Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02585606 2007-04-27
WO 2006/052227 PCT/US2004/032785
TITLE OF THE INVENTION
HIGH-DOSAGE EXTENDED-RELEASE FORMULATION OF GEPIRONE
BACKGROUND OF THE INVENTION
Field of the Invention
The present invention relates to high-dosage extended-release formulation of
gepirone
and methods of treating major depression by administering the same to a
subject in need
thereof. More specifically, the present invention relates to a high-dosage
extended-release
tablet form of gepirone. The present invention also relates to a method of
treating depression
in mammals by administering to a subject in need thereof an effective amount
the high-
dosage extended-release formulation of gepirone in accordance with the present
invention.
Discussion of the Back rg ound
Gepirone (also known as 4,4-dimethyl-l- [4- [4-(2-pyrimidinyl)- 1 -
piperazinyl] -butyl] -
2,6-piperidinedione hydrochloride) can be obtained by the process described in
Example 7 of
Temple, US patent 4,423,049, (which is incorporated herein in its entirety by
reference) and
has the following structure:
0
N
N
ONN \
N
Gepirone and its salts are known to possess antidepressant and anxiolytic
properties,
presumably by serving as an agonist of the 5-HT1A receptor. They are typically
used to treat
depression, dysthymia, impulse disorders, panic attacks and the like. However,
in
immediate-release formulations, gepirone has a short half-life. The time to
maximum drug
concentration of gepirone in the bloodstream (Tmax) is about 1 hour and its
T50 (i.e., time until
50% of the drug has been released under controlled in vitro conditions) is
about 2.5-3 hours
(see U.S. 5,478,572, incorporated herein in its entirety by reference).
-1-
CA 02585606 2007-04-27
WO 2006/052227 PCT/US2004/032785
Owing to its rapid metabolism, gepirone has been administered in the past in
several
small dosages--e.g., 5 to 10 mg doses, 2 to 3 times per day. However, this
multiple dosing
scheme can lead to compliance problems. Moreover, failure to take the second
or third dose
results in unacceptably low plasma levels of gepirone. Further, studies
indicate that, for 15 to
20 hours after administration, oral immediate release gepirone formulations
can yield
significant variations in human plasma concentrations. (For as further
discussion see
D.S.Robinson et al, Clinical Therapeutics, 1618-1633 (2003)).
However, as with any medicinal compound, each patient requires a different
dosage
level to specifically tailor the therapy to that patient's physical make-up,
age, and disease
complexity. Therefore, even with the low-dosage extended-release form of
gepirone
disclosed in U.S. Pat. No. 5,478,572, multiple dosing may be required (i.e., 4
tablets of a 20-
mg dosage as opposed to a single high dosage tablet), which is inconvenient
and expensive.
And, as such, there remains a critical need for high-dosage extended-release
form of gepirone
having a dissolution profile analogous to the low-dosage extended-release form
of gepirone
disclosed in U.S. Pat. No. 5,478,572, while maintaining the same relative
bioequivalence.
Heretofore, it was believed that it was not possible to formulate a high-
dosage
extended-release form of gepirone that would not result in an unsatisfactory
increase in the
amount of 1-(2-pyrimidinyl)piperazine (gepirone's principal metabolite, which
is believed to
be responsible for adverse side effects, including dizziness, nausea, headache
and drowsiness)
or a decrease in the relative efficacy. This skepticism was borne of the fact
that in order to
sufficiently control the release of gepirone the binder content would have to
be increased to
such a degree such that the resultant tablet for oral administration would end
up being a
"horse pill" (i.e., prohibitively large). Alternatively, the dosage
administration would have to
be formulated for delivery by a less desirable means (e.g., intravenous
administration).
SUMMARY OF THE INVENTION
It is an object of the present invention to provide a pharmaceutical
composition,
containing:
(1) from about 14 to about 25.wt% gepirone, or bioactive metabolite thereof,
as a
free base or a pharmaceutically acceptable salt thereof;
(2) from about 70 to about 85 wt% of a pharmaceutically acceptable cellulosic
polymer matrix; and
-2-
CA 02585606 2007-04-27
WO 2006/052227 PCT/US2004/032785
(3) suitable amounts of one or more pharmaceutically acceptable excipients,
wherein the release rate of gepirone from the dosage form is such that about
18 to 24 hours are required to attain from about 90 to about 95% absorption
of gepirone.
In this object of he invention, (3) further comprises at least one
pharmaceutically
acceptable excipient selected from the group consisting of colorant,
microcrystalline
cellulose, colloidal silica and magnesium stearate; (2) is
hydroxypropylmethylcellulose
having a viscosity of from about 15,000 cps to about 100,000 cps; and (a)
ranges from about
15.5 to about 18.7 wt%.
Another object of the present invention is to provide a composition
containing:
(a) from about 14.0 to about 24.4 wt% gepirone, or bioactive metabolite
thereof,
as a free base or a pharmaceutically acceptable salt thereof,
(b) from 70.5 to 82.1 wt% hydroxypropylmethylcellulose having a viscosity of
from about 15,000 to about 100,000 cps.,
(c) 0 to about 1 wt% colorant,
(d) about 8.0 to about 16.7 wt% microcrystalline cellulose,
(e) about 0.39 to about 0.47 wt% colloidal silica, and
(f) about 0.29 to about 1.0 wt% magnesium stearate.
In another object of the present invention is to provide a composition
containing:
(a) about 15.6 wt% of gepirone hydrochloride
(b) about 75.3 wt% hydroxypropylmethylcellulose,
(c) about 0.31 wt% yellow ferric oxide,
(d) about 8.0 wt% microcrystalline cellulose,
(e) about 0.42 wt% colloidal silica, and
(f) about 0.31 wt% magnesium stearate.
In another object of the present invention is to provide a composition
containing:
(a) about 19.5 wt% of gepirone hydrochloride
(b) about 70.7 wt% hydroxypropylmethylcellulose,
(c-1) about 0.24 wt% yellow ferric oxide,
(c-2) about 0.61 wt% red ferrid oxide
(d) about 8.2 wt% microcrystalline cellulose,
(e) about 0.39 wt% colloidal silica, and
(f) about 0.29 wt% magnesium stearate.
-3-
CA 02585606 2007-04-27
WO 2006/052227 PCT/US2004/032785
In each of the aforementioned objects it is yet a further object of the
present invention
to provide a tablet form of the high-dosage extended-release forms of
gepirone.
