Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02549094 2006-06-12
WO 2005/058500 PCT/GB2004/005334
1
S-, sy tem
The present invention relates to a microfluidic separation system for
separating
particles from a fluid sample, and to an assay system for detecting the amount
and/or
presence of an analyte in a fluid sample, or for the determination of a
property of a
fluid sample. In particular this invention relates to the separation of cells
from a
sample, for example the separation of red blood cells from a sample of whole
blood.
Diagnostic assay devices for the measurement of the presence and/or amount of
analytes present in a fluid sample have been developed for use at the point of
care or
in the home setting. Such devices may be used by the healthcare professional
or non-
specialist personnel alike, such as the patient for self-monitoring.
Consequently, such
devices are designed to be easy to use, to require small volumes of fluid
sample and
penorm the measurement rapidly. Small volume samples are desirable as they may
be
less painful to collect from the patient, for example when obtaining a sample
of
capillary blood by application of a lancet or finger-sticle to the skin.
Typically it will
be a one-step device, the user having simply to apply a fluid sample to the
device
without the need to perform any further sample manipulation steps in order to
obtain a
result. Such systems will typically be portable and have minimal or no moving
parts.
Typically, a microfluidic channel or a porous carrier is employed to move
sample into
and/or through the device by capillary action avoiding the need to actively
move the
fluid sample within the device.
When performing a diagnostic assay measurement on a fluid sample, it may be
desirable or necessary to remove components from the sample that may interfere
with
the assay. For example, it may be necessary to remove red-blood cells from
whole
blood where the testing regimen requires a sample or plasma. Red-blood cells
may
also interfere with the assay measurement, for example by absorbing light of a
particular wavelength. Red-blood cells are conventionally removed from whole
blood
by centrifugation or by allowing the red-blood cells to settle. However such
methods
are ill-suited for the purposes of conducting a rapid diagnostic assay on a
fluid sample
of a volume ranging from typically 100p,1 to less than lp,l.
CA 02549094 2006-06-12
WO 2005/058500 PCT/GB2004/005334
The separation of particles from a fluid medium may be effected by the use of
a filter
such as a non-woven fabric with the appropriate pore-size. However, for the
purposes
of red-blood cell separation from whole blood, the use of such a filter is
inappropriate,
due to the tendency of the filter to block which results in low yields of
plasma filtrate.
Also, the time for plasma yield to take place increases. This is clearly
disadvantageous in a flow system such as a microfluidic device that may be
reliant
only on capillary forces to drive the fluid sample.
There therefore exists a need to provide a separation system that results in
rapid
separation of small volumes of sample, with high levels of yield.
According to a first aspect of the present invention there is provided a
microfluidic
separation system for separating fluid sample medium from cells provided in a
sample, the system comprising a microfluidic structure and a cell aggregation
agent,
the microfluidic structure comprising one or more microfluidic channels
operable to
separate aggregated cells from fluid sample medium by size exclusion.
The team aggregation agent is intended to encompass any agent that can cause
aggregation and/or agglutination of cells, as well as agents that can cause
rosetting of
cells such as red blood cells and/or promote the formation of roleaux in cells
such as
red blood cells.
The provision of an aggregation agent causes cells in the sample to aggregate
together
thereby assisting the separation of the sample medium from cells. The
aggregation
agent may be provided adjacent to or remote to the microfluidic structure. The
aggregation agent may be mixed with a reagent that aids its release from an
internal
surface of the microfluidic system into the fluid sample. A plurality of
aggregation
agents may be provided.
The microfluidic structure may comprise a size exclusion element having one or
more
size exclusion spacings which enable the aggregated cells to be filtered from
the
CA 02549094 2006-06-12
WO 2005/058500 PCT/GB2004/005334
3
sample medium. The size exclusion spacing thereby enables the microfluidic
channels
to separate aggregated cells from fluid sample medium. The size of the
exclusion
spacing may be chosen from any suitable size that enables aggregated cells to
be
separated or substantially separated from the sample medium and will be
determined
upon the dimensions of the system, the amount of time that the aggregation
agent
interacts with the fluid sample and the nature of the sample itself. A
suitable size of
exclusion spacing may be determined by routine experimentation. The optimum
sizes
of exclusion spacings may be determined on the basis of the efficiency of
aggregate
separation and are also most easily determined by routine experimentation.
