Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02623405 2013-08-26
Title: METHODS AND COMPOSITION TO GENERATE UNIQUE SEQUENCE DNA
PROBES LABELING OF DNA PROBES AND THE USE OF THESE PROBES
Inventors: Mark Carle Connelly, Brad Foulk; Michael Kagan, Joost F.
Swennenhuis; Leon
W.M.M. Terstappen, Arjan G.J. Tibbe, and John Verrant
BACKGROUND OF THE INVENTION
Field of the Invention
The invention relates generally to the field of identification of DNA
sequences, genes or
chromosomes.
Generation of DNA probes
Human genomic DNA is a mixture of unique sequences and repetitive sequences
that are
present in multiple copies throughout the genome. In some applications,
nucleic acid
hybridization probes to detect repetitive sequences are desirable. These
probes have shown
utility in the fields of fetal cell diagnostics, oncology, and cytogenetics.
In other applications
it is desirable to generate hybridintion probes that anneal only to unique
sequences of
interest on a chromosome. Preparation of unique sequence probes is confounded
by the
presence of numerous classes of repetitive sequences throughout the genome of
the organism
(Hood et al., Molecular Biology of Eucalyotic Cells (Benjamin/Cummings
Publishing
Company, Menlo Park, CA 1975). The presence of repetitive sequences in
hybridization
probes will reduce the specificity of the probes because portions of the probe
will bind to
other repetitive sequences found outside the sequence of interest. Thus, to
ensure binding of
hybridization probes to a specific sequence of interest, efforts must be made
to ensure that
repetitive sequences in the probe do not anneal to the target DNA outside the
sequence of
interest.
1
CA 02623405 2008-03-20
WO 2007/053245 PCT/US2006/036656
Recent contributions have addressed this question by inhibiting hybridization
of the
repetitive sequences with the use of unlabeled blocking nucleic acids (US
5,447,841 and US
6,596,479). Use of blocking nucleic acids in hybridizations is expensive, does
not
completely prevent hybridization of the repetitive sequences, and can distort
genomic
hybridization patterns (Newkirk et al., "Distortion of quantitative genomic
and expression
hybridization by Cot-1 DNA: mitigation of this effect," Nucleic Acids Res. vol
33 (22):e191
(2005)). Thus, methods that prevent hybridization of repeat sequences without
the use of blocking
DNA are necessary for optimal hybridization.
One means to achieve this is to remove unwanted repeat segments from the
hybridization
probes prior to hybridization. Techniques involving the removal of highly
repetitive
sequences have been previously described. Absorbents, like hydroxyapatite,
provide a means
to remove highly repetitive sequences from extracted DNA. Hyroxyapatite
chromatography
fractionates DNA on the basis of duplex re-association conditions, such as
temperature, salt
concentration, or other stringencies. This procedure is cumbersome and varies
with different
sequences. Repeat DNA can also be removed by hybridization to immobilized DNA
(Brison
et al., "General Methods for Cloning Amplified DNA by Differential Screening
with
Genomic Probes," Molecular and Cellular Biology, Vol. 2, pp. 578-587 (1982)).
In all of
these procedures, the physical removal of the repetitive sequences will depend
upon the strict
optimization of conditions with inherent variations based upon the base
composition of the
DNA sequence.
Several other methods to remove repetitive sequences from hybridization probes
have
been described. One method involves using a cross-linking agent to cross-link
repetitive
sequences either to directly prevent hybridization of repetitive sequences or
to prevent
amplification of repeat sequences in a PCR reaction. (US 6,406,850). Another
method uses
PCR assisted affinity chromatography to remove repeats from hybridization
probes (US
6,569,621). Both of these methods rely on the use of labeled DNA to remove
repeat
sequences which makes these processes complex and difficult to reproduce.
Further, both
methods are time consuming, requiring multiple rounds of repeat removal to
produce
functional probes, suitable for use in fluorescent in situ hybridization
(FISH) or other
hybridization reactions requiring high target specificity.
The use of duplex specific nucleases which preferentially cleave double
stranded
deoxyribonucleic acid molecules has been described for sequence variant
detection
applications such as single nucleotide polymorphisms (US 2005/0164216; US
6,541,204).
The ability of the enzyme to preferentially cleave perfectly matched nucleic
acid duplex
2
CA 02623405 2008-03-20
WO 2007/053245 PCT/US2006/036656
polynucleotides as compared to single stranded provides a means for removing
non-target
double stranded DNA from the sample mixture.
The ability of these nucleases to specifically digest the duplex form of
polynucleotides
was discovered in the instant invention to provide substantial benefit in
manufacturing unique
target specific probes that do not require blocking DNA, thus eliminating the
costs and
interfering affect of blocking DNA, and providing a means for rapid, efficient
and cost
effective production of high specificity probes.
Detection of specific sequences in a genome makes use of the fact that DNA
consists of a
helix of two DNA strands and that this double strand is most stable when these
two strands
are homologues. The DNA consists of a phosphate-sugar phosphate backbone and
to every
sugar one of four different nitrogenous bases, cytosine guanine thymine or
adenine, might be
present. Homologue strands pair every cytosine with a guanine and every
thymine with an
adenine. When a labeled homologue sequence is added to a genome and the DNA is
made
single stranded, these labeled sequences will hybridize, under the right
circumstances, to the
specific homologue sequence in the genome. For this in situ hybridization, a
number of
probes are available for different detections purposes and applications.
Whole chromosome I paint probes (WCP)
WCP incorporates labeled DNA material, homologous to a specific chromosome.
The
material is obtained by flow sorting of metaphase chromosomes or by laser
dissection from a
metaphase spread which is amplified by PCR or a related technique. After
labeling and
applying it to a properly prepared nucleus, it will stain the target
chromosome. However,
such labeled probes will in addition stain other non-target chromosomes
because of structural
or repetitive sequence elements that are shared among some or all chromosomes.
Accordingly in order to stain only those sequences originating from the
intended
chromosome of interest, these common repetitive elements are usually inhibited
by
hybridization with blocking DNA or other methods that block or remove non-
specific
interactions.
Multiple chromosome paints are also applied to a single nucleus. WCP are
labeled by
different fluorochromes or with a combination of fluorochromes, providing no
limit to the
amount of WCP's applied in a single hybridization. WCP's are mainly used for
karyotyping
and to study translocations of large fragments, regions and subregions of
chromosomes which
are best observed in a metaphase spread of a nucleus.
3
CA 02623405 2008-03-20
WO 2007/053245 PCT/US2006/036656
Centromere probes
Centromere probes are targeted to a 171 bp sequence that occurs in repetitive
order in
every centromeric region of the human chromosomes. All chromosomes have a
slightly
different sequence and because of this all chromosomes are detected separately
when the
right hybridization stringency is used. Only two chromosome pairs, 13 with 21
and 14 with
22, share the same repo/ and cannot be detected independently. Generally,
centromeric
probes are produced from plasmids containing an insert from one or a few
copies of the 171
pb repeat. These probes are able to be hybridized without the addition of
blocking DNA
because the 171bp sequences do not occur outside the regions of interest.
Teloinere probes
Human telomeres consist of an array of short repetitive sequences (i.e.
TTAGGG). This
is repeated several times in different amounts for every chromosome and
individual test
subject age. This repetitive sequence is used as a probe that will stain all
chromosomes
although not every chromosome will stain equally strong. To detect the
telomeric end of
chromosomes, mostly a sub-telomeric bacterial artificial chromosome (BAC)
clone is used.
This BAC clone contains repetitive sequences which should be blocked or
removed during or
before hybridization.
Comparative Genomic Hybridization (CGH) probes
CGH is a process that involves hybridizing a test genome to a reference
genome. The
reference genome may take the form of a metaphase chromosome spread from a
healthy
individual or may be array based using probe sequences that represent all or
part of a
genome. Microarray probes made using BAC clones contain repetitive sequences
which
must be blocked prior to hybridization. However, blocking has the potential to
cause a
deviation in the results when compared to repeat depleted probes (Knol and
Rogan, Nucleic
Acids Research, 2005, Vol. 33, No. 22). Further, the blocking step increases
the cost of
hybridization assays. If the probe sequences are depleted of repetitive
sequences, the
blocking step of the labeled genomic DNA is not necessary, resulting in a
reduction in the
cost and removal of any variation.
Gene specific probes
Gene specific probes are designed to detect a region of the genome containing
a target
gene or group of genes. These probes are used to detect amplifications or
deletions of
4
CA 02623405 2008-03-20
WO 2007/053245 PCT/US2006/036656
specific genomic areas which correlated to the expression level of the
specific gene of
interest. The coding sequence of the gene(s) itself is not large enough to
generate a detection
signal for the probe that is visible using standard fluorescence microscope.
Therefore such
gene specific probes are not limited to just the coding gene sequences (exons)
but also
involve non-coding (introns), regulatory or other sequences around the gene.
Because of the
large sequences encompassed within even a gene specific probe design they
often suffer from
the undesirable inclusion of unwanted repetitive sequences. When such material
is then
either labeled and used in hybridizations or used in hybridizations and then
labeled, the
unwanted sequences must be blocked or removed from the probe to be able to
detect the gene
area specifically.
Microarray probes
Similar to CGH, microarray probes are fixed to a carrier. In general,
automated robotic
techniques are used to spot cDNA-PCR products or synthetic oligonucleotides on
a slide or
similar fixed surface. Also, techniques exist to synthesize sequences directly
on a slide
(Affimetrix, Inc, Santa Clara). The slides are hybridized with labeled cDNA or
RNA in
combination with different labeled cDNA or RNA as controls.
