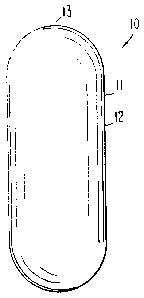Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02575042 2007-02-12
67696-219D
1
EFFECTIVE DOSAGE FORM FOR ANTIEPILEPTIC DRUGS
FiFLD OF THE INVENTION
This invention pertains to novel and unobvious dosage forms for
administering a drug effective in the therapy of the epilepsies. The
invention concerns also a pharmaceutical composition comprising an
s antiepileptic drug and a pharmaceutical carrier. The invention relates
further to the manufacture of a dosage form for administering a drug
useful for treating epilepsies. Additionally the invention pertains to a
method for producing antiepifeptic therapy in a patient in need of
antiepileptic therapy.
~o BA(',~KGROUND OF THE INVENTION
The term epiiepsies is a collective designation far a group of central
nervous system disorders having in common the repeated occurrence of
sudden and transitory episodes of abnormal phenomena of motor,
convuision, sensory, autonomic, or psychic origin. The seizures are
~s nearly always correlated with abnormal and excessive discharges in the
brain which can be recorded by an electroencephalogram.
Epilepsy afflicts millions of people worldwide, and the disease is more
common in children than in adults. For the purposes of drug treatment,
it is useful to classify patients according to the type of seizure the patient
zo experiences. The generally accepted classification of epileptic seizures
comprises partial seizures consisting of focal and local seizures, and
generalized seizures consisting of convulsive or nonconvulsive seizures.
Partial seizures are classified further as simple partial seizures, complex
partial seizures, and partial seizures secondarily generalized. Generalized
zs seizures are classified further as absence seizures, atypical absence
CA 02575042 2007-02-12
v o ~~~ ~~~«,; Pcr z-s~~~,ns~m
2
seizures, myoclonia seizures, cfonic seizures, tonic seizures, tonic-clonic
and atonic seizures. The epilepsies are presented in The Pharmacological
Basis of Therapeutics, 8th Ed, Chapter 19 ( 19901, Editors Giiman and
Rall, Pergamon Press.
s Antiepileptic drugs are available for treating epilepsies, as disclosed in
Pharmaceutical SciP~,nces, Remington's, 18th Ed., pp 1072-1081 ( 1990?
published by Mack Publishing Co., and while the drugs are useful for
treating the epilepsies, there are many shortcomings associated with
these drugs. For instance, the drugs often are poorly soluble in aqueous
~ o and biological fluids, which property makes it difficult to both provide
and
dispense the drugs from a dosage form in a known dose over and
extended time. The drugs also can be extremely hygroscopic and they
may liquify rapidly, which physical-chemical characteristic dictates
against their delivery from a dosage form at a controlled rate over a
~s prolonged period of time. Then too, many drugs exhibit a short half-life
that can lead to fluctuations in blood antiepileptic drug levels. These
properties can interfere with manufacture and the release of the drugs
from dosage form and from pharmaceutical compositions; and these
shortcomings are serious drawbacks in the management of epilepsies.
zo Prior to this invention, the prior art administered an antiepifeptic drug
in conventional forms like a standard nonrate tablet or a common
dose-dumping capsule at repetitive dosing intervals. The prior art modes
of therapy Isads to a drug concentration in the blood during the dosing
interval, followed by a decrease in drug concentration as a result of drug
zs absorption, distribution, metabolism, and elimination. The concentration
difference in dosing intervals is related to the presence and to the
absence of administered drug, which is a major disadvantage associated
with conventional dosage forms. Conventional dosage forms and their
mode of operation are discussed in Pharmac~utiear~ciences, Remington,
CA 02575042 2007-02-12
WO 9~:'2966~ Pcr:, ~E,~-t
3
i 8th Ed., pp i 676-1686 ( 1990), Mack Publishing Co.;
The Pharmacological and Clinical Phari~nacokinetics, 3rd Ed., pp 1-28
1984), published by Lea & Febiger, Philadelphia, PA; and in United
States Patent Nos. 3,598, 7 22 and 2,598, i 23, both issued to Zaffaroni.
The above presentation dictates of the critical need for a dosage form
that overcomes the shortcomings of conventional dosage forms, including
tablets, capsules, elixirs and suspensions. These conventional dosage
forms produce peaks and valley patterns, and they do not provide for
dosage-regulated drug therapy over an extended period of time. The
~ o drug, as delivered by the prior art is dosed twice or thrice a day, which
does not lend itself to controlled and sustained therapy. This prior art
pattern of drug administration speaks of the need for a dosage form that
can administer the drug in a rate-controlled pattern over an extended time
to provide constant therapy and thereby eliminate the peaks and valleys
~ s and eliminate the need for multiple uncontrolled dosing of the drug.
The prior art provided controlled-release dosage forms that can
administer a drug continuously over time for controlled-rate therapy,
as in, for example, United States Patent No. 4,327,725 issued to Cortese
and Theeuwes, and in United States Patent Nos. 4,612,008; 4,765,989;
Zo and 4,783,337 issued to Wong, Barclay, Deters and Theeuwes.
The dosage forms disclosed in these patents provide a controlled-rate
drug delivery over an extended time to provide constant drug therapy and
thereby eliminate the need for multiple dosing of the drug. These dosage
forms can deliver many drugs for their intended therapy, but there are
a5 certain drugs that are not readily manufactured and delivered from
dosage forms. For example, phenytoin sodium converts to practically
insoluble phenytoin in the gastrointestinal pH range of 1 to 8 and the
release of unprotected drug in this range is incomplete and this abstracts
from acceptable therapy.
CA 02575042 2007-02-12
wo ~o "~«.~- PcTn:s~~~;n~c3a
4
It is immediately apparent, in the light of the above presentation, that
an urgent need exists for a dosage form endowed with controlled-release
delivery for the administration of an antiepileptic drug for antiepiieptic
therapy. The need exists for this dosage form for delivering an
antiepileptic drug in a controlled-sustained dose in a therapeutic
antiepileptic range and for simultaneously providing extended therapy.
!t will be appreciated by those versed in the dispensing antiepileptic drug
art, that such a dosage form that can administer an antiepileptic drug in a
controlled-rate dose over time, and it would be a major advancement in
~o the therapy of the epilepsies.
Accordingly, in view of the above presentation, it is an immediate
object of this invention to provide a dosage form for delivering an
antiepileptic drug for treating epilepsies that overcomes the shortcomings
~ s known to the prior art.
Another object of the present invention is to provide a dosage form
that delivers an antiepileptic drug in a continuous-release dose over time.
Another object of the present invention is to provide a dosage form for
administering an antiepileptic drug as a controlled-rate in a therapeutic-
Zo dose over an extended period of time.
Another object of the present invention is to provide a dosage form
that delivers an antiepileptic drug in the gastrointestinal tract, by a
process selected from osmotic, diffusion, bioerosion, or ion-exchanged
kinetics.
CA 02575042 2007-02-12
v0 9~%2966' PCT 16.i-t
Another object of the invention is to provide an antiepileptic drug
formulation in a controlled-continuous-release dose to a patient for
maintaining an essentially constant antiepileptic level in the blood as a
function of a prolonged-release system.
s Another object of the invention is to provide an antiepileptic
continuous-release dosage form that provides a slow-release of an
antiepileptic formulation over an extended time.
Another object of the invention is to provide a dosage form that
substantially reduces and/or substantially eliminates the unwanted
~ o influences of a gastrointestinal environment on the delivery of an
antiepileptic formulation in the gastrointestinal tract.
Another object of the present invention is to provide an improvement
in a dosage form that administers an antiepileptic drug formulation,
wherein the improvement comprises delivering the antiepileptic drug
~ s formulation in a continuous-release dose form the dosage form for
predictable and improved therapy.
Another object of the invention is to provide a dosage form that
delivers an antiepileptic drug formulation orally to a patient in need of
antiepileptic therapy.
zo Another object of the present invention is to provide both a fast-
release prompt delivery and a slow-release extended antiepileptic drug
formulation from a single dosage form comprising a first and a second
antiepileptic drug formulation for administering to a patient experiencing
epilepsies.
CA 02575042 2007-02-12
W'O')~':')6ti~ PCT'L~S9~~111G.i1
6
Another object of the present invention is to provide a method for the
prevention and for the control of epileptic seizures.
Another object of the present invention is to provide a method of add-
on antiepifeptic drug therapy for patients taking other epilepsy
s medication, thereby providing adjunctive therapy in epilepsy patients.
Another object of the present invention is to provide a dosage form
that delivers by a process selected from the group consisting of osmotic,
diffusion, bioerosion and ion-exchange a therapeutic dose of an
antiepileptic drug formulation over an extended time for dosage form
~o governed antiepileptic therapy.
Another object of the invention is to provide a method for
administering an antiepileptic drug by orally administering the antiepileptic
drug in a dose per unit time over an extended time to a patient in need
of antiepileptic therapy.
~s Another object of the present invention is to provide a method for
administering an antiepileptic drug formulation in a therapeutic range
while simultaneousty-avoiding a toxic range of the antiepileptic drug
formulation.
Another object of the invention is to provide a method for
zo administering an antiepileptic drug formulation by administering a dosage
form that administers by osmotic, diffusion, bioerosion or ion-exchange
an antiepileptic drug formulation as a dosage form governed rate over an
extended time.
