Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02576714 2007-01-29
WO 2006/024731 PCT/FR2005/001867
1
Procédé de mesure du rapport du débit volumique de gaz au débit volumique
d'un mélange d'hydrocarbures multiphasigue.
La gestion et la régulation des installations de production pétrolière
nécessitent la connaissance de nombreux paramètres de transport par conduites
(pipelines) de mélanges d'hydrocarbures multiphasiques en écoulement dans ces
dernières, ces mélanges multiphasiques constituant, pour les explôitants, un
fluide de production sur lequel les paramètres de régulation et de gestion
précités
doivent être appliqués.
En particulier, une régulation et une gestion rationalisées de ces fluides
de production, et de la production finalement atteinte, impliqLle la
connaissance
avec un degré de précision suffisant de paramètres spécifiques de ces mélanges
multiphasiques, tels que, notamment, le facteur de glissement de phase (slip
ratio) intervenant dans ces mélanges en écoulement. On rappelle que le facteur
de glissement de phase peut être défini comme le rapport entre la vitesse
d'écoulement du gaz de la phase gazeuse à celle du liquide de la phase
liquide,
lorsqu'on considère un mélange diphasique en écoulement dans une conduite.
Les vitesses d'écoulement de chaque phase sont définies elles-mêmes
comme le rapport entre le débit volumique de chacune d'elles à leûr section de
passage dans la conduite, encore désignée section d'occupation (hold up).
Les méthodes actuellement connues et utilisées de détermination du
rapport GVF du débit volumique de gaz au débit volumique d'un mélange
d'hydrocarbures multiphasique mettent en uvre le processus, succession
d'étapes, ci-après :
- mesure de la section d'occupation de la phase gazeuse, en valeur
relative vis-à-vis de la section totale de la conduite ;
- détermination du rapport de glissement S par mesure physique ;
- détermination du GVF à partir du rapport de glissement S mesuré.
Parmi les méthodes actuellement utilisées, on peut citer celle décrite
par S. JAYAWARDANE SPE, Society of Petroleum Engineers Inc. et B.C.
THEUVENY, Schlumberger, présentée par leurs auteurs au 2002 Annual
Technical Conference and Exhibition, San Antonio, Texas, USA du 29 Septembre
au 2 Octobre 2002 et publiée dans le document SPE 77405.
La méthode précitée mettant en ceuvre les étapes précédemment
décrites nécessite la mise en oruvre d'un modèle de rapport de glissement,
basé
CA 02576714 2007-01-29
WO 2006/024731 PCT/FR2005/001867
2
sur un modèle de mécanique des fluides, exprimant la valeur du rapport de
glissement en fonction du rapport de la section d'occupation, de la densité de
la
phase liquide et de la phase gazeuse et de la viscosité du liquide. La valeur
du
GVF est ensuite obtenue à partir de la fonction inverse représentative du
modèle
et de la valeur du rapport de glissement mesuré S.
La méthode précitée donne satisfaction. Toutefois, la mise en oeuvre
de cette dernière nécessite d'importants moyens de calcul permettant de
calculer
les paramètres essentiels tels que les densité du gaz et du liquide, eau et
hydrocarbures, à partir des conditions de pression P volume V et Température
dans la conduite et de corrélations empiriques (EOS).
La mise en eeuvre efficace des corrélations (EOS) précitées nécessite
en outre la prise en compte d'analyses de composition du fluide en écoulement,
ce qui alourdit de manière non négligeable le coût en moyens et temps de
calcul
de la mise en oeuvre de la méthode précitée.
La présente invention a pour objet de réduire, sinon supprimer, les
inconvénients de méthodes de l'art antérieur, par simplification et diminution
corrélative des moyens et des temps de calcul.
En particulier, un objet de la présente invention est la mise en oeuvre
d'un procédé permettant de déterminer directement le rapport du débit
volumique
de gaz au débit volumique d'un mélange d'hydrocarbures multiphasique, GVF, à
partir d'une mesure du rapport de la section d'occupation de la phase gazeuse
et
d'un modèle thermodynamique.
