Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02596799 2007-08-01
WO 2006/084116 PCT/US2006/003817
AMINE DERIVATIVE CONTAINING COMPOSITIONS AND METHODS FOR
THE TREATMENT OF VIRAL INFECTIONS RELATED TO THE ETIOLOGY
OF CANCER
By Vladimir Khazak
Erica A. Golemis
Sanjay R. Menon
Lutz Weber
FIELD OF THE INVENTION
The present invention relates to the fields of virology and cellular
signaling.
More specifically, the invention provides compositions and methods useful for
the
treatment of viral infections, particularly those caused by herpes viruses.
BACKGROUND OF THE INVENTION
Several publications and patent documents are cited throughout the
specification
in order to describe the state of the art to which this invention pertains.
Each of these
citations is incorporated herein by reference as though set forth in full.
HHV-8 is a recently identified virus that has been associated with Kaposi's
sarcoma (KS) and possibly with a type of cancer called body cavity lymphoma (a
tumor
that arises from the lymph tissue) (1). KSHV is an important pathogen capable
of
causing disease that affects all age groups worldwide (2). Kaposi's sarcoma is
an unusual
slcin tumor that is seen primarily in HIV-infected men. HHV-8 has also been
isolated in
the semen of HIV infected individuals. Because of these factors, it is
believed that HHV-
8 may cause a sexually transmitted infection.
From the beginning of the AIDS epidemic, it was suspected that there might be
another infectious agent besides HIV that causes KS. In-the 1980's, around 30-
40% of
homosexual men with AIDS developed KS at some point in their illness. In
contrast, KS
was a rare occurrence in women or hemophiliacs with HIV. This suggested that
there was
an additional factor among gay and bisexual men that increased their chances
of
developing KS. In 1994, researchers identified a previously unkn.own virus in
KS
biopsies (3). This virus was named human herpesvirus 8 (also known as Kaposi's
sarcoma-associated herpesvirus-KSHV). It belongs to the important family of
human
1
CA 02596799 2007-08-01
WO 2006/084116 PCT/US2006/003817
heipesviruses that includes varicella-zoster (chiclcenpox/shingles), Epstein-
Barr virus
(mononucleosis), and herpes simplex 1 and 2 (oral and genital herpes). After
identification of HHV-8, researchers have been able to detect this virus in
virtually all
types of Kaposi's sarcoma tumors, including those seen before the AIDS
epidemic.
Analysis of the complete HHV-8 genome sequence reveals similarities to other
gammaherpesviruses, including heipesvirus saimiri (HVS), murine
gammaherpesvirus 68
(MIIV68) and Epstein-Barr Virus (HHV-4). The -1651cb genome contains over 80
open
reading frames arranged in a long unique region flanked by multiple 801bp
terminal
repeat units of high G+C content. The long unique region contains blocks of
conserved
genes found in most herpesviruses, interspersed with blocks of non-homologous
genes
that are specific for HHV-8 and related viruses.
The pathogenic mechanism by which KSHV induces tumorigenicity is presently
unknown. However, KSHV encodes a G-protein coupled receptor (GPCR) that acts
as an
oncogene, the expression of which causes malignant transformation of rodent
fibroblast
cells, and produces tumors in nude mice. Transgenic mice expressing the KSHV-
GPCR
develop highly vascular endothelial tumors (4-6). Such tumorigenicity relates
to the
ability of the KSHV-GPCR to constitutively activate the extracellular signal-
regulated
kinase (ERK) signal-transduction cascade (7),(8). One of the main activators
of the ERK
cascade is the Ras-Raf-MEK1/2-ERK signaling axis (9). Further, in some cases,
GPCRs
are known to signal through Ras (10-12). Considering that a number of viruses
utilize
this signaling axis to establish infection, agents which disrupt this pathway
should prove
efficacious against viruses which include, for example, Epstein Bar Virus and
Hepatitis B
virus.
Clearly a need exists for compositions and methods useful for treating viral
infections, including KSHV. The present application provides such compositions
and
methods.
SUMMARY OF THE INVENTION
In accordance with the present invention, a method for the treatment of viral
infection in a patient in need thereof is provided. An exemplary method
entails
2
CA 02596799 2007-08-01
WO 2006/084116 PCT/US2006/003817
administration to a patient of a therapeutically effective amount of a
compound having
the foimula
R3
~
Y
(
N R2 (I)
x U
I
R1
wherein U is (CHZ),,, CO, SO2 or CONH;
nis0, 1,2,3,4or5;
X is CH2, CO, SO2 or CONH;
Y is CH2, CO, SO2 or CONH;
R1 is an optionally substituted aryl, aralkyl, heteroaryl or heteroarylalkyl;
R2 is an optionally substituted heteroalkyl, aryl, arallcyl, heteroaryl,
heteroaralleyl,
cycloalkyl, heterocycloallcyl or heteroalkylcycloalkyl and
R3 is an optionally substituted allcyl, alkenyl, allcinyl, heteroalkyl,
cycloalkyl,
alkylcycloalkyl, heterocycloalkyl, heteroalkylcycloalkyl, aryl, heteroaryl,
heteroarylalkyl
or aralkyl;or a pharmacologically acceptable salt, solvate, hydrate or
formulation thereof.
A preferred embodiment of the invention comprises administration of the
compound disclosed above in combination with a MAPK pathway inhibitor and/or
an
antiproliferative agent. Suitable inhibitors and agents are provided in Table
I
hereinbelow. The methods of the invention should prove efficacious in the
treatment of
viral infection caused by HHV-8, HHV-4 and hepatitis B virus.
In yet another embodiment of the invention, a pharmaceutical composition for
treating or inhibiting viral infection is provided. An exemplary composition
comprises
the above-mentioned compound and at least one agent or inhibitor selected from
the
group provided in Table I in a pharmaceutically acceptable carrier.
3
CA 02596799 2007-08-01
WO 2006/084116 PCT/US2006/003817
Another embodiment of the invention comprises a method for prophylaxis of
viral
infection in a host susceptible to said infection. One such method comprises
administration of a therapeutically effective amount of the above mentioned
compound
and optionally at least one MAPK pathway inhibitor and/or an antiproliferative
agent.
BRIEF DESCRIPTION OF THE DRAWINGS
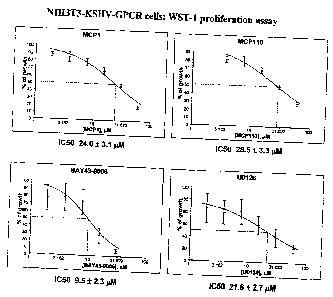
Figure 1. Inhibition of anchorage-dependent growth of NIH3T3 cells
transformed witli KSHV-GPCR oncogene by MCP compounds. IC50 values in a
WST-1 proliferation assay were established in KSHV-GPCR transformed NIH3T3
cells
treated with MCP1, MCP110 or control compounds BAY43-9006 and U0126.
Figure 2. Inhibition of anchorage-independent growth of NIH3T3 cells
transformed
with KSHV-GPCR oncogene by MCP compounds. IC50 values in a colony formation
assay were established in KSHV-GPCR transformed NIH3T3 cells treated with
MCP1,
MCP1 10 or control compounds BAY43-9006 and U0126.
Figure 3. Inhibition of anchorage-independent growth: combination effect of
MCP110 at 10 M with BAY43-9006, U0126 and staurosporine. A. NIH3T3 cells
transformed with K-Ras(G12V) were treated with the DMSO control, 10 M of
MCP110, compounds at concentrations indicated, or combinations of MCP110 and
compounds, and grown in soft agar for 21 days, then colonies scored. B. HLR-
Elkl cells
were incubated with DMSO control, 5 M of MCP110, compounds at concentrations
indicated, or combinations of MCP110 and compounds for 6 hours (see Materials
and
Methods). Percent of residual luciferase activity was determined in comparison
with
DMSO treated cells. C. Experiment as in A., except with staurosporine. Solid
black bar -
DMSO treated cells; solid white bars - cells treated only with inhibitors;
black dotted
bars - cells treated with combinations of MCP110 and the compounds. Results
reflect the
average of at least 3 independent experiments with 3 repetitions in each
assay.
4
CA 02596799 2007-08-01
WO 2006/084116 PCT/US2006/003817
Figure 4. Inhibition of anchorage-independent and dependent growth:
combination
of MCPI or MCP114 with taxol. A. IC50 values for taxol were established in
Raf22W,
K-Ras(V12) and KSHV-vGPCR-transformed NIH3T3 cells, for growth in soft agar,
or
by WST-1 proliferation assay. B. IC50 values for taxol inhibition of NIH3T3-K-
Ras(V12) cell growth in soft agar were re-assessed in the presence of MCPl,
MCP110,
BAY43-9006, or U0126 at the concentrations as indicated. C. IC50 values for
taxol
inhibition of NIH3T3-K-Ras(V 12) cell proliferation were re-assessed in the
presence of
MCP1 10, BAY43-9006, or U0126 at the concentrations as indicated. D. IC50
values for
taxol inhibition of NIH3T3-KSHV-GPCR cell growth in soft agar were re-assessed
in the
presence of MCP1, MCP110, BAY43-9006, or U0126 at the concentrations as
indicated.
E. IC50 values for taxol inhibition of EC-vGPCR cell growth in WST-1
proliferation
assay were determined in the presence of MCP110, BAY43-9006, or U0126 at the
concentrations as indicated.
Figure 5. Cell cycle profile induced by MCP11O and taxol in K-Ras(V12) versus
cRaf22W NIH3T3 transformed cells. Cells were treated with compounds at the
concentrations shown for 72 hours then fixed, stained with propidium iodide
and
analyzed by FACS. The software program Ce1lQuest was used to establish the
frequency
of G1 versus G2/M cells.
Figure 6. Induction of cell death by MCP11O and taxol in K-Ras(V12) versus
cRaf22W NIH3T3 transformed cells. Cells were treated with compounds at the
concentrations as indicated for 72 hours, then fixed and stained with annexin
V and
analyzed by FACS. The frequency of annexin V - positive cells is shown.
Figure 7. Schematic of the signaling cascade that leads to cellular
transformation
associated with KSHV infection.
Figure 8. Schematic of the signaling cascade that leads to cellular
transformation
associated with HHV-4/Epstein Barr virus infection.