Coupling reporter molecules to DNA probes
DNA probes are visualized by coupled reporter molecules. These molecules need
to be
incorporated in or attached to the DNA probe. One method utilizes a reporter
molecule,
having nucleotides linked to enzymatic reactions. Examples include
incorporation by nick
translation or a random prime reaction. Further, an amine coupled nucleotide,
built in this
way, is subsequently coupled directly or indirectly to reporter molecules.
Coupling is done
by chemical labeling of the DNA. An example is the coupling of a reporter
molecule linked
to a platinum group which forms a coordinative bond to the N7 position of
guanine as used in
ULS labeling (Kreatech Diagnostics, Amsterdam) and described in US 5,580,990;
US
5,714,327; US 5,985,566; US 6,133,038; US 6,248,531; US 6,338,943; US
6,406,850; and
US 6,797,818. Reporter molecules can be radioactive isotope, non-isotopic
labels,
digoxygenin, enzymes, biotin, avidin, streptavidin, luminescent agents such as
radioluminescent, chemiluminescent, bioluminescent, and photoluminescent,
(including
fluorescent and phosphorescent), dyes, haptens, and the like.
Sample preparation
CA 02623405 2008-03-20
WO 2007/053245 PCT/US2006/036656
To be able to detect the labeled probes bound to interphase chromosomes, the
nucleus
should maintain morphology during and after the FISH procedures. Using
fixation, cells or
nuclei are attached to a solid layer such as a microscope slide. Fixation
before during or after
attachment to the solid layer, provide reference for identification. Depending
on the type of
cell or tissue, the nuclei have to be accessible for probe DNA, usually by pre-
treating with
proteolytic enzymes, heat, alcohols, denaturants, detergent solutions or a
combination of
treatments. Probe and nucleic DNA are made single stranded by heat or alkali
treatment and
then allowed to hybridize.
Use of DNA probes
Microarrays
One common use of microarrays is to determine the RNA expression profile of a
suspect
tissue, tumor, or microbe. By analyzing the RNA expression profile, a
prognosis for the
treatment and survival of the patient is proposed. The prognostic value of RNA
microarrays
for clinical usage has yet to be determined. Another common use of microarrays
are array
based CGH. With this technique an entire genome can be screened for
amplifications and/or
deletions of chromosomal regions
Microscopy
Cytogenetic analysis in pre and post natal testing is used to determine
whether or not a
fetus has a cytogenetic abnormality in a cell population from the fetus.
Samples are
frequently obtained through amniocenthesis, conducted in pregnant women who
are
considered to have an increased risk for cytogenetic abnormalities.
Accordingly, these cells
are investigated for cytogenetic abnormalities. The same type of
investigations are
performed to confirm cytogenetic abnormalities or investigate suspect
cytogenetic
abnormalities in cell populations obtained after delivery.
Assessing Fetal Cells in Maternal Blood
During pregnancy, fetal cells may enter into the maternal blood with increases
in the
number of these fetal cells found with trauma, (pre)-ecclampsy and abnormal
pregnancies. In
routine assessments of fetomaternal hemorrhages, the frequently used Kleihauer-
Betke test is
based on the detection of red blood cells expressing fetal hemoglobin. For
detection of
cytogenetic abnormalities, nucleated cells from maternal blood are needed. The
frequency of
these cells is considerably lower and are estimated to be in the range of 1-
10 fetal cells per
6
CA 02623405 2008-03-20
WO 2007/053245 PCT/US2006/036656
mL of maternal blood. Nucleated red blood cells, trophoblast cells and the
presence of
hematopoeitic progenitors that are of fetal origin provide a target for
isolation and probe
hybridization in the detection of cytogenetic abnormalities early in the
pregnancies. To date
a reliable and reproducible method to identify and assess the cytogenetic
composition of
these cells is not available. One of the main problems with this analysis is
the loss of fetal
cells at various steps throughout the procedure, resulting in inconsistent or
inconclusive
information.
Oncology
FISH is used to detect various kinds of chromosomal aberrations like
translocations,
deletions, amplifications, inversions, and duplications. These aberrations are
detected in all
types of cells and tissue. In leukemia, cells are isolated from blood or bone
marrow for
subsequent FISH analysis. In bladder cancer, cells are isolated from urine.
Cells from solid
tumors are obtained by puncture or excision of the tumor itself. Also, cells
that are released
by solid tumors are isolated from the blood and analyzed by FISH. The latter
gives the
opportunity to monitor tumor treatment closely in order to detect a
chromosomal change in
the tumor. In some types of cancer, FISH provides a prognosis of tumor
progression or
predicts the efficacy of specific medication. Commercially, the most used FISH
tests are the
BCR-ABL translocation FISH in Chronic myelogenous leukemia and the her2/neu
gene
amplification FISH in breast cancer.
Disseminated tumor cells
Methods for the characterization of not only tumor cells, but also rare cells,
or other
biological entities from biological samples have been previously described (US
6,365,362).
This two stage method requires efficient enrichment to ensure acquisition of
target cells while
eliminating a substantial amount of debris and other interfering substances
prior to analysis,
allowing for cellular examination by imaging techniques. The method combines
elements of
immunomagnetic enrichment with multi-parameter flow cytometry, microscopy and
immunocytochemical analysis in a uniquely automated way. The combination
method is
used to enrich and enumerate epithelial cells in blood samples, thus providing
a tool for
measuring cancer.
The two stage method has applications in cancer prognosis and survival for
patients with
metastatic cancer (WO 04076643). Based on the presence of morphologically
intact
circulating cancer cells in blood, this method is able to correlate the
presence of circulating
7
CA 02623405 2013-08-26
cancer cells of metastatic breast cancer patients with time to disease
progression and survival.
More specifically, the presence of five (5) or more circulating tumor cells
per 7.5 milliliters
provides a predictive value at the first follow-up, thus providing an early
prognostic indicator
of patient survival.
The specificity of the assay described above increases with the number of
cells detected
and is not sufficient in cases were only few (generally less than 5
circulating tumor cells) are
detected. One solution to this problem is to provide detailed genetic
information about
suspected cancer cells. Accordingly, a method that would incorporate
enrichment of a blood
sample with multi-parametric image cytometry and multi-parametric genetic
analysis on an
individual suspect cancer cell would provide a complete profile and
confirmatory mechanism
to significantly improve current procedures for patient screening, assessing
recurrence of
disease, or overall survival.
Fluorescent in situ hybridization (FISH) has been described as a single mode
of analysis
in rare cell detection after enrichment as described in WO 00/60119; Meng et
al. PNAS 101
(25): 9393-9398 (2004); Fehm et al. Clin Can Res 8: 2073-2084 (2002) .
After epithelial cell enrichment, captured cells are screened by known
hybridization methods and imaged on a microscope slide. Because of inherent
technical
variations and a lack of satisfactory confirmation of the genetic information,
the hybridization
pattern alone does not provide a level of clinical confidence that would be
necessary for
sensitive analysis, as in assessing samples with less than 5 target cells.
Further, this method
for FISH analysis is difficult to automate.
Coupling hybridization-based methods with immtmocytochemistry in the analysis
of
individual cells has been previously described (US 6,524,798). Simultaneous
phenotypic and
genotypic assessment of individual cells requires that the phenotypic
characteristics remain
stable after in situ hybridization preparatory steps and are limited in the
choice of detectable
labels. Typically, conventional in situ hybridization assays require the
following steps: (1)
denaturation with heat or alkali; (2) an optional step to reduce nonspecific
binding; (3)
hybridization of one or more nucleic acid probes to the target nucleic acid
sequence; (4)
removal of nucleic acid fragments not bound; and (5) detection of the
hybridi7ed probes. The
reagents used to complete one or more of these steps (i.e. methanol wash) will
alter antigen
recognition in subsequent immunocytochemistry, cause small shifts in the
position of target
cells or completely removes the target cells, which introduces the possibility
of
mischaracterization of suspect cells.
8
CA 02623405 2013-08-26
Probe sets and methods for multi-parametric FISH analysis has been described
in lung
cancer (US 20030087248). A 3 probe combination resulting in 95% sensitivity
for detecting
bladder cancer in patients has also been described, see US 6,376,188; US
6,174,681. These
methods lack the specificity and sensitivity for assessing small numbers of
target cells, and
thus a confirmatory assessment for early detection of disease state. They also
do not provide
a means for convenient automation.
One aspect of the present invention provides a confirmatory assay in the
analysis of rare
circulating cells by combining phenotypic and genotypic multiparametic
analysis of an
individually isolated target cell, resulting in a clinically significant level
of sensitivity and,
therefore, assurance to the clinician of any quantitative information
acquired. Relevant
disease states are assessed using extremely small (1, 2, 3, or 4) numbers of
circulating tumor
cells (CTC's) and provide a confirmation for early disease detection.
SUMMARY OF THE INVENTION
Generation of repeat depleted DNA probes
In one embodiment, there is provided a method for producing nucleic acid
probes
complementary to a target sequence comprising the steps of: a. obtaining
double strand
polynucleotides known to contain complementary target sequences and repetitive
sequences; b.
fragmenting said double strand polynucleotides into fragments; c. denaturing
said fragments into
single strands; d. hybridizing said repetitive sequences with a non-specific
competitor of
repetitive sequences to form a mixture of double strands and single strands;
e. cleaving said
double strands, wherein the cleaving step is performed by enzymatic digestion
specific to double
stranded DNA; and f. amplifying said single strands wherein said single
strands are
complementary to said target sequences.