Un autre objet de la présente invention est en outre, le GVF du fluide
multiphasique ayant été obtenu grâce à la mise en oeuvre de la méthode
précitée, une utilisation de cette méthode pour déterminer la valeur du
facteur de
glissement S d'une phase telle que la phase gazeuse, vis-à-vis d'au moins une
phase de référence, telle que la phase liquide.
Un autre objet de la présente invention est enfin la mise en oeuvre d'un
dispositif d'analyse d'un mélange d'hydrocarbures multiphasique permettant non
seulement la mise en oeuvre du procédé de mesure du GVF d'un mélange
d'hydrocarbures multiphasique selon l'invention, mais également de déterminer
des paramètres essentiels à la gestion de ce fluide multiphasique tels que,
notamment, le facteur de glissement entre une phase et une phase de référence,
CA 02576714 2007-01-29
WO 2006/024731 PCT/FR2005/001867
3
le débit massique d'au moins une phase de ce fluide multiphasique en
écoulement.
Le procédé de mesure du rapport du débit volumique de gaz au débit
volumique d'un mélange d'hydrocarbures multiphasique, comprenant au moins
une phase gazeuse et une phase liquide en écoulement dans une conduite, objet
de l'invention, est remarquable en ce qu'il consiste au moins à
échantillonner,
dans les conditions de température et de pression statique de la conduite, le
gaz
et le liquide et mesurer au moins, dans l'écoulement du mélange, le rapport de
la
section d'occupation relative de la phase gazeuse à la section totale de
l'écoulement ; analyser la composition des échantillons de gaz et de liquide
pour
déterminer les paramètres de la composition du mélange, tels que la fraction
massique ou molaire des composés carbonés contenus dans ces échantillons,
déterminer, à partir de ces paramètres de composition, par simulation d'un
équilibre thermodynamique, les propriétés intensives, paramètres thermo-
dynamiques locaux, de chaque phase, ces paramètres thermodynamiques
locaux comportant au moins la densité de la phase liquide respectivement de la
phase gazeuse, établir, à partir de ces paramètres thermodynamiques locaux et
du rapport de la section d'occupation relative de la phase gazeuse, la densité
mesurée du mélange, calculer, à partir d'un processus adaptatif d'évaluation
de
la densité calculée du mélange et de comparaison par itération successive de
cette densité calculée du mélange à cette densité mesurée du mélange, les
propriétés intensives telles que la densité locale de la phase liquide, de la
phase
gazeuse et du mélange, et, sur égalité de la densité calculée du mélange et de
la
densité mesurée du mélange, les propriétés intensives du mélange étant
établies
et le mélange physiquement analysé et analytiquement reconstitué, calculer le
rapport du débit volumique de gaz et de mélange, exprimé comme le rapport du
produit du débit volumique de gaz et du débit molaire de gaz à la somme du
produit du débit volumique du gaz et du produit du débit molaire du gaz et du
produit du débit volumique du liquide et du débit molaire du liquide.
Le procédé objet de l'invention trouve application à la gestion de
l'exploitation de la production d'hydrocarbures en temps réel, notamment à la
gestion de la production des puits en aval des têtes de puits, et, plus
généralement à la gestion des conduites (pipelines) de mélanges
d'hydrocarbures multiphasiques.
CA 02576714 2007-01-29
WO 2006/024731 PCT/FR2005/001867
4
Le procédé objet de l'invention sera mieux compris à la lecture de la
description et à l'observation des dessins ci-après dans lesquels :
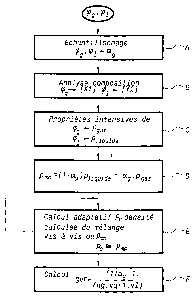
- la figure la représente, à titre illustratif, un organigramme général des
étapes de mise en oeuvre du procédé objet de l'invention ;
- la figure 1 b représente, à titre purement illustratif, un schéma
spécifique de mise en oeuvre de l'étape d'échantillonnage représentée en
figure
1a;
- la figure 1 c représente, à titre illustratif, un mode spécifique non
limitatif de mise en ceuvre de l'étape de calcul adaptatif de la densité
calculée du
mélange et de comparaison de cette densité calculée du mélange à la densité
mesurée de ce mélange obtenue à l'étape D de la figure 1 a;
- la figure 1 d représente une variante de mise en oeuvre de l'étape E1
de la figure 1 c pour une phase liquide comportant de l'eau et des
hydrocarbures ;
- la figure 2 représente, à titre purement illustratif, une utilisation
particulièrement remarquable du procédé objet de l'invention, appliqué à la
détermination du facteur de glissement d'une phase d'un mélange
d'hydrocarbures multiphasique, comprenant au moins une phase gazeuse et une
phase liquide en équilibre.
