Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
NSAID Compositions Exhibiting Clinical Superiority
CROSS-REFERENCE TO RELATED APPLlCATIONS
10001a This is a continuation-in-part of U.S. Serial No.10/119,313, filed
April 10,
2002; and U.S. Serial No. 10/119,303, filed April 10, 2002; and U.S. Serial
No.
10/166,050, filed June 11, 2002, and U.S. Serial No. 60/624,806, filed
November 3,
2004.
BACKGROUND OF THE INVENTION
FIELD OF THE INVENTION
[0002] The present invention is directed to solid NSAID formulations having
increased absorption in rate suppressed vagal systems. One of the primary
NSAIDs,
(d)-2-(4-lsobutylphenyi) propionic acid, ibuprofen, is a potent and well
tolerated
anti-inflammatory, analgesic, and anti-pyretic compound.
DESCRIPTION OF RELATED ART
[0003] In the treatment of acute pain rapid absorption of orally administered
analgesics is desirable. For non-steroidal anti-infiammatory drugs (NSAIDs),
such as
ibuprofen, meloxicam, and ketoprofen, there appears to be a positive
relatiohship
between plasma drug concentration and analgesic activity. Any delay in
absorption or
reduction in the circulating drug concentration may result In treatment
failure or in
reduced activity of the analgesic. After oral administration of regular-
release tablet
preparations to healthy subjects, ibuprofen is almost completely absorbed from
the
gastrointestinal tract. it is, therefore, expected that, typically, peak serum
concentrations and maximal analgesic effect normally occur within I to 2 hours
of
administration. One skilled in the art readily recognizes that analgesic
formulations with
enhanced absorption rates are expected to be more effective in treating acute
pain.
[0004] However, none of the widely available solid dosage forms of NSAIDs have
been claimed to be superior over the products of the same drug with respect to
onset of
action. This is despite differences in apparent rate of absorption usually
measured in
--1--
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
healthy volunteers. It appears that rapid absorption observed in healthy
subjects does
not necessarily result in a quick onset of action in patients experiencing
pain.
[0005] Jamall & Kunz, BritJ. Clin. PharmacoL, 47:391-396 (1999) have reported
that, using denta( surgery as a pain marker, pain or its associated trauma
causes
reduced rate of absorption of ibuprofen. Surgery resulted in a two hour delay
In the
mean time to peak concentration, significant decreases in serum ibuprofen
concentrations following both doses, and a fall to sub-optimal serum
concentrations.
[0006] The observed reduced absorption Is believed to be caused by suppression
of the vagal nervous system. The vagus nerve, nervus vagus, is the 10t'
cranial nerve;
suppressing the activity of the vagus nerve causes reduced gastric juice
secretion and
motility, both of which are associated with decreased absorption of NSAIDs.
Sufficient
fluid and a rather quick exit from stomach (hence entry to small intestine,
the major site
of absorption) is needed for efficient absorption.
[0007] If an active agent does not dissolve readily or cannot penetrate the
epithelial membrane (e.g., if it is highly ionized and polar), residence time
at the
absorption site may be insufficient. In such cases, bloavaiiabiiity tends to
be highly
variable as well as low. The physicochemical properties of a drug govem its
absorptive
potential, but the properties of the dosage form (which partly depend on its
design and
manufacture) can also largely determine drug bloavailability. Differencesr in
bioavallabiiity among formulations of a given drug can have clinical
signi6cance. Thus,
the concept of equivalence among drug products is important in making clinical
decisions.
[0008] The problem of decreased absorption in vagally suppressed mammals is
further exacerbated by the low solubility of NSAIDs in an aqueous or gastric
(acidic)
environment. Finally, there is growing evidence that these conditions, namely,
reduction in stomach motility, stomach secretion diminution, and reduced
absorption
appear to be present also in the elderly, or what shail be termed herein, the
geriatric
stomach.
[0009] Some prior art formulations, such as U.S. Patents 6,197,336 and
4,834,966, dissolve the ibuprofen formulation prior to administering the
composition.
--2--
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
Other prior art formulations, e.g., PCT/EP97100841, incorporate an alkali
metal
bicarbonate into the ibuprofen formulation to enhance the compressibility of
the solid
dosage form. These formulations Include ibuprofen as the active agent, the
bicarbonate
as a compressibility enhancer, a compressible filler, and a disintegrant
(preferably
croscarmellose sodium or sodium starch glycollate).
[00101 Alkali metal carbonates and bicarbonates are solubie materials which
have previously been proposed for use in effervescent tablets, for example to
react with
the acid component in an effervescent couple (see for example WO 94/10994) or
to
prevent initiation of the effervescent reaction e.g. during storage.
Effervescent tablets
disintegrate by means of the reaction between acid and base particularly in
the
presence of water leading to the production of carbon dioxide. The system of
disintegration of non-effervescent dosage forms according to the present
Invention,
which are arranged to be swallowed and for which an effervescent reaction is
not
desired, is different to that of effervescent formulations. In the present
dosage form the
soluble acidic component is not intended to react with the alkali metal
carbonate or
bicarbonate to produce an effervescent reaction prior to administering the
tablet. That
is, the solid dosage form of the present invention does not need to be
dissolved in water
prior to taking the tablet.
[0011] US Patent 5,681,583 describes a multilayer tablet in which one layer is
designed to accelerate release of active ingredients. The patent discloses
compounds
that "can produce effervescence and disintegration". As stated in claim 1,
however, the
same layer contains several other excipients that are included to facilitate
dissolution
and disintegration independent of the effervescent. In contrast, the present
invention
relies on the acid-base reaction to enhance disintegration and dissolution.
[0012] The '583 patent also teaches the need, in the first layer, for coating
reagents. The present deals wfth an uncoated compressed tablet. Indeed, the
coating
action will be detrimental to a rapid disintegration and dissolution in the
stomach of a
patient under pain due to low agitation, emptying and fluid. Acute pain
suppresses
gastric emptying, agitation and juice.
--3--
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
SUMMARY OF THE INVENTION
10013] It is desirable to provide an NSAID and other analgesic formulation
that
can deliver drugs into the blood stream despite a suppressed vagal system.
[0014] It would be advantageous to provide a composition having enhanced
absorption of NSAIDs and other drugs that tend to be poorly water soluble, as
well as
providing an improved concentration of the drug at the cellular level at the
site of its
action.
[0015]. It would also be advantageous to provide a,method and composition for
increasing the absorption rate of such poorly water-soluble active agents by
increasing
the disintegration efficiency of the composition in tablet form, by
accelerating the time
and speed of the tablet disintegrating into molecules in solution, and by
increasing the
speed by which active agent is available in solution for absorption.
[0016] Ibuprofen Is a relatively weak acid (pka 4.4) and has poor solubility
in
water: less than I part of drug will dissolve in 10,000 parts of water.
However, it is fairly
soluble in simple organic solvents. lbuprofen solubility is particularly low
in acidic
environment where it wiil have a reiatively long residence time when the
patient is in
pain. This slows down absorption.
[0017] In comparison with standard ibuprofen tablet formulations, which
contain
the active ingredient as the free acid, liquid formulations or faster
dissolving ibuprofen
salts (e.g. lysine or arginine salts) demonstrate faster absorption and higher
peak serum
concentrations when. given to healthy subjects.
[0018] NSAIDs are weak organic acids and are not ionized in the acidic
environment of the stomach. Therefore, NSAIDs are able to diffuse freely into
the
mucosal cells. Once inside the mucosal cell, the more neutral intracellular pH
causes
NSAIDs to ionize, resulting in a diminished ability to passively diffuse back
through the
mucosal cells. Mucosal cell ion trapping causes NSAIDs to accumulate,
resulting in
elevated intracellular concentrations and direct cellular injury. .
[0019] Since the cell membranes on the stomach wall contain lipids, they offer
little resistance to the lipid-soluble NSAID. The NSAID acts against the cell
membrane,
increasing its permeability. This results in cell swelling and death. The
local acid effect
..4..
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
of NSAIDs has been reduced by enteric-coating the drug, delaying dissolution
until later
in the digestive process. However, not all NSAIDs are enteric-coated as it
increases
cost. In addition, enteric-coating does little rimore than improve the
symptoms of upset
stomach.
[0020] In healthy subjects, ibuprofen is well absorbed and extensively bound
to
plasma proteins in vivo. It is prescribed for rheumatic arthritis and other
musculoskeletal
disorders, as well as acute gout. The dosage of the drug is typically from 600
to 1200
mg daily in divided doses, with 2,400,mg per day being the maximum.
[0021] lbuprofen is also indicated for use in the treatment of rheumatoid
arthritis,
osteoarthritis, ankylosing spondylitis, seronegative arthropathies,
periarticular disorders
and soft tissue injuries. Ibuprofen may also be used in the treatment of
postoperative
pain, postpartum pain, dental pain, dysmenorrhoea, headache, musculoskeletal
pain or
the pain or discomfort associated with the following: respiratory infections,
colds or
influenza, gout or moming stiffness.
