Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02550080 2006-06-08
WO 2005/058852 PCT/FR2004/003288
Procédé de synthèse de dérivés de cycloalkyl-hydrazines exocycliques et de
dérivés d'hétérocycloalkyl-hydrazines exocycliques
La présente invention concerne un nouveau procédé de synthèse de dérivés de
cycloalkyl-hydrazines exocycliques et de dérivés d'hétérocycloalkyl-hydrazines
exocycliques.
Les dérivés de cycloalkyl-hydrazines et d'hétérocycloalkyl-hydrazines
exocycliques,
notamment la N-aminopipéridine, sont utilisés très fréquemment comme
intermédiaires dans la fabrication de médicaments.
A l'heure actuelle, les méthodes de synthèse de dérivés de cycloalkyl-
hydrazines et
d'hétérocycloalkyl-hydrazines exocycliques décrites dans la littérature
scientifique font
appel à l'urée et aux nitrosamines. Dans le cas de la synthèse de la N-
aminopipéridine
par exemple, une première méthode de synthèse réalisée en trois étapes
consiste à
préparer la 1-pipéridyl urée suivie d'une oxydation par l'hypochlorite de
sodium. La
1-pipéridyl-3-chlorourée formée est ensuite transformée en N-aminopipéridine
sous
l'action d'une solution concentrée en soude (R. Ohme, H. Preuschhof, J. Prakt.
Chem.
312, 349 (1970)). Une seconde méthode consiste en une nitrosation de la
pipéridine
suivie d'une hydrogénation chimique (LiA1H4) ou catalytique (Zn/AcOH) du
dérivé
nitrosé (1-nitrosopipéridine) (Allen &.Hanburys Ltd. (1965), 74, 3693-4). Dans
tous
les cas, le composé nitrosé doit être purifié par distillation. Cette méthode
conduit à
d'assez bons rendements (75%). Cependant, le produit issu de la première étape
doit
être manipulé avec beaucoup de précautions à cause de sa toxicité (composé
hautement
cancérogène), ce qui pose industriellement des problèmes de mise en
exploitation. De
plus, l'utilisation de LiA1H4 impose l'absence de traces d'eau, de réacteurs
étanches et
de solvants anhydres (éther diéthylique), ce qui a pour effet d'augmenter les
risques
d'inflammation du mélange réactionnel.
D'autre part, il est reconnu que pour la préparation des différentes
hydrazines, on fait
souvent appel à la réaction dite de Raschig qui consiste à synthétiser la
monochloramine par réaction de l'ammoniac sur une solution d'hypochlorite de
sodium et ensuite faire réagir la monochloramine formée sur une amine pour
obtenir
l'hydrazine correspondante. Ce procédé nécessite deux étapes distinctes, la
première
CA 02550080 2011-07-06
85279-21
2
réalisée à froid pour la synthèse de la monochloramine et la deuxième réalisée
à chaud,
pendant laquelle est effectuée la synthèse proprement dite de l'hydrazine. Par
ailleurs, la
monochloramine doit se trouver en présence d'un excès suffisant d'amine dans
les
solutions intermédiaires de manière à éviter des réactions secondaires de
dégradation, et
par la suite le procédé exige des quantités très importantes de solutions à
traiter.
Cependant, ce procédé ne peut pas être appliqué pour la préparation de toutes
les alkyl- et
hétéroalkyl-hydrazines exocycliques et surtout pas pour la préparation
d'hydrazines
organiques, thermodégradables à température d'ébullition élevée. En
particulier, le
traitement des solutions de synthèse nécessite l'extraction de l'eau puis de
l'amine, ce qui
1o exige des opérations onéreuses.
Le brevet EP 0 277 267 décrit un procédé de synthèse en continu du N-amino aza-
3
bicyclo [3,3,0] octane, caractérisé en ce que l'on fait réagir une solution
d'hydroxyde
d'ammonium et de chlorure d'ammonium avec une solution aqueuse d'hypochlorite
de
sodium à une température comprise entre -15 C et 7 C en milieu alcalin, et
ensuite, que
l'on fait réagir la monochloramine ainsi formée avec l'aza-3 bicyclo [3,3,0]
octane, en milieu
biphasique dans un réacteur approprié muni d'un agitateur coaxial à ailettes,
à une
température comprise entre 30 C et 90 C et en milieu alcalin, puis que l'on
sépare du
milieu réactionnel l'ammoniac et ensuite l'aza-3 bicyclo [3,3,0] octane qui
n'a pas réagi par
distillation pour le recycler, puis que l'on isole par démixtion une solution
concentrée de
N-amino aza-3 bicyclo [3,3,0] octane par addition d'hydroxyde de sodium au
milieu
réactionnel, et que l'on purifie l'hydrazine ainsi obtenue, si on le désire,
par distillation.