Une description plus détaillée du procédé de mesure du rapport du
débit volumique de gaz au débit volumique d'un mélange d'hydrocarbures
multiphasique conforme à l'objet de la présente invention sera maintenant
donnée en liaison avec la figure la.
Ainsi que représenté sur la figure précitée, on considère un mélange
d'hydrocarbures multiphasique en écoulement dans une conduite dans des
conditions normales d'exploitation. Ce mélange est réputé comprendre au moins
une phase gazeuse et une phase liquide notée ~9 respectivement 9,, en
écoulement dans la conduite précitée.
Le procédé objet de la présente invention consiste, ainsi que
représenté sur la figure la, à échantillonner en une étape A, dans les
conditions
de température et de pression de la conduite, le gaz et le liquide et à
mesurer au
moins dans l'écoulement du mélange le rapport de la section d'occupation
relative de la phase gazeuse à la section totale de l'écoulement. Ce rapport
est
noté a9.
CA 02576714 2007-01-29
WO 2006/024731 PCT/FR2005/001867
L'étape A d'échantillonnage est alors suivie d'une étape B consistant à
analyser la composition des échantillons de gaz et de liquide pour déterminer
les
paramètres de composition du mélange, tels que la composition massique ou
molaire des composés carbonés contenus dans ces échantillons.
5 D'une manière plus spécifique, on indique que la composition
massique ou molaire précitée est notée [X;] pour la phase gazeuse
respectivement [Y;] pour la phase liquide, l'indice i désignant des indices de
référence des composés carbonés précités.
D'une manière générale, on indique que les composés carbonés sont
désignés par leur désignation en référence au nombre d'atomes de carbone C5-
C8 par exemple, ou des indices supérieurs, i désignant en fâit l'indice des
composés carbonés correspondants.
L'étape B est alors suivie d'une étape C consistant à déterminer, à
partir des paramètres de composition [X;] et [Yi] de la phase gazeuse
respectivement de la phase liquide, les propriétés intensives, c'est-à-dire
les
paramètres thermodynamiques locaux de chaque phase liquide respectivement
gazeuse, à partir des paramètres de composition précités par simulation d'un
équilibre thermodynamique.
D'une manière plus spécifique, on indique que les paramètres
thermodynamiques locaux comportent au moins la densité de la phase liquide,
notée plIquide et la densité de la phase gazeuse notée p9a~ .
Les étapes A, B et C permettent ensuite de conduire une étape D
consistant à établir, à partir des paramètres thermodynamiques locaux, c'est-à-
dire essentiellement la densité de la phase liquide respectivement de la phase
gazeuse et du rapport de la section d'occupation relative de la phase gazeuse,
c'est-à-dire le paramètre ag précédemment mentionné, la densité mesurée du
mélange notée pmm, la densité mesurée du mélange vérifiant la relation :
Pmm -~~ ~ ag) Pfiquide + ag Pgaz '
L'étape D est alors suivie d'une étape E consistant à calculer, à partir
d'un processus adaptatif, l'évaluation de la densité calculée du mélange,
notée
pC , et de comparaison par itération successive de la densité calculée du
mélange
p, à la densité mesurée du mélange pmm à l'étape D précédente, les propriétés
CA 02576714 2007-01-29
WO 2006/024731 PCT/FR2005/001867
6
intensives du mélange précité, telles que la densité locale de la phase
liquide, de
la phase gazeuse et du mélange.