CA 02575042 2007-02-12
67696-219D
6a
According to one aspect of the present invention,
there is provided a pharmaceutical composition for delivery
from an osmotic dosage form, adapted for oral
administration, of an antiepileptic drug in a
gastrointestinal tract having a pH of 1 to 8 of a patient,
said pharmaceutical composition comprising 0.5 wt% to 90 wt%
of an antiepileptic drug, and a pharmaceutically acceptable
carrier comprising 10 wt% to 75 wt% of a
carboxymethylcellulose and 0.1 wt% to 25 wt% of a
polyvinylpyrrolidone, said pharmaceutical composition
additionally comprising means for protecting the
antiepileptic drug from the gastrointestinal tract pH of
1 to 8, whereby upon release of a dose of antiepileptic drug
to the patient, an antiepileptic level of the drug is
provided in blood of the patient, as a function of the
osmotic dosage form, which lessens the incidence of
epilepsy.
According to another aspect of the present
invention, there is provided use of a composition
comprising: 0.5 wt% to 90 wt% of an antiepileptic drug
selected from the group consisting of phenytoin,
mephenytoin, phenobarbital, primidone, carbamazepine,
ethosuximide, methsuximide, phensuximide, trimethadione,
clonazepam, clorazepate, phenacemide, paramethadione,
primaclone, clobazam, felbamate, flunarizine, lamotrigine,
progabide, vigabatrin, eterobarb, gabapentin, oxcarbazepine,
ralitoline, tiagabine, sulthiame, and tioridone;
10 wt% to 75 wt% of a dispensing polymer compatible with the
antiepileptic drug that aids in delivery of the
antiepileptic drug in a therapeutic dose from the dosage
form; and 0 wt% to 10 wt% of a pharmaceutically acceptable
surfactant; with the total weight of all ingredients in the
composition equal to 100 wt%, wherein the composition is
CA 02575042 2007-02-12
67696-219D
6b
surrounded by a subcoat comprising a nontoxic, nonionic
polymer that prevents the antiepileptic drug from converting
from a soluble to an insoluble antiepileptic drug in the
gastrointestinal pH, in the manufacture of a medicament for
continuous oral administration of an antiepileptic drug to
the gastrointestinal tract of a human.
According to still another aspect of the present
invention, there is provided use of a composition
comprising: 0.5 wt% to 90 wt% of an antiepileptic drug
selected from the group consisting of phenytoin,
mephenytoin, Phenobarbital, primidone, carbamazepine,
ethosuximide, methsuximide, phensuximide, trimethadione,
clonazepam, clorazepate, phenacemide, paramethadione,
primaclone, clobazam, felbamate, flunarizine, lamotrigine,
progabide, vigabatrin, eterobarb, gabapentin, oxcarbazepine,
ralitoline, tiagabine, sulthiame, and tioridone;
10 wto to 75 wto of a dispensing polymer compatible with the
antiepileptic drug that aids in delivery of the
antiepileptic drug in a therapeutic dose from the dosage
form; and 0 wto to 10 wto of a pharmaceutically acceptable
surfactant; with the total weight of all ingredients in the
composition equal to 100 wto, wherein the composition is
surrounded by a subcoat comprising a nontoxic, nonionic
polymer that prevents the antiepileptic drug from converting
from a soluble to an insoluble antiepileptic drug in the
gastrointestinal pH, for continuous oral administration of
an antiepileptic drug to the gastrointestinal tract of a
human.
According to yet another aspect of the present
invention, there is provided use of an osmotic dosage form
that maintains its integrity in the gastrointestinal tract,
comprising: a composition comprising: 0.5 wto to 90 wto of
an antiepileptic drug selected from the group consisting of
CA 02575042 2007-02-12
67696-219D
6c
phenytoin, mephenytoin, phenobarbital, primidone,
carbamazepine, ethosuximide, methsuximide, phensuximide,
trimethadione, clonazepam, clorazepate, phenacemide,
paramethadione, primaclone, clobazam, felbamate,
flunarizine, lamotrigine, progabide, vigabatrin, eterobarb,
gabapentin, oxcarbazepine, ralitoline, tiagabine, sulthiame,
and tioridone; 10 wto to 75 wto of a dispensing polymer
compatible with the antiepileptic drug that aids in delivery
of the antiepileptic drug in a therapeutic dose from the
dosage form; and 0 wto to 10 wts of a pharmaceutically
acceptable surfactant; with the total weight of all
ingredients in the composition equal to 200 wto, which
composition is surrounded by a subcoat comprising a
nontoxic, nonionic polymer that prevents the antiepileptic
drug from converting from a soluble to an insoluble
antiepileptic drug in the gastrointestinal pH; a wall
permeable to fluid and impermeable to an antiepileptic drug
that surrounds the subcoat; and, an exit in the wall for
delivery of the antiepileptic drug from the dosage form over
an extended period of time, in the manufacture of a
medicament for continuous oral administration of an
antiepileptic drug to the gastrointestinal tract of a human.
According to a further aspect of the present
invention, there is provided use of an osmotic dosage form
that maintains its integrity in the gastrointestinal tract,
comprising: a composition comprising: 0.5 wt% to 90 wto of
an antiepileptic drug selected from the group consisting of
phenytoin, mephenytoin, phenobarbital, primidone,
carbamazepine, ethosuximide, methsuximide, phensuximide,
trimethadione, clonazepam, clorazepate, phenacemide,
paramethadione, primaclone, clobazam, felbamate,
flunarizine, lamotrigine, progabide, vigabatrin, eterobarb,
gabapentin, oxcarbazepine, ralitoline, tiagabine, sulthiame,
CA 02575042 2007-02-12
67696-219D
6d
and tioridone; 10 wt% to 75 wt% of a dispensing polymer
compatible with the antiepileptic drug that aids in delivery
of the antiepileptic drug in a therapeutic dose from the
dosage form; and 0 wt% to 10 wt% of a pharmaceutically
acceptable surfactant; with the total weight of all
ingredients in the composition equal to 100 wt%, which
composition is surrounded by a subcoat comprising a
nontoxic, nonionic polymer that prevents the antiepileptic
drug from converting from a soluble to an insoluble
antiepileptic drug in the gastrointestinal pH; a wall
permeable to fluid and impermeable to an antiepileptic drug
that surrounds the subcoat; and, an exit in the wall for
delivery of the antiepileptic drug from the dosage form over
an extended period of time, for continuous oral
administration of an antiepileptic drug to the
gastrointestinal tract of a human.
According to yet a further aspect of the present
invention, there is provided use of a dosage form comprising
10 nanograms to 1000 milligrams of an antiepileptic drug,
which dosage form is adapted to release the antiepileptic
drug from the dosage form by controlled-release kinetics,
and wherein the dosage form is characterized by a nonionic
polymer film in the dosage form that substantially protects
the antiepileptic drug from fluid of the gastrointestinal
environment that contacts the dosage form, in the
manufacture of a medicament for administration of an
antiepileptic drug to the gastrointestinal tract of a human.
According to still another aspect of the present
invention, there is provided use of a dosage form
comprising 10 nanograms to 1000 milligrams of an
antiepileptic drug, which dosage form is adapted to release
the antiepileptic drug from the dosage form by controlled-
release kinetics, and wherein the dosage form is
CA 02575042 2007-02-12
67696-219D
6e
characterized by a nonionic polymer film in the dosage form
that substantially protects the antiepileptic drug from
fluid of the gastrointestinal environment that contacts the
dosage form, for administration of an antiepileptic drug to
the gastrointestinal tract of a human.
According to another aspect of the present
invention, there is provided a dosage form adapted for oral
administration of an antiepileptic drug selected from the
group consisting of phenytoin, sodium phenytoin, potassium
phenytoin, mephenytoin, phenobarbital, primidone,
carbamazepine, ethosuximide, methsuximide, phensuximide,
trimethadione, clonazepam, clorazepate, phenacemide,
paramethadione, primaclone, clobazam, felbamate,
flunarizine, lamotrigine, progabide, vigabatrin, eterobarb,
gabapentin, oxcarbazepine, ralitoline, tiagabine, sulthiame,
and tioridone to a patient in need of the antiepileptic
drug, wherein the dosage form comprises: (a) a dosage amount
of the antiepileptic drug in the dosage form; (b) a
composition comprising a member selected from the group
consisting of a polyalkylene and a carboxyalkylcellulose in
the dosage form; (c) an internal coat that surrounds at
least the dosage amount of the antiepileptic drug for aiding
in maintaining the integrity of the dosage form and for
protecting the antiepileptic drug in the dosage form; (d) an
external coat that contacts the internal coat and surrounds
and defines the dosage form for use in antiepileptic
therapy; and, (e) an exit in the dosage form for release of
the antiepileptic drug from the dosage form to the patient
in need of antiepileptic therapy.