Après formation de N-amino aza-3 bicyclo [3,3,0] octane et refroidissement, la
solution
réactionnelle subit un dégazage pour éliminer l'ammoniac et l'amino-3 bicyclo
[3,3,0]
CA 02550080 2011-12-20
85279-21
2a
octane qui n'a pas réagi est séparé du milieu réactionnel par simple
distillation sous
pression atmosphérique et à une température environ 90 à 100 C. Sous ces
conditions,
l'amine est obtenue sous forme d'une solution aqueuse concentrée à 30% en aza-
3 bicyclo
[3,3,0] octane. Cette solution est recyclée.
RÉSUMÉ DE L'INVENTION
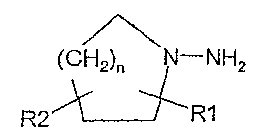
Selon un premier aspect, la présente invention vise un procédé de synthèse de
dérivés de
cycloalkyl-hydrazines et d'hétérocycloalkyl-hydrazines exocycliques de formule
(I)
(CH2)/ õ N-NH2
R2 R1
dans laquelle un des atomes de carbone du cycle est éventuellement remplacé
par un
1o hétéroatome, l'hétéroatome étant un atome d'azote ou d'oxygène, RI et R2,
identiques ou
différents, représentent un atome d'hydrogène ou un radical alkyle en Cl-C6 ou
RI et R2
forment ensemble un radical cycloalkyl en C3-C8 et n vaut 1 à 3, comprenant
les étapes
successives suivantes : a) synthétiser le dérivé de cycloalkyl-hydrazine ou
d'hétérocycloalkyl-hydrazine exocyclique dans un réacteur approprié en faisant
réagir en
milieu alcalin et à une température comprise entre 30 et 60 C une
monochloramine avec
une amine hétérocyclique; b) démixter la solution obtenue suite à l'étape. a)
en une phase
organique et une phase aqueuse par l'ajout d'hydroxyde de sodium anhydre sous
refroidissement afin que la température ne dépasse pas la température
d'ébullition des
composés; et c) le cas échéant, isoler, par distillation de la phase organique
ainsi obtenue,
le dérivé de cycloalkyl-hydrazine ou d'hétérocycloalkyl-hydrazine exocyclique.
CA 02550080 2011-12-20
85279-21
2b
Selon un deuxième aspect, la présente invention vise le procédé tel que décrit
plus haut et
plus bas, dans lequel à l'étape a), le rapport molaire amine
hétérocyclique/monochloramine
est compris entre 4 et 10.
BREVE DESCRIPTION DES FIGURES
Figure 1. illustre un exemple de mise en oeuvre du procédé selon la présente
invention.
Figure 2. illustre un exemple de mise en oeuvre du procédé selon la présente
invention.
Les inventeurs ont maintenant découvert un nouveau procédé de synthèse de
dérivés de
cycloalkyl-hydrazines exocycliques et de dérivés d'hétérocycloalkyl-hydrazines
exocycliques, notamment de la N-aminopipéridine. Ce procédé mis en oeuvre en
CA 02550080 2006-06-08
WO 2005/058852 PCT/FR2004/003288
3
continu est en partie basé sur une transposition du procédé Raschig, et il
consiste à
préparer la monochloramine par action de l'hypochlorite de sodium sur
l'ammoniac à
base température, et ensuite à faire agir la monochloramine ainsi produite sur
une
amine hétérocyclique en milieu homogène ou, selon la température, en milieu
hétérogène, puis à extraire l'hydrazine formée. L'amine de départ est
recyclée, puis le
cas échéant réinjectée directement sur la monochloramine sans aucun traitement
supplémentaire.
Dans le cadre de la présente invention, on pourra désigner, dans un souci de
simplification, le ou les dérivés de cycloalkyl-hydrazines exocycliques et le
ou les
dérivés d'hétérocycloalkyl-hydrazines exocycliques par le terme hydrazine(s)
.