On conçoit, en particulier, que le calcul adaptatif précité peut être
effectué tant que la densité calculée du mélange p,, est suffisamment
distincte de
la densité mesurée du mélange pmr~, les méthodes de mesure et d'analyse des
paramètres thermodynamiques locaux n'étant pas suffisamment proches, mais
qu'au contraire, sur égalité de la densité calculée du mélange p. et de la
densité
mesurée du mélange pmm, les propriétés intensives du mélange sont établies et
le mélange est alors physiquement analysé et analytiquement reconstitué grâce
à
la mise en oruvre du procédé objet de la présente invention.
Sur la figure la, le caractère adaptatif du calcul .de la densité calculée
du mélange Rc, vis-à-vis de la densité mesurée du mélange pmm, est représenté
par la flèche de retour, le calcul adaptatif étant conduit tant que l'égalité
de la
densité calculée du mélange et de la densité mesurée du mélange n'est pas
satisfaite.
On indique toutefois que la notion d'égalité est une notion d'égalité
numérique, c'est-à-dire une notion d'égalité compte tenu d'une valeur de seuil
inhérente à, la précision des mesures, cette valeur de seuil pouvant être
prise
égale à quelques pour_cent.
Lorsque l'égalité dans les conditions précédemment mentionnées de la
densité calculée du mélange p~ et de la densité mesurée du mélange Pmm est
atteinte, l'étape E est alors suivie d'une étape F consistant à calculer le
rapport
du débit volumique de gaz et du mélange, ce rapport étant exprimé comme le
rapport du produit du débit volumique du gaz et du débit molaire du gaz à la
somme du produit du débit volumique du gaz et du débit molaire du gaz et du
produit du débit volumique du liquide et du débit molaire du liquide selon la
relation :
a9-1 =
GVF 1
n9.vg+1.v1~
CA 02576714 2007-01-29
WO 2006/024731 PCT/FR2005/001867
7
Dans la relation précédente, on indique que a9 désigne le rapport de la
section d'occupation relative de la phase gazeuse à la section totale de
l'écoulement, ng désigne le débit volumique de gaz, le débit volumique de
liquide
ni étant pris égal à 1, ce débit étant normé à la valeur unitaire par
convention.
En outre vg désigne le débit molaire de gaz et vl désigne le débit
molaire du liquide.
Différentes indications seront maintenant données quant à la mise en
uvre des étapes A à F du procédé objet de la présente invention tel que
représenté en figure la.
D'une manière générale, on indique que l'étape A d'échantillonnage de
la phase liquide et de la phase gazeuse consiste ainsi à isoler par séparation
du
liquide et du gaz du mélange en écoulement, pour en tirer une analyse de
composition.
Dans ce but, ainsi que représenté en figure lb, on place par exemple
de part et d'autre d'une cellule de mesure physique permettant d'effectuer la
mesure du paramètre de rapport de la section d'occupation relative de la phase
gazeuse à la section totale de l'écoulement, c'est-à-dire du paramètre o9 deux
chambres de capture, c'est-à-dire de diffusion du gaz de la phase gazeuse
respectivement de décantation du liquide de la phase liquide, ces chambres de
capture CH1 et CHZ étant désignées dans le domaine technique comme des
bottes. Elles permettent, de manière connue en tant que telle, au gaz de la
phase
gazeuse respectivement au liquide de la phase liquide de s'accumuler dans les
conditions de température et de pression de la conduite.
Le prélèvement des échantillons précités est justifié en raison du fait
que les deux échantillons prélevés de liquide respectivement de gaz sont
caractéristiques des phases en présence dans la cellule de mesure. Cette
hypothèse est justifiée bien que, dans l'absolu, il existe certaines pertes de
charge où certains phénomènes mécaniques, lesquels tendent à enrichir ou
appauvrir le gaz ou le liquide en composés volatils. L'échantillonnage précité
est
réalisé dans les bottes et donc à l'état statique, mais les produits
échantillonnés
sont représentatifs des composants des phases liquide respectivement gazeuse
en écoulement. Sur cette même figure TBP pour True Boiling Point en
anglais
désigne le point d'ébullition réel du composant échantillonné.
