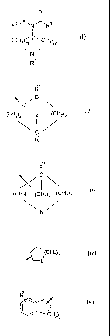Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
1
Novel Compounds
This invention relates to novel amide compounds which inhibit monoamine re-
uptake, to processes for
their preparation, to pharmaceutical compositions containing them and to their
use in medicine.
The compounds of the invention exhibit activity as serotonin and/or
noradrenaline re-uptake inhibitors and
therefore have utility in a variety of therapeutic areas. For example, the
compounds of the invention are
of use in the treatment of disorders in which the regulation of monoamine
transporter function is
implicated, more particularly disorders in which inhibition of re-uptake of
serotonin or noradrenaline is
implicated. Furthermore, the compounds of the invention are of use in
disorders in which inhibition of both
serotonin and noradrenaline is implicated, such as urinary incontinence.
Additionally, the compounds of
the invention are of use in disorders in which it may be desired to inhibit
preferentially the reuptake of one
of noradrenaline or serotonin compared with the other, such as pain,
depression, premature ejaculation;
ADHD or fibromyalgia.
According to a first aspect, the invention provides a compound of Formula (I)
O
R2iX, N'J~ R3
(CHZ n ' ' CHZ)m
N
i, I
R
and pharmaceutically and/or veterinarily acceptable derivatives thereof,
wherein:
R' is -H, -Ci-6alkyl, -C(A)Y, -C3$cycloalkyl, -aryl, -het, aryl-CI-4alkyl- or
het-C,-4alkyl-, wherein the
cycloalkyl, aryl or het groups are optionally substituted by at least one
substituent independently selected
from B;
A is S or 0;
Y is -H, -Cl-6alkyl, -aryl, -het, aryl-C,-aalkyl- or het-Cl-4alkyl-;
aryl is independently selected from phenyl, naphthyl, anthracyl or
phenanthryl;
het is independently selected from an aromatic or non-aromatic 4-, 5- or 6-
membered
heterocycle which contains at least one N, 0 or S heteroatom, optionally fused
to a 5- or 6-
membered carbocyclic group or a second 4-, 5- or 6-membered heterocycle which
contains at
least one N, 0 or S heteroatom;
B represents CI$alkyl-, CI$alkoxy-, -OH, -halo, -CF3, -CHF2, -OCF3, -OCHF2, -
SCF3, hydroxy-C,_
6alkyl-, C,4alkoxy-C,-6alkyl- and C1.aalkyl-S-C1_aalkyl-;
R2 is aryl' or het', each optionally substituted by at least one substituent
independently selected from D;
aryl' is independently selected from phenyl, naphthyl, anthracyl, phenanthryl,
or indanyl;
het' is an aromatic 5 to 10 membered heterocyclic ring system which contains
at least one N, 0
or S heteroatom, optionally containing an aryl group;
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
2
D represents C,$alkyl-, C,$alkoxy-, -OH, -halo, -CF3, -CHF2, -OCF3, -OCHF2, -
SCF3, hydroxy-Cl_
salkyl-, C, aalkoxy-C,-6alkyl-, -SC,$alkyl, C14alkyl-S-C,4alkyl-, -ary 12, -
het2, -Oaryl2, -Ohet2, -
Sary lZ, -Shet2, -CF2CF3, -CH2CF3, -CF2CH3, -OCF2CHF2, C3-acycloalkyl-,
C3.scycloalkyl-Cl4alkyl-,
C3-6cycloalkyl-Claalkoxy-, CMcycloalkyl-O-Cl4alkyl-, C3-.6cycloalkyl-Cl4alkoxy-
C,4alkyl-, -OC3_
6cycloalkyl, -OCj_8alkyl-C3$cycloalkyl and -SC3-6cycloalkyl, wherein the ary
lZ and het2 groups are
optionally substituted by at least one group selected from E;
ary lZ is independently selected from phenyl, naphthyl, anthracyl or
phenanthryl;
het2 is independently selected from an aromatic or non-aromatic 4-, 5- or 6-
membered
heterocycle which contains at least one N, 0 or S heteroatom, optionally fused
to a 5- or
6- membered carbocyclic group or a second 4-, 5- or 6-membered heterocycle
which
contains at least one N, 0 or S heteroatom;
E represents C,-6alkyl-, C3-6cycloalkyl-, CI-6alkoxy-, -OC3-6cycloalkyl, -
halo, -CN, -OH, -
CF3, -CHF2, -OCF3, -OCHF2, hydroxyC,-6alkyl-, CI-4alkoxy-Cl-4alkyl-, -SC,-
6alkyl and -SCF3i
R3 is -H, Cl$alkyl-, C3-8cycloalkyl-, Cmcycloalkyl-C,.6alkyl-,
C,$alkylSCI$alkyl-, -het3, or het3-CI-4alkyl-,
wherein the alkyl, cycloalkyl and het3 groups are each optionally substituted
by at least one substituent
independently selected from G;
het3 is a non-aromatic 4-, 5- or 6- membered heterocycle which contains at
least one N, 0 or S
heteroatom, optionally fused to a 5- or 6- membered carbocyclic group or a
second 4-, 5- or 6-
membered heterocycle which contains at least one N, 0 or S heteroatom;
G represents C,$alkyl-, CI-6alkoxy-, -OH, -halo, -CF3, -OCHF2, -OCF3, -SCF3, -
CN, -CF2CF3, -
CFZ-C,4alkyl, hydroxy-C,-6alkyl-, Cj4alkoxy-Cj_6alkyl- and C,4alkyl-S-Cl4alkyl-
; and the alkyl
groups being optionally substituted by at least one substituent independently
selected from J;
J represents C1_6alkoxy-, -OH, -halo, -CF3, -OCHF2, -OCF3, -SCF3, -CN, -
CF2CF3, -CF2-
C,_4alkyl, hydroxy-C,.6alkyl-, C14alkoxy_-C,_6alkyl- and CI.4alkyl-S-C,4alkyl-
;
or R3 is (CHZ)a.K, wherein a' is 0, 1 or 2 and K is a group selected from:
(i)
R
C
(CHz)a z (CHZ)b
30 R
wherein:
Z is O, S, NR12, (CHz)õ or a bond;
ais1,2,3or4;
bis1,2or3;
35 visl or2;
Rt0 and R" are each independently -H or C14 alkyl-;
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
3
R12 is -H, C,-s alkyl-, -C(O)C1 -6 alkyl, -SOZ-C,-6 alkyl;
and wherein one or more pairs of hydrogen atoms on adjacent carbon or nitrogen
atoms
may be replaced by a corresponding number of double bonds, provided the ring
system
is not aromatic;
(ii) a carbocyclic spiro group containing 6 to 12 carbon atoms;
(iii)
R
C
(CH2)c (CH2)e (CH2)d
N
10 wherein:
cis1,2,3or4;
dis1,2or3;
e is 1 or 2; and
R30 is -H or C,-4 alkyl-;
15 and wherein one or more pairs of hydrogen atoms on adjacent carbon atoms
may be
replaced by a corresponding number of double bonds, provided the ring system
is not
aromatic;
(iv)
~(CHZ)f
20 L
wherein:
fis0,1,2or3;
L is SO, SO2 or NR40; and
R40 is -H, CI.6 alkyl-, -C(O)C1 -6 alkyl, -SO2-C1-6 alkyl;
25 and wherein one or more pairs of hydrogen atoms on adjacent carbon atoms
may be
replaced by a corresponding number of double bonds, provided the ring system
is not
aromatic;
(v)
30 (CHZ)g
wherein:
gis0,1,2or3;and
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
4
R50 is -H, C,$alkyl-, C,-8alkoxy-, -OH, -halo, -CF3, -OCHF2, -OCF3, -SCF3,
hydroxy-C,_
6alkyl-, Cl.4alkoxy-C,-6alkyi- and C14aikyI-S-C,_4alkyl-; and
(vi) -CH(cyclopropane)2;
X is a covalent bond, CI$alkyl- or Cmcycloalkyl-, wherein if X is
C3$cycloalkyl, then R2-X may form a
fused aryl-cycloalkyl ring system; and
n is 1 or 2, provided that:
when n is 1, m is 0 or 1; and
when n is 2, m is 0;
wherein if m is 0, then * represents a chiral centre.
In an embodiment of the invention, R' is -H.
In a further embodiment of the invention, m is 0. Where m is 0, * represents
the R or S enantiomeric
configuration. Thus, in a further embodiment, m is 0; and * represents the S
enantiomer. In a yet further
embodiment, n is 1 and m is 0 or 1.
In a yet further embodiment, X represents a covalent bond and Cl-8alkyl-;
preferably C,$alkyl represents
C,-6alkyi, more preferably C14 alkyl, more preferably C1_2alkyl, and most
preferably represents -CH2-.
In a further embodiment, aryl' represents phenyl, naphthyl, and indanyl.
In a yet further embodiment, het' represents furyl, thienyl, oxazolyl,
thiazolyl, pyrazolyl, isoxazolyl,
isothiazolyi, pyridinyl, pyridazinyl, pyrimidinyl, pyrazinyl, benzoxazolyl,
benzothiazinyl, benzofuranyl,
quinolinyl, isoquinolinyl, quinazolinyl, quinoxalinyl, benzothiazolyl,
cinnolinyl, phthalzinyl, indolyl and
isoindolyl; preferably it represents quinolinyl, isoquinolinyl or pyridinyl;
preferably it represents quinolinyl.
In a further embodiment R2 is aryl' or het', each optionally substituted by
between one and three
substituents independently selected from D.
In a yet further embodiment D represents -halo, Cl$alkyl-, SC,$aikyl-, C,-
8alkyloxy-, C,-4aikoxy-C,-6alkyl-,
-ary 12, -Oary 12, -het2, C3$cycloalkyl-, -OCI$alkyl-C3-8cycioalkyl, -CF3, -
SCF3, -OCHF2, -CHF2, -OCF2CHF2,
and -OCF3; preferably halo represents fluoro, chloro, and bromo; preferably
C,$alkyl represents C14alkyl;
preferably CI$alkyloxy represents methoxy, ethoxy, and propoxy; preferably
aryl2 represents phenyl;
preferably Oary 12 represents OPh; preferably het2 represents pyridinyl;
preferably SCI$alkyl represents
SMe and SEt; preferably C3-8cycloalkyl represents cyclopropyl, cyclobutyl and
cyclopentyl; preferably C,_
4alkoxy-C,-6alkyl represents CHZOMe; and preferably OC,$alkyI-C3$cycloalkyl
represents OCH2-
cyclopropyl.
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
In a further embodiment D represents -halo, C,$alkyl-, SC,_aalkyl-,
C,$alkyloxy-, C,4aIkoxy-C,.6alkyl-, -
ary 12, -Oary 12, C3$cycloalkyl-, -OC,$alkyI-C3$cycloalkyI, -CF3, -SCF3, -
OCF2CHF2, and -OCF3; preferably
halo represents fluoro, chloro, and bromo; preferably C,$alkyl represents
C1_3alkyl; preferably C1$alkyloxy
represents methoxy, ethoxy, and propoxy; preferably ary 12 represents phenyl;
preferably Oary 12
5 represents OPh; preferably SC1$alkyl represents SMe and SEt; preferably
C3$cycloalkyl represents
cyclopropyl, cyclobutyl and cyclopentyl; preferably Cl.4alkoxy-CI.6alkyl
represents CH2OMe; and
preferably OCj$alkyl-C3-8cycloalkyl represents OCHZ-cyclopropyl.
In a yet further embodiment D represents -halo, C,$alkyl-, -SC,$alkyl,
Cl.8alkyloxy-, -ary 12, -Oary 12, C3_
acycloalkyl-, -OC,$alkyl-C3$cycloalkyl, -CF3, _OCF2CHF2, and -OCF3; preferably
halo represents fluoro,
chloro, and bromo; preferably C,$alkyl represents C,_3alkyl; preferably
Cl$alkyloxy represents methoxy
and ethoxy; preferably ary 12 represents phenyl; preferably Oary lZ represents
OPh; preferably SC,$alkyl
represents SMe and SEt; preferably C3-8cycloalkyl represents cyclopropyl; and
preferably OC1$alkyl-C3_
8cycloalkyl represents OCH2-cyclopropyl.
In a further embodiment E represents halo; preferably chloro and fluoro;
preferably fluoro.
In a still further embodiment R3 represents CI-salkyl-, C3$cycloalkyl-, C3-
8cycloalkyl-Cj_6alkyI-, and Cl_
8aIkyISC,-8alkyl-; preferably C1$alkyl. represents C1_6alkyl; preferably
C3$cycloalkyl represents C3_
scycloalkyl; preferably C3$cycloalkyl-C,-6alkyl represents cyclopentylmethyl;
and preferably C,$alkylSC,_
8alkyl represents CH2SMe.
In a yet further embodiment, G represents Cl-6alkoxy-, -halo, -OH, and -CF3;
preferably halo represents
fluoro; preferably CI$alkoxy represents methoxy and ethoxy. In a further
embodiment, G represents CF3.
In a further embodiment het3 represents a non-aromatic 6- membered heterocycle
which contains at least
one N, 0 or S heteroatom; preferably it represents a non-aromatic 6- membered
heterocycle which
contains at least one 0 heteroatom; preferably it represents
tetrahydropyranyl.
According to an alternative aspect, the invention provides a compound of
Formula (I')
O
2 ~( ~ 3
R~ ~N R
(CHZ n = ' CHZ)m
N~
R
and pharmaceutically and/or veterinarily acceptable derivatives thereof,
wherein:
R' is -H, C,-6alkyl-, -C(A)Y, C3-8cycloalkyl-, -aryl, -het, aryl-C,4alkyl- or
het-C,-4alkyl-, wherein the
cycloalkyl, aryl or het groups are optionally substituted by at least one
substituent independently selected
from C,$alkyl-, C,$alkoxy-, -OH, -halo, -CF3, -CHF2, -OCF3, -OCHF2, -SCF3,
hydroxy-C,-6alkyl-, Cl_
4alkoxy-C,-6alkyl- and C,_4alkyl-S-C,4alkyl-;
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
6
R2 is aryl or heteroaryl, each optionally substituted by at least one
substituent independently selected
from C,$alkyl-, C,$alkoxy-, -OH, -halo, -CF3, -CHF2, -OCF3, -OCHF2, -SCF3,
hydroxy-C,-6alkyl-, Cl_
4alkoxy-C,-6alkyl-, Cl-4alkyl-S-Cl-4alkyl-, -aryl', -het', -Oaryl', -Ohet', -
Saryl', -Shet', -CF2CF3, -CH2CF3, -
CF2CH3, C3$cycloalkyl-, C3-6cycloalkyl-C1_4alkyl-, C3-6cycloalkyl-C,.4alkoxy-,
C3-6cycloalkyl-O-CI_4aIkyl-, C3-
ficycloalkyl-C1-4alkoxy-C,-4alkyl-, -OC3-6cycloalkyl and -SC3-6cycloalkyl,
wherein the aryl' and het' groups
are optionally substituted by at least one group selected from C,-6alkyl-, C3-
6cycloalkyl-, C,-6alkoxy-, -OC3-
6cycloalkyl, -halo, -CN, -OH, -CF3, -CHF2, -OCF3, -OCHF2, hydroxyC,.6alkyl-,
C,-4alkoxy-CI.4alkyl-, -SCj_
6alkyl and -SCF3;
R3 is -H, C,-8alkyl-, C3$cycloalkyl-, C3$cycloalkyl-Cl-6alkyl-, -heterocycle,
or heterocycle-C,-4alkyl-,
wherein the cycloalkyl and heterocycle groups are each optionally substituted
by at least one substituent
independently selected from C,$alkyl-, CI-6alkoxy-, -OH, -halo, -CF3, -OCHF2, -
OCF3, -SCF3, -CN, -
CF2CF3, -CF2-C,-4alkyl, hydroxy-CI-6alkyl-, C,.4alkoxy-CI-6alkyl- and
C1_4alkyl-S-C1_4alkyl-; and the alkyl
groups are optionally substituted by at least one substituent independently
selected from C,-6alkoxy-, =
OH, -halo, -CF3, -OCHF2, -OCF3, -SCF3, -CN, -CF2CF3, -CF2-CI-4alkyl, hydroxy-
C,.6alkyl-, C,-4alkoxy-Cl_
salkyl- and C1_4alkyl-S-C1_4alkyl-;
or R3 is (CH2)a,E, wherein a' is 0, 1 or 2 and E is a group selected from:
(i)
R
C
(CH2)a z (CHZ)b
/
õ
R
wherein:
Z is 0, S, NR12, (CH2), or a bond;
ais1,2,3or4;
bis1,2or3;
v is 1 or 2;
R10 and R" are each independently -H or C1_4 alkyl-; and
R12 is -H, Cl-6 alkyl-, -C(O)C1 -6 alkyl, -SO2-CI-6 alkyl;
and wherein one or more pairs of hydrogen atoms on adjacent carbon or nitrogen
atoms
may be replaced by a corresponding number of double bonds, provided the ring
system
is not aromatic;
(ii) a carbocyclic spiro group containing 6 to 12 carbon atoms;
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
7
(iii)
R
I
(CH2)a (CHz)r (CH2)e
N
wherein:
ais1,2,3or4;
5 bis1,2or3;
c is 1 or 2; and
R30 is -H or CI-4 alkyl-;
-and wherein one or more pairs of hydrogen atoms on adjacent carbon atoms may
be
replaced by a corresponding number of double bonds, provided the ring system
is not
10 aromatic;
(iv)
~(CH2)d
J
wherein:
15 d.is0,1,2or3;
J is SO, SOZ or NR40; and
R40 is -H, C,, alkyl-, C(O)CI.6 alkyl-, -SO2-CI$ alkyl;
and wherein one or more pairs of hydrogen atoms on adjacent carbon atoms may
be
replaced by a corresponding number of double bonds, provided the ring system
is not
20 aromatic;
(v)
(CHZ)e
wherein:
25 e is 0, 1, 2 or 3; and
R50 is -H, C,$atkyl-, C,$alkoxy-, -OH, -halo, -CF3, -OCHF2, -OCF3, -SCF3,
hydroxy-C,_
6alkyl-, C,4alkoxy-CI-6alkyl- and C,4alkyl-S-C,4alkyl-; and
(vi) -CH(cyclopropane)2;
X is a covalent bond, C,-8alkyl- or C3$cycloalkyl-, wherein if X is
3$cycloalkyl, then R2-X may form a fused
aryl-cycloalkyl ring system;
AisSor0;
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
8
Y is -H, CI.6alkyl-, -aryl, -het, aryl-C,-4alkyl- or het-C,-aalkyl-;
n is 1 or 2, provided that when n is 1, m is 0 or 1 and when n is 2, m is 0,
wherein if m is 0, then "
represents a chiral centre;
aryl and aryl' are each independently selected from phenyl, riaphthyl,
anthracyl or phenanthryl;
heteroaryl is an aromatic 5- or 6- membered heterocycle which contains at
least one N, 0 or S
heteroatom, optionally fused to an aryl group;
heterocycle is a non-aromatic 4-, 5- or 6- membered heterocycle which contains
at least one N, 0 or S
heteroatom, optionally fused to a 5- or 6- membered carbocyclic group or a
second 4-, 5- or 6-membered
heterocycle which contains at least one N, 0 or S heteroatom; and
het and het' are each independently selected from an aromatic or non-aromatic
4-, 5- or 6- membered
heterocycle which contains at least one N, 0 or S heteroatom, optionally fused
to a 5- or 6- membered
carbocyclic group or a second 4-, 5- or 6-membered heterocycle which contains
at least one N, 0 or S
heteroatom.
In a yet further embodiment of the invention, there is provided a compound of
Formula II'
O
4 X-N R 3
R~
CJCH2)i II,
H
and pharmaceutically and/or veterinarily acceptable derivatives thereof,
wherein:
R3 is as defined above in any embodiment;
R4 is phenyl, naphthyl, or quinolinyl, each optionally substituted by at least
one substituent independently
selected from C1$alkyl-, CI$alkoxy-, -OH, -halo, -CF3, -CHF2, -OCF3, -OCHF2, -
SCF3, hydroxy-C,-6alkyl-,
C,_4alkoxy-Cj_~.alkyl-, Cl-4alkyl-S-Cl-4alkyl-, -aryl', -het', -Oaryl', -
Ohet', -Saryl', -Shet', -CF2CF3, -
CH2CF3, -CF2CH3, C3-6cycloalkyl-, C3-6cycloalkyl-C,.4alkyl-, C3-6cycloalkyl-Cl-
4alkoxy-, C3.6cycloalkyl-O-C,_
4alkyl-, C3-6cycloalkyl-C,-4alkoxy-C,-4alkyl-, -OC3-6cycloalkyl and -SC3-
6cycloalkyl, wherein the aryl' and
het' groups are optionally substituted by at least one group selected from
C1_6alkyl, C3-6cycloalkyl, Cl_
6alkoxy, OC3-6cycloalkyl, halo, CN, OH, CF3, CHF2, OCF3, OCHF2,
hydroxyC,.6alkyl, C,-4alkoxy-C,-4alkyl,
SC1.6alkyl and SCF3;
X is a covalent bond, C,$alkyl- or C3$cycloalkyl-, wherein if X is 3-
8cycloalkyl, then R4-X may form a fused
aryl-cycloalkyl ring system; and
m is 0 or 1, wherein if m is 0, then * represents the R or S enantiomer.
In a further embodiment, R4 is phenyl, 1-naphthyl or 2-naphthyl, each
optionally'substituted by at least
one substituent independently selected from C,$alkyl-, C, aalkoxy-, -OH, -
halo, -CF3, -CHF2, -OCF3, -
OCHF2, -SCF3, hydroxy-C,-salkyl-, C,-4alkoxy-CI-6alkyl-, C,-4alkyl-S-Cl4alkyl-
, -aryl', -het', -Oaryl', -Ohet',
-Saryl', -Shet', -CF2CF3, -CH2CF3, -CF2CH3, C3-6cycloalkyl-, C3-6cycloalkyl-C,-
4alkyl-, C3.6cycloalkyl-C,_
4alkoxy-, C3-6cycloalkyl-O-Cl.4alkyl-, C3-6cycloalkyl-C,4alkoxy-CI.4alkyl-, -
OC3-6cycloalkyl and -SC3_
6cycloalkyl, wherein the aryl' and het' groups are optionally substituted by
at least one group selected
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
9
from C,-6alkyl-, C3-6cycloalkyl-, C,-6alkoxy-, -OC3-6cycloalkyl, -halo, -CN, -
OH, -CF3, -CHF2, -OCF3, -
OCHF2, hydroxyC,-6alkyl-, C,-4alkoxy-CI-4alkyl-, -SC,-6alkyl and -SCF3. The
phenyl or naphthyl groups
may be substituted by one, two or three substituents.
In a yet still further embodiment, m is 0. In this embodiment, * represents
the R or S enantiomer. In a
further embodiment, m is 0 and * represents the S enantiomer.
In a still further embodiment, there is provided a compound of Formula III'
O 3
FP-N1~1 R
N
H III'
and pharmaceutically and/or veterinarily acceptable derivativesthereof,
wherein:
R3 is as defined above in any embodiment;
R6 is phenyl, naphthyl or quinolinyl, each optionally substituted by at least
one substituent independently
selected from C,.8alkyl-, C,$alkoxy-, -OH, -halo, -CF3, -CHF2, -OCF3, -OCHF2, -
SCF3, hydroxy-C,-6alkyl-,
C,4alkoxy-CI-6alkyl-, C,4alkyl-S-Cl4aikyl-, -aryl', -het', -Oaryl', -Ohet', -
Saryl', -Shet', -CF2CF3, -
CH2CF3, -CF2CH3, C3-6cycloalkyl-, C3-6cycloalkyl-C,_4alkyl-, C3-6cycloalkyl-
C,_4alkoxy-, C3.6cycloalkyl-O-Cj_
4alkyl-, C3-6cycloalkyl-CI-4alkoxy-C,-4alkyl-, -OC3-6cycloalkyl and -SC3-
6cycloalkyl, wherein the aryl' and
het' groups are optionally substituted by at least one group selected from
C,.salkyl-, C3-6cycloalkyl-, C,_
salkoxy-, -OC3-6cycloalkyl, -halo, -CN, -OH, -CF3, -CHF2, -OCF3, -OCHF2,
hydroxyC,.6alkyl-, CI-4alkoxy-Cl_
4alkyl-, -SC,-6alkyl and -SCF3;
X is a covalent bond, C1$alkyl- or C3$cycloalkyl-, wherein if X is
3$cycloalkyl, then R6-X may form a fused
aryl-cycloalkyl ring system; and
* represents the R or S enantiomer.