Where a
plurality of size exclusion spacings are used, they may be of the same size or
of
differing sizes. The upper limit of the exclusion spacings may be determined
by the
size of the aggregates and the lower limit determined by the speed and
efficiency of
separation.
The microfluidic structure may define a capillary pathway. The capillary
pathway
may define one or more size exclusion spacings to separate aggregated cells
from
fluid sample medium.
The dimensions of the microfluidic channels) of the microfluidic structure may
correspond to a size exclusion spacing. The microfluidic channels) may be of a
dimension which varies to define the size exclusion spacing. The microfluidic
channels) may be in fluidic communication with other microfluidic channels of
different or varying dimensions.
The microfluidic structure may comprise at least one first microfluidic
channel, the at
least one first channel having a base with extending side walls, the channel
being in
fluid communication along a longitudinal side thereof with one or more
passages
having a depth less than the depth of the channel, the depth of the passage
defining a
size exclusion spacing.
CA 02549094 2006-06-12
WO 2005/058500 PCT/GB2004/005334
4
The depth of the passages) defining the size exclusion spacing is such that
sample
medium, can pass into the passage from the first channel. Therefore aggregated
cells
can be separated from the sample medium by size exclusion.
The, or each passage may be in further fluid communication with a further
microfluidic channel. Therefore the, or each passage may operate as a
connecting
region between the first and further channels which defines a size exclusion
spacing.
The passages) may be provided with one or more step formations to vary the
size
exclusion spacing. The, or each step formation may provide the passage with a
size
exclusion spacing.
The passages) and/or further channels) may be in fluid communication with a
sample medium collecting region, wherein the sample medium separated from
aggregated cells flows to said collecting region. The collecting region may be
a
further conduit or may comprise a chamber in the microfluidic structure.
Alternatively, the capillary pathway may comprise one or more microfluidic
channels
in which is provided one or more microstructures that define gaps
corresponding to
the desired size exclusion spacing(s). The microstructures may be configured
to
separate aggregated cells from the sample. The microstructures may define size
exclusion spacings in the region of about l~.m to about 50~,m. A plurality of
groups
of microstructures maybe provided of the same size or of different sizes and
may be
arranged in any particular configuration with respect to each other. Each
group may
define size exclusion spacings different to other group(s). The groups of
microstructures may be in an ordered configuration so that the sample flows
through
the group defining larger size exclusion spacings before flowing through the
groups)
having smaller size exclusion spacings, wherein the sample medium is separated
from
aggregated cells. A collecting region may be provided downstream of the
microstructures, the sample medium flowing to the collecting region after
flowing
through the size exclusion spacings and being separated.
CA 02549094 2006-06-12
WO 2005/058500 PCT/GB2004/005334
The microstructures may be in the form of grooved surfaces, pillars, or any
form that
defines size exclusion spacings.
The dimensions of the size exclusion spacing(s) in the capillary pathway may
be less
than or equal to about 50~,m, less than or equal to about 40p,m, less than or
equal to
about 30~,m, less than or equal to about 20p,m, less than or equal to about
15~,m, less
than or equal to about 10~.m, less than or equal to about 5~,m, or may be less
than or
equal to about 2p,m.
The capillary pathway may define a tortuous path.
The system may further comprise a conduit in fluid communication with the
microfluidic structure to supply the microfluidic structure with the sample.
The supply
conduit may also supply the aggregation agent to the microfluidic structure.
The
aggregation agent may be provided on one or more surfaces of the conduit
and/or to
one or more surfaces of the capillary pathway of the microfluidic structure
upstream
of the size exclusion spacing(s).
The conduit may have a Reynolds number of less than 3000. Alternatively the
conduit
may have a Reynolds number of less than 100. The conduit is preferably a
capillary.
Reynolds number can be calculated using the formula:
Re = pVd/r~
Where Re = Reynolds number, p = Fluid density, V = Fluid velocity, d = length
scale,
r~ = dynamic density. A Reynolds number of 2000 or less will cause the conduit
(which may be considered to be a microstructure or microchannel) to be filled
passively by surface tension (capillarity) alone.
The sample may be applied to the conduit. The sample may be applied via a
sample
inlet port in fluidic connection with the conduit.