CA 02576714 2007-01-29
WO 2006/024731 PCT/FR2005/001867
8
Toutefois, une analyse de sensibilité, telle que le facteur de glissement
de l'écoulement, a permis de constater qu'une erreur dans l'évaluation de
composition des composés intermédiaires, typiquement, notamment, des
composés carbonés C5-C8, erreur due à un mauvais échantillonnage, n'a que
peu d'impact sur le calcul du facteur de glissement précité.
La raison physique en est que la concentration de ces composés
intermédiaires est généralement négligeable en comparaison des composés qui
sont déterminés avec une bonne précision dans les fluides ou mélanges en
écoulement dans les lignes de production de produits pétroliers.
D'une manière plus spécifique, on indique que les chambres de
capture CH1, CH2 représentées en figure 1 b peuvent également être remplacées
par des piquages de liquide respectivement de gaz effectués au même niveau
sur la ligne. Dans tous les cas, la seule contrainte est d'éviter les pertes
de
charge trop importantes, lesquelles seraient susceptibles d'avoir un impact
sur
les propriétés intensives des fluides échantillonnés, qui doivent rester
compatibles avec celles de la cellule de mesure.
En référence à la figure lb, on indique que la cellule de mesure du
rapport de la section d'occupation relative de la phâse gazeuse à la section
totale
de l'écoulement peut être constituée par une cellule de mesure par absorption
d'un rayonnement, tel qu'un rayonnement y par exemple, permettant de
déterminer par discrimination de l'absorption réalisée par la section
d'occupation
de la phase gazeuse par rapport à la phase liquide et finalement par rapport à
la
totalité de la section de la conduite, le rapport ag correspondant.
En ce qui concerne la mise en oruvre de l'étape B d'analyse de la
composition des échantillons de gaz et de liquide, on indique que cette
opération
peut être réalisée pour le gaz, encore désigné produit léger ou light ends
, par
chromatographie.
Une analyse poussée jusqu'aux composés carbonnés C7 à C8 a pu
s'avérer amplement suffisante, en particulier en considérant une température
de
conduite excédant rarement les 90 à 100 C.
On rappelle que la température d'ébullition du composé carboné C7 est
de 120 C à pression atmosphérique et que la marge de température jusqu'à
l'ébullition de ce dernier est tout à fait acceptable.
CA 02576714 2007-01-29
WO 2006/024731 PCT/FR2005/001867
9
En ce qui concerne la mise en oeuvre de l'analyse de la composition du
liquide et en particulier, des produits tels que l'huile encore désignée
heavy
ends ou produit lourd, cette analyse peut être effectuée par une
distillation
physique.
Toutefois, pour un gain de temps et une conduite du procédé objet de
l'invention sensiblement en temps réel, on préfèrera une distillation simulée,
en
particulier, selon la norme IASTN 2887 proposée par l'American Society For
Thermodynamics, laquelle préconise un intervalle d'analyse compris entre les
composés carbonés C5 et C44 qui peut s'avérer suffisant pour caractériser les
produits lourds.
On rappelle, toutefois, que la distillation simulée précitée à haute
température permet aujourd'hui d'atteindre le composé carboné C120 mais que la
mise en oeuvre de celle-ci, pour des composés carbonés à indice plus élevé que
C44, suppose des moyens de calcul et des temps de traitement plus importants.
A la fin de l'étape B, on rappelle que les paramètres de composition
sont notés :
- [X;] pour la phase gazeuse (P9
-[Y;] pour la phase liquide i désignant dans les deux cas l'indice
des composés carbonés correspondants.
L'étape C peut alors être effectuée à partir des paramètres de
composition précités, lesquels permettent de déterminer par simulation d'un
équilibre thermodynamique les propriétés intensives, c'est-à-dire les
paramètres
thermodynamiques locaux de chaque phase.
Les propriétés intensives de chaque phase sont notamment la densité
Aliquiaedu liquide de la phase liquide et la densité Pgaz de la phase gazeuse.