CA 02575042 2007-02-12
67696-219D
6f
According to yet another aspect of the present
invention, there is provided a dosage form for use in
adjunctive epilepsy therapy, wherein the dosage form
comprises 10 nanograms to 1000 milligrams of a first
antiepileptic drug selected from group (A) consisting of
phenytoin, mephenytoin, phenobarbital, primidone,
carbamazepine, ethosuximide, methsuximide, phensuximide,
trimethadione, clonazepam, clorazepate, phenacemide,
paramethadione, primaclone, clobazam, felbamate,
flunarizine, lamotrigine, progabide, vigabatrin, eterobarb,
gabapentin, oxcarbazepine, ralitoline, tiagabine, sulthiame,
and tioridone, and 10 nanograms to 1000 milligrams of a
second and different antiepileptic drug selected from group
(A) which is a different drug (B) for providing adjunctive
antiepileptic (A) and (B) therapy, and wherein the
adjunctive antiepileptic drugs are adapted for oral
administration to a patient in need of adjunctive therapy
from a dosage form by a process selected from the group
consisting of osmotic diffusion, erosion, and ion exchange
over an extended period of time.
CA 02575042 2007-02-12
V'O 95;296(~ PC'i
Another object of the present invention is to provide a therap _:;;.
composition comprising an antiepileptic drug blended with an anaepileptc
pharmaceutically acceptable carrier.
Another object of the ir~~~ention is to provide a dosage form comprising
s an antiepileptic drug formulation, which dosage form passes the
pharmacokinetic property fc~~ delivering substantially 100% of the
antiepileptic drug formulation in a programmable and controlled manner
thereby substantially avoiding inherent drug residual in the dosage form.
Another object of the invention is to provide a laminate comprising a
~o first lamina comprising an awtiepileptic drug formulation and a second
lamina initially-tree of an ant~epileptic drug formulation.
Another object of the present invention is to provide a dosage form for
buccaily or sublingually adm;nistering an antiepileptic drug.
Other objects, features and advantages of the invention will be more
,s apparent to those versed in the dispensing art from the accompanying
detailed specification, taken in conjunction with the drawings and the
claims.
In the drawing figures, which are not drawn to scale, but one set forth
Zo to illustrate various embodiments of the invention, the drawing figures
are as follows:
Drawing Figure 1 is a general view of a dosage form designed and
shaped for oral administration of a drug for the therapy of epilepsies at a
CA 02575042 2007-02-12
W'O 9~ »i6~ PCTIC 59~iI1.163J
8
continuous-release rate over time to a patient in need of therapy for the
management of epilepsies;
Drawing Figure 2 is an opened view of drawing Figure 1 for depicting
an embodiment of the dosage form comprising a pharmaceutical
comprising a drug indicated for the management of epiiepsies and a
composition comprising means for pushing the pharmaceutical
composition from the dosage form;
Drawing Figure 3 is an opened view of drawing Figure 1 for depicting
an embodiment of the dosage form comprising an internal subcoat
~o positioned between the internal surface of the wall of the dosage form
and the pharmaceutical composition and the composition for pushing the
pharmaceutical composition from the dosage form;
Drawing Figure 4 is a view of a dosage from provided by the
invention, which dosage form comprises a prompt-release coat
Is comparison, a drug for the therapy of the epilepsies on the exterior
surface of the dosage form;
Drawing Figure 5 is an opened view of a dosage form provided by the
invention, which dosage form comprises a single composition in the
dosage form comprising a drug for treating epilepsies and means for
zo delivering the single composition from the dosage form;
Drawing Figures 6A and 6B depicts the antiepileptic drug release rate
for two dosage forms over two different times;
Drawing Figure 7 depicts the release pattern for the dosage form in an
acid and alkaline fluid environment;
CA 02575042 2007-02-12
67696-219
9
Drawing Figure 8 depicts a drug release curve illustrating for this
invention the delivery rate is independent of the size of the passageway;
Drawing Figure 9 illustrates the dosage form has substantially identical
release patterns in vivo and in vitro; and
s Drawing Figure 10 illustrates the internal coat protect cracking in an
external wall.
In the drawing figures and in the specification, like parts in related
drawing figures are identified by like numbers. The terms appearing
earlier in the specification and in the description of the drawing figures,
~o as well as embodiments thereof, are further described elsewhere in the
specification.
Turning now to the drawing figures in detail, which drawing figures
are examples of dosage forms provided by this invention and which
~s examples are not to be construed as limiting, one example of a dosage
form is seen in drawing Figure 1. In drawing Figure 1, a dosage form 10
is seen comprised of a body member 11, which body member i 1
comprises a wall 12, that surrounds and forms an internal area, not seen
in drawing Figure 7. Drawing Figure 1 comprises at least one exit 13
zo that connects the exterior of dosage form 10 with the interior of dosage
form 7 0. The dosage form 10 of drawing Figure 1 illustrates a
controlled-release dosage form that delivers an antiepileptic drug over an
extended time. The dosage form comprises controlled-release properties
provided by this invention is successful at maintaining substantially
is therapeutic antiepileptic levels in the blood or in body tissue. The dosage
form, as seen in drawing Figure 1, embraces the shape of a vertically
CA 02575042 2007-02-12
WO 95:')fiG~ PCT'L~S95if1a634
model tablet manufactured as a dosage form, comprises continuous-
release, extended release and prolonged-release forms. These dosage
forms provide antiepileptic blood levels and targeted tissue levels within a
therapeutic range optionall~~ below side-effect levels over time.
5 An extended period of time, as used for the purpose of this invention
includes a prolonged period of time, and a continuous-controlled release
period of time. The extended, prolonged and continuous time denotes a
duration of antiepiieptic drug deliver time over that achieved by
conventional delivery forms such as noncontrolled tablets and
ro noncontrolled capsules.
In drawing Figure 2, dosage form i 0 as seen in opened section.
In drawing Figure 2, dosage form 10 comprises a body 1 1, a wall 12 that
surrounds and defines an internal compartment 14. Internal compartment
14 communicates through exit port 13 with the exterior of dosage form
10. Wall 12 of dosage form 7 0 comprises totally or in at least a part a
composition that is permeable to the passage of an exterior fluid, such as
an aqueous fluid or a biological fluid present in the gastrointestinal tract.
Wal( 12 is nontoxic and it is substantially impermeable to the passage of
an antiepiieptic drug 15, represented by dots, present in lumen-
zo compartment 14. Wall 12 is substantially inert, and it maintains its
physical and chemical integrity during the dispensing life of antiepiieptic
drug 15. The phrase, maintains its physical and chemical integrity means
wall 12 does not lose its structure and it does not undergo chemical
change during the dispensing of antiepiieptic drug 15. In drawing
zs Figure 2, dosage form 10 comprises means 27 for providing protection
for antiepileptic drug 15 from the pH of 1 to 8 of the gastrointestinal
environment, thereby providing means 27 for ensuring antiepileptic drug
can give its full therapeutic benefit to an epileptic patient. Means 27,
also gives support as a subcoat to external wall 12 to give wall 12
so support against the stress and the strain of a fluid moving
gastrointestinal
CA 02575042 2007-02-12
WO 9e~29665 PCT .Jt.~~
11
tract. Means 27 comprises a nonionic, nontoxic fluid pervious pe
In drawing Figure 2, means 27 surrounds the internal area of
compartment 14 housing drug 1 5. An exit passageway 13 in means 27
is present to enable the delivery of drug 15 from dosage form 10.
s An opening 28 in means 27 distant from exit passageway 13 is an
internal channel for an expandable layer 26 to move towards exit
passageway 13 for contributing in the delivery of drug 15 from dosage
form 10.
Wall 12 comprises a composition that does not adversely effect an
~o animal, a human, or components of the dosage form. Compositions for
forming wall 12, are in one embodiment, comprise a member selected
from the group consisting of a cellulose ester polymer, a cellulose ether
polymer, and a cellulose ester-ether polymer. These cellulosic polymers
have a degree of substitution, D.S. on the anhydroglucose unit, from
~s greater than 0 up to 3 inclusive. By degree of substitution is meant the
average number of hydroxyl group originally present on the
anyhydroglucose unit comprising the cellulose polymer that are replaced
by a substituting group. Representative wall 12 polymers comprise a
member selected from the group consisting of cellulose acylate, cellulose
zo diacylate, cellulose triacylate, cellulose acetate, cellulose diacetate,
cellulose triacetate, mono-, di- and triceiiulose aikanylates, mono-,
di-, and tricellulose aroyiates, mono-, di-, and tricellulose alkenylates, and
mono-, di-, and tricellulose alkinylates. Exemplary polymers include
cellulose acetate having a D.S. up to 1 and an acetyl content up to 21 %;
zs cellulose acetate having a D.S. of 1 to 2 and an acetyl content of 21 to
35%; cellulose acetate having a D.S. of 2 to 3 and an acetyl content of
35 to 44.8%, and the like. More specific cellulosic polymers comprise
cellulose propionate having a D.S. of 1.8 and a propyl content of 39.2 to
45% and a hydroxyl content of 2.8 to 5.4 cellulose acetate butyrate
3o having a D.S. of 1.8, an acetyl content of 13 to 15% and a butynyl
CA 02575042 2007-02-12
WO "~ v'. .~6CO PCT'L S~>;,~?.1G3.1
12
content of 34 to 39%; cellulose acetate butyrate having an acetyl
content of 2 to 29%, a butyryi content of 17 to 53% and a hydroxyl
content of 0.5 to 4.7; cellulose triacyiates having a D.S. of 2.9 to 3 such
as cellulose trivalerate, cellulose trilaurate, cellulose tripalmitate,
cellulose
s trisuccinate, and cellulose trioctanoate; celluloses diacylate having a D.S.
of 2.2 to 2.6 such as cellulose disuccinate, dipalmitate, cellulose
dioctanoate, cellulose dipentanoate, co-esters of cellulose such as
cellulose acetate butyrate, and cellulose acetate propionate.
