Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
PROCÉDÉ DE PRODUCTION DE DISTILLATS MOYENS PAR HYDROISOMERISATION ET
HYDROCRAQUAGE DE
CHARGES ISSUES DU PROCÉDÉ FISCHER-TROPSCH UTILISANT UN CATALYSEUR DOPE A BASE
D'ALUMINE-SILICE
MESOPOREUSE A TENEUR CONTROLEE EN MACROPORES
La présente invention concerne un procédé de traitement avec hydrocraquage et
hydroisomérisation, de charges issues du procédé Fischer-Tropsch, permettant
d'obtenir des distillats
moyens (gazole, kérosène) mettant en oeuvre un catalyseur dopé comportant une
silice-alumine
particulière.
Dans le procédé Fischer-Tropsch, le gaz de synthèse (CO+H2) est transformé
catalytiquement
en produits oxygénés et en hydrocarbures essentiellement linéaires sous forme
gazeuse, liquide ou
solide. Ces produits sont généralement exempts d'impuretés hétéroatomiques
telles que, par exemple,
le soufre, l'azote ou des métaux. Ils ne contiennent également pratiquement
peu ou pas
d'aromatiques, de naphtènes et plus généralement de cycles en particulier dans
le cas de catalyseurs
au cobalt. Par contre, ils peuvent présenter une teneur non négligeable en
produits oxygénés qui,
exprimée en poids d'oxygène, est généralement inférieure à 5% poids environ et
également une
teneur en insaturés (produits oléfiniques en général) généralement inférieure
à 10% en poids.
Cependant, ces produits, principalement constitués de normales paraffines, ne
peuvent être utilisés
tels quels, notamment à cause de leurs propriétés de tenue à froid peu
compatibles avec les
utilisations habituelles des coupes pétrolières. Par exemple, le point
d'écoulement d'un hydrocarbure
linéaire contenant 20 atomes de carbone par molécule (température d'ébullition
égale à 340 C environ
c'est à dire souvent comprise dans la coupe distillat moyen) est de +37 C
environ ce qui rend son
utilisation impossible, la spécification étant de -15 C pour le gasoil. Les
hydrocarbures issus du
procédé Fischer-Tropsch comprenant majoritairement des n-paraffines doivent
être transformés en
produits plus valorisables tels que par exemple le gazole, kérosène, qui sont
obtenus, par exemple,
après des réactions catalytiques d'hydroisomérisation.
Le brevet EP-583,836 décrit un procédé pour la production de distillats moyens
à partir de
charge obtenue par la synthèse Fischer-Tropsch. Dans ce procédé, la charge est
traitée dans sa
globalité, tout au plus on peut enlever la fraction C4 moins et obtenir la
fraction C5+ bouillant à près de
100 C. Ladite charge est soumise à un hydrotraitement puis à une
hydroisomérisation avec une
conversion (de produits bouillant au-dessus de 370 C en produits à point
d'ébullition inférieur) d'au
moins 40% poids. Un catalyseur utilisable en hydroconversion est une
formulation platine sur silice-
alumine. Les conversions décrites dans les exemples sont d'au plus 60% poids.
Le brevet EP-321,303 décrit également un procédé de traitement desdites
charges en vue de
produire des distillats moyens et éventuellement des huiles. Dans un mode de
réalisation, des distillats
moyens sont obtenus par un procédé consistant à traiter la fraction lourde de
la charge, c'est à dire à
point d'ébullition initial compris entre 232 C et 343 C, par
hydroisomérisation sur un catalyseur fluoré
contenant un métal du groupe VIII et de l'alumine et présentant des
caractéristiques physico-chimiques
particulières. Après hydroisomérisation, l'effluent est distillé et la partie
lourde est recyclée en
hydroisomérisation. La conversion en hydroisomérisation des produits 370 C+
est donnée comme
comprise entre 50-95% pds et les exemples vont jusqu'à 85-87%.
Tous les catalyseurs utilisés actuellement en hydroisomérisation sont du type
bifonctionnels
associant une fonction acide à une fonction hydrogénante. La fonction acide
est apportée par des
supports de grandes surfaces (150 à 800 m2.g-1 généralement) présentant une
acidité superficielle,
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
2
telles que les alumines halogénées (chlorées ou fluorées notamment), les
alumines phosphorées, les
combinaisons d'oxydes de bore et d'aluminium, les silices-alumines amorphes et
les silice-alumines.
La fonction hydrogénante est apportée soit par un ou plusieurs métaux du
groupe VIII de la
classification périodique des éléments, tels que fer, cobalt, nickel,
ruthénium, rhodium, palladium,
osmium, iridium et platine, soit par une association d'au moins un métal du
groupe VI tels que chrome,
molybdène et tungstène et au moins un métal du groupe VIII.
L'équilibre entre les deux fonctions acide et hydrogénante est l'un des
paramètres qui régissent
l'activité et la sélectivité du catalyseur. Une fonction acide faible et une
fonction hydrogénante forte
donnent des catalyseurs peu actifs et sélectifs envers l'isomérisation alors
qu'une fonction acide forte
et une fonction hydrogénante faible donnent des catalyseurs très actifs et
sélectifs envers le craquage.
Une troisième possibilité est d'utiliser une fonction acide forte et une
fonction hydrogénante forte afin
d'obtenir un catalyseur très actif mais également très sélectif envers
l'isomérisation. Il est donc
possible, en choisissant judicieusement chacune des fonctions d'ajuster le
couple activité/sélectivité du
catalyseur.
La présente invention concerne donc un procédé pour la production de
distillats moyens. Ce
procédé permet :
- d'améliorer fortement les propriétés à froid des paraffines issues du
procédé Fisher-Tropsch et
ayant des points d'ébullition correspondants à ceux des fractions gazole et
kérosène, (encore
appelées distillats moyens) et notamment d'améliorer le point de congélation
des kérosènes.
- d'augmenter la quantité de distillats moyens disponibles par hydrocraquage
des composés
paraffiniques les plus lourds, présents dans l'effluent de sortie de l'unité
Fischer-Tropsch, et qui
ont des points d'ébullition supérieurs à ceux des coupes kérosène et gazole,
par exemple la
fraction 380 C+.
et ce procédé met en oeuvre une silice-alumine particulière permettant
d'obtenir des catalyseurs très
sélectifs et actifs.
Plus précisément l'invention concerne un procédé de production de distillats
moyens à partir d'une
charge paraffinique produite par synthèse Fischer-Tropsch mettant en oeuvre un
catalyseur
d'hydrocraquage/hydroisomérisation particulier comprenant :
- au moins un élément hydro-déshydrogénant choisi dans le groupe formé par les
éléments du
groupe VIB et du groupe VIII de la classification périodique,
- de 0.01 à 6% de phosphore comme élément dopant, (en combinaison
éventuellement avec bore
et/ou silicium),
- et un support non zéolitique à base d'alumine-silice
ladite alumine-silice présentant les caractéristiques suivantes :
- un pourcentage de silice compris entre 5 et 95 % poids, de préférence entre
10 et 80%, de
manière plus préférée entre 20 et 60 % et de manière très préférée entre 30 et
50%,
- un contenu en sodium inférieur à 0.03% poids,
- un volume poreux total mesuré par porosimétrie au mercure compris entre 0.45
et 1.2 ml/g,
- une porosité telle que :
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
3
i) le volume des mésopores avec un diamètre compris entre 40 et 150 A et un
diamètre moyen
poreux compris entre 80 et 140 A (de préférence entre 80 et 120 A) représente
30-80% du volume
poreux total mesuré par porosimétrie au mercure
ii) le volume des macropores avec un diamètre supérieur à 500 A représente 20-
70% du
volume poreux total mesuré par porosimétrie au mercure
- une surface spécifique BET comprise entre 100 et 550 m2/g , de préférence
comprise entre
150 et 500 m2/g, de manière préférée inférieure à 350 m2/g et de manière
encore plus préférée
inférieure à 250 m2/g,
- un diagramme de diffraction X qui contient au moins les raies principales
caractéristiques d'au
moins une des alumines de transition comprise dans le groupe composé par les
alumines alpha, rhô,
khi, êta, gamma, kappa, thêta et delta.
Description détaillée de l'invention
Techniques de caractérisation
Dans l'exposé qui suit de l'invention, on entend par surface spécifique, la
surface spécifique
B.E.T. déterminée par adsorption d'azote conformément à la norme ASTM D 3663-
78 établie à partir
de la méthode BRUNAUER-EMMETT-TELLER décrite dans le périodique The Journal
of American
Society", 60, 309, (1938).
Dans l'exposé qui suit de l'invention, on entend par volume mercure des
supports et des
catalyseurs, le volume mesuré par intrusion au porosimètre à mercure selon la
norme ASTM D4284-
83 à une pression maximale de 4000 bar, utilisant une tension de surface de
484 dyne/cm et un angle
de contact pour les supports alumine-silice amorphe de 140 . Une des raisons
pour lesquelles il est
préférable d'utiliser le support comme base pour définir la distribution
poreuse tient dans le fait que
l'angle de contact du mercure varie après imprégnation des métaux et ceci en
fonction de la nature et
de type de métaux. L'angle de mouillage a été pris égal à 140 en suivant les
recommandations de
l'ouvrage "Techniques de l'ingénieur, traité analyse et caractérisation, P
1050-5, écrits par Jean
Charpin et Bernard Rasneur".
Afin d'obtenir une meilleure précision, la valeur du volume mercure en ml/g
donnée dans le
texte qui suit correspond à la valeur du volume mercure total (volume poreux
total mesuré par
intrusion au porosimètre à mercure) en ml/g mesurée sur l'échantillon moins la
valeur du volume
mercure en ml/g mesurée sur le même échantillon pour une pression
correspondant à 30 psi (environ
2 bars). On définit également le diamètre moyen mercure comme étant un
diamètre tel que tous les
pores de taille inférieure à ce diamètre constituent 50% du volume poreux
total mercure.
Afin de mieux caractériser la distribution poreuse, on définit enfin les
critères de distribution poreuse
suivants en mercure: le volume V1 correspond au volume contenu dans les pores
dont le diamètre
est inférieur au diamètre moyen moins 30 Å. Le volume V2 correspond au volume
contenu dans les
pores de diamètre supérieur ou égal au diamètre moyen moins 30 Å et inférieur
au diamètre moyen
plus 30 Å. Le volume V3 correspond au volume contenu dans les pores de
diamètre supérieur ou
égal au diamètre moyen plus 30 Å. Le volume V4 correspond au volume contenu
dans les pores
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
4
dont le diamètre est inférieur au diamètre moyen moins 15 Å. Le volume V5
correspond au volume
contenu dans les pores de diamètre supérieur ou égal au diamètre moyen moins
15 Å et inférieur au
diamètre moyen plus 15 Å. Le volume V6 correspond au volume contenu dans les
pores de diamètre
supérieur ou égal au diamètre moyen plus 15 A.
La distribution poreuse mesurée par adsorption d'azote" a été déterminée par
le modèle
Barrett-Joyner-Halenda (BJH). L'isotherme d'adsorption - désorption d'azote
selon le modèle BJH est
décrit dans le périodique "The Journal of American Society" , 73, 373, (1951)
écrit par E.P.Barrett,
L.G.Joyner et P.P.Halenda. Dans l'exposé qui suit de l'invention, on entend
par volume adsorption
azote, le volume mesuré pour P/Po= 0,99, pression pour laquelle il est admis
que l'azote a rempli tous
les pores. On définit le diamètre moyen désorption azote comme étant un
diamètre tel que tous les
pores inférieurs à ce diamètre constituent 50% du volume poreux (Vp) mesuré
sur la branche de
désorption de l'isotherme azote.
Par surface adsorption, on entend la surface mesurée sur la branche de
l'isotherme
d'adsorption. On se reportera par exemple à l'article de A. Lecloux "Mémoires
Société Royale des
Sciences de Liège, 6ème série, Tome I, fasc.4, pp.169-209 (1971)".
La teneur en sodium a été mesurée par spectrométrie d'absorption atomique.
La diffraction X est une technique pouvant être utilisée pour caractériser les
supports et
catalyseurs selon l'invention. Dans l'exposé qui suit, l'analyse des rayons X
est réalisée sur poudre
avec un diffractomètre Philips PW 1830 opérant en réflexion et équipé d'un
monochromateur arrière
en utilisant la radiation CoKalpha (? Kat = 1.7890 A, A,IK,2 = 1.793 Å,
rapport d'intensité Ka1/ K,2 = 0,5).
Pour le diagramme de diffraction X de l'alumine gamma, on se reportera à la
base de données ICDD,
fiche 10-0425. En particulier, les 2 pics les plus intenses sont situés à une
position correspondant à un
d compris entre 1,39 et 1,40 Å et un d compris entre 1,97 A à 2,00 A. On
appelle d la distance inter-
réticulaire qui est déduite de la position angulaire en utilisant la relation
dite de Bragg (2 d (hkl) * sin (D)
= n * l Par alumine gamma, on entend dans la suite du texte entre autres par
exemple une alumine
comprise dans le groupe composé des alumines gamma cubique, gamma pseudo-
cubique, gamma
tétragonale, gamma mal ou peu cristallisée, gamma grande surface, gamma basse
surface, gamma
issue de grosse boehmite, gamma issue de boehmite cristallisée, gamma issue de
boehmite peu ou
mai cristallisée, gamma issue d'un mélange de boehmite cristallisée et d'un
gel amorphe, gamma
issue d'un gel amorphe, gamma en évolution vers delta . Pour les positions des
pics de diffraction des
alumines êta, delta et thêta, on peut se référer à l'article de B.C. Lippens,
J.J. Steggerda, dans
Physical and Chemical aspects of adsorbents and catalysts, E.G. Linsen (Ed.),
Academic Press,
London. 1970, p.171-211.
Pour les supports et catalyseurs selon l'invention, le diagramme de
diffraction X met en
évidence un pic large caractéristique de la présence de silice amorphe.
Par ailleurs, dans l'ensemble du texte qui suit, le composé d'alumine peut
contenir une fraction
amorphe difficilement détectable par les techniques de DRX. On sous-entendra
donc par la suite que
les composés d'alumine utilisés ou décrits dans le texte peuvent contenir une
fraction amorphe ou mal
cristallisée.
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
Les supports et catalyseurs selon l'invention ont été analysés par RMN MAS du
solide de 27AI
sur un spectromètre de la firme Brüker de type MSL 400, en sonde 4 mm. La
vitesse de rotation des
échantillons est de l'ordre de 11 kHz. Potentiellement, la RMN de l'aluminium
permet de distinguer
trois types d'aluminium dont les déplacements chimiques sont reportés ci-après
:
Entre 100 et 40 ppm, aluminiums de type tétra-coordinés, notés AI iv,
Entre 40 et 20 ppm, aluminiums de type penta-coordinés, notés Alv,
Entre 20 et - 100 ppm, aluminiums de type hexa-coordinés, notés Alvi.
L'atome d'aluminium est un noyau quadripolaire. Dans certaines conditions
d'analyse (champs de
radiofréquence faible : 30 kHz, angle d'impulsion faible : ir/2 et échantillon
saturé en eau), la technique
de RMN de rotation à l'angle magique (MAS) est une technique quantitative. La
décomposition des
spectres RMN MAS permet d'accéder directement à la quantité des différentes
espèces. Le spectre
est calé en déplacement chimique par rapport à une solution 1M de nitrate
d'aluminium. Le signal
d'aluminium est à zéro ppm. Nous avons choisi d'intégrer les signaux entre 100
et 20 ppm pour les AI
iv et Alv, ce qui correspond à l'aire 1, et entre 20 et -100 ppm pour Alv,, ce
qui correspond à l'aire 2.