Au sens de la présente invention, l'expression dérivé de cycloalkyl-
hydrazine ou
d'hétérocycloalkyl-hydrazines exocyclique doit se lire dérivé de
cycloalkyl-
hydrazine exocyclique ou dérivé d'hétérocycloalkyl-hydrazine exocyclique .
D'une manière similaire, on pourra désigner la(les) amine(s) hétérocyclique(s)
par le
terme amine(s) .
La présente invention concerne ainsi un procédé de synthèse de dérivés de
cycloalkyl-
hydrazines exocycliques et de dérivés d'hétérocycloalkyl-hydrazines
exocycliques
caractérisé en ce qu'il comprend les étapes successives suivantes :
a) synthétiser le dérivé de cycloalkyl-hydrazine ou d'hétérocycloalkyl-
hydrazine exocyclique dans un réacteur approprié en faisant réagir en milieu
alcalin et à une température comprise entre 30 et 60 C une monochloramine
avec une amine hétérocyclique ; puis
b) démâter la solution obtenue suite à l'étape a) en une phase organique et
une
phase aqueuse par l'ajout d'hydroxyde de sodium anhydre sous refroidissement
afin que la température ne dépasse pas la température d'ébullition des
composés ; et
c) le cas échéant, isoler, par distillation de la phase organique ainsi
obtenue, le
dérivé de cycloalkyl-hydrazine ou d'hétérocycloalkyl-hydrazine exocyclique.
Le dérivé de cycloalkyl-hydrazine exocyclique ou d'hétérocycloalkyl-hydrazine
exocyclique est avantageusement de formule (I)
CA 02550080 2006-06-08
WO 2005/058852 PCT/FR2004/003288
4
(CH2)n N-NH
R2 R1
dans laquelle un des atomes de carbone du cycle est éventuellement remplacé
par un
hétéroatome choisi parmi un atome d'azote ou d'oxygène, R1 et R2, identiques
ou
différents, représentent un atome d'hydrogène ou un radical alkyle en C1-C6 ou
Rl et
R2 forment ensemble un cycloalkyle en C3-C8 et n vaut 1 à 3.
Le dérivé de cycloalkyl-hydrazine ou d'hétérocycloalkyl-hydrazine exocyclique
est
encore plus avantageusement choisi dans le groupe constitué par la N-
aminopipéridine,
la N-aminomorpholine, la N-amino-2,6-diméthyl-pipéridine, la N-
aminopyrrolidine, la
N-aminoazépine, la N-amino-4-méthyl-pipérazine.
Le réacteur de l'étape a) est avantageusement placé sous atmosphère inerte,
notamment sous balayage d'argon ou d'azote. Ledit réacteur approprié de
l'étape a) est
avantageusement un réacteur tubulaire agité. Le réacteur tubulaire permet
d'éviter un
contact entre l'hydrazine naissante et la monochloramine et ainsi il permet
d'éviter une
réaction d'oxydo-réduction entre ces deux réactifs. Le front réactionnel se
déplace le
long du tube et l'hydrazine n'est plus en contact avec la monochloramine
injectée à la
base du réacteur.
Selon une variante avantageuse de l'invention, la concentration en ions
hydroxyles
dans le milieu de réaction de l'étape a) est comprise entre 0,3 et 0,8 mol.1-
1.
Le rapport des concentrations molaires de l'amine hétérocyclique sur la
monochloramine doit avantageusement être supérieur ou égal à 4 et inférieur ou
égal à
10. Le temps de réaction est variable est dépend de la température à laquelle
s'effectue
la réaction et du rapport des concentrations des réactifs. Par exemple, dans
le cas de la
synthèse de la N-aminopipéridine et dans la gamme des rapports de
concentrations
donnée, le temps de réaction est de l'ordre de 20 secondes à 2 minutes à 25 C
et de
l'ordre de 4 secondes à 30 secondes à 60 C.
Selon un mode avantageux de la présente invention, la monochloramine est
alcalinisée
préalablement à l'étape a) dans un mélangeur par ajout d'une solution d'une
base forte
telle que l'hydroxyde de sodium de telle sorte que le titre massique en
hydroxyde de
sodium soit compris entre 2 et 6%. Ledit mélangeur est avantageusement
maintenu à
une température comprise entre -10 et 5 C.
CA 02550080 2006-06-08
WO 2005/058852 PCT/FR2004/003288
La réaction de la monochloramine avec l'amine hétérocyclique s'effectue ainsi
avantageusement en présence d'une solution aqueuse d'hydroxyde de sodium à
chaud.