In a preferred object of the present invention, the tablet has an ovoid-
rectangular
shape with either biconvex or flat faces. In this object, the tablet has an
overall dimension of
0.400 0.05 inches by 0.325 0.05 inches with a thickness of 0.240 0.025
inches
(biconvex faces) or a thickness of 0.195 0.025 inches (flat faces). Also
within this object,
the total weight of said tablet preferably ranges from 350 to 450 mg.
In yet another object of the present invention is to provide a method of
ameliorating
depression, anxiety, or psychological disorders (in particular depression) by
administering to
a patient (in particular a human) in need thereof an effective amount of the
compositions or
tablet form of the compositions in accordance with the present invention.
Another object of the present invention is to provide methods of making the
aforementioned tablet forms of the compositions of the present invention.
The above objects higlilight certain aspects of the invention. Additional
objects,
aspects and embodiments of the invention are found in the following detailed
description of
the invention.
BRIEF DESCRIPTION OF THE FIGURES
A more complete appreciation of the invention and many of the attendant
advantages
thereof will be readily obtained as the same becomes better understood by
reference to the
following Figures in conjunction with the detailed description below.
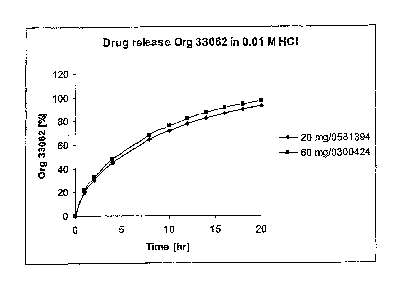
Figure 1 shows the comparative dissolution profile of 20 mg and 60 mg tablets
described in the Preparation Example under in 0.01 M HCI (see Example 1).
Figure 2 shows the comparative dissolution profile of 20 mg and 60 mg tablets
described in the Preparation Example under in water (see Example 1).
Figure 3 shows the comparative dissolution profile of 20 mg and 60 mg tablets
described in the Preparation Example under in a pH 4.5 media (see Example 1).
Figure 4 shows the comparative dissolution profile of 20 mg and 60 mg tablets
described in the Preparation Example under in a pH 6.8 media (see Example 1).
-4-
CA 02585606 2007-04-27
WO 2006/052227 PCT/US2004/032785
DETAILED DESCRIPTION OF THE INVENTION
Unless specifically defined, all technical and scientific terms used herein
have the
same meaning as commonly understood by a skilled artisan in biochemistry,
chemistry,
pharmacology, and the medical sciences.
All methods and materials similar or equivalent to those described herein can
be used
in the practice or testing of the present invention, with suitable metliods
and materials being
described herein. All publications, patent applications, patents, and other
references
mentioned herein are incorporated by reference in their entirety. In case of
conflict, the
present specification, including definitions, will control. Further, the
materials, methods, and
examples are illustrative only and are not intended to be limiting, unless
otherwise specified.
The present invention is based, in part, on the inventor's discovery that a
high-dosage
extended-release form of gepirone can be obtained wherein the dosage form has
a
comparable dissolution profile and bioequivalence (for 1-PP, 3'-OH gepirone,
and gepirone)
to that of the low-dosage extended-release form of gepirone disclosed in U.S.
Pat. No.
5,478,572, despite a reduction in the overall ratio between active ingredients
and the release-
controlling polymer (binder).
Extended release drug forms (especially gepirone) offer several advantages
over
immediate-release systems. Patient compliance is better because the extended
release dosage
forms need be taken only once in a 24-hour period. Thus, plasma concentration
levels do not
vary unacceptably--i.e., give high initial drug levels that are associated
with the incidence of
unwanted side effects, as well as having rapid drops in drug levels to below
therapeutic
levels--when the ER dosage forms are administered. In addition, in the case of
gepirone, due
to the slow time-release 1-PP levels are maintained at a satisfactory level
thus avoiding
significant complications arising from this metabolite.
In a general embodiment, the present invention provides a pharmaceutical
composition for making an oral extended release gepirone dosage form
comprising:
(1) from about 14 to about 25 wt % gepirone, or bioactive metabolite thereof,
as a free
base or a pharmaceutically acceptable salt thereof;
(2) from about 70 to about 85 wt % of a pharmaceutically acceptable cellulosic
polymer matrix; and
(3) suitable amounts of one or more pharmaceutically acceptable excipients,
wherein
the release rate of gepirone from the dosage form is such that about 18 to 24
hours
are required to attain from about 90 to about 95% absorption of gepirone.
-5-
CA 02585606 2007-04-27
WO 2006/052227 PCT/US2004/032785
In a particularly preferred embodiment of the present invention is an extended-
release
oral dosage form of gepirone, containing, expressed in weight percent:
(a) about 14.0 to about 24.4% (preferably, about 15.5 to about 18.7%)
gepirone, or
bioactive metabolite thereof, as a free base or a pharmaceutically acceptable
salt
thereof, for example a hydrochloride salt,
(b) about 70.5 to about 82.1% hydroxypropylmethylcellulose having a viscosity
of
from about 15,000 to about 100,000 cps.,
(c) 0 to about 1% (preferably 0 to about 0.3%) iron oxide,
(d) about 8.0 to about 16.7% microcrystalline cellulose,
(e) about 0.39 to about 0.47% (preferably about 0.42 to about 0.47%) colloidal
silica,
and
(f) about 0.29 to about 1.0% magnesium stearate.
Although the aforementioned embodiment is defined in terms of particular
compounds for each component, it is to be understood that within the scope of
the present
invention these components may be replaced individually or in various
combinations as
described below. For each of the components below it is to be understood that
the recited
weight percentage reflects the total concentration of each component. For
example, if there is
a mixture of compounds that fall within the scope of a defined component group
the recited
weight percentage reflects the total for that mixture of compounds.
Component (a):
In a preferred embodiment, component (a) is present in the extended-release
oral
dosage form in a weight percentage of about 14.0 to about 24.4%, more
preferably about 15.5
to about 18.7%. It is particularly preferred that component (a) be gepirone or
a
pharmaceutically acceptable salt thereof.
In vivo, gepirone is metabolized resulting in to major pharmacologically
active
metabolites: 1-(2-pyrimidinyl)piperazine (1-PP) and 3'-OH gepirone (D.S.
Robinson et al,
Clinical Therapeutics, pp. 1618-1633 (2003)). Two additional metabolites that
may also
posses bioactivity are also formed in vivo: 5-OH gepirone and 3',5-dihydroxy
gepirone.
In humans, the release of 1-PP, a common azapirone metabolite, is believed to
be
responsible for adverse side effects, including dizziness, nausea, headache
and drowsiness.