Dans l'exposé qui suit de l'invention, on entend par proportion des Alvi
octaédriques le rapport suivant:
aire 2/ (aire 1 + aire 2).
L'environnement du silicium des alumines-silice est étudié par la RMN de 29Si.
Les tables de
déplacements chimiques en fonction du degré de condensation ont été déduites
de l'ouvrage de
G.Engelhardt et D.Michel : High resolution solid-state NMR of silicates and
zeolites (Wiley), 1987.
La RMN 29 Si montre les déplacements chimiques des différentes espèces de
silicium telles que Q4 (-
105ppm à - 120 ppm), Q3 (-90ppm à -102 ppm) et Q2 (-75ppm à - 93 ppm). Les
sites avec un
déplacement chimique à -102 ppm peuvent être des sites de type Q3 ou Q4, nous
les appelons sites
Q34 . Les définitions des sites sont les suivantes :
sites Q4 : Si lié à 4Si (ou AI),
sites Q3 : Si lié à 3 Si(ou AI) et 1 OH
sites Q2 : Si lié à 2 Si(ou AI) et 2 OH;
Les alumines-silice de l'invention sont composées de silicium de types Q2, Q3,
Q3-4 et Q4. De
nombreuses espèces seraient de type Q2, approximativement de l'ordre de 10 à
80%, de préférence
20 à 60 % et manière préférée de 20 à 40%. La proportion des espèces Q3 et Q34
est également
importante, approximativement de l'ordre de 5 à 50 % et de manière préférée de
10 à 40% pour les
deux espèces.
L'environnement des siliciums a été étudié par RMN CP MAS 1H->29Si, (300 MHz,
vitesse de rotation :
4000 Hz). Dans ce cas, seul le silicium lié à des liaisons OH doit répondre.
La table des déplacements
chimiques utilisés est celle de Kodakari et al. , Langmuir, 14, 4623-4629,
1998. Les attributions sont
les suivantes : -108 ppm (Q4), -99 ppm (Q3/Q4(1 AI)), -91 ppm (Q3/Q3(1Al)) , -
84 ppm (Q2/Q3(2AI), -
78 ppm (Q2/Q3(3AI) et -73 ppm Q1/Q2 (3 AI).
Les alumines-silice de l'invention se présentent sous la forme de
superposition de plusieurs massifs.
Le pic principal de ces massifs est généralement situé à -110 ppm.
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
6
Une méthode de caractérisation des supports et catalyseurs selon l'invention
pouvant être
utilisée est la microscopie électronique par transmission (MET). Pour cela on
utilise un microscope
électronique (du type Jeol 2010 ou Philips Tecnai20F éventuellement avec
balayage) équipé d'un
spectromètre à dispersion d'énergie (EDS) pour l'analyse des rayons X (par
exemple un Tracor ou un
Edax). Le détecteur EDS doit permettre la détection des éléments légers.
L'association de ces deux
outils, MET et EDS, permet de combiner l'imagerie et l'analyse chimique locale
avec une bonne
résolution spatiale.
Pour ce type d'analyse, les échantillons sont finement broyés à sec dans un
mortier ; la poudre
est ensuite incluse dans de la résine pour réaliser des coupes ultrafines
d'épaisseur 70 nm environ.
Ces coupes sont recueillies sur des grilles de Cu recouvertes d'un film de
carbone amorphe à trous
servant de support. Elles sont ensuite introduites dans le microscope pour
observation et analyse sous
vide secondaire. En imagerie, on distingue alors aisément les zones
d'échantillon des zones de résine.
On procède ensuite à un certain nombre d'analyses, 10 au minimum, de
préférence comprises entre
15 et 30, sur différentes zones de l'échantillon industriel. La taille du
faisceau électronique pour
l'analyse des zones (déterminant approximativement la taille des zones
analysées) est de 50 nm de
diamètre au maximum, de préférence de 20 nm, de manière encore plus préférée
10, 5, 2 ou 1nm de
diamètre. En mode balayé, la zone analysée sera fonction de la taille de la
zone balayée et non plus de
la taille du faisceau généralement réduit.
Le traitement semi quantitatif des spectres X recueillis à l'aide du
spectromètre EDS permet
d'obtenir la concentration relative de AI et de Si (en % atomique) et le
rapport Si/AI pour chacune des
zones analysées. On peut alors calculer la moyenne Si/Alm et l'écart type ^ de
cet ensemble de
mesures. Dans les exemples non limitatifs de l'exposé qui suit de l'invention,
la sonde de 50 nm est la
sonde utilisée pour caractériser les supports et catalyseurs selon l'invention
sauf mention contraire.
La densité de remplissage tassée (DRT) est mesurée de la manière décrite dans
l'ouvrage
Applied Heterogenous Catalysis " de J.F. Le Page, J. Cosyns, P. Courty, E.
Freund, J-P. Franck, Y.
Jacquin, B. Juguin, C. Marcilly, G. Martino, J. Miquel, R. Montarnal, A.
Sugier, H. Van Landeghem,
Technip, Paris, 1987. Un cylindre gradué de dimensions acceptables est rempli
de catalyseur par
additions successives; et entre chaque addition, le catalyseur est tassé en
secouant le cylindre jusqu'à
atteindre un volume constant. Cette mesure est généralement réalisée sur 1000
cm3 de catalyseur
tassé dans un cylindre dont le ratio hauteur sur diamètre est proche de 5:1.
Cette mesure peut être,
de manière préférée, réalisée sur des appareils automatisés tels que Autotap
commercialisé par
Quantachrome .
L'acidité de la matrice est mesurée par spectrométrie Infra-Rouge (IR). Les
spectres IR sont
enregistrés sur un interféromètre Nicolet de type Nexus-670 sous une
résolution de 4 cm-1 avec une
apodisation de type Happ-Gensel. L'échantillon (20 mg) est pressé sous la
forme d'une pastille auto-
supportée, puis est placé dans une cellule d'analyse in-situ (25 C à 550 C,
four déporté du faisceau
IR, vide secondaire de 10-6 mbar). Le diamètre de la pastille est de 16 mm.
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
7
L'échantillon est prétraité de la façon suivante afin d'éliminer l'eau
physisorbée et de déshydroxyler
partiellement la surface du catalyseur afin d'obtenir une image représentative
de l'acidité du catalyseur
en fonctionnement :
- montée en température de 25 C à 300 C en 3 heures
- palier de 10 heures à 300 C
- descente de température de 300 C à 25 C en 3 heures
La sonde basique (pyridine) est ensuite adsorbée à pression saturante à 25 C
puis thermo-
désorbée selon les paliers suivants :
- 25 C pendant 2 heures sous vide secondaire
- 100 C 1 heure sous vide secondaire
- 200 C 1 heure sous vide secondaire
- 300 C 1 heure sous vide secondaire
Un spectre est enregistré à 25 C à la fin du prétraitement et à chaque palier
de désorption en
mode transmission avec un temps d'accumulation de 100 s. Les spectres sont
ramenés à iso-masse
(donc supposés à iso-épaisseur) (20 mg exactement). Le nombre de sites de
Lewis est proportionnel à
la surface du pic dont le maximum se situe vers 1450 cm-1, tout épaulement
étant inclus. Le nombre
de sites de Bronsted est proportionnel à la surface du pic dont le maximum se
situe vers 1545 cm-1.
Le rapport du nombre de sites de Bronsted /nombre de sites de Lewis B/L est
estimé égal au rapport
des surfaces de deux pics décrits ci-dessus. On utilise généralement la
surface des pics à 25 C. Ce
rapport B/L est de manière générale calculé à partir du spectre enregistré à
25 C à la fin du
prétraitement.
Lorsqu'un élément dopant, P et éventuellement B et/ou Si, est introduit, sa
répartition et sa
localisation peuvent être déterminées par des techniques telles que la
microsonde de Castaing (profil
de répartition des divers éléments), la microscopie électronique par
transmission couplée à une
analyse X des composants du catalyseurs, ou bien encore par l'établissement
d'une cartographie de
répartition des éléments présents dans le catalyseur par microsonde
électronique. Ces techniques
permettent de mettre en évidence la présence de ces éléments exogènes ajoutés
après la synthèse
de l'alumine-silice selon l'invention.
La composition globale du catalyseur peut être déterminée par fluorescence X
sur le catalyseur
à l'état pulvérulent ou par absorption atomique après attaque acide du
catalyseur.
La mesure de la composition locale à l'échelle du micron, par opposition à la
composition
globale du catalyseur, peut s'effectuer par microsonde électronique Cette
mesure peut s'effectuer en
déterminant les teneurs en métal sur des zones de quelques microns cubes le
long du diamètre d'une
particule de catalyseurs que l'on appelle unités de mesures. Cette mesure
permet d'évaluer la
répartition macroscopique des éléments à l'intérieur des particules. Elle peut
être complétée
éventuellement à l'échelle du nanomètre par STEM (Scanning Transmission
Electron Microscopy
(Microscopie électronique de transmission à balayage)).
CA 02612856 2012-07-27
8
Les analyses sont conduites sur une microsonde électronique CAMECA SX100
(équipée de 5
spectromètres à dispersion de longueur d'onde)(appareillage préféré) ou
éventuellement sur JEOL
8800R(4 spectromètres) Les paramètres d'acquisition sont les suivants :
tension d'accélération 20 kV,
courant 80 ou 200 nA et temps de comptage 10 s ou 20 s selon le niveau de
concentration. Les
particules sont enrobées dans la résine puis polies jusqu'à leur diamètre.
On notera que l'appellation diamètre ne se réfère pas uniquement à une forme
en bille ou en
extrudé, mais plus généralement à toute forme de particules ; est dénommée
diamètre en fait la
longueur représentative de la particule sur laquelle est effectuée la mesure.
Les mesures sont effectuées sur un échantillon représentatif du lit ou du lot
de catalyseur qui
sera utilisé sur un lit catalytique. On a considéré que les analyses devraient
être faites sur au moins 5
particules avec au moins 30 mesures par particule, uniformément réparties le
long du diamètre.
On appelle CMo, CNi, CW et CP les concentrations locales (exprimées en %)
respectivement en
Molybdène, Nickel, Tungstène et phosphore
On pourrait tout aussi bien exprimer les concentrations en % atomique, les
fluctuations relatives étant
les mêmes.
Il est intéressant de préparer des catalyseurs présentant des concentrations
homogènes
CMo, CNi, CW et CP le long de l'extrudé. Il est également intéressant de
préparer des catalyseurs
présentant des concentrations CMo, CNi, CW et CP au coeur et en périphérie
différente. Ces
catalyseurs présentent des profils de répartition dits en cuvette ou en
dôme . Un autre type
de répartition est celle en croûte où les éléments de la phase active sont
répartis en surface.
Description de l'invention
La présente invention concerne un procédé de production de distillats moyens à
partir d'une charge
paraffinique produite par synthèse Fischer-Tropsch, comprenant une étape de
fractionnement ou de
séparation de la charge, une étape d'hydrotraitement d'au moins une partie
d'une fraction issue de
l'étape de fractionnement ou de séparation, au moins une étape d'hydrocraquage
/
hydroisomérisation, et une étape de distillation des fractions hydrocraquées /
hydroisomérisées,
procédé dans lequel ladite étape d'hydrocraquage / hydroisomérisation utilise
un catalyseur
d'hydrocraquage/hydroisomérisation qui comporte :
- au moins un élément hydro-déshydrogénant choisi dans le groupe formé
par les éléments du groupe VIB et du groupe VIII de la classification
périodique,
- de 0.01 à 6% de phosphore comme élément dopant,
- et un support non zéolitique à base d'alumine-silice
CA 02612856 2012-07-27
9
ladite alumine-silice présentant les caractéristiques suivantes:
- un pourcentage de silice compris entre 5 et 95 % poids,
- un contenu en sodium inférieur à 0.03% poids,
- un volume poreux total mesuré par porosimétrie au mercure compris entre
0.45 et 1.2 ml/g, une porosité telle que :
ii) le volume des mésopores avec un diamètre compris entre
40 et 150 A et un diamètre moyen poreux compris entre 80 et 140 A
représente 30-80% du volume poreux total mesuré par porosimétrie au
mercure
ii) le volume des macropores avec un diamètre supérieur à 500
A représente 20-70% du volume poreux total mesuré par porosimétrie au
mercure
- une surface spécifique BET comprise entre 100 et 550 m2/g ,
- un diagramme de diffraction X qui contient au moins les raies principales
caractéristiques d'au moins une des alumines de transition comprise dans
le groupe composé par les alumines alpha, rhô, khi, êta, gamma, kappa,
thêta et delta.
Caractéristiques du catalyseur utilisable dans le procédé selon l'invention
Le catalyseur selon l'invention comprend donc :
- de préférence une teneur en impuretés cationiques inférieure à 0,1%poids, de
manière préférée
inférieure à 0,05% poids et de manière encore plus préférée inférieure à
0,025% poids. On entend
par teneur en impuretés cationiques la teneur totale en alcalins.
- de préférence une teneur en impuretés anioniques inférieure à 1% poids, de
manière préférée
inférieure à 0,5% poids et de manière encore plus préférée inférieure à 0,1%
poids.
- au moins un élément hydro-déshydrogénant choisi dans le groupe formé par les
éléments du
groupe VIB et du groupe VIII (de préférence groupe VIII noble) de la
classification périodique,
- de préférence une teneur massique en métal(aux) du groupe VIB, sous forme
métallique ou sous
forme oxyde comprise entre 1 et 50 % en poids, de manière préférée entre 1,5
et 35 %, et de
manière encore plus préférée entre 1,5 et 30%,
- de préférence une teneur massique en métaux du groupe VIII, sous forme
métallique ou sous
forme oxyde comprise entre 0,1 et 30 % en poids, de manière préférée entre 0,2
et 25 % et de
manière encore plus préférée entre 0,2 et 20%,
CA 02612856 2012-07-27
9a
- de 0.01 à 6% de phosphore comme élément dopant déposé sur le catalyseur (on
entend par
élément dopant un élément introduit après la préparation du support alumino-
silicate décrit
précédemment), éventuellement en combinaison avec le bore et/ou le silicium.
Ainsi on pourra
utiliser comme éléments dopants une combinaison phosphore et bore ou une
combinaison
phosphore, bore et silicium. Lorsque les éléments bore et/ou silicium sont
présents sur le
catalyseur, les teneurs massiques en bore et silicium, calculées sous leur
forme oxyde, sont
comprises entre 0,01 et 6%, de préférence entre 0,1 et 4%, de manière plus
préférée entre 0.2 et
2.5%.
- éventuellement au moins un élément du groupe VIIB (manganèse par exemple et
de préférence),
et une teneur pondérale comprise entre 0 et 20%, de préférence entre 0 et 10 %
du composé
sous forme oxyde ou métal.