5
CA 02596799 2007-08-01
WO 2006/084116 PCT/US2006/003817
Figure 9. Schematic of the signaling cascade that leads to cellular
transformation
associated with Hepatitis B infection.
DETAILED DESCRIPTION OF THE INVENTION
Recently, a new class of protein-protein interaction inhibitors, represented
by
MCP1, N-(4-Benzyloxy-3-methoxybenzyl)-N-(2-pyridin-2-ylethyl)-2-
chlorobenzamide,
and MCP 110, N-(4-Benzyloxy-3-methoxybenzyl)-N-(2-pyridin-2-ylethyl)-5-
phenylpentanoic acid amide, were described that block interaction between the
human
Ras and Raf oncoproteins, and inhibit various oncogenic phenotypes associated
with
activated Ras (13, PCT/EP02/1222 (WO 03/037865)). Based on the implication of
Ras in
KSHV-GPCR cell transforination, we evaluated these compounds for efficacy
against
KSHV-GPCR-transformed cells. We determined that these compounds were able to
inhibit anchorage dependent and independent growth of murine fibroblasts
transformed
with GPCR oncogene from HHV-8 (KSHV) virus, and produce strong additive
effects in
growth inhibition of transformed cells when they were combined with the MAPK
pathway inhibitors U0126 and BAY43-9006, as well as the cytotoxic agent taxol.
The compounds described herein block the interaction between Ras and Raf and
thus should have demonstrable efficacy as antiviral agents. Based on
previously
identified signaling cascades, such viruses include without limitation, HHV-8,
HHV-4
and hepatitis B virus. Furthermore, the compounds used in the practice of this
invention
can be combined with other lcnown anti-viral or anti-proliferative agents in
methods of
treating or preventing viral infection.
Compounds useful in the practice of the present invention include those of
Formula (I):
6
CA 02596799 2007-08-01
WO 2006/084116 PCT/US2006/003817
R3
~
Y
N R2 (I)
x U
I
Ri
wherein U is (CHZ)n, CO, SO2 or CONH;
nis0,1,2,3,4or5;
X is CH2, CO, SO2 or CONH;
Y is CH2, CO, SO2 or CONH;
R1 is an optionally substituted aryl, aralkyl, heteroaryl or heteroarylalkyl;
R2 is an optionally substituted heteroalkyl, aryl, aralkyl, heteroaryl,
heteroaralkyl,
cycloalkyl, heterocycloalkyl or heteroallcylcycloallcyl and
R3 is an optionally substituted alkyl, alkenyl, allcinyl, heteroalkyl,
cycloalkyl,
allcylcycloalkyl, heterocycloalkyl, heteroalkylcycloalkyl, aryl, heteroaryl,
heteroarylalkyl
or aralkyl; or a pharmacologically acceptable salt, solvate, hydrate or
formulation thereof.
The term alkyl refers to a saturated or unsaturated (i. e. allcenyl and
alkinyl)
straight or branched chain alkyl group, containing from one or two to ten
carbon atoms,
preferably from one or two to six carbon atoms, e.g. 1 or 2 to 6 carbon atoms,
for
example methyl, ethyl, propyl, iso-propyl, butyl, iso-butyl, tert.-butyl, n-
pentyl, n-hexyl,
2,2-dimethylbutyl, n-octyl, ethenyl (vinyl), propenyl, iso-propenyl, butenyl,
isoprenyl or
hexa-2-enyl; ethinyl, propinyl or butinyl groups.
The teims alkenyl and alkinyl refer to unsaturated straight or branched chain
allcyl
groups, containing from two to ten carbon atoms, preferably from two to six
carbon
atoms, e.g. 2 to 6 carbon atoms, for example ethenyl (vinyl), propenyl, iso-
propenyl,
butenyl, isoprenyl or hexa-2-enyl; ethinyl, propinyl or butinyl groups.
7
CA 02596799 2007-08-01
WO 2006/084116 PCT/US2006/003817
The term heteroalkyl refers to an alkyl, alkenyl or allcinyl group as defined
herein
where one or more carbon atoms are replaced by an oxygen, nitrogen,
phosphorous or
sulphur atom, for example an alkoxy group containing from one to ten carbon
atoms,
preferably from one to six carbon atoms, e.g. 1 to 4 carbon atoms, such as
methoxy,
ethoxy, propoxy, iso-propoxy, butoxy or tert.-butoxy, a(1-4C)alkoxy(1-4C)alkyl
group
such as methoxymethyl, ethoxymethyl, 1-methoxyethyl, 1-ethoxyethyl, 2-
methoxyethyl
or 2- ethoxyethyl; or a cyano group; or a 2,3-dioxyethyl group. The term
heteroalkyl
furtheimore refers to a group derived from a carboxylic acid or carboxylic
acid amide
containing from one to ten carbon atoms, preferably from one to six carbon
atoms, e.g. 1
to 4 carbon atoms, and may, for example, be acyl containing from one to ten
carbon
atoms, preferably from one to six carbon atoms, e.g. 1 to 4 carbon atoms, such
as acetyl,
propionyl, butyryl or pivaloyl; acyloxy containing from one to ten carbon
atoms,
preferably from one to six carbon atoms, e.g. 1 to 4 carbon atoms such as
acetyloxy,
propionyloxy, butyrlyoxy or pivaloyloxy; carboxyalkyl containing from one to
ten carbon
atoms, preferably from one to six carbon atoms, e.g. 1 to 4 carbon atoms such
as
carboxymethyl, carboxyethyl, carboxypropyl, carboxybutyl, carboxyalkyl ester
containing from one to ten carbon atoms, preferably from one to six carbon
atoms, e.g. 1
to 4 carbon atoms, such as carboxyalkyl methyl ester, carboxyalkyl ethyl
ester,
carboxyalkyl propyl ester, carboxyalkyl isopropyl ester, carboxyalkyl butyl
ester or
carboxyalkyl tert.-butyl ester, carboxyalldyl aminde or alkylcarbamoyl such as
N-(1-
4C)alkylcarbamoyl or N, N'-(1-4C)dialkylcarbamoyl) containing from one to ten
carbon
atoms, preferably from one to six carbon atoms, e.g. 1 to 4 carbon atoms such
as N-
methylcarbamoyl, N-ethylcarbamoyl, N-propylcarbamoyl, N, N'-dimethylcarbamoyl,
N-
ethyl-N-methylcarbamoyl or N, N'-dipropylcarbamoyl, alkoxycarbonyl containing
from
one to ten carbon atoms, preferably from one to six carbon atoms, e. g. 1 to 4
carbon
atoms, such as methoxycarbonyl, ethoxycarbonyl, propoxycarbonyl,
isopropoxycarbonyl,
butoxy- or tert.-butoxycarbonyl or alkoxycarbonyloxy containing from one to
ten carbon
atoms, preferably from one to six carbon atoms, e.g. 1 to 4 carbon atoms such
as
methoxycarbonyloxy, ethoxycarbonyloxy, propoxycarbonyloxy,
isopropoxycarbonyloxy,
butoxycarbonyloxy, tert.-butoxycarbonyloxy.
8
CA 02596799 2007-08-01
WO 2006/084116 PCT/US2006/003817
The teim cycloalkyl refers to a saturated or partially unsaturated cyclic
group,
having one or more rings, formed by a skeleton that contains from three to 14
carbon
atoms, preferably from three, four, five or six to nine or ten'carbon atoms,
for example
cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, tetralin, cyclopentenyl or
cyclohex-2-
enyl groups.
The term heterocycloalkyl refers to a cycloalkyl group as defined herein where
one or more carbon atoms are replaced by one or more oxygen, nitrogen,
phosphorous or
sulphur atoms. Specific examples for heterocyclalkyl are piperidino,
morpholino, N-
methyl-piperazino or N-phenyl-piperazino groups.
The term aryl refers to an aromatic cyclic group, having one or more rings,
formed by a skeleton that contains from five to 14 carbon atoms preferably
from five or
six to nine or ten carbon atoms, for example phenyl, inden or naphthyl groups.
Specific
examples are a benzyl, tolyl, phenethyl, biphenyl, xylyl, cumyl, 2-, 3-or 4-
methoxyphenyl, 2-, 3- or 4- ethoxyphenyl, 4-carboxyphenyl alkyl or a 4-
hydroxyphenyl
group.
The term heteroaryl refers to an aryl group as defined herein where one or
more
carbon atoms are replaced by an oxygen, nitrogen, phosphorous or sulphur atom,
for
example 4- pyridyl, 2-imidazolyl, 3-pyrazolyl, quinolinyl, isoquinolinyl,
pyrrolyl,
oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, 1,2, 3-triazolyl, 1,2, 4-
triazolyl, oxadiazolyl,
thiadia- zolyl, indolyl, indazolyl, tetrazolyl, pyrazinyl, pyridinyl,
pyrimidinyl and
pyridazinyl groups.
The terms aralkyl and heteroarylalkyl refer to groups that comprise both aryl
or,
respectively, heteroaryl as well as alkyl, alkenyl, allcinyl and/or
heteroalkyl (for example
alkoxy groups in case of aralkyloxy) and/or cycloalkyl and/or heterocycloalkyl
ring
systems as defined herein. Examples of such groups are arylalkyl-,
arylallcenyl-,
arylalkinyl-, arylheteroalkyl-, arylheteroallcenyl-, arylheteroalkinyl-,
heteroarylheteroallcyl-, heteroarylheteroallcenyl-, heteroarylheteroallcinyl-,
arylcycloalkyl,
heteroaryl- cycloalkyl-, arylheterocycloallcyl-, heteroarylheterocycloalkyl-,
arylcycloallcenyl-, heteroarylcycloallcenyl-, arylcycloalkinyl-,
heteroarylcycloalkinyl-,
arylheteroallcenyl-, heteroarylheteroallcenyl-, arylheteroalkinyl-,
heteroarylheteroalkinyl-,
heteroarylalkyl-, heteroalkenyl- and heteroarylakinyl-groups, wherein the
cyclic groups
9
CA 02596799 2007-08-01
WO 2006/084116 PCT/US2006/003817
can be saturated or once, twice or three-times unsaturated. Examples are the
tetrahydroisoquinolinyl, benzyl, benzyloxy, 2-or 3-ethyl-indolyl or 4-
methylpyridino
groups.
