Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-1-
N-SUBSTITUTED PIPERIDINE AND PIPERAZINE DERIVATIVES
BACKGROUND OF THE INVENTION
This invention relates to N-substituted piperidine and piperazine derivatives,
pharmaceutical compositions containing them and their use for the treatment of
schizophrenia and other central nervous system (CNS) disorders or conditions.
The
N-substituted piperidine and piperazine derivatives of this invention exhibit
activity as
antagonists of dopamine D2 receptors and of serotonin 2A (5HT2A) receptors.
Other heterocyclic piperazine derivatives that are useful for the treatment of
schizophrenia are referred to in United States patent 5,350,747, which issued
on
September 27, 1994, and in United States patent 6,127,357, which issued on
October 3, 2000. These patents are incorporated herein by reference in their
entirety.
Other piperazine and piperidine derivatives that have been stated to be useful
as antipsychotic agents are those referred to in PCT patent publication WO
93/04684, which published on March 18, 1993, and European patent application
EP
402644A, which was published on December 19, 1990. These patent applications
are incorporated herein by reference in their entirety.
SUMMARY OF THE INVENTION
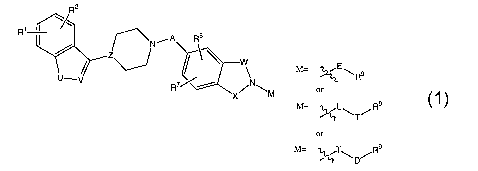
The present invention relates to compounds of formula 1
R2
R~ /~' a
I ~NiA
W
M= E\Rs
RW/ X~N~M or
M= ~L\ /Rs
T
or
M= T R9
wherein
U is sulfur, oxygen, SO, S02, CH2 or NR3;
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-2-
V is nitrogen or carbon;
Z is nitrogen or carbon;
A is -(CH2)m0-; -(CH2)mNR4-; or -(CH2)mC(R5R6)-, wherein R5 and R6 are
independently selected from hydrogen, (C1-C4) alkyl optionally substituted
with from
one to three fluorine atoms, (Ci-C4) alkoxy optionally substituted with from
one to
three fluorine atoms, hydroxy, and aminoalkyl; or R5 and R6 together form a
carbonyl, and wherein m is an integer from one to four;
R' and R2 are independently selected from hydrogen, (C~-C4) alkyl optionally
substituted with from one to three fluorine atoms, (Ci-C4) alkoxy optionally
substituted with from one to three fluorine atoms, halogen, nitro, cyano,
amino, (C1-
C4) alkylamino and di-(C~-C4) alkylamino;
R3 and R4 are independently selected from hydrogen, (C1-C4) alkyl optionally
substituted with from one to three fluorine atoms and (C1-C4) alkoxy
optionally
substituted with from one to three fluorine atoms;
or, when U is NR3, one of Ri and R2 can form, together with the carbon to
which it is attached, and together with R3 and the nitrogen to which it is
attached, a
heterocyclic ring containing from four to seven ring members of which from one
to
three ring members can be heteroatoms selected from nitrogen, oxygen and
sulfur,
and of which the remaining ring members are carbon, with the proviso that when
R3
forms a ring with one of R1 and R2, the other of R' and R2 is absent.
X is -[C(R")(R'2)]o , wherein Rii and R'2 are independently selected from
hydrogen and (C,-Ca) alkyl optionally substituted with from one to three
fluorine
atoms, and wherein o is an integer from zero to three, with the proviso that
when W
is absent, o must be greater than or equal to two;
W is -~C(R13)(R14)]p-, wherein R'3 and R14 are independently selected from
hydrogen and (C1-C4) alkyl optionally substituted with from one to three
fluorine
atoms, and wherein p is an integer from zero to four, with the proviso that
when X is
absent, p is greater than or equal to two;
R' and R$ are independently selected from halo, Ri and -ORi; or R', when
attached to a carbon adjacent to one of the carbon atoms shared by both the
phenyl
ring to which R' is attached and the ring containing W, N and X, forms,
together with
a carbon atom of X or a carbon atom of W, a saturated carbocyclic ring
containing
from three to six carbon atoms;
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-3-
R9 is selected from phenyl, phenoxy, benzyloxy, and phenylamino, wherein
the phenyl moieties of said phenyl, phenoxy, benzyloxy, and phenylamino are
optionally substituted with from one to three substituents independently
selected
from halo, (C1-C3) alkyl optionally substituted with from one to three
fluorine atoms,
(C1-C3) alkoxy optionally substituted with from one to three fluorine atoms,
nitro,
cyano, amino, and (C~-C3) alkylamino; or
R9 is a pyrrolidine, piperidine or morpholine ring wherein the point of
attachment to D, T or E is the ring nitrogen, and wherein said pyrrolidine,
piperidine
or morpholine ring can be optionally substituted with one or two substituents
independently selected from methyl, amino, (Ci-C4) alkylamino, and di-(C~-C4)
alkylamino; or
R9 is a furan, thiophene, or pyrazole ring optionally substituted with one or
two
(C1-C4) alkyl groups; or
R9 is (C1-C6) straight or branched alkyl or (C3-Cs) cycloalkyl, wherein said
straight, branched and cyclic alkyl moieties are be optionally substituted
with from
one to three halo atoms or (Ci-C4) alkoxy optionally substituted with from one
to
three fluorine atoms; or
R9 is halogen, vitro, cyano, amino, (Ci-C4) alkylamino, di-(C1-C4) alkylamino
or OR', wherein the alkyl moieties of (C1-C4) alkylamino and di-(Cy-C4)
alkylamino
are optionally substituted with an amino, (C1-C4) alkylamino, or di-(C1-C4)
alkylamino
group;
E is -C(O)-, -S(O)- or -S02-;
T is -C(O)- or -C02-;
L is -(CH2)" wherein n is an integer from zero to three;
D is -(CHR'°)q , wherein q is an integer from one to three, or
NR'o;
Ri° is hydrogen or straight or branched (Ci-C3) alkyl;
and pharmaceutically acceptable salts thereof.
Specific embodiments of the invention include compounds of the formula 1
wherein: U is sulfur;
U is sulfur, oxygen or NR3;
V is nitrogen;
Z is nitrogen;
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-4-
A is -(CH2)mC(R5R6)-, wherein R5 and R6 are hydrogen and m is one or
two;
A is ethylene or propylene;
R' and R2 are independently selected from hydrogen, (C1-C4) alkyl,
(Ci-C4) alkoxy and halogen;
R' and R2 are hydrogen;
X is absent;
W is [C(R'3)(R~4)]p -, wherein R13 and R'4 are independently selected
from hydrogen and (C1-C4) alkyl, and wherein p is two to four;
W is ethylene or propylene;
R' and R$ are independently selected. from hydrogen, chloro and
methyl;
R8 is chloro or methyl;
M is C(O)R9 and R9 is (C1-C4) alkyl;
M is ER9, E is -C(O)- and R9 is (Ci-C4) alkyl;
M is ER9, E is -C(O)- and R9 is optionally substituted phenyl or
optionally substituted phenoxy; or
M is TDR9, T is -C(Q)- and R9 is optionally substituted phenyl or
optionally substituted phenoxy.
Examples of specific embodiments of this invention include the following
compounds and their pharmaceutically acceptable salts:
1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-6-chloro-3,3-
dimethyl-
2,3-dihydro-indol-1-yl}-ethanone;
1-{5-[2-(4-Benzo[d]isoxazol-3-yl-piperazin-1-yl)-ethyl]-6-chloro-3,3-dimethyl-
2,3-dihydro-indol-1-yl}-ethanone;
1-~7-[2-(4-Benzo[d]isoxazol-3-yl-piperazin-1-yl)-ethyl]-5,5-dimethyl-2,3,4,5-
tetrahydro-benzo[b]azepin-1-yl}-ethanone;
1-{5-[3-(4-Benzo[d]isothiazol-3-yl-piperazi n-1-yl)-propyl]-3,3-diethyl-2,3-
dihydro-indol-1-yl}-ethanone;
1-{5-[3-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-propyl]-6-chloro-3,3-
dimethyl-2,3-dihydro-indol-1-yl}-ethanone;
1-{6-[3-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-propyl]-3,3-dimethyl-3,4-
dihydro-2H-quinolin-1-yl}-ethanone;
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-5-
1-{6-[3-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-propyl]-7-fluoro-4,4-
dimethyl-
3,4-dihydro-2H-quinolin-1-yl}-ethanone;
1-{6-[3-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-propyl]-4,4-dimethyl-3,4-
dihydro-2H-quinolin-1-yl}-ethanone;
1-(7-{2-[4-(7-Fluoro-benzo[d]isothiazol-3-yl)-piperazin-1-yl]-ethyl}-2,3,4,5-
tetrahydro-benzo[b]azepin-1-yl)-ethanone;
1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-6-chloro-2,3-dihydro-
indol-1-yl}-2-pyrrolidin-1-yl-ethanone;
3-(4-{2-[ 1-(4, 5-D i hyd ro-oxazo I-2-yl)-2, 3-d i hyd ro-1 H-i nd o I-5-yl]-
ethyl}-p i p a razi n-
1-yl)-benzo[d]isothiazole;
1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-1,3-dihydro-isoindol-
2-
yl}-propan-1-one;
1-{5-[3-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-propyl]-1,3-dihydro-
isoindol-2-
yl}-2,2-dimethyl-propan-1-one;
1-{5-[3-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-propyl]-1,3-dihydro-
isoindol-2-
yl}-2-(3-dimethylamino-pyrrolidin-1-yl)-ethanone;
1-{6-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-3,4-dihydro-2H-
quinolin-1-yl}-ethanone;
1-{7-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-.yl)-ethyl]-2,3,4,5-tetrahydro-
benzo[b]azepin-1-yl}-propan-1-one;
7-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-1-p ropyl-2,3,4,5-
tetrahydro-1 H-benzo[b]azepine; and
1-{7-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3,4,5-tetrahydro-
benzo[b]azepin-1-yl}-2-methyl-propan-1-one.
DETAILED DESCRIPTION OF THE INVENTION
The term "alkyl", unless otherwise indicated, includes saturated monovalent
hydrocarbon radicals having straight, branched or cyclic moieties or
combinations
thereof. Examples of "alkyl" groups include, but are not limited to, methyl,
ethyl,
propyl, isopropyl, butyl, iso- sec- and tert-butyl, pentyl, hexyl, heptyl, 3-
ethylbutyl,
cyclopropyf, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, norbornyl, and
the like.
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-6-
The term "alkoxy", unless otherwise indicated, means "alkyl-O=', wherein
"alkyl" is as defined above. Examples of "alkoxy" groups include, but are not
limited
to, methoxy, ethoxy, propoxy, butoxy and pentoxy.
The term "alkenyl", unless otherwise indicated, includes unsaturated
hydrocarbon radicals having one or more double bonds connecting two carbon
atoms, wherein said hydrocarbon radical may have straight, branched or cyclic
moieties or combinations thereof. Examples of "alkenyl" groups include, but
are not
limited to, ethenyl, propenyl, butenyl, pentenyl.
The term "one or more substituents", refers to a number of substituents that
equals from one to the maximum number of substituents possible based on the
number of available bonding sites.
The terms "halo" and "halogen", unless otherwise indicated, include, fluoro,
chloro, bromo and iodo.
The compounds of formula 1 and their pharmaceutically acceptable salts are
also referred to herein, collectively, as the "compounds of this invention"
and the
"active compounds of this invention".
This invention also relates to a pharmaceutical composition comprising a
compound of formula 1 and a pharmaceutically acceptable carrier. Additionally,
this
invention relates to a pharmaceutical composition comprising a therapeutically
effective amount of a compound of formula 1, or a pharmaceutically acceptable
salt
thereof, and a pharmaceutically acceptable carrier.
Compounds of formula 1 may contain chiral centers and therefore may exist
in different enantiomeric and diastereomeric forms. This invention relates to
all
optical isomers and all stereoisomers of compounds of the formula 1, both as
racemic mixtures and as individual enantiomers and diastereoisomers of such
compounds, and mixtures thereof, and to all pharmaceutical compositions and
methods of treatment defined above that contain or employ them, respectively.
Individual isomers can be obtained by known methods, such as optical
resolution,
optically selective reaction, or chromatographic separation in the preparation
of the
final product or its intermediate. Individual enantiomers of the compounds of
formula
1 may have advantages, as compared with the racemic mixtures of these
compounds, in the treatment of various disorders or conditions.
In so far as the compounds of formula 1 of this invention are basic
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-7-
compounds, they are all capable of forming a wide variety of different salts
with
various inorganic and organic acids. Although such salts must be
pharmaceutically
acceptable for administration to animals, it is often desirable in practice to
initially
isolate the base compound from the reaction mixture as a pharmaceutically
unacceptable salt and then simply convert to the free base compound by
treatment
with an alkaline reagent and thereafter convert the free base to a
pharmaceutically
acceptable acid addition salt. The acid addition salts of the base compounds
of this
invention are readily prepared by treating the base compound with a
substantially
equivalent amount of the chosen mineral or organic acid in an aqueous solvent
or in
a suitable organic solvent, such as methanol or ethanol. Upon careful
evaporation of
the solvent, the desired solid salt is readily obtained. The acids which are
used to
prepare the pharmaceutically acceptable acid addition salts of the
aforementioned
base compounds of this invention are those which form non-toxic acid addition
salts,
i.e., salts containing pharmaceutically acceptable anions, such as the
hydrochloride,
hydrobromide, hydroiodide, nitrate, sulfate or bisulfate, phosphate or acid
phosphate,
acetate, lactate, citrate or acid citrate, tartrate or bi-tartrate, succinate,
maleate,
fumarate, gluconate, saccharate, benzoate, methanesulfonate, ethanesulfonate,
benzenesulfonate, p-toluenesulfonate and pamoate (i.e., 1,1'-methylene-bis-(2-
hydroxy-3-naphthoate)) salts.
The present invention also includes isotopically labeled compounds, which
are identical to those recited in formula 1, but for the fact that one or more
atoms are
replaced by an atom having an atomic mass or mass number different from the
atomic mass or mass number usually found in nature. Examples of isotopes that
can be incorporated into compounds of the present invention include isotopes
of
hydrogen, carbon, nitrogen, oxygen, phosphorous, sulfur, fluorine and
chlorine, such
as 2H, 3H,'3C, "C,'4C,'SN,'$O, "O, 3'P, 32P, 35S,'$F, and 36C1, respectively.
Compounds of the present invention, prodrugs thereof, and pharmaceutically
acceptable salts of said compounds or of said prodrugs which contain the
aforementioned isotopes and/or other isotopes of other atoms are within the
scope of
this invention. Certain isotopically labeled compounds of the present
invention, for
example those into which radioactive isotopes such as 3H and'4C are
incorporated,
are useful in drug and/or substrate tissue distribution assays. Tritiated,
i.e., 3H, and
carbon-14, i.e.,'4C, isotopes are particularly preferred for their ease of
preparation
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
_g_
and detectability. Further, substitution with heavier isotopes such as
deuterium, i.e.,
2H, can afford certain therapeutic advantages resulting from greater metabolic
stability, for example increased in vivo half-life or reduced dosage
requirements and,
hence, may be preferred in some circumstances. Isotopically labeled compounds
of
formula 1 of this invention and prodrugs thereof can generally be prepared by
carrying out the procedures disclosed in the Schemes and/or in the Examples
below,
by substituting a readily available isotopically labeled reagent for a non-
isotopically
labeled reagent.
The compounds of formula 1 of this invention have useful pharmaceutical and
medicinal properties.
The term "treating", refers to reversing, alleviating, inhibiting the progress
of,
or preventing the disorder or condition to which such term applies, or
preventing one
or more symptoms of such condition or disorder. The term "treatment", refers
to the
act of treating, as "treating" is defined immediately above.
This invention also relates to a method of treating a disorder or condition
selected from the group consisting of single episodic or recurrent major
depressive
disorders, dysthymic disorders, depressive neurosis and neurotic depression,
melancholic depression including anorexia, weight loss, insomnia, early
morning
waking or psychomotor retardation; atypical depression (or reactive
depression)
including increased appetite, hypersomnia, psychomotor agitation or
irritability,
seasonal affective disorder and pediatric depression; bipolar disorders or
manic
depression, for example, bipolar I disorder, bipolar II disorder and
cyclothymic
disorder; conduct disorder; disruptive behavior disorder; behavioral
disturbances
associated with mental retardation, autistic disorder, and conduct disorder;
anxiety
disorders such as panic disorder with or without agoraphobia, agoraphobia
without
history of panic disorder, specific phobias, for example, specific animal
phobias,
social anxiety, social phobia, obsessive-compulsive disorder, stress disorders
including post-traumatic stress disorder and acute stress disorder, and
generalized
anxiety disorders; borderline personality disorder; schizophrenia and other
psychotic
disorders, for example, schizophreniform disorders, schizoaffective disorders,
delusional disorders, brief psychotic disorders, shared psychotic disorders,
psychotic
disorders with delusions or hallucinations, psychotic episodes of anxiety,
anxiety
associated with psychosis, psychotic mood disorders such as severe major
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
_g_
depressive disorder; mood disorders associated with psychotic disorders such
as
acute mania and depression associated with bipolar disorder; mood disorders
associated with schizophrenia; delirium, dementia, and amnestic and other
cognitive
or neurodegenerative disorders, such as Parkinson's disease (PD), Huntington's
disease (HD), Alzheimer's disease, senile dementia, dementia of the
Alzheimer's
type, memory disorder, vascular dementia, and other dementias, for example,
due to
HIV disease, head trauma, Parkinson's disease, Huntington's disease, Pick's
disease, Creutzfeldt-Jakob disease, or due to multiple aetiologies; movement
disorders such as akinesias, dyskinesias, including familial paroxysmal
dyskinesias,
spasticities, Tourette's syndrome, Scott syndrome, PALSYS and akinetic-rigid
syndrome; extra-pyramidal movement disorders such as medication-induced
movement disorders, for example, neuroleptic-induced Parkinsonism, neuroleptic
malignant syndrome, neuroleptic-induced acute dystonia, neuroleptic-induced
acute
akathisia, neuroleptic-induced tardive dyskinesia and medication-induced
postural
tremour; chemical dependencies and addictions (e.g., dependencies on, or
addictions to, alcohol, heroin, cocaine, benzodiazepines, nicotine, or
phenobarbitol)
and behavioral addictions such as an addiction to gambling; and ocular
disorders
such as glaucoma and ischemic retinopathy in a mammal, including a human,
comprising administering to the mammal in need of such treatment an amount of
a
compound of the formula 1, or a pharmaceutically acceptable salt thereof, that
is
effective in treating such disorder or condition.
This invention also relates to a pharmaceutical composition for treating a
disorder or condition selected from the disorders and conditions as defined in
the
paragraph directly above, in a mammal in need of such treatment, including a
human, comprising an amount of a compound of the formula 1, or a
pharmaceutically acceptable salt thereof, that is effective in treating such
disorder or
condition, and a pharmaceutically acceptable carrier.
A more specific embodiment of this invention relates to the above method and
composition wherein the disorder or condition that is being treated is
selected from
major depression, single episode depression, recurrent depression, child abuse
induced depression, postpartum depression, dysthymia, cyclothymia and bipolar
disorder.
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-10-
Another more specific embodiment of this invention relates to the above method
and composition wherein the disorder or condition that is being treated is
selected from
schizophrenia, schizoaffective disorder, delusional disorder, substance-
induced
psychotic disorder, brief psychotic disorder, shared psychotic disorder,
psychotic
disorder due to a general medical condition, and schizophreniform disorder.
Another more specific embodiment of this invention relates to the above method
and composition wherein the disorder or condition that is being treated is
selected from
autism, pervasive development disorder, and attention deficit hyperactivity
disorder.
Another more specific embodiment of this invention relates to the above method
and composition wherein the disorder or condition that is being treated is
selected from
generalized anxiety disorder, panic disorder, obsessive-compulsive disorder,
post-
traumatic stress disorder, and phobias, including social phobia, agoraphobia,
and
specific phobias.
Another more specific embodiment of this invention relates to the above method
and composition wherein the disorder or condition that is being treated is
selected from
movement disorders such as akinesias, dyskinesias, including familial
paroxysmal
dyskinesias, spasticities, Tourette's syndrome, Scott syndrome, PALSYS and
akinetic-rigid syndrome; and extra-pyramidal movement disorders such as
medication-induced movement disorders, for example, neuroleptic-induced
Parkinsonism, neuroleptic malignant syndrome, neuroleptic-induced acute
dystonia,
neuroleptic-induced acute akathisia, neuroleptic-induced tardive dyskinesia
and
medication-induced postural tremour.
Another more specific embodiment of this invention relates to the above method
and composition wherein the disorder or condition that is being treated is
selected from
delirium, dementia, and amnestic and other cognitive or neurodegenerative
disorders, such as Parkinson's disease (PD), Huntington's disease (HD),
Alzheimer's
disease, senile dementia, dementia of the Alzheimer's type, memory disorder,
vascular dementia, and other dementias, for example, due to HIV disease, head
trauma, Parkinson's disease, Huntington's disease, Pick's disease, Creutzfeldt-
Jakob
disease, or due to multiple aetiologies.
Another more specific embodiment of this invention relates to the above method
and composition wherein the compound of formula 1 is administered to a human
for the
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-11-
treatment of any two or more comorbid disorders or conditions selected from
those
disorders and conditions referred to in any of the above methods.
For the treatment of depression, anxiety, schizophrenia or any of the other
disorders and conditions referred to above in the descriptions of the methods
and
pharmaceutical compositions of this invention, the compounds of this invention
can
be used in conjunction with one or more other antidepressants or anti-anxiety
agents. Examples of classes of antidepressants that can be used in combination
with the active compounds of this invention include norepinephrine reuptake
inhibitors, selective serotonin reuptake inhibitors (SSRIs), NK-1 receptor
antagonists,
monoamine oxidase inhibitors (MAOIs), reversible inhibitors of monoamine
oxidase
(RIMAs), serotonin and noradrenaline reuptake inhibitors (SNRIs),
corticotropin
releasing factor (CRF) antagonists, a-adrenoreceptor antagonists, and atypical
antidepressants. Suitable norepinephrine reuptake inhibitors include tertiary
amine
tricyclics and secondary amine tricyclics. Suitable tertiary amine tricyclics
and
secondary amine tricyclics include amitriptyline, clomipramine, doxepin,
imipramine,
trimipramine, dothiepin, butripyline, iprindole, lofepramine, nortriptyline,
protriptyline,
amoxapine, desipramine and maprotiline. Suitable selective serotonin reuptake
inhibitors include fluoxetine, fluvoxamine, paroxetine and sertraline.
Examples of
monoamine oxidase inhibitors include isocarboxazid, phenelzine, and
tranylcyclopramine. Suitable reversible inhibitors of monoamine oxidase
include
moclobemide. Suitable serotonin and noradrenaline reuptake inhibitors of use
in the
present invention include venlafaxine. Suitable CRF antagonists include those
compounds described in International Patent Application Nos. WO 94/13643, WO
94/13644, WO 94/13661, WO 94!13676 and WO 94/13677. Suitable atypical anti-
depressants include bupropion, lithium, nefazodone, trazodone and viloxazine.
Suitable NK-1 receptor antagonists include those referred to in World Patent
Publication WO 01/77100.
Suitable classes of anti-anxiety agents that can be used in combination with
the active compounds of this invention include benzodiazepines and serotonin
IA (5-
HT,A) agonists or antagonists, especially 5-HT,A partial agonists, and
corticotropin
releasing factor (CRF) antagonists. Suitable benzodiazepines include
alprazolam,
chlordiazepoxide, clonazepam, chlorazepate, diazepam, halazepam, lorazepam,
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-12-
oxazepam, and prazepam. Suitable 5-HT,A receptor agonists or antagonists
include
buspirone, flesinoxan, gepirone and ipsapirone.
This invention also relates to a method of treating a disorder or condition
selected from single episodic or recurrent major depressive disorders,
dysthymic
disorders, depressive neurosis and neurotic depression, melancholic depression
including anorexia, weight loss, insomnia, early morning waking or psychomotor
retardation; atypical depression (or reactive depression) including increased
appetite, hypersomnia, psychomotor agitation or irritability, seasonal
affective
disorder and pediatric depression; bipolar disorders or manic depression, for
example, bipolar I disorder, bipolar II disorder and cyclothymic disorder;
conduct
disorder; disruptive behavior disorder; behavioral disturbances associated
with
mental retardation, autistic disorder, and conduct disorder; anxiety disorders
such as
panic disorder with or without agoraphobia, agoraphobia without history of
panic
disorder, specific phobias, for example, specific animal phobias, social
anxiety,
social phobia, obsessive-compulsive disorder, stress disorders including post-
traumatic stress disorder and acute stress disorder, and generalized anxiety
disorders; borderline personality disorder; schizophrenia and other psychotic
disorders, for example, schizophreniform disorders, schizoaffective disorders,
delusional disorders, brief psychotic disorders, shared psychotic disorders,
psychotic
disorders with delusions or hallucinations, psychotic episodes of anxiety,
anxiety
associated with psychosis, psychotic mood disorders such as severe major
depressive disorder; mood disorders associated with psychotic disorders such
as
acute mania and depression associated with bipolar disorder; mood disorders
associated with schizophrenia; delirium, dementia, and amnestic and other
cognitive
or neurodegenerative disorders, such as Parkinson's disease (PD), Huntington's
disease (HD), Alzheimer's disease, senile dementia, dementia of the
Alzheimer's
type, memory disorder, vascular dementia, and other dementias, for example,
due to
HIV disease, head trauma, Parkinson's disease, Huntington's disease, Pick's
disease, Creutzfeldt-Jakob disease, or due to multiple aetiologies; movement
° disorders such as akinesias, dyskinesias, including familial
paroxysmal dyskinesias,
spasticities, Tourette's syndrome, Scott syndrome, PALSYS and akinetic-rigid
syndrome; extra-pyramidal movement disorders such as medication-induced
movement disorders, for example, neuroleptic-induced Parkinsonism, neuroleptic
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-13-
malignant syndrome, neuroleptic-induced acute dystonia, neuroleptic-induced
acute
akathisia, neuroleptic-induced tardive dyskinesia and medication-induced
postural
tremour; chemical dependencies and addictions (e.g., dependencies on, or
addictions to, alcohol, heroin, cocaine, benzodiazepines, nicotine, or
phenobarbitol)
and behavioral addictions such as an addiction to gambling; and ocular
disorders
such as glaucoma and ischemic retinopathy in a mammal in need of such
treatment,
including a human, comprising administering to said mammal:
(a) a compound of the formula 1 or a pharmaceutically acceptable salt
thereof; and
(b) another pharmaceutically active compound that is an antidepressant or
anti-anxiety agent, or a pharmaceutically acceptable salt thereof;
wherein the active compounds "a" and "b" are present in amounts that render
the combination effective in treating such disorder or condition.
This invention also relates to a pharmaceutical composition for treating a
disorder or condition selected from the disorders and conditions as defined in
the
paragraph directly above, in a mammal in need of such treatment, including a
human, comprising:
(a) a compound of the formula 1 or a pharmaceutically acceptable salt
thereof;
(b) another pharmaceutically active compound that is an antidepressant or
anti-anxiety agent, or a pharmaceutically acceptable salt thereof; and
(c) a pharmaceutically acceptable carrier;
wherein the active compounds "a" and "b" are present in amounts that render
the composition effective in treating such disorder or condition.
A more specific embodiment of this invention relates to the above method and
composition wherein the disorder or condition that is being treated is
selected from
major depression, single episode depression, recurrent depression, child abuse
induced depression, postpartum depression, dysthymia, cyclothymia and bipolar
disorder.
Another more specific embodiment of this invention relates to the above method
and composition wherein the disorder or condition that is being treated is
selected from
schizophrenia, schizoaffective disorder, delusional disorder, substance-
induced
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-14-
psychotic disorder, brief psychotic disorder, shared psychotic disorder,
psychotic
disorder due to a general medical condition, and schizophreniform disorder.
Another more specific embodiment of this invention relates to the above method
and composition wherein the disorder or condition that is being treated is
selected from
autism, pervasive development disorder, and attention deficit hyperactivity
disorder.
Another more specific embodiment of this invention relates to the above method
and composition wherein the disorder or condition that is being treated is
selected from
generalized anxiety disorder, panic disorder, obsessive-compulsive disorder,
post-
traumatic stress disorder, and phobias, including social phobia, agoraphobia,
and
specific phobias.
Another more specific embodiment of this invention relates to the above method
and composition wherein the disorder or condition that is being treated is
selected from
movement disorders such as akinesias, dyskinesias, including familial
paroxysmal
dyskinesias, spasticities, Tourette's syndrome, Scott syndrome, PALSYS and
akinetic-rigid syndrome; and extra-pyramidal movement disorders such as
medication-induced movement disorders, for example, neuroleptic-induced
Parkinsonism, neuroleptic malignant syndrome, neuroleptic-induced acute
dystonia,
neuroleptic-induced acute akathisia, neuroleptic-induced tardive dyskinesia
and
medication-induced postural tremour.
Another more specific embodiment of this invention relates to the above method
and composition wherein the disorder or condition that is being treated is
selected from
delirium, dementia, and amnestic and other cognitive or neurodegenerative
disorders, such as Parkinson's disease (PD), Huntington's disease (HD),
Alzheimer's
disease, senile dementia, dementia of the Alzheimer's type, memory disorder,
vascular dementia, and other dementias, for example, due to HIV disease, head
trauma, Parkinson's disease, Huntington's disease, Pick's disease, Creutzfeldt-
Jakob
disease, or due to multiple aetiologies.
Another more specific embodiment of this invention relates to the above method
and composition wherein the compound of formula 1 and the additional
antidepressant
or anti-anxiety agent are administered to a human for the treatment of any two
or more
comorbid disorders or conditions selected from those disorders and conditions
referred
to in any of the above methods.
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-15-
The compounds of formula 1 of the present invention may be prepared as
described in the following reaction schemes. Unless otherwise indicated, in
the
reaction schemes and discussion that follow, R' through R'4, A, n, m, o, p, q,
U, V, L,
W, D, E, T, Z and X are defined as above.
SCHEME A-1
R$
o Q
+ ~ Q ~(C
s i
R~ ~X X~ ~C~"~2)m
2
3
[In compounds 2 and 3, one of the -CH2- groups of X or W is replaced by -
C(O)-]
The above scheme illustrates a method for preparing compounds of the
formula 3 by reacting a compound of the formula 2 with a compound of formula
X'CO(CH2)mQ, wherein X' is either a halogen or OH and Q is either a halogen,
mesylate, or tosylate. When X1 is represented by a halogen, the reaction is
typically
carried out in the presence of a Lewis acid such as aluminum bromide (AIBr3),
aluminum chloride (AICI3), gallium trichloride (GaCl3), ferric chloride
(FeCl3), zinc
chloride (ZnCl2), antimony pentachloride (SbCl5), zirconium tetrachloride
(ZrCl4), tin
tetrachloride (SnCl4), boron trichloride (BC13), boron trifluoride (BF3), or
antimony
trichloride (SbCl3). The reaction can be carried out in nonpolar solvents such
as
chloroform, dichloromethane, dichloroethane, or carbon disulfide, or in polar
solvents
such as nitrobenzene, or may be run neat in the presence of excess Lewis acid.
The
reaction is typically carried out at a temperature of 25°C to about
120°C for a period
of about 1 hour to 45 hours preferably, 1 hour to 6 hours. Where X' is
represented
by OH, the reaction is typically carried out in the presence of a proton acid
such as
polyphosphoric acid or sulfuric acid.
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-16-
SCHEME A-2
Q H2
~(CI Q~
(CH2)m
3 R
4
[In compounds 3 and 4, one of the -CH2- groups of X or W is replaced by -
C(O)_l
The above scheme illustrates a method for preparing compounds of the
formula 4. In compounds of the formulas 3 and 4, Q is defined as it is defined
above
in the description of Scheme A-1. The reaction illustrated in Scheme A-2 can
be
carried out using triethylsilane in trifluoroacetic acid at a temperature from
about rt to
the reflux temperature of the solvent for a period of up to about 24 hours.
Alternatively, the reaction may be carried out using borane-tert-butylamine in
the
presence of a Lewis acid such as aluminum chloride or by using borane-
dimethylamine in the presence of a Lewis acid such as titanium tetrachloride
in an
inert solvent such as dichloromethane, chloroform, or nitrobenzene under
temperatures described.
20
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-17-
SCHEME A-3
Ri
Hz Ra
y is ~/. \
~CH2)m
/NH
R~~~ X
4
H
W ~N\
R
[In compounds 4, 5 and 6, one of the -CH2- groups of X or W is replaced by -
C(O)-}
5 The above scheme illustrates a method for preparing compounds of the
formula 6, by reacting a compound of the formula 4, as described in Scheme A-
2,
with a compound of formula 5. The reaction is typically run in the presence of
a base
such as potassium carbonate, sodium carbonate, triethylamine, or
diisopropylethylamine. The solvent used may be water, acetonitrile, dioxane,
benzene, toluene, tetrahydrofuran, methyl isobutyl ketone, or a combination of
two of
the formerly mentioned solvents. Inorganic salts such as a sodium or potassium
halide (e.g., sodium iodide or potassium iodide) may be employed as catalysts
in the
reaction. The temperature of the reaction may vary from ambient to reflux
temperature of the solvent used, preferably from about 80°C to
120°C, for a period of
about 1 hour to about 96 hours, preferably from about 12 hours to 48 hours.
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
_18_
SCHEME A-4
H H
/ l
W ~N~ W ~N~
R X RB~~X
R' ~ ~R
\ ' ., ~R
'~ ~N'(CH2)m'CH2 R Cf / ~N'~CH~m'CH2
Rz ~Z Rz U
~~V
6
7
[In compound 6, one of the -CHz- groups of W or X is replaced by -C(O)-]
The above scheme illustrates a method for preparing compounds of the
formula 7 by reducing the amide carbonyl in the compound of the formula 6 with
a
reducing agent such as borane THF, or borane dimethyl sulfide. The reaction
above can be carried out in a solvent such as methylene chloride,
tetrahydrofuran,
dichloroethane, benzene, or toluene. This reaction is typically carried out at
a
temperature from about -78 °C to about the reflux temperature of the
solvent,
preferably from about-20 °C to about 50 °C, for a period of
about 5 minutes to about
48 hours, preferably from about 0.5 to about 16 hours. The reaction is
typically
quenched with methanol, water, or a dilute base such as sodium carbonate or
sodium bicarbonate. Preferably, the reaction is quenched with methanol or 10%
sodium carbonate and the complexes are broken up by heating the reaction
mixture
to a temperature from about 30 °C to about the reflux temperature of
the solvent,
preferably to about 90 °C, for about 0.5 to about 20 hours, preferably
for about 2
hours.
SCHEME A-5
H M
W ~N\
a X
R'-
R ~1
~ \ ~ '
/ ~N'iCHz)m~CHz R
Rz
~-V
formula 1
The above scheme illustrates a method for preparing compounds of the
formula 1 by reacting compounds of the formula 7 with a compound of the
formula
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-19-
R9-G wherein G is -COCI, an acid or a suitably activated acid derivative such
as the
mixed anhydride, -OCOCI, -N=C=O, or -S02CI, or wherein R9-G is CIS02N(Me)2.
This reaction may be carried out in an inert solvent such as methylene
chloride,
dichloromethane, dichloroethane, benzene, toluene, tetrahydrofuran, or
pyridine,
preferably methylene chloride. Typically, it is carried out at a temperature
from about
-78 °C to about the reflux temperature of the solvent, preferably from
about 0 °C to
about 25 °C, for a period of about 5 minutes to 72 hours, preferably
from about 0.5 to
about 16 hours. This reaction is generally performed in the presence of
organic
base such as diisopropylethylamine, pyridine, or triethylamine, preferably
triethylamine, or in the presence of a polymer supported base such as tris-(2-
aminoethyl)amine polystyrene.
SCHEME B-1
Rs Ra
(~ (Chic 2 ~~ Q-(CH2)ni CH2 ~~ W
I N_H ~ ~/N_H
x/
R~ R~
[In compound 4, one of the -CH2- groups of W or X is replaced by -C(O)-]
The above scheme illustrates a method for preparing compounds of the
formula 8 by reducing the amide carbonyl in a compound of the formula 4 with a
reducing agent such as borane THF, or borane dimethyl sulfide. The reaction
above can be carried out in a solvent such as methylene chloride,
tetrahydrofuran,
dichloroethane, benzene, or toluene. This reaction is typically carried out at
a
temperature from about -78°C to about the reflux temperature of the
solvent,
preferably from about -20°C to about 50°C, for a period of about
5 minutes to about
48 hours, preferably from about 0.5 to about 16 hours. The reaction is
typically
quenched with methanol, water, or a dilute base such as sodium carbonate or
sodium bicarbonate. Preferably, the reaction is quenched with methanol or 10%
sodium carbonate and the complexes are broken up by heating the reaction
mixture
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-20-
to a temperature from about 30°C to about the reflux temperature of the
solvent,
preferably to about 90°C, for about 0.5 to about 20 hours, preferably
for about 2
hours.
SCHEME B-2
Ra Re
Q'(CH~m CH2 ~~ W' Q-(CH~m CH2 ~~_ W
N_H > \N_IV
x°
R~ R~
The above scheme illustrates a method for preparing compounds of the
formula 9 by reacting compounds of the formula 8 with a compound of the
formula
R9-G wherein G is -COCI, an acid or a suitably activated acid derivative such
as the
mixed anhydride, -OCOCI, -N=C=O, or -S02C1, or wherein R9-G is CIS02N(Me)2:
This reaction may be carried out in an inert solvent such as methylene
chloride,
dichloroethane, benzene, toluene, tetrahydrofuran, or pyridine, preferably
methylene
chloride. Typically, it is carried out at a temperature from about -
78°C to about the
reflux temperature of the solvent, preferably from about 0°C to about
25°C, for a
period of about 5 minutes to 48 hours, preferably from about 0.5 to about 16
hours.
This reaction is generally performed in the presence of organic base such as
diisopropylethylamine, pyridine, or triethylamine, preferably triethylamine,
or in the
presence of a polymer supported base such as tris-(2-aminoethyl)amine
polystyrene.
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-21-
SCHEME B-3
Q-W
Rz
K V
9 5
~'-V
(CHz~Crn R,
Formula 1
The above scheme illustrates a method for preparing compounds of the
formula 1 wherein A is -(CH2)mCH2- by reacting a compound of the formula 9, as
described in scheme B-2, with a compound of formula 5. The reaction is
typically
run in the presence of a base such as potassium carbonate, sodium carbonate,
triethylamine, or diisopropylethylamine. The solvent used may be water,
acetonitrile,
dioxane, benzene, toluene, tetrahydrofuran, methyl isobutyl ketone, or a
combination
of two of the formerly mentioned solvents. Inorganic salts such as a sodium or
potassium halide (e.g., sodium iodide or potassium iodide) may be employed as
catalysts in the reaction. The temperature of the reaction may vary from
ambient to
reflux temperature of the solvent used, preferably from about 80°C to
120°C, for a
period of about 1 hour to about 96 hours, preferably from about 12 hours to 48
hours.
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-22-
SCHEME C-1
Rs Ra
/w '/.
NH ~ ~ / ~N-M
R~ R
11
The above scheme illustrates a method for preparing compounds of the
formula 11 by reacting compounds of the formula 10 with a compound of the
formula
5 R9-G wherein G is -COCI, an acid or a suitably activated acid derivative
such as the
mixed anhydride, -OCOCI, -N=C=O, or -S02C1, or wherein R9-G is CISO2N(Me)2.
This reaction may be carried out in an inert solvent such as methylene
chloride,
dichloroethane, benzene, toluene, tetrahydrofuran, or pyridine, preferably
methylene
chloride. Typically, it is carried out at a temperature from about -78
°C to about the
10 reflux temperature of the solvent, preferably from about 0 °C to
about 25 °C, for a
period of about 5 minutes to 48 hours, preferably from about 0.5 to about 16
hours.
This reaction is generally performed in the presence of organic base such as
diisopropylethylamine, pyridine, or triethylamine, preferably triethylamine,
or in the
presence of a polymer supported base such as tris-(2-aminoethyl)amine
polystyrene.
SCHEME C-2
RB
R
O ~N-M
\N-M . ~ ~CH2)m /
/ + ~ ~Q Q
R~ x~ ~CH2)m I R
12
11
The above scheme illustrates a method for preparing compounds of the
formula 12 by reacting a compound of the formula 11 with a compound of formula
XiCO(CH2)mQ, X' is either a halogen or OH and Q is either a halogen, mesylate,
or
tosylate. When X1 is represented by a halogen, the reaction is typically
carried out in
the presence of a Lewis acid such as aluminum bromide (AIBr3), aluminum
chloride
(AICIs), gallium trichloride (GaCls), ferric chloride (FeCl3), zinc chloride
(ZnCl2),
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-23-
antimony pentachloride (SbClS), zirconium tetrachloride (ZrCl4), tin
tetrachloride
(SnCl4), boron trichloride (BCl3), boron trifluoride (BF3), or antimony
trichloride
(SbCl3). The reaction can be carried out in nonpolar solvents such as
chloroform,
dichloromethane, dichloroethane, or carbon disulfide, or in polar solvents
such as
nitrobenzene, or may be run neat in the presence of excess Lewis acid. The
reaction is typically carried out at a temperature of 25°C to about
120°C for a period
of about 1 hour to 6 hours. Where X is represented by OH, the reaction is
typically
carried out in the presence of a proton acid such as polyphosphoric acid or
sulfuric
acid.
SCHEME C-3
R1
-M
(CH;
~H
12
5
M
~(CH
N
Z'
13
The above scheme illustrates a method for preparing compounds of formula
13, by reacting a compound of the formula 12 with a compound of formula 5~HCL.
The reaction is typically run in the presence of a base such as potassium
carbonate,
sodium carbonate, triethylamine, or diisopropylethylamine. The solvent used
may be
water, acetonitrile, dioxane, benzene, toluene, tetrahydrofuran, methyl
isobutyl
ketone, or a combination of two of the formerly mentioned solvents. Inorganic
salts
such as a sodium or potassium halide (e.g., sodium iodide or potassium iodide)
may
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-24-
be employed as catalysts in the reaction. The temperature of the reaction may
vary
from ambient to reflux temperature of the solvent used, preferably from about
80°C
to 120°C, for a period of about 1 hour to about 96 hours, preferably
from about 12
hours to 48 hours.
SCHEME C-4
Ra
Ra
z I ~Iw N-M R2 I ~/~N-M
R WCHz) ~ ~/ ~~ NWCH2)
I ~ )~'N
R ~/ ~ C1
Z HO
R
R ~ U-V
U V
14 15
The above scheme illustrates a method for preparing compounds of the
formula 15 by reacting compounds of the formula 14. The reaction is typically
run in
the presence of a chlorinating reagent such as toluenesulfonyl chloride,
methanesulfonyl chloride, carbon tetrachloride, or hydrogen chloride. The
solvent
used may be methylene chloride, dichloroethane, toluene, tetrahydrofuran,
chloroform, or a combination of two of the formerly mentioned solvents. The
temperature of the reaction may vary from -20°C to reflux temperature
of the solvent
used, preferably from about 0°C to ambient, for a period of about 1
hour to about 96
hours, preferably from about 2 hours to 12 hours.
25
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-25-
SCHEME C-5
R$
z
R (CHz)m I ~N-M
Ri ~~ ~N ~ R~
ZJ C1
U_V
R$
/.
/R2 (CHz)m I ~~N-M
Rt ~ ~N R~
ZJ ~ CH3S03H
U-V
16
The above scheme illustrates a method for preparing compounds of the
formula 16 by reacting compounds of the formula 15. The reaction is typically
run in
the presence of a reducing reagent such as trialkyltin hydride, or triaryltin
hydride.
The reaction is typically run in the presence of a radical initiator such as
2,2-
azobisisobutyronitrile or light. The solvent used may be benzene, toluene,
tetrahydrofuran, or a combination of two of the formerly mentioned solvents.
The
temperature of the reaction may vary from 0°C to reflux temperature of
the solvent
10 used, preferably from about 40°C to reflux, for a period of about 1
hour to about 96
hours, preferably from about 1 hour to 4 hours.
The preparation of other compounds of the formula 1 not specifically
described in the foregoing experimental section can be accomplished using
combinations of the reactions described above that will be apparent to those
skilled
15 in the art.
In each of the reactions discussed or illustrated above, pressure is not
critical
unless otherwise indicated. Pressures from about 0.5 atmospheres to about 5
atmospheres are generally acceptable, and ambient pressure, i.e., about 1
atmosphere, is preferred as a matter of convenience.