15
25
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
éventuellement au moins un élément du groupe VB (niobium par exemple et de
préférence), et
une teneur pondérale comprise entre 0 et 40%, de préférence entre 0 et 20% du
composé sous
forme oxyde ou métal,
un support non zéolitique à base d'alumine-silice, ladite alumine-silice
présentant les
caractéristiques suivantes :
un pourcentage de silice compris entre 5 et 95 % poids, de préférence entre 10
et
80%, de manière plus préférée entre 20 et 60 % et de manière très préférée
entre 30 et 50%,
un contenu en sodium inférieur à 0.03% poids,
un volume poreux total mesuré par porosimétrie au mercure compris entre 0.45
et 1.2
m 1/g,
- une porosité telle que :
il Le volume des mésopores dont le diamètre est compris entre 40Å et 150Å, et
dont le
diamètre moyen poreux varie entre 80 et 140 Å (de préférence entre 80 et 120
A) représente
entre 30 et 80% du volume poreux total précédemment défini et de préférence
entre 40 et
70%.
ii/ Le volume des macropores, dont le diamètre est supérieur à 500 Å, et de
préférence
compris entre 1000 Å et 10000 Å représente entre 20 et 70% du volume poreux
total et de
préférence entre 30 et 60% du volume poreux total et de manière encore plus
préférée le
volume des macropores représente au moins 35% du volume poreux total.
- une surface spécifique BET comprise entre 100 et 550 m2/g , de préférence
comprise entre
150 et 500 m2/g, de manière préférée inférieure à 350 m2/g et de manière
encore plus
préférée inférieure à 250 m2/g,
- un diagramme de diffraction X qui contient au moins les raies principales
caractéristiques d'au
moins une des alumines de transition comprises dans le groupe composé par les
alumines
rhô, khi, kappa, êta, gamma, thêta et delta, de manière préférée qui contient
au moins les
raies principales caractéristiques d'au moins une des alumines de transition
compris dans le
groupe composé par l'alumine gamma, êta, thêta et delta, et de manière plus
préférée qui
contient au moins les raies principales caractéristiques de l'alumine gamma et
êta, et de
manière encore plus préférée qui contient les pics à un d compris entre 1,39 à
1,40 Å et à un
d compris entre 1,97 Å à 2,00 A.
De manière préférée, l'alumine-silice comprend de 30 à 50% de sites Q2, dans
lesquels un
atome de Si est lié à deux atomes de Si ou AI et à deux groupes OH et comprend
également 10-30 %
de sites Q3 dans lesquels un atome de Si est lié à trois atomes de Si ou AI ou
à un groupe OH.
Selon un mode préféré de l'invention, le support de catalyseur est constitué
d'alumine-silice
seule.
Selon un autre mode de réalisation de l'invention, le support comprend de 1 à
40% en poids de
liant. Le support peut alors résulter du mélange de l'alumine-silice et d'au
moins un liant choisi dans le
groupe formé par la silice, l'alumine, les argiles, l'oxyde de titane, l'oxyde
de bore et la zircone.
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
11
Dans le catalyseur, la proportion des Alvi octaédriques déterminée par
l'analyse des spectres
RMN MAS du solide de 27AI est généralement supérieure à 50%.
Le catalyseur peut également contenir une proportion mineure d'au moins un
élément
promoteur choisi dans le groupe formé par la zircone et le titane.
De préférence, le catalyseur est soumis à un traitement hydrothermal après la
synthèse, comme
décrit ultérieurement.
De préférence, le catalyseur est soumis à un traitement de sulfuration, comme
décrit
ultérieurement.
Test standard d'activité : évaluation des catalyseurs selon l'invention
L'acidité et la performance en hydrogénation des catalyseurs selon l'invention
peuvent être évaluées
par un test catalytique d'un mélange de molécules modèles : l'hydrogénation du
toluène et l'isomérisation du
cyclohexane.
Le test catalytique permettant de contrôler l'hydrogénation et l'acidité des
catalyseurs est réalisé
selon le protocole suivant :
Les catalyseurs sont sulfurés in situ en dynamique dans un réacteur tubulaire
à lit fixe traversé d'une
unité pilote de type catatest (constructeur Vinci Technologies), les fluides
circulant de haut en bas. Les
mesures d'activité hydrogénante et isomérisante sont effectuées immédiatement
après
la suifuration sous pression sans remise à l'air avec la charge
d'hydrocarbures qui a servi à sulfurer
les catalyseurs.
La charge de suifuration et de test est composée de 5,8 % de diméthyl
disulfure (DMDS), 20 % de
toluène et 74,2 % de cyclohexane en poids. On mesure ainsi les activités
catalytiques stabilisées de volumes
égaux de catalyseurs dans la réaction d'hydrogénation du toluène. Le suivi de
l'isomérisation du cyclohexane,
diluant du toluène, permet d'estimer l'acidité des catalyseurs.
Les conditions de mesure d'activité sont les suivantes (en considérant une
vaporisation totale et la loi
des gaz parfaits) :
Pression totale : 6,0 MPa
Pression de toluène : 0,38 MPa
Pression de cyclohexane : 1,55 MPa
Pression d'hydrogène : 3,64 MPa
Pression d'H2S : 0,22 MPa
Volume de catalyseur : 40 cc
Débit de charge : 80 cc/h
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
12
Vitesse spatiale horaire : 2 I/l/h-1
Débit d'hydrogène : 361/h
Température de suifuration et de test : 350 C (3 C/min).
Des prélèvements de l'effluent liquide sont analysés par chromatographie en
phase gazeuse, La
détermination des concentrations molaire en toluène non converti (T) et des
concentrations des produits
d'hydrogénation : le méthyle cyclohexane (MCC6), l'éthyle cyclopentane (EtCC5)
et les diméthyle
cyclopentane (DMCC5) permettent de calculer un taux d'hydrogénation de toluène
XHYD défini par :
XHYD (%) = 100 * (MCC6 + EtCC5 + DMCC5) / (T + MCC6 + EtCC5 + DMCC5)
Le taux d'isomérisation du cyclohexane XISO est calculé de la même façon à
partir des concentrations
en cyclohexane non converti et de son produit de réaction, le méthyl
cyclopentane. La réaction
d'hydrogénation du toluène et d'isomérisation du cyclohexane étant d'ordre 1
dans nos conditions de test et
le réacteur se comportant comme un réacteur piston idéal, on calcule
l'activité hydrogénante AHYD et
isomérisante AISO des catalyseurs en appliquant la formule : Ai = ln(100/(100-
Xi))
Avantageusement, le catalyseur selon l'invention possède dans le test standard
d'activité une
activité AHYD > 0. 7 et une activité A,soM > 0.1, de manière préférée AHYD >
0.9 et AisoM > 0.12, de
manière plus préférée AHYp > 1.2 et AisoM > 0.13 et de manière encore plus
préférée AHYp > 1.4 et
AisoM > 0.13.
Le rapport d'activité hydrogénante sur activité isomérisante H/A est égal à
AHYD/AISO.
Le rapport d'activité hydrogénante sur activité isomérisante H/A est
avantageusement compris
entre 6,5 et 30, de manière préférée entre 7 et 30, de manière très préférée
entre 7,5 et 25, de
manière plus préférée entre 8,5 et 20 et de manière encore plus préférée entre
9,5 et 15.
Lorsque l'élément dopant est le phosphore, la teneur en phosphore est
avantageusement
comprise entre 0.01 et 4 %poids d'oxyde, de manière très préférée comprise
entre 0.01 et 2.5% poids
d'oxyde.
De préférence, le catalyseur d'hydrocraquage/hydroisomérisation est à base de
platine et/ou
palladium.
De manière très préférée, le catalyseur d'hydrocraquage/hydroisomérisation
contient de 0,05 à
% de métal noble du groupe VIII.
Un catalyseur préféré selon l'invention comprend l'association platine-
palladium et une teneur en
phosphore comprise entre 0,01 et 4% poids d'oxyde.
Un catalyseur très préféré selon l'invention comprend l'association platine-
palladium et une
teneur en phosphore comprise entre 0,01 et 2,5% poids d'oxyde.
Le catalyseur peut également contenir une proportion mineure d'au moins un
élément stabilisant
choisi dans le groupe formé par la zircone et le titane.
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
13
Procédés de préparation
Les catalyseurs selon l'invention peuvent être préparés selon toutes les
méthodes bien connues
de l'homme du métier.
Un procédé préféré de préparation du catalyseur selon la présente invention
comprend les
étapes suivantes :
Selon un mode de préparation préféré, le précurseur est obtenu par mise en
forme directe de
l'alumine-silice seule ou par mise en forme de l'alumine-silice avec au moins
un liant, puis séchage et
calcination. Les éléments des groupes VIB et/ou VIII, et éventuellement ceux
choisis parmi le
phosphore, le bore, le silicium et éventuellement les éléments des groupes VB,
et VIIB, sont alors
éventuellement introduits par toute méthode connue de l'homme du métier, avant
ou après la mise en
forme et avant ou après la calcination du précurseur ou du catalyseur.
L'élément hydrogénant peut être introduit à toute étape de la préparation, de
préférence lors du
mélange, ou de manière très préférée après mise en forme. La mise en forme est
suivie d'une
calcination, l'élément hydrogénant peut également être introduit avant ou
après cette calcination. La
préparation se termine généralement par une calcination à une température de
250 à 600 C. Une
autre des méthodes préférées selon la présente invention consiste à mettre en
forme l'alumine-silice
sans liant après un malaxage de cette dernière, puis passage de la pâte ainsi
obtenue au travers
d'une filière pour former des extrudés de diamètre compris entre 0,4 et 4 mm.
La fonction
hydrogénante peut être alors introduite en partie seulement (cas, par exemple,
des associations
d'oxydes de métaux des groupes VIB et VIII) ou en totalité, au moment du
malaxage. Elle peut
également être introduite par une ou plusieurs opérations d'échange ionique
sur le support calciné
constitué d'au moins une alumine-silice, éventuellement mise en forme avec un
liant, à l'aide de
solutions contenant les sels précurseurs des métaux choisis lorsque ceux-ci
appartiennent au groupe
VIII. Elle peut aussi être introduite par une ou plusieurs opérations
d'imprégnation du support mis en
forme et calciné, par une solution des précurseurs des oxydes des métaux des
groupes VIII
(notamment le cobalt et le nickel) lorsque les précurseurs des oxydes des
métaux du groupe VIB
(notamment le molybdène ou le tungstène) ont été préalablement introduits au
moment du malaxage
du support. Elle peut enfin également être introduite, de façon très préférée
par une ou plusieurs
opérations d'imprégnation du support calciné constitué d'au moins une alumine-
silice selon l'invention
et éventuellement d'au moins un liant, par des solutions contenant les
précurseurs des oxydes de
métaux des groupes VI et/ou VIII, les précurseurs des oxydes de métaux de
groupe VIII étant de
préférence introduits après ceux du groupe VIB ou en même temps que ces
derniers.
D'une façon préférée, le support est imprégné par une solution aqueuse.
L'imprégnation du
support est de préférence effectuée par la méthode d'imprégnation dite "à sec"
bien connue de
l'homme du métier. L'imprégnation peut être effectuée en une seule étape par
une solution contenant
l'ensemble des éléments constitutifs du catalyseur final.
Le catalyseur de la présente invention peut donc renfermer au moins un élément
du groupe VIII
tel que fer, cobalt, nickel, ruthénium, rhodium, palladium, osmium, iridium ou
platine. Parmi les métaux
du groupe VIII on préfère employer un métal choisi dans le groupe formé par le
fer, le cobalt, le nickel,
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
14
le platine , le palladium et le ruthénium. Le catalyseur selon l'invention
peut également renfermer au
moins un élément du groupe VIB, de préférence le tungstène et le molybdène.
D'une manière
avantageuse on utilise les associations de métaux suivantes : nickel-
molybdène, cobalt-molybdène,
fer-molybdène, 'fer-tungstène, nickel-tungstène, cobalt-tungstène, platine-
palladium, les associations
préférées sont : nickel-molybdène, cobalt-molybdène, cobalt-tungstène et
encore plus
avantageusement platine-palladium et nickel-tungstène. Il est également
possible d'utiliser des
associations de trois métaux par exemple nickel-cobalt-molybdène, nickel-
molybdène-tungstène,
nickel-cobalt-tungstène. D'une manière avantageuse on utilise les associations
de métaux suivantes :
nickel-niobium-molybdène, cobalt-niobium-molybdène, fer-niobium-molybdène,
nickel-niobium-
tungstène, cobalt-niobium-tungstène, fer-niobium-tungstène, les associations
préférées étant : nickel-
niobium-molybdène, cobalt-niobium-molybdène. Il est également possible
d'utiliser des associations de
quatre métaux par exemple nickel-cobalt-niobium-molybdène. On peut également
utiliser des
associations contenant un métal noble tel que ruthénium-niobium-molybdène, ou
encore ruthénium-
nickel-niobium-molybdène.
L'un au moins des éléments suivants : phosphore et éventuellement bore et/ou
silicium et
éventuellement l'(les) élément(s) choisi(s) dans le(s) groupe(s) VIIB et VB,
sont introduits dans le
catalyseur à tout niveau de la préparation et selon toute technique connue de
l'homme du métier.
Une méthode préférée selon l'invention consiste à déposer le ou les éléments
dopants choisis
sur le précurseur calciné ou non, de préférence calciné. Pour le dépôt de bore
par exemple, on
prépare une solution aqueuse d'au moins un sel de bore tel que le biborate
d'ammonium ou le
pentaborate d'ammonium en milieu alcalin et en présence d'eau oxygénée et on
procède à une
imprégnation dite à sec, dans laquelle on remplit le volume des pores du
précurseur par la solution
contenant par exemple le bore. Dans le cas où l'on dépose par exemple
également du silicium, on
utilisera par exemple une solution d'un composé du silicium de type silicone
ou émulsion d'huile
silicone.
Le dépôt de bore et de silicium peut aussi être réalisé de manière simultanée
en utilisant par
exemple une solution contenant un sel de bore et un composé du silicium de
type silicone. Ainsi, par
exemple dans le cas où le précurseur est un catalyseur de type nickel-
tungstène supporté sur alumine-
silice, il est possible d'imprégner ce précurseur par de la solution aqueuse
de biborate d'ammonium et
de silicone Rhodorsil E1 P de la société Rhodia de procéder à un séchage par
exemple à 120 C, puis
d'imprégner par une solution de fluorure d'ammonium, de procéder à un séchage
par exemple à
120 C, et de procéder à une calcination par exemple et de façon préférée sous
air en lit traversé, par
exemple à 500 C pendant 4 heures.
L'élément dopant choisi dans le groupe formé par le phosphore, le silicium et
le bore ainsi que
les éléments des groupes VIIB, VB, peuvent être introduits par une ou
plusieurs opérations
d'imprégnation avec excès de solution sur le précurseur calciné.
Lorsqu'au moins un élément dopant, P et éventuellement B et/ou Si, est
introduit, sa répartition
et sa localisation peuvent être déterminées par des techniques telles que la
microsonde de Castaing
(profil de répartition des divers éléments), la microscopie électronique par
transmission couplée à une
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
analyse X des composants du catalyseurs, ou bien encore par l'établissement
d'une cartographie de
répartition des éléments présents dans le catalyseur par microsonde
électronique. Ces techniques
permettent de mettre en évidence la présence de ces éléments exogènes ajoutés
après la synthèse
de l'alumine-silice selon l'invention.
Il est intéressant de préparer des catalyseurs présentant des concentrations
homogènes CMo,
CN;, CW et CP le long de l'extrudé. Il est également intéressant de préparer
des catalyseurs présentant
des concentrations CMo, CNi, CW et CP au coeur et en périphérie différente.
Ces catalyseurs présentent
des profils de répartition dits en cuvette ou en cc dôme . Un autre type
de répartition est celle en
croûte où les éléments de la phase active sont répartis en surface.
De façon générale, le rapport coeur/bord des concentrations CMo, CN,, CW et CP
est compris entre
0.1 et 3. Dans une variante de l'invention, il est compris entre 0.8 et 1.2.