In a further embodiment, R 6 is phenyl, 1-naphthyl or 2-naphthyl, each
optionally substituted by at least
one substituent independently selected from C,$alkyl-, C,$alkoxy-, -OH, -halo,
-CF3, -CHF2, -OCF3, -
OCHF2, -SCF3, hydroxy-C,.6alkyl-, C14alkoxy-C,-6alkyl-, C14alkyl-S-Cl.aalkyl-,
-aryl', -het', -Oaryl', -Ohet',
-Saryl', -Shet', -CF2CF3, -CH2CF3, -CF2CH3, C3-6cycloalkyl-, C3-6cycloalkyl-
C,_4alkyl-, C3-6cycloalkyl-C,_
4alkoxy-, C3-6cycloalkyl-O-Cl.4alkyl-, C3-6cycloalkyl-C,-4alkoxy-Cl4alkyl-, -
OC3-6cycloalkyl and -SC3_
6cycloalkyl, wherein the aryl' and het' groups are optionally substituted by
at least one group selected
from C,-salkyl-, Cmcycloalkyl-, C,-6alkoxy-, -OCmcycloalkyl, -halo, -CN, -OH,.-
CF3, -CHF2, -OCF3, -
OCHF2, hydroxyC,-6alkyl-, C1.4alkoxy-C, 4alkyl-, -SCI-6alkyl and -SCF3.
In a yet further embodiment, * represents the S enantiomer.
In a further embodiment, the invention provides a compound selected from:
N-(Biphenyl-2-ylmethyl)-2-methyl-N-[(3S)-pyrrolidin-3-yl]propanamide;
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
N-(2,4-Dichlorobenzyl)-3-methyl-N-[(3S)-pyrrolidin-3-yl]butanamide;
N-(2,4-Dichlorobenzyl)-2-methyl-N-[(3S)-pyrrolidin-3-yl]propanamide;
N-(2,3-Dichlorobenzyl)-2-methyl-N-[(3S)-pyrrol idin-3-yl]propanamide;
N-(2-Naphthylmethyl)-N-[(3S)-pyrrolidin-3-yl]acetamide;
5 N-[(2'-fl uorob i phenyl -2-yl)m ethyl] -2-methyl-N-[(3S)-pyrrol i di n-3-
yl] propanam ide;
N-[(3'-fluorobiphenyl-2-yl)methy-]-2-methyl-N-[(3S)-pyrrolidin-3-yl]propanam
ide;
N-(biphenyl-2-ylmethyl)-N-[(3S)-pyrrolidin-3-yl]butanamide;
N-(biphenyl-2-ylmethyl)-3-methyl-N-[(3S)-pyrrolidin-3-yl]butanamide;
N-[(4'-chlorobiphenyl-2-yl)methyl]-2-methyl-N-[(3S)-pyrrolidin-3-
yl]propanamide;
10 N-(biphenyl-2-ylmethyl}N-[(3S)-pyrrolidin-3-yl]propanamide;
N-[2-(ethylthio)benzyl]-2-m ethyl-N-[(3S)-pyrrol idi n-3-yl]propanam ide;
N-(4-chloro-2-methoxybenzyl )-2-methyl-N-[(3S)-pyrrolidin-3-yl]propanamide;
2-m ethyl-N-(2-phenoxybenzyl )-N-[(3S)-pyrrol idi n-3-yl] propanam ide;
N-[(4'-fluorobiphenyl-2-yl )methyl]-2-methyl-N-[(3S)-pyrrolidin-3-
yl]propanamide;
N-[(2',4'-difluorobiphenyl-2-yl)methyi]-2-methyl-N-[(3S)-pyrrolidin-3-
yl]propanamide;
2-methyl-N-[(3S)-pyrrolidin-3-yl]=N-[2-(1,1,2,2-
tetrafluoroethoxy)benzyl]propanamide;
N-(2-bromobenzyl )-2-methyl-N-[(3S)-pyrrolid in-3-yl]propanamide;
N-(4-chloro-2-methoxybenzyl )-3-methyl-N-[(3S)-pyrrolidin-3-yl]butanamide;
2-methyl-N-[(3S)-pyrrolidin-3-yl]-N-[2-(trifluoromethyl)benzyl]propanamide;
2-methyl-N-[(3S)-pyrrolidin-3-yl]-N-(2,3,4-trichlorobenzyl)propanamide;
2-methyl-N-[2-(methylthio)benzyl]-N-[(3S)-pyrrolidin-3-yl]propanam ide;
N-[(3'-fluorobiphenyl-2-yl)methyl]-2-methyl-N-[(3R)-pyrrolidin-3-
yl]propanamide;
2-methyl-N-(3-phenoxybenzyl )-N-[(3S)-pyrrol idin-3-yl]propanam ide;
N-[(3'-chlorobiphenyl-2-yl)methyl]-2-methyl-N-[(3S)-pyrrolidin-3-
yl]propanamide;
N-(2,4-dichlorobenzyl)-N-[(3S)-pyrrolidin-3-yl]cyclopentanecarboxamide;
N-(2-cyclopropyl benzyl )-2-methyl-N-[(3S)-pyrrol idin-3-yl]propanamide;
N-(2-bromobenzyl)-N-[(3S)-pyrrolidin-3-yl]cyclopropanecarboxamide;
N-[(3S)-pyrrolidin-3-yl]-N-[2-(trifluoromethyl)benzyl]cyclopropanecarboxamide;
N-[(3',4'-difluorobiphenyl-2-yl)methyl]-N-[(3S)-pyrrolidin-3-
yl]cyclobutanecarboxamide;
N-(2,3-dichlorobenzyl)-N-[(3S)-pyrrolidin-3-yl]cyclobutanecarboxamide;
N-[(3S)-pyrrol idin-3-yl]-N-[2-(trifluoromethyl
)benzyl]cyclopentanecarboxamide;
N-[2-(cyclopropylmethoxy)benzyl]-2-methyl-N-[(3S)-pyrrolidin-3-yl]propanamide;
N-(4-chloro-2-ethoxybenzyl )-3-methyl-N-[(3S)-pyrrol idin-3-yl]butanamide;
N-(2,4-dichlorobenzyl)-2-methyl=N-[(3S)-pyrrolidin-3-yl]butanamide;
N-(2,4-dichlorobenzyl)-2-ethyl-N-[(3S)-pyrrolidin-3-yl]butanamide;
N-(4-chloro-2-methylbenzyl )-3-methyl-N-[(3S)-pyrrolidin-3-yl]butanamide;
N-(2,4-dichlorobenzyl)-N-[(3S)-pyrrolidin-3-yl]cyclohexanecarboxamide;
N-(2,4-dichlorobenzyl )-N-[(3S)-pyrrolidin-3-yl]butanamide;
N-(2,4-dichlorobenzyl)-3,3-dimethyl-N-[(3S)-pyrrolidin-3-yl]butanamide;
N-(2,3-dichlorobenzyl)-3-methyl-N-[(3S)-pyrrolidin-3-yl]butanamide;
N-(2,3-dichlorobenzyl)-N-[(3S)-pyrrolidin-3-yl]pentanamide;
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
11
N-(2,4-dichlorobenzyl)-N-[(3S)-pyrrolidin-3-yl]pentanamide;
2-cyclopropyl-N-(2,4-dichlorobenzyl)-N-[(3S)-pyrrolidin-3-yl]acetamide;
N-(2,4-dichlorobenzyl)-4-methyl-N-[(3S)-pyrrolidin-3-yl]pentanamide;
2-methyl-N-[(3S)-pyrrolidin-3-yl]-N-[3-(trifluoromethoxy)benzyl]propanamide;
2-methyl-N-[(3S)-pyrrolidin-3-yl]-N-(2,3,5-trichlorobenzyl)propanamide;
N-(2-naphthylmethyl)-N-[(3R)-pyrrolidin-3-yl]acetamide;
N-(2-naphthylmethyl)-N-[(3R)-pyrrolidin-3-yl]cyclohexa necarboxam ide;
N-(2-naphthylmethyl)-N-[(3R)-pyrrolidin-3-yl]propanamide;
N-(2-naphthylmethyl)-N-[(3R)-pyrrolidin-3-yl]cyclobutanecarboxamide;
3-methyl-N-(2-naphthylmethyl)-N-[(3R)-pyrrolidin-3-yl]butanamide;
N-(2-naphthylmethyl)-N-[(3S)-pyrrolidin-3-yl]cyclopentanecarboxam ide;
N-(2-naphthylmethyl)-N-[(3S)-pyrrolidin-3-yl]propanam ide;
N-(2-naphthylmethyl)-N-[(3S)-pyrrolidin-3-yl]cyclobutanecarboxamide;
3-methyl-N-(2-naphthylmethyl)-N-[(3S)-pyrrolidin-3-yl]butanamide;
N-(2-naphthylmethyl)-N-[(3S)-pyrrolidin-3-yl]cyclohexanecarboxamide;
2-methyl-N-(2-naphthylmethyl)-N-[(3S)-pyrrolidin-3-yl]propanamide;
N-[(1-methyl-2-naphthyl)methyl]-N-[(3S)-pyrrolidin-3-yl]acetamide;
N-[(1-methyl-2-naphthyl)methyl]-N-[(3S)-pyrrolidin-3-yl]propanamide;
N-[(6-fluoro-l-methyl-2-naphthyl) methyl]-N-[(3R)-pyrrolidin-3-yl]acetamide;
N-[(6-fluoro- 1 -methyl-2-na p hthyl)m ethyl]- N-[(3R)-pyrrolidin-3-
yl]propanam ide;
N-[(6-fluoro-1-methyl-2-naphthyl)methyl]-N-[(3S)-pyrrolidin-3-yl]acetamide;
N-[(6-fluoro-1 -methyl-2-naphthyl)methyl]-N-[(3S)-pyrrolidin-3-yl]propanamide;
2-(methylthio)-N-(2-naphthylmethyl)-N-[(3R)-pyrrolidin-3-yl]acetamide;
N-[(3R)-pyrrolidin-3-yl]-N-(quinolin-6-ylmethyl)pentanamide;
N-[(3S)-pyrrolidin-3-yl]-N-(quinolin-6-ylmethyl)pentanamide;
N-[(3S)-pyrrolidin-3-yl]-N-(quinolin-6-ylmethyl)butanamide;
N-(2,4-dichlorobenzyl)-N-[(3S)-pyrrolidin-3-yl]cyclopropanecarboxamide;
N-(2,4-dichlorobenzyl)-N-[(3S)-pyrrolidin-3-yl]propanam ide;
4,4,4-trifluoro-N-[(3R)-pyrrolidin-3-yl]-N-(quinolin-6-ylmethyl)butanamide;
4,4,4-trifluoro-N-[(3S)-pyrrolidin-3-yl]-N-(quinolin-6-ylmethyl)butanamide;
N-(2,4-dimethylbenzyl)-N-[(3S)-pyrrolidin-3-yl]cyclobutanecarboxamide;
N-(4-chloro-2-fluorobenzyl)-N-[(3S)-pyrrolidin-3-yl]cyclobutanecarboxamide;
N-(3-chloro-2-methylbenzyl)-N-[(3S)-pyrrolidin-3-yl]cyclopropanecarboxamide;
N-(2,4-dim ethyl benzyl)-N-[(3 S)-pyrrolid i n-3-yl]cyclopropanecarboxam ide;
N-(2,4-dichlorobenzyl)-N-[(3S)-pyrrolidin-3-yl]cyclobutanecarboxamide;
N-(2,3-dimethylbenzyl)-N-[(3S)-pyrrolidin-3-yl]cyclopropanecarboxamide;
N-(3-chloro-4-methylbenzyl)-N-[(3S)-pyrrolidin-3-yl]cyclobutanecarboxamide;
N-[2-fluoro-4-(trifluoromethyl)benzyl]-2-methyl-N-[(3S)-pyrrolidin-3-
yl]propanamide;
N-(2-chloro-4-fl uorobenzyl)-2-m ethyl-N-[(3 S)-pyrro lid i n-3-yl] propa nam
ide;
N-(2,3-dichlorobenzyl)-2-methyl-N-[(3R)-pyrrolidin-3-yl]propanamide;
N-(4-chloro-2-ethoxybenzyl)-2-methyl-N-[(3S)-pyrrolidin-3-yl]propanamide;
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
12
N-[2-methoxy-4-(trifluoromethyl)benzyl]-2-methyl-N-[(3S)-pyrrolidin-3-
yl]propanamide;
N-(5-chloro-2,3-dihydro-1 H-inden-1-yl)-2-methyl-N-[(3S)-pyrrolidin-3-
yl]propanamide;
N-(2-naphthylmethyl)-N-piperidin-4-ylacetamide;
N-[(1-methyl-2-naphthyl)methyl]-N-piperidin-4-ylacetamide;
N-(2-naphthylmethyl)-N-piperidin-4-ylpropanamide;
N-(2-naphthylmethyl)-N-piperidin-4-ylbutanamide;
3-methyl-N-(1-naphthylmethyl)-N-piperidin-4-ylbutanamide;
N-piperidin-4-yl-N-(quinolin-6-ylmethyl)propanamide;
N-piperidin-4-yl-N-(quinolin-6-ylmethyl)butanamide;
2-methyl-N-piperidin-4-yl-N-(quinolin-6-ylmethyl)propanamide;
N-[(1-ethyl-2-naphthyl )methyl]-N-piperidin-4-ylacetam ide;
N-[(7-methoxy-1-methyl-2-naphthyl)methyl]-N-piperidin-4-ylacetamide;
N-[(7-methoxy-1 -methyl-2-naphthyl)methyl]-N-piperidin-4-ylpropanamide;
2-hydroxy-N-(2-naphthylmethyl)-N-piperidin-4-ylpropanamide;
N-(2,3-dichlorobenzyl)-2-methyl-N-piperidin-4-ylpropanamide;
N-(2,4-dichlorobenzyl)-2-methyl-N-piperidin-4-ylpropanamide;
and pharmaceutically and/or veterinarily acceptable derivatives thereof.
The above described embodiments of the invention may be combined with one or
more further
embodiments such that further embodiments are provided wherein two or more
variables are defined
more specifically in combination. For example, within the scope of the
invention is a further embodiment
wherein the variables R', RZ, R3, X, m and n all have the more limited
definitions assigned to them in the
more specific embodiments described above. All such combinations of the more
specific embodiments
described and defined above are within the scope of the invention
By pharmaceutically and/or veterinarily acceptable derivative it is meant any
pharmaceutically or
veterinarily acceptable salt, solvate, ester or amide, or salt or solvate of
such ester or amide, complex,
polymorph, stereoisomer, geometric isomer, tautomeric form, or isotopic
variation, of the compounds of
formula (I), (I'), (II) or (III) or any other compound which upon
administration to the recipient is capable of
.30 providing (directly or indirectly) a compound of formula (I), (I'), (II')
or (III') or an active metabolite or
residue thereof. Preferably, pharmaceutically acceptable derivatives are
salts, solvates, esters and
amides of the compounds of formula (I), (I'), (II') or (III'). More
preferably, pharmaceutically acceptable
derivatives are salts and solvates.
For pharmaceutical or veterinary use, the salts referred to above will be the
pharmaceutically or
veterinarily acceptable salts, but other salts may find use, for example in
the preparation of compounds of
formula (I), (I'), (II') or (III') and the pharmaceutically or veterinarily
acceptable salts thereof.
The aforementioned pharmaceutically or veterinarily acceptable salts include
the acid addition and base
salts thereof.
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
13
Suitable acid addition salts are formed from acids which form non-toxic salts.
Examples include the
acetate, aspartate, benzoate, besylate, bicarbonate/carbonate,
bisulphate/sulphate, camsylate, citrate,
hemicitrate, edisylate, hemiedisylate, esylate, fumarate, gluceptate,
gluconate, glucuronate, hibenzate,
hydrochloride/chloride, hydrobromide/bromide, hydroiodide/iodide, isethionate,
lactate, malate, maleate,
malonate, mesylate, methylsulphate, 2-napsylate, nicotinate, nitrate, orotate,
pamoate,
phosphate/hydrogen phosphate/dihydrogen phosphate, saccharate, stearate,
succinate, tartrate and
tosylate salts.
Suitable base salts are formed from bases which form non-toxic salts. Examples
include the aluminium,
arginine, benzathine, calcium, choline, diethylamine, diolamine, glycine,
lysine, magnesium, meglumine,
olamine, potassium, sodium, tromethamine and zinc salts.
For a review on suitable salts, see "Handbook of Pharmaceutical Salts:
Properties, Selection, and Use"
by Stahl and Wermuth (Wiley-VCH, Weinheim, Germany, 2002).
A pharmaceutically acceptable salt of a compound of formula (I), (I'), (II')
or (III') may be readily prepared
by mixing together solutions of the compound and the desired acid or base, as
appropriate. The salt may
precipitate from solution and be collected by filtration or may be recovered
by evaporation of the solvent.
The degree of ionisation in the salt may vary.from completely ionised to
almost non-ionised.
Pharmaceutically acceptable solvates in accordance with the invention include
hydrates and solvates of
the compounds of formula (I), (I'), (II') or (III').
Also within the scope of the invention are complexes such as clathrates, drug-
host inclusion complexes
wherein, in contrast to the aforementioned solvates, the drug and host are
present in stoichiometric or
non-stoichiometric amounts. Also included in this invention are complexes of
the pharmaceutical drug
which contain two or more organic and/or inorganic components which may be in
stoichiometric or non-
stoichiometric amounts. The resulting complexes may be ionised, partially
ionised, or non-ionised. For a
review of such complexes, see J Pharm Sci, 64 (8), 1269-1288 by Haleblian
(August 1975).
The compounds of formula (I), (I'), (II') or (III') may be modified to provide
pharmaceutically or veterinarily
acceptable derivatives thereof at any of the functional groups in the
compounds. Examples of such
derivatives are described in: Drugs of Today, Volume 19, Number 9, 1983, pp
499 - 538; Topics in
Chemistry, Chapter 31, pp 306 - 316; and in "Design of Prodrugs" by H.
Bundgaard, Elsevier, 1985, Chapter
1 (the disclosures in which documents are incorporated herein by reference)
and include: esters, carbonate
esters, hemi-esters, phosphate esters, nitro esters, sulfate esters,
sulfoxides, amides, sulphonamides,
carbamates, azo-compounds, phosphamides, glycosides, ethers, acetals and
ketals.
It will be further appreciated by those skilled in the art, that certain
moieties, known in the art as "pro-
moieties", for example as described by H. Bundgaard in "Design of Prodrugs"
(ibid) may be placed on
appropriate functionalities when such functionalities are present within
compounds of the invention.
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
14
The compounds of formula (I), (I'), (II') or (III') may contain one or more
chiral centres, by virtue of the
asymmetric carbon atom defined by certain meanings of the "R" groups (e.g. s-
butyl), or the value of the
integer m. Such compounds exist in a number of stereoisomeric forms (e.g. in
the form of a pair of optical
isomers, or enantiomers). It is to be understood that the present invention
encompasses all isomers of
the compounds of the invention, including all geometric, tautomeric and
optical forms, and mixtures
thereof (e.g. tautomeric or racemic mixtures).
The compounds of the invention may exist in one or more tautomeric forms. AII
tautomers and rriixtures
thereof are included in the scope of the present invention. For example, a
claim to 2-hydroxypyridinyl
would also cover its tautomeric form a-pyridonyl.
It is to be understood that the present invention includes radio labelled
compounds of formula (I), (I'), (II')
or (III').
The compounds of formula (I), (I'), (II') or (III') and their pharmaceutically
and veterinarily acceptable
derivatives may also be able to exist in more than one crystal form, a
characteristic known as
polymorphism. AII such polymorphic forms ("polymorphs") are encompassed within
the scope of the
invention. Polymorphism generally can occur as a response to changes in
temperature or pressure or
both, and can also result from variations in the crystallisation process.
Polymorphs can be distinguished
by various physical characteristics, and typically the x-ray diffraction
patterns, solubility behaviour, and
melting point of the compound are used to distinguish polymorphs.
Unless otherwise indicated, any alkyl group may be straight or branched and is
of 1 to 8 carbon atoms,
such as 1 to 6 carbon atoms or 1 to 4 carbon atoms, for example a methyl,
ethyl, n-propyl, i-propyl, n-
butyl, i-butyl, s-butyl or t-butyl group. Where the alkyl group contains more
than one carbon atom, it may
be unsaturated. Thus, the term C,-6 alkyl includes C2_6 alkenyl and C2.6
alkynyl. Similarly, the term Cl$
alkyl includes C2$ alkenyl and C2$ alkynyl, and the term Cl-4 alkyl includes
C2_4 alkenyl and C2_4 alkynyl.
The term halogen is used to represent fluorine, chlorine, bromine or iodine.
Unless otherwise indicated, the term het includes any aromatic, saturated or
unsaturated 4-, 5- or 6-
membered heterocycle which contains up to 4 heteroatoms selected from N, 0 and
S. Examples of such
heterocyclic groups included furyl, thienyl, pyrrolyl, pyrrolinyl,
pyrrolidinyl, imidazolyl, dioxolanyl, oxazolyl,
thiazolyl, imidazolyl, imidazolinyl, imidazolidinyl, pyrazolyl, pyrazolinyl,
pyrazolidinyl, isoxazolyl,
isothiazolyl, oxadiazolyl, triazolyl, thiadiazolyl, pyranyl,
tetrahydropyranyl, pyridyl, piperidinyl, dioxanyl,
morpholino, dithianyl, thiomorpholino, pyridazinyl, pyrimidinyl, pyrazinyl,
piperazinyl, sulfolanyl, tetrazolyl,
triazinyl, azepinyl, oxazapinyl, thiazepinyl, diazepinyl and thiazolinyl. In
addition, the term heterocycle
includes fused heterocyclyl groups, for example benzimidazolyl, benzoxazolyl,
imidazopyridinyl,
benzoxazinyl, benzothiazinyl, oxazolopyridinyl, benzofuranyl, quinolinyl,
quinazolinyl, quinoxalinyl,
dihydroquinazdinyl, benzothiazolyl, phthalimido, benzodiazepinyl, indolyl and
isoindolyl.
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
For the avoidance of doubt, unless otherwise indicated, the term substituted
means substituted by one or
more defined groups. In the case where groups may be selected from a number of
alternative groups, the
selected groups may be the same or different. Further, the term independently
means that where more
than one substituent is selected from a number of possible substituents, those
substituents may be the
5 same or different.
Hereinafter, the compounds of formula (I), (I'), (II') or (III') and their
pharmaceutically and veterfnarily
acceptable derivatives, the radio labelled analogues of the foregoing, the
isomers of the foregoing, and
the polymorphs of the foregoing, are referred to as "compounds of the
invention".
In one embodiment of the invention, the compounds of the invention are the
pharmaceutically and
veterinarily acceptable derivatives of compounds of formula (I), (I'), (II')
or (III'), such as the
pharmaceutically or veterinarily acceptable salts or solvates of compounds of
formula (I), (I'), (II') or (III'),
(e.g. pharmaceutically or veterinarily acceptable salts of compounds of
formula (1), (i'), (11') or (ill')),
In a still further embodiment of the invention, there is provided a compound
of the invention which is an
inhibitor of serotonin and/or noradrenaline monoamine re-uptake, having SRI or
NRI ICSo/Ki values of
500nM or less, preferably 400nM or less, more preferably 200nM or less. In a
further embodiment, the
compound has SRI and/or NRI IC50/Ki values of lOOnM or less. In a yet further
embodiment, the
compound has SRI and/or NRI IC50/Ki values of 5OnM or less. In a still yet
further embodiment, the
compound has SRI and/or NRI IC5o/Ki values of 25nM or less.