The terms alkylcycloallcyl and heteroalkylcycloalkyl refer to groups that
comprise
both cycloalkyl or, respectively, heterocycloalkyl as well as alkyl, allcenyl,
alkinyl and/or
lieteroalkyl (for example alkoxy groups in case of aralkyloxy) groups as
defined herein.
Examples of such groups are alkylcycloalkyl, alkenylcycloalkyl,
allcinylcycloalkyl, al-
kylheterocycloalkyl, alkenylheterocycloalkyl, alkinylheterocycloallcyl,
heteroalkylcyclo-
alkyl, heteroalkenylcycloalkyl, heteroalltinylcycloalkyl,
heteroalkylheterocycloalkyl,
heteroalleenylheterocylcloalkyl, heteroallcinylheterocycloallcyl, which cyclic
groups can
be saturated or once, twice or three-times unsaturated.
Any alkyl, alkenyl, allcinyl, heteroalkyl, cycloalkyl, heterocycloalkyl, aryl,
heteroaryl aralkyl or heteroarylalkyl groups as defined herein may be
substituted with one
or more halogen atoms, NH2, SH, NO2 or OH groups or unsubstituted alkyl,
heteroalkyl,
aryl, aralkyl, aralkyloxy, heteroaryl, cycloalkyl or heterocycloalkyl groups
as defined
herein.
The term "optionally substituted" refers to groups wherein one or more
hydrogen
atoms may be replaced by a halogen atom, a NH2, SH, NOZ or OH group or by an
unsubstituted allcyl, heteroalkyl, aryl, aralkyl, aralkyloxy, heteroaryl,
cycloalkyl or
heterocycloalkyl group as defined herein.
PrefeiTed are compounds of Formula (I), wherein U is (CH2)n and n is 0, 1 or
2.
Further preferred are compounds of Formula (I), wherein R1 is an optionally
substituted phenyl ring; moreover preferred the phenyl ring is substituted by
a benzyloxy
group.
Also preferred are compounds of Formula (I), wherein R2 is heterocycloalkyl or
heteroaryl (especially preferred nitrogen containing heterocycloalkyl or
heteroaryl
groups).
Further preferred are compounds of Formula (I), wherein R2 is a pyridyl or a
piperidyl group.
Further preferred are compounds of Formula (I), wherein R3 is aryl or aralkyl.
CA 02596799 2007-08-01
WO 2006/084116 PCT/US2006/003817
Also preferred are compounds of formula (I), wherein R1 is a group of the
formula
R5
R4
wherein R4 is H, alkyloxy or aralkyloxy (more preferred H, methoxy or
benzyloxy) and
R5 is F, Cl, alkyl, heteroalkyl, aryl, aralkyl, heteroaryl or heteroarylalkyl
(preferred Cl,
methoxy or benzyloxy; more preferred benzyloxy).
Further preferred are compounds of Formula (I) wherein R3 is a Cl-C6 alkyl
group
(especially an isopropyl group).
Moreover preferred are compounds of Formula (I), wherein X is CO or SOZ and
Y is CH2.
Further preferred are compounds of Formula (I), wherein X is CH2 _ and Y is CO
or SO2.
Also preferred are compounds of Formula (II)
Y /R3
Het
R4
R5
11
CA 02596799 2007-08-01
WO 2006/084116 PCT/US2006/003817
wherein Het is a pyridyl group; n is 0,1 or 2; X is CH2 ; Y is CO or SOZ ; R3
is aryl or
aralkyl ; R4 is H, alkyloxy or aralkyloxy (more preferred H, methoxy or
benzyloxy) and
R5 is F, Cl, alkyl, heteroalkyl, aryl, aralkyl, heteroaryl or heteroarylalkyl
(preferred Cl,
methoxy or benzyloxy; more preferred benzyloxy).
Further preferred are compounds of Formula (lI) wherein Het is a pyridyl
group;
n is 0,1 or 2; X is CO or SO2 ; Y is CH2 ; R3 is aryl or aralkyl ; R4 is H,
alkyloxy or
aralkyloxy (preferred H, methoxy or benzyloxy) and R5 is F, Cl, alkyl,
heteroalkyl, aryl,
aralkyl, heteroaryl or heteroarylalkyl (preferred Cl, methoxy or benzyloxy;
more
preferred benzyloxy).
Also preferred are compounds of Formula (II), wherein Het is a piperidyl
group; n
is 0,1 or 2; X is CH2 ; Y is CO or SOZ ; R3 is aryl or aralkyl ; R4 is H,
alkyloxy or
aralkyloxy (more preferred H, methoxy or benzyloxy) and R5 is F, Cl, alkyl,
heteroalkyl,
aryl, aralkyl, heteroaryl or heteroarylalkyl (preferred Cl, methoxy or
benzyloxy; more
preferred benzyloxy).
Further preferred are compounds of Formula (II) wherein Het is a piperidyl
group;
n is 0, 1 or 2; X is CO or SO2 ; Y is CH2 ; R3 is aryl or aralkyl ; R4 is H,
alkyloxy or
aralkyloxy (preferred H, methoxy or benzyloxy) and R5 is F, Cl, alkyl,
heteroalkyl, aryl,
aralkyl, heteroaryl or heteroarylalkyl (preferred Cl, methoxy or benzyloxy;
more
preferred benzyloxy).
Further preferred are compounds of Formulas (I) or (II) wherein R3 is a group
of
the Formula (CH2)mPh wherein m is 0,1, 2,3, 4 or 5 (more preferred m is 2,3 or
4) and
wherein the phenyl group may be optionally substituted.
The compounds of Formula (I) or (II) do not include N-(4-Benzyloxy-3-methoxy-
benzyl)-N-(2-pyridin-2-yl-ethyl)-2-chloro-benzamide.
Especially preferred are the following compounds:
- 1V-(4-Benzyloxy-3-methoxy-benzyl)-N-(2-pyridin-4-yl-ethyl)-
benzenesulfonamide,
- 1V-(2-Chloro-benzyl)-N-(2-pyridin-2-yl-ethyl)-(4-Benzyloxy-3-methoxy)-
benzamide,
- 1V-(4-Benzyloxy-3-methoxy-benzyl)-N-(2-piperidin-2-yl-ethyl)-4-phenyl-
butyric
acid amide,
12
CA 02596799 2007-08-01
WO 2006/084116 PCT/US2006/003817
- N-(4-Benzyloxy-3-methoxy-benzyl)-N-(2-pyridin-2-yl-ethyl)-5-phenyl-pentanoic
acid amide.
It should be appreciated that certain compounds of Formula (I) or (II) may
have
tautomeric forms from which only one might be specifically mentioned or
depicted in the
following description, different geometrical isomers (which are usually
denoted as
cis/trans isomers or more generally as (E) and (Z) isomers) or different
optical isomers as
a result of one or more asymmetric or chiral carbon atoms (which are usually
nomenclatured under the Cahn-Ingold-Prelog or R/S system). Further, some
compounds
may display polymorphism. Unless expressly excluded, all these tautomeric
forms,
geometrical or optical isomers (as well as racemates and diastereomers) and
polymorphous forms are included in the invention.
The present invention also relates to pharmacologically acceptable salts, or
solvates and hydrates, respectively, and to compositions and formulations of
compounds
of Formula (1) or (II). The pharmaceutical compositions according to the
present
invention contain at least one compound of Formula (I) or (II) as the active
agent and
optionally carriers and/or diluents and/or adjuvants. Examples of such
pharmacologically
acceptable salts of sufficiently basic compounds of Formula (I) or (II) are
salts of
physiologically acceptable mineral acids like hydrochloric, hydrobromic,
sulfuric and
phosphoric acid; or salts of organic acids like methanesulfonic, p-
toluenesulfonic, lactic,
acetic, trifluoroacetic, citric, succinic, fumaric, maleinic and salicylic
acid. Further, a
sufficiently acid compound of Formula (I) or (II) may forin alkali or earth
alkaline metal
salts, for example sodium, potassium, lithium, calcium or magnesium salts;
ammonium
salts; or organic base salts, for example methylamine, dimethylamine,
trimethylamine,
triethylamine, ethylenediamine, ethanolamine, choline hydroxide, N-methyl-D-
aminomethane (meglumin), piperidine, morpholine, tris-(2-hydroxyethyl) amine,
lysine
or arginine salts. Compounds of Formula (I) or (II) may be solvated,
especially hydrated.
The hydration can occur during the process of production or as a consequence
of the
hygroscopic nature of the initially water free compounds of Formula (1) or
(II). The
compounds of Formula (I) or (II) contain asymmetric C-atoms and may be present
either
as achiral compounds, mixtures of diastereomers, mixtures of enantiomers or as
optically
pure compounds.
13
CA 02596799 2007-08-01
WO 2006/084116 PCT/US2006/003817
The present invention also relates to pro-drugs which are composed of a
compound of Formula (I) or (II) and at least one pharmacologically acceptable
protective
group which will be cleaved off under physiological conditions, such as an
allcoxy-,
aralkyloxy-, acyl- or acyloxy group as defined herein, e. g. ethoxy,
benzyloxy, acetyl or
acetyloxy.
As mentioned above, therapeutically useful agents that contain compounds of
Formula (I) or (II), their solvates, salts and formulations are also comprised
in the scope
of the present invention. In general, compounds of Formula (I) or (II) will be
administered by using the lcnown and acceptable modes lcnown in the art,
either alone or
in combination with any other therapeutic agent. Such therapeutically useful
agents can
be administered by one of the following routes: oral, e. g. as tablets,
dragees, coated
tablets, pills, semisolids, soft or hard capsules, for example soft and hard
gelatine
capsules, aqueous or oily solutions, emulsions, suspensions or syrups,
parenteral
including intravenous, intramuscular and subcutaneous injection, e. g. as an
injectable
solution or suspension, rectal as suppositories, by inhalation or
insufflation, e. g. as a
powder formulation, as microcrystals or as a spray (e. g. liquid aerosol),
transdermal, for
example via an transdermal delivery system (TDS) such as a plaster containing
the active
ingredient or intranasal. For the production of such tablets, pills,
semisolids, coated
tablets, dragees and hard, e. g. gelatine, capsules the therapeutically useful
product may
be mixed with pharmaceutically inert, inorganic or organic excipients e. g.,
lactose,
sucrose, glucose, gelatin, malt, silica gel, starch or derivatives thereof,
talc, stearinic acid
or their salts, dried skim milk, and the like. For the production of soft
capsules one may
use excipients e. g., vegetable, petroleum, animal or synthetic oils, wax,
fat, polyols. For
the production of liquid solutions, emulsions or suspensions or syrups one may
use
excipients e. g., water, alcohols, aqueous saline, aqueous dextrose, polyols,
glycerin,
vegetable, petroleum, animal or synthetic oils. For suppositories one may use
excipients
e.g., vegetable, petroleum, animal or synthetic oils, wax, fat and polyols.