A la fin de la réaction de synthèse du dérivé de cycloalkyl-hydrazine ou
d'hétérocycloalkyl-hydrazine exocyclique, c'est-à-dire en sortie du réacteur
de l'étape
5 a), la concentration en hydroxyde de sodium dans le milieu réactionnel est
d'environ
0,3 mol.L-1. La concentration en soude ne doit pas être trop élevée sinon le
mélange
réactionnel risque de démixter par relargage. En cas de relargage, il faudrait
alors faire
intervenir un réacteur du type réacteur piston agité.
Lors de la réaction de synthèse de l'hydrazine, de l'acide chlorhydrique est
également
formé, or il faut éviter toute protonation locale de l'amine au moment du
mélange afin
d'éviter la formation d'une chloramine substituée. Par exemple, dans le cas de
la
synthèse de la N-aminopipéridine, la pipéridine protonée par l'acide
chlorhydrique
(pipéridinium) peut réagir avec la monochloramine pour former la 1-
chloropipéridine,
qui peut alors réagir avec les ions hydroxyles pour former la
2,3,4,5-tetrahydropyridine, qui risque ensuite de se trimériser.
L'alcalinisation de la
monochloramine, c'est-à-dire l'ajout d'une base forte telle que la soude,
permet ainsi
de neutraliser l'acide formé. La quantité de base forte ajoutée doit être
suffisante pour
neutraliser tout l'acide formé. De plus, la vitesse de formation de
1'hydrazine augmente
avec l'alcalinité du milieu, ce qui n'est pas le cas pour la vitesse des
réactions de
dégradation, telles que par exemple l'oxydation de 1'hydrazine naissante par
la
chloranine.
Lors de l'étape b), la quantité d'hydroxyde de sodium anhydre ajoutée est
telle que le
titre massique en hydroxyde de sodium soit compris entre 10 et 35%, de
préférence
environ égal à 30%. Dans ces conditions, le milieu démixte en deux phases dont
l'une
concentre le dérivé de cycloalkyl-hydrazine ou d'hétérocycloalkyl-hydrazine
exocyclique formé dans la phase légère (phase organique). Ce traitement à
l'hydroxyde
de sodium permet par démixtion d'éliminer l'eau présente dans le milieu
réactionnel et
d'extraire les sels et le cas échéant l'ammoniac dans la phase inférieure
(phase
aqueuse).
Dans le cas de la synthèse de la N-aminopipéridine par exemple, la température
du
milieu de démixtion de l'étape b) ne doit pas dépasser 80 C.
CA 02550080 2011-12-20
85279-21
6
Selon une première variante avantageuse de l'invention, l'amine hétérocyclique
est
introduite, à l'étape a), sous forme d'une amine hétérocyclique anhydre.
La monochloramine, avantageusement alcalinisée, et l'amine hétérocyclique
sont de manière avantageuse introduites simultanément dans le réacteur. Les
débits
d'addition de l'amine hétérocyclique et de la monochloramine sont tels que le
rapport
des concentrations molaires de l'amine hétérocyclique, sur la monochloramine
soit avantageusement compris entre 4 et 10, les bornes pouvant être incluses.
Une
partie de la réaction de synthèse du dérivé de cycloalkyl-hydrazine et
d'hétérocycloalkyl-hydrazine exocyclique peut être effectuée en milieu
hétérogène.
L'ajout d'hydroxyde de sodium anhydre lors de l'étape b), avantageusement de
telle
sorte que le titre massique en hydroxyde de sodium soit compris entre 15 et 35
,
permet de démâter le milieu en deux phases dont l'une, la phase supérieure ou
la
phase organique, concentre la quasi-totalité des organiques, à savoir le
dérivé de
cycloalkyl-hydrazines ou d'hétérocycloalkyl-hydrazine exocyclique et l'amine
hétérocyclique.
L'avantage de ce traitement permet, en une seule étape, d'éliminer 80.à 90% en
poids
de l'eau présente dans le milieu réactionnel, selon le caractère organique
(nombre
d'atomes de carbones) des molécules d'amine et d'hydrazine, et d'extraire
l'ammoniac
avec les sels dans la phase inférieure (phase aqueuse).