Without wishing to be bound by theory, it is believed that for certain of the
diseases or conditions for
which the compounds of the invention are indicated it is useful for the
compound to be a more potent
inhibitor of the reuptake of one of serotonin or noradrenaline than the other.
Thus, in an embodiment of
the invention, the reuptake of noradrenaline is inhibited to greater degree
than the reuptake of serotonin.
In an alternative embodiment, the reuptake of serotonin is inhibited to a
greater degree than the reuptake
of noradrenaline. For example, in the treatment of pain, it is believed that
compounds of the invention
which inhibit the reuptake of noradrenaline have good efficacy. Thus, an
embodiment of the invention
provides a method of treating pain which comprises administering to a patient
in need of such treatment a
therapeutically effective amount of a compound according to the invention
which is capable of inhibiting
the reuptake of noradrenaline. In this embodiment, the compound of the
invention may selectively inhibit
the reuptake of noradrenaline or it may inhibit the reuptake of noradrenaline
preferentially to the inhibition
of serotonin reuptake or it may inhibit the reuptake of serotonin
preferentially to the inhibition of
noradrenaline reuptake. In a further embodiment of the invention, there
provided compounds which are
more potent noradrenalin reuptake inhibitors than serotonin reuptake
inhibitors. Accordingly, such an
embodiment of the invention provides a method of treating pain which comprises
administering to a
patient in need of such treatment a therapeutically effective amount of a
compound according to the
invention which is capable of inhibiting the reuptake of noradrenaline to a
greater extent than the reuptake
of serotonin.
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
16
According to Scheme 1, compounds of Formula (I) may be prepared from compounds
of Formula (I11) by
reaction with an aldehyde, R2CHO (or a suitable ketone), in the presence of a
reducing agent, followed by
reaction with an acid or anhydride or acid chloride R3COX', where X' is OH or
halo or OCOR3, or
alternatively with an acid mixed anhydride, and deprotection.
0 O
NH2 H R2/'-, NH R3J1x, R2/~NRs
O1~'RZ (X'=0H or halo or OCOR3)
n(HzC~ N ) CH2)m (a) n(H2C CHZ)m (b) n(H2C~ J(CHZ)m
N N
PG PG PG
III IV V
O
Rz~\N~R
deprotection
I n(HZC ~ N ) CHz)m (c)
~
i
H
Scheme I
In the above scheme, R3, R2, m and n are as defined above, X is CH2 and PG is
a protecting group.
(a) - Reductive Amination
The reaction of the 1 amine (III) with the aldehyde R2CHO to form the 2
amine (IV) is a reductive
amination reaction, in which the dehydration of the amine and the aldehyde is
followed by reduction of the
formed imine by a metal hydride reagent or hydrogenation, in a suitable
solvent at room temperature.
In this reaction, equimolar amounts of amine and aldehyde are typically
treated with either sodium
triacetoxyborohydride (STAB), NaBH3CN or NaBH4, in a suitable solvent (e.g.
DCM, THF) at room
temperature for 1 to 24 hours. Alternatively, an excess of a reducing agent
(e.g. NaBH4, LiAIH4, STAB) in
a suitable solvent (e.g. THF, MeOH, EtOH, toluene) is added after the amine
and aldehyde have been
mixed for 1-18 hours, optionally in the presence of a drying agent (e.g.
molecular sieve) or with the
removal of water using Dean-Stark apparatus with a suitable solvent (e.g.
toluene, xylene). A further
alternative involves catalytic hydrogenation in the presence of a palladium or
nickel catalyst (e.g. Pd/C,
Raney Ni) under an atmosphere of H2, optionally at elevated temperature and
pressure, in a suitable
solvent (e.g. EtOH).
A more specific example of the reductive amination involves treatment of the
amine with the aldehyde in
the presence of either 10% Pd/C; optionally in the presence of triethylamine,
in ethanol under about 415
kPa (about 60psi) of hydrogen at room temperature for 18 hours.
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
17
Another more specific example of the reductive amination is treatment of the
amine with the aldehyde in
toluene at reflux under Dean-Stark conditions for 18 hours and then after
concentration, treatment with an
excess of sodium borohydride in methanol at room temperature for 3 hours.
Suitable aldehydes are either, known and available from commercial sources,
or, are derivable from
commercially available materials using known techniques; for example, 7-
methoxy-l-methyl-naphtalene-
2-carbaldehyde can be prepared using the preparation described in WO
2004/111003.
(b) - Amide Formation
The formation of an amide bond between the acid or acid halide or anhydride
(R3COX') and the amine
(IV) may be undertaken by using either:
(i) the acyl halide or anhydride and the amine (IV), with an excess of acid
acceptor in a suitable solvent;
or
(ii) the acid, optionally with a conventional coupling agent, and the amine
(IV), optionally in the presence
of a catalyst, with an excess of acid acceptor in a suitable solvent.
Examples of such reactions are as follows:
(a) An acid chloride (optionally generated in situ) or anhydride is reacted
with an excess of the amine
(IV), optionally with an excess of 30 amine (such as Et3N, Hunig's base,
pyridine or NMM), in
DCM (or dioxane), optionally at an elevated temperature for between 1 and 24
hours;
(b) An acid, WSCDI (or DCCI, or TBTU) and HOBT (or HOAT) is reacted with an
excess of amine
(IV) and an excess of NMM ( or Et3N, or Hunig's base) in THF (or DCM, or
EtOAc), at room
temperature for between 4 and 48 hours; or
(c) An acid and PYBOP (or PyBrOP , or Mukaiyama's reagent) is reacted with an
excess of amine
(IV) and an excess of NMM (or Et3N, or Hunig's base) in THF (or DCM, or
EtOAc), at room
temperature for between 4 and 24 hours.
Where the acid halide is an acid chloride (i.e. X'=Cl), this may be generated
in situ by standard
methodology and then reacted with the amine (IV) in the presence of
triethylamine in dichloromethane at
room temperature for 1 hour.
Suitable acids, anhydrides and acid chlorides are either, known and available
from commercial sources,
or, are derivable from commercially available materials using known
techniques.
(c) - Deprotection
Where PG is a suitable amine-protecting group, preferably BOC,
trifluoroacetate, benzyloxycarbonyl (Bz)
or benzyl (Bn), the removal of PG from (V), to forrri the unprotected amine
(I), is performed by a method
selective to the protecting group as detailed in "Protective Groups in Organic
Synthesis", 3rd edition, by
TW Greene and PGM Wuts. John Wiley and Sons, Inc., 1999, incorporated herein
by reference.
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
18
Examples of such deprotection reactions are as follows:
When PG is BOC, the deprotection involves treatment of (V) with an excess of
strong acid (e.g. HCI, TFA)
at room temperature in a suitable solvent (e.g. DCM, EtOAc,. dioxane).
When PG is trifluoroacetate, the deprotection involves treatment of (V) with a
base (e.g. K2CO3, Na2CO3,
NH3, Ba(OH)2) in an alcoholic solvent (e.g. MeOH, EtOH), optionally with water
and optionally at elevated
temperature. More specifically, when PG is trifluoroacetate, the deprotection
involves treatment with
KZC03 in methanol:water mixture (5:1 to 10:1) at room temperature for 18 hours
(WO 2004110995).
When PG is Bn or Bz, the deprotection involves either transfer hydrogenation
with a transition metal or
transition metal salt hydrogenation catalyst (e.g. Pd/C, Pd(OH)2) in the
presence of a hydrogen donor
(e.g. NH4+HC02 ) in a polar solvent (e.g. THF, EtOH, MeOH) optionally at
elevated temperature and/or
pressure, or catalytic hydrogenation in the presence of a palladium or nickel
catalyst (e.g. Pd/C, Raney -
Ni) under an atmosphere of H2, optionally at elevated temperature and
pressure, in a suitable solvent.
More specifically:
When PG is BOC, the deprotection involves treatment with either an excess of
4M hydrogen chloride in
dioxane for 18 hours at room temperature or with TFA in DCM for 20 hours at
room temperature.
When PG is trifluoroacetate, the deprotection involves treatment with K2CO3 in
methanol:water mixture
(5:1 to 10:1) at room temperature for 18 hours.
When PG is Bn or Bz, the deprotection involves treatment with NH4+HC02 and 10%
Pd/C in ethanol
under gentle reflux for between 4 and 20 hours.
When PG is trifluoroacetate, the deprotection involves treatment of (V) with a
base (e.g. K2CO3, Na2CO3,
NH3, Ba(OH)2) in an alcoholic solvent (e.g. MeOH, EtOH), optionally with water
and optionally at an
elevated temperature.
More specifically, when PG is trifluoroacetate, the deprotection involves
treatment with K2CO3 in
methanol:water mixture (5:1 to 10:1) at room temperature for 18 hours
(examples of such deprotection
are described in WO 2004110995).
According to Scheme 2, compounds of Formula (I) may be prepared from compounds
of Formula (III) by
reaction with R2-X-L, where L is a leaving group, under suitable conditions.
The resulting compound of
Formula (VI) may then be converted to a compound of Formula (I) by amide
formation and deprotection in
a manner analogous to that described above in relation to Scheme 1.
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
19
OII O
NH2 R2~X~NH R3JlX, R2iX.N~Ra
R2.,X. 1 (X'=OH or halo or OCOR3)
n(HZC ( L n(H2C n(H2T ~ CHZ)m
JCH2)m JCH2)m N PG PG PG
III VI VII
O
R2,-X, N1~, R3 deprotection
I n(H2C CH2)m
~N
I
H
Scheme 2
In the above scheme, RZ, R3, X, m and n are as defined above, PG is a suitable
protecting group and L is
a leaving group, whose meaning will depend, inter alia, on the nature of the
reaction and the specific
reaction conditions employed. Suitable leaving groups will be readily apparent
to the skilled person and
are described in many standard organic chemistry texts, for example: "Advanced
Organic Chemistry",
Jerry March, Third Edition, Wiley (1985), page 587, incorporated herein by
reference; they include
halogen (e.g. Br) and sulfonate esters (e.g. methanesulfonate or
trifluoromethanesulfonate).
Conveniently, R2 is an aryl group, X is alkyl, L is Br and reaction (d) is
carried out in a suitable solvent, at
elevated temperatures, in the presence of a 3 amine (such as Et3N, or HUnig's
base, or NMM, or an
inorganic base).
In a more specific example of a process according to Scheme 2, amine (III) is
treated with the
arylalkylbromide in acetonitrile under gentle reflux for between 1 and 20
hours in the presence of
potassium carbonate.
Alternatively, where R 2 is an aryl group, X is a covalent bond and L is Br,
reaction (d) may be carried out
in a suitable solvent, at elevated temperatures, in the presence of a
palladium catalyst. Such palladium
mediated aryl amination reactions are well known to those skilled in the art.
More specifically, this involves the treatment of an aryl bromide with an
amine of Formula (III) in the
presence of tris(dibenzylideneacetone)dipalladium, 2,2'-bis(diphenylphosphino)-
1,1'-binaphthyl and
sodium tert-butoxide in toluene at 100 C for 18 hours.
Suitable alkyl halides are either, known and available from commercial
sources, or are derivable from
commercially available materials using known techniques.
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
According to Scheme 3, compounds of Formula (VI) may be prepared from a ketone
of Formula (VIII) by
reaction with a primary amine R2-X-NHZ in the presence of a reducing agent,
under suitable conditions.
The resulting compound of Formula (VI) may then be converted to a compound of
Formula (I) by amide
formation and deprotection in a manner analogous to that described above in
relation to Scheme 1.
O
0 R2iX.NH R3~X. RZ~X,N R3
RZ~X NH (X'=0H or halo or OCOR3)
n(HZC~ N j CHZ)m 2 n(HZC CH2)m n(HZC~ JCHZ)m
e -- ~ ~ N N
PG PG PG
VIII VI VII
O
RZ,-X, N~R3
deprotection
I n(HzCl CH2)m
N Jf
1
5 H
In the above scheme, R3, RZ, X, m and n are as defined above and PG is a
suitable protecting group.
Scheme 3
The reaction (e) of the primary amine R2-X-NH2 with the ketone (VIII) may
conveniently be a reductive
10 amination reaction in which the dehydration of the amine and the ketone is
followed by reduction of the
resultant imine, for example by a metal hydride reagent or hydrogenation,
under suitable conditions.
Conveniently, the reaction of the amine and the ketone is carried out in the
presence of titanium (IV)
tetraisopropoxide in THF at room temperature for 18 hours, followed by
reduction by an excess of sodium
15 borohydride in methanol at room temperature for 5 hours.
Suitable amines are either, known and available from commercial sources, or
are derivable from
commercially available materials using known techniques.
0 O
~ z
j)N Z R= x. Z H N R
(x'=OH or halo) HN R BH3
( x) (Y) N
PG N\ PG
PG
III IV
20 Scheme 4.
In the above scheme, R 2 is as defined above and PG is a protecting group.
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
21
According to Scheme 4, compounds of Formula (IV) may be prepared from a 1
amine of Formula (III) by
reaction with a carboxylic acid or acid halide, optionally prepared in situ,
RZCOX' (where X' is OH or halo),
followed by reaction with a reducing agent, such as borane.
The formation of an amide bond between the acid or acid halide and the 1
amine (III) may be undertaken
by using either:
(i) the acyl halide (or the acid or acid anhydride) and the amine (III), with
an excess of acid acceptor in a
suitable solvent, or
(ii) the acid, optionally with a conventional coupling agent, and the amine
(III), optionally in the presence
of a catalyst, with an excess of acid acceptor in a suitable solvent.
Examples of such reactions are as follows:
(a) An acid chloride (optionally generated in situ) is reacted with an excess
of the amine (III),
optionally with an excess of 3 amine (such as Et3N, Hunig's base or NMM), in
DCM (or dioxane),
optionally at an elevated temperature for between 1 and 24 hours;
(b) An acid, WSCDI (or DCCI, or TBTU) and HOBT (or HOAT) is reacted with an
excess of amine
(III) and an excess of NMM (or Et3N, or Hunig's base) in THF (or DCM, or
EtOAc), at room
temperature for between 4 and 48 hours; or
(c) An acid and 1-propyl phosphonic ester cyclic anhydride (or PYBOP , or
PyBrOP , or
Mukaiyama's reagent) is reacted with an excess of amine (III) and an excess of
NMM (or Et3N, or
Hunig's base) in THF (or DCM, or EtOAc), at room temperature for between 1 and
24 hours.
A more specific example of the amide formation involves treatment of the acid
with the amine in the
presence of 1-propyl phosphonic ester cyclic anhydride and in the presence of
triethylamine in DCM at
room temperature for 1 hour.
Where the acid halide is an acid chloride (i.e. X'=CI), this may be generated
in situ by standard
methodology and then reacted with the amine (III) and triethylamine in
dichloromethane at 70 C for 90
minutes
The reaction (y) is a reduction of the amide to amine (IV) for example by a
hydride reducing agent under
suitable conditions.
Conveniently, the reduction of the amide is carried out in the presence of
Borane in THF at reflux for 2
hours, followed by addition of methanol and optionally of aqueousammonium
chloride and further reflux
for 4 hours before isolation of the amine (IV).
The skilled person is able to select the most appropriate synthetic route to
the desired compound
according to Formula (I), (II) or (III). The above schemes may of course be
modified as appropriate in
accordance with the common general knowledge of those skilled in the art.
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
22
For example, the skilled person will of course appreciate that the hydrogen
attached to the piperidine or
pyrrolidine nitrogen (depending upon the value of m) of the deprotected amide
(I) can be replaced with
alternative groups as desired to form an alternative compound of Formula (I)
where n is 1 and m is 0 or 1
by the use of conventional synthetic methodologies.
In addition, compounds of Formula (I) where n is 2 and m is 0 can be prepared
by analogous processes
to those described above using the appropriate starting materials.
It will be appreciated by those skilled in the art that one or more sensitive
functional groups may need to
be protected and deprotected during the synthesis of a compound of Formula
(I), (II) or (III). This may be
achieved by conventional techniques, for example as described in "Protective
Groups in Organic
Synthesis", 3rd edition, by TW Greene and PGM Wuts. John Wiley and Sons, Inc.,
1999, incorporated
herein by reference, which also describes methods for the removal of such
groups.
It will be apparent to those skilled in the art that certain protected
derivatives of compounds of the
invention, which may be made prior to a final deprofection stage, may not
possess pharmacological
activity as such, but may, in certain instances, be administered orally or
parenterally and thereafter
metabolized in the body to form compounds of the invention which are
pharmacologically active. Such
derivatives may therefore be described as prodrugs. Further, certain compounds
of the invention may act
as prodrugs of other compounds of the invention.
According to a further aspect of the invention, there is provided a process
for preparing compounds of
Formula (I), (I'), (II') or (III'), which comprises reacting a compound of
formula (X):
H, N"Xl- R
(CH2 ~ CH2)m X
~N~
I
Y
wherein R2, n and m are as defined above and Y is R' or a protecting group,
with an acid or acyl halide:
R3COX, wherein X is OH or halo, or an acid anhydride: (R3CO)20, and
deprotecting if necessary.
Where X is CH2, then the compound of Formula (X) may be prepared by reacting a
compound of Formula
(XXI) with an aldehyde: R2CHO in the presence of a reducing agent.
NH2
(CH2 n ' CHZ)m XXI
~
Y
Alternatively, the compound of Formula (X) may be prepared by reacting a
compound of Formula (XXI)
with a compound R2-X-L, where L is a leaving group, optionally selected from
halide, methanesulfonate
and trifluoromethanesulfonate.
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
23
Furthermore, the compound of Formula (X) may be prepared by reacting a
compound of Formula (XXII)
with a compound RZ-X-NHZ in the presence of a reducing agent.
O
(CH2_' (CH2)m XXII
~~\ NJ
Y
Certain intermediates described above are novel compounds and it is to be
understood that all novel
intermediates herein form further aspects of the present invention.
Racemic compounds may be separated either using preparative HPLC and a column
with a chiral
stationary phase, or resolved to yield individual enantiomers utilizing
methods known to those skilled in
the art. In addition, racemic chiral intermediate compounds may be resolved
and used to prepare
enantio-enriched chiral compounds of the invention.
According to a further aspect of the invention, there is provided one or more
metabolites of the
compounds of the invention when formed in vivo.
The compounds of the invention may have the advantage that they are more
potent, have a longer duration
of action, have a broader range of activity, are more stable, have fewer side
effects or are more selective, or
have other more useful properties than the compounds of the prior art.
The compounds of the invention are useful because they have pharmacological
activity in mammals,
including humans. Thus, they are useful in the treatment or prevention of
disorders in which the
regulation of monoamine transporter function is implicated, more particularly
disorders in which inhibition
of re-uptake of serotonin or noradrenaline is implicated. Furthermore, the
compounds of the invention are
of use in disorders in which inhibition of both serotonin and noradrenaline is
implicated, such as urinary
incontinence. Additionally, the compounds of the invention are of use in
disorders in which it may be
desired to inhibit preferentially the reuptake of one of noradrenaline or
serotonin compared with the other,
such as pain.
Accordingly the compounds of the invention are useful in the treatment of
urinary incontinence, such as
genuine stress incontinence (GSI), stress urinary incontinence (SUI) or
urinary incontinence in the elderly;
overactive bladder (OAB), including idiopathic detrusor instability, detrusor
overactivity secondary to
neurological diseases (e.g. Parkinson's disease, multiple sclerosis, spinal
cord injury and stroke) and
detrusor overactivity secondary to bladder outflow obstruction (e.g. benign
prostatic hyperplasia (BPH),
urethral stricture or stenosis); nocturnal eneuresis; urinary incontinence due
to a combination of the
above conditions (e.g. stress incontinence associated with overactive
bladder); and lower urinary tract
symptoms, such as frequency and urgency. The term OAB is intended to encompass
both OAB wet and
OAB dry.
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
24
In view of their aforementioned pharmacological activity the compounds of the
invention are also useful in
the treatment of depression, such as major depression, recurrent depression,
single episode depression,
subsyndromal symptomatic depression, depression in cancer patients, depression
in Parkinson's
patients, postmyocardial infarction depression, paediatric depression, child
abuse induced depression,
depression in infertile women, post partum depression, premenstrual dysphoria
and grumpy old man
syndrome.
In view of their aforementioned pharmacological activity the compounds of the
invention are also useful in
the treatment of cognitive disorders such as dementia, particularly
degenerative dementia (including
senile dementia, Alzheimer's disease, Pick's disease, Huntingdon's chorea,
Parkinson's disease and
Creutzfeldt-Jakob disease) and vascular dementia (including multi-infarct
dementia), as well as dementia
associated with intracranial space occupying lesions, trauma, infections and
related conditions (including
HIV infection), metabolism, toxins, anoxia and vitamin deficiency; mild
cognitive impairment associated
with ageing, particularly age associated memory impairment (AAMI), amnestic
disorder and age-related
cognitive decline (ARCD); psychotic disorders, such as schizophrenia and
mania; anxiety disorders, such
as generalised anxiety disorder, phobias (e.g. agoraphobia, social phobia and
simple phobias), panic
disorder, obsessive compulsive disorder, post traumatic stress disorder, mixed
anxiety and depression;
personality disorders such as avoidant personality disorder and attention
deficit hyperactivity disorder
(ADHD); sexual dysfunction, such as premature ejaculation, male erectile
dysfunction (MED) and female
sexual dysfunction (FSD) (e.g. female sexual arousal disorder (FSAD));
premenstrual syndrome;
seasonal affective disorder (SAD); eating disorders, such as anorexia nervosa
and bulimia nervosa;
obesity; appetite suppression; chemical dependencies resulting from addiction
to drugs or substances of
abuse, such as addictions to nicotine, alcohol, cocaine, heroin, phenobarbital
and benzodiazepines;
withdrawal syndromes, such as those that may arise from the aforementioed
chemical dependencies;
cephalic pain, such as migraine, cluster headache, chronic paroxysmal
hemicrania, headache associated
with vascular disorders, headache associated with chemical dependencies or
withdrawal syndromes
resulting from chemical dependencies, and tension headache; pain; Parkinson's
diseases, such as
dementia in Parkinson's disease, neuroleptic-induced Parkinsonism and tardive
dyskinesias); endocrine
disorders; such as hyperprolactinaemia; vasospasm, such as in the cerebral
vasculature; cerebellar
ataxia; Tourette's syndrome; trichotillomania; kleptomania; emotional
lability; pathological crying; sleep
disorder (cataplexy); and shock.
In view of their aforementioned pharmacological activity the compounds of the
invention are also useful in
the treatment of a number of other conditions or disorders, including
hypotension; gastrointestinal tract
disorders (involving changes in motility and secretion) such as irritable
bowel syndrome (IBS), ileus (e.g.
post-operative ileus and ileus during sepsis), gastroparesis (e.g. diabetic
gastroparesis), peptic ulcer,
gastroesophageal reflux disease (GORD, or its synonym GERD), flatulence and
other functional bowel
disorders, such as dyspepsia (e.g. non-ulcerative dyspepsia (NUD)) and non-
cardiac chest pain (NCCP);
and fibromyalgia syndrome.
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
The compounds of the invention, being serotonin and/or noradrenaline reuptake
inhibitors are potentially
useful in the treatment of a range of disorders, including pain.
Physiological pain is an important protective mechanism designed to warn of
danger from potentially
5 injurious stimuli from the external environment. The system operates through
a specific set of primary
sensory neurones and is activated by noxious stimuli via peripheral
transducing mechanisms (see Millan,
1999, Prog. Neurobiol., 57, 1-164 for a review). These sensory fibres are
known as nociceptors and are
characteristically small diameter axons with slow conduction vefocities.
Nociceptors encode the intensity,
duration and quality of noxious stimulus and by virtue of their
topographically organised projection to the
10 spinal cord, the location of the stimulus. The nociceptors are found on
nociceptive nerve fibres of which
there are two main types, A-delta fibres (myelinated) and C fibres (non-
myelinated). The activity
generated by nociceptor input is transferred, after complex processing in the
dorsal horn, either directly,
or via brain stem relay nuclei, to the ventrobasal thalamus and then on to the
cortex, where the sensation
of pain is generated.
