Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02587749 2007-05-17
Description
PIPERIDINE DERIVATIVE AND PROCESS FOR PRODUCING THE SANE
Technical Field
[0001]
The present invention relates to a process for
producing a 2-cyano-4-fluoropyrrolidine derivative known
as a medicament, particularly a dipeptidylpeptidase-IV
(hereinafter referred to as DPP-IV) inhibitor, and an
intermediate thereof, as well as a process for producing
the intermediate.
Background Art
[0002]
4-Fluoro-i-({[4-methyl-l-(methanesulfonyl)piperidin-
4-yl]amino}acetyl)pyrrolidine-2-carbonitrile (hereinafter
referred to as Compound A) is a compound known as a DPP-IV
inhibitor (Patent Document 1) and is a compound known to
be useful for treatment and/or prevention of type 1
diabetes, type 2 diabetes, insulin-resistant diseases,
obesity, and the like.
1
CA 02587749 2007-05-17
[Chem 1]
F
Ms, N
M~ N (IV)
N i1
H 0 CN
[0003]
With regard to Compound A, the following production
process X is specifically known, wherein 4-methyl-l-
(methanesulfonyl)piperidin-4-amine (hereinafter referred
to as Compound B) or a hydrochloride salt thereof is used
as a starting compound (Patent Document 1).
[0004]
(a) Production Process of Compound A[Production Process
X]
[Chem 2]
Ms.N CI O CN Ms, N
F
~~Me Me
NH2 K2CO3 CH3CN \H~
O CN
B A
or a hydrochloride thereof
wherein Ms represents methanesulfonyl and Me represents
methyl; the same shall apply hereinafter.
[0005]
Moreover, Compound B or a hydrochloride salt thereof
as a starting compound in the production process X is
2
CA 02587749 2007-05-17
known to be produced by the following production process Y
or production process Z (Patent Document 1).
[0006]
(b) Production Process of Hydrochloride Salt of Compound B
[Production Process Y]
[Chem 3]
Ms, HCI
HNMe Et3N, MsCI NMe 4M HCI-EtOAc Ms~N Me
NH NH EtOAc
CH2CI21 DMF NH
Boc Boc Z
Y-1 Y-0 B
wherein Boc represents tert-butyloxycarbonyl, Et3N
represents triethylamine, MsCl represents methanesulfonyl
chloride, CH2C12 represents methylene chloride, DMF
represents N,N-dimethylformamide, 4M HC1-EtOAc represents
an ethyl acetate solution of 4 mol/L hydrochloric acid,
and EtOAc represents ethyl acetate; the same shall apply
hereinafter.
[0007]
(c) Production Process of Compound B[Production Process
Z]
[Chem 4]
Ms~
HNMe Et3N, MsCI NMe 10% Pd/C, H2 Ms, N
Me
NH CH2CIZ, DMF NH MeOH NH
Cbz Cbz 2
Z-1 Z-0 B
3
CA 02587749 2007-05-17
wherein Cbz represents benzyloxycarbonyl, 10% Pd/C
represents 10% palladium supported on carbon, and MeOH
represents methanol; the same shall apply hereinafter.
[0008]
The starting compounds used in the above production
process Y and production process Z are known compounds but
are compounds which are not easily commercially and
inexpensively available. In order to obtain these
starting compounds, it is necessary to produce them
separately by the following known production processes and
the production steps are as follows.
[0009]
(d) Production Process of Compound Y-1 (Patent Document 2,
Non-Patent Document 1, Non-Patent Document 2)
4
CA 02587749 2007-05-17
[Chem 5]
HNa O
1 Y=8
\ Me
Bn, N Y=9
BnNH
O 2
Y=7 HCHO
Bn. Bn, N Me N Bn, NNH
OH Me
Ac
Y-6 Y-5 Y-4
Bn~N Me~ Bn\N Me. HN Me NHZ NH NH
Boc Boc
Y-3 Y-2 Y-1
wherein Bn represents benzyl, BnNH2 represents
benzylamine, and Ac represents acetyl; the same shall
apply hereinafter.