For aerosol
formula tions one may use compressed gases suitable for this purpose, e.g.,
oxygen,
nitrogen and carbon dioxide. The phaimaceutically useful agents may also
contain
additives for conservation, stabilisation, e.g., UV stabilizers, emulsifiers,
sweetener,
14
CA 02596799 2007-08-01
WO 2006/084116 PCT/US2006/003817
aromatisers, salts to change the osmotic pressure, buffers, coating additives
and
antioxidants.
Combinations with other therapeutic agents may include other therapeutically
useful agents, e.g., those used to prevent or treat cancer (see Table I.).
A detailed description of methods used for the synthesis of compounds of
Foimula (I) and (II), above, is provided in PCT/EP02/12222, published May 8,
2003
(WO 03/037865 Al), the entire disclosure of which is incorporated by reference
herein.
The following examples are provided to illustrate various embodiments of the
invention. They are not intended to limit the invention in any way.
EXAMPLE I
The following materials and methods are provided to facilitate the practice of
Example I.
Cell lines. NIH3T3 cells stably transfected with K-Ras(G12V), N-terminally
truncated,
constitutively active Raf-1 (Raf22w) (13), (kindly provided by Drs. Channing
Der and
Janiel M. Shields), or KSHV-GPCR (7), EC-vGPCR endothelial cells (80) (kindly
provided by Dr. Silvio Gutlcind) were maintained in Dulbecco's modified
Eagle's
medium (DMEM) supplemented with 10% (vlv) calf serum and antibiotics (100
units/ml
penicillin, 100 ,ug/mi streptomycin, and 2.5 ,ug/ml puromycin or 500 g/ml G-
418) in 5%
CO2 at 37 C. HeLa cells stably transfected with a yeast Ga14 binding site-
luciferase
transcriptional reporter gene and GAL4-DNA binding domain fusions with Ellcl
(HLR-
Elkl, Stratagene) were maintained according to supplier recommendations.
Compounds. The amine derivatives N-(4-Benzyloxy-3-methoxybenzyl)-N-(2-pyridin-
2-
ylethyl)-2-chlorobenzamide] (MCP1) and N-(4-Benzyloxy-3-methoxybenzyl)-N-(2-
pyridin-2-ylethyl)-5-phenylpentanoic acid amide (MCP110) were synthesized by a
protocol described in (14). Bay43-9006 N-(4-chloro-3-(trifluoromethyl)phenyl)-
N'-(4-(2-
CA 02596799 2007-08-01
WO 2006/084116 PCT/US2006/003817
(N-methylcarbamoyl))-4-pyridyloxy)phenyl)urea and U0126 1,4-Diamino-2,3-
dicyano-
1,4-bis(2-aminophenylthio)-butadiene were purchased from Calbiochem and
Promega,
respectively. Taxol and staurosporine were obtained from Biomol.
Proliferation assays. 5000 cells were plated in each well of 96-well flat
bottom plates,
and incubated overnight at 37 C in 5% CO2. The growth of plated cells was
determined
by adding 7.5 l of a prepared WST-1 reagent (Roche Applied Sciences, Germany)
to 3
control wells and measuring ODG50 and OD450 absorbances with a SpectraMax250
plate
reader. If the OD650-OD450 values were above 0.5, the remainder of the plate
was used for
incubation with the above mentioned amine derivatives, other pharmacological
agents or
solvent control for 48 hours. After this incubation, WST-1 reagent was added
to the wells
and OD65o-OD450 values were calculated as before. Triplicate wells were
assayed for each
conditions and standard deviation was determined: all experiments were
performed
independently at least three times.
Anchorage-independent growth assays. Anchorage-independent growth assays were
performed as described in (15) witll minor modifications. Cells were
resuspended in
DMEM media supplemented with 10% calf serum and antibiotics, and mixed with
0.5%
agar to a final concentration of 6,000 to 25,000 cells/ml in 0.33% agar. 0.3
ml of this cell
suspension was layered over a 0.5% agar base in 24 well plates and incubated
for 2 to 4
hours at 37 C. After solidification of the top agar layer, compounds in DMSO,
or DMSO
diluted to 1% final concentration, were added, followed by incubation for 12 -
21 days.
To visualize colonies, 40 l of MTT (5mg/ml thiazolyl blue tetrazolium
bromide) and 60
,ul of PBS was added to each well, followed by overnight incubation under
normal culture
conditions. Stained colonies with sizes above 600 m were scored using a
Nilcon
SMZ1500 dissecting microscope coupled with a Roper Scientific Inc. Cool Snap
color
cooled charge coupled device (CCD) camera driven by Image Pro-Plus software
(Media
Cybernetics; Silver Spring, MD). For each compound, the survival curve was
based on a
minimum of six concentration points. Each concentration was assayed by at
least three
16
CA 02596799 2007-08-01
WO 2006/084116 PCT/US2006/003817
separate experiments, with each assay performed in triRlicate. IC50 values
were
determined by using XLfit software from Activity Base suite package (ID
Business
Solutions, Inc., NJ, USA).
Elk1-Luciferase transcription assay. HLR-Ellcl cells were plated (30,000 cells
per
well) in a 96 well plate. After 20 hours medium was replaced with 90 1 per
well of
starvation medium containing no serum. After 72 hours, compound or DMSO
solvent
was added. After one more hour, the cells were induced with 10 ng/ml EGF. As a
negative control (minimal reporter assay), reference cells were not induced
with EGF.
After an additional five hours of incubation, the cells were lysed and
luciferase activity
was measured by using the Bright-Glo Luciferase Assay lcit (Promega Co.,
Madison,
WI).
Cell cycle analysis. Cell cycle compartmentalization was examined by flow
cytometry.
Cells were trypsinized, collected by centrifugation, washed twice and
resuspended in
sterile PBS, and fixed with 70% ethanol at -20 C for 2-12 hours. Cells were
again
centrifuged, washed twice with cold PBS and resuspended in 100 .l of PBS.
Cell
suspensions were mixed with 0.5 ml of 30 mM sodium citrate, 20 g/ml propidium
iodide, and 1 l of 10 mg/ml DNase-free RNase A. The cells were incubated for
1 hour
at 37 C in the dark, then DNA content from 10,000 cells was determined using
the
Becton Dickinson FACScan, equipped with argon-ion laser tuned to 488 nm wave
length,
and CellQuest program (Immunochemistry Systems, San Jose, CA). . Results were
expressed as a histogram representing the relative cell number with a given
DNA content.
Apoptosis assays. To estimate apoptosis of stably transfected cell lines
treated with the
above mentioned amine derivatives and/or taxol, 500,000 cells were seeded in
six-well
plates. Cells were treated for 72h with various concentrations of the amine
derivatives
and/or taxol and collected for flow cytometry or used to prepare lysates.
Cells were
collected by trypsinization and, labeled with annexin V-FITC and propidium
iodide
according to the manufacturer's recommendations (Clontech) for fluorescence-
activated
cell sorting analysis (Becton Dickinson).
17
CA 02596799 2007-08-01
WO 2006/084116 PCT/US2006/003817
RESULTS AND DISCUSSION
MCPl and MCP110 strongly inhibit proliferation and soft-agar colony formation
in
I6HV-GPCR-transformed NIH3T3 fibroblasts. MCP1 and MCP110 were tested for
their ability to inhibit anchorage dependent and independent growth of the
NIH3T3
murine fibroblast cell line stably transfoimed with the KSHV-GPCR. Both MCPl
and
MCP110 efficiently inhibited growth of NIH3T3-KSHV-GPCR cells in a WST-1 assay
of cell proliferation,'with IC50 of 24 and 28 M, respectively (Figure 1). The
MCP1 and
MCP1 10 compounds also effectively inhibited soft-agar colony formation by the
same
cells, with IC50 values of 16 and 14 M, respectively (Figure 2). To further
test the idea
that KSHV-GPCR relays signals through Ras/Raf/MEK/ERK signaling module, we
also
evaluated whether other relevant pathway inhibitors, including the Raf kinase
catalytic
inhibitor BAY43-9006, and the Mek kinase inhibitor U0126, would also be able
to inhibit
the proliferation of NIH3T3-KSHV-GPCR cells. Both BAY43-9006 and U0126 also
inhibited growth of NIH3T3-KSHV-GPCR cells, with IC50 values comparable to the
MCP compounds (Figures 1, 2).
Combination effect of MCP110 with BAY43-9006 and U0126. We have shown that
MCP1 and MCP110 reduce signaling through the ERK signal transduction pathway
by
modulating Ras and Raf interaction, but do not result in 100% inhibition of
ERK
signaling, as judged by phosphorylation of endogenous ERK, or transcriptional
activation
of ERK-dependent promoters such as ELK1(16). We considered the possibility
that
combination of two different inhibitors of this Ras-dependent signaling axis
might have
an additive or synergistic effect in inhibiting ERK and on cell growth. As a
first step, we
evaluated the degree to which MCP compounds in combination with BAY43-9006 Raf
kinase and U0126 MEK kinase inhibitors would inhibit the soft agar growth and
proliferation of a NIH3T3 model cell line transformed with the constitutively
activated
K-Ras(G12V) oncogene. A potent synergistic effect was observed following the
combination of MCP110 with each of these inhibitors in soft-agar colony
formation assay
(Figure 3A). These results were well supported by the observation that the
combination
18
CA 02596799 2007-08-01
WO 2006/084116 PCT/US2006/003817
effect was also determined between MCP110 and BAY43-9006 and to a lesser
extent
with U0126 in an Elkl-luciferase reporter assay in HeLa cells (Figure 3B).