Pain may generally be classified as acute or chronic: Acute pain begins
suddenly and is short-lived
(usually in twelve weeks,or less). It is usually associated with a specific
cause such as a specific injury
and is often sharp and severe. It is the kind of pain that can occur after
specific injuries resulting from
surgery, dental work, a strain or a sprain. Acute pain does not generally
result in any persistent
psychological response. In contrast, chronic pain is long-term pain, typically
persisting for more than three
months and leading to significant psychological and emotional problems. Common
examples of chronic
pain are neuropathic pain (e.g. painful diabetic neuropathy, postherpetic
neuralgia), carpal tunnel
syndrome, back pain, headache, cancer pain, arthritic pain and chronic post-
surgical pain.
When a substantial injury occurs to body tissue, via disease or trauma, the
characteristics of nociceptor
activation are altered and there is sensitisation in the periphery, locally
around the injury and centrally
where the nociceptors terminate. These effects lead to a hightened sensation
of pain. In acute pain these
mechanisms can be useful, in promoting protective behaviours which may better
enable repair processes
to take place. The normal expectation would be that sensitivity returns to
normal once the injury has
healed. However, in many chronic pain states, the hypersensitivity far
outlasts the healing process and is
often due to nervous system injury. This injury often, leads to abnormalities
in sensory nerve fibres
associated with maladaptation and aberrant activity (Woolf & Salter, 2000,
Science, 288, 1765-1768).
Clinical pain is present when discomfort and abnormal sensitivity feature
among the patient's symptoms.
Patients tend to be quite heterogeneous and may present with various pain
symptoms. Such symptoms
include: 1) spontaneous pain which may be dull, burning, or stabbing; 2)
exaggerated pain responses to
noxious stimuli (hyperalgesia); and 3) pain produced by normally innocuous
stimuli (allodynia - Meyer et
al., 1994, Textbook of Pain, 13-44). Although patients suffering from various
forms of acute and chronic
pain may have similar symptoms, the underlying mechanisms may be different and
may, therefore,
require different treatment strategies. Pain can also therefore be divided
into a number of different
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
26
subtypes according to differing pathophysiology, including nociceptive,
inflammatory and neuropathic
pain.
Nociceptive pain is induced by tissue injury or by intense stimuli with the
potential to cause injury. Pain
afferents are activated by transduction of stimuli by nociceptors at the site
of injury and activate neurons
in the spinal cord at the level of their termination. This is then relayed up
the spinal tracts to the brain
where pain is perceived (Meyer et al., 1994, Textbook of Pain, 13-44). The
activation of nociceptors
activates two types of afferent nerve fibres. Myelinated A-delta fibres
transmit rapidly and are responsible
for sharp and stabbing pain sensations, whilst unmyelinated C fibres transmit
at a slower rate and convey
a dull or aching pain. Moderate to severe acute nociceptive pain is a
prominent feature of pain from
central nervous system trauma, strains/sprains, burns, myocardial infarction
and acute pancreatitis, post-
operative pain (pain following any type of surgical procedure), posttraumatic
pain, renal colic, cancer pain
and back pain. Cancer pain may be chronic pain such as tumour related pain
(e.g. bone pain, headache,
facial pain or visceral pain) or pain associated with cancer therapy (e.g.
postchemotherapy syndrome,
chronic postsurgical pain syndrome or post radiation syndrome). Cancer pain
may also occur in response
to chemotherapy, immunotherapy, hormonal therapy or=radiotherapy. Back pain
may be due to herniated
or ruptured intervertabral discs or abnormalities of the lumber facet joints,
sacroiliac joints, paraspinal
muscles or the posterior longitudinal ligament. Back pain may resolve
naturally but in some patients,
where it lasts over 12 weeks, it becomes a chronic condition which can be
particularly debilitating.
Neuropathic pain is currently defined as pain initiated or caused by a primary
lesion or dysfunction in the
nervous system. Nerve damage can be caused by trauma and disease and thus the
term 'neuropathic
pain' encompasses many disorders with diverse aetiologies. These include, but
are not limited to,
peripheral neuropathy, diabetic neuropathy, post herpetic neuralgia,
trigeminal neuralgia, back pain,
cancer neuropathy, HIV neuropathy, phantom limb pain, carpal tunnel syndrome,
central post-stroke pain
and pain associated with chronic alcoholism, hypothyroidism, uremia, multiple
sclerosis, spinal cord
injury, Parkinson's disease, epilepsy and vitamin deficiency. Neuropathic pain
is pathological as it has no
protective role. It is often present well after the original cause has
dissipated, commonly lasting for years,
significantly decreasing a patient's quality of life (Woolf and Mannion, 1999,
Lancet, 353, 1959-1964). The
symptoms of neuropathic pain are difficult to treat, as they are often
heterogeneous even between
patients with the same disease (Woolf & Decosterd, 1999, Pain Supp., 6, S141-
S147; Woolf and
Mannion, 1999, Lancet, 353, 1959-1964). They include spontaneous pain, which
can be continuous, and
paroxysmal or abnormal evoked pain, such as hyperalgesia (increased
sensitivity to a noxious stimulus)
and allodynia (sensitivity to a normally innocuous stimulus).
The inflammatory process is a complex series of biochemical and cellular
events, activated in response to
tissue injury or the presence of foreign substances, which results in swelling
and pain (Levine and Taiwo,
1994, Textbook of Pain, 45-56). Arthritic pain is the most common inflammatory
pain. Rheumatoid
disease is one of the commonest chronic inflammatory conditions in developed
countries and rheumatoid
arthritis is a common cause of disability. The exact aetiology of rheumatoid
arthritis is unknown, but
current hypotheses suggest that both genetic and microbiological factors may
be important (Grennan &
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
27
Jayson, 1994, Textbook of Pain, 397-407). It has been estimated that almost 16
million Americans have
symptomatic osteoarthritis (OA) or degenerative joint disease, most of whom
are over 60 years of age,
and this is expected to increase to 40 million as the age of the population
increases, making this a public
health problem of enormous magnitude (Houge & Mersfelder, 2002, Ann
Pharmacother., 36, 679-686;
McCarthy et al., 1994, Textbook of Pain, 387-395). Most patients with
osteoarthritis seek medical
attention because of the associated pain. Arthritis has a significant impact
on psychosocial and physical
function and is known to be the leading cause of disability in later life.
Ankylosing spondylitis is also a
rheumatic disease that causes arthritis of the spine and sacroiliac joints. It
varies from intermittent
episodes of back pain that occur throughout life to a severe chronic disease
that attacks the spine,
peripheral joints and other body organs.
Another type of inflammatory pain is visceral pain which includes pain
associated with inflammatory bowel
disease (IBD). Visceral pain is pain associated with the viscera, which
encompass the organs of the
abdominal cavity. These organs include the sex organs, spleen and part of the
digestive system. Pain
associated with the viscera can be divided into digestive visceral pain and
non-digestive visceral pain.
Commonly encountered gastrointestinal (GI) disorders that cause pain include
functional bowel disorder
(FBD) and inflammatory bowel disease (IBD). These GI disorders include a wide
range of disease states
that are currently only moderately controlled, including, in respect of FBD,
gastro-esophageal reflux,
dyspepsia, irritable bowel syndrome (IBS) and functional abdominal pain
syndrome (FAPS), and, in
respect of IBD, Crohn's disease, ileitis and ulcerative colitis, all of which
regularly produce visceral pain.
Other types of visceral pain include the pain associated with dysmenorrhoea,
cystitis and pancreatitis and
pelvic pain.
It should be noted that some types of pain have multiple aetiologies and thus
can be classified in more
than one area, e.g. back pain and cancer pain have both nociceptive and
neuropathic components.
Other types of pain include:
= pain resulting from musculo-skeletal disorders, including myalgia,
fibromyalgia, spondylitis, sero-
negative (non-rheumatoid) arthropathies, non-articular rheumatism,
dystrophinopathy, glycogenolysis,
polymyositis and pyomyositis;
= heart and vascular pain, including pain caused by angina, myocardical
infarction, mitral stenosis,
pericarditis, Raynaud's phenomenon, scleredoma and skeletal muscle ischemia;
= head pain, such as migraine (including migraine with aura and migraine
without aura), cluster
headache, tension-type headache mixed headache and headache associated with
vascular
disorders; and
= orofacial pain, including dental pain, otic pain, burning mouth syndrome and
temporomandibular
myofascial pain.
Disorders of particular interest include urinary incontinence, such as mixed
incontinence, GSI and USI;
pain; depression; anxiety disorders, such as obsessive-compulsive disorder and
post traumatic stress
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
28
disorder; personality disorders, such as ADHD; sexual dysfunction; and
chemical dependencies and
withdrawal syndromes resulting from chemical dependencies.
Thus, according to further aspects, the invention provides:
i) a compound of the invention for use in human or veterinary medicine;
ii) a compound of the invention for use in the treatment of a disorder in
which the regulation of
monoamine transporter function is implicated, such as urinary incontinence;
iii) the use of a compound of the invention in the manufacture of a medicament
for the treatment of a
disorder in which the regulation of monoamine transporter function is
implicated;
iv) a compound of the invention for use in the treatment of a disorder in
which the regulation of
serotonin or noradrenaline is implicated;
v) the use of a compound of the invention in the manufacture of a medicament
for the treatment of a
disorder in which the regulation of serotonin or noradrenaline is implicated;
vi) a compound of the invention for use in the treatment of a disorder in
which the regulation of
serotonin and noradrenaline is implicated;
vii) the use of a compound of the invention in the manufacture of a medicament
for the treatment of a
disorder in which the regulation of serotonin and noradrenaline is implicated;
viii) a compound of the invention for use in the treatment of pain or urinary
incontinence;
ix) the use of a compound of the invention in the manufacture of a medicament
for the treatment of
pain or urinary incontinence;
x) a method of treatment of a disorder in which the regulation of monoamine
transporter function is
implicated which comprises administering a therapeutically effective amount of
a compound of the
invention to a patient in need of such treatment;
xi) a method of treatment of a disorder in which the inhibition of the
reuptake of serotonin or
noradrenaline is implicated which comprises administering a therapeutically
effective amount of a
compound of the invention to a patient in need of such treatment;
xii) a method of treatment of a disorder in which the inhibition of. the
reuptake of serotonin and
noradrenaline is implicated which comprises administering a therapeutically
effective amount of a
compound of the invention to a patient in need of such treatment; and
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
29
xiii) a method of treating pain or urinary incontinence, such as GSI or USI,
which comprises
administering a therapeutically effective amount of a compound of the
invention to a patient in need
of such treatment.
It is to be appreciated that all references herein to treatment include
curative, palliative and prophylactic
treatment, unless explicitly stated otherwise.
The compounds of the invention may be administered alone or as part of a
combination therapy. If a
combination of therapeutic agents is administered, then the active ingredients
may be administered either
sequentially or simultaneously in separate or combined pharmaceutical
formulations.
Examples of suitable agents for adjunctive therapy include:
= an opioid analgesic, e.g. morphine, heroin, hydromorphone, oxymorphone,
levorphanol,
levallorphan, methadone, meperidine, fentanyl, cocaine, codeine,
dihydrocodeine, oxycodone,
hydrocodone, propoxyphene, nalmefene, nalorphine, naloxone, naltrexone,
buprenorphine,
butorphanol, nalbuphine or pentazocine;
= a nonsteroidal antiinflammatory drug (NSAID), e.g. aspirin, diclofenac,
diflusinal, etodolac,
fenbufen, fenoprofen, flufenisal, flurbiprofen, ibuprofen, indomethacin,
ketoprofen, ketorolac,
meclofenamic acid, mefenamic acid, meloxicam, nabumetone, naproxen,
nimesulide,
nitroflurbiprofen, olsalazine, oxaprozin, phenylbutazone, piroxicam,
sulfasalazine, sulindac,
tolmetin or zomepirac;
. a barbiturate sedative, e.g. amobarbital, aprobarbital, butabarbital,
butabital, mephobarbital,
metharbital, methohexital, pentobarbital, phenobartital, secobarbital,
talbutal, theamylal or
thiopental;
= a benzodiazepine having a sedative action, e.g. chlordiazepoxide,
clorazepate, diazepam,
flurazepam, lorazepam, oxazepam, temazepam or triazolam;
= an H, antagonist having a sedative action, e.g. diphenhydramine, pyrilamine,
promethazine,
chlorpheniramine or chlorcyclizine;
= a sedative such as glutethimide, meprobamate, methaqualone or
dichloralphenazone;
. a skeletal muscle relaxant, e.g. baclofen, carisoprodol, chlorzoxazone,
cyclobenzaprine,
methocarbamol or orphrenadine;
. an NMDA receptor antagonist, e.g. dextromethorphan ((+)-3-hydroxy-N-
methylmorphinan) or its
metabolite dextrorphan ((+)-3-hydroxy-N-methylmorphinan), ketamine, memantine,
pyrroloquinoline quinine, cis-4-(phosphonomethyl)-2-piperidinecarboxylic acid,
budipine, EN-3231
(MorphiDex , a combination formulation of morphine and dextromethorphan),
topiramate,
neramexane or perzinfotel including an NR2B antagonist, e.g. ifenprodil,
traxoprodil or (-)-(R)-6-
{2-[4-(3-fluorophenyl)-4-hydroxy-1-piperidinyl]-1-hydroxyethyl-3,4-dihydro-2(1
H)-quinolinone;
= an alpha-adrenergic, e.g. doxazosin, tamsulosin, clonidine, guanfacine,
dexmetatomidine,
modafinil, phentolamine, terazasin, prazasin or 4-amino-6,7-dimethoxy-2-(5-
methane-
sulfonamido-1,2,3,4-tetrahydroisoquinol-2-yl)-5-(2-pyridyl) quinazoline;
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
= a tricyclic antidepressant, e.g. desipramine, imipramine, amitriptyline or
nortriptyline;
= an anticonvulsant, e.g. carbamazepine, lamotrigine, topiratmate or
valproate;
= a tachykinin (NK) antagonist, particularly an NK-3, NK-2 or NK-1 antagonist,
e.g. (aR,9R)-7-[3,5-
bis(trifluoromethyl)benzyl]-8,9,1 0,11 -tetrahydro-9-methyl-5-(4-methylphenyl)-
7H-
5 [1,4]diazocino[2,1-g][1,7]-naphthyridine-6-13-dione (TAK-637), 5-[[(2R,3S)-2-
[(1R)-1-[3,5-
bis(trifluoromethyl)phenyl]ethoxy-3-(4-fl uorophenyl)-4-morpholinyl]-methyl]-
1,2-dihydro-3H-1,2,4-
triazol-3-one (MK-869), aprepitant, lanepitant, dapitant or 3-[[2-methoxy-5-
(trifluoromethoxy)phenyl]-niethylamino]-2-phenylpiperidine (2S,3S);
= a muscarinic antagonist, e.g oxybutynin, tolterodine, propiverine, tropsium
chloride, darifenacin,
10 solifenacin, temiverine and ipratropium;
= a COX-2 selective inhibitor, e.g. celecoxib, rofecoxib, parecoxib,
valdecoxib, deracoxib,
etoricoxib, or lumiracoxib;
= a coal-tar analgesic, in particular paracetamol;
= a neuroleptic such as droperidol, chlorpromazine, haloperidol, perphenazine,
thioridazine,
15 mesoridazine, trifluoperazine, fluphenazine, clozapine, olanzapine,
risperidone, ziprasidone,
quetiapine, sertindole, aripiprazole, sonepiprazole, blonanserin, iloperidone,
perospirone,
raclopride, zotepine, bifeprunox, asenapine, lurasidone, amisulpride,
balaperidone, palindore,
eplivanserin, osanetant, rimonabant, meclinertant, Miraxion or sarizotan;
= a vanilloid receptor agonist (e.g. resinferatoxin) or antagonist (e.g.
capsazepine);
20 = a beta-adrenergic such as propranolol;
= a local anaesthetic such as mexiletine;
= a corticosterpid such as dexamethasone;
= a 5-HT receptor agonist or antagonist, particularly a 5-HTIBõo agonist such
as eletriptan,
sumatriptan, naratriptan, zolmitriptan or rizatriptan;
25 ' = a 5-HT2A receptor antagonist such as R(+)-alpha-(2,3-dimethoxy-phenyl)-
1-[2-(4-
fluorophenylethyl)]-4-piperidinemethanol (MDL-100907);
= a cholinergic (nicotinic) analgesic, such as ispronicline (TC-1734), (E)-N-
methyl-4-(3-pyridinyl)-3-
buten-l-amine (RJR-2403), (R)-5-(2-azetidinylmethoxy}2-chloropyridine (ABT-
594) or nicotine;
= Tramadol ;
30 = a PDEV inhibitor, such as 5-[2-ethoxy-5-(4-methyl-l-piperazinyl-
sulphonyl)phenyl]-1-methyl-3-n-
propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (sildenafil), (6R,12aR)-
2,3,6,7,12,12a-
hexahydro-2-methyl-6-(3,4-methylenedioxyphenyl)-pyrazino[2',1':6,1 ]-
pyrido[3,4-b] indole-1,4-
dione (IC-351 or tadalafil), 2-[2-ethoxy-5-(4-ethyl-piperazin-1-yl-1-
sulphonyl)-phenyl]-5-methyl-7-
propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one (vardenafil), 5-(5-acetyl-2-
butoxy-3-pyridinyl)-3-ethyl-
2-(1 -ethyl -3-azetidinyl)-2,6-d ihyd ro-7H-pyrazol o[4,3-d] pyrim idi n-7-
one, 5-(5-acetyl-2-propoxy-3-
pyridinyl)-3-ethyl-2-(1-isopropyl-3-azetidinyl}2,6-dihydro-7H-pyrazolo[4,3-
d]pyrimidin-7-one, 5-[2-
ethoxy-5-(4-ethyl piperazi n-1-yl sul phonyl )pyridi n-3-yl]-3-ethyl-2-[2-
methoxyethyl]-2,6-d i hydro-7 H-
pyrazolo[4,3-d]pyrimidin-7-one, 4-[(3-chloro-4-methoxybenzyl)amino]-2-[(2S)-2-
(hydroxymethyl)pyrrolidin-1-yl]-N-(pyrimidin-2-ylmethyl)pyrimidine-5-
carboxamide, 3-(1-methyl-7-
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
31
oxo-3-propyl-6,7-dihydro-1 H-pyrazolo[4,3-d]pyrimidin-5-yl)-N-[2-(1-
methylpyrrolidin-2-yl)ethyl]-4-
propoxybenzenesulfonamide;
= an alpha-2-delta ligand such as gabapentin, pregabalin, 3-methylgabapentin,
(1a,3a,5a)(3-
amino-methyl-bicyclo[3.2.0]hept-3-yl)-acetic acid, (3S,5R)-3-aminomethyl-5-
methyl-heptanoic
acid, (3S,5R)-3-amino-5-methyl-heptanoic acid, (3S,5R)-3-amino-5-methyl-
octanoic acid,
(2S,4S)-4-(3-chlorophenoxy)proline, (2S,4S)-4-(3-fluorobenzyl)-proline, [(1
R,5R,6S)-6-
(aminomethyl)bicyclo[3.2.0]hept-6-yl]acetic acid, 3-(1-aminomethyl-
cyclohexylmethyl)-4H-
[1,2,4]oxadiazol-5-one, C-[1 -(1 H-tetrazol-5-ylmethyl)-cycloheptyl]-
methylamine, (3S,4S)-(1-
aminomethyl-3,4-dimethyl-cyclopentyl)-acetic acid, (3S,5R)-3-aminomethyl-5-
methyl-octanoic
acid, (3S,5R)-3-amino-5-methyl-nonanoic acid, (3S,5R)-3-amino-5-methyl-
octanoic acid,
(3R,4R,5R)-3-amino-4,5-dimethyl-heptanoic acid and (3R,4R,5R)-3-amino-4,5-
dimethyl-octanoic
acid;
= a cannabinoid;
= metabotropic glutamate subtype 1 receptor (mGIuR1) antagonist;
= a serotonin reuptake inhibitor such as sertraline, sertraline metabolite
demethylsertraline,
fluoxetine, norfluoxetine (fluoxetine desmethyl metabolite), fluvoxamine,
paroxetine, citalopram,
citalopram metabolite desmethylcitalopram, escitalopram, d,I-fenfluramine,
femoxetine, ifoxetine,
cyanodothiepin, litoxetine, dapoxetine, nefazodone, cericlamine and trazodone;
= a noradrenaline (norepinephrine) reuptake inhibitor, such as maprotiline,
lofepramine,
mirtazepine, oxaprotiline, fezolamine, tomoxetine, mianserin, buproprion,
buproprion metabolite
hydroxybuproprion, nomifensine and viloxazine (Vivalan ), especially a
selective noradrenaline
reuptake inhibitor such as reboxetine, in particular (S,S)-reboxetine;
= a dual serotonin-noradrenaline reuptake inhibitor, such as venlafaxine,
venlafaxine metabolite O-
desmethylvenlafaxine, clomipramine, clomipramine metabolite
desmethylclomipramine,
duloxetine, milnacipran and imipramine;
= an inducible nitric oxide synthase (iNOS) inhibitor such as S-[2-[(1-
iminoethyl)amino]ethyl]-L-
homocysteine, S-[2-[(1-iminoethyl)-amino]ethyl]-4,4-dioxo-L-cysteine, S-[2-[(1-
iminoethyl)amino]ethyl]-2-methyl-L-cysteine, (2S,5Z)-2-amino-2-methyl-7-[(1-
iminoethyl)amino]-5-
heptenoic acid, 2-[[(1 R,3S)-3-amino-4- hydroxy-1-(5-thiazolyl)-butyl]thio]-5-
chloro-3-
pyridinecarbonitrile; 2-[[(1 R,3S}3-amino-4-hydroxy-l-(5-thiazolyl)butyl]thio]-
4-chlorobenzonitrile,
(2S,4R)-2-amino-4-[[2-chloro-5-(trifluoromethyl)phenyl]thio]-5-
thiazolebutanol,
2-[[(1 R,3S)-3-amino-4-hydroxy-l-(5-thiazolyl) butyl]thio]-6-(trifluoromethyl)-
3 pyridinecarbonitrile,
2-[[(1 R,3S)-3- amino-4-hydroxy- 1 -(5-thiazolyl)butyl]thio]-5-
chlorobenzonitrile, N-[4-[2-(3-
chlorobenzylamino)ethyl]phenyl]thiophene-2-carboxamidine, or
guanidinoethyidisulfide;
= an acetylcholinesterase inhibitor such as donepezil;
= a prostaglandin E2 subtype 4 (EP4) antagonist such as N-[({2-[4-(2-ethyl-4,6-
dimethyl-1H-
imidazo[4,5-c]pyridin-l-yl)phenyl]ethyl}amino)-carbonyl]-4-
methylbenzenesulfonamide or 4-[(1 S)-
1-({[5-chloro-2-(3-fluorophenoxy)pyridin-3-yl]carbonyl}amino)ethyl]benzoic
acid;
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
32
= a leukotriene B4 antagonist; such as 1-(3-biphenyl-4-ylmethyl-4-hydroxy-
chroman-7-yl)-
cyclopentanecarboxylic acid (CP-105696), 5-[2-(2-Carboxyethyl)-3-[6-(4-
methoxyphenyl)-5E-
hexenyl]oxyphenoxy]-valeric acid (ONO-4057) or DPC-1 1870,
= a 5-lipoxygenase inhibitor, such as zileuton, 6-[(3-fluoro-5-[4-methoxy-
3,4,5,6-tetrahydro-2H-
pyran-4-yl])phenoxy-methyl]-1-methyl-2-quinolone (ZD-2138), or 2,3,5-trimethyl-
6-(3-
pyridylmethyl),1,4-benzoquinone (CV-6504);
= a sodium channel blocker, such as lidocaine;
= a 5-HT3 antagonist, such as ondansetron, granisetron, tropisetron,
azasetron, dolasetron or
alosetron;
= an oestrogen agonist or selective oestrogen receptor modulator (e.g. HRT
therapies or
lasofoxifene);
= an alpha-adrenergic receptor agonist, such as phenylpropanolamine or R-450;
= a dopamine receptor agonist (e.g. apomorphine, teachings on the use of which
as a
pharmaceutical may be found in US-A-5945117), including a dopamine D2 receptor
agonist (e.g.
premiprixal, Pharmacia Upjohn compound number PNU95666; or ropinirole);
= a PGE1 agonist (e.g. alprostadil);
and the pharmaceutically acceptable salts and solvates thereof.
