed.
1997,
29(2), 231; Arch. Pharm. (Weinheim, Ger.) 1997, 330(3), 63 - 66 ; Tetrahedron
Lett.
1994, 35(50), 9323 ; Tetrahedron Lett. 1994, 35(50), 9323 ; Synthesis, 1993,
290 ; J.
Chem. Educ. 1983, Vol. 60, No. 3, 244 ; Tetrahedron Lett. 30, 1992, 5983 ;
Synthesis, 1998, S. 633 ; Chem. Commun. 1999, 1113 ; Tetrahedron, 1985, Vol.
41,
5229 ; Angewandte Chemie 1979, S. 76 ; Tetrahedron Lett. 35, 50, 1994, 9323 -
9326.
The starting products of general formulae V and VI are known in the literature
and
partly commercially available or can be prepared in accordance witih methods
well
known to a person skilled in the art.
The following examples are given: J. Chem. Soc. 1947, 437, 441 ; JACS 1939,
61,
183 ; Bioorg. Med. Chem. Lett. 1999, 2583 ; Chem. Commun. 2, 2002, 180 ; Chem.
Ber. 23, 1890, 2272 ; US 2647050 (1949 Du Pont) ; Zh. Obshch. Khim., 26, 1956,
3388, 3390 ; J. Chem. Soc. 1949, 355, 361 ; Synthetic Commun. 26, 19, 1996,
3535
- 3542 ; J. Chem. Soc. 1966, 1980 -1983 ; JACS 75, 1953, 6237 ; JAGS, 75,
1953,
2770 ; J. Chem. Soc. Perkin Trans. 1974, 903, 908 ; Zh. Obshch. Khim., 32,
1962,
1581 ; Chem. Pharm. Bull., 14, 1966, 375, 381.
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
17
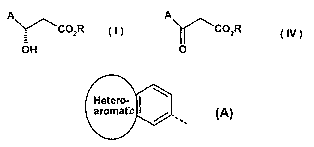
Most of the processes for (3-ketoesters that are described in literature have,
however,
the disadvantage that large amounts of by-products such as the compounds A and
B
A O
A
A 'C02R
'COZR HO
O C02R
(A) (B)
are formed, which interferes with the quantitative crystallization of the
desired
product.
Even in the preparation of (3-ketoesters described by X. Wang in Tetrahedron
Letters,
vol. 35, 50, 1994, 9323, in which DBU (diazobicycloundecane) is used in the
deprotonation step, large amounts of the byproducts A and B are observed in
scaling-up.
A further object of the present invention is to provide for a novel process
which uses
tert-butylates and/or tert-amylates for deprotonation, which avoids the
formation of
by-products A and B.
The following one-pot sequence (Scheme 3) starting from the acids of the
general
formula VII:
A OH
(VII)
O
wherein A has the same meaning as indicated above under general formula I,
proved
to be especially advantageous.
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
18
Scheme 3
A R
'OH O O
(VII) (Vlll)
O
O-K+
O
~,N
A ,!
1
Starting from the acid of the general formula VII, the imidazolide is prepared
according to methods known to a person skilled in the art. More specifically,
N,N-
carbodiimidazole (Staab's reagent) in a non-erotic solvent such as THF, 2-
methyl-
THF, dioxane, dichloromethane, toluene, dimethylformamide, optionally with the
addition of dimethylaminopyridine (0.001 - 3 eq.) is preferably reacted with
the acid
VII at temperatures from 0-70°C for 1-10 hours.
After the completed reaction, the imidazolide can be isolated. However, it is
preferred
to add the obtained solution to a second solution (preparation: reaction of
malonic
acid semiester potassium salt VIII in an aprotic solvent such as THF, 2-methyl-
THF,
dioxane, dichloromethane, toluene, dimethylformamide with
trimethylsilylchloride to
silylester, reaction time 1 - 10 hours at temperatures of -10 to 30°C).