L'étape c) comprend alors avantageusement les étapes successives suivantes :
i) isoler l'amine hétérocyclique qui n'a pas réagi et une solution concentrée
de
dérivé de cycloalkyl-hydrazine ou d'hétérocycloalkyl-hydrazine exocyclique
par distillation de la phase organique obtenue suite à l'étape b) ; puis
ii) le cas échéant, rectifier par distillation sous pression réduite ladite
solution
concentrée du dérivé de cycloalkyl-hydrazine et d'hétérocycloalkyl-hydrazine
exocyclique.
Lors de l'étape i), la distillation s'effectue avantageusement dans une
colonne de
distillation unique sous pression atmosphérique ou réduite, . en fonction de
la
température d'ébullition de l'amine de départ. Dans un premier temps, on
recueille en
tête de colonne une solution concentrée d'amine hétérocyclique ou le cas
échéant une
solution azéotropique eau-amine hétérocyclique jusqu'à épuisement de l'eau,
puis
l'amine hétérocyclique anhydre.
CA 02550080 2006-06-08
WO 2005/058852 PCT/FR2004/003288
7
L'amine hétérocyclique anhydre ainsi récupérée peut être réinjectée
directement dans
le réacteur de l'étape a) où a lieu la synthèse de l'hydrazine. L'amine
hétérocyclique
obtenue sous forme de solution concentrée ou de solution azéotropique avec
l'eau peut
être récupérée, puis éventuellement réinjectée après un traitement approprié.
Le cas échéant, une rectification sous pression réduite, avantageusement vers
115 min de Hg, du produit obtenu en pied de colonne à l'étape i) permet de
recueillir le
dérivé de cycloalkyl-hydrazine ou d'hétérocycloalkyl-hydrazine exocyclique
avec un
titre supérieur à 99%, avantageusement supérieur à 99,9%.
Ledit dérivé de cycloalkyl-hydrazine ou d'hétérocycloalkyl-hydrazine
exocyclique doit
ensuite être stocké sous atmosphère inerte, tel que sous argon par exemple, de
manière
à éviter toute réaction d'oxydation par l'oxygène.
Cette variante de l'invention est particulièrement avantageuse dans le cadre
d'une
préparation en batch du dérivé de cycloalkyl-hydrazine exocyclique ou
d'hétérocycloalkyl-hydrazine exocyclique.
Selon une seconde variante avantageuse de l'invention, l'amine hétérocyclique
est
introduite, à l'étape a), sous forme d'une solution aqueuse concentrée d'amine
hétérocyclique ou d'une solution azéotropique eau-amine hétérocyclique.
L'étape a)
s'effectue alors en milieu homogène ou hétérogène (dans le cas d'une amine
très
lourde).
En fonction de l'amine considérée, la solution aqueuse concentrée d'amine
hétérocyclique peut être sous la forme d'un azéotrope eau-amine
hétérocyclique.
La monochloramine, avantageusement alcalinisée, et la solution d'amine
hétérocyclique sont de manière avantageuse introduites simultanément dans le
réacteur. Les débits d'addition de la solution aqueuse concentrée d'amine
hétérocyclique, ou de la solution azéotropique eau-amine hétérocyclique, et de
la
monochloramine sont tels que le rapport des concentrations molaires de la
solution
d'amine hétérocyclique sur la monochloramine soit avantageusement compris
entre 4
et 10, les bornes pouvant être incluses.
Lors de l'étape a), le mélange réactionnel peut le cas échéant subir une ou
plusieurs
étape(s) de dégazage pour éliminer l'ammoniac contenu dans ledit mélange.
CA 02550080 2006-06-08
WO 2005/058852 PCT/FR2004/003288
8
Dans cette seconde variante de l'invention, suite à l'étape a) et
préalablement à l'étape
b), le procédé comprend les étapes suivantes :
P) éliminer l'ammoniac présent dans la solution obtenue suite à l'étape a) par
stripping ; puis
ii') isoler une solution comprenant le dérivé de cycloalkyl-hydrazine ou
d'hétérocycloalkyl-hydrazine exocyclique formé et une solution concentrée
d'amine hétérocyclique qui n'a pas réagi, ou le cas échéant une solution
azéotropique eau-amine hétérocyclique qui n'a pas réagi, par distillation de
la
solution obtenue suite à l'étape i') sous pression atmosphérique ou réduite à
une température comprise entre 50 et 180 C ; et
iii') réinjecter dans le réacteur de l'étape a) ladite solution aqueuse
concentrée
ou azéotropique d'amine hétérocyclique obtenue suite à l'étape ii').
Au sens de la présente invention, on entend par l'expression stripping
l'élimination
d'un produit très volatil, en l'occurrence l'ammoniac, par simple chauffage du
mélange.
