Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
CHEMOTHERAPEUTIC FORMULATIONS OF ZOSUQUIDAR
TRIHYDROCHLORIDE AND MODIFIED CYCLODEXTRINS
RELATED APPLICATION
This application claims priority to U.S. Provisional Application No.
60/696,939
filed July 6, 2005, U.S. Application No. 11/417,958 filed May 3, 2006, U.S.
Application
No. 11/418,400 filed May 3, 2006, U.S. Application No. 11/418,324 filed May 3,
2006,
U.S. Provisional Application No. 60/696,756 filed July 6, 2005, U.S.
Application No.
11/416,833 filed May 3, 2006, U.S. Application No. 11/416,992 filed May 3,
2006, U.S.
Provisional Application No. 60/696,930 filed July 6, 2005 U.S. Application No.
11/416,832 filed May 3, 2006, U.S. Application No. 11/416,829 filed May 3,
2006, and
U.S. Application No. 11/416,571 filed May 3, 2006, which are incorporated by
reference
herein in their entirety, and which are hereby made a part of this
specification.
FIELD OF THE INVENTION
The present invention relates to a method of treating patients with leukemias,
solid
tumors, and other malignancies using chemotherapeutic agents in combination
with
zosuquidar that has been solubilized by a modified cyclodextrin, such as
sulfobutylcyclodextrin or hydroxypropyl cyclodextrin. The invention is also
directed to
pharmaceutical formulations comprising zosuquidar in combination with a
modified
cyclodextrin.
BACKGROUND OF THE INVENTION
The field of oncology is in the midst of a major evolution. In the past, the
treatment of cancer has been dominated by empiric, "one-size-fits-all"
treatments based
on types and stages of tumors. Toxic chemotherapy drugs have dominated the
treatment
landscape despite a very low cure rate, particularly against the most common
cancers and
those with known metastatic disease.
Now, treatments in development are targeted against specific proteins. Such
targeting is based on a more robust knowledge of cancer mechanisms, which
often crosses
over many tumor types. These treatments are designed to work in defined
subsets of
patients, typically based on expression and function of the target protein
rather than the
type of tumor, and often in combination with standard chemotherapies. Advances
in the
molecular analysis of cancers will enable the identification of such susbsets
of patients
and the coupling of targeted therapeutics to novel diagnostic approaches.
-1-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
The future of oncology lies in defining the disease in molecular terms (i.e.,
genetics, genomics, proteomics) and tailoring therapies according to
individual tumor and
normal cell properties. This new paradigm will predetermine likely responders,
assess
responses earlier, and adjust treatment based on continued molecular analyses
of tumors.
Drug resistance is one of the most difficult problems that must be overcome in
order to achieve successful treatment of human tumors with chemotherapy.
Clinically,
drug resistance, a characteristic of intrinsically resistant tumors (for
example, colon, renal,
and pancreas) or other malignancies, may be evident at the onset of therapy.
Alternatively, acquired drug resistance results when tumors or malignancies
initially
respond to therapy but become refractory to subsequent treatments. Once a
tumor or
malignancy has acquired resistance to a specific chemotherapeutic agent, it is
common to
observe collateral resistance to other structurally similar agents.
Multidrug resistance (MDR), the ability of cancer cells to become resistant to
the
agent(s) actively used for therapy, as well as other drugs that are
structurally and
functionally unrelated, is a particularly insidious form of drug resistance.
Zosuquidar, a
10, 11 -methanobenzosuberane derivative, is useful in enhancing the efficacy
of existing
cancer chemotherapeutics and for treating multidrug resistance. However,
zosuquidar has
limited solubility in aqueous solution, such that the formulation
concentration is limited,
resulting in a large number of vials to contain doses in the potentially
efficacious range
2p (e.g., a clinical formulation of zosuquidar without solubility enhancers of
50 mg per 30
mL vial that requires 11 units to provide 550 mg of zosuquidar).
SUMMARY OF THE INVENTION
Dosage forms and treatment regimens for treating solid tumors, leukemias such
as
acute myelogenous leukemia (AML) and other malignancies that result in
increased rates
of complete remission and increased cancer-free survival rates are desirable.
Also
desirable are intravenous zosuquidar formulations having a greater zosuquidar
concentration and increased content per dosage unit. Zosuquidar formulated
with a
modified cyclodextrin to enhance its solubility provides an improved
formulation that can
offer such advantages. Hydroxypropylcyclodextrins and sulfobutylcyclodextrins
are
particularly preferred modified cyclodextrins for use in zosuquidar
formulations.