Subsequently,
deprotonation takes place with 1 - 4 eq. of a base such as potassium tent-
butylate,
sodium tert-butylate, lithium tert-butylate, potassium-O-CH2C(CHs)s (in the
case of
reactions on an industrial scale, the inorganic bases are preferably added,
dissolved
in a solvent such as THF, at temperatures of from -10 to +30°C,
subsequent stirring
time 10 minutes to 5 hours). The addition time is 30 minutes to 10 hours, and
it is
A
~COZR l IV )
I IO
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
19
possible to meter a cold solution (0°C) or a solution having a
temperature of up to
70°C. After the addition has been completed, renewed stirring is
carried out for 1 to
24 hours, preferably at temperatures of from 0 to 50°C.
The addition 1 to 5 mole equivalents of lithium chloride or lithium bromide
prior to the
addition of the base proved to be advantageous in some cases in order to
improve
the stirrability of the batch. This stirrability is of special importance in
view of scaling-
up to the pilot plant, i.e. industrial scale which could involve the risk of
breaking the
stirrer.
Water is added for reprocessing the reaction solution, adjustment of the pH
value is
carried out with a mineral acid such as HCI, sulphuric acid or phosphoric acid
(pH 1.5
- 8) and the product is isolated by means of extraction (e.g. acetic acid
ethyl ester,
MTB, etc.). After drying of the organic phase over a desiccant (MgS04 or
Na2S04) or
by means of azeotropic distillation (pilot plant), redistillation to the final
solvent used
for crystallization is carried out.
Since the ketoesters of general formula IV are obtained as crystalline solids,
they can
be easily purified by means of crystallization. Isolation is carried out by
means of
filtration, rewashing with the previously-used solvent and subsequent drying
(vacuum
or circulating air).
The t3-ketoesters of general formula IV prepared as described above are
obtained in
a high yield (approx. 91 - 93%, starting from the acid) and purity.
The sodium andlor lithium salt can also be used instead of the potassium salt
of the
malonic acid semi-ester.
For the sake of comparison of the novel process of the present invention with
examples from the previously-mentioned patent literature, it can be said that
starting
from acids known in the literature, the respectively required key
intermediates can be
prepared with a few steps in high purity and high yields (without
chromatography).
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
The synthesis according to the invention can be further illustrated by means
of the
following two examples:
1st example: Chiral methylbenzimidazole aldehyde
One-pot
COOH \ ( CO=Et
a
Asymmehic TBSC1 f Imfdazole
hydrogenation \
oryeastreduction ~ ~ / CO~Et
OH
N
/ DIBALH
CO,Et ~ H \ ~ H'
OTBS ~TBS v
2nd example: China! methylbenzothiazole ethyl ester
S As
\ One-~ ~ ~ \ hydrog nation
/ COOH \ / CO=Et or yeast reduction
U
TBSCI f Imidazote ,~ ~
/ COlEt '~~COlEt
OH
OTBS
The reactions described above are preferably carried-out under conditions
analogous
to those given in the following examples. The following examples are intended
to
illustrate the invention without being intended to restrict the scope of the
invention:
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
21
Examples
I3-Ketoester Synthesis (general specification for the preparation of compounds
of general formula Il~
Preparation of Solution A:
260 mmole of the acid (A-COOH; compound of the general formula Formel Vli) are
suspended in 300 ml of THF at room temperature and 950 mg of
dimethylaminopyridine are added. A solution consisting of 285 mmole 1,1-
carbodiimidazole in 500 ml of THF are added dropwise at 40°C and
stirring is carried
out at 50°C for 7 hours.
Preparation of Solution 8:
777 mmole of malonic acid semiester potassium salt (ROOC-CH2-COOK; compound
of the general formula VIII) are suspended in 250m1 of THF at 20°C.
Subsequently,
777 mmole of chlorotrimethylsilane are added d ropwise and stirring is again
carried
out for 7 hours. Cooling to 0°C is carried out, 1000 mmole of lithium
chloride are
optionally added and a solution of 1.365 mole of potassium tert. butylate,
dissolved in
300 ml of THF is added dropwise (under counter-cooling). Subsequently,
stirring is
carried out again for 30 minutes at 0°C.