Dans une autre variante de
l'invention, le rapport coeur/bord des concentrations C est compris entre 0.3
et 0.8.
La source de phosphore préférée est l'acide orthophosphorique H3PO4, mais ses
sels et esters
comme les phosphates d'ammonium conviennent également. Le phosphore peut par
exemple être
introduit sous la forme d'un mélange d'acide phosphorique et un composé
organique basique
contenant de l'azote tels que l'ammoniaque, les amines primaires et
secondaires, les amines
cycliques, les composés de la famille de la pyridine et des quinoléines et les
composés de la famille du
pyrrole. Les acides tungsto-phosphorique ou tungsto-molybdique peuvent être
employés.
La teneur en phosphore est ajustée, sans que cela limite la portée de
l'invention, de telle
manière à former un composé mixte en solution et/ou sur le support par exemple
tungstène -
phosphore ou molybdène-tungstène-phosphore. Ces composés mixtes peuvent être
des
hétéropolyanions. Ces composés peuvent être des hétéropolyanions d'Anderson,
par exemple. La
teneur massique en phosphore calculée sous la forme P205 est comprise entre
0,01 et 6%, de
préférence entre 0,01 et 4%, de manière très préférée entre 0.01 et 2.5%.
La source de bore peut être l'acide borique, de préférence l'acide
orthoborique H3BO3, le
biborate ou le pentaborate d'ammonium, l'oxyde de bore, les esters boriques.
Le bore peut par
exemple être introduit sous la forme d'un mélange d'acide borique, d'eau
oxygénée et un composé
organique basique contenant de l'azote tels que l'ammoniaque, les amines
primaires et secondaires,
les amines cycliques, les composés de la famille de la pyridine et des
quinoléines et les composés de
la famille du pyrrole. Le bore peut être introduit par exemple par une
solution d'acide borique dans un
mélange eau/alcool.
De nombreuses sources de silicium peuvent être employées. Ainsi, on peut
utiliser l'orthosilicate
d'éthyle Si(OEt)4, les siloxanes, les polysiloxanes, les silicones, les
émulsions de silicones, les
silicates d'halogénures comme le fluorosilicate d'ammonium (NH4)2SiF6 ou le
fluorosilicate de sodium
Na2SiF6. L'acide silicomolybdique et ses sels, l'acide silicotungstique et ses
sels peuvent également
être avantageusement employés. Le silicium peut être ajouté par exemple par
imprégnation de silicate
d'éthyle en solution dans un mélange eau/alcool. Le silicium peut être ajouté
par exemple par
imprégnation d'un composé du silicium de type silicone ou l'acide silicique
mis en suspension dans
l'eau.
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
16
Les métaux du groupe VIB et du groupe VIII du catalyseur de la présente
invention peuvent être
présents en totalité ou partiellement sous forme métallique et/ou oxyde et/ou
sulfure.
Par exemple, parmi les sources de molybdène et de tungstène, on peut utiliser
les oxydes et
hydroxydes, les acides molybdiques et tungstiques et leurs sels en particulier
les sels d'ammonium tels
que le molybdate d'ammonium, l'heptamolybdate d'ammonium, le tungstate
d'ammonium, l'acide
phosphomolybdique, l'acide phosphotungstique et leurs sels, l'acide
silicomolybdique, l'acide
silicotungstique et leurs sels.
Les sources d'éléments du groupe VIII qui peuvent être utilisées sont bien
connues de l'homme du
métier. Par exemple, pour les métaux non nobles on utilisera les nitrates, les
sulfates, les hydroxydes, les
phosphates, les halogénures par exemple, chlorures, bromures et fluorures, les
carboxylates par exemple
acétates et carbonates. Pour les métaux nobles on utilisera les halogénures,
par exemple les chlorures, les
nitrates, les acides tels que l'acide chloroplatinique, les oxychlorures tels
que l'oxychlorure ammoniacal de
ruthénium.
De préférence, on n'ajoute pas d'halogènes autres que celui introduit à
l'imprégnation, cet halogène
étant de préférence le chlore.
Préparation du support
Le support peut être constitué d'alumine-silice pure ou résulte du mélange
avec ladite alumine-
silice d'un liant tel que la silice (SiO2), l'alumine (AI2O3), les argiles,
l'oxyde de titane (T102), l'oxyde de
bore (B2O3) et la zircone (ZrO2) et tout mélange des liants précédemment
cités. Les liants préférés
sont la silice et l'alumine et de manière encore plus préférée l'alumine sous
toutes ces formes connues
de l'homme du métier, par exemple l'alumine gamma. La teneur pondérale en
liant dans le support du
catalyseur est comprise entre 0 et 40%, plus particulièrement entre 1 et 40%
et de manière encore
plus préférée entre 5% et 20%. Cependant, les catalyseurs selon l'invention
dont le support est
constitué uniquement d'alumine-silice sans aucun liant sont préférés.
Le support peut être préparé par mise en forme de l'alumine-silice en présence
ou en absence de liant
par toute technique connue de l'homme du métier.
Dans l'ensemble des méthodes précitées, il peut être éventuellement
souhaitable d'ajouter, lors
d'une étape quelconque de la préparation, une proportion mineure d'au moins un
élément promoteur choisi
dans le groupe formé par la zircone et le titane.
Mise en forme des supports et catalyseurs
Le support peut être obtenu par mise en forme de l'alumine-silice par toute
technique connue
de l'homme du métier. La mise en forme peut être réalisée par exemple par
extrusion, par pastillage,
par la méthode de la coagulation en goutte (oil-drop), par granulation au
plateau tournant ou par toute
autre méthode bien connue de l'homme du métier.
La mise en forme peut également être réalisée en présence des différents
constituants du
catalyseur et extrusion de la pâte minérale obtenue, par pastillage, mise en
forme sous forme de billes
au drageoir tournant ou au tambour, coagulation en goutte, oil-drop, oil-up,
ou tout autre procédé
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
17
connu d'agglomération d'une poudre contenant de l'alumine et éventuellement
d'autres grédients
choisis parmi ceux mentionnés ci-dessus. _--
Les catalyseurs utilisés selon l'invention ont la forme de sphères ou
d'extrudés. Il est toutefois
avantageux que le catalyseur se présente sous forme d'extrudés d'un diamètre
compris entre 0,5 et 5
mm et plus particulièrement entre 0,7 et 2,5 mm. Les formes sont cylindriques
(qui peuvent être
creuses ou non), cylindriques torsadés, multilobées (2, 3, 4 ou 5 lobes par
exemple), anneaux. La
forme cylindrique est utilisée de manière préférée, mais toute autre forme
peut être utilisée.
Par ailleurs, ces supports mis en oeuvre selon la présente invention peuvent
avoir été traités
ainsi qu'il est bien connu de l'homme de l'art par des additifs pour faciliter
la mise en forme et/ou
améliorer les propriétés mécaniques finales des supports alumino-silicates. A
titre d'exemple d'additifs,
on peut citer notamment la cellulose, la carboxyméthyl-cellulose, la carboxy-
ethyl-cellulose, du tail-oil,
les gommes xanthaniques, des agents tensio-actifs, des agents flocculants
comme les
polyacrylamides, le noir de carbone, les amidons, l'acide stéarique, l'alcool
polyacrylique, l'alcool
polyvinylique, des biopolymères, le glucose, les polyéthylènes glycols, etc.
Le réglage de la porosité caractéristiques des supports de l'invention est
opéré partiellement
lors de cette étape de mise en forme des particules de supports.
La mise en forme peut être réalisée en utilisant les techniques de mise en
forme des
catalyseurs, connues de l'homme de l'art, telles que par exemple: extrusion,
dragéification, séchage
par atomisation ou encore pastillage.
On peut ajouter ou retirer de l'eau pour ajuster la viscosité de la pâte à
extruder. Cette étape
peut être réalisée à tout stade de l'étape de malaxage. Dans le cas de
supports alumino-silicates, il
peut être avantageux de diminuer la quantité d'eau de la pâte afin d'accroître
la puissance mécanique
fournie à la pâte. Cette action se traduit généralement par une diminution du
volume total pour une
teneur en acide optimale.
Pour ajuster la teneur en matière solide de la pâte à extruder afin de la
rendre extrudable, on
peut également ajouter un composé majoritairement solide et de préférence un
oxyde ou un hydrate.
On utilisera de manière préférée un hydrate et de manière encore plus préférée
un hydrate
d'aluminium. La perte au feu de cet hydrate sera supérieure à 15%.
La teneur en acide ajouté au malaxage avant la mise en forme est inférieure à
30%, de
préférence comprise entre 0,5 et 20% poids de la masse anhydre en silice et
alumine engagée dans la
synthèse.
L'extrusion peut être réalisée par n'importe quel outil conventionnel,
disponible
commercialement. La pâte issue du malaxage est extrudée à travers une filière,
par exemple à l'aide
d'un piston ou d'une mono-vis ou double vis d'extrusion. Cette étape
d'extrusion peut être réalisée par
toute méthode connue de l'homme de métier.
Les extrudés de support selon l'invention ont généralement une résistance à
l'écrasement d'au moins
70 N/cm et de manière préférée supérieure ou égale à 100 N/cm.
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
18
Calcination du support
Le séchage est effectué par toute technique connue de l'homme du métier.
Pour obtenir le support de la présente invention, il est préférable de
calciner de préférence en
présence d'oxygène moléculaire, par exemple en effectuant un balayage d'air, à
une température
inférieure ou égale à 1100 C. Au moins une calcination peut être effectuée
après l'une quelconque
des étapes de la préparation. Ce traitement par exemple peut être effectué en
lit traversé, en lit
léché ou en atmosphère statique. Par exemple, le four utilisé peut être un
four rotatif tournant ou
être un four vertical à couches traversées radiales. Les conditions de
calcination: température et
durée dépendent principalement de la température maximale d'utilisation du
catalyseur. Les
conditions préférées de calcination se situant entre plus d'une heure à 200 C
à moins d'une
heure à 1100 C. La calcination peut être opérée en présence de vapeur d'eau.
La calcination
finale peut être éventuellement effectuée en présence d'une vapeur acide ou
basique. Par
exemple, la calcination peut être réalisée sous pression partielle
d'ammoniaque.
Traitements post-synthèse
.Des traitements post-synthèse peuvent être effectués, de manière à améliorer
les propriétés du
support, notamment son homogénéité telle que définie précédemment.
Selon un mode de réalisation préféré, le traitement post-synthèse est un
traitement
hydrothermal. Le traitement hydrothermal est effectué par toute technique
connue de l'homme du
métier. Par traitement hydrothermal, on entend mise en contact à n'importe
quel étape de l'élaboration
du support mixte avec de l'eau en phase vapeur ou en phase liquide. Par
traitement hydrothermal, on
peut entendre notamment mûrissement, steaming (traitement à la vapeur),
autoclavage, calcination
sous air humide, réhydratation. Sans que cela réduise la portée de
l'invention, un tel traitement a pour
effet de rendre mobile le composant silice.
Selon l'invention, le mûrissement peut avoir lieu avant ou après la mise en
forme. Selon un mode
préféré de l'invention, le traitement hydrothermal se fait par steaming
(traitement à la vapeur) dans un four en
présence de vapeur d'eau. La température pendant le steaming (traitement à la
vapeur) peut être comprise
entre 600 et 1100 C et de préférence supérieure à 700 C pendant une période
de temps comprise entre 30
minutes et 3 heures. La teneur en vapeur d'eau est supérieure à 20 g d'eau par
kg d'air sec et de préférence
supérieure à 40 g d'eau par kg d'air sec et de manière préférée supérieure à
100 g d'eau par kg d'air sec. Un
tel traitement peut, le cas échéant, remplacer totalement ou en partie le
traitement de calcination.
Le support peut ainsi être avantageusement soumis à un traitement hydrothermal
en atmosphère
confinée. On entend par traitement hydrothermal en atmosphère confinée un
traitement par passage à
l'autoclave en présence d'eau sous une température supérieure à la température
ambiante.
Au cours de ce traitement hydrothermal, on peut traiter de différentes
manières l'alumine-silice mise
en forme. Ainsi, on peut imprégner l'alumine silice d'acide, préalablement à
son passage à l'autoclave,
l'autoclavage de l'alumine-silice étant fait soit en phase vapeur, soit en
phase liquide, cette phase vapeur ou
liquide de l'autoclave pouvant être acide ou non. Cette imprégnation,
préalable à l'autoclavage, peut être
acide ou non. Cette imprégnation, préalable à l'autoclavage peut être
effectuée à sec ou par immersion de
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
19
l'alumine-silice dans une solution aqueuse acide. Par imprégnation à sec, on
entend mise en contact de
l'alumine avec un volume de solution inférieur ou égal au volume poreux total
de l'alumine traitée. De
préférence, l'imprégnation est réalisée à sec.
L'autoclave est de préférence un autoclave à panier rotatif tel que celui
défini dans la demande
brevet EP-A- 0 387 109.
La température pendant l'autoclavage peut être comprise entre 100 et 250 C
pendant une période de
temps comprise entre 30 minutes et 3 heures.
Modes de réalisation du procédé selon l'invention
L'invention est ci-après décrite dans des modes préférés de réalisation et à
partir des figures 1 à
6.
Un mode de réalisation de l'invention comprend les étapes suivantes :
a) séparation d'une seule fraction dite lourde à point d'ébullition initial
compris entre 120-200 C,
b) hydrotraitement d'une partie au moins de ladite fraction lourde,
c) fractionnement en au moins 3 fractions :
- au moins une fraction intermédiaire ayant un point d'ébullition initial T1
compris entre 120 et
200 C, et un point d'ébullition final T2 supérieur à 300 C et inférieur à 410
C,
- au moins une fraction légère bouillant au-dessous de la fraction
intermédiaire,
- au moins une fraction lourde bouillant au-dessus de la fraction
intermédiaire.
d) passage d'une partie au moins de ladite fraction intermédiaire dans un
procédé selon l'invention
sur un catalyseur non zéolitique d'hydroisomérisation I hydrocraquage,
e) passage dans un procédé selon l'invention sur un catalyseur non zéolitique
d'hydroisomérisation /
hydrocraquage d'une partie au moins de ladite fraction lourde,
f) distillation des fractions hydrocraquées / hydroisomérisées pour obtenir
des distillats moyens, et
recyclage de la fraction résiduelle bouillant au-dessus desdits distillats
moyens dans l'étape (e) sur
le catalyseur traitant la fraction lourde.
La description de ce mode de réalisation sera faite en se référant à la figure
1 sans que la figure
1 limite l'interprétation.
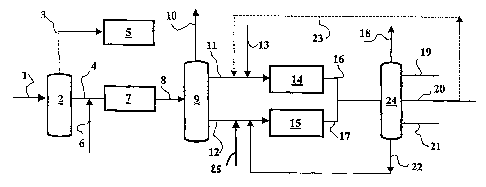
Etape a
L'effluent issu de l'unité de synthèse Fischer-Tropsch arrivant par la
conduite 1 est fractionné
(par exemple par distillation) dans un moyen de séparation (2) en au moins
deux fractions : au moins
une fraction légère et une fraction lourde à point d'ébullition initial égal à
une température comprise
entre 120 et 200 C et de préférence entre 130 et 180 C et de manière encore
plus préférée à une
température d'environ 150 C, en d'autres termes le point de coupe est situé
entre 120 et 200 C. La
fraction légère de la figure 1 sort par la conduite (3) et la fraction lourde
par la conduite (4).