[0010]
(e) Production Process of Compound Z-1 (Patent Document 3)
[Chem 6]
HN Boc,N Boc'N
~ -~ Me
C02Et COZEt COZEt
Z-6 Z-5 Z-4
Boc, N Boc, N HN
Me Me Me
~
C02H NH NH
6bz 'Cbz
Z-3 Z-2 Z-1
5
CA 02587749 2007-05-17
wherein CO2Et represents ethoxycarbonyl; the same shall
apply hereinafter.
[0011]
Namely, in order to produce Compound B which is a
useful intermediate in the production of Compound A using
an inexpensive and easily available compound as a starting
material, it is necessary to produce Compound B by a
process of (A), (B), or (C) shown below and total number
of steps and overall yields in individual processes are as
follows.
(A) A process of producing B or a salt thereof via Y-5 and
Y-1 using Y-8 as a starting material
Total number of steps: 9 steps
Overall Yield: Among the total 9 steps, steps wherein
yields can specifically be derived from the description in
the precedent technical document are four steps. Overall
yield is 30.0% through only these four steps and thus it
is obvious that the yield is 30.0% or less when remaining
five steps whose specific yields are unknown are included.
(B) A process of producing B or a salt thereof via Y-5 and
Y-1 using Y-9 as a starting material
Total number of steps: 7 steps
Overall Yield: Among the total 7 steps, steps wherein
yields can specifically be derived from the description in
the precedent technical document are five steps. Overall
yield is 17.7% through only these five steps and thus it
6
CA 02587749 2007-05-17
is obvious that the yield is 17.7% or less when remaining
two steps whose specific yields are unknown are included.
(C) A process of producing B or a salt thereof via Z-1
using Z-6 as a starting material
Total number of steps: 7 steps
Overall Yield: among total 7 steps, steps wherein yields
can specifically be derived from the description in the
precedent technical document are four steps. Overall
yield is 47.6% through only these four steps and thus it
is obvious that the yield is 47.6% or less when remaining
three steps whose specific yields are unknown are
included.
[0012]
Patent Document 1: W02004/009544
Patent Document 2: JP-A-7-165754
Patent Document 3: W099/40070
Non-Patent Document 1: Journal of American Chemical
Society, 1985, Vol. 107, pp. 1768-1769
Non-Patent Document 2: Tetrahedron, 1970, Vol. 26, pp.
5519-5527
Disclosure of the Invention
Problems that the Invention is to Solve
[0013]
Accordingly, it has been desired to develop a
process for producing Compound B or a salt thereof wherein
7
CA 02587749 2007-05-17
total number of steps is shortened and overall yield is
improved using an inexpensive and easily available
compound as a starting material on industrial production.
Means for Solving the Problems
[0014]
As a result of the extensive studies on alternative
production processes of Compound A useful for treatment
and/or prevention of type 1 diabetes, type 2 diabetes,
insulin-resistant diseases, and obesity as a DPP-IV
inhibitor, the present inventors have found that Compound
A can be efficiently produced by the processes of the
following production process 1 and production process 2
and thus they have accomplished the invention.
[0015]
Namely, according to the invention, in the adoption
of a process for producing Compound A shown in the
production process 2 described below, there is provided a
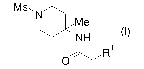
piperidine derivative represented by the formula (I):
[0016]
[Chem 7]
Ms, N
Me
NH
~R
O
wherein R1 represents -H, halogeno, or -OH,
8
CA 02587749 2007-05-17
or a salt thereof.
In this connection, R1 is preferably halogeno; more
preferably chloro or bromo.
[0017]
Moreover, there is provided a process for producing
Compound B represented by the formula (II):
[Chem 8]
Ms,N
Me (II)
NH2
which comprises eliminating an acetyl group having R' from
a piperidine derivative represented by the above formula
(I) or a salt thereof.
In the production process, the reaction of
eliminating an acetyl group having R' is preferably a
hydrolysis reaction with an acid. In particular, a
hydrolysis reaction with one or more acids selected from
the group consisting of hydrochloric acid, hydrobromic
acid, trifluoroacetic acid, sulfuric acid, methanesulfonic
acid, and p-toluenesulfonic acid and hydrates thereof is
preferred.