The compounds of the formula 1, and the intermediates shown in the above
reaction schemes can be isolated and purified by conventional procedures, such
as
recrystallization or chromatographic separation.
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-26-
The compounds of the formula 1 and their pharmaceutically acceptable salts
can be administered to mammals via either the oral, parenteral (such as
subcutaneous, intraveneous, intramuscular, intrasternal and infusion
techniques),
rectal; buccal or intranasal routes. In general, these compounds are most
desirably
administered in doses ranging from about 3 mg to about 600 mg per day, in
single or
divided doses (i.e., from 1 to 4 doses per day), although variations will
necessarily
occur depending upon the species, weight and condition of the patient being
treated,
the patient's individual response to said medicament, the nature and severity
of the
particular disorder being treated, as well as on the type of pharmaceutical
formulation
chosen and the overall time period and intervals over which such
administration is
carried out. However, a dosage level that is in the range of about 25 mg to
about 100
mg per day is most desirably employed. In some instances, dosage levels below
the
lower limit of the aforesaid range may be more than adequate, while in other
cases still
larger doses may be employed without causing any harmful side effects,
provided that
such higher dose levels are first divided into several small doses for
administration
throughout the day.
The compounds of the present invention may be administered alone or in
combination with pharmaceutically acceptable carriers or diluents by any of
the routes
previously indicated, and such administration may be carried out in single or
multiple
doses. More particularly, the therapeutic agents of this invention can be
administered
in a wide variety of different dosage forms, i.e., they may be combined with
various
pharmaceutically acceptable inert carriers in the form of tablets, capsules,
lozenges,
troches, hard candies, suppositories, jellies, gels, pastes, ointments,
aqueous
suspensions, injectable solutions, elixirs, syrups, and the like. Such
carriers include
solid diluents or fillers, sterile aqueous media and various non-toxic organic
solvents,
etc. Moreover, oral pharmaceutical compositions can be suitably sweetened
and/or
flavored. In general, the weight ratio of the compounds of this invention to
the
pharmaceutically acceptable carrier will be in the range from about 1:6 to
about 2:1,
and preferably from about 1:4 to about 1:1,
For oral administration, tablets containing various excipients such as
microcrystalline cellulose, sodium citrate, calcium carbonate, dicalcium
phosphate
and glycine may be employed along with various disintegrants such as starch
(and
preferably corn, potato or tapioca starch), alginic acid and certain complex
silicates,
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-27-
together with granulation binders like polyvinylpyrrolidone, sucrose, gelatin
and
acacia. Additionally, lubricating agents such as magnesium stearate, sodium
lauryl
sulfate and talc are often very useful for tabletting purposes. Solid
compositions of a
similar type may also be employed as fillers in gelatin capsules; preferred
materials
in this connection also include lactose or milk sugar as well as high
molecular weight
polyethylene glycols. When aqueous suspensions andlor elixirs are desired for
oral
administration, the active ingredient may be combined with various sweetening
or
flavoring agents, coloring matter or dyes, and, if so desired, emulsifying
and/or
suspending agents as well, together with such diluents as water, ethanol,
propylene
glycol, glycerin and various like combinations thereof.
For parenteral administration, solutions of a compound of the present
invention in either sesame or peanut oil or in aqueous propylene glycol may be
employed. The aqueous solutions should be suitably buffered (preferably pH
greater
than 8) if necessary and the liquid diluent first rendered isotonic. These
aqueous
solutions are suitable for intravenous injection purposes. The oily solutions
are
suitable for intra-articular, intra-muscular and subcutaneous injection
purposes. The
preparation of all these solutions under sterile conditions is readily
accomplished by
standard pharmaceutical techniques well known to those skilled in the art.
This invention relates to methods of treating anxiety, depression,
schizophrenia
and the other disorders referred to in the description of the methods of the
present
invention, wherein a compound of this invention and one or more of the other
active
agents referred to above (e.g., an NK1 receptor antagonist, tricyclic
antidepressant,
5HT1 D receptor antagonist, or serotonin reuptake inhibitor) are administered
together,
as part of the same pharmaceutical composition, as well as to methods in which
such
active agents are administered separately as part of an appropriate dose
regimen
designed to obtain the benefits of the combination therapy. The appropriate
dose
regimen, the amount of each dose of an active agent administered, and the
specific
intervals between doses of each active agent will depend upon the subject
being
treated, the specific active agent being administered and the nature and
severity of the
specific disorder or condition being treated. In general, the compounds of
this
invention, when used as a single active agent or in combination with another
active
agent, will be administered to an adult human in an amount from about 3 mg to
about
600 mg per day, in single or divided doses, preferably from about 25 to about
100 mg
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-28-
per day. Such compounds may be administered on a regimen of up to 6 times per
day, preferably 1 to 4 times per day, especially 2 times per day and most
especially
once daily. Variations may nevertheless occur depending on the species, weight
and
condition of the patient being treated, the patient's individual response to
said
medicament, the nature and severity of the particular disorder being treated,
as well as
on the type of pharmaceutical formulation chosen and the overall time period
and
intervals over which such administration is carried out. In some instances,
dosage
levels below the lower limit of the aforesaid range may be more than adequate,
while
in other cases still larger doses may be employed without causing any harmful
side
effect, provided that such larger doses are first divided into several small
doses for
administration throughout the day.
A proposed daily dose of a 5HT reuptake inhibitor, preferably sertraline, in
the
combination methods and compositions of this invention, for oral, parenteral
or
buccal administration to the average adult human for the treatment of the
conditions
referred to above, is from about 0.1 mg to about 2000 mg, preferably from
about 1
mg to about 200 mg of the 5HT reuptake inhibitor per unit dose, which could be
administered, for example, 1 to 4 times per day. A proposed daily dose of a
5HT1 D
receptor antagonist in the combination methods and compositions of this
invention,
for oral, parenteral, rectal or buccal administration to the average adult
human for the
treatment of the conditions referred to above, is from about 0.01 mg to about
2000
mg, preferably from about 0.1 mg to about 200 mg of the 5HT1 D receptor
antagonist
per unit dose, which could be administered, for example, 1 to 4 times per day.
For intranasal administration or administration by inhalation, the compounds
of the invention are conveniently delivered in the form of a solution or
suspension
from a pump spray container that is squeezed or pumped by the patient or as an
aerosol spray presentation from a pressurized container or a nebulizer, with
the use
of a suitable propellant, e.g., dichlorodifluoromethane,
trichlorofluoromethane,
dichlorotetrafluoroethane, carbon dioxide or other suitable gas. In the case
of a
pressurized aerosol, the dosage unit may be determined by providing a valve to
deliver a metered amount. The pressurized container or nebulizer may contain a
solution or suspension of the active compound. Capsules and cartridges (made,
for
example, from gelatin) for use in an inhaler or insufflator may be formulated
containing a powder mix of a compound of the invention and a suitable powder
base
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-29-
such as lactose or starch. Formulations of the active compounds of this
invention for
treatment of the conditions referred to above in the average adult human are
preferably prepared so that each metered dose or "puff" of aerosol contains 20
~g to
1000,ug of active compound. The overall daily dose with an aerosol will be
within the
range 100,ug to 10 mg. Administration may be several times daily, for example
2, 3,
4 or 8 times, giving for example, 1, 2 or 3 doses each time.
The ability of the compounds of this invention to bind to the dopamine D2 and
serotonin 2A (5HT2A) receptors can be determined using conventional
radioligand
receptor binding assays. All receptors can be heterologously expressed in cell
lines
and experiments conducted in membrane preparations from the cell lines using
procedures outlined below. ICSO concentrations can be determined by nonlinear
regression of concentration-dependent reduction in specific binding. The Cheng-
Prussoff equation can be used to convert the IC5o to Ki concentrations.
Dopamine D2 Receptor Binding_
[3H]Spiperone binding to a membrane preparation from CHO-hD2L cells is
carried out in 250 ~I of 50 mM Tris-HCI buffer containing 100 mM NaCI, 1 mM
MgCl2
and 1 % DMSO at pH 7.4. Duplicate samples containing (in order of addition)
the
test compounds, 0.4 nM [3H]spiperone and approximately l2,ug protein are
incubated for 120 minutes at rt. Bound radioligand is separated by rapid
filtration
under reduced pressure through Whatman GF/B glass fiber filters previously
treated
with 0.3% polyethyleneimine. Radioactivity retained on the filter is
determined by
liquid scintillation spectrophotometry.
Certain of the title compounds of Examples 1-164 were tested using the
above assay, in which specific binding determined in the presence of 1 mM
haloperidol was 95%. The results of the testing are shown in Table A below.
Serotonin 2A Binding:
[3H] Ketanserin binding to Swiss-h5HT2A cell membranes can be carried out
in 250,u1 of 50 mM Tris-HCI buffer pH 7.4. Duplicate samples containing (in
order of
addition) test compounds, 1.0 nM [3H]ketanserin, and approximately 75 ~ug
protein
are incubated for 120 minutes at rt. Bound radioligand is separated by rapid
filtration
under reduced pressure through Whatman GF/B glass fiber filters previously
treated
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-30-
with 0.3% polyethyleneimine. Radioactivity retained on the filter is
determined by
liquid scintillation spectrophotometry.
Certain of the compounds of Examples 1-164 were tested using the above
assay, in which specific binding determined in the presence of 1 mM ketanserin
was
90%. The results of the testing are shown in Table A below.
TABLE A
jExampleCompound Name D2 5I=IT2A
Kl Kl
1 {5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-3,3-dimethyl-
2,3-95.70
dihydro-indol-1-yl}-(4-tluoro-phenyl)-methanone
2 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-3,3-dimethyl-
103.87
2,3-dihydro-indol-1-yl}-2-(3-methoxy-phenyl)-ethanone
3 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-3,3-dimethyl-
92.41
2,3-dihydro-indol-1-yl}-2-thiophen-2-yl-ethanone
4 1-{5-[2-(4-Benzo[d] isothiazol-3-yl-piperazin-1-yl)-ethyl]-3,3-
dimethyl-
2,3-dihydro-indol-1-yl}-2-phenoxy-propan-1-one
{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-3,3-dimethyl-2,3-54.80
dihydro-indol-1-yl}-(2,5-dimethyl-2H-pyrazol-3-yl)-methanone
6 {5-[2-(4-Benzo[d]isothiaz0l-3-yl-piperazin-1-yl)-ethyl]-3,3-dimethyl-
2,3-65.00
dihydro-indol-1-yl}-(2,5-dimethyl-2H-pyrazol-3-yl)-methanone
7 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-3,3-dimethyl-
55.10
2,3-dihydro-indol-1-yl}-2-methyl-propan-1-one
g {5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-3,3-dimethyl-
2,3-
dihydro-indol-1-yl}-m-tolyl-methanone
g 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-3,3-dimethyl-
2,3-dihydro-indol-1-yl}-2-(4-chloro-phenoxy)-ethanone
1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-3,3-dimethyl-
2,3-dihydro-indol-1-yl}-3-phenyl-propan-1-one
11 ' 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-3,3-
dimethyl-
2,3-dihydro-indol-1-yl}-2-(3,4-dimethoxy-phenyl)-ethanone
12 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-3,3-dimethyl-
2,3-dihydro-indol-1-yl}-2-(4-chloro-phenyl)-ethanone
13 {5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-3,3-dimethyl-
2,3-45.00
dihydro-indol-1-yl)-(4-methoxy-phenyl)-methanone
14 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-3,3-dimethyl-
68.83
2,3-dihydro-indol-1-yl}-2-phenyl-ethanone
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-31-
ExampleCompound Name D2 5HT2A K1
K1
15 1-{5-[2-(4-Benzo[d]isoth iazol-3-yl-piperazi
n-1-yl)-ethyl]-3,3-dimethyl-
2,3-dihydro-indol-1-yl}-2-(2,5-dimethoxy-phenyl)-ethanone
16 5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-3,3-dimethyl-2,3-
dihydro-indole-1-carboxylic acid phenyl
ester
17 {5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-3,3-dimethyl-
2,3-52.001.10
dihydro-indol-1-yl}-furan-2-yl-methanone
1g 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-3,3-dimethyl-
2,3-dihydro-indol-1-yl}-3-methyl-butan-1-one
19 {5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-3,3-dimethyl-
2,3-
dihydro-indol-1-yl}-cyclopentyl-methanone
20 1-{5-[2-(4-Benzo[d]isoth iazol-3-yl-p iperazin-1-yl)-ethyl]-3,3-
dimethyl-
2,3-dihydro-indol-1-yl}-2-benzyloxy-ethanone
21 {5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-3,3-dirnethyl-
2,3-83.00
dihydro-indol-1-yl}-phenyl-methanone
22 1-{5-[2-(4-Benzo[d]isoth iazol-3-yl-piperazin-1-yl)-ethyl]-3,3-dimethyl-
2,3-dihydro-indol-1-yl}-2-cyclopentyl-ethanone
23 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-3,3-dimethyl-
16.730.11
2,3-dihydro-indol-1-yl}-ethanone
25 1-{6-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2a,3,4,5-
13.420.55
tetrahydro-2H-benzo[cd]indol-1-yl}-ethanone
26 {6-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethylJ-2a,3,4,5-
66.451.10
tetrahydro-2H-benzo[cd]indol-1-yl}-cyclopropyl-methanone
27 1-{5-[2-(4-Benzo[d]isoth iazol-3-yl-piperazin-1-yl)-ethyl]-3,3-dimethyl-
2,3-dihydro-indol-1-yl}-ethanone
27 1-{6-[2-(4-Benzo(d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2a,3,4,5-
59.042.15
tetrahydro-2H-benzo[cd]indol-1-yl}-propan-1-one
2g 5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-3,3-dimethyl-2,3-
29.390.30
dihydro-indole-1-carboxylic acid isopropylamide
2g 1-{6-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2a,3,4,5-
106.000.90
tetrahydro-2H-benzo[cd]indol-1-yl}-2,2-dimethyl-propan-1-one
29 1-{6-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2a,3,4,5-
64.311.85
tetrahydro-2H-benzo[cd]indol-1-yl}-pentan-1-one
30 1-{6-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2a,3,4,5-
1.40
tetrahydro-2H-benzo[cd]indol-1-yl}-3-methyl-butan-1-one
31 1-{6-[2-(4-Benzo(dJisothiazol-3-yl-piperazin-1-yl)-ethylJ-2a,3,4,5-
29.800.80
tetrahydro-2H-benzo[cd]indol-1-yl}-2-methyl-propan-1-one
32 1-{6-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2a,3,4,5-
44.600.60
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-32-
ExampleCompound Name D2 5HT2A
Kl K1
tetrahydro-2H-benzo[cd]indol-1-yl}-butan-1-one
33 {6-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2a,3,4,5-
tetrahydro-2H-benzo[cd]indol-1-yl}-phenyl-methanone
34 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-6-chloro-2,3-
6.48 0.32
dihydro-indol-1-yl}-ethanone
34 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-6-chloro-2,3-
dihydro-indol-1-yl}-ethanone
34 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-6-chloro-2,3-
dihydro-indol-1-yl}-ethanone
35 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-6-chloro-2,3-
26.38 2.00
dihydro-indol-1-yl}-propan-1-one
36 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-6-chloro-2,3-
47.99 1.05
dihydro-indol-1-yl}-butan-1-one
37 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-6-chloro-2,3-
39.97 1.60
dihydro-indol-1-yl}-2-methyl-propan-1-one
3g 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-6-chloro-2,3-
78.50
dihydro-indol-1-yl}-pentan-1-one
39 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-6-chloro-2,3-
81.31
dihydro-indol-1-yl}-3-methyl-butan-1-one
40 {5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-6-chloro-2,3-
68.82
dihydro-indol-1-yl}-cyclopentyl-methanone
41 {5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-6-chloro-2,3-
dihydro-indol-1-yl}-cyclohexyl-methanone
42 3-{4-[2-(6-Chloro-1-methanesulfonyl-2,3-dihydro-1H-indol-5-yl)-ethyl]-
15.49 1.65
piperazin-1-yl}-benzo[d]isothiazole
43 5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-6-chloro-2,3-
8.94 0.15
dihydro-indole-1-carboxylic acid methylamide
44 5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-6-chloro-2,3-
9.00 0.09
dihydro-indole-1-carboxylic acid ethylamide
45 5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-6-chloro-2,3-
16.49 3.48
dihydro-indole-1-carboxylic acid propylamide
46 5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-6-chloro-2,3-
62.40 2.70
dihydro-indole-1-carboxylic acid isopropylamide
47 5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-6-chloro-2,3-
1.15
dihydro-indole-1-carboxylic acid tert-butylamide
4g 5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-6-chloro-2,3-
2.40
dihydro-indole-1-carboxylic acid cyclopentylamide
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-33-
Example Compound Name D2 5HT2A I~1
Kl
49 5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-6-chloro-2,3-
5.10
dihydro-indole-1-carboxylic acid phenylamide
50 1-{5-[3-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-propyl]-6-chloro-
2,3-23.454.95
dihydro-indol-1-yl}-ethanone
51 1-{6-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-4,4,8-
trimethyl-30.940.65
3,4-dihydro-2H-quinolin-1-yl}-ethanone
52 1-{6-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-7-chloro-
4,4,8-8.77 0.30
trimethyl-3,4-dihydro-2H-quinolin-1-yl}-ethanone
53 1-{6-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-7-chloro-
4,4,8-24.370.70
trimethyl-3,4-dihydro-2H-quinolin-1-yl}-propan-1-one
54 {6-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-7-chloro-4,4,8-
24.002.20
trimethyl-3,4-dihydro-2H-quinolin-1-yl}-cyclopropyl-methanone
55 1-{7-[2-(4-Benzo[d]isoxazol-3-yl-piperazin-1-yl)-ethyl]-2,3,4,5-
20.493.92
tetrahydro-benzo[b]azepin-1-yl}-ethanone;
compound with
methanesulfonic acid
56 1-(7-{2-[4-(5-Fluoro-benzo[d]isothiazol-3-yl)-piperazin-1-yl]-ethyl}-
36.378.77
2,3,4,5-tetrahydro-benzo[b]azepin-1-yl)-ethanone;
compound with
methanesulfonic acid
57 1-(7-{2-[4-(7-Fluoro-benzo[d]isothiazol-3-yl)-piperazin-1-yl]-ethyl}-
38.880.01
2,3,4,5-tetrahydro-benzo[b]azepin-1-yl)-ethanone;
compound with
methanesulfonic acid
{7-[2-(4-Benzo[d]isoxazol-3-yl-piperazin-1-yl)-ethyl]-5,5-dimethy1-59.295.00
2,3,4,5-tetrahydro-benzo[b]azepin-1-yl}-(4-fluoro-phenyl)-methanone;
compound with methanesulfonic acid
61 1-{7-[2-(4-Benzo[d]isoxazol-3-yl-piperazin-1-yl)-ethyl]-5,5-dimethyl-
16.492.30
2,3,4,5-tetrahydro-benzo[b]azepin-1-yl}-propan-1-one;
compound with
methanesulfonic acid
62 1-(6-{2-[4-(1 H-Indazol-3-yl)-piperazin-1-yl]-ethyl}-3,3-dimethyl-3,4-
8.08 1.30
dihydro-2H-quinolin-1-yl)-ethanone
63 1-{6-[2-(4-Benzo[d]isoxazol-3-yl-piperazin-1-yl)-ethyl]-7-chloro-4,4-
19.971.35
dimethyl-3,4-dihydro-2H-quinolin-1-yl}-ethanone;
compound with
methanesulfonic acid
64 1-{6-[2-(4-Benzo[d]isoxazol-3-yl-piperazin-1-yl)-ethyl]-7-chloro-4,4-
25.981.70
dimethyl-3,4-dihydro-2H-quinolin-1-yl}-2-methyl-propan-1-one;
compound with methanesulfonic acid
65 1-{7-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3,4,5-5.43
0.55
tetrahydro-benzo[b]azepin-1-yl}-ethanone;
compound with
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-34-
ExampleCompound Name D2.K1 5HT2A
Kl
methanesulfonic acid
66 1-{5-[3-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-propyl]-3,3-dimethyl-
7.42 1.20
2,3-dihydro-indol-1-yl}-ethanone
73 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-1,3-dihydro-
23.16 0.42
isoindol-2-yl}-ethanone; compound with methanesulfonic
acid
74 1-{5-[2-(4-Benzo[d]isoxazol-3-yl-piperazin-1-yl)-ethyl]-1,3-dihydro-
1.83
isoindol-2-yl}-ethanone; compound with methanesulfonic
acid
g1 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-1,3-dihydro-
77.00 1.35
isoindoi-2-yl}-2-morpholin-4-yl-ethanone
g3 1-{5-[3-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-propyl]-1,3-dihydro-
70.94 3.10
isoindol-2-yl}-ethanone
85 1-{5-[3-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-propyl]-1,3-dihydro-
49.70 4.20
isoindol-2-yl}-2-dimethylamino-ethanone
g6 1-{5-[3-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-propyl]-1,3-dihydro-
2.40
isoindol-2-yl}-2-piperidin-1-yl-ethanone
g7 1-{5-[3-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-propyl]-1,3-dihydro-
56.48 2.90
isoindol-2-yl}-2-morpholin-4-yl-ethanone
8$ 1-{5-[3-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl}-propyl]-1,3-dihydro-
55.50 4.67
isoindol-2-yl}-2-[(2-dimethylamino-ethyl)-methyl-amino]-ethanone
gg 1-{5-[3-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-propyl]-1,3-dihydro-
67.45 9.79
isoindol-2-yl}-2-diethylamino-ethanone .
91 5-[3-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-propyl]-1,3-dihydro-
65.19 1.25
isoindole-2-carboxylic acid (4-fluoro-phenyl)-amide
g5 {5-[3-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-propyl]-1,3-dihydro-
2.55
isoindol-2-yl}-phenyl-methanone
g6 1-{7-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-3,4-dihydro-
1H-18.97 0.85
isoquinolin-2-yl}-2,2,2-trifluoro-ethanone
g7 {6-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-3,4-dihydro-2H-
51.61
quinolin-1-yl}-(4-fluoro-phenyl)-methanone
g7 {6-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl}-ethyl]-3,4-dihydro-2H-
quinolin-1-yl}-(4-fluoro-phenyl)-methanone
gg {6-[2-(4-Benzo[d]isoxazol-3-yl-piperazin-1-yl)-ethyl]-3,4-dihydro-2H-
57.92 4.35
quinolin-1-yl}-(4-fluoro-phenyl)-methanone
gg {6-[2-(4-Benzo[d]isoxazol-3-yl-piperazin-1-yl)-ethyl]-3,4-dihydro-2H-
quinolin-1-yl}-(4-fluoro-phenyl)-methanone
99 1-{5-[2-(4-Benzo[d]isoxazol-3-yl-piperazin-1-yl)-ethyl]-4-fluoro-2,3-
80.11 1.32
dihydro-indol-1-yl}-ethanone
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-35-
ExampleCompound Name D2 5HT2A
Kl Kl
100 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-4-fluoro-2,3-
20.00 0.34
dihydro-indol-1-yl}-ethanone
101 1-{5-[2-(4-Benzo[d]isoxazol-3-yl-piperazin-1-yl)-ethyl]-4-chloro-2,3-
20.45 0.64
dihydro-indol-1-yl}-ethanone
102 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-4-chloro-2,3-
16.83 0.12
dihydro-indol-1-yl}-ethanone
103 1-{5-[2-(4-Benzo[d]isoxazol-3-yl-piperazin-1-yl)-ethyl]-4-chloro-2,3-
56.44 1.46
dihydro-indol-1-yl}-ethanone
104 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-6-fluoro-2,3-
13.96 0.50
dihydro-indol-1-yl}-ethanone
105 1-{5-[2-(4-Benzo[d]isoxazol-3-yl-piperazin-1-yl)-ethyl]-6-fluoro-2,3-
50.48 3.38
dihydro-indol-1-yl}-ethanone
106 1-(6-Chloro-5-{2-[4-(6-fluoro-benzo[d]isothiazol-3-yl)-piperazin-1-yl]-
2.71 0.30
ethyl}-2,3-dihydro-indol-1-yl)-ethanone
107 1-(6-Chloro-5-{2-[4-(5-methoxy-benzo[d]isothiazol-3-yl)-piperazin-1-yl]-
24.50
ethyl}-2,3-dihydro-indol-1-yl)-ethanone
108 1-(6-Chloro-5-{2-[4-(7-fluoro-benzo[d]isothiazol-3-yl)-piperazin-1-yl]-
8.05
ethyl}-2,3-dihydro-indol-1-yl)-ethanone
109 1-(6-Chloro-5-{2-[4-(7-methoxy-benzo[d]isothiazol-3-yl)-piperazin-1-yl]-
20.50
ethyl}-2,3-dihydro-indol-1-yl)-ethanone
110 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperidin-1-yl)-ethyl]-6-chloro-2,3-
2.45 0.45
dihydro-indol-1-yl}-ethanone
111 1-(6-Chloro-5-{2-(4-(5-fluoro-benzo[d]isothiazol-3-yl)-piperazin-1-yl]-
83.67 21.00
ethyl}-2,3-dihydro-indol-1-yl)-ethanone
112 1-(6-Chloro-5-{2-[4-(6-fluoro-benzo[d]isoxazol-3-yl)-piperidin-1-yl]-
13.98 2.15
ethyl}-2,3-dihydro-indol-1-yl)-ethanone
113 1-{5-[2-(4-Benzo[d]isoxazol-3-yl-piperazin-1-yl)-ethyl]-6-chloro-2,3-
12.63 0.55
dihydro-indol-1-yl}-ethanone
114 1-(6-Chloro-5-{2-[4-(5-fluoro-benzo[d]isoxazol-3-yl)-piperazin-1-yl]-
23.00
ethyl}-2,3-dihydro-indol-1-yl)-ethanone
115 1-(6-Chloro-5-{2-[4-(5-chloro-benzo[d]isoxazol-3-yl)-piperazin-1-yl]-
26.00
ethyl}-2,3-dihydro-indol-1-yl)-ethanone
116 1-(6-Chloro-5-{2-[4-(6-fluoro-benzo[d]isoxazol-3-yl)-piperazin-1-yl]-
18.98 2.65
ethyl}-2,3-dihydro-indol-1-yl)-ethanone
117 1-(6-Chloro-5-{2-[4-(6-methyl-benzo[d]isoxazol-3-yl)-piperazin-1-yl]-
20.93 6.90
ethyl}-2,3-dihydro-indol-1-yl)-ethanone
118 1-(6-Chloro-5-{2-[4-(7-methyl-benzo[d]isoxazol-3-yl)-piperazin-1-yl]-
19.50
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-36-
ExampleCompound Naive D2 5HT2A
K1 Kl
ethyl}-2,3-dihydro-indol-1-yl)-ethanone
119 1-{5-[2-(4-Benzo[b]thiophen-3-yl-piperazin-1-yl)-ethyl]-6-chloro-2,3-
48.76 14.00
dihydro-indol-1-yl}-ethanone
120 1-(6-Chloro-5-{2-[4-(1H-indazol-3-yl)-piperazin-1-yl]-ethyl}-2,3-
dihydro-g,95 1.55
indol-1-yl)-ethanone
121 1-(5-{2-[4-(6-Fluoro-benzo[d]isothiazol-3-yl)-piperazin-1-yl]-ethyl}-
2,3-10.39 0.10
dihydro-indol-1-yl)-ethanone
122 1-(5-{2-[4-(5-Methoxy-benzo[d]isothiazol-3-yl)-piperazin-1-yl]-ethyl}-
2,3-62.63 8.05
dihydro-indol-1-yl)-ethanone
123 1-(5-{2-[4-(7-Fluoro-benzo[d]isothiazol-3-yl)-piperazin-1-yl]-ethyl}-
2,3-93.00 1.85
dihydro-indol-1-yl)-ethanone
124 1-(5-{2-[4-(7-Methoxy-benzo[d]isothiazol-3-yl)-piperazin-1-yl]-ethyl}-
2,3- 30.50
dihydro-indol-1-yl)-ethanone
125 1-(5-{2-[4-(5-Fluoro-benzo[d]isothiazol-3-yl)-piperazin-1-yl]-ethyl}-
2,3-67.35 2.20
dihydro-indol-1-yl)-ethanone
126 1-{5-[2-(4-Benzo[d]isoxazol-3-yl-piperazin-1-yl)-ethyl]-2,3-dihydro-
indol-88,00 1.15
1-yl}-ethanone
127 1-(5-{2-[4-(5-Fluoro-benzo[d]isoxazol-3-yl)-piperazin-1-yl]-ethyl}-2,3-
18.50
dihydro-indol-1-yl)-ethanone
128 i -(5-{2-[4-(5-Chloro-benzojd]isoxazol-3-yl)-piperazin-1-yl]-ethyl}-2,3-
78.00 5.85
dihydro-indol-1-yl)-ethanone ,
129 1-(5-{2-[4-(6-Fluoro-benzo[d]isoxazol-3-yl)-piperazin-1-yl]-ethyl}-2,3-
1.50
dihydro-indol-1-yl)-ethanone
130 1-(5-{2-[4-(7-Methyl-benzo[d]isoxazol-3-yl)-piperazin-1-yl]-ethyl}-2,3-
14.35
dihydro-indol-1-yl)-ethanone
131 1-t5-[2-(4-Benzo[b]thiophen-3-yl-piperazin-1-yl)-ethyl]-2,3-dihydro-
indol-20.35 2.20
1-yl}-ethanone
132 1-(5-{2-[4-(1 H-Indazol-3-yl)-piperazin-1-yl]-ethyl}-2,3-dihydro-indol-
1-yl)-62.16 1.30
ethanone
134 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3-dihydro-
21.35 0.60
indol-1-y1}-ethanone
135 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3-dihydro-
12.41 0.90
indol-1-yl}-propan-1-one
136 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3-dihydro-
29.98 1.50
indol-1-yl}-butan-1-one
137 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3-dihydro-
36.66 0.07
indo!-1-yl}-2-methyl-propan-1-one
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-37-
ExampleCompound Name, D2 5HT2A
Kl Kl
138 {5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3-dihydro-
indol-15.97 0.65
1-yl}-cyclopropyl-methanone
139 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3-dihydro-
55.23 0.20
indol-1-yl}-pentan-1-one
140 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3-dihydro-
53.96 0.35
indol-1-yl}-3-methyl-butan-1-one
141 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3-dihydro-
22.49 0.20
indol-1-yl}-2,2-dimethyl-propan-1-one
142 {5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3-dihydro-
indol-36.00 0.40
1-yl}-cyclopentyl-methanone
143 {5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3-dihydro-
indol-75.94 0.45
1-yl}-cyclohexyl-methanone
144 {5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3-dihydro-
indol-34.99 0.55
1-yl}-phenyl-methanone
145 3-{4-[2-(1-Methanesulfonyl-2,3-dihydro-1H-indol-5-yl)-ethyl]-piperazin-
12.49 0.06
1-yl}-benzo[d]isothiazole
146 5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3-dihydro-
indole-60.50 0.15
1-carboxylic acid methylamide
147 5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3-dihydro-
indole-74.00 0.35
1-carboxylic acid ethylamide
148 5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3-dihydro-
indole-56.00 0.35
1-carboxylic acid propylamide
149 5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3-dihydro-
indole- 0.20
1-carboxylic acid isopropylamide
150 5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3-dihydro-
indole- 0.30
1-carboxylic acid tert-butylamide
151 5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3-dihydro-
indole-69.00 0.55
1-carboxylic acid cyclopentylamide
152 5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3-dihydro-
indole- 2.75
1-carboxylic acid phenylamide
154 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3-dihydro-
10.39 1.35
indol-1-yl}-2-pyrrolidin-1-yl-ethanone
155 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3-dihydro-
35.33 1.85
indol-1-yl}-2-diethylamino-ethanone
156 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3-dihydro-
24.90 2.08
indol-1-yl}-2-dimethylamino-ethanone
157 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3-dihydro-
77.48 1.14
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-38-
ExampleCompound Name D2 5HT2A
K1 I~1
indol-1-yl}-2-morpholin-4-yl-ethanone
158 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3-dihydro-
23.54 2.48
indol-1-yl}-2-piperidin-1-yl-ethanone
160 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3-dihydro-
18.57 0.65
indol-1-yl}-3-pyrrolidin-1-yl-propan-1-one
161 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3-dihydro-
18.57 0.75
indol-1-yl}-3-diethylamino-propan-1-one
162 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3-dihydro-
12.40 2.08
indol-1-yl}-3-d imethylam ino-propan-1-one
163 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3-dihydro-
27.71 2.77
indol-1-yl}-3-morpholin-4-yl-propan-1-one
164 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3-dihydro-
20.90 2.13
indol-1-yl}-3-piperidin-1-yl-propan-1-one
The following Examples illustrate the preparation of the compounds of the
present invention. Melting points are uncorrected. NMR data are reported in
parts
per million and are referenced to the deuterium lock signal from the sample
solvent.
EXAMPLES
PREPARATION 1
5-(2-Chloroacetyll-3,3-dimethyl-1,3-dihydroindol-2-one
A 12.5 L 4-neck flask equipped with a mechanical stirrer, reflux condenser
and two stoppers and heating mantle, was charged with AICI3 (633.29 g, 4.75
mol), 2
L of carbon disulfide and chloroacetyl chloride (87 mL, 1.09 mol) and this was
stirred
at room temperature (rt) during the portionwise addition of 3,3-dimethyl-1,3-
dihydro-
indol-2-one (123.5 g, 0.766 mol). This mixture was then heated to reflux for 3
hours,
then cooled overnight. The solvent was decanted and the reaction was quenched
with addition of ice and water (8 L). The suspension was stirred vigorously
for 1.5
hours, followed by filtration. The solids were washed with water (4.2 L) and
then
dried overnight in a vacuum oven at 50 degrees Celsius (°C) (192.19 g
of 5-(2
chloroacetyl)-3,3-dimethyl-1,3-dihydroindol-2-one). 29.54 g of this material
was
taken up in hot acetone and purified by flash chromatography (250 g silica
gel)
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-39-
eluting with acetone which provided >96% pure 5-(2-chloroacetyl)-3,3-dimethyl-
1,3-
dihydroindol-2-one by HPLC. Yield = 15.54 g (52%); MS (APCI), (M + 1)+= 238.
Anal. calculated for C12H12CINO2: C, 60.64; H, 5.09; N, 5.89. Found: C, 61.04;
H,
5.15; N, 5.38%.
PREPARATION 2
S-(2-Chloroethyl)-3,3-dimethyl-1,3-dihydroindol-2-one
A 5 L 4-neck flask equipped with a mechanical stirrer, 1 L addition funnel and
two stoppers were charged with 5-(2-chloroacetyl)-3,3-dimethyl-1,3-
dihydroindol-2-
one (162.65 g, 0.684 mol) and this was taken up in trifluoroacetic acid (700
mL).
The solution was cooled in an icelwater bath, followed by addition of
triethylsilane
(260 mL) over a 1 hour period. The reaction was stirred at rt overnight. The
mixture
as poured into a 12.5 L flask containing 8 L of rapidly stirring water. The
reaction
flask was washed with 1.5 L of water and 2 L of heptanes, both added to the
12.5 L
flask. The mixture was again stirred overnight. The suspension was vacuum
filtered
and the solids were washed with water (2 L) and heptanes (2 L) and dried
overnight
in a vacuum oven (73.2 g). The solid was purified by flash chromatography (550
g
silica gel) eluting with acetone (2 L). Yield = 68.38g (45%); MS (APCI), (M +
1 )+ _
224. Anal. calculated for C12H12CIN02: C, 64.43; H, 6.31; N, 6.29. Found: C,
64.35;
H, 6.36; N: 5.84.
PREPARATION 3
5-f2-(4-Benzofdlisothiazol-3-yl-piperazin-1-yl)-ethyll-3,3-dimethyl-1,3-
dihydro
indol-2-one
A 500 mL round bottom (RB) flask was charged with 3-piperazin-1-yl-
benzo[dJisothiazole (12.5 g, 5.0 mol), 5-(2-chloroethyl)-3,3-dimethyl-1,3-
dihydroindol-
2-one (11.5 g, 5.0 mol) and sodium carbonate (10.5 g, 10.0 mol), diluting with
water
(200 mL). The stirring reaction was warmed to reflux for 24 hours. The
reaction was
slowly cooled with vigorous stirring and a solid formed. The tan solid was
filtered
washed with ether and dried in a vacuum oven. Yield = 19.49 g (96%); 1 H NMR
(400
MHz, DMSO-d6) S ppm 1.20 (s, 6 H) 2.61 (m, 8 H) 3.42 (m, 5 H) 6.73 (d, J=7.82
Hz,
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-40-
1 H) 7.00 (dd, J=7.82 Hz, 1 H) 7.14 (d, 1 H) 7.41 (t, J--7.69 Hz, 1 H) 7.53
(t, J--7.45
Hz, 1 H) 8.03 (d, J=9.04 Hz, 2 H) 10.21 (s, 2 H)
PREPARATION 4
3-~'4-f2-(3,3-Dimethyl-2,3-dihydro-1 H-indol-5-yl)-ethyll-piperazin-1-yl~-
benzofd~isothiazole
A solution of 5-[2-(4-benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-3,3-
dimethyl-
1,3-dihydro-indol-2-one (810 mg, 2.0 mmol) in toluene (15 mL) with stirring at
rt was
treated with borane dimethyl sulfide in toluene (2 mL, 4 mmol). The reaction
was
warmed to reflux for 1 hour. The reaction was cooled and treated with a 10%
aqueous solution of sodium carbonate (10 mL) and warmed to reflux for 20
hours.
The reaction was cooled and the layers were separated. The aqueous layers were
extracted with ethyl acetate (2 x 20 mL). The combined organics were dried
over
magnesium sulfate, filtered and the filtrate concentrated. The crude product
was
eluted through a flash column (silica gel 40, 230-400 mesh, methylene chloride
(CH2Cl2) to 8% ethanol (EtOH) and 1% ammonium hydroxide (NH40H in CH2CI2) to
give the title compound as a brown oily solid, yield = 620 mg (79%). 'H-NMR
(CDCI3, 8): 7.91 (d, J=8.30 Hz, 1 H) 7.81 (d, J=8.30 Hz, 1 H) 7.47 (t, J--7.56
Hz, 1 H)
7.35 (t, J--7.56 Hz, 1 H) 6.89 (m, 2 H) 6.59 (d, J 8.30 Hz, 1 H) 3.61 (m, 4 H)
3.30 (s,
2 H) 2.69 (m, 8 H) 1.30 (s, 6 H).
EXAMPLE 1
~5-f2-(4-Benzofdlisothiazol-3-yl-piperazin-1-yl)-ethyll-3,3-dimethyl-2,3-
dihydro
indol-1-yf1~-(4-fluoro-phenyl)-methanone
3-{4-[2-(3,3-Dimethyl-2,3-dihydro-1 H-indol-5-yl)-ethyl]-piperazin-1-yl~-1,2-
benzo[d]isothiazole was diluted to 0.20 M with anhydrous dichloromethane, then
delivered to an 8 mL vial via pipette (0.20 mmol). To the amine solution was
added
polystyrene-N-Methylmorpholine resin (PS-N-Methylmorpholine resin) (0.40
mmol).
Isoxazole-4-fluoro-benzoyl chloride (0.40 mmol) was diluted to 0.20 M with
dichloromethane, and added at rt. The solution was shaken overnight at rt.
Polyamine scavenging resin (0.5 mmol) was added. The solution was shaken
overnight at rt, then filtered into an 8 mL vial. The filtrate was evaluated
by MS, then
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-41-
concentrated via HT-12 GeneVac. Crude was purified by HPLC (30x100 mm ODS-A
C(18) 5u column). 4-[2{5-[2-(4-1,2-Benzisothiazol-3-yl-piperazin-1-yl)-ethyl]-
3,3-
dimethyl-2,3-dihydro-indol-1-yl}-(4-fluoro-phenyl)-methanone was isolated in
98%
purity @ 254 nm, LCMS (APCI) 515 [M+H]+.
The amides of Examples 2-22 were synthesized in combinatorial library
format following the steps outlined in Example 1 on a 0.2 mmol scale using 3-
{4-[2-
(3,3-dimethyl-2,3-dihydro-1 H-indol-5-yl)-ethyl]-piperazin-1-yl}-1,2-
benz[o]isothiazole
with appropriate acid chloride starting materials and PS-N methylmorpholine.
The
crude products were purified by HPLC (30x100 mm ODS-A C(18) 5u column).
EXAMPLE a COMPOUND NAME ' ~ DATA
= } A
~ ~
#2 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-Isolated in 100% purity
@
254
piperazin-1-yl)-ethyl]-3,3-dimethyl-2,3-nm; LCMS (APCI) 541
[M+H]+
dihydro-indol-1-yl }-2-(3-methoxy-phenyl)-
ethanone
#3 1-{5-[2-(4-Benzo[d]isothiazol-3-ylIsolated in 100% purity
@ 254
piperazin-1-yl)-ethyl]-3,3-dimethyl-2,3nm; LCMS (APCI) 517
[M+H]+
dihydro-indol-1-yl}-2-thiophen-2-yl
ethanone
#4 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-Isolated in 96% purity
@ 254
piperazin-1-yl)-ethyl]-3,3-dimethyl-2,3-nm; LCMS (APCI) 541
[M+H]+
dihydro-indol-1-yl }-2-phenoxy-propan-1-one
#5 {5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-Isolated in 100% purity
@ 254
1-yl)-ethyl]-3,3-dimethyl-2,3-dihydro-indol-nm; LCMS (APCn 515[M+H]+
1-yl }-(2,5-dimethyl-2H-pyrazol-3-yl)-
methanone
#6 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-Isolated in 100% purity
@ 254
piperazin-1-yl)-ethyl]-3,3-dimethyl-2,3-nm; LCMS (APCI) 463
[M+H]+
dihydro-indol-1-yl }-butan-1-one
#7 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-Isolated in 100% purity
@ 254
piperazin-1-yl)-ethyl]-3,3-dimethyl-2,3-nm; LCMS (APCI) 463
[M+H]+
dihydro-indol-1-yl }-2-methyl-propan-1-one
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-42-
EXAMPLE COMPOUND NAME DATA
#8 {5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-Isolated in 98% purity
@ 254
1-yl)-ethyl]-3,3-dimethyl-2,3-dihydro-indol-nm; LCMS (APCI) 511
[M+H]+
1-yl }-m-tolyl-methanone
#9 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-Isolated in 100% purity
@ 254
piperazin-1-yl)-ethyl]-3,3-dimethyl-2,3-nm; LCMS (APCI) 562
[M+H]+
dihydro-indol-1-yl } -2-(4-chloro-phenoxy)-
ethanone
#10 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-Isolated in 97% purity
@ 254
piperazin-1-yl)-ethyl]-3,3-dimethyl-2,3-nm; LCMS (APC)) 525
[M+H]+
dihydro-indol-1-yl } -3-phenyl-propan-1-one
#11 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-Isolated in 100% purity
@ 254
piperazin-1-yl)-ethyl]-3,3-dimethyl-2,3-nm; LCMS (APCI) 571
[M+H]+
dihydro-indol-1-yl }-2-(3,4-dimethoxy-
phenyl)-ethanone
#12 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-Isolated in 96% purity
@ 254
piperazin-1-yl)-ethyl]-3,3-dimethyl-2,3-nm; LCMS (APCI) 546
[M+H]+
dihydro-indol-1-yl}-2-(4-chloro-phenyl)-
ethanone
#13 {5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-Isolated in 100% purity
@ 254
1-yl)-ethyl]-3,3-dimethyl-2,3-dihydro-indol-nm; LCMS (APCI) 527
[M+H]+
1-yl }-(4-methoxy-phenyl)-methanone
#14 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-Isolated in 100% purity
@ 254
piperazin-1-yl)-ethyl]-3,3-dimethyl-2,3-nm; LCMS (APCI) 511
[M+H]+
dihydro-indol-1-yl }-2-phenyl-ethanone
#15 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-Isolated in 100% purity
@ 254
piperazin-1-yl)-ethyl]-3,3-dimethyl-2,3-nm; LCMS (APC)] 571
[M+H]+
dihydro-indol-1-yl }-2-(2,5-dimethoxy-
phenyl)-ethanone
#16 5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-Isolated in 90% purity
@ 254
yl)-ethyl]-3,3-dimethyl-2,3-dihydro-indole-1-nm; LCMS (APCI) 513
[M+H]+
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-43-
EXAMPLE COMPOUND NAME - DATA
carboxylic acid phenyl ester
#17 {5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-Isolated in 100% purity
@ 254
1-yl)-ethyl]-3,3-dimethyl-2,3-dihydro-indol-nm; LCMS (APCI) 487
[M+H]~
1-yl}-furan-2-yl-methanone
#18 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-Isolated in 100% purity
@ 254
piperazin-1-yl)-ethyl]-3,3-dimethyl-2,3-nm; LCMS (APCI) 477
[M+H]+
dihydro-indol-1-yl}-3-methyl-butan-1-one
#19 {5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-Isolated in 100% purity
@ 254
1-yl)-ethyl]-3,3-dimethyl-2,3-dihydro-indol-nm; LCMS (APCI) 489
[M+H]+
1-yl }-cyclopentyl-methanone
#20 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-Isolated in 100% purity
@ 254
piperazin-1-yl)-ethyl]-3,3-dimethyl-2,3-nm; LCMS (APCI) 541
[M+H]+
dihydro-indol-1-yl }-2-benzyloxy-ethanone
#21 {5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-Isolated in 100% purity
@ 254
1-yl)-ethyl]-3,3-dimethyl-2,3-dihydro-indol-nm; LCMS (APCI) 497
[M+H]~
1-yl }-phenyl-methanone
#22 1-{5-[2-(4-Benzo[d]isothiazol-3-yl-Isolated in 100% purity
@ 254
piperazin-1-yl)-ethyl]-3,3-dimethyl-2,3-nm; LCMS (APCI) 503
[M+H]+
dihydro-indol-1-yl }-2-cyclopentyl-ethanone
EXAMPLE 23
1-f 5-f 2-(4-Benzof dlisothiazoi-3-yl-piperazin-1-yl)-ethyll-3,3-dimethyl-2,3
dihydro-indol-1-yl~-ethanone
3-{4-[2-(3,3-Dimethyl-2,3-dihydro-1 H-indol-5-yl)-ethyl]-piperazin-1-yl)-1,2
benz[o]isothiazole (0.81 mmol, 313 mg) was diluted with anhydrous THF (5 mL)
and
triethylamine (0.225 mL) then treated with acetyl chloride (0.71 i ml) and
allowed to
stir for 72 hours (h or hr). The reaction was filtered and the filtrate was
concentrated.