Moreover, 1-PP is a presynaptic a-2-adrenoceptor antagonist and it has been
reported that it
did not exhibit antidepressant-like characteristics in pre-clinical tests
(D.S. Robinson et al,
-6-
CA 02585606 2007-04-27
WO 2006/052227 PCT/US2004/032785
Clinical Therapeutics, pp. 1618-1633 (2003)). In contrast, the bioactive
metabolites (3'-OH
gepirone, in particular) have significant affinity for 5-HT1A receptors. 3'-OH
gepirone has
been demonstrated to modify 5-HT neurotransmission in a comparable manner to
gepirone
even though 3'-OH has been found to exhibit full agonism at post-synaptic
receptors in the
hippocampus, whereas gepirone is a partial agonist (D.S. Robinson et al,
Clinical
Therapeutics, pp. 1618-1633 (2003)).
In view of the foregoing, in an embodiment of the present invention one or
more of
the bioactive gepirone metabolites 3'-OH gepirone, 5-OH gepirone, and 3',5-
dihydroxy
gepirone, or a pharmaceutically acceptable salt thereof, may be used in place
of gepirone.
Further, although the pharmaceutically acceptable salt form is preferred, it
is
contemplated that gepirone or the bioactive gepirone metabolites may be in a
hydrate form,
an enantiomeric form or mixture, or crystal form.
The pharmaceutical compounds suitable for administration in the present
invention
may be hydrochloride salts, but the free bases and other pharmaceutically
acceptable salts are
also suitable. The term "pharmaceutically acceptable salt" is well known in
the art and the
artisan is directed to S. M. Berge, et al. (J. Pharmaceutical Sciences, 66: 1-
19 (1977),
incorporated herein by reference) for a further description. Suitable
pharmaceutically
acceptable salts for administration in the present invention include acid
addition salts. The
acid addition salt may be formed by mixing a solution of the compound with a
solution of a
pharmaceutically acceptable non-toxic acid such as hydrochloric acid,
hydrobromic acid,
fumaric acid, maleic acid, succinic acid, acetic acid, citric acid, tartaric
acid, carbonic acid,
phosphoric acid, perchloric acid, sulphuric acid, oxalic acid, or malonic
acid. Where the
compound carries an acidic group, for example a carboxylic acid group, the
present invention
also contemplates salts thereof, preferably non-toxic pharmaceutically
acceptable salts
thereof, such as the sodium, potassium and calcium salts thereof.
Other pharmaceutically acceptable salts include adipate, alginate, ascorbate,
aspartate,
benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate,
camphorsulfonate,
citrate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate,
formate,
fumarate, glucoheptonate, glycerophosphate, gluconate, hemisulfate,
heptanoate, hexanoate,
hydroiodide, 2-hydroxy-ethanesulfonate, lactobionate, lactate, laurate, lauryl
sulfate, malate,
maleate, malonate, methanesulfonate, 2-naphthalenesulfonate, nicotinate,
nitrate, oleate,
oxalate, palmitate, pamoate, pectinate, persulfate, 3-phenylpropionate,
phosphate, pictate,
pivalate, propionate, stearate, succinate, sulfate, tartrate, thiocyanate, p-
toluenesulfonate,
-7-
CA 02585606 2007-04-27
WO 2006/052227 PCT/US2004/032785
undecanoate, valerate salts, and the like. Representative alkali or alkaline
earth metal salts
include sodium, lithium, potassium, calcium, magnesium, and the like. Further
pharmaceutically acceptable salts include, when appropriate, salts of amine
groups. Salts of
amine groups may also comprise the quaternary ammonium salts in which the
amino nitrogen
atom carries an alkyl, alkenyl, alkynyl or aralkyl group, nontoxic ammonium,
quaternary
ammonium, and amine cations formed using counterions such as halide,
hydroxide,
carboxylate, sulfate, phosphate, nitrate, loweralkyl sulfonate and aryl
sulfonate.
Component (b):
In a preferred embodiment, component (b) is present in the extended-release
oral
dosage form in a weight percentage of about 70.5 to about 82.1%. Preferably,
component (b)
is hydroxypropylmethylcellulose having a viscosity of from about 15,000 to
about 100,000
cps, which is a release-controlling polymer & binder (a.k.a., sustaining
agent).
For oral formulations and dosage fonns, the use of a polymeric cellulose
matrix, or
sustaining agent, is preferred. Suitable matrixes include
hydroxyalkylsubstituted
alkylcelluloses having viscosities of about 15,000 cps to about 100,000 cps.
Examples of
acceptable hydroxymethyl propylcellulose (HPMC) samples include grades K15M
and
K100M (i.e., 15,000 and 100,000 cps, respectively).
HPMC may be replaced (all or in part) or added to in the present invention,
the
replacement of some or the entire HPMC matrix may be with dicalcium phosphate
or lactose,
each of which generally increases dissolution rates.
In addition to the binder of component (b), the formulation of the present
invention
may also contain auxiliary binding agents, such as syrup, acacia, gelatin,
sorbitol, tragacanth,
or polyvinyl pyrrolidone. Examples of suitable fillers or extenders also
include starches (e.g.,
maize-starch), lactose, sorbitol, glycine, sucrose, glucose, mannitol, and
silicic acid, sodium
citrate and dicalcium phosphate. Examples of other suitable binders may
include chitosan,
alginates, gelatin, polyvinylpyrrolidinone, sucrose, acacia, and mixtures
thereof.
In an embodiment of the present invention the ratio of gepirone to binder
(gepirone:binder) ranges from 1:3.5 to 1:14.5. Preferably, the binder is
hydroxymethyl
propylcellulose (HPMC).
Component (c):
-8-
CA 02585606 2007-04-27
WO 2006/052227 PCT/US2004/032785
In a preferred embodiment, component (c) is present in the extended-release
oral
dosage form in a weight percentage of 0 to about 1%, preferably 0 to about
0.3%. Although
component (c) may be any colorant, it is preferred that the colorant be an
iron oxide. In a
particularly preferred embodiment the iron oxide is red ferric oxide, yellow
ferric oxide, or
mixtures thereof.
Additional exemplary colorants that may be used in place of or in addition to
the
foregoing include, but are not limited to, FD&C and D&C lakes, titanium
dioxide, iron
oxides, natural pigments, or dyes approved for ingestion by the U.S. Federal
Drug
Administration, or combinations thereof.
-9-
CA 02585606 2007-04-27
WO 2006/052227 PCT/US2004/032785
Component (d):
In a preferred embodiment, component (d) is present in the extended-release
oral
dosage form in a weight percentage of about 8.0 to about 16.7%. Although
component (d)
may be any diluent and/or compression aid, it is preferred that the
diluent/compression aid be
microcrystalline cellulose.