In one specific embodiment, the hybridizing of said repetitive sequences is
with with a
non-specific competitor of repetitive sequences selected from a group
consisting of sonicated
salmon sperm DNA, COT-1 DNA, E.coli tRNA, placental total genomic DNA, cloned
Alu, and
combinations thereof.
Methods and compositions to eliminate repetitive sequences from DNA are
provided.
Any double stranded DNA is a suitable source in the application of the methods
of the present
DOCSTOR: 2793405\1
9
CA 02623405 2013-08-26
invention. To obtain single stranded DNA, devoid of any repetitive sequences,
first an
amplified whole genome library is made from the
source DNA according to standard procedures. The library obtained consists of
randomly
selected fragments ranging in size from approximately 200 to 500 base pairs.
Each fragment
consists of double stranded DNA, having PCR primer sequences at each end of a
target
sequence. Generally, this library is representative of the source DNA. Other
methods that
results in modified fragments of DNA to permit amplification are also
considered in this
invention with no limit to the size of the fragments. These include, but are
not limited to,
degenerate oligonucleotide primed polymerase chain reaction (DOP PCR), rolling
circles and
isothermal amplification methods. Double stranded DNA fragments are denatured
by heating
up to 95 C or other means to obtain single stranded DNA fragments. The
resulting single
stranded DNA fragments contain repetitive sequences, unique sequences or a
combination of
unique and repetitive sequences. An excess of Cot DNA or other appropriate
subtractor
DNA that binds to repetitive sequences is added. Subsequent lowering of the
temperature
results in the formation of double stranded DNA for only those fragments that
contain
repetitive sequences. Duplex Specific Nuclease (DSN) is added to allow
digestion of double
stranded DNA. In one embodiment, the DSN enzyme is added for 2 hours at 65 C.
The
resulting composition contains mostly single stranded DNA, having only unique
sequences,
and digested DNA. The unique sequence, now single stranded DNA with PCR
primers at
both ends, is used as a template to generate large amounts of the unique
sequence for use in
probe production. When BAC clones containing a desired unique sequence is used
as source
DNA, the template generated by this method contains only that unique sequence.
When the
boundary sequences are known, this method is useful in obtaining probes that
cover the
nucleotides between the boundary sequences in genomic DNA. Further, the
present
invention includes methods of use and compositions, resulting from the
production of these
DNA sequences after elimination of their repetitive sequences. These repeat
depleted DNA
sequences function as hybridization probes without the use of a blocking DNA
in any
appropriate application requiring disabling or blocking of undesired DNA
sequences.
DOCSTOR. 2793405\1
CA 02623405 2013-08-26
Also disclosed are a system, apparatus, and
methods in the preservation of immunomagnetically labeled cells for subsequent
FISH
analysis. This aspect permits the reanalysis of individual cells, utilizing
the same or similar
reporter molecules previously used to identify them. Accordingly after
immunomagnetic
selection and initial fluorescent labeling, the cells of interest are
identified and their location
is recorded. The cells are fixed in position followed by appropriate
processing.
Alternatively, the cells are fixed in position and stored for processing at a
later point in time.
For FISH applications, the sample is heated above the melting temperature of
DNA, resulting
in the loss of reporter molecules used to initially identify the target cells.
After completing
FISH in which the fluorescent FISH probes are hybridized and the nuclear
material is again
fluorescently labeled, the sample is reintroduced in an analyzer which locates
the cells of
interest to examine fluorescent signals from the FISH probes.
Also disclosed are methods for the reanalysis of
immunornagnetically labeled cells as a confirmation in identifying rare
circulating cells such
as circulating tumor cells (CTC's). Thus, methods and techniques for the
further processing
of cells after enrichment, immunofluorescent labeling and subsequent
confirmatory analysis,
using in situ hybridization, as a means to increase specificity and thereby
confirm the identity
of suspect CTC's in patients as being cancer cells. Cytogenetic abnormalities
detected in
morphologically suspect CTC's, detected in metastatic carcinoma patients, have
a prognosis
similar to patients with morphologically obvious CTCs or having an abundance
of CTCs.
One embodiment of the present invention considers confirmation assays in
patients diagnosed
with carcinomas and having CTCs, or disseminated tumor cells (DTC's) in bone
marrow,
where there is an increased risk for recurrence. In addition, the methods of
the present
invention are applicable when there is a need to assess for the presence or
absence of drug
targets in CTC such as, but not limited to, Hen, Her2, Androgen Receptor (AR),
cMyc, or
P10.
DOCSTOR: 2793405\1
11
CA 02623405 2008-03-20
WO 2007/053245 PCT/US2006/036656
BRIEF DESCRIPTION OF THE DRAWINGS
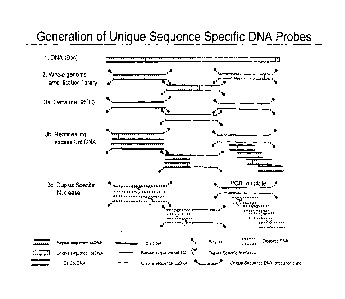
Figure 1: Schematic representation depicting the generation of repeat depleted
DNA probes
from BAC starting DNA. A fragmented whole genome amplification library is
denatured and
allowed to re-anneal in the presence of excess Cot DNA. DSN digestion of the
double strand
DNA results in a mixture of single strand unique sequence, available as a
template for probe
production.
Figure 2: Schematic representation depicting the cleavage of a specific DNA
sequence from
a DNA source and production of clones thereof. Double stranded DNA from an
appropriate
source is denatured and specific DNA sequence is allowed to hybridize. DSN
digestion of
the double strand DNA followed by separation of single strand DNA by size
results in
isolation of the desired single strand DNA. After synthesis of the second
strand, the desired
DNA is cloned into an appropriate vector for production.
Figure 3: Comparison using repeat-depleted probes in FISH analysis. White
blood cells
hybridized with a Her-2 probe containing repeats and no blocking DNA. In the
absence of
blocking DNA, the probe labels the entire nucleus after hybridization with
repeat regions.
Panel A shows white blood cells hybridized with a Her-2 FISH probe containing
repeats and
no blocking DNA. Panel B shows the same cells after labeling the nucleus with
DAPI. Panel
C shows the overlay of the two signals and the lack of Her-2 resolution. Panel
D shows FISH
analysis on white blood cells using repeat-depleted a Her-2 probe. Arrows
indicate locations
of unique chromosome sequence for Her-2. Panel E shows the same cells after
labeling the
nucleus with DAPI. Panel F shows the overlay of panel D and E, visualizing the
location of
the Her-2 site within the cell nucleus.
Figure 4: Panel A shows a chromosome spread hybridized with a P16 (CDKN2A)
labeled
repeat free probe targeting 9p21 frequently used to characterize melanoma. Two
chromosomes show the presence of 9p21 and are illustrated by arrows. Panel B
shows a
chromosome spread hybridized with MLL labeled repeat free probe targeting
11q23 used to
identify a specific type of leukemia. Two chromosomes show the presence of
11q23 as
illustrated by arrows.
12
CA 02623405 2008-03-20
WO 2007/053245 PCT/US2006/036656
Figure 5: Schematic representation of the fixation and hybridization device
used to prepare
samples for FISH analysis. A compact and portable device for preparing a
sample for FISH
analysis after immunomagnetic enrichment and initial fluorescent imaging.
Shown are the
control panel, electronics, pump, and poser supply in relation to the sample
cartridge.
Figure 6: Schematic representation of the basic steps for FISH after initial
fluorescent
imaging. Shown are cross-sectional images of the sample cartridge and the
presence of
magnetic support (black wedges). Panel 1 shows the cells arranged along the
internal surface
of the imaging face of the cartridge after initial fluorescent imaging using
CellTracks System.
Panel 2 shows the simultaneous replacement of the buffer solution with a
fixative for FISH.
In Panel 3, the fixative is aspirated to remove fluids from the cartridge.
Panel 4 shows the
addition of forced air to dry the cartridge. In Panel 5, the cartridge is
inverted and enough
FISH probe is added to cover the cells. Panel 6 shows the cartridge on a heat
source to allow
hybridization. In Panel 7, the FISH reagents are washed to allow rescanning
and analysis of
the FISH signals in Panel 8.
Figure 7: Schematic representation of the stopper with probe extension for
FISH cartridge
for reducing the chamber volume of the cartridge..
Figure 8: Schematic representation of the cartridge. A cross-sectional
illustration depicts
the cartridge inverted with the location of the cells and FISH reagents
illustrating a low
reagent volume distribution the lower face of the chamber, thus allowing
enough reagents to
only cover the cells along entire lower surface.
Figure 9: Representative image of a tumor cell initially identified through
immunocytochemistry (ICC) with subsequent FISH analysis for the presence of
chromosome
1, 7, 8 and 17. Panel A shows a list of CTC candidates identified by the
software on basis of
their ICC signature. Panel B shows the acquired fluorescent ICC images
acquired. Panel C
shows the corresponding FISH signals for chromosomes 1, 7, 8 and 17
demonstrating the
aneuploid signature of the same cell tumor cell.
Figure 10: Shown are five fluorescence images at different focal planes
through the cell
using excitation/emission filters for 5 different fluorochromes. Panel A shows
the images for
a cell using PE. Panel B shows images for the same cell using DAPI. Panel C
shows images
13
CA 02623405 2008-03-20
WO 2007/053245 PCT/US2006/036656
for the same cell using APC. Panel D shows images for the same cell using
FITC. Panel E
shows images for the same cell using Dy415.
Figure 11: Results from ICC and FISH analysis to confirm a CTC. Panel A shows
ICC
images of the ICC scan on which a suspect CTC was identified. Panel B shows
the
corresponding fluorescence signals, using FISH probes. Corresponding counts of
the signals
for each probe are shown next to each image; 4 count for PE, 1 count for APC,
4 count for
FITC and 2 count for Dy415.
