Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-1-
FCC PROCESS USING MESOPOROUS CATALYST
FIELD OF THE INVENTION
[0001] The invention relates to a catalyst composition or catalyst, a method
for
making a catalytic catalyst precursor, a method for making a catalyst
composition
or catalyst, and a catalytic cracking process.
[0002] Particularly but not exclusively, the invention relates to mesoporous
catalytic cracking catalyst, a process for the production of such catalysts,
and a
process utilizing such catalysts in cracking operations, and more in
particular, the
invention relates a mesoporous fluidized catalytic cracking catalyst selective
for
minimizing the production of coke and light gas, a process for the production
of
such catalyst, and a process utilizing such catalyst in fluidized catalytic
cracking
operations.
BACKGROUND OF THE INVENTION
[0003] Catalytic cracking, notably fluidized catalytic cracking ("FCC"), is a
conventional (i.e., well known) process for converting higher average
molecular
weight, higher boiling hydrocarbons to more valuable, lower average molecular
weight, lower boiling hydrocarbons. The products are useful as fuels for
transportation, heating, etc. In the process, the conversion step is usually
conducted
by contacting a hydrocarbon feedstock, e.g., a heavy gas oil, with a moving
bed of
particulate catalyst in the substantial absence of hydrogen at elevated
temperatures.
[0004] The FCC process is cyclic and includes, for example, separate zones for
catalytic feedstock conversion, steam stripping, and catalyst regeneration. In
the
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-2-
cycle, feedstock is blended with the FCC catalyst in a catalytic reactor,
typically a
riser reactor, for catalytic conversion into products. Lower boiling products
are
separated from the catalyst in a separator, e.g., a cyclone separator, and
deactivated
catalyst is conducted to a stripper and contacted with steam to remove
entrained
hydrocarbons; the latter can be combined with vapors from the cyclone
separator,
and both can be conducted away from the process. Stripped deactivated catalyst
contains a carbonaceous residue, called "coke". Stripped catalyst recovered
from
the stripper is conducted to a regenerator, e.g., a fluidized bed regenerator,
and
contacted with a combusting gas, e.g., air, at elevated temperature to bum off
the
coke and reactivate the catalyst. Regenerated catalyst is then blended with
the
feedstock entering the riser, completing the cycle.
[0005] In continuous, cyclic operation, exothermic coke combustion in the
regenerator provides at least a portion of the heat required to balance the
endothermic feedstock cracking in the reactor. However, the presence of coke
beyond that necessary for heat balance is undesirable since converting
feedstock
hydrocarbon into catalyst coke diminishes the quantity of hydrocarbon products
obtained from the feedstock. There is therefore a need for catalysts that
selectively
make a greater quantity of hydrocarbon products but less catalytic coke.
[0006] Mesoporous FCC catalysts, such as those described in U.S. Patent No.
5,221,648 are effective for feedstock conversion into high value hydrocarbon
products, such as light olefins. Such catalysts have the desirable property
that
undesirably high catalyst coke levels are avoided in FCC operation. However,
such
catalysts contain a mesoporous silica-alumina matrix formed from silica sols
that
undesirably add to the expense of catalyst production. Moreover, conventional
sols
are acidic, and, consequently, can undesirably affect catalytic constituents,
such as
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-3-
zeolite, during catalyst synthesis. There is therefore a need for improved
mesoporous catalysts.
SUMMARY OF THE INVENTION
[0007] According to embodiments of the invention there are provided a catalyst
composition or catalyst, a method for making a catalytic catalyst precursor, a
method for making a catalyst composition or catalyst, and a catalytic cracking
process as defined in any of the accompanying claims.
[0008] In an embodiment, the invention relates to a composition or catalyst
composition or catalyst, comprising at least one amorphous, porous matrix,
each
matrix having pores ranging in diameter from 1 A to 10 A and pores ranging in
diameter from 40 A to at least 500 A, wherein in the pore range from 50 A to
250
A, there is a single maximum in differential pore volume distribution over the
50 A
to 250 A range. The matrix may be a single amorphous entity, or may be a blend
of
two or more individual amorphous matrices provided that each matrix
individually
meets the above-noted differential pore volume distribution requirement.
[0009] The catalyst composition of the invention is highly selective in the
production of liquids, notably olefins, during fluid catalytic cracking
operations,
and coke make is low whereas the attrition resistance of these catalysts is
quite high
in comparison to conventional mesoporous FCC catalysts.
[0010] In an embodiment, the composition's matrix has a differential pore
volume as a function of matrix pore diameter, and this function has a maximum
between 50 A and 250 A, preferably between 50 A and 150 A. The integrated
differential pore volume for matrix pores having a diameter between 1 A and 10
A
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-4-
cannot be distinguished from the pore volume in zeolites typically used in the
application. Thus, it is not feasible to estimate the pore volumes for pores
below 10
A because one cannot distinguish between the pore volume of the matrix and
that
of the zeolite. The integrated maximum pore volume for the volume of matrix
pores
having a diameter between 10 A and 40 A is less than 0.03 cc/g, preferably
less
than 0.01 cc/g, more preferably less than 0.006 cc/g.
[00111 In another embodiment, the invention relates to a method for making a
cracking catalyst precursor comprising:
(a) combining water, at least one molecular sieve, at least one aluminum
hydroxide or aluminum oxyhydroxide, at least one clay, at least one
urea compound having the formula
X
I I
R1R2N - C - NR3R4
where R1, R2, R3, and R4 are individually H or C 1 to C4 alkyl and X is
sulfur or oxygen, and at least one phosphate to form a first mixture;
(b) combining the first mixture with sufficient aqueous alkaline silicate
solution to form a slurry having a pH sufficient to prevent gellation of
the aqueous alkaline silicate solution;
(c) drying the slurry at a drying temperature to remove water to form a
first solid, said solid preferably comprising ammonium silicate, alkali
silicate and alkali carbonate, urea compound, clay, at least one
aluminum hydroxide or aluminum oxyhydroxide and molecular sieve;
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-5-
(d) combining the first solid with water and an ion exchange composition
comprising one or more mineral acid, preferably sulfuric acid,
aluminum salts of mineral acids such as aluminum sulfate, and/or
ammonium salts of mineral acids such as ammonium sulfate, to form
the catalyst precursor, the catalyst precursor having a lower
concentration of alkali metal compared to the first solid.
[0012] In another embodiment, the invention relates to making catalyst from
the
catalyst precursor comprising the further steps of:
(e) combining the catalyst precursor with water and a second,
independently selected ion exchange composition comprising one or
more mineral acid such as sulfuric acid, aluminum salts of mineral
acids such as aluminum sulfate, and/or ammonium salts of mineral
acids such as ammonium sulfate, to form an ion-exchanged catalyst
precursor having a lower concentration of alkali metal compared to
the first and second solids;
(f) calcining the ion-exchanged catalyst precursor at a temperature
ranging from 250 C to 850 C for a calcination time to make a
calcined, ion-exchanged catalyst precursor; and
(g) contacting the calcined, ion-exchanged catalyst precursor with steam
at a temperature ranging from 650 C to 850 C for a steaming time. A
preferred steaming time is 4 to 48 hours. The steaming deactivates
the cracking catalyst and simulates the deactivation in a commercial
FCC unit which runs at significantly lower water pressures for a
much longer time.
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-6-
[0013] In yet another embodiment, the invention relates to a catalytic
cracking
process, comprising contacting a hydrocarbon feedstock with a catalytically
effective amount of a cracking catalyst under catalytic conversion conditions,
wherein the cracking catalyst comprises zeolite and an amorphous, porous
matrix
having pores ranging in diameter from 1 A to 10 A and pores ranging in
diameter
from 40 A to at least 500 A, wherein in the pore range from 50 A to 250 A,
there is
a single maximum in differential pore volume distribution over the 50 A to 250
A
range.