The invention thus provides, in a further aspect, a combination comprising a
compound of the invention
together with a further therapeutic agent.
For human use the compounds of the invention can be administered alone, but in
human therapy will
generally be administered in admixture with a suitable pharmaceutical
excipient, diluent or carrier
selected with regard to the intended route of administration and standard
pharmaceutical practice.
For example, the compounds of the invention, can be administered orally,
buccally or sublingually in the
form of tablets, capsules (including soft gel capsules), ovules, elixirs,
solutions or suspensions, which
may contain flavouring or colouring agents, for immediate-, delayed-, modified-
, sustained-, dual-,
controlled-release or pulsatile delivery applications. The compounds of the
invention may also be
administered via intracavernosal injection. The compounds of the invention may
also be administered via
fast dispersing or fast dissolving dosage forms.
Such tablets may contain excipients such as microcrystalline cellulose,
lactose, sodium citrate, calcium
carbonate, dibasic calcium phosphate, glycine, and starch (preferably corn,
potato or tapioca starch),
disintegrants such as sodium starch glycollate, croscarmellose sodium and
certain complex silicates, and
granulation binders such as polyvinylpyrrolidone, hydroxypropylmethylcellulose
(HPMC),
hydroxypropylcellulose (HPC), sucrose, gelatin and acacia. Additionally,
lubricating agents such as
magnesium stearate, stearic acid, glyceryl behenate and talc may be included.
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
33
Solid compositions of a similar type may also be employed as fillers in
gelatin capsules. Preferred
excipients in this regard include lactose, starch, a cellulose, milk sugar or
high molecular weight
polyethylene glycols. For aqueous suspensions and/or elixirs, the compounds of
the invention, and their
pharmaceutically acceptable salts, may be combined with various. sweetening or
flavouring agents,
colouring matter or dyes, with emulsifying and/or suspending agents and with
diluents such as water,
ethanol, propylene glycol and glycerin, and combinations thereof.
Modified release and pulsatile release dosage forms may contain excipients
such as those detailed for
immediate release dosage forms together with additional excipients that act as
release rate modifiers,
these being coated on and/or included in the body of the device. Release rate
modifiers include, but are
not exclusively limited to, hydroxypropylmethyl cellulose, methyl cellulose,
sodium
carboxymethylcellulose, ethyl cellulose, cellulose acetate, polyethylene
oxide, Xanthan gum, Carbomer,
ammonio methacrylate copolymer, hydrogenated castor oil, carnauba wax,
paraffin wax, cellulose acetate phthalate, hydroxypropylmethyl cellulose
phthalate, methacrylic acid copolymer and mixtures thereof.
Modified release and pulsatile release dosage forms may contain one or a
combination of release rate
modifying excipients. Release rate modifying excipients may be present both
within the dosage form i.e.
within the matrix, and/or on the dosage form, i.e. upon the surface or
coating.
Fast dispersing or dissolving dosage formulations (FDDFs) may contain the
following ingredients:
aspartame, acesulfame potassium, citric acid, croscarmellose sodium,
crospovidone, diascorbic acid,
ethyl acrylate, ethyl cellulose, gelatin, hydroxypropylmethyl cellulose,
magnesium stearate, mannitol,
methyl methacrylate, mint flavouring, polyethylene glycol, fumed silica,
silicon dioxide, sodium starch
glycolate, sodium stearyl fumarate, sorbitol, xylitol. The terms dispersing or
dissolving as used herein to
describe FDDFs are dependent upon the solubility of the drug substance used
i.e. where the drug
substance is insoluble a fast dispersing dosage form can be prepared and where
the drug substance is
soluble a fast dissolving dosage form can be prepared.
The compounds of the invention can also be administered parenterally, for
example, intravenously, intra-
arterially, intraperitoneally, intrathecally, intraventricularly,
intraurethrally, intrasternally, intracranially,
intramuscularly or subcutaneously, or they may be administered by infusion
techniques. For such
parenteral administration they are best used in the form of a sterile aqueous
solution which may contain
other substances, for example, enough salts or glucose to make the solution
isotonic with blood. The
aqueous solutions should be suitably buffered (preferably to a pH of from 3 to
9), if necessary. The
preparation of suitable parenteral formulations under sterile conditions is
readily accomplished by
standard pharmaceutical techniques well known to those skilled in the art.
For oral and parenteral administration to human patients, the daily dosage
level of the compounds of the
invention or salts or solvates thereof will usually be from 10 to 500 mg (in
single or divided doses).
Thus, for example, tablets or capsules of the compounds of the invention or
salts or solvates thereof may
contain from 5 mg to 250 mg of active compound for administration singly or
two or more at a time, as
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
34
appropriate. The physician in any-event will determine the actual dosage which
will be most suitable for
any individual patient and it wiil vary with the age, weight and response of
the particular patient. The
above dosages are exemplary of the average case. There can, of course, be
individual instances where
higher or lower dosage ranges are merited and such are within the scope of
this invention. The skilled
person will also appreciate that, in the treatment of certain conditions
(including PE), compounds of the
invention may be taken as a single dose on an "as required" basis (i.e. as
needed or desired).
Example Tablet Formulation
In general a tablet formulation could typically contain between about 0.01mg
and 500mg of a compound
according to the present invention (or a salt thereof) whilst tablet fill
weights may range from 50mg to
1000mg. An example formulation for a 10mg tablet is illustrated:
Ingredient %w/w
Free base or salt of compound 101000*
Lactose 64.125
Starch 21.375
Croscarmellose Sodium 3.000
Magnesium Stearate 1.500
* This quantity is typically adjusted in accordance with drug activity and is
based on the weight of the free
base.
The compounds of the invention can also be administered intranasally or by
inhalation and are
conveniently delivered in the form of a dry powder inhaler or an aerosol spray
presentation from a
pressurised container, pump, spray or nebulizer with the use of a suitable
propellant, e.g.
dichlorodifluoromethane, trichlorofluoromethane, dichlorotetra- fluoro-ethane,
a hydrofluoroalkane such
as 1,1,1,2-tetrafluoroethane (HFA 134A [trade mark]) or 1,1,1,2,3,3,3-
heptafluoropropane (HFA 227EA
[trade mark]), carbon dioxide or other suitable gas. In the case of a
pressurised aerosol, the dosage unit
may be determined by providing a valve to deliver a metered amount. The
pressurised container, pump,
spray or nebulizer may contain a solution or suspension of the active
compound, e.g. using a mixture of
ethanol and the propellant as the solvent, which may additionally contain a
lubricant, e.g. sorbitan
trioleate. Capsules and cartridges (made, for example, from gelatin) for use
in an inhaler or insufflator
may be formulated to contain a powder mix of a compound of the invention and a
suitable powder base
such as lactose or starch.
Aerosol or dry powder formulations are preferably arranged so that each
metered dose or "puff' contains
from 1 to 50 mg of a compound of the invention for delivery to the patient.
The overall daily dose with an
aerosol will be in the range of from 1 to 50 mg which may be administered in a
single dose or, more
usually, in divided doses throughout the day.
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
The compounds of the invention may also be formulated for delivery via an
atomiser. Formulations for
atomiser devices may contain the following ingredients as solubilisers,
emulsifiers or suspending agents:
water, ethanol, glycerol, propylene glycol, low molecular weight polyethylene
glycols, sodium chloride,
fluorocarbons, polyethylene glycol ethers, sorbitan trioleate, oleic acid.
5
Alternatively, the compounds of the invention can be administered in the form
of a suppository or pessary,
or they may be applied topically in the form of a gel, hydrogel, lotion,
solution, cream, ointment or dusting
powder. The compounds of the invention may also be dermally or transdermally
administered, for
example, by the use of a skin patch. They may also be administered by the
ocular, pulmonary or rectal
.0 routes.
For ophthalmic use, the compounds can be formulated as micronized. suspensions
in isotonic, pH
adjusted, sterile saline, or, preferably, as solutions in isotonic, pH
adjusted, sterile saline, optionally in
combination with a preservative such as a benzylalkonium chloride.
Alternatively, they may be
[5 formulated in an ointment such as petrolatum.
For application topically to the skin, the compounds of the invention can be
formulated as a suitable
ointment containing the active compound suspended or dissolved in, for
example, a mixture with one or
more of the following: mineral oil, liquid petrolatum, white petrolatum,
propylene glycol, polyoxyethylene
?0 polyoxypropylene compound, emulsifying wax and water. Alternatively, they
can be formulated as a
suitable lotion or cream, suspended or dissolved in, for example, a mixture of
one or more of the
following:.mineral oil, sorbitan monostearate, a polyethylene glycol, liquid
paraffin, polysorbate 60, cetyl
esters, wax, cetearyl alcohol, 2-octyldodecanol, benzyl alcohol and water.
2_ 5 The compounds of the invention may also be used in combination with a
cyclodextrin. Cyclodextrins are
known to form inclusion and non-inclusion complexes with drug molecules.
Formation of a drug-
cyclodextrin complex may modify the solubility, dissolution rate,
bioavailability and/or stability property of
a drug molecule. Drug-cyclodextrin complexes are generally useful for most
dosage forms and
administration routes. As an alternative to direct complexation with the drug
the cyclodextrin may be
30 used as an auxiliary additive, e.g. as a carrier, diluent or solubiliser.
Alpha-, beta- and gamma-
cyclodextrins are most commonly used and suitable examples are described in WO-
A-91/11172, WO-A-
94/02518 and WO-A-98/55148.
For oral or parenteral administration to human patients the daily dosage
levels of compounds of formula
35 (I), (1'), (II'), (III'), and their pharmaceutically acceptable salts, will
be from 0.01 to 30 mg/kg (in single or
divided doses) and preferably will be in the range 0.01 to 5 mg/kg. Thus
tablets will contain 1 mg to 0.4g
of compound for administration singly or two or more at a time, as
appropriate. The physician will in any
event determine the actual dosage which will be most suitable for any
particular patient and it will vary
with the age, weight and response of the particular patient. The above dosages
are, of course only
exemplary of the average case and there may be instances where higher or lower
doses are merited, and
such are within the scope of the invention.
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
36
Oral administration is preferred.
For veterinary use, a compound of the invention is administered as a suitably
acceptable formulation in
accordance with normal veterinary practice and the veterinary surgeon will
determine the dosing regimen
and route of administration which will be most appropriate for a particular
animal.
Thus according to a further aspect, the invention provides a pharmaceutical
formulation containing a
compound of the invention and a pharmaceutically acceptable adjuvant, diluent
or carrier.
The combinations referred to above may also conveniently be presented for use
in the form of a
pharmaceutical formulation and thus pharmaceutical formulations comprising a
combination as defined
above together with a pharmaceutically acceptable adjuvant, diluent or carrier
comprise a further aspect
of the invention. The individual components of such combinations may be
administered either '
sequentially or simultaneously in separate or combined pharmaceutical
formulations.
When a compound of the invention is used in combination with a second
therapeutic the dose of each
compound may differ from that when the compound is used alone. Appropriate
doses will be readily
appreciated by those skilled in the art.
The invention is illustrated by the following non-limiting examples in which
the following abbreviations and
definitions may be used:
APCI Atmospheric pressure chemical ionisation
Arbocel filter agent
br Broad
BOC tert-butoxycarbonyl
CDI carbonyldiimidazole
b chemical shift
d doublet
dd doublet of doublets
A heat
DCCI dicyclohexylcarbodiimide
DCM dichloromethane
DMF N,N-dimethylformamide
DMSO dimethylsulfoxide
ES+ electrospray ionisation positive scan
ES- electrospray ionisation negative scan
h hours
HOAT 1 -hydroxy-7-azabenzotriazole
HOBT 1 -hydroxybenzotriazole
HPLC high pressure liquid chromatography
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
37
LRMS Low resolution mass spectrum
m/z mass spectrum peak
min minutes
NMM N-methyl morpholine
NMR nuclear magnetic resonance
q quartet
s singlet
t triplet
TBTU 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate
Tf trifluoromethanesulfonyl
TFA, trifluoroacetic acid
THF tetrahydrofuran
TLC thin layer chromatography
WSCDI 1-(3-d im ethyl am i nopropyl)-3-ethyl carbodi im ide hydrochloride
The Preparations and Examples that follow illustrate the invention but do not
limit the invention in any
way. All temperatures are in C. Flash column chromatography was carried out
using Merck silica gel 60
(9385). Solid Phase Extraction (SPE) chromatography was carried out using
Varian Mega Bond Elut (Si)
cartridges (Anachem) under 15mmHg vacuum. Thin layer chromatography (TLC) was
carried out on
Merck silica gel 60 plates (5729). Melting points were determined using a
Gallenkamp MPD350
apparatus and are uncorrected. NMR was carried out using a Varian-Unity Inova
400MHz NMR
spectrometer or a Varian Mercury 400MHz NMR spectrometer. Mass spectroscopy
was carried out using
a Finnigan Navigator single quadrupole electrospray mass spectrometer or a
Finnigan aQa APCI mass
spectrometer.
Conveniently, compounds of the invention are isolated following work-up in the
form of the free base, but
pharmaceutically acceptable acid addition salts of the compounds of the
invention may be prepared using
conventional means. Solvates (e.g. hydrates) of a compound of the invention
may be formed during the
work-up procedure of one of the aforementioned process steps.
Where it is stated that compounds were prepared in the manner described for an
earlier Preparation or
Example, the skilled person will appreciate that reaction times, number of
equivalents of reagents and
reaction temperatures may be modified for each specific reaction, and that it
may nevertheless be
necessary or desirable to employ different work-up or purification conditions.
Preparation 1: tert-Butyl (3S)-3-[(biphenyl-2-ylmethyl)amino]pyrrolidine-l-
carboxylate
, I N H3C CH3
\ QNKCH3
O
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
38
Biphenyl-2-carbaldehyde (270mg, 1.45mmol) was added to a solution of tert-
butyl (3S)-3-
aminopyrrolidine-l-carboxylate (270mg, 1.45mmol) in methanol (5ml), under
nitrogen, at room
temperature. The reaction mixture was stirred at room temperature for 16
hours, and sodium borohydride
(110mg, 2.90mmol) was then added. The reaction mixture was stirred at room
temperature for a further 3
hours, after which time it was quenched with a saturated sodium bicarbonate
solution (25m1), diluted with
water (25m1) and extracted into ethyl acetate (2x100ml). The organic extracts
were combined, washed
with brine (50m1), dried over magnesium sulphate and concentrated in vacuo to
yield the title compound
(448mg, 87%) as a colourless gum.
1H-NMR (CDCI3, 400MHz): 1.44(s, 9H), 1.56(m, 1H), 1.83(m, 1H), 2.91(m, 0.5H),
3.01(m, 0.5H), 3.15(m,
1 H), 3.24(m, 1 H), 3.31-3.38(m, 2H), 3.76(s, 2H), 7.33-7.42(m, 8H), 7.48(m, 1
H); LRMS APCI+ m/z 353
[M H]+.
Preparation 2: tert-Butyl (3S)-3-[(bi phenyl -2-ylmethyl)(isobutyryl)am i no]
pyrrol id i ne- 1 -carboxyl ate
0 CH3
N CH3
CN ~~C CH3
~ CH
O 3
Isobutyryl chloride (220pl, 2.1mmol) was added to a solution of the amine
described in preparation 1
(448mg, 1.27mmol), and triethylamine (480N1, 3.44mmol), in dioxane (15m1). The
reaction mixture was
heated at 70 C for 2 hours, after which time it was concentrated in vacuo. The
resulting product was
taken up in ethyl acetate and washed with water, 2M hydrochloric acid, 1 M
sodium hydroxide and brine.
The organic extracts were dried over magnesium sulphate, filtered and
concentrated in vacuo to yield the
title compound (608mg, 100%) as a gum.
' H-NMR (CDCI3, 400MHz): 1.03-1.06(m, 6H), 1.41(s, 9H), 1.72(m, 1 H), 1.93(m,
1 H), 2.50(m, 1 H), 2.93(m,
1 H), 3.20(m, 1 H), 3.37(m, 1 H), 3.54(m, 1 H), 4.27-4.35(m, 2H), 5.04(m, 1
H), 7.15(m, 1 H), 7.27-7.46(m,
8H); LRMS APCI' m/z 323 [(M-BOC)H]+.
Preparation 3: tert-Butyl (3S)-3-[(2,4-dichlorobenzyl)amino]pyrrolidine-1-
carboxylate
CI H
N ~~C CH3
~
cl O CH3
2,4-Dichlorobenzaldehyde (1.88g, 10.74mmol) was added to a solution , of tert-
butyl (3S)-3-
aminopyrrolidine-l-carboxylate (2g, 10.74mmol) in toluene (50m1). The reaction
mixture was heated at
reflux under Dean-Stark conditions for 18 hours, under nitrogen. It was then
concentrated in vacuo and
the residue was taken up in methanol (50m1). The mixture was cooled down to 0
C and then sodium
borohydride (812mg, 21.48mmol) was added portionwise. The solution was stirred
at 0 C for 30 minutes
and then at room temperature for 1.5 hours. It was then quenched with water
(10mi) and concentrated in
vacuo. The resulting residue was partitioned between water (40ml) and
dichloromethane (40m1). The
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
39
layers were separated and the aqueous layer was further extracted with
dichloromethane (2xlOml). The
organic extracts were combined, dried over magnesium sulphate and concentrated
in vacuo. The crude
product was purified by column chromatography on silica gel eluting with ethyl
acetate:pentane (1:5 to 1:3
by volume) to yield the title compound (3.58g, 96%).
'H-NMR (CDCI3, 400MHz): 1.45(s, 9H), 1.58(brs, 1H), 1.76(m, 1 H), 2.03(m, 1H),
3.11-3.20(brm, IH),
3.32(brs, 2H), 3.44-3.56(m, 2H), 3.86(s, 2H), 7.23(d, 1H), 7.35-7.38(m, 2H);
LRMS APCI+ mlz 289 [MH-
isobutylene]+.
Preparation 4: tert-Butyl (3S)-3-[(2,4-dichlorobenzyl)(3-
methylbutanoyl)amino]pyrrolidine-l-carboxylate
H3C
CI CH3
N
CI 63C ~CH3
0 CH3
The title compound (208mg, 84%) was prepared by a method similar to that
described in ,preparation 2
using the amine described in preparation 3 and isovaleryl chloride (3-
methylbutanoyl chloride).
'H-NMR (CDCI3, 400MHz, rotamers): 0.99(d, 6H), 1.43(s, 9H), 1.96-2.03(m, 2H),
2.11(m, 1H), 2.23(d,
2H), 3.03(m, 1 H), 3.26(m, 1 H), 3.45(m, 1 H), 3.59(m, 1 H), 4.47-4.58(brm,
2H), 5.16(m, 1 H), 6.95-7.05(m,
1 H), 7.17-7.27(m, 1 H), 7.35-7.43(m, 1 H); LRMS APCI+ m/z 329 [MH-BOC]+.
Preparation 5: te-t-Butyl (3S)-3-[(2,4-d ichlorobenzyl)(isobutyryl)am i no]
pyrrol idine- 1 -carboxyl ate
~CH3
CI
N CH3
ON OC CH3
CI y
O CH3
The title compound (458mg, 94%) was prepared by a method similar to that
described in preparation 2
using the amine described in preparation 3 and isobutyryl chloride.
'H-NMR (CDCI3, 400MHz, rotamers): 1.08(d, 3H), 1.19(d, 3H), 1.42(s, 9H),
1.76(m, 1H), 1.98(m, 1 H),
2.39(m, 1H), 3.01(m, 1H), 3.26(m, 1H), 3.45(m, 1H), 3.58(m, 1H), 4.47(brs,
1.5H), 4.61(brs, 0.5H),
5.18(m, 1H), 6.86(d, 0.5H), 7.01(d, 0.5H), 7.16(d, 0.5H), 7.28(m, 0.5H),
7.35(s, 0.5H), 7.42(s, 0.5H);
LRMS APCI' m/z 315 [MH-BOC]+.
Preparation 6: tert-Butyl (3S)-3-[(2,3-dichlorobenzyl)amino]pyrrolidine-l-
carboxylate
H
CI N
HdC CH3
CI ~ ~ CN~
'~' CH3
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
The title compound (1.55g, 85%).was prepared by a method similar to that
described in preparation 3
using tert-butyl (3S)-3-aminopyrrolidine-l-carboxylate and 2,3-
dichlorobenzaldehyde.
' H-NMR (CDCI3, 400MHz): 1.45(s, 9H), 1.64(brs, 1H, NH), 1.76(m, 1 H), 2.04(m,
1 H), 3.10-3.20(brm, 1 H),
3.33(brs, 2H), 3.44-3.56(m, 2H), 3.92(s, 2H), 7.18(t, 1 H), 7.32(brs, 1H),
7.38(d, 1H); LRMS APCI+ m/z
5 345 [MH]+.
Preparation 7: tert-Butyl (3S)-3-[(2,3-d ichlorobenzyl)(isobutyryl)am i no]
pyrrol idi ne- 1 -carboxyl ate
cH3
O~-ICH3
CI ~C CH3
CI ~ ~ CN ~
~ CH3
3
The title compound (592mg, 100%) was prepared by a method similar to that
described in preparation 2
10 using the amine described in preparation 6 and isobutyryl chloride.
'H-NMR (CDCI3, 400MHz, rotamers): 1.08(d, 4H), 1.23-1.27(m, 2H), 1.42(s, 9H),
1.76(m, 1H), 1.98(m,
1H), 2.39(m, 1 H), 3.02(m, 1 H), 3.26(m, 1H), 3.45(m, 1H), 3.59(m, 1 H), 4.52
(brs, 1.5H), 4.64 (m, 0.5H),
5.20(m, IH), 6.83(d, 0.5H), 7.01(d, 0.5H), 7.13(t, 0.5H), 7.23(m, 0.5H),
7.33(d, 0.5H), 7.43(d, 0.5H):
LRMS APCI+ m/z 415 [MH]+.
Preparation 8: (3S)-1-Benzyl-N-(2-naphthylmethyl)pyrrolidin-3-amine
H
N
The title compound (4.1g, 100%) was prepared by a method similar to that
described in preparation 1
using (3S)-1-benzylpyrrolidin-3-amine and 2-naphthaldehyde.
' H-NMR (CDCI3, 400MHz): 1.69(m, IH), 1.80(brs, IH, NH), 2.17(m, 1 H), 2.48(m,
1H), 2.56(m, 1 H),
2.70(m, 1H), 2.78(m, 1 H), 3.42(m, IH), 3.64(q, 2H), 3.91(s, 2H), 7.26(m, 1H),
7.31-7.33(m, 4H), 7.44-
7.46(m, 3H), 7.74(s, 1 H), 7.79-7.82(m, 3H): LRMS APCI' m/z 317 [MH]+.
Preparation 9: N-[(3S}1-Benzylpyrrolidin-3-yl]-N-(2-naphthylmethyl)acetamide
O
N CH3
Acetic anhydride (270p1, 2.84mmol) was added to a solution of the amine
described in preparation 8
(300mg, 0.948mmol) and pyridine (460N1, 5.68mmol), in dichloromethane (5ml).
The reaction mixture
was stirred at room temperature for 16 hours, after which time it was washed
with water and brine. The
organic extracts were dried over magnesium sulphate, filtered and concentrated
in vacuo. The crude
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
41
product was purified by column chromatography on silica gel eluting with
dichloromethane:methanol
(100:0 to 98:2, by volume) to yield the title compound (330mg, 97%).
LRMS ESI' m/z 359 [MH]+.