Accordingly, in a first aspect, a stable chemotherapeutic composition
comprising
zosuquidar in combination with a modified cyclodextrin is provided.
-2-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
In an embodiment of the first aspect, the modified cyclodextrin is a
hydroxypropyl-(3-cyclodextrin.
In an embodiment of the first aspect, the modified cyclodextrin is a
sulfobutylcyclodextrin, e.g., a polyanionic B-cyclodextrin derivative with a
sodium
sulfonate salt separated from a lipophilic cavity by a butyl ether spacer
group.
In an embodiment of the first aspect, the composition is in lyophilized form.
In an embodiment of the first aspect, the composition is in solution form.
In an embodiment of the first aspect, the composition is in liquid unit dosage
form,
comprising from about 10 mg/mL to about 30 mg/mL zosuquidar and from about 100
mg/mL to about 200 mg/mL sulfobutylcyclodextrin.
In an embodiment of the first aspect, the composition is in liquid unit dosage
form,
comprising from about 20 mg/mL to about 25 mg/mL zosuquidar and from about 125
mg/mL to about 175 mg/mL sulfobutylcyclodextrin.
In an embodiment of the first aspect, the composition is in liquid unit dosage
form,
comprising about 22.5 mg/mL zosuquidar and about 150 mg/mL
sulfobutylcyclodextrin.
In an embodiment of the first aspect, the composition is in lyophilized forin,
comprising zosuquidar and sulfobutylcyclodextrin in a weight ratio of
zosuquidar to
sulfobutylcyclodextrin of from about 1:5.7 to about 1:7.4.
In an embodiment of the first aspect, the composition is in lyophilized form,
comprising zosuquidar and sulfobutylcyclodextrin in a weight ratio of
zosuquidar to
sulfobutylcyclodextrin of from about 1:6 to about 1:7.
In an embodiment of the first aspect, the composition is in lyophilized form,
comprising zosuquidar and sulfobutylcyclodextrin in a weight ratio of
zosuquidar to
sulfobutylcyclodextrin of about 1:6.73.
In an embodiment of the first aspect, the solution is a dextrose solution.
In a second aspect, a pharmaceutical kit is provided, the kit comprising at
least one
container containing a stable chemotherapeutic composition comprising
zosuquidar in
combination with a modified cyclodextrin; and directions for administering the
chemotherapeutic composition to treat a malignancy that expresses P-
glycoprotein.
In an embodiment of the second aspect, the modified cyclodextrin is
hydroxypropyl-(3-cyclodextrin.
In an embodiment of the second aspect, the modified cyclodextrin is
sulfobutylcyclodextrin.
-3-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
In an embodiment of the second aspect, the malignancy is acute myelogenous
leukemia.
In an embodiment of the second aspect, the kit further comprises at least one
container containing daunorubicin and at least one container containing
cytarabine, and
directions for administering the daunorubicin and cytarabine to treat newly
diagnosed
acute myelogenous leukemia.
In an embodiment of the second aspect, the kit further comprises at least one
container containing Mylotarg, and directions for administering the Mylotarg
to treat
relapsed acute myelogenous leukemia.
In a third aspect, a pharmaceutical kit is provided, the kit comprising at
least one
vial containing a stable chemotherapeutic lyophilized composition, comprising
about 275
mg/vial zosuquidar and about 1850 mg/vial sulfobutylcyclodextrin; and
directions for
reconstituting the lyophilized coinposition with a 15 mL of a 5% dextrose
solution and
administering the reconstituted solution to a patient to treat acute
myelogenous leukemia.
In a fourth aspect, a method of treating cancer in a patient exhibiting
positive P-
glycoprotein expression or positive P-glycoprotein function is provided, the
method
coinprising administering to the patient a chemotherapeutic agent that is a
substrate for P-
glycoprotein efflux and a stable chemotherapeutic composition comprising
zosuquidar in
combination with a modified cyclodextrin, whereby the cancer is treated.
In an embodiment of the fourth aspect, the modified cyclodextrin is a
hydroxypropyl-(3-cyclodextrin.
In an embodiment of the fourth aspect, the modified cyclodextrin is a
sulfobutylcyclodextrin, e.g., a polyanionic 13-cyclodextrin derivative with a
sodium
sulfonate salt separated from a lipophilic cavity by a butyl ether spacer
group.