Since all these
compounds were shown to modulate signaling through MAPK signal transduction
pathway and thus regulate expression of the Elkl-luciferase reporter, the
possible
mechanism of synergistic potentiation in soft-agar assay could be explained by
more
complete inhibition of MAPK signaling. In contrast, combination of MCP110 with
staurosporine (a protein kinase C (PKC) inhibitor) did not result in
synergistic inhibition
of growth (Figure 3C), demonstrating that MCP110 is not promiscuously
cytotoxic in
combined application.
Combination effect of MCP110 with taxol: synergy in inhibition of anchorage-
independent growth. Taxol (paclitaxel) is widely used in the treatment of
lung, ovarian,
and breast carcinomas (17). Recently, the FDA approved taxol as a second line
treatment
of Kaposi's sarcoma in AIDS patients (18). Although a primary mode of action
of taxol is
as a cytotoxic agent that disrupts microtubule dynamics, and taxol does not
directly affect
signaling through the MAPK pathway, taxol has also been implicated as a
compound that
both modulates Raf-1 activation, and has differing activity dependent on Raf-1
status (19-
25), making it of interest to analyze the interaction of MCP compounds with
taxol. We
first established IC50 for inhibition of anchorage dependent or independent-
growth by
taxol in NIH3T3 cells transformed with K-Ras(V12), cRaf22W (constitutively
activated
Raf, independent of interaction with Ras), and KSHV-GPCR cells. In a WST-1
proliferation assay, the IC50 value of taxol was determined as 0.9 M in K-
Ras(V12)
transformed cells, 0.2 M in Raf22W transformed cells, and 0.75 M in KSHV-
GPCR
transformed cells (Figure 4A). In a soft agar colony formation assay, the IC50
for taxol
was established at 49.7, 8.7 and 63.3 nM, in K-Ras(V12), Raf22W and KSHV-GPCR,
respectively (Figure 4A). We then determined the taxol IC50 values in the
presence of
U0126, BAY43-9006, or MCP110 at concentrations of 1 or 10 M in a soft agar
colony
foimation assay performed with NIH3T3-K-Ras(V12) cells. Strikingly, in the
presence of
1 M MCP1 or MCPI10, the taxol IC50 was reduced from 49.7 nM to 10.7 nM; with
10
gM MCP1 or MCP110, the taxol IC50 values were reduced to 1-1.5 nM (Figure 4B).
This reduction in IC50 values was specific for the MCP compounds, as only a
minimal
19
CA 02596799 2007-08-01
WO 2006/084116 PCT/US2006/003817
effect was obtained following combination of U0126 or BAY43-9006 with taxol
(Figure
4B). Similar results were obtained between the MCP1 10 and taxol combinations
in
inhibiting the anchorage-dependent proliferation of NIH3T3-K-Ras(V12) cells,
although
BAY43-9006 has some efficacy in these cells (Figure 4C).
We next examined the effect of combining the above-mentioned amine
derivatives, BAY43-9006, and U0126 compounds in KSHV-GPCR transformed NIH3T3
cells. In this case, strong enhancement of inhibition of anchorage-independent
growth
was seen, similar to the results with Ras-transformed cells (Figure 4D).
Together, these
data suggest that the synergistic effect of the above-mentioned amine
derivatives with
taxol may be beneficial for the treatment of HEIV-8 related diseases, and the
mechanism
of such effect may be related to that observed in K-Ras(V12) transformed
cells.
MCP110 induces the G2/M arrest and apoptosis of taxol-treated NIH3T3-K-
Ras(V12) cells. Since a strong dose dependent combination effect was
established for the
above-mentioned amine derivatives and taxol in K-Ras(V12) and KSHV-GPCR, the
possible mechanism of the observed synergism was investigated. Previously, we
have
shown that MCP110 used at its IC50 concentration promotes cell cycle arrest in
Gl (16).
Dependent on the DNA damage and spindle checkpoint status of cells, taxol
causes
mitotic arrest leading to cell death (25, 26).
We examined the cell cycle compartmentalization of K-Ras(V12) and cRaf22W
transformed NIH3T3 cells treated with low concentrations of MCP1 10 or taxol,
alone or
in combination, as well as a DMSO control (Figure 5). We first established the
critical
concentration necessary for taxol-dependent arrest of each cell line in G2/M
(1 M in
each case). We next established that the cell cycle profiles of cells treated
with 1 M
MCP110 were identical to those of cells grown with a DMSO control. Similarly,
cells
treated with an IC5 dose of taxol (100 nM in K-Ras(V12) cells, 10 nM in
cRaf22W cells)
did not differ from controls. However, the combination of 1 M MCP1 10 with
IC5
concentrations of taxol resulted in a substantial shift of cells from the G1
to the G2/M
compartment in K-Ras(V12) cells (from 18-19% cells in G2/M to 86% G2/M), while
no
such effect was observed in cRaf22W cells (Figure 5). To determine whether
this cell
cycle shift was coupled with increased cellular apoptosis, we used annexin V
and
CA 02596799 2007-08-01
WO 2006/084116 PCT/US2006/003817
propidium iodide co-staining in FACS analysis to identify apoptotic or
necrotic cells. As
shown (Figure 6), the combination of 1 M MCP110 and taxol at IC5 doses led to
a
significant increase in the level of annexin V-positive cells in K-Ras(V12),
but not
cRaf22W-transformed cells. Moreover, in control experiments, U0126 at
concentrations
up to 20 M had no effect on induction of apoptosis or cell cycle arrest in
either of the
cell lines tested (results not shown), arguing that the pro-apoptotic effect
was induced at a
level upstream of the Mek1,2 kinases in response to treatment of cells with
taxol and
MCP110.
The taxol combination results, together with the demonstrated ability of amine
derivatives described herein, to inhibit proliferation and anchorage-
independent growth
properties of KSHV-GPCR transformed NIH3T3 cells, provide logical support for
the
use of the MCP compound class as anti-KSHV agents. In addition, the results
suggest
that the combination of the above-mentioned amine derivatives (or Raf-
catalytic
inhibitors such as BAY43-9006) with taxol, or combination of MCP compounds
with
BAY43-9006, may be a particularly potent means of inhibiting KSHV- or Ras-
transformed cells.
Indeed, the following Table I lists a variety of anti-proliferative and
cytotoxic
agents that, when used in combination with the amine derivatives described
herein,
should have therapeutic efficacy for the treatment of viral infection.
TABLEI
Compound Target Phenotype/Assay Reference Source
FTI277 Farnesyl transferase Proliferation (WST-1) (27) Calbiochem
p-MAPK1,21eve1
Bay43-9006 Raf-1, Proliferation (28) Calbiochem
B-Raf kinases p-MAPK1,21eve1
U0126 Mekl,2 kinases Proflferation (29) Promega
p-MAPK1,21eve1
CI-1040 Mekl,2 kinases Proliferation (30) Pfizer
p-MAPKI,2 level
AA-COCF3 cPLA2 Proliferation (WST-1) (31) Biomol
p-MAPK1,21eve1
Bryostatin PKC Proliferation (WST-1) (32,33) Biomol
p-MAPK1,21eve1
UCN-01 PKC, Chkl Proliferation (WST-1) (34) NCI
p-MAPK1,21eve1
Iressa EGFR Proliferation (WST-1) (35) FCCC
p-MAPK1,2 level
21
CA 02596799 2007-08-01
WO 2006/084116 PCT/US2006/003817
Glivec BCR/ABL, Proliferation (WST-1) (36) Novartis
c-Kit, PDGFR
LY294002 PY3K Proliferation (WST-1) (37) Cell Signaling
p-AICT1,2 level
Herceptin Her-2 Proliferation (WST-1) (38) FCCC
Sirolimus mTor Proliferation (WST-1) (39) FCCC
(CCI-779)
SP600125 JNK Proliferation (WST-1 (40) Tocris Cookson
p-c-JUN level Bristol, UK
Gemcitabine DNA syntliesis Proliferation (WST-1) (38) Eli Lilly
Paclitaxel Anti-mitotic, Proliferation (WST-1) (41) Biomol
Tubulin polymerization
While the previous example is directed to inhibition of HI3V-8 infection, it
is
known that a number of viruses co-opt cellular signaling pathways to promote
their
replication, and in some cases may also induce tumorigenesis. A description of
some of
these viruses (e.g., HHV-8, HHV-4 and hepatitis B virus) follows.
1) As mentioned in the example above, Kaposi's sarcoma, caused by HHV-8, is a
multifocal angioproliferative neoplasm induced following long-term infection
with
Kaposi's sarcoma herpesvirus/human helpesvisrus 8(KSHV/HI-iV-8). Development
of
this neoplasm strictly depends upon the availability of multiple angiogenic
growth factors
and cytokines, which act in combination from virally encoded oncogenic signals
provided
by such proteins as the KSHV-encoded viral G-protein coupled receptor (vGPCR).
As
shown in Figure 7, vGPCR induction of transformation of KSHV-infected cells
involves
direct and indirect autocrine/paracrine mechanisms, which requires enhanced
expression
and secretion of number of angiogenic factors and cytolcines. These factors
include
VEGF, IL-8, IL-6, Gro a(42-44), and potentiate vGPCR signaling by enhancing
vGPCR
direct transformation effect in autocrine fashion (45,46). Recently, the KSHV-
vGPCR
was implicated in immortalization of human endothelial HUVEC cells via
activation of
their VEGF receptor-2/KDR protein (47). Finally, vGPCR induces expression of
the
cytokines and growth factors by activation of key transcription factors,
including AP-1,
NF-xB and NF-AT (48,7), through activation of p21-activated kinase-1 (Palcl)
that
forwards the signaling on Raf-1 and IKK lcinases (7). Since MCP compounds
regulate
22
CA 02596799 2007-08-01
WO 2006/084116 PCT/US2006/003817
activation of AP-1 and NF-icB transcription factors induced by oncogenic Ras
growth
factors and TNF (50), and because Raf and Ras signaling has been shown to be
relevant
to signal transduction induced by cellular GPCRs (51,52), it is reasonable to
believe that
interruption of the Ras-Raf interaction will interrupt KSHV-vGPCR-dependent
functions.