Preparation 10: Ethyl 4-chloro-2-ethoxybenzoate
0
O
-CH3
O
CI \-CH3
4-Chloro-2-hydroxybenzoic acid (5g, 29mmol), iodoethane (11.7m1, 145mmol) and
potassium carbonate
(20g, 145mmol) were combined in acetone (100m1) and the reaction mixture was
heated at reflux for 18
hours. The acetone was then evaporated in vacuo. The resulting residue was
partitioned between water
(100mI) and ethyl acetate (150m1). The layers were separated and the aqueous
layer was further
extracted with ethyl acetate (150m1). The organic extracts were combined,
dried over sodium sulphate,
filtered and evaporated in vacuo. The title compound was obtained as an orange
solid (6.66g, 100%).
'H-NMR (CDCI3i 400MHz) S: 1.18 (t, 3H), 1.23 (t, 3H), 4.10 (q, 2H), 4.38 (q,
2H), 6.95 (m, 2H), 7.75 (d,
1 H).
Preparation 11: (4-Ch loro-2-ethoxyphenyl)m ethanol
OH
O
Ci ~CH3
A 1M solution of lithium aluminium hydride (5ml) was added dropwise to a
solution of the compound
described in preparation 10 (1.14g, 5mmol) in tetrahydrofuran (10m1), at room
temperature, under
nitrogen. The reaction mixture was stirred at room temperature for 3 hours,
after which time it was
quenched with 2M hydrochloric acid (20m1). The solution was extracted with
ethyl acetate (2x30m1), and
the combined organic extracts were combined, dried over magnesium sulphate and
evaporated in vacuo
to yield the title compound (780mg, 84%).
'H-NMR (CDCI3, 400MHz) 5: 1.43 (t, 3H), 2.32 (bs, 1H), 4.05(q, 2H), 4.62 (s,
2H), 6.84 (s, 1H), 6.92 (d,
1 H), 7.20 (d, 1 H).
Preparation 12: 4-Chloro-2-ethoxybenzaldehyde
H
0
e-(\-CH3
CI Manganese dioxide (1.8g, 21.2mmol) was added to a solution of 4-chloro-2-
ethoxyphenyl)methanol
(described in preparation 11) (1g, 5.3mmol) in dichloromethane (15m1), and the
reaction mixture was
heated at reflux for 72 hours. It was then filtered through arbocel and the
filtrate was evaporated in
vacuo. The crude material was purified by column chromatography using an ISCO
silica cartridge,
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
42
eluting with a solvent gradient of pentane changing to pentane: ethyl acetate
(80:20 by volume). The title
compound was obtained as a yellow solid (965mg, 5.2mmol, 98%). .
'H-NMR (CDCI3, 400MHz) $:1.45 (t, 3H), 4.17 (q, 2H), 6.99 (m, 2H), 7.78 (d,
1H), 10.42 (s, 1H).
Preparation 13: 3'-Fluorobiphenyl-2-carbaldehyde
/ H 0
\ I
F ~ I
\
2-Bromo-benzaldehyde (1.24g, 6.7mmol), 3-fluorophenylboronic acid (1.12g,
8.04mmol),
tris(dibenzylideneacetone)dipalladium (0) (92mg, 0.1mmol),
tricyclohexylphosphine (85mg, 0.30mmol)
and potassium phosphate (2.84g, 13.4mmol) were combined in toluene (20m1) and
the reaction mixture
was heated at 100 C for 18 hours. The suspension was then filtered through
arbocel and the filtrate was
evaporated in vacuo. The crude material was purified by column chromatography
over silica gel eluting
with a solvent gradient of pentane changing to pentane: ethyl acetate (90:10
by volume). The title
compound was obtained as a yellow oil (756mg, 3.7mmol, 55%).
'H-NMR(CDCI3, 400MHZ) S: 7.12 (m, 3H), 7.42 (m, 2H), 7.51 (t, 1 H), 7.64 (t, 1
H), 8.03 (d, 1 H), 10.00 (s,
1H).
Preparation 14: 2'-Fluorobiphenyl-2-carbaldehyde
H 0
\ I /
F
2-Bromobenzaldehyde (1g, 5.4mmol), 2-fluorophenylboronic acid (909mg,
6.5mmol),
tri s(d i benzyl ideneacetone)d i pal ladi um (0) (73mg, 0.08mmol),
tricyclohexylphosphine (67mg, 0.24mmol)
and potassium phosphate (2.3g, 10.8mmol) were combined in toluene (20m1) and
the reaction mixture
was heated at 100 C for 18 hours. The suspension was filtered through arbocel
and the filtrate was
evaporated in vacuo. The residue was dissolved in diethyl ether, washed with 1
M sodium hydroxide,
dried over magnesium sulphate, filtered and evaporated in vacuo. The title
compound was obtained as a
yellow oil (1.1g, 5.5mmol, 84%).
1H-NMR (CDCI3, 400MHz) 5; 7.18 (t, 1 H), 7.26 (t, 1 H), 7.34 (t, 1 H), 7.43
(m, 2H), 7.54 (t, 1 H), 7.67 (t, 1 H),
8.04 (d, 1 H), 9.92 (s, 1 H); LRMS APCI' m/z 201 [MH]+.
Preparation 15: 2-(ethylthio)benzoic acid
H3C 0
\--5 OH
e
A 1M sodium hydroxide solution (10ml) was added to 2-mercaptobenzoic acid (1g,
6.4mmol) in ethanol
(10m1), followed by iodoethane (1g, 6.4mmol). The reaction mixture was stirred
for 72 hours, after which
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
43
time the ethanol was evaporated under reduced pressure. The reaction mixture
was then cooled in an
ice bath and acidified to pH 1 with 2N aqueous hydrochloric acid. The
resulting precipitate was collected
by filtration, washed with water and dried under reduced pressure to afford
the title compound, (1.09g,
98%).
' HNMR(400MHz, CD3OD) b: 1.32(t, 3H), 2.9(q, 2H), 7.13(t, 1H), 7.38(d, 1H),
7.43 (t, 1H), 7.88 (d, 1H);
LRMS APCI m/z 181 [M-H]-
Preparation 16: Tert-butyl (3S)-3-{[2-(ethylthio)benzoyl]amino}pyrrolidine-1-
carboxylate
O H
H3C\~
__S N CN _ %H3
'
/ 3
o
~
A 50% solution of 2,4,6-tripropyl-1,3,5,2,4,6-trioxatriphosphinane 2,4,6-
trioxide (T3P ) (50% solution in
ethyl acetate by weight, 9.7m1, 16.5mmol) was added dropwise to a solution of
2-(ethylthio)benzoic acid
(described in preparation 15) (3.0g, 16.5mmol), tert-butyl (3S)-3-
aminopyrrolidine-l-carboxylate (2.79g,
15mmol) and triethylamine (5.2m1, 33.5mmol) in dichloromethane (75m1). The
reaction mixture was
stirred at room temperature for 18 hours. An aqueous solution of potassium
carbonate (50ml) was then
added and the reaction mixture was stirred at room temperature for a further
18 hours. The layers were
separated and the organic phase was washed with an aqueous solution of
potassium carbonate, dried
over magnesium sulphate, filtered and evaporated in vacuo. The title compound
was obtained as a brown
gum (4.02g, 11.6mmol, 77%).
LRMS APCI+ m/z 351 [MH]'.
Preparation 17: Tert-butyl (3S)-3-{[2-(ethylthio)benzyl]amino}pyrrolidine-l-
carboxylate
H
H3C~S N% H8C~CH3
~ CN~ CH3
\I 0
A 1 M solution of borane in tetrahydrofuran (38m1, 38.3mmol) was added
dropwise to a solution of the
compound described in preparation 16 (4.02g, 11.6mmol) in tetrahydrofuran
(35m1). The reaction
mixture was heated at reflux for 2.5 hours after which time it was allowed to
cool to room temperature.
The mixture was quenched with methanol and the solvent was evaporated in
vacuo. The resulting
residue was dissolved in methanol (80m1) and the solution was heated at reflux
for 4 hours. The methanol
was then evaporated in vacuo and the crude material was purified by column
chromatography over silica
gel eluting with a solvent gradient of pentane: ethyl acetate (90:10 by
volume) changing to pentane: ethyl
acetate (50:50, by volume). The title compound was obtained as a colourless
oil (2.34g, 6.96mmol,
60%).
'H-NMR (CDCI3, 400MHz): 5:1.31 (t, 3H), 1.44 (s, 9H), 1.76 (m, 2H), 2.02 (m,
1H), 2.94 (q, 2H), 3.07-3.23
(m, 1 H), 3.32 (s, 2H), 3.40-3.60 (m, 2H), 3.87 (s, 2H), 7.14 (t, 1 H), 7.22
(t, 1 H), 7.28-7.34 (m, 2H); LRMS
APCI+ m/z 337 [M H]'.
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
44
Preparation 18: 1-Bromo-2-(bromomethyl)naphthalene
Br
C
N-bromosuccinimide (4.4g, 24.8mmol) was added to a solution of 1-bromo-2-
methylnaphthalene (5.0g,
22.6mmol) in trichloroethane (100m1). Benzoyl peroxide (44mg, 0.18mmol) was
then added, and the
reaction mixture was heated at 85 C for 21 hours. It was then quenched with a
saturated aqueous
solution of sodium hydrogen carbonate (100m1). The organic phase was
separated, washed with brine
(150m1), dried over magnesium sulphate, filtered and evaporated in vacuo. The
yellow solid was
triturated with pentane to yield the tile compound (4.42g, 14.7mmol, 65%).
'H-NMR (CDCI3, 400MHz) S. 4.85 (s, 2H), 7.50-7.65 (m, 3H), 7.80 (m, 2H), 8.38
(d, I H).
Preparation 19: Tert-butyl 4-{[(1-bromo-2-naphthyi)methyl]amino}piperidine-1-
carboxyiate
Br H
N
bN
CH3>==0
H3C--/- O
H3C
The compound described in preparation 18 (200mg, 0.66mmol), tert-butyl 4-
aminopiperidine-l-
carboxylate (200mg, lmmol) and potassium carbonate (182mg, 1.31mmol) were
combined in acetonitrile
(4ml), and the reaction mixture was heated at reflux for 18 hours. The solvent
was evaporated in vacuo
and the resulting residue was partitioned between water (50m1) and ethyl
acetate (50ml). The organic
layer was washed with brine (30mi), dried over magnesium sulphate, filtered
and evaporated in vacuo.
The crude material was purified by column chromatography over silica gel
eluting with a solvent gradient
of pentane: ethyl acetate (50:50 by volume) changing to pentane: ethyl acetate
(10:90 by volume) to yield
the title compound (220mg, 0.52mmol, 78%).
'H-NMR (CDCI3, 400MHz) 5; 1.40 (m, 2H), 1.45 (s, 9H), 1.90 (m, 2H), 2.74 (m,
IH), 2.82(m, 2H), 4.05
(brs, 2H), 4.15 (s, 2H), 7.50 (m, 1 H), 7.60 (m, 2H), 7.80 (t, 2H), 8.30 (d, 1
H); LRMS ESI' m/z 421 [MH]t.
Preparation 20: Tert-butyl 4-{[(1-methyl-2-naphthyl)methyl]amino}piperidine-l-
carboxylate
H3C
ZDN
H3~0
H3C-~'0
H3C
The compound described in preparation 19 (215mg, 0.51mmoi), potassium
carbonate (283mg,
2.05mmol), tetrakis(triphenylphosphine)palladium (0) (58mg, 0.05mmol) and
trimethylboroxin (0.110ml,
0.79mmol) were combined in 1,4- dioxane (2ml). The reaction mixture was heated
at 100 C for 18 hours.
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
It was then filtered through arbocel and the filtrate was extracted from
water (30m1) into ethyl acetate,
which was dried over magnesium sulphate, filtered and evaporated in vacuo. The
crude material was
purified by column chromatography over silica gel eluting with a solvent
gradient of pentane: ethyl acetate
(50:50 by volume) changing to pentane: ethyl acetate (10:90 by volume). The
title compound was
5 obtained as a colourless oil (110mg, 0.31mmol, 61%).
'H-NMR (CDCI3, 400MHz) &:1.40 (m, 2H), 1.48 (s, 9H), 1.92 (m, 2H), 2.70 (s,
3H), 2.75-2.95 (m, 3H),
4.00 (s, 2H), 4.05 (m, 2H), 7.40-7.55 (m, 3H), 7.68 (d, 1 H), 7.82 (d, 1 H),
8.06 (d, 1 H).
Preparation 21: tert-Butyl 4-{[(1-ethyl-2-naphthyl)methyl]amino}piperidine-1-
carboxylate
CH3 N
H3C CH
N C3
O~O
Diethyl zinc bromide (3.6m1 of a 1M solution in hexane, 3.6mmol) was added
dropwise to a mixture of the
compound described in preparation 19 (500mg, 1.19mmol) and
[1,3bis(diphenylphosphino)propane]
dichloronickel(II) (96 mg, 0.18 mmol) in tetrahydrofuran (3 ml) at 0 C under
nitrogen. The reaction
mixture was allowed to warm to room temperature and stirred then for 1.5
hours, before being cooled to
0 C and quenched by the addition of aqueous ammonium chloride (10mL). The
mixture was diluted with
ethyl acetate (30m1) and filtered through Celite before being washed with
brine, dried (MgSO4) and
evaporated. The residue was purified by column chromatography over silica gel
eluting with a solvent
gradient of ethyl acetate:pentane 2:3 by volume, changing to ethyl
acetate:pentane 3:2 by volume to give
the title compound (130mg, 29%) as an oil.
LRMS APCI+ m/z 369 [MH]'.
Preparation 22: 1 -Bromo-2-(bromomethyl )-6-fl uoronaphthalene
F / \
- Br
A mixture of 1,1'-azobis(cyclohexanecarbonitrile) (VAZO catalyst 88) (200mg,
0.8mmol), 1-bromo-6-
fluoro-2-methylnaphthalene (prepared according to J. Med. Chem., 1993, 36,
2485) (4.12g, 17.2mmol)
and N-bromosuccinimide (3.1g, 17.2mmol) in carbon tetrachloride (35m1) was
heated at reflux for 6 hours
then was stirred at room temperature for a further 18 hours. The reaction
mixture was diluted with
dichloromethane, washed with water, dried over sodium sulphate and evaporated
in vacuo. The residue
was triturated with pentane to yield the title compound as an off white solid
(2.96g,.54%)
'H-NMR (CDCI3, 400MHz): S 4.85 (s, 2H), 7.38 (m, 1 H), 7.45 (dd, 1 H), 7.53
(d, 1 H), 7.74 (d, 1 H), 8.36
(dd, 1 H).
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
46
Preparation 23: (3R)-N-[(1-Bromo-6-fluoro-2-naphthyl)methyl]-1-
(trifluoroacetyl)pyrrolidin-3-amine
H
N F
5:31 O
F
A mixture of the compound described in preparation 22 (2.75g, 8.65mmol), (3R)-
1-
(trifluoroacetyl)pyrrolidin-3-amine (2.27g, 10.4mmol) and potassium carbonate
(2.38g, 17.2mmol) in
acetonitrile (30m1) was heated at reflux for 16 hours. The solvent was then
removed in vacuo and the
resulting residue was applied directly to a silica column. Elution with
dichloromethane:methanol (100:0
increasing polarity to 97:3 by volume) gave the title compound (2.22g, 61%) as
an oil.
LRMS APCI+ m/z 419, 421 [MH]'.
Preparation 24: (3 R)-N-[(6-Fl uoro- 1 -m ethyl -2-naphthyl)methyl]- 1 -(trifl
uoroacetyl)pyrrol id i n-3-ami ne
H
H3C
F
N
F
O
F
A mixture of the compound described in preparation 23 (2.21g, 5.27mmol),
trimethylboroxine (1.32g,
11.4mmol), potassium carbonate (2.91g, 21.1mmol) and
tetrakis(triphenylphosphine)palladium(0) (1.22g,
1.06mmol) in 1,4-dioxane (30ml) was heated at reflux under nitrogen for 3
hours. The reaction mixture
was then cooled to room temperature and partitioned between ethyl acetate (100
ml) and water (100 ml).
The organic layer was washed with brine, dried (MgSO4) and evaporated. The
resulting residue was
purified by column chromatography (silica, eluting with dichloromethane,
increasing polarity to
dichloromethane:methanol 95:5 by volume) to give the title compound (1.87g,
100%) as an oil.
LRMS APCI+ m/z 355 [MH]+.
Preparation 25: tert-butyl (3S)-3-[(5-chloro-2,3-dihydro-1 H-inden-1 -
yl)amino]pyrrolidine-1 -carboxylate
H
N
HC CH3
O CH3
CI
The title compound (0.252g, 70%) was prepared as a mixture of diastereoisomers
by a method similar to
that described in preparation 1 using tert-butyl (3S)-3-aminopyrrolidine-l-
carboxyfate and 5-chloroindan-
1-one.
Modified work-up and purification procedure: The reaction mixture was quenched
with water (40m1) and
extracted into dichloromethane (5x3Oml). The organic extracts were combined,
dried over magnesium
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
47
sulphate, filtered and evaporated in vacuo. The crude material was purified by
column chromatography
over silica gel eluting with 50:50 ethyl acetate: pentane to give the title
compound as an oil.
LRMS EI+ m/z 337 [MH]'.
Example 1: N-(Bi phenyl-2-yl methyl)-2-m ethyl -N-[(3 S)-pyrrol id i n-3-yl]
propanam ide hydrochloride
CCH3
CH3
HCI
NH
The Boc protected amine described in preparation 2 (608mg, 1.3mmol) was
dissolved in
dichloromethane (15ml) under nitrogen, and the mixture was treated with
trifluoroacetic acid (10m1). The
reaction mixture was then stirred at room temperature under nitrogen for 20
hours. It was then
concentrated in vacuo and the resulting residue was taken up in
dichloromethane (50m1) and washed with
1 M sodium hydroxide solution (25m1). The organic phase was separated, dried
over magnesium sulphate
and concentrated in vacuo. The resulting residue was suspended in diethyl
ether (10mI) and 1M
hydrogen chloride in diethyl ether (5ml) was added. The mixture was
concentrated in vacuo and dried
under high vacuum to yield the title compound (388mg, 83%) as a white foam.
'H-NMR (CD3OD, 400MHz): 0.96-0.98(m, 6H), 2.09(m, 1H), 2.18(m, 1 H), 2.55(m,
1H), 3.07(q, 1H),
3.24(m, 1H), 3.37(m, 1H), 3.60(m, 1H), 4.06(m, 1H), 4.61(q, 2H), 7.27-7.34(m,
4H), 7.39-7.49(m, 5H);
LRMS APCI+ m/z 323 [MH]+.
Example 2: N-(2,4-Dichlorobenzyl)-3-methyl-N-[(3S}pyrrolidin-3-yl]butanamide
hydrochloride
CI O CH3
~ NCH3
I /
CI
ON HCI
H
A solution of 4M hydrogen chloride in dioxane (2.5m1) was added to a solution
of the Boc protected amine
described in preparation 4 (208mg, 0.48mmol), in dichloromethane (2ml), and
the reaction mixture was
stirred at room temperature for 18 hours. It was then concentrated in vacuo
and the resulting residue was
azeotroped with dichloromethane (x3) and diethyl ether (x3). The resulting gum
was dissolved in diethyl
ether:isopropanol (20:1, 2ml) at 30 C then cooled in a solid carbon dioxide
bath for 5 minutes, until a solid
precipitated. This solid was triturated with another 2ml of diethyl
ether:isopropanol (20:1), the solution
was concentrated and the resulting product was dried under high vacuum to
yield the title compound
(124mg, 70%) as a solid.
'H-NMR (CD3OD, 400MHz): 0.94(d, 6H), 2.13-2.28(m, 5H), 3.12(q, 1 H), 3.35(m,
1H), 3.53(m, 1H),
3.67(m, 1 H), 4.15(m, 1 H), 4.72(s, 2H), 7.27(d, 1 H), 7.42(d, 1 H), 7.56(s, 1
H); LRMS APCI+ mlz 329 [MH]+;
Microanalysis: Found: C, 52.51; H, 6.32; N, 7.57%. C16H22N20C12.HCI requires
C, 52.55; H, 6.34; N,
7.66%.
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
48
Example 3: N-(2,4-Dichlorobenzyl)-2-methyl-N-[(3S)-pyrrolidin-3-yl]propanamide
hydrochloride
CI 0
\ CIJ)iJCH3
/ CH3
CN)
HCI
A solution of 4M hydrogen chloride in dioxane (1.5ml) was added to a solution
of the Boc protected amine
described in preparation 5 (450mg, 1.08mmol), in dichloromethane (3ml), and
the reaction mixture was
stirred at room temperature for 2 hours. It was then concentrated in vacuo,
and the resulting residue was
partitioned between 2M sodium hydroxide and dichloromethane. The layers were
separated and the
aqueous layer was extracted twice with dichloromethane. The organic extracts
were combined, dried
over magnesium sulphate and concentrated in vacuo. The crude product was
purified by column
chromatography on silica gel eluting with dichloromethane:methanol:ammonia
(95:5:0.5, by volume) to
yield the title product as the free base. This was taken up in
dichloromethane, and 1 M hydrogen chloride
in diethyl ether was added. The reaction mixture was stirred at room
temperature for 2 hours and then
concentrated in vacuo. The solid was azeotroped twice with diethyl ether to
yield the title compound
(331 mg, 86%) as an off-white solid.
' H-NMR (CD3OD, 400MHz): 1.11(d, 6H), 2.14-2.30(m, 2H), 2.79(m, 1H), 3.12(q,
1H), 3.34(m, 1H),
3.49(m, 1 H), 3.67(m, 1 H), 4.16(m, 1 H), 4.76(s, 2H), 7.27(d, 1 H), 7.43(d, 1
H), 7.55(s, 1 H); LRMS APCI+
m/z 315 [MH]+; Microanalysis: Found: C, 49.58; H, 6.14; N, 7.72%.
C15H20N2OCI2.HCI.2/3H20 requires C,
49.55; H, 6.19; N, 7.70%.
Example 4: N-(2,3-Dichlorobenzyl)-2-methyl-N-[(3S}pyrrolidin-3-yl]propanamide
hydrochloride
CI O
CH3
CI N__1 /
~ / - ~C"
ON H3
H HCI
The title compound was prepared by a method similar to that described in
example 2 using the Boc
protected amine described in preparation 7. Trituration with diethyl ether
yielded the title compound
(487mg, 100%) as a solid.
'H-NMR (CD3OD, 400MHz): 1.11 (d, 6H), 2.16-2.31 (m, 2H), 2.75 (m, 1 H), 3.12
(q, 1 H), 3.36 (m, 1 H),
3.52 (m, 1 H), 3.67 (m, 1 H), 4.19 (m, 1 H), 4.81(s, 2H), 7.21 (d, 1 H), 7.39
(t, 1 H), 7.55 (d, 1 H); LRMS
APCI+ m/z 315 [MH]+; Microanalysis: Found : C, 50.33; H, 6.21; N, 7.68%.
C15H2ONzOCI2.HCl.1/3H20
requires C, 50.38; H, 6.10; N, 7.83%.
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
49
Example 5: N-(2-Naphthylmethyl)-N-[(3S)-pyrrolidin-3-yl]acetamide
hydrochloride
O
0yNACH3
ON HCI
H
A solution of the benzyl protected product described in preparation 9 (320mg,
0.893mmol), ammonium
formate (56.3mg, 8.93mmol) and 10% Pd/C (40mg) in ethanol (5ml) was heated at
reflux under nitrogen
for 4.5 hours. The reaction mixture was then filtered through Arbocel , washed
through with ethanol and
the filtrate was concentrated in vacuo. The crude product was purified by
column chromatography on
silica gel eluting with dichloromethane:methanol:0.88 ammonia (100:0:0 to
90:10:1, by volume). The free
base of the product was dissolved in dichloromethane, minimal volume of 1 M
hydrogen chloride in diethyl
ether was added and the mixture was concentrated in vacuo. The solid was
triturated in diethyl ether (x3)
and dried under vacuum to yield the title compound (80mg, 28%).