[0014] In another embodiment the catalytic conversion conditions include a
temperatures of from 450 C to 700 C, a hydrocarbon partial pressure of from 10
to
40 psia (69 to 276 kPa), a cracking catalyst to feedstock (wt/wt) ratio of
from 3 to
100, where catalyst weight is total weight of the cracking catalyst, a
pressure
ranging from atmospheric pressure to 45 psig (411 kPa), and a feedstock
residence
time of from 0.1 to 20 seconds.
[0015] In a further embodiment, the cracking catalyst is made by:
(a) combining water, at least one molecular sieve, at least one aluminum
hydroxide, at least one clay, urea compound, and at least one
phosphate to form a first mixture, said urea compound having the
formula:
X
I I
R1R2N - C - NR3R4
where R1, R2, R3, and R4 are individually H or C1 to C4 alkyl and X is
sulfur or oxygen;
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-7-
(b) combining the first mixture with sufficient aqueous alkaline silicate
solution to form a slurry having a pH sufficient to prevent gellation of
the aqueous alkaline silicate solution;
(c) drying the slurry at a drying temperature to remove water to form a
first solid;
(d) combining the first solid with water and an ion exchange composition
comprising one or more of sulfuric acid, aluminum sulfate, and/or
ammonium sulfate, to form a catalyst precursor, the catalyst precursor
having a lower concentration of alkali metal compared to the first
solid;
(e) combining the catalyst precursor with water and a second,
independently selected ion exchange composition comprising one or
more of sulfuric acid, aluminum sulfate, and/or ammonium sulfate, to
form an ion-exchanged catalyst precursor having a lower
concentration of alkali metal compared to the first solid and catalyst
precursor;
(f) calcining the ion-exchanged catalyst precursor at a temperature
ranging from 250 C to 850 C for a calcination time to make a
calcined, ion-exchanged catalyst precursor; and
(g) contacting the calcined, ion-exchanged catalyst precursor with steam
at a temperature ranging from 650 C to 850 C for a steaming time in
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-8-
order to make the cracking catalyst, wherein the preferred steaming
time is 4 to 48 hours.
BRIEF DESCRIPTION OF THE DRAWINGS
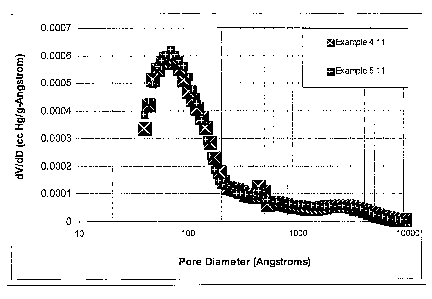
[0016] Figure 1 is a plot of the differential mercury pore volume (dV/dD) vs.
pore diameter for catalyst matrix between 40 and 10000 Angstroms.
[0017] Figure 2 is a plot of integrated differential mercury pore volume vs.
pore
diameter between 40 and 500 Angstroms for catalyst matrix.
[0018] Figure 3 is a plot of dV/dD vs. pore diameter showing the extrapolation
of pore volume for pores in the 10 to 40 Angstrom range.
[0019] Figure 4a is a plot showing a comparison of coke make vs. 221 C- plus
coke make for the base case comparative catalysts vs. catalysts of the
invention.
[0020] Figure 4b is a plot showing a comparison of coke make vs. 221 C- plus
coke make for commercially available catalysts vs. catalysts of the invention.
[0021] Figure 5 is a plot showing a comparison of the coke yield normalized to
remove the influence of conversion vs. 221 C- plus coke make for the base case
comparative catalysts vs. catalysts of the invention.
[0022] - Figure 6a is a plot showing a comparison of dry gas make vs. 221 C- +
coke make for the base case comparative catalyst vs. catalysts of the
invention.
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-9-
[0023] Figure 6b is a plot showing a comparison of dry gas make vs. 221 C- +
coke make for the commercially available catalysts vs. catalysts of the
invention.
[0024] Figure 7a is a plot showing a comparison of propene make vs. 221 C +
coke conversion for catalysts of the invention vs. base case comparative
catalysts.
[0025] Figure 7b is a plot showing a comparison of propene make vs. 221 C +
coke conversion for the commercially available catalysts vs. catalysts of the
invention.
[0026] Figure 8a is a plot of dV/dD vs. pore diameter showing that catalysts
of
the invention have a maximum occurring at pore diameters above 50 Angstroms.
[0027] Figure 8b is a plot of dV/dD vs. pore diameter showing that the
commercially available catalysts have a local maximum in the dV/dD plot below
60
Angstroms regardless of the severity of the steaming.
[0028] Figure 8c is a plot of dV/dD vs. pore diameter showing that
commercially available catalyst when blended with other commercially available
catalysts from the same manufacturer has a local maximum in the dV/dD plot
below 60 Angstroms regardless of the severity of the steaming.
[0029] Figure 8d is a plot of dV/dD vs. pore diameter showing that catalysts
of
this invention have a maximum in the dV/dD plot above 60 Angstroms and below
80 Angstroms unlike the commercially available catalysts which have a local
maximum below 60 Angstroms in all cases and may have a local maximum above
80 Angstroms in some cases.
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-10-
DETAILED DESCRIPTION OF THE INVENTION
(I) The Catalytic Cracking Catalyst
[0030] In an embodiment, the invention relates to a catalytic cracking
catalyst
composition comprising cracking catalyst (generally in particle form) and,
optionally, other reactive and non-reactive components. More than one type of
catalyst may be present in the composition. Typically, the catalyst comprises
matrix and at least one crystalline molecular sieve, said matrix comprising at
least
one clay, at least one aluminum hydroxide or oxyhydroxide, and binder
colloids.
The molecular sieve can be an aluminosilicate, such as zeolite, having an
average
pore diameter between 3 and 15 Angstroms. The pore diameter also sometimes
referred to as effective pore diameter can be measured using standard
adsorption
techniques and hydrocarbons of known minimum kinetic diameters. See Breck,
Zeolite Molecular Sieves, 1974, and Anderson et al., J. Catalysis 58, 114
(1979),
and the Atlas of Zeolite Structure Types, eds. W. H. Meier and D. H. Olson,
Butterworth-Heineman, Third Edition, 1992. More than one type of catalyst may
be present in the composition. For example, individual catalyst particles may
contain large pore zeolite, shape selective zeolite, and mixtures thereof. In
addition
to catalyst particles, the composition may also include fines, inert
particles,
particles containing a metallic species such as platinum and compounds
thereof.
[0031] In addition to matrix and molecular sieve, the catalyst can further
comprise metals such as platinum, promoter species such as phosphorous-
containing species, and species for imparting additional catalytic
functionality
(additional to the cracking functionality) such as bottoms cracking and metals
passivation. Such an additional catalytic functionality may be provided, for
example, by aluminum-containing species.
CA 02569123 2010-06-04
-11-
[0032] The inorganic matrix is a porous inorganic oxide matrix component for
(i)
binding the components together so that the catalyst is attrition resistant
enough to
survive inter-particle and reactor wall collisions (i.e., attrition
resistance), and (ii) to
provide a degree of size selectivity with respect to molecules capable of
cracking on
or in the molecular sieve. The inorganic oxide matrix may be made from, e.g.,
an
inorganic oxide sol, which is then dried. Conventional sols can be used.
Examples of
conventional sols include silica sols derived from the reaction of sodium
silicate and
sulfuric acid/aluminum sulfate solutions, silica sols prepared through an ion-
exchange
process typified by materials with trade names such as LudoxTMand NyacolTM 5/6
basic aluminum chlorhydroxide typified by materials such as ChlorhydrolTM and
peptized alumina slurries such as those that can be made from the reaction of
acid
with materials such as the VersalTM series of aluminum pseudoboehmites. The
matrix
itself may possess catalytic properties, generally of an acidic nature, but
matrix
catalytic activity is not required. In an embodiment, the matrix comprises
oxides of
silicon and aluminum. The matrix can comprise more than one oxide phase, for
example, aluminum oxyhydroxides-y-alumina, boehmite, diaspore, and
transitional
aluminas such as a-alumina, (3-alumina, y-alumina, 8-alumina, c-alumina, x-
alumina,
and p-alumina can be employed. In a related embodiment, the alumina species is
an
aluminum hydroxide such as gibbsite, bayerite, nordstrandite, or doyelite. The
matrix
material may contain phosphorous or aluminum phosphate, and while generally
undesirable, a small amount of sodium. The matrix may also comprise clays such
as
kaolin, bentonite, attapulgite, montmorillonite, hectorite and pyrophyllite.