[0022] A critical factor relating to the use of ibuprofen to treat the above
disorders
concerns, as noted above, improving the onset of action of ibuprofen,
particularly in the
treatment of pain. This issue partially concems improving the amount and speed
of
achieving a certain blood.serum level of ibuprofen. It is believed that
ideally a rapid
disintegration of a formulation, beginning in the mouth, but primarily In the
stomach in
which 100% of the formulation is disintegrated results in the release the drug
into the
body more quickly, thereby leading to a more rapid onset of therapeutic
action, as
compared with a standard dosage form or with dosage forms calibrated against
healthy
individuals. Accordingly, it is desired to produce a solid dosage form for
oral
administration adapted to disintegrate quickly in the gastro-intestinal tract.
it is also
preferred that the dosage form is manufactured by compression on standard
tabletting
machines.
[0023] In accordance with one embodiment of the present Invention, the
composition contains an NSAID, preferably ibuprofen (hereinafter referred to
as IB); a
disintegration and dissolution agent, such as a bicarbonate, preferably sodium
bicarbonate; tartaric acid as an additional excipient; and neusilin as a
matrix. The
composition may optionally also include starch. These ingredients are formed
into a
..g..
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
tablet, caplet, or solid form, these forms having enhanced disintegration into
particles
and subsequently enhanced dissolution of the particles into dispersed
molecules in
solution.
[0024] In accordance with the present invention, the bicarbonate is a
disintegrator
or disintegrating agent that increases the solubility of the NSAID. One of the
important
effects of sodium bicarbonate ingestion is that the acid-base reaction takes
place in the
stomach. This results is production of carbon dioxide in a bubbling fashion.
This motion
results in rapid disintegration of the formulation and the rapid drug
dissolution in
aqueous environments.
100251 Mere exposure of ibuprofen with bicarbonate in the aqueous environment
of stomach results in the acid-base reaction. However, this reaction is
accelerated with
the addition of a soluble acid such as tartaric acid.
[0026] The acid-base reaction results in the break down of the formulation
integrity and exposes the contained maize to water. This results a bursting
effect that
further accelerates disintegration and dissolution of the drug thereiri.
[0027] While not intending to be limited to a particular mechanism of action,
the
inventor believes that the bicarbonate increases solubility by promoting the
formation of
sodium ibuprofen, a salt that is readily converted to ibuprofen; ibuproÃen
precipitates
under gastric conditions, so the anti-precipitation agent prevents
precipitation by
increasing the solubility of the ibuprofen in the gastric environment. This is
achieved by
addition of solubilizing agents such,as Neusilin.
[0028] The compositions and methods of the present Invention achieve
chemically what happens biologically when NSAIDS are administered and absorbed
in
healthy subjects. Biologically, the stomach has a certain amount of movement
or
motility, as well as gastric juice that contribute to a tablet disintegrating
into particles,
and then dissolving into molecules.
[0029] The compositions and methods of the present invention provide a
formulation whereby the solid dosage form provides it own movement through the
acid-
base reaction and is disintegrated in the stomach at a faster rate, it
subsequently
presents itself in the stomach as a solubie salt of ibuprofen leading in part
to quicker
absorption, thereby, faster onset of action.
--6--
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
[0030] In a vagally suppressed human, i.e., a human in pain, both the motility
and
gastric juice extraction are reduced. Also in the geriatric population stomach
emptying
and gastrointestinal transit time are delayed. These conditions result in
delayed
absorption, . The present invention accelerates the time line of
disintegration Into
particle form by chemically mimicking the agitation provided by the motility
function, by
initiating the disintegration from tablet form into particles as soon as the
tablet is
exposed to a very limited amount of fluid. In the presence of some moisture,
the
incorporated bicarbonate starts reacting with acids. The result is breaking
down of the
larger solid particles, enhancing solubility, and providing a greater amount
of active
agent earlier in the process, thereby accelerating the absorption rate, and
thereby
providing more relief, faster.
[0031] The compositions and methods of the present invention achieve this
result
by surrounding, capturing, or formulating active agent particles, such as
ibuprofen, in a.
matrix or the like of a metasilicate, such as Neusilin. The composition may
further
include a disintegrating agent that, upon exposure to an aqueous environment,
promotes the break-up of the tablet into smaller particles of active agent,
thereby
increasing the availability of the active agent for absorption.
[0032] The solid dosage forms according to the invention are adapted for
direct
administration to a patient to obtain the desired therapeutic 'effect. They
are not -
intended to be dissolved or dispersed in water prior to administration.
Furthermore, the
compressed dosage forms according to the present invention need no further
processing, e.g. coating, after compression of a composition comprising a
mixture of the
ingredients to produce a solid dosage form.
[0033] The accompanying drawings show illustrative embodiments of the
invention from which these and other of the objectives, novel features and
advantages
will be readily apparent.
DESCRIPTION OF THE DRAWINGS
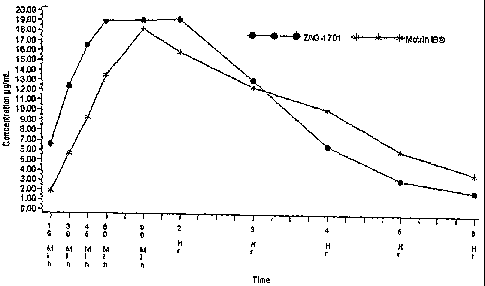
Figure 1 shows the mean serum concentration versus time for S-ibuprofen
generated by a formula of the present invention (n=12 patients) compared to
Motrin
(n=14 patients).
..7..
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
Figure 2 shows mean serum concentration for R-ibuprofen generated by a
formula of the present invention (n=12 patients) compared to Motrin (n=14
patients).
Figure 3 shows the pharmacokinetic parameters for total (S+R) concentration of
ibuprofen generated by a formula of the present invention (n=12 patients) R-
ibuprofen
compared to Motrin (n=14 patients).
Figure 4 shows the mean incremental area under the curve (AUC) for S-
lbuprofen in plasma concentrations following administration of either a
formula of the
-present invention (n=12 patients) or Motrin (n+14 patients).
Figure 5 shows the mean incremental area under the curve (AUC) for R-
ibuprofen in plasma concentrations following administration of either a
formula of the
present invention or Motrin.
Figure 6 shows the mean incremental area under the curve (AUC) for total
ibuprofen In plasma concentrations following administration of either a
formula of the
present invention or Motrin.
Figure 7 shows the mean cumulative (incremental) AUC of ibuprofen (o-1 h).
following administration of the formula (open bars) or Motrin (closed bars) to
patients
after dental surgery.
Figure 8.shows the mean cumulative (incremental) AUC of ibuprofen (0-6 h)
following administration of the formula (open bars) or Motrin (closed bars) to
patients
after dental surgery.
Figure 9 shows the man pain intensity score (PIS) over time using a
categorical
pain intensity scale for patients after dental surgery following administering
either the
formula or Motrin. There was no statistically significant differences between
the two
products due to the small number of subjects tested. However, there was
consistent
trend for greater P!S for Motrin IB.
Figure 10 shows the mean pain intensity difference (PID) between the two
products over time using a visual analogue scale (VAS) after dental surgery
following
administering either ZAG-1701 or Motrin.
Figure 11 shows the percent of patients who recorded meaningful relief
following
a dose of either Motrin or ZAG-1701.
-.g..
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
Figure 12 shows the comparative dissoiution profiles among lbuprofen alone;
ibuprofen and sodium bicarbonate; and ibuprofen, sodfum bicarbonate, and
Geiucire .
DETAILED DESCRIPTION OF THE INVENTION
[0034] The present invention Is a method of treating pain and{for inliammation
comprising administering a solid dosage formulation comprising an active agent
in a
matrix, and a first and second disintegrant; wherein said active agent is an
NSAID;
wherein said matrix Is a metasiiicate, wherein the first disintegrant
comprises sodium
bicarbonate; and wherein said second disintegrant comprises tartaric acid.
100351 The present invention is also a method of treating pain and or
inflammation by administering a solid dosage formulation having a fast onset
of action,
wherein the solid dosage formulation comprises a formulation of the present
invention.
[00361 The present invention is also a method of increasing the onset of
action of
a solid formulation, e.g., a tablet, comprising blending tartaric acid and a
diluent to form
a first blend; adding at least one diluent and a binder to said first blend to
form a second
blend; blending an active agent and a metasilicate matrix to form a third
blend; adding
sodium bicarbonate to said third blend to form a fourth blend; and compressing
said
fourth blend into a tabiet.
[0037] The present invention is a. compositian cflntaining an NSAID as an
active
agent, said composition having increased absorption and onset of action in
vagaiiy
suppressed systems. The composition comprises an NSAID and a disintegration
and
dissolution agent, such as a bicarbonate, in a matrix or carrier, such as a
metasilicate.