D'une façon plus spécifique, on indique en outre que les densités du
gaz et du liquide peuvent également être mesurées physiquement de façon à
permettre, par recalage vis-à-vis des valeurs calculées de densité pl.quide et
p9az
précédemment mentionnées à l'étape C, d'effectuer un affinage du modèle
thermodynamique utilisé par une méthode de recalage. Cette méthode de
recalage consiste, par exemple, à partir des paramètres calculés de densité de
liquide respectivement du gaz ~ pliquide et p9aZ de réintroduire les valeurs
mesurées
CA 02576714 2007-01-29
WO 2006/024731 PCT/FR2005/001867
localement, c'est-à-dire au niveau des échantillons de gaz et de liquide pour
effectuer un recalage du calcul de densité p,iq,,de et pgaZ , à partir des
données de
composition précitées.
Le processus de recalage ne sera pas décrit en détail car il correspond
5 à des méthodes physiques connues en tant que telles.
L'étape C peut alors être suivie selon un aspect particulièrement
remarquable du procédé objet de l'invention par l'étape B consistant à
calculer, à
partir des paramètres thermodynamiques locaux p~~qu~de et p9az , la densité
mesurée du mélange en intégrant le paramètre de rapport de la section
10 d'occupation relative de la phase gazeuse à la section totale de
l'écoulement,
paramètre ag selon la relation :
Pmm (1 ag)pliquide + ag. pgaz
L'étape D peut alors être suivie de l'étape E représentée sur la figure
1a, cette étape étant avantageusement conduite sur la base d'un débit unitaire
exprimé en kilomoles/heure ou en Lbmole/heure pour livremole/heure ou toute
unité dérivée pour le liquide.
Dans ces conditions, on cherche à évaluer le débit de gaz également
exprimé dans l'unité correspondante pour arriver à une convergence et à une
égalité sensiblement de la densité du mélange mesuré pmm obtenu à l'étape D
avec la densité du mélange calculé pC .
Lorsque l'égalité entre la densité du mélange mesuré pmm et la densité
du mélange calculé pc est sensiblement atteinte dans les conditions
mentionnées précédemment, alors les propriétés intensives du mélange, c'est-à-
dire les propriétés de paramètres thermodynamiques locaux du mélange, sont
établies et le mélange est physiquement analysé et analytiquement reconstitué.
Lorsque l'échantillonnage du gaz et du liquide de la phase gazeuse
respectivement de la phase liquide est suffisamment représentatif du mélange
en
écoulement dans la conduite, la température du mélange est sensiblement la
même que la température d'échantillonnage.
Ceci permet de confirmer l'hypothèse selon laquelle les phases liquide
et gazeuse prélevées dans les chambres de capture sont les mêmes que celles
CA 02576714 2007-01-29
WO 2006/024731 PCT/FR2005/001867
11
évoluant dans la cellule de mesure du rapport de la section d'occupation
relative
de la phase gazeuse à la section totale d'écoulement dans la conduite,
L'étape E a ainsi permis de reconstituer analytiquement le fluide, c'est-
à-dire le mélange en écoulement dans son ensemble qui, bien entendu, valide la
méthode de reconstruction analytique de ce fluide polyphasique, même en
présence d'un facteur de glissement.
La connaissance des propriétés intensives du mélange telles que les
volumes molaires et les proportions respectives de liquide et de gaz calculés
par
méthode du flash enthalpique à l'étape E, permet alors d'obtenir le rapport du
débit volumique de gaz au débit volumique de mélange d'hydrocarbures
multiphasique, rapport dénommé GVF, ainsi que mentionné précédemment dans
la description.
Une description plus détaillée de l'étape E de calcul adaptatif de la
densité calculée du mélange pc, vis-à-vis de la densité mesurée du mélange
Rmm, sera maintenant donnée en liaison avec la figure Ic.
Pour mettre en oeuvre l'étape E de la figure la, on dispose des
résultats d'analyse de composition des composés carbonés de la- phase gazeuse
respectivement de la phase liquide, notée [X;] respectivement [Y;], obtenus à
l'étape B, de la valeur de la pression dans la conduite mesurée, ainsi que de
la
température d'échantillonnage T. mesurée au niveau des chambres de capture
du gaz respectivement du liquide.
L'ensemble de ces données constïtue l'étape de départ Eo de la figure
1 c.
Suite à l'étape de départ précitée, on procède en une étape El en une
estimation de ng désignant le débit molaire de gaz ramené à un débit molaire
de
liquide unitaire, ainsi que mentionné précédemment dans la description.