[0033] The catalyst in the composition will now be described in more detail.
The catalyst comprises matrix, said matrix comprising at least one clay, at
least one
aluminum hydroxide or oxyhydroxide, and binder colloids, in an amount ranging
from 5 wt. percent to 100 wt. percent, preferably from 8 wt. percent to 95 wt.
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-12-
percent, based on the total weight of the catalyst, within which is dispersed
a
crystalline molecular sieve. In an embodiment, the molecular sieve is a
crystalline
aluminosilicate, i.e., zeolite, natural or synthetic, typically having a
silica-to-
alumina mole ratio (Si/Al) of 2, and greater, and uniform pores with diameters
ranging from 3 A to 15 A. The zeolite content of the catalyst ranges from 0
percent
to 95 percent by weight, preferably from 5 percent to 92 percent, and more
preferably from 10 percent to 60 percent, based on the total weight of the
catalyst.
[0034] Under the IUPAC, microporous refers to pores in the 2 to 20 A range and
mesoporous in the 20 to 500 A range. As defined in this invention, the
respective
ranges are 1 A to 10 A for micropores and 40 A to at least 500 A, preferably
between 40 A and 250 A for mesopores. A functional definition of "mesoporous"
as used herein is that porosity which extends above the range normally
associated
with the adsorption of mid-distillate in FCC, in particular the porosity in
pores
which have larger diameters than those associated with the standard commercial
FCC zeolite, structure type FAU as set forth in the Atlas of Zeolite Structure
Types,
ed. W.M.Meier, D.H. Olson and Ch. Baerlocher, Elsevier, 1996.
[0035] The differential pore volume for matrix pores has a maximum at a
diameter between 40 A and 250 A as illustrated in Figure 1. This Figure shows
that
in the pore range from 50 A to 250 A, there is a single maximum in the
differential
mercury pore volume over the 50 A to 250 A range.
[0036] The pore volume measured with mercury for matrix pores with diameters
less than 250 Angstroms comprises between 60 and 80% of the pore volume
measured by mercury below 500 Angstroms as illustrated in Figure 2.
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-13-
[0037] Mercury is not capable of measuring pore volumes below 35 Angstroms
and while gas phase adsorption done under very specific conditions may be able
to
capture the pore volume in this range, interference from pores associated with
the
zeolites contained in the system precludes accurate measurement of two
different
types of pores within this range.
[0038] The matrix is substantially free of pores ranging in diameter between
10
A and 40 A, i.e., these pore diameters are substantially absent from the
matrix pore
distribution. By "substantially free of is meant that the integrated maximum
pore
volume for the volume of matrix pores having a diameter between 10 A and 40 A
is
less than 0.03 cc/g, preferably less than 0.01 cc/g, more preferably less than
0.006
cc/g. It has been discovered that an adequate indication of the pore volume
below
35 Angstroms is given by the slope of the differential mercury porosimetry as
is
indicated in Figure 3. When lines tangent to the differential mercury
intrusion
curve at points below 50 Angstroms intercept the pore diameter axis with a
positive
slope at a value not less than 0 Angstroms and most preferentially at not less
than
Angstroms when the value of dV/dD is 0.0000, catalysts of this invention give
lower coke yields. The following plots in Figure 3 for two different materials
of
this invention show tangent lines which intercept the pore diameter at values
of 10
and 25 Angstroms respectively.
[0039] For Figure 3, which is a plot of dV/dD vs. pore diameter, the maximum
volume of the pore volume of pores in the 10 - 40 A is equal to 0.0004 ccHg/(g-
Angstrom) times 30 Angstroms divided by 2 = 0.006 cc/g.
[0040] In an embodiment, the matrix is an amorphous, porous silica-alumina
matrix having pores ranging in size from 1 A to 10 A and from 40 A to 500 A,
but
substantially free of pores ranging in size from 1 0A to 40A, provided that in
the
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-14-
pore range from 50 A to 250 A, there is a single maximum in differential pore
volume distribution over the 50 A to 250 A range.
[0041] In a related embodiment, the composition's matrix has a differential
pore volume as a function of matrix pore diameter, and this function has a
maximum between 50 A and 150 A. The integrated differential pore volume for
matrix pores having a diameter between 1 A and 10 A cannot be distinguished
from
the pore volume in zeolites typically used in the catalyst. The integrated
maximum
pore volume for the volume of matrix pores having a diameter between 40 A and
500 A ranges from 0.06 cc/g to 0.12 cc/g, and the integrated pore volume for
matrix
pores having a diameter between 10 A and 40 A is less than 0.03 cc/g,
preferably
less than 0.01 cc/g.
[0042] Catalysts of these types are highly selective in the production of
liquids,
notably olefins, during fluid catalytic cracking operations, and coke make is
low.
The attrition resistance of these catalysts is quite high, as indicated by the
low
Davison Indices ranging from 1 to 8, most often and preferably from 1 to 5
measured in terms of the Davison Index. See "Advances in Fluid Catalytic
Cracking," Catalytica, Mountain View, Calif., Part 1, 1987. p. 355.
[0043] A preferred catalyst particle comprises (a) amorphous, porous solid
acid
matrix, such as alumina, silica-alumina, silica-magnesia, silica-zirconia,
silica-
thoria, silica-beryllia, silica-titania, silica-alumina-rare earth and the
like; and (b) a
zeolite such as faujasite. The matrix can comprise ternary compositions, such
as
silica-alumina-thoria, silica-alumina-zirconia, magnesia and silica-magnesia-
zirconia. The matrix may also be in the form of a cogel. Silica-alumina is
particularly preferred for the matrix, and can contain 10 to 40 wt.% alumina.
As
discussed, promoters can be added.
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-15-
[0044] In an embodiment, the catalyst's zeolite includes zeolites which are
iso-
structural to zeolite Y. These include the ion-exchanged forms such as the
rare-
earth hydrogen and ultrastable (USY) form. The zeolite may range in
crystallite
size from 0.1 to 10 microns, preferably from 0.3 to 3 microns. The relative
concentrations of zeolite component and matrix on an anhydrous basis may vary
widely, with the zeolite content ranging from 1 to 100, preferably 10 to 99,
more
usually from 10 to 80, percent by weight of the dry composition.
[0045] The amount of zeolite component in the catalyst particle will generally
range from 1 to 60 wt.%, preferably from 5 to 60 wt.%, and more preferably
from
to 50 wt.%, based on the total weight of the catalyst. As discussed, the
catalyst
is typically in the form of a catalyst particle contained in a composition.
When in
the form of a particle, the catalyst particle size will range from 10 to 300
microns in
diameter, with an average particle diameter of 60 microns. The surface area of
the
matrix material after artificial deactivation in steam at pressures higher
than in
commercial operations [i.e. at pressures of 1 atmosphere (101.3 kPa)] will be
<_ 350
m2/g, preferably 50 to 200 m2/g, more preferably from 50 to 100 m2/g. While
the
surface area of the catalysts will be dependent on such things as type and
amount of
zeolite and matrix components used, it will usually be less than 500 m2/g,
preferably from 50 to 300 m2/g, more preferably from 50 to 250 m2/g, and most
preferably from 100 to 250 m2/g.
[0046] Another preferred catalyst contains a mixture of zeolite Y and a second
zeolite such as zeolite beta. The first and second zeolite may be on the same
catalyst particle, on different particles, or some combination thereof.