[0018]
Furthermore, there is provided a process for
producing a piperidine derivative represented by the
formula (I) or a salt thereof which is a compound of the
9
CA 02587749 2007-05-17
invention, which comprises using a 4-hydroxypiperidine
derivative represented by the formula (III):
[Chem 9]
Ms, N
Me (III)
OH
or a salt thereof.
As the reaction to be applied in the production
process, it is preferred to use a reaction of treatment
with a compound having a cyano group selected from the
group consisting of chloroacetonitrile, bromoacetonitrile,
acetonitrile, hydrogen cyanide, and glycolonitrile under
an acidic condition. In particular, it is preferred to
react an acetonitrile derivative selected from the group
consisting of chloroacetonitrile, bromoacetonitrile, and
acetonitrile with the compound of the formula (III) in the
presence of an acid selected from the group consisting of
methanesulfonic acid and sulfuric acid.
[0019]
In addition, there is provided a process for
producing Compound A represented by the formula (IV):
[Chem 10]
F
Ms, N
M~ /N (IV)
N ~
H O CN
CA 02587749 2007-05-17
which comprises using Compound B which has been produced
by the production process comprising eliminating an acetyl
group having R' from a piperidine derivative represented
by the above formula (I) or a salt thereof.
[0020]
(1) Production Process 1
[Chem 11]
HCI
HN OH First Step MS'N Second Step
OH O
1-2 1-1
Ms, N
Ms~ N Me Third Step Me
NH
OH O~R~
(III) (I)
[0021]
The present production process is a process for
producing the compound represented by the formula (I),
which is a compound of the invention, via the compound
represented by the formula (III).
(First Step)
The present step is a step of adding a
methanesulfonyl group to the amino group of the compound
1-2.
The reaction can be carried out by a method obvious
to those skilled in the art or by a method described in
11
CA 02587749 2007-05-17
Greene and Wuts, "Protective Groups in Organic Synthesis
(third edition)".
The compound 1-1 can be also produced by the method
described in W098/57862.
(Second Step)
The present step is a step of adding a methyl group
to the carbonyl group of the compound 1-1.
As a reaction reagent, there may be mentioned a
methylmagnesium halide such as methylmagnesium chloride or
methylmagnesium bromide, methyllithium, trimethylaluminum,
lithium dimethylcuprate, methyltrichlorotitanium, or the
like. Moreover, in the case of using a methylmagnesium
halide or methyl lithium, enolization of the compound 1-1
is suppressed by the addition of cerium trichloride or the
like and thereby improvement of yields can be expected.
The reaction can be carried out under cooling, under
cooling to under room temperature, or under room
temperature to under heating in the presence of a solvent
inert to the reaction, such as an aromatic hydrocarbon
such as toluene or xylene; an ether such as ethyl ether,
isopropyl ether, di-n-butyl ether, tetrahydrofuran (THF),
or dioxane; a halogenated hydrocarbon such as
dichloromethylene, chloroform, dichloroethane,
trichloroethane, or carbon tetrachloride; or a mixed
solvent thereof, although it varies depending on the
12
CA 02587749 2007-05-17
reaction reagent used. The reaction temperature may be
suitably selected depending on the reaction conditions.
(Third Step)
The present step is a step of adding a compound
having a cyano group to the compound (III) under an acidic
condition.
As a reaction reagent, there may be mentioned
chloroacetonitrile, bromoacetonitrile, acetonitrile,
hydrogen cyanide, or glycolonitrile. Moreover, as an acid
to be used, there may be mentioned a sulfonic acid such as
methanesulfonic acid or p-toluenesulfonic acid or a
hydrate thereof; sulfuric acid; trifluoroacetic acid;
perchloric acid; phosphoric acid; polyphosphoric acid;
formic acid; or a Lewis acid such as boron trifluoride
etherate or trimethylsilyl triflate. The reaction can be
carried out without any solvent or in a solvent inert to
the reaction although it varies depending on the reaction
reagent used. As the solvent inert to the reaction, there
may be mentioned acetic acid; acetic anhydride; an ether;
an aliphatic hydrocarbon such as hexane, pentane, or
heptane; a halogenated hydrocarbon; nitrobenzene; or the
like. The reaction temperature may be suitably selected
depending on the reaction conditions and the reaction can
be carried out under cooling, under cooling to under room
temperature, or under room temperature to under heating.