The crude solid was washed with saturated sodium carbonate solution (10 mL)
and
extracted with methylene chloride (25 mL) dried and concentrated to oily
solid. The
solid was crystallized from ether to yield pure 1-{5-[2-(4-benzo[d]isothiazol-
3-yl-
piperazin-1-yl)-ethyl]-3,3-dimethyl-2,3-dihydro-indol-1-yl}-ethanone : Yield:
182 mg
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-44-
(52%). 1H NMR (400 MHz,CDCl3) 5 ppm 1.34 (s, 6 H) 2.19 (s, 3 H) 2.76 (m, 8 H)
3.60 (s, 4 H) 3.75 (s, 2 H) 6.98 (s, 1 H) 7.05 (d, J--8.06 Hz, 1 H) 7.34 (t, J-
-7.57 Hz, 1
H) 7.46 (t, J=7.57 Hz, 1 H) 7.80 (d, J--8.30 Hz, 1 H) 7.90 (d, J--8.30 Hz, 1
H) 8.08 (d,
J=8.06 Hz, 1 H) MS (APCI) = 435.2 [M+H]+.
EXAMPLE 24
5-f2-(4-Benzo~dlisothiazol-3-yl-piperazin-1-yl)-ethyll-3,3-dimethyl-2,3-
dihydro
indole-1-carboxylic acid isopropylamide
A solution of 3-f4-[2-(3,3-dimethyl-2,3-dihydro-1 H-indol-5-yl)-ethyl]-
piperazin-
1-yl}-1,2-benz[o]isothiazole (0.4 mmols, 160 mg) in THF (5mL) was treated by
dropwise addition with isopropyl isocyanate at rt (RT or rt) and allowed to
stir for 4
days. The reaction was concentrated to dryness, diluted with H20 and extracted
with
CH2CI2. The organics were placed through a phase separator and dried down in a
100X16mm tube, resulting in a solid. The solid was then recrystallized from
acetonitrile to yield pure 5-[2-(4-benzo[d]isothiazol-3-yl-piperazin-1-yl)-
ethyl]-3,3-
dimethyl-2,3-dihydro-indole-1-carboxylic acid isopropylamide. Yield: 112 mg
(58%).
1H NMR (400 MHz, DMSO-d6) 8 ppm 1.07 (d, J--6.59 Hz, 6 H) 1.22 (s, 6 H) 2.60
(m,
8 H) 3.40 (s, 4 H) 3.57 (s, 2 H) 3.80 (m, 1 H) 6.14 (d, J=7.81 Hz, 1 H) 6.90
(d, J--8.05
Hz, 1 H) 6.98 (s, 1 H) 7.38 (t, J--7.69 Hz, 1 H) 7.51 (t, J--7.81 Hz, 1 H)
7.65 (d,
J--8.05 Hz, 1 H) 8.00 (d, J--8.78 Hz, 2 H).
PREPARATION 5
6-(2-Chloro-acetyl)-2a,3,4,5-tetrahydro-1 H-benzo~cdlindol-2-one
A RB flask was charged with 60 mL dichloroethane and AIC13 (11.5 g, 86
mmol, 3 eq) and cooled on an ice-bath. To this solution was added,
portionwise, 5.0
g (29 mmol, 1 eq) of the known 2a,3,4,5-tetrahydro-1 H-benzo[cd]indol-2-one
(Protiva, M.; Sedivy, Z.; Holubek, J.; Svatek, E.; Nemec, J. Collection of
Czechoslovak Chemical Communications 1985, 50, 1888-98, Clark, R. D.;
Muchowski, J. M.; Fisher, L. E.; Flippin, L. A.; Repke, D. B.; Souchet, M.
Synthesis
1991, 871-8). After 20 min, 2.8 mL (34 mmol, 1.2 eq) of chloroacetylchloride
was
added and the solution was allowed to warm slowly to rt. After 30 hours at rt
the
solution was poured carefully into 500 mL of iced-water and the resulting
precipitate
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-45-
was filtered and washed with chloroform. The white solid was oven dried
overnight
at 60 °C under vacuum to give 6.0 g (83 % yield) of the desired product
as a beige
solid. 1H NMR (400 MHz, DMSO-d6) 8 ppm 1.1 (m, J--12.3, 12.3, 12.1, 3.3 Hz, 1
H)
1.7 (m, 1 H) 2.0 (m, J=10.4, 7.2, 3.7, 1.7 Hz, 1 H) 2.1 (ddd, J--12.0, 8.7,
3.4 Hz, 1 H)
2.7 (m, 1 H) 3.1 (dd, J--19.0, 7.1 Hz, 1 H) 3.3 (s, 1 H) 3.4 (dd, J--12.1, 5.2
Hz, 1 H)
5.0 (m, 2 H) 6.7 (d, J=8.3 Hz, 1 H) 7.8 (dd, J--8.3, 1.0 Hz, 1 H) 10.6 (s, 1
H). MS
(APCI), (M+1 )+ = 250.
PREPARATION 6
6-(2-Chloro-ethyl)-2a,3,4,5-tetrahydro-1 H-benzofcdlindol-2-one
A RB flasle was charged with 5.85 g (23 mmol, 1 eq) 6-(2-chloro-acetyl)-
2a,3,4,5-tetrahydro-1 H-benzo[cd]indol-2-one, 18 mL (10 eq) trifluoroacetic
acid and
11 mL (3 eq) triethylsilane and heated to 60 °C for 4 hr. The reaction
mixture was
then quenched carefully into 300 mL of iced water and extracted with 400 mL of
CH2C12. The aqueous layer was extracted with an additional 200 mL CH2C12,
dried
over MgS04 and concentrated in vacuo to give 11 g of a semi-solid. This
material
was triturated with 200 mL of 1:1 diethylether:hexanes and the resulting solid
dried in
vacuo to give 4.68 g (86 % yield) of the desired material as a pale yellow
solid. ' H
NMR (400 MHz, DMSO-d6) S ppm 1.1 (m, 1 H) 1.8 (m, 1 H) 2.0 (m, 1 H) 2.1 (m, 1
H)
2.5 (m, 1 H) 2.7 (dd, J--17.6, 7.3 Hz, 1 H) 2.9 (td, J=14.8, 6.6 Hz, 2 H) 3.3
(m, 1 H)
3.7 (m, 2 H) 6.5 (d, J--7.6 Hz, 1 H) 6.9 (d, J--7.8 Hz, 1 H) 10.1 (s, 1 H). MS
(APCI),
(M+1 )+ = 236.
PREPARATION 7
6-f2-(4-Benzo[dlisothiazol-3-yl-piperazin-1-yl)-ethyll-2a,3,4,5-tetrahydro-1H-
benzofcdlindol-2-one
A 20 mL reaction vial was charged with 0.47 g (2 mmol, 1 eq) 6-(2-chloro-
ethyl)-2a,3,4,5-tetrahydro-1 H-benzo[cd]indol-2-one, 0.51 g 3-piperazin-1-yl-
benzo[d]-
iso-thiazole hydrochloride, and 5 mL of 1 M aqueous sodium carbonate (Na2C03)
and heated to 100°C for 60 h. The solid was filtered, washed with water
and dried in
vacuo overnight. The resulting solid was purified by medium pressure liquid
chromatography (MPLC) (ethyl acetate (EtOAc) eluent) to give 0.36 g (43 %
yield) of
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-46-
the desired material as a white solid. 1H NMR (400 MHz, CDCI3) 8 ppm 1.3 (qd,
J--12.3, 3.5 Hz, 1 H) 1.9 (m, 1 H) 2.2 (m, 1 H) 2.4 (dt, J--12.2, 4.4 Hz, 1 H)
2.6 (m, 3
H)2.8(m,6H)3.3(dd,J--11.8,5.OHz,IH)3.6(s,4H)6.6(d,J=7.6Hz,lH)7.0
(d, J--7.8 Hz, 1 H) 7.3 (m, 1 H) 7.5 (m, 1 H) 7.8 (d, J--8.1 Hz, 1 H) 7.9 (d,
J=8.1 Hz, 1
H) 8.2 (s, 1 H). MS (APCI), (M+1 )+ = 292, 419.
PREPARATION 8
6-f2-(4-Benzofdlisothiazol-3-yl-piperazin-1-yl)-ethyl~-1,2,2a,3,4,5-hexahydro-
benzofcdlindole
A RB flask was charged with 5.0 g (12 mmol, 1 eq) 6-[2-(4-benzo[d]isothiazol-
3-yl-piperazin-1-yl)-ethyl]-2a,3,4,5-tetrahydro-1 H-benzo[cd]indol-2-one and
50 mL
tetrahydrofuran (THF) and cooled on an ice bath. The reaction was treated
dropwise
with 48 mL (48 mmol, 4 eq) 1 M borane-THF (BH3-THF) in THF over 0.5 h and then
allowed to warm slowly to rt. After 24 h the reaction was quenched with 20 mL
methanol (MeOH) (gas evolution) and heated at 50°C for 15 h. The
reaction was
cooled, partitioned between CH2CI2 and brine. The organic extracts were dried
over
magnesium sulfate (MgS04), filtered, and concentrated to give 4.7 g of a
yellow foam
that was a mixture of indole and indoline products by'H NMR. This mixture was
taken on without purification.
A RB flask was charged with 4.38 g of the above mixture and 43 mL acetic
acid (HOAc). This mixture was treated with 1.8 g (27 mmol, 2.5 eq) sodium
cyanoborohydride (NaCNBH3) and stirred at rt for 15 h. The reaction solvent
was
removed in vacuo and the resulting solid was dissolved in 200 mL CH2CI2 and
washed with 1 M sodium bicarbonate (NaHCOs). The aqueous layer was back-
extracted with 100 mL CH2C12. The combined organic layers were washed with
brine, separated, and dried over MgS04. Concentration in vacuo gave 4.0 g of a
yellow foam that was purified by MPLC to give 1.65 g (33 % yield) of the
desired
material as a slightly yellow foam. 1H NMR (400 MHz, CDC13) S ppm 1.3 (d, J--
10.5
Hz, 1 H) 1.7 (s, 1 H) 2.1 (m, 2 H) 2.6 (dd, J--10.3, 5.4 Hz, 4 H) 2.8 (m, 7 H)
3.1 (m, 2
H) 3.6 (m, 5 H) 6.5 (d, J--7.6 Hz, 1 H) 6.8 (d, J--7.6 Hz, 1 H) 7.3 (dd, J--
8.1, 7.1 Hz, 1
H) 7.5 (m, 1 H) 7.8 (m, 1 H) 7.9 (d, J--8.3 Hz, 1 H). MS (APCI), (M+1 )+ =
403.
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-47-
EXAMPLE 25
1-d6-f2-(4-Benzofdlisothiazol-3-yl-piperazin-1-yl)-ethyll-2a,3,4,5-tetrahydro-
2H
benzofcdlindol-1-yl)-ethanone
A solution of 1.0 g (2.0 mmol, 1 eq) 6-[2-(4-benzo[d]isothiazol-3-yl-piperazin-
1-yl)-ethyl]-1,2,2a,3,4,5-hexahydro-benzo[cd]indole was treated with 0.689 mL
(4.9
mmol, 2 eq) triethylamine (Et3N) and then 0.263 mL (3.7 mmol, 1.5 eq) acetyl
chloride and stirred at rt for 20 h. The reaction was quenched with 1 M
NaHC03,
filtered through 5 p.m PTFE (phase-separating filter), and concentrated in
vacuo to
give 1.19 g (quantitative yield) of a pale-yellow foam. 1H NMR (400 MHz,
CDCI3)
(Integration does not take into account a 3:1 mixture of rotamers, the
presence of
which is most pronounced at the peaks labeled major/minor.) ppm 1.1 (m, 3 H)
1.2
(m, 1 H) 1.3 (m, 1 H) 1.8 (m, 1 H) 2.1 (m, 5 H) 2.4 (s, 1 H) 2.6 (m, 5 H) 2.8
(m, 7 H)
2.9 (t, J=7.3 Hz, 1 H) 3.3 (m, 1 H) 3.5 (dd, J--11.2 Hz, 1 H) 3.6 (m, 4 H) 4.2
(major, t,
J--9.2 Hz, 1 H) 4.6 (minor, 1 H) 6.8 (minor, d, J--8.1 Hz, 1 H) 7.0 (major, m,
1 H) 7.3
(m, 1 H) 7.4 (t, J=7.6 Hz, 1 H) 7.8 (d, J--8.1 Hz, 2 H) 7.9 (d, J--8.1 Hz, 1
H). MS
(APCI), (M+1 )+ = 447.
The title compounds of Examples 26 through 33 were prepared from 6-[2-(4-
benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-1,2,2a,3,4,5-hexahydro-
benzo[cd]indole
in a fashion similar to that reported above. Rt (min) reported is for the
following high
pressure liquid chromatography (HPLC) conditions: 60:40 [H2O:MeCN]+0.1 % TFA,
1.5 mUmin, 250x4.6 mm LUNA Cps. Purity for each compound reported is > 90% by
UV (254 nM).
EXAMPLE COMPOUND NAME ~ DATA
#26 { 6-[2-(4-Benzo[d]isothiazol-3-yl-HPLC: Rt = 3.005;
MS
piperazin-1-yl)-ethyl]-2a,3,4,5-tetrahydro-(APCI), (M+1)+ = 473
2H-benzo [cd]indol-1-yl }-cyclopropyl-
methanone
#27 1-{6-[2-(4-Benzo[d]isothiazol-3-yl-HPLC: Rt = 2.898;
MS
piperazin-1-yl)-ethyl]-2a,3,4,5-tetrahydro-(APCI), (M+1)+ = 461
2H-benzo[cd]indol-1-yl}-propan-1-one
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-48-
#28 1-{6-[2-(4-Benzo[d]isothiazol-3-yl-HPLC: Rt - 3.885;
MS
piperazin-1-yl)-ethyl]-2a,3,4,5-tetrahydro-(APCI), (M+1)+ =
489
2H-benzo[cd]indol-1-yl }-2,2-dimethyl-
propan-1-one
#29 1-{6-[2-(4-Benzo[d]isothiazol-3-yl-HPLC: Rt = 3.935;
MS
piperazin-1-yl)-ethyl]-2a,3,4,5-tetrahydro-(APCZ], (M+1)+ =
489
2H-benzo [cd] indol-1-yl }
-pentan-1-one
#30 1-{6-[2-(4-Benzo[d]isothiazol-3-yl-HPLC: Rt = 3.753;
MS
piperazin-1-yl)-ethyl]-2a,3,4,5-tetrahydro-(APCl7, (M+1)+ =
489
2H-benzo [cd] indol-1-yl }
-3-methyl-butan-
1-one
#31 1-{6-[2-(4-Benzo[d]isothiazol-3-yl-HPLC: Rt - 3.218;
MS
piperazin-1-yl)-ethyl]-2a,3,4,5-tetrahydro-(APCn, (M+1)+ = 475
2H-benzo[cd]indol-1-yl}-2-methyl-propan-
1-one
#32 1-{6-[2-(4-Benzo[d]isothiazol-3-yl-HPLC: Rt = 3.302;
MS
piperazin-1-yl)-ethyl]-2a,3,4,5-tetrahydro-(APCn, (M+1)+ = 475
2H-benzo[cd]indol-1-yl}-butan-1-one
#33 {6-[2-(4-Benzo[d]isothiazol-3-yl-HPLC: Rt = 3.441;
MS
piperazin-1-yl)-ethyl]-2a,3,4,5-tetrahydro-(APCl7, (M+1)+= 509
2H-benzo[cd]indol-1-yl}-phenyl-
methanone
PREPARATION 9
3-f4-f2-(6-Chloro-2,3-dihydro-1 H-indol-5-yl)-ethyll-piperazin-1-yl'f-
benzo~dlisothiazole
A solution of borane dimethyl sulfide in THF (9.6 mL, 19.2 mmol) was added
dropwise to a suspension of 3.17g (7.7 mmols) of known 5-[2-(4-
benzo[d]isothiazol-
3-yl-piperazin-1-yl)-ethyl]-6-chloro-1,3-dihydro-indol-2-one (Howard, Harry
R.; Lowe,
John A. III; Seeger, Thomas F.; Seymour, Patricia A.; Zorn, Stevin H.;
Maloney,
Patrick R.; Ewing, Frank E.; Newman, Michael E.; Schmidt, Anne W.; et al., "3-
Benzisothiazolylpiperazine Derivatives as Potential Atypical Antipsychotic
Agents",
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-49-
Journal of Medicinal Chemistry 1996, 39(7), 143-8) in THF (15 mL), with
stirring at
0°C. The reaction was warmed to reflux for 2 hours. The reaction was
cooled and
treated with a 30% aqueous solution of sodium carbonate (10 mL). The layers
were
separated. The aqueous layer was extracted with ethyl acetate (2 x 20 mL). The
combined organics were dried over magnesium, filtered and the filtrate
concentrated.
The crude product was eluted through a flash column (silica gel 40, 230-400
mesh,
CH2C12 to 8% EtOH and 1 % NH40H in CH2CI2) to give the title compound as a
solid,
yield = 348 mg (11%). 1H NMR (400 MHz, DMSO-d6) 8 ppm 3.01 (m, 2 H) 3.16 (m, 2
H) 3.31 (m, 4 H) 3.51 (m, 4 H) 3.67 (d, J=11.72 Hz, 2 H) 4.07 (d, J--13.68 Hz,
2 H)
7.04 (s, 1 H) 7.28 (s, 1 H) 7.45 (t, J--8.30 Hz, 1 H) 7.56 (t, J--8.06 Hz, 1
H) 8.10 (m, 2
H) 11.32 (s, 1 H): MS (APCI), (M+1 )+ = 399Ø
FXAMPI F Rd
1-d5-f 2-(4-Benzof dlisothiazol-3-yl-pi perazi n-1-yl)-ethyll-6-chloro-2,3-di
hydro-
indol-1-yl~-ethanone
A solution of 3-{4-[2-(6-chloro-2,3-dihydro-1H-indol-5-yl)-ethyl]-piperazin-1-
y1}-
benzo[d]isothiazole (500 mg, 1.25 mmols) in THF (10 mL) with triethylamine
(0.262
mL, 1.88 mmols) was treated with acetyl chloride 0.088 mL, 1.25 mmols) and
stirred
for 16 hours at rt. The reaction was quenched with water, extracted with
methylene
chloride and filtered through 5 ~,m PTFE (phase-separating filter), and
concentrated
in vacuo, followed by a crystallization from isopropyl alcohol to yield : 510
mg (93%)
of 1-{5-[2-(4-benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-6-chloro-2,3-
dihydro-indol-
1-yl}-ethanone. ' H NMR (400 MHz, DMSO-d6) 8 ppm 2.91 (m, 2 H) 3.07 (m, 2 H)
3.23 (m, 1 H) 3.49 (t, J--8.43 Hz, 1 H) 3.84 (m, 2 H) 4.49 (t, J--8.55 Hz, 1
H) 7.64 (s,
1 H) 7.81 (m, 1 H) 7.94 (m, 1 H) 8.43 (m, 1 H); mp = 160.2-162.3 °C: MS
(APCI),
(M+1 )+ = 441.1.
EXAMPLE 35
1-f5-f 2-(4-Benzofdlisothiazol-3-yl-piperazin-1-yl)-ethyll-6-chloro-2,3-
dihydro-
indol-1-yl'~-propan-1-one
A solution of 3-{4-[2-(6-chloro-2,3-dihydro-1 H-indol-5-yl)-ethyl]-piperazin-1-
yl}-
benzo[d]isothiazole (375 mg, 0.94 mmols) in THF (2.OmL) with triethylamine
(0.196
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-50-
mL, 1.41 mmols) was treated with propionyl chloride (0.083 mL, 0.95 mmols) and
stirred for 16 hours at rt. The reaction was quenched with sodium hydroxide (1
N, 5
mL), extracted with methylene chloride and filtered through 5 p,m PTFE (phase-
separating filter), and concentrated in vacuo, followed by a crystallization
from
isopropyl alcohol to yield: 207 mg (48%) Isolated in 100% purity C 254 nm;
LCMS
(APCI) 455.2 [M+H]+.
The title compounds of Examples 36 through 42 were prepared from 3-{4-[2-
(6-chloro-2,3-dihydro-1H-indol-5-yl)-ethyl]-piperazin-1-yl}-
benzo[d]isothiazole in a
fashion similar to that reported above using the appropriate commercially
available
acid chloride.
EXrAMPLE ACll~ CHLORff~,ECOMPOU~TD~'NAME DATA;
; ~,
s
#36 butyryl chloride1-{ 5-[2-(4- Yield: 265
mg (60%)
Benzo[d]isothiazol-3-yl-Isolated in 100%
purity @
piperazin-1-yl)-ethyl]-6-254 nm; LCMS (APCI)
chloro-2,3-dihydro-indol-1-469.2 [M+H]+.
yl}-butan-1-one
#37 isobutyryl 1-{5-[2-(4- Yield: 136 mg (31%)
chloride
benzo[d]isothiazol-3-yl-Isolated in 100%
purity @
piperazin-1-yl)-ethyl]-6-254 nm; LCMS (APCl~
chloro-2,3-dihydro-indol-1-469.3 [M+H]+.
y1 }-2-methyl-propan-1-one
#38 valeryl chloride1-{5-[2-(4- Yield: 310 mg (67%)
Benzo[d]isothiazol-3-yl-Isolated in 100%
purity @
piperazin-1-yl)-ethyl]-6-254 nm; LCMS (APCI)
chloro-2,3-dihydro-indol-1-483.3 [M+H]+.
y1 }-pentan-1-one
#39 isovaleryl 1-{5-[2-(4- Yield: 198 mg (44%)
chloride
Benzo[d]isothiazol-3-yl-Isolated in 100%
purity
piperazin-1-yl)-ethyl]-6-254 nm; LCMS (APCI)
chloro-2,3-dihydro-indol-1-483.2 [M+H]+.
y1 }-3-methyl-butan-1-one
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-51-
#40 cyclopentane {5-[2-(4-Benzo[d]isothiazol-Yield: 332 mg (72%)
carbonyl chloride3-yl-piperazin-1-yl)-ethyl]-6-Isolated in 100%
chloro-2,3-dihydro-indol-1-purity @
yl}-cyclopentyl methanone254 nm; LCMS (APCI)
495.2 [M+H]+.
#41 cyclohexane 5-[2-(4-Benzo[d]isothiazol-Yield: 166 mg (35%)
carbonyl chloride3-yl-piperazin-1-yl)-ethyl]-6-Isolated in 100%
purity
chloro-2,3-dihydro-indol-1-254 nm; LCMS (APCI)
yl}-cyclohexyl- methanone509.3 [M+H]+.
#42 methane sulfonyl3-{4-[2-(6-Chloro-1-Yield: 83 mg (18%)
Isolated
chloride methanesulfonyl-2,3-in 100% purity @
254 nrn;
dihydro-1H-indol-5-yl)-LCMS (APCI) 477.2
ethyl]-piperazin-1-yl}-[M+H]+.
benzo[d]isothiazole
EXAMPLE 43
5-f2-(4-Benzofdlisothiazol-3-yl-piperazin-1-yl)-ethyl~-6-chloro-2,3-dihydro
indole-1-carboxylic acid methylamide
A solution of the product of Preparation 9, 3-{4-[2-(6-chloro-2,3-dihydro-1 H-
indol-5-yl)-ethyl]-piperazin-1-yl}-benzo[d]isothiazole (160 mg, 0.4 mmols in
THF
(5mL) was treated by dropwise addition with above methyl isocyanate at rt and
allowed to stir for 72 hours. The reaction was concentrated to dryness,
diluted with
H20 and extracted with methylene chloride and filtered through 5 p,m PTFE
(phase-
, separating filter), and concentrated in vacuo, followed by a crystallization
from
acetonitrile to yield: 164 mg (90%). Rt (min) reported is for the following
HPLC
conditions: 65:35 [H20:MeCN]+0.1 % TFA, 1.5 mUmin, 250x4.6 mm LUNA Cis.
Isolated in 100% purity @ 254 nm HPLC: Rt = 4.856; MS (APCI), (M+1 )+ = 456.1
The title compounds of Examples 44-49 were prepared from 3-{4-[2-(6-chloro-
2,3-dihydro-1 H-indol-5-yl)-ethyl]-piperazin-1-yl}-benzo[d]isothiazole in a
fashion
similar to that reported above using the appropriate commercially available
isocyanate.
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-52-
EXAMPLE lSOCYANATE COMPOUND NAME DATA
#44 ethyl isocyanate5-[2-(4- Yield: 170 mg (90%)
Benzo[d]isothiazol-3-yl-Isolated in 100%
purity @
piperazin-1-yl)-ethyl]-6-254 nm HPLC: Rt =
chloro-2,3-dihydro-indole-6.216; MS (APCI),
(M+1 )+
1-carboxylic acid = 470.1.
ethylamide
#45 n-propyl 5-[2-(4- Yield: 178 mg (92%)
isocyanate Benzo[d]isothiazol-3-yl-Isolated in 100%
purity @
piperazin-1-yl)-ethyl]-6-254 nm HPLC: Rt=
chloro-2,3-dihydro-indole-8.880; MS (APCI),
(M+1)+
1-carboxylic acid = 484.1.
propylamide
#46 isopropyl 5-[2-(4- Yield: 100 mg (52%)
isocyanate Benzo[d]isothiazol-3-yl-Isolated in 100%
purity @
piperazin-1-yl)-ethyl]-6-254 nm HPLC: Rt =
chloro-2,3-dihydro-indole-8.862; MS (APCI),
(M+1 )'~
1-carboxylic acid = 484.1.
isopropylamide
#47 t-butyl 5-[2-(4- Yield: 94 mg (47%)
isocyanate Benzo[d]isothiazol-3-yl-Isolated in 100%
purity @
piperazin-1-yl)-ethyl]-6-254 nm HPLC: Rt =
chloro-2,3-dihydro-indole-17.330; MS (APCI),
1-carboxylic acid (M+1 )+ = 498.2.
tert-
butylamide
#48 cyclopentyl 5-[2-(4- Yield: 200 mg (98%)
isocyanate Benzo[d]isothiazol-3-yl-Isolated in 100%
purity @
piperazin-1-yl)-ethyl]-6-254 nm HPLC: Rt =
chloro-2,3-dihydro-indole-15.499; MS (APCI),
1-carboxylic acid (M+1)+ = 510.1.
cyclopentylamide
#49 , phenyl 5-[2-(4- Yield: 120 mg (58%)
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-53-
isocyanate Benzo[d]isothiazol-3-yl-Isolated in 100%
purity ~
piperazin-1-yl)-ethyl]-6-254 nm HPLC: Rt=
chloro-2,3-dihydro-indole-13.190; MS (APCI),
1-carboxylic acid (M+1 )+ = 518.1.
phenylamide
PREPARATION 10
6-Chloro-5-(3-chloro-propionyl)-1,3-dihydro-indol-2-one
A 500mL RB flask equipped with a stirrer, reflux condenser and heating
mantle, was charged with aluminum chloride (AICI3) (14.76 g, 0.11 mol), 75 mL
of
carbon disulfide and 3-chloropropionyl chloride (2.21 mL, 0.023 mol) and this
was
stirred at rt during the portionwise addition of 6-chloro oxindole (3.0 g,
0.0179 mol).
This mixture was then heated to reflux for 3 hours, then cooled. The solvent
was
decanted and the reaction was quenched with addition of ice and water. The
suspension was stirred vigorously for 0.5 hours, followed by filtration. The
solids
were washed with water and then dried overnight in a vacuum oven. Yield = 4.23
g
(92%); MS (APCI), (M + 1 )+ = 259.1.
PREPARATION 11
6-Chloro-5-(3-chloro-propyl)-1,3-dihydro-indol-2-one
6-Chloro-5-(3-chloro-propyl)-1,3-dihydro-indol-2-one was prepared in a similar
fashion to that of Preparation 2 from scheme A-2. Yield = 1.15 g (82%); MS
(APCI),
(M + 1 )+ = 244.1.
PREPARATION 12
5-f3-(4-Benzot'dlisothiazol-3-yl-piperazin-1-yl)-propyll-6-chloro-1,3 dihydro-
indol-2-one
5-[3-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-propyl]-6-chloro-1,3-dihydro-
indol-2-one was prepared in a similar fashion to that of Preparation 3 from
scheme
A-3. Yield: 155 mg (16%) MS (APCI), (M + 1 )+ = 427.1.
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-54-
PREPARATION 13
3-(4-f3-(6-Chloro-2,3-dihydro-1 H-indol-5-yl)-propyll-piperazin-1-vl~
benzof dlisothiazole
A solution of 5-[3-(4-benzo[d]isothiazol-3-yl-piperazin-1-yl)-propyl]-6-chloro-
1,3-dihydro-indol-2-one (1.0 g, 2.34 mmol) in THF (40 mL) with stirring at
0°C was
treated with BH3~THF complex (9.4 mL, 9.4 mmol). The reaction was warmed to
reflux for 16 hours. The reaction was cooled and treated with a 10% aqueous
solution of sodium carbonate (10 mL) and warmed to reflux for 5.0 hours. The
reaction was cooled and the layers were separated. The aqueous layers were
extracted with ethyl acetate. The combined organics were dried over magnesium
sulfate, filtered and the filtrate concentrated. The crude product was eluted
through
a flash column (1:1 ethyl acetate:methylene chloride) to give the title
compound.
Yield = 70 mg (7%). MS (APCI), (M + 1 )+ = 413.1.
EXAMPLE 50
1-(5-f3-(4-Benzofdlisothiazol-3-yl-piperazin-1-yl)-propyll-6-chloro-2,3-
dihydro-
indol-1-yl~-ethanone
A solution of 3-(4-[3-(6-chloro-2,3-dihydro-1 H-indol-5-yl)-propyl]-piperazin-
1-
yl}-benzo[d]isothiazole (69 mg) in THF (1.0 mL) with triethylamine (0.03 mL)
was
treated with acetic anhydride (0.03 ml) and stirred for 4 hours at reflux. The
reaction
was quenched with water, extracted with ethyl acetate and filter and
concentrated in
vacuo. Yield: 34 mg (45%) MS (APCI), (M+1 )+ = 455.1.
PREPARATION 14
3-Methyl-but-2-enoic acid o-tolylamide
To a cold 0.25 M solution of o-toluidine (5.0 mL, 46.85 mmole, 1 eq) in dry
THF and pyridine (2 eq) was added dropwise neat 3,3-dimethyl-acryloyl chloride
and
stirred vigorously. The reaction was filtered and the filtrate diluted with
EtOAc (equal
volume) and washed with H20 (3x), 1 N HCI (2x), sat. Na2C03 (1x), brine (1x),
dried
(MgS04), and concentrated to a solid. A mixture of the titled product and its
terminal
olefin isomer were isolated as a 1:1 mixture. MS (APCI) = 190.1 [M+H]+.
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-55=
PREPARATION 15
4,4,8-trimethyl-3,4-dihydro-1 H-auinolin-2-one
To a solution of 3-methyl-but-2-enoic-acid o-tolylamide (7.27 g, 38.41 mmole,
1 eq) in 1,2-dichlorobenzene (50 mL) was added AICI3 (30.73 g, 230.49 mmole, 6
eq) and the whole mixture heated to 50-70°C. As the reaction reached
about 50 °C
vigorous gaseous hydrogen chloride (HCI(g)) was released. After the HCI
evolution
appeared to cease, the reaction was allowed to continue for an additional
l0min
before cooling. The reaction was cooled and poured into cold H20. The
heterogeneous mix was extracted with CH2CI2 (3 x 100 mL), dried (MgS04) and
concentrated to an orange oil which was purified by MPLC (30% EtOAc/hexanes)
to
give the above titled compound (5.357 g, 28.31 mmole, 74% yield).'H NMR (400
MHz, CDCI3) S 8.43 (s, 1 H), 7.16 (d, J= 7.5Hz, 1 H), 7.04 (d, J= 7.5Hz, 1 H),
6.96 (t,
J = 7.5Hz, 1 H), 2.48 (s, 2H), 2.30 (s, 3H), 1.32 (s, 6H).
PREPARATION 16
6-(2-Chloro-acetyl)-4,4,8-trimethyl-3,4-dihydro-1 H-auinolin-2-one
To a solution of 4,4,8-trimethyl-3,4-dihydro-1 H-quinolin-2-one (3.545 g,
18.71
mmole, 1 eq) in CS2 (200 mL) was added chloroacetyl chloride (2.23 mL, 28.06
mmole, 1.5 eq), followed by aluminum chloride (9.98 g, 74.84 mmole, 4 eq) in
one
portion. The reaction was heated to reflux for 1 h after which thin layer
chromatography (TLC) and mass spectroscopy (MS) indicated complete reaction.
After cooling, the solvent was decanted and the remaining residue was
carefully
hydrolyzed with cold H2O. The resulting precipitate was filtered and dried at
50°C
under high vacuum to give titled compound as a tan solid (4.79 g, 18.03 mmole,
96%
yield). 100% purity at 254nm; LCMS (APCI) 266.3 [M+H]+; 1H NMR (400 MHz,
CDCIs) 8 7.89 (bs, 1 H), 7.81 (s, 1 H), 7.67 (s, 1 H), 4.65 (s, 2H), 2.52 (s,
2H), 2.32 (s,
3H), 1.36 (s, 6H).
PREPARATION 17
6-(2-Chloro-ethyl)-4,4,8-trimethyl-3,4-dihydro-1 H-auinolin-2-one
To a solution of 6-(2-chloro-acetyl)-4,4,8-trimethyl-3,4-dihydro-1 H-quinolin-
2-
one (4.79 g, 18.03 mmole, 1.0 eq) in trifluoroacetic acid (100 mL) was added
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-56-
triethylsilane (7.20 mL, 45.08 mmole, 2.5 eq) and the whole mixture heated to
60°C.
After 2 h TLC (30% EtOAc/hexanes) and MS indicated complete reaction. The
reaction was cooled and poured over ice. After extracting with CH2C12 (3 x 100
mL),
drying (MgS04) and concentrating to an oil, the crude was purified by MPLC
(30%
EtOAc/hexanes) to give the titled compound as a white solid (3.23 g, 12.84
mmole,
71% yield). 100% purity at 254nm; LCMS (APCI) 252.2 [M+H]+;'H NMR (400 MHz,
CDCI3) b 7.41 (bs, 1 H), 6.99 (s, 1 H), 6.89 (s, 1 H), 3.67 (t, J = 7.3Hz,
2H), 2.98 (t, J =
7.3Hz, 2H), 2.46 (s, 2H), 2.21 (s, 3H), 1.30 (s, 6H).
PREPARATION 18
6-f2-(4-Benzofdlisothiazol-3-yl-piperazin-1-yl)-ethyll-4,4,8-trimethyl-3,4-
dihydro-
1 H-auinolin-2-one
A heterogeneous mix of 6-(2-chloro-ethyl)-4,4,8-trimethyl-3,4-dihydro-1 H-
quinolin-2-one (2.200 g, 8.739 mmole, 1.0 eq), sodium carbonate (1.158 g,
10.924
mmole, 1.25 eq), and added 3-piperazin-1-yl-benzo[d]isothiazole hydrochloride
(3.353 g, 13.110 mmole, 1.5 eq) in water (20 mL) was heated to 175°C
under
microwave assistance for 10 min. The reaction was diluted with H20 (100mL),
CH2C12 (1 OOmL) and the layers separated. The aqueous layer was extracted with
CH2C12 (2x 50mL) and the organic dried (MgS04), concentrated, and the residue
purified by MPLC (25% EtOAc/CH2C12 ----- 50% EtOAc gradient over 20min and
hold
for 20min ---- 100% EtOAc gradient over 20min). Titled product was obtained as
a
white crystalline solid in 63% yield. 100% purity at 254 nm; LCMS (APCI) 435.2
[M+H]+;'H NMR (400 MHz, CDCI3) s 7.90 (d, 1 H, J= 7.94Hz), 7.80 (d, 1 H, J=
7.94Hz), 7.46 (t, 1 H, J = 7.94Hz), 7.34 (t, 1 H, J = 7.94Hz), 7.02 (s, 1 H),
6.91 (s, 1 H),
4.78 (s, 1 H), 3.69-3.55 (m, 4H), 2.86-2.59 (m, 8H), 2.45 (s, 2H), 2.21 (s,
3H), 1.30 (s,
6H).
PREPARATION 19
6-f2-(4-Benzo~dlisothiazol-3-yl-piperazin-1-yl)-ethyll-4,4,8-trimethyl-1,2,3,4
tetrahydro-auinoline
To a stirring solution of 6-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-
ethyl]-
4,4,8-trimethyl-3,4-dihydro-1 H-quinolin-2-one (0.500 g, 1.150 mmole) in dry
THF (50
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-57-
mL) was added Borane THF complex (1 M in THF, 2 eq) and the reaction heated to
reflux for 3 h. Upon cooling the reaction was quenched with MeOH and 1 mL
acetic
acid. The reaction was concentrated to a residue and stripped from MeOH (3x 50
mL). The residue was taken up in CH2CI2, washed with water and the organics
dried
(MgS04) and concentrated to a residue. The residue was taken up in dioxane and
the HCI salt was precipitated from HCI dioxane treatment. The salt was
collected by
filtration and then converted to its free base by treatment with NaOH and
extraction
with EtOAc to give titled product (0.421 g, 1.000 mmole, 87% yield). 100%
purity at
254nm; LCMS (APCI) 421.2 [M+H]+;'H NMR (400 MHz, CDC13) 8 ppm 1.29 (s, 6 H)
1.73(m,2H)2.06(s,3H)3.05(m,6H)3.28(m,2H)3.35(m,2H)3.72(d,J=13.43
Hz, 2 H) 3.98 (m, 2 H) 6.74 (s, 1 H) 6.93 (d, J--0.98 Hz, 1 H) 7.37 (t, J--
7.69 Hz, 1 H)
7.48 (t, J--7.57 Hz, 1 H) 7.84 (dd, J=11.72, 8.30 Hz, 2 H).
EXAMPLE 51
1-f6-f2-(4-Benzo~dlisothiazol-3-yl-piperazin-1-yl)-ethyl~-4,4,8-trimethyl-3,4-
dihydro-2H-quinolin-1-yl'f-ethanone hydrochloride
To a solution of 6-[2-(4-benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-4,4,8-
trimethyl-1,2,3,4-tetrahydro-quinoline (0.499 g, 1.000 mmole) in dry THF (5
mL) was
added sodium hydride (NaH) (60% dispersed in oil, 1.1 eq). After stirring for
10 min.
at rt, acetyl chloride was added dropwise. The reaction was stirred for 30
min. and
was then diluted with H20lEtOAc and the layers separated. The organics were
dried
and concentrated and the residue subjected to MPLC (EtOAc). The HCI salt was
precipitated by treatment of an ether solution of free base with 1 N HCI in
ethyl ether
(Et20) to give titled product (0.125 g, 0.270 mmole). 100% purity at 254nm;
LCMS
(APCI) 463.2 [M+H]+;'H NMR (400 MHz,CDCl3) 8 ppm 1.14 (s, 2 H) 1.25 (s, 2 H)
1.32 (d, J--8.30 Hz, 3 H) 1.55 (ddd, J--13.31, 7.57, 7.45 Hz, 1 H) 1.90 (s, 3
H) 2.11
(s, 1 H) 2.21 (s, 2 H) 2.27 (s, 1 H) 2.68 (m, 2 H) 2.80 (m, 7 H) 3.02 (ddd, J--
13.00,
7.75, 5.37 Hz, 1 H) 3.59 (m, 4 H) 4.66 (m, 1 H) 6.95 (d, J--15.14 Hz, 1 H)
7.03 (dd,
J--7.82, 1.71 Hz, 1 H) 7.34 (td, J--7.57, 0.98 Hz, 1 H) 7.45 (m, 1 H) 7.79 (m,
1 H)
7.90 (d, J--8.06 Hz, 1 H).
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-58-
PREPARATION 20
3-Methyl-but-2-enoic acid (3-chloro-2-methyl-phenyl)-amide
3,3-Diethylacryoyl chloride (21.0 mL, 0.189 mol) was slowly added to a
solution of 3-chloro-2-methylaniline (20.0 mL, 0.167 mol) and pyridine (17.0
mL,
0.210 mol) in dichloromethane (210 mL) at 0 °C. After 1.5 h, the
reaction was
quenched by slow addition of saturated sodium bicarbonate solution (60 mL).
The
solution was transferred to a separatory funnel and the layers separated. The
aqueous layer was back-extracted with dichloromethane (2 x 100 mL). The
combined organic extracts were dried over anhydrous sodium sulfate, filtered
and
concentrated in vacuo. The resulting purple solid was used directly without
purification. MS (APCI): (M+1 )+ = 224.1.
PREPARATION 21
7-Chloro-6-(2-chloro-acetyl)-4,4,8-trimethyl-3,4-dihydro-1 H-duinolin-2-one
To a solution of 3-methyl-but-2-enoic acid (3-chloro-2-methyl-phenyl)-amide in
CH2CI2 (167 mL) was slowly added aluminum chloride (91.5 g, 0.686 mol) at a
rate
to maintain gentle reflux. Upon complete addition of the aluminum chloride, a
reflux
condenser was attached and the reaction was heated to reflux. After 1.5 h, TLC
showed no remaining starting material. Chloroacetyl chloride (20.0 mL, 0.250
mol)
was slowly added and the mixture was refluxed for an additional 4 h. The
reaction
mixture was cooled to ambient temperature, poured into ice water (1000 mL) and
extracted with CH2C12 (4 x 300 mL). The organic extracts were combined, washed
with saturated sodium chloride solution (200 mL), dried over anhydrous sodium
sulfate, filtered and the solvent removed under reduced pressure. The
resulting solid
was used directly without purification. MS (APCI): (M+1 )+ = 300.1, (M+3)+ =
302.1.
PREPARATION 22
7-Chloro-6-(2-chloro-ethyl)-4,4,8-trimethyl-3,4-dihydro-1 H-guinolin-2-one
To a solution of 7-chloro-6-(2-chloro-acetyl)-4,4,8-trimethyl-3,4-dihydro-1 H-
quinolin-2-one in trifluoroacetic acid (168.0 mL) was added triethylsilane
(59.0 mL,
0.369 mol). The reaction mixture was heated to 60 °C under nitrogen.
After 5.5 h, the
reaction was cooled to ambient temperature and the reaction was stirred
overnight.
The reaction mixture was poured into ice water (350 mL). The reaction flask
was
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-59-
rinsed with methanol (50 mL). The mixture was vigorously stirred resulting in
formation of a precipitate. The solid was filtered and then triturated with
hexanes.
The solid was recrystallized from hot methyl-tert butyl ether (MTBE) (600 mL)
to
product as a light tan solid. Yield: 36.0345 g (0.126 mol, 75% yield over four
steps).