Solution A with a temperature of 50°C is added to solution B dropwise
under vigorous
stirring within 30 minutes (the temperature is kept at 0°C by means of
counter-
cooling). After the addition has been completed, stirring is carried out at
0°C for 30
minutes and subsequently at 20°C for 15 hours.
Processing:
1000 ml of acetic acid ethyl ester is added and a pH of 2 is adjusted by
adding 920
ml of 2N hydrochloric acid (when doing so, the solution clears up, two phases
are
formed). The organic phase is separated and washed twice with 750 ml of
saturated
aqueous sodium hydrogen carbonate solution. Subsequently, the organic phase is
washed with 500 ml of saturated sodium chloride solution. After drying over a
desiccant (sodium sulfate or magnesium sulfate) 7 g of activated carbon are
added
and stirred is carried out at 20°C for 30 minutes. After filtering off
of the activated
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
22
carbon, the filfirate is concentrated in vacuo and then the solvent for the
final
crystallization is added.
Recrystallization is carried out from the respectively most advantageous
solvent (cf.
table). Mostly, further crystal fractions can be obtained from the mother
liquors. The
product is dried in a vacuum-drying cabinet or under circulating air (20 to
50°C).
The following examples were implemented in accordance with the process
described
above (product = compound of the general formula IV):
Example 1
Product Yield Solvent of Elementary analysis
of the theorycrystallizationFound l calc.
C 69.51 / 69.12
91 Cyclohexane
H 5.58 / 5.39
(~ N 5.64 / 5.76
Example 2
Product Yield Solvent of Elementary analysis
of the theorycrystallizationFound / calc.
C 67.90 l 68.11
91 Cyclohexane/H 5.05 / 4.84
I
~ ~ MTBE
N 6.00 / 6.11
0 0
Example 3
Product Yield Solvent of Elementary analysis
of the theorycrystallizationFound / calc.
C 68.89 / 69.12
92 Cyclohexane
~ H 5.53 / 5,39
i i
N 5.58 / 5.76
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
23
Example 4
Product Yield Solvent of Elementary analysis
of the theorycrystallizationFound / cafc.
C 69.88 / 70.02
92 Heptane/acetic
H 6.10 I 5.88
acid ethyl
ester N 5.51 / 5.44
0
Example 5
Product Yield Solvent of Elementary analysis
of the theorycrystallizationFound / calc.
C 70.21 / 70.02
N ~ ~ ~ 91 Cyclohexane
I ~ ~ H 6.09 / 5.88
o ~1 N 5.31 / 5.44
Example 6
Product Yield Solvent of Elementary analysis
of the theorycrystallizationFound / calc.
C 71.70 / 71.56
92 Cyclohexane H 6.85 / 6.71
N ~
I i i o N 4.80 / 4.91
0
Example 7
Product Yield Solvent of Elementary analysis
of the theorycrystallizationFound / calc.
C 69.85 / 70.02
NI % % ~ 91 Cyclohexane H 6.05 / 5.88
o ~I N 5.37/5.44
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
24
Example 8
Product Yield Solvent of Elementary analysis
of the theorycrystallizationFound / calc.
C 61.90 / 62.06
93 Cyclohexane/ H 5.35 / 5.21
0 MTBE N 11.91 / 12.06
Example 9
Product Yield Solvent of Elementary analysis
of the theorycrystallizationFound / calc.
C 63.59 / 63.40
N~N~ 93 Cyclohexane H 5.90 / 5.73
\ / N 11.21 / 11.38
II
0 0
Example 10
Product Yield Solvent of Elementary analysis
of the theorycrystallizationFound / calc.
C 63.31 / 63.15
92 Cyclohexane H 5.49 / 5.30
N a
N. 5.51 / 5.66
Example 11
Product Yield Solvent of Elementary analysis
of the theorycrystallizationFound / calc.
C 65.35 / 65.44
93 Cyclohexane H 6.39 / 6.22
N 4.98 / 5.09
\ /
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
Example 12
Product Yield Solvent of Elementary analysis
of the theorycrystallizationFound / calc.
C 59.41 / 59.30
s
93 Hexane H 5.12 / 4.98
I a N 5.21 / 5.32
S 12.03 / 12.78
Example 13
Product Yield Solvent of Elementary analysis
of the theorycrystallizationFound / calc.