13
CA 02587749 2007-05-17
[0022]
(2) Production Process 2
[Chem 12]
Ms, N First MS, Second Ms" F
aMe Step aNH Me Step N Me
NH N
~R ZN~
O H p CN
(~) (11) (IV)
[0023]
The present production process is a process for
producing Compound B represented by the formula (II) and a
process for producing Compound A represented by the
formula (IV) using the compound of the invention
represented by the formula (I).
(First Step)
The present step is a step of eliminating an acetyl
group having R' from the compound (I) of the invention.
Preferably, a hydrolysis reaction with an acid may
be mentioned. As the acid, there may be mentioned
hydrochloric acid, hydrobromic acid, trifluoroacetic acid,
sulfuric acid, methanesulfonic acid, or p-toluenesulfonic
acid or a hydrate thereof and one acid or two or more
acids thereof can be used. The reaction can be carried
out under cooling, under cooling to under room
temperature, or under room temperature to under heating in
a mixed solvent of an alcohol such as methanol (MeOH),
ethanol (EtOH), 1-propanol, 2-propanol (iPrOH), or 1-
14
CA 02587749 2007-05-17
butanol (nBuOH) and water (water originally contained in
concentrated hydrochloric acid, sulfuric acid, or the like
may be used for the water) although it varies depending on
the substituent represented by R1 in the compound (I). The
reaction temperature may be suitably selected depending on
the reaction conditions.
(Second Step)
The present step is a step of condensing the
compound (II) with 1-chloroacetyl-4-fluoropyrrolidine-2-
carbonitrile or an analog thereof (e.g., 1-bromoacetyl-4-
fluoropyrrolidine-2-carbonitrile can be mentioned) which
is produced by the method described in W02004/009544 or a
method in accordance therewith.
The reaction can be advantageously effected by
adding a base or by using the compound (II) in excess. As
the base, there may be mentioned an organic base such as
triethylamine, N-ethyldiisopropylamine, or pyridine; and
an inorganic base such as potassium carbonate, cesium
carbonate, sodium hydrogen carbonate, or potassium
hydroxide. The reaction can be carried out without any
solvent or with a solvent although it varies depending on
the reactivity and the like of the compound to be
condensed with the compound (II). As the solvent to be
used, there may be mentioned acetonitrile, N,N-
dimethylformamide, dimethyl sulfoxide, an aromatic
hydrocarbon, an ether, an alcohol, a halogenated
CA 02587749 2007-05-17
hydrocarbon, a ketone such as methyl ethyl ketone or
acetone, water, or a mixed solvent thereof. The reaction
can be carried out under cooling, under cooling to under
room temperature, or under room temperature to under
heating. The reaction temperature may be suitably
selected depending on the reaction conditions.
Effect of the Invention
[0024]
Namely, according to the production process of the
invention, the compound (II) can be produced within only
four steps from piperidine-4-one hydrochloride hydrate,
which is an inexpensive and easily available starting
compound. Moreover, as shown in Examples 4 to 6 described
below, overall yield thereof is 43.7%.
On the other hand, at the production of the compound
(II), the known production processes are as described in
the above (A) ,(B) , and (C). In view of the total number
of steps, the compound can be produced within four steps
according to the production process of the invention while
nine steps are necessary in the known production process
(A) and seven steps are necessary in the known production
processes (B) and (C). It is well known that the
shortening of the total number of steps results in very
advantageous effects in view of economy, efficiency,
stable supply, and the like on industrial production.
16
CA 02587749 2007-05-17
Moreover, with regard to overall yields, each of the
known production processes (A) ,(B) , and (C) includes
steps whose yields are unknown. In the known production
process (A), the overall yield is 30.0% even when yields
of the five steps whose yields are unknown are assumed to
be all 100%. In the known production process (B), the
yield is 17.7% even when yields of the two steps whose
yields are unknown are assumed to be all 100%.
Furthermore, with regard to the known production process
(C), there are three steps whose yields are unknown.