Ce fractionnement peut être réalisé par des méthodes bien connues de l'homme
du métier
telles que le flash, la distillation etc... A titre d'exemple non limitatif,
l'effluent issu de l'unité de synthèse
Fischer-Tropsch sera soumis à un flash, une décantation pour éliminer l'eau et
une distillation afin
d'obtenir au moins les 2 fractions décrites ci-dessus.
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
La fraction légère n'est pas traitée selon le procédé de l'invention mais peut
par exemple
constituer une bonne charge pour la pétrochimie et plus particulièrement pour
une unité (5) de
vapocraquage. La fraction lourde précédemment décrite est traitée selon le
procédé de l'invention.
Etape (b)
Cette fraction est admise en présence d'hydrogène (conduite 6) dans une zone
(7) contenant un
catalyseur d'hydrotraitement qui a pour objectif de réduire la teneur en
composés oléfiniques et
insaturés ainsi que d'hydrotraiter les composés oxygénés (alcools) présents
dans la fraction lourde
décrite ci-dessus.
Les catalyseurs utilisés dans cette étape (b) sont des catalyseurs
d'hydrotraitement non
craquants ou peu craquants comportant au moins un métal du groupe VIII et/ou
du groupe VI de la
classification périodique des éléments.
Avantageusement, au moins un élément choisi parmi P, B, Si est déposé sur le
support.
Ces catalyseurs peuvent être préparés par toutes les méthodes connues de
l'homme de l'art ou bien
peuvent être acquis auprès de sociétés spécialisées dans la fabrication et la
vente de catalyseurs.
Dans le réacteur d'hydrotraitement (7), la charge est mise. en contact en
présence d'hydrogène et du
catalyseur à des températures et des pressions opératoires permettant de
réaliser
l'hydrodeoxygénation (HDO) des alcools et l'hydrogénation des oléfines
présents dans la charge. Les
températures réactionnelles utilisées dans le réacteur d'hydrotraitement sont
comprises entre 100 et
350 C, de préférence entre 150 et 300 C, de façon encore plus préférée entre
150 et 275 C et mieux
encore entre 175 et 250 C. La gamme de pression totale utilisée varie de 5 à
150 bars, de préférence
entre 10 et 100 bars et de manière encore plus préférée entre 10 et 90 bars.
L'hydrogène qui alimente
le réacteur d'hydrotraitement est introduit à un débit tel que le rapport
volumique
hydrogène/hydrocarbures soit compris entre 100 à 3000 NI/I/h, de préférence
entre 100 et 2000NI/l/h et
de façon encore plus préférée entre 250 et 1500 NI/I/h. Le débit de charge est
tel que la vitesse
volumique horaire est comprise entre 0,1 et 10h-1, de préférence entre 0,2 et
5h-1 et de manière encore
plus préférée entre 0,2 et 3h"1. Dans ces conditions, la teneur en molécules
insaturées et oxygénées
est réduite à moins de 0,5% et à environ moins de 0,1% en général. L'étape
d'hydrotraitement est
conduite dans des conditions telles que la conversion en produits ayant des
points d'ébullition
supérieurs ou égaux à 370 C en des produits ayant des points d'ébullition
inférieurs à 370 C est
limitée à 30% poids, de préférence est inférieure à 20% et de façon encore
plus préférée est inférieure
à 10%.
Etape c)
L'effluent issu du réacteur d'hydrotraitement est amené par une conduite (8)
dans une zone de
fractionnement (9) où il est fractionné en au moins trois fractions :
- au moins une fraction légère (sortant par la conduite 10) dont les composés
constituants ont des
points d'ébullition inférieurs à une température Ti comprise entre 120 et 200
C, et de préférence
entre 130 et 180 C et de manière encore plus préférée à une température
d'environ 150 C. En
d'autres termes le point de coupe est situé entre 120 et 200 C.
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
21
au moins une fraction intermédiaire (conduite 11) comportant les composés dont
les points
d'ébullition sont compris entre le point de coupe Ti, précédemment défini, et
une température T2
supérieure à 300 C, de manière encore plus préférée supérieure à 350 C et
inférieure à 410 C ou
mieux à 370 C.
au moins une fraction dite lourde (conduite 12) comportant les composés ayant
des points
d'ébullition supérieurs au point de coupe T2 précédemment défini.
Le fractionnement est obtenu ici par distillation, mais il peut être réalisé
en une ou plusieurs
étapes et par d'autres moyens que la distillation.
Ce fractionnement peut être réalisé par des méthodes bien connues de l'homme
du métier telles que
le flash, la distillation etc...
Les fractions intermédiaires et lourdes précédemment décrites sont traitées
selon le procédé de
l'invention.
Etape (d)
Une partie au moins de ladite fraction intermédiaire est alors introduite
(conduite 11), ainsi
qu'éventuellement un flux d'hydrogène, (conduite 13) dans la zone (14)
contenant le catalyseur
d'hydroisomérisation / d'hydrocraquage du procédé selon la présente invention.
Les conditions opératoires dans lesquelles est effectuée cette étape (d) sont
:
La pression est maintenue entre 2 et 150 bars et de préférence entre 5 et 100
bars et
avantageusement de 10 à 90 bars, la vitesse spatiale est comprise entre 0,1
h"1 et 10 h'1 et de
préférence entre 0,2 et 7h"1 est avantageusement entre 0,5 et 5,0h"1. Le taux
d'hydrogène est compris
entre 100 et 2000 Normaux litres d'hydrogène par litre de charge et par heure
et préférentiellement
entre 150 et 1500 litres d'hydrogène par litre de charge.
La température utilisée dans cette étape est comprise entre 200 et 450 C et
préférentiellement de
250 C à 450 C avantageusement de 300 à 450 C, et encore plus avantageusement
supérieure à
320 C ou par exemple entre 320-420 C.
L'étape (d) d'hydroisomérisation et d'hydrocraquage est avantageusement
conduite dans des
conditions telles que la conversion par passe en produits à points
d'ébullition supérieurs ou égaux à
150 C en des produits ayant des points d'ébullition inférieurs à 150 C est la
plus faible possible, de
préférence inférieure à 50%, de manière encore plus préférée inférieure à 30%,
et permet d'obtenir
des distillats moyens (gazole et kérosène) ayant des propriétés à froid (point
d'écoulement et de
congélation) suffisamment bonnes pour satisfaire aux spécifications en vigueur
pour ce type de
carburant.
Ainsi dans cette étape (d), on cherche à favoriser l'hydroisomérisation plutôt
que l'hydrocraquage.
Etaee
Une partie au moins de ladite fraction lourde est introduite via la ligne (12)
dans une zone (15)
où elle est mise, en présence d'hydrogène (25), au contact d'un catalyseur
d'hydroisomérisation/hydrocraquage selon le procédé de la présente invention
afin de produire une
coupe distillat moyen (kérosène + gazole) présentant de bonnes propriétés à
froid.
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
22
Le catalyseur utilisé dans la zone (15) de l'étape (e) pour réaliser les
réactions
d'hydrocraquage et d'hydroisomérisation de la fraction lourde, définie selon
l'invention, est du même
type que celui présent dans le réacteur (14). Cependant, il est à noter que
les catalyseurs mis en
oeuvre dans les réacteurs (14) et (15) peuvent être identiques ou différents.
Durant cette étape (e) la fraction entrant dans le réacteur subit au contact
du catalyseur et en
présence d'hydrogène essentiellement des réactions d'hydrocraquage qui,
accompagnés de réactions
d'hydroisomérisation des n-paraffines, vont permettre d'améliorer la qualité
des produits formés et plus
particulièrement les propriétés à froid du kérosène et du gazole, et également
d'obtenir de très bons
rendements en distillats. La conversion en produits ayant des points
d'ébullition supérieurs ou égal à
370 C en produits à points d'ébullition inférieurs à 370 C est supérieure à
80% poids, souvent d'au
moins 85% et de préférence supérieure ou égal à 88%. Par contre, les
conversions des produits à
point d'ébullition supérieurs ou égaux à 260 C en produits à points
d'ébullition inférieurs à 260 C est
d'au plus 90% poids, généralement d'au plus 70% ou 80%, et de préférence d'au
plus 60% poids.
Etape f
Les effluents en sortie des réacteurs (14) et (15) sont envoyés par les
conduites (16) et (17) dans un
train de distillation, qui intègre une distillation atmosphérique et
éventuellement une distillation sous
vide, et qui a pour but de séparer d'une part les produits légers
inévitablement formés lors des étapes
(d) et (e) par exemple les gaz (Ci-C4) (conduite 18) et une coupe essence
(conduite 19), et de distiller
au moins une coupe gazole (conduite 21) et kérosène (conduite 20). Les
fractions gazole et kérosène
peuvent être recyclées (conduite 23) en partie, conjointement ou de façon
séparée, en tête du réacteur
(14) d'hydroisomérisation /hydrocraquage étape (d).
Il est également distillé une fraction (conduite 22) bouillant au-dessus du
gazole, c'est à dire
dont les composés qui la constituent ont des points d'ébullition supérieurs à
ceux des distillats moyens
(kérosène + gazole). Cette fraction, dite fraction résiduelle, présente
généralement un point d'ébullition
initial d'au moins 350 C, de préférence supérieure à 370 C. Cette fraction est
avantageusement
recyclée en tête du réacteur (15) via la conduite (26) d'hydroisomérisation
/hydrocraquage de la
fraction lourde (étape e).
Il peut être également avantageux de recycler une partie du kérosène et/ou du
gazole dans
l'étape (d), l'étape (e) ou les deux. De façon préférée, l'une au moins des
fractions kérosène et/ou
gazole est recyclée en partie dans l'étape (d) (zone 14). On a pu constater
qu'il est avantageux de
recycler une partie du kérosène pour améliorer ses propriétés à froid.
Avantageusement et dans le même temps, la fraction non hydrocraquée est
recyclée en partie dans
l'étape (e) (zone 15).
Il va sans dire que les coupes gazole et kérosène sont de préférence
récupérées séparément,
mais les points de coupe sont ajustés par l'exploitant en fonction de ses
besoins.
Sur la figure 1, on a représenté une colonne (24) de distillation, mais deux
colonnes peuvent
être utilisées pour traiter séparément les coupes issues de zones (14) et
(15).
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
23
Sur la figure 1, on a représenté seulement le recyclage du kérosène sur le
catalyseur du
réacteur (14). Il va sans dire qu'on peut aussi bien recycler une partie du
gazole (séparément ou avec
le kérosène) et de préférence sur le même catalyseur que le kérosène.
Un autre mode de réalisation de l'invention comprend les étapes suivantes :
a) séparation d'au moins une fraction légère de la charge de façon à obtenir
une seule fraction dite
lourde à point d'ébullition initial compris entre 120-200 C,
b) éventuel hydrotraitement de ladite fraction lourde, éventuellement suivi
d'une étape
c) d'enlèvement d'au moins une partie de l'eau,
d) passage dans un procédé selon l'invention d'une partie au moins de ladite
fraction éventuellement
hydrotraitée, la conversion sur le catalyseur
d'hydroisomérisation/hydrocraquage des produits à
points d'ébullition supérieurs ou égaux à 370 C en produits à points
d'ébullition inférieures à 370 C
est supérieure à 80% pds,
e) distillation de la fraction hydrocraquée/hydroisomérisée pour obtenir des
distillats moyens, et
recyclage dans l'étape d) de la fraction résiduelle bouillant au-dessus
desdits distillats moyens.
La description de ce mode de réalisation sera faite en se référant à la figure
2 sans que la figure 2
limite l'interprétation.
Etape (a)
L'effluent issu de l'unité de synthèse Fischer-Tropsch arrivant par la
conduite 1 est fractionné
(par exemple par distillation) dans un moyen de séparation (2) en au moins
deux fractions : au moins
une fraction légère et une fraction lourde à point d'ébullition initial égal à
une température comprise
entre 120 et 200 C et de préférence entre 130 et 180 C et de manière encore
plus préférée à une
température d'environ 150 C, en d'autres termes le point de coupe est situé
entre 120 et 200 C. La
fraction légère de la figure 1 sort par la conduite (3) et la fraction lourde
par la conduite (4).
Ce fractionnement peut être réalisé par des méthodes bien connues de l'homme
du métier
telles que le.flash, la distillation etc...
La fraction légère n'est pas traitée selon le procédé de l'invention mais peut
par exemple
constituer une bonne charge pour la pétrochimie et plus particulièrement pour
une unité (5) de
vapocraquage. La fraction lourde précédemment décrite est traitée selon le
procédé de l'invention.
Etape b
Eventuellement, cette fraction est admise en présence d'hydrogène (conduite 6)
dans une zone
(7) contenant un catalyseur d'hydrotraitement qui a pour objectif de réduire
la teneur en composés
oléfiniques et insaturés ainsi que d'hydrotraiter les composés oxygénés
(alcools) présents dans la
fraction lourde décrite ci-dessus.
Les catalyseurs utilisés dans cette étape (b) sont des catalyseurs
d'hydrotraitement non
craquants ou peu craquants comportant au moins un métal du groupe VIII et/ou
du groupe VI de la
classification périodique des éléments.
Avantageusement, au moins un élément choisi parmi P, B, Si est déposé sur le
support.
Ces catalyseurs peuvent être préparés par toutes les méthodes connues de
l'homme de l'art ou
bien peuvent être acquis auprès de sociétés spécialisées dans la fabrication
et la vente de catalyseurs.
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
24
Dans le réacteur d'hydrotraitement (7), la charge est mise en contact en
présence d'hydrogène
et du catalyseur à des températures et des pressions opératoires permettant de
réaliser
l'hydrodeoxygénation (HDO) des alcools et l'hydrogénation des oléfines
présents dans la charge. Les
températures réactionnelles utilisées dans le réacteur d'hydrotraitement sont
comprises entre 100 et
350 C, de préférence entre 150 et 300 C, de façon encore plus préférée entre
150 et 275 C et mieux
encore entre 175 et 250 C. La gamme de pression totale utilisée varie de 5 à
150 bars, de préférence
entre 10 et 100 bars et de manière encore plus préférée entre 10 et 90 bars.
L'hydrogène qui alimente
le réacteur d'hydrotraitement est introduit à un débit tel que le rapport
volumique
hydrogène/hydrocarbures soit compris entre 100 à 3000 NIA/h, de préférence
entre 100 et 2000NI/l/h et
de façon encore plus préférée entre 250 et 1500 NI/I/h. Le débit de charge est
tel que la vitesse
volumique horaire est comprises entre 0,1 et 10h"1, de préférence entre 0,2 et
5h'1 et de manière
encore plus préférée entre 0,2 et 30. Dans ces conditions, la teneur en
molécules insaturées et
oxygénées est réduite à moins de 0,5% et à environ moins de 0,1% en général.
L'étape
d'hydrotraitement est conduite dans des conditions telles que la conversion en
produits ayant des
points d'ébullition supérieurs ou égaux à 370 C en des produits ayant des
points d'ébullition inférieurs
à 370 C est limitée à 30% pds, de préférence est inférieure à 20% et de façon
encore plus préférée
est inférieure à 10%.
Etape c
L'effluent (conduite 8) issu du réacteur (7) d'hydrotraitement est
éventuellement introduit dans
une zone (9) d'enlèvement d'eau qui a pour but d'éliminer au moins en partie
l'eau produite lors des
réactions d'hydrotraitement. Cette élimination d'eau peut s'effectuer avec ou
sans élimination de la
fraction gazeuse C4 moins qui est généralement produite lors de l'étape
d'hydrotraitement. On entend
par élimination de l'eau, l'élimination de l'eau produite par les réactions
d'hydrodeoxygénation (HDO)
des alcools mais on peut aussi y inclure l'élimination au moins en partie de
l'eau de saturation des
hydrocarbures. L'élimination de l'eau peut être réalisée par toutes les
méthodes et techniques connues
de l'homme du métier, par exemple par séchage, passage sur un dessicant,
flash, décantation...