C 59.17 / 59.30
~ 91 Cyclohexane
s H 5.18 / 4.98
N 5.18 / 5.32
S 12.01 / 12.18
b
Example 14
Product Yield Solvent of Elementary analysis
of the theorycrystallizationFound / calc.
C 58.01 / 57.82
s
93 Hexane H 4.58 / 4.45
I N 5.71 / 5.62
S 12.78 / 12.86
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
26
Example 15
Product Yield Solvent of Elementary analysis
of the theorycrystallizationFound / calc.
C 56,27 / 56.16
~s 91 Cyclohexane H 4.0213.86
N 5.89 / 5.95
\ /
S 13. 57 / 13.63
0 0
The above-mentioned compounds of general formula IV illustrated in examples '1
to
15 all form part of the subject matter of the present invention.
In the following, the processes used for asymmetric reduction are in each case
generally described:
Chiral Reduction Methods
A ) General description for a microbiological reduction
A 500 ml Erlenmeyer flask which contains 100 ml of a nutrient solution of 5%
glucose
and 2% corn steep liquor (pH 6.0-6.5), which was sterilized in an autoclave at
121 °C
for 20 minutes, is inoculated with an oblique tube culture of the Pichia
wickerhamii
strain (1F0 1278) and shaken on a rotation shaker at 28°C for 48 hours.
Two 2 I
Erlenmeyer flasks are inoculated with 50 ml each of this culture, which are
charg ed
with 500 ml of sterile medium of the same composition as described for the
culture.
After a growth phase of 6 hours at 28°C, a solution of 5 mmole f3-
ketoester of the
general formula IV in 15 ml DMF is added to each Erlenmeyer flask.
Subsequently,
shaking is continued at 28°C.
After a contact time of 114 hours, the combined culture broths are extracted
twi ce
with 1 I of acetic acid ethyl ester. The combined organic phases are dried,
filtered
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
27
over Celite (or a silica gel layer) and concentrated in vacuo. The residue is
recrystallized from a solvent of choice.
B ) Asymmetric hydrogenation with chiral metal catalysts
The asymmetric hydrogenation was carried out in accordance with methods
described in literature. If an acid was used for hydrogenation, this is
indicated in the
examples.
Processing of the hydrogenation batches: Concentration by evaporation is
carried
out, the residue is taken up with an nonpolar solvent (e.g. dichloromethane,
MTBE)
and filtering off is carried out over a short silica gel layer. The filtrate
is evaporated to
dryness in vacuo and the residue is recrystallized from a suitable solvent.
C) Example of a transfer hydrogenation
1 mmole of dichloro(pentamethylcyclopentadienyl)rhodium(Ill) dimer is added to
a
solution consisting of 4 mmole of (R,R) -Tos-DPEN in 1 I of isopropanol under
a
nitrogen atmosphere and stirred 20 minutes at 80°C until an orange red,
homogeneous solution is obtained. Subsequently, 100 ml of potassium
isopropylate
(0.12 m solution = 120 mmole) are added. Then 200 mole of f3-ketoester of the
general formula IV (dissolved in 500 m1 of isopropanol) are added and stirring
is
carried out at 50°C (1 to 20 hours) and the course of the reaction is
pursued by
means of DC.
After completion of the reaction, the reaction mixture is evaporated to
dryness in
vacuo and the residue is filtered over a layer of silica gel (solvent:
mixtures of hexane
acetic acid ethyl ester). The reaction mixture is evaporated to dryness in
vacuo and
recrystallization from a suitable solvent is carried out.
D) Asymmetric reduction with complex hydrides (NaBH~ /LiBH~)
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
28
Reductions with the chiral auxiliary component (R,R')-N,N'-dibenzoylcysteine
are
carried out in accordance with the specifiication from J. Chem. Soc. Chem.
Commun.
1985, 138.
Reductions with the chiral auxiliary component (2R,3R) - tartaric acid are
carried out
in accordance with the specification from J. Chem. Soc. Perkin Trans. 1, 1990,
1826.