'H-NMR (DMSO-D6, 400MHz): 1.90-1.97(m, 2H), 2.98-3.13(m, 2H), 3.24-3.34(m,
2H), 3.81(s, 3H),
4.71(m, 1H), 4.80(s, 2H), 7.36(m, 1 H), 7.46-7.49(m, 2H), 7.63-7.69(m, 1H),
7.84(m, 1H), 7.81-7.93(m,
2H); MS APCI+ m/z 269 [MH]'; Microanalysis: Found: C, 63.41; H, 7.20; N,
8.64%. C17H2oN20.HCI.H20
requires C, 63.24; H, 7.18; N, 8.68%.
Examples 6-84
The following tabulated examples were made using analogous methods to those
described in the
Examples and Preparations described above.
O
R2,-~~ N~R3
N
H
Eg R R
F
6 N-[(2'-fluorobiphenyl-2-yl)methyl]-2-methyl-N- APCI m/z:
[(3S)-pyrrolidin-3-yl]propanamide 341 (MH+)
7 N-[(3'-fluorobiphenyl-2-yl)methyl]-2-methyl-N- APCI m/z:
[(3S)-pyrrolidin-3-yl]propanamide F 341 (MH+)
8 N-(biphenyl-2-ylmethyl}N-[(3S)-pyrrolidin-3-yl] ~_~~ APCI m/z:
butanamide hydrochloride 323 (MH+)
9 N-(biphenyl-2-ylmethyl}3-methyl-N-[(3S)- APCI m/z:
pyrrolidin-3-yl]butanamide hydrochloride 335 (MH+)
10 N-[(4'-chlorobiphenyl-2-yl)methyl]-2-methyl-N- ~a APCI m/z:
[(3S)-pyrrolidin-3-yl]propanamide 357 (MH+)
11 N-(biphenyl-2-ylmethyl)-N-[(3S)-pyrrolidin-3-yl] q-o APCI m/z:
propanamide hydrochloride 309 (MH+)
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
APCI m/z:
12 N-[2-(ethylthio)benzyl]-2-methyl-N-[(3S)-
307 (MH+)
pyrrolidin-3-yl]propanamide hydrochloride
o-
13 N-(4-chloro-2-methoxybenzyl)-2-methyl-N-[(3S)- -\ APCI m/z:
pyrrolidin-3-yl]propanamide hydrochloride c' ~ 311 (MH+)
14 2-methyl-N-(2-phenoxybenzyl)-N-[(3S)- 0-06 APCI m/z:
pyrrolidin-3-yl]propanamide hydrochloride /~ 339 (MH+)
15 N-[(4'-fluorobiphenyl-2-yl)methyl]-2-methyl-N- F APCI m/z:
[(3S)-pyrrolidin-3-yl]propanamide 341 (MH+)
16 N-[(2',4'-d ifl uorobi phenyl-2-yl)m ethyl] -2-m ethyl - APCI m/z:
N-[(3S)-pyrrolidin-3-yl]propanamide 359 (MH+)
FF
17 2-methyl-N-[(3S)-pyrrolidin-3-yl]-N-[2-(1,1,2,2- APCI m/z:
tetrafluoroethoxy)benzyl]propanamide F 363 (MH+)
APCI m/z:
2-bromobenzyl)-2-methyl-N-[(3S)-pyrrolidin- & 325,327
e 18 N-(
3-yl]propanamide hydrochloride (MH+)
-
-\ APCI m/z:
N-(4-chloro-2-methoxybenzyl)-3-methyl-N-[(3S.)- o
19 pyrrolidin-3-yl]butanamide hydrochloride c,-~325 (MH+)
2-methyl-N-[(3S)-pyrrolidin-3-yl]-N-[2- c F
\ / 339 APCI m/z:
20 (trifluoromethyl)benzyl]propanamide
(MH+)
hydrochloride
21 2-methyl-N-[(3S)-pyrrolidin-3-yl]-N-(2,3,4- ci~ \ci APCI m/z:
trichlorobenzyl)propanamide hydrochloride 349 (MH+)
22 2-methyl-N-[2-(methylthio)benzyl]-N-[(3S)- -S APCI m/z:
pyrrolidin-3-yl]propanamide hydrochloride 293 (MH+)
23 N-[(3'-fluorobiphenyl-2-yl)methyl]-2-methyl-N- ~-\ \ ~ APCI m/z:
[(3R)-pyrrolidin-3-yl]propanamide F /~ 341 (MH+)
24 2-methyl-N-(3-phenoxybenzyl)-N-[(3S)- APCI m/z:
pyrrolidin-3-yl]propanamide hydrochloride ~ /~ 339 (MH+)
25 N-[(3'-chlorobiphenyl-2-yl)methyl]-2-methyl-N- 0 0 APCI m/z:
[(3S)-pyrrolidin-3-yl]propanamide G 357 (MH+)
ci
26 N-(2,4-dichlorobenzyl)-N-[(3S)-pyrrolidin-3-yl] APCI m/z:
cyclopentanecarboxamide hydrochloride 341 (MH+)
27 N-(2-cyclopropylbenzyl)-2-methyl-N-[(3S)- APCI m/z:
pyrrolidin-3-yI]propanamide hydrochloride 287 (MH+)
28 N-(2-bromobenzyl)-N-[(3S)-pyrrolidin-3-yl] APCI m/z:
cyclopropanecarboxamide hydrochloride 323, 325
M H+
29 N-[(3S)-pYrrolidi n-3-YI]-N-[2-(trifluorometh YI) cF' _ APCI m/z:
benzyl]cyclopropanecarboxamide hydrochloride \ 313 (MH+)
30 N-[(3',4'-difluorobiphenyl-2-yl)methyl]-N-[(3S)- APCI m/z:
pyrrolidin-3-yl]cyclobutanecarboxamide 371 (MH+)
ci ci
31 N-(2,3-dichlorobenzyl)-N-[(3S)-pyrrolidin-3-yl] APCI m/z:
cyclobutanecarboxamide hydrochloride 327 (MH+)
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
51
~ 1-0 32 N-[(3S)-pyrrol idi n-3-yl]-N-[2-(trifl uoromethyl) CF APCI m/z:
benzyl]cyclopentanecarboxamide hydrochloride 341 (MH+)
33 N-[2-(cyclopropylmethoxy)benzyl]-2-methyl-N- -o _ APCI m/z:
[(3S)-pyrrolidin-3-yl]propanamide hydrochloride ~ / 317 (MH+)
0
34 N -(4-chl oro-2-ethoxybenzyl)-3-m ethyl -N-[(3S)- a C 1 APCI m/z:
pyrrolidin-3-yl]butanamide hydrochloride 339 (MH+)
a
35 N-(2,4-dichlorobenzyl)-2-methyl-N-[(3S)- /-~ Ar, APCI m/z:
pyrrolidin-3-yl]butanamide hydrochloride c' 329 (MH+)
36 N-(2,4-dichlorobenzyl)-2-ethyl-N-[(3S)- APCI m/z:
pyrrolidin-3-yl]butanamide hydrochloride c' 343 (MH+)
Me
37 N-(4-chloro-2-methylbenzyl)-3-methyl-N-[(3S)- ~ APCI m/z:
pyrrolidin-3-yl]butanamide hydrochloride c, 309 (MH+)
38 N-(2,4-dichlorobenzyl)-N-[(3S)-pyrrolidin-3-jrl] APCI m/z:
cyclohexanecarboxamide hydrochloride c' G 355 (MH+)
a
39 N-(2,4-dichlorobenzyl)-N-[(3S)-pyrrolidin-3-yl] APCI m/z:
butanamide hydrochloride c' 315 (MH+)
G
40 N-(2,4-dichlorobenzyl)-3,3-dimethyl-N-[(3S)- / ~ APCI m/z:
pyrrolidin-3-yl]butanamide hydrochloride c' 343 (MH+)
41 N-(2,3-dichlorobenzyl)-3-methyl-N-[(3S)- ci ci APCI m/z:
pyrrolidin-3-yl]butanamide hydrochloride L5-4 A-~, 329 (MH+)
42 N-(2,3-dichlorobenzyl)-N-[(3S)-pyrrolidin-3-yl] cl ci APCI m/z:
pentanamide hydrochloride 6 329 (MH+)
43 N-(2,4-dichlorobenzyl)-N-[(3S)-pyrrolidin-3-yl] G /-~ APCI m/z:
pentanamide hydrochloride c' 329 (MH+)
/-~ APCI m/z:
44 2-cyclopropyl-N-(2,4-dichlorobenzyl)-N-[(3S)- G
pyrrolidin-3-yl]acetamide hydrochloride c' 327 (MH+)
45 N-(2,4-d i chlorobenzyl)-4-m ethyl -N -[(3S Y APCI m/z:
pyrrolidin-3-yl]pentanamide hydrochloride c' a 343 (MH+)
2-methyl-N-[(3S)-pyrrolidin-3-yl]-N-[3- F
46 (trifluoromethoxy)benzyl]propanamide F _ APCI m/z:
F~o / 331 (MH+)
hydrochloride
ci
2-methyl-N-[(3S)-pyrrolidin-3-yl]-N-(2,3,5- G APCI m/z:
47 trichlorobenzyl)propanamide hydrochloride c~ ~ ~ /~ 349 (MH+)
48 N-(2-naphthylmethyl)-N-[(3R)-pyrrolidin-3-yl] APCI m/z:
acetamide hydrochloride Me 269 (MH+)
49 N-(2-naphthylmethyl)-N-[(3R)-pyrrolidin-3-yl] APCI m/z:
cyclohexanecarboxamide hydrochloride 337 (MH+)
50 N-(2-naphthylmethyl)-N-[(3R)-pyrrolidin-3-yl] APCI m/z:
propanamide hydrochloride 283 (MH+)
51 N-(2-naphthylmethyl)-N-[(3R)-pyrrolidin-3-yl] APCI m/z:
cyclobutanecarboxamide hydrochloride 309 (MH+)
52 3-methyl-N-(2-naphthylmethyl}N-[(3R)- APCI m/z:
pyrrolidin-3-yl]butanamide hydrochloride ~ 311 (MH+)
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
52
53 N-(2-naphthylmethyl)-N-[(3S)-pyrrolidin-3-yl] APCI m/z:
cyclopentanecarboxamide hydrochloride 323 (MH+)
54 N-(2-naphthylmethyl)-N-[(3S)-pyrrolidin-3-yl] APCI m/z:
propanamide hydrochloride 283 (MH+)
55 N-(2-naphthylmethyl)-N-[(3S)-pyrrolidin-3-yl] APCI m/z:
cyclobutanecarboxamide hydrochloride 309(MH+)
56 3-methyl-N-(2-naphthylmethyl)-N-[(3S)- APCI m/z:
pyrrolidin-3-yl]butanamide hydrochloride 311 (MH+)
57 N-(2-naphthylmethyl)-N-[(3S)-pyrrolidin-3-yl] APCI m/z:
cyclohexanecarboxamide hydrochloride ~-O 337 (MH+)
58 2-methyl-N-(2-naphthylmethyl)-N-[(3S)- APCI m/z:
pyrrolidin-3-yl]propanamide hydrochloride 297 (MH+)
59 N-[(1-methyl-2-naphthyl)methyl]-N-[(3S)- Me APCI m/z:
pyrrolidin-3-yl]acetamide hydrochloride 283 (MH+)
60 N-[(1-methyl-2-naphthyl)methyl]-N-[(3S)- APCI m/z:
pyrrolidin-3-yl]propanamide hydrochloride 297 (MH+)
61 N-[(6-fluoro-1-methyl-2-naphthyl)methyl]-N- Me APCI m/z:
[(3R)-pyrrolidin-3-yl]acetamide hydrochloride 301 (MH+)
62 N-[(6-fluoro-l-methyl-2-naphthyl)methyl]-N- APCI m/z:
[(3R)-pyrrolidin-3-yl]propanamide hydrochloride 315 (MH+)
63 N-[(6-fluoro-1-methyl-2-naphthyl)methyl]-N- Me APCI m/z:
[(3S)-pyrrolidin-3-yl]acetamide hydrochloride 301 (MH+)
6', N-[(6-fluoro-1 -methyl-2-naphthyl)methyl]-N- F APCI m/z:
[(3S)-pyrrolidin-3-yl]propanamide hydrochloride 315 (MH+)
65 2-(methylthio)-N-(2-naphthylmethyl)-N-[(3R)- El m/z: 315
pyrrolidin-3-yl]acetamide hydrochloride (MH+)
CI m/z:
AP
N-[(3 R)-pyrrol idi n-3-yl]-N-(qui noli n-6-ylm ethyl) V
66 pentanamide hydrochloride ~N 312 (MH+)
N-[(3S)-pyrrol idi n-3-yl]-N-(qui noli n-6-ylm ethyl) APCI m/z:
67 pentanamide hydrochloride ~N 312 (MH+)
N-[(3S)-pyrrol idi n-3-yl]-N-(qui nol i n-6-yl m ethyl) APCI m/z:
68 butanamide hydrochloride ~N 298 (MH+)
G
69 N-(2,4-dichlorobenzyl)-N-[(3S)-pyrrolidin-3-yl] APCI m/z:
cyclopropanecarboxamide hydrochloride c~ 313 (MH+)
ci
70 N-(2,4-dichlorobenzyl)-N-[(3S)-pyrrolidin-3-yl] APCI m/z:
propanamide hydrochloride c' 301 (MH+)
71 4,4,4-trifluoro-N-[(3R)-pyrrolidin-3-yl]-N- ~ F F El m/z: 352
(MH+)
(quinolin-6-ylmethyl)butanamide L-(+)-tartrate ~N ~ F
72 4,4,4-trifluoro-N-[(3S)-pyrrolidin-3-yl]-N- ~F APCI m/z:
(quinolin-6-ylmethyl)butanamide L-(+)-tart rate N ~ F 352 (MH+)
73 N-(2,4-dimethylbenzyl)-N-[(3S)-pyrrolidin-3-yl] APCI m/z:
cyclobutanecarboxamide hydrochloride 287 (MH+)
CI
74 N-(3-chl oro-2-m ethyl benzyl)-N-[(3S)-pyrrol idi n- 6-4 APCI m/z:
3-yl]cyclopropanecarboxamide hydrochloride 293(MH+)
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
53
75 N -(2,4-d im ethyl benzyl)-N-[(3S)-pyrrol idi n-3-yl] APCI m/z:
cyclopropanecarboxamide hydrochloride 273 (MH+)
ci
76 N-(2,4-dichlorobenzyl)-N-[(3S)-pyrrolidin-3-yl] APCI m/z:
cyclobutanecarboxamide hydrochloride 327 (MH+)
77 N-(2,3-dimethylbenzyl)-N-[(3S)-pyrrolidin-3-yl] APCI m/z:
cyclopropanecarboxamide hydrochloride 273 (MH+)
ci
78 N -(3-chl oro-4-m ethyl benzyl)-N -[(3S)-pyrrol idi n- El m/z: 307
3-yl]cyclobutanecarboxamide hydrochloride (MH+)
N-[2-fluoro-4-(trifluoromethyl)benzyl]-2-methyl- F
79 N-[(3S)-pyrrolidin-3-yl]propanamide APCI m/z:
333 (MH+)
hydrochloride
ci
80 N-(2-chloro-4-fluorobenzyl)-2-methyl-N-[(3S)- APCI m/z:
pyrrolidin-3-yl]propanamide hydrochloride F 299 (MH+)
ci a
81 N-(2,3-dichlorobenzyl)-2-methyl-N-[(3R)- APCI mlz:
pyrrolidin-3-yl]propanamide hydrochloride 6-4 315 (MH+)
82 N-(4-chloro-2-ethoxybenzyl)-2-methyl-N-[(3S)- ci 1 c~ APCI m/z:
pyrrolidin-3-yl]propanamide hydrochloride 326 (MH+)
N-[2-methoxy-4-(trifluoromethyl)benzyl]-2- cF, 11 ll APCI mlz:
83 methyl-N-[(3S)-pyrrolidin-3-yl]propanamide ~ I 345 (MH+)
hydrochloride
Example 84
The following tabulated example was made using analogous methods to those
described in the Examples
and Preparations described above.
0
R, NR3
N
H
Eg R R
N-(5-chloro-2,3-dihydro-1 H-inden-1-yl)-2- APCI m/z:
84 methyl-N-[(3S)-pyrrolidin-3-yl]propanamide 307 (MH+)
hydrochloride c1 ~ I
Examples 85-98
The following tabulated examples were made using analogous methods to those
described in the
Examples and Preparations described above.
0
R2~~N~R3
N
I
H
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
54
Eg R R
85 N-(2-naphthylmethyl)-N-piperidin-4-ylacetamide Me APCI m/z:
hydrochloride 283 (MH+)
86 N-[(1-methyl-2-naphthyl)methyl]-N-piperidin-4-yl Me ES m/z:
acetamide hydrochloride 297 (MH+)
87 N-(2-naphthylmethyl)-N-piperidin-4-yl El m/z: 297
propanamide hydrochloride (MH+)
88 N-(2-naphthylmethyl)-N-piperidin-4-ylbutanamide El m/z: 311
hydrochloride (MH+)
89 3-methyl-N-(1-naphthylmethyl)-N-piperidin-4-yl APCI m/z:
butanamide oxalate 325 (MH+)
90 N-piperidin-4-yl-N-(quinolin-6-ylmethyl) El m/z: 298
propanamide hydrochloride N (MH+)
91 N-piperidin-4-yl-N-(quinolin-6-ylmethyl) EI m/z: 312
butanamide hydrochloride N (MH+)
92 2-methyl-N-piperidin-4-yl-N-(quinolin-6-ylmethyl) V,-- EI m/z: 312
propanamide hydrochloride ~N (MH+)
93 N-[(1-ethyl-2-naphthyl)methyl]-N-piperidin-4-yl Me APCI m/z:
acetamide hydrochloride 311 (MH+)
-o
94 N-[(7-methoxy-1 -methyl-2-naphthyl)methyl]-N- Me APCI mlz:
piperidin-4-ylacetamide hydrochloride 327 (MH+)
-o
95 N-[(7-methoxy-1-methyl-2-naphthyl)methyl]-N- - ~ APCI m/z:
piperidin-4-ylpropanamide hydrochloride 341 (MH+)
96 2-hydroxy-N-(2-naphthylmethyl)-N-piperidin-4-yI OH APCI m/z:
propanamide hydrochloride '3- 313 (MH+)
ci ci
97 N-(2,3-dichlorobenzyl)-2-methyl-N-piperidin-4-yl b-4 APCI m/z:
propanamide hydrochloride 11~j- 329 (MH+)
98 N-(2,4-dichlorobenzyl)-2-methyl-N-piperidin-4-yl ~~c~ APCI m/z:
propanamide hydrochloride c' ~ 329 (MH+)
The compounds of the invention were evaluated for biological activity by
measuring the functional
inhibition of monoamine reuptake by the cognate human monoamine transporter
protein in a whole cell
assay (Method 1). Alternatively, the activity of a compound was determined by
measuring its affinity for
the human monoamine transporter protein as a function of its ability to bind
and hence displace a specific
ligand (Method 2).
Method 1:
The NRI and SRI IC50 values of the exemplified compounds were determined as
described below. A
selection of the results is set out below in Table 1. AII of the exemplified
compounds exhibited an NRI
IC50 value and/or an SRI IC50 value of less than 100 nM; a selection are
characterized in table 1.
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
SRI IC50 (nM) NRI IC50 (nM)
Example 1 >400 14
Example 2 93 31
Example 3 14 28
Example 4 12 23
Example 5 2.6 243
Table 1.
The compounds were tested for biological activity by their ability to inhibit
the uptake of serotonin and/or
noradrenaline by human serotonin and/or noradrenaline transporters as follows.
5 (i) Cell Culture
Human embryonic kidney cells (HEK-293), stably transfected with the human
recombinant cDNA
encoding either the human serotonin transporter (hSERT, TRAN0105) or the
noradrenaline
transporter (hNET, TRAN0107), were cultured under standard cell culture
techniques.
Specifically, cells were grown at 37 C and 5% COz in Dulbecco's Modified
Eagle's Medium
10 (DMEM) culture media supplemented with 10% dialysed foetal calf serum
(FCS), 2mM L-
giutamine and 250 g/ml geneticin. Prior to assay, cells were harvested by
utilising cell
dissociation solution (Sigma) and centrifugation, and resuspended in standard
assay buffer (see
below) at a viable cell density of 750,000 cells/ml.
15 (ii) Determination of inhibitor potency
All test compounds were dissolved in 100% DMSO at 4mM and diluted down in 1%
DMSO in
water to give appropriate test concentrations. Assays were carried out in 96-
well filter'bottom
plates. Cells expressing the appropriate human transporter protein (75,000
cells/assay well)
were pre-incubated at 25 C in standard assay buffer containing either test
compound, a standard
20 inhibitor (positive control) or compound vehicle (DMSO in water; final DMSO
concentration was
0.1% in each assay well) for 5 minutes. Reactions were started by addition of
either 3H-
serotoninor 3H-noradrenaline substrates. All reactions were carried out at 25
C in a shaking
incubator. Incubation times were 5 minutes for the hSERT and 15 minutes for
the hNET assay.
Reactions were terminated by addition of ice-cold wash buffer (see below),
followed by filtration
T' of the assay mixture using a vacuum manifold and rapid washing with ice-
cold wash buffer. The
quantity of 3H-substrate incorporated into the cells was then
quantified.Filtered/washed assay
plates were dried at 45 C for 1 hour, scintillation fluid added, and
radioactivity measured by
scintillation counting. Potency of test compounds was quantified as IC5o
values (concentration of
test compound required to inhibit the specific uptake of radiolabelled
substrate into the cells by
3 50% relative to maximum (compound vehicle only) and minimum (complete
inhibition by standard
inhibitor) responses).
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
56
(iii) Standard Assay Buffer Composition:
Tris (hydroxymethyl) amino methane hydrochloride (26mM)
NaCI (124mM)
KCI (4.5mM)
KH2PO4 (1.2mM)
MgC12.6H20 (1.3mM)
Ascorbic acid (1.136mM)
Glucose (5.55mM)
pH 7.40
CaC12 (2.8mM)
Pargyline (100 M)
Note: The pH of the buffer was adjusted to 7.40 with 1 M NaOH before addition
of CaCIZ and
pargyline.
Wash Buffer Composition:
Tris (hydroxymethyl) methylamine (26mM)
NaCI (124mM)
KCI (4.5mM)
KH2PO4 (1.2mM)
MgCI2.6H20 (1.3mM)
Ascorbic acid (1.136mM)
pH 7.40 at 4'C with 6M HCI
(iv) Summary of Assay Parameters
hSERT hNET
Assay Assay
Cells per assay well. 75,000 75,000
73 -
H-5HT (50nM) H-Noradrenaline (200nM)
Substrate Concentration.
Incubation time (minutes) 5 15
Method 2:
The NRI and SRI Ki values of the exemplified compounds were determined as
described below. A
selection of the results is set out below in Table 2. AII of the exemplified
compounds exhibited an NRI Ki
value and/or an SRI Ki value of less than 100 nM; a selection are
characterized below.
Compound SRI Ki (nM) NRI Ki (nM)
Example 1 960 5.8
Example 2 14 44
Example 4 9.5 52
Table 2
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
57
The compounds were tested for biological activity by their ability to inhibit
binding of selective tritiated
radioligands at the human serotonin and noradrenaline transporters (SERT and
NET, respectively), using
scintillation proximity assay (SPA) technology. The SPA binding was performed
using cellular membranes
prepared from cell lines expressing human cDNA encoding either SERT or NET
(hSERT, hNET), using
the radioligands 3H-citalopram and 3H-nisoxetine respectively.
i) Cell culture methodology
Human embryonic kidney cells (HEK-293) expressing each transporter were
maintained as a continuous
culture, using standard cell culture techniques, in 50 mL of growth medium
(see Media and Buffers for
composition) in 225 cm2 flasks, at 37 C in a humidified atmosphere with 5 %
CO2 present. Cells were
passaged from a 90 % confluent monolayer at a ratio of approximately 1:3.