The preferred NSAID is ibuprofen. The composition may further include an anti-
precipitation agent e.g. Gelucire .
[0038] The composition may further Include tartaric acid as an additional
disintegrating agent, Examples of a porous carrier include, for instance,
aluminum
magnesium metasilicate (available from Fuji Chemicai Industry Co., Ltd.; Trade-
name:
NEUSILIN).
(0039] The present invention Is also a composition comprising ibuprofen on a
matrix comprising a metasilicate, and a disintegration and dissolution agent,
such as a
-9-
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
bicarbonate. The invention also includes a method of treating inflammation or
alleviating pain comprising administering a composition as described in this
paragraph.
[0040] The present invention is also a composition comprising ibuprofen and a
matrix comprising a metasilicate, a disintegration and dissolution agent, such
as a
bicarbonate. Such a composition is characterized by having earlier onset of
action and
increased absorption of the active agent, as compared to other compositions
when the
comparison assesses the absorption of the active agent under pain conditions.
The
invention also includes a method of treating inflammation or alleviating pain
comprising
administering a composition as described in this paragraph.
[0041] The present invention is also any of the above compositions, further
comprising one or more lubricating agents, one or ore binders, one or more
additional
disintegrating agents, one or more flow aids, one or more preservatives,
and/or one or
more colorants and/or flavorants.
[0042] The present invention is also a method for increasing the absorption of
an
NSAID-containing composition, said method comprising providing a composition, -
such
as one of the compositions described above, whose ingredients are specifically
formulated to increase absorption under pain conditions, i.e;, In a vagaily
suppressed
system.
[0043] The present invention is also a method of treating acute pain in humans
comprising administering a composition according to the present invention.
[0044] It will be appreciated that the present invention provides a method of
treating inflammation, pain and pyrexia by administration of a pharmaceutical
composition comprising racemic ibuprofen, together with a pharmaceutically
acceptable
carrier to a mammal, e.g. a human, in need thereof. Preferably the ibuprofen
is present
in one or more of its well known forms, namely, ibuprofen, its S(+) and R(-)
enantiomers, including different enantiomeric ratios thereof, salts, hydrates,
and other
derivatives. The preferred form is a dihydrate. The most preferred form is the
acid
form.
[0045] The ibuprofen may be also present in the form of any salt or other
derivative of ibuprofen or its enantiomers. If necessary, the ibuprofen may
comprise one
or more ibuprofen active ingredients such as racemic ibuprofen and S(+)-
ibuprofen in
--10--
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
combination. However, we prefer that the ibuprofen comprises a single
ibuprofen active
ingredient. The (buprofen active agent may also be present in different
degrees of
hydration. The present invention applies to both anhydrous and hydrated forms,
for
example the monohydrate or the dihydrate. The most stable anhydrous or
hydrated
form wil) generally be used. Preferably, the ibuprofen is in the form of a
salt of racemic
or S(+)-ibuprofen. Representative examples include alkali metal salts, for
example the
sodium or potassium salts of ibuprofen; aikaline earth metal salts, e.g. the
calcium or
magnesium salts of ibuprofen; metal salts, e.g. the aluminum salt of
ibuprofen; amino
acid salts for example the lysine or arginine salts of ibuprofen: or amine
salts, e.g. the
meglumine salt of ibuprofen. Preferably the ibuprofen is a single salt
selected from alkali
metal salts, amino acid salts and amine salts.
[0046] These soluble ibuprofen salts also have the advantage that, as they are
more soluble in an aqueous medium, on release from the formulation they have
improved absorption, thus leading to an improved onset of action compared to
the
substantially insoluble forms of. ibuprofen. The sodium salt of ibuprofen is
particulariy
preferred, especially the sodium salt of racemic ibuprofen. It has been found
that the
dihydrate of the sodium salt of racemic ibuprofen is a particularly stable
hydrated form,
accordingly we prefer to use the sodium salt dihydrate in a compressed dosage
form
according to the present invention.
10047] The compositions and methods of the present invention are particularly
suited to forming non-aqueous granulations and to solid dosage formulations,
preferably
dosage formulations that are non-effervescent prior to administration or
prior to in vivo delivery. In preferred embodiments of the invention, one or
more of the
disintegrants burst the tablet or caplet apart in the stomach, thereby
presenting a
greater amount of active agent.
[0048) The present invention further relates to tablets and granules,
formulated in
accordance with the present invention, that are fast dissolving and fast
acting. The.
granulation and tabletting composition also includes normal excipients useful
for the
preparation of tablets.
[0049] The present invention is also a composition and method of treatment
comprising an NSAID as an active agent, blended with a metasilicate matrix and
a
-11--
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
bicarbonate as a disintegrating agent. The composition may further comprise
one or
more of the fo(lowing: one or more disintegrating agents, preferably sodium
bicarbonate
and/or tartaric acid; one or more diluents or fillers; one or more binders or
adhesives;
one or more additional disintegrating agents; one or more lubricating agents;
one or
more preservatives, preferably propyl gailate; and one or more miscellaneous
adjuncts,
such as colorants and/or flavorants, any of said adjuncts being well known to
those
skilled in the art.
[0050] Any number of pharmaceutically active agents may be employed in the
formulations of the present invention. These active agents may exist as either
solids or
liquids at standard temperature and pressure. Exemplary pharmaceutically
active
agents suitable for use herein include, but are not limited to, the non-
steroidal anti-
inflammatory agents such as piroxicam, indomethacin, fenoprofen, meloxicam,
and
ibuprofen. In a preferred embodiment of the fnvention, the composition and
method
includes ibuprofen as the active agent.
[0051] The compositions of the invention may include about 15 to about 99% by
weight of an active agent, such as lbuprofen, preferably up to about 80% by
weight,
more preferably from about 15% to about 70% by weight; about 10% to about 60%
by
weight of a first disintegrant, such as a bicarbonate, preferably between
about 12% and
30%, and more preferably between about 12% and 20%.
[0052] The compositions of the present invention may include up to about 30%
by weight of a second disintegrant, such as tartaric acid, preferably up to
about 15%,
more preferably between about 1% and about 10%.
[0053] The compositions of the present invention may include up to about 30%
by weight of a matrix, such as a metasilicate, preferably up to about 2%, more
preferably between about 1% and about 15%.
[0054] The compositions of the present invention may Include up to about 80%
by weight of one or more diluents, such as microcrystalline cellulose and
sodium starch
glycolate, preferably up to about 40%, more preferably between about 5% and
about
20%.
[0055] The compositions of the invention are generally prepared in unit dosage
form. The unit dosage of ibuprofen is in the range.of 5-1200 mg in a pre-
calculated
--12-
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
amount to provide doses which are equivalent by weight to doses of for example
700
mg, 200 mg, 400 mg or 800 mg of ibuprofen. The amount of an NSAID substance in
a
quick release, sustained release, or modified release composition according to
the
invention may be selected so that is corresponds to about I mg, 2 mg, 3 mg, 4
mg, 5
mg, 8 mg, 10 mg, 12 mg, 16 mg, 20 mg, 24 mg, 25 mg, 30 mg, 32 mg, 50 mg, 60
mg,
100 mg, 200 mg, 300 mg, 400 mg, 500 mg, 600 mg, 700 mg, 800 mg, 900 mg, I g,
1.1
g, 1:2 g, 1.3 g or 1.6 g of NSAID substance which are dosages generally known
in the
art.
[0056] The preferred dosage form according to the invention is In the form of
a
capsule, tablet, sachet etc. The size of the dosage form may be adapted to the
amount
of the active drug substance contained in the composition.
[00371 The above suggested dosage amounts should not be regarded as a
limitation of the scope of the invention as It is obvious for the skilled
person that any
desired amount of the active drug substance may be appiied and is only limited
by the
size.of the composition and the type bf the active drug substance;
[00581 The bicarbonate is preferably an alkali metal carbonate, more
preferably
any bicarbonate salt that is pharmaceutically acceptable, preferably sodium.or
potassium bicarbonate. The alkali metal carbonates may be supplied anhydrous
or in
varying degrees of hydration, e.g., the monohydrate and decahydrate. Any of
these
forms may be used.
[0059] In therapeutic use, the composltions of the present invention may be
administered orally, rectally, or topically, preferably orally or topicaliy.
In the present
invention, the preferred mode of administration is orally, i.e. as a solid
tablet easily
dissolved in the gastric environment. Suitably the therapeutic compositions of
the
present invention may take the form of any of the known pharmaceutical
compositions
for oral, rectal, or topical administration.
[0060) Solid compositions for oral administration are preferred compositions
of
the invention and there are known pharmaceutical forms for such
administration, for
example tablets, capiets, and capsules.