Zeolite
amount and matrix type and properties are as set forth in the description of
the Y
zeolite catalyst. In a related embodiment the second zeolite is a shape-
selective
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-16-
zeolite species such as ZSM-5. Alternatively, the shape-selective zeolite can
be
used in the catalyst without the first zeolite. The Y zeolite, shape-selective
zeolite,
or both can be on the same catalyst particle, on different particles, or some
combination thereof.
[0047] Shape-selective zeolite species useful in the invention include medium
pore size zeolites generally having a pore size from 0.5 nm, to 0.7 nm. Such
zeolites include, for example, MFI, MFS, MEL, MTW, EUO, MTT, HEU, FER,
and TON structure type zeolites (IUPAC Commission of Zeolite Nomenclature).
Non-limiting examples of such medium pore size zeolites, include ZSM-5, ZSM-
12, ZSM-22, ZSM-23, ZSM-34, ZSM-35, ZSM-38, ZSM-48, ZSM-50, silicalite,
and silicalite 2. The most preferred is ZSM-5, which is described in U.S.
Patent
Nos. 3,702,886 and 3,770,614. ZSM-11 is described in U.S. Patent No.
3,709,979;
ZSM-12 in U.S. Patent No. 3,832,449; ZSM-21 and ZSM-38 in U.S. Patent No.
3,948,758; ZSM-23 in U.S. Patent No. 4,076,842; and ZSM-35 in U.S. Patent No.
4,016,245.
[0048] While the shape-selective species has been described in terms of
zeolite,
it can be a shape-selective (i.e., medium pore size) molecular sieve. In an
embodiment, suitable medium pore size molecular sieve includes the
silicoaluminophosphates (SAPO), such as SAPO-4 and SAPO-11 which is
described in U.S. Patent No. 4,440,871; chromium silicates; gallium silicates;
iron
silicates; aluminum phosphates (ALPO), such as ALPO-11 described in U.S.
Patent
No. 4,310,440; titanium aluminosilicates (TASO), such as TASO-45 described in
EP-A No. 229,295; boron silicates, described in U.S. Patent No. 4,254,297;
titanium aluminophosphates (TAPO), such as TAPO-11 described in U.S. Patent
No. 4,500,651; and iron aluminosilicates.
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-17-
[00491 The large pore (e.g., zeolite Y) and shape-selective zeolites in the
catalytic species can include "crystalline admixtures" which are thought to be
the
result of faults occurring within the crystal or crystalline area during the
synthesis
of the zeolites. Examples of crystalline admixtures of ZSM-5 and ZSM-11 are,
for
example, disclosed in U.S. Patent No. 4,229,424. The crystalline admixtures
are
themselves medium pore, i.e., shape-selective, size zeolites and are not to be
confused with physical admixtures of zeolites in which distinct crystals of
crystallites of different zeolites are physically present in the same catalyst
composition or hydrothermal reaction mixtures.
(II) Process for the Preparation of the Catalytic Cracking Catalyst
[00501 The catalyst of this invention comprises a catalytically active
molecular
sieve dispersed in a mesoporous inorganic matrix. In an embodiment, a
crystalline
aluminosilicate zeolite, or zeolite, suitably a USY or high silica USY
zeolite, is
admixed, preferably with water, urea compound, a phosphate, a clay, e.g.,
kaolin
and an aluminum hydroxide, e.g. gibbsite, and these solids slurried. An
aqueous
silica solution, e.g., a silica sol (a binder colloid), is added to the
aqueous slurry.
The sol should not be allowed to gel. The slurry of blended components is
dried,
ion-exchanged to remove sodium, calcined, and then steamed to form the
catalyst.
[00511 A catalyst precursor may be made by:
(a) combining molecular sieve with (i) an aqueous solution containing
alkali silicate, e.g., sodium silicate; (ii) urea; (iii) a phosphate, such
as alkali metal phosphate, ammonium phosphate, or both; and (iv) a
clay component such as bentonite, kaolin, or both to form a slurry;
(b) spray drying the slurry; and
(c) removing sodium in an ion exchange operation.
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-18-
[0052] Gibbsite may be added to the catalyst precursor as a component in part
(a) above. The order of addition of the components (a) may be varied. A
catalyst
can be made from the precursor by additional ion exchange, if necessary to
further
remove sodium, and then calcining and steaming. The calcining and steaming can
be conventional. Calcining can take place at temperatures in the range of 250
C to
850 C. The time of calcining depends on the temperature chosen but is
typically
greater than 1 hr. Steaming is preferably done for 4 to 48 hours at
temperatures of
650 C'to 850 C.
[0053] The matrix, after steaming, can be characterized by pore size
distribution,
as measured by mercury porosimetry (Structure of Metallic Catalysts, J. R.
Anderson, 1975, Chapter 6, Pages 384-385; theta=140 degrees, Hg surface
tension
equals 474 ergs/cm2). Under the IUPAC, microporous refers to pores in the 2 to
20
A range.
[0054] The urea compound has the general formula
X
R1R2N - C - NR3R4
where R1, R2, R3, and R4 are individually H or C 1 to C4 alkyl, preferably H
and X
are sulfur or oxygen, preferably oxygen. The preferred urea compound is urea,
i.e.,
H2NCONH2.
[0055] Urea may be added to the slurry in an amount which is stoichiometric
based on the reaction of urea with the sodium in sodium silicate to form
sodium
carbonate and ammonia. The amount of urea may vary from 0.8 to 1.2 times the
stoichiometric amount based on the amount of sodium silicate. The addition of
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-19-
urea to the slurry generally leads to an increase in the pore volume of the
catalysts
according to the invention.
[0056] The phosphates are water soluble phosphate salts, typically sodium or
ammonium phosphate salt, preferably sodium phosphate. The salts may be
primary, secondary or tertiary salts such as NaH2PO4, (NH4)2HP04, (NH4)3PO4,
Na2HPO4, Na3 P04, as well as polyphosphates such as (NaPO3),,, Na4P207 and the
like. The amount of phosphate is preferably less than that required to react
with all
the aluminum present.
[0057] The clays used in the slurry may be kaolin, bentonite, attapulgite,
montmorillonite, hectorite and pyrophyllite. The preferred clay is kaolin or
bentonite,
especially kaolin. In an embodiment, zeolite, clay, phosphate, sodium
silicate, at
least one aluminum hydroxide or aluminum oxyhydroxide and urea are added
together or in sequence, in any order, and slurried at ambient temperature in
a
limited, controlled, and amount of water. In general, it has been found that
the
weight ratio of water: solids in the slurry can range between 1:1 to 4:1,
preferably
between 1.5:1 to 3:1. A weight ratio of water: solids approximating 2:1 has
been
found highly successful in forming high quality catalysts. When the weight
ratio of
water: solids is less than 1:1, the viscosity of the slurry is too high to
spray dry,
while weight ratios of water: solids exceeding 4:1 may lead under some
circumstances to a loss in the attrition-resistance of the catalyst. The pH of
the
slurry at this time ranges between 10 and 12 in order to avoid gellation of
the silica
sol. In an embodiment, the silica in the sol ranges from 1.0 rim (nanometers)
to 22.0
nm, preferably from 1.5 nm to 15.0 nm average diameter. Silica sols are
described
in "The Chemistry of Silica: Solubility, Polymerization, Colloid and Surface
Properties, and Biochemistry," by Ralph K. Her, A Wiley Interscience
Publication,
1979. Water may be added to the sodium silicate sol to maintain the
water:solids
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-20-
weight ratio between 1:1 and 3:1. The preferred solids contents are between 28
and
45 wt.%, based on catalyst precursor. The density of the slurry, on completing
the
addition of the starting materials, preferably ranges from 1.2 to 1.4, and
more
preferably from 1.20 to 1.35. Preferably also, the viscosity of the slurry at
this time
ranges from 60 to 300 cP (60000 to 300000 Pa-s), more preferably from 80 to
200
cP (80000 to 200000 Pass) at 22 C.