DETAILED DESCRIPTION OF THE INVENTION
Generation of repeat depleted DNA probes
DNA contains unique as well as repetitive sequences. The repetitive sequences
occur
throughout the chromosomes and have the potential to interfere with
hybridization reactions,
such as with in situ hybridization, targeted toward specific regions or unique
sequences
outside these repetitive sequences. To identify the presence, amount and
location of specific
sequences on chromosomes, genes or DNA sequences it is important that the
hybridization
probes hybridize only at the location of interest. The presence of repetitive
sequences in the
hybridization probe mixture reduces the specificity of the binding, requiring
methods to
either remove the repetitive sequences from the probes or prevent the probes
from
hybridizing to the repetitive sequences on the target. For example, Cot-1 DNA
is often added
during hybridization to prevent binding of the probes to the repetitive
sequences (US
5,447,841 and US 6,596,479).
Recent contributions have addressed this question by disabling the repetitive
sequences.
The use of Cot-1 DNA relies on the ability of Cot-1 DNA to form a duplex
structure with
available single strand repeat sequences, and thereby minimize non-specific
binding
interaction of this portion of the sequence with the unique target sequence.
Blocking the
repetitive DNA, either during a hybridization step with the unique target
sequence or prior as
in a pre-association step, results in a mixture having repetitive segments
forming duplex
structures with their complementary sequence and a single strand form of the
target probe,
available for hybridization to its unique target segment. Unfortunately, the
presence of this
duplex in a subsequent amplification or labeling reaction affects the signal
through the
introduction of non-specific noise, especially in situations where the signal
is very weak. An
14
CA 02623405 2013-08-26
alternative to blocking the repetitive sequence is to remove the unwanted
repeat segments
from the reaction mix.
Generation of repeat-depleted DNA probes of this prevention is depicted in
Figure 1.
One embodiment of the present invention makes use of duplex specific nucleases
(DSN)
which preferentially cleave deoxyribonucleic acid molecules (US 2005/0164216
and US
6,541,204). The ability of the enzyme to preferentially cleave
nucleic acid duplex polynucleotides as compared to single strand DNA provides
a means for
removing non-target double stranded DNA from the sample mixture. The ability
of these
nucleases to preferentially digest the duplex form of polynucleotides provides
potential use in
manufacturing an unique target specific probe, eliminating the interfering
affect of blocking
DNA, and providing a means for their rapid, efficient and cost effective
production.
Starting DNA used in the practice of this invention is typically in the form
of one or more
DNA sequences which contain a multiplicity of DNA segments. The initial source
of
individual starting material in the production of the probe composition has
been described in
the production of direct-labeled probes (US 6,569,626). Optimally the source
of the starting
polynucleotide is purified from tissue and fragmented into 150 kb to 200 kb
segments, using
any known technique such as, but not limited to, enzyme treatment (restriction
enzymes),
polym.erase, limited DNase I digestion, limited mung bean nuclease digestion,
sonication,
shearing of DNA, and the like. Some of these segmental fragments will be
complementary to
at least a portion of one or more DNA segments in the particular unique target
sequence.
The individual DNA segments are propagated by commonly known methods, such as
cloning
into a plasmid construct and then transfected into bacteria. After propagating
the cloned
fragments, individual colonies representing isolated fragments are identified
as containing at
least a portion of the sequence of interest. Identification is accomplished by
known
techniques such as hybridization, PCR, or searching established databases of
commercially
available libraries. Each chosen colony is grown to obtain an isolated plasmid
construct
having a unique fragment, at least partially complementary to a segment of the
target
sequence on the chromosome. Exemplary target sequences include BER-2, IGF-1,
MUC-1,
EGFR, and AR and may be available through commercial vendors (i.e. BAC
clones).
Once the cloned fragments of interest are propagated and isolated, they are
depleted of their
repetitive polynucleotide sequences. Using whole gene amplification (WGA), the
fragments
are amplified as 200 to 500bp segments from the isolated plasmid constructs.
Commercially
available DOP PCR is considered as one embodiment to this portion of the
procedure.
CA 02623405 2008-03-20
WO 2007/053245 PCT/US2006/036656
Cot-1 DNA is combined with the WGA library pool after amplification by first
heating to
95 C to denature the double-strand polynucleotide into a single strand state
and then cooling
to 65 C to allow selective re-annealing of the repeat sequences. Duplex
specific nucleases
(DSN) under optimized DSN conditions are then added to preferentially cleave
deoxyribonucleic acid molecules containing perfectly matched nucleic acid
duplexes while
not affecting any remaining single stranded segments. Selectively cleaving the
duplex
nucleic acids is accomplished by enzymatic digestion of DNA-DNA duplexes and
DNA-
RNA duplexes. Specific embodiments of the present invention include DSN
isolated from
the Kamchatka crab (US 10/845,366) or shrimp (US 6,541,204), but any enzymatic
removal
of duplex structure is considered in the present invention. The use of
endonuclease-specific
nucleases hydrolyzes a phosphodiester bond in the duplex DNA backbone,
providing the
advantage of not being nucleotide sequence-specific and therefore applicable
to most targets
of interest. DSN digestion provides for the removal of a substantial amount of
the nucleic
acid duplex for subsequent amplification of the remaining single-strand
polynucleotide. One
embodiment of the present invention is a 2 hour DSN digestion at 65 C. The
resulting
composition contains single stranded DNA, corresponding to portions of the
unique target
sequence on the chromosome, some amount of undigested double-strand DNA, and
digested
base pairs. Preferably, the undigested DNA is separated from the digested DNA
and the
DSN by centrifugation (i.e. spin column chromatography). The mixture is used
immediately
or stored at 80 C, either before or after amplification of the purified
composition for
subsequent utilization such as labeling and use for in situ hybridization.
After amplification,
the resulting target probe sequence is amplified by PCR yielding 90% to 99%
pure target
probe sequence, and designated repeat-depleted DNA.
The use of DSN in the enrichment and isolation of a single strand
polynucleotide from
double strand is applicable in the production of any single strand
polynucleotide wherein
separation of the single strand entity from double strand contaminants is
desirable. This is
particularly relevant, although not limited, in the production of labeled
probes for gene or
chromosome identification, karyotype or panning a pool of single strand and
double strand
polynucleotides.
The resulting probes, both composition and production, are incorporated in the
subject
matter embodied in the present invention. Repeat-depleted DNA, as described in
the present
invention, is useful for in situ hybridization, including FISH, and all other
nucleic acid
hybridization assays. The requirement for competitive binding is eliminated
using the repeat-
16
CA 02623405 2008-03-20
WO 2007/053245 PCT/US2006/036656
depleted probes described in this invention, resulting in increased
specificity of the reaction
and a reduction in the amount of probes necessary for binding.
The Duplex Specific Nuclease Method
To make a hybridization probe toward a target sequence, DNA containing the
sequence of
interest is obtained. Methods to obtain DNA containing sequences of interest
will be known
to those skilled in the art and include, without limitation, isolation of
genomic DNA from
tissues or cells, flow sorting of chromosomes, and screening libraries of
cloned fragments of
chromosomes by hybridization, electronically, or PCR.
Starting DNA used in the practice of this invention is purified from a source
by any
method. Typically the starting DNA consists of genomes, chromosomes, portions
of
chromosomes, or cloned fragments of chromosomes. Flow ¨sorted chromosomes and
Bacterial Artificial Chromosomes (BAC) known to contain target sequences of
cancer related
genes make the present invention particularly applicable. Exemplary target
sequences
include HER-2, IGF-1, MYC, EGFR, and AR. BAC clones containing these sequences
are
available through commercial vendors.
Once the DNA containing sequences of interest are identified and obtained,
they are
depleted of repetitive polynucleotide sequences. This process begins by
fragmenting and
preparing a library containing the sequence of interest. One method is the
GenomePlex
Whole Genome Amplification (WGA) method (GenomePlex is a trademark of Rubicon
Genomics, Inc.) that randomly cleaves the cloned fragments into 200-500 bp
fragments and
attaches linker sequences which can then be used to amplify and re-amplify the
library using
PCR. In this example the fragmented, amplified library is considered the
source DNA.
To remove the repetitive sequences, the source DNA is denatured to a single
stranded
state and then cooled under conditions that selectively allow repetitive
sequences to anneal to
form double stranded molecules and unique sequences to remain single stranded.
A duplex
specific nuclease (DSN) is then added which preferentially clea,Ves the double
stranded
repetitive fragments while not cleaving the single stranded unique sequences.
The resulting
mixture contains single stranded DNA, corresponding to portions of the unique
target
sequence on the chromosome, some amount of undigested double-strand DNA, and
digested
base pairs. Preferably, the undigested DNA is separated from the digested DNA
and the
DSN by spin column chromatography, phenol chloroform extraction or some other
similar
method, but separation is not a requirement. Then, the repeat-depleted library
is used as a
hybridization probe or re-amplified using PCR to prepare larger amounts of
probe DNA.
17
CA 02623405 2013-08-26
After amplification, the resulting target probe sequence is 90% to 99% pure
target probe
sequence, and designated Repeat-depleted DNA.
The library fragmentation and amplification methods described above are not
intended to
be limiting but rather serve as an example of how one fragmentation and
amplification
method is used to make repeat-depleted probes. There are numerous methods of
fragmenting
and amplifying nucleic acids including linker-adapter PCR, DOP PCR, rolling
circle
amplification, transcription-mediated amplification and all other methods are
considered this
invention. It is expected that some modification of the above method to
prepare repeat-
depleted DNA will be necessary to accommodate the different methods of library
fragmentation and amplification and these modifications are also included in
the present
invention.