[0061] Within the context of the present description the identity of the
components and amounts thereof refer to the weight and identity of the
starting
-13--
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
materials used in preparing the composition. It is possibie that during
preparation of the
cornposition and/or tablets, some interaction or reaction may occur between
two or
more components. To the extent that such interaction or reaction occurs the
present
invention is intended to cover such occurrences.
[0062] Normai excipients useful in the preparation of the tabiets include, but
are
not limited to: lubricants such as magnesium stearate, sodium stearyl fumarate
and
sodium benzoate; anti-adherents such as taic and polyethylenglycoi; glidants
such as
colloidal silica; diiuents such as dicalcium phosphate, cellulose (for example
microcrystalline cellulose) and its derivatives, carbohydrates and
polyalcohols such as
saccharose, xylitol and lactose; disintegrants such as crosslinked vinylic
polymers (such
as crosslinked PVP), derivatives of starch and of cellulose such as sodium
carboxymethyl-starch and sodium croscarmelose; wetfing agents such as TWEEN 80
-(Trademark registered by ICI of Americas for polysorbate) and sodium lauryl
sulphate.
[0063] Suitable exciplents and their amounts can be readily determined by
those
of ordinary skill in the art according to the methods normally used in
pharmaceutical
technology. However, in the present invention, it is important to avoid
excipients that
would cause a significant decrease in tablet dissolution rate.
[0064] Further, excipients should allow a good workability of the tablet. In
preparing the tablet of the present invention, simple mixing may be
sufficient. One
skilled in the art will readily recognize that a number of mixing and
tabletting protocols
may be used. For example, it may be desirable to prepare an IB granulate, to
mix it
with the bicarbonate and the excipients, and then to compress. An exempiary
method of
preparing a composition of the present invention comprises dissolving
ibuprofen in an
alkaline solution; mix with neusitin; change the pH (e.g., by adding glacier
acid) so that
the ibuprofen re-crystallizes; and remove the soiution. The ibuprofen is
thereby loaded
on the neusilin matrix. It should then be dried, e.g., overnight. Other
processes that
lead to a workable tablet are shown in the Examples.
[0065] Preferably the diluent includes lactose, calcium phosphate, dextrin,
microcrystalline cellulose, sucrose, starch, calcium sulphate, sodium
bicarbonate, or
mixtures thereof.
..~ 4~-
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
[0066] Preferably the lubricating agent includes magnesium stearate, stearic
acid, calcium stearate, sodium bicarbonate, or mixtures thereof. More
preferably the
lubricating agent is magnesium stearate or stearic acid.
[0067] Preferably the disintegrating agent includes microcrystalline
cellulose,
maize starch, sodium starch glycolate, low substituted hydroxypropyl
cellulose, alginic
acid or croscarmelose sodium, sodium bicarbonate, or mixtures thereof.
[0068] Preferably the binder includes polyvinyl, pyrrolidone, gelatin,
Gelucire ,
hydroxypropylmethyl cellulose, starch, or mixtures thereof.
[0069] Suitable flow aids include, but are not limited to talc and colloidal
silicon
dioxide.
[0070] The compositions of the present invention may additionally comprise a
taste masking component for example a sweetener, a flavoring agent, arginine,
sodium
carbonate or sodium bicarbonate.
[0071] Solid non-effervescent compositions are preferred compositions of the
present invention. The preferred compositions are preferably formed into a
tablet. In
the most preferred compositions and methods, disintigarion occurs in stomach
through
the acid/base reaction.
[0072] In the compositions of the present invention the NSAID, such as
ibuprofen, may, if desired, be associated with other compatible
pharmacologically active
ingredients and/or enhancing agents. Thus, for example, ibuprofen may be
combined
with any ingredient commonly used in a cough or cold remedy, for example, an
antihistamine, caffeine or another xanthine deryvative, a cough suppressant, a
decongestant, an expectorant, a muscle relaxant, or combinations thereof.
Exemplary
compatible pharmacologically active ingredients include, but are not limited
to codeine,
oxycodone, hydrocodone, and/or hydromorphone.
[0073] Suitable antihistamines which are preferably non-sedating include
acrivastine, astemizole, azatadine, azelastine, bromodiphenhyrdramine,.
brompheniramine, carbinoxamine, cetirizine, chlorpheniramine, cyproheptadine,
dexbrompheniramine, dexchlorpheniramine, diphenhydramine, ebastine, ketotifen,
lodoxamide, loratidine, levocubastine, rnequitazine, oxatomide, phenindamine,
phenyftoloxamine, pyrilamine, sefastine, tazifylline, temelastine,
terfenadine,
.w~ ~~
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
tripelennamine or triproildine. Suitable cough suppressants include
caramiphen,
codeine or dextromethorphan. Suitable decongestants include pseudoephedrine,
phenylpropanolamine and phenylephrine. Suitable expectorants include
gualfensin,
potassium citrate, potassium gualacoisulphonate, potassium sulphate and terpin
hydrate.
[0074] The disintegration time of the tablet formed in accordance with the
present
invention is less than 30 minutes as measured by the method described in the
European Pharmacopoeia 1986, Ref V .5.1.1 (updated 1995) (A. Disintegration
Test for
Tablets and Capsules). Preferred disintegration times are less than 6 minutes
(e.g. 1-10
minutes), more preferably less than 5 minutes (e.g. 1-5 minutes) and most
preferably 3
minutes or less (e.g. 1-3 minutes). Consequently the release is faster with
respect to the
commercially available ibuprofen based analgesic tablets (see the examples
below).
[0075] in the present invention the tablet size Is between 100-700 mg
preferably
between 400-600 mg (in the Examples, the typical size of a tablet of the
present
invention is 570 mg) for a tablet containing 200 mg racemic IB. Considering
the need for
acid/base reaction, this size tablet is favorable due to the ease of
swallowing.
[0076] As used herein, a diluent or filler is used in its conventional
pharmacological definition, and refers to an ingredient that adds necessary
bulk to a
formulation to prepare tablets of a desired slze.
[0077] As used herein, a binder or adhesive is used In its conventional
pharmacological definition, and refers to an ingredient that promotes the
adhesion of the
particles of the formulation.
[0078] As used herein, a disintegrator or disintegrating agent is used in its
conventional pharmacological definition, and refers to an ingredient that
promotes the
post-administration break-up of the tablets into smaller particles for more
ready drug
availability.
[0079] As used herein, a lubricant or lubricating agent is used. in its
conventional
pharmacological definition, and refers to an ingredient that enhances the flow
of the
tabletting material into the tablet dies, and prevents the tabletting material
from sticking
to punches and dies.
..16..
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
[0080] - As used herein, enhanced absorption or similar terms and phrases
relating to the relative speed, rate, and/or quantity of the bioavailability
of the active
agent. In accordance with the present invention, enhanced absorption Is
measured in
reference to the standard in the industry, Motrin. In essence, the
compositions of the
present invention provide, to a patient in pain, a greater concentration of
active agent
faster, as compared to the bioavailability curve for Motrin. For example, see
Figures 1-
3. In graphical or mathematical terms, enhanced absorption may be determined
or
quantified by using the area under the curve (AUC). As shown in Figures 4-6,
the
extent and rate of absorption, as represented by the AUC, for the formulations
of the
present invention, delivers a greater amount of active agent in a shorter time
frame as
compared to Motrin. In accordance with the teachings of the present invention,
it is
'important to determine enhanced absorption of a particular composition as it
applies to
a patient in pain, or data obtained from a patient or subject in pain.
100813 In therapeutic use the dosage forms of the present invention are
administered orally, thus the therapeutic dosage forms are presented in solid
dosage
form, preferably as a tablet. The dosage forms may be coated with a sugar or
film
coating, which dissolves substantially Immediately the dosage form comes into
contact
with an aqueous medium. The composition may aiso be compressed onto a solid
core
of another material to form a solid formulation with a quick reiease outer
coating.
Alternatively, the compressed composition may be present in one or more layers
of a
multi-layer solid dosage Ãorm. In such formulations the remaining layers or
core may
comprise standard excipients to provide conventional, fast or slow release and
are well
within the knowledge of a person skilled in the art (e,g., see Remington's
Pharmaceutical Sciences, t7th Edition, Ed Gennaro et al; or Ansel's
"Introduction to
Pharmaceutical Dosage Forms", 2"d edition, Henry Kimpton Publishers).
[0082] Although not wishing to be held to any particular theory of action, the
inventor believes that a tablet or capiet of the present invention functions
as follows:
after swallowing a capiet with a glass of water, the sodium bicarbonate reacts
with
tartaric acid and (buprofen in the stomach. This appears to provide stomach
agitation or
movement that breaks the caplet into finer pieces and solubilizes the
ingredients. A
weak resultant solution of sodium ibuprofen may be formed, which may
eventually react
--17--
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
with stomach acid resulting in its conversion to crystallized ibuprofen. To
prevent
crystaliization and precipitation, Neusilin acts as a solubilizer due to its
lingering alkaline
nature.
j00833 The following Examples illustrate specific formulations comprehended by
the present invention, and methods for their preparation. The Examples are not
intended to be limiting to the scope of the invention in any respect and
should not be so
construed.