Example 16
Reduction method: C
e.e. of the product after crystailizaton: > 98
Literature: R. Noyori, Acc. Res. 1997, 30, 97-102
K. Murata, JOC 1999, 64, 2186-2187
Product Yield Solvent of Elementary analysis
of the theory crystallizationFound ! calc.
C 68.41 / 68.56
95 Cyclohexane/H 6.31 / 6.16
MTBE N 5.63 / 5.71
OH O
Example 17
Reduction method: B
Solvent: methanol
Pressure: 1200 psi
Temperature: 25°C
Reaction time: 14 hours
e.e. of the product after crystallization: > 98
Literature: JACS, vol. 121, No. 30, 1999, page 7061 (compound 43)
Product Yield Solvent of Elementary analysis
of the theory crystallizationFound / calc.
C 67.47 / 67.52
94 Cyclohexane/H 5.78 / 5.67
off o MTBE N 5.89 / 6.06
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
29
Example 18
Reduction method: B
Solvent: ethanol
Pressure: 1300 psi
Temperature: 40 °C
Reaction time: 18 hours
e.e. of the product after crystallization: > 99
Literature: as indicated in example 17
Product Yield Solvent of Elementary analysis
of the theory crystallizationFound / calc.
C 68.47 / 68.56
93 Cyclohexane H 6.2916.16
N 5.89 / 5.71
OH O
Example 19
Reduction method: A
e.e. of the product after crystallization: > 98
Strain: Pichia wickerhamii (1F0 1278)
Product Yield Solvent of Elementary analysis
of the theory crystallizationFound / calc.
C 69,37 / 69.48
94 HeptanelaceticH 6.78 / 6.61
i
acid ethyl N 5.27 /. 5.40
off ester
Example 20
Reduction method: C
e.e. of the product after crystallization: > 98
Literature: R. Noyori, Acc. Res. 1997, 30, 97-102
K. Murata, JOC 1999, 64, 2186-2187
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
Product Yield Solvent of Elementary analysis
of the theory crystallizationFound / calc.
C 69.61 / 69.48
95 Cyclohexane H 6.84 / 6.61
I
~ ~ N 5.27 / 5.40
OH O
Example 21
Reduction method: B / Ru - (R) - Me0-Bipheg (catalyst)
Solvent: EtOH
Pressure: atmospheric pressure H2
Temperature: 50 °C
Reaction time: 50 hours
e.e. of the product after crystallization: > 98
Literature: Tetrahedron Letters, vol. 36, No. 27, 4801 - 4804
Product Yield Solvent of Elementary analysis
of the theory crystallizationFound / calc.
C 70.91 / 71.06
96 Cyclohexane H 7.48 / 7.37
N
I N 4.93 / 4.87
, , o
OH
Example 22
Reduction method: B / Ru - (R) - Me0-BIHEP (catalyst)
Solvent: MeOH
Pressure: 10 bar
Temperature: 80 °C
Reaction time: 40 hours
e.e. of the product after crystallization: > 98
Literature: Tetrahedron, 57 (2001 ), 2563 - 2568
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
31
Product Yield Solvent of Elementary analysis
of the theory crystallizationFound / calc.
C 69.57 / 69.48
I ~ ~ ~ 95 Cyclohexane H 6.75 / 6.61
N / /
N 5.28 l 5.40
OH
Example 23
Reduction method: B
Solvent: EtOH
Pressure: 50 bar
Temperature: 60°C
Reaction time: 18 hours
e.e. of the product after crystallization: > 99
Literature: Angewandte Chemie 1999, 111, page 3397
Product Yield Solvent of Elementary analysis
of the theory crystallizationFound / calc.
C 61.68 / 61.53
93 Cyclohexane/ H 6.10 / 6.02
MTBE N 11.87 / 11.96
off a
Example 24
Reduction method: B
Solvent: methanol
Pressure: 120 psi
Temperature: 60 °C
Reaction time: 18 hours
e.e. of the product after crystallization:
Literature: see example 17
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
32
Product Yield Solvent of Elementary analysis
of the theory crystallizationFound / talc.