CA 02549094 2006-06-12
WO 2005/058500 PCT/GB2004/005334
6
Suitable non-limiting examples of aggregation agents are those that cause
aggregation
of red blood cells, such as dextran. Alternatively the aggregation agent may
cause
agglutination of red blood cells, such as lectin. As yet a further
alternative, the
aggregation agent may comprise one or more antibodies to red blood cells.
Alternatively still, the aggregation agent may promote rosetting of red blood
cells, or
may promote the formation of roleaux in red blood cells.
The microfluidic system may comprise further microfluidic elements such as
internal
microstructures, time gates, fluid mixing chambers, sample collections
chambers,
wells, channels, baffles, constrictions, a sample application port, etc. The
microfluidic
elements may be of a regular or irregular shape and may be in the same plane
or in
different planes. The microfluidic channels) may be of a capillary dimension
which
varies along its length, and may be in fluidic communication with other
microfluidic
elements of different or varying capillary dimensions.
The time which a sample is allowed to interact with the aggregation agent
before
reaching the size exclusion spacing(s) may be influenced for example by where
in the
system the aggregation agent is positioned, the dimensions of the upstream
fluid
conduit and speed with which the fluid sample travels along the fluid conduit.
Where
necessary, means to ensure that the aggregation agent has interacted for a
sufficiently
long enough time with the fluid sample may be provided, such as a chamber or
time
gate which serves to slow down the rate of passage of fluid sample between the
supply conduit and the size exclusion spacing(s).
Typical dimensions of the microfluidic structure elements are those having a
cross-
sectional dimension, such as a cross-sectional diameter, of between 0.1 and
500p,m,
more typically having a cross-sectional dimension of between 1 and 100~,m.
Preferably the cross-sectional dimensions are chosen to be of a size such that
fluid is
able to be transported along, through or into the various elements of the
system by
capillary action. As an alternative or in addition, fluid may be transported
through one
or more of the elements of the system by external forces such as by
electroleinetic
CA 02549094 2006-06-12
WO 2005/058500 PCT/GB2004/005334
7
pumping. In such cases the cross-sectional dimension may exceed the capillary
dimension.
The substrate from which the system is prepared may be any suitable such as
polycarbonate. In the case where the substrate is hydrophobic, the surfaces
may be
treated to render them hydrophilic by methods known in the art, such as for
example
by treatment with an oxygen plasma. As well as providing hydrophilic surfaces,
other
types of reagents, immobilised or otherwise may be provided on the internal
surfaces.
The system may be prepared for example by providing a planar first substrate
layer
onto which are provided walls which serve to define the depth of the
microfluidic
channels in the structure, followed by provision of a second planar substrate
layer
which is disposed onto the upper surfaces of the walls. Suitable adhering
means such
as adhesives may be used to join the various structures as appropriate. Other
means of
preparing the structure are for example screen-printing, or the provision of
multi-
laminated systems having an upper and lower laminated surface which serve as
upper
and lower surfaces of the system, as well as intermediate laminates having
structures
which serve to define the elements of the microfluidic structure.
The fluid sample medium to be separated is preferably whole blood. The system
may
therefore enable aggregated red blood cells to be separated from the sample
medium.
The microfluidic separation system may be disposable.
According to a second aspect of the present invention there is provided an
assay
system for conducting an assay on a fluid sample, the assay system comprising
a
microfluidic separation system according to the first aspect in fluid
communication
with an analyte detection zone
The assay system may comprise a sample entry port for the application of fluid
sample in fluid connection with the microfluidic structure of the separation
system.
The detection zone may be provided downstream from the microfluidic structure,
into
CA 02549094 2006-06-12
WO 2005/058500 PCT/GB2004/005334
g
which fluid sample may flow from the microfluidic structure after separation.
Reagents either specific or non-specific to the analyte of interest may be
provided
within the assay system and may be provided on the inner surface of the
detection
zone. The assay system may further comprise an interferent zone that serves to
neutralise or remove molecules from the sample that may interfere with the
binding
interactions in the assay system, or with the signal generation and detection.
Yet
further zones may be provided within the assay system in fluidic communication
with
one or more of the other zones and may comprise wash zones, time gates, pre-
mixing
zones, reaction zones and the like.