Among the steps, when a replication experiment is
conducted three times on the step of producing Z-0 from Z-
1, an average yield thereof was found to be 68.7% and when
a replication experiment is conducted twice on the step of
producing B from Z-0, an average yield thereof was found
to be 98.5%. Therefore, when results of these replication
experiments are considered with regard to the overall
yield of the known production process (C), although one
step whose yield is unknown still remains, overall yield
is 32.2% even when the yield of the step is assumed to be
100%. Since the overall yield of the production process
of the invention is 43.7%, the overall yield is actually
remarkably improved in the production process of the
invention as compared with the known production processes
(A), (B), and (C). It is well known that the improvement
in overall yield results in very advantageous effects in
17
CA 02587749 2007-05-17
view of economy, efficiency, stable supply, and the like
on industrial production.
Additionally, in the known production process (C),
lithium diisopropylamide is used in the introduction of a
methyl group on the 4-position of the piperidine but the
reagent requires a reaction at a very low temperature, and
a low temperature of -78 C is necessary in Patent Document
3. In industrial production, control of such a very low
temperature frequently involves difficulty and thus it is
unsuitable for industrial production in view of economy
and efficiency. Furthermore, in the known production
process (C), so-called Curtius rearrangement reaction is
used in the conversion of the carboxyl group on the 4-
position of the piperidine into a benzyloxycarbonylamino
group. However, since an intermediate generated in the
reaction system is easily decomposed with water, the
reaction necessitates strict control of water in the
reaction system and is unsuitable for industrial
production in view of economy and efficiency.
[0025]
Therefore, the production process of the invention
is an excellent production process and very useful and
convenient production process since (1) total number of
steps is remarkably shortened, (2) overall yield is
remarkably improved, and (3) reactions which are not so
suitable for industrial production, such as reaction
18
CA 02587749 2007-05-17
necessitating a very low reaction temperature and a
reaction whose progress may be inhibited by the presence
of moisture, are not required and the process is composed
of only reactions suitable for industrial production in
comparison with the known production processes. Namely,
at the production of the compound (II) and further the
production of the compound (IV), it is extremely
advantageous to adopt the compound (I) of the invention as
a production intermediate and the advantages are as
described above.
Best Mode for Carrying Out the Invention
[0026j
The following will further explain the present
invention.
Herein, "halogeno" refers to chloro, bromo, iodo,
and fluoro and is preferably chloro or bromo.
Moreover, the salt in "piperidine derivative
represented by the formula (I) or a salt thereof", "4-
aminopiperidine derivative represented by the formula (II)
or a salt thereof", "4-hydroxypiperidine derivative
represented by the formula (III) or a salt thereof", or
"2-cyano-4-fluoropyrrolidine derivative or a salt thereof"
means an acid addition salt of the piperidine derivative
represented by the formula (I), the 4-aminopiperidine
derivative represented by the formula (II), the 4-
19
CA 02587749 2007-05-17
hydroxypiperidine derivative represented by the formula
(III), or the 2-cyano-4-fluoropyrrolidine derivative. As
the acid, there may be mentioned an mineral acid such as
hydrochloric acid, hydrobromic acid, hydroiodic acid,
sulfuric acid, nitric acid, or phosphoric acid; a sulfonic
acid such as methanesulfonic acid, ethanesulfonic acid,
benzenesulfonic acid, toluenesulfonic acid, or
trifluoromethanesulfonic acid; an organic acid such as
formic acid, acetic acid, propionic acid, oxalic acid,
malonic acid, succinic acid, fumaric acid, maleic acid,
lactic acid, malic acid, tartaric acid, or citric acid; an
acidic amino acid such as aspartic acid or glutamic acid;
or the like. Moreover, the "salt thereof" includes
various hydrates, solvates, and crystal polymorphs of each
compound and a salt thereof.
Furthermore, since the compound (IV) has an
asymmetric carbon, the following four optical isomers are
present but preferred is (2S,4S)-isomer represented by the
formula (IV-1).