C 62.99 / 62.89
94 Cyclohexane H 6.71 / 6.50
N 11.13 / 11.28
OH
Example 25
Reduction method: B
Solvent: ethanol
Pressure: 1300 psi
Temperature: 60 °C
Reaction time: 24 hours
e.e. of the product after crystallization: > 99
Literature: see example 17
Produkt Yield Solvent of Elementary analysis
of the theory crystallizationFound / talc.
C 62.57 / 62.64
96 Cyclohexane H 6.19 / 6.07
~N
N 5.49 / 5.62
OH O
Example 26
Reduction method: C
e.e. of the product after crystallization: > 98
Literature: R. Noyori, Acc. Res. 1997, 30, 97-102
K. Murata, JOC 1999, 64, 2186-2187
Product Yield Solvent of Elementary analysis
of the theory crystallizationFound / talc.
Cyclohexane/ C 64.91 / 64.97
94 acetic acid H 7.05 / 6.91
ethyl ester N 4.98 / 5.05
OH
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
33
Example 27
Reduction method: A
e.e. of the product after crystallization: > 99
Strain : Pichia wickerhamii (1F0 1278)
Product Yield Solvent of Elementary analysis
of the theorycrystallizationFound / calc.
C 58.81 / 58.85
I~
95 Hexane H 5.85 / 5.70
" Y ~ N 5.17/5.28
OH O
S 11.93 / 12.08
Example 28
Reduction method: A
e.e. of the product after crystallization: > 99
Strain: Pichia virickerhamii (1F0 1278)
Product Yield Solvent of Elementary analysis
of the theory crystallization Found / calc.
C 58.94 / 58.85
94 Cyclohexane H 5.85 / 5.70
~ s
_ N 5.17 / 5.28
\ / S 11.93 / 12.08
OH
Example 29
Reduction method: C
e.e. of the product after crystallization: > 98
Literature: R. Noyori, Acc. Res. 1997, 30, 97-102
K. Murata, JOC 1999, 64, 2186-2187
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
34
Product Yield Solvent of Elementary analysis
of the theorycrystallizationFound l talc.
C 57.28 / 57.35
94 Hexane H 5.35 / 5.21
(
i
N 5.48 / 5.57
O
H
S 12.63 / 12.76
Example 30
Reduction method: B
Solvent: Me OH
Pressure: 50 bar
Temperature: 50 °C
Reaction time: 24 hours
e.e, of the product after crystallization: > 99
Literature: see example 23
Product Yield Solvent of Elementary analysis
of the theorycrystallizationFound / calc.
C 55.67 / 55.68
~s 96 Cyclohexane H 4.80 / 4.67
N 5.95 / 5.90
S 13.40 / 13.51
OH I
The aforementioned compounds of general formula I of examples 16 to 30 all
form
part of the subject matter of the present invention.
The use of the novel intermediate products according to the invention for the
preparation of epothilone intermediates known in literature is shown in the
following
examples:
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
General Specification for the Preparation of the TBDMS Ethers
(intermediate compounds from WO 00/66589, Schering AG)
100 mmole of the hydroxy ester of the general formula (I) and 150 mmole of
imidazole are dissolved in 150 ml of dimethylformamide and 125 mmole of tent.
butyl
dimethylsilyl chloride (TBDMS-CI) are added at 0°C. Stirring is carried
out at room
temperature for 12 hours. 70 mmole of methanol are added to destroy the excess
of
TBDMS-CI and stirring is carried out at room temperature for further 2 hours.
5 ml of
water and 50 ml of n-hexane are added and vigorous stirring is carried out for
10
minutes. The hexane phase is separated and rejected. 1000 ml of water is added
to
the DMF phase and extraction is carried out twice with 150 ml acetic acid
ethyl ester
each. The combined organic phases are separated and concentrated to dryness in
vacuo.
It proved to be advantageous in the case of a consecutive reaction with DIBAH
or
DIBAHIBuLi to extract with dichloromethane or toluene and to carry out the
further
reaction directly with the solution.
Example 31
Product Yield Solvent for Elementary analysis
of the theory extraction Found / calc.