Additional diluents may include, but are not limited to, water or other
solvents,
solubilizing agents and emulsifiers such as ethyl alcohol, isopropyl alcohol,
ethyl carbonate,
ethyl acetate, benzyl alcohol, benzyl benzoate, propylene glycol, 1,3-butylene
glycol,
dimethylformamide, oils (in particular, cottonseed, groundnut, corn, germ,
olive, castor, and
sesame oils), glycerol, tetrahydrofurfuryl alcohol, polyethylene glycols and
fatty acid esters
of sorbitan, and mixtures thereof.
In yet another embodiment of the present invention, the ratio of gepirone to
diluent
(gepirone:diluent) ranges from 1:3 to 2.5:1. Preferably, the diluent is
microcrystalline
cellulose.
Component (e):
In a preferred embodiment, component (e) is present in the extended-release
oral
dosage form in a weight percentage of about 0.39 to about 0.47%, preferably
about 0.42 to
about 0.47%. Although component (e) may be any glidant, it is preferred that
the glidant be
colloidal silica (colloidal silicon dioxide).
Additional glidants include, but are not limited to, cornstarch, talc, or
stearic acid, or
combinations thereof.
-10-
CA 02585606 2007-04-27
WO 2006/052227 PCT/US2004/032785
Component (f):
In a preferred embodiment, component (f) is present in the extended-release
oral
dosage form in a weight percentage of about 0.29 to about 1.0%. Although
component (f)
may be any lubricant, it is preferred that the lubricant be magnesium
stearate.
Additional lubricants may include, but are not limited to cellulose, talc,
polyethyleneglycol, silicas, sodium lauryl sulfate, calcium stearate, and
mixtures thereof.
Alternative com onents:
It is contemplated in the present invention that the extended-release oral
dosage form
may also contain, in addition to components (a) through (f), various
additional components.
Based on the description above and Remington's Pharmaceuticals Sciences, 18th
Edition
(incorporated herein by reference), especially Part 8 therein, "Pharmaceutical
Preparations
and Their Manufacture, it would be well within the purview of the skilled
artisan to add new
compounds to the preferred embodiments described above.
In an embodiment of the present invention, the extended-release oral dosage
form
may include one or more additional pharmaceutically acceptable carriers. As
used herein, the
term "pharmaceutically acceptable carrier" means a non-toxic, inert solid,
semi-solid or liquid
filer, diluent, encapsulating material or formulation auxiliary. Some examples
of materials
which can serve as pharmaceutically acceptable carriers are already mentioned
above,
therefore, it is understood that in this aspect of the invention,
pharmaceutically acceptable
carriers may be any type of filer, diluent, encapsulating material or
formulation auxiliary not
previously mentioned.
In another embodiment, the extended-release oral dosage form may include may
also
include one or more releasing agents, coating agents, sweetening, flavoring
and perfuming
agents, preservatives and antioxidants.
The flavorant(s), which is used primarily for taste- and/or odor-masking, may
be
vanillin, sodium citrate, citric acid, mint, orange, lemon oil, or any other
pharmaceutically
approved flavorant or tastemasking agent, and combinations thereof.
Shape And Physical State Of The Hi h-g Dosage Extended-Release Form Of
Gepirone
The present invention contemplates extended-release oral dosage forms that are
suitable for orally administration. These forms include tablets, capsules,
caplets, lozenges,
powders, suspensions, syrups and the like are suitable forms. Preferentially,
the tablet may
-11-
CA 02585606 2007-04-27
WO 2006/052227 PCT/US2004/032785
be mentioned including those having a convex shape, a spherical (i.e., round)
shape, or a
capsule shape.
In a preferred embodiment, the extended-release oral dosage form of gepirone
is a
tablet. Preferably, the tablet has oval shape, which enhances their surface
area and improves
the release of gepirone therefrom. More preferably, the tablet has an ovoid-
rectangular
shape, which may have either flat or biconvex faces.
When the extended-release oral dosage form of gepirone is a tablet having an
ovoid-
rectangular shape, it is preferred that the tablets have an overall dimension
of 0.400 0.05
inches by 0.325 0.05 inches witli a thickness of 0.240 0.025 inches
(biconvex faces) or
0.195 0.025 inches (flat faces).
In another embodiment of the present invention the overall tablet weight
ranges from
350 to 450 mg, preferably from 375 to 425 mg, more preferably from 385 to 410
mg.
Further, within the context of the present invention, the tablet weight range
of 350 to 450 mg,
preferably from 375 to 425 mg, more preferably from 385 to 410 mg correspond
to the total
weight of the unit dose form.
Therefore, the pharmaceutical compositions (i.e., extended-release oral dosage
form
of gepirone) of the present invention preferably contain between about 55 to
100 mg
(preferably from 60 to 80 mg, more preferably 60 or 80 mg) of the active
ingredient per unit
dose.
The tablets may, if desired, be coated using known methods and excipients that
may
include enteric coating using for example hydroxypropylmethylcellulose
phthalate. Such
tablets may, if desired, be provided with enteric coatings by known methods,
for example by
the use of cellulose acetate phthalate. They may optionally contain opacifying
agents and can
also be of a composition that they release the active ingredient(s) only, or
preferentially, in a
certain part of the intestinal tract, optionally, in a delayed manner.
Examples of embedding
compositions that can be used include polymeric substances and waxes.
Similarly, capsules, for example hard or soft gelatin capsules, containing the
active
compound with or without added excipients, may be prepared by known methods
and, if
desired, provided with enteric coatings in a known manner. The contents of the
capsule may
be formulated using known methods so as to give sustained release of the
active compound.
In such solid dosage forms the active compound may be admixed with at least
one inert
diluent such as sucrose, lactose or starch. Such dosage forms may also
comprise, as is
normal practice, additional substances other than inert diluents, e.g.,
tableting lubricants and
-12-
CA 02585606 2007-04-27
WO 2006/052227 PCT/US2004/032785
other tableting aids such a magnesium stearate and microcrystalline cellulose.
In the case of
capsules, tablets and pills, the dosage forms may also comprise buffering
agents. They may
optionally contain opacifying agents and can also be of a composition that
they release the
active ingredient(s) only, or preferentially, in a certain part of the
intestinal tract, optionally,
in a delayed manner. Examples of embedding compositions that can be used
include
polymeric substances and waxes.
Solid compositions of a similar type may also be employed as fillers in soft
and hard-
filled gelatin capsules using such excipients as lactose or milk sugar as well
as high
molecular weight polyethylene glycols and the like.