[0058] After blending zeolite, clay(s), at least one aluminum hydroxide or
aluminum oxyhydroxide, urea, sodium silicate, and phosphate, with adjustment
of
the water content, density, and preferably also the pH and viscosity, the
slurry can
be dried in conventional process equipment, e.g., a spray drier, to form
catalyst
precursor. `
[0059] In an embodiment, the slurry, preferably at/or below ambient
temperature
is conducted to a drier, preferably a spray drier, at a temperature sufficient
to
remove the water and form microspheres of average particle diameter ranging
from
microns to 200 microns, preferably from 60 microns to 100 microns. The
temperature is sufficiently high to dry the slurry and form a rigid structure,
but
insufficiently high as to cause alkali metal components (e.g., sodium from the
sodium silicate) to be occluded within the zeolite and prevent it from being
washed,
ion- exchanged, and removed from the zeolite. Typically, the slurry is fed to
a drier,
preferably a spray drier at an average inlet temperature ranging from 250 C to
500 C, and an outlet temperature ranging from 125 C to 225 C.
[0060] Following drying, catalyst precursor, preferably in the form of a
powder
of microspherical particles, are washed with deionized water, e.g., between
ambient
temperature and 100 C. The washed precursor is then ion-exchanged for a time
sufficient to remove the alkali metal, e.g., sodium, from the zeolite. In an
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-21-
embodiment, one or more of sulfuric acid, aluminum sulfate, and ammonium
sulfate, are used. Preferably, aluminum sulfate hydrate and ammonium sulfate
are
used. Preferably, a stoichiometric amount of aluminum sulfate hydrate to
ammonium sulfate is used, based on the amount of sodium present. Preferably, a
2/3 atomic ratio of A13+/NH4+ in sulfate salts is used. Data in Tables 2.3 and
2.5
following indicate that the optimum A13+/ NH4+ molar ratio lies at this atomic
ratio
for the removal of sodium from the catalyst precursor.
[0061] When necessary, a second ion-exchange step can be used to further
lower the amount of sodium. The ion-exchanged particles are generally again
washed, e.g., between ambient temperature and 100 C. The zeolite portion of
the
catalyst, after ion-exchange, and washing, typically contains less than 0.4
percent
alkali metal, based on the weight of the catalyst. It is believed that a small
amount
of aluminum from the aluminum sulfate hydrate is incorporated into the
catalyst
during ion exchange.
[0062] While not wishing to be bound by any theory or model, the presence of
phosphate in the slurry is believed to affect the matrix microporosity. It is
believed
that the phosphate interacts with aluminum species in the slurry to make
aluminum
phosphate. Since aluminum phosphate has an isoelectric point similar to
silica's,
particles in the slurry appear to have a similar charge and agglomeration is
avoided.
Agglomeration, it is believed, would lead to a degradation in the
microporosity
characteristics of the catalyst. Other factors, which are believed to lead to
the
unusual pore distribution of the catalysts of the invention, relate to the use
of
sodium silicate in the binder system in combination with urea as the directing
compound.
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-22-
(III) The Catalytic Cracking Process
[0063] In yet another embodiment the invention relates to a catalytic cracking
process. The catalytic cracking process may be carried out in a fixed bed,
moving
bed, ebullated bed, slurry, transfer line (dispersed phase) or fluidized bed
operation.
Suitable hydrocarbon feedstocks (i.e., the primary feed) for the catalytic
cracking
process described herein include natural and synthetic hydrocarbonaceous oils
boiling in the range of 430 F to 1050 F (221.11 C to 565.56 C), such as gas
oil;
heavy hydrocarbonaceous oils comprising materials boiling above 1050 F
(565.56 C); heavy and reduced petroleum crude oil; petroleum atmospheric
distillation bottoms; petroleum vacuum distillation bottoms; pitch, asphalt,
bitumen, other heavy hydrocarbon residues; tar sand oils; shale oil; liquid
products
derived from coal liquefaction processes, naphtha, and mixtures thereof.
[0064] In an embodiment, the catalytic cracking process is performed in one or
more FCC process units. Each unit comprises a reaction zone, usually a riser
reaction zone, a stripping zone, a catalyst regeneration zone, and at least
one
separation zone. In an FCC process, the feedstock is conducted to and injected
into
the reaction zone wherein the primary feed contacts a flowing source of hot,
regenerated catalyst. The hot catalyst vaporizes and cracks the feed at a
temperature from 450 C to 650 C, preferably from 500 C to 600 C. The cracking
reaction deposits carbonaceous hydrocarbons, or coke, on the catalyst, thereby
deactivating the catalyst. The cracked products may be separated from the
coked
catalyst and a portion of the cracked products may be conducted to a
separation
zone such as a fractionator. Fractions such as a naphtha fraction can be
separated
from the cracked products in the separation zone and conducted away from the
process.
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-23-
[0065] FCC process conditions in the riser reactor's reaction zone include
temperatures from 450 C to 700 C, hydrocarbon partial pressures from 10 to 40
psia (69 to 276 kPa), preferably from 20 to 35 psia (138 to 241 kPa); and a
catalyst
to primary feed (wt/wt) ratio from 3 to 100, where catalyst weight is total
weight of
the catalyst composition. The total pressure is from atmospheric to 45 psig
(411
kPa). Though not required, it is also preferred that steam be concurrently
introduced with the feedstock into the reaction zone, with the steam
comprising up
to 50 wt.%, preferably 2 to 10 wt.% of the primary feed. Also, it is preferred
that
the feedstock's residence time in the reaction zone be less than 20 seconds,
preferably from 0.1 to 20 seconds, and more preferably from 1 to 5 seconds.
[0066] The present process and catalyst provides both economic and technical
advantages over state of the art commercial FCC catalysts. There are three
major
binders currently used to make FCC catalysts. These are the acidified silica
sol
binder, aluminum chlorhydrol, and peptized alumina. These three binding
systems
are acidic, and this acidity can adversely affect the physical properties of
acid,
susceptible active materials. Previous mesoporous catalysts based on silica
used
Ludox as the silica source. This is a more expensive ingredient than basic
sodium silicate.
[0067] In the present process, sodium silicate (a basic system) can
successfully
bind FCC catalysts. Furthermore, the incorporation of urea with sodium
silicate
into the spray drier feed produces catalysts which are more mesoporous and
more
selective than catalysts which have not been so treated. Neutralization of the
sodium contained in the sodium silicate is done with the ammonium salt of an
acid
which is stronger than silicic acid. The cheapest acid source is sulfuric
acid.
Neutralization with sulfuric acid requires careful control or gellation will
occur
with consequent loss in catalyst strength and integrity. Carbonic acid is a
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-24-
somewhat stronger acid than silicic acid and as such can also be used to
neutralize
sodium silicate. However, neutralization with carbonic acid leads to gellation
because the carbonic acid forms a silica sol at a pH at which gellation
readily
occurs. Urea is the anhydride of diammonium carbonate and hydrolyzes slowly to
form ammonia and sodium carbonate in basic solutions. Incorporation of urea in
the binder system allows reaction to take place after drying so that a silica
gel does
not form prior to drying leading to weaker products. Incorporation of urea
also
may also assist in the formation of the present mesoporous pore structures,
which
improve product selectivity. Alkali metal salts of phosphate seem to be
especially
efficacious in forming pore structure, which leads to improved selectivity to
products other than coke. Comparative Examples 1, 2, and 3 following show that
materials made with ammonium phosphate fail to produce beneficial product
selectivities. Figures 4 and 5 show the selectivity to products other than
coke on
conversion to 221 C- is poorer for materials made with ammonium phosphate,
viz.
materials labeled "1.11 ", "2.11 ", "3.11". 'I 1.12", "2.12", and "3.12", than
for those
made with alkali metal phosphates, viz. materials labeled "4.11" and "5.11 ".
Figure
8 shows the porosity in the region below 50 Angstroms is larger for the
materials
made with ammonium phosphate.
[0068] The following examples are presented to illustrate the invention and
should not be considered limiting in any way.
CA 02569123 2010-06-04
-25-
Comparative Example 1: No Urea, Diammonium Hydrogen Phosphate
[0069] This is a comparative (base case) example using sodium silicate as the
silica source in the binder but without adding urea during catalyst
preparation.