The assay may be chosen from any that is able to determine the presence and/or
amount of an analyte of interest. The assay may be a binding assay, such as a
specific
binding assay in which a specific binding event takes place between a specific
binding
pair, one of the binding partners being the analyte of interest, the other
being chosen
from any compound or composition capable of recognising a particular spatial
or
polar orientation of a molecule, e. g., epitopic or determinant site. Examples
of
suitable binding pairs are an antibody and antigen, biotin and avidin,
carbohydrates
and lectins, complementary nucleotide sequences, complementary peptide
sequences,
effector and receptor molecules, enzyme cofactors and enzymes, enzyme
inhibitors
and enzymes, a peptide sequence and an antibody specific for the sequence or
the
entire protein, polymeric acids and bases, dyes and protein binders, peptides
and
specific protein binders and so on. Where the binding partner is an antibody
it may be
monoclonal or polyclonal, or it may be a fragment thereof. Fragments thereof
may
include Fab, Fv and F(ab') 2, Fab', and the like. The assay may involve a
specific
reaction which talces place between the analyte and an enzyme, Suitable
enzymes are
ones which employ FAD/FADH2, NAD/NADH2 or NADP/NADPHa systems.
One of the specific binding partners may be provided with a detectable label.
"Label"
refers to any substance that is capable of producing a signal that is
detectable by
visual or instrumental means. Examples of suitable labels include enzymes and
substrates, chromogens, catalysts, fluorescent compounds, chemiluminescent
CA 02549094 2006-06-12
WO 2005/058500 PCT/GB2004/005334
9
compounds, radioactive labels as well as particulate colloidal metallic
particles such
as gold, or particulate dyed organic substances such as polyurethane.
The binding assay may be either heterogeneous or homogeneous.
Where the assay is homogeneous it is desirable that the label, which is
attached to the
specific binding partner, is able to undergo some detectable physical or
chemical
change upon binding to form a specific binding pair such that the bound
species may
be distinguished from the unbound species. Examples of such binding assays
which
involve an energy transfer or result in a change in wavelength are given in
US5705622 and US6215560.
The sample medium of interest may be whole blood, but may be chosen from any
sample medium where separation of particulate matter that is able to be
aggregated by
an aggregation agent is required, such as white blood cells.
Analytes of interest include, but are not limited to, toxins, organic
compounds,
proteins, peptides, microorganisms, bacteria, viruses, amino acids, nucleic
acids,
carbohydrates, hormones, steroids, vitamins, drugs of abuse, pollutants,
pesticides,
and metabolites of or antibodies to any of the above substances. Specific
examples
thereof are specific cardiac markers including troponin T and troponin I,
CI~MB, C-
reactive protein (CRP), natriuretic peptides such as ANP and BNP as well as
their N-
terminal fragments. Other analytes of interest include human chorionic
gonadotrophin
(hCG), luteinising hormone (LH) and follicle stimulating hormone (FSH) as well
as
marlcers of bone resorption.
The assay may be a binding assay, which has an optical emission and may be a
luminescent oxygen channelling immunoassay. Such an immunoassay can comprise:
a. donor particles able to generate singlet oxygen when irradiated with light;
b. acceptor particles containing emission means activated by the singlet
oxygen to
emit detectable light;
CA 02549094 2006-06-12
WO 2005/058500 PCT/GB2004/005334
c. the donor and acceptor particles being adapted to recognise and bind to the
analyte, wherein on binding of both the donor and acceptor particles to the
analyte, generated singlet oxygen activates the emission means on the acceptor
particles to emit detectable light; and
5 d. a detector to detect light emitted by the acceptor particle.
The donor and acceptor particles recognise the analyte through antibodies
provided on
the surfaces of the particles. The emission means comprise a dissolved dye
that can be
activated by the singlet oxygen to produce chemiluminescent emission. The
10 chemiluminescent emission activates fluorophores in the acceptor particle
causing
emission of light. The light may be emitted at 520-620nm. Singlet oxygen may
be
emitted when irradiated with light of wavelength 680nm. The donor particles
may
comprise dissolved phthalocyanine, the phthalocyanine generating the singlet
oxygen
when irradiated.
The assay system may further comprise a transduction system. The transduction
system may be optical, magnetic, electrochemical, radiological or may involve
measurement of a change of mass, frequency or energy state, depending upon the
signal of interest to be detected. Where the signal is an optical one, it may
be of any
particular detectable wavelength or wavelength range and includes fluorescent
and
chemiluminescent signals.
The assay system may alternatively or in addition be used to determine a
particular
property of a fluid sample such as the coagulation time or prothrombin time.