Treatment methods:
Gepirone can be orally administered in once-a-day extended release dosage
forms,
which contain gepirone hydrochloride (or a bioactive metabolite thereof), a
cellulosic
polymer matrix and suitable amounts of pharmaceutical excipients. The
resultant gepirone
formulation yields oral products the take about 19 to about 24 hours to
release 90 to 95% of
the active agent.
Therefore, in yet another embodiment of the present invention is a method of
treating
a patient diagnosed as suffering from major (primary or general) depression,
with or without
generalized anxiety disorder, comprising administering to said patient an
effective amount of
the extended-release oral dosage form gepirone as described in the present
application.
In this embodiment, the patient (preferably a human, but inclusive of all
mammals) in
need of treatment is first diagnosed as suffering from a particular depressive
condition. For a
discussion of patient evaluation, diagnostic criteria, etc. of anxiety and
depression, the skilled
artisan is referred to: DSM-IV-TR (Diagnostic and Statistical Manual ofMental
Disorders,
American Psychiatric Association, Washington, D.C., 2000; Depressive
Disorders: 369-428,
and Anxiety Disorders: 429-484).
To this end, the gepirone extended-release composition and dosage forms of the
invention are designed to deliver an effective anti-depressive and/or
anxiolytic amount of
gepirone or a pharmaceutically acceptable salt thereof to a mammal, preferably
a human
patient.
"Effective" doses as used herein refer to an amount of about 0.01 to 40 mg/kg
body
weight/day are contemplated, preferably 0.05 to 20 mg/kg body weight/day, more
preferably
0.1 to 2 mg/kg of body weight/day. For certain central nervous system
disorders, 15 to 90
-13-
CA 02585606 2007-04-27
WO 2006/052227 PCT/US2004/032785
mg/day, preferably 30-60 mg/day, are recommended. See U.S. Pat. No. 4,771,053
and U.S.
Pat. No. 5,478,572. Alternatively, the effective dose or delivery system
should result in
plasma concentrations in the range of about 1 ng/ml to about 20 ng/ml,
preferably about 1
ng/ml to about 5 ng/ml.
As described above, in an embodiment of the present invention, the extended-
release
oral dosage form of gepirone for use in the inventive method contains,
expressed in weight
percent:
(a) about 14.0 to about 24.4% (preferably, about 15.5 to about 18.7%)
gepirone, or
bioactive metabolite thereof, as a free base or a pharmaceutically acceptable
salt
thereof, for example a hydrochloride salt,
(b) about 70.5 to about 82.1% hydroxypropylmethylcellulose having a viscosity
of
from about 15,000 to about 100,000 cps.,
(c) 0 to about 1% (preferably 0 to about 0.3%) iron oxide,
(d) about 8.0 to about 16.7% microcrystalline cellulose,
(e) about 0.39 to about 0.47% (preferably about 0.42 to about 0.47) colloidal
silica,
and
(f) about 0.29 to about 1.0% magnesium stearate.
As stated herein above, gepirone in the extended-release oral dosage form of
gepirone
also embraces the bioactive gepirone metabolites selected from the group
consisting of 3'-OH
gepirone, 3',5-dihydroxy gepirone, and 5-OH gepirone. The method may employ
any one of
these compounds. However, combinations of these metabolites, or combinations
of the
metabolites with other active or inert ingredients, are also contemplated.
The present invention further provides methods for ameliorating depression,
anxiety,
or psychological disorders in a patient (preferably human, but inclusive of
all mammals) in
need of such treatment, by administering to the patient an effective amount or
dose of
gepirone or a bioactive gepirone metabolite such as 3'-OH gepirone, 3',5-
dihydroxy
gepirone, and 5-OH gepirone. As used herein, the administration of gepirone or
a bioactive
gepirone metabolite includes the administration of any active salt form,
hydrate form,
enantiomeric form or mixture, or crystal form of the compound.
Also within the scope of the present invention are methods in which the
extended-
release oral dosage form of gepirone is administered (sequentially or
simultaneously) with
one or more anti-depressive or anxiolytic agents in a combination therapy for
treatment of
depression and/or anxiety.
-14-
CA 02585606 2007-04-27
WO 2006/052227 PCT/US2004/032785
Production methods:
The extended-release oral dosage form of gepirone of the present invention may
be
made by the following procedure: (i) admixing all or a part of components (a)
through (f), (ii)
blending the mixture, (iii) adding a part of or the remainder (if any) of
components (a)
through (f), (iv) blending the mixture (if remainder added during (iii)), (v)
slugging, (vi)
milling, (vii) adding the remainder of the lubricant (component (f); if part
reserved during (i)
and (iv)), (viii) blending the mixture; (ix) compressing the final finished
tablet blend.
Of course it is to be understood that the aforementioned steps may be
rearranged or
substituted so long as the rearrangement or substitution does not
substantially alter the
pharmacological efficacy of the resultant dosage form from the process
described above. In
this regard, the artisan is referred to Remington's Pharmaceuticals Sciences,
18th Edition
(incorporated herein by reference), for example Part 8 therein,
"Pharmaceutical Preparations
and Their Manufacture.
Further, the artisan is directed to the processing procedures described in
U.S. Pat. No.
4,423,049 and 5,478,572 (incorporated herein by reference) as providing
relevant examples
of the extended-release oral dosage form of gepirone production process
described above.
In a preferred embodiment, the following procedure is followed to make the
inventive
extended-release oral dosage form of gepirone:
i) admixing all of component (a), all of component (c), all of component (e),
and
20% of the total concentration of component (b);
ii) blending the mixture from (i);
iii) adding to the blend from (ii) all of component (d), half of the total
concentration
of component (f), and the remaining 80% of the total concentration of
component (b);
iv) blending the mixture from (iii);
v) slugging the blend from (iv);
vi) milling the slug from (v);
vii) adding the remaining 50% of component (f) to the milled slug from (vi);
viii) blending the mixture from (vii);
ix) compressing the blend from (viii) into the final desired form.
Typically, the resultant blends are compressed in step (ix) into tablets or
into
micropellets. If micropellets are made, they are optionally overcoated with
conventional
coating adjuvant(s) and then tableted or filled into capsules.
-15-
CA 02585606 2007-04-27
WO 2006/052227 PCT/US2004/032785
In the case where an enteric coating is desired, this coating would typically
be added
following step (ix).
It is to be understood that each of the above-described manufacturing
processes may
be scaled-up to an industrially applicable scale and is in no way limited to
individual dosage
preparation methods. It is also contemplated that alterations to each step
(individually or
collectively) may be made to facilitate the production of the inventive
extended-release oral-
dosage form of gepirone, so long the modifications do not alter the
composition of the final
product beyond the tolerance limits described above or the bioefficacy of the
resultant
product.