La solution réactionnelle contenant l'hydrazine, récupérée à l'étape ii'), est
ensuite
traitée par ajout d'une base forte, telle que la soude (étape b)). Cette
opération permet
la séparation du dérivé de cycloalkyl-hydrazine ou d'hétérocycloalkyl-
hydrazine
exocyclique dans une phase organique, ayant un titre compris entre 70 et 90%
en
hydrazine, selon le caractère organique de la molécule d'hydrazine. Suivant
les
spécifications d'emploi, la solution concentrée de l'hydrazine ainsi obtenue
peut être
utilisée directement ou distillée sous pression réduite (étape c)).
Le recyclage de l'amine, lors de l'étape de distillation ii'), s'effectue sans
entraînement
de l'hydrazine et à une température inférieure au point d'ébullition de
l'amine, ce qui
évite sa thermo-dégradation. Compte tenu de l'absence de traces d'hydrazine
dans la
solution aqueuse contenant l'amine hétérocyclique, éventuellement sous la
fonne d'un
azéotrope, celle-ci peut être injectée, sans traitement supplémentaire,
directement au
niveau du réacteur de l'étape a), où se forme l'hydrazine.
Cette variante de l'invention est particulièrement avantageuse dans le cadre
d'une
préparation en continu du dérivé de cycloalkyl-hydrazine exocyclique ou
d'hétérocycloalkyl-hydrazine exocyclique.
CA 02550080 2006-06-08
WO 2005/058852 PCT/FR2004/003288
9
Le procédé selon la présente invention permet donc non seulement la synthèse
du
dérivé de cycloalkyl-hydrazine ou d'hétérocycloalkyl-hydrazine exocyclique en
continu, sans formation d'aucun intermédiaire toxique, mais il permet aussi
l'obtention
de ladite hydrazine avec un coût peu élevé.
En effet, la synthèse classique de Raschig nécessite en général un grand excès
d'amine,
ce qui constitue un inconvénient considérable quand les amines utilisées comme
matière première pour la préparation des hydrazines correspondantes ont un
coût très
élevé. Le procédé de la présente invention, grâce à la récupération et au
recyclage
d'une solution concentrée d'amine hétérocyclique, éventuellement sous la forme
d'un
azéotrope, permet l'obtention du dérivé de cycloalkyl-hydrazine ou
d'hétérocycloalkyl-hydrazine exocyclique correspondant avec un coût très peu
élevé
par rapport aux autres procédés connus. L'isolement de l'amine sous forme
d'une
solution aqueuse, éventuellement azéotropique, à relativement basse
température
constitue aussi une autre originalité ainsi qu'un avantage économique
considérable du
procédé selon l'invention.
La monochloramine introduite à l'étape a) est avantageusement préparée selon
un
procédé comprenant les étapes successives suivantes :
a) préparer une solution aqueuse d'hypochlorite de sodium ayant un degré
chlorométrique compris entre 36 et 100 éventuellement par dilution d'une
solution d'hypochlorite ayant un degré chlorométrique compris entre 100 et
120 ; puis
(3) faire réagir une solution d'hydroxyde d'ammonium et de chlorure
d'ammonium avec la solution aqueuse d'hypochlorite de sodium obtenue
suite à l'étape cx), en milieu faiblement alcalin, à une température comprise
entre -15 et -7 C, pour former ladite monochloramine.
Au sens de la présente invention, on entend par l'expression milieu
faiblement
alcalin un milieu dont la valeur de pH est d'environ 10 1.
Le rapport molaire solution d'hydroxyde d'ammonium et de chlorure
d'ammonium/solution aqueuse d'hypochlorite de sodium est avantageusement
compris
entre 2,5 et 3, les bornes étant incluses.
CA 02550080 2006-06-08
WO 2005/058852 PCT/FR2004/003288
Le rapport molaire chlorure d'ammonium/hydroxyde d'ammonium est
avantageusement compris entre 0,1 et 1,75, les bornes étant incluses, plus
avantageusement il est d'environ 0,65.
Dans le cas où le réactif chloré mis en oeuvre à l'étape a) est obtenu par
dilution d'une
5 solution d'hypochlorite haut titre à 100-120 chlorométrique, cette dilution
présente
l'avantage de diminuer de 40% la teneur en chlorure de sodium. Ce traitement,
favorable pour l'environnement, permet un refroidissement de la solution d'eau
de
Javel, sans risque de cristallisation jusqu'à -15 C.