[0070] To 3000 g of water in a 2-gallon (7570 m) plastic bucket was added:
1319.3 g of zeolite Z-14G NaUSY, 12.5 g diammonium hydrogen phosphate, 728 g
TM TM
of Hydrite UF kaolin clay, and 560 g of Spacerite S-11 Gibbsite. The resulting
mixture was stirred 30 minutes with a Cowles mixer.
[0071] 1742.2 g of "N" brand Sodium Silicate was diluted with 1700 g of
deionized water and added to the water/zeolite/diammonium hydrogen
phosphate/clay slurry and then colloid milled. The pH of colloid-milled slurry
was
10.8 at 22 C. The viscosity of the slurry was 188 cP (188000 Pass) at 100 rpm,
and
the density of the slurry was 1.322 g/cc.
[0072] The slurry was spray dried in a Bowen M#1 Tower Spray Drier [rated at
7/35 kg/hr with an airflow of 250 cfin (7.08 m3/min.) at 80 C] with an exit
temperature of 121 C. 787 g of solids with 50% above 74.4 microns were
collected
from the Main Tower Pot. The properties of the slurry and product are
summarized
in Table 1.1.
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
26
O
N \p N M
O
00
~ O l~ M d'
C 00 0 00
O N O ~ --~
cd
I r O
00
0
0 0
z
~, N d' N M
4; N N I M
O O
00 r- 00 t~
heio0 d
c 0 0 0
j U
N d N N
- dN N N
O M t~ l~
c) kn W)
kn p d N N -1
N P-~
Q ~ O
o rrV
'd
rx ~'
O b
coo
0 du
H Nzx~~
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-27-
[0073] To ion-exchange this material, the following solution of ammonium
sulfate and aluminum sulfate was used:
360 g A12(S04)3'16H20 was dissolved in 2040 g DI water
106.2 g (NH4)2SO4 was dissolved in 2294 g DI water.
[0074] To 120 g of the spray drier product was added 200 g of the
A12(S04)3'16H20 solution and 200 g of the (NH4)2SO4 solution. This mixture was
shaken at 80 C in a shaker bath at 260 rpm for 1 hour, cooled and then washed
3
times with 500 g of DI (deionized) water at 80 C on the filter and air dried.
The
product from this first ion exchange, Example 1.1, was analyzed. The remaining
sample was added to 200 g of the dilute aluminum sulfate solution and then 200
g
of the dilute ammonium sulfate solution was added. This was shaken for 1 hour
at
80 C at 260 rpm, washed 3 times with 500 g of deionized water, dried at 120 C
for
2 hours and calcined to give Example 1.2. Table 1.2 contains the analyses for
these
samples.
TABLE 1.2
Example 1.1 Example 1.2
Si02 (wt%) 62.04 63.39
A1203 wt% 36.99 36.29
F Na (wt%) 0.72 0.24
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-28-
Comparative Example 2: Urea, Diammonium Hydrogen Phosphate
[0075] This is a further comparative example. Unlike standard commercial FCC
catalysts, the preparation takes place in a basic environment. It is assumed
that 1
mole of urea neutralizes 2 moles of sodium by decomposing to form ammonium
silicate and sodium carbonate.
[0076] To 2500 g of water in a 2-gallon (7570 m3) plastic bucket was added:
1319.3 g of zeolite Z-14G NaUSY, 12.5 g diammonium hydrogen phosphate, 728 g
of Hydrite UF kaolin clay, and 560 g of Spacerite S-11 Gibbsite. The resulting
mixture was stirred 30 minutes with a Cowles mixer.
[0077] 1742.2 g of "N" brand Sodium Silicate was diluted with 2275 g of
deionized water and added to the water/zeolite/diammonium hydrogen
phosphate/clay slurry and then colloid milled. The pH of colloid-milled slurry
was
11.0 at 21 C. The viscosity of the slurry was 247 cP (247000 Pass) at 100
rpm.
The slurry was spray dried in a Bowen #1 Tower Spray Drier [rated at 7/35
kg/hr
with an airflow of 250 cfm (7.08 m3/min.) at 80 C] with an exit temperature of
145 C.
[0078] 1436 g of solids was collected from the Main Tower Pot. The
composition of the slurry and product are summarized in Table 2.1.
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
29
01 Vn tfi N r-+
N N ,-~ O
'd O lp lp M N
N ~t O l~
cl~ 00 O r M O d
N O O '- 00
N O
MO 00 O O
O O O
O
z
N d' N M O
N N d.-+ M
O O O O
t - -
0 0 N 0o O
O
ri Urs1
a
N --~ d l~ ~O l~ M O
E~ N N N - L C) "o N
M
fl Ll
p 0 d0 N N ,v- 00
0 'o
P-4 C,
'd b 0 b
O '-' 0
A 0 O 0
0
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-30-
[0079] 52.4 g H2SO4 was dissolved in 1548 g of DI water to make a dilute
sulfuric acid solution.
[0080] 270 g A12(SO4)3'16H2O was dissolved in 1530 g of DI water to make a
dilute aluminum sulfate solution with 4.5 X 10-4 moles A13+/g solution.
[0081] 70.8 g (NH4)2SO4 was dissolved in 1529.2 g of DI water to make a dilute
ammonium sulfate solution with 6.7 X 10-4 moles NH4+/g solution.
[0082] To 100 g of spray drier product was added solutions according to Table
2.2.
TABLE 2.2
Example 2.1 Example 2.2 Example 2.3 Example 2.4 Example 2.5
200 g dilute 100 g dilute 200 g dilute 100 g dilute 200 g dilute
sulfuric acid sulfuric acid aluminum aluminum ammonium
solution solution / sulfate sulfate sulfate
100 g dilute solution solution / solution
aluminum 100 g dilute
sulfate ammonium
solution sulfate
solution
[0083] These were shaken at 80 C for 1 hour; filtered, washed 3 times with 400
g of DI water at 80 C for 0.5 hours on the shaker bath and warmed to 120 C at
1 C/minute and dried at 120 C for 6 hours.
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-31-
[0084] The elemental analyses on these samples are contained in Table 2.3.
TABLE 2.3
Example Example Example Example Example
2.1 2.2 2.3 2.4 2.5
Si02 (wt%) 64.82 63.67 63.22 62.00 63.01
A1203 (wt%) 33.88 35.34 36.00 37.35 34.84
Na (wt%) 0.96 0.74 0.57 0.49 1.59
[0085] Note that the Al203/SiO2 weight ratio is 0.60 with example 2.4, the
NH4+/A13+ preparation, and only 0.55 with example 2.5, the NH4+preparation.
This
indicates that the system is incorporating roughly 0.60/0.55 -1 = 9.1% more
alumina or that.091 x 35 = 3.2 % alumina (1.6 g alumina) has been added to the
original weight of the catalyst. This alumina comes from the 100/1800 x 270 x
102/666 = 2.3 g in the exchange solution.
[0086] To further demonstrate the specific nature of this interaction, the
spray
dried product of Example 2 was exchanged in a second series in which only the
dilute aluminum sulfate solution and the dilute ammonium sulfate solution were
used as outlined in Table 2.4. To 50 g of spray drier product was added
solutions
according to Table 2.4:
TABLE 2.4
Example 2.6 Example 2.7 Example 2.8
150 g dilute aluminum 100 g dilute aluminum 50 g dilute aluminum
sulfate solution / sulfate solution / sulfate solution /
50 g dilute ammonium 100 g dilute ammonium 150 g dilute ammonium
sulfate solution sulfate solution sulfate solution
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-32-
[0087] These were shaken at 80 C for 1 hour; filtered, washed 3 times with 400
g of DI water at 80 C for 0.5 hours on the shaker bath and warmed to 120 C at
1 C/minute and dried at 120 C for 6 hours. Table 2.5 clearly shows that for
these
sodium silicate bound catalysts, sodium removal is most efficient with a
combination of an aluminum salt and an ammonium salt in a specific ratio,
namely,
2 moles of Al/3 moles of ammonia.