The above written description of the invention provides a manner and process
of
making and using it such that any person skilled in this art is enabled to
make and use the
same, this enablement being provided in particular for the subject matter of
the appended
claims, which inake up a part of the original description.
As used above, the phrases "selected from the group consisting of," "chosen
from," and
the like include mixtures of the specified materials.
Where a numerical limit or range is stated herein, the endpoints are included.
Also, all
values and subranges within a numerical limit or range are specifically
included as if
explicitly written out. In this regard, where the term "about" is recited,
this term is
understood to include the recited value, as well as values that are within 1%
of the recited
value (either above or below depending upon whether "about" defines an upper
or lower
boundary), preferably with 0.5% of the recited value.
The above description is presented to enable a person skilled in the art to
make and use
the invention, and is provided in the context of a particular application and
its requirements.
Various modifications to the preferred embodiments will be readily apparent to
those skilled
in the art, and the generic principles defined herein may be applied to other
embodiments and
applications without departing from the spirit and scope of the invention.
Thus, this
invention is not intended to be limited to the embodiments shown, but is to be
accorded the
widest scope consistent with the principles and features disclosed herein.
Having generally described this invention, a further understanding can be
obtained by
reference to certain specific examples, which are provided herein for purposes
of illustration
only, and are not intended to be limiting unless otherwise specified.
-16-
CA 02585606 2007-04-27
WO 2006/052227 PCT/US2004/032785
EXAMPLES
Preparation Example
Following the procedure described above and in U.S. Pat. No. 5,478,572, tablet
forms
of the extended-release oral dosage form of gepirone were prepared. Generally,
this
procedure entailed (i) admixing all of gepirone hydrochloride, all of the
colorant, all of the
colloidal silicon dioxide, and 20% of the total mass of
hydroxypropylmethylcellulose; (ii)
blending the mixture from (i) for 15 minutes and subsequently delumping using
a Fitzmill
#0020 plate; (iii) adding to the blend from (ii) all of microcrystalline
cellulose, 50% of the
total mass of magnesium stearate, and the remaining 80% of the total mass of
hydroxypropylmethylcellulose; (iv) blending the mixture from (iii) for 28
minutes; (v)
slugging the blend from (iv) using a rotary tablet press; (vi) milling the
slug from (v) using a
Fitzmill #0093 plate; (vii) adding the remaining 50% of magnesium stearate to
the milled
slug from (vi); (viii) lubricating/blending the mixture from (vii) for 7
minutes; and (ix)
compressing the blend from (viii) into the final desired tablet form.
In this manner ovoid-rectangular shaped tablets having biconvex faces were
obtained.
The overall dimensions of the tablets were 0.405 inches (102.87 mm) by 0.338
inches (85.85
m) with a cup depth of 0.050 inches (12.70 mm). The tablet weights and
component
quantity/unit are given in Table 1 below:
-17-
CA 02585606 2007-04-27
WO 2006/052227 PCT/US2004/032785
Table 1:
Quantity per unit (units in mg)
Ingredients Function 20 mg 40 mg 60 mg 80 mg
tablets tablets tablets tablets
Gepirone HCl Active 20.0 (18.1)1 40.0 (36.2)1 60.0 (54.3)1 80.0 (72.4)1
ingredient
Colloidal Glidant 1.6 1.6 1.6 1.6
silicon dioxide
Yellow ferric Colorant 0.4 0.08 1.2 1.0
oxide
Red ferric oxide Colorant 2.5
Hydroxypropyl Release 290.0 290.0 290.0 290.0
methylcellulose controlling
polymer &
binder
Microcrystalline Diluent & 61.8 52.12 31.0 33.7
cellulose compression
aid
Magnesium Lubricant 1.2 1.2 1.2 1.2
stearate
Total Tablet 375.0 385.0 385.0 410.0
Weight
indicates the free base content
Example 1: Dissolution profiles for the 60-mg formulation vs. the 20-mg
formulation
Comparative dissolution profiles for 20 mg and 60 mg tablets in all SUPAC-MR
dissolution media are presented in Table 2 below.
-18-
CA 02585606 2007-04-27
WO 2006/052227 PCT/US2004/032785
Table 2:
Time (Hours)
Medium Strength 1 2 4 8 10 12 14 16 18 20
0.01 M 20 mg 20.2 30.4 45.1 64.7 71.8 77.8 82.8 86.9 90.5 93.4
HCl
0.01 M 60 mg 22.0 32.8 48.2 68.6 76.0 82.0 87.1 91.2 94.5 97.3
HCl
water 20 mg 19.8 29.8 44.0 63.1 70.1 76.0 80.9 85.0 88.4 91.3
water 60 mg 21.9 32.7 48.2 68.6 75.8 81.7 86.6 90.5 93.7 96.2
pH = 4.5 20 mg 17.7 26.6 39.5 57.2 63.8 69.4 74.3 78.6 82.3 85.6
pH = 4.5 60 mg 20.2 30.0 43.8 62.2 68.8 74.3 79.1 83.1 86.5 89.4
pH = 6.8 20 mg 15.9 24.1 36.4 53.7 60.5 66.4 71.5 76.1 80.2 83.8
pH = 6.8 60 mg 17.5 26.4 39.6 57.6 64.4 70.2 75.1 79.5 83.3 86.6
The results above are graphically represented in Figures 1-4.
The fl and f2 calculation (see Table 3) for the comparative dissolution
profiles
demonstrates equivalence between the tablets in all 4 media
-19-
CA 02585606 2007-04-27
WO 2006/052227 PCT/US2004/032785
Table 3:
Medium Fl' F21
0.01MHC1 5.2 71.2
Water 6.7 66.1
pH=4.5 6.8 70.1
pH = 6.8 5.3 76.3
Criteria2 0-15 50-100
1 Fl and F2 provide insight and/or a measure of the distribution of
results among batches.
2 The criteria corresponds to acceptable Fl and F2 value ranges.
Example 2: Bioequivalence of the 80-mg formulation vs. the 20-mg formulation
Heretofore, efficacy and pharmacokinetic data for extended release gepirone
tablets
have been primarily obtained by administering one or multiple 20-mg extended-
release
gepirone tablets (similar to that described in the Preparation Example above).
Therefore,
studies were undertaken to demonstrate the bioequivalence of 40-mg (data not
shown) and
80-mg extended-release gepirone tablets (as described in the Preparation
Example above)
versus multiple 20-mg extended-release gepirone tablets.