CA 02587749 2007-05-17
[Chem 13]
F F
Ms, N Ms, N
N-' YN N
H N
0 CN H 0 CN
(IV-1) (IV-2)
F F
Ms, N Ms, N =
Me Me
N ND
H0 CN H0 CN
(IV-3) (IV-4)
Examples
[0027]
The following will specifically explain the
invention with reference to Examples but the invention is
not limited by these Examples.
[0028]
Example 1
A solution of 150.0 g of 4-[1-
(methanesulfonyl)]piperidone dissolved in 1950 mL of THF
was added to 847.0 g of a 2 mol/L THF solution of
methylmagnesium chloride at 10 C or lower with stirring,
followed by addition of 75 ml of THF with washing. After
4 hours of stirring, 75 mL of water, 1000 mL of 20% (w/v)
aqueous ammonium chloride solution, and 750 mL of toluene
were added thereto and an organic layer was separated.
The organic layer was washed with 900 mL of 20% (w/v)
21
CA 02587749 2007-05-17
aqueous sodium chloride solution and the solution was
concentrated under reduced pressure. An operation of
adding 1050 mL of toluene and concentrating the solution
under reduced pressure was further repeated twice. A
residue was added with 450 mL of toluene and the whole was
heated at 80 C for dissolution. Under stirring, the
solution was cooled to 0 C and precipitated crystals were
collected by filtration. The crystals were washed with
toluene and dried under reduced pressure to obtain 127.1 g
of 4-methyl-l-(methanesulfonyl)piperidin-4-ol as white
crystals.
1H-NMR (DMSO-d6) : 1.14 (s, 3H) , 1.47-1.58 (m, 4H) , 2.83 (s,
3H), 3.00 (dt, 2H), 3.25 (dt, 2H), 4.37 (s, 1H).
FAB-MS m/z: 194 (M+1).
[0029]
Example 2
To 740.5 g of methanesulfonic acid was added 200.0 g
of 4-methyl-l-(methanesulfonyl)piperidin-4-ol, followed by
addition of 148.1 g of methanesulfonic acid with washing.
To the resulting mixture was added 78.1 g of
chloroacetonitrile, and the whole was stirred under room
temperature overnight. To the reaction solution was added
1200 mL of ethyl acetate (EtOAc), and the resulting
solution was gradually added dropwise to 2700 mL of 27%
(w/v) aqueous potassium carbonate solution with stirring.
An organic layer was separated and washed with 600 mL of
22
CA 02587749 2007-05-17
10% (w/v) aqueous potassium hydrogen carbonate solution
and 600 mL of water. The solvent was removed by
evaporation under reduced pressure to obtain 264.7 g of 2-
chloro-N-[4-methyl-l-(methanesulfonyl)piperidin-4-
yl]acetamide as pale yellow crystals.
1H-NMR (CDC13) : 1.45 (s, 3H), 1.74-1.81 (m, 2H), 2.25-2.29
(m, 2H), 2.81 (s, 3H), 2.97-3.04 (m, 2H), 3.47-3.52 (m,
2H), 4.00 (S, 2H), 6.27 (brs, 1H) .
FAB-MS m/z: 269 (M+1).
[0030]
Example 3
To 15 mL of acetic acid was added 10.00 g of 4-
methyl-l-(methanesulfonyl)piperidin-4-ol, followed by
cooling to 10 C. Thereto were added 10 mL of sulfuric acid
and 5.08 g of chloroacetonitrile at 20 C with stirring,
followed by 9 hours of stirring. The reaction solution
was added to a mixed liquid of 150 mL of water, 22.00 g of
lithium carbonate, and 100 mL of isopropyl acetate at 35 C
or lower. An organic layer was separated and washed with
20 mL of water and the solvent was removed by evaporation
under reduced pressure.
To a concentration residue containing 2-chloro-N-[4-
methyl-l-(methanesulfonyl)piperidin-4-yl]acetamide was
added 8 mL of water and 8 mL of concentrated hydrochloric
acid, and the whole was stirred at 100 C for 24 hours. The
mixture was cooled under room temperature and 50 mL of 2-
23
CA 02587749 2007-05-17
propanol was added thereto to precipitate crystals. Under
stirring, the resulting mixture was cooled to 0 C and the
crystals were collected by filtration. After washed with
2-propanol, the crystals were dried under reduced pressure
to obtain 4-methyl-l-(methanesulfonyl)piperidin-4-amine
monohydrochloride monohydrate as white crystals.