Etape (d)
La fraction lourde (éventuellement hydrotraitée) ainsi séchée est alors
introduite (conduite 10)
ainsi qu'éventuellement un flux d'hydrogène (conduite 11), dans la zone (12)
contenant le catalyseur
d'hydroisomérisation / hydrocraquage. Une autre éventualité du procédé aussi
selon l'invention
consiste à envoyer la totalité de l'effluent sortant du réacteur
d'hydrotraitement (sans séchage) dans le
réacteur contenant le catalyseur d'hydroisomérisation / hydrocraquage et de
préférence en même
temps qu'un flux d'hydrogène.
Avant utilisation dans la réaction, le métal contenu dans le catalyseur doit
être réduit. Une des
méthodes préférées pour conduire la réduction du métal est le traitement sous
hydrogène à une
température comprise entre 150 C et 650 C et une pression totale comprise
entre 0,1 et 25 Mpa. Par
exemple, une réduction consiste en un palier à 150 C de 2 heures puis une
montée en température
jusqu'à 450 C à la vitesse de 1 C/min puis un palier de 2 heures à 450 C ;
durant toute cette étape de
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
réduction, le débit d'hydrogène est de 1000 litres hydrogène/ litre
catalyseur. Notons également que
toute méthode de réduction ex-situ est convenable.
Les conditions opératoires dans lesquelles est effectuée cette étape (d) sont
:
La pression est maintenue entre 2 et 150 bars et de préférence entre 5 et 100
bars et
avantageusement de 10 à 90 bars, la vitesse spatiale est comprise entre 0,1
h"1 et 10 h-1 et de
préférence entre 0,2 et 7h"1 est avantageusement entre 0,5 et 5,0h"1. Le taux
d'hydrogène est compris
entre 100 et 2000 Normaux litres d'hydrogène par litre de charge et par heure
et préférentiellement
entre 150 et 1500 litres d'hydrogène par litre de charge.
La température utilisée dans cette étape est comprise entre 200 et 450 C et
préférentiellement
de 250 C à 450 C avantageusement de 300 à 450 C, et encore plus
avantageusement supérieure à
320 C ou par exemple entre 320-420 C.
L'étape d'hydroisomérisation et d'hydrocraquage est conduite dans des
conditions telles que la
conversion par passe en produits à points d'ébullition supérieurs ou égaux à
370 C en des produits
ayant des points d'ébullition inférieurs à 370 C est supérieure à 80% poids,
et de façon encore plus
préférée d'au moins 85%, de préférence supérieure à 88%, de manière à obtenir
des distillats moyens
(gazole et kérosène) ayant des propriétés à froid suffisamment bonnes (point
d'écoulement, point de
congélation) pour satisfaire aux spécifications en vigueur pour ce type de
carburant.
Les deux étapes, hydrotraitément et hydroisomérisation-hydrocraquage, peuvent
être réalisées
sur les deux types de catalyseurs dans deux ou plusieurs réacteurs différents,
ou/et dans un même
réacteur.
Etape (e)
L'effluent (fraction dite hydrocraquée / hydroisomérisée) en sortie du
réacteur (12), étape (d),
est envoyé dans un train de distillation (13), qui intègre une distillation
atmosphérique et
éventuellement une distillation sous vide, qui a pour but de séparer les
produits de conversion de point
d'ébullition inférieur à 340 C et de préférence inférieur à 370 C et incluant
notamment ceux formés
lors de l'étape (d) dans le réacteur (12), et de séparer la fraction
résiduelle dont le point initial
d'ébullition est généralement supérieur à au moins 340 C et de préférence
supérieur ou égal à au
moins 370 C. Parmi les produits de conversion et hydroisomérisés, il est
séparé outre les gaz légers
C1-C4 (conduite 14) au moins une fraction essence (conduite 15), et au moins
une fraction distillat
moyen kérosène (conduite 16) et gazole (conduite 17). La fraction résiduelle
dont le point initial
d'ébullition est généralement supérieur à au moins 340 C et de préférence
supérieur ou égal à au
moins 370 C est recyclée (conduitel8) en tête du réacteur (12)
d'hydroisomérisation et
d'hydrocraquage.
Il peut être également avantageux de recycler (conduite 19) dans l'étape (d)
(réacteur 12) une
partie du kérosène et/ou du gazole ainsi obtenus.
Un autre mode de réalisation de l'invention comprend les étapes suivantes :
a) Fractionnement (étape a) de la charge en au moins 3 fractions :
- au moins une fraction intermédiaire ayant un point d'ébullition initial Ti
compris entre 120 et
200 C, et un point d'ébullition final T2 supérieur à 3000C et inférieur à 410
C,
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
26
- au moins une fraction légère bouillant au-dessous de la fraction
intermédiaire,
- au moins une fraction lourde bouillant au-dessus de la fraction
intermédiaire.
b) Hydrotraitement (étape b) d'au moins une partie de ladite fraction
intermédiaire, puis passage
(étape d) dans un procédé de traitement d'au moins une partie de la fraction
hydrotraitée sur un
catalyseur d'hydrocraquage/hydroisomérisation.
f) Passage (étape f) dans un procédé de traitement d'une partie au moins de
ladite fraction lourde
sur un catalyseur d'hydrocraquage/hydroisomérisation avec une conversion des
produits 370 C+ en
produits 370 C moins supérieure à 80% poids.
e) et g) Distillation (étapes e et g) d'au moins une partie des fractions
hydrocraquées /
hydroisomérisées pour obtenir des distillats moyens.
et l'un au moins desdits procédés de traitement des étapes d) et f) est le
procédé de l'invention.
La description de ce mode de réalisation sera faite en se référant à la figure
3 sans que la
figure 3 limite l'interprétation.
Etape (a)
L'effluent issu de l'unité de synthèse Fischer-Tropsch arrivant par la
conduite (1) est fractionné
dans un zone de fractionnement (2) en au moins trois fractions :
- au moins une fraction légère (sortant par la conduite 3) dont les composés
constituants ont des
points d'ébullition inférieurs à une température Ti comprise entre 120 et 200
C, et de préférence
entre 130 et 180 C et de manière encore plus préférée à une température
d'environ 150 C. En
d'autres termes le point de coupe est situé entre 120 et 200 C.
- au moins une fraction intermédiaire (conduite 4) comportant les composés
dont les points
d'ébullition sont compris entre le point de coupe Ti, précédemment défini, et
une température T2
supérieure à 300 C, de manière encore plus préférée supérieure à 350 C et
inférieure à 410 C ou
mieux à 370 C.
- au moins une fraction dite lourde (conduite 5) comportant les composés ayant
des points
d'ébullition supérieurs au point de coupe T2 précédemment défini.
Une coupe entre un point d'ébullition Ti comprise entre 120 - 200 C et T2
supérieure à 300 C et
inférieure à 370 C est préférée. La coupe à 370 C est encore plus préférée
c'est à dire la fraction
lourde est une coupe 370 C+.
Le fait de couper à 370 C permet de séparer au moins 90% pds des oxygénés et
des oléfines, et
le plus souvent au moins 95% pds. La coupe lourde à traiter est alors purifiée
et une élimination des
hétéroatomes ou insaturés par hydrotraitement n'est alors pas nécessaire.
Le fractionnement est obtenu ici par distillation, mais il peut être réalisé
en une ou plusieurs
étapes et par d'autres moyens que la distillation.
Ce fractionnement peut être réalisé par des méthodes bien connues de l'homme
du métier telles
que le flash, la distillation etc...
La fraction légère n'est pas traitée selon le procédé de l'invention mais peut
par exemple
constituer une bonne charge pour une unité pétrochimique et plus
particulièrement pour un
vapocraqueur (installation 6 de vapocraquage).
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
27
Les fractions plus lourdes précédemment décrites sont traitées selon le
procédé de l'invention.
Etape b
Ladite fraction intermédiaire est admise via la ligne (4), en présence
d'hydrogène amené par la
tubulure (7), dans une zone d'hydrotraitement (8) contenant un catalyseur
d'hydrotraitement. L'objectif
de cet hydrotraitement est de réduire la teneur en composés oléfiniques et
insaturés ainsi que
d'hydrotraiter les composés oxygénés (alcools) présents.
Les catalyseurs utilisés dans cette étape (b) sont des catalyseurs
d'hydrotraitement non
craquants ou peu craquants comportant au moins un métal du groupe VIII et/ou
du groupe VI de la
classification périodique des éléments.
Avantageusement, au moins un élément choisi parmi P, B, Si est déposé sur le
support.
Ces catalyseurs peuvent être préparés par toutes les méthodes connues de
l'homme de l'art
ou bien peuvent être acquis auprès de sociétés spécialisées dans la
fabrication et la vente de
catalyseurs.
Dans le réacteur d'hydrotraitement (8), la charge est mise en contact en
présence d'hydrogène
et du catalyseur à des températures et des pressions opératoires permettant de
réaliser
l'hydrodeoxygénation (HDO) des alcools et l'hydrogénation des oléfines
présents dans la charge. Les
températures réactionnelles utilisées dans le réacteur d'hydrotraitement sont
comprises entre 100 et
350, de préférence entre 150 et 300 C, de façon encore plus préférée entre 150
et 275 C et mieux
encore entre 175 et 250 C. La gamme de pression totale utilisée varie de 5 à
150 bars, de préférence
entre 10 et 100 bars et de manière encore plus préférée entre 10 et 90 bars.
L'hydrogène qui alimente
le réacteur d'hydrotraitement est introduit à un débit tel que le rapport
volumique
hydrogène/hydrocarbures soit compris entre 100 à 3000 NI/l/h, de préférence
entre 100 et 2000NI/l/h et
de façon encore plus préférée entre 250 et 1500 NI/I/h. Le débit de charge est
tel que la vitesse
volumique horaire est comprises entre 0,1 et 10h"1, de préférence entre 0,2 et
5h"1 et de manière
encore plus préférée entre 0,2 et 3h"1. Dans ces conditions, la teneur en
molécules insaturées et
oxygénées est réduite à moins de 0,5% et à environ moins de 0,1% en général.
L'étape
d'hydrotraitement -est conduite dans des conditions telles que la conversion
en produits ayant des
points d'ébullition supérieurs ou égaux à 370 C en des produits ayant des
points d'ébullition inférieurs
à 370 C est limitée à 30% pds, de préférence est inférieure à 20% et de façon
encore plus préférée
est inférieure à 10%.
Etape c
L'effluent issu du réacteur d'hydrotraitement est éventuellement introduit
dans une zone (9)
d'enlèvement d'eau qui a pour but d'éliminer au moins une partie de l'eau
produite lors des réactions
d'hydrotraitement. Cette élimination d'eau peut s'effectuer avec ou sans
élimination de la fraction
gazeuse C4 moins qui est généralement produite lors de l'étape
d'hydrotraitement. On entend par
élimination de l'eau, l'élimination de l'eau produite par les réactions
d'hydrodeoxygénâtion (HDO) des
alcools, mais on peut aussi y inclure l'élimination au moins en partie de
l'eau de saturation des
hydrocarbures. L'élimination de l'eau peut être réalisée par toutes les
méthodes et techniques connues
de l'homme du métier, par exemple par séchage, passage sur un dessicant,
flash, décantation....
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
28
Etape (d)
La fraction ainsi éventuellement séchée est alors introduite (conduite 10),
ainsi
qu'éventuellement un flux d'hydrogène, (conduite 11) dans la zone (12)
contenant le catalyseur
d'hydroisomérisation / d'hydrocraquage. Une autre éventualité du procédé aussi
selon l'invention
consiste à envoyer la totalité de l'effluent sortant du réacteur
d'hydrotraitement (sans séchage) dans le
réacteur contenant le catalyseur d'hydroisomérisation / d'hydrocraquage et de
préférence en même
temps qu'un flux d'hydrogène,
Les conditions opératoires dans lesquelles est effectuée cette étape (d) sont
:
La pression est maintenue entre 2 et 150 bars et de préférence entre 5 et 100
bars et
avantageusement de 10 à 90 bars, la vitesse spatiale est comprise entre 0,1
h"1 et 10 h'' et de
préférence entre 0,2 et 7h"1 est avantageusement entre 0,5 et 5,0h"1. Le taux
d'hydrogène est compris
entre 100 et 2000 Normaux litres d'hydrogène par litre de charge et par heure
et préférentiellement
entre 150 et 1500 litres d'hydrogène par litre de charge.
La température utilisée dans cette étape est comprise entre 200 et 450 C et
préférentiellement
de 250 C à 450 C avantageusement de 300 à 450 C, et encore plus
avantageusement supérieure à
320 C ou par exemple entre 320-420 C.
Les deux étapes, hydrotraitement et hydroisomérisation-hydrocraquage, peuvent
être réalisées
sur les deux types de catalyseurs dans deux ou plusieurs réacteurs différents,
ou/et dans un même
réacteur.
L'étape (d) d'hydroisomérisation et d'hydrocraquage est avantageusement
conduite dans des
conditions telles que la conversion par passe en produits à points
d'ébullition supérieurs ou égaux à
150 C en des produits ayant des points d'ébullition inférieurs à 150 C est la
plus faible possible, de
préférence inférieure à 50%, de manière encore plus préférée inférieure à 30%,
et permet d'obtenir
des distillats moyens (gazole et kérosène) ayant des propriétés à froid (point
d'écoulement et de
congélation) suffisamment bonnes pour satisfaire aux spécifications en vigueur
pour ce type de
carburant.
Ainsi dans cette étape (d), on cherche à favoriser l'hydroisomérisation plutôt
que
l'hydrocraquage.
Etape f
Ladite fraction lourde dont les points d'ébullition sont supérieurs au point
de coupe T2,
précédemment défini, est introduite via la ligne (5) dans une zone (13) où
elle est mise, en présence
d'hydrogène (26), au contact d'un catalyseur non zéolitique
d'hydroisomérisation/hydrocraquage afin
de produire une coupe distillat moyen (kérosène + gazole) présentant de bonnes
propriétés à froid.
Le catalyseur utilisé dans la zone (13) de l'étape (f) pour réaliser les
réactions d'hydrocraquage
et d'hdydroisomérisation de la fraction lourde, définie selon l'invention, est
du même type que celui
présent dans le réacteur (12). Cependant, il est à noter que les catalyseurs
mis en couvre dans les
réacteurs (12) et (13) peuvent être identiques ou différents.
Durant cette étape (f) la fraction entrant dans le réacteur subit au contact
du catalyseur et en
présence d'hydrogène essentiellement des réactions d'hydrocraquage qui,
accompagnés de réactions
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
29
d'hydroisomérisation des n-paraffines, vont permettre d'améliorer la qualité
des produits formés et plus
particulièrement les propriétés à froid du kérosène et du gazole, et également
d'obtenir de très bons
rendements en distillats. La conversion en produits ayant des points
d'ébullition supérieurs ou égal à
370 C en produits à points d'ébullition inférieurs à 370 C est supérieure à
80% poids, souvent d'au
moins 85% et de préférence supérieure ou égal à 88%. Par contre, les
conversions des produits à
point d'ébullition supérieurs ou égaux à 260 C en produits à points
d'ébullition inférieurs à 260 C est
d'au plus 90% poids, généralement d'au plus 70% ou 80%, et de préférence d'au
plus 60% poids.