Additional semipermeabie polymers comprise acetaldehyde
~o dimethylcellulose acetate, cellulose acetate ethylacarbamate, cellulose
acetate methyfcarbamate, cellulose diacetate propylcarbamate, cellulose
acetate diethylaminoacetate, semipermeable polyamide; semipermeable
polyurethane; semipermeable sutfonated polystyrene; semipermeabie
cross-linked selective polymer formed by the coprecipitation of a
~ s polyanion and polycation as disclosed in United States Patent
Nos. 3,173,876; 3,276,586; 3,541,005; 3,541,006; and 3,546,142;
semipermeable polymers as disclosed by Loeb and Sourirajan in
United States Patent No. 3,133,132; semipermeabte tightly cross-linked
polystyrenes; semipermeable cross-linked polyisodium styrene sulfonate);
zo semipermeabls cross-linked poly (vinytbenzyltrimethyl ammonium
chloride); semipermeable polymers possessing a fluid permeability of
2.5 x 10'8 to 2.5 x '4 (cm2/hr~ atm) expressed per atmosphere of
hydrostatic or osmotic pressure difference across the semipermeable
wall. The polymers are known to the polymer art in United States
zs Patent Nos. 3,845,770; 3,916,899; and 4,160,020; and in ,~iandbook of
Common Polymers by Scott, J.R. and Roff W.J., 1971, published by CRC
Press, Cleveland, OH.
Compartment 14 comprises a drug 15 effective in the therapy of the
epilepsies. The antiepileptic drug 15 comprises a member selected from
CA 02575042 2007-02-12
WO 95.:966 PCT .tt~3-1
13
the group consisting of hydantoins, barbiturates, deoxybarbiturate
iminostilbenes, suocinimedes, oxazolidinediones, and benzodiazepi: es.
The antiepileptic drug 15 for treating all types of epilepsy comprise a
member selected from the group consisting of phenytoin, phenytoin
sodium, phenytoin potassium, mephenytoin, ethytoin, Phenobarbital,
Phenobarbital sodium, Phenobarbital potassium, primidone,
carbamazepine, ethosuximide, methsuximide, phensuximide,
trimexhadione, clonazepam, clorazepate, phenacemide, paramethadione,
primaclone, clobazam, felbamate, flunarizine, lamotrigine, progabide,
~o vigabatrin, eterobarb, gabapentin, excarbazepine, ralitone, tiagabine,
suithiame, and tioridone. The antiepileptic drug 15 are disclosed in
Pharmaceuticaj_S~jences, by Remington, 18th Ed., pp 1072-1081
( 1990), Mark Publishing Co., Easton, PA; and The Ph~~ac~jogical Basis
of 1 eraQ~utics, by Gilman and Rall, 8th Ed., pp 436-462 (i 990),
~s Pergamon Press, New York, NY. The dosage amount of antiepileptic
drug 15 is 10 nanograms (ng) to 2000 milligrams (mg) that is delivered
over an extended period of 30 hours. The antiepiieptic drug 15 is present
in individual doses of 5, 30, 50, 75, 100, 130, 150, 200, 250, 300,
350, 400, 500, 625, 700, 1000 to 2000 mgs of antiepileptic drug 15.
zo The antiepileptic drug 15 is delivered by dosage form 10 over a period of
immediate delivery of time up to 30 hours. The antiepileptic drug 15 can
be administered for adjunctive therapy with a different antiepileptic drug
15 in epilepsy patients. Representative of adjunctive antiepileptic drugs
15 that can be administered from dosage form 10 comprise phenytoin
zs and phenobarbitone, phenytoin and carbamazepine; phenytoin and
primidone, phenobarbitone and carbamazepine, carbamazepine and
primidone, felbamate and phenytoin, feibamate and carbamazepine,
felbamate and gabapentin, phenytoin and gabapentin, and carbamazepine
and gabapentin. The dosage amount of adjunctive-antiepileptic drug 15
ao for each adjunctive drug 15 is 10 ng to 1000 mg with the total dosage
for the adjunctive pain is 10 ng to 2000 mg.
CA 02575042 2007-02-12
y0 9~ .',')(iti~ PCT:MS95;I1J63J
14
Antiepileptic drug 1 5 is present in compartment 14 in antiepileptic
drug 1 5 formulation 16. The antiepileptic drug 1 5 formulation 16
comprises 0.5 wt % to 90 wt % of antiepileptic drug 15, a drug which
dispensing polymer is compatible with antiepileptic drug 15 and aids in
delivering antiepileptic drug 15 in a known dose from dosage form 10.
Dispensing polymer 17 comprises a member selected from the group
consisting of an osmopolymer possessing a 15,000 to 4,500,000
molecular weight, a polyalkyline oxide possessing a 175,000 to 225,000
molecular weight, a polyalkyline oxide possessing a 275,000 to 325,000
,o molecular weight, and a carboxyalkylcellulose possessing a 15,000 to
175,000 molecular weight. Representative members comprise a
polyethylene oxide of 200,000 molecular weight, a polyethylene oxide of
300,000 molecular weight and an alkali including sodium and potassium
carboxymethylcellulose of 40,000 to 1,000,000 molecular weight, as
~ s represented by dashes 17. Layer 16 comprises additionally 0 wt % to
20 wt % of an osmotically effective solute also known as an osmagent
18 for contributing to the delivery kinetics of antiepileptic drug 15.
Representative of osmagent 18, represented by vertical dashes 18,
comprises a member selected from the group consisting of magnesium
zo sulfate, magnesium chloride, sodium chloride, potassium chloride, lithium
chloride, potassium sulfate, sodium sulfate, mannitol, sorbitol, inositol,
urea, sucrose, glucose, glucitol, polyhydride alcohol and osmagents
exhibiting an osmotic pressure gradient across semipermeable wall 12 of
5 atmospheres to 500 atmospheres. Layer 16 comprises 0.1 wt % to
zs 25 wt % of a polyvinyl pyrrofidone of 5,000 to 150,000 as a suspending
and hydropumping agent, represented by slanted lines 19; 0 wt % to
5 wt % of a lubricant 20 selected from the group consisting of sodium
stearate, magnesium stearate, stearic acid, calcium stearate, calcium
oleate, oleic acid and caprylic acid as represented by dashes 20; and
so 0 wt % to 10 wt % of a surfactant 21 as represented by a nonionic
surfactant to prevent sticking to the wall of the dosage form, as
CA 02575042 2007-02-12
67696-219
represented by polyethylene glycol stearate, propylene glycol
monolaurate, polyethylene glycol sorbitol, and polyethylene glycol sorbitol
lanolin. The total weight of all ingredients in layer 16 is equal to
100 wt %, wherein wt % denotes weight percent.
s Compartment 14 comprises an expandable layer 26 that cooperates
with antiepileptic drug layer 16 to deliver antiepileptic drug 15 from
dosage form 10. Expandable layer 26 comprises 30 wt % to 70 wt % of
an expandable polymer 22 as represented by a polyalkylene oxide
comprising a 3,000,000 to 7,500,000 molecular weight, which is a
,o different polyalkylene oxide than the polyalkylne oxide in an antiepileptic
drug layer 16, a carboxyalkylcellulose comprising a 250,000 to
3,250,000 molecular weight that is a different carboxyalkylcellulose than
the carboxyalkylcellulose in layer 16; 5 wt % to 50 wt % of an osmagent
23; 0 wt % to 25 wt % of a hydroxypropyl alkylcellulose 24 possessing
~ s a 9,000 to 375,000 molecular weight; 0 wt % to 3 wt % of ferric oxide;
Owt%to5wt%ofalubricant;andOwt%tol5wt%ofa
hydroxyalkylcellulose 25 comprising a 7,000 to 250,000. Representative
of a polyalkylene oxide is polyethylene oxide; representative of
hydroxypropylalkylcellulose are hydroxypropylmethylcellulose,
zo hydroxypropylethylcellulose, hydroxypropylisopropyicellulose,
hydroxypropylbutylcellulose and hydroxypropyipentylcellulose;
representative of an osmagent comprise a member selected from the
group consisting of an inorganic salt, organic salt, acid, ester, ether,
carbohydrate, oxide, magnesium sulfate, magnesium chloride, sodium
zs chloride, lithium chloride, potassium chloride, potassium sulfate, sodium
sulfate, sodium sulfite, lithium sulfate, potassium lactate, mannitol, urea,
magnesium succinate, tartaric acid, raffinose, sorbitol, sucrose, fructose,
and glucose; representative of lubricant comprise a member selected from
the group consisting of stearic acid, magnesium stearate, calcium
3o stearate, magnesium oleate, calcium oleate, oleic acid, caprylic acid,
CA 02575042 2007-02-12
y'O ~~~ ::966 PCT,'t-595~(1.i63.J
16
magnesium pafmitate, and calcium lactate; and representative of
carboxyalkylcellulose comprise a member selected from the group
;.insisting of alkalcarboxyalkylcellulose, sodium carboxymethyl- cellulose,
potassium carboxymethylcellulose and sodium carboxyethylcellulose.
s The total weight of all ingredients in the expandable layer 26 is equal to
100 wt %.