To this end, an open label, randomized, four-way crossover, single dose study
design
was used with a washout phase of one week between different treatments. During
each study
period, the subjects were hospitalized from the afternoon before until 36
hours after gepirone
administration. Following discharge, blood sample collections were taken at
48, 60, and 72
hours post-administration. During the study period, gepirone was administered
to the thirty
two (32) subjects (average age 30.9 ~ 6.3 years; average weight 73.8 8.3 kg;
average height
178.1 5.6; and average BMI 23.3 ~ 2.3 kg/m2) as a single oral dose of 40 mg
or 80 mg. To
this end, the following treatments were given in a randomized order (tablet
formulation
shown in the Preparation Example above):
A) 1 x 40-mg extended-release gepirone tablet (data not shown);
B) 2 x 20-mg extended-release gepirone tablets (data not shown);
-20-
CA 02585606 2007-04-27
WO 2006/052227 PCT/US2004/032785
C) 1 x 80-mg extended-release gepirone tablet;
D) 4 x 20-mg extended-release gepirone tablets.
After each treatment, blood samples were obtained from the subjects at regular
intervals for 72 hours for determining the phaimacokinetics of gepirone, 1-PP,
and 3'-OH
gepirone.
To determine the bioequivalence of one 80-mg extended-release gepirone tablet
versus four 20-mg extended-release gepirone tablets the following
pharmacokinetic
parameter were calculated for each subject and treatment:
- tma, (TMAX)
The actual time of occurrence of Cm~,.
- Cma, (CMAX)
The measured maximum concentration in plasma after single dosing.
- dn-Cmax (DCMAX)
The dose-normalized Cm,,x was calculated as Cma,,/Dose.
- kZ (LZ) and t1/2 (THALF)
The slope ((3) of the terminal log-linear phase of the concentration-versus-
time
curve was determined by linear regression. The data had to be fit to the
function:
1og0Ci =1ogoCinterc - Pti
starting with the last 3(Ci, ti) data pairs for which Ci > LOQ; Cinterc is the
intercept
with the concentration axis at zero time. The procedure continued, adding
preceding data points one at a time and fitting the regression equation, until
Crõax
occurring at the tmax was reached. The terminal log-linear portion is then
defined
by the data yielding the smallest mean square error (MSE) term in the
regression
analysis. The terminal elimination rate constant (kZ) was defined as -(3 from
which the elimination half-life (t1/2) was calculated as logo2/kZ.
Concentrations
lower than LOQ in the elimination phase were ignored.
- AUCo-tlaast (AUCT)
The AUC from zero to tiast (AUCo-tiast) was calculated by means of the linear
trapezoidal rule, where t1aSt represented the last time point with a
measurable
concentration within a subject.
- dn-AUCo_tIast (DUCT)
The dose-normalized AUCo_tiast was calculated as AUCo_tlast/Dose.
- AUCo_,, (AUC)
-21-
CA 02585606 2007-04-27
WO 2006/052227 PCT/US2004/032785
The AUC from zero to infinity was calculated as AUCo_. = AUCo_tiast + AUCtlast-
oo.
For AUo_tlast see above ad AUCtlast_. = Ctjast/Xz, where Ctlast was the fitted
concentration at time tlast using the regression line for which kz had been
calculated.
- dn-AUCo_. (DAUC)
The dose-normalized AUCo_,,,, was calculated as AUCo_./Dose.
- CLapp (CLAPP)
The true total plasma clearance (CL) after a single oral dose equals f=
Dose/AUCo-. where f is the fractional absolute bioavailability of the oral
preparation. Since f cannot be calculated from oral data alone, CL/F = Dose/
AUCo_. was calculated and denoted as "apparent clearance" (CLapp). The
administered dose of gepirone was expressed as a free base. The amount free
bases of gepirone was also used for CLapp calculation of the 1-PP and 3'-OH
gepirone metabolites.
- wn-CLapp (WCLAPP)
The weight-normalized CLapp was calculated as CLapp divided by the body weight
(kg).
- VZ,app (VZAPP)
The apparent volume of distribution during the terminal phase after a single
oral
dose was defined as Vz,app = CLapp/XZ.
- wn-VZ,app (WVZAPP)
The weight-normalized VZ,app was calculated as Vz,app divided by the body
weight
(kg).
For dn-Cma,,, dn-AUCo-., and dn-AUCo_tiast, 0.80-1.25 was used as acceptance
range.
The formulations were declared bioequivalent with respect to the tested
parameters if the
90% confidence interval was fully contained within the acceptance range. This
was done for
gepirone, 1-PP and 3'-OH gepirone. For all other pharmacokinetic parameters,
classical
hypothesis testing was performed on the loge-transformed values using the same
ANOVA
models as for the bioequivalence testing.
The following mean (std. dev.) pharmacokinetic parameters were calculated for
gepirone, 1-PP, and 3'-OH gepirone (see Table 4):
-22-
Table 4:
Gepirone (n=32) 1-PP (n=32) 3'-OH gepirone (n=32)
Parameter Units 1 x 80 mg 4 x 20 mg 1 x 80 mg 4 x 20 mg 1 x 80 mg 4 x 20 mg
Cmax (ng/mL) 11.2 (5.36) 10.2 (7.05) 14.2 (10.0) 13.1 (7.05) 26.9 (7.24) 25.1
(6.99)
dn-Cmax (ng/mL/mg) 0.140 (0.067) 0.128 (0.088) 0.178 (0.125) 0.164 (0.088)
0.337 (0.091) 0.314 (0.087)
tR,~l (h) 5.5 (2.0 -12.0) 6.0 (0.5 -12.0) 5.0 (3.0 -18.0) 6.0 (2.0 -12.0) 6.0
(4.0-12.0) 8.0 (4.0 -12.0)
~
AUC0 _U.t (ng.h/mL) 151 (69.2) 149 (89.8) 250 (214) 260 (200) 551 (170) 557
(181) o
Ln
O
Ln
N dn-AUCo_tiazt (ng.h/mL/mg) 1.89 (0.865) 1.86 (1.12) 3.12 (2.67) 3.25 (2.50)
6.89 (2.13) 6.96 (2.27) o
O
AUCaoo (ng.h/mL) 158 (73.4) 155 (88.1) 259 (213) 268 (202) 558 (170) 562 (183)
0
0
dn-AUCo.oo (ng.h/mL/mg) 1.98 (0.917) 1.94 (1.10) 3.24 (2.66) 3.35 (2.52) 6.97
(2.13) 7.03 (2.29)
t~, (h) 7.93 (8.26) 8.22 (9.12) 9.43 (6.06) 7.41 (3.34) 8.14 (3.30) 7.40
(2.13)
CLapp (L/h) 692 (804) 625 (386) 417 (249) 396 (242) 146 (61.7) 144 (53.9)
wn-CLaPp (L/h/kg) 9.70 (12.7) 8.61 (5.95) 5.73 (3.78) 5.41 (3.54) 2.01 (0.971)
1.98 (0.836)
V2;apP (L) 6416 (6156) 7334 (9108) 5531 (4982) 4168 (3289) 1672 (806)
1437(346) Wn-Vz,app (L/kg) 86.1 (80.8) 102 (137) 74.0 (62.1) 57.0 (44.7) 22.8
(11.0) 19.8 (5.92)
median range
CA 02585606 2007-04-27
WO 2006/052227 PCT/US2004/032785
The statistical analyses concerning the average bioequivalence testing for
gepirone, 1-
PP, and 3'-OH gepirone are given in Table 5 below.