The detection zone may comprise a microfluidic channel or may comprise a well.
Detection means may be provided as an integral part of the detection zone. An
excitation means may also be integrally provided as part of this zone.
Examples of an
excitation means and a detection means are respectively a light emitting diode
and a
photodetector. There may be one of more detection zones and one or more
excitation
and detector means. The detector means and excitation means may be chosen to
be a
size and shape as is convenient and will preferably be chosen such as to
maximise the
CA 02549094 2006-06-12
WO 2005/058500 PCT/GB2004/005334
11
capture efficiency of the signal to be measured. The excitation means and
detector
means will typically be located on a surface exterior to the fluid sample in
the vicinity
of the detection zone.
The substrate of the detection zone may be chosen from any suitable material
or
materials depending upon the purpose. Examples of suitable substrates are
plastics
such as polycarbonate. In the region of the detection zone where the signal to
be
detected is an optical signal, the substrate may chosen from one that is able
to transmit
the optical signal such as a suitable optically transparent plastics material.
Additionally or alternatively the plastics material may incorporate a filter
to remove
light of undesired wavelength. As yet a further alternative, the filter may be
pxesent on
a surface of the assay substrate. The optically transparent substrate may also
be a
lens, either converging or diverging onto which an excitation source or
detector may
be positioned. Light may then be either converged or diverged as desired. The
substrate may also be partially transparent or have a surface roughness thus
allowing
for a diffuse source of light.
The assay system may be configured so that the sample flows into the assay
detection
zone after the interferent zone. The interferent zone may comprise one or more
agents
to influence the pH of the fluid sample or to remove or solubilise particular
species
such as lipids by for example solubilising them with surfactants, or by
selectively
binding them with a lipid-binding agent. The interferent zone may be time-
gated so
that only fully treated sample passes into the assay.
The assay system may measure analyte levels in bodily fluid which may be whole
blood. Plasma may have been separated from aggregated red blood cells by the
microfluidic separation system of the assay system.
The assay system may be disposable and may be designed to be used in
combination
with a meter that is able to display the results of the assay. The meter may
comprise a
display, a power source, and the appropriate circuitry. The meter may also
comprise a
CA 02549094 2006-06-12
WO 2005/058500 PCT/GB2004/005334
12
1~0
light source and detector. Alternatively the assay system and meter may be
wholly
integrated into a disposable system. The assay system may be integrated with a
fluid
sampling and collection system such as a lancet, such that transfer of sample
from the
site of collection to the assay system may be avoided.
According to a further aspect of the present invention there is provided a
method of
separating fluid sample medium from cells provided in a fluid sample,
comprising
applying a fluid sample to a microfluidic separation system according to the
first
aspect of the present invention.
A sample of less than or equal to 100.1 may be applied.
The method may be for separating plasma from red blood cells in whole blood.
The sample may be mixed with a cell aggregation agent before addition to the
microfluidic structure of the microfluidic separation system.
According to a further aspect of the present invention there is provided a
method of
detecting an analyte in a fluid sample, the method comprising applying a fluid
sample
to an assay system according to the second aspect of the present invention.
The sample may be mixed with a cell aggregation agent before addition to the
microfluidic structure of the microfluidic separation system.
2.5 Preferred features of each aspect of the present invention are as for each
other aspect
mutatis mutandis.
The present invention will now be described by way of example only, and with
reference to the accompanying figures, in which
Figure 1 illustrates a microfluidic separation system of the present
invention;
CA 02549094 2006-06-12
WO 2005/058500 PCT/GB2004/005334
13
Figure 2 illustrates an assay of the present invention incorporating a
microfluidic
separation system of the present invention;
Figure 3 illustrates further the assay system of Figure 2: Figure 3a is a plan
view of
the system; Figure 3b is a cross section taken along line X-X in Figure 3a;
Figure 3c
is an enlargement of the region A in Figure 3b; and Figure 3d is an
enlargement of the
region B in Figure 3c;
Figure 4 further illustrates the microfluidic separation system in the assay
system of
Figures 2 and 3: Figure 4a is a perspective view of the system; and Figure 4b
is an
enlargement of the region A illustrating the microstructures;
Figure 5 illustrates a further assay system in which an alternative
nucrofluidic
separation system is provided: Figure 5a is a perspective view of the assay
system;
and Figure 5b is an enlargement of the region A illustrating the separation
system;
Figure 6 provides further illustration of the alternative separation system in
Figure 5:
Figure 6a is a plan view of the assay system of Figure 5; Figure 6b is a cross
section
taken along line X-X in Figure 6a; Figure 6c is an enlargement of the region A
in
Figure 6b; and Figure 6d is an enlargement of the region B in Figure 6a
illustrating
the separation system;
Figure 7a and b illustrate an assay system;
Figure 8 illustrates the sensitivity of the assay system;
Figure 9 also illustrates the sensitivity of the assay system; and
Figure 10 illustrates a standard curve relating to the assay system.