C 66.93 / 66.81
99 Acetic acid H 8.30 / 8.13
ethyl
N
ester N 3.76 / 3.90
~S~~o
Si 7.67 / 7.81
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
36
Example 32
Product Yield Solvent for Elementary analysis
of the theory extraction Found / calc.
C 65.93 / 66.05
I 98 DichloromethaneH 7.97 / 7.88
~
N 3.92 / 4.05
~S~~o 0
Si 8.01 / 8.13
Example 33
Product Yield Solvent for Elementary analysis
of the theory extraction Found / calc.
C 66.93 / 66.81
99 Toluene H 8.27 / 8.13
N 3.76 / 3.90
~S~~o 0
Si 7.67 / 7.81
Example 34
Product Yield Solvent for Elementary analysis
of the theory extraction Found / calc.
C 67.69 / 67.52
98 Acetic acid H 8.51 / 8.36
ethyl
ester N 3.67 / 3.75
~S~~o
Si 7.34 / 7.52
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
37
Example 35
Product Yield Solvent for Elementary analysis
of the theory extraction Found / calc.
C 67.65 / 67.52
N \ \ ~ 99 Acetic acid H 8.45 / 8.36
I ethyl
~
ester N 3.68 / 3.75
~S~ 0 0
\ Si 7.43 / 7.52
Example 36
Product ~ Yield Solvent for Elementary analysis
of the theory extraction Found / calc.
C 68.91 / 68.78
100 DichloromethaneH 8.87 / 8.78
N \ \
o N 3.35 / 3.49
~s~~ o
Si 6.87 / 6.99
Example 37
Product Yield Solvent for Elementary analysis
of the theory extraction Found / calc.
C 67.65 / 67.52
I \ \ ~ 99 Acetic acid H 8.43 / 8.36
N / / ethyl
ester N 3.68 / 3.75
,~ o
Si 7.48 / 7.52
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
38
Example 38
Product Yield Solvent for Elementary analysis
of the theory extraction Found / calc.
C 62.12 / 62.03
98 Acetic acid H 8.30 / 8.10
ethyl
ester N 7.92 / 8.04
o Si 7.94 / 8.06
Example 39
Product Yield Solvent for Elementary analysis
of the theory extraction Found / calc.
C 63.09 / 62.95
N~N~ 99 Toluene H 8.43 / 8.34
\ / N 7.65 / 7.73
Si 7.65 / 7.75
0
s
~
~
Example 40
Product Yield Solvent for Elementary analysis
of the theory extraction Found / calc.
C 62.87 / 62.78
I 99 Toluene H 8.16 / 8.04
~
j( N 3.76 / 3.85
~S~~o 0
\ Si 7.65 / 7.73
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
39
Example 41
Product Yield Solvent for Elementary analysis
of the theory extraction Found l calc.
C 64.61 / 64.41
~ 100 DichloromethaneH 8.62 / 8.49
-' -
O
N N 3.45 / 3.58
\ / Si 7.03/7.17
Example 42
Product Yield Solvent for Elementary analysis
of the theoryextraction Found / calc.
C 60.23 / 60.12
99 Acetic acid ethylH 7.82 / 7.70
ester N 3.57 / 3.69
S 8.34 / 8.45
W
Si 7.30 / 7.40
Example 43
Product Yield Solvent for Elementary analysis
of the theory extraction Found . / calc.
C 60.26 / 60.12
N~s 99 Acetic acid ethyl H 7.8517.70
ester N 3.59 / 3.69
S 8.33 / 8.45
\S',o of Si 7.31 / 7.40
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
Example 44
Product Yield Solvent for Elementary analysis
of the theoryextraction Found / calc.
C 59.23 / 59.14
98 Dichloromethane H 7.51 / 7.44
o ~ N 3.72 / 3.83
,
\ S 8.66/8.77
Si 7.60 / 7.68
Example 45
Product Yield Solvent for Elementary analysis
of the theoryextraction Found / calc.