MS (APCI): (M+1)+= 288.1. 1H NMR (400 MHz, CDC13)8 7.50 (br s, 1 H), 7.06 (s,
1
H), 3.71 (t, J--7.2 Hz, 2 H), 3.16 (t, J--7.2 Hz, 2 H), 2.45 (s, 2 H), 2.30
(s, 3 H), 1.30
(s, 6 H).
PREPARATION 23
6-f2-(4-Benzo~dlisothiazol-3-yl-piperazin-1-yl)-ethyll-7-chloro-4,4,8-
trimethyl-
3,4-dihydro-1 H-auinolin-2-one
A mixture of 7-chloro-6-(2-chloro-ethyl)-4,4,8-trimethyl-3,4-dihydro-1 H-
quinolin-2-one (5.0016 g, 17.476 mmol), 3-piperazin-1-yl-benzo[d]isothiazole
hydrochloride (4.4811 g, 17.520 mmol), potassium carbonate (4.8299 g, 34.946
mmol} and potassium iodine (0.2903 g, 1.749 mmol) were reacted in acetonitrile
(29.0 mL) in a microwave reactor for 1 h at 200 °C. The reaction was
cooled to rt,
diluted with H20 and filtered. The solid was washed with H20 and hexanes. The
resulting solid was eluted through a flash column (silica gel 60, 230-400
mesh, 0-3%
MeOH in CH2CI2 gradient over 1 h) to give an off-white solid. Yield: 5.6591 g
(12.065 mmol, 69%). Anal.: calculated for C25H29CIN40S~0.02CH2CI2: C, 63.84;
H,
6.22; N, 11.90. Found: C, 63.49, H, 6.13; N, 11.72. LC-MS (APCI): (M+1 )+ =
471Ø
1H NMR (400 MHz, CDCI3) 8 7.90 (d, J--8.2 Hz, 1 H), 7.80 (d, J=8.0 Hz, 1 H),
7.58 (s,
1 H), 7.45 (ddd, J--8.0, 7.1, 1.0 Hz, 1 H), 7.34 (ddd, J--8.2, 7.1, 1.0 Hz, 1
H), 7.08 (s,
1 H), 3.61 (m, 4 H), 2.98 (m, 2 H), 2.80 (s, 4 H), 2.68 (m, 2 H), 2.44 (s, 2
H), 2.31 (s,
3 H), 1.30 (s, 6 H).
PREPARATION 24
6-f2-(4-Benzofdlisothiazol-3-yl-piperazin-1-yl)-ethyll-7-chloro-4,4,8-
trimethyl
1,2,3,4-tetrahydro-duinoline
To a solution of 6-[2-(4-benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-7-
chloro-
4,4,8-trimethyl-3,4-dihydro-1 H-quinolin-2-one (2.5024 g, 5.335 mmol) in
anhydrous
THF (18 mL) under a nitrogen atmosphere was added borane-THF complex (1.0 M,
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-60-
6.4 mL, 6.4 mmol). The reaction was refluxed overnight. Additional borane-THF
complex (1.0 M, 6.4 mL, 6.4 mmol) was added and the reaction was refluxed for
an
additional 4 h then cooled to ambient temperature. The excess reagent was
quenched by slow addition of methanol (MeOH, 20.0 mL). The reaction mixture
was
heated to reflux overnight then cooled to ambient temperature. The organic
solvents
were removed in vacuo to give a white residue. The residue was dissolved in
MeOH
(20 mL) and removed in vacuo. The residue was dissolved in methylene chloride
(CH2CI2) and washed with saturated sodium bicarbonate solution (NaHC03, 2 x 30
mL) and the organic layer was dried over anhydrous sodium sulfate (Na2S04),
filtered and concentrated to an oil. The oil was eluted through a flash column
(silica
gel 60, 230-400 mesh, 0-5% MeOH in CH2C12 gradient over 1 h) to give an off-
white
solid. Yield = 1.6876 g (3.709 mmol, 70%).Anal.: calculated for
C25H3~CIN4S~0.05CH2CI2: C, 65.50; H, 6.82; N, 12.20. Found: C, 65.14, H, 6.91;
N,
11.91. MS (APCI), (M+1 )+ = 456. ' H NMR (400 MHz, CDCI3) 8 7.91 (d, J--8.2
Hz, 1
H), 7.80 (d, J=8.2 Hz, 1 H), 7.45 (t, J--7.4 Hz, 1 H), 7.34 (t, J--7.4 Hz, 1
H), 7.00 (s, 1
H), 3.76 (br s, 1 H), 3.60 (m, 4 H), 3.36 (m, 2 H), 2.90 (m, 2 H), 2.80 (m, 4
H), 2.65
(m, 2 H), 2.17 (s, 3 H), 1.71 (m, 2 H), 1.28 (s, 6 H).
FX~MPI F ~9
1-f6-f 2-(4-Benzo(dlisothiazol-3-yl-piperazin-1-yl)-ethyll-7-chloro-4,4,8-
trimethyl-
3,4-dihydro-2H-auinolin-1-yl)-ethanone methane sulfonate
To a stirred solution of 6-[2-(4-benzo[d]isothiazol-3-yl-piperazin-1-yl)-
ethyl]-7-
chloro-4,4,8-trimethyl-1,2,3,4-tetrahydro-quinoline (0.4497 g, 0.988 mmol) in
THF (10
mL) under a nitrogen atmosphere was added acetyl chloride (77.4 p,L, 1.089
mmol).
The reaction was stirred overnight. Additional acetyl chloride (0.0250 mL,
0.351
mmol) was added to try to drive the reaction to completion. After 0.5 h, there
was no
change in TLC. The reaction was quenched by slow addition of sat. NaHC03
solution
(10 mL) and ethyl acetate (EtOAc, 10 mL). The layers were separated and the
organic layer was dried over anhydrous Na2S04, filtered and concentrated in
vacuo.
The residue was eluted through a flash column (silica gel 60, 230-400 mesh, 25-
75%
EtOAc in hexanes gradient over 1 h) to a clear oil. Attempts to crystallize
using
CH2CI2 and CH2C12/hexanes were unsuccessful. The oil was taken up in THF (9.5
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-61-
mL) and heated to 40 °C. Methanesulfonic acid (61.5 p,L, 0.948 mmol)
was added
and after 5 min, the reaction was allowed to cool to ambient temperature. The
product was allowed to crystallize overnight. Hexanes were added to the
reaction
mixture and the solid was filtered and washed with hexanes. The wet solid was
dried
in a vacuum oven at 60 °C to give a white/off-white crystalline solid
as the mesylate
salt. Yield: 0.4576 g (0.771 mmol, 78%). Anal: calculated for
C2~H33CIN4OS'CH4OsS'O.34H2O: C, 56.11; H, 6.34; N, 9.35. Found: C, 55.72, H,
6.01; N, 8.97. MS (APCI), (M+1)+=498.'H NMR (400 MHz, CDCI3) & 11.60 (brs,
0.6 H), 11.47 (br s, 0.4 H), 7.84 (m, 2 H), 7.51 (m, 1 H), 7.40 (m, 1 H), 7.35
(s, 0.6
H), 7.32 (s, 0.4 H), 4.65 (m, 0.6 H), 4.16 (m, 1.4 H), 3.97 (m, 2 H), 3.70 (m,
1 H),
3.32 (m, 4 H), 3.01 (m, 1 H), 2.90 (s, 3 H), 2.28 (s, 1.2 H), 2.25 (s, 1.8 H),
2.11 (s, 1
H), 1.90 (m, 1.2 H), 1.85 (s, 2,4 H), 1.81 (s, 3.6 H), 1.75 (m, 1.2 H), 1.55
(m, 0.6 H),
1.35 (s, 1.8 H), 1.33 (s, 1.2 H), 1.25 (s, 1.2 H), 1.14 (s, 1.8 H).
EXAMPLE 53
1-(6-f2-(4-Benzofdlisothiazol-3-yi-piperazin-1-yl)-ethyll-7-chloro-4,4,8-
trimethVl
3 4-dihydro-2H-auinolin-1-yl~-propan-1-one methane suifonate
To a stirred solution of 6-[2-(4-benzo[d]isothiazol-3-yl-piperazin-1-yl)-
ethyl]-7
chloro-4,4,8-trimethyl-1,2,3,4-tetrahydro-quinoline (0.4500g, 0.989 mmol) in
THF (10
mL) under a nitrogen atmosphere was added propionyl chloride (94.6 p,L, 1.089
mmol). The reaction was stirred overnight. Additional propionyl chloride (31.0
wL,
0.357 mmol) was added to try to drive the reaction to completion. After 0.5 h,
there
was no change in TLC. The reaction was quenched by slow addition of sat.
NaHC03
solution (10 mL) and EtOAc (10 mL). The layers were separated and the organic
layer was dried over anhydrous Na2S04, filtered and concentrated in vacuo. The
residue was eluted through a flash column (silica gel 60, 230-400 mesh, 25-75%
EtOAc in hexanes gradient over 1 h) to a clear oil. Attempts to crystallize
using
CH2Cl2 and CH2Cl2/hexanes were unsuccessful. The oil was taken up in THF (9.0
mL) and heated to 40 °C. Methanesulfonic acid (59.0 p,L, 0.909 mmol)
was added
and after 5 min, the reaction was allowed to cool to ambient temperature. The
product was allowed to crystallize overnight. Hexanes were added to the
reaction
mixture and the solid was filtered and washed with hexanes. The wet solid was
dried
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-62-
in a vacuum oven at 60 °C to give a white/off-white crystalline solid
as the mesylate
salt. Yield: 0.4484 g (0.738 mmol, 75%). Anal. calculated for
C28H35CIN4OS~CH4O3S:
C, 57.36; H, 6.47; N, 9.23. Found: C, 57.01, H, 6.23; N, 8.90. MS (APCI), (M+1
)+ _
512.'H NMR (400 MHz, CDCI3) 8 11.58 (br s, 0.6 H), 11.45 (br s, 0.4 H), 7.84
(m, 2
H), 7.51 (m, 1 H), 7.40 (m, 1 H), 7.34 (s, 0.6 H), 7.31 (s, 0.4 H), 4.66 (m,
0.6 H), 4.16
(m, 1.4 H), 3.98 (m, 2 H), 3.71 (m, 1 H), 3.36 (m, 2 H), 3.26 (m, 1 H), 3.00
(m, 0.6 H),
2.90 (s, 3 H), 2.60 (m, 0.4 H), 2.48 (m, 0.4 H), 2.23 (s, 1.8 H), 2.16 (m, 0.6
H), 2.08
(s, 1.2 H), 1.89 (m, 1.4 H), 1.75 (s, 6 H), 1.54 (m, 0.6 H), 1.34 (s, 1.8 H),
1.32 (s, 1.2
H), 1.24 (s, 1.2 H), 1.23 (t, J--7.5 Hz, 1.2 H), 1.13 (s, 1.8 H), 1.04 (t, J--
7.5 Hz, 1.8 H).
EXAMPLE 54
f 6-f 2-(4-Benzof dlisoth iazol-3-yl-piperazi n-1-yl)-ethyl~-7-ch loro-4,4,8-
trimethyl-
3,4-dihydro-2H-auinolin-1-yl)-cyclopropylmethanone
To a stirred solution of 6-[2-(4-benzo[d]isothiazol-3-yl-piperazin-1-yl)-
ethyl]-7-
chloro-4,4,8-trimethyl-1,2,3,4-tetrahydro-quinoline (0.4496g, 0.988 mmol) in
THF (10
mL) under a nitrogen atmosphere was added cyclopropanecarbonyl chloride (98.8
~L, 1.089 mmol). The reaction was stirred overnight. Additional propionyl
chloride
(31.0 ~L, 0.342 mmol) was added to try to drive the reaction to completion.
After 0.5
h, there was no change in TLC. The reaction was quenched by slow addition of
sat.
NaHC03 solution (10 mL) and EtOAc (10 mL). The layers were separated and the
organic layer was dried over anhydrous sodium sulfate (Na2S04), filtered and
concentrated in vacuo. The residue was eluted through a flash column (silica
gel 60,
230-400 mesh, 25-75% EtOAc in hexanes gradient over 1 h) to give a white foam.
The foam was dried overnight under high vacuum. Yield: 0.4477 g (0.856 mmol,
87%). Anal. calculated for C29H35CIN4OS~O.O7CH2C12: C, 65.99; H, 6.69; N,
10.49.
Found: C, 65.87, H, 6.50; N, 10.19. MS (APCI), (M+1)+= 524. 1H NMR (400 MHz,
CDC13) 8 7.91 (d, J=8.1 Hz, 1 H), 7.81 (d, J=8.1 Hz, 1 H), 7.46 (ddd, J=8.1,
7.0, 1.0
Hz, 1 H), 7.35 (ddd, J=8.1, 7.0, 1.0 Hz, 1 H), 7.14 (s, 0.9 H), 7.08 (s, 0.1
H), 4.65 (m,
1 H), 3.60 (m, 4 H), ) 3.04 (m, 2 H), 2.81 (m, 4 H), 2.71 (m, 2 H), 2.34 (s,
2.7 H), 2.10
(s, 0.3 H), 1.91 (m, 1 H), 1.58 (m, 3 H), 1.37 (m, 3 H), 1.21 (s, 0.3H), 1.17
(s, 2.7 H),
1.05 (m, 2 H), 0.83 (m, 1 H), 0.55 (m, 1 H).
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-63
PREPARATION 25
7-f2-(4-Benzof dlisoxazol-3-yl-piperazin-1-yl)-ethyll-1,3,4,5-tetrahydro
benzof blazepi n-2-one
A. 1,3,4,5-tetrahydro-benzof blazepin-2-one
Beilstein Registry Number 137258; CAS Registry Number 4424-80-0
Horning, E. C.; Stromberg, V. L.; Lloyd, H. A. J. Am. Chem. Soc. 1952, 74,
5153-
5155.
B. 7-(2-Chloro-acetyl)-1,3,4,5-tetrahydro-benzo('blazepin-2-one
To a 1-L round-bottom flask was added AICI3 (83.4 g, 626 mmol), CS2 (500 mL),
chloroacetyl chloride (12.0 mL, 156.4 mmol), and azepinone (from step A)
[16.79 g,
104.3 mmol]. The reaction was stirred to a thick gummy deposit, refluxed for 3
h and
cooled to 0°C. The solvent was decanted and ice water (300 mL) was
added very
slowly (CAUTION: exotherm and HCI gas produced) until a solid suspension was
formed. -The solid was collected by filtration and washed with H20 (50 mL) to
produce a brown solid. This material was recrystallized in methanol/H20 to
give the
title compound (19.2 g, 78%) as a dark tan solid: 'H NMR (300 MHz, CDC13) 8
8.50
(s, 1 H), 7.87-7.81 (m, 2 H), 7.10 (d, J = 9.0 Hz, 1 H), 4.68 (s, 2 H), 2.89
(t, J = 7.2
Hz, 2 H), 2.42 (t, J = 7.2 Hz, 2 H), 2.36-2.25 (m, 2 H).
C. 7-(2-Chloro-ethyl)-1,3,4,5-tetrahydro-benzo[blazepin-2-one
Into a 250-mL round-bottom flask was placed the ketone from step B (5.00 g,
21.1
mmol) and TFA (25 mL). The solution was cooled to 0 °C and
triethylsilane (10.2
mL, 63.2 mmol) was added dropwise over a 5 min period. The reaction was warmed
to 50 °C and stirred for 18 h. The mixture was cooled to rt, diluted
with H20 (100
mL) and extracted with ethyl acetate (3 x 50 mL). The combined organic layers
were
dried over Na2S04, and concentrated under high vacuum to produce the title
compound (5.1 g, >99%): 1H NMR (300 MHz, CDCI3) 8 9.19 (s, 1 H), 7.17-7.12 (m,
2
H),7.00(d,J=9.OHz,1 H),3.72(t,J=9.OHz,2H),3.06(t,J=7.2Hz,2H),2.80
(t, J = 7.2 Hz, 2 H), 2.41-2.28 (m, 4 H).
D. 7-f 2-(4-Benzof dlisoxazol-3-yl-piperazin-1-yl)-ethyll-1,3,4,5-tetrahydro-
benzo~blazepin-2-one
A suspension of the compound from step C (1.00 g, 4.48 mmol) in CH3CN (40 mL)
was treated with 3-piperazin-1-yl-benzo[dJisoxazole ~ HCI (1.20 g, 4.98 mmol),
Nal
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-64-
(744 mg, 4.96 mmol), and potassium carbonate (K2C03) (1.87 g, 13.5 mmol). The
mixture was heated to reflux under nitrogen (N2) for 44 h, then allowed to
cool. The
mixture was diluted with H20 (100 mL) and extracted with CH2CI2 (4 x 100 mL).
The
combined organic layers were dried over Na2S04, decanted, and the solvent was
removed in vacuo. The residue was purified by flash column chromatography
(silica
gel, EtOAc) to give the title compound (0.95 g, 54%) as a white powder: mp 199-
200
°C; 1H NMR (300 MHz, CDC13) 8 7.70 (d, J= 8.1 Hz, 1 H), 7.45-7.52 (m, 2
H), 7.19
7.25 (m, 1 H), 7.18 (s, 1 H), 7.08-7.11 (m, 2 H), 6.88 (d, J = 8.4 Hz, 1 H),
3.61-3.64
(m, 4 H), 2.65-2.86 (m, 10 H), 2.35 (t, J= 7.5 Hz, 2 H), 2.23 (m, 2 H); ESI MS
m/z
391 ~C23H26N4O2 + H]+~ Rt 0.41 (silica gel, 95:5 EtOAc/MeOH); HPLC 97.0%
(AUC),
tR = 11.36 min. Anal. Calc'd for C23H26N4O2: C, 70.75; H, 6.71; N, 14.35.
Found: C,
70.71; H, 6.72; N, 14.28.
EXAMPLE 55
1-f7-[2-(4-Benzo[dlisoxazol-3-yl-piperazin-1-yl)-ethyll-2,3,4,5-tetrahydro-
benzo[blazepin-1-yl~-ethanone methanesulfonate
A. 7-[2-(4-Benzo[dlisoxazol-3-yl-piperazin-1-yl)-ethyll-2,3,4,5-tetrahydro-1 H
benzo[blazepine
A suspension of the title compound of Preparation 25 (0.36 g, 0.92 mmol) in
THF (8
mL) was treated with a solution of BH3 in THF (4.0 mL, 1.5 M, 6.0 mmol). The
resulting clear solution was heated to reflux for 3 h, then allowed to cool.
The
reaction was quenched with 6N HCI until gas evolution subsided. The mixture
was
heated to reflux for 1 h, allowed to cool, and then made basic (pH 8) with
solid
sodium hydroxide (NaOH) and a 1 N NaOH solution. The biphasic mixture was
diluted with H20 (50 mL) and extracted with CH2C12 (3 x 50 mL). The combined
organic layers were dried over Na2S04, filtered, and the solvent was removed
in
vacuo. The residue was purified by flash column chromatography (silica gel,
2:1
hexanes/EtOAc) to give the title compound (0.22 g, 63%) as a colorless oil: 1H
NMR
(300 MHz, CDCI3) S 7.70 (d, J= 8.1 Hz, 1 H), 7.44-7.52 (m, 2 H), 7.19-7.25 (m,
1 H),
6.96 (d, J = 1.7 Hz, 1 H), 6.89 (dd, J = 7.9, 2.0 Hz, 1 H), 6.67 (d, J = 7.8
Hz, 1 H),
3.71 (bs, 1 H), 3.61-3.64 (m, 4 H), 3.02 (t, J = 5.3 Hz, 2 H), 2.62-2.78 (m,
10 H),
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-65-
1.76-1.83 (m, 2 H), 1.60-1.67 (m, 2 H); ESI MS m/z 377 [C23H28N4O + H]+; Rf
0.52
(silica gel, 1:1 hexanes/EtOAc).
B. 1-f 7-~2-(4-Benzof dlisoxazol-3-yl-piperazi n-1-yl)-ethyll-2,3,4,5-
tetrahydro-
benzofblazepin-1-yl)-ethanone methanesulfonate
A solution of the title compound from step A (0.22 g, 0.58 mmol) in CH2CI2 (12
mL)
was treated with acetic anhydride (Ac20) (55 ~,L, 0.58 mmol). After stirring
at rt
under N2 for 15 h, the reaction was quenched with saturated NaHCO$ (30 mL) and
extracted with CH2CI2 (3 x 50 mL). The combined organic layers were dried over
Na2S04, decanted, and the solvent was removed in vacuo. The residue was
purified
by flash column chromatography (silica gel, 2:1 EtOAc/hexanes) to give a white
solid
residue (0.21 g, 0.50 mmol, 86°l°). The residue was dissolved in
EtOAc (15 mL) and
treated with methanesulfonic acid (CH3S03H) (2 M in Et2O, 0.25 mL, 1 mmol).
After
stirring for 5 min, the resulting precipitate was isolated by filtration,
washed with Et2O
(3 x 6 mL), and dried in a vacuum oven at 50 °C for 4 days to give the
title compound
(228 mg, 88%) as a white powder: mp 234-235 °C (dec);'H NMR (300 MHz,
CDCI3)
b 11.99 (s, 1 H), 7.51-7.63 (m, 3 H), 7.27-7.33 (m, 1 H), 7.13-7.17 (m, 2 H),
7.08 (d, J
= 7.8 Hz, 1 H), 4.68 (m, 1 H), 4.20 (d, J = 14.5 Hz, 2 H), 3.99 (t, J = 12.3
Hz, 2 H),
3.72 (d, J = 12.2 Hz, 2 H), 3.08-3.33 (m, 6 H), 2.90 (s, 3 H), 2.68-2.74 (m, 2
H), 2.50-
2.59 (m, 1 H), 1.74-1.99 (m, 3 H), 1.83 (s, 3 H), 1.32-1.40 (m, 1 H); ESI MS
m/z419
[C25H30N4O2 + H]+; Rf 0.47 (silica gel, 95:5 EtOAc/MeOH); HPLC >99% (AUC), tR
=
12.12 min. Anal. Calc'd for C25H3oN4O2~CH3SO3H: C, 60.68; H, 6.66; N, 10.89.
Found: C, 60.62; H, 6.57; N, 10.82.
PREPARATIONS 26A and 26B
7-(2-f4-(5-Fluoro-benzo~dlisothiazol-3-yl)-piperazin-1-yll-ethyll-1,3,4,5-
tetrahydro-benzofblazepin-2-one (Prep. 26A)
A suspension of 7-chloroethylazepin-2-one (1.71 g, 7.64 mmol) and 5-fluoro-3-
piperazin-1-yl-benzo[d]isothiazole (1.99 g, 8.39 mmol) in CH3CN (80 mL) was
treated
with Nal (1.27 g, 8.47 mmol), and K2C03 (2.12 g, 15.3 mmol). The mixture was
heated to reflux under N2 for 3 days, then allowed to cool. The mixture was
diluted
with H20 (200 mL) and extracted with CH2CI2 (3 x 200 mL). The combined organic
layers were dried over Na2S04, filtered, and the solvent was removed in vacuo.
The
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-66-
residue was purified by flash column chromatography (silica gel, 2:1
EtOAc/hexanes)
to give an off-white solid (2.58 g, 79°l°). The solid was
recrystallized from
EtOAc/hexanes to give the title compound as a white fluffy solid: mp 168-169
°C;'H
NMR (300 MHz, CDCI3) S 7.75 (dd, J= 8.9, 4.6 Hz, 1 H), 7.54 (dd, J= 9.3, 2.2
Hz, 1
H), 7.23-7.29 (m, 2 H), 7.09-7.12 (m, 2 H), 6.89 (d, J = 8.4 Hz, 1 H), 3.54-
3.57 (m, 4
H), 2.67-2.85 (m, 10 H), 2.34-2.36 (m, 2 H), 2.21-2.26 (m, 2 H); ESI MS m/z
425
[C23H25FN4~S + H]+; Rf 0.31 (silica gel, EtOAc); HPLC >99% (AUC), tR =12.28
min.
Anal. Calcd for C23H25FN4OS: C, 65.07; H, 5.94; N, 13.20. Found: C, 64.94; H,
5.97;
N, 13.13.
7-~2-f4-(7-Fluoro-benzofdlisothiazol-3-yl)-piperazin-1-yll-ethyl)-1,3,4,5-
tetrahydro-benzo~blazepin-2-one (Prep. 26B) ,
A suspension of 7-chloroethylazepin-2-one (1.49 g, 6.66 mmol) and 7-fluoro-3-
piperazin-1-yl-benzo[dJisothiazole ~ HCI (2.00 g, 7.31 mmol) in CH3CN (70 mL)
was
treated with Nal (1.10 g, 7.34 mmol), and K2C03 (2.77 g, 20.0 mmol). The
mixture
was heated to reflux under N2 for 3 days, then allowed to cool. The mixture
was
diluted with H20 (200 mL) and extracted with CH2C12 (3 x 200 mL). The combined
organic layers were dried over Na2S04, filtered, and the solvent was removed
in
vacuo. The residue was purified by flash column chromatography (silica gel,
4:1
EtOAc/hexanes) to give the title compound (2.54 g, 90%) as an off-white solid:
mp
180-181 °C; 1H NMR (300 MHz, CDC13) 8 7.70 (d, J= 8.1 Hz, 1 H), 7.31-
7.37 (m, 1
H), 7.09-7.17 (m, 4 H), 6.88 (d, J = 8.5 Hz, 1 H), 3.59-3.62 (m, 4 H), 2.66-
2.87 (m, 10
H), 2.34-2.36 (m, 2 H), 2.21-2.25 (m, 2 H); ESI MS m/z 425 [C23H25FN4OS + H]+;
Rf
0.22 (silica gel, EtOAc); HPLC >99% (AUC), tR = 12.79 min. Anal. Calc'd for
C23H2sFNaOS: C, 65.07; H, 5.94; N, 13.20. Found: C, 64.86; H, 5.86; N, 13.01
EXAMPLE 56
1-(7-f 2-f4-(5-Fluoro-benzo~dlisothiazol-3-yl)-piperazin-1-yll-ethyl)-2,3,4,5
tetrahydro-benzofblazepin-1-yl)-ethanone: compound with methane sulfonic
acid
A. 7-(2-f4-(5-Fluoro-benzo f dlisothiazol-3-yl)-piperazin-1-yll-ethyl~-2,3,4,5-
tetrahydro-1 H-benzo~blazepine
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-67-
A suspension of the title compound'of Preparation 26A (1.46 g, 3.44 mmol) in
THF
(10 mL) was treated dropwise with a solution of BH3 in THF (22 mL, 1.0M, 22
mmol).
The mixture was heated at reflux for 3 h, then allowed to cool. The reaction
was
quenched with 6N HCI until gas evolution subsided. The mixture was heated at
reflux for 30 min, allowed to cool, and then made basic (pH ~8-9) with solid
NaOH
and 1 N NaOH. The biphasic mixture was extracted with CH2CI2 (3 x 100 mL). The
combined organic layers were dried over Na2S04, filtered, and the solvent was
removed in vacuo. The residue was purified by flash column chromatography
(silica
gel, gradient from 2:1 to 1:1 hexanes/EtOAc) to give the title compound (0.53
g,
38%) as a colorless oil: 'H NMR (300 MHz, CDCI3) 8 11.56 (bs, 1 H), 7.82-7.86
(m,
2 H), 7.49-7.54 (m, 1 H), 7.40-7.43 (m, 1 H), 6.88 (s, 1 H), 6.75 (s, 1 H),
4.03-4.11
(m, 4 H), 3.46-3.70 (m, 2 H), 3.04-3.36 (m, 8 H), 2.91-2.97 (m, 4 H), 2.89 (s,
3 H),
2.69-2.73 (m, 2 H), 1.86 (bm, 2 H), 1.70 (bm, 2 H); ESI MS m/z419 ~C25~H3pNaS
+
H]+; Rf 0.34 (silica gel, 1:1 EtOAc/hexanes).
B. 1-(7-f2-f4-(5-Fluoro-benzofdlisothiazol-3-yl)-piperazin-1-yll-ethyl)-
2,3,4,5-
tetrahydro-benzofblazepin-1-yl)-ethanone: compound with methane sulfonic
acid
A solution of the title compound from step A (0.53 g, 1.29 mmol) in CH2C12 (15
mL)
was treated with acetic anhydride (0.13 mL, 1.4 mmol) under N2. After stirring
at rt
overnight, saturated NaHC03 (30 mL) was added, and the mixture was extracted
with CH2C12 (3 x 50 mL). The combined organic layers were dried over Na2S04,
filtered, and the solvent was removed in vacuo. The residue was purified by
flash
column chromatography (silica gel, gradient from 2:1 to 3:1 EtOAc/hexanes) to
give
a white semi-solid (0.53 g, 91 %). The semi-solid (0.53 g, 1.2 mmol) was
dissolved in
EtOAc (30 mL) and treated with CH3S03H (2M in Et20, 0.59 mL, 1.2 mmol). After
stirring for 30 minutes (min), the resulting precipitate was isolated by
filtration,
washed with Et20 (3 x 10 mL), and dried in a vacuum oven at 55 °C
overnight to give
the title compound (0.49 g, 76%) as a white powder: mp 188-191 °C
(dec);'H NMR
(300 MHz, CDCI3) S 11.86 (s, 1 H), 7.50-7.64 (m, 3 H), 7.28-7.33 (m, 2 H),
7.15 (dd,
J = 7.9, 1.9 Hz, 1 H), 7.01 (d, J = 7.9 Hz, 1 H), 4.73-4.77 (m, 1 H), 4.21 (d,
J = 14.6
Hz, 2 H), 3.95-4.04 (m, 2 H), 3.74 (d, J = 11.4 Hz, 2 H), 3.10-3.35 (m, 6 H),
2.91 (s, 3
H), 2.47-2.57 (m, 1 H), 2.14-2.26 (m, 2 H), 1.85-1.95 (m, 1 H), 1.45-1.74 (m,
3 H),
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-68-
1.42 (s, 3 H), 1.17 (s, 3 H), 1.07 (t, J = 7.4 Hz, 3 H); ESI MS m/z 461
[C2gHggNq.O2 -+-
H]+; Rf 0.66 (silica gel, 2:1 EtOAc/hexanes); HPLC 98.4% (AUC), tR = 13.67
min.
Anal. Calc'd for C28H36N402~CH3S03H~1.2H20: C, 60.23; H, 7.39; N, 9.69. Found:
C, 60.24; H, 7.40; N, 9.49.
EXAMPLE 57
1-f 7-f 2-f 4-(7-F1 uoro-benzo f dlisothiazol-3-yl)-piperazi n-1-yl~-ethyl)-
2,3,4,5
tetrahydro-benzofblazepin-1-yl)-ethanone; compound with methanesulfonic
acid
A. 7-f2-f4-(7-Fluoro-benzofdlisothiazol-3-yl)-piperazin-1-yll-ethyl)-2,3,4,5-
tetrahvdro-1 H-benzofblazepine
A suspension of the title compound from Preparation 26B (1.02 g, 2.40 mmol) in
THF
(10 mL) was treated dropwise with a solution of BH3 in THF (15 mL, 1.0 M, 15
mmol). The mixture was heated at reflux for 2.5 h, then allowed to cool. The
reaction was quenched with 1 N HCI until gas evolution subsided. The mixture
was
heated at reflux for 30 min, allowed to cool, and then made basic (pH ~8-9)
with 1 N
NaOH. The biphasic mixture was extracted with CH2C12 (3 x 100 mL). The
combined organic layers were dried over Na2S04, filtered, and the solvent was
removed in vacuo. The residue was purified by flash column chromatography
(silica
gel, 1:1 hexanes/EtOAc) to give the title compound (0.79 g, 80%) as a
colorless oil:
1H NMR (300 MHz, CDCI3) 8 11.56 (bs, 1 H), 7.82-7.86 (m, 2 H), 7.49-7.54 (m, 1
H),
7.40-7.43 (m, 1 H), 6.88 (s, 1 H), 6.75 (s, 1 H), 4.03-4.11 (m, 4 H), 3.46-
3.70 (m, 2
H), 3.04-3.36 (m, 8 H), 2.91-2.97 (m, 4 H), 2.89 (s, 3 H), 2.69-2.73 (m, 2 H),
1.86
(bm, 2 H), 1.70 (bm, 2 H); ESI MS m/z 419 [C25H3oN4S + H]+; Rf 0.34 (silica
gel, 1:1
EtOAc/hexanes).
B. 1-(7-('2-f4-(7-Fluoro-benzofdlisothiazol-3-yl)-piperazin-1-yll-ethyl)-
2,3,4,5-
tetrahydro-benzofblazepin-1-yl)-ethanone; compound with methanesulfonic
acid
A solution of the title compound from step A (0.72 g, 1.75 mmol) in CH2C12 (20
mL)
was treated with acetic anhydride (0.17 mL, 1.8 mmol) under N2. After stirring
at rt
overnight, the reaction mixture was diluted with CH2CI2 (50 mL) and washed
with
saturated NaHC03 (50 mL). The aqueous layer was extracted with CH2C12 (2 x 50
mL), and the combined organic layers were dried over Na2SOa., filtered, and
the
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-69-
solvent was removed in vacuo. The residue was purified by flash column
chromatography (silica gel, 4:1 EtOAc/hexanes) to give a white semi-solid
(0.60 g,
76%). The semi-solid (0.60 g, 1.3 mmol) was dissolved in EtOAc (20 mL) and
treated with CH3S03H (2M in Et20, 0.66 mL, 1.3 mmol). After stirring for 10
min, the
clear solution was diluted with Et20 (20 mL) to give a gummy precipitate.
After
stirring for 10 min, the solvent was removed in vacuo to give a gummy residue.
The
residue was suspended in Et20 (40 mL), and the mixture was stirred until the
residue
became particulate. The solid was isolated by filtration, washed with Et20 (4
x 20
mL), and dried in a vacuum oven at 45 °C overnight to give the title
compound (0.62
g, 85%) as a white powder: mp 188-191 °C (dec);'H NMR (300 MHz, CDCI3)
S
11.86 (s, 1 H), 7.50-7.64 (m, 3 H), 7.28-7.33 (m, 2 H), 7.15 (dd, J = 7.9, 1.9
Hz, 1 H),
7.01 (d; J = 7.9. Hz, 1 H), 4.73-4.77 (m, 1 H), 4.21 (d, J = 14.6 Hz, 2 H),
3.95-4.04 (m,
2 H), 3.74 (d, J = 11.4 Hz, 2 H), 3.10-3.35 (m, 6 H), 2.91 (s, 3 H), 2.47-2.57
(m, 1 H),
2.14-2.26 (m, 2 H), 1.85-1.95 (m, 1 H), 1.45-1.74 (m, 3 H), 1.42 (s, 3 H),
1.17 (s, 3
H), 1.07 (t, J= 7.4 Hz, 3 H); ESI MS m/z461 [C28H36N4O2 + H]+; Rf0.66 (silica
gel,
2:1 EtOAc/hexanes); HPLC 98.4% (AUC), tR = 13.67 min. Anal. Calcd for
C28H36N402~CH3S03H~1.2H20: C, 60.23; H, 7.39; N, 9.69. Found: C, 60.24; H,
7.40;
~' N, 9.49.
PREPARATION 27
7-f 2-(4-Benzo~dlisoxazol-3-yl-piperazin-1-yl)-ethyll-5,5-dimethyl-1,3,4,5-
tetrahydro-benzofblazepin-2-one
A. 4,4-Dimethyl-3,4-dihydro-2H naphthalen-1-one
Beilstein Registry Number 1818110; CAS Registry Number 2979-69-3
Endo, Y.; Takehana, S.; Ohno, M.; Driedger, P. E.; Stabel, S.; Mizutani, M.
Y.;
Tomioka, N.; Itai, A.; Shudo, K. J. Med. Chem. 1998, 41, 1476-1496.
B. 4,4-Dimethyl-3,4-dihydro-2H-naphthalen-1-one oxime
Beilstein Registry Number 1818110; CAS Registry Number 2979-69-3
Woods, G. F.; Heying, T. L.; Schwartzman, L. H.; Grenell, S. M.; Gasser, W.
F.;
Rowe, E. W.; Bolgiano, N. C. J. Org. Chem. 1954, 19, 1290-1295. Into a 1-L
round-
bottom flask was placed the tetralone from step A (8.94 g, 51.4 mmol),
hydroxylamine hydrochloride (4.29 g, 61.7 mmol), sodium acetate (8.43 g, 103
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-70-
mmol), and 50% aqueous ethanol (350 mL). The mixture was refluxed for 16 h,
cooled to rt and made alkaline by the addition of 10% aqueous NaHC03. The
reaction was extracted with CH2CI2 (3 x 100 mL) and the combined organic
layers
were dried over Na2S04 and concentrated under vacuum to produce the title
oxime
(8.37 g, 84%) as an orange solid.
C. 5,5-Dimethyl-1,3,4,5-tetrahydro-benzo~blazepin-2-one
Into a 500-mL round-bottom flask equipped with a mechanical stirrer was placed
polyphosphoric acid (90 g). The reaction was heated to 125°C, the title
compound
from step B (8.37 g, 44.3 mmol) was added in one portion, and the reaction was
stirred for 5 min. The mixture was poured into ice water (300 mL), stirred
until the
polyphosphoric acid was dissolved and extracted with CH2CI2 (3 x 100 mL). The
combined organic layers were dried over Na2S04 and concentrated under vacuum
to
produce the title benzazepinone (7.76 g, 93%) as a tan solid: 'H NMR (300 MHz,
CDCI3) 5 7.45 (bs, 1 H), 7.42 (dd, J = 7.7, 1.7 Hz, 1 H), 7.13-7.25 (m, 2 H),
6.91 (dd,
J = 7.6, 1.6 Hz, 1 H), 2.40 (t, J = 7.0 Hz, 2 H), 2.11 (t, J = 7.0 Hz, 2 H),
1.42 (s, 6 H);
ESI MS m/z 190 [C12H15NO + H]+.
D. 7-(2-Chloro-acetyl)-5,5-dimethyl-1,3,4,5-tetrahydro-benzo~blazepin-2-one
Into a 500-mL round-bottom flask equipped with a mechanical stirrer was placed
aluminum chloride (33.6 g, 252 mmol), anhydrous dichloromethane (230 mL),
chloroacetyl chloride (4.87 mL, 61.05 mmol), and the title compound from step
C
(7.69 g, 40.7 mmol). The reaction was slowly heated to reflux and stirred for
15 h.
The mixture was cooled to 0°C and ice water (200 mL) was slowing
added
(CAUTION: exotherm). The two layers were separated and the aqueous layer was
extracted with dichloromethane (3 x 100 mL). The combined organic layers were
washed with H20 (150 mL), dried over Na2SOa, concentrated under vacuum, and
chromatographed (silica, 4:1 hexanes/ethyl acetate) to produce the title
compound
(3.82 g, 35%) as a yellow solid: 'H NMR (300 MHz, CDCI3) 8 9.07 (s, 1 H), 8.09
(d, J
=1.9 Hz, 1 H), 7.80 (dd, J = 8.2, 2.0 Hz, 1 H), 7.10 (d, J = 8.3 Hz, 1 H),
4.68 (s, 2 H),
2.49(t,J=6.5Hz,2H),2.15(t,J=8.3Hz,2H),1.46(s,6H).
E. 7-(2-Chloro-ethyl)-5,5-dimethyl-1,3,4,5-tetrahydro-benzo~blazepin-2-one
Into a 100-mL round-bottom-flask was placed the ketone from step D (2.23 g,
8.4
mmol) and TFA (16 mL). The solution was cooled to 0°C and
triethylsilane (4.07 mL,
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-71-
25.2 mmol) was added dropwise over a 5 min period. The reaction was warmed to
50°C and stirred for 15 h. The mixture was cooled to rt, diluted with
H20 (100 mL)
and extracted with ethyl acetate (3 x 50 mL). The combined organic layers were
dried over Na2S04, and concentrated under high vacuum and chromatographed
(silica, 4:1 hexanes/ethyl acetate) to produce the title compound (1.58 g,
75%) as a
white solid: 'H NMR (300 MHz, CDCI~) 8 7.83 (s, 1 H), 7.24 (d, J= 1.8 Hz, 1
H), 7.08
(dd, J = 8.0, 2.0 Hz, 1 H), 6.89 (d, J = 8.0 Hz, 1 H), 3.71 (t, J = 7.3 Hz, 2
H), 3.06 (t, J
=7.3Hz,2H),2.39(t,J=6.8Hz,2H),2.11 (t,J=7.4Hz,2H),1.40(s,6H).
F. 7-f2-(4-Benzo~dlisoxazol-3-yl-aiperazin-1-yl)-ethyll-5,5-dimethyl-1,3,4,5-
tetrahydro-benzofblazepin-2-one
A suspension of the title compound from step E (2.55 g, 10.1 mmol) in CH3CN
(100
mL) was treated with 3-piperazin-1-yl-benzo[d]isoxazole ~ HCI (2.68 g, 11.2
mmol),
Nal (1.68 g, 11.2 mmol), and K2C03 (4.22 g, 30.5 mmol). The mixture was heated
to
reflux under N2 for 87 h, then allowed to cool. The mixture was diluted with
H20 (250
mL) and extracted with CH2CI2 (4 x 200 mL). The combined organic layers were
dried over Na2S04, decanted, and the solvent was removedin vacuo. The residue
was purified by flash column chromatography (silica gel, EtOAc) to give the
title
compound (3.09 g, 73%) as a white powder: mp 193-194°C (dec);'H NMR
(300
MHz, CDCI3) S 7.70 (d, J = 8.0 Hz, 1 H), 7.44-7.52 (m, 2 H), 7.20-7.28 (m, 2
H), 7.15
(s, 1 H), 7.08 (dd, J = 7.9, 1.9 Hz, 1 H), 6.82 (d, J = 7.9 Hz, 1 H), 3.61-
3.65 (m, 4 H),
2.82-2.87 (m, 2 H), 2.73-2.76 (m, 4 H), 2.65-2.70 (m, 2 H), 2.39 (t, J = 7.0
Hz, 2 H),
2.10 (t, J = 7.0 Hz, 2 H), 1.41 (s, 6 H); ESI MS m/z 419 [C25H3oN4O2 + H]+; Rf
0.55
(silica gel, EtOAc); HPLC 96.4% (AUC), tR =12.12 min. Anal. Calc'd for
C25H30N4~2~~~2H2O: C, 71.13; H, 7.26; N, 13.27. Found: C, 71.11; H, 7.30; N,
13.06.
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-72
EXAMPLE 60
f7-f2-(4-Benzofdlisoxazol-3-yl-piperazin-1-yl)-ethyll-5,5-dimethyl-2,3,4,5
tetrahydro-benzo~blazepin-1-yl)-(4-fluoro-phenyl)-methanone; compound with
methanesulfonic acid
A. 7-f2-(4-Benzofdlisoxazol-3-yl-piperazin-1-yl)-ethyll-5,5-dimethyl-2,3,4,5-
tetrahydro-1 H benzofblazepine
A suspension of 7-[2-(4-Benzo[dJisoxazol-3-yl-piperazin-1-yl)-ethyl]-5,5-
dimethyl-
1,3,4,5-tetrahydro-benzo[b]azepin-2-one (3.09 g, 7.38 mmol) in THF (50 mL) was
treated with borane (1.5 M in THF, 35.0 mL, 52.5 mmol) over 10 min. The
resulting
clear solution was heated to reflux under N2 for 3.5 h, then allowed to cool.
The
reaction was quenched with 6N HCI until gas evolution subsided. The mixture
was
heated to reflux for 45 min, allowed to cool, and then made basic with solid
NaOH
and a 1 N NaOH solution (50 mL). The biphasic mixture was extracted with
CH2C12
(4 x 50 mL), and the combined organic layers were washed with brine, dried
over
Na2S04, filtered, and the solvent was removed in vaeuo. The residue was
purified
by flash column chromatography (silica gel, 2:1 hexanes/EtOAc) to give the
title
compound (2.42 g, 81%) as a colorless oil: 'H NMR (300 MHz, CDCI3) 87.70 (d,
J=
8.0 Hz, 1 H), 7.45-7.48 (m, 2 H), 7.18-7.24 (m, 1 H), 7.16 (d, J = 1.9 Hz, 1
H), 6.88
(dd, J = 7.8, 2.0 Hz, 1 H), 6.62 (d, J = 7.8 Hz, 1 H), 3.61-3.64 (m, 4 H),
3.00 (t, J =
5.6 Hz, 2 H), 2.62-2.81 (m, 8 H), 1.82-1.90 (m, 2 H), 1.60-1.64 (m, 2 H), 1.38
(s, 6
H); ESI MS m/z 405 [C25H~2N40 + H]+; Rf 0.39 (silica gel, 2:1 hexanes/EtOAc).