The prelizninary data provided herein support this proposed mechanism for KSHV-
vGPCR-dependent cell transformation.
HHV-4/Epstein-Barr virus (EBV) belongs to the same group of gammaherpes
viruses as KSHV. Epstein Barr virus is associated with a number of
malignancies of
lymphoid and epithelial origin, including endemic Burlcitt's lymphoma, T-cell
lymphomas, Hodgkin's disease, undifferentiated nasopharyngeal carcinoma and
several
other carcinomas (53-56). Like KSHV, the EBV genome is frequently found in the
lymphomas and lymphoproliferative disorders of immunocompromised transplant
patients and individuals with AIDS (57,58). LMP1, an integral membrane protein
expressed by EBV during type 2 and type 3 latent infections, is the only EBV
protein that
produces a classic oncogenic effect in Rat-1 and NIH3T3 cells, and in B cells
(56, 59).
An animal model for LMP1 exists, as LMP1 transgenic mice develop lymphomas at
increased frequency (60). Significantly, LMP1 produces its effect through
activation of
two major signaling cascades, the NF-icB pathway and the Ras-MAPK signaling
pathway
(61-63) (see Figure 7). The induction of these signaling cascades occurs
through
interaction of cytoplasmic domain of LMP1 with TRAF and TRADD proteins (64,65)
which subsequently signal through the NIK and IKK kinases towards I7cB
repressor (66).
Significantly, the Raf ldnase inhibitor protein (RKIP) has been found to also
function as a
negative regulator of the NFicB pathway (67), and may provide a physical
bridge between
the two complexes: in any case, this and other data from non-viral systems
(68) indicate
that the MAPK and NFxB signaling cascades are engaged in significant cross-
talk that
may be important in specifying the course of EBV infection.
Hepatitis B(HBV) virus is small hepatotropic pararetrovirus that causes
persistent
liver infection and cirrhosis, and is strongly associated with development of
primary liver
cancer (hepatocellular carcinoma, HCC) (69). HCC is one of the most prevalent
forms of
human cancer worldwide, with extremely limited treatment options (70). In this
regard,
many studies have focused on identification of potential viral oncogenes. HBV
encodes
23
CA 02596799 2007-08-01
WO 2006/084116 PCT/US2006/003817
two transcriptional activators, HBx (71) and the pre-S2/S region of LHB/NII3B
proteins
(72), which activate cellular targets and promote the transformation of
infected cells(73).
See Figure 9. The HBx protein of HBV is essential for infection and is also
thought to be
an essential cofactor in HCC development. The mechanism mediating HBx-
dependent
activator function includes the Ras-dependent activation of c-Raf-1/MEK/Erk2,
PI3K/Akt and MEKKl/JNK cascades, causing induction of several major
transcription
factors, including AP-1 and NF-xB (74, 75). The HBx protein activates Ras-Raf-
MAPK
signaling pathway, most likely through activation of the Pyk2 (76), c-Src and
Fyn kinases
(77). Moreover, the activity and cellular localization of HBx protein are
tightly regulated
through phosphorylation by ERKI/2 lcinase, causing the HBx protein to shuttle
to the
nucleus, where it induces the transcription of genes critical for HBV
replication and cell
transformation. Thus, the phosphorylation of HBx protein creates a possible
feedback
regulatory circuit between HBX and MAPK signaling pathway (78), indicating
that the
MAPK pathway may be a suitable target for developing novel anti-HBV
therapeutic
agents.
Combination effect of MCP11O with taxol: synergy in inhibition of anchorage-
dependent growth in EC-vGPCR endothelial cells.
As KSHV-induced cancers are marked by action of the vGPCR in promotion of
vascular
endothelial growth factor (VEGF)-driven angiogenesis, this endothelial model
has a
higher physiological relevance than a fibroblast model. While EC-vGPCR cells
do not
form colonies in soft-agar, MCP110 was as effective in these cells as it was
in NIH3T3-
KSHV-GPCR cells in inhibiting proliferation. The proliferation was measured by
WST-1
assay with IC50 value of 22 M. Similar to NIH-3T3-KSHV-GPCR cells, MCP110
produced a strong synergistic effect with taxol in the endothelial cells. The
IC50 of taxol
in EC-vGPCR cells was markedly reduced by addition of 1-10 M MCP110.
Moreover,
similar synergies were observed in combination of the BAY43-9006 Raf lcinase
inhibitor
with taxol, and to a lesser degree with the MEKl kinase inhibitor U0126
(Figure 4E).
This observation is compatible with reports in the scientific literature
indicating Raf
activity is required for pro-survival signals involving Akt and NF-kappaB (79-
81), and
24
CA 02596799 2007-08-01
WO 2006/084116 PCT/US2006/003817
indicating that KSHV transformation is associated with and requires activation
of Akt
signaling (82).
REFERENCES
1. Ablashi, D. V., Chatlynne, L. G., Whitman, J. E., Jr., and Cesarman, E.
Spectrum
of Kaposi's sarcoma-associated herpesvirus, or human herpesvirus 8, diseases.
Clin Microbiol Rev, 15: 439-464, 2002.
2. Gallo, R. C. The enigmas of Kaposi's sarcoma. Science, 282: 1837-1839,
1998.
3. Chang, Y. C., Ethel; Pessin, Melissa S.; Lee, Frank; Culpepper, Janice;
Knowles,
Daniel M.; Moore, Patrick S. Identification of Herpesvirus-Like DNA Sequences
in AIDS-Associated Kaposi's Sarcoma. Science, 266: 1865-1869, 1994.
4. Arvanitalcis, L., Geras-Raaka, E., Varma, A., Gershengorn, M. C., and
Cesarman,
E. Human herpesvirus KSHV encodes a constitutively active G-protein-coupled
receptor linked to cell proliferation. Nature, 385: 347-350, 1997.
5. Montaner, S., Sodhi, A., Molinolo, A., Bugge, T. H., Sawai, E. T., He, Y.,
Li, Y.,
Ray, P. E., and Gutlcind, J. S. Endothelial infection with KSHV genes in vivo
reveals that vGPCR initiates Kaposi's sarcomagenesis and can promote the
tumorigenic potential of viral latent genes. Cancer Cell, 3: 23-36, 2003.
6. Jenner, R. G. and Boshoff, C. The molecular pathology of Kaposi's sarcoma-
associated herpesvirus. Biochim Biophys Acta, 1602: 1-22, 2002.
7. Dadlee, D., Fryer, B. H., Golemis, E. A., and Field, J. Activation of p21-
activated
kinase 1-nuclear factor kappaB signaling by Kaposi's sarcoma-associated herpes
virus G protein-coupled receptor during cellular transformation. Cancer Res,
63:
8837-8847, 2003.
8. Cannon, M., Philpott, N. J., and Cesarman, E. The Kaposi's sarcoma-
associated
herpesvirus G protein-coupled receptor has broad signaling effects in primary
effusion lymphoma cells. J Virol, 77: 57-67, 2003.
9. Robinson, M. J. and Cobb, M. H. Mitogen-activated protein kinase pathways.
Curr Opin Cell Biol, 9: 180-186, 1997.
10. van Corven, E. J., Hordijk, P. L., Medema, R. H., Bos, J. L., and
Moolenaar, W.
H. Pertussis toxin-sensitive activation of p2lras by G protein-coupled
receptor
agonists in fibroblasts. Proc Natl Acad Sci U S A, 90: 1257-1261, 1993.
11. Yart, A., Roche, S., Wetzker, R., Laffargue, M., Tonks, N., Mayeux, P.,
Chap, H.,
and Raynal, P. A function for phosphoinositide 3-kinase beta lipid products in
coupling beta gamma to Ras activation in response to lysophosphatidic acid. J
Biol Chem, 277: 21167-21178, 2002.
CA 02596799 2007-08-01
WO 2006/084116 PCT/US2006/003817
12. Kranenburg, O. and Moolenaar, W. H. Ras-MAP kinase signaling by
lysophosphatidic acid and other G protein-coupled receptor agonists. Oncogene,
20: 1540-1546, 2001.
13. Stanton, V. P., Jr., Nichols, D. W., Laudano, A. P., and Cooper, G. M.
Definition
of the human raf amino-terminal regulatory region by deletion mutagenesis. Mol
Cell Biol, 9: 639-647, 1989.
14. Lu, Y.; Sakamuri, S.; Chen, Q.-Z.; Keng, Y.-F.; Khazak, V.; Illgen, K.;
Schabbert, S.; Weber, L.; Menon, S. R. Solution phase parallel synthesis of
MAPK inhibitory activities of close structural analogues of a Ras pathway
modulator. Bioorg. Med. Chem. Lett., 14: 3957-3962, 2004.
15. Cox, A. D. and Der, C. J. Biological assays for cellular transformation.
Methods
Enzymol, 238: 277-294, 1994.
16. Kato-Stanlciewicz, J., Haldmi, I., Zhi, G., Zhang, J., Serebriiskii, I.,
Guo, L.,
Edamatsu, H., Koide, H., Menon, S., Eckl, R., Salcamuri, S., Lu, Y., Chen, Q.
Z.,
Agarwal, S., Baumbach, W. R., Golemis, E. A., Tamanoi, F., and Khazak, V.
Inhibitors of Ras/Raf-1 interaction identified by two-hybrid screening revert
Ras-
dependent transformation phenotypes in human cancer cells. Proc Natl Acad Sci
U S A, Vol. 99, pp. 14398-14403, 2002.
17. Mekhail, T. M. and Marlcman, M. Paclitaxel in cancer therapy. Expert Opin
Pharmacother, 3: 755-766, 200218.
18. Tulpule, A., Groopman, J., Saville, M. W., Harrington, W., Jr., Friedman-
Kien,
A., Espina, B. M., Garces, C., Mantelle, L., Mettinger, K., Scadden, D. T.,
and
Gill, P. S. Multicenter trial of low-dose paclitaxel in patients with advanced
AIDS-related Kaposi sarcoma. Cancer, 95: 147-154, 2002.