EXAMPLES
Example 1. Description of Manufacturing Process
Due to the sensitivity of the product to moisture, the relative humidity
during the
manufacturing process should be maintained below about 40%.
Milling
1. Pass tartaric acid (98.0 g) and microcrystalline cellulose (98.0 g) through
the
Quadro Comil at 1800:L50 rpm speed into a labeled container lined with double
polyethylene bags.
Pre-Blend
2. Place the milled blend, microcrystalline cellulose (98.0 g), hydroxypropyl
cellulose
(279.3 g), and sodium starch glycolate (420.0 g) into a 16 qt Gallay Blender.
3. Blend for 5 minutes at 25 rpm.
4. Add ibuprofen, colloidal silicon dioxide (39.9 g), and Neusilin UFL2 (245.0
g) into a 16
qt. Gallay Blender.
5. Purge the 16 qt. Gailay Blender with Nitrogen gas for 30 seconds and blend
for 10
minutes at 25 rpm.
6. Discharge the Pre-Blend into appropriate containers lined with double
polyethylene
bags.
7. Pass the pre-blend through 30-mesh hand screen into labeled containers
lined with
double polyethylene bags.
Screened Blend
8. Place screened blend and sodium bicarbonate (728.0 g) into 16 qt. Gallay
Blender.
--18-
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
Purge blender with nitrogen gas for 30 seconds. Blend for 10 minutes at 25
rpm.
9. Add talc (59.$5 g) and magnesium stearate (39.9 g) to the blender and purge
w'ith
nitrogen gas for 30 seconds before blending for 2 minutes at 25 rpm.
10. Discharge the final blend into a labeled container with double
polyethylene bags,
place 15 desiccants between the bags and tightly seal each bag with a twist-
tie.
Tablet.Compression
11. Ecisure correct tooling set-up (Upper and Lower Punch: 0.5650" x 0.2671",
capsule
shape, plain) and set up Tablet Deduster in-line with the tablet press.
12. Compress blend according to the specifications.
13. Store the finished tablets in tared, labeled containers lined with double
polyethylene
bags. Place 15 desiccants between the bags and tightly seal each bag with a
twist tie.
In-Process Controls '
Table 1: Tablet Compression Specifications
Parameter Specification
Average weight of 10 Target: 5.70 g
tablets Range: 5.50-5.90 g
Check average weight of 10 tablets every 10 minutes
Individual weight Target: 570.0 mg
Range: 541.5-598.5 mg
At start-up check (n+5) tablets, when n= no. of stations. During
run check 10 tablets every 20 minutes
Hardness Target: 10kp
Range: 5-11 kp* (Target 8 kp)
At start-up check (n+5) tablets, when n= no. of stations. During
run check 10 tablets every 20 minutes.
*Guidefine only: Adjust hardness to achieve friability NMT 0.7%
in 4 minutes
Thickness Cornpiie ait thickness data and record minimum and maximum as
range.
At start-up check (n+5) tablets, when n= no. of stations. During
run check* 10 tablets every 20 minutes.
Friabiiity NMT 0.7% in 4 minutes.
(Determine at start-up and compietlon of run)
Disintergration NMT 15 minutes In water at 37 C without disks.(Determine at
start-up and completion of run)
.-1 g-.
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
Example 2. Exemplary Process for producing 200 mg tablets
Pass tartaric acid and microcrystalline cellulose through Quadro Comil at
1800 1:50 rpm. Add microcrystalline cellulose, hydroxypropyi cellulose and
sodium starch glycolate; blend for 5 minutes at 25 rpm. Add ibuprofen,
colloidal
silicon dioxide, and Neusilln UFI.2; blend for 10 minutes at 25 rpm. Screen
blend
through 30 mesh hand screen. Add sodium bicarbonate and blend for 10
minutes at 25 rpm. Add talc and magnesium stearate; blend for 2 minutes at 25
rpm. Compress into tablets.
Example 3. The ZAG - 1701 formulation
Drug Product Components and Composition (Per Tablet and Per Batch)
Component Quantity per unit % Quantity per batch
ibuprofen (active agent) 200,00 mg 35.1 1400.0 g
Sodium Bicarbonate (disintegrant) 104.00 mg 18.2 728.0 g
Tartaric Acid (disintegrant) 14.00 mg 2.5 98.0 g
Microcrystalline Cellulose 200 97.15 mg 17.0 680.1 g
(diluent)
Hydroxypropyl cellulose EXF 39.90 mg 7.0 279.3 g
(binder)
Sodium Starch glycolate (diluent) 60.00 mg 10:5 420.0 g
Colloidal silicon dioxide (300) 5.7 mg 1.0 39.9 g
(glidant)
Neusilin UFL2 (mat(x) 35.00 mg 6.1 245.0 g
Talc (lubricant and glidant) 8.55 mg 1.5 59.85 g
Magnesium Sterate (non-bovine) 5.7 mg 1.0 39.9 g
(iubricant)
Total 570.00 mg 100 3.99 kg
--20--
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
Specification and Analyticat Methods for Inactive Ingredients
Listed in the table below is each excipient with its compendial status. With
the exception of Neusifin UFL2, all exciplents meet current USP/NF
requirements.
Inactive Ingredient Compendial Funcfiion
Desi nation
Sodium Bicarbonate USP Disintegrant
Tartaric Acid NF Disintegrant
Microcrystalline cellulose USP/NF/EP/JP Diluent
200
Hydroxypropyi cellulose USP Binder
EXF
Sodium starch glypplate USP/NF Diluent
Colloidal silicon dioxide NF Hydrophilic water-insoluble
(300) iidant
Neusilin UFL2 JP Absorbent and disintegrant
Talc USP Hydrophilic water-insoluble
lubricant and glidant
Magnesium Stearate (non- USP/NF/EP/JP Hydrophilic water-insoluble
bovine) lubricant
The specification for the inactive ingredient Neusilin UFL2 is as follows:
Test
Specification
Desc(tion White powder or granules
identification 1) The solution responds to the
qualitative tests for aluminum
salt.
2) The filtrate responds to the
qualitative tests for magnesium
salt.
3) The residue becomes blue.
Soluble saifi The residue weighs NMT 0.020g.
Aikali The solution is colourless.
Chloride - NMT 0.053%
Sulfate NMT 0.480%
Heavy metais NMT 30 ppm
Iron The solution has no more colour than
the control solution.
Arsenic NMT 5 m
Loss on D in NMT 20.0%
..29..
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
Test
Specification
Acid-consuming capacity The amount of HCI consumed is not
less than 210 mL per g of Magnesium
Aluminometasilicate on dried basis).
Assay 1) Sificon Dioxide: 29.2-35.6%
2) Aluminum Oxide: 29.1-35.5%
3) Magnesium Oxide: 11.4-14.0%
(All values calculated on dried basis)
Example 4.
The purpose of this experiment is to produce tablets that disintegrate in
15-30 min with no external agitation.
(mg)
Ibuprofen 200
Sodiurm bicarbonate 104.3
Getucire@ 44114 12.0
Maize 62.9
Results: Acceptable disintegration time for small batches and when tablets
were prepared under low compression pressure. Increased compression
pressure required for optimal friability test resulted in unacceptable
dissolution
times.
Example S.
(mg)
lbuprofen 200
Sodium bicarbonate 104.3
Gelucire 44114 12.0
Maize 62.9
Tartaric acid . 14.3
The above ingredients were mixed according to the procedure described
in examples I or 2, and were then subjected to direct compression.
Results: Acceptable disintegration time (15 min) for small batches even
under optimal compression pressure. The mixer was found unsuitable for larger
batches. It was unable to properly chop and mix GelucireO.
..22..
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
Examate B.
(mg)
(burpofen 200
Sodium bicarbonate 104.3
Getucire 44114 12.0
Maize 62.9
Tartaric Acid 14.3
Mg-Stearate 4.3
The above ingredients were mixed according to the procedure described
in examples I or 2, and were then subjected to direct compression.
Results: Problems were noticed during compression due to poor powder
flow and sticking to punch and die.
Exampte 7:
(mg)
Ibuprofen 200
Sodium bicarbonate 104.3
Gelucire 44/14 12.0
Maize 62.9
Tartaric acid 14.3
Mg-Stearate 4.3
Neusilin 8.6
The above ingredients were mixed according to the procedure described
in examples I or 2, and were then subjected to direct compression.
Results: Problems were noticed during compression due to melting of
Gefucire .
Example 8:
(mg)
Ibuprofen 200
Sodium bicarbonate 104.3
Gelucire 44/14 12.0
Maize 62.9
Tartaric acid 14.3
Mg-Stearate 4.3
Neusilin 8.6
The above ingredients were mixed according to the procedure described
--23--
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
in examples I or 2, and were then subjected to direct compression. The bulk
was kept at 4 C to cool down before compression.