One consideration in this invention is the use of an enzyme that is capable of
cleaving
double stranded DNA while not cleaving single stranded DNA. Enzymes included
in this
invention may cleave double stranded DNA in any way, including lysis of the
sugar-
phosphate backbone, removal of one or both strands in a DNA duplex or removal
of
nitrogenous bases to form apurinic/apyrimidinic sites. Non-limiting examples
of these
enzymes include endonucleases, exonucleases, restriction enzymes, nicking
enzymes, DNA
repair enzymes, topoisomerases, DNA gyrases, and enzymes involved in
homologous
recombination. Specific embodiments of the present invention include DSN
isolated from
the Kamchatka crab (US 10/845,366), shrimp (US 6,541,204), T7 Endonuclease 1,
and E.coli
exonuclease )11,.
Enzyme concentration, time of digestion, and buffer conditions such as salt
and
magnesium ion concentration are factors that can affect the specificity of DSN
toward double
stranded DNA. Optimization of these conditions is necessary to get efficient
repeat-
depletion.
Efficiency and specificity of repeat removal in this invention are dependent
on the
reaction conditions used to denature and re-anneal as well as the conditions
present during
digestion with the DSN. Denaturation is accomplished by alkali or heating. The
degree of
DNA re-annealing is dependent on the concentration of DNA present in the
samples and the
time allowed for re-annealing. In order for selective re-annealing of
repetitive sequences to
occur, the repeat sequences must be present in a higher concentration than the
unique
sequences. The ratio of repeat to unique sequences within a particular clone
will vary from
region to region throughout the genome. To standardize the depletion process
across regions
with varying numbers of repeat sequences, an excess of subtractor DNA is added
to the
18
CA 02623405 2008-03-20
WO 2007/053245 PCT/US2006/036656
reaction. The mass of subtractor DNA added varies depending on the desired
amount of
repeat removal and is preferably 10-50 times the mass of the source DNA. The
present
invention considers that a subtractor is any nucleic acid or nucleic acid
analogue containing
sequences sufficiently homologous in nucleotide sequence to the repetitive
sequences as to
allow hybridization between subtractor sequences and a portion of the
sequences in the
source DNA, making the subtractor sequence useful. One embodiment of the
present
invention includes Cot-1 DNA as a subtractor DNA which is used to remove
repetitive
sequences from source DNA.
The stringency for re-annealing is another component in the present invention.
Salt
concentration and temperature are factors that determine the stringency of any
re-annealing
step. The degree of repeat removal is controlled by adjusting stringency
conditions for this
step. Adjusting the stringency conditions to allow some degree of annealing
between
sequences that are not 100% homologous improves the degree of repeat removal.
Salt
concentrations range from 5 millimolar to 1000 millimolar NaC1 with annealing
temperatures
range from 15 C to 80 C.
In one embodiment, the repeat-depletion process is performed such that re-
annealing and
DSN digestion occur sequentially. Accordingly, the DNA is denatured and
allowed to cool
for a period of time under conditions optimized for annealing. Then, the
reaction conditions
are changed to conditions that optimize the specificity and activity of DSN
digestion. In
another embodiment, the re-annealing and DSN digestion take place
simultaneously, under
the same conditions.
Also within the scope of this invention, the source or subtractor DNA is
treated with an
agent, either chemical or physical, before or during enzymatic digestion to
alter the
specificity of an enzyme toward either the single stranded or double stranded
fractions within
the mixture. For example, E.coli RecA protein is added to a mixture of single
stranded and
double stranded DNA. This protein coats the single stranded DNA in the mix and
protects
the single strand DNA from E.coli RecBC DNase while allowing the double
stranded DNA
in the mixture to be digested ("Escherichia coli RecA protein protects singles
stranded DNA
or Gapped Duplex DNA from degradation by RecBC DNase". Williams, JGK, Shibata,
T.
Radding, CM Journal of Biological Chemistry V246 no.14 pp 7573-7582). It is
also possible
to generate source or subtractor DNA using modified nucleotides which alter
the specificity
of an enzyme toward the single strand or double strand DNA fractions.
The use of DSN in the enrichment and isolation of a single strand
polynucleotide from
double strand is applicable in the production of any single strand
polynucleotide wherein
19
CA 02623405 2008-03-20
WO 2007/053245 PCT/US2006/036656
separation of the single strand entity from double strand contaminants is
desirable. This
includes removal of any undesirable sequence from a source DNA. These
undesirable
sequences include without limitation, repetitive sequences, unique sequences,
and vector
sequences. This method is particularly relevant in the production of labeled
probes for gene
or chromosome identification, karyotyping, or panning a pool of single strand
and double
strand polynucleotides.
The Selective Binding Method
By denaturing source DNA and selectively allowing repetitive sequences to
anneal, any
agent that binds preferentially to a single or double stranded DNA structure
is used to remove
repeat sequences from source DNA. Examples of these agents include, without
limitation,
DNA or RNA polynucleotides, enzymes, antibodies, DNA binding proteins,
combinations of
antibodies and DNA binding agents, and natural or synthetic compounds and
molecules.
DNA binding agents may be linked directly or indirectly to a solid support
which allows for
positive or negative chromatographic selection of unique or repetitive
sequences. One
example includes separation of single and double strand DNA using biotinylated
antibodies
toward single strand or double strand DNA. The desired population is separated
using
streptavidin-coated paramagnetic particles. Alternately, a biotinylated
antibody toward a
DNA binding agent that preferentially binds single or double strand DNA is
used in the same
fashion.
Other Single or Double Strand Specific Enzymes
The present invention also embodies any enzyme that preferentially acts on
single or
double strand DNA in modifying source or subtractor DNA in facilitating repeat
removal.
One non-limiting example is to selectively ligate a DNA linker to the double
stranded DNA
population after denaturation and selective re-annealing the repeats. This
linker is hybridized
to a homologous oligonucleotide attached to a magnetic (or paramagnetic)
particle to remove
repetitive sequences. A second example is to use a single strand DNA/RNA
ligase to
selectively circularize single stranded DNA present after denaturation and
selective re-
annealing the source DNA. The resulting circles are then be amplified and
enriched by
rolling circle amplification.
Structure Specific Separation of Repetitive Sequences
CA 02623405 2008-03-20
WO 2007/053245 PCT/US2006/036656
In addition to specific probe production methods and based on separation of
single
stranded DNA from double strand DNA, the present invention considers any
method known
in the art whereby separation of repeat sequences from unique sequences occurs
with the
establishment some detectable DNA structure in either the repeat sequences or
the unique
sequences and this detectable structure is used to separate one population
from the other.
Some examples of detectable DNA structures include without limitation, triple
or quadruple
stranded DNA, hairpins, panhandles, flaps, Z-DNA, Holliday junctions and other
structures
formed during recombination. These structures may be naturally occurring
within the
sequences of interest or they may be induced by modifying either or both the
source nucleic
acid or the subtractor nucleic acid.
Selective Digestion of Repeat Sequences
Another method to remove repeat sequences from source DNA is to digest a
fragmented
and amplifiable DNA library with a restriction enzyme whose recognition
sequence is known
to exist in repetitive DNA sequences. When the digested source DNA is re-
amplified by
PCR, the remaining library will be enriched for unique sequences and depleted
of sequences
that contain repeats.
Digestion and Selective Ligation
Another method is to prepare repeat-depleted probes is to digest target DNA
with two
restriction enzymes that leave different overhangs on the digested sequence.
The first
restriction enzyme is preferably an enzyme that cuts within the repeat
sequences and the
second is an enzyme that the does not cut within the repeat sequences.
Following digestion,
linkers are selectively attached to the ends of the sequences cut by the
second restriction
enzyme. These linker sequences are then used to PCR amplify a library,
depleted of repeat
sequences. The resulting repeat-depleted DNA, both composition and production,
are
incorporated in the present invention. Repeat-depleted DNA, as described in
the present
invention, is useful as probes for any type of hybridization assay where
specific binding of
target sequences is desired. These techniques include, without limitation,
ISH, FISH, CGH,
spectral karyotyping, chromosome painting, Southern blot, Northern blot, and
microarrays.
Production of hybridization probes that only contain unique sequence is one
embodiment of
the present invention. Consequently, the requirement for competitive binding
is eliminated,
resulting in an increase in the specificity of the reaction, and reducing the
amount of probes
necessary for binding.
21
CA 02623405 2013-08-26
=
Use of Duplex Specific Nuclease to Cleave DNA at a Desired Location.
The use of duplex specific nucleases has utility in cleaving specific
sequences from
DNA.. Figure 2 show a schematic representation of this embodiment. Single
strand DNA is
obtained from a DNA source, containing the desired sequence. Oligonucleotides
identifying
both ends of the desired sequence are added, and duplex specific nucleases
introduced to cut
the desired DNA probe from the source DNA. After separating the desired DNA
probe by
size exclusion, second strand DNA is synthesized and cloned to provide a
source for DNA
probes. Thus, duplex specific nucleases are used to cleave DNA sequences at
specific
regions. This method is most useful in cloning of fragments of interest that
are too large to
amplify by FCR and when the fragments lack appropriate restriction enzyme
sites. These
fragments may then be used as hybridization probes, sequenced, or used for any
other
purpose. In Figure 2, two oligonucleotides are designed to anneal to one or
both strands of
the DNA, and flank the sequence of interest. DNA containing a sequence of
interest is
denatured and allowed to re-anneal in the presence of an excess of the
flanking
oligonucleotides. Digestion with a duplex specific nuclease selectively cuts
the DNA at the
site where the oligonucleotides are annealed. The remaining single strand DNA
molecules
are fractionated by size to obtain the sequence of interest. The sequence of
interest is made
double stranded using a DNA polymerase and cloned into a plasmid. Thus making
possibilities to clone or subclone sequences of interest from a larger DNA
polynucleotides.