19. Britten, R. A., Perdue, S., Opoku, J., and Craighead, P. Paclitaxel is
preferentially
cytotoxic to human cervical tumor cells with low Raf-1 lcinase activity:
implications for paclitaxel-based chemoradiation regimens. Radiother Oncol,
48:
329-334, 1998.
20. Britten, R. A., Perdue, S., Eshpeter, A., and Merriam, D. Raf-1 kinase
activity
predicts for paclitaxel resistance in TP53mut, but not TP53wt human ovarian
cancer cells. Oncol Rep, 7: 821-825, 2000.
21. Rasouli-Nia, A., Liu, D., Perdue, S., and Britten, R. A. High Raf-1
lcinase activity
protects human tumor cells against paclitaxel-induced cytotoxicity. Clin
Cancer
Res, 4: 1111-1116, 1998.
22. Lee, M., Koh, W. S., and Han, S. S. Down-regulation of Raf-1 lcinase is
associated with paclitaxel resistance in human breast cancer MCF-7/Adr cells.
Cancer Lett, 193: 57-64, 2003.
23. Blagoslclonny, M. V., Schulte, T., Nguyen, P., Trepel, J., and Neckers, L.
M.
Taxol-induced apoptosis and phosphorylation of Bcl-2 protein involves c-Raf-1
and represents a novel c-Raf-1 signal transduction pathway. Cancer Res, 56:
1851-1854, 1996.
24. Blagoslclonny, M. V., Giannakakou, P., el-Deiry, W. S., Kingston, D. G.,
Higgs,
P. I., Neckers, L., and Fojo, T. Raf-1/bcl-2 phosphorylation: a step from
microtubule damage to cell death. Cancer Res, 57: 130-135, 1997.
26
CA 02596799 2007-08-01
WO 2006/084116 PCT/US2006/003817
25. Blagosklonny, M. V. Sequential activation and inactivation of G2
checkpoints for
selective killing of p53-deficient cells by microtubule-active drugs.
Oncogene,
Vol. 21, pp. 6249-6254, 2002.
26. Rowinsky, E. K. and Donehower, R. C. Paclitaxel (taxol). N Engl J Med,
332:
1004-1014, 1995.
27. Lemer, E. C., Qian, Y., Blaskovich, M. A., Fossum, R. D., Vogt, A., Sun,
J., Cox,
A. D., Der, C. J., Hamilton, A. D., and Sebti, S. M. Ras CAAX peptidomimetic
FTI-277 selectively blocks oncogenic Ras signaling by inducing cytoplasmic
accumulation of inactive Ras-Raf complexes. J Biol Chem, 270: 26802-26806,
1995.
28. Lyons Jf Fau - Wilhelm, S., Wilhelm S Fau - Hibner, B., Hibner B Fau -
Bollag,
G., and Bollag, G. Discovery of a novel Raf lcinase inhibitor. Endocr Relat
Cancer, 8: 219-225., 2001.
29. Mirza, A. M., Gysin, S., Malek, N., Nakayama, K., Roberts, J. M., and
McMahon,
M. Cooperative regulation of the cell division cycle by the protein lcinases
RAF
and AKT. Mol Cell Biol, 24: 10868-10881, 2004.
30. Allen, L. F., Sebolt-Leopold, J., Meyer, M. B. CI-1040 (PD184352), a
targeted
signal transduction inhibitor of MBK (MAPKK). Semin Oncol, 30: 105-116,
2003.
31. Bhalla, U. S., Ram, P. T., and Iyengar, R. MAP kinase phosphatase as a
locus of
flexibility in a mitogen-activated protein kinase signaling network. Science,
297:
1018-1023, 2002.
32. Hofmann, J. Modulation of protein kinase C in antitumor treatment. Rev
Physiol
Biochem Pharmacol,142: 1-96, 2001.
33. Wang, X., Wang, Q., Hu, W., and Evers, B. M. Regulation of phorbol ester-
mediated TRAF1 induction in human colon cancer cells through a
PKC/RAFlERK/NF-kappaB-dependent pathway. Oncogene, 23: 1885-1895,
2004.
34. Dai, Y., Yu, C., Singh, V., Tang, L., Wang, Z., Mclnistry, R., Dent, P.,
Grant, S.
Pharmacological inhibitors of the mitogen-activated protein kinase (MAPK)
kinase/MAPK cascade interact synergistically with UCN-01 to induce
mitochondrial dysfunction and apoptosis in human leukemia cells. Cancer Res.,
61:5106-15, 2001.
35. Herbst, R. S., Fukuoka, M., and Baselga, J. Gefitinib--a novel targeted
approach
to treating cancer. Nat Rev Cancer, 4: 956-965, 2004.
36. Attoub S Fau - Rivat, C., Rivat C Fau - Rodrigues, S., Rodrigues S Fau -
Van
Bocxlaer, S., Van Bocxlaer S Fau - Bedin, M., Bedin M Fau - Bruyneel, E.,
Bruyneel E Fau - Louvet, C., Louvet C Fau - Kornprobst, M., Kornprobst M Fau -
Andre, T., Andre T Fau - Mareel, M., Mareel M Fau - Mester, J., Mester J Fau -
Gespach, C., and Gespach, C. The c-kit tyrosine kinase inhibitor ST1571 for
colorectal cancer therapy. Cancer Res, 62: 4879-4883., 2002.
37. Murphy, G. A., Graham, S. M., Morita, S., Reks, S. E., Rogers-Graham, K.,
Vojtek, A., Kelley, G. G., and Der, C. J. Involvement of phosphatidylinositol3-
kinase, but not Ra1GDS, in TC21/R-Ras2-mediated transformation. J Biol Chem,
277: 9966-9975, 2002.
27
CA 02596799 2007-08-01
WO 2006/084116 PCT/US2006/003817
38. Safran, H., Iannitti, D., Ramanathan, R., Schwartz, J. D., Steinhoff, M.,
Nauman,
C., Hesketh, P., Rathore, R., Wolff, R., Tantravahi, U., Hughes, T. M., Maia,
C.,
Pasquariello, T., Goldstein, L., King, T., Tsai, J. Y., and Kennedy, T.
Herceptin
and gemcitabine for metastatic pancreatic cancers that overexpress HER-2/neu.
Cancer Invest, 22: 706-712, 2004.
39. Boni, J. P., Leister, C., Bender, G., Fitzpatrick, V., Twine, N., Stover,
J., Dorner,
A., Immermann, F., and Burczynslci, M. E. Population pharmacokinetics of CCI-
779: coirelations to safety and pharmacogenomic responses in patients with
advanced renal cancer. Clin Pharmacol Ther, 77: 76-89, 2005.
40. Pruitt, K., Pruitt, W. M., Bilter, G. K., Westwick, J. K., and Der, C. J.
Raf-
independent Deregulation of p38 and JNK Mitogen-activated Protein Kinases Are
Critical for Ras Transformation. J Biol Chem, 277: 31808-31817, 2002.
41. Safran, H., DiPetrillo, T., Nadeem, A., Steinhoff, M., Tantravahi, U.,
Rathore, R.,
Wanebo, H., Hughes, M., Maia, C., Tsai, J. Y., Pasquariello, T., Pepperell, J.
R.,
Cioffi, W., Kennedy, T., Reeder, L., Ng, T., Adrian, A., Goldstein, L., Chak,
B.,
and Choy, H. Trastuzumab, paclitaxel, cisplatin, and radiation for
adenocarcinoma of the esophagus: a phase I study. Cancer Invest, 22: 670-677,
2004.
42. Polson, A. G., Wang, D., DeRisi, J., and Ganem, D. Modulation of host gene
expression by the constitutively active G protein-coupled receptor of Kaposi's
sarcoma-associated herpesvirus. Cancer Res, 62: 4525-4530, 2002.
43. Schwarz, M. and Murphy, P. M. Kaposi's sarcoma-associated herpesvirus G
protein-coupled receptor constitutively activates NF-kappa B and induces
proinflammatory cytokine and chemokine production via a C-terminal signaling
determinant. J Immuno1,167: 505-513, 2001.
44. Shepard, L. W., Yang, M., Xie, P., Browning, D. D., Voyno-Yasenetskaya,
T.,
Kozasa, T., and Ye, R. D. Constitutive activation of NF-kappa B and secretion
of
interleulcin-8 induced by the G protein-coupled receptor of Kaposi's sarcoma-
associated herpesvirus involve G alpha(13) and RhoA. J Biol Chem, 276: 45979-
45987, 2001.
45. Gershengorn, M. C., Geras-Raaka, E., Varma, A., and Clark-Lewis, I.
Chemokines activate Kaposi's sarcoma-associated herpesvirus G protein-coupled
receptor in mammalian cells in culture. J Clin Invest, 102: 1469-1472, 1998.
46. Montaner, S., Sodhi, A., Pece, S., Mesri, E. A., and Gutkind, J. S. The
Kaposi's
sarcoma-associated herpesvirus G protein-coupled receptor promotes endothelial
cell survival through the activation of Alct/protein lcinase B. Cancer Res,
61:
2641-2648, 2001.
47. Bais, C., Van Geelen, A., Eroles, P., Mutlu, A., Chiozzini, C., Dias, S.,
Silverstein, R. L., Rafii, S., and Mesri, E. A. Kaposi's sarcoma associated
herpesvirus G protein-coupled receptor immortalizes human endothelial cells by
activation of the VEGF receptor-21 KDR. Cancer Cell, 3: 131-143, 2003.
48. Montaner, S., Sodhi, A., Servitja, J. M., Ramsdell, A. K., Barac, A.,
Sawai, E. T.,
and Gutkind, J. S. The small GTPase Rac1 links the Kaposi sarcoma-associated
herpesvirus vGPCR to cytolcine secretion and paracrine neoplasia. Blood, 104:
2903-2911, 2004.
28
CA 02596799 2007-08-01
WO 2006/084116 PCT/US2006/003817
49. Dadlce, D., Fryer, B. H., Golemis, E. A., and Field, J. Activation of p21-
activated
lcinase 1-nuclear factor kappaB signaling by Kaposi's sarcoma-associated
herpes
virus G protein-coupled receptor during cellular transformation. Cancer Res,
63:
8837-8847, 2003.