B. 1-f 7-f 2-(4-Benzo f dlisoxazol-3-yl-piperazi n-1-yl)-ethyll-5,5-d imethyl-
2,3,4,5-tetrahydro-benzofblazepin-1-yl)-ethanone methanesulfonate
A solution of the title compound from step A (0.90 g, 2.2 mmol) in CH2CI2 (20
mL)
was treated with Ac20 (0.21 mL, 2.2 mmol). After stirring at rt under N2 for
18 h, the
reaction was quenched with saturated NaHCOs (30 mL) and extracted with CH2CI2
(3 x 50 mL). The combined organic layers were dried over Na2S04, filtered, and
the
solvent was removed in vacuo. The residue was purified by flash column
chromatography (silica gel, 2:1 EtOAc/hexanes to 3:1 EtOAc/hexanes to EtOAc)
to
give a white solid residue (0.58 g, 1.30 mmol, 58%). The residue was dissolved
in
EtOAc (6.5 mL) and treated with CH3S03H (2M in Et20, 0.65 mL, 1.3 mmol). After
stirring for 45 min, the oily, precipitous mixture was diluted with EtOAc (6.5
mL).
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-73-
After stirring for an additional 19 h, the resulting precipitate was isolated
by filtration,
washed with Et20 (3 x 20 mL), and dried in a vacuum oven at 60°C
overnight to give
the title compound (595 mg, 84%) as a white powder: mp 227-229°C (dec);
1H NMR
(300 MHz, CDCI3) 8 11.94 (s, 1 H), 7.62 (d, J= 8.0 Hz, 1 H), 7.50-7.59 (m, 2
H),
7.27-7.33 (m, 2 H), 7.16 (dd, J = 7.9, 1.9 Hz, 1 H), 7.02 (d, J = 7.9 Hz, 1
H), 4.71-
4.75 (m, 1 H), 4.21 (d, J = 14.6 Hz, 2 H), 3.96-4.04 (m, 2 H), 3.74 (d, J =
11.7 Hz, 2
H), 3.12-3.35 (m, 6 H), 2.90 (s, 3 H), 2.48-2.52 (m, 1 H), 2.15-2.20 (m, 1 H),
1.86 (s,
3 H), 1.47-1.74 (m, 3 H), 1.43 (s, 3 H), 1.20 (s, 3 H); ESI MS m/z 447
~C27H34N4O2 -+-
H]+; Rf 0.64 (silica gel, EtOAc); HPLC >99% (AUC), tR = 12.97 min. Anal.
Calc'd for
C2~H34N402~CH3S03H~0.3H20: C, 61.36; H, 7.10; N, 10.22. Found: C, 61.42; H,
7.13; N, 10.17.
C. ~7-f2-(4-Benzofdlisoxazol-3-yl-piperazin-1-yl)-ethyll-5.5-dimethy1-2.3,4,5-
tetrahydro-benzo~blazepin-1-yl}-(4-fluoro-phenyl)-methanone
methanesulfonate
A solution of the title compound from step B (0.90 g, 2.2 mmol) in CH2C12 (20
mL)
under N2 was cooled in an ice bath, treated Et3N (0.34 mL, 2.42 mmol) and p-
fluorobenzoyl chloride (0.26 mL, 2.2 mmol), and warmed to rt. After stirring
at rt for
17 h, the reaction was quenched with saturated NaHCOs (30 mL) and extracted
with
CH2CI2 (3 x.50 mL). The combined organic layers were dried over Na2S04,
filtered,
and the solvent was removed in vacuo. The residue was purified by flash column
chromatography (silica gel, 7:3 hexanes/EtOAc) to give a colorless residue
(0.74 g,
1.40 mmol, 63%). The residue (0.71 g, 1.35 mmol) was dissolved in EtOAc (13
mL)
and treated with CH3S03H (2M in Et20, 0.67 mL, 1.34 mmol). After stirring for
21 h,
the clear solution was treated with Et20 (20 mL). After stirring for 45 min,
the oily,
precipitous mixture was treated with Et20 (10 mL) and stirred for an
additional 27 h.
The mixture was concentrated in vacuo, and the resulting oily solid was
suspended
in Et20. The solid was isolated by filtration, washed with Et20, and dried in
a
vacuum oven at 60 °C for 4 d to give the title compound (629 mg, 75%)
as a white
powder: mp 135-140 °C; 1H NMR (300 MHz, CDC13) S 11.91 (s, 1 H), 7.50-
7.62 (m,
4 H), 7.21-7.36 (m, 3 H), 6.78-6.85 (m, 3 H), 6.50 (d, J = 7.9 Hz, 1 H), 5.02-
5.06 (m,
1 H),4.19(d,J=13.9Hz,2H),3.98(t,J=13.4Hz,2H),3.69(d,J=11.2Hz,2H),
3.05-3.26 (m, 6 H), 2.88 (s, 3 H), 2.72 (t, J = 11.7 Hz, 1 H), 2.23-2.28 (m, 1
H), 1.77-
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-74-
1.90 (m, 2H), 1.55-1.64 (m, 1 H), 1.50 (s, 3H), 1.49 (s, 3H); ESI MS m/z 527
[C32H35FN4O2 + H]+; Rf 0.77 (silica gel, 2:1 EtOAc/hexanes); HPLC >99% (AUC),
tR =
15.23 min. Anal. Calc'd for C32Ha5FN402~CH3S03H~H20: C, 61.86; H, 6.45; N,
8.74.
Found: C, 61.89; H, 6.40; N, 8.67.
EXAMPLE 61
1-f 7-f2-(4-Benzof dlisoxazol-3-yl-piperazi n-1-yl)-ethyll-5,5-d imethyl-
2,3,4,5
tetrahydro-benzofblazepin-1-yl)-propan-1-one; compound with
methanesulfonic acid
A solution of the product of Example 60, step A, 7-[2-(4-Benzo[d]isoxazol-3-yl
piperazin-1-yl)-ethyl]-5,5-dimethyl-2,3,4,5-tetrahydro-1 H benzo[b]azepine
(0.45 g,
1.1 mmol) in CH2C12 (10 mL) under N2 was cooled in an ice bath, treated with
Et3N
(0.17 mL, 1.2 mmol) and propionyl chloride (0.10 mL, 1.1 mmol), and warmed to
rt.
After stirring at rt for 23 h, additional propionyl chloride (0.10 mL, 1.1
mmol) was
added. After stirring for an additional 8 h, the reaction was puenched with
saturated
NaHC03 (20 mL) and extracted with CH2C12 (3 x 20 mL). The combined organic
layers were dried over Na2S04, filtered, and the solvent was removed in vacuo.
The
residue was purified by flash column chromatography (silica gel, 2:1
EtOAc/hexanes)
to give a colorless residue (0.40 g, 0.87 mmol, 79%). The residue (0.40 g,
0.87
mmol) was dissolved in EtOAc (10 mL) and treated with CH3S03H (2 M in Et20,
0.43
mL, 0.86 mmol). After stirring for 15 min, the clear solution was treated with
Et20 (30
mL). After stirring for 20 min, the oily, precipitous mixture was concentrated
in
vacuo, and the resulting oily solid was suspended in Et20 (15 mL). After
stirring for
16 h, the solid was isolated by filtration, washed with Et20 (3 x 10 mL), and
dried in a
vacuum oven at 60 °C for 24 h to give the title compound (416 mg, 86%)
as a white
powder: mp 188-191 °C (dec); 1H NMR (300 MHz, CDCI3) 8 11.86 (s, 1 H),
7.50-
7.64 (m, 3 H), 7.28-7.33 (m, 2 H), 7.15 (dd, J = 7.9, 1.9 Hz, 1 H), 7.01 (d, J
= 7.9 Hz,
1 H), 4.73-4.77 (m, 1 H), 4.21 (d, J = 14.6 Hz, 2 H), 3.95-4.04 (m, 2 H), 3.74
(d, J =
11.4 Hz, 2 H), 3.10-3.35 (m, 6 H), 2.91 (s, 3 H), 2.47-2.57 (m, 1 H), 2.14-
2.26 (m, 2
H), 1.85-1.95 (m, 1 H), 1.45-1.74 (m, 3 H), 1.42 (s, 3 H), 1.17 (s, 3 H), 1.07
(t, J = 7.4
Hz, 3 H); ESI MS mlz 461 [C28H36N4~2 "~ H]+~ Rf 0.66 (silica gel, 2:1
EtOAc/hexanes);
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-75-
HPLC 98.4% (AUC), tR = 13.67 min. Anal. Calc'd for C28H36N402~CH3S03H~1.2H20:
C, 60.23;.H, 7.39; N, 9.69. Found: C, 60.24; H, 7.40; N, 9.49.
PREPARATION 28
6-(2-Chloroacetyl)-3,3-dimethyl-3,4-dihydro-1 H-auinolin-2-one
3,3-Dimethyl-3,4-dihydro-1 H-quinolin-2-one was prepared according to the
procedure in J. Med. Chem., 1986, 29, 1832, and underwent a Friedel-Crafts
acylation with chloroacetyl chloride according to the procedure described in
Preparation 1 to give the title compound as a solid. MS (APCI): (M + 1)~ =
252.
PREPARATION 29
6-(2-Chloro-ethyl)-3,3-dimethyl-3,4-dihydro-1 H-auinoiin-2-one
To a mixture of the title compound of Preparation 28 (6.52 g, 0.026 mol) and
trifluoroacetic acid (20 ml, 0.26 mol), cooled to 0°C under nitrogen,
was added
portionwise triethylsilane (9.57 ml, 0.059 mol). The reaction mixture was
heated at
40-45 °C for 20 minutes and then stirred at rt for 16 hours. The
solution was poured
into ice water layered with hexane and vigorously stirred for several hours.
The
precipitate that formed was collected and washed with water and hexanes to
give the
title compound as a solid. MS (APCI): (M + 1)+= 238.
PREPARATION 30
6-(2-Chloro-ethyl)-3,3-dimethyl-1,2,3,4-tetrahydro-auinoline
To a solution of 6-(2-chloroethyl)-3,3-dimethyl-3,4-dihydro-1 H-quinolin-2-one
(1.0 g, 4.21 mmol) in anhydrous THF (75 ml) under nitrogen was added 1.0 M
borane-THF complex (14.3 ml, 14.3 mmol). The reaction mixture was refluxed for
1.5 hours and cooled to ambient temperature. The excess reagent was quenched
with water and the organic solvent was removed in vacuo. The aqueous residue
was extracted with methylene chloride and the organic extract was dried over
magnesium sulfate, filtered, and concentrated to an oil. Yield = 942 mg
(100%); MS
(APCI), (M + 1 )f = 224. ~ H-NMR (CDCl3) S 6.80 (d, J = 8.1 Hz, 1 H), 6.75 (s,
1 H),
6.43 (d, J = 8.1 Hz, 1 H), 3.87 (br s, 1 H), 3.62 (t, J = 7.6, 8.1 Hz, 2 H),
2.88 (m, 4 H),
2.45 (s, 2 H), 0.99 (s, 6 H).
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-76-
PREPARATION 31
1-f 6-(2-Chloro-ethyl)-3,3-dimethyl-3,4-di hydro-2H-auinoli n-1-yll-ethanone
A mixture of 6-(2-chloroethyl)-3,3-dimethyl-1,2,3,4-tetrahydroquinoline (942
mg, 4.21 mmol) and acetyl chloride (0.317 ml, 4.44 mmol) in dry acetone (15
ml) was
refluxed for 2 hours. The reaction mixture was poured into dilute aqueous HCI
(100
ml) and the whole extracted with chloroform. The organic extract was dried
over
magnesium sulfate, filtered, and concentrated. The product was washed with
hexane to give an off-white, crystalline solid. Yield = 791 mg (71 %); MS
(APCI), (M
+ 1 )+ = 266. ' H NM R (CDC13) S 7.25 (s, 1 H), 7.00 (d, J = 7.1 Hz, 1 H),
6.94 (s, 1 H),
3.68(t,J=7.6,7.3Hz,2H),3.52(brs,2H),3.00(t,J=7.3,7.3Hz,2H),2.57(s,2
H), 2.27 (s, 3 H), 0.99 (s, 6 H).
EXAMPLE 62
1-(6-(2-(4-(1 H-Indazol-3-yll-piperazin-1-yl)-ethyl'i-3,3-dimethyl-3,4-dihydro-
2H-
puinolin-1-yl)-ethanone
A mixture of 3-piperazin-1-yl-1 H-indazole hydrochloride (520 mg, 2.60 mmol),
1-[6-(2-chloroethyl)-3,3-dimethyl-3,4-dihydro-2H-quinolin-1-yl]-ethanone (791
mg,
2.98 mmol), anhydrous potassium carbonate (791 mg, 5.70 mmol) and potassium
iodide (75 mg) in acetonitrile (50 ml) was refluxed for 72 hours. The reaction
mixture
was concentrated and the residue was partitioned between water and methylene
chloride. The organic layer was dried over magnesium sulfate, filtered, and
concentrated. The crude product was eluted through a flash column (silica gel
60,
230-400 mesh, 5% MeOH in EtOAc) and taken up in methylene chloride. Treatment
with 4.0 N HCI solution in dioxane precipitated the dihydrochloride salt.
Yield = 496
mg (38%); Anal. calculated for C26HssNsO~2HCI: C, 61.90; H, 6.99; N, 13.88.
Found:
C, 61.50: H, 7.27; N, 13.45. MS (APCI), (M + 1 )+ = 432; (M -1 )+ = 430. ' H-
NMR
(DMSO-d6) 812.18 (s, 1 H), 10.75 (br s, 1 H), 7.75 (d, J= 8.3 Hz, 1 H), 7.34
(m, 3
H), 7.02 (m, 3 H), 3.92 (br d, H =10.5 Hz, 2 H), 3.61-3.22 (m, 10 H), 3.00 (m,
2 H),
2.46 (s, 2 H), 2.16 (s, 3 H), 0.89 (s, 6 H).
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-77-
EXAMPLE 63
1- 6- 2-(4-Benzo d~isvxazol-3-yi-piperazin-1-yl)-ethyll-7-chloro-4,4-dimethyl-
3,4
dihydro-2H-auinolin-1-yl)-ethanone; compound with methanesulfonic acid
A. 7-Chloro-6-(2-chloroethyl)-4,4-dimethyl-3,4-dihydro-1 H auinolin-2-one
Triethylsilane (10.0 mL, 62.6 mmol) was added to a stirred solution of 7-
chloro-6-(2-
chloroacetyl)-4,4-dimethyl-3,4-dihydro-1 H-quinolin-2-one (5.03 g, 17.6 mmol)
in
trifluoroacetic acid (25 mL) at 0 °C under nitrogen (N2). After
stirring for 5 min, the
cooling bath was replaced with a heating bath, and the thermostat set for 50
°C.
After stirring for 15 hours (h), the mixture was allowed to cool, poured into
H20 (150
rnL), then extracted twice with EtOAc (150 mL). The extracts were combined,
washed with H20 (150 mL), saturated NaHCO~, and saturated sodium chloride
(NaCI), dried over Na2S0~., filtered, and the solvent removed in vacuo. The
residue
was purified by column chromatography (silica gel (100 g), eluting with 800 mL
of
10% EtOAc/hexanes to remove the triethylsilane (Et3SiH) (collected as a single
fraction), then eluting with 1.5 L of 40% EtOAc/hexanes to elute the product)
to give
the title compound (1.93 g, 40%) as an off-white amorphous solid. Due to the
low
mass recovery, the initial column wash was examined and determined to contain
more of the product. The initial column wash was concentrated in vacuo to give
a
mixture of solid and liquid. The liquid was decanted, and the solid washed and
decanted twice with hexanes. The solid was dried under vacuum to give more of
the
title compound (1.82 g, 38%) as an off-white amorphous solid: 'H NMR (300 MHz,
CDCI3) 8 8.46 (br s, 1 H), 7.17 (s, i H), 6.84 (s, 1 H), 3.72 (t, J= 7.3 Hz, 2
H), 3.15 (t,
J = 7.3 Hz, 2 H), 2.49 (s, 2 H), 1.32 (s, 6 H); ESI MS mlz 272 [C13H15CI2NO +
H]+.
B. 7-Chloro-6-(2-chloroethyl)-4,4-dimethyl-1,2,3,4-tetrahydroauinoline
Borane (25 mL, 38 mmol, 1.5 M in THF) was added portionwise over 5 min to a
stirred solution from step A (1.93 g, 7.09 mmol) in anhydrous THF (40 mL)
under N2.
The mixture was heated to reflux for 3 h, then allowed to cool. The mixture
was
quenched by adding (slowly at first) 6 M HCI (25 mL) with stirring. The
mixture was
heated to reflux for 30 min, then allowed to cool. The mixture was diluted
with H20
(100 mL), then NaOH pellets were added until the mixture was strongly
alkaline.
The organic phase was separated, and the aqueous phase extracted twice with
chloroform (CHCI3) (75 mL). The organic phases were combined, dried over
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
_78_
Na2S04, filtered, and the solvent removed in vacuo. The residue was purified
by
column chromatography (silica gel (100 g), 10% EtOAc/hexanes) to give the
title
compound (1.92 g, 1.83 g theoretical) as a clear, light yellow oil: 'H NMR
(300 MHz,
CDC13) 8 7.02 (s, 1 H), 6.46 (s, 1 H), 3.92 (br s, 1 H), 3.65 (t, J = 7.7 Hz,
2 H), 3.27-
3.32 (m, 2 H), 3.04 (t, J= 7.7 Hz, 2 H), 1.68-1.73 (m, 2 H), 1.27 (s, 6 H);
ESI MS mlz
258 [C13H1~CI2N + H]+.
C. 1-f7-Chloro-6-(2-chloroethyl)-4,4-dimethyl-3,4-dihydro-2H auinolin-1-
yllethanone
Acetic anhydride (1.0 mL, 11 mmol) was added to a stirred solution from step B
(918
mg, 3.56 mmol) and Et3N (1.5 mL, 11 mmol) in anhyd CH2CI2 (10 mL) under N2.
After stirring for 23 h, MeOH (1 mL) was added to quench the excess reagent.
After
stirring for 1.25 h, the solvents were removed in vacuo. The residue was
purified by
column chromatography (silica gel (50 g), 10-30% EtOAc/hexanes) to give the
title
compound (934 mg, 87% from the product of step A) as a pale yellow oil: 'H NMR
(300 MHz, CDCI3) 8 7.33 (br s, 1 H), 7.20 (s, 1 H), 3.79 (t, J= 6.4 Hz, 2 H),
3.73 (t, J
= 7.3 Hz, 2 H), 3.16 (t, J = 7.3 Hz, 2 H), 2.27 (s, 3 H), 1.77 (t, J = 6.3 Hz,
2 H), 1.29
(s, 6 H); ESI MS m/z 300 [C15H19CI2NO + H]+.
D. 1-d6-f2-(4-Benzof dlisoxazol-3-ylpiperazin-1-yl)ethyll-7-chloro-4,4-
dimethyl-3,4-dihydro-2H auinolin-1-yl)ethanone methanesulfvnate
Anhydrous acetonitrife (15 mL) was added to a flask containing the title
compound
from step C (929 mg, 3.09 mmol), 3-piperazin-1-yl-benzo[d]isoxazole
hydrochloride
(821 mg, 3.43 mmol), potassium carbonate (K2C03) (883 mg, 6.39 mmol), and
sodium iodide (Nal) (486 mg, 3.24 mmol) under N2. The mixture was stirred,
giving a
suspension. The mixture was heated to reflux overnight (16 h). TLC analysis
'S indicated low conversion, so tetrabutylammonium iodide (Bu4Nl) (3.34 g,
9.04 mmol)
was added and the mixture heated to reflux for 4 d, then allowed to cool. TLC
analysis indicated moderate conversion. The mixture was diluted with EtOAc
(200
mL), then washed twice with H20 (200 mL) and saturated (saturated) NaCI (75
mL).
The combined aqueous phases were reextracted with EtOAc (100 mL), and the
0 extract was washed with H20 (100 mL) and saturated NaCI (50 mL). The organic
phases were combined, dried over Na2S04, filtered, and the solvent removed in
vacuo. The residue was purified by column chromatography (silica gel (50 g),
30-
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
_79_
50% EtOAclhexanes containing 1% Et3N) to give the title compound as a free
base
(945 mg, 65%) as a colorless sticky oil. The product was dissolved in EtOAc
(20
mL), then CH3S03H (120 p,L, 1.85 mmol) was added dropwise with stirring to
give a
clear solution. After stirring a couple more minutes, a white precipitate
began to
form. After stirring for 2 h, the precipitate was collected by suction
filtration washing
with EtOAc, then dried in a vacuum oven at 50 °C for 20 h to give the
title compound
(772 mg, 68%) as a white amorphous solid: mp 174-177 °C; 1 H NMR (300
MHz,
DMSO-d6) 8 9.95 (br s, 1 H), 8.08 (d, J = 8.1 Hz, 1 H), 7.75 (br s, 1 H), 7.60-
7.68 (m,
2 H), 7.44 (s, 1 H), 7.31-7.41 (m, 1 H), 4.21 (br d, J= 10.8 Hz, 2 H), 3.68-
3.79 (m, 4
H), 3.27-3.50 (m + H20), 3.09-3.19 (m, 2 H), 2.34 (s, 3 H), 2.22 (s, 3 H),
1.70-1.78
(m, 2 H), 1.26 (s, 6 H); ESI MS mlz467 [C26H31CIN4O2 + H]+; HPLC >99% (AUC),
tR
= 13.71 min. Anal. Calc'd for C26H31CIN4O2~CH3SO3H: C, 57.59; H, 6.26; N,
9.95.
Found: C, 57.68; H, 6.20; N, 9.74.
EXAMPLE 64
1-~6-[2-(4-Benzo[dlisoxazol-3-yl-piperazi n-1-VI)-ethyll-7-chloro-4,4-d
imethyl-3,4-
dihydro-2H-auinolin-1-yl)-2-methyl-propan-1-one; compound with
methanesulfonic acid
A. 1-[7-Chloro-6-(2-chloroethyl)-4,4-dimethyl-3,4-dihydro-2H auinolin-1-yll-
2-methylpropan-1-one
Isobutyric anhydride (1.7 mL, 10 mmol) was added to a stirred solution of the
title
compound of step B of Example 63 (1.00 g, 3.87 mmol) and triethylamine (Et3N)
(2.0
mL, 14 mmol) in anhydrous CH2CI2 (10 mL) under N2. After stirring for 13 h,
TLC
analysis indicated only starting material, so the acylation catalyst 4-
dimethylaminopyridine (353 mg, 2.89 mmol) was added. After stirring for 17 h,
TLC
and HPLC analysis indicated low (15%) conversion. The mixture was heated to
reflux overnight (21 h), at which point HPLC analysis indicated 50%
conversion. The
mixture was heated to reflux for another 3 days, during which time most of the
solvent evaporated, then allowed to cool. The residue was partitioned between
EtOAc (200 mL) and H20 (200 mL). The organic phase was washed with 0.5 M HCI
(100 mL), H20 (100 mL), saturated NaHC03 (50 mL), and saturated NaCI (50 mL).
The aqueous phase was reextracted with EtOAc (100 mL) and the extract washed
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-80-
as before. (Note: All the extractions and washings were slowed by emulsions.)
The
organic phases were combined, dried over Na2SOa., filtered, and the solvent
was
removed in vacuo. The residue was purified by column chromatography (silica
gel
(50 g), 5-15% EtOAc/hexanes) to give the title compound (298 mg, 23%) as a
dark
yellow, viscous oil: 1H NMR (300 MHz, CDC13) 8 7.31 (br s, 1 H), 7.21 (s, 1
H), 3.78
(t,J=6.3Hz,2H),3.73(t,J=7:3Hz,2H),3.16(t,J=7.3Hz,2H),3.06-3.17(m,1
H), 1.76 (t, J = 6.3 Hz, 2 H), 1.29 (s, 6 H), 1.21 (d, J = 7.0 Hz, 3 H), 1.17
(d, J = 6.7
Hz, 3 H); ESI MS m/z 328 [C17H23CI2NO + H]+.
B. 1-f6-f2-(4-Benzo~d~isoxazol-3-ylpiperazin-1-yl)ethyll-7-chloro-4,4-
dimethyl-3,4-dihydro-2H ctuinolin-1-yl~-2-methylpropan-1-one
methanesulfonate
Anhydrous acetonitrile (10 mL) was added to a flask containing the product
from step
A (293 mg, 0.893 mmol), 3-piperazin-1-yl-benzo[d]isoxazole hydrochloride (243
mg,
1.01 mmol), K2COa (271 mg, 1.96 mmol), and sodium iodide (Nal) (459 mg, 3.06
mriiol) under N2. The mixture was stirred to give a suspension, then heated to
reflux.
After 2 d at reflux, HPLC analyses indicated approximately 50% conversion.
More
anhydrous acetonitrile (10 mL) was added to replace solvent that had escaped,
then
the mixture was heated to reflux overnight (23 h). HPLC analysis indicated
approximately 1:2 SM/Pdt. After heating to reflux overnight (total reaction
time = 4
d), the mixture was allowed to cool. HPLC analysis indicated approximately 1:3
SM/Pdt. The mixture was diluted with EtOAc, washed twice with H20, once with
saturated NaCI, dried over Na2S04, filtered, and the solvent was removed in
vacuo.
The residue was purified by column chromatography (silica gel (18 g), 30-50%
EtOAc/hexanes containing 1 % Et3N) to give the free base of the title compound
(234
mg, 53%) as a dark yellow oil. The product was dissolved in EtOAc (10 mL),
then
methanesulfonic acid (CH3S03H) (30 p.L, 1.0 equiv.) was added dropwise with
stirring. After stirring for 5 min, hexanes (5 mL) was added to the stirred
solution.
After stirring another 5 min, a precipitate began to form. After stirring for
2 h, the
precipitate was collected by suction filtration washing with EtOAc, then dried
in a
vacuum oven at 50°C for 22 h to give the title compound (134 mg, 48%)
as a light
brown amorphous solid: mp 153-157 °C dec; ' H NMR (300 MHz, DMSO-ds) b
9.91
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-81-
(br s, 1 H), 8.08 (d, J = 8.1 Hz, 1 H), 7.56-7.68 (m, 3 H), 7.45 (s, 1 H),
7.33-7.39 (m,
1 H), 4.12-4..27 (m, 2 H), 3.69-3.80 (m, 4 H), 3.24-3.48 (m + H20), 3.02-3.19
(m, 2
H), 2.32 (s, 3 H), 1.69-1.77 (m, 2 H), 1.27 (s, 6 H), 1.06 (d, J= 6.6 Hz, 6
H); ESI MS
m/z 495 [C28H35CINa.O2 + H]~; HPLC >99% (AUC), tR = 15.37 min. Anal. Calc'd
for
C2sH35CIN4O2~CHsSO3H: C, 58.92; H, 6.65; N, 9.48. Found: C, 58.92; H, 6.76; N,
9.33.
EXAMPLE 65
1-(7-(2-(4-Benzo(dlisothiazol-3-yl-i~iperazin-1-yl)-ethyl~-2,3,4,5-tetrahydro
benzo(blazepin-1-yl)-ethanone~ compound with methanesulfonic acid
A. 7-(2-Chloro-ethyl)-2,3,4,5-tetrahydro-1 H benzo~blazepine
A solution of 7-(2-chloroethyl)-1,3,4,5-tetrahydro-benzo[b]azepin-2-one (784
mg,
3.50 mmol) (Example 36 of US Patent No. 5,350,747) in THF (15 mL) was added
dropwise to a solution of BHP in THF (15.0 mL, 1.5 M, 22.5 mmol). The mixture
was
heated to reflux for 3 h, then allowed to cool. The reaction was quenched with
6N
HCI until gas evolution subsided. The mixture was heated to reflux for 1 h,
allowed
to cool, and then made basic with solid NaOH. The biphasic mixture was
extracted
with CH2CI2 (3 x 50 mL), and the combined organic layers were dried over
Na2S04,
filtered, and the solvent was removed in vacuo. The residue was purified by
flash
column chromatography (silica gei, 9:1 hexanes/EtOAc) to give the title
compound
(0.55 g, 75%) as a colorless oil: 1H NMR (300 MHz, CDC13) 8 6.95 (d, J=1.9 Hz,
1
H), 6.88 (dd, J = 7.8, 2.1 Hz, 1 H), 6.68 (d, J = 7.8 Hz,.1 H), 3.76 (bs, 1
H), 3.66 (t, J
= 7.7 Hz, 2 H), 3.01-3.04 (m, 2 H), 2.96 (t, J= 7.7 Hz, 2 H), 2.72-2.76 (m, 2
H), 1.75-
1.83 (m, 2 H), 1.59-1.67 (m, 2 H); ESI MS m/z 210 [Cl2HisCIN + H]+; Rf 0.62
(silica
gel, 2:1 hexanes/EtOAc).
B. 1-(7-(2-Chloro-ethyl)-2,3,4,5-tetrahydro-benzo(blazepin-1-yll-ethanone
A solution of the title compound from step A (0.55 g, 2.6 mmol) in CH2CI2 (20
mL)
was treated with acetic anhydride (Ac20) (0.25 mL, 2.6 mmol). After stirring
at rt
under N2 for 7.5 h, the reaction was quenched with saturated sodium
bicarbonate
(NaHCOs) (50 mL) and extracted with CH2CI2 (4 x 50 mL). The combined organic
layers were dried over Na2S0~, decanted, and the solvent was removed in vacuo.
The residue was purified by flash column chromatography (silica gel, 2:1
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-82-
hexanes/EtOAc) to give the title compound (602 mg, 91 %) as a white solid: 1H
NMR
(300 MHz, CDCI3) S 7.09 (s, 1 H), 7.08 (s, 2 H), 4.66-4.71 (m, 1 H), 3.73 (t,
J= 7.3
Hz, 2 H), 3.06 (t, J = 7.3 Hz, 2 H), 2.58-2.76 (m, 3 H), 1.79-1.97 (m, 3 H),
1.86 (s, 3
H), 1.30-1.45 (m, 1 H); ESI MS m/z252 [C14Hi8CINO + H]+; Rf0.30 (silica gel,
2:1
hexanes/EtOAc).
C. 1-(7-[2-(4-Benzo[dlisothiazol-3-yl-piperazin-1-yl)-ethyll-2,3,4,5-
tetrahydro-benzofblazepin-1-yl'[-ethanone methanesulfonate
A solution of the title compound from step B (602 mg, 2.39 mmol) in CH3CN (20
mL)
was treated with 3-piperazin-1-yl-benzo[d]isothiazole ~ HCI (683 mg, 2.67
mmol), Nal
(406 mg, 2.71 mmol), and K2C03 (1.09 g, 7.86 mmol). The mixture was heated to
reflux under N2 for 43 h, then allowed to cool. The mixture was diluted with
H20 (100
mL) and extracted with CH2CI2 (3 x 100 mL). The combined organic layers were
dried over Na2SOa., decanted, and the solvent was removed in vaeuo. The
residue
was purified by flash column chromatography (silica gel, EtOAc) to give a
white solid
residue (430 mg, 0.99 mmvl, 41 %). The residue was dissolved in EtOAc (10 mL)
and treated with a solution of CH3S03H in Et20 (0.5 mL, 2M, 1 mmol). After
stirring
for 5 min, the resulting precipitate was isolated by filtration, washed with
Et20 (3 x 10
mL), and dried in a vacuum oven at 50 °C for 3 d to give the title
compound (465 mg,
89%) as a white powder: mp 189-190 °C (dec);'H NMR (300 MHz, CDCI3)
811..76
(s, 1 H), 7.85 (t, J=7.8 Hz, 2 H), 7.51-7.56 (m, 1 H), 7.39-7.45 (m, 1 H),
7.14-7.18
(m, 2 H), 7.08 (d, J = 7.8 Hz, 1 H), 4.66-4.70 (m, 1 H), 4.11-4.20 (m, 2 H),
3.95-4.03
(m, 2 H), 3.68 (d, J = 11.3 Hz, 2 H), 3.13-3.34 (m, 6 H), 2.91 (s, 3 H), 2.68-
2.78 (m, 2
H), 2.51-2.59 (m, 1 H), 1.74-2.00 (m, 3 H), 1.83 (s, 3 H), 1.32-1.40 (m, 1 H);
ESf MS
m/z 435 [C25H3oN~.OS + H]+; R f 0.35 (silica gel, 95:5 EtOAc/MeOH); HPLC) >99%
(AUC), tR = 12.68 min. Anal. Calc'd for C25HaoNa.OS~CH3SO3H: C, 58.84; H,
6.46; N,
10.56. Found: C, 58.56; H, 6.49; N, 10.31.
PREPARATION 32
5-(3-Chloro-propionyl)-3,3-dimethyl-1,3-dihydro-indol-2-one
The title compound was prepared using a procedure similar to that described
in Preparation 28 using chloro propionyl chloride as the acylating agent.
Yield = 3.05
g (98%); MS (APCI), (M + 1 )~ = 252.
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-83-
PREPARATION 33
5-(3-Chloro-propel)-3,3-dimethyl-1,3-dihydro-indol-2-one
The title compound was prepared using a procedure similar to that described
in Preparation 29. Yield = 2.87g (100%); MS (APCI), (M + 1 )+ = 238.
PREPARATION 34
5-(3-Chloro-propel)-3,3-dimethyl-2,3-dihydro-1 H-indole
The title compound was prepared using a procedure similar to that described
in Preparation 30. Yield = 0.172g (7%); MS (APCI), (M + 1 )+ = 224.
PREPARATION 35
1-~5-(3-Chloro-propel)-3;3-dimethyl-2,3-dihydro-indol-1-ell-ethanone
A solution of 5-(3-chloro-propyl)-3,3-dimethyl-2,3-dihydro-1 H-indole (172 mg)
in THF (2.0 mL) with triethylamine (0.145 mL) was treated with acetic
anhydride
(0.145 mL) and stirred for 14 hours at reflux. The reaction was quenched with
water,
extracted with ethyl acetate and filter and concentrated in vacuo. Yield: 159
mg
(78%) MS (APCI), (M+1 )+ = 266.
EXAMPLE 66
1-~'5-f3-(4-Benzof dlisothiazol-3-yl-piperazin-1-yl)-propyll-3,3-d imethyl-2,3-
dihydro-indol-1-yl)-ethanone
To a stirring solution of 1-[5-(3-chloro-propyl)-3,3-dimethyl-2,3-dihydro-
indol-1-
yl]-ethanone (159 mg) in acetonitrile (20 mL) was added 3-piperazin-1-yl-
benzo[dJisothiazole (263 mg), potassium carbonate (332 mg) and water (20 mL)
the
reaction was warmed to reflux for 72 hours. The reaction cooled and
precipitate was
filtered off. Yield: 114 mg (45%) MS (APCI), (M+1 )+ = 449.1.
PREPARATION 36
2 3-Dihydro-1 H isoindole
Beilstein Registry Number 111921; CAS Registry Number 496-12-8
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-84-
PREPARATION 37
1-(1,3-dihydroisoindol-2-yl)ethanone
Beilstein Registry Number 131840; CAS Registry Number 18913-38-7
PREPARATION 38
1-(2-Acetyl-2,3-dihydro-1 H-isoindol-5-yl)-2-chloroethanone
Anhydrous CS2 (15 mL) and chloroacetyl chloride (0.75 mL, 9.4 mmol) were added
to a stirred (mechanical stirrer) mixture of the title compound of Preparation
37 (1.00
g, 6.20 mmol) and AIC13 (3.3 g, 4.0 mmol) under N2. The mixture was heated to
reflux for 3 h, then allowed to cool, to give a dark oil with very little CS2
remaining
over it due to evaporation/leakage. Some ice was added to the stirred oil to
quench
the excess reagent. After stirring for 5 min, 6 M HCI (25 mL) was added. After
stirring for 1 h, the solid precipitate was collected by suction filtration
washing with
water, then dried in vacuo at 55 °C for 15 h to give the title compound
(1.19 g, 81%)
as a brown amorphous solid: 'H NMR (500 MHz, C6D6; low solubility, but this
solvent gave the best spectral dispersion of the aromatic signals; Note: The
spectrum shows two sets of signals due to rotational isomers) ~ 7.58 (d, J =
7.7 Hz,
0.5 H), 7.37 (s, 0.5 H), 7.29 (d, J = 7.9 Hz, 0.5 H), 7.25 (s, 0.5 H), 6.55
(d, J = 7.9 Hz,
1 H), 4.53 (br s, 2 H), 3.91 (s, 1 H), 3.89 (s, 1 H), 3.65 (br s, 2 H), 1.654
(s, 1.5 H),
1.646 (s, 1.5 H);'H NMR (300 MHz, CDC13; Note: The spectrum shows two sets of
signals due to rotational isomers) 8 7.88-7.95 (m, 2 H), 7.44 (d, J = 8.6 Hz,
0.5 H),
7.40 (d, J = 8.0 Hz, 0.5 H), 4.88 (br s, 2 H), 4.86 (br s, 2 H), 4.71 (s, 1
H), 4.70 (s, 1
H), 2.20 (s, 1.5 H), 2.19 (s, 1.5 H); Variable Temperature 1H NMR (500 MHz,
DMSO-
d6) spectra at 25°C and 90°C showed differences consistent with
rotational
isomerization; ESI MS m/z 238 [C12H12CIN02 + H]+; HPLC 98.5% (AUC), tR = 13.09
min.
EXAMPLE 67
1-(2-Acetyl-2,3-dihydro-1 H isoindol-5-yl)-2-(4-benzo~dlisothiazol-3-
ylpiperazin-
1-yllethanone
A mixture (suspension) of the title compound of Preparation 38 (2.10 g, 8.84
mmol),
3-piperazin-1-yl-benzo[c~isothiazole hydrochloride (2.49 g, 9.72 mmol), K2C03
(3.63
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-85-
g, 26.3 mmol), and Nal (1.40 g, 9.34 mmol) in anhyd CH3CN (90 mL) under N2 was
stirred at 25 °C for 20 h, then the solvent was removed in vacuo. The
residue was
suspended in H20, then extracted twice with EtOAc, however a solid remained
undissolved in the aqueous phase. The solid was collected by suction
filtration,
washing and triturating with H20, then dried in a vacuum oven at 50°C
for 3 d to give
the title compound (2.68 g, 72%) as a light brown amorphous solid. Both TLC
and
'H NMR analyses indicated that the product was of high purity: 'H NMR (300
MHz,
CDC13) 8 8.02 (d, J = 8.1 Hz, 1 H), 7.98 (br s, 1 H), 7.90 (d, J = 8.2 Hz, 1
H), 7.82 (d,
J = 8.0 Hz, 1 H), 7.48 (t, J = 7.5 Hz, 1 H), 7.32-7.43 (m, 2 H), 4.87 (br s, 2
H), 4.85
(br s, 2 H), 3.91 (s, 1 H), 3.90 (s, 1 H), 3.59-3.67 (m, 4 H), 2.81-2.89 (m, 4
H), 2.20
(s, 1.5 H), 2.19 (s, 1.5 H); ESI MS m/z421 [C23H24N4O2S + H]+.
EXAMPLE 68
1-(2-Acetyl-2,3-dihydro-1 H isoindol-5-yl)-2-(4-benzofdlisoxazol-3-ylpiperazin-
1-
yl)ethanone
The title compound was prepared from the title compound of Preparation 38
(2.18 g,
9.17 mmol) and 3-piperazin-1-yl-benzo[dJisoxazole hydrochloride (2.40 g, 10.0
mmol) by the procedure used to prepare the title compound of Example 67 (3.03
g,
82%) as an off-white amorphous solid: 1H NMR (300 MHz, CDCI3) 8 7.99 (d, J=
8.1
Hz, 1 H), 7.95 (br s, 1 H), 7.69 (d, J = 8.1 Hz, 1 H), 7.35-7.53 (m, 3 H),
7.18-7.26
(m, 1 H), 4.87 (br s, 2 H), 4.85 (br s, 2 H), 3.91 (s, 1 H), 3.90 (s, 1 H),
3.62-3.70 (m,
4 H), 2.80-2.87 (m, 4 H), 2.20 (s, 1.5 H), 2.19 (s, 1.5H); ESI MS m/z 405
~C23H24N-
4~3 + H]+~
EXAMPLE 69
1-f 5-f2-(4-Benzo('d~isoth iazol-3-yl piperazi n-1-yl)-1-hydroxyethyll-1,3-
dihydroisoindol-2-yl)ethanone
Sodium borohydride (0.20 g, 5.3 mmol) was added to a stirred solution of the
title
compound of Example 67 (2.67 g, 6.35 mmol) in 1:1 MeOH/CHCI3 (130 mL) at
0°C.
The mixture was allowed to warm to rt while stirring overnight. The solvents
were
removed in vacuo, and the residue was partitioned between CHC13 (200 mL) and
H20 (100 mL). The aqueous phase was reextracted with CHCI3 (50 mL). The
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-86-
combined organic phases were washed with saturated NaCI (50 mL), dried over
Na2S04, filtered, and the solvent removed in vacuo to give the title compound
(2.92 g
crude; 2.68 g theoretical) as a light brown amorphous solid (foam): 'H NMR
(300
MHz, CDCI3) s 7.91 (d, J = 8.1 Hz, 1 H), 7.83 (d, J = 8.0 Hz, 1 H), 7.49 (t, J
= 7.4 Hz,
1 H), 7.22-7.41 (m, 4 H), 4.77-4.87 (m, 5 H), 4.05 (d, J = 1.9 Hz, 1 H), 3.54-
3.69 (m,
4 H), 2.96-3.06 (m, 2 H), 2.49-2.77 (m, 4 H), 2.18 (s, 3 H); ESI MS m/z423
~C23H26N402S + H]+.
EXAMPLE 70
1-f 5-f2-(4-Benzo~d~isoxazol-3-ylpiperazi n-1-yl)-1-hydroxyethyll-1,3-
dihvdroisoindol-2-yl)ethanone
The title compound was prepared from the title compound of Example 68 (2.97 g,
7.34 mmol) using the procedure used to prepare the title compound of Example
69
(3.18 g crude; 2.98 g theoretical) as a light brown amorphous solid (foam): 'H
NMR
(300 MHz, CDC13) 8 7.70 (d, J= 8.0 Hz, 1 H), 7.44-7.54 (m, 2 H), 7.20-7.38 (m,
4
H), 4.77-4.87 (m, 5 H}, 3.96 (br s, 1 H), 3.58-3.72 (m, 4 H), 2.92-3.02 (m, 2
H),
2.48-2.74 (m, 4 H), 2.18 (s, 3 H); ESI MS m/z407 ~C23Hp6N4O3 + H]+.
EXAMPLE 71
1-f 5-f2-(4-Benzo~dlisoth iazol-3-ylpiperazi n-1-yl)-1-chloroethyll-1,3-
dihydroisoindol-2-yl)ethanone
Methanesulfonyl chloride (0.80 mL, 10 mmol) was added to a stirred solution of
the
title compound of Example 69 (2.92 g crude, 6.35 mmol theoretical) and
triethylamine (2.0 mL, 14 mmol) in anhydrous CH2CI2 (200 mL) at 0°C
under N2.
After stirring for 10 min, the ice-water bath was removed. After stirring for
2 h, TLC
analysis indicated that no starting material was remaining. The solution was
washed
twice with H20, once with saturated NaCI, dried over Na2S04, filtered, and the
solvent removed in vacuo to give the title compound (2.75 g, 98% from the
ketone
product of Example 102) as a light brown amorphous solid (foam): 'H NMR (300
M Hz, CDC13) 8 7.87 (d, J = 8.2 Hz, 1 H), 7.81 (d, J = 8.1 Hz, 1 H), 7.47 (t,
J = 7.5 Hz,
1 H), 7.22-7.39 (m, 4 H), 5.02 (t, J = 7.0 Hz, 1 H), 4.774..85 (m, 4 H), 3.47-
3.53 (m,
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
_87_
4 H), 3.13 (ddd, J = 13.5, 7.4, 1.5 Hz, 1 H), 2.94 (dd, J = 13.5, 6.8 Hz, 1
H), 2.66-
2.82 (m, 4 H), 2.180 (s, 1.5 H), 2.178 (s, 1.5 H); ESI MS m/z 441
[C2sH25CIN4OS +
H]~.