A second example of site specific cleavage is the recovery of cloned fragments
from plasmid
vectors by selectively digesting the vectors. In one example, the plasmid
containing cloned
DNA fragment is denatured and allowed to re-anneal in the presence of excess
plasmid,
lacking cloned DNA. Addition of a duplex specific nuclease cleaves the plasmid
sequences
and leaves the single strand cloned DNA intact. The remaining single strand
DNA is then be
used for any application known in the art including, but not limited to,
sequencing or
subcloning.
Example 1-Detection of Chromosomes or Portions of Chromosomes using Repeat-
Depleted DNA Probes
BAC clone CTD-2019C10 was selected to be used as a probe for the Her-2 gene by
electronically screening the human genome using the UCSC Genome Browser
software
and clones were obtained from Invitrogen
(Carlsbad, CA). BAC DNA was isolated using the Large Construct Kit from Qiagen
22
CA 02623405 2013-08-26
(Valencia, CA). Source DNA was prepared using 10 nanograms of purified BAC and
the
Genomeplex Complete Whole Genome Amplification Kit (Sigma-Aldrich St. Louis,
MO)
according to the manufacturer's directions. Depletion mixes were prepared
containing 2
micrograms of Cot-1 DNA, lx Duplex Specific Nuclease buffer (Ewogen, Moscow,
Russia),
0.3 molar NaC1, and 66 nanograms of source DNA. The depletion mixes were
denatured for
minutes at 95 C, placed on ice for 10 seconds and 1 unit of Duplex Specific
Nuclease
(Evrogen, Moscow, Russia) added. Samples were incubated at 65 C for 90
minutes. Five
microliters of the reaction were purified using the GenelutTPCR Clean-Up Kit
(Sigma-
Aldrich St. Louis, MO) and the purified DNA was eluted in 50 microliter
aliquots. Fifteen
microliters of the depleted samples were then re-amplified by PCR using the
Whole Genome
Re-amplification kit (Sigma-Aldrich St. Louis, MO). PCR reactions were
purified as
described and quantified based upon their A260. Ten nanograms of the first re-
amplification
mixture was used as template in a second re-amplification, purified as
described. This
material was sonicated to an average molecular weight of 200-500 base pairs,
ethanol
precipitated, and resuspended in distilled H20. The resulting DNA was
fluorescently labeled
using the ICreatech ULS Platinum Bright Red/Orange Kit (Kreatech, Amsterdam,
Netherlands). For comparison, probes with repeats were also made from the
source DNA
which was used in the depletion process..
Phytohemagglutinin-stimulated white blood cells were prepared for FISH by
fixation in
75%methanol, 25% acetic acid and spotted on slides using standard techniques.
Repeat-
depleted probes and source DNA probes were hybridized at 2ng/ 1 without Cot
blocking
DNA in a hybridization buffer consisting of 50% formamide, 10% dextran
sulfate, and lx
SSC. Slides and probe were co-denatured at 80 C for 3 minutes and hybridized
overnight at
37 C. Following hybridization, samples were washed for five minutes at 50 C in
0.5x SSC,
0.001% SDS. Samples were counterstained in 0.5 Rg,/m1DAPI for 5 minutes and
mounted in
50% glycerol. Images were acquired using a Leica DM-RXA fluorescent
microscope(Leica
Microsystems, Bannockburn, IL) equipped with filters appropriate for rhodamine
and DAPI.
Images were acquired with a Photometrics SynSYnsviblack and white digital
camera
(Photometrics, Tucson, AZ). DAPI signals were enhanced and overlay images were
generated using Leica FW 4000 software. Her-2 images are unedited and were
captured using
identical camera settings comparison purposes. Figure 3 depicts a comparison
of the images.
Panel A shows that when source DNA containing repeats is used as a
hybridization probe, the
probes stain the entire nucleus and no Her-2 specific signals are visible.
When repeat-
23
CA 02623405 2008-03-20
WO 2007/053245 PCT/US2006/036656
depleted DNA is used as a probe (Panel D) specific signals that correspond to
the Her-2 gene
are clearly detectable (arrows).
Example 2- DNA probes depleted from repeat sequences according to this
invention
improves the visualization of fluorescently labeled DNA probes as compared to
traditional DNA probes that contain repeats which are blocked during the
procedure.
The signal to noise ration of fluorescently labeled probes is significantly
improved when
employing repeat depleted DNA probes obtained according to the invention. For
this
comparison, DNA probes targeting 9p21 and 11q23 were used as they are known to
those
skilled in the art. These signals are problematic in that they are relatively
small signals and
difficult to discern. Probes were depleted from repeat sequences according to
the invention
and fluorescent reporter molecule linked to a platinum group which forms a
coordinative
bond t the N7 position of guanine was used to fluorescently label the probes
(ULS labeling,
Kreatech, Amsterdam). Figure 4 Panels A and B show a chromosome spread
hybridized with
rhodamine labeled 9p21 and dGreen labeled 11q23 probe respectively. Clear
signals from
the repeat free probes can be discerned with the repeat free probes as
indicated by arrows in
the figures. Thus, the visualization of the presence of these probes is
superior to those that
are obtained using probes that are obtained through traditional methods. This
improvement
in visualization provides a more accurate differential diagnosis of melanoma
(Panel A,
9p21, P16 (CDKN2A) and leukemia (Panel B, 11q23, MLL).
Preservation of Immunomagnetically-Labeled Cells for Subsequent Analysis
During immunocytochemistry (ICC) image analysis the cells are magnetically
held to the
optically transparent surface of the cartridge by magnetic forces applied by
an external
magnet (US 5,466,574). The calculated holding force of the device is
approximately 10-9
Newtons. This holding force is dependant upon several variables including but
not limited to
the number of ferrofluid particles on the cell, the size of the magnetic
particles and the
magnetic field gradient applied by the external magnet. In order to fix the
cells to the glass
surface, the buffer solution must be removed and replaced by a cell fixative
solution, such as
methanol, acetone, acetic acid, other agents known in the art and combinations
of these.
Aspiration of the buffer solution must be carefully completed so as to not
displace or remove
the cells to be analyzed. So as fluid is aspirated from the sample chamber,
the meniscus of
the fluid applies shear forces on the magnetically held cells. These shear
forces can '1:0
greater than the magnetic holding forces (calculated at greater than 10-9
Newtons). In such
24
CA 02623405 2008-03-20
WO 2007/053245 PCT/US2006/036656
situations, the cells will either be moved within the cartridge or displaced
such that they are
aspirated from the cartridge along with the buffer solution. Fluid shear
forces are a function
of the rate at which the meniscus moves across the glass portion of the
cartridge, the distance
between the aspiration probe and the glass surface, the velocity and viscosity
of the fluid
being aspirated and other parameters. Additionally after aspiration, any
drying of the glass
surface before the fixation solution is added can have a negative effect on
the cells within the
cartridge. Plus, the addition of a fixation solution into an empty cartridge
will further disturb
the distribution of the cells.
Accordingly, one aspect of the present invention address these issues by
providing a
method for replacing the buffer solution with the fixation fluid without
subjecting the cells to
fluid shear forces caused by the meniscus. Fixation solution is dispensed into
the bottom of
the cattlidge with the simultaneous aspiration of the displaced buffer
solution from the top of
the caitiidge. While some mixing of fixative and buffer will take place at the
interface of the
two fluids, sufficient fixation solution will be dispensed to complete the
required cell fixation
to the glass surface. This fluid displacement will occur with minimal shear
forces applied to
the cells in the cartridge by balancing the flow between the dispensed
fixative solution and
aspiration of the displaced fluid, in addition to the magnetic holding force
retaining the
immunomagnetic attached cells to the surface of the glass. One preferred
embodiment of the
present invention utilizes the entry area of sample chambers described in US
6,861,259; US
10/988,057; and US 7,011,794; US 11/294,012 in displacing approximately 100
microliters
of fluid within the cartridge without spilling out of the cartridge. The
opening port of the
cartridge is sufficient to allow an aspiration probe to remove buffer solution
as the fixative
solution is being dispensed to displace the buffer solution. Once the cells
have been fixed in
place by the fixation fluid, the fluid may be removed without risk of cell
disturbance. This
procedure allows for automated processing of samples for subsequent FISH or
other analysis
with minimal operator interaction that could introduce variability into the
preparation
process.
The present invention describes an automated device which allows for complete
and
consistent fixation of cells in the cartridge after ICC imaging in a bench-top
device, and
incorporates all the steps in the preparation of target cells after ICC for
subsequent FISH
image analysis. Figure 5 depicts a schematic view of the apparatus showing the
relative
locations of the individual components. Accordingly, the cartridge containing
the ICC
imaged sample is placed into the device for buffer removal and fixation. A
syringe and
syringe pump in combination with a pipette aspirates the buffer and dispenses
the fixative.
CA 02623405 2008-03-20
WO 2007/053245 PCT/US2006/036656
Figure 6 is a schematic representation of the steps involved in the fixation
and hybridization
of the cells. In one embodiment of the invention the buffer removal and
addition of fixative
are performed simultaneously to minimize cell movement through the forces
exhibited by the
fluid removal and addition. The fixation is completed by removal of all fluids
from the
car tiidge followed by drying of the cartridge by a forced air flow inside
the cartridge using
the same pipette as used for the addition and removal of fixation reagents.