Dans cette étape (f), on cherchera donc à favoriser l'hydrocraquage, mais de
préférence en limitant le
craquage du gazole.
Eta e
L'effluent en sortie du réacteur (12), étape (d) est envoyé dans un train de
distillation, qui
intègre une distillation atmosphérique et éventuellement une distillation sous
vide, et qui a pour but de
séparer d'une part les produits légers inévitablement formés lors de l'étape
(d) par exemple les gaz
(Ci-C4) (conduite 14) et une coupe essence (conduite 19), et de distiller au
moins une coupe gazole
(conduite 17) et kérosène (conduite 16). Les fractions gazole et kérosène
peuvent être recyclées
(conduite 25) en partie, conjointement ou de façon séparée, en tête du
réacteur (12)
d'hydroisomérisation /hydrocraquage étape (d).
L'effluent en sortie de l'étape (f), est soumis à une étape de séparation dans
un train de
distillation de manière à séparer d'une part les produits légers
inévitablement formés lors de l'étape (f)
par exemple les gaz (Ci-C4) (conduite 18) et une coupe essence (conduite 19),
à distiller une coupe
gazole (conduite 21) et kérosène (conduite 20) et à distiller la fraction
(conduite 22) bouillant au-
dessus de gazole, c'est à dire dont les composés qui la constituent ont des
points d'ébullition
supérieurs à ceux des distillats moyens (kérosène + gazole). Cette fraction,
dite fraction résiduelle,
présente généralement un point d'ébullition initial d'au moins 350 C, de
préférence supérieure à
370 C. Cette fraction non hydrocraquée est avantageusement recyclée en tête du
réacteur (conduite
13) d'hydroisomérisation /hydrocraquage étape (f).
Il peut être également avantageux de recycler une partie du kérosène et/ou du
gazole dans
l'étape (d), l'étape (f) ou les deux. De façon préférée, l'une au moins des
fractions kérosène et/ou
gazole est recyclée en partie (conduite 25) dans l'étape (d) (zone 12). On a
pu constater qu'il est
avantageux de recycler une partie du kérosène pour améliorer ses propriétés à
froid.
Avantageusement et dans le même temps, la fraction non hydrocraquée est
recyclée en partie dans
l'étape (f) (zone 13).
Il va sans dire que les coupes gazole et kérosène sont de préférence
récupérées séparément,
mais les points de coupe sont ajustés par l'exploitant en fonction de ses
besoins.
Sur la figure 3, on a représenté 2 colonnes (23) et (24) de distillation, mais
une seule peut être
utilisée pour traiter l'ensemble des coupes issues de zones (12) et (13).
- Sur la figure 3, on a représenté seulement le recyclage du kérosène sur le
catalyseur du
réacteur (12). Il va sans dire qu'on peut aussi bien recycler une partie du
gazole (séparément ou avec
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
le kérosène) et de préférence sur le même catalyseur que le kérosène. On peut
également recycler
une partie du kérosène et/ou du gazole produits dans les lignes (20) (21).
Un autre mode de réalisation de l'invention comprend les étapes suivantes :
a) éventuel fractionnement de la charge en au moins une fraction lourde à
point d'ébullition initial
compris entre 120 et 200 C, et au moins une fraction légère bouillant en-
dessous de ladite fraction
lourde,
b) éventuel hydrotraitement d'une partie au moins de la charge ou de la
fraction lourde,
éventuellement suivi d'une étape c)
c) élimination d'au moins une partie de l'eau,
d) passage d'une partie au moins de l'effluent ou de la fraction
éventuellement hydrotraité dans un
procédé de traitement sur un premier catalyseur d'hydroisomérisation f
hydrocraquage contenant
au moins un métal noble du groupe VIII,
e) distillation de l'effluent hydroisomérisé / hydrocraqué pour obtenir des
distillats moyens (kérosène,
gasoil) et une fraction résiduelle bouillant au-dessus des distillats moyens,
f) sur un second catalyseur d'hydroisomérisation I hydrocraquage contenant au
moins un métal
noble du groupe Vlil, passage dans un procédé de traitement d'au moins une
partie de ladite
fraction lourde résiduelle et/ou d'une partie desdits distillats moyens, et
distillation de l'effluent
résultant pour obtenir des distillats moyens,
et l'un au moins desdits procédés de traitement de l'étape d) et f) est celui
de l'invention.
La description de ce mode de réalisation sera faite en se référant aux figures
4 et 5, sans que
ces figures limitent l'interprétation.
Eta e a
Lorsque cette étape est mise en oeuvre, l'effluent issu de l'unité de synthèse
Fischer-Tropsch
est fractionné (par exemple par distillation) en au moins deux fractions : au
moins une fraction légère
et au moins une fraction lourde à point d'ébullition initial égal à une
température comprise entre 120 et
200 C et de préférence entre 130 et 180 C et de manière encore plus préférée à
une température
d'environ 150 C, en d'autres termes le point de coupe est situé entre 120 et
200 C.
La fraction lourde présente généralement des teneurs en paraffines d'au moins
50% poids.
Ce fractionnement peut être réalisé par des méthodes bien connues de l'homme
du métier
telles que le flash, la distillation etc... A titre d'exemple non limitatif,
l'effluent issu de l'unité de synthèse
Fischer-Tropsch sera soumis à un flash, une décantation pour éliminer l'eau et
une distillation afin
d'obtenir au moins les 2 fractions décrites ci-dessus.
La fraction légère n'est pas traitée selon le procédé de l'invention mais peut
par exemple
constituer une bonne charge pour la pétrochimie et plus particulièrement pour
une unité de
vapocraquage. Au moins une fraction lourde précédemment décrite est traitée
selon le procédé de
l'invention.
Etape b
Eventuellement, cette fraction ou une partie au moins de la charge initiale,
est admise via la
ligne (1) en présence d'hydrogène (amené par la conduite (2)) dans une zone
(3) contenant un
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
31
catalyseur d'hydrotraitement qui a pour objectif de réduire la teneur en
composés oléfiniques et
insaturés ainsi que d'hydrotraiter les composés oxygénés (alcools) présents
dans la fraction lourde
décrite ci-dessus.
Les catalyseurs utilisés dans cette étape (b) sont des catalyseurs
d'hydrotraitement non
craquants ou peu craquants comportant au moins un métal du groupe VIII et/ou
du groupe VI de la
classification périodique des éléments.
Avantageusement, au moins un élément choisi parmi P, B, Si est déposé sur le
support.
Ces catalyseurs peuvent être préparés par toutes les méthodes connues de
l'homme de l'art
ou bien peuvent être acquis auprès de sociétés spécialisées dans la
fabrication et la vente de
catalyseurs.
Dans le réacteur d'hydrotraitement (3), la charge est mise en contact en
présence d'hydrogène
et du catalyseur à des températures et des pressions opératoires permettant de
réaliser
l'hydrodeoxygénation (HDO) des alcools et l'hydrogénation des oléfines
présents dans la charge. Les
températures réactionnelles utilisées dans le réacteur d'hydrotraitement sont
comprises entre 100 et
350, de préférence entre 150 et 300 C, de façon encore plus préférée entre 150
et 275 C et mieux
encore entre 175 et 250 C. La gamme de pression totale utilisée varie de 5 à
150 bars, de préférence
entre 10 et 100 bars et de manière encore plus préférée entre 10 et 90 bars.
L'hydrogène qui alimente
le réacteur d'hydrotraitement est introduit à un débit tel que le rapport
volumique
hydrogène/hydrocarbures soit compris entre 100 à 3000 NI/l/h, de préférence
entre 100 et 2000Ni/l/h et
de façon encore plus préférée entre 250 et 1500 NI/I/h. Le débit de charge est
tel que la vitesse
volumique horaire est comprises entre 0,1 et 10h'1, de préférence entre 0,2 et
5h-1 et de manière
encore plus préférée entre 0,2 et 3h"1. Dans ces conditions, la teneur en
molécules insaturées et
oxygénées est réduite à moins de 0,5% et à environ moins de 0,1% en général.
L'étape
d'hydrotraitement est conduite dans des conditions telles que la conversion en
produits ayant des
points d'ébullition supérieurs ou égaux à 370 C en des produits ayant des
points d'ébullition inférieurs
à 370 C est limitée à 30% pds, de préférence est inférieure à 20% et de façon
encore plus préférée
est inférieure à 10%.
Eta e c)
L'effluent (conduite 4) issu du réacteur (3) d'hydrotraitement est
éventuellement introduit dans
une zone (5) d'enlèvement d'eau qui a pour but d'éliminer au moins en partie
l'eau produite lors des
réactions d'hydrotraitement. Cette élimination d'eau peut s'effectuer avec ou
sans élimination de la
fraction gazeuse C4 moins qui est généralement produite lors de l'étape
d'hydrotraitement. On entend
par élimination de l'eau, l'élimination de l'eau produite par les réactions
d'hydrodéoxygénation (HDO)
des alcools mais on peut aussi y inclure l'élimination au moins en partie de
l'eau de saturation des
hydrocarbures. L'élimination de l'eau peut être réalisée par toutes les
méthodes et techniques connues
de l'homme du métier, par exemple par séchage, passage sur un dessicant,
flash, décantation....
Eta e d)
Une partie au moins et de préférence la totalité de la fraction hydrocarbonée
(une partie au
moins de la charge ou une partie au moins de la fraction lourde de l'étape a)
ou une partie au moins de
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
32
la fraction ou de la charge hydrotraitée et éventuellement séchée) est alors
introduite (conduite 6) ainsi
qu'éventuellement un flux d'hydrogène (conduite 7) dans la zone (8) contenant
ledit premier catalyseur
d'hydroisomérisation / hydrocraquage. Une autre éventualité du procédé aussi
selon l'invention
consiste à envoyer une partie ou la totalité de l'effluent sortant du réacteur
d'hydrotraitement (sans
séchage) dans le réacteur contenant le catalyseur d'hydroisomérisation /
d'hydrocraquage et de
préférence en même temps qu'un flux d'hydrogène.
Avant utilisation dans la réaction, le métal contenu dans te catalyseur doit
être réduit. Une des
méthodes préférées pour conduire la réduction du métal est le traitement sous
hydrogène à une
température comprise entre 150 C et 650 C et une pression totale comprise
entre 0,1 et 25 Mpa. Par
exemple, une réduction consiste en un palier à 150 C de 2 heures puis une
montée en température
jusqu'à 450 C à la vitesse de 1 C/min puis un palier de 2 heures à 450 C ;
durant toute cette étape de
réduction, le débit d'hydrogène est de 1000 litres hydrogène/ litre
catalyseur. Notons également que
toute méthode de réduction ex-situ est convenable.
Les conditions opératoires dans lesquelles est effectuée cette étape (d) sont
:
La pression est maintenue entre 2 et 150 bars et de préférence entre 5 et 100
bars et
avantageusement de 10 à 90 bars, la vitesse spatiale est comprise entre 0,1
h"1 . et 10 h" et de
préférence entre 0,2 et 7h" est avantageusement entre 0,5 et 5,0h"1. Le taux
d'hydrogène est compris
entre 100 et 2000 Normaux litres d'hydrogène par litre de charge et par heure
et préférentiellement
entre 150 et 1500 litres d'hydrogène par litre de charge.
La température utilisée dans cette étape est comprise entre 200 et 450 C et
préférentiellement
de 250 C à 450 C avantageusement de 300 à 450 C, et encore plus
avantageusement supérieure à
320 C ou par exemple entre 320-420 C.
Les deux étapes, hydrotraitement et hydroisomérisation-hydrocraquage, peuvent
être réalisées
sur les deux types de catalyseurs dans deux ou plusieurs réacteurs différents,
ou/et dans un même
réacteur.
Etape e)
L'effluent hydroisomérisé / hydrocraqué en sortie du réacteur (8), étape (d),
est envoyé dans
un train de distillation (9) qui intègre une distillation atmosphérique et
éventuellement une distillation
sous vide qui a pour but de séparer les produits de conversion de point
d'ébullition inférieur à 340 C et
de préférence inférieur à 370 C et incluant notamment ceux formés lors de
l'étape (d) dans le réacteur
(8), et de séparer la fraction résiduelle dont le point initial d'ébullition
est généralement supérieur à au
moins 340 C et de préférence supérieur ou égal à au moins 370 C. Parmi les
produits de conversion
et hydroisomérisés il est séparé, outre les gaz légers C1-C4 (conduite 10) au
moins une fraction
essence (conduite 11), et au moins une fraction distillat moyen kérosène
(conduite 12) et gazole
(conduite 13).
Etape f)
Le procédé selon l'invention utilise une seconde zone (16) contenant un
catalyseur
d'hydroisomérisation / hydrocraquage (dit second catalyseur). Il passe sur ce
catalyseur, en présence
d'hydrogène (conduite 15) un effluent choisi parmi une partie du kérosène
produit (conduite 12), une
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
33
partie du gazole (conduite 13) et la fraction résiduelle et de préférence, la
fraction résiduelle dont le
point initial d'ébullition est généralement supérieur à au moins 370 C.
Le catalyseur présent dans le réacteur (16) de l'étape (f) du procédé selon
l'invention est de la même
façon que pour l'étape d), de type acide non zéolitique et à base d'au moins
un métal noble du groupe
VIII ; cependant il peut être identique ou différent de celui de l'étape d).
Durant cette étape la fraction entrant dans le réacteur (16) subit au contact
du catalyseur et en
présence d'hydrogène des réactions d'hydroisomérisation et/ou d'hydrocraquage
qui vont permettre
d'améliorer la qualité des produits formés et plus particulièrement les
propriétés à froid du kérosène et
du gazole, et d'obtenir des rendements en distillat amélioré par rapport à
l'art antérieur.
Le choix des conditions opératoires permet d'ajuster finement la qualité des
produits (distillats
moyens) et en particulier les propriétés à froid.
Les conditions opératoires dans lesquelles est effectuée cette étape (f) sont
:
La pression est maintenue entre 2 et 150 bars et de préférence entre 5 et 100
bars et
avantageusement de 10 à 90 bars, la vitesse spatiale est comprise entre 0,1 h-
1 et 10 h-' et de
préférence entre 0,2 et 7h-1 est avantageusement entre 0,5 et 5,0h"1. Le taux
d'hydrogène est compris
entre 100 et 2000 Normaux litres d'hydrogène par litre de charge et par heure
et préférentiellement
entre 150 et 1500 litres d'hydrogène par litre de charge.
La température utilisée dans cette étape est comprise entre 200 et 450 C et
préférentiellement
de 250 C à 450 C avantageusement de 300 à 450 C, et encore plus
avantageusement supérieure à
320 C ou par exemple entre 320-420 C.
L'exploitant ajustera les conditions opératoires sur le premier et second
catalyseur
d'hydrocraquage/hydroisomérisation de façon à obtenir les qualités de produits
et les rendements
souhaités.
Ainsi, de façon générale, sur le premier catalyseur, la conversion par passe
en produits à
points d'ébullition supérieurs ou égaux à 150 C en des produits à points
d'ébullition inférieurs à 150 C
est inférieure à 50%pds, de préférence inférieure à 30% pds. Ces conditions
permettent au particulier
d'ajuster le rapport kérosène/gazole produits ainsi que les produits à froid
des distillats moyens, et plus
particulièrement du kérosène.
Egalement de façon générale, sur le second catalyseur, lorsque la fraction
résiduelle est
traitée, la conversion par passe en produits à points d'ébullition supérieurs
ou égaux à 370 C en
produits à points d'ébullition inférieurs à 370 C, est supérieure à 40% pds,
de préférence supérieure à
50% pds, ou mieux à 60% pds. Il peut même s'avérer avantageux d'avoir des
conversions d'au moins
80% pds.