[0031]
Example 4
To 2500 mL of water were added 500.0 g of piperidin-
4-one monohydrochloride monohydrate, 674.8 g of potassium
carbonate, and 2400 mL of acetonitrile, followed by
cooling to 10 C or lower. Thereto was added 559.3 g of
methanesulfonyl chloride at 35 C or lower with stirring,
followed by addition of 100 mL of acetonitrile with
washing. After 24 hours of stirring, the mixture was
neutralized by adding 45.0 g of potassium carbonate and
then extracted by adding 2500 mL of toluene. After an
aqueous layer was separated, 500 mL of acetonitrile and
1500 mL of toluene were further added, followed by
extraction. The resulting organic layer was mixed and the
solvent was removed by evaporation under reduced pressure.
Further, 2500 mL of toluene was added thereto and the
solvent was removed by evaporation under reduced pressure.
Then, 2500 mL of toluene was added to the resulting
residue and the whole was heated at 85 C for dissolution.
Under stirring, the mixture was cooled to 0 C and
24
CA 02587749 2007-05-17
precipitated crystals were collected by filtration. After
washed with toluene, the crystals were dried under reduced
pressure to obtain 488.7 g (yield 84.7%) of 4-[1-
(methanesulfonyl)]piperidone as white crystals.
'H-NMR (DMSO-d6) : 2.46 (t, 4H) , 2.97 (s, 3H) , 3.49 (t, 4H) .
FAB-MS m/z: 178 (M+1)
[0032]
Example 5
A solution of 2.00 g of 4-[1-
(methanesulfonyl)]piperidone dissolved in 26 mL of THF was
added to 21.02 g of a 1 mol/L THF solution of
methylmagnesium chloride at 10 C or lower with stirring,
followed by addition of 1 ml of THF with washing. After
24 hours of stirring at room temperature, 1 mL of water
and 13 mL of 20% (w/v) aqueous ammonium chloride solution
were added thereto and an organic layer was separated.
The organic layer was washed with 11 mL of 20% (w/v)
aqueous sodium chloride solution. The organic layer was
concentrated under reduced pressure to a volume of 10 mL.
An operation of adding 14 mL of toluene and concentrating
the solution under reduced pressure to a total volume of
14 mL was repeated four times. Precipitated crystals were
collected by filtration and dried under reduced pressure
to obtain 1.84 g (yield 84.4%) of 4-methyl-l-
(methanesulfonyl)piperidin-4-ol as white crystals.
CA 02587749 2007-05-17
[0033]
Example 6
To 15 mL of acetic acid was added 10.00 g of 4-
methyl-l-(methanesulfonyl)piperidin-4-ol, followed by
cooling to 10 C. Thereto were added 10 mL of sulfuric acid
and 5.08 g of chloroacetonitrile at 20 C or lower with
stirring, followed by 3 days of stirring. The reaction
solution was added to a mixed liquid of 150 mL of water,
21.00 g of lithium carbonate, and 100 mL of isopropyl
acetate at 30 C or lower. An organic layer was separated
and washed with 20 mL of water and the solvent was removed
by evaporation under reduced pressure.
To a concentration residue containing 2-chloro-N-[4-
methyl-l-(methanesulfonyl)piperidin-4-yl]acetamide was
added 60 mL of nBuOH, 10 mL of water, and 10 mL of
concentrated hydrochloric acid, followed by 13 hours of
stirring at 100 C. The mixture was cooled under room
temperature and 200 mL of iPrOH was added thereto. The
white crystals of Example 3 were seeded and crystals were
precipitated. Then, the resulting mixture was cooled to
0 C under stirring. After the crystals were collected by
filtration and washed with iPrOH, the crystals were dried
under reduced pressure to obtain 7.81 g (yield 61.2%) of
4-methyl-l-(methanesulfonyl)piperidin-4-amine
monohydrochloride monohydrate as white crystals.