CA 02549094 2006-06-12
WO 2005/058500 PCT/GB2004/005334
14
The present invention relates to a microfluidic separation system for
separating
sample medium from cells, an assay system for detecting and measuring an
analyte in
a sample, and/or for measuring analyte in a sample.
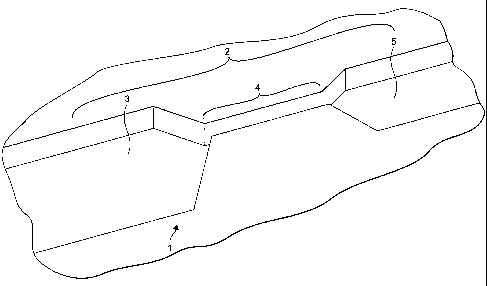
A microfluidic separation system I is illustrated in figure 1. The system 1
includes a
microfluidic structure 2 and a cell aggregation agent (not shown). The
microfluidic
structure 2 defines a capillary pathway. The capillary pathway includes a
channel 3.
The dimensions of the channel 3 varies along the length thereof. In particular
at
region 4 the channel narrows to provide a size exclusion spacing having
dimensions
less than lOp.m. This size exclusion spacing is designed to prevent cells
aggregated by
the aggregation agent from flowing further downstream from this region 4. The
aggregation agent can be provided in the channel 3 upstream of the size
exclusion
spacing or may be applied to the sample before the sample is applied to the
microfluidic structure 2. Alternatively the aggregation agent can be
immobilised onto
one or more surfaces of the channel upstream of the size exclusion spacing.
The
channel widens at region 5 to provide a sample collecting region into which
sample
medium without cells can flow. The system is also provided with a lid (not
shown)
defining an upper surface of the channel 3. The sample flows through the
capillary
pathway by capillary forces. To assist this flow, the surfaces of the
capillary pathway
are coated with a hydrophilic coating.
Figure 2 of the accompanying drawings illustrates an assay system 10. The
system 10
incorporates a microfluidic separation system 12 and an assay detection zone
18. The
system 10 also includes an interferent zone 14, a pre-treatment zone 16, and a
sample
application region 20. The assay system 10 is for use in the detection of an
analyte in
a sample applied to the system. A sample of less than 100p.1 can be applied to
the
region 20 before flowing down a conduit 24 towards the microfluidic separation
system 12. The conduit 24 can be a microchannel and could have a Reynolds
number
of less than 3000, or less than 100 (in the case of a capillary). The sample
flows
passively by capillary action, or actively through the application of a
pressure
differential to the system 10. The assay system IO is arranged so that the
sample
travels uni-directionally.
CA 02549094 2006-06-12
WO 2005/058500 PCT/GB2004/005334
The microfluidic separation system 12 comprises a rnicrofluidic structure 13
defining
a capillary pathway. The structure is arranged to separate sample medium from
cells
provided in the sample by size exclusion. Before the sample passes through the
5 structure 13, the sample is mixed with a cell aggregation agent of the
system to cause
aggregation of cells. The cell aggregation agent can be added to the sample
before
applying the sample to region 20. Alternatively the agent can be provided in
the
region 20, or in the conduit 24. The agent can be provided on at least one
surface of
the region 20 or conduit 24.
The microfluidic structure 13 is best illustrated in Figure 3c and Figure 4b .
As can
been seen the conduit 24 splits into a capillary pathway having a number of
channels
26. Each of the channels 26 is in fluid communication along a longitudinal
side
thereof with a respective further channel in the form of a passage 28, which
has a
depth that is smaller than the depth of the respective channel 26. The depth
of the
passage 28 is configured so that aggregates of cells are unable to pass there
through
whereas the sample medium can, due to size exclusion. Therefore the passage
depth
provides a size exclusion spacing. The channels 26 and passages 28 have an
upper
surface defined by a lid 22 (see Figure 3c).