Lorsque une partie du kérosène et/ou du gazole est traitée sur le second
catalyseur, la
conversion par passe en produits à points d'ébullition supérieurs ou égaux à
150 C en des produits à
points d'ébullition inférieurs à 150 C est inférieure à 50% pds, de préférence
inférieure à 30% pds.
De façon générale les conditions opérations appliquées dans les réacteurs (8)
et (16) peuvent
être différentes ou identiques. De façon préférée les conditions opératoires
utilisées dans les 2
réacteurs d'hydroisomérisation / hydrocraquage sont choisies différentes en
termes de pression
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
34
opératoire, température, temps de contact (vvh) et rapport H2/charge. Ce mode
de réalisation permet à
l'exploitant d'ajuster les qualités et/ou rendements en kérosène et gazole.
L'effluent issu du réacteur (16) est ensuite envoyé via la ligne (17) dans le
train distillation de
manière à séparer les produits de conversion, essence, kérosène et gazole.
Sur la figure 4, il est représenté un mode de réalisation avec la fraction
résiduelle (conduite 14)
passant dans la zone (16) d'hydroisomérisation / hydrocraquage (étape f),
l'effluent obtenu étant
envoyé (conduite 17) dans la zone (9) de séparation.
Avantageusement, dans le même temps, le kérosène et/ou le gazole peut être en
partie
recyclé (conduite 18) dans la zone (8) d'hydroisomérisation / hydrocraquage
(étape d) sur le premier
catalyseur.
Sur la figure 5, une partie du kérosène et/ou du gazole produits passent dans
la zone (16)
d'hydroisomérisation / hydrocraquage (étape f), l'effluent obtenu étant envoyé
(conduite 17) dans la
zone (9) de séparation.
Dans le même temps, la fraction résiduelle (conduite 14) est recyclée dans la
zone (8)
d'hydroisomérisation / hydrocraquage (étape d) sur le premier catalyseur.
On a pu constater qu'il est avantageux de recycler une partie du kérosène sur
un catalyseur
d'hydrocraquage / hydroisomérisation pour améliorer ses propriétés à froid.
Sur les figures 4 et 5, on a représenté seulement le recyclage du kérosène. Il
va sans dire qu'on
peut aussi bien recycler une partie du gazole (séparément ou avec le kérosène)
et de préférence sur le
même catalyseur que le kérosène.
L'invention peut également être utilisée dans d'autres modes de réalisation
pour produire des
distillats moyens.
Par exemple, un mode de réalisation inclut l'hydrotraitement d'un effluent
issu de la synthèse
Fischer-Tropsch en totalité ou après séparation de la fraction C4 moins (de
préférence la conversion
des produits ayant des points d'ébullition supérieurs à 370 C est inférieure à
20%), une éventuelle
séparation de la fraction C4 moins de l'effluent d'hydrotraitement et au moins
une partie de l'effluent
résiduel est traité par le procédé selon l'invention (la conversion étant de
préférence d'au moins 40%).
Dans un autre mode de réalisation, l'effluent issu de la synthèse Fischer-
Tropsch est séparé en
une fraction lourde (de préférence bouillant au-dessus de 260 C) et au moins
une fraction légère (de
préférence bouillant en-dessous de 260 C), la fraction lourde est traitée par
le procédé selon
l'invention, au moins une fraction légère est hydrotraitée puis
hydrocraquée/isomérisée de préférence
selon le procédé de l'invention.
Dans un autre mode de réalisation de l'invention, le procédé peut être utilisé
pour la production
de distillats moyens, essentiellement en l'absence de composés organiques
oxygénés, à partir d'un
mélange synthétique d'hydrocarbures partiellement oxygénés issus du procédé de
synthèse Fischer -
Trospch, substantiellement linéaires, contenant au moins 20 % en poids d'une
fraction ayant une
température de distillation supérieure à 370 C. Le procédé comprend alors les
étapes suivantes :
a) séparation dudit mélange en au moins une fraction à faible température
d'ébullition (B)
(généralement avec un point d'ébullition maximum allant de 150 à 380 C,
préférentiellement de 260 à
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
370 C) plus riche en composés oxygénés et au moins une fraction à haute
température d'ébullition (A)
moins riche en composés oxygénés (généralement avec un point d'ébullition
supérieur à 370 C,
éventuellement comprenant au moins une partie d'une coupe gazole type) ;
b) soumission de ladite fraction (B) à un traitement hydrogénant dans des
conditions de nature à
éviter toute variation substantielle dans son poids moléculaire moyen pour
obtenir un mélange
hydrogéné d'hydrocarbures substantiellement non oxygénés;
c) recombinaison d'au moins une partie dudit mélange hydrogéné selon l'étape
(b) avec ladite
fraction (A) pour former un mélange (C) d'hydrocarbures linéaires avec un
contenu réduit en
hydrocarbures oxygénés et soumission dudit mélange (C) à un traitement
d'hydrocraquage selon
l'invention en présence d'un catalyseur d'hydroisomérisation/hydrocraquage,
pour convertir au moins
% de ladite fraction à haut point d'ébullition en une fraction d'hydrocarbures
pouvant être distillés à
une température inférieure à 370 C;
d) séparation d'au moins une fraction d'hydrocarbures du produit obtenu à
l'étape (c) dont la
température de distillation est dans le domaine des distillats moyens.
La description de ce mode de réalisation sera faite en se référant à la figure
(6), sans que cette
figure limite l'interprétation.
Un flux synthétique d'hydrocarbures substantiellement linéaires, partiellement
oxygénés ou
essentiellement exempts de soufre obtenus par un procédé de type Fischer-
Tropsch, de préférence de
type "non-shifting" est ôté du réacteur de synthèse préalablement subdivisé en
une fraction bouillant à
haut point d'ébullition (A), avec un point d'ébullition initial allant de 250
C à 450 C, et une fraction à bas
point d'ébullition (B), avec un point d'ébullition final allant de 200 C à 450
C. Le rapport massique
(B)/(A) entre les deux fractions est de manière préférée compris entre 0,5 et
2,0, de manière plus
préférée entre 0,8 et 1,5 et si nécessaire, la composition des deux fractions
peut partiellement
coïncider, avec une coupe d'hydrocarbures présente dans les deux fractions,
préférentiellement en
quantité allant de 0,1 à 20% en poids par rapport au poids total de chaque
fraction.
La fraction à bas point d'ébullition (B) est chargée, par une ligne 1 dans
l'unité d'hydrogénation
(HDT) pour effectuer l'étape (b) du présent mode de réalisation, dans laquelle
il est mis en contact
avec de l'hydrogène (ligne 2) en présence d'un catalyseur adéquat, dans des
conditions de nature à
minimiser ou exclure la réaction d'hydrocraquage.
Une fraction d'hydrocarbures produite dans l'étape d'hydrogénation ayant un
contenu en
oxygène inférieur à 0,001 % poids (dont la fraction d'hydrocarbures gazeux C5-
) est avantageusement
séparée et enlevée, par l'intermédiaire de la ligne 5. Cette fraction ne
représente cependant pas plus
de 5%, de préférence pas plus de 3% en poids de la fraction totale (B).
Une fraction à bas point d'ébullition est ainsi obtenue, essentiellement
constituée d'un mélange
d'hydrocarbures saturés, de préférence partiellement isomérisés, qui est au
moins en partie, de
préférence complètement ajoutée par l'intermédiaire de la ligne 4 à la
fraction (A) (ligne 3)
d'hydrocarbures à haut point d'ébullition à faible contenu en oxygène pour
former une charge (C) qui
alimente une unité d'hydrocraquage (HCK) selon l'étape (c) du présent mode de
réalisation de
l'invention.
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
36
Les flux suivants sont introduits dans l'unité d'hydrocraquage (HCK) :
- la charge (C), obtenue par l'adjonction de la fraction (A) et de la fraction
résultant du
prétraitement d'hydrogénation de la fraction (B), par l'intermédiaire de la
ligne 4;
-la fraction à haut point d'ébullition recyclée par la ligne 12, ayant de
préférence un point
d'ébullition supérieur à 360 C, formant le résidu de la séparation du
distillat moyen, dans un rapport de
masse compris préférentiellement entre 1 et 40%, de manière plus préférée
entre 5 et 15% par rapport
à ladite charge (C);
-une quantité suffisante d'hydrogène par l'intermédiaire de la ligne 6.
Le produit de l'étape d'hydrocraquage, composé d'un mélange d'hydrocarbures
ayant un degré
d'isomérisation (hydrocarbures non-linéaires / masse du mélange) de préférence
supérieur à 50%, de
manière plus préférée supérieur à 70% est introduit par l'intermédiaire de la
ligne 7 dans une étape de
séparation par distillation (DIST), de préférence dans une colonne opérant à
pression atmosphérique
ou légèrement au-dessus, dont les distillats visés sont retirés par
l'intermédiaire des lignes 10
(kérosène) et 11 (gazole). Dans la figure 6, les produits suivants sont aussi
obtenus dans l'unité de
distillation : une fraction gazeuse C1-C5, relativement insignifiante, par
l'intermédiaire de la ligne 8, et
une fraction d'hydrocarbures légers, par l'intermédiaire de la ligne 9, de
préférence avec un point
d'ébullition inférieur à 150 C (naphta) qui est formé à l'étape (c).
Les produits obtenus
Le(s) gazole(s) obtenu (s) présente(nt) un point d'écoulement d'au plus 0 C,
généralement
inférieur à -10 C et souvent inférieur à -15 C. L'indice de cétane est
supérieur à 60, généralement
supérieur à 65, souvent supérieur à 70.
Le(s) kérosène(s) obtenu(s) présente(nt) un point de congélation d'au plus -35
C, généralement
inférieur à -40 C. Le point de fumée est supérieur à 25 mm, généralement
supérieur à 30 mm. Dans
ce procédé, la production d'essence (non recherchée) est la plus faible
possible. Le rendement en
essence sera toujours inférieur à 50% poids, de préférence inférieur à 40%
poids, avantageusement
inférieur à 30% poids ou encore à 20% pds ou même à 15% poids.
Exemple 1 : Caractéristiques du support alumine-silice (SA1)
Le support SA1 est une alumine-silice qui a une composition chimique en poids
de 60% de A12O3 et
40% de SiO2. Son rapport SI/AI est de 0.6. Sa teneur en sodium est de l'ordre
de 100-120 ppm en
poids. Les extrudés sont cylindriques de diamètre 1.6 mm. Sa surface
spécifique est de 320 m2/g. Son
volume poreux total, mesuré par porosimétrie au mercure est 0.83 cm3/g. La
distribution poreuse est
bimodale. Dans le domaine des mésopores, un large pic entre 4 et 15 nm avec un
maximum à 7 nm
est observé. Pour le support, les macropores, dont le diamètre est plus grand
que 50 nm
représentent environ 40% du volume poreux total.
Exemple 2 : Préparation des catalyseurs d'hydrocraquage utilisables dans le
procédé selon
l'invention (Cl conforme, C2 et C3 non conformes)
Le catalyseur Cl est obtenu par imprégnation à sec du support SA-1 (sous forme
d'extrudés),
préparé dans l'exemple 1 par une solution aqueuse d'acide phosphorique H3P04
et par une solution
d'acide hexachloroplatinique H2PtC16 dissous dans un volume de solution
correspondant au volume
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
37
poreux total à imprégner. Les extrudés imprégnés sont ensuite calcinés à 550 C
sous air pendant 4
heures. La teneur en platine est de 0,48% poids et sa dispersion mesurée par
titrage H2-02 est de 82%
et sa répartition est uniforme dans les extrudés. La teneur en % P205 est 1 %.
Le catalyseur C2 est obtenu par imprégnation à sec du support SA-1 (sous forme
d'extrudés),
préparé dans l'exemple 1 par une solution d'acide hexachloroplatinique H2PtCI6
dissous dans un
volume de solution correspondant au volume poreux total à imprégner. Les
extrudés imprégnés sont
ensuite calcinés à 550 C sous air pendant 4 heures. La teneur en platine est
de 0,48% poids et sa
dispersion mesurée par titrage H2-02 est de 82% et sa répartition est uniforme
dans les extrudés. Le
catalyseur C2 ne contient pas d'élément dopant.
Le catalyseur C3 est obtenu par imprégnation à sec du support SA-1 (sous forme
d'extrudés),
préparé dans l'exemple 1 par une solution aqueuse d'acide phosphorique H3PO4
puis par une solution
dichlorure de platine tétramine Pt(NH3)4CI2 dissous dans un volume de solution
correspondant au
volume poreux total à imprégner. Les extrudés imprégnés sont ensuite calcinés
à 550 C sous air
pendant 4 heures. La teneur en platine est de 0,57% poids et sa dispersion
mesurée par titrage H2-02
est de 62% et sa répartition est uniforme dans les extrudés. La teneur en %
P205 du catalyseur C3 est
7%.
Exemple 3 : Evaluation des catalyseurs Cl, C2, C3 en h drocra ua e d'une
charge paraffinigue
issue de la synthèse Fischer-Tropsch
Les catalyseurs dont la préparation est décrite dans l'exemple 2 sont utilisés
pour réaliser
l'hydrocraquage d'une charge paraffinique issue d'une unité Fischer-Tropsch
dont les principales
caractéristiques sont données ci-après :
Densité à 20 C 0,79
Distillation simulée DS
DS : Point initial 170
DS : 10%p C 197
DS : 50%p C 310
DS : 90%p C 495
DS : Point final C 590
Teneur en fraction 370 C+ (%pds) 33
Les catalyseurs Cl, C2, C3 sont mis en oeuvre selon le procédé de l'invention
en utilisant une
unité pilote comportant 1 réacteur à lit fixe traversé, les fluides circulent
de bas en haut (up-flow).
Préalablement au test d'hydrocraquage, les catalyseurs sont réduits à 50 bars,
à 450 C sous
hydrogène pur.
Après réduction, le test catalytique s'effectue dans les conditions suivantes
:
Pression totale 50 MPa T=320 C
Rapport H2 sur charge de 800 normaux litres/litre de charge
La vitesse spatiale (WH) est égale à 1 h-1.
CA 02612856 2007-12-13
WO 2007/003793 PCT/FR2006/001567
38
Les performances catalytiques sont mesurées par le dosage des n-paraffines.
Les
performances catalytiques sont exprimées par la conversion nette en C22+.
La conversion nette C22+est prise égale à :
CN C22+ = [(% de C22+ effluents)- (% de C22+ charge)]/ [100- (% de C22+
charge)]]
Les rendements sont exprimés en C5-C9, C10-C22 et C22+.
Les performances catalytiques obtenues sont données dans les tableaux 1 et 2
ci-après.
Conversion nette C22+
Cl 72%
C2 68%
C3 65%
Tableau 1 : Conversions nettes de la faction C22+-
Rendements Rendements Rendements (% poids)
(% poids) (% poids) C22+.
C5-C9 C10-C22
Cl 11.6 79 9.3
C2 11.8 77.5 10.6
C3 12 76 11.7
Tableau 2: Rendements des produits formés :
Ces résultats, montrent (tableaux 1 et 2) que l'utilisation des catalyseurs
selon l'invention et
dans un procédé selon l'invention permettent par hydrocraquage d'une charge
paraffinique issue du
procédé de synthèse Fischer-Tropsch d'obtenir de très bons rendements en
distillats moyens, coupes
150-250 (kérosène) et 250-370 C (gazole).