26
CA 02587749 2007-05-17
[0034]
Namely, when the compound of the invention is
adopted as a production intermediate and the compound (II)
is produced by the production process of the invention,
from the results of Examples 4 to 6, the compound (II) can
be produced within only four steps in a overall yield of
43.7% using piperidin-4-one hydrochloride hydrate which is
an inexpensive and easily available starting compound as a
starting material.
[0035]
Example 7
To 2.85 g of 4-methyl-l-(methanesulfonyl)piperidin-
4-ol was added 8.5 mL of acetonitrile, followed by cooling
to 10 C or lower. Thereto was added 5.7 mL of sulfuric
acid at 20 C or lower with stirring, followed by 3 hours
of stirring. The reaction solution was poured into 100 mL
of ice-cooled water and neutralized by adding 12.7 g of
sodium carbonate. The mixture was extracted with 80 mL of
EtOAc and 50 mL of EtOAc, respectively. The resulting
organic layer was concentrated under reduced pressure to
remove the solvent by evaporation. The resulting residue
was purified by silica gel column chromatography
(chloroform:MeOH=50:1) to obtain 3.56 g of N-[4-methyl-l-
(methanesulfonyl)piperidin-4-yl]acetamide as a yellow oil.
27
CA 02587749 2007-05-17
1H-NMR (DMSO-d6) : 1.27 (s, 3H) , 1 .45-1 .52 (m, 2H) , 1.81 (s,
3H), 2.15-2.18 (m, 2H), 2.85 (s, 1H), 2. 88-2 . 97 (m, 2H),
3.20-3.27 (m, 2H), 7.36 (brs, 1H).
FAB-MS m/z: 235 (M+1).
[0036]
Example 8
A reaction was carried out in the same manner as in
Example 6 using bromoacetonitrile instead of
chloroacetonitrile to obtain 4-methyl-l-
(methanesulfonyl)piperidin-4-amine monohydrochloride
monohydrate.
1H-NMR (DMSO-d6) : 1.33 (s, 3H) , 1 .73-1 .77 (m, 2H) , 1.81-
1.84 (m, 2H), 2.90 (s, 3H), 3.03-3.09 (m, 2H), 3.39-3.45
(m, 2H) , 8.33 (brs, 3H)
FAB-MS m/z: 193 (M+1).
[0037]
Example 9
To 2.00 g of (2S,4S)-1-(chloroacetyl)-4-
fluoropyrrolidine-2-carbonitrile which had been produced
by the method described in W02004/009544 were added 14 mL
of acetonitrile, 2.88 g of 4-methyl-i-
(methanesulfonyl)piperidin-4-amine monohydrochloride
monohydrate, 5.4 mL of N-ethyldiisopropylamine, followed
by 14 hours of heating at 80 C under stirring. The solvent
was removed by evaporation under reduced pressure, 14 mL
of EtOH was added, and further the solvent was removed by
28
CA 02587749 2007-05-17
evaporation. Thereto were added 2 mL of water, 50 mL of
EtOH, and 0.54 mL of N-ethyldiisopropylamine, followed by
heating for dissolution. Under stirring, the mixture was
cooled to precipitate crystals. It was cooled to 0 C and
crystals were collected by filtration. After washed with
EtOH, the crystals were dried under reduced pressure to
obtain 3.04 g of (2S,4S)-4-fluoro-l-({[4-methyl-l-
(methanesulfonyl)piperidin-4-yl]amino}acetyl)pyrrolidine-
2-carbonitrile as white crystals.
1H-NMR (CDC13): 1.12, 1.14 (s, 3H), 1.66 (brs, 4H), 2.26-
2.72 (m, 2H), 2.78 (s, 3H), 3.16-4.05 (m, 6H), 3.33 (ABq,
2H), 4.89, 4.95 (d, 1H), 5.36, 5.44 (dt, iH).
FAB-MS m/z: 347 (M+1).
Industrial Applicability
[0038]
According to the present invention, there are
provided an efficient process for producing Compound A or
a salt thereof useful as a medicament, particularly a DPP-
IV inhibitor, Compound B which is an intermediate
particularly in the case where the production process is
adopted or a salt thereof, and a process for producing the
same.
29