The passages 28 can be provided with step formations 30 that define a
shallower
depth. This is illustrated in Figure 3d. The provision of step formations 30
provides
for the initial passage depth acting as a pre-filter with the step formations
30 defining
a size exclusion spacing functioning as the main filter separating the sample
medium
from the aggregated cells. The step formations 30 can be in the form of
pillars or
raised surfaces.
The depth of the channels 26 is in the region of 100p.m. The depth of the
passages 28
is in the region of 20pm, with the depth provided by the step formations 30
being in
the region of l0p,m.
CA 02549094 2006-06-12
WO 2005/058500 PCT/GB2004/005334
16
The cell medium passing into the passages 28 is channelled towards a
collecting
region 29 of the capillary pathway in the microfluidic structure 13.
An alternative microfluidic separation system 34 is provided in the assay
system 32
illustrated in Figures 5 and 6. Figure 5a provides a perspective view of the
system 32.
The system 32 includes an interferent zone 36, a pre-treatment zone 38, an
assay
system 40, and a sample application region 42.
The microfluidic separation system 34 has a aggregation agent and also a
microfluidic structure that defines a capillary pathway. The capillary pathway
comprises a first channel 48 in fluid communication with a second channel 50.
A
number of microstructures in the form of pillars 46 are provided between the
channels
48, 50. The pillars 46 define a number of gaps 44. The gaps 44 are of a size
to enable
sample medium to flow there through whereas aggregated cells cannot. The gaps
are
size exclusion spacings. The second channel 50 is provided with an outlet 54
to a pre-
treatment zone 38. As with the system 10, a lid 52 is provided.
The aggregation agent is provided in the first channel 48 and it may be
immobilised
on one or more surfaces of the channel. Alternatively, the aggregation agent
is
contacted with the sample before the sample flows into the capillary pathway
of the
microfluidic structure of the separation system 34.
The channel 48 is deeper than the channel 50. Indeed the channel 48 has a
depth of
100p.m and the channel 50 has a depth of between 10 - 20p,m. The gaps defined
by
the pillars 46 are in the region of IOpm.
As with the microfluidic system 10, the system can work actively (for example
using
a pressure differential or electrol~inetic pumping),or passively (through
capillary
action and surface tension).
CA 02549094 2006-06-12
WO 2005/058500 PCT/GB2004/005334
17
The microfluidic separation systems 1, 12, 34 described above can be used to
separate
plasma fxom red blood cells in whole blood. In this example, a whole blood
sample
after application flows towards the respective microfluidic structures of the
separation
systems 1, 12, 34 . A red blood cell aggregation agent is provided upstream of
the
xnicrofluidic structures of system 1, 12, 34. Alternatively the agent can be
provided in
the microfluidic structure of the system 1, 12, 34 and may be immobilised onto
one or
more surfaces upstream of the size exclusion spacing(s). The microfluidic
structures
of the separation systems 1,12, 34 are configured to separate the plasma from
the
aggregated red blood cells using size exclusion. The size exclusion spacings
defined
in the structures allow the plasma to flow there through.
The assay systems 10, 32 also include homogeneous assay/detection systems
24, 40 to detect and measure analyte in a sample of less than or equal to
50,1. An
example of an assay system is illustrated in Figures 7a and 7b. This system
depends
on a combination of latex agglutination and chemi-luminescent signal. In this
regard,
an analyte molecule brings together two beads producing a cascade of chemical
reactions to greatly amplify the signal such that in principle attomolar
concentrations
of analyte can be detected. The system provides a highly sensitive homogenous
immunoassay that takes place in a highly efficient light capturing detection
chamber.
Incubation of the sample with assay components is achieved through a time-
gated
structure.