Dosage form 10 as seen in drawing Figure 3 depicts another dosage
form provided by this invention. Dosage form 10 comprises an exterior
wail 12, internal compartment 14 comprising antiepileptic drug 15,
~o in antiepileptic drug 15 formulation 16, osmopolymer 17, osmagent 18,
polyvinylpyrrolidone, 19, lubricant 20 and surfactant 27 ; and expandable
layer 26 comprising expandable polymer 22, osmagent 23,
hydroxypropyiaikylcellulose 24 and hydroxyalkylcellulose 25. Dosage
form 10 of drawing Figure 3 comprises further an interior wall 27, which
~s interior wall 27 is in contracting relation with exterior wall 12,
antiepileptic drug formulation 16 and expandable formulation 26. Interior
wall 27 is positioned between wall i 2 and antiepiieptic drug formulation
16 and expandable formulation 26 and it surrounds antiepileptic drug
formulation 16 and expandable formulation 26, except for exit orifice 13.
zo The dual walls 12 and 27 provides unexpected advantages as wall 12
and wall 27 in combination protect a hydroscopic antiepileptic drug 15
from the unwanted influences of aqueous and biological fluids,
they shield an antiepiieptic drug 15 from converting from a soluble to an
insoluble antiepileptic drug 15 in the gastrointestinal pH range of 1 to 8.
zs The combination of wail 12 and wall 27 provides for both fast-release
and slow-release of antiepileptic drug 15. A fast-release of antiepileptic
drug 15 can be effected by providing wall i 2 thinner than wall 27.
A thin wall 12 lets an increased fluid flux through wall 12 thereby
providing a greater volume in compartment 14 for aiding in delivery of
so antiepileptic drug 15 from dosage form 10. A slow-release of
CA 02575042 2007-02-12
V O 9~;=96ti~ PCT
17
antiepileptic drug 15 is effected by providing wall 12 thicker than
27, as a thicker wall restricts the flux into compartment 14. The
presents of wall 27 provides structural support for wall 12. In pr:,,viding
support for wall 12, wall 2? substantially prevents andlor lessens the
incidence of cracking of wail 12. Wall 27 also in cooperation with wall
12 substantially maintains the integrity and the performance of dosage
form 10.
Dosage form 10, as seer .n drawing Figure 4, depicts another
manufactured provided by invention. Dosage form 10 comprises an
external coat 28 on the exterior surface of dosage form 10. Exterior coat
28 is a therapeutic composition comprising a member selected from the
group consisting of alkyl cellulose, methyl cellulose,
hydroxyalkylcellulose, hydroxypropylcellulose, hydroxypropylmethyl-
cellulose, hydroxypropylethylcellulose, and acacia. External coat 28
~ s optionally comprises 0 to 5 wt % of polyethylene glycol, or 0 to 5 wt
acetylated triglyceride. Coat 28 provides antiepileptic drug 15 therapy
immediately as coat 28 dissolves or undergoes dissolution in the
presence of gastrointestinal fluid and concurrently therewith delivers
antiepileptic drug 15 to an antiepiieptic drug 15 receiving patient. Coat
zo 28 provides antiepileptic drug 15 on entrance into the gastrointestinal
tract for immediate antiepileptic drug 15 therapy.
fn drawing Figure 5, dosage from i 0 is seen in opened view.
In drawing Figure 5, dosage form 10 comprises body 11, wall 12, wall
27, exit port 13 and internal compartment 14 as identified in previous
zs drawing Figures 2 and 3. Internal compartment 14 comprises a single
homogenous composition comprising 0.5 wt to
80 wt % of antiepileptic drug 15; from 5 wt % to 50 wt °r6 of a
polyethylene oxide comprising a 150,000 to 725,000 molecular weight;
from 0 wt % to 40 wt % of a cellulose either 29 selected from the group
CA 02575042 2007-02-12
WO " 'r6G~ PCT:~T'S9~i0.J63.1
18
consisting of hydroxypropylalkycellulose, hydroxypropymethyl-
cellulose,hydroxypropylethylcellulose, hydroxypropylisopropylcellulose,
hydroxypropylbutylcellulose, hydroxypropylpentylcellulose, and
hydroxypropyihexylceilulose possessing a 9,000 to 240,000 molecular
s weight; 0 wt % to 20 wt % of an osmotically effective solute 30
selected from the group consisting of an inorganic salt, an organic salt,
acid, ester, carbohydrate, oxide, and osmotically effective solutes that
exhibit an osmotic pressure gradient across wall 12; and 0 wt % to
3.5 wt % of lubricant 31. The tots! weight of ail ingredients in single
,o core 32 is equal to 100 wt %.
The antiepileptic drugs 15 selected from the group consisting of
hydantoins, barbiturates, deoxybarbiturates, iminostilbenes, succinimides,
oxazolidinediones and benzodiazepines for the purpose of this invention
can be administered from a dosage form selected from the group
~ s consisting of bioerodible dosage form, diffusion dosage form, and ion-
exchanged dosage forms.
The bioerodible dosage form 10 comer ises a bioerodible polymer
matrix comprising 1 mg to 1200 mg of an antiepileptic drug selected
from the group consisting of phenytoin, phenytoin sodium, phenytoin
Zo potassium, mephenytoin, ethotin, Phenobarbital, Phenobarbital sodium,
Phenobarbital potassium, primidone, carbamazepine, ethosuximide,
methsuximide, phensuximide, trimethadione, clonazepam, clorazepate,
phenacemide, paramethadione, primaclone, clobazam, felbamate,
flunariizine, Iamotrigine, progabide, vigabatin, eterobarb, gabapentin,
is oxcarbazepine, ralitoline, tiagabine, sulthiame and tioridone in 1 mg to
1200 mg of a polymer matrix that delivers the said drug to a drug
receptor at a rate of release controlled by the bioeroding polymer matrix
thirty minutes to seven days. The bioerodible polymers for forming the
dosage form containing the antiepileptic drug include a member selected
CA 02575042 2007-02-12
67696-219
19
from the group consisting of poly(ester), poly (amine), poly(lactide
poly(glycolide), poly(lactide-co-glycolide), poly(caprolactone),
polylhydroxybutyric acid), poiy(orthoester), poly(orthocarbonate),
poly(acetate), poly(carbohydrate), poly(peptide), poly(acetal) and
s poiy(dihydropyron).
The diffusion-dosage-form that release a drug under the influence of
fluid flux mechanism comprise a membrane-controlled diffusion consisting
of diffusion through a nonporous polymer membrane or through a porous-
poiymer membrane. The diffusion-operated dosage form structurally
~o includes a polymer matrix with an antiepileptic drug therein, that is
released by the process of diffusion and, a reservoir or depot of an
antiepileptic drug therein that is released therefrom by the process of
diffusion through a contacting polymer rate-governing membrane.
Representative diffusional polymers for providing a differsional-dosage
,s form comprising 1 mg to 1200 mg of antiepileptic drug with 7 mg to
1200 mg of a polymer selected from the group consisting of a
poly(olefin), poly(vinyl), poly(carbohydrate), poly(peptide), poly(additon),
poly(conden-sation), poly(rubber) and polylsilicone) polymers.
Representative of specific polymers are a mernber selected from this
zo group consisting of poly(ethylene), poly(propylene), copoly(ethylenevinyl
acetatel, poly(isobutyiene), poly (isobutylethylene), poly (vinylacetate),
cross-linked polyvinyl alcohol), poly(methacryiate), polylamide),
poly(ester), and polylsilicone).
Dosage form 10 comprising an antiepileptic drug 15 can be
zs manufactured as an ion-exchange dosage form 10 which comprises a
water-insoluble cross-linked polymers with an antiepileptic drug bound to
an ion-exchange resin. In dosage form 10, an antiepiieptic drug 15 is
released at a rate controlled by the antiepileptic drug 15 resin complex by
the ionic environment within the gastrointestinal tract. The antiepileptic
CA 02575042 2007-02-12
67696-219
drug 15 attached to the resins are released at a rate controlled by the
exchanging-rate with a charged ion in the gastrointestinal tract. This
ion-exchange dosage form 10 comprises catron-exchange resins
containing electronegative charges and anion-exchange resins containing
s electropositive charges. This cation-exchange resins include strong-acid
or weak-acid resins as with suifonic acid, carboxylic acid, and phosphoric
acid; and the anion-exchange resins include strong-base and weak-base
resins with quaternary ammonium, secondary amine, tertiary amine,
aromatic and tertiary amine aliphate resins. Examples include acidic
~o ion-exchange resins such as AmberliteT"" IR-120, basic ion-exchange resins
such as Amberlite IR-400, and weak basic ion-exchange resins such as
Amberlite IR-45.
Dosage form i 0, as further provided by this invention, and as seen in
the above drawing figures can be manufactured for administering an
~s antiepileptic drug 15 by oral route. Dosage form 10 comprising exterior
and interior antiepileptic drug 15 can be sized and shaped for
administering antiepileptic drug 7 5 by the sublingual or the buccal routes.
The sublingual and buccat routes can be used for quicker therapy and
they can be used when a small dose of antiepileptic drug i 5 is needed
zo for therapy. The buccal and sublingual routes can be used as a by-pass
of the first pass of hepatic metabolism antiepileptic drug 15.
The sublingual or buccal routes can be used for administering the first
dose of antiepileptic drug 15 followed by permitting dosage form 10
entrance into the gastrointestinal tract for antiepileptic 7 5 delivery.