TABLE 2.5
Example 2.6 Example 2.7 Example 2.8
Si02 (wt%) 63.29 62.24 61.89
A1203 (wt%) 36.04 37.14 37.11
Na (wt%) 0.50 0.46 0.74
[0088] To demonstrate that successive ion exchanges complete the removal of
sodium from the catalyst, 120 g of the spray drier product of Example 2 was
added
to 200 g of the dilute aluminum sulfate solution and then 200 g of the dilute
ammonium sulfate solution was added. This was shaken for 1 hour at 80 C at 260
rpm, washed 3imtes with 500 g of deionized water, dried at 120 C for 4 hours
to
give Example 2.9. The remaining sample was added to 200 g of the dilute
aluminum sulfate solution and then 200 g of the dilute ammonium sulfate
solution
was added. This was shaken for 1 hour at 80 C at 260 rpm, washed 3 times with
500 g of deionized water, dried at 120 C for 2 hours and calcined to give
Example
2.10. The volatile free analyses for examples 1.9 and 1.10 are contained in
Table
2.6.
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
- 33 -
TABLE 2.6
Example 2.9 Example 2.10
Si02 wt% 61.55 62.68
A1203 (wt%) 37.54 37.01
Na (wt%) 0.68 0.23
Comparative Example 3: 2X Urea, Diammonium Hydrogen Phosphate
[0089] This comparative example illustrates the removal of sodium by ion
exchange from the product from the spray drier which is prepared as follows.
To
3000 g of water in a 2-gallon (7570 m) plastic bucket was added 1319.3 g of
zeolite NaUSY, 12.5 diammonium hydrogen phosphate, 728 g of Hydrite UF
kaolin clay, 403.5 g of urea 560 g of Spacerite S-11 Gibbsite. The resulting
mixture was stirred 30 minutes with a Cowles mixer.
[0090] 1742.2 g of "N" brand Sodium Silicate was diluted with 1700 g of
deionized water and added to the water/zeolite/diammonium hydrogen
phosphate/clay slurry and then colloid milled. The pH of colloid-milled slurry
was
10.79 at 17 C. The viscosity of the slurry was 199 cP (199000 Pa's) at 100
rpm,
and the density of the slurry was 1.293 g/cc.
[0091] The slurry as spray dried in a Bowen #1 Tower Spray Drier [rated at
7/35 kg/hr with an airflow of 250 cfm (7.08 m3/min.) at 80 C] with an exit
temperature of 160 C. After drying, collection and weighing of the solids from
the
bottom of the main tower and the solids from the bottom of the cyclone yielded
1129 g of solids with 50% above 66.0 microns. The nominal slurry and product
compositions are shown in Table 3.1.
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
34
O r+ O O l~
^ 01v~~v~~
N N N O
O I'0 \0 M N
O
JD
O l~ M d'
rn 00 O 00
y N O ~ ~
N O
z
o oo O
z
N d' N M O
r-I O p p O
00 00
00 kr) 110
N 00
O O O
U cD
110 ~o It O d d-
N r+ d: r- O
01 N 00 O~ M
kr) \0
d N
M N N Vf tr)
O
0 N N tf) O
0
y O
Q ~ N
rc)
rig ~
H NZxco
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-35-
[0092] 120 g of the spray drier product of Example 3 was added to 200 g of the
dilute aluminum sulfate solution and then 200 g of the dilute ammonium sulfate
solution was added. This was shaken for 1 hour at 80 C at 260 rpm, washed 3
times
with 500 g of deionized water, dried at 120 C for 4 hours to give Example 3.1.
The
remaining sample was added to 200 g of the dilute aluminum sulfate solution
and
then 200 g of the dilute ammonium sulfate solution was added. This was shaken
for
1 hour at 80 C at 260 rpm, washed 3 times with 500 g of deionized water, dried
at
120 C for 2 hours and calcined to, give Example 3.2. The'volatile free
analyses for
examples 3.1 and 3.2 are contained in Table 3.2.
TABLE 3.2
Example 3.1 Example 3.2
Si02 (wt.%) 61.46 62.04
A1203 (wt%) 37.73 37.69
Na (wt%) 0.60 0.20
Example 4: Urea, Disodium Hydrogen Phosphate
[0093] This example is a catalyst of this invention. In its preparation, an
alkali
phosphate salt, urea, and sodium silicate were employed to make a spray dried
product which was ion-exchanged using the optimum mix of aluminum and
ammonium salts to make a finished catalyst. When this catalyst was then
deactivated using steam, it had a pore structure according to the catalyst and
process of the invention. To 3000 g of water in a 2-gallon (7570 m) plastic
bucket
was added: 13.4 g disodium hydrogen phosphate, 200 g of urea, 373 g Alcoa C-33
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-36-
gibbsite, 1319.3 g of zeolite NaUSY, 874 g of Hydrite UF kaolin clay. The
resulting mixture was stirred with a Cowles mixer until it flowed smoothly.
[0094] 1742.2 g of "N" brand Sodium Silicate was diluted with 2400 g of
deionized water and to this was added the water/disodium hydrogenphosphate/
urea/gibbsite/zeolite/clay slurry. This slurry was then colloid milled twice.
The pH
of colloid-milled slurry was 10.8 at 22 C. The viscosity of the slurry was 188
cP
(188000 Pass) at 100 rpm, and the density of the slurry was 1.288 g/cc. The
slurry
was spray dried in a Bowen #1 Tower Spray Drier [rated at 7/35 kg/hr with an
airflow of 250 cfm (7.08 m3/min.) at 80 C] with an exit temperature of 150 C.
939
g of solids were collected from the Main Tower Pot.
[0095] 120 g of the spray drier product of Example 4 was added to 200 g of the
dilute aluminum sulfate solution and then 200 g of the dilute ammonium sulfate
solution was added. This was shaken for 1 hour at 80 C at 260 rpm, washed
3times
with 200 g of deionized water. The wet cake was added to 200 g of the dilute
aluminum sulfate solution and then 200 g of the dilute ammonium sulfate
solution
was added. This was shaken for 1 hour at 80 C at 260 rpm, washed 3 times with
200 g of deionized water, dried at 150 C for 1 hour and calcined at 760 C for
1
hour to give Example 4.1. Table 4.1 contains the analyses:
TABLE 4.1
Example 4.1
Si02 (wt.%) 61.22
A1203 (wt%) 38.38
Na (wt%) 0.29
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-37-
Example 5: Urea, Tetrasodium Pyrophosphate
[0096] This example is a catalyst according to the catalyst and process of the
invention. To 3000 g of water in a 2-gallon (7570 m) plastic bucket was added:
21.0 g tetrasodium pyrophosphate, 200 g of urea, 373 g Alcoa C-33 gibbsite,
1319.3 g of zeolite NaUSY, 874 g of Hydrite UF kaolin clay. The resulting
mixture was stirred with a Cowles mixer until it flowed smoothly.
[0097] 1742.2 g of "N" brand Sodium Silicate was diluted with 2400 g of
deionized water and to this was added the water/ tetrasodium pyrophosphate/
urea/gibbsite/zeolite/clay slurry. This slurry was then colloid milled twice.
The pH
of colloid-milled slurry was 11.0 at 22 C. The viscosity of the slurry was 189
cP
(189000 Pass) at 100 rpm, and the density of the slurry was 1.26 g/cc. The
slurry
as spray dried in a Bowen #1 Tower Spray Drier [rated at 7/35 kg/hr with an
airflow of 250 cfm (7.08 m3/min.) at 80 C] with an exit temperature of 150 C.
980
g of solids were collected from the Main Tower Pot.