50. Kato-Stanlciewicz, J., Halcimi, I., Zhi, G., Zhang, J., Serebriislcii, I.,
Guo, L.,
Edamatsu, H., Koide, H., Menon, S., Eclcl, R., Salcamuri, S., Lu, Y., Chen, Q.
Z.,
Agarwal, S., Baumbach, W. R., Golemis, E. A., Tamanoi, F., and Khazak, V.
Inhibitors of Ras/Raf-1 interaction identified by two-hybrid screening revert
Ras-
dependent transformation phenotypes in human cancer cells. Proc Nati Acad Sci
U S A, 99: 14398-14403, 2002.
51. Yang, C. M., Lin, M. I., Hsieh, H. L., Sun, C. C., Ma, Y. H., and Hsiao,
L. D.
Bradylcinin-induced p42/p44 MAPK phosphorylation and cell proliferation via
Src, EGF receptors, and PI3-K/Akt in vascular smooth muscle cells. J Cell
Physiol, 2004.
52. Beom, S., Cheong, D., Torres, G., Caron, M. G., and Kim, K. M. Comparative
studies of molecular mechanisms of dopamine D2 and D3 receptors for the
activation of extracellular signal-regulated lcinase. J Biol Chem, 279: 28304-
28314, 2004.
53. Fahraeus, R., Fu, H. L., Ernberg, I., Finke, J., Rowe, M., Klein, G.,
Falk, K.,
Nilsson, E., Yadav, M., Busson, P., and et al. Expression of Epstein-Barr
virus-
encoded proteins in nasopharyngeal carcinoma. Int J Cancer, 42: 329-338, 1988.
54. Klein, G. Viral latency and transformation: the strategy of Epstein-Barr
virus.
Cell, 58: 5-8, 1989.
55. Pallesen, G., Hamilton-Dutoit, S. J., Rowe, M., and Young, L. S.
Expression of
Epstein-Barr virus latent gene products in tumour cells of Hodglcin's disease.
Lancet, 337: 320-322, 1991.
56. Su, I. J., Hsieh, H. C., Lin, K. H., Uen, W. C., Kao, C. L., Chen, C. J.,
Cheng, A.
L., Kadin, M. E., and Chen, J. Y. Aggressive peripheral T-cell lymphomas
containing Epstein-Barr viral DNA: a clinicopathologic and molecular analysis.
Blood, 77: 799-808, 1991.
57. Karp, J. E., and and Broders, S. Acquired immunodeficiency syndrom and non-
Hodgkin's lymphomas. Cancer Res., 51: 4743-4756, 1991.
58. Ho, M., Miller, G., Atchison, R. W., Breinig, M. K., Dummer, J. S.,
Andiman,
W., Starzl, T. E., Eastman, R., Griffith, B. P., Hardesty, R. L., Bahnson, H.
T.,
Hakala, T. R., and and Rosenthal, J. T. Epstein-Barr virus infections and DNA
hybridization studies in posttransplant lymphoma and lymphoproliferative
lesions: The role of primary infection. J. Infect. Dis., 152: 876-886, 1985.
59. Wang, D., Liebowitz, D., and Kieff, E. An EBV membrane protein expressed
in
immortalized lymphocytes transforms established rodent cells. Cell, 43: 831-
840,
1985.
60. Kulwichit W, Edwards R. H., and M., D. E. Expression of the Epstein-Barr
virus
latent membrane protein 1 induces B cell lymphoma in transgenic mice. Proc
Natl
Acad Sci U S A, 95: 11963-11968, 1998.
61. Lo, A. K., Liu, Y., Wang, X. H., Huang, D. P., Yuen, P. W., Wong, Y. C.,
and
Tsao, G. S. Alterations of biologic properties and gene expression in
29
CA 02596799 2007-08-01
WO 2006/084116 PCT/US2006/003817
nasopharyngeal epithelial cells by the Epstein-Barr virus-encoded latent
meinbrane protein 1. Lab Invest, 83: 697-709, 2003.
62. Roberts, M. L. and Cooper, N. R. Activation of a ras-MAPK-dependent
pathway
by Epstein-Barr virus latent membrane protein 1 is essential for cellular
transformation. Virology, 240: 93-99, 1998.
63. Kaye, K. M., Devergne, 0., Harada, J. N., Izumi, K. M., Yalamanchili, R.,
Kieff,
E., and Mosialos, G. Tumor necrosis factor receptor associated factor 2 is a
mediator of NF-kappa B activation by latent infection membrane protein 1, the
Epstain-Barr virus transforming protein. Proc Natl Acad Sci U S A, 93: 11085-
11090, 1996.
64. Izumi, K. M. and Kieff, E. D. The Epstein-Barr virus oncogene product
latent
membrane protein 1 engages the tumor necrosis factor receptor-associated death
domain protein to mediate B lymphocyte growth transformation and activate NF-
kappaB. Proc Natl Acad Sci U S A, 94: 12592-12597, 1997.
65. Mosialos, G., Birkenbach, M., Yalamanchili, R., VanArsdale, T., Ware, C.,
and
Kieff, E. The Epstein-Barr virus transforming protein LMP1 engages signaling
proteins for the tumor necrosis factor receptor family. Cell, 80: 389-399,
1995.
66. Sylla, B. S., Hung, S. C., Davidson, D. M., Hatzivassiliou, E., Malinin,
N. L.,
Wallach, D., Gilmore, T. D., Kieff, E., and Mosialos, G. Epstein-Barr virus-
transforming protein latent infection membrane protein 1 activates
transcription
factor NF-lcappaB through a pathway that includes the NF-kappaB-inducing
kinase and the IkappaB kinases IKKalpha and IKKbeta. Proc Natl Acad Sci U S
A, 95: 10106-10111, 1998.
67. Yeung, K. C., Rose, D. W., Dhillon, A. S., Yaros, D., Gustafsson, M.,
Chatterjee,
D., McFerran, B., Wyche, J., Kolch, W., and Sedivy, J. M. Raf kinase inhibitor
protein interacts with NF-kappaB-inducing kinase and TAK1 and inhibits NF-
kappaB activation. Mol Cell Biol, 21: 7207-7217, 2001.
68. Jones, W. K., Brown, M., Ren, X., He, S., and McGuinness, M. NF-kappaB as
an
integrator of diverse signaling pathways: the heart of myocardial signaling?
Cardiovasc Toxicol, 3: 229-254, 2003.
69. Ganem, D. and Varmus, H. E. The molecular biology of the hepatitis B
viruses.
Annu Rev Biochem, 56: 651-693, 1987.
70. Beasley, R. P., Hwang, L. Y., Lin, C. C., and Chien, C. S. Hepatocellular
carcinoma and hepatitis B virus. A prospective study of 22 707 men in Taiwan.
Lancet, 2: 1129-1133, 1981.
71. Twu, J. S. and Schloemer, R., H. Transcriptional trans-activating function
of
hepatitis B virus. J. Virol., 61: 3448-3453, 1987.
72. Kelcule, A. S., Lauer, U., Meyer M., Caselmann, W. H., Hofschneider, P.
H., and
R., K. The preS2/S region of integrated hepatitis B virus DNA encodes a
trancriptional transactivator. Nature, 343: 457-461, 1990.
73. Stocld, L., Berting, A., Mallcowslci, B., Foerste, R., Hofschneider, P.
H., and
Hildt, E. Integrity of c-Raf-1/MEK signal transduction cascade is essential
for
hepatitis B virus gene expression. Oncogene, 22: 2604-2610, 2003.
74. Benn, J. and Schneider, R. J. Hepatitis B virus HBx protein activates Ras-
GTP
complex formation and establishes a Ras, Raf, MAP kinase signaling cascade.
Proc Natl Acad Sci U S A, 91: 10350-10354, 1994.
CA 02596799 2007-08-01
WO 2006/084116 PCT/US2006/003817
75. Chung, T. W., Lee, Y. C., and Kim, C. H. Hepatitis B viral HBx induces
matrix
metalloproteinase-9 gene expression through activation of ERK and PI-3K/AKT
pathways: involvement of invasive potential. Faseb J, 18: 1123-1125, 2004.
76. Bouchard, M. J., Wang, L. H., and Schneider, R. J. Calcium signaling by
HBx
protein in hepatitis B virus DNA replication. Science, 294: 2376-2378, 2001.
77. Klein, N. P. and Schneider, R. J. Activation of Src family kinases by
hepatitis B
virus HBx protein and coupled signaling to Ras. Mol Cell Bio1,17: 6427-6436,
1997.
78. Noh, E. J., Jung, H. J., Jeong, G., Choi, K. S., Parlc, H. J., Lee, C. H.,
and Lee, J.
S. Subcellular localization and transcriptional repressor activity of HBx on
p21(WAF1/Cipl) promoter is regulated by ERK-mediated phosphorylation.
Biochem Biophys Res Commun, 319: 738-745, 2004.
79. Sinha, D., Bannergee, S., Schwartz, J. H., Lieberthal, W., and Levine, J.
S.
Inhibition of ligand-independent ERK1/2 activity in lcidney proximal tubular
cells
deprived of soluble survival factors up-regulates Akt and prevents apoptosis.
J
Biol Chem, 279: 10962-10972, 2004.
80. Sulciner, D. J., Irani, K., Yu, Z. X., Ferrans, V. J., Goldschinidt-
Clermont, P., and
Finkel, T. racl regulates a cytolcine-stimulated, redox-dependent pathway
necessary for NF-lcappaB activation. Mol Cell Bio1,16: 7115-7121, 1996.
81. Romashlcova, J. A. and Makarov, S. S. NF-kappaB is a target of AKT in anti-
apoptotic PDGF signalling. Nature, 401: 86-90, 1999.
82. Sodhi, A., Montaner, S., Patel, V., Gomez-Roman, J. J., Li, Y., Sausville,
E. A.,
Sawai, E. T., and Gutlcind, J. S. Alct plays a central role in sarcomagenesis
induced by Kaposi's sarcoma herpesvirus-encoded G protein-coupled receptor.
Proc Natl Acad Sci U S A, 101: 4821-4826, 2004.
While cei-tain preferred embodiments of the present invention have been
described and specifically exemplified above, it is not intended that the
invention be
limited to such enlbodiments. Various modifications may be made thereto
without
departing from the scope and spirit of the present invention, as set forth in
the following
claims.
31