R~~ESS FOR PROV~INC THE DOSAGE FORM
Dosage form 7 0, when manufactured as an osmotic controlled-release
dosage form comprises at least one passageway 13. The phrase
controlled-release as used herein, indicates that control is exercised over
CA 02575042 2007-02-12
PCT~'L ~ti,
ii'u 9s.'2966~
21
both the duration and the profile of the antiepileptic-release pattern
The expression passageway, as used for the purpose of this invent..,n,
includes aperture, orifice, bore, pore, porous element through whic~ the
antiepileptic drug 15 can be pumped, diffuse, travel or migrate a hollow
s fiber, capillary tube, porous overlay, porous insert, microporous member,
and porous composition. The expression also includes a compound that
erodes or is leached from wall 12 in the fluid environment of use to
produce at least one passageway 13 in dosage form 10. Representative
compounds suitable for forming at least one passageway, or a multiplicity
~o of passageways, includes an erodible poly(glycoiic) acid or poly(lactic)
acid member in the wail; a gelatinous filament; a water-removable
polyvinyl) alcohol); teachable compounds such as fluid removable
pore-forming polysaccharides, acid, salts, or oxides. A passageway or a
plurality of passageways can be formed by leaching a compound such as
~s sorbitol, sucrose, lactose, maltose, fructose, or the like, from wall 12 to
provide a controlled-release dimensioned pore-passageway.
The passageway can have any shape such as round, triangular, square,
elliptical, and the like, for assisting in the controlled-metered release of
antiepileptic drug 15 from dosage form 10. Dosage form 10 can be
zo constructed with one or passageways in spaced apart relation to one or
more surfaces of a dosage form 10. Passageway 13 and equipment for
forming passageways are disclosed in United States Patent
Nos. 3.845,770 and 3,916,899 by Theeuwes and Higuchi; in United
States Patent No. 4,063,064 by Sounders et al; and in United States
zs Patent No. 4,08$,864 by Theeuwes et al. Passageways comprising
controlled releasing dimension, sized, shaped and adapted as a
releasing-pore formed by aqueous leaching to provide a releasing-pore of
controlled release-rate are disclosed in United States Patent
No. 4,200,098 by Ayer and Theeuwes; and in United States Patent
so No. 4,285,987 by Ayer and Theeuwes.
CA 02575042 2007-02-12
pC'1'L~S~~rn.~f3y
~V O ''
22
Wall 12 is manufactured in one process, comprises an air suspension
process. This procedure consists in suspending and tumbling a
compressed drug core comprising a single layer or a bilayer core, in a
current of air and wall forming composition until a wall is applied to the
s drug-core or the drug-push compartment. The air suspension procedure
is well-suited for independently forming the wall. The air suspension
procedure is described in United States Patent No., 2,799,241;
J Amer Pharm Assoc, Vol 48, pp 451-454 ( i 959); and j~j.d, Vol 49,
pp 82-84 (1960). Dosage form 10 can be coated also with a
~o wall-forming composition in a Wurster° air suspension coater, using
methylene dichloride-methanol cosolvent, for example, 80:20, wt:wt,
an ethanol-water, or acetone-water cosolvent, for example, 95:5 wt:wt
using 2.5 to 4% solids. An Aeromatic° air suspension coater using a
methylene dichloride-methanol cosolvent for example, 80:20 wt:wt,
~s can be used for applying wall 12. Other wail forming techniques such as
a pan-coating system, wherein wail forming compositions are deposited
by successive spraying of the composition on the drug-core or drug
bilayer to provide a compartment, accompanied by tumbling in a rotating
pan. Finally, the wall coated compartments are dried in a forced air aver
zo at 30 ° C to 50 ° C for up to a week to free dosage form 10
of solvent.
Generally, the walls formed by these techniques have a thickness of 1 to
30 mils (0.0254 mm to 0.762 mml.
Dosage form 10 of the invention is manufactured by standard
manufacturing techniques. For example, in one manufacture the drug
zs and other core-forming ingredients comprising a single drug layer or
bilayer core with drug facing the exit means 13 are blended and pressed
into a solid layer, or a solid bilayer. The drug and other ingredients can
be dry-blended or blended with a solvent and mixed into a solid or
semisolid formed by conventional methods such as ball-milling,
ao calendaring, stirring, roll-milling or churning and then pressed into a
CA 02575042 2007-02-12
ii O 9~Z966~ PCT' li,..
23
preselected shape. The layer possesses dimensions that correspoc
the internal dimensions of the area the layer is to occupy in the do~~ge
form and in a bilayer it also possesses dimensions corresponding to the
second layer for forming a contacting arrangement therewith. Next, in a
s bilayer core, the push layer is placed in contact with the drug layer.
The push layer is manufactured using techniques for providing the drug
layer. The layering of the drug layer and the push layer can be fabricated
by convention press-layering techniques. Finally, a single layer or the
two layer compartment forming members are surrounded and coated with
~o an outer wall. A passageway is laser, leached, or mechanically drilled
through the wall to contact the drug layer. When the passageway is
formed by a laser, the dosage form is optically-oriented automatically by
the laser equipment for forming the passageway on the preselected
surface for forming the passageway.
~s In another manufacture, dosage form 10 is manufactured by the wet
granulation technique. In the wet granulation technique, for example,
the drug and the ingredients comprising the drug-forming core or the
drug-forming layers are blended using a solvent, such as ethyl
alcohol-water 98:2 v:v tvolume:voiume) as the granulation fluid.
zo Other granulating fluid, such as denatured alcohol 100%, can be used for
this purpose. The ingredients forming the drug core or layers are
individually passed through a 20 mesh screen and then thoroughly
blended in a mixer. Next, other ingredients comprising the core or layers
are dissolved in a portion of the granulation fluid, such as the cosolvent
25 described above. Then, the latter prepared we blend is slowly added to
the drug blend with continual mixing in the blender. The granulating fluid
is added until a wet blend is produced, which wet mass then is forced
through a 20 mesh screen onto oven trays. The blend ~s dried for
18 to 24 hours at 30°C to 50°C. The dry granules are sized then
with a
so 20 mesh screen. Next, a lubricant is passed through screen, such as an
CA 02575042 2007-02-12
WO :96(,~ PCTT~S~)~ WG3a
24
80-mesh screen, and added to the dry screen granule blend.
The granulation is placed in a blender and blended for 1 to 1 5 minutes.
A push layer is made by the same wet granulation techniques. The
compositions are pressed into their individual layers in a HATA~ layer
s press.
Another manufacturing process that can be used for providing the
compartment-forming composition core or layers comprises blending the
powdered ingredients for each core or layers independently in a fluid bed
granulator. After the powdered are dry blended in the granulator, a
~o granulating fluid, for example, poly(vinyl)pyrrolidone) in water, or in
denatured alcohol, or in 95:5 ethyl alcohollwater, or blends of ethanol
and water, is sprayed on the powders. Optionally, the ingredients can be
dissolved or suspended in the granulating fluid. The coated powders are
then dried in a granulator. This process granulates a!I the ingredients
~ 5 present therein while adding the granulating fluid. After the granules are
dried, a lubricant such as stearic acid or magnesium stearate is added to
the granulator. The granules for each separate core or layers are pressed
then in the manner described below.
Dosage form 10 of the invention can be manufactured by mixing a
Zo drug with composition-forming ingredients and pressing the composition
into a layer possessing dimensions that correspond to the internal
dimensions of the compartment of the dosage form. in another
manufacture the drug and other drug composition-forming ingredients and
a solvent are mixed into a solid, or a semisolid, by conventional methods
zs such as ballmilling, shaking, calendaring, tumbling, stirring or
rollmilling,
and then pressed into a preselected layer-forming shape. Next, a layer of
a composition comprising an osmopolymer and an optional osmagent are
placed in contact with the drug layer. The layering of the first layer
comprising the drug and the second layer comprising the osmopolymer
CA 02575042 2007-02-12
~1'O 9~i2966~ PCT.'S. ~(~_
and optional osmagent composition can be accomplished by using
conventional layer-press technique. The wall can be applied by mc~ding,
brushing, spraying or dipping the pressed bilayer's shapes with
wall-forming materials. Another and preferred technique that can be used
s for applying the wall is the air-suspension coating procedure.
This procedure consists in suspending and tumbling the two contacting
layers in current of air until the wall-forming composition surrounds the
layers. The air suspension procedure is described in United States Patent
No. 2,799,241; J Amer Pharm Assoc, Vol 48 pp 451-454 (1979); and,
~o j~j,~, Vol 49 pp 82-84 (1960). Other standard manufacturing procedures
are described in IVlodern Plastics Encycl~, Vol 46, pp 62-70 ( 1969);
and in Pharmaceutical Science, by Remington, 14th Ed, pp 1626-1678
( 1970), Mack Publishing Co., Easton, PA.