For cell harvesting, the growth medium was removed from the monolayer and the
cells were incubated
with cell dissociation solution (Sigma) until signs of dissociation were
observed. The cells were
subsequently knocked from the base of the flask and pelleted by centrifugation
for storage (frozen at - 80
C) prior to further use.
ii) Cellular membrane preparation
Cell pellets were thawed on ice and resuspended in 3 mL of membrane
preparation buffer (see Media
and Buffers for composition) per 1 mL of packed cell volume, using a vortex
mixer to disperse the cell
pellet.
After incubation on ice for 10 minutes, the suspension was homogenised for
four individual 10 second
intervals using a hand-held homogeniser. The homogenate was then centrifuged
at 1,075 x g for 20
minutes at 4 C.
The supernatants were then collected and retained. Initial cell & nuclei
pellets (P1) were subsequently
rehomogenised and centrifuged using the conditions cited above, and the
supernatants collected and
pooled with those retained from the first spin.
The pooled supernatants were centrifuged at 35,000 x g for 30 minutes at 4 C,
and the supernatants
discarded. The pellets (P2) were then resuspended in 1 mL of membrane
preparation buffer per I mL of
the original packed cell volume. Protein concentrations were subsequently
measured and the membrane
suspension was finally frozen in aliquots of set volume and stored at - 80 C
prior to use in assays.
iii) Assay methodology
A. Determination of Optimal Assay Conditions for Individual Membrane Batches
The specific SPA bead type differed for each transporter studied, wheat germ
agglutinin-coated yttrium
silicate (YSi WGA) SPA beads were used for hSERT and WGA-coated
polyvinyltoluene (PVT WGA) SPA
beads for hNET assays. For each batch of membrane used, optimal concentrations
of bead and
membrane were determined.
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
58
Tritiated radioligands specific to each transporter (3H-citalopram for hSERT
and 3H-nisoxetine for hNET)
were used. The assay free radioligand concentration was expressed as a
percentage of the total free
radioligand concentration to give an estimate of the radioligand depletion.
The radioligand depletion in
assays for both transporters was less than 30% to ensure that there was
sufficient radioligand available
for binding. The ligand depletion value was also used for selecting the
optimal assay conditions when
using new batches of membranes.
The affinity of the specific radioligand for the respective transporter was
determined for each membrane
batch at the selected protein and bead concentrations. This was achieved by
the determination of the KD,
the concentration of free radioligand at which 50 % of the transporter binding
sites were occupied. The
mean Kpfor a radioligand at a batch of membranes was determined from data from
a minimum of three
separate assays. The mean KD was subsequently used for all assays using the
membrane batch profiled
to enable determination of K; values of compounds studied using the method
determined by Cheng and
Prussoff (Cheng YC and Prusoff WH. Relationship between the inhibition
constant (K;) and the
concentration of inhibitor which causes 50% inhibition of an enzymatic
reaction. Biochem Pharmacol
1973; 22:2099-3108.)
B. Assay protocol
Bead/membrane complex preparation
The required amount of membrane was thawed on ice and added to a pre-
determined volume of bead
suspension in assay buffer. The beads were then pre-coupled by incubating the
predetermined protein
quantity per mg of bead on a shaker at a temperature of 4 C for 2 hours.
Subsequently, the
bead/membrane complex was spun down at 865 x g for 5 minutes. The resulting
pellet was resuspended
in assay buffer and this spin/wash step was then repeated. The final pellet
was then resuspended in
assay buffer at the specific concentration required for the final assay.
Ligand preparation
An aliquot of [3H]-radioligand stock was diluted in assay buffer to give a pre-
determined final assay
concentration less than the equilibrium dissociation constant (KD) value.
Compound plate preparation
All test compounds were prepared at a concentration of 4 mM in 100 % dimethyl
sulphoxide (DMSO) from
dry samples. Compounds were diluted in 0.75 % DMSO in ddHZO to give
appropriate test concentrations
in a 384 well plate to give a final volume of 20 pL.
The same volume of assay buffer was added to specific wells of the plate to
enable subsequent
measurement of total radioligand binding. Furthermore, 20 pL of a high
concentration of compound
specific to each transporter assay was subsequently added to pre-determined
wells to determine non-
specific binding (NSB). Fluoxetine (10pM final assay concentration) was used
for hSERT and
desipramine (40pM final assay concentration) for hNET.
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
59
For each individual transporter assay, 20 pL of the prepared specific
radioligand was added to each well
of the final assay plates (containing compound solutions). Subsequently, 20 NL
of the corresponding
bead/membrane complex was added to each well of the final assay plate,
ensuring that the suspension
was mixed well. The plates were then sealed and incubated, with shaking, for 1
hour at room
temperature. The plates were subsequently incubated for an additional 6 hours,
with dark adaptation,
prior to reading.
C. Data analysis
The assay window (specific binding) per plate was calculated by subtracting
the mean NSB readings (in
counts per minute, or cpm) from the mean of total binding readings.
Subsequently the cpm read per well
(with mean NSB subtracted) were expressed as a percentage of the plate window
to determine the
amount of radioligand bound to the transporter.
These values were plotted against the concentration of the compound tested and
a sigmoidal inhibitory
concentration effect curve was fitted to the data using a four-parameter
logisitic equation and free-fitting
parameters to give an IC50 value (the concentration of compound required to
inhibit 50% of the specific
binding at the neurotransmitter transporter).
The inhibitory dissociation constant (K;) value was then calculated from the
IC50 value using the Cheng-
Prusoff equation.
Following determination of individual K; values for compounds tested, an
overall geometric mean was
calculated together with 95% confidence intervals and n values, where n is the
total number of individual
K; values.
iv) Media and Buffers
hSERT and hNET Cell Growth Medium
DMEM, 10 % (w/v) dialysed FCS
2 mM L-glutamine (diluted from 200 mM stock)
25 mM HEPES (diluted from 1 M stock)
250 pg/mL genetecin
MEMBRANE PREPARATION BUFFER
20 mM HEPES (diluted from 1 M stock with ddHZO), pH 7.4 at room temperature,
stored at 4 C. Prior to
use, one complete protease inhibitor tablet was dissolved per 50 mL of buffer.
Assay Buffer (1.5 x final assay concentration)
30 mM HEPES (diluted from 1 M stock with ddH2O) and 180 mM NaCI (diluted from
5 M stock with
ddHZO), pH 7.4 at room temperature, stored at 4 C.
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
The compounds can also be tested in specific disease models, such as the pain
models as follows:
6.2 Neuropathic pain
The activity of a compound in the treatment of neuropathic pain may be
measured according to the
5 following test protocol.
Animals: Male Sprague Dawley rats are housed in appropriately sized groups.
All animals are kept under
a 12h light/dark cycle (lights on at 07h 00min) with food and water ad
libitum. AII experiments are carried
out by an observer blind to the treatments and in accordance with the Home
Office Animals (Scientific
10 Procedures) Act 1986.
Chronic constriction iniury (CCI) rat model of neuropathic pain
The CCI of sciatic nerve is performed as previously described by Bennett and
Xie (Bennett GJ, Xie YK. A
peripheral mononeuropathy in rat that produces disorders of pain sensation
like those seen in man.
15 Pain:33:87-107, 1988). Animals are anaesthetised with a 2% isofluorane/02
mixture. The right hind thigh
is shaved and swabbed with 1% iodine. Animals are then transferred to a
homeothermic blanket for the
duration of the procedure and anaesthesia maintained during surgery via a nose
cone. The skin is cut
along the line of the thighbone. The common sciatic nerve is exposed at the
middle of the thigh by blunt
dissection through biceps femoris. About 7mm of nerve is freed proximal to the
sciatic trifurcation, by
20 inserting forceps under the nerve and the nerve gently lifted oLit of the
thigh. Suture is pulled under the
nerve using forceps and tied in a simple knot until slight resistance is felt
and then double knotted. The
procedure is repeated until 4 ligatures (4-0 silk) are tied loosely around the
nerve with approx 1mm
spacing. The incision is closed in layers and the wound treated with topical
antibiotics.
Streptozocin (STZ)-induced diabetes neuropathy in the rat
25 Diabetes is induced by a single intraperitoneal injection of streptozotocin
(50mg/kg) freshly dissolved in
0.9% sterile saline. Streptozotocin injection induces a reproducible
mechanical allodynia within 3 weeks,
lasting for at least 7 weeks (Chen and Pan, (Chen SR and Pan HL.
Hypersensitivity of Spinothalamic
Tract Neurons Associated With Diabetic Neuropathic Pain in Rats. J
Neurophysiol 87: 2726-2733, 2002).
30 Assessment of static and dynamic allodynia
Static allodynia.
Animals are habituated to wire bottom test cages prior to the assessment of
allodynia. Static allodynia is
evaluated by application of von Frey hairs (Stoelting, Wood Dale, Illinois,
USA.) in ascending order of
35 force (0.6, 1, 1.4, 2, 4, 6, 8, 10, 15 and 26 grams) to the plantar surface
of hind paws. Each von Frey hair
is applied to the paw for a maximum of 6 sec, or until a withdrawal response
occurred. Once a withdrawal
response to a von Frey hair is established, the paw is re-tested, starting
with the filament below the one
that produced a withdrawal, and subsequently with the remaining filaments in
descending force sequence
until no withdrawal occurrs. The highest force of 26g lifts the paw as well as
eliciting a response, thus
40 represented the cut off point. Each animal has both hind paws tested in
this manner. The lowest amount
of force required to elicit a response is recorded as paw withdrawal threshold
(PWT) in grams. Static
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
61
allodynia is defined as present if animals respond to a stimulus of, or less
than, 4g, which is innocuous in
naive rats (Field MJ, Bramwell S, Hughes J, Singh L. Detection of static and
dynamic components of
mechanical allodynia in rat models of neuropathic pain: are they signalled by
distinct primary sensory
neurones? Pain,1999;83:303-11).
Dynamic allodynia
Dynamic allodynia is assessed by lightly stroking the plantar surface of the
hind paw with a cotton bud. To
avoid recording general motor activity, care is taken to perform this
procedure in fully habituated rats that
are not active. At least two measurements are taken at each time point, the
mean of which represents the
paw withdrawal latency (PWL). If no reaction is exhibited within 15 sec the
procedure is terminated and
animals are assigned this withdrawal time. A pain withdrawal response is often
accompanied with
repeated flinching or licking of the paw. Dynamic allodynia is considered to
be present if animals respond
to the cotton stimulus within 8 sec of commencing stroking (Field et al,
1999).
6.3 Nociceptive pain
The activity of a compound in the treatment of nociceptive pain may be
measured according to the
following test protocols.
Hotplate
Experimental Procedure: Male Sprague Dawley rats are placed on a hot plate
(Ugo Basile, Italy)
maintained at 55 5 C. The time between placement of the animal on the hot
plate and occurrence of
either licking of fore or hind paw, shaking or jumping off the surface is
measured. Baseline
measurements are made and animals reassessed following drug administration.
The cut off time for hot
plate latencies is set at 20 seconds to prevent tissue damage.
Ovariohysterectomy (OVX)
Experimental Procedure: Female Sprague Dawley rats are placed into an
anaesthetic chamber and
anaesthetised with a 2% isofluorane 02 mixture. During surgery, anaesthesia is
maintained via a nose
cone. OVX is performed via a midline incision (2cm in length) in the linea
alba, whilst the animal is on a
heat blanket. The ovarian ligaments and cervix are ligated with 5-0 silk,
using a single clamp technique.
The ovaries and uterus are then removed. The abdominal wall is closed using 4
simple interrupted
sutures and the skin closed using 4 wound clips. Immediately after surgery
animals are placed in
individual plexiglass chambers. Once the 'animal has recovered from the
anaesthetic the abdominal body
postures are recorded in 30 min bins at various time points. Postures scored
are humpback position,
contraction of the muscle of the abdomen associated with inward movements of
the hind limb, stretching
of the body and squashing of the lower abdomen against the floor. Each of
these behaviours is scored as
one posture.
Brennan
Experimental Procedure: Male Sprague Dawley rats are placed into an
anaesthetic chamber and
anaesthetised with a 2% isofluorane 02 mixture. During surgery, anaesthesia is
maintained via a nose
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
62
cone. The plantar aspect of the right hind paw is cleaned with 50% ethanol. A
1cm long longitudinal
incision is made with a number 11 blade through the skin and fascia of the
plantar aspect of the foot,
starting 0.5cm from the proximal edge of the heel and extending toward the
toes. The plantaris muscle is
elevated using forceps and incised longitudinally, the muscle origin and
insertion remain intact. After
haemostasis with gentle pressure, the skin is closed with two simple sutures
of braided silk.
Mono-lodoacetate (MIA)-induced OA model
Male 6 weeks-old Sprague-Dawley (SD, Japan SLC or Charles River Japan) rats
are anesthetized with
pentobarbital. Injection site is shaved and cleaned with 70% ethanol. 25 NI of
MIA solution or saline is
injected in the right knee joint using a 29G needle. 7, 14, 19 and 20 days
after the MIA injection, train rats
to measure the weight bearing (WB) without their stress. 21 days after the MIA
injection, the WB on two
of each hind paw is measured and the WB deficit is calculated. Define the WB
deficit value as "pre
value". Arrange for experimental group evenly in consideration of pre value
and prepre value. After the
administration of test compounds or vehicle, the WB on two of each hind paw
was, measured.
Cancer pain model
These experiments use adult male C3H/HeN mice (Nihon SLC, Shizuoka, Japan).
The mice are housed
in accordance with National Institutes of Health guidelines in a vivarium
maintained at 22 C with a 12-
hour alternating light-dark cyde, and were given food and water ad libitum.
The sarcoma injection
protocol which is used has been described. After induction of general
anesthesia with an inhalation of
isofluran (2%), a superficial incision is made in the skin overlying the
patella, using Mora scissors. The
patellar ligament is then cut, exposing the condyles of the distal femur. A 30-
gauge needle is inserted at
the level of the intercondylar notch and into the medullary canal to create an
initial core pathway. After the
initial core is made, a 29-gauge needle is used to make the final pathway into
the bone. A 0.5-mm
depression is then made using a half-round bur in a pneumatic dental high
speed handpiece, to serve as
mechanical retention for the dental resin plug. Then, 20 l a-minimum
essential media (Sigma; sham
injection) or 20 wl media containing 1 X10 5 2472 osteolytic sarcoma cells
(American Type Culture
Collection, Rockville, Maryland; sarcoma injection) is injected using a 29-
gauge needle and a .25 cc
syringe. To prevent leakage of cells outside the bone, the injection site is
closed with dental resin,
followed by copious irrigation with filtered water. Wound closure is achieved
using auto wound clips
(Becton Dickinson, San Jose, California). Wound clips are removed at day 5 to
prevent interference with
behavioral testing.
Assessment of static and dynamic allodynia
Static allodynia.
Animals are habituated to wire bottom test cages prior to the assessment of
allodynia. Static allodynia is
evaluated by application of von Frey hairs (Stoelting, Wood Dale, Illinois,
USA.) in ascending order of
force (0.6, 1, 1.4, 2, 4, 6, 8, 10, 15 and 26 grams) to the plantar surface of
hind paws. Each von Frey hair
is applied to the paw for a maximum of 6 sec, or until a withdrawal response
occurrs. Once a withdrawal
response to a von Frey hair is established, the paw is re-tested, starting
with the filament below the one
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
63
that produces a withdrawal, and subsequently with the remaining filaments in
descending force sequence
until no withdrawal occurrs. The highest force of 26g lifts the paw as well as
eliciting a response, thus
represents the cut off point. Each animal has both hind paws tested in this
manner. The lowest amount
of force required to elicit a response is recorded as paw withdrawal threshold
(PWT) in grams. Static
allodynia is defined as present if animals respond to a stimulus of, or less
than,. 4g, which is innocuous in
naive rats (Field MJ, Bramwell S, Hughes J, Singh L. Detection of static and
dynamic components of
mechanical allodynia in rat models of neuropathic pain: are they signalled by
distinct primary sensory
neurones? Pain,1999;83:303-11).
Dynamic allodynia
Dynamic allodynia is assessed by lightly stroking the plantar surface of the
hind paw with a cotton bud. To
avoid recording general motor activity, care is taken to perform this
procedure in fully habituated rats that
are not active. At least two measurements are taken at each time point, the
mean of which represents the
paw withdrawal latency (PWL). If no reaction is exhibited within 15 sec the
procedure is terminated and
animals are assigned this withdrawal time. A pain withdrawal response is often
accompanied with
repeated flinching or licking of the paw. Dynamic allodynia is considered to
be present if animals respond
to the cotton stimulus within 8 sec of commencing stroking (Field et al,
1999).
Radiant heat paw withdrawal
Experimental procedure: Thermal paw withdrawal is assessed using the rat
plantar test (Ugo Basile, Italy)
following a modified method of Hargreaves et al., 1988. Rats are habituated to
the apparatus that
consists of three individual perspex boxes on an elevated glass table. A
mobile radiant heat source is
located under the table and focused onto the hind paw and paw withdrawal
latencies (PWL) are recorded.
There is an automatic cut off point of 22.5 s to prevent tissue damage. PWL
are taken 2-3 times for both
hind paws of each animal, the mean of which represents baselines for right and
left hind paws. The
apparatus is calibrated to give a PWL of approximately 10 s.
Weight bearing
Experimental procedure: Animals are examined for hypersensitivity in the
weight-bearing test, using an
"incapacitance tester" (Linton Instruments, Diss, Norfolk, U.K.). Rats were
positioned with their fore limbs
up on a perspex slope and hind limb weight distribution was measured via force
transducers under each
of the hind paws. Each animal is placed in the apparatus and the weight load
exerted by the hind paws is
noted. The difference in weight bearing is calculated by subtracting the
ipsilateral (injured) paw from the
contralateral paw (normal) and this constitutes the raw data.
6.4 Inflammatory pain
The activity of compound in the treatment of inflammatory pain may be measured
according to the
following test protocol.
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
64
CFA-induced weight bearing deficits in rats
Male 7-week-old SD rats are fasted overnight. CFA (300 pg of Mycobacterium
Tuberculosis H37 RA
(Difco Laboratories) in 100 pL of liquid paraffin (Wako)) is injected into the
rat's right hind footpad. Two
days after the administration of CFA, the changes in hind paw weight
distribution between the left
(ipsilateral) and the right (contralateral) limbs are measured as an index of
pain by using Linton
Incapacitance tester (Linton Instrumentation, UK). The test compound suspended
in 0.1% MC (Wako) is
administered orally in a volume of 1 mL per 100 g body weight. Each animal is
placed in the apparatus
and the weight load exerted by the hind paws is measured before, 1, 2 and 4
hours after drug
administration.
Carrageenin-induced mechanical hyperalgesia in rats
Male 4-week-old SD rats are fasted overnight. Hyperalgesia is induced by
intraplantar injection of
Lambda-carrageenin (0.1 ml of 1% w/v solution in saline, Zushikagaku). The
test compound (1 ml of 0.1 %
methylcellulose/100g body weight) is given orally at 5.5 hours after the
carrageenin injection. The paw
withdrawal threshold (gram) is measured by analgesimeter (Ugo Basile) at 3.5,
4.5, 6.5 and 7.5 hours
after the carrageenin injection. (Randall L.O. & Selitto I.J., Arch. Int.
Pharmacodyn. 111, 409-419, 1957)
Carrageenan-Induced Thermal Hyperalpesia (CITH) in the Rat
Thermal hyperalgesia is assessed using the rat plantar test (Ugo Basile,
Comerio, Italy), according to a
method modified by Hargreaves et al. (1988). Briefly, rats are habituated to
the apparatus that consists of
three individual Perspex boxes on a glass table. A mobile radiant heat source
is located under the table
and focused onto the desired paw. Paw withdrawal latencies (PWLs) are recorded
three times for both
hind paws of each animal, the mean of which represents baseline for left and
right hind paws. The
apparatus is calibrated to give a PWL of approximately 10 s in nai've rats. To
prevent tissue damage of
the plantar zone, a 22.5 sec cut-off is observed. Lambda carrageenan is
injected intraplantarly (100 l,
20 mg/ml) the right hind paw and baseline recordings of PWT are taken 2 hr
post administration.
6.5 Visceral pain
The activity of a compound in the treatment of visceral pain may be measured
according to the following
test protocols.
Several models are available to determine if a compound is effective in
treating disorders of the viscera.
These models include a LPS model (Eutamene H et al, J Pharmacol Exp Ther 2000
295 (1):162-7), a
TNBS model (Diop L. et al, Gastroenterology 1999, 116, 4(2): A986), a IBD
model (Clerriett D, Markham
A, Drugs 2000 Apr;59(4):929-56), a pancreatic pain model (Isla AM, Hosp Med
2000 Jun;61(6):386-9)
and a visceral non digestive pain model (Boucher M et al, J Uro12000
Jul;164(1):203-8).
TNBS-induced chronic visceral allodynia in rats
In this experimental model of colonic distension in awake rats, previous
injection of
trinitrobenzenesulfonic acid (TNBS) into the proximal colon lowered the
visceral pain threshold.
CA 02591415 2007-06-15
WO 2006/064351 PCT/IB2005/003791
Materials and methods: Male Sprague-Dawley rats are used. The animals are
housed 3 per cage in a
regulated environment (20 1 C, 50 5 % humidity, with light 8:00 am to 8:00
pm). At day 0, under
anesthesia (ketamine 80 mg/kg i.p.; acepromazine 12 mg/kg i.p.), the injection
of TNBS (50 mg/kg in
ethanol 30 %), or saline (1.5 ml/kg) for control rats, is performed into the
proximal colon wall (1 cm from
5 the cecum). After the surgery, animals are individually housed in
polypropylene cages and kept in a
regulated environment (20 1 C, 50 5 % humidity, with light 8:00 a.m. to
8:00 p.m.) during 7 days. At
day 7 after TNBS administration, a balloon (5-6 cm length) is inserted by
anus, and kept in position (tip of
balloon 5 cm from the anus) by taping the catheter to the base of the tail.
Oral administration of the test
compound is performed 1 h before the colonic distension cycle: the balloon is
progressively inflated by
10 steps of 5 mm Hg (0.667 kPa), from 0 to 75 mm Hg, each step of inflation
lasting 30 s. Each cycle of
colonic distension is controlled by a standard barostat. The threshold (mm Hg)
corresponds to the
pressure which produced the first abdominal contraction, and the cycle of
distension is then discontinued.
The colonic threshold is determined after performance of four cycles of
distension on the same animal.
15 LPS-induced rectal hypersensitivity in rats
Intraperitoneal injection of bacterial lipo-polysaccharide (LPS) has been
shown to induce rectal
hyperalgesia in awake rats.
Materials and methods: Animals are surgically prepared for electromyography:
rats are anaesthetized by
20 intraperitoneal injection of acepromazine (0.6 mg/kg) and ketamine (120
mg/kg). Three groups of three
electrodes are implanted in the abdominal external oblique musculature, just
superior to the inguinal
ligament. Electrodes are exteriorized on the back of the neck and protected by
a glass tube attached to
the skin. Animals are individually housed in polypropylene cages and kept in a
temperature-controlled
room (21 C). Food (UAR pellets, Epinay, France) and water are provided ad
libitum.
Electromyographic recordings begin five days after surgery. The electrical
activity of abdominal striated
muscles is recorded with an electroencephalograph machine (Mini VIII Alvar,
Paris, France) using a short
time constant (0.03 s) to remove low-frequency signals (< 3 Hz) and a paper
speed of 3.6 cm/min. Spike
bursts are recorded as an index of abdominal contractions.
Distension procedure: Rats are placed in plastic tunnels (6 cm diameter x 25
cm long), where they cannot
move, escape, or turn around, in order to prevent damage to the balloon.
Animals are accustomed to this
procedure for four days before rectal distension in order to minimize stress
reactions during experiments.
The balloon used for distension is an arterial embolectomy catheter (Fogarty,
Edwards Laboratories Inc.).
Rectal distension is performed by insertion of the balloon (2 mm diameter x 2
cm long) into the rectum, at
1 cm from the anus, and catheter is fixed at the base of the tail. It is
inflated progressively with tepid water
by steps of 0.4 ml, from 0 to 1.2 ml, each step of inflation lasting 5 min. To
detect possible leakage, the
volume of water introduced in the balloon is checked by complete removal with
a syringe at the end of the
distension period.