Results: Compression was satisfactory. Clinical batch was prepared.
Problem: After storage for a few days tablets failed the disintegration test
(15
min). Exposure to moisture was blamed for the failure.
Example 9:
(mg)
lbuprofen 200
Sodium bicarbonate 104.3
Maize (sodium starch giycoiiate) 60.0
Tartaric acid 14.3
Mg-Stearate 5.4
Neusilin 34.9
The above ingredients were mixed according to the procedure described
in examples I or 2, and were then subjected to direct compression. The bulk
was compressed directly to form tablets.
Results: Compression was satisfactory, Clinical batch was prepared. After
storage for six months, tablets failed due to lack of stability and loss of
potency.
This was unexpected for ibuprofen, as based on previous experiences the
method of dry mixing and granulation was expected to yield a stable
formulation.
The presence of alkaline materials or exposure to humidity might have caused
degradation.
Example 10:
(mg)
ibuprofen 200
Sodium bicarbonate 104.3
Maize. (sodium starch glycolate) 60.0
Tartaric acid 14.3
Mg-Stearate 5.4
Neusilin 34.9
Propyl gallate 11.4
The above ingredients were mixed according to the procedure described
in examples I or 2, and were then subjected to direct compression.
Results: Compression was satisfactory. Tablets had satisfactory stability.
--24--
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
Example 11:
(mg)
Ibuprofen 200
Sodium bicarbonate 104.3
Maize (sodium starch glycolate) 60.0
Tartaric acid 14.3
Mg-Stearate 5.4
Neusilin 34.9
EDTA 0.6
The above ingredients were mixed according to the procedure described
in examples I or 2, and were then subjected to direct compression.
Results: Compression was satisfactory. A batch was prepared. The stability of
the tablet did not improve to a satisfactory level.
Examale 12:
(mg)
ibuprofen 200
Sodium bicarbonate 104.3
Maize (sodium starch glycolate) 60.0
Tartaric acid 14.3
Mg-Stearate 5.4
Neusilin 34.9
Propyl gallate 11.4
EDTA 0.6
The above Ingredients were mixed according to the procedure described
in examples I or 2, and were then subjected to direct compression.
Results: Compression was satisfactory. The tablets have satisfactory
stability.
Examples 10 and 12 are suitable for manufacturing in bulk quantities.
Example 13. Accelerated stability test:
An accelerated stability test was carried out on tablets containing the
ingredients described below. Tablets of each formulation were piaced at 22 C,
45 C, and 60 C. Three tablets of each formulation were sampled after 0, 3, 7,
14
days,1, 3 and 6 months. Samples were analyzed using an HPLC method (Paul.
A. Asmus, J. of Chromatogr. 331 (1) 1985, P: 169-176) with minor
modifications.
--25-- .
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
The potency of ibuprofen and impurity was determined simultaneously. The
impurity of interest due to thermal degradation of lbuprofen was 4-
isobutyiacetophenone.
Formula A:
(mg)
Ibuprofen 200
Sodium bicarbonate 104.3
sodium starch glycolate 60.0
Tartaric acid 14.3
Mg-Stearate 5.4
Neusilin 8.6
Formula B:
(m9)
lbuprofen 200
Sodium bicarbonate 104.3
sodium starch glycolate 60.0
Tartaric acid 14.3
Mg-Stearate 5.4
Neusilin 34.9
Propyl gailate 11.4
Formula C:
(mg)
lbuprofen 200
Sodium bicarbonate 104.3
sodium starch glycolate, 60.0
Tartaric acid 14.3
Mg-Stearate 5.4
Neusilin 34.9
EDTA 0.6
The result of accelerated stability test during the first 3 months and
extrapolating assessment indicate that the formulation without any
antioxidants,
Formula A, is only stable for 20 days. However, formulations with propyl
gailate
(Formula B) and EDTA (Formula C) will be stable at least for 2.8 years and 243
days, respectively.
Example 14. in vitro dissolution test
Using the U.S. Pharmacoipoeia Apparatus ii, the dissolution rates of
ibuprofen alone, ibuprofen plus sodium bicarbonate (1:1 molar based), and
.-26--
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
ibuprofen plus sodium bicarbonate (1:1 molar based) plus Geluclre (5% total
weight) were assessed. The apparatus contained 2 g of NaCI and 7 mL of
concentrated HCI (pH 1.2) in 900 mL water. The medium was kept at 37 C, and
was stirred with a rotating paddle at 75 rounds per minute. lbuprofen was
detected at 232 nm. The amount dissolved per unit time is shown in Figure 12.
Examale 15. Formulation having improved dissolution rate and absorption of
ibuprofen.
In this example, the solid dispersion and physical mixture of ibuprofen was
tested for dissolution rate and improved rate of bioavailability. The
Influence of
the carrier and two other excipients (sodium bicarbonate and tartaric acid) in
solid dispersion and the physical mixture as they affect drug dissolution
behavior
were also studied. Gelucire 44/14, sodium bicarbonate, tartaric acid, and
ibuprofen were made into a physical mixture and solid dispersion. The soiid
dispersion was prepared by the fusion method at 60 C, then cooled, then ground
into granules of suitable size, then compressed as a tablet.
After optimization of the formulation, solid disperslon and physical mixture
formulations were compared with a marketed product in healthy and treated
rats.
(an animal model for pain; see f'CT/IB02/01139, incorporated herein by
reference) in terms of rapid absorption and early exposure of the lbuprofen to
the
systemic circulation.
ln vitro dissolution test results showed that the rate of dissolution of the
ibuprofen
was considerably improved when formulated in solid dispersion or physical
mixture with Gelucire 44/14, as compared to pure ibuprofen. In vivo studies
showed that the early exposure of ibuprofen (AUC 0-0.5) was significantly
higher
in the physical mixture and solid dispersion than the brand formulation.
Examale % i=ormulation with tartaric acid.
A composition was formed using ibuprofen (1 mole), sodium bicarbonate
(1.8 mole), tartaric acid (0.2 mole), and Gelucire 44/14 (5% of total mass).
First, the exact amount of Gelucire was weighed and dissolved in a small
amount of isopropyl alcohol. The remaining ingredients were mixed together,
..27N
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
then mixed with the Geiucire solution to produce a fluffy paste. Using a
sieve
(mesh 20), granules with the size of 841 mm were produced. The granules were
dried overnight at room temperature, then used to produce tablets using a
single
punch compressing machine with 500 kg (punch id no. 3/16N) of compressing
pressure. It is preferred that the compression force should be minimal.
The formulation in this example may also include starch, and a portion of
the sodium bicarbonate may optionally be used as a lubricant. In another
alternative, the Gelucire dissolved in the isopropyl alcohol may be sprayed
over
the remaining mixture of ingredients.
Example 17. Clinical Trials
The objective of this trial was to compare the rate of absorption on
ibuprofen administered as ZAG 1701 (the present invention) with a commercialiy
available product (Motrin IB). This study was conducted in patients after
dental
surgery, a FDA accepted test of analgesia.
This was tow-arm, doubie-biind study consisting of patients who needed
dental extraction. Subjects were randomly'divided into two groups (n = 12-
14/group), Dental extraction was carried out under local anesthesia. When a
patient complained of pain and asked for analgesics, a single oral dose of 400
mg (2X200 mg) ibuprofen (the formulation shown- in Example 3) or Motrin !B
were administered randomly with a glass of water. Serial blood samples were
collected for pharmacokinetic analysis; pain Intensity and relief were also
measured. In addition, the time to meaningful pain relief and the request for
rescue medications were recorded.
Plasma was separated from blood and ibuprofen enantiomers (S and R)
were measured using an HPLC method. Plasma concentrations were plotted
versus time, and the area under curve (AUC) for ibuprofen concentration over
time was calculated. Incremental (cumulative) AUC values from the
administration time (zero) to each subsequent collection time were calculated.
Incremental AUC during the absorption phase is the most reliable measure of
absorption rate under conditions such those used in this trial.
All the avaiiable data collected for pharmacokinetic analyses from the 26
--2g-.
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
subjects who completed the study were used In the analyses. All
pharmacokinetic calculations were performed using SAS (PC version 6.12). Any
sample concentration reported less than the assay limit of quar<titation was
set to
zero for use in the pharmacokinetic and statistical analyses. Pharmacokinetic
and statistical analyses were not conducted for samples labeled "pain relief',
"onset and "prior to rescue". No concentration estimates were calculated for
missing values.
Pharmacokinetic Procedures
Pharmacokinetic parameters (areas, times to peak and elimination rates)
were calculated using the actual, rather than the scheduled, times of sample
collection. Graphical presentations of individual subject results also used
the actual
times of sample collection. Graphical presentations of mean results used the
schedufed times. Pharmacokinetic analyses were conducted on the concentrations
of R-ibuprofen, S-ibuprofen and total ibuprofen (sum of R-ibuprofen and S-
ibuprofen). See Figure 3.