Exemplary solvents suitable for manufacturing the wall, a single layer
~s and a bilayer core include inert inorganic and organic solvents final
laminated wall. The solvents broadly include members selected for the
group consisting of aqueous solvents, alcohols, ketones, esters, ethers,
aliphatic hydrocarbons, haiogenated solvents, cyclaliphatics, aromatics,
hetercyciic solvents and mixtures thereof. Typical solvents include
2o acetone, diacetone, alcohol, methanol, ethanol, ispropyl alcohol, butt'(
alcohol, methyl acetate, ethyl acetate, isopropyl acetate, n-butyl acetate,
methyl isobutyl ketone, methyl propyl ketone, n-hexane, n-heptane
ethylene glycol monoethyl ether, ethylene glycol monoethyl acetate,
methylene dichloride, ethylene dichloride, propylene dichloride, carbon
zs tetrachloride, chloroform, nitroethane, nitropropane, tetrachloroethane,
ethyl ether, isopropyl ether, cyclohexane, cyclooctane, benzene, toluene,
naptha, tetrahydrofuran, diglyme, aqueous and nonaqueous mixtures
thereof, such as acetone and water, acetone and methanol, acetone and
ethyl alcohol, methyiene dichloride and methanol, and ethylene dichloride
ao and methanol.
CA 02575042 2007-02-12
yQ x9665 PCT,'L-595~O.Jfi3.~
26
The following examples are merely illustrative of the present invention
and they should not be considered as limiting the scope of the invention
in any way as these examples and other equivalents thereof will become
s apparent to those versed in the art in the light of the present disclosure,
the drawings and accompanying claims.
A dosage form for delivering the antiepileptic drug phenytoin is made
as follows: first an antiepileptic drug layer is prepared by blending
~o phenytoin, polyoxyethylene stearate, sodium carboxymethylcellulose,
sorbitol and polyvinylpyrrolidone are blended into a homogenous mass.
Then, anhydrous, denatured ethyl alcohol is added to the freshly prepared
mass, with blending to produce a wet mass. Next, the ethyl alcohol is
evaporated to yield a dry composition, and followed by the addition of
~s magnesium stearate and the ingredients blended again to yield an
antiepileptic drug composition.
Next, a displacement layer is prepared by blending into a homogenous
blend sodium carboxymethylcellulose possessing a higher molecular than
the sodium carboxymethyicellulose in the drug composition, sodium
zo chloride, hydroxypropyimethylcellufose, ferric oxide and
hyroxypropylcellulose are blended to yield an osmotic displacement
composition. Then, water is added to the composition to produce a fluid
bed granulate, followed by evaporating the water and then milling the dry
blend accompanied by the addition of magnesium stearate.
is The antiepileptic drug composition is next pressed in layered
arrangement against the osmotic displacement layer, to provide a
CA 02575042 2007-02-12
PCT!L-595i!)~b~-~
«'O 95;966~
27
compressed bilayer core. The core next is coated with a subcoat
comprising hydroxypropylcellulose, hydroxypropylmethylcellufose and
water to coat the bilayer core. The water is removed by evaporation to
provide the subcoated bilayered core. Then, a semi permeable wall is
s coated around the subcoated bilayer core. The semipermeable wall is
coated from a wall-forming composition comprising cellulose acetate,
polyethylene glycol, polyvinylpyrrolidone and cosolvent acetone and
methanol to apply the semipermeable wall. The cosolvent is removed by
evaporation and an orifice is drilled through the wall and the subcoat to
~o connect the antiepileptic layer with the exterior of the dosage form.
The procedure of Example 1 is followed to provide a dosage form
comprising the following: a drug layer comprising 50 wt % phenytoin,
28.5% wt % sodium carboxymethylcellulose comprising a 90,000
~ s molecular weight, 9 wt % sorbitol, 3 wt % polyethylene glycol stearate,
9 wt % polyvinyipyrrolidone and 0.5 mg magnesium stearate;
a displacement layer comprising 58,75 wt °Yo sodium
carboxymethylcellulose comprising a 300,000 molecular weight,
30 wt °~ sodium chloride, 5 wt % hydroxypropylmethylcellulose
zo comprising a 9,200 molecular weight, 5 wt °~ hydroxypropyiceilulose
comprising a 12,300 molecular weight, 1 wt % ferric oxide and
0.25 wt °r6 magnesium stearate. The drug-osmotic bilayer core
comprises a subcoat of 70 wt % hydroxypropylceliulose comprising a
38,000 molecular weight and 30 wt % hydroxypropylmethylcellulose
~s comprising a 11,200 molecular weight; and comprises a semipermeable
wall comprising 80 wt % cellulose acetate comprising an acetyl content,
and 20 wt % polyethylene glycol comprising a 3350 molecular weight.
The dosage form comprised a 0.76 mm exit port.
CA 02575042 2007-02-12
WO ~?~ ,9ti~i~ PCT~L'S9S;~LJC3.1
28
doles 3 and 4
Two dosage forms are prepared according to the invention, wherein
both dosage forms comprise 276 mg of phenytoin. One dosage form is
manufactured with a slow rate of release that release 90% of the
s phenytoin in 14.7 hours at a release rate of 21 mg/h as seen in drawing
Figure 6A; and a fast reieas~ dosage form that release 90% of the
phenytoin in 5.7 hours at a release rate of 50 mg/h, as seen in drawing
Figure 6B. The slow release dosage form of drawing Figure 6A
comprised a semipermeable wall 0.101 mm thick and the fast release
,o dosage form of drawing Fio«re 6B comprised a semipermeable wall
0.025 mm thick. Each of the dosage forms are identical, except for the
thickness of the semipermea~ale wall.
The dosage forms of the invention provides protection against an acid
environment and against the alkaline environment of the gastrointestinal
tract. The protection provided substantially lessens or substantially
reduces the conversion of a drug from one therapeutically form to
another therapeutically inactive form. The dosage form substantially
eliminates a change of a drug from an active to an inactive farm.
zo In drawing Figure 7, the protection for phenytoin against the effects of
artificial gastric fluid is seen in the curved line with black circles and the
protection against the effects of artificial intestinal fluid is seen in the
curved line with clear circles.
zs The procedures of the above examples are followed in this example to
provide four dosage forms for dispensing an antiepiieptic drug, wherein
CA 02575042 2007-02-12
WO 95; 29666 PCT;'I: 595/0.,
29
the dosage forms are identical except for the size and the number of the
exit passageways. The dosage forms are made comprising one
passageway of 1.016 mm diameter, a dosage form with on 0.559 mm
passageway, a dosage form comprising two 0.055 mm in diameter
s passageways, and a dosage form comprising three 0.559 mm
passageways. The accompanying drawing Figure 8 shows the
cumulative amount of drug released from the dosage form for the
different sized passageways and for the different number of passageways
is independent of the environment of and free of the influence of fluid in
~ o the environment that contacts a passageway during operation of a
dosage form. The dosage form of the invention prevents, for example,
an alkali salt, such as a sodium salt of phenytoin, from a premature
release from the dosage form coupled with the conversion of an alkali
salt to a practically insoluble form in the gastrointestinal pH range of
~ s 1 to 8.
The procedure of the above examples are followed in this example to
provide dosage forms of different geometries and to provide dosage
forms having a high cumulative amount of drug release from the dosage
zo form. The dosage forms provided are as follows: a dosage form
comprising an oval shape with a surface area of 4.2 cm, a wall thickness
of 0.14 mm and a T90 release rate of 13.2 hours; a dosage form
comprising a solid vertical shape, a surface area of 4.1 cm, a wall
thickness of 0.14 mm and a T90 release rate of 11.8 hours; and a
zs dosage form comprising a round shape, a surface area of 4.0 cm, a wall
thickness of 0.14 mm and a T90 release rate of 14 hours. The amount
of drug, residual drug, remaining int he dosage form at the termination of
the delivery period is for a dosage form comprising an oval shape 1 .54%;
for a dosage form comprising a round shape 0.85%; and for a dosage
CA 02575042 2007-02-12
WO 9512966~ PCT-'L-S9~;f)l~,sa
form comprising a vertical shape possessing a lengthwise axis larger than
its cross-section, cross-sectional or perpendicular thereto is 0.12%.
The results demonstrate a dosage form provided by the invention delivers
substantially of its drug over time.
s Examples 8 and 9
Drawing Figure 8, demonstrates the in vivo and in vitro drug release
rate from a dosage from comprising the same structure and the same
drug dose are substantially identical over a prolonged time. In the
drawing figure, the clear squares depict the in vivo release rate
~o determined by measuring the dose of phenytoin released at various time
intervals from a dosage from as it moves through the gastrointestinal
tract of a laboratory animal. The black squares indicate the dose of
phenytoin released at a corresponding time interval measured in a distilled
water bath. Drawing Figure 9 demonstrates the invention comprising
~s means for maintaining the integrity of the wall of the dosage form and
correspondingly substantially lessening and/or preventing wall cracking
during operation of the dosage form as the dosage form osmotically and
hydrodynamically pumps a drug in a water bath. In drawing Figure 9, the
black squares indicate a dosage form made with a single wall without a
Zo subcoat, which wall appeared to crack at one hour that resulted in the
loss of osmotic and hydrodynamic pressure in the dosage form.
The white squares depict the release rate for a dosage form wherein the
wall is suppbrted by a subcoat that enables the semipermeable wall to
keep its integrity and maintain an osmotic and hydrodynamic pressure in
25 the dosage form during the life of the dosage form which results in
substantially all the drug delivered from the dosage form.
CA 02575042 2007-02-12
W~O 95;?956~ PCT~'L'S95i~.1v
31
exam Ip a 10
A dosage form is prepared as follows: first 250 mg of carbamazepine,
a white practically insoluble in water antiepileptic drug, is passed through
a 40 mesh screen, and then rescreened with sodium
s carboxymethylcellulose, polyvinylpyrrolidone, and sorbitol.