10 L'exemple 1 donne, à titre non limitatif, une description détaillée de la
mise en oeuvre
du procédé de l'invention, procédé dont le schéma de principe est représenté
figure 1.
Signification des abréviations utilisées :
RI: réacteur 1 M : mélangeur R2 : réacteur 2
CD I : colonne de distillation n 1
CD2: colonne de distillation n 2
CD3 : colonne de distillation n 3
1 : solution de pipéridine
2: N-amino-pipéridine
3 : Solution eau+NaCI+NaOH
4: résidus
L'exemple 2 donne, à titre non limitatif, une description détaillée de la mise
en oeuvre
du procédé de l'invention, procédé dont le schéma de principe est représenté
figure 2.
Signification des abréviations utilisées :
Ri : réacteur 1 M : mélangeur R2: réacteur 2
CD l' : colonne de distillation n l'
CD2' : colonne de distillation n 2'
V: pipéridine anhydre
1" : solution de pipéridine à 66% en poids (azéotrope eau-pipéridine)
2': N-ainino-pipéridine
Y: Solution eau+NH3+NaCI+NaOH
CA 02550080 2006-06-08
WO 2005/058852 PCT/FR2004/003288
11
EXEMPLE 1 : Préparation de la N-amino pipéridine en continu
Toutes les quantités indiquées correspondent à une unité en régime et sont
rapportées à
un litre d'hypochlorite injecté.
Un litre d'une solution d'hypochlorite de sodium élaborée par dilution de 50%
d'une
solution d'hypochlorite de haut titre (100 à 120 chlorométrique, soit
[NaOCI]=2,14
mol.L"1 ; [NaCl]=0,85 mol.L-1) et un litre de solution ayant une concentration
en
ammoniac de 3,60 mol.L"1 et en chlorure d'ammonium de 2,38 mol.L-1 sont
introduits
en continu dans un réacteur agité (RI) à raison de 5 mL.min 1 chacun (soit 6
ghnin de
solution d'hypochlorite à 48 chlorométrique et 5,05 g/min du mélange
ammoniacal
NH3 + NH4C1).
La température au sein du réacteur est maintenue entre -8 C et -11 C, et le pH
de la
réaction est voisin de 10. A la sortie de R1, on obtient une solution de
monochloramine
de titre supérieur à 1 mol.L-1, ce qui correspond à un rendement proche de
100% par
rapport à l'hypochlorite de sodium.
A la sortie de R1, la solution de monochloramine obtenue ci-dessus (2 litres)
est
alcalinisée par introduction en continu d'une solution concentrée d'hydroxyde
de
sodium (0,37 litre à 30% en poids) au sein d'un mélangeur M à double enveloppe
maintenu à basse température entre -9 C et -11 C. L'homogénéisation est
assurée par
entraînement magnétique.
La synthèse de la N-aminopipéridine est effectuée en milieu monophasique dans
un
réacteur tubulaire (R2) agité, sous balayage d'argon ou d'azote.
Le mélange NH2CI / NaOH obtenu (2,37 litres) et la solution de pipéridine
(2,36 litres
à 66% en poids) sont introduits simultanément (sous argon ou azote) en continu
dans le
réacteur R2 avec un débit adéquat pour avoir un rapport molaire pipéridine sur
monochloramine environ égal à 8 et un titre en hydroxyde de sodium dans le
milieu
réactionnel en fin de réaction égal à 0,3 mol.L-1. La température de la
réaction est
maintenue à environ 55 C. Après 30 secondes de réaction, le mélange
réactionnel subit
ensuite une opération de dégazage pour éliminer l'ammoniac contenu dans la
solution.
La solution réactionnelle est tout d'abord débarrassée de l'ammoniac par
stripping
(colonne de distillation CD I, on récupère en tête de colonne environ 62 g
d'ammoniac)
puis environ 4,6 kg de la solution débarrassée de l'ammoniac sont distillés à
92,2 C
CA 02550080 2006-06-08
WO 2005/058852 PCT/FR2004/003288
12
sous pression atmosphérique (colonne de distillation CD2) pour éliminer
l'amine qui
n'a pas réagi, la pipéridine. Ainsi, après cette étape de distillation, on
obtient en tête de
colonne la pipéridine sous la forme d'une solution aqueuse de composition
d'environ
66% en poids en amine (environ 2 kg). Cette solution est ensuite recyclée et
ré-injectée
immédiatement au sein de R2, sans traitement supplémentaire (figure 1 : flèche
en
pointillé).