[0098] 120 g of the spray drier product of Example 5 was added to 200 g of the
dilute aluminum sulfate solution and then 200 g of the dilute ammonium sulfate
solution was added. This was shaken for 1 hour at 80 C at 260 rpm, and washed
3
times with 200 g of deionized water. The wet cake was added to 200 g of the
dilute
aluminum sulfate solution and then 200 g of the dilute ammonium sulfate
solution
was added,. This was shaken for 1 hour at 80 C at 260 rpm, washed 3 times with
200 g of deionized water, dried at 150 C for 1 hour and calcined at 760 C for
1
hour to give Example 5.1. Table 5.1 contains the analyses:
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-38-
TABLE 5.1
Example 5.1
Si02 (wt%) 62.28
A1203 (wt%) 37.32
Na (wt%) 0.29
Example 6
[00991 Examples 1, 2, 3, 4, and 5 were calcined at 760 C. Examples 1, 2, and 3
were then steamed at a temperature of 760 C for 16 hours to give Examples
1.11,
2.11, and 3.11 in Table 6, then at a temperature of 788 C for 16 hours to
produce
examples 1.12, 2.12, and 3.12'respectively, in Table 6. Examples 4 and 5 were
steamed at 788 C, 16 hours to produce the catalysts 4.11 and 5.11 for
evaluation in
an ACE FCC. An ACE unit is a commercially available unit made for FCC
laboratory evaluations and is manufactured by Xytel Co., Elk Grove Village,
IL.
The properties of the steamed catalysts are set forth in Table 6.
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
39
N
~~ \ N N N 00 N z O O O O O
z
N 00 N
00
9 M M m M
z
cd o N M 00 to N
O v
z
a)
110 00
CIS u
a dN- N ~O m m
a)
cd
LP
cd bap O 00 d: O 00 01 l0 M
O O o kr)
O
a)
N
a)
cH
~bA M \O C O O M N O
r/] N O N d 00 00
y N N N ,-~ ,~ .--+
O
H
cd
W W W W W W W W
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-40-
[0100] Examples 1.11, 2.11, 3.11, 1.12, 2.12, and 3.12 as comparison catalysts
and 4.11 and 5.11 of this invention were then evaluated by injecting a vacuum
gas
oil with the following physical properties over the catalyst in a fixed
fluidized bed
reactor whose operations are described in the open literature. The conditions
under
which the unit operated follow:
REACTOR INIT TEMP, F ( C) 1030 (554.44)
REACTOR MIN TEMP, F ( C) 1010 (543.33)
FLUID BED REGEN TEMP, F ( C) 1250 (676.67)
CAT STRIP TIME, SEC 330-610s
LIQ STRIP TIME, SEC 350-1050 s
N2 DURING RXN TOP FEED, SCCM 20
N2 DURING RXN TOP FLUID, SCCM 20
N2 DURING RXN BTM FLUID, SCCM 100
N2 DURING RXN TOT PURGE, SCCM 41
N2 DURING REM LIQ STRIP, SCCM 100
CAT TO OIL RATIO, WT/WT 3.0-9.0
CATALYST CHARGE WT, GMS 9.0
OIL CHARGE WT, GMS 1-3
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-41-
[0101] The feedstock used was a Gulf Coast vacuum gas oil having the
following properties:
GRAVITY, API 23.9
CARBON 85.81
HYDROGEN 12.55
NITROGEN, SYRINGE INLET (ppm) 909
N (basic) wppm 313
SULFUR IN OILS 0.968
Ni (wppm) 0.42
V (wppm) 0.37
CARBON. RESIDUE (MICRO)! 0.22
TEMP. @ 5.0 WT%, F ( C) 658.4 (348)
TEMP. @ 10.0 WT%, OF ( C) 701.7 (372.06)
TEMP. @ 20.0 WT%, F ( C) 755.7 (402.06)
TEMP. @ 30.0 WT%, F ( C) 795.7 (424.28)
TEMP. @ 40.0 WT%, OF ( C) 829 (442.78)
TEMP. @ 50.0 WT%, OF ( C) 860.1 (460.06)
TEMP. @ 60.0 WT%, OF ( C) 892.9 (478.28)
TEMP. @ 70.0 WT%, OF ( C) 931.2 (499.56)
TEMP. @ 80.0 WT%, OF ( C) 970.7 (521.5)
TEMP. @ 90.0 WT%, F ( C) 1014 (545.56)
TEMP. @ 95.0 WT%, OF ( C) 1033.1 (556.17)
SATS 55.4-56.56
1 RING AROM 20.4-20.9
2 RING AROM 11.7-11.1
3 RING AROM 5.7-5.5
4 RING AROM 3.1-3.1
POLARS 3.8-2.9
SATS (UV CORES) 0.04
1 RING AROM (UV CORES) 3.3-3.0
2 RING AROM (UV CORES) 4.4-3.7
3 RING AROM (UV CORES) 3.4-2.9
4 RING AROM (UV CORES) 2.1-2.2
POLARS (UV CORES) 1.9-1.3
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-42-
[0102] Consistent with the observed differences in the pore size distribution
of
the steamed, artificially deactivated catalysts, the base case and the two
examples
of this invention show significantly different selectivities for coke in the
cracking
of a Vacuum Gas Oil in a small captive fixed fluidized bed unit (ACE). Figure
4a
is a graph of coke make vs. 430 F- (221 C-) + coke make for base case
comparative
catalysts vs. catalysts of the invention. Figure 4b shows that the catalysts
of this
invention are more coke selective than the average coke selectivity seen from
commercially available state of the art catalysts.
[0103] Figure 4a shows that the catalysts of this invention (Examples 4 and 5)
differ from the similar comparative catalysts in that they produce less coke
for a
given conversion level than other catalysts made with ammonium phosphates. In
Figure 5, the same data are normalized for conversion which removes the slopes
for
coke vs. conversion seen in Figure 4.
[0104] If light hydrocarbon moieties (such as methyl groups) associated with
heavy polynuclear aromatics in coke are cracked off as a result high
temperature,
coke yields can fall while light gas yields rise. Since either coke or light
gas can
constrain unit operations, trading off coke for light gas is not a clear win.
With the
catalysts of this invention, it appears that both coke and light gas are lower
than the
base case comparative catalysts as shown in Figure 6a which is a plot of dry
gas
make vs. 430 F- (221 C-) + coke make. Figure 6b shows that the catalysts of
this
invention make less dry gas than commercially available state of the art
catalysts.
[0105] Figure 7a shows that the catalysts of this invention make more valuable
propene than do similar base case comparative catalysts when they achieve the
same conversion. Figure 7b shows that the catalysts of this invention make
more
propene than commercially available state of the art catalysts.
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-43-
[01061 Figure 8a shows that the catalysts of this invention (Examples 4 and 5)
differ from the similar base case comparative catalysts in that :
1. The maximum in the dV/dD vs. pore diameter plot occurs at pore
diameter greater than 50 Angstroms for the catalysts of this invention.
2. The tangents to the dV/dD vs. pore diameter curves below 50
Angstroms for steamed catalysts of this invention are positive and
intercept the pore diameter axis (dV/dD = 0) at greater than 10
Angstroms.
3. The catalysts of this invention are made with sodium silicate, urea,
and a sodium salt of phosphate as well as a faujasite and and alumina.
[0107] Figure 8b shows that the commercially available state of the art
catalysts
have a local maximum in the dV/dD plot below 60 Angstroms (50 Angstroms)
regardless of the severity of the steaming, and that above 60 Angstroms, there
may
be more than one maximum in the 60 to 200 Angstrom range.
[0108] Figure 8c is a plot of dV/dD vs. pore diameter showing that
commercially available state of the art catalysts when blended with other
catalysts
from the same manufacturer have a local maximum in the dV/dD plot below 60
Angstroms regardless of the severity of the steaming.
[0109] Figure 8d is a plot of dV/dD vs. pore diameter showing that catalysts
of
this invention have a maximum in the dV/dD plot above 60 Angstroms and below
80 Angstrom unlike the commercially available state of the art catalysts which
have
CA 02569123 2006-11-29
WO 2005/123252 PCT/US2005/018019
-44-
a local maximum in the dV/dD plot below 60 Angstroms regardless of the
severity
of the steaming and may have one or more maxima above 80 Angstroms.