The ingredients are blended on a blender for 15 minutes then transferred
to a granulation bowl. With constant stirring, ethanol is added to the
continuous blend with blending continued until a homogenous blend is
produced in the granulator. Then, the blend is passed through a 20 mesh
~o screen. The screened granules are spread over a tray and placed in an
oven to a moisture content of 2%. Then, the dried granulation is passed
through a 20 mesh screen and transferred to a blender.
Next, magnesium stearate is passed through a 60 mesh screen, added to
the blender and mixed for two minutes.
~ s Next, hydroxypropyiceliulose is added to distilled water and blended
for two hours. Then, sodium chloride is screened through a 20 mesh
screen and blended with sodium carboxymethylcellulose possessing a
higher molecular weight, hydroxypropyimethyiceilulose and ferric oxide,
and all the ingredients blended for 5 minutes. The blend is screened and
2o transferred to a granulation bowl and prescreened magnesium stearate is
added to the mixing bowl, followed by mixing for seven to eight minutes.
Then, 550 mg of the carbamazepine composition and 220 mg of the
displacement push composition are transferred into a vertical die
possessing a lengthwise axis longer than the cross-section axis and the
25 layers pressed under one ton of pressure for each layer, to yield a solid
capsule-shaped two layer core.
CA 02575042 2007-02-12
' WO 9~ =9cici~ PCT'/: 59~(1363.i
32
Next, hydroxypropylcellulose and hydroxypropylmethylcellulose are
blended to provide a 70/30 ratio, respectively. Then, in a mixing vessel,
distilled water is added to give a 6% solid content, with constant stirring
to give a smooth homogenous solution, which is an suspension
homogenous solution, which is air suspension coated around the core to
yield the subcoated core. Then, cellulose acetate, polyvinyl-pyrrolidone
and polyethylene glycol are mixed with acetone and methanol in a ratio of
80/20 (wt/wt) to achieve a solid content of 5 °,% and the subcoated
core
is overcoated in an air suspension machine with a semipermeable wall.
,o then the dosage forms are dried to substantially free the dosage form of
solvents. Next, a 30 mil (0.76 mm) exit port is drilled through the
semipermeable wall and the subcoat to connect the carbamazepine drug
layer with the exterior of the dosage form.
~ s The procedure of Example 10 is followed to provide a dosage form as
follows: an antiepiieptic drug layer composition comprising 250 mg of
ethotoin, 157 mg of sodium carboxymethylcellulose of 80,000 molecular
weight, 50 mg of polyvinylpyrrolidone, 50 mg of sorbitol, 17 mg of
polyoxystearate, and 3 mg of magnesium stearate; a displacement layer
Zo composition comprising 130 mg of sodium carboxymethylcellulose of
300,000 molecular weight, 67 mg of sodium chloride, 1 1 mg of
hydroxypropylmethylcellulose of 1 1,200 molecular weight, 11 mg of
hydroxypropylcellulose of 28,000 molecular weight, 2 mg of ferric oxide
and 0.6 mg of magnesium stearate. The dosage form comprises a
25 subcoat of 21 mg hydroxypropylcellulose and 9 mg
hydroxypropyfmethylcellulose; and a semipermeable wall comprising
58.8 mg of cellulose acetate and 14.7 mg of polyethylene glycol.
The dosage forms provided by this example comprises additional a
semipermeable wall of 44 mg of cellulose acetate and 1 1 mg of
CA 02575042 2007-02-12
PCT,'L~S95~fO,
V1-'O 95!2966
33
polyethylene glycol. The dosage form comprising the higher amount of
cellulose acetate is a slow release dosage form and the dosage form
comprising the lesser amount of cellulose acetate is a fast release dosage
form.
Exam 12e 12
The procedures of the above examples are followed to provide a
dosage form comprising an antiepileptic drug selected from the group
consisting of mephenytoin, Phenobarbital, primidone, ethosuxirnide,
methosuximide, phensuximide, trimethadione, clonazepam, clorazepate,
~o clobazam, felbamate, vigabatin, gabapentin and tioridone.
A dosage form is provided by following the above examples to provide
a dosage form comprising a drug layer comprising 45 wt % phenytoin,
46.5 wt % polyethylene glycol of 300,000 molecular weight, 3 wt % of
~s polyvinylpyrrolidone of 30,000 molecular weight, 0.50 wt % calcium
stearate, and 5 wt % of polyethylene glycol monolaurate; an osmotic
layer comprising 58.75 wt % of a polyethylene oxide having 7,500,000
molecular weight, 30 wt % of sodium chloride, 5 wt % of hydroxy-
propylmethylcellulose possessing a 9,200 molecular weight, 1 wt % of
2o ferric oxide, 0.25 wt % of calcium stearate and 5 wt % of
hydroxypropylcellulose possessing a 30,000 molecular weight, an internal
coat that enrobes the drug and osmotic layers, which enrobing coat
comprises 95% wt % hydroxyethylcellulose a nonionic water soluble
polymer and 5 wt % of polyethylene glycol; and an outer semipermeable
zs wall comprising 85 wt % cellulose acetate having 39.8% acetyl content,
and 15 wt % polyethylene glycol. The dosage form had a mean release
CA 02575042 2007-02-12
WO 9~i?9665 PCr:'I: s95iila",.~
34
rate of 23,745 mg; hr, one 1 mm passageway, and a T90 of 12.9 hours.
The semipermeable wall of this dosage form is 0.15 mm thick.
The procedure of Example 13 is followed in this example, with all
s procedures as set forth previously except that in this example the dosage
form comprises a semi-permeable wall 0.025 mm thick, a T90 of
6 hours, and a mean release rate of 46.19 mg/hour.
An exterior, quick lease coat comprising the antiepileptic drug
~o carbamazepine as adjunct therapy to slow release phenytoin from the
interior of the dosage form comprises blending carbamazepine with a
member selected from the group consisting of a water-binder,
water-soluble film-former polymer selected from the group consisting of
hydroxyethylcellulose, hydroxypropylcellulose and hydroxymethylcellulose
~ s are added to a fluid bed granuiator and the materials blended in a moving
current of air. Then, a granulating fluid is sprayed onto the fluidizing
powders until the powders are agglomerated. then, the fluidizing process
is continued until the granulation is dry. The prompt release coat is
compressed or air sprayed around the external surface of the
zo semipermeable wall to yield a prompt release coat of antiepileptic drug
and a stow release antiepileptic drug from a single dosage form.
The procedure of Example 15 is followed to yield a single dosage form
comprising an antiepileptic drug combination with one antiepileptic drug
is in releasable contact with the exterior surface of the dosage form and a
CA 02575042 2007-02-12
PcT!L s9s~rob..-.
W'O 9~i:966~
different antiepileptic drug in extended releases in the interior of the
dosage form. Examples of ~ntiepileptic combinations comprises
phenytoin and phenobarbitone; phenytoin and carbamazepine,
phenobarbitone and carbamazepine, felbamate and carbamazepine,
s phenobarbitone and primidone, carbamazepine and primidone,
carbamazepine and clonazepam, carbamazepine and clorazepate,
phenytoin and clonazepam, phenytoin and clorazepate, phenytoin and
felbamate, phenytoin and vigabatron, and phenytoin and gabapentin.
METHOD 0~ USING THE INVENTION FOR
~o ANT'EeJ,LE~T1C THERAPY
An embodiment of the invention pertains to a method for delivering an
antiepileptic drug orally to a patient in need of antiepileptic therapy,
which method comprises the steps of (AI admitting into the patient a
dosage form comprising (11 an antiepileptic drug layer comprising a
~s dosage amount of an antiepiieptic therapeutic program; (2) a push layer
comprising means for imbibir~.g fluid for expanding for pushing the
antiepiieptic layer from the dosage form; (3) an internal coat for
maintaining the structural integrity of the dosage form and for
maintaining an osmotic and hydrodynamic pressure surrounding the
zo antiepileptic drug layer and the push layer; (4) a semipermeabie wall
surrounding the internal coat with semipermeable wall is permeable to
fluid flux and impervious to the flux of an antiepileptic drug;
(5) a passageway in the dosage form for releasing the antiepileptic drug
from the dosage form; (B) imbibing fluid through the semipermeable wall
zs at a rate determined by the permeability of the semipermeable wall and
the osmotic pressure gradient across the semipermeable wall causing the
push layer to expand; and (C) deliver the antiepileptic drug from the
dosage form through the passageway to the patient over a prolonged
period of time. The method comprises further positioning the dosage
CA 02575042 2007-02-12
«'O 9~'?966~ PCT~t.'S9~;t1.16,i.1
36
form buccally or sublingually for buccal antiepileptic therapy or sublingual
antiepileptic therapy.
In summary, it will be appreciated the present invention contributes to
the antiepileptic art an unobvious dosage form that possess a practical
s utility, can administer an antiepileptic drug in a prompt dose and in a
known dose released per unit time over time. While the invention has
been described and pointed out in detail with reference to operative
embodiments thereof, it will be understood to those skilled in the
antiepileptic art that various changes, modifications, substitutions and
~o omissions can be made without departing from the spirit of the invention.
It is intended, therefore, that the invention embrace those equivalents
within the scope of the claim which follow.