In an embodiment of the fourth aspect, the stable chemotherapeutic composition
is
in lyophilized form.
In an embodiment of the fourth aspect, the stable chemotherapeutic composition
is
in solution form.
In an embodiment of the fourth aspect, the stable chemotherapeutic composition
is
in liquid unit dosage form, comprising from about 10 mg/mL to about 30 mg/mL
zosuquidar and from about 100 mg/mL to about 200 mg/mL sulfobutylcyclodextrin.
-4-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
In an embodiment of the fourth aspect, the stable chemotherapeutic composition
is
in liquid unit dosage form, comprising from about 20 mg/mL to about 25 mg/mL
zosuquidar and from about 125 mg/mL to about 175 mg/hnL
sulfobutylcyclodextrin.
In an embodiment of the fourth aspect, the stable chemotherapeutic composition
is
in liquid unit dosage form, comprising about 22.5 mg/mL zosuquidar and about
150
mg/mL sulfobutylcyclodextrin.
In an einbodiment of the fourth aspect, the stable chemotherapeutic
composition is
in lyophilized form, comprising zosuquidar and sulfobutylcyclodextrin in a
weight ratio of
zosuquidar to sulfobutylcyclodextrin of from about 1:5.7 to about 1:7.4.
In an embodiment of the fourth aspect, the stable chemotherapeutic composition
is
in lyophilized form, comprising zosuquidar and sulfobutylcyclodextrin in a
weight ratio of
zosuquidar to sulfobutylcyclodextrin of from about 1:6 to about 1:7.
In an embodiment of the fourth aspect, the stable chemotherapeutic composition
is
in lyophilized form, comprising zosuquidar and sulfobutylcyclodextrin in a
weight ratio of
zosuquidar to sulfobutylcyclodextrin of about 1:6.73.
In an embodiment of the fourtli aspect, the stable chemotherapeutic
composition is
a dextrose solution.
In an embodiment of the fourth aspect, the cancer is acute myelogenous
leukemia.
In an embodiment of the fourth aspect, the cancer is a carcinoma, e.g., breast
cancer or ovarian cancer.
In an embodiment of the fourth aspect, the cancer is a sarcoma.
In an enlbodiment of the fourth aspect, the cancer is a hematologic
malignancy,
e.g., acute lymphoblastic leukemia, chronic myeloid leukemia, plasma cell
dyscrasias,
lymphoma, and myelodysplasia.
In an embodiment of the fourth aspect,, the chemotherapeutic agent is an
anthracycline, e.g., doxorubiciri, daunorubicin, epirubicin, idarubicin, or
mitoxantrone.
In an embodiment of the fourth aspect, the chemotherapeutic agent is a
Topoisomerase-II inhibitor, e.g., etoposide or teniposide.
In an embodiment of the fourth aspect, the chemotherapeutic agent is a vinca,
e.g.,
vincristine, vinblastine, vinorelbine, or vindesine.
In an embodiment of the fourth aspect, the chemotherapeutic agent is a taxane,
e.g., paclitaxel or docetaxel.
-5-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
In an embodiment of the fourth aspect, the chemotherapeutic agent is selected
from the group consisting of gleevec, dactinomycin, bisantrene, mitoxantrone,
actinomyocin D, mithomycin C, mitramycin, methotrexate, adriamycin, mitomycin,
mithramycin, anthracene, and epipodophyllo-toxin.
In an embodiment of the fourth aspect, the chemotherapeutic agent comprises
daunorubicin and cytarabine, and the cancer is newly diagnosed acute
myelogenous
leukemia.
In an embodiment of the fourth aspect, the chemotherapeutic agent comprises
Mylotarg, and the cancer is relapsed acute myelogenous leukemia.
In a fifth aspect, a method of administering a therapeutic agent that is a
substrate
for P-glycoprotein efflux to a patient in need thereof is provided, wherein
the patient
exhibits positive P-glycoprotein expression or P-glycoprotein function, the
method
comprising administering the therapeutic agent to the patient; and
administering a stable
P-glycoprotein efflux pump inhibiting composition comprising zosuquidar in
combination
with a modified cyclodextrin to the patient.
In an embodiment of the fifth aspect, the modified cyclodextrin is a
hydroxypropyl-(3-cyclodextrin.