L'étape El est alors suivie d'une étape E2 consistant à calculer la
composition molaire du mélange selon la relation :
1 n ([XI] + ns[Yi]) =
[Zil= +
9
Dans la relation précédente, on indique que la composition molaire est
ainsi obtenue pour tous les composés carbonés d'indice i correspondants.
CA 02576714 2007-01-29
WO 2006/024731 PCT/FR2005/001867
12
L'étape E2 est alors suivie d'une étape E3 consistant à calculer
l'enthalpie du mélange selon la relation :
Hm = 1 + n (Hi +n9. Hg),
9
Dans la relation précédente, on rappelle que :
- Hm désigne l'enthalpie du mélange ;
- Hi désigne l'enthalpie du liquide ;
- Hg désigne l'enthalpie du gaz.
L'étape E3 est alors suivie d'une étape E4 consistant à calculer la
température du mélange par simulation d'un équilibre flash enthalpique à
partir
de la valeur de pression P, de la composition molaire obtenue à l'étape E2 et
de
l'enthalpie du mélange Hm obtenu à l'étape E3. Cette opération permet de
calculer la densité calculée p, du mélange.
A la suite de l'étape E4, l'appel de la valeur de la densité du mélange
mesuré pmm obtenue à l'étape D permet ensuite à l'étape E6 de soumettre la
valeur de la densité calculée p. à l'étape E4 àï un test d'égalité avec la
valeur
mesurée de la densité du mélange pmm '
La notion d'égalité a été définie préalablement dans la description.
Sur réponse négative au test E6 précité, la densité calculée du
mélange n'étant pas suffisamment proche de la densité mesurée du mélange, un
retour à l'étape de calcul de la composition molaire E2 est effectué par
l'intermédiaire d'une étape E7 permettant d'ajuster en fait la valeur de
l'estimation
de ng.
Le processus d'ajustage de la valeur de n9 peut être effectué soit, à
partir de la méthode mathématique dite sécante, soit à partir d'une méthode
plus
générale de Raphson-Newton, par exemple. Ces méthodes mathématiques
mises en eeuvre par des algorithmes et des programmes de calcul disponibles
dans le commerce ne seront pas décrites en détail pour cette raison. Elles
permettent d'assurer la convergence à partir du réajustement de la valeur de
ng,
de la valeur de densité calculée du mélange p~,avec la valeur de densité
mesurée du mélange pmm .
CA 02576714 2007-01-29
WO 2006/024731 PCT/FR2005/001867
13
Sur réponse positive au test de comparaison E6, on retient en une
étape E$ pour composition molaire les valeurs de composition molaire finale
[Z;]f
calculées à-la dernière itération provoquée par le retour E7, la valeur de
pression
P et la valeur de température du mélange final Tmf.
Le procédé objet de l'invention, tel que décrit en liaison avec les figures
la, lb et Ic, doit être compris comme un procédé relatif à la mesure du
rapport
du débit volumique de gaz au débit volumique d'un mélange d'hydrocarbures
multiphasiques.
En particulier, en référence à la figure la, bien que l'étape D décrite en
liaison avec cette figure fasse appel à une relation exprimée dans le cas d'un
mélange diphasique, l'ensemble des relations précitées peut être mis en oruvre
pour un mélange multiphasique dans les conditions ci-après.
Pour une phase liquide comportant de l'eau et des hydrocarbures
mélangés, l'étape El de la figure 1 c consistant à estimer le débit molaire du
gaz
de la phase gazeuse, vis-à-vis du débit molaire du liquide de la phase
liquide,
peut avantageusement être remplacée par une étape consistant à fixer l'un des
composants du liquide comme constituant une phase de référence (Pix =Çor a
un débit molaire unitaire, en une étape Elo représentée en figure 1d, puis à
estimer en une étape El, le débit molaire relatif du gaz respectivement de
l'autre
composant du liquide vis-à-vis du débit molaire unitaire de la phase de
référence.
Ceci permet d'obtenir un vecteur de débit molaire relatif constitué par
l'ensemble des composants de débit molaire relatif de chaque phase, vis-à-vis
de
la phase de référence.