EXAMPLE 72
1-f 5-f2-(4-Benzof dlisoxazol-3-ylpiperazin-1-yl)-1-chloroethyll-'1,3
dihydroisoindol-2-yl'~ethanone
The title compound was prepared from the title compound of Example 70 (3.18 g
crude, 7.34 mmol theoretical) using the procedure used to prepare the title
compound of Example 71 (2.96 g, 95% from the ketone product of Example 103) as
a light brown amorphous solid (foam): ' H NMR (300 MHz, CDCI3) S 7.66 (d, J =
8.0
Hz, 1 H), 7.41-7.52 (m, 2 H), 7.17-7.38 (m, 4 H), 5.01 (t, J = 7.0 Hz, 1 H),
4.78-4.86
(m, 4 H), 3.49-3.58 (m, 4 H), 3.10 (ddd, J = 13.5, 7.4, 1.4 Hz, 1 H), 2.93
(dd, J =
13.5, 6.6 Hz, 1 H), 2.63-2.80 (m, 4 H), 2.18 (s, 3 H); ESI MS m/z 425
[C2sH25CIN4O2
+ H]+.
EXAMPLE 73
1-f5-f 2-(4-Benzof dlisothiazol-3-yf-piperazin-1-of)-ethyll-1,3-
dihydroisoindol-2
yl'~-ethanone methanesulfonate
A stirred solution of the title compound of Example 71 (2.74 g, 6.21 mmol) and
tributyl tin hydride (Bu3SnH) (2.5 mL, 9.3 mmol) in anhydrous toluene (170 mL)
was
degassed by bubbling argon through the solution for 30 min. 2,2'-
Azobisisobutyronitrile (AIBN) (0.15 g, 0.91 mmol) was added and the flask was
heated with a preheated 80 °C oil bath for 1 h. After allowing to cool,
H20 (10 mL)
was added. After stirring for 20 min, the solvents were removed in vacuo. The
residue was dissolved in CHCI3 (250 mL), then washed with H20 (100 mL) and
saturated NaCI (50 mL), dried over Na2S04, filtered, and the solvent removed
in
vacuo. The residue was purified by column chromatography (silica gel (100 g),
1:1:98 MeOHlEt3N/CHCI3) to give the free base of the title compound (1.28 g,
51 °I°;
plus slightly impure fractions: 0.69 g, 27%). The free base was dissolved in
warm
EtOAc-MeOH, then CHsSOsH (0.20 mL, 3.1 mmol, 1.0 equiv.) was added dropwise
with stirring. After stirring for 15 min, the solution was diluted with
hexanes to
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-88-
precipitate the salt, which was collected by suction filtration washing with
hexanes,
then dried overnight (18 h) in a vacuum oven at 50 °C to give the title
compound
(1.36 g, 87% yield for salt formation, 44% yield from the title compound of
Example
71 ) as a light brown amorphous solid: mp 219-222 °C dec; 1 H NMR (300
MHz,
DMSO-d6) b 9.81 (br s, 1 H), 8.16 (d, J = 8.2 Hz, 1 H), 8.12 (d, J = 8.2 Hz, 1
H), 7.61
(t, J = 7.2 Hz, 1 H), 7.49 (t, J = 7.2 Hz, 1 H), 7.22-7.38 (m, 3 H), 4.81 (br
s, 2 H),
4.60 (br d, J = 5.3 Hz, 2 H), 4.14 (br d, J = 9.2 Hz, 2 H), 3.68-3.75 (m, 2
H), 3.02-
3.12 (m, 2 H), 2.34 (s, 3 H), 2.072 (s, 1.5 H), 2.066 (s, 1.5 H); IR (KBr)
3437, 2959,
2920, 1643, 1448, 1423, 1207, 1035, 973, 774, 558 cm~'; ESI MS m/z407
[C23H2sN40S + H]+; HPLC 95.7% (AUC), tR = 11.78 min. Anal. Calc'd for
C2sH2sNa.OS~CH3S03H~0.1 CH30H~0.25 H20: C, 56.81; H, 6.07; N, 11.03. Found:
C, 56.65; H, 6.22; N, 10.71.
EXAMPLE 74
1-~5-f2-(4-Benzof dlisoxazol-3-yl-piperazin-1-yl)-ethyll-1,3-dihydroisoindol-2-
yl~-
ethanone methanesulfonate
The title compound was prepared from the title compound of Example 72 (2.95 g,
6.94 mmol) using the procedure used to prepare the title compound of Example
73
(1.44 g, 43%) as a white amorphous solid: mp 217-220 °C dec; 1H NMR
(300 MHz,
DMSO-ds) ~ 9.87 (br s, 1 H), 8.07 (d, J = 8.1 Hz, 1 H), 7.59-7.67 (m, 2 H),
7.22-7.39
(m, 4 H), 4.81 (br s, 2 H), 4.60 (br d, J = 4.8 Hz, 2 H), 4.19 (br d, J = 11.5
Hz, 2 H),
3.71 (br d, J= 10.3 Hz, 2 H), 3.01-3.11 (m, 2 H), 2.34 (s, 3 H), 2.07 (s, 1.5
H), 2.06
(s, 1.5 H); IR (KBr) 3438, 1631, 1529, 1449, 1198, 1058 cm'1; ESI MS m/z391
[C2sH2sNa.02 + H]+; HPLC 96.3% (AUC), tR =11.16 min. Anal. Calc'd for
C23H26N402~CH3S03H~0.1 CH30H~H20: C, 57.00; H, 6.43; N, 11.03. Found: C,
57.17; H, 6.49; N, 10.92.
PREPARATION 39
3-~4-f2-(2,3-Dihydro-1 H-isoindol-5-yl)-ethyll-piperazin-1-yl~-
benzofdlisothiazole
The free base of Example 74, 1-f5-[2-(4-Benzo[d]isothiazol-3-ylpiperazin-1-
yl)ethyl]-
1,3-dihydro-isoindol-2-yl} ethanone, (6.6 g, 1.60 mmol) was dissolved in 630
mL of
EtOH and 630 mL of cone. NCI and refluxed for 77 h. After reaction, the
solvent was
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
_89_
removed to afford the title compound (7.4 g) as a light brown solid. m.p.: 112-
121
°C.'H NMR (400 MHz, CD30D) b 8.12-7.93 (dd, 2 H), 7.60-7.40 (m, 5 H),
4.60 (d, 4
H), 4.20 (d, 2 H), 3.80 (d, 2 H), 3.62-3.40 (m, 6 H), 3.30-3.20 (m, 2 H). MS
m/z365
[M+1 ].
EXAMPLE 75
~5-f 2-(4-Benzof dlisothiazol-3-yl-~iperazin-1-yl)-ethyll-1,3-d i hydroisoi
ndol-2-yl)
acetic acid methyl ester
To a solution of the title compound from Preparation 39 (4.0 g, 0.91 mmol) in
200 mL of chloroform were slowly added methyl bromoacetate (1.67 g, 1.09 mmol)
and triethylamine (7.6 mL, 5.47 mmol). The reaction mixture was stirred at rt
overnight, washed with water, dried over sodium sulphate and concentrated. The
crude residue was subjected to chromatography (silica gel, EtOAc/MeOH/Et3N,
98/1!1 ) to afford the title compound (2.5 g, 63%) as yellow oil, which
darkens in the
air.'H NMR (400 MHz, CDCI3) ~ 7.94-7.79 (dd, 2 H), 7.50-7.32 (dt, 2 H), 7.16-
7.04
(m, 3 H), 4.08 (s, 4 H), 3.78 (s, 2 H), 3.62-3.58 (m, 6 H), 2.88-2.60 (m, 8
H). MS m/z
437 [M+1 ].
EXAMPLE 76
20, ~5-f2-(4-Benzo[dlisothiazol-3-yl-piperazin-1-yl)-ethyll-1,3-
dihydroisoindol-2-yl~-
acetic acid
A solution of the title compound from Example 75 (2.5 g, 0.57 mmol) in
THF/H20 (100/10 mL) was treated with LiOH~H20 (0.36 g, 0.86 mmol). The
reaction
mixture was stirred at rt overnight. The solvent was removed under reduced
pressure. The crude product was diluted with water, and neutralized with 0.5 N
HCI
to pH = 7. The solution was extracted with DCM, dried over sodium sulphate and
concentrated to give the title compound (2.49 g) as green solid. The crude
product is
pure in NMR spectrum, but is 75 % purity in HPLC analysis. The title compound
was
further purified by chromatography (silica gel, MeOH/CH2CI2/acetic acid,
40/60/0.1 ).
Yellow oil was obtained, but it darkens quickly. It is 85 % purity in HPLC
analysis. 'H
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-90-
NMR (400 MHz, CDC13) ~ 7.95-7.79 (dd, 2 H), 7.50-7.30 (dt, 2 H), 7.16-7.10 (m,
3
H), 4.70-4.50 (br s, 4 H), 3.80 (s, 2 H), 3.62-3.58 (br s, 4 H), 2.90-2.60 (m,
8 H). MS
m/z 423 [M+1 ].
EXAMPLE 77
2-~5-f 2-(4-Benzo~d~isothiazol-3-yl-piperazin-1-yl)-ethyll-1,3-d i hydroisoi
ndol-2-
yl'~-1-f3-dimethylamino-pyrrolidin-1-yll-ethanone
Oxalyl chloride (0.42 g, 3.30 mmol) was added dropwise to a stirred solution
of the title compound from Example 76 (0.70 g, 1.65 mmol) and DMF (2 drops) in
CH2CI2 (20 mL). The reaction mixture was stirred at rt for 3 h. The solvent
was
removed under reduced pressure and the residue was suspended in CH2C12 (15
mL), which was added dropwise to a stirred solution of 3-
(dimethylamino)pyrrolidine
(0.28 g, 2.47 mmol) and triethylamine ( 1.8 mL, 13.25 mmol). The reaction
mixture
was stirred at rt for 3 h, 100 mL of CH2C12 was added. The solution was washed
with
water, dried over sodium sulphate and concentrated. The crude residue was
subjected to chromatography (MeOH/CH2C12/Et3N, 2/98/0.5) to provide the title
compound (0.27 g, 31 %) as a light brown oil, which was treated with 4 mL of
2M
solution of hydrogen chloride in ether to provide the title compound as
hydrochloride
salt. mp 181-188 °C. 1H NMR (400 MHz, DMSO-d6) b 11.90-11.50 (m, 3 H),
8.10
(m, 2 H), 7.60-7.23 (m, 5 H), 4.90 (m, 2 H), 4.60 (m, 4 H), 4.10 (d, 2 H),
4.00-3.00
(m, 15 H), 2.72 (br s, 6 H), 2.40-2.20 (m, 2 H). MS m/z 519 [M+1 ]. Anal.
Calcd for
C29H38N60S~3HCI~4H20: C, 49.75; H, 7.05; N, 12.00. Found: C, 49.06; H, 6.32;
N,
11.01.
EXAMPLE 78
2-(5-f2-(4-Benzofdlisothiazol-3-yl-piperazin-1-yl)-ethyll-1,3-dihydroisoindol-
2-
yl~-N-(2-dimethylamino-ethyl)-N-methyl-acetamide
Oxalyl chloride (0.42 g, 3.30 mmol) was added dropwise to a stirred solution
of the title compound from Example 76 (0.70 g, 1.65 mmol) and DMF (2 drops) in
CH2CI2 (20 mL). The reaction mixture was stirred at rt for 3 h. The solvent
was
removed under reduced pressure and the residue was suspended in CH2CI2 (15
mL), which was added dropwise to a stirred solution of amine (0.25 g, 2.47
mmol)
and triethylamine ( 1.8 mL, 13.25 mmol). The reaction mixture was stirred at
rt for 3
h, 100 mL of CH2C12 was added. The solution was washed with water, dried over
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-91-
sodium sulphate and concentrated. The crude residue was subjected to
chromatography (MeOHI CH2C12/Et3N, 2/98/0.5) to provide the title compound
(0.28
g, 34 %) as a light brown oil, which was treated with 4 mL of 2 M solution of
hydrogen chloride in ether to provide the title compound as hydrochloride
salt. mp
152-160 °C. 1H NMR (400 MHz, DMSO-d6) b 11.79 (br s, 1 H), 11.35 (br s,
1 H),
10.86 (br s, 1 H), 8.15 (m, 2 H), 7.62-7.28 (m, 5 H), 5.00-4.30 (m, 10 H),
4.10 (d, 2
H), 3.80-3.10 (m, 11 H), 2.99 (s, 2 H), 2.75 (d, 6 H). MS mlz 507 [M+1 ].
Anal. Calcd
for C2~H36N60S~3HCI~5H20: C, 47.62; H, 7.28; N, 11.90. Found: C, 47.28; H,
6.86;
N, 11.06.
PREPARATION 40
N-chloroacetyl-morpholine
To a solution of chloroacetyl chloride (2.0 mL, 2.5 mmol) in 20 mL CH2CI2 was
slowly added a solution of morpholine (2.2 mL, 5.0 mmol) in 20 mL CH2CI2 at -
78 °C.
The reaction mixture was warmed to rt and stirred for 3 h. A white suspension
solution resulted. The white precipitate was filtered off. The filtrate was
washed with
1 N HCI, dried over sodium sulphate and concentrated to give the title
compound
(3.11 g, 78 %) as colorless oil. 1H NMR (400 MHz, CDCI3) ~ 4.12 (s, 2 H), 3.74
(br s,
4 H), 3.61 (br s, 2 H), 3.50 (br s, 2 H). MS m/z 164 [M+1 ].
EXAMPLE 79
2-f 5-f2-(4-Benzof dlisothiazol-3-yl-piperazin-1-yl)-ethyll-1,3-
dihydroisoindol-2
yl)-1 morpholin-4-yl-ethanone
A mixture of the title compound from Preparation 39 (0.40 g, 0.091 mmol), the
title compound from Preparation 40 (0.15 g, 0.092 mmol), potassium carbonate
(0.38
g, 0.28 mmol) and sodium iodide ( 0.15 g, 0.10 mmol) was suspended in 40 mL of
acetonitrile and stirred under reflux overnight, cooled to rt, solvent was
removed, and
water was added. The mixture was extracted with CH2C12, dried over sodium
sulphate and concentrated. The crude residue was subjected to chromatography
(MeOH/CH2CI2/acetic acid, 3197/0.1) to provide the title compound (0.27 g, 60
%) as
light yellow oil, which darken quickly in the air. 1H NMR (400 MHz, CDCI3) b
7.93-
7.80 (dd, 2 H), 7.50-7.37 (dt, 2 H), 7.12-7.06 (m 3 H), 4.01 (s, 4 H), 3.71-
3.58 (m 14
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-92-
H), 2.90-2.65 (m, 8 H). MS m/z 292 [M+1 ]. Anal. Calcd for C27H33N5O2S~1.5H20:
C,
62.52; H, 7.00; N, 13.50. Found: C, 62.63; H, 6.48; N, 13.03.
EXAMPLE 80
2-(5-f2-(4-Benzo(dlisothiazol-3-yl-piperazin-1-yl)-ethyll-1,3-dihydroisoindol-
2-
y1) N-(2-methoxy-ethyl)-acetamide hydrochloride
A mixture of the title compound from Preparation 39 (1.00 g, 2.28 mmol), 2-
chloro-1-(methoxyethylamino)ethanone (0.34 g, 2.28 mmol), potassium carbonate
(0.94 g, 6.84 mmol) and sodium iodide (0.37 g, 2.50 mmol) was suspended in 100
mL of acetonitrile and stirred under reflux overnight, cooled to rt. The
solvent was
removed, and water was added. The mixture was extracted with CH2CI2, dried
over
sodium sulphate and concentrated. The crude residue was subjected to
chromatography (MeOH/CH2CI2, 3/97) to provide the title compound (0.53 g, 49
%)
as a light brown oil, which was treated with 4 mL of 2 M solution of hydrogen
chloride
in ether to provide the title compound as a hydrochloride salt. mp 149-154
°C. 1H
NMR (400 MHz, DMSO-d6) S 11.70 (br s, 1 H), 11.40 (br s, 1 H), 8.68 (br s, 1
H),
8.13 (m, 2 H), 7.62-7.20 (m, 5 H), 4.90-4.80 (m, 2 H), 4.50 (br s, 2 H), 4.13
(br s, 2
H), 4.10 (d, 2 H), 3.65-3.43 (m, 4 H), 3.46-3.05 (m, 13 H). MS m/z 480 [M+1 ].
Anal.
Calcd for C26H3sNs02S~2HC1~2H20: C, 53.06; H, 6.68; N, 11.90. Found: C, 53.01;
H,
5.80; N, 11.34.
PREPARATION 41
Morpholin-4-yl-acetic acid methyl ester
To a solution of morpholine (5.46 mL, 39.2 mmol) and triethylamine (1.71 mL,
19.6 mmol) in THF (100 mL), methyl bromoacetate (1.86 mL, 19.6 mmol) was
added. The reaction mixture was stirred at rt for 5 h. The solvent was removed
under
reduced pressure. Sodium bicarbonate solution was added. The mixture was
extracted with EtOAc, dried over sodium sulphate and concentrated to provide
the
title compound (2.7 g, 87 %) as a colorless oil. 'H NMR (400 MHz, CDC13) ~
3.75 (t,
4 H), 3.73 (s, 3 H), 3.23 (s, 2 H), 2.58 (t, 4 H). MS m/z 160 [M+1 ].
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-93-
PREPARATION 42
Morpholin-4-yl-acetic acid lithium salt
A mixture of morpholin-4-yl-acetic acid methyl ester (2.7 g, 16.9 mmol) and
lithium hydroxide (1.06g, 2.54 mmol) in THF (100 mL) and water (10 mL) was
stirred
at rt overnight. Light yellow solution resulted. The solvent was removed under
reduced pressure to provide the title compound (3.6 g, quant.) as a lithium
salt.'H
NMR (400 MHz, DMSO-d6) b 3.62 (t, 4 H), 3.13 (s, 2 H), 2.62 (t, 4 H). MS m/z
146
[M+1 ].
EXAMPLE 81
1-~-f 2-(4-Benzof dlisothiazol-3-yl-pi perazin-1-yl)-ethyl~-1,3-d ihydroisoi
ndol-2-
yl)-2-morpholin-4-yl-ethanone
A mixture of the title compound of Preparation 39, 3-{4-[2-(2,3-dihydro-1 H-
isoindol-5-yl)-ethyl]-piperazin-1-yl}-benzo[d]isothiazole dichloride (0.50 g,
1.14
mmol), the product of Preparation 42 (0.30g, 1.37 mmol), HBTU (0.86 g, 2.28
mmol),
HOBt (0.31 g, 2.28 mmol) and diisopropylethylamine (1.0 mL, 6.84 mmol) in 7 mL
of
DMF was stirred at rt under argon overnight, 200 mL of EtOAc was added. The
solution was washed with water (4 x 100 mL). The organic layer was
concentrated.
The crude residue was subjected to chromatography (MeOH/CH2CI2/EtOAc/Et3N,
1/20120/0.1) to provide the title compound (0.12 g, 23 %) as a light yellow
oil, which
was treated with 4 mL of 2M solution of hydrogen chloride in ether to provide
the title
compound as hydrochloride salt. mp 183-187 °C. 1H NMR (400 MHz, DMSO-
ds} ~
11.42 (br s, 1 H), 10.42 (br s, 1 H), 8.15 (m, 2 H), 7.60 (m, 1 H), 7.45 (m, 1
H), 7.40-
7.28 (m, 3 H), 4.83-4.70 (dd, 4 H), 4.39 (br s, 2 H), 4.10 (d, 2 H), 4.00-3.15
(m, 18
H). MS m/z 492 [M+1 ]. Anal. Calcd for C2~H33N502S~2HC1~3H~0: C, 52.42; H,
6.68;
N, 11.32. Found: C, 52.84; H, 6.36; N, 10.96.
EXAMPLE 82
3-~4-f2-(2-Methanesulfonyl-2,3-dihydro-1 H-isoindol-5-yl)-ethyll-piperazin-1-
yl'~-
benzofdlisothiazole hydrochloride
3-{4-[2-(2,3-Dihydro-1 H-isoindol-5-yl)-ethyl]-piperazin-1-yl}-
benzo[d]isothiazole (0.55 g, 1.25 mmol) and triethylamine (0.35 ml, 2.50 mmol)
were
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-94-
dissolved in 10 mL of CH2CI2 at 0°C. Methanesulfonyl chloride (0.17 g,
1.50 mmol)
was added slowly. The reaction mixture was warmed to rt and stirred for 2 h,
50 mL
of CH2CI2 was added. The solution was washed with water, dried over sodium
sulphate and concentrated. The crude residue was subjected to chromatography
(MeOH/CH2CI2, 2/98) to provide the title compound (0.32 g, 58 %) as a light
yellow
oil, which was treated with 5 mL of 2 M solution of hydrogen chloride in ether
to
provide the title compound as hydrochloride salt. mp 179 °C (dec).'H
NMR (400
MHz, DMSO-ds): b 11.00 (br s, 1 H), 8.10 (m, 2 H), 7.62 (dt, 1 H), 7.45 (dt, 1
H), 7.30
(m, 3 H), 4.60 (br s, 4 H), 4.10 (d, 2 H), 3.61 (d, 2 H), 3.59-3.10 (m, 8 H),
2.98 (s, 3
H). MS m/z 443 [M+1 ]. Anal. Calcd for C22H2sN402S2~HCI: C, 55.16; H, 5.68; N,
11.69. Found: C, 54.52; H, 5.43; N, 11.13.
EXAMPLE 83
1-f'5-f3-(4-Benzof dlisothiazol-3-yl-pi perazi n-1-yl)-propyll-1,3-di
hydroisoi ndol-2-
yl)-ethanone
A. 2,3-Dihydro-1 H isoindole
Beilstein Registry Number 111921; CAS Registry Number 496-12-8
B. 1-(1,3-dihydroisoindol-2-yl)ethanone
Beilstein Registry Number 131840; CAS Registry Number 18913-38-7
C. 1-(2-Acetyl-2,3-dihydro-iH-isoindol-5-yl)-3-chloro-propan-1-one
Anhydrous CS2 (15 mL) and chloropropionyl chloride (0.81 mL, 9.4 mmol) were
added to a stirred (mechanical stirrer) mixture of the product of step B (1.00
g, 6.20
mmol) and AIC13 (3.3 g, 4.0 mmol) under N2. The mixture was heated to reflux
for 3
h, then allowed to cool, to give a dark oil with very little CS2 remaining
over it due to
evaporationheakage. Some ice was added to the stirred oil to quench the excess
reagent. After stirring for 5 min, 6M HCI (25 mL) was added. After stirring
for 1 h,
the solid precipitate was collected by suction filtration washing with water,
then dried
in vacuo at 55 °C for 15 h to give the title product (1.29 g, 83%) as a
brown solid: ESI
MS m/z 251 [M+1 ] ; HPLC (Method A) 98.5% (AUC), tR = 13.27 min.
D. 1-(2-Acetyl-2,3-dihydro-1 H-isoindol-5-yl)-3-(4-benzo~dlisothiazol-3-yl-
piperazin-1-yl)-propan-1-one
A mixture (suspension) of 1-(2-Acetyl-2,3-dihydro-1 H-isoindol-5-yl)-3-chloro-
propan-
1-one (2.14 g, 8.84 mmol), 3-piperazin-1-yl-benzo[djisothiazole hydrochloride
(2.49
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-95-
g, 9.72 mmol), K2C03 (3.63 g, 26.3 mmol), and Nal (1.40 g, 9.34 mmol) in
anhydrous
CH3CN (90 mL) under N2 was stirred at 25 °C for 20 h, then the
solvent was
removed in vacuo. The residue was suspended in H20, then extracted twice with
EtOAc, however a solid remained undissolved in the aqueous phase. The solid
was
collected by suction filtration, washing and triturating with H20, then dried
in a
vacuum oven at 50 °C for 3 d to give the titled product (2.74 g, 71 %)
as a light brown
amorphous solid. ESI MS m/z 435 [M+1 ].
E. 1-f5-f3-(4-Benzof dlisothiazol-3-yl-piperazin-1-yl)-1-hydroxy-propyll-1,3-
dihydro-isoindol-2-yl'f-ethanone
Sodium borohydride (0.20 g, 5.3 mmol) was added to a stirred solution of the
product
of step D (2.69 g, 6.35 mmol) in 1:1 MeOH/CHCI3 (130 mL) at 0 °C. The
mixture
was allowed to warm to room temperature while stirring overnight. The solvents
were removed in vacuo, and the residue was partitioned between CHCIs (200 mL)
and H20 (100 mL). The aqueous phase was reextracted with CHCI3 (50 mL). The
combined organic phases were washed with sat'd NaCI (50 mL), dried over
Na2SO4,
filtered, and the solvent removed in vacuo to give the title product (2.7 g)
as a light
brown solid: ESI MS m/z 437 [M+1 ].
F. 1-~'5-f 3-(4-Benzof dlisothiazol-3-yl-piperazi n-1-yl)-1-chloro-propyll-1.3-
dihydro-isoindol-2-yl)-ethanone
Methanesulfonyl chloride (0.80 mL, 10 mmol) was added to a stirred solution of
the
product of step E (2.7 g, 6.35 mmol) and triethylamine (2.0 mL, 14 mmol) in
anhydrous CH2CI2 (200 mL) at 0 °C under N2. After stirring for 10 min,
the ice-water
bath was removed. After stirring for 2 h, TLC analysis indicated that no
starting
material was remaining. The solution was washed twice with H20, once with
saturated NaCI, dried over Na2S04, filtered, and the solvent removed in vacuo
to
give the title product (2.75 g) as a light brown solid: ESI MS m/z455 [M+1].
G. 1-~5-f3-(4-Benzofdlisothiazol-3-yl-piperazin-1-yl)-propyll-1,3-dihydro-
isoindol-2-yl)-ethanone
A stirred solution of the product of step F (2.74 g, 6.21 mmol) and Bu3SnH
(2.5 mL,
9.3 mmol) in anhyrous toluene (170 mL) was degassed by bubbling argon through
the solution for 30 min. AIBN (0.15 g, 0.91 mmol) was added and the flask was
heated with a preheated 80 °C oil bath for 1 h. After allowing to cool,
H20 (10 mL)
was added. After stirring for 20 min, the solvents were removed in vacuo. The
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-96-
residue was dissolved in CHC13 (250 mL), then washed with H20 (100 mL) and
saturated NaCI (50 mL), dried over Na2S04, filtered, and the solvent removed
in
vacuo. The residue was purified by column chromatography (silica gel (100 g),
1:1:98 MeOHlEt3NlCHCl3) to give the free base of the title compound (2.1 g,
87%) as
a light brown solid: ESI MS m/z 421 [M+1 ].
H. 3-(4-f3-(2,3-Dihydro-1 H-isoindol-5-yl)-propyll-piperazin-1-yl'~-
benzofdlisothiazole
The product of step G (6.6 g, 1.60 mmol) was dissolved in 630 mL of EtOH and
630
mL of conc. HCI and refluxed for 77 h. After reaction, the solvent was removed
to
afford 3-{4-[3-(2,3-Dihydro-1 H-isoindol-5-yl)-propyl]-piperazin-1-yl}-
benzo[d]isothiazole (7.4 g) as a light brown solid. MS m/z 379 [M+1 ].
I. 1-~5-f3-(4-Benzof dlisoth iazol-3-yl-piperazin-1-yl)-propyll-1,3-
dihydroisoindol-2-yl'~-ethanone
Acetic anhydride (0.62 mL, 6.6 mmol) was added to a solution containing 3-{4-
[2-
(2,3-Dihydro-1 H-isoindol-5-yl)-propyl]-piperazin-1-yl}-benzo[d]isothiazole
(1.0 g, 2.6
mmol), triethylamine (1.62 mL, 11.6 mmol), 4-dimethylamino- pyridine (0.08 g,
0.65
mmol), and anhydrous dichloromethane (40 mL) at rt. The reaction mixture was
stirred overnight at rt. The resulting solution was washed with NaHCOs, dried
over
Na2S04, and evaporated. The residue thus obtained was purified over silica gel
column (230 - 400 mesh, 2.5 x 12 cm), using ethyl acetate:methanol:acetic acid
(88:10:2) solvent mixture as eluent to obtain the acetylated amine product.
The
product was dissolved in 40 mL of anhydrous ethyl acetate and to which was
added
1 M hydrogen chloride solution in ether (5.0 mL, 5 mmol) with stirring. A
white
precipitate of hydrogen chloride salt was obtained, which was filtered off,
washed
with ether (2 x 10 mL), and dried under vacuum. Yield: 0.884 g, 73.2 %. mp
130.0
-133.0 °C. HPLC: Purity 98.10% (retention time: 9.918 min.; mobile
phase: 0.1%
H3P0~/MeCN gradient; column: ACE_C18 5p,m_4-6 x 150 mm). 1H NMR (400
MHz, DMSO-d6): 510.9 (br s, 1 H), 7.88 (d, 1 H), 7.83 (d, 1 H), 7.48 (t, 1 H),
7.36 (t,
1 H), 7.21 (t, 1 H), 7.14 (m, 2 H), 4.79 (m, 4 H), 3.67 (m, 4 H), 2.89 (m, 4
H), 2.67 (t,
2 H), 2.63 (t, 2 H), 2.18 (s, 3 H), 1.93 (t, 2 H). ES-MS m/z 420.58
(C24H28N4OS + 1 )+.
Analysis calculated for C24H28N40S~HCI: C, 63.07; H, 6.40; N, 12.26. Found C,
63.03; H, 6.51; N, 12.08.
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-97-
EXAMPLE 84
~(5-[3-(4-Benzof dlisothiazol-3-yl-pi aerazin-1-yl)-propyll-1,S-
dihydroisoindol-2
yl~-acetic acid
St_-e~ A:
Methyl bromoacetate (0.28 mL, 2.9 mmol) was added to a suspension containing 3-
{4-[2-(2,3-Dihydro-1 H-isoindol-5-yl)-propyl]-piperazin-1-yl}-
benzo[d]isothiazole (1.0 g,
2.6 mmol), potassium carbonate (0.44 g, 3.2 mmol) and anhydrous acetonitrile
(50
mL) at rt and stirred overnight. Solvent was evaporated and the residue was
distributed between chloroform (50 mL) and water (30 mL). The organic layer
was
separated, dried over Na2S04, and evaporated. The residue was purified over
silica
gel column (230 - 400 mesh, 2.5 x 18 cm) using methanoi:ethyl acetate
(1.5:98.5)
solvent mixture as eluent to obtain pure {5-[3-(4-Benzo[d]isothiazol-3-yl-
piperazin-1-
yl)-propyl]-1,3-dihydro-isoindol-2-yl}-acetic acid methyl ester. Yield: 0.12
g, 10.08%.
Using triethylamine as a base, the yield was improved to 67.8%. 'H NMR (400
MHz,
CDCI3): ~ 7.91 (d, 1 H), 7.80 (d, 1 H), 7.47 (t, 1 H), 7.36 (t, 1 H), 7.11 (d,
1 H), 7.08
(d, 1 H), 4.09 (s, 4 H), 3.76 (s, 2 H), 3.62 (s, 3 H), 3.56 (m, 4 H), 2.67 (m,
4 H), 2.64
(m, 2 H), 2.44 (t, 2 H), 1.85 (m, 2 H). ES-MS mlz 451.08 (C25H3pN4O2S + 1 )~.
Step B:
Lithium hydroxide monohydrate (0.1 g, 2.4 mmol) was added to a solution
containing
(5-[3-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-propyl]-1,3-dihydro-isoindol-
2-yl}-
acetic acid methyl ester (0.6 g, 1.3 mmol), tetrahydrofuran (30 mL), and water
(4
mL), and stirred overnight. The solution was evaporated to dryness and the
residue
was dissolved in 3 mL of water, and pH was adjusted to 6 using 1 M HCI. The
resulting solution was evaporated to dryness, and washed with ether (2 x 10
mL) and
tetrahydrofuran (2 x 10 mL). As the elemental analysis results were not
satisfactory,
the product was further purified using HP20 Diaion column (Supelco product).
The
impure product was treated with 1 mL of triethylamine and then loaded aver
HP20
column. After washing the column with methanol:water (1:1 ), elution with
acetonitrile
yielded pure product containing fractions, which were evaporated, and dried
under
vacuum. Yield: 0.44 g, 72.7 %. m.p. 78.0 - 80.0 °C. HPLG: Purity 94.45%
(retention time: 11.601 min.; mobile phase: 0.1 °!° H3P0~/MeCN
gradient; column:
ACE C18 5p.m 4-6 x 150 mm), iH NMR (400 MHz, CDC13): S 7.90 (d, 1 H), 7.79 (d,
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
_98_
2 H), 7.46 (t, 1 H), 7.37 (t, 1 H), 7.13 (m, 2 H), 7.07 (s, 1 H), 4.58 (s, 4
H), 3.77 (s, 2
H), 3.58 (m, 4 H), 2.70 (m, 4 H), 2.67 (m, 2 H), 2.47 (m, 2 H), 1.86 (m, 2 H).
ES-MS
m/z 437.09 (C24H~$N402S + 1 )+. Analysis calculated for
C24H28N402S.~0.5Et3N~H20:
C, 64.19; H, 7.50; ,N 12.48. Found: C, 65.26; H, 6.94; N, 12.41.
EXAMPLE 85
1-f 5-f 3-(4-Benzof dlisoth iazol-3-yl-pi perazi n-1-yl)-propyll-1,3-di hydro-
isoi ndol-2
yl~-2-dimethylamino-ethanone
Chloroacetylchloride (0.15 mL, 1.9 mmol) was added to a solution containing 3-
(4-[2-
(2,3-Dihydro-1H-isoindol-5-yl)-propyl]-piperazin-1-yl}-benzo[d]isothiazole
(0.5 g, 1.1
mmol), 4-dimethylaminopyridine (0.014 g, 0.01 mmol), triethylamine (0.7 mL,
5.0
mmol) and dichloromethane (50 mL) at rt. The reaction mixture was stirred
overnight
at rt and later an aliquot was examined by NMR, which indicated the absence of
starting material and formation of 1-{5-[3-(4-Benzo[d]isothiazol-3-yl-
piperazin-1-yl)-
propyl]-1,3-dihydro-isoindol-2-yl}-2-chloro-ethanone. The resultant solution
was
washed with NaHCOs, brine, dried over Na2S04, and evaporated to obtain crude 1-
{5-[3-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-propyl]-1,3-dihydro-isoindol-
2-yl}-2-
chloro-ethanone. This was dissolved in acetonitrile (50 mL), and to which was
added dimethylamine (1.11 mL of 2 M MeOH solution, 2.2 mmol), potassium
carbonate (0.18 g, 1.3 mmol), and sodium bromide (0.028 g, 0.30 mmol) at rt,
and
stirred overnight. The resulting suspension was filtered and the precipitate
was
washed with chloroform. The filtrate was evaporated, and the residue obtained
was
purified over silica gel column (230 - 400 mesh, 2.5 x 12 cm), using
triethylamine:ethyl acetate:methanol (1:94:5) solvent mixture as eluent to
obtain the
title compound. The product was dissolved in 5 mL of anhydrous diethylether
and to
which was added 1 M hydrogen chloride solution in ether (2.0 mL, 2.0 mmol)
with
stirring. A white precipitate of hydrogen chloride salt was obtained, which
was
filtered off, washed with ether (3 x 15 mL), and dried under vacuum. Yield:
0.40 g,
66.9 °l°. mp 90.0 °C turned brown. HPLC: Purity 97.59%
(retention time: 11.683
min.; mobile phase: 0.1% H3P0~/MeCN gradient; column: ACE C18 5~m 4-6 x 150
mm). 1H NMR (400 MHz, DMSO-d6): S 11.62 (br s, 1 H), 10.02 (br s, 1 H), 8.12
(m,
2 H), 7.60 (M, 1 H), 7.49 (m, 1 H), 7.36 (m, 1 H), 7.32 (m, 1 H), 7.23 (m, 1
H), 4.81
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
_99_
(m, 2 H), 4.73 (m, 2 H), 4.33 (s, 2 H), 4.05 (m, 2 H), 3.57 (m, 4 H), 3.30 (m,
2 H),
3.15 (m, 2 H), 2.88 (s, 6 H), 2.72 (m, 2 H), 2.11 (m, 2 H). ES-MS m/z 464.18
(C26H33N5OS + 1 )+. Analysis calculated for C26HssNsOS~2HCI~H20: C, 56.31; H,
6.72; N, 12.63. Found: C, 56.25; H, 6.70; N, 12.32.
EXAMPLE 86
1-~5-f3-(4-Benzo~dlisothiazol-3-yl-piperazin-1-yl)-propyll-1,3-di hydro-
isoindol-2-
yl'~-2-piperidin-1-yl-ethanone
Chloroacetylchloride (0.15 mL, 1.9 mmol) was added to a solution containing 3-
{4-[2-
(2,3-Dihydro-1 H-isoindol-5-yl)-propyl]-piperazin-1-yl)-benzo[d]isothiazole
(0.6 g, 1.6
mmol), 4-dimethylaminopyridine (0.020 g, 0.02 mmol), triethylamine (0.97 mL,
6.9
mmol) and acetonitrile (40 mL) at rt. The reaction mixture was stirred
overnight at rt
and later an aliquot was examined by NMR, which indicated the absence of
starting
material and formation of 1-(5-[3-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-
propyl]-
1,3-dihydro-isoindol-2-yl]-2-chloro-ethanone. Solvent was evaporated to afford
an
oil, which was diluted with 50 mL of chloroform, washed with NaHC03, brine,
dried
over Na2S04, and evaporated to obtain crude 1-{5-[3-(4-Benzo[d]isothiazol-3-yl-
piperazin-1-yl)-propyl]-1,3-dihydro-isoindol-2-yl}-2-chloro-ethanone. This was
dissolved in acetonitrile (50 mL), and to which was added piperidine (0.33 mL,
3.3
mmol), potassium carbonate (0.26 g, 1.9 mmol), and sodium bromide (0.040 g,
0.39
mmol) at rt, and stirred overnight. The resulting suspension was filtered and
the
precipitate was washed with chloroform. The filtrate was evaporated, and the
residue obtained was purified over silica gel column (230 - 400 mesh, 2.5 x 12
cm),
using triethylamine:ethyl acetate:methanol (1:93:6) solvent mixture as eluent
to
obtain the title compound. The product was dissolved in 5 mL of anhydrous
ethyl
acetate and to which was added 1 M hydrogen chloride solution in ether (2.1
mL, 2.1
mmol) with stirring. A white precipitate of hydrogen chloride salt was
obtained, which
was filtered off, washed with ether (3 x 5 mL), and dried under vacuum. Yield:
0.39
g, 43.6 %. mp 98.0 °C turned brown. HPLC: Purity 94.40% (retention
time: 11.987
min.; mobile phase: 0.1 % H3P0~/MeCN gradient; column: ACE C18 5~,m 4-6 x
150 mm). 1H NMR (400 MHz, DMSO): S 11.58 (br s, 1 H), 9.78 (br s, 1 H), 8.12
(m,
2 H), 7.60 (m, 1 H), 7.49 (m, 1 H), 7.36 (m, 1 H), 7.30 (m, 1 H), 7.25 (m, 1
H), 4.83
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
100-
(m, 2 H), 4.72 (m, 2 H), 4.31 (s, 2 H), 4.02 (m, 4 H), 3.60 (m, 4 H), 3.47 (m,
2 H),
3.30 (m, 2 H), 3.19 (m, 2 H), 3.15 (m, 2 H), 2.70 (br s, 2 H), 2.11 (br s, 2
H), 1.81 (br
s, 2 H), 1.68 (br s, 2 H). ES-MS m/z 504.17 (C2gH37N5OS + 1 )+. Analysis
calculated
for C29H3~N50S~2HCI~0.5H20: C, 59.47; H, 6.90; N, 11.96. Found: C, 59.64; H,
6.89;
N, 11.49.
EXAMPLE 87
1-(5-f 3-(4-Benzof dlisoth iazol-3-yl-piperazin-1-yl)-propyll-1,3-d ihydro-
isoindol-2
yl'f-2-morpholin-4-yl-ethanone
Chloroacetylchloride (0.14 mL, 1.7 mmol) was added to a solution containing 3-
{4-[2-
(2,3-Dihydro-1 H-isoindol-5-yl)-propyl]-piperazin-1-yl}-benzo[d]isothiazole
(0.6 g, 1.6
mmol), potassium carbonate (0.26 g, 1.9 mmol), 4-dimethylaminopyridine (0.050
g,
0.04 mmol), and acetonitrile (40 mL) at rt. The reaction mixture was stirred
overnight
at rt and later an aliquot was examined by NMR, which indicated the absence of
starting material and formation of compound 1-{5-[3-(4-Benzo[d]isothiazol-3-yl-
piperazin-1-yl)-propyl]-1,3-dihydro-isoindol-2-yl}-2-chloro-ethanone. To the
same pot
was added morpholine (0.28 mL, 3.2 mmol), potassium carbonate (0.4 g, 2.9
mmol),
and sodium bromide (0.048 g, 0.47 mmol) at rt, and stirred overnight. The
resulting
suspension was diluted with 100 mL of chloroform, washed with brine, dried
over
Na2S04, and evaporated. The residue thus obtained was purified over silica gel
column (230 - 400 mesh, 2.5 x 14 cm), using ethyl acetate:methanol:acetic acid
(88:10:2) solvent mixture as eluent to obtain the title compound. The product
was
dissolved in 20 mL of anhydrous ethyl acetate and to which was added 1 M
hydrogen chloride solution in ether (5.0 mL, 5 mmol) with stirring. A white
precipitate
of hydrogen chloride salt was obtained, which was filtered off, washed with
ether (2 x
10 mL), and dried under vacuum. Yield: 0.66 g, 71.9 %. mp 179.0 -183.0
°C.
HPLC: Purity 94.29% (retention time: 11.665 min.; mobile phase: 0.1 %
H3P0~/MeCN
gradient; column: ACE C18_5p.m 4-6 x 150 mm). 1H NMR (400 MHz, DMSO-ds): 8
11.58 (br s, 1 H), 10.58 (br s, 1 H), 8.12 (m, 2 H), 7.62 (m, 1 H), 7.48 (m, 1
H), 7.30
(m, 3 H), 4.83 (m, 2 H), 4.72 (m, 2 H), 4.42 (s, 2 H), 4.03 (m, 4 H), 3.84 (m,
2 H), 3.6
(m, 4 H), 3.54 (m, 2 H), 3.30 (m, 4 H), 3.16 (m, 2 H), 2.70 (m, 2 H), 2.11 (m,
2 H).
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-101-
ES-MS m/z 379.08 (C28H35N5O2S + 1 )+. Analysis calculated for
C28H35NsO2S~2HC1:
C, 58.12; H, 6.45; N, 12.10. Found: C, 58.18; H, 6.51; N, 11.84.
EXAMPLE 88
1-(5-f3-(4-Benzofdlisothiazol-3-yl-piperazin-1-yl)-propyll-1,3-dihydro-
isoindol-2-
yl'f-2-f (2-d i methylami no-ethyl)-methyl-am inol-ethanone
Chloroacetylchloride (0.23 mL, 2.9 mmol) was added to a solution containing 3-
{4-[2-
(2,3-Dihydro-1 H-isoindol-5-yl)-propyl]-piperazin-1-yl}-benzo[d]isothiazole
(1.0 g, 2.2
mmol), 4-dimethylaminopyridine (0.010 g, 0.002 mmol), triethylamine (1.4 mL,
9.9
mmol) and dichloromethane (50 mL) at rt. The reaction mixture was stirred
overnight
at rt and later an aliquot was examined by NMR, which indicated the absence of
starting material and formation of 1-~5-[3-(4-Benzo[d]isothiazol-3-yl-
piperazin-1-yl)-
propyl]-1,3-dihydro-isoindol-2-yl}-2-chloro-ethanone. The resultant solution
was
washed with NaHC03, brine, dried over Na2S04, and evaporated to obtain crude 1-
{5-[3-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-propyl]-1,3-dihydro-isoindol-
2-yl}-2-
chloro-ethanone. About half of the crudel-{5-[3-(4-Benzo[d]isothiazol-3-yl-
piperazin-
1-yl)-propyl]-1,3-dihydro-isoindol-2-yl}-2-chloro-ethanone was dissolved in
acetonitrile (50 mL), and to which was added N,N,N trimethylethylenediamine
(0.29
mL, 2.2 mmol), potassium carbonate (0.18 g, 1.3 mmol), and sodium bromide
(0.028
g, 0.27 mmol) at rt, and stirred overnight. The resulting suspension was
filtered and
the precipitate was washed with chloroform (2 x 10 mL). The filtrate was
evaporated
and the residue obtained was purified over silica gel column (230 - 400 mesh,
2.5 x
11 cm), using triethylamine:ethyl acetate:methanol (1:94:5) solvent mixture as
eluent
to obtain the crude title compound. The product was dissolved in 5 mL of
anhydrous
ethyl acetate and to which was added 1 M hydrogen chloride solution in ether
(4.4
mL, 4.4 mmol) with stirring. A white precipitate of hydrogen chloride salt was
obtained, which was filtered off, washed with ether (2 x 10 mL), and dried
under
vacuum. Yield: 0.33 g, 42.9 %. mp 85.1 - 87.3 °C. HPLC: Purity 95.42%
(retention
time: 11.097 min.; mobile phase: 0.1 % H3P0~/MeCN gradient; column:
ACE C18 5p,m 4-6 x 150 mm). 1H NMR (400 MHz, DMSO-d6): S 11.64 (br s, 1 H),
11.16 (br s, 1 H), 8.14 (m, 2 H), 7.61 (m, 1 H), 7.49 (m, 1 H), 7.36 (m, 1 H),
7.33 (m,
1 H), 7.26 (m, 1 H), 4.84 (m, 2 H), 4.73 (m, 2 H), 4.49 (brs, 2 H), 4.21 (brs,
4 H), 4.05
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-102-
(m, 2 H), 3.74 (m, 2 H), 3.41 (m, 4 H), 3.28 (m, 2 H), 3.07 (m, 2 H), 2.98 (s,
3 H),
2.85 (s, 6 H), 2.70 (br s, 2H), 2.11 (b.s, 2H). ES-MS m/z 521.26 (C29H4oN6OS +
1)+.