In an embodiment of the fifth aspect, the modified cyclodextrin is a
sulfobutylcyclodextrin, e.g., a polyanionic B-cyclodextrin derivative with a
sodium
sulfonate salt separated from a lipophilic cavity by a butyl ether spacer
group.
In an embodiment of the fifth aspect, the stable chemotherapeutic composition
is
in lyophilized form.
In an embodiment of the fiftli aspect, the stable chemotherapeutic composition
is
in solution form.
In an embodiment of the fifth aspect, the stable chemotherapeutic composition
is
in liquid unit dosage form, comprising from about 10 mg/mL to about 30 mg/mL
zosuquidar and from about 100 mg/mL to about 200 mg/mL sulfobutylcyclodextrin.
In an embodiment of the fifth aspect, the stable chemotherapeutic composition
is
in liquid unit dosage form, comprising from about 20 mg/mL to about 25 mg/mL
zosuquidar and from about 125 mg/mL to about 175 mg/mL sulfobutylcyclodextrin.
In an embodiment of the fifth aspect, the stable chemotherapeutic composition
is
in liquid unit dosage form, comprising about 22.5 mg/mL zosuquidar and about
150
mg/mL sulfobutylcyclodextrin.
-6-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
In an embodiment of the fifth aspect, the stable chemotherapeutic composition
is
in lyophilized form, comprising zosuquidar and sulfobutylcyclodextrin in a
weight ratio of
zosuquidar to sulfobutylcyclodextrin of from about 1:5.7 to about 1:7.4.
In an embodiment of the fifth aspect, the stable chemotherapeutic composition
is
in lyophilized form, comprising zosuquidar and sulfobutylcyclodextrin in a
weight ratio of
zosuquidar to sulfobutylcyclodextrin of from about 1:6 to about 1:7.
In an embodiment of the fifth aspect, the stable chemotherapeutic composition
is
in lyophilized form, comprising zosuquidar and sulfobutylcyclodextrin in a
weight ratio of
zosuquidar to sulfobutylcyclodextrin of about 1:6.73.
In an embodiment of the fifth aspect, the stable chemotherapeutic composition
is a
dextrose solution.
In an embodiment of the fifth aspect, the therapeutic agent comprises an
immunosuppressant, e.g., cyclosporine, cyclosporine A, and tacrolimus.
In an embodiment of the fifth aspect, the therapeutic agent comprises a
steroid,
e.g., dexamethasone, hydrocortisone, corticosterone, triamcinolone,
aldosterone, and
methylprednisolone.
In an embodiment of the fifth aspect, the therapeutic agent comprises an
antiepileptic, e.g., phenytoin.
In an embodiment of the fifth aspect, the therapeutic agent comprises an
antidepressant, e.g., citalopram, thioperidone, trazodone, trimipramine,
amitriptyline, and
phenothiazines.
In an embodiment of the fifth aspect, the therapeutic agent comprises an
antipsychotic, e.g., fluphenazine, haloperidol, thioridazine, and
trimipramine.
In an embodiment of the fifth aspect, the therapeutic agent comprises a
protease
inhibitor, e.g., amprenavir, indinavir, lopinavir, nelfinavir, ritonavir, and
saquinavir.
In an embodiment of the fifth aspect, the therapeutic agent comprises a
calcium
blocker, e.g., bepridil, diltiazem, flunarizine, lomerizine, secoverine,
tamolarizine,
verapamil, nicardipine, prenylamine, and fendiline.
In an embodiment of the fifth aspect, the therapeutic agent comprises a
cardiac
drug, e.g., digoxin, diltiazem, verapamil, and talinolol.
In an embodiment of the fifth aspect, the therapeutic agent comprises
daunorubicin and cytarabine, and the patient is newly diagnosed with acute
myelogenous
leukemia.
-7-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
In an embodiment of the fifth aspect, the therapeutic agent comprises
Mylotarg,
and the patient is diagnosed with relapsed acute myelogenous leukemia.
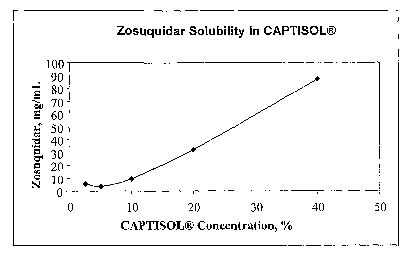
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1 illustrates the increase of zosuquidar concentration in solution as a
function of sulfobutylcyclodextrin concentration.
DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENT
The following description and examples illustrate a preferred embodiment of
the
present invention in detail. Those of skill in the art will recognize that
there are numerous
variations and modifications of this invention that are encompassed by its
scope.
Accordingly, the description of a preferred embodiment should not be deemed to
limit the
scope of the present invention.
Zosuqguidar
U.S. Pat. Nos. 5,643,909 and 5,654,304 disclose a series of 10,11-
methanobenzosuberane derivatives useful in enhancing the efficacy of existing
cancer
chemotherapeutics and for treating multidrug resistance. One such derivative
having
good activity, oral bioavailability, and stability, is zosuquidar, a compound
of formula
(2R)-anti-5-
3-[4-(10,11-difluoromethanodibenzosuber-5-yl)piperazin-l-yl]-2-hydroxypropoxy)
quinoline.
HO N
H~
F
1N H O
NN
F
.~
~'
H
Zosuquidar
Given the limitations of previous generations of MDR modulators, three
preclinical critical success factors were identified and met for zosuquidar:
1) it is a potent
-8-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
inhibitor of P-glycoprotein; 2) it is selective for P-glycoprotein; and 3) no
pharmacokinetic interaction with co-administered chemotherapy is observed.
Zosuquidar is extremely potent in vitro (K; = 59 nM) and is among the most
active
modulators of P-gp-associated resistance described to date. Zosuquidar has
also
demonstrated good in vivo activity in preclinical animal studies. In addition,
the
compound does not appear to be a substrate for P-gp efflux, resulting in a
relatively long
duration of reversal activity in resistant cells even after the modulator has
been
withdrawn.
Another significant attribute of zosuquidar as an MDR modulator is the minimal
pharmacokinetic (PK) interactions with several oncolytics tested in
preclinical models.
Such minimal PK interaction permits normal doses of oncolytics to be
administered and
also a more straightforward interpretation of the clinical results.
The zosuquidar employed in formulations of preferred embodiments can be
administered in the form of a pharmaceutically acceptable salt, e.g., the
trihydrochloride
salt. The terms "pharmaceutically acceptable salts" and "a pharmaceutically
acceptable
salt thereof' as used herein in regard to therapeutic agents are broad terms
and are used in
their ordinary sense, including, without limitation, to refer to salts
prepared from
pharmaceutically acceptable, non-toxic acids (e.g., as for zosuquidar) or
bases (for other
therapeutic agents capable of forming a salt with a base). Suitable
pharmaceutically
acceptable salts include metallic salts, e.g., salts of aluminum, zinc, alkali
metal salts such
as lithium, sodium, and potassium salts, alkaline earth metal salts such as
calcium and
magnesium salts; organic salts, e.g., salts of organic acids (e.g.,
benzenesulfonate,
mesylate, fumarate, citrate), lysine, N,N'-dibenzylethylenediamine,
chloroprocaine,
choline, diethanolamine, ethylenediamine, meglumine (N-methylglucamine),
procaine,
and tris; salts of free acids and bases; inorganic salts, e.g., sulfate,
hydrochloride, and
hydrobromide; and other salts which are currently in widespread pharmaceutical
use and
are listed in sources well known to those of skill in the art, such as, for
example, The
Merck Index. Any suitable constituent can be selected to make a salt of
zosuquidar or
other therapeutic agents discussed herein, provided that it is non-toxic and
does not
substantially interfere with the desired activity. In addition to salts,
pharmaceutically
acceptable precursors and derivatives of the compounds can be employed.
Pharmaceutically acceptable amides, lower alkyl esters, protected derivatives,
and
chelates can also be suitable for use in compositions and methods of preferred
-9-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
embodiments. Also suitable for administration are selected therapeutic agents
in hydrated
form, selected enantiomeric forms of certain therapeutic agents, racemic
mixtures of
certain therapeutic agents, and the like.
Zosuquidar is generally administered in the form of the trihydrochloride salt.
Conventional zosuquidar trihydrochloride formulations include those containing
zosuquidar (50 mg as free base), glycine (15 mg), and mannitol (200 mg)
dissolved in
enough water for injection, to yield a free base concentration of 5 mg/mL. The
formulation is filled into vials and lyophilized to give a vial containing 50
mg of free
base. For such formulations, a 30 mL vial size is necessary to contain 50 mg
of the
zosuquidar formulation. For a typical >200 mg dose of zosuquidar, multiple 50
mg vials
are needed to contain the formulation, greatly increasing manufacturing costs
and
- reducing convenience for the end user (e.g., a pharmacist).