Peak concentration (Cmax) was the observed maximum value during the
collection period of 0 (baseline) to 6 hours. The time to peak concentration
(Tmax)
was the time at which Cmax was observed (or first observed, if the peak value
occurred at more than one time).
The apparent first-order elimination rate constant (Ke) was estimated as
the negative value of the slope of the regression line for the terminal log-
linear
concentration-time values. A minimum of three terminal values was required to
obtain an estimate. The values included in the regression analyses were
determined by examination of the individual subject plots of natural logarithm
of
concentration against time. Whenever the terminal concentration-time values
were
not log-linear or the estimated rate would have been physiologically
implausible
(Ke<0.01), no elimination rate was estimated. Elimination half-life (T%2) was
estimated as 1oge(2)/Ke.
Areas under the curve were calculated from time 0 to the scheduled
time of each sample collection through the 6-hour collection sample. Area
under
the curve was calculated by the linear trapezoidal method. Area to infinite
time
--29w
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
(AUCinf) was calculated by extrapolating AUC 0-6, by the addition of the
quantity: Cg / Ke, where Ce is the concentration at the 6-hour collection
time.
Area to infinity could only be calculated when an elimination rate constant
had
been estimated.
Statistical analyses
Analysis of variance (ANOVA) was performed using the General Linear Models
(GLM) procedure of the SAS statistical program (PC version 6.12).
The statistical model contained a main effect of treatment. F-ratios for
testing treatment effect were constructed using the mean square term for
treatment as the numerator and the mean square error term from the ANOVA as
the denominator. Hypothesis testing was conducted at a=0.05. ANOVA was
performed for each pharmacokinetic parameter estimate
The intra-subject coefflcient of variation was estimated from the mean
square error term (MSE) of the ln-transformed (loge) results as:
100% * SQRT(eMSE -1)
Confidence Intervals (95%) for the area and peak concentration
comparisons were calculated by the t-test approach (2,1-sided) at a= 0.05
overall,
a = 0.025 each side:
Interval Lower Limit =(XT - XR )- Se * t./2
Interval Upper Limit =(XT - XR )+ Se * t,,,j2
Where:
XT and XR are the formulation of example 3[ZAG-1701] and Motrin IB
treatment least-squares means, respectively; Se is the standard error of the
estimated difference between means from the SAS estimate statement; and t',2
is
the critical value from the t-distribution with degrees of freedom that of the
error
term and a = 0.05.
For In-transformed data the interval was calculated from the ANOVA
. --30--
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
results on the transformed values and then exponentiated to convert to the non-
transformed scale:
Interval Limit = e{imtransf0r(ned lnterval Ilmit)
The Intervals were computed for the "true" mean treatment differences,
expressed as a percent of the reference mean, and true geometric mean ratios
(from logarithmic transformation).
Results
Table 2 summarizes the statistical analyses comparing 200 mg ZAG-
1701 capiets to 200 mg Motrin IB caplets with regard to incremental areas
under
the curve and time to peak. Incremental areas from time 0 to the time of the
early
sample collections are known to reflect the rate of drug absorption. Time of
peak
concentration is a direct function of this rate. ZAG-1701 caplets had
statistically
significantly (p<0.05) greater mean (up to 2.8 fold) incremental areas under
the
curve than Motrin iB through the first hour post-dose. This was true for R-
ibuprofen, S-ibuprofen and Total ibuprofen. The mean time of peak for [ZAG-
1701a
was less than 60% that of Motrin iB . These results indicate that the rate of
ibuprofen absorption for [ZAG-1701] is faster than that for Motrin !B .
Figures 1 and 2 show that during the first hour post-dose, when analgesics
are most needed, the formula provides a significantly greater concentration of
the
drug into the blood stream thereby to the site of action. S-ibuprofen is the
pharmacologically active enanitomer of racemic ibuprofen. R-ibuprofen converts
S-ibuprofen once in the body. Figure 4-6 show that the extent of absorption is
significantly greater following administration of the formula as compared with
Motrin IB during the first hour post-dose when the analgesic effect is most
needed. Figure 7 shows that there are significant differences between the two
products were noticed during the first 60 minutes following a complaint of
pain.
The formula demonstrated 2.8 fold greater extent of absorption during the
first 15
minutes as compared with Motrin 1B. Figure 8 shows that signifcant differences
between the two products were noticed during the first 60 minutes following a
--31-
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
complaint of pain. The extent of absorption becomes equal for the two products
in 6 hours. Figure 9 shows that there were no statistically significant
differences
between the two products due to the small number of subjects tested. However,
there was a consistent trend for greater PIS for Motrin IB. Figure 10 shows
that
there was no statistically significant difference between the two prodUcts due
to
the small number of subjects tested. However, there was a consistent trend for
greater PIS for Motrin IB after correction for the baseline pain. Figure 11
shows
that in the first 80 minutes, over 90% of the patients recorded relief after
taking
the formula of the present invention, as compared with over 60% after Motrin
!B.
Conclusion
The rate of ibuprofen absorption from ZAG-1701 is significantly faster than
that for Motrin IB . During the absorption phase (up to about 90 minutes after
administration) the incremental extent of absorption of ibuprofen was up to
2.8
fold greater from ZAG-1701 as compared with Motrin. ZAG-1701 and Motrin IB
have comparable total ibuprofen absorption.
--32-
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
Table 2: Statisticat Comparisons to Evaluate Rates of ibuprofen Absorption
Summary of statistical comparisons of arithmetic mean Incremental areas
under the curve and time to peak for ZAG-1701 and Motrin IB (ZAG-1701: n=12
subjects, Motrin IB : n=14 subjects).
Arithmetic Means
R-lbuprofen S-Ibuprofen Total Ibuprofen
Parameter Ratio Significance Ratio Significance Ratio Si nificance
z ;. e W '_ .~q
= .s ~. ~.v
AUC 0-0.25
( g-hr/mL) 2.867* 0.0438 2.744 0.0544 2.811* 0.0481
AUC Q-0.5
( g-hr/mL) 2.772* 0.0127 2.685* 0.0185 2.733* 0.0150
( g hrg/mL)5 2.398* 0.0056 2.294* 0.0123 2.352* 0.0079
~g hr0/mL) 2.052* 0.0058 1.948* 0.0140 2.006* 0.0085
AUC 0-1.5
(pg-hr/mL) 1.468 0.0752 1.380 0.1396 1.429 0.0972
AUC 0-2 1.368 0.0796 1.276 0.1691 1.327 0.1070
( g-hr/mL)
Al1C 0-3
(pg-hr/mL) 1.274 0.1163 1.173 0.2673 1.230 0.1554
AUC 0-4
( g-hr/mL) 1.181 '0.2219 1.094 0.4398 1.143 0.2742
AUC 0-5
( g-hrlmL) 1.096 0.4453 1.022 0.8196 1.063 0.5519
AUC 0-6
(pg-hr/mL) 1.049 0.6613 0.971 0.7166 1.013 0.8843
Tmax
0.569* 0.0390 0.575* 0.0448 0.575* 0.0448
* Comparison between products was detected as statistically sighificant by
ANOVA (a=0.05).
--33-
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
Example 18. Secondary Pharmacokinetic Parameters for Bioequivaience
Evaluations
Summary of statistical comparisons for secondary pharmacokinetic
parameters related to the determination of bloequivalence of EquiTech's 200 mg
ZAG-1701 caplets and 200 mg Motrin IB caplets-when each was administered as
a single 400 mg (2 X 200 mg) dose.
R-ibu rofen
Arithmetic Mean
Ratio
Parameters ZAG-1701 Motrin IB
(ho~ 1.26 -2.22 0,569*
K/hour 0.6254 Ø6998 0.894
hour 1.14 1.03 1.110
S-1bu rofen
Arithmetic Mean
Ratio
Parameters ZAG-1701 Motrin lB
houX 1.26 2.19 0.575*
8
(1lhaur 0.3088 0.3245 0.952
hour 2.48 2.38 1.046
Total ibu rofen
Arithmetic Mean
Parameters ~-G-1701 Motrin IB Ratio
max
hour 1.26 2.19 0.575*
1%hour 0.4526 0.4697 0.964
T'/z 1.57 1.51 1.034
hour
* Comparison detected as statistically significant by ANOVA (a=0.05).
=-34-.
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
Facamale 19.
Patients who require rescue medication due to lack of response to either
ZAG-1701 or Motrin. There were 12-14 patients/group. The time of request for
the rescue medication and the corresponding S-ibuprofen serum concentration
were recorded.
Rescue Medication
mg/L
Subject Treatment (S) at, min
7 ZAG 2.63 60
8 MOT 0 120
25 MOT 2.66 180
37 MOT 0.67 240
Example 20. Analytical Methods for Drug Product
Two analyfical testing procedures, Dissolution Assay of Ibuprofen Release in
ZAG-1701 Tablets, and ADetermination of (buprofen and lmpurities in Ibuprofen
Drug Product by HPLC are summarized below.