Analysis calculated for C29H4oN60S~4HC1~H20: C, 50.88; H, 6.77; N, 12.28.
Found:
C, 50.50; H, 7.17; N, 11.67.
EXAMPLE 89
1-f 5-f3-(4-Benzof dlisothiazol-3-yl-piperazin-1-yl)-propyll-1 3-di hydro-isoi
ndol-2
yl~-2-diethylamino-ethanone
Chloroacetylchloride (0.23 mL, 2.9 mmol) was added to a solution containing 3-
{4-[2
(2,3-Dihydro-1H-isoindol-5-yl)-propyl]-piperazin-1-yl}-benzo[d]isothiazole
(1.0 g, 2.2
~mmol), 4-dimethylaminopyridine (0.01 D g, 0.002 mmol), triethylamine (1.4 mL,
9.9
mmol) and dichloromethane (50 mL) at rt. The reaction mixture was stirred
overnight
at rt and later an aliquot was examined by NMR, which indicated the absence of
starting material and formation of 1-{5-[3-(4-Benzo[d]isothiazol-3-yl-
piperazin-1-yl)-
propyl]-1,3-dihydro-isoindol-2-yl}-2-chloro-ethanone. The resultant solution
was
washed with NaHC03, brine, dried over Na2SO4, and evaporated to obtain crude 1-
{5-[3-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-propyl]-1,3-dihydro-isoindol-
2-yl}-2-
chloro-ethanone. About half of the crude 1-{5-[3-(4-Benzo[d]isothiazol-3-yl-
piperazin-1-yl)-propyl]-1,3-dihydro-isoindol-2-yl}-2-chloro-ethanone~was
dissolved in
acetonitrile (50 mL), and to which was added diethylamine (0.6 mL, 5.7 mmol),
potassium carbonate (0.18 g, 1.3 mmol), and sodium bromide (0.025 g, 0.28
mmol)
at rt, and stirred overnight. The resulting suspension was filtered and the
precipitate
was washed with chloroform (2 x 10 mL). The filtrate was evaporated, and the
residue obtained was purified over silica gel column (230 - 400 mesh, 2.5 x 11
cm),
using triethylamine:ethyl acetate:methanol (1:93:6) solvent mixture as eluent
to
obtain the title compound. The product was dissolved in 5 mL of anhydrous
diethylether and to which was added 1 M hydrogen chloride solution in ether
(3.3
mL, 3.3 mmol) with stirring. A white precipitate of hydrogen chloride salt was
obtained, which was filtered off, washed with ether (3 x 10 mL), and dried
under
vacuum. Yield: 0.40 g, 58.3 %. m.p. 90.0 °C turned waxy. HPLC: Purity
95.82%
(retention time: 11.936 min.; mobile phase: 0.1 % H3P0~/MeCN gradient; column:
ACE C18_5p,m 4-6 x 150 mm). 1H NMR (400 MHz, DMSO-d6): S 11.76 (br s, 1 H),
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-103-
9.66 (br s, 1 H), 8.18 (m, 2 H), 7.66 (m, 1 H), 7.55 (m, 1 H), 7.39 (m, 1 H),
7.36 (m, 1
H), 7.31 (m, 1 H), 4.94 (m, 2 H), 4.78 (m, 2 H), 4.36 (s, 2 H), 4.10 (m, 2 H),
3.64 (m,
4 H), 3.36 (m, 4 H), 3.24 (m, 4 H), 2.76 (br s, 2 H), 2.18 (br s, 2rH), 1.29
(m, 6 H).
ES-MS m/z 492.22 (C28H3~N50S + 1 )+. Analysis calculated for
C28H3~N50S~3HC1~H20: C, 54.32; H, 6.84; N, 11.31. Found: C, 54.45; H, 7.25; N,
11.11.
EXAMPLE 90
~5-f 3-(4-Benzo(dI isothiazol-3-yl-piperazin-1-yl)-propyll-'1,3-d i hyd roisoi
ndol-2-
yl'i-(3-dimethylamino-pyrrolidin-1-yl)-methanone
Chloroacetylchloride (0.14 mL, 1.7 mmol) was added to a solution containing 3-
{4-[2-
(2,3-Dihydro-1 H-isoindol-5-yl)-propyl]-piperazin-1-yl}-benzo[d]isothiazole
(0.6 g, 1.6
mmol), potassium carbonate (0.26 g, 1.9 mmol), 4-dimethylaminopyridine (0.050
g,
0.04 mmol), and acetonitrile (40 mL) at rt. The reaction mixture was stirred
overnight
at rt and later an aliquot was examined by NMR, which indicated the presence
of
starting material. Hence, added triethylamine (1.0 mL, 7.2 mmol) and stirred
for an
hour. Now, the starting material completely disappeared. The resulting
suspension
was diluted with 100 mL of chloroform, washed with NaHC03, brine, dried over
Na2S04, and evaporated to obtain crude 1-{5-[3-(4-Benzo[d]isothiazol-3-yl-
piperazin-
1-yl)-propyl]-1,3-dihydro-isoindol-2-yl}-2-chloro-ethanone. This was dissolved
in
acetonitrile (50 mL), and to which was added (S)-2 dimethylaminopyrrolidine
(0.36 g,
3.2 mmol), potassium carbonate (0.4 g, 2.9 mmol), and sodium bromide (0.048 g,
0.47 mmol) at rt, and stirred overnight. The resulting suspension was diluted
with
100 mL of chloroform, washed with brine, dried over Na2S04, and evaporated.
The
residue thus obtained was purified over silica gel column (230 - 400 mesh, 2.5
x 12
cm), using chloroform:methanol (88:12) solvent mixture as eluent to obtain the
title
compound. The product was dissolved in 20 mL of anhydrous tetrahydrofuran and
to which was added 1 M hydrogen chloride solution in ether (6.8 mL, 6.8 mmol)
with
stirring. A white precipitate of hydrogen chloride salt was obtained, which
was
filtered off, washed with ether (3 x 10 mL), and dried under vacuum. Yield:
0.73 g,
68.8 %. mp 166.0 -170.0 °C. HPLC: Purity 94.60% (retention time: 11.032
min.;
mobile phase: 0.1 % H3P0~/MeCN gradient; column: ACE C18_5~m 4-6 x 150
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-104-
mm). 1H NMR (400 MHz, DMSO-ds): S 11.99 (br d,~ 1 H), 11.66 (br s, 1 H), 11.09
(br
s, 1 H), 8.10 (m, 2 H), 7.59 (m, 1 H), 7.47 (m, 1 H), 7.34 (m, 2 H), 7.23 (m,
1 H), 4.82
(br s, 2rH), 4.71 (br s, 2 H), 4.57 (br m, 2 H), 4.43 (br m, 4 H), 4.04 (m, 4
H), 3.72 (m,
2 H), 3.60 (m, 4 H), 3.28 (m, 2 H), 3.14 (m, 2 H), 2.79 (br s, 6 H), 2.40 (br
m, 1 H),
2.12 (m, 2 H). ES-MS m/z 533.22 (CsoH4oNsOS + 1)+. Analysis calculated for
CsoHaoNsOS~3HCI~1.5H2O: C, 53.84; H, 6.94; N, 12.56. Found: C, 53.84; H, 7.39;
N,
11.85.
EXAMPLE 91
5-f3-(4-Benzofdlisothiazol-3-yl-piperazin-1-yl)-propyll-1,3-dihydro-isoindole-
2-
carboxylic acid (4-fluoro-phenyl)-amide
4-Fluorophenylisocyanate (0.15 mL, 1.3 mmol) was added to a solution
containing 3-
(4-[2-(2,3-Dihydro-1 H-isoindol-5-yl)-propyl]-piperazin-1-yl}-
benzo[d]isothiazole (0.5 g,
1.1 mmol), triethylamine (0.46 mL, 3.3 mmol) and tetrahydrofuran (40 mL) at
rt. The
reaction mixture was stirred for 2 h at rt and later evaporated. The residue
was
dissolved in 30 mL of dichloromethane, washed with NaHC03, dried over Na2S04,
and evaporated to obtain crude product. Purification over silica gel column
(230 -
400 mesh, 2.5 x 12 cm), using triethylamine:ethyl acetate (1:99) solvent
mixture as
eluent yielded pure title compound. The product was dissolved in 5 mL of
anhydrous
ethyl acetate and to which was added 1 M hydrogen chloride solution in ether
(2.0
mL, 2.0 mmol) with stirring. A white precipitate of hydrogen chloride salt of
the title
compound was obtained, which was filtered off, washed with ether (3 x 15 mL),
and
dried under vacuum. Yield: 0.46 g, 75.2 %. mp 145.0 -150.0 °C. HPLC:
Purity
96.34% (retention time: 15.080 min.; mobile phase: 0.1 % H3P0~/MeCN gradient;
column: ACE_C18 5pm 4-6 x 150 mm). 1H NMR (400 MHz, DMSO-d6): b 11.71 (br
s, 1 H), 8.46 (s, 1 H), 8.10 (m, 2 H), 7.59 (m, 3 H), 7.45 (m, 1 H), 7.30 (m,
1 H), 7.26
(m, 1 H), 7.20 (m, 1 H), 7.12 (m, 2 H), 4.74 (s, 4 H), 4.02 (m, 2 H), 3.56 (m,
2 H),
3.49 (m, 2 H), 3.31 (m, 2 H), 3.16 (m, 2 H), 2.71 (m, 2 H), 2.12 (m, 2 H). ES-
MS m/z
516.20 (C29H3oFN50S + 1)+. Analysis calculated for C29H3oFN50S~HCI~0.5H20: C,
62.07; H, 5.76; N, 12.48. Found: C, 61.83; H, 5.61; N, 12.23.
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-105-
EXAMPLE 92
5-f3-(4-Benzofd~isothiazol-3-yl-piperazin-1-yl)-propyll-1,3-dihydro-isoindole-
2
carbothioic acid cyclohexylamide
Cyclohexylisothiocyanate (0.31 mL, 2.2 mmol) was added to a solution
containing 3-
(4-[2-(2,3-Dihydro-1 H-isoindol-5-yl)-propyl]-piperazin-1-yl}-
benzo[d]isothiazole (0.5 g,
1.1 mmol), triethylamine (0.46 mL, 3.3 mmol) and tetrahydrofuran (40 mL) at
rt. The
reaction mixture was stirred for 2 h at rt and later evaporated. The residue
was
washed with hexane to remove CyNCS. Purification over silica gel column (230 -
400 mesh, 2.5 x 10 cm) using ethyl acetate as eluent yielded pure title
compound as
an oil, which was washed with dry ether (2 x 10 mL) to obtain white solid.
Yield: 0.35
g, 60.8 %. mp 194.2 -195.3 °C. HPLC: Purity 98.95% (retention time:
16.571 min.;
mobile phase: 0.1% H3PO~lMeCN gradient; column: ACE C18 5g,m 4-6 x 150
mm). 1 H NMR (400 MHz, CDCI3): S 7.91 (m, 1 H), 7.82 (m, 1 H), 7.47 (m, 1 H),
7.37
(rn, 1 H), 7.19 (m, 1 H), 7.15 (m, 2H), 5.14 (m, 2H), 4.82 (b.s, 2H), 4.37 (m,
1 H), 3.58
(b.s, 4H), 2.68 (m, 4H), 2.67 (b.s, 2H), 2.45 (m, 2H), 2.17 (m; 2H), 1.88 (m,
2H), 1.76
(m, 2H), 1.68 (m, 2H), 1.50 (m, 2H), 1.44 (m, 2H), 1.24 (m, 2H). ES-MS m/z
520.20
(C2gH37N5S2 + 1 )+. Analysis calculated for C2gH37N5S2: C, 67.01; H, 7.18; N,
13.47.
Found: C, 66.54; H, 7.00; N, 13.18.
EXAMPLE 93
3-f4-f3-(2-Methanesulfonyl-2,3-dihydro-1 H-isoindol-5-yl)-propyll-piperazin-1-
yl)-
benzofdlisothiazole hydrochloride
Methanesulfonylchloride (0.16 mL, 2.2 mmol) was added to a solution containing
3-
(4-[2-(2,3-Dihydro-1H-isoindol-5-yl)-propyl]-piperazin-1-yl)-
benzo[d]isothiazole (0.8 g,
1.8 mmol), triethylamine (4.0 mL, 28.7 mmol) and anhydrous chloroform (50 mL)
at
5°C. After 2 h stirring at rt, the reaction mixture was washed with
NaHC03, dried
over Na2S04, and evaporated to obtain crude product. Purification over silica
gel
column (230 - 400 mesh, 2.5 x 12 cm) using ethyl acetate as eluent yielded
pure title
compound. The product was dissolved in 20 mL of anhydrous tetrahydrofuran and
to which was added 1 M hydrogen chloride solution in ether (4.3 mL, 4.3 mmol)
with
stirring. A white precipitate of hydrogen chloride salt of the title compound
was
obtained, which was filtered off, washed with ether (3 x 6 mL), and dried
under
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-106-
vacuum. Yield: 0.79 g, 90.4 %. m.p. 201.0- 203.0 °C. HPLC: Purity
97.50%
(retention time: 14.072 min.; mobile phase: 0.1 % H3P04/MeCN gradient; column:
ACE C18 5~m 4-6 x 150 mm). 'H NMR (400 MHz, DMSO-d6): ~ 11.48 (br s, 1 H),
8.14 (m, 1 H), 7.60 (m, 1 H), 7.47 (m, 1 H), 7.29 (m, 1 H), 7.27 (m, 1 H),
7.20 (m, 1
H), 4.61 (s, 4 H), 4.03 (d, 2 H), 3.59 (m, 4 H), 3.30 (m, 2 H), 3.25 (m, 2 H),
2.98 (s, 3
H), 2.69 (m, 2 H), 2.09 (m, 2 H). ES-MS m/z 457.06 (C23H28N4O2S2 + 1 )+.
Analysis
calculated for C2sH28N4O2S2~HCI~1.5H20: C, 53.12; H, 6.21; N, 10.78. Found: C,
53.38; H, 5.61; N, 10.33.
EXAMPLE 94
3-f4-~3-(2-Benzenesulfonyl-2,3-dihydro-1 H-isoindol-5-yl)-propyll-piperazin-1-
yl)-benzofdlisothiazole hydrochloride
Benzenesulfonylchloride (0.14 mL, 1.06 mmol) was added to a solution
containing 3-
~4-[2-(2,3-Dihydro-1 H-isoindol-5-yl)-propyl]-piperazin-1-yl)-
benzo[d]isothiazole (0.4 g,
0.88 mmol), triethylamine (0.43 mL, 3.1 mmol) and anhydrous dichloromethane
(40
mL) at 5 °C. After 2 h stirring at rt, the reaction mixture was
evaporated to obtain a
residue, which was washed with hexane (3 x 10 mL), and dried. The residue was
purified over silica gel column (230 - 400 mesh, 2.5 x 12 cm) using ethyl
acetate as
eluent to obtain pure title compound. The product was dissolved in 10 mL of
anhydrous ethyl acetate and to which was added 1 M hydrogen chloride solution
in
ether (2.0 mL, 2.0 mmol) with stirring. A white precipitate of hydrogen
chloride salt of
the title compound was obtained, which was filtered off, washed with ether (3
x 5
mL), and dried under vacuum. Yield: 0.286 g, 62.2 %. mp 130.2 -133.9
°C.
HPLC: Purity 96.92% (retention time: 11.078 min.; mobile phase: 0.1 %
H3P0~/MeCN
gradient; column: ACE C18_5~,m 4-6 x 150 mm). 'H NMR (400 MHz, DMSO-ds): S
10.68 (br s, 1 H), 8.11 (m, 2 H), 7.88 (m, 2 H), 7.69 (m, 1 H), 7.65 (m, 3 H),
7.59 (m,
1 H), 7.45 (m, 1 H), 7.19 (m, 2 H), 4.55 (s, 4 H), 4.05 (d, 2 H), 3.56 (d, 2
H), 3.44 (m,
2 H), 3.26 (m, 2 H), 3.12 (m, 2 H), 2.63 (m, 2 H), 2.02 (m, 2 H). ES-MS m/z
519.23
(C28H3oN4O2S2 + 1 )+. Analysis calculated for C28H3oN4O2S2~HCI~H2O: C, 58.67;
H,
5.80; N, 9.77. Found: C, 58.84; H, 5.54; N, 9.60.
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-107-
EXAMPLE 95
f5-[3- 4-Benzo[dlisothiazol-3-yl-piperazin-i-yl)-propyll-1,3-dihydro-isoindol-
2
yl'[-phenyl-methanone hydrochloride
Benzoic anhydride (0.50 g, 2.2 mmol) was added to a solution containing 3-{4-
[2-
(2,3-dihydro-1H-isoindol-5-yl)-propyl]-piperazin-1-yl}-benzo[d]isothiazole
(0.4 g, 0.88
mmol), triethylamine (0.43 mL, 3.1 mmol), 4-dimethyl-aminopyridine (0.001 g,
0.008
mmol), and anhydrous dichloromethane (40 mL) at rt. The reaction mixture was
stirred overnight at rt. The resulting solution was washed with NaHC03, dried
over
Na2SOa, and evaporated. The residue thus obtained was purified over silica gel
column (230 - 400 mesh, 2.5 x 12 cm), using ethyl acetate:methanol solvent
mixture
(95:5) as eluent to obtain the title product. The product was dissolved in 10
mL of
anhydrous ethyl acetate and to which was added 1 M hydrogen chloride solution
in
ether (2.0 mL, 2 mmol) with stirring. A white precipitate of hydrogen chloride
salt
was obtained, which was filtered off, washed with ether (2 x 6 mL), and dried
under
vacuum. Yield: 0.342 g, 74.4 %. mp 120.1 -123.2 °C. HPLC: Purity 97.45%
(retention time: 14.707 min.; mobile phase: 0.1 % H3P0~/MeCN gradient; column:
ACE C18 5~,m 4-6 x 150 mm). 1H NMR (400 MHz, DMSO-d6): b 11.27 (br s, 1 H),
8.13 (m, 2 H), 7.58 (m, 3 H), 7.49 (m, 4 H), 7.34 (m, 1 H), 7.19 (m, 2 H),
4.84 (m, 2
H), 4.74 (m, 2 H), 4.02 (m, 2 H), 3.86 (br s, 1 H), 3.59 (m, 4 H), 3.15 (m, 4
H), 2.68
(m, 2 H), 2.08 (m, 2 H), ES-MS m/z 482.65 (C29HsoN4OS + 1 )~. Analysis
calculated
for C29H3oN40S~HCI~H20: C, 64.85; H, 6.19; N, 10.43. Found: C, 64.39; H, 5.96;
N,
10.07.
PREPARATION 43
1-(3,4-Dihydro-1 H-isoe~uinolin-2-yl)-2,2,2-trifiluoro-ethanone
To a stirred solution of 1,2,3,4-tetrahydroisoquinoline (10.0 mL, 79.886 mmol)
in anhydrous CH2CI2 (200 mL) and pyridine (7.2 mL, 89.021 mmol) under a
nitrogen
atmosphere was added trifluoroacetic anhydride (12.4 mL, 87.791 mmol). The
reaction was stirred overnight at ambient temperature. The reaction was
quenched
by slow addition of sat. NaHCOs solution (50 mL) and transferred to a
separatory
funnel. The layers were separated and the organic layer was extracted with
brine (50
mL), dried over anhydrous Na2S04, filtered and concentrated in vacuo. The
resulting
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-108-
yellow oil was eluted through a flash column (silica gel 60, 230-400 mesh, 0-
3%
MeOH in CH2CI2 gradient over 1 h) to give a yellow oil. Yield: 17.1459 g
(74.808,
94%). MS (APCI), (M+1 )+ = 230. ' H NMR (400 MHz, CDCI3) 8 7.22 (m, 2 H), 7.15
(m, 2 H), 4.78 (s, 1.3 H), 4.73 (s, 0.7 H), 3.88 (t, J--6.0 Hz, 0.7 H), 3.83
(m, 1.3 H),
2.95 (q, J--6.0 Hz, 2 H).
PREPARATION 44
1-f7-(2-Chloro-acetyl)-3,4-dihydro-1 H-isoauinolin-2-yll-2,2,2-
trifluoro-ethanone
To a stirred solution of 1-(3,4-dihydro-1 H-isoquinolin-2-yl)-2,2,2-trifluoro-
ethanone (16.7121 g, 72.915 mmol) in anhydrous CH2CI2 (182 mL) was added
chloroacetyl chloride (7.0 mL, 87.515 mmol). The reaction was heated to 40
°C (oil
bath). Aluminum chloride (38.90 g, 291.735 mmol) was slowly added in portions.
The
process was slightly exothermic. A reflux condenser was attached and the
reaction
1~ was heated to reflux. After 2.5 h, the reaction was cooled to ambient
temperature
and slowly poured into an ice bath and stirred vigorously. The mixture was
transferred to a separatory funnel and the aqueous layer extracted with
additional
CH2CI2. The organic portions were combined and extracted with sat. NaHCOs
solution, passed through a phase separator and then concentrated in vacuo to a
yellow solid. The solid was eluted through a flash column (silica gel 60, 230-
400
mesh, 2% MeOH in CH2CI2) to give an impure yellow solid. The solid was
repurified
by flash column under the same condition and then recrystallized from
EtOAc/hexanes to give pure product as a yellow solid. Yield: 10.5912 g (34.648
mmol, 48%). MS (APCI, (M-1)' = 304.'H-NMR (400 MHz, CDCI3) 8 7.78 (m, 2 H),
7.30 (m, 1 H), 4.85 (s, 1.3 H), 4.80 (s, 0.7 H), 4.66 (d, J=2.69 Hz, 2 H),
3.89 (m, 2 H),
3.02 (m, 2 H).
PREPARATION 45
1-f7-(2-Chloro-ethyl)-3,4-dihydro-1 H-isoauinolin-2-yll-2,2,2
trifluoro-ethanone
To a stirred solution of 1-[7-(2-chloro-acetyl)-3,4-dihydro-1 H-isoquinolin-2-
yl]-
2,2,2-trifluoro-ethanone (10.5851 g, 34.628 mmol) in boron trifluoride
etherate
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-109-
(BF3~Et20, 26.4 mL, 0.208 mol) in a sealed tube was added triethylsilane (33.2
mL,
0.208 mol). The tube was sealed and then placed into a preheated 80 °C
oil bath.
After 4 h, the reaction was cooled to ambient temperature and poured into an
ice
bath and extracted with CH2CI2 (3 x 50 mL). The combined organic layers were
dried
over anhydrous Na2S04, filtered and concentrated in vacuo to give a brown oil.
The
oil was eluted through a flash column (silica gel 60, 230-400 mesh, 10-25%
EtOAc in
hexanes gradient over 1 h) to give a brown oil. Yield: 3.5199 g (12.067 mmol,
35%).
MS {APCI), (M+1 )+ = 292. ' H-NMR (400 MHz, CDCIs,) 8 7.09 (m, 2 H), 6.99 (m,
1 H),
4.77 (s, 1.3 H), 4.72 (s, 0.7 H), 3.85 (m, 2 H), 3.70 (m, 2 H), 3.03 (m, 2 H),
2.93 (m, 2
H).
EXAMPLE 96
1-f7-[2-(4-Benzofdlisothiazol-3-VI-piperazin-1-yl)-ethyll-3,4-dihydro-1 H
isoauinolin-2-yl1-2,2,2-trifluoro-ethanone methane sulfonate
A mixture of 3-piperazin-1-yl-benzo[d]isothiazole hydrochloride (0.4056 g,
1.417 mmol), 1-[7-(2-chloro-ethyl)-3,4-dihydro-1 H-isoquinolin-2-yl]-2,2,2-
trifluoro-
ethanone (0.3755 g, 1.287 mmol), anhydrous sodium carbonate (0.3019 g, 2.848
mmol) and potassium iodide (0.0259 g, 0.156 mmol) in acetonitrile (10 mL) was
allowed to react at 175 °C for 0.5 h in a microwave reactor. The
reaction was cooled
to ambient temperature. CH2CI2 and H20 were added and the solution was mixed
well then poured into a phase separator. The organic layer was concentrated in
vacuo to give an oil. The oil was eluted through a flash column (silica gel
60, 230-
400 mesh, 30-100% EtOAc in CH2CI2 gradient over 1 h) to give a white
waxyfgummy
solid. Yield: 0.4106 g (0.865 mmol, 67 %). The solid (0.405 g, 0.853 mmol) was
taken up in THF (8.5 mL) and heated to 40 °C. Methanesulfonic acid
(55.5 ~L, 0.855
mmol) was added and after 5 min, the reaction was allowed to cool to ambient
temperature. The product was allowed to crystallize overnight. Hexanes were
added
to the reaction mixture and the solid was filtered and washed with hexanes.
The wet
solid was dried in a vacuum oven at 50 °C to give a white/off-white
crystalline solid
as the mesylate salt. Anal. calculated for C24H25FsN4OS~CHa.O3S: C, 52.62; H,
5.12;
N, 9.82. Found: C, 52.36, H, 4.98; N, 9.69. 1H NMR (400 MHz, CDCI3) S 11.66
(s, 1
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-110-
H), 7.84 (d, J--7.42 Hz, 2 H), 7.52 (m, 2 H), 7.41 (m, 1 H), 7.12 (m, 2 H),
4.76 (m, 4
H), 4.17 (m, 2 H), 4.00 (m, 4 H), 3.82 (m, 2 H), 3.27 (m, 3 H), 2.91 (m, 6 H).
EXAMPLES 97 AND 98
A. 1,2,3,4-Tetrahydroquinoline
Beilstein Registry Number 116149; CAS Registry Number 635-46-1
B. (3,4-Dihydro-2H guinolin-1-yl)(4-fluorophenyl)methanone
CAS Registry Number 313276-23-2
C. 2-Ch loro-1-f 1-(4-fluorobenzoyl)-1,2,3,4-tetrahydroqui nol in-6-
yllethanone
Carbon disulfide (100 mL) and chloroacetyl chloride (6.0 mL, 75 mmol) were
added
sequentially to a mechanically stirred mixture of the compound identified in
Step B
(13 g crude, 48 mmol theoretical) and AIC13 (24 g, 180 mmol) under N2. The
mixture
was heated to reflux for 3 h, then allowed to cool. After sitting at rt
overnight, the
clear liquid top phase was decanted (by pipet) off of the dark oil lower
phase. Ice
water (250 mL) was added cautiously to the stirred dark oil. Then, 6 M HCI
(150 mL)
was added to the stirred mixture. After stirring for 30 min, the solid was
collected by
suction filtration washing several times with H20. The solid was dried in a
vacuum
oven at 50 °C for 3 d to give the title compound (14.9 g, 93% from
1,2,3,4-
tetrahydroquinoline) as a brown amorphous solid: ~H NMR (300 MHz, CDC13) 8
7.81
(d, J = 1.8 Hz, 1 H), 7.49 (dd, J = 8.6, 2.0 Hz, 1 H), 7.37-7.46 (m, 2 H),
6.96-7.06
(m, 2 H), 6.83 (d, J = 8.6 Hz, 1 H), 4.63 (s, 2 H), 3.92 (t, J = 6.4 Hz, 2 H),
2.93 (t, J =
6.6 Hz, 2 H), 2.01-2.13 (m, 2 H); ESI MS m/z332 [C1aH15CIFNO2 + H]+.
D. [6-(2-Chloroethyl)-3,4-dihydro-2H quinolin-1-yl~(4-
fluoroahenyl)methanone
Triethylsilane (7.0 mL, 44 mmol) was added portionwise over 10 min to a
stirred solution of the product of Step C (5.03 g, 15.2 mmol) in
trifluoroacetic acid (20
mL) under N2. The mixture was heated to 50 °C for 17 h, then allowed to
cool. The
mixture (a dark brown solution) was poured into a stirred mixture of 1 M NaOH
(300
mL) and ice (100 mL). The two-phase mixture was stirred for 1 h, during which
time
the dark oil turned into a brown solid. The mixture was extracted with EtOAc
(200
mL), which dissolved the brown solid. The organic phase was washed with H20
(200 mL) and saturated NaCI (100 mL), dried over Na2S04, filtered, and the
solvent
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-111-
was removed in vacuo. The residue (brown solid and clear, colorless oil) was
purified by column chromatography (silica gel (120 g), 10-40% EtOAc/hexanes)
to
give the title compound (4.35 g, 90%) as a yellow waxy solid: 'H NMR (300 MHz,
CDCI3): 8 7.32-7.41 (m, 2 H), 7.01 (br s, 1 H), 6.91-7.00 (m, 2 H), 6.74 (dd,
J= 8.2,
1.7 Hz, 1 H), 6.63 (d, J = 8.1 Hz, 1 H), 3.89 (t, J = 6.5 Hz, 2 H), 3.66 (t, J
= 7.4 Hz, 2
H), 2.97 (t, J= 7.4 Hz, 2 H), 2.83 (t, J= 6.6 Hz, 2 H), 1.98-2.10 (m, 2 H);
ESI MS
m/z 318 [C18H1~CIFNO + H]+.
E. f6-f2-(4-Benzo(dlisothiazol-3-yl-piperazin-1-yl)-ethyl)-3,4-dihydro-2H
auinolin-1-yl)-(4-fluorophenyl)-methanone (97)
A stirred mixture of the product of Step D (2.00 g, 6.29 mmol), 3-piperazin-1-
yl-benzo[dJisothiazole hydrochloride (9, 1.82 g, 7.12 mmol), K2C03 (2.34 g,
16.9
mmol), and Nal (1.00 g, 6.67 mmol) in anhyd CH3CN (60 mL) under N2 was heated
to reflux for 3 d, then allowed to cool. The mixture was diluted with EtOAc
(300 mL),
then washed twice with H20 (300 mL), once with saturated NaCI (100 mL), dried
over Na2SOa, filtered, and the solvent was removed in vacuo. The residue was
purified by column chromatography (silica gel (150 g), 40-60% EtOAclhexanes
containing 1% Et3N) to give the title compound (2.75 g, 87%) as a sticky oil
and solid
mixture. The product was dissolved in warm EtOAc (60 mL), then allowed to cool
with stirring. A small amount of precipitate was observed after 30 min. The
mixture
was diluted with hexanes (120 mL) portionwise over 2 h. After stirring an
additional
hour, the precipitate was collected by suction filtration washing with
hexanes, then
dried in vacuo at 46 °C for 3 d to give the title compound (1.71 g,
54%) as a white
amorphous solid: mp 126-129 °C;'H NMR (300 MHz, CDCI3) 8 7.91 (d, J=
8.1 Hz,
1 H), 7.82 (d, J = 8.1 Hz, 1 H), 7.47 (td, J = 7.6, 1.0 Hz, 1 H), 7.32-7.41
(m, 3 H),
7.02 (br s, 1 H), 6.91-7.00 (m, 2 H), 6.76 (dd, J= 8.2, 1.5 Hz, 1 H), 6.60 (br
d, J=
7.7 Hz, 1 H), 3.89 (t, J = 6.5 Hz, 2 H), 3.54-3.63 (m, 4 H), 2.60-2.87 (m, 10
H), 2.05
(p, J = 6.6 Hz, 2 H); IR (ATR) 2947, 2835, 1634, 1600, 1504, 1374, 1271, 1227
cm'';
ESI MS m/z 501 [C29H29FN40S + H]+; HPLC >99% (AUC), tR = 14.77 min. Anal.
calcd. for C29H29FN40S: C, 69.57; H, 5.84; N, 11.19. Found: C, 69.36; H, 5.86;
N,
11.03.
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-112-
F. f6-f2-(4-Benzofdlisoxazol-3-yl-piperazin-1-yl)-ethyll-3,4-dihydro-2H
auinolin-1-VI'~-(4-fluoro-phenyl)-methanone (98)
A stirred mixture of the product of Step E (2.30 g, 7.24 mmol), 3-piperazin-1-
yl-benzo[dJisoxazole hydrochloride (2.03 g, 8.47 mmol), K2C03 (2.66 g, 19.2
mmol),
and Nal (1.24 g, 8.27 mmol) in anhyd CH3CN (75 mL) under N2 was heated to
reflux
for 3 d, then allowed to cool. The mixture was diluted with EtOAc (300 mL),
then
washed twice with H20 (300 mL). The organic phase was diluted with more EtOAc
(200 mL) to dissolve some solid particles, then washed with saturated NaCI
(100
mL), dried over Na2S04, filtered, and the solvent was removed in vacuo. The
residue was dissolved in hot 10% MeOH/EtOAc (220 mL), then allowed to cool
with
stirring. After stirring for 4 h, no precipitate had formed. The mixture was
diluted
portionwise with hexanes (200 mL) over the next 2 h to promote precipitation
of the
product. After stirring overnight, the precipitate was collected by suction
filtration
washing with 50% EtOAc/hexanes, then hexanes, then dried in vacuo at 49
°C for 20
h to give the title compound (1.65 g, 47%) as a white amorphous solid: mp 170-
172
°C; 1H NMR (300 MHz, CDCl3) 8 7.70 (d, J= 8.1 Hz, 1 H), 7.43-7.53 (m, 2
H), 7.33-
7.41 (m, 2 H), 7.18-7.26 (m, 1 H), 7.01 (br s, 1 H), 6.96 (t, J= 8.7 Hz, 2 H),
6.75 (dd,
J = 8.2, 1.5 Hz, 1 H), 6.60 (br d, J = 7.8 Hz, 1 H), 3.89 (t, J = 6.5 Hz, 2
H), 3.58-3.65
(m, 4 H), 2.58-2.86 (m, 10 H), 2.05 (p, J= 6.5 Hz, 2 H); IR (ATR) 1630, 1602,
1527,
1498, 1445, 1385, 1230 cm-'; ESI MS m/z485 [C2sH29FN4O2 + H]~; HPLC 98.8%
(AUC), tR = 14.16 min. Anal. calcd, for C29H29FN402: C, 71.88; H, 6.03; N,
11.56.
Found: C, 71.75; H, 6.08; N, 11.38.
PREPARATION 46
1-(4-Fluoro-2,3-dihydro-indol-1-yl)-ethanone
A 10 mL flask equipped with a magnetic stir bar was charged with 4-fluoro-
1 H-indole (1.0g, 7.4 mmol). The solid was dissolved in glacial acetic acid
(lOmL).
Sodium cyanoborohydride (932 mg, 14.8 mmol) was added portion-wise, and the
reaction stirred at ambient temperature while being monitored by TLC. After 72
hours, the reaction was quenched by drop-wise addition of H20 and the pH was
adjusted to ~8 with 1 N NaOH. The aqueous layer was extracted with CH2CI2
(3x).
The organic extracts were combined, dried over MgS04, filtered, and
concentrated in
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-113-
vacuo. A yellow oil weighing 1.14g was obtained. The yellow oil was dissolved
in
THF (50 mL). Triethylamine (1.7 mL, 12.5 mmol) was added with stirring,
followed
by acetyl chloride (688 ~,L, 9.9 mmol). The reaction stirred overnight for 15
hours
and was then quenched by drop-wise addition of H20. The aqueous phase was
extracted with CH2C12 (3x), the combined organic extracts were dried over
MgS04,
filtered, and concentrated in vacuo to yield 1.58 g of a light yellow solid.
The yellow
solid was recrystallized from 2-propanol to yield 1-(4-fluoro-2,3-dihydro-
indol-1-yl)-
ethanone as white needle crystals weighing 858 mg (65%, 2 steps).
PREPARATION 47
1-(1-Acetyl-4-fluoro-2,3-dihydro-1 H-indol-5-yl)-2-chloro-ethanone
A 50 mL flask was equipped with a magnetic stir bar, charged with 1-(4-fluoro-
2,3-dihydro-indol-1-yl)-ethanone (358 mg, 2.0 mmol) and aluminum chloride (400
mg, 3 mmol). The flask was fitted with a nitrogen bubbler, purged with
nitrogen gas
and cooled to 0 °C in an ice water bath. Chloro-acetyl chloride (242
p,L, 3.0 mmol)
was added drop-wise to the stirring solution, and the reaction was allowed to
gradually warm to ambient temperature. The reaction stirred at rt for two
hours, and
was then fitted with a condenser and heated to reflux. After an additional two
hours,
an additional 1.5 equivalents of AICI3 (400 mg) were added and the reaction
stirred
overnight at reflux. When all starting material had disappeared by TLC, the
reaction
was quenched by drop-wise addition of H20. The contents of the flask were
extracted with CH2C12 (3x) and the combined organic extracts were dried over
MgS04, filtered, and concentrated in vacuo to yield 368 mg of a brown solid
that was
shown to be the desired product (66%).
PREPARATION 48
1-['S-(2-Chloro-ethyl)-4-fl uoro-2,3-di hydro-i ndol-1-yll-ethanone
A 25 mL flask equipped with a magnetic stir bar was charged with 1-(1-acetyl-
4-fluoro-2,3-dihydro-1 H-indol-5-yl)-2-chloro-ethanone (368 mg, 1.44 mmol),
and the
contents were dissolved in trifluoroacetic acid (3.2 mL). The flask was fitted
with a
rubber septum and purged with nitrogen gas. After drop-wise addition of
triethylsilane (690 ~.L, 4.32 mmol), the reaction was heated to 50 °C
in an oil bath.
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-114-
Stirring continued for 4 hours, after which time no starting material was
visible by
TLC. The contents of the flask were poured into a seperatory funnel containing
water and extracted with dichloromethane (3x). The organic extracts were dried
over
MgS04, filtered, and concentrated in vacuo to yield 334 mg of a brown solid
shown
to be the desired product (89%).
EXAMPLE 99
1-f5-f 2-(4-Benzofdlisoxazol-3-yl-piperazi n-1-yl)-ethyll-4-fl uoro-2,3-
dihydro
indol-1-yl'f-ethanone
A microwave vessel equipped with a magnetic stir bar was charged with 1-[5-
(2-chloro-ethyl)-4-fluoro-2,3-dihydro-indol-1-yl]-ethanone (121 mg, 0.5 mmol),
3-
piperazin-1-yl-benzo[d]isoxazole (180 mg as the HCI salt, 0.75 mmol), and
sodium
carbonate (106 mg, 1 mmol). The contents of the vessel were diluted with 2.5
mL
H20, the vessel was sealed, placed in a CEM Discover microwave, and heated to
175 °C for a duration of 10 minutes. After cooling to rt, the contents
were diluted
with 2 mL of an 8:1 solution of ethanoI:NH40H and extracted three times with
dichloromethane. The combined organic extracts were dried over MgS04,
filtered,
and concentrated in vacuo to yield 367 mg of a crude brown solid. The solid
was
purified on a column of silica gel (15g) using a slow elution gradient of
CH2C12 to
100:8:1 CH2CI2:ethanoI:NH40H over the course of an hour. The isolated brown
solid
weighed 148 mg (73%).
EXAMPLE 100
1-f5-f2-(4-Benzofdlisothiazol-3-yl-piperazin-1-yl)-ethyll-4-fluoro-2,3-dihydro-
indol-1-yl)-ethanone
1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-4-fluoro-2,3-dihydro-
indol-1-yl}-ethanone was prepared in a similar fashion as decribed above in
Example
99 starting with 1-[5-(2-chloro-ethyl)-4-fluoro-2,3-dihydro-indol-1-yl]-
ethanone and 3-
piperazin-1-yl-benzo[d]isothiazole. Yield: 35 mg (39%), Isolated in 100%
purity C
254 nm; LCMS (APCI) 425.1 [M+H]+.
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-115-
EXAMPLE 101
1-(4-Fluoro-5-~'2-f4-(5-fluoro-benzofdlisoxazol-3-yl)-piperazin-1-yll-ethyl)-
2,3
dihydro-indol-1-yl)-ethanone
1-(4-Fluoro-5-{2-[4-(5-fluoro-benzo[d]isoxazol-3-yl)-piperazin-1-yl]-ethyl}-
2,3-
dihydro-indol-1-yl)-ethanone was prepared in a similar fashion as decribed
above in
Example 99 starting with 1-[5-(2-chloro-ethyl)-4-fluoro-2,3-dihydro-indol-1-
yl]-
ethanone and 5-fluoro-3-piperazin-1-yl-benzo[ d]isoxazole. Yield: 50 mg (40%),
Isolated in 100% purity @ 254 nm; LCMS (APCI) 427.1 [M+H]+.
PREPARATION 49
1-(4-Chloro-2,3-dihydro-indol-1-yl)-ethanone
1-(4-Chloro-2,3-dihydro-indol-1-yl)-ethanone was prepared in a similar fashion
as decribed above in Preparation 46 for 1-(4-fluoro-2,3-dihydro-indol-1-yl)-
ethanone
starting with 4-chloroindole yield: 923 mg (71%). 1H NMR (400 MHz, CDC13) ~
ppm
2.20 (s, 3 H} 3.18 (t, J--8.55 Hz, 2 H) 4.07 (t, J=8.55 Hz, 2 H) 6.97 (d, J--
8.06 Hz, 1
H) 7.11 (d, J--8.06 Hz, 1 H) 8.08 (d, J--8.06 Hz, 1 H).
PREPARATION 50
1-(1-Acetyl-4-chloro-2,3-dihydro-1 H-indol-5-yl)-2-chloro-ethanone
1-(1-Acetyl-4-chloro-2,3-dihydro-1 H-indol-5-yl)-2-chloro-ethanone was
prepared in a similar fashion as decribed above in Preparation 47 for 1-(1-
acetyl-4-
fluoro-2,3-dihydro-1 H-indol-5-yl)-2-chloro-ethanone starting with 1-(4-chloro-
2,3-
dihydro-indol-1-yl)-ethanone] yield: 180 mg (26%). 'H NMR (400 MHz, CDCI3) b
ppm 2.24 (s, 3 H) 3.25 (t, J--8.67 Hz, 2 H) 4.14 (m, 2 H) 4.68 (s, 2 H) 7.57
(d, J--8.30
Hz, 1 H) 8.14 (d, J=8.30 Hz, 1 H).
PREPARATION 51
1-~'4-Chloro-5-(2=ch loro-ethyl)-2,3-di hydro-i ndol-1-yll-ethanone
1-[4-Chloro-5-(2-chloro-ethyl)-2,3-dihydro-indol-1-yl]-ethanone was prepared
in a similar fashion as decribed above in Preparation 48 for 1-[5-(2-chloro-
ethyl)-4-
fluoro-2,3-dihydro-indol-1-yl]-ethanone starting with 1-(1-acetyl-4-chloro-2,3-
dihydro-
1 H-indol-5-yl)-2-chloro-ethanone yield: 168g (84%). 1H NMR (400 MHz, CDC13) 8
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-116-
ppm 2.20 (s, 3 H) 3.17 (m, 4 H) 3.68 (t, J=7.45 Hz, 2 H) 4.09 (t, J=8.55 Hz, 2
H) 7.09
(d, J--8.06 Hz, 1 H) 8.03 (m, J--8.06 Hz, 1 H).
EXAMPLE 102
1-f5-f2-(4-Benzo f dlisothiazol-3-yl-pi perazi n-1-yl)-ethyll-4-chloro-2,3-d i
hydro-
indol-1-yl)-ethanone
1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-4-chloro-2,3-dihydro-
indol-1-yl}-ethanone was prepared in a similar fashion as decribed above in
Example
99 starting with 1-[5-(2-chloro-ethyl)-4-chloro-2,3-dihydro-indol-1-yl]-
ethanone and 3-
piperazin-1-yl-benzo[d]isothiazole. Yield: 117 mg (83°l°),
Isolated in 100% purity
254 nm; LCMS (APCI) 441.1 [M+H]+.