The assay illustrated in Figure 7a and 7b is a luminescent oxygen channelling
immunoassay. In more detail, photosensitiser particles (Donor particles)
containing
dissolved phthalocyanine generate singlet oxygen when irradiated with light of
wavelength 680nm, Figure 7a. The singlet oxygen produced has a very short half-
life,
circa 4 microseconds and hence decays rapidly to a ground state. As such it
can only
diffuse to a distance of a few hundred microns from the surface of the
particles before
it decays to ground state. However, it can survive long enough to enter any
paired
adjacent particle, Figure 7b. The paired adjacent particles (Acceptor
particles)
contain a dissolved dye that is activated by the singlet oxygen received to
produce
chemiluminescent emission. This chemiluminescent emission activates further
CA 02549094 2006-06-12
WO 2005/058500 PCT/GB2004/005334
1g
fluorophores contained in the same bead, subsequently causing emission of
light at
520-620 nm. The reagents can be lyophilised in a well with an optimised
geometry
and low optical absorbance to ensure maximum excitation and light capturing
efficiency. The donor and acceptor particles can recognise the analyte through
antibodies provided on the surfaces of the particles, such that the particles
are brought
together by the analyte.
The sensitivity of detection of the assay was determined in two ways, Figure 8
and
Figure 9. A first experiment in a 384 well microtiter plate, Figure 8,
demonstrated the
linear behaviour between the total number of Unibeads and assay output signal.
Unibeads are a pre-conjugated single bead that incorporates both the acceptor
and
donor particles. As such the quantum efficiency of the transfer of the singlet
oxygen
can be neglected due to the proximity of the particles. In this experiment the
total
number of beads was varied from 100 to 100000.
In Figure 9, reducing the volume to that likely to be used in the final chip
configuration, 2~1, the signal can be seen to flattens off as the
concentration is
reduced. This is due to the auto-fluorescence of the unibeads. The minimum
number
of particles that can be reliably detected is approximately 400 in 2~1 of
sample. In
developing the assay, Figure 10, a standard curve obtained from a biotinylated
- DIG
assay using 384 microtiter plate with 25 ~l assay volume was obtained.
A sensitive homogeneous assay has been successfully designed and manufactured
based on a micro fluidic system. The assay has been successfully transferred
from a
microtiter plate to a chip format and has generated a dose response curve
whereby 1.6
x 10-1° Molar of analyze can be detected in 2 ~tl of plasma.
The assay system does not necessarily have to be in a form of a luminescent
oxygen
channelling immuno-assay.
As discussed above the assay systems 10, 32 also comprise an interference zone
14,
36 for solid phase extraction of molecules that can interfere with the binding
CA 02549094 2006-06-12
WO 2005/058500 PCT/GB2004/005334
19
interactions in the detection zones 18, 40, or with a signal generation and
detection.
The interFerent zone 14, 36, may be provided before the sample encounters the
cell
separation system 12, 34, or before the sample flows into the assay detection
zone 25,
41. The zone 14, 36 includes a number of agents to neutralise or remove
molecules
from the sample. The interferent zones 14, 36 can be time gated to ensure that
only
fully treated sample passes into the assay system 18, 40.
Example
A sample of whole blood from a human patient (45% hematocrit) was drawn into
an
EDTA tube and 4~,1 of the blood sample was mixed in a ratio of 1:1 with 2~,1
of Lectin
PHA-E (Sigma) dissolved in phosphate buffer solution (PBS) at a concentration
of 5
mgs/ ml. The sample was subsequently incubated at room temperature for 1
minute to
allow the red-blood cells to agglomerate prior to application to the
microfluidic
separation system described above with reference to Figures 2, 3 and 4.
The conduit 24 has dimensions of 200~.m (width) by 100~,m (height). The system
was prepared from a polycarbonate substrate by injection moulding of a base
substrate
to form the base or Iower part of the device as well as the microfluidic
elements
followed by ultrasonic welding of a one-piece injection moulded substrate to
the base
substrate to form the lid or upper surface of the system. Prior to assembly,
the
substrates were treated with an oxygen plasma to render the surfaces
hydrophilic. The
degree of hydrophilicity of the surfaces was measured and the surface contact
angle
found to be 20 degrees.
Fluid sample (4~.1) was applied to the sample application region 20 and
subsequently
moved towards the microfluidic structure 13. The agglomerated cells were
substantially unable to pass through the microfluidic structure thus resulting
in
separation of the plasma/buffer filtrate from the agglomerated red cells.
CA 02549094 2006-06-12
WO 2005/058500 PCT/GB2004/005334
The total plasma/ buffer extracted was ZOOnI in a time of less than 10
minutes. The
efficiency of the plasma separation was calculated as being 11% of the total
available
plasma.
Preferred features of each aspect of the present invention are as for each
other aspect
jnutatis nauta~zdis.