C 59.15 / 59.08
98 Dichloromethane H 7.25 / 7.17
N 3.87 / 3.98
S 9.01 / 9.12
~
~s~~o Si 7.88 / 7.99
General Production Specification for the Reduction of the Silyl Esters
(Intermediate stages from US 6,387,927 and PCTIEP99/10129, NOVARTIS )
20 ml of a 1 M solution of DIBAH in dichloromethane are added dropwise at -
78°C
(under nitrogen) for 30 minutes to a solution of silyl ester, 10 mmole,
dissolved in 100
ml of dichloromethane, which was produced in examples 31 to 46 (with the
exception
of the tert. butyl esters). Stirring is carried out for 3 hours at -
78°C. Further 5 ml of
DIBAH solution are added and stirring is continued for further 2 hours. 6 ml
of
methanol are added dropwise and the mixture is allowed to reach room
temperature.
ml of dichloromethane and 50 ml of water are added and the suspension is
filtered
over Celite. The organic phase is separated, washed with water and
concentrated to
dryness in vacuo.
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
41
Example 46
Product Yield Elementary analysis
of the theory Found / calc.
C 68.65 / 68.53
97 H 8.12 / 7.99
N / / H
N 4.31 / 4.44
Ws~ o
Si8.75 / 8.90
Example 47
Product Yield Elementary analysis
of the theory Found / calc.
C 68.62 / 68.53
N w w 98 H 8.09 / 7.99
I
/ / H
N 4.32 / 4.44
Si8.80 / 8.90
Example 48
Product Yield Elementary analysis
of the theory Found / calc.
C 68.60 / 68.53
96 H 8.11 / 7.99
/ / H
N 4.31 / 4.44
ws~ o
Si 8.82 / 8.90
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
42
Example 49
Product Yield Elementary analysis
of the theory Found / calc.
C 68.62 / 68.53
w w 98 H 8.14 / 7.99
H
N 4.36 / 4.44
~S~~o 0
Si 8.81 / 8.90
Example 50
Product Yield Elementary analysis
of the theory Found / calc.
C 69.32 / 69.26
w 97 H 8.41 / 8.26
/ H
N 4.12 / 4.25
Si8.44/8.52
W
Example 51
Product Yield Elementary analysis
of the theory Found / calc.
C 69.35 / 69.26
96 H 8.40 / 8.26
/ H N 4.17 / 4.25
~S~~o 0
Si8.40 / 8.52
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
43
Example 52
Product Yield Elementary analysis
of the theory Found l calc.
C 64.15 / 64.11
98 H 8.34 / 8.23
H N 8.67 / 8.80
o Si 8.69 / 8.82
Example 53
Product Yield Elementary analysis
of the theory Found / calc.
C 64.19 / 64.11
N~N~ 97 H 8.37 / 8.23
N 8.64 / 8.80
/
H
Si 8.66 / 8.82
,0 0
s
~
~
Example 54
Product Yield Elementary analysis
of the theory Found / calc.
C 63.99 / 63.91
~ 96 H 8.01 / 7.89
I
N 4.34 / 4.38
~s~ o
Si 8.68 / 8.79
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
44
Example 55
Product Yield Elementary analysis
of the theory Found / calc.
C 60.93 / 60.85
98 H 7.66 / 7.51
~
I /
N N 4.08/4.17
_
i
~s~ o
S 9.44 / 9.56
Si 8.25 / 8.37
Example 56
Product Yield Elementary analysis
of the theory Found / calc.
C 60.96 / 60.85
~ 97 H 7.68 / 7.51
N i 'S
N 4.09 / 4.17
S 9.47 / 9.56
o Si 8.27 / 8.37
Example 57
Product Yield Elementary analysis
of the theory Found / calc.
C 59.85 / 59.77
96 H 7.34 / 7.21
/ H
N 4.27 / 4.36
ws~ o
S 9.88 / 9.97
Si8.66/8.74
CA 02546981 2006-05-23
WO 2005/064006 PCT/EP2004/014753
Example 58
Product Yield Elementary analysis
of the theory Found / calc.
C 59.88 l 59.77
ms's 97 H 7.32 / 7.21
N 4.28 / 4.36
\ l
S 9.86 / 9.97
~S~~o 0
Si 8.6918.74