EXAMPLE 103
1-f5-f2-(4-Benzo~dlisoxazol-3-yl-pi perazi n-1-yl)-ethyll-4-ch loro-2,3-di
hydro-
indol-1-yl)-ethanone
1-{5-[2-(4-Benzo[d]isoxazol-3-yl-piperazin-1-yl)-ethyl]-4-chloro-2,3-dihydro-
i'ndol-1-yl}-ethanone was prepared in a similar fashion as decribed above in
Example
99 starting with 1-[5-(2-chloro-ethyl)-4-chloro-2,3-dihydro-indol-1-yl]-
ethanone and 3-
piperazin-1-yl-benzo[d]isoxazole. Yield: 75 mg (59%), Isolated in 100% purity
C 254
nm; LCMS (APCI) 425.0 [M+H]+.
PREPARATION 52
1-(6-Fluoro-2,3-dihydro-indol-1-yl)-ethanone
1-(6-Fluoro-2,3-dihydro-indol-1-yl)-ethanone was prepared in a similar fashion
as decribed above in Preparation 46 for 1-(4-fluoro-2,3-dihydro-indol-1-yl)-
ethanone
starting with 6-fluoroindole yield: 1.85 g (70%). 'H NMR (400 MHz, CDCI3) 8
ppm
2.20 (s, 3 H) 3.14 (t, J--8.55 Hz, 2 H) 4.08 (m, 2 H) 6.68 (t, J--8.55 Hz, 1
H) 7.05 (m,
1 H) 7.94 (d, J=10.75 Hz, 1 H).
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-117-
PREPARATION 53
1-(1-Acetyl-6-fluoro-2,3-dihydro-1 H-indol-5-yl)-2-chloro-ethanone
1-(1-Acetyl-6-fluoro-2,3-dihydro-1 H-indol-5-yl)-2-chloro-ethanone was
prepared in a similar fashion as decribed above in Preparation 47 for 1-(1-
acetyl-4-
fluoro-2,3-dihydro-1H-indol-5-yl)-2-chloro-ethanone starting with 1-(6-fluoro-
2,3-
dihydro-indol-1-yl)-ethanone yield: 665 mg (52%). 1H NMR (400 MHz, CDCI3) 8
ppm 2.25 (s, 3 H) 3.20 (t, J--8.43 Hz, 2 H) 4.15 (t, J--8.43 Hz, 2 H) 4.70 (d,
J--2.93
Hz, 2 H) 7.76 (d, J=6.84 Hz, 1 H) 7.97 (d, J=13.19 Hz, 1 H).
PREPARATION 54
1-f 5-(2-Chloro-ethyl)-6-fluoro-2,3-dihydro-i ndol-1-yll-ethanone
1-[5-(2-Chloro-ethyl)-6-fluoro-2,3-dihydro-indol-1-yl]-ethanone was prepared
in a similar fashion as decribed above for Preparation 48 starting with 1-(1-
acetyl-6-
fluoro-2,3-dihydro-1 H-indol-5-yl)-2-chloro-ethanone yield: 168g (84%). 1H NMR
(400 MHz, CDCI3) 8 ppm 2.20 (s, 3 H) 3.01 (t, J--7.08 Hz, 2 H) 3.14 (t, J--
8.30 Hz, 2
H) 3.67 (t, J--7.08 Hz, 2 H) 4.07 (t, J--8.55 Hz, 2 H) 6.98 (d, J=7.33 Hz, 1
H) 7.92 (d,
J=11.72 Hz, 1 H).
EXAMPLE 104
1-f5-f2-(4-Benzofdlisothiazol-3-yl-piperazin-1-yl)-ethyll-6-fluoro-2,3-dihydro-
indol-1-yl)-ethanone
1-{5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-6-fluoro-2,3-dihydro-
indol-1-yl}-ethanone was prepared in a similar fashion as decribed above in
Example
99 starting with 1-[5-(2-chloro-ethyl)-6-fluoro-2,3-dihydro-indol-1-yl]-
ethanone and 3-
piperazin-1-yl-benzo[d]isothiazole. Yield: 146 mg (57%), Isolated in 100%
purity @
254 nm; LCMS (APCI) 425.1 [M+H]+.
EXAMPLE 105
1-(5-f2-(4-Benzo~dlisoxazol-3-yl-piperazin-1-yl)-ethyll-6-fluoro-2,3-dihydro
indol-1-yl)-ethanone
1-{5-[2-(4-Benzo[d]isoxazol-3-yl-piperazin-1-yl)-ethyl]-6-fluoro-2,3-dihydro-
indol-1-yl}-ethanone was prepared in a similar fashion as decribed above in
Example
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-118-
99 starting with 1-[5-(2-chloro-ethyl)-6-fluoro-2,3-dihydro-indol-1-yl]-
ethanone and 3-
piperazin-1-yl-benzo[d]isoxazole. Yield: 123mg (50%), Isolated in 100% purity
@ 254
nm; LCMS (APCI) 409.1 [M+H]+.
PREPARATION 55
1-(6-Chloro-2,3-di hydro-i ndol-1-yl)-ethanone
1-(6-Chloro-2,3-dihydro-indol-1-yl)-ethanone was prepared in a similar fashion
as decribed above in Preparation 46 for 1-(4-fluoro-2,3-dihydro-indol-1-yl)-
ethanone
starting with 6-chloroindole yield: 2.05 g (80%). 'H NMR (400 MHz, DMSO-d~) 8
ppm 2.10 (s, 3 H) 3.06 (t, J=8.42 Hz, 2 H) 4.06 (m, 2 H) 6.97 (dd, J--7.93,
2.07 Hz, 1
H) 7.18 (d, J--8.05 Hz, 1 H) 7.99 (d, J=2.20 Hz, 1 H).
PREPARATION 56
1-(1-Acetyl-6-chloro-2,3-d ihydro-1 H-i ndol-5-yl)-2-ch loro-ethanone
1-(1-Acetyl-6-chloro-2,3-dihydro-1 H-indol-5-yl)-2-chloro-ethanone was
prepared in a similar fashion as decribed above in Preparation 47 for 1-(1-
acetyl-4-
fluoro-2,3-dihydro-1 H-indol-5-yl)-2-chloro-ethanone starting with 1-(6-chloro-
2,3-
dihydro-indol-1-yl)-ethanone] yield: 2.8 g (100%). 'H NMR (400 MHz, CDCI3) 8
ppm
2.25 (s, 3 H) 3.21 (t, J--8.54 Hz, 2 H) 4.14 (t, J--8.54 Hz, 3 H) 4.75 (s, 2
H) 7.49 (s, 1
H) 8.29 (s, 1 H).
PREPARATION 57
1-f6-Chloro-5-(2-chloro-ethyl)-2,3-dihydro-indol-1-yl1-ethanone
1-[6-Chloro-5-(2-chloro-ethyl)-2,3-dihydro-indol-1-yl]-ethanone was prepared
in a similar fashion as decribed above in Preparation 48 for 1-[5-(2-chloro-
ethyl)-4-
fluoro-2,3-dihydro-indol-1-yl]-ethanone starting with 1-(1-acetyl-6-chloro-2,3-
dihydro-
1 H-indol-5-yl)-2-chloro-ethanone yield: 1.87g (96%). 'H NMR (400 MHz, DMSO-
d6)
8ppm2.10(s,3H)3.03(m,4H)3.74(t,J--7.08Hz,2H)4.06(t,J--8.54Hz,2H)
7.22 (s, 1 H) 8.00 (s, 1 H).
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-119-
EXAMPLE 106
1-(6-Chloro-5-(2-~4-(6-fluoro-benzo(dlisoth iazol-3-yl)-piperazin-1-yll-ethyl)-
2,3
dihydro-indol-1-yl)-ethanone
1-(6-Ch loro-5-{2-[4-(6-filuoro-benzo[d]isoth iazol-3-yl)-piperazin-1-yl]-
ethyl}-2,3-
dihydro-indol-1-yl)-ethanone was prepared in a similar fashion as decribed
above in
Example 99 starting with 1-[6-chloro-5-(2-chloro-ethyl)-2,3-dihydro-indol-1-
yl]-
ethanone and 6-filuoro-3-piperazin-1-yl-benzo[d]isothiazole. Yield: 47 mg
(51%). 'H
NMR (400 MHz, DMSO-d6) 8 ppm 2.12 (s, 3 H) 2.52 (m, 2 H) 2.65 (m, 4 H) 2.81
(m,
2 H) 3.07 (m, 2 H) 3.41 (m, 4 H) 4.07 (t, J--8.55 Hz, 2 H) 7.22 (s, 1 H) 7.27
(m, 1 H)
7.93 (m, 1 H) 8.00 (s, 1 H) 8.07 (m, 1 H). Isolated in 97°!°
purity @ 254 nm; LCMS
(APCI) 459 [M+H]+.
The following compounds were prepared from 1-[6-chloro-5-(2-chloro-ethyl)-
2,3-dihydro-indol-1-yl]-ethanone and the appropriate piperazine or piperidine
in a
fashion similar to that reported above.
E'.- ~A P1R0 . . COMPOUND NAME ~ >DATA 7 ~''
zEXAMPL fATE~~: ~
x P ~?R' -
~
' a
PIF'ERAZINE ' r
OR
' '~. ~z" ~'
DIN'E ~ '
E
. ,
PIP ,G' ~$ . w
j ~R ~: ~ 4 ro!-
'~' :' '~ ~ .....
4.. a ~ V
#107 5-methoxy-3- 1-(6-Chloro-5-{2-[4-(5-Yield: 52 mg (55%),
Isolated
piperazin-1-yl-methoxy- in 100% purity @ 254
nm;
benzo[d]isothiazolebenzo[d]isothiazol-3-yl)-LCMS (APCI) 471 [M+H]+.
piperazin-1-yl]-ethyl}-
2,3-dihydro-indol-1-yl)-
ethanone
#108 7-filuoro-3- 1-(6-Chloro-5-(2-[4-(7-Yield: 64 mg (70%),
Isolated
piperazin-1-yl-filuoro- in 94% purity @ 254
nm;
benzo[d]isothiazolebenzo[d]isothiazol-3-yl)-LCMS (APCI) 459 [M+H]+.
piperazin-1-yl]-ethyl}-
2,3-dihydro-indol-1-yl)-
ethanone
#109 7-methoxy-3- 1-(6-Chloro-5-{2-[4-(7-Yield: 49 mg (52%),
Isolated
piperazin-1-yl-methoxy- in 100% purity @ 254
nm;
benzo[d]isothiazole.benzo[d]isothiazol-3-yl)-LCMS (APCI) 471 [M+H]+.
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-120-
piperazin-1-yl]-ethyl}-
2,3-dihydro-indol-1-yl)-
ethanone
#110 3-piperidin-4-yl-1-{5-[2-(4- Yield: 54 mg (61 %),
Isolated
benzo[d]isothiazoleBenzo[d]isothiazol-3-yl-in 95% purity @ 254
nm;
piperidin-1-yl)-ethyl]-6-LCMS (APCI) 440 [M+H]+.
chloro-2,3-dihydro-indol-
1-yl}-ethanone
#111 5-fluoro-3- 1-(6-Chloro-5-{2-[4-(5-Yield: 52 mg (57%),
Isolated
piperazin-1-yl-fluoro- in 100! purity @ 254
nm;
benzo[d]isothiazolebenzo[d]isothiazol-3-yl)-LCMS (APCI) 459 [M+H]+.
piperazin-1-yl]-ethyl}-
2,3-dihydro-indol-1-yl)-
ethanone
#112 6-fluoro-3-piperidin-1-(6-Chloro-5-{2-[4-(6-Yield: 29 mg (31%),
Isolated
4-yl- fluoro-benzo[d]isoxazol-in 100% purity @ 254
nm;
benzo[d]isoxazole3-yl)-piperidin-1-yl]-LCMS (APCI) 458 [M+H]+.
ethyl}-2,3-dihydro-indol-
1-yl)-ethanone
#113 3-piperazin-1-yl-1-{5-[2-(4- Yield: 14 mg (16%),
Isolated
benzo[d]isoxazoleBenzo[d]isoxazol-3-yl-in 100% purity @ 254
nm;
piperazin-i-yl)-ethyl]-6-LCMS (APCI) 425 [M+H]+.
chloro-2,3-dihydro-indol-
1-yl}-ethanone
#114 5-fluoro-3- 1-(6-Chloro-5-{2-[4-(5-Yield: 40 mg (45%),
Isolated
piperazin-1-yl-fluoro-benzo[d]isoxazol-in 100% purity @ 254
nm;
benzo[d]isoxazole3-yl)-piperazin-1-yl]-LCMS (APCI) 443 [M+H]+.
ethyl}-2,3-dihydro-indol-
1-yl)-ethanone
#115 5-chloro-3- 1-(6-Chloro-5-{2-[4-(5-Yield: 38 mg (41%),
Isolated
piperazin-1-yl-chloro-benzo[d]isoxazol-in 91% purity @ 254
nm;
benzo[d]isoxazole3-yl)-piperazin-1-yl]-LCMS (APCI) 459 [M+H]+.
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-121-
ethyl}-2,3-dihydro-indol-
1-yl)-ethanone
#116 6-fluoro-3- 1-(6-Chloro-5-{2-[4-(6-Yield: 40 mg (44%),
Isolated
piperazin-1-yl- fluoro-benzo[d]isoxazol-in 100% purity ~ 254
nm;
benzo[d]isoxazole.3-yl)-piperazin-1-yl]-LCMS (APCI) 443 [M+H]+
ethyl}-2,3-dihydro-indol-
1-yl)-ethanone
#117 6-methyl-3- 1-(6-Chloro-5-{2-[4-(6-Yield: 40 mg (46%),
Isolated
piperazin-1-yl- methyl-benzo[d]isoxazol-in 91% purity C 254
nm;
benzo[d]isoxazole3-yl)-piperazin-1-yl]-LCMS (APCI) 439 [M+H]+.
ethyl}-2,3-dihydro-indol-
1-yl)-ethanone
#118 7-methyl-3- 1-(6-Chloro-5-(2-[4-(7-Yield: 43 mg (49%),
Isolated
piperazin-1-yl- methyl-benzo[d]isoxazol-in 100% purity @ 254
nm;
benzo[d]isoxazole3-yl)-piperazin-1-yl]-LCMS (APCI) 439 [M+H]+.
ethyl}-2,3-dihydro-indol-
1-yl)-ethanone
#119 1- ' 1-{5-[2-(4- Yield: 79 mg (86%),
Isolated
benzo[b]thiophen-Benzo[b]thiophen-3-yl-in 100% purity @ 254
nm;
3-yl-piperazine piperazin-1-yl)-ethyl]-6-LCMS (APCI) 458 [M+H]+.
chloro-2,3-dihydro-indol-
1-yl}-ethanone
#120 3-piperazin-1-yl-11-(6-Chloro-5-(2-[4-(1Yield: 35 mg (41 %),
H- H- Isolated
indazole indazol-3-yl)-piperazin-1-in 100% purity @ 254
nm;
yl]-ethyl}-2,3-dihydro-LCMS (APCI) 424 [M+H]+.
indol-1-yl)-ethanone
PREPARATION 58
1-(2,3-Dihydro-indol-1-yl)-ethanone
A solution of 11.2 mL (0.1 mol) of indoline (commercially available from
Aldrich Chemical company) in THF (200mL) was treated with triethyl amine
(15.33
mL, 0.11 mol) followed by dropwise addition of acetyl chloride (7.82 mL, 0.11
mol).
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-122-
The reaction was stirred at room temperature for 20 hours, quenched with water
(50
mL) fiollowed by concentration in vacuo. White solid was collected and washed
with
water. Yield: 15.7 g (97.5°f°). 'H NMR (400 MHz, DMSO-d6) b ppm
2.11 (s, 3 H)
3.09 (t, J=8.55 Hz, 2 H) 4.03 (m, 2 H) 6.95 (m, 1 H) 7.10 (s, 1 H) 7.19 (d, J--
6.84 Hz,
1 H) 8.01 (d, J--7.82 Hz, 1 H).
PREPARATION 59
1-(1-Acetyl-2,3-dihydro-1 H-indol-5-yl)-2-chloro-ethanone
1-(1-Acetyl-2,3-dihydro-1H-indol-5-yl)-2-chloro-ethanone was prepared in a
similar fashion as decribed above in Preparation 47 starting with 1-(2,3-
dihydro-
indol-1-yl)-ethanone] yield: 11.85 g (100%). 'H NMR (400 MHz, CDCI3) 8 ppm
2.26
(d,J--3.90Hz,3H)3.25(t,J--8.42Hz,2H)4.13(t,J=8.54Hz,2H)4.67(s,2H)
7.80 (m, 2 H) 8.26 (d, J=8.30 Hz, 1 H).
PREPARATION 60
1-[5-(2-Ch Toro-ethyl)-2,3-di hydro-indol-1-yll-ethanone
1-[5-(2-Chloro-ethyl)-2,3-dihydro-indol-1-yl]-ethanone was prepared in a
similar fashion as decribed above in Preparation 48 starting with 1-(1-acetyl-
2,3-
dihydro-1 H-indol-5-yl)-2-chloro-ethanone yield: 2.85 g (85%). ' H NMR (400
MHz,
DMSO-d6) s ppm 2.11 (s, 3 H) 2.92 (t, J--7.08 Hz, 2 H) 3.08 (t, J--8.55 Hz, 2
H) 3.76
(t, J=7.20 Hz, 2 H) 4.04 (t, J=8.55 Hz, 2 H) 7.00 (d, J=8.30 Hz, 1 H) 7.11 (s,
1 H)
7.92 (d, J--8.30 Hz, 1 H).
EXAMPLE 121
1-(5-(2-[4-(6-Fluoro-benzo[dlisothiazol-3-yl)-piperazin-1-yll-ethyf[-2,3-d
ihydro-
indol-1-yl)-ethanone
1-(5-{2-[4-(6-Fluoro-benzo[d]isothiazol-3-yl)-piperazin-1-yl]-ethyl}-2,3-
dihydro-
indol-1-yl)-ethanone was prepared in a similar fashion as decribed above in
Example
99 starting with 1-[5-(2-chloro-ethyl)-2,3-dihydro-indol-1-yl]-ethanone and 6-
fluoro-3-
piperazin-1-yl-benzo[d]isothiazole. The crude products were purified by HPLC
(30x100 mm ODS-A C(18) 5u column). Yield: 20 mg (24%). Rt (min) reported is
for
the following HPLC conditions: 70:30 [H20:MeCN]+0.1% TFA, 1.5 mUmin, 250x4.6
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-123-
mm LUNA Cl8.Isolated in 100% purity @ 254 nm HPLC: Rt = 9.693; MS (APCI),
(M+1 )+ = 425.1.
The following compounds were prepared from 1-[5-(2-chloro-ethyl)-2,3-
dihydro-indol-1-yl]-ethanone and the appropriate piperazine or piperidine in a
fashion
similar to that reported above. The crude products were purified by HPLC
(30x100
mm ODS-A C(18) 5u column).
Example. Appropriate Compound Name Data-'
Piperazine
or
,.- Piperidirie,i:
,, t -
#122 5-methoxy-3- 1-(5-(2-[4-(5-Methoxy-Yield: 37 mg (42%),
, Isolated in
piperazin-1-yl-benzo[d]isothiazol-3-yl}-95% purity @ 254 nm
HPLC: Rt
benzo[d]isothiazolepiperazin-1-yl]-ethyl}-2,3-= 16.04; MS (APCI),
(M+1 )+ _
dihydro-indol-1-yl)-ethanone437.3.
#123 7-fluoro-3-piperazin-1-(5-{2-[4-(7-Fluoro-Yield: 23 mg (27%),
Rt(min)
1-yl- benzo[d]isothiazol-3-yl)-reported is for the
following
benzo[d]isothiazolepiperazin-1-yl]-ethyl}-2,3-HPLC conditions: 60:40
dihydro-indol-1-yl)-ethanone[H2O:MeCN]+0.1% TFA,
1.5
mUmin, 250x4.6 mm LUNA
C18. Isolated in 100%
purity
254 nm HPLC: Rt = 5.725;
MS
(APCI), (M+1 )+ = 425.2.
#124 7-methoxy-3- 1-(5-{2-[4-(7-Methoxy-Yield: 23 mg (26%),
Rt(min)
piperazin-1-yl-benzo[d]isothiazol-3-yl)-reported is for the
following
benzo[d]isothiazolepiperazin-1-yl]-ethyl}-2,3-HPLC conditions: 60:40
dihydro-indol-1-yl)-ethanone[H20:MeCN]+0.1% TFA,
1.5
mUmin, 250x4.6 mm LUNA
Cis. Isolated in 100%
purity @
254 nm HPLC: Rt = 5.275;
MS
(APCI), (M+1 )+ = 437.1.
#125 5-fluoro-3-piperazin-1-(5-(2-[4-(5-Fluoro-Yield: 52 mg (57%),
Rt(min)
1-yl- benzo[d]isothiazol-3-yl)-reported is for the
following
benzo[d]isothiazolepiperazin-1-yl]-ethyl}-2,3-HPLC conditions: 70:30
dihydro-indol-1-yl)-ethanone[H20:MeCN]+0.1% TFA,
1.5
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-124-
mUmin, 250x4.6 mm LUNA
C,B. Isolated in 100%
purity @
254 nm HPLC: Rt =14.59;
MS
(APCI), (M+1 )+ = 425.2.
#126 3-piperazin-1-yl-1-{5-[2-(4-Benzo[d]isoxazol-Yield: 21 mg (27%),
R, (min)
benzo[d]isoxazole3-yl-piperazin-1-yl)-ethyl]-reported is for the
following
2,3-dihydro-indol-1-yl}-HPLC conditions: 70:30
ethanone [H20:MeCN]+0.1 % TFA,
1.5
mUmin, 250x4.6 mm LUNA
Cie. Isolated in i00%
purity @
254 nm HPLC: Rt = 7.87;
MS
(APCI), (M+1 )+ = 391.1.
#127 5-fluoro-3-piperazin-1-(5-{2-[4-(5-Fluoro-Yield: 15 mg (18%),
Rt(min)
1-yl- benzo[d]isoxazol-3-yl)-reported is for the
following
benzo[d]isoxazolepiperazin-1-yl]-ethyl}-2,3-HPLC conditions: 70:30
dihydro-indol-1-yl)-ethanone[H20:MeCN]+0.1 % TFA,
1.5
mUmin, 250x4.6 mm LUNA
Cis. Isolated in 100%
purity @
254 nm HPLC: R~ = 9.699;
MS
(APCI), (M+1 )~ = 409.2.
#128 5-chloro-3-piperazin-1-(5-{2-[4-(5-Chloro-Yield: 18 mg (21 %),
Rt (min)
1-yl- benzo[d]isoxazol-3-yl)-reported is for the
following
benzo[d]isoxazolepiperazin-1-yl]-ethyl}-2,3-HPLC conditions: 70:30
dihydro-indol-1-yl)-ethanone[H20:MeCN]+0.1 % TFA,
1.5
mUmin, 250x4.6 mm LUNA
C18. Isolated in 100%
purity @
254 nm HPLC: Rt=15.722;
MS
(APCI), (M+1 )+ = 425.1.
#129 6-fluoro-3-piperazin-1-(5-{2-[4-(6-Fluoro-Yield: 19 mg (23%),
Rt(min)
1-yl- benzo[d]isoxazol-3-yl)-reported is for the
following
benzo[d]isoxazole.piperazin-1-yl]-ethyl}-2,3-HPLC conditions: 60:40
dihydro-indol-1-yl)-ethanone[H~O:MeCN]+0.1%TFA,
1.5
mUmin, 250x4.6 mm LUNA
Cis. Isolated in 100%
purity @
254 nm HPLC: Rt = 9.971;
MS
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-125-
(APCI), (M+1 )+ = 409.2.
#130 7-methyl-3- 1-(5-{2-[4-(7-Methyl-Yield: 18 mg (22%),
Rt(min)
piperazin-1-yl-benzo[d]isoxazol-3-yl)-reported is for the
following
benzo[d]isoxazolepiperazin-1-yl]-ethyl}-2,3-HPLC conditions: 70:30
dihydro-indol-1-yl)-ethanone[HZO:MeCN]+0.1 % TFA,
1.5
mUmin, 250x4.6 mm LUNA
C18. Isolated in 100%
purity C
. 254 nm HPLC: Rt = 12.705;
MS
(APCI), (M+1 )+ = 405.2.
#131 1-benzo[b]thiophen-1-{5-[2-(4-Benzo[b]thiophen-Yield: 24 mg (30%),
Rt(min)
3-yl-piperazine3-yl-piperazin-1-yl)-ethyl]-reported is for the
following
2,3-dihydro-indol-1-yl}-HPLC conditions: 60:40
ethanone [H20:MeCN]+0.1 % TFA,
1.5
mUmin, 250x4.6 mm LUNA
C18. Isolated in 100%
purity ~
254 nm HPLC: Rt = 5.760;
MS
(APCI), (M+1 )+ = 406.1.
#132 3-piperazin-1-yl-11-(5-{2-[4-(1 H-Indazol-3-yl)-Yield: 32 mg (41 %),
H- Rt (min)
indazole piperazin-1-yl]-ethyl}-2,3-reported is for the
following
dihydro-indol-1-yl)-ethanoneHPLC conditions: 60:40
[H20:MeCN]+0.1% TFA,
1.5
mUmin, 250x4.6 mm LUNA
C~B. Isolated in 100%
purity C
254 nm HPLC: Rt = 2.902;
MS
(APC1), (M+1 )+ = 391.
PREPARATION 61
5-(2-Chloro-ethyl)-2,3-dihydro-indole-1-carboxylic acid tert-butyl ester
ci~.~~~
~ N
~0~~
To a suspension 30 g (0.286 mol) of known 5-(2-chloro-ethyl)-1,3-dihydro-
indol-2-one (Lowe, John A., III; Seeger, Thomas F.; Nagel, Arthur A.; Howard,
Harry
R.; Seymour, Patricia A.; Heym, James H.; Ewing, Frank E.; Newman, Michael E.;
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-126-
Schmidt, Anne W.; et al. 1-Naphthylpiperazine derivatives as potential
atypical
antipsychotic agents. J. Med. Chem. 1991, 34(6), 1860-6) in anhydrous toluene
(600
mL) was added Borane methyl sulfide complex (2.0 M in toluene, 230 mL) and the
resulting mixture was refluxed for 5 hours. The mixture was cooled and
saturated
sodium bicarbonate (300 mL) was added. The mixture was then heated to reflux
for
an additional 5 hours. The organic solvent was removed in vacuo. To the
aqueous
residue was added 1,4 dioxane (300 mL), di-tert-butyl dicarbonate (42 g, 0.192
mol)
and the resulting mixture was stirred for 60 hours at rt. The reaction was
diluted with
water, extracted with ethyl acetate, dried, concentrated and the residue was
purified
via flash chromatography (heptane-ethyl acetate/4:1) to afford solid. Yield:
41.6 g
(96%). MS (APCI), (M+1 )+ = 225.9.
EXAMPLE 133
5-f 2-(4-Benzof dlisoth iazol-3-yl-piperazi n-1-yl)-ethyll-2,3-di hydro-i
ndole-1-
carboxylic acid tert-butyl ester
A mixture of 5-(2-chloro-ethyl)-2,3-dihydro-indole-1-carboxylic acid tent-
butyl
ester (27.7 g, 0.098 mol), 3-piperazin-1-yl-benzo[dJisothiazole hydrochloride
(20.2g,
0.079 mol) and sodium carbonate (18.6 g, 0.175 mol) in 1,4-dioxane-water (320
+
560 mL) was stirred at reflux for 48 hours. Additional cesium carbonate (18 g,
0.055
mol) was added and the mixture was heated at reflux for an additional 6 hours.
Mixture was cooled, diluted with water and extracted with ethyl acetate (2 x 1
L),
dried and concentrated, purified via flash chromatography (heptane-ethyl
acetate-
triethyl amine/2:1:0.01) to provide a white powder. Yield: 26.2 g (67%). MS
(APCI),
(M+1 )+ = 465.2.
PREPARATION 62
3-('4-f2-(2,3-Dihydro-1 H-indol-5-vl)-ethvll-piperazin-1-yl~-
benzofdlisothiazole
5-[2-(4-Benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3-dihydro-indole-1-
carboxylic acid tert-butyl ester was dissolved in 1,4-dioxane (300 mL) and
cooled to
10 °C before 1,4-dioxane-HCI (4.0N, 700mL) was added and the resulting
mixture
was stirred at rt overnight. The resulting white precipitate was collected via
filtration.
Yield: 29.5 g (95%). 1H NMR (400 MHz, D20) 8 ppm 3.02 (m, 2 H) 3.14 (t, J--
7.69
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-127-
Hz, 2 H) 3.25 (d, J--10.01 Hz, 4 H) 3.33 (m, 2 H) 3.56 (m, 2 H) 3.71 (t, J--
7.69 Hz, 2
H) 3.96 (d, J--10.75 Hz, 2 H) 7.19 (m, 1 H) 7.28 (m, 3 H) 7.42 (t, J=7.69 Hz,
1 H)
7.80 (dd, J=16.97, 8.18 Hz, 2 H).
EXAMPLE 134
1-f5-f 2-(4-Benzo~dlisothiazol-3-yl-piperazi n-1-yl)-ethyll-2,3-dihydro-i ndol-
1-yl'f -.
ethanone
A solution of 3-{4-[2-(2,3-dihydro-1 H-indol-5-yl)-ethyl]-piperazin-1-yl)-
benzo[d]isothiazole (364 mg, 1.0 mmols) in THF (4.0 mL) with triethylamine
(0.200
mL, 1.5 mmols) vvas treated with acetyl chloride (0.078 mL, 1.1 mmols) and
stirred
for 16 hours at rt. The reaction was puenched with sodium hydroxide (1 N, 5
mL),
extracted with methylene chloride and filtered through 5 pm PTFE (phase-
separating
filter), and concentrated in vacuo, followed by a crystallization from
isopropyl alcohol
to yield: 253 mg (62%) Isolated in 100% purity @ 254 nm; LCMS (APCI) 406.9
[M+H]+.
The title compounds of Examples 135 through 145 were prepared from 3-{4-
[2-(2,3-dihydro-1 H-indol-5-yl}-ethyl]-piperazin-1-yl}-benzo[d]iso-thiazole in
a parallel
fashion to that reported above using the appropriate commercially available
acid
chloride.
~EXANIPLE~ACID CHLORIDE COMPOUND NAME DATA
V 4
1
~
#1 propionyl chloride1-{5-[2-(4- Yield:
35 301
mg
(72%)
Benzo[d]isothiazol-3-yl-Isolated in 100% purity
@
piperazin-1-yl)-ethyl]-2,3-254 nm; LCMS (APCI)
dihydro-indol-1-yl}-propan-420.9 [M+H]+.
1-one
#136 butyryl chloride1-{5-[2-(4- Yield: 240 mg (55%)
Benzo[d]isothiazol-3-yl-Isolated in 100% purity
@
piperazin-1-yl)-ethyl]-2,3-254 nm; LCMS (APCI)
dihydro-indol-1-yl}-butan-1-435.0 [M+H]+.
one
#137 isobutyryl chloride1-{5-[2-(4- Yield: 165 mg (38%)
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-128-
Benzo[d]isothiazol-3-yl-Isolated in 98.4%
purity ~
piperazin-1-yl)-ethyl]-2,3-254 nm; LCMS (APCI)
dihydro-indol-1-yl}-2-methyl-435.0 [M+H]+.
propan-1-one
#138 cyclopropane {5-[2-(4-Benzo[d]isothiazol-Yield: 311 mg (72%)
carbonyl chloride3-yl-piperazin-1-yl)-ethyl]-Isolated in 100% purity
@
2,3-dihydro-indol-1-yl}-254 nm; LCMS (APCI)
cyclopropyl-methanone432.9 [M+H]+.
#139 valeryl chloride1-{5-[2-(4- Yield: 158 mg (35%)
Benzo[d]isothiazol-3-yl-Isolated in 100% purity
G
piperazin-1-yl)-ethyl]-2,3-254 nm; LCMS (APCI)
dihydro-indol-1-yl]-pentan-448.9 [M+H]+.
1-one
#140 isovaleryl chloride1-{5-[2-(4- Yield: 270 mg (60%)
Benzo[d]isothiazol-3-yl-Isolated in 100% purity
C~
piperazin-1-yl)-ethyl]-2,3-254 nm; LCMS (APCI)
dihydro-indol-1-yl}-3-methyl-448.9 [M+H]+.
butan-1-one
#141 trimethyl acetyl1-{5-[2-(4- Yield: 142 mg (32%)
chloride Benzo[d]isothiazol-3-yl-Isolated in 96% purity
C
piperazin-1-yl)-ethyl]-2,3-254 nm; LCMS (APCI)
dihydro-indol-1-yl}-2,2-448.9 [M+H]+.
dimethyl-propan-1-one
#142 cyclopentane {5-[2-(4-Benzo[d]isothiazol-Yield: 320 mg (70%)
carbonyl chloride3-yl-piperazin-1-yl)-ethyl]-Isolated in 100% purity
C~
2,3-dihydro-indol-1-yl)-254 nm; LCMS (APCI)
cyclopentyl-methanone460.9 [M+H]+.
#143 cyclohexane {5-[2-(4-Benzo[d]isothiazol-Yield: 358 mg (76%)
carbonyl chloride3-yl-piperazin-1-yl)-ethyl]-Isolated in 100% purity
Q
2,3-dihydro-indol-1-yl}-254 nm; LCMS (APCI)
cyclohexyl-methanone 474.9 [M+H]+.
#144 benzoyl chloride{5-[2-(4-Benzo[d]isothiazol-Yield: 352 mg (75.2%)
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-129-
3-yl-piperazin-1-yl)-ethyl]-Isolated in 100% purity
@
2,3-dihydro-indol-1-yl}-254 nm; LCMS (APCI)
phenyl-methanone 468.9 [M+H]+.
#145 methane sulfonyl3-{4-[2-(1-Methanesulfonyl-Yield: 321 mg (73%)
chloride 2,3-dihydro-1 H-indol-5-yl)-Isolated in 100% purity
@
ethyl]-piperazin-1-yl}-254 nm; LCMS (APCI)
benzo[d]isothiazole 442.9 [M+H]+.
EXAMPLE 146
5-f 2-(4-Benzof dlisoth iazol-3-yl-piperazin-1-yl)-ethyll-2,3-d i hydro-i
ndole-1-
carboxylic acid methylamide
A solution of the product of Preparation 62, 3-{4-[2-(2,3-dihydro-1 H-indol-5-
yl)-
ethyl]-piperazin-1-yl}-benzv[d]isothiazole (150 mg, 0.4 mmols in THF (5mL))
was
treated by dropwise addition with above methyl isocyanate (0.03 mL) at rt and
allowed to stir for 72 hours. The reaction was concentrated to dryness,
diluted with
H20 and extracted with methylene chloride and filtered through 5 p,m PTFE
(phase-
separating filter), and concentrated in vacuo, followed by a crystallization
from
acetonitrile to yield: 124 mg (74%). Rt (min) reported is for the following
HPLC
conditions: 65:35 [H20:MeCN]+0.1 % TFA, 1.5 mUmin, 250x4.6 mm LUNA C,s.
Isolated in 100% purity @ 254 nm HPLC: Rt = 3.70; MS (APCI), (M+1 )+ = 422.2.
The following compounds were prepared from 3-(4-[2-(2,3-dihydro-1 H-indol-5-
yl)-ethyl]-piperazin-1-yl}-benzo[d]isothiazole in a fashion similar to that
reported
above using the appropriate commercially available isocyanate.
EXAMPLE ISOCYANATE COMPOUND NAME DATA
#147 ethyl 5-[2-(4-Benzo[d]isothiazol-3-yl-Yield: 120 mg (69%)
isocyanate piperazin-1-yl)-ethyl]-2,3-Isolated in 100% purity
@
dihydro-indole-1-carboxylic254 nm HPLC: Rt =
acid 4.474;
ethylamide MS (APCI), (M+1 )+
_
436.3.
#148 n-propyl 5-[2-(4-Benzo[d]isothiazol-3-yl-Yield: 118 mg (66%)
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-130-
isocyanate piperazin-1-yl)-ethyl]-2,3-Isolated in 100% purity
@
dihydro-indole-1-carboxylic254 nm HPLC: Rt =
acid 5.864;
propylamide MS (APCI), (M+1 )+
_
450.3.
#149 isopropyl 5-[2-(4-Benzo[d]isothiazol-3-yl-Yield: 128 mg (71
%)
isocyanate piperazin-1-yl)-ethyl]-2,3-Isolated in 100% purity
@
dihydro-indole-1-carboxylic254 nm HPLC: Rt =
acid 5.834;
isopropylamide MS (APCI), (M+1 )+
_
450.2.
#150 t-butyl 5-[2-(4-Benzo[d]isothiazol-3-yl-Yield: 109 mg (59%)
isocyanate piperazin-1-yl)-ethyl]-2,3-Isolated in 100% purity
@
dihydro-indole-1-carboxylic254 nm HPLC: Rt =
acid 9.459;
tert-butylamide MS (APCI), (M+1 )+
_
464.3.
#151 cyclopentyl 5-[2-(4-Benzo[d]isothiazol-3-yl-Yield: 179 mg (94%)
isocyanate piperazin-1-yl)-ethyl]-2,3-Isolated in 100% purity
@
dihydro-indole-1-carboxylic254 nm HPLC: Rt =
acid 8.975;
cyclopentylamide MS (APCI), (M+1 )+
476.2.
#152 phenyl 5-[2-(4-Benzo[d]isothiazol-3-yl-Yield: 63 mg (32%)
isocyanate piperazin-1-yl)-ethyl]-2,3-Isolated in 100% purity
@
dihydro-indole-1-carboxylic254 nm HPLC: Rt =
acid 13.091;
phenylamide MS (APCI), (M+1 )+
_
484.1.
EXAMPLE 153
1-~5-f2-(4-Benzofdlisothiazol-3-yl-piperazin-1-yl)-ethyll-2,3-dihydro-indol-1-
yl'~-
2-chloro-ethanone
Chloroacetyl chloride was added dropwise to a solution of the product of
Preparation 62, 3-{4-[2-(2,3-dihydro-1 H-indol-5-yl)-ethyl]-piperazin-1-yl}-
benzo[d]isothiazole in THF at 0 °C and allowed to warm to RT with
stirring. The
reaction was allowed to stir for 16 hours. The reaction was quenched with
water and
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-131-
concentrated to dryness. The crude was suspended in CH2C12 (500mL) and washed
with brine (2X 100mL). The organics were dried with MgS04 and filtered. The
sample was concentrated to dryness resulting in an off-white solid Yield: 4.2
g (95%)
MS (APCI), (M+1 )+ = 441.1.
EXAMPLE 154
1-f5-f2-(4-Benzo~dlisothiazol-3-yl-piperazin-1-yl)-ethyll-2,3-dihydro-indol-1-
yl~
2-pyrrolidin-1-yl-ethanone
To a solution of 1-f5-[2-(4-benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3-
dihydro-indol-1-yl}-2-chloro-ethanone (475 mg, 1.1 mmol) in DMF (8 mL) was
added
sodium carbonate (200 mg, 2.0 mmol) and pyrrolidine (0.125 mL, 1.5 mmol) and
the
reaction was warmed to 65 °C for 4.5 hours. The reacton was poured into
water and
extracted with methylene chloride, dried, concentrated and purified via medium
pressure chromatography (15 g silica cartridge) elution with methylene
chloride to
methylene chloride:ethanol:ammonium hydroxide (100:8:1 over 1 hour).
Recrystallization from acetonitrile yielded 181 mg (35%). 'H NMR (400 MHz,
DMSO-ds) 8 ppm 1.67 (s, 4 H) 2.54 (m, 4 H) 2.64 (m, 4 H} 2.70 (m, 2 H) 3.07
(t,
J=8.43Hz,2H)3.31 (m,4H)3.42(m,4H)4.10(t,J=8.55Hz,2H)6.99(d,J=8.55
Hz, 1 H) 7.09 (s, 1 H) 7.41 (t, J=7.69 Hz, 1 H) 7.53 (t, J--7.57 Hz, 1 H) 7.93
(d,
J--8.06 Hz, 1 H) 8.03 (d, J--8.06 Hz, 2 H), MS (APCI), (M+1 )''~ = 476.2.
The following compounds were prepared from 1-~5-[2-(4-benzo[d]isothiazol-3-
yl-piperazin-1-yl)-ethyl]-2,3-dihydro-indol-1-yl}-2-chloro-ethanone in a
fashion similar
to that reported above using the appropriate commercially available amine.
EXAMPLE AMINE COMPOUND NAME DATA
#155 diethyl amine 1-{5-[2-(4- Yield: 137 mg (29%)
Isolated
Benzo[d]isothiazol-3-yl-in 97% purity @ 254
nm;
piperazin-1-yl)-ethyl]-LCMS (APCI) 478.1 [M+H]+.
2,3-dihydro-indol-1-yl}-2-
diethylamino-ethanone
#156 dimethyl amine1-{5-[2-(4- Yield: 158 mg (35%)
Isolated
Benzo[d]isothiazol-3-yl-in 100% purity @ 254
nm;
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-132-
piperazin-1-yl)-ethyl]-LCMS (APCI) 450.1 [M+H]+.
2,3-dihydro-indol-1-yl}-2-
dimethylamino-ethanone
#157 morpholine 1-{5-[2-(4- Yield: 380 mg (77%)
Isolated
Benzo[d]isothiazol-3-yl-in 100% purity @ 254
nm;
piperazin-1-yl)-ethyl]-LCMS (APCI) 492.1 [M+H]+.
2,3-dihydro-indol-1-yl}-2-
morpholin-4-yl-ethanone
#158 piperidine 1-{5-[2-(4- Yield: 150 mg (31%)
Isolated
Benzo[d]isothiazol-3-yl-in 100% purity @ 254
nm;
piperazin-1-yl)-ethyl]-LCMS (APCI) 490.2 [M+H]+.
2,3-dihydro-indol-1-yl}-2-
piperidin-1-yl-ethanone
EXAMPLE 159
1-f5-f 2-(4-Benzo~dlisoth iazol-3-yl-piperazi n-1-yl)-ethyll-2,3-di hydro-
indol-1-yl'>~-
3-ch loro-~ropan-1-one
Sample prepared in a same fashion to example above (1-{5-[2-(4-
benzo[d]isothiazol-3-yl-piperazin-1-yl)-ethyl]-2,3-dihydro-indol-1-yl}-2-
chloro-
ethanone) treating with chloropropionyl chloride. Yield: 4.5 g (99%) MS
(APCI),
(M+1 )+ = 450Ø
The following compounds were prepared from 1-{5-[2-(4-benzo[d]isothiazol-3-
yl-piperazin-1-yl)-ethyl]-2,3-dihydro-indol-1-yl}-3-chloro-propan-1-one in
afashion
similar to that reported above using the appropriate commercially available
amine.
EXAMPLE AMINE COMPOUND NAME DATA
#160 pyrrolidine1-{5-[2-(4-Benzo[d]isothiazol-Yield: 224 mg (42%)
3-yl-piperazin-1-yl)-ethyl]-2,3-Isolated in 100% purity
@
dihydro-indol-1-yl}-3-pyrrolidin-254 nm; LCMS (APCI)
1-yl-propan-1-one 490.1 [M+H]+.
#161 diethyl 1-{5-[2-(4-Benzo[d]isothiazol-Yield: 220 mg (45%)
amine 3-yl-piperazin-1-yl)-ethyl]-2,3-Isolated in 100% purity
@
CA 02551346 2006-06-22
WO 2005/066165 PCT/IB2004/004239
-133-
dihydro-indol-1-yl)-3- 254 nm; LCMS (APCI)
diethylamino-propan-1-one492.1 [M+H]+.
#162 dimethyl 1-{5-[2-(4-Benzo[d]isothiazol-Yield: 114 mg (18%)
amine 3-yl-piperazin-1-yl)-ethyl]-2,3-Isolated in 100%
purity @
dihydro-indol-1-yl}-3- 254 nm; LCMS (APCI)
dimethylamino-propan-1-one464.1 [M+H]w
#163 morpholine1-{5-[2-(4-Benzo[d]isothiazol-Yield: 257 mg (51%)
3-yl-piperazin-1-yl)-ethyl]-2,3-Isolated in 100%
purity @
dihydro-indol-1-yl}-3- 254 nm; LCMS (APCI)
morpholin-4-yl-propan-1-one506.1 [M+H]+
#164 piperidine1-{5-[2-(4-Benzo[d]isothiazol-Yield: 205 mg (41%)
3-yl-piperazin-1-yl)-ethyl]-2,3-Isolated in 100%
purity @
dihydro-indol-1-yl]-3-piperidin-254 nm; LCMS (APCI)
1-yl-propan-1-one 504.4 [M+H]+