Après séparation de la pipéridine, la solution réactionnelle contenant
l'hydrazine
(récupérée en bas de colonne CD2, environ 2,7 kg) est traitée par addition
d'hydroxyde
de sodium solide sous refroidissement et sous balayage d'argon, pour séparer
la N-
aminopipéridine dans une phase organique titrant près de 70 à 80% en hydrazine
à
80 C. Le titre massique de soude anhydre injectée se situe de préférence entre
15 et
30% en poids. On récupère ainsi ladite phase organique titrant près de 92% en
hydrazine et une phase aqueuse comprenant l'eau et les sels (NaCI, NaOH).
Suivant les
spécifications d'emploi, la solution concentrée de l'hydrazine (phase
organique) peut
ensuite être utilisée directement ou distillée sous pression réduite (colonne
de
distillation CD3).
Après distillation sous pression réduite, on obtient la N-aminopipéridine avec
un degré
de pureté supérieur à 99,5%.
Le rendement en hydrazine par rapport à la pipéridine consommée est supérieur
à 92%.
EXEMPLE 2: Préparation de la N-amino pipéridine en batch
Toutes les quantités indiquées correspondent à une unité en régime et sont
rapportées à
un litre d'hypochlorite injecté.
Le procédé est caractérisé en ce que l'on fait réagir une solution d'hydroxyde
d'ainrnonium et de chlorure d'ammonium ([NH3] = 3,60 mol.-L-1 ; [NH4C1] = 2,38
mol.L"' ; 5 mLhnin) avec une solution aqueuse d'hypochlorite de sodium (débit
de 5
mLhnin) à une température comprise entre -15 C et -7 C en milieu alcalin dans
un
réacteur agité continu R1.
Le fluide réactionnel issu de R1 (2 litres) de titre supérieur à 1 mol.L-' en
monochloramine est introduit dans un mélangeur M alimenté en continu par une
CA 02550080 2006-06-08
WO 2005/058852 PCT/FR2004/003288
13
solution d'hydroxyde de sodium à 30% (débit de 1,75 mL/min). Un enveloppe
thermostatique permet de fixer la température, au sein du mélangeur, à -10 C.
La synthèse de la N-aminopipéridine est effectuée au moyen d'un agitateur
tubulaire
R2 agité et sous balayage d'argon. La monochloramine alcalinisée (2,35
litres), issue
de l'enceinte du mélange M, et le réactif aminé sont introduits simultanément
à la base
du réacteur au moyen de pompes doseuses. On ajoute 1,69 litres, soit 1,455 kg
car la
densité est de 0,861, de pipéridine anhydre. Le débit de la pipéridine anhydre
est de
8,47 mL/min et une partie de la réaction est effectuée en milieu hétérogène à
55 C. La
concentration finale en NaOH au sortir de R2 est de 0,3 inol.L-1.
La présente variante est caractérisée en ce que l'on ajoute, à la liqueur
réactionnelle
homogène (4,04 litres), une quantité en hydroxyde de sodium comprise entre 13
et
30% sous refroidissement de manière que la température ne dépasse pas 60 C.
Dans
ces conditions, on obtient deux phases dont l'une, légère (1,8 kg), contient
la totalité
des organiques, c'est-à-dire la N-aminopipéridine et la pipéridine en excès
qui titre
environ entre 15 et 20% en poids. Ce traitement permet ainsi d'éliminer entre
80 et
85% de l'eau présente dans les solutions de synthèse.
L'obtention de la N-aminopipéridine nécessite ensuite deux étapes successives
:
- récupération de la pipéridine qui n'a pas réagi par distillation de la phase
organique à la pression atmosphérique sous argon. On récupère, dans un premier
temps, environ 1 kg d'une solution concentrée d'amine à 66% en poids (l") à
une
température de 92,2 C jusqu'à épuisement de l'eau puis, environ 600 g de
pipéridine
anhydre (1') à une température de 105 C (colonne de distillation CD l').
- Rectification sous 115 mm de Hg de la solution obtenue en pied de colonne
(colonne de distillation CD2').
Après distillation sous pression réduite, on obtient la N-aminopipéridine avec
un degré
de pureté supérieur à 99,5%.
Le rendement en hydrazine par rapport à la pipéridine consommée est supérieur
à 90%.