Modified Cyclodextrins
Cyclodextrins are cyclic oligomers of glucose; these compounds form inclusion
complexes with any drug whose molecule can fit into the lipophile-seeking
cavities of the
cyclodextrin molecule. See U.S. Pat. No. 4,727,064 for a description of
various
cyclodextrin derivatives. Cyclodextrins of preferred embodiments can include a-
, 0-, and
x-cyclodextrins. The a-cyclodextrins include six glucopyranose units, the P-
cyclodextrins include seven glucopyranose units, and the x-cyclodextrins
include eight
glucopyranose units. The (3-cyclodextrins are generally preferred as having a
suitable
cavity size for zosuquidar. Cyclodextrin can be in any suitable form,
including
amorphous and crystalline forms, with the amorphous form generally preferred.
Cyclodextrins suitable for use in the formulations of preferred embodiments
include the
hydroxypropyl, hydroxyethyl, glucosyl, maltosyl, and maltotrosyl derivatives
of (3-
cyclodextrin, carboxyamidomethyl-(3-cyclodextrin, carboxymethyl-(3-
cyclodextrin, and
diethylamino-p-cyclodextrin.
Pharmaceutical complexes including various cyclodextrins and cyclodextrin
derivatives are disclosed in the following United States patents: U.S. Pat.
No. 4,024,223;
U.S. Pat. No. 4,228,160; U.S. Pat. No. 4,232,009; U.S. Pat, No. 4,351,846;
U.S. Pat. No.
4,352,793; U.S. Pat. No. 4,383,992; U.S. Pat. No. 4,407,795; U.S. Pat. No.
4,424,209;
U.S. Pat. No. 4,425,336; U.S. Pat. No. 4,438,106; U.S. Pat. No. 4,474,881;
U.S. Pat. No.
4,478,995; U.S. Pat. No. 4,479,944; U.S. Pat. No. 4,479,966; U.S. Pat. No.
4,497,803;
-10-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
U.S. Pat. No. 4,499,085; U.S. Pat. No. 4,524,068; U.S. Pat. No. 4,555,504;
U.S. Pat. No.
4,565,807; U.S. Pat. No. 4,575,548; U.S. Pat. No. 4,598,070; U.S. Pat. No.
4,603,123;
U.S. Pat. No. 4,608,366; U.S. Pat. No. 4,659,696; U.S. Pat. No. 4,623,641;
U.S. Pat. No.
4,663,316; U.S. Pat. No. 4,675,395; U.S. Pat. No. 4,728,509; U.S. Pat. No.
4,728,510;
and U.S. Pat. No. 4,751,095.
Chemically modified and substituted a-, P-, and x-cyclodextrins are generally
preferred over unmodified a-, (3-, and x-cyclodextrins due to improved
toxicity and
solubility properties. The degree of substitution of the hydroxyl groups of
the
glucopyranose units of the cyclodextrin ring can affect solubility. In
general, a higher
average degree of substitution of substituent groups in the cyclodextrin
molecule yields a
cyclodextrin of higher solubility.
Typically, only one guest molecule interacts with the cavity of the
cyclodextrin to
become entrapped. In order to form a complex with a cyclodextrin, a stable
association is
necessary. A variety of non-covalent forces, such as van der Waal forces,
hydrophobic
interaction, dipole moment and other forces are responsible for formation of a
stable
complex. In the case of some low molecular weight guest molecules, more than
one guest
molecule may fit into the cavity. In the case of some high molecular weight
guest
molecules, more than one molecule of cyclodextrin might bind to the guest
molecule.
Only a portion of the molecule must fit into the cavity to form a complex. As
a result, a
one-to-one molar ratio is not always achieved, especially with high or low
molecular
weight guest molecules. The guest molecule associates with the cyclodextriri
so that the
hydrophobic portion of the guest interacts with the hydrophobic cavity of the
cyclodextrin. This interaction is an equilibrium reaction, with the direction
of the
equilibrium dependent upon the guest molecule. For some guest molecules, the
complex
is predominant while for other guest molecules, the free state might be
preferred. In order
to reduce the probability of free guest molecules self-associating to form an
insoluble
precipitate, excess cyclodextrin is frequently used to increase the
probability of the guest
molecule associating with the cavity of the cyclodextrin rather than
associating with other
guest molecules. For most modified cyclodextrins, a moderate excess of the
cyclodextrin
is generally desirable. However, in certain embodiments, a molar ratio of
zosuquidar to
the cyclodextrin approaching one-to-one may be preferred.