Table 5:
Gepirone Comparison: 1 x 80 mg (test) versus 4 x 20 mg (reference)
Geom.
Geom. Point 90%
mean
Parameter mean test Estimate Confidence Conclusion
reference
formulation test/ref. interval
formulation
dn-C,,,,,, 0.123 0.105 1.17 1.02 -1.33 Indeterminant
dn-AUCo-oo 1.71 1.68 1.02 0.89 - 1.16 Bioequivalent
dn-AUCo-tiast 1.63 1.59 1.03 0.90 -1.18 Bioequivalent
1-PP Comparison: 1 x 80 mg (test) versus 4 x 20 mg (reference)
Geom.
Geom. Point 90%
mean
Parameter mean test Estimate Confidence Conclusion
reference
formulation test/ref. interval
formulation
dn-C,,,a. 0.151 0.146 1.03 0.94-1.13 Indeterminant
dn-AUCo-oo 2.60 2.74 0.95 0.87 - 1.04 Bioequivalent
dn-AUCo-tlast 2.47 2.62 0.94 0.86 -1.03 Bioequivalent
3'-OH Gepirone Comparison: 1 x 80 mg (test) versus 4 x 20 mg (reference)
Geom. Geom. Point 90%
Parameter mean test mean Estimate Confidence Conclusion
formulation reference test/ref. interval
-24-
CA 02585606 2007-04-27
WO 2006/052227 PCT/US2004/032785
formulation
dn-Cmax 0.325 0.303 1.07 0.98 -1.18 Bioequivalent
dn-AUCo-0o 6.62 6.67 0.99 0.90 - 1.09 Bioequivalent
dn-AUCo-tlast 6.54 6.61 0.99 0.90 - 1.09 Bioequivalent
As can be seen from Table 5, for all compounds the 1 x 80 mg tablet was found
average bioequivalent to four 20 mg tablets with respect to dn-AUC. This is
also the case for
dn-C,,,a,, of 1-PP and 3'-OH gepirone. For gepirone an inconclusive result was
found for dn-
Cmax.
Since the average bioequivalence testing leads to indeterminate conclusions in
some
cases for gepirone, despite the fact that the point estimate of the ratio of
the geometric means
of the test and reference treatments lies within the acceptance range 0.80 -
1.25. Therefore,
an additional statistical analysis using the population approach as supportive
evidence was
performed. The results are shown in Table 6 where
ST2 : total sample variance (i.e. sum of within- and between-subject
variances) of the test treatment based on loge 4ransformed parameters.
SR2 . total sample variance (i.e. sum of within- and between-subject
variances) of the reference treatment based on loge transformed parameters.
Sd2 : sample variance of the intra-subject differences.
01, (0.50): the 50% quantile of the bioequivalence criterion of 0 1 (= median
for
0,).
01 (0.95): the 95% quantile of the bioequivalence criterion of 0 1 (= upper
bound
of 95% confidence interval for 01).
Since for all analyses S R 2 > 6T02 (= 0.22 = 0.04), the reference-scaled
method was
applied.
-25-
CA 02585606 2007-04-27
WO 2006/052227 PCT/US2004/032785
Table 6:
Gepirone Comparison: 1 x 80 mg (test) versus 4 x 20 mg (reference)
Parameter ST2 SR2 Sa2 01 (0.50) 01 (0.95) Conclusion*)
dn-C,,,. 0.3318 0.3755 0.2699 -0.033 0.401 Bioequivalent
dn-AUCo_oo 0.4063 0.3114 0.2747 0.268 0.876 Bioequivalent
dn-AUCo_tl.t 0.4154 0.3449 0.3233 0.193 0.768 Bioequivalent
1-PP Comparison: 1 x 80 mg (test) versus 4 x 20 mg (reference)
Parameter STZ SR2 Sd2 01 (0.50) 01(0.50) Conclusion*)
dn-Cm,,, 0.2467 0.2435 0.0763 0.018 0.269 Bioequivalent
dn-AUCo_oo 0.4128 0.4108 0.0883 0.007 0.253 Bioequivalent
dn-AUCo_uast 0.4338 0.4383 0.0935 -0.007 0.251 Bioequivalent
3'-OH Gepirone Comparison: 1 x 80 mg (test) versus 4 x 20 mg (reference)
Parameter STZ SR2 Sd2 01 (0.50) 01(0.50) Conclusion*)
dn-Cmax 0.0684 0.0648 0.1069 0.108 0.632 Bioequivalent
dn-AUCo_oo 0.1132 0.1054 0.1333 0.078 0.710 Bioequivalent
dn-AUCo_tiast 0.1137 0.1058 0.1360 0.077 0.714 Bioequivalent
Bioequivalent if 01i (0.95) < 6 P(= 1.745)
The results obtained with the population approach showed bioequivalence for
Cm,,,,
AUCo_oa, and AUCo_tlast of gepirone, 1-PP and 3'-OH gepirone with respect to
one 80-mg
gepirone extended release tablet and four 20 mg gepirone extended-release
tablets (the same
was observed for one 40-mg gepirone extended release tablet and two 20 mg
gepirone
-26-
CA 02585606 2007-04-27
WO 2006/052227 PCT/US2004/032785
extended-release tablets). It was further determined that a comparison between
the 40 mg
tablet and the 80 mg tablet showed no statistically significant differences
for gepirone or its
metabolites. This finding indicates dose proportionality of gepirone in the 40-
80 mg dose
range.
Numerous modifications and variations on the present invention are possible in
light
of the above teachings. It is, therefore, to be understood that within the
scope of the
accompanying claims, the invention may be practiced otherwise than as
specifically
described herein.
-27-