After fixation and
drying the cartridge is stored or used immediately for FISH or other
additional analysis.
Optimal mixtures for the fixative differ depending on the target entity (i.e.
DNA, RNA,
protein).
Fixation Protocol for FISH
To fix the cells on the upper surface and leave them intact and accessible for
FISH
probes, the following protocol is developed and implemented in the automated
bench-top
device:
1. Dispense 250 microliters of fixative from the bottom of the cartridge
(cartridge in up-
right position).
2. Aspirate 250 microliters from the top and dispose..
3. Repeat the dispense 250 microliters new fixative from the bottom of the
cartridge.
4. Aspirate all fluid from the top of the cartridge.
5. Dry the cartridge by flowing air through the cartridge. Pipette used for
aspiration/dispensing is used for air flow as well.
Volume Reduction
As a consequence of the expense of antibodies or polynucleotide probes and the
requirement to use them in high concentrations, reactions are carried out in
very small closed
volumes (for example 5 microliters to 25 microliters) so the cost of using a
high
concentration is offset by having to use very small volumes of reagents.
Sample cartridges as
described in US 6,861,259; 10/988,057; and US 7,011,794; 11/294,012 are used
as the
reaction vessel after immobilization in the chamber. In these cartridges, the
immediate
volume of the chamber where the cells are immobilized is 320 microliters.
Thus, there is a
need to analyze immobilized cells by in situ hybridization, but the adding 320
microliters of a
high concentration of most probes are expensive and impractical.
26
CA 02623405 2013-08-26
_
To address this problem, a uniform distribution of the probe mixture across
the surface of
the optically transparent surface of the cartridge where the cells are
immobilized is needed,
while reducing the volume of the added probe. This method is obtained by the
following:
1. Inserting an object inside the cartridge to reduce the volume.
2. Using a volume that is large enough, but smaller that the 320 microliters
across the
entire surface where the cells are immobilized when the cartridge is in a
horizontal
position and the surface with the cells is downside (optical viewing surface
on
bottom).
3. Use a small volume plus a fluid with a density that is lower than the
density of the
reagents. The low density fluid floats on top of the reagent and allows the
reagents to
spread uniformly across the entire surface.
As an example of the first possibility, an extension is introduced at the
probe end of the
stopper so that a portion extends the full length of the chamber (Figure 7).
The extension
consumes approximately 1/3 of the volume of the chamber. The extension
diameter is
dimensioned such that it will slide through the chamber opening, 2.36 mm
diameter. The
extension is made by molding an entire new plug over the molding on the
existing plug, or
inserting a solid metal rod through the center line of the plug. The meterial
must be inert to
TM
the reagents as, for example, 316 stainless steel, polypropylene or Inconel
625. The over
molding of the upper portion of the plug with a thermal plastic is necessary
to ensure the
proper plastic durometer for maintaining shape during insertion and when
positioned in the
chamber to maintain liquid seal and locking of the plug. Further, the plug and
extension
optionally has an access hole through the center for monitoring temperature
within the
chamber during processing. The plug is further designed to be removed and re-
used after
proper cleaning.
A second embodiment is depicted in Figure 8. A volume of 50 microliters of
FISH
reagents is sufficient to cover the whole upper surface of the cartridge. This
volume needed
to ensure complete reactions and is dependent on the viscosity and
hydrophobicity of the
reagents. After addition of the reagents, the cartridge is placed in a
horizontal position to
further ensure exposure to the reagents.
One other embodiment incorporates the second embodiment with a further
reduction in
reagent volume. After injecting of the reagents as described, an extra fluid
with a lower
density is injected. As the lower density fluid floats on top of the reagents
of the reagents,
there is a more complete reaction over the entire surface. Depending on the
components of
the reagents, a volume of 25 microliters reagents is obtained.
27
CA 02623405 2013-08-26
Reanalysis of Immunomagnetically-Labeled Cells
Chromosomal aneuploidy is associated with genetic disorders, particularly
cancer.
Diagnostic methods are available that provide for the detection of these
chromosomal
abnormalities particularly with the use of in situ hybridization (ISH). The
application of ISH
and immunocytochemistry (ICC) on tissue or cell samples has been well
established, but
there is a clear need to establish a diagnostically effective method for the
simultaneous
analysis of ISH and ICC on a single cell. One aspect of the present invention
provides for the
detection of these chromosomal abnormalities on individual cells as they
relate to the
confirmation of morphologically suspect cancer cells through a Cost effective
and highly
specific means.
One aspect of the present invention provides for the further processing of
rare cells after
enrichment and immunocytochemical (ICC) analysis. For example, circulating
rare cells
such as epithelial cells are identified as suspect cancer cells (US 6,365,362;
US 6,645,731;
and US 11/202,875). Suspect cells are identified through
specific cellular antigens and nucleic acid labeling. Confirmation of these
suspect cells are
subsequently determined by the expression of specific unique target sequences,
defining
either a chromosome and/or gene, used to assess chromosomal changes (i.e.
aneuploidy)
within the identified suspect cell. Accordingly, one embodiment of the present
invention
includes the combination of ICC staining and subsequent confirmation by
fluorescent in situ.
hybridization (FISH) on a group of selected chromosomes which define a CTC.
The cancer confirmatory assay provides an increased specificity after
hrnnunomagnetic
enrichment and fluorescent imaging of circulating tumor cells as provided by
the
CellTracks AutoPrep and CellTracks Analyzer II Systems (Immunicon
Corporation)
and further described in US 6,365,362. A confirmatory test permits the
designation of 1 or
more CTC's as a cancer cell regardless of the stage of the disease and thus
lowers the
threshold for calling a sample positive for CTCs. One embodiment of the
present invention is
assessing aneuploidy in chromosomes 1, 7, 8 and/or 17 to confirm ICC-
determined suspect
CTC's. A further embodiment includes the detection of individual genes such
as, but not
limited to, HER-2, IGF-1, MYC, EGFR, and the androgen receptor (AR) to detect
the
presence or absence of therapeutic targets and thus provides a means to make
the correct
choice of treatment.
Accordingly, an automated and standardized method for blood sample processing
provides identification of circulating epithelial cells by ICC. Aspirated
plasma from a
28
CA 02623405 2008-03-20
WO 2007/053245 PCT/US2006/036656
partitioned blood sample is combined with a ferrofluid reagent conjugated to
antibodies
specific for a target cell population (i.e. EpCAM positive). These cells are
immunomagnetically collected through an externally applied magnetic field,
allowing for
separation and removal of unlabeled cells.
Once the target cells are separated, they are dispensed into a disposable
cartridge for
image analysis using an image presentation device (US 6,790,366 and US
6,890,426). The
device is designed to exert a magnetic field that orients the labeled cells
along the optically
transparent surface of the chamber for subsequent ICC imaging.
After ICC imaging, suspect cells are identified using appropriate algorithms.
Images of
the suspected cells are presented to the user who makes the final decision
about the identity
of the presented suspect cells. Images of the suspect cells and their relative
position along the
optically transparent viewing surface of the chamber are recorded and archived
for later use.
Since ICC imaging alone lacks the specificity to assess the clinical
significance of blood
samples with less than 5 CTC's or to provide detailed genetic information
about suspected
cancer cells, subsequent analysis using multiparametric genetic profiling on
individual
suspect cells is needed to provide a complete profile and establish a
confirmatory mechanism
that can be used in diagnostic analysis, including screening, assessing
recurrance of disease,
and overall survival. One embodiment of the present invention utilizes
fluorescent in situ
hybridization (FISH) as a multiparametric genetic analysis, but other profile
assessments are
considered. This provides both phenotypic and genotypic profile assessment for
an
individual cell present along the viewing surface
FISH requires temperatures above the melting temperature of DNA as well a
reagents that
are not compatible with the ICC labeling. Most of the ICC and DNA labels do
not survive
the FISH procedure with any signals lost in processing. Thus, a cell that was
identified as
being an interesting cell for FISH analysis can not be traced back on its
position. Therefore
there is a need to have a detection method that once the ICC image is
obtained, the cell
position along the optically transparent viewing surface is maintained for
subsequent
multiparametric genetic analysis (FISH) or other types of analysis in which
the ICC labels are
lost. This is achieved, in part, by fixing the cells on the optically
transparent surface after the
ICC image is obtained without a loss of cells or any substantial movement
along the surface.
Accordingly after addition of the FISH reagents, the cartridge is placed on a
hotplate having
the surface with the immobilized cells in contact with the hotplate. Depending
on the type of
assay the hotplate is programmed with different temperature cycles that run
between 2 and 48
hours. After the temperature cycles are completed, the excess FISH reagents
are removed
29
CA 02623405 2008-03-20
WO 2007/053245 PCT/US2006/036656
from the cartridge. The cartridge is filled with a buffer solution containing
a DNA label to
visualize the nuclei of immobilized cells. Depending on the DNA label used,
the label
remains in the cartridge or is washed out of the cartridge after staining.
Next, the cartridge is placed back in the CellTracks Analyzer II System for a
second
scan. Because cells present on the upper surface during the first ICC image
analysis were
immobilized, the same cells are still in the same relative location inside the
cartridge. To
assess the shift of the cartridge relative to the imaging system (CellTracks
Analyzer II
System), the locations of the nuclei in the images of the second scan are
compared to the
location of the nuclei in the images of the first ICC scan. The shift of these
images with
respect to each other is determined using convolution algorithms. After this
shift has been
determined a specific cell of interest, based on its ICC image, can be
selected from a list and
be relocated on the surface of cartridge after FISH in the second scan. Next
fluorescent
images of the different FISH probes are acquired.