HPLC Parameters:
Column: Agilent Zorbax Eclipse XDB-C8, 5,um, 150 mm x 4.6 mm
Flow rate:0.8 mi/min
I n jection volume: 15 pL
Wavelength: 254 nm
Column temperature: Ambient
Mobile phase: i% chioroacetate buffer pH 3.0)/Acetonitrile 40/60 (v/v)
Mobile Phase and Diiuent Preparation:
1% chloroacetate buffer solution (pH 3.0): Dissolve 10.0 g chioroacetic acid
in
1000 ml of dionized watet. Adjust the pH to 3.0 with ammonium hydroxide
solution.
Mobile phase: Combine 400 mL of 1% chioroactetate buffer (pH and 600 mL of
acetonitrile and mix well. Filter the soiution through 0.45,um Nylon membrane
filter and degas before use, Diluent (0.2% chioroacetate buffer, pH
Acetonitriie,
--35--
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
40/60 v/v): Dissolve 20.0 g of chioracetic acid in 1000 mL of deionized water.
Adjust the pH to 3.0 with ammonium hydroxide solution. Mix 400 rni of 02%
chioroactate buffer with 600 mL of CAN.
Standard Solution Preparation
ibuprofen working Standard (4 mgJmL): Accurately weigh approximately
100 mg of ibuprofen reference standard, and transfer into a 25 mL volumetric
flask. Dissolve in approximately 15 mL of the diluent with shaking, dilute to
volume with diluent and mix well.
Resolution Solution Preparation
Isobutylacetophenone stock solution (0.20 mglmL): Weigh approximately
20 mg of 4- lsobutyiacetophenone reference standard, and transfer into a 100
mL volumetric flask. Dissolve in approximately 80 mL of acetonitrile with
shaking,
dilute to volume with acetonitrile and mix well
Resolution standard solution: Weigh approximately 100 mg of ibuprofen
reference standard into a 25 mL volumetric flask. Pipette 1,0 mL of 4-
isobutylacetophenone stock standard solution into the 25 mL volumetric flask.
Add approxirriately 15 mL of the diluent into the flask and shake until the
standard is completely dissolved. Dilute to volume with the djluent and mix.
Sample Solution Preparation
Potency and Related Substances: Accurately weigh 10 intact tablets and
record the tablets weight. Transfer the 10 tablets into a 500 mL volumetric
flask.
Add approximately 350 mL of the diluent Into the flask and shake mechanically
for 30 minutes. Sonicate the mixture for 30 minutes with occasional shaking.
Allow to cool to room temperature. Dilute to volume with the diluent and mix
well.
Centrifuge a portion of the resulting solution at about 3500 rpm for 10
minutes.
Content Uniformity: Weigh 10 individual intact tablets and record each
tablet weight. Transfer each tablet into a 50 mL volumetric flask. Add
approximately 30 mL of the diluent into the flask and shake mechanically for
30
minutes. Sonicate the mixture for 30 minutes with occasional shaking. Allow to
cool to room temperature. Dilute to volume with diluent and mix well.
Centrifuge a
portion of the resulting solution at about 3500 rpm for 10 minutes.
--36--
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
Blend Uniformity: Accurately weigh and record the weight of the container
with in process blend. This is gross weight. Depending on the weight of
contents,
transfer the entire contents into an appropriate volumetric flask. Add
appropriate
diluent in each volume flask, and shake mechanically for 30 minutes. Sonicate
the mixture for 30 minutes with occasional shaking. Allow cooling to room
temperature. Dilute to volume with the diluent and mix well. Centrifuge a
portion
of the resulting solution at about 3500 rpm for 10 minutes.
Identification: Accurately weigh 13 intact tablets and record the
tablets weight. Transfer the 10 tablets into a 500 mL volumetric flask. Add
approximately 350 mL of the diluent into the flask and shake mechanically
for 30 minutes. Sonicate the mixture for 30 minutes with occasional
shaking. Allow cooling to room temperature. Dilute to volume with the
diluent and mix well, Centrifuge a portion of the resulting solution, at about
3500 rpm for 10 minutes.
Example 21. Dissolution Assay of Ibuprofen Release in Tablets
Dissolution Parameters:
Dissolution Apparatus: USP Apparatus 2 (paddle)
Temperature: 37 ~ 0.5 C
Dissolution volume: 900 mL
Rotation speed: 50 rpm
Dissolution medium: 1% SDS in deionized water
Sampling time: For release testing: sampling at 30 minutes only
For stability testing: sampling at 5, 10, 20 and 30 minutes
Sampling volume: 10 mL with dissolution medium replacement
HPLC Parameters:
Column: Agilent Zorbax Eclipse XDB-C8, 5Nm, 150 mm x 4.6 mm
Flow rate: 0.8 mUmin
Injection volume: 15,uL
Wavelength: 254 nm
Column temperature: Ambient
--37--
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
Mobile phase: 1% chloroacetate buffer (pH 3.0)lAcetonitrile 40/60 (v/v)
Preparation of Dissolution Medium
Weigh and transfer approximately 10g of SDS into a suitable container,
add 1000 mL of deionized water to dissolve and mix well. Degas by sonicating
for 15 minutes before use.
Preparation of Diluent
For the preparation of about one liter of diluent, combine 500 mL of mobile
phase with 500 mL of dissolution medium and mix well.
Preparation of pH 3.0 Buffer
For the preparation of one liter of pH 3.0 buffer, weigh approximately 10.0 g
of
chioroacetic acid and dissolve in 950 rnL of deionized water, adjust pH with
NH4OH to
3Ø Dilute to 1000 mL with deionized water and mix thoroughly.
Preparation of Mobile Phase
For the preparation of about one liter of mobile phase, combine 400 mL of pH
3.0
buffer with 600 mL of acetonitrile and mix well. Filter the mobile phase
through a 0.45
,um nylon membrane filter and degas prior to use.
Preparation of Ibuprofen Standard Solution (110 ualmL
Accurately weigh approximately 22 mg of lbuprofen reference standard and
transfer into a 200 mL volumetric flask. Add about 150 mL of diluent to
dissolve,
sonicate if necessary. Dilute to volume with diluent once it is cooled to room
temperature and mix well. Filter a portion of this solution through the 0.45
pm PVDF
syringe, discarding the first 2 mL, and collect the fiitrate for analysis.
Preparation of Dissolution Sample Solution
1. According to the dissolution conditions, place 900 mL of degassed
dissolution
medium Into each of six dissolution vessels, assemble the apparatus and
equilibrate the
dissolution medium to 37 0.5 C.
2. Weigh each of six tablets and record the individual tablet weight. Place
each
tablet Into a dissolution vessel, taking care to exclude air bubbles from the
surface of
the dosage-form unit and immediately operate the apparatus at a rotation speed
of 50
rpm.
3. Sampling (For release) c Using a syringe fitted with a stainless steel
cannula,
..38..
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
withdraw 10 mL of the solution from a zone midway between the surface of the
dissolution medium and the top of the paddle, not less than 1 cm from the
vessel wall at
30 minutes.
4. Sampling (For stability) . Using a syringe fitted with a stainless steel.
cannufa,
withdraw 10 mL of the solution from a zone midway between the surface of the
dissolution medium and the top of the paddle, not less than 1 cm from the
vessel wall at
5, 10, 20, and 30 minutes time points for stability analysis, replace 10 mL of
dissolution
medium pre-wanned to 37 0.5 C back to dissolution vessel after every
sampling point.
5. Filter the sample solution through the 0.45,um PVDF syringe filter and
collect
the filtrate, discarding the first 2 mL. Dilute the filtrate 1:1 with mobile
phase and
mix thoroughly,
Test Specification Results
physical appearance capiet shape, white to off-white conforms
tablets
potency 90-110% of label claim 108.1%
content uniformity meets USP<905> requirements 103.8 B 113.0%
Mean: 108:3%
%RSD: 2.3
related substances
1) 4-isobutylacetophenone NMT 0.25% 0:00%
2) individual unknown NMT 0.2% 0.05% (RRT 0.64)
related substances 0.06% (RRT 0.82)
3) total related substances 0.11%
dissolution Q380% at 30 minutes 99.6%
(% dissolved 385%) %RSD = 2.0
loss on drying 2.9%
disintegration all tablets completely 2 min.13 sec.
disintegrated in NMT 15 minutes
..39...
CA 02589079 2007-05-02
WO 2006/056042 PCT/CA2005/001687
Although the present invention has been described in terms of particular
preferred embodiments, it is not limited to those embodiments. Alfiemative
embodiments, examples, and modifications which would still be encompassed by
the
invention may be made by those skiiled in the art, particularly in light of
the foregoing
teachings.
-40--