-11-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
3uiio nmicycioaextrin
Sulfobutyl-(3-cyclodextrin is a particularly preferred modified cyclodextrin
for
solubilizing zosuquidar. This cyclodextrin is marketed by CyDex, Inc.,
(Lenexa, KS)
under the trade name CAPTISOL . CAPTISOL cyclodextrins are polyanionic B-
cyclodextrin derivatives with a sodium sulfonate salt separated from the
lipophilic cavity
by a butyl ether spacer group, or sulfobutylether (SBE).
Sulfobutylcyclodextrin may
provide a beneficial and protected environrrient for zosuquidar in its
lipophilic cavity
while its hydrophilic surface contributes good water solubility, improving
both solubility
and stability. Interaction of the zosuquidar with sulfobutylcyclodextrin may
reduce
decomposition by protecting the labile region from potential reactants in the
aqueous
environment. The inherent pharmacokinetics and pharmacodynamics of zosuquidar
are
unaffected by sulfobutylcyclodextrin. Upon administration, the zosuquidar -
sulfobutylcyclodextrin complex rapidly disassociates, releasing zosuquidar.
Hydroxypropylcyclodextrin
Hydroxypropyl-(3-cyclodextrin is also a preferred modified cyclodextrin for
solubilizing zosuquidar. This cyclodextrin is marketed by RDI Division of
Fitzgerald
Industries Intl., (Concord, MA). Hydroxypropyl-(3-cyclodextrin is produced
from (3-
cyclodextrin by hydroxpropylation of the hydroxyl groups of the cyclodextrin.
It is a
partially substituted poly(hydroxpropyl) ether of beta cyclodextrin (BCD). The
structure
of a hydroxypropyl-(3-cyclodextrin, wherein R= CH2CH(OH)CH3 or H, is as
follows:
-12-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
CH2OR
ROH2C O
O O
OR RO
OR
O RO Q CH2OR
O OR RO
ROCH2 O
OR OR
O O
OR OR
O CH2OR
OR
O OR O
ROCH2 OR OR 0
O ZOR O OR
O
CH2OR
ROCH2
The basic closed circular structure of (3-cyclodextrin is maintained in
hydroxypropyl-p-cyclodextrin. The glycosidic oxygen forming the bond between
the
adjacent glucose monomers and the hydrogen atoms lining the cavity of the
cyclodextrin
impart an electron density and hydrophobic character to the cavity. Organic
compounds,
such as zosuquidar, interact with the walls of the cavity to form inclusion
complexes. The
hydroxyl groups and the hydroxypropyl groups are on the exterior of the
molecule and
interact with water to provide the increased aqueous solubility of the
hydroxypropyl-(3-
cyclodextrin and the complexes made with the hydroxypropyl-P-cyclodextrin.
The hydroxypropyl groups are randomly substituted onto the hydroxyl groups of
the (3-cyclodextrin and the amount of substitution is reported as average
degree of
substitution or number of hydroxypropyl groups per (3-cyclodextrin. In bulk
hydroxypropyl-(3-cyclodextrin, some molecules will have more substituents than
the
average number of substituents and some less. The result is a mixture of many
molecules
varying with respect to the number and location of substitutions around the
ring of the (3-
cyclodextrin. Substitution can have an effect on the binding of guest
molecules to the
hydroxypropyl-(3-cyclodextrin. At low degrees of substitution, binding is very
similar to
that of the unmodified B-cyclodextrin. Increasing substitution can lead to
weakened
binding due to steric hindrance. The effect is dependent upon the particular
guest
-13-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
molecule, but it is possible to obtain increased binding due to an increase in
surface area
to which the guest molecule can bind. With most guest molecules, these
differences in
binding with degree of substitution are small. A preferred average degree of
substitution
of hydroxypropyl-(3-cyclodextrin when employed in combination with zosuquidar
is from
about 4 or 5 to about 6, 7, or 8.
Hydroxypropyl-(3-cyclodextrin is very soluble in water, with substitution of
the
hydroxyl groups of the (3-cyclodextrin disrupting the networlc of hydrogen
bonding around
the rim of the (3-cyclodextrin. As a result of disruption of the hydrogen-
bonding network