Figure 9 shows a representative image of a tumor cell, identified by ICC and
probed for
the presence of chromosome 1, 7, 8 and 17. Panel A shows a list of CTC
candidates
identified by the software as cytokeratin (cytoskeletal protein present in
cells of epithelial
origin) positive and DAPI (nucleic acid stain) positive. The corresponding
images of the
highlighted event were identified as a CTC by the user as it confirmed the CTC
definition
(cytokeratin positive, CD45 negative, DAPI positive event with the
morphological
appearance of a cell). Four images taken with a 10X objective are shown in
Panel B. The
The top left image shows the DAPI staining of the nucleus and the bottom left
image the
cytokeratin staining of the cytoplasm. CD45 staining and FITC staining are
lacking as
illustrated by the lack of positive staining. After the cells were preserved
and probed for the
centromeric probes for chromosome 1, 7, 8 and 17, images of the upper surface
of the
cartridge were reacquired and the fluorescent signals of the probes for
chromosome 1, 7, 8
and 17 are shown in Panel C for the same cell shown in B. Two copies of
chromosome 1,
three copies of chromosome 7, four copies of chromosome 8 and two copies of
chromosome
17 are clearly visible demonstrating that the cell is aneuploid and confirming
that the cell
indeed is a cancer cell.
Images displayed in Figure 10 are acquired using a 10X, NA 0.5 plan achromat
objective.
Although the resolution is sufficient for most centromeric probes, it is not
sufficient for the
gene specific probes, for example HER-2 and EGFR FISH probes. For this reason
the 10X,
NA 0,5 objective, the objective used for ICC image acquisition, is replaced by
a 40X, NA 0.6
objective, corrected for the optical thickness of the transparent upper
surface of the cartridge.
CA 02623405 2008-03-20
WO 2007/053245 PCT/US2006/036656
...
The use of high NA objectives allows for 3D imaging of the cell of interest
allowing for a
confident determination of the correct number of copies for FISH probe labeled
sequence in a
selected cell. Multiple images at different focal planes along the optical
axis of a specific cell
of interest are acquired followed by 3D reconstruction of the cell. Figure 10
shows 5 such
slices through the cell using excitation/emission filters for 5 different
fluorochromes. In
Panel A, five slices for PE are shown. In slice # 2, only two signals are
visible whereas in
slice # 3 three signals are visible. Panel B shows 5 slices of the DAPI
staining. In the APC
slices of Panel C, # 2 slice shows 1 signal. In the FITC slices of Panel D,
slice #3 shows two
signals and slice #4 shows two different signals, making the total for this
probe 4. In Panel E,
Dy415 slices show 1 signal in slice #2 and two signals in slice #3. From the
images, it is
clear that the probes are located in different parts of the nucleus and that
using only one focal
plane the counting of the signals would not be correct. In Figure 11 Panel A
fluorescence
ICC images of the CTC, presented as a stack of images from Figure 10. In panel
B, the slices
for each fluorochrome are added and the average intensity is presented thereby
scaling the
images to use the full range of intensity levels which facilitates
enumeration. The count for
the number of signals with each probe are shown next to each image (i.e. 4 for
PE, 1 for
APC, 4 for FITC and 2 for dy415).
It is understood that the subject matter of this invention is not limited to
the detection of
cancer cells but can also be used to characterize other cell types. One cell
type frequently
pursued for detection of cytogenetic abnormalities is fetal cells in maternal
blood. To enrich
for such cells, markers need to be targeted that are present at high frequency
on the fetal cells
and in low frequency on the maternal cells. One cell type that is frequently
pursued is the
nucleated red blood cells. A marker that is present on all nucleated red blood
cells is, for
example, the transferin receptor (CD71). When coupled to ferrofluids,
nucleated red blood
cells are reproducibly enriched from whole blood with the CellTracks Autoprep
System.
The enriched cells contain fetal nucleated red blood cells, maternal nucleated
red blood cells,
activated T-lymphocytes, immature reticulocytes and other cells that have been
carried over
by the immunomagnetic enrichment. The enriched cell population is now be
stained with
markers that discriminate between cells of fetal and maternal origin. One such
panel of
markers is the use of CD45 to eliminate leukocytes from the analysis in
combination with
Hemoglobin F that is present in fetal red blood cells but only rarely in
maternal red blood
cells, carbonic anhydrase that is only present in adult red blood cells and
DAPI to identify the
nucleus of the cells. The CellTracks Autoprep System is used to stain the
cells in a
reproducible manner. As some of the antigens are intracellular the cells,
cells need to be
31
CA 02623405 2008-03-20
WO 2007/053245 PCT/US2006/036656
permeabilized for the antibodies to pass the cell membrane. The agents used
for
permeabilization also lyse the immature reticulocytes, specifically selected
by the use of
CD71 and the remaining erythrocytes that were carried over through the
procedure. After the
staining of the cells with the probes that have different reporter molecules,
the cartridge
containing the stained cells are placed in the CellTracks Analyzer II System.
The system
identifies fetal nucleated red blood cell candidates as DAPI+, CD45-, Fetal
Hemoglobulin+,
Carbonic Anhydrase- events. The user can confirm that these events indeed have
all the
characteristics typical for fetal nucleated red blood cells. After the system
remembers the
location of the fetal nucleated red blood cells, the cartridge is emptied as
described above and
the cells are hybridized with probes for cytogenetic analysis. Probes that are
typically used to
identify relatively frequent cytogenetic abnormalities are those that
recognize chromosome
X, Y, 13, 18 and 21. After the cells have been stained, the cartridge is
reinserted in the
CellTracks 0 Analyzer II System, because the cells that were present on the
upper surface
during the first ICC image analysis were immobilized the same cells are still
on the same
location inside the cartridge. The system returns to the events and takes
images of the
fluorochromes used to identify chromosomes X, Y, 13, 18 and 21. The user than
assesses
whether the copy number of each of the chromosomes and determines the sex of
the fetus and
whether or not the copy number of the chromosomes suggest the presence of
cytogenetic
abnormalities.
Example 1- Detection of Cytogenetic Aberrations after CTC Identification.
CTCs from 7.5 mL of blood were identified as cytokeratin+, CD45- nucleated
cells after
immunomagnetic enrichment targeting the EpCAM antigen using the CellSearch
System
(Veridex, LLC). CTCs are identified by the CellTracks Analyzer (Immunicon
Corporation)
where the cells are magnetically held along the upper surface of a cartridge.
For cytogenetic
analysis, the fluid in the cartridge was removed and the cells fixed while
maintaining their
original position. Fluorescently labeled probes for chromosome 1, 7, 8 and 17
were
introduced into the cartridge and hybridized to the cells. The fixation and
hybridization
process removes the fluorescent labels used for CTC identification. After
hybridization the
cartridges were again placed on the CellTracks Analyzer and analyzed for a
second time.
The fluorescent images of the CTC's identified in the first scan are then
combined with the
fluorescent images from each of the four chromosomes labels obtained in the
second scan.
The number of chromosomes 1, 7, 8 and 17 were enumerated for each CTC that was
identified in the first scan. The number of chromosomes detected in leukocytes
that
32
CA 02623405 2008-03-20
WO 2007/053245 PCT/US2006/036656
surrounded the CTC's were used as internal controls. In 7.5 mL of blood from 8
patients
with metastatic carcinoma, 1 to 7 CTC's were identified. Greater than or less
than two copies
of chromosome 1, 7, 8 or 17 were detected in all 8 patients. Heterogeneity in
the
chromosomal abnormalities were not only detected between CTC's of different
patients but
also among CTCs of the same patient. Of the 21 CTCs examined, 77% showed
chromosomal
abnormalities and a majority showed an increase in the number of copies of the
chromosomes. In contrast, more than 80% of the leukocytes examined showed two
copies of
the chromosomes and none showed an increase in chromosome copy number.
Conclusions: Cytogenetic composition of CTC's can be assessed after they have
been
identified. The presence of aneusomic CTC's provides information to the
outcome of patient
conditions and provides a prognostic indicator of clinical outcome. Further,
gene alterations
in CTC's provide indices to current and future cancer therapies.
Example 2- Evaluation of Anti-Cancer Targets on CTC's to Predict Therapeutic
Success.
The CellSearch SystemTM has been used in multi-center prospective studies to
demonstrate that presence of tumor cells in blood of patients with metastatic
carcinomas is
associated with poor survival prospects. Failure to eliminate Circulating
Tumor Cells (CTCs)
after one cycle of therapy in these studies strongly suggests that these
patients are on a futile
therapy. Assessment of the presence of therapeutic targets on the tumor should
enable the
appropriate choice of therapy. Anti-cancer targets are identified on CTCs
before initiation of
therapy. Cells from 7.5 mL of blood are identified as cytokeratin(CK)+, CD45-
and
nucleated after EpCAM immunomagnetic selection. Suspect CTCs are identified
and
localized at the upper surface of a cartridge where they are held by a
magnetic field.
Fluorescently labeled antibodies that recognize treatment targets associated
with known
therapies such as HER2, IGF-1, Bc1-2 and EGFR are assessed on the CTCs.
Subsequently,
CTCs are preserved for cytogenetic analysis. After the fluid in the cartridge
is removed, the
cells are fixed and maintain their original position for probe hybridization.
Since the system
knows their original position, the cells can be reexamined for the presence of
probes of
interest. The results show a CTC and a leukocyte before and after
hybridization with
chromosome 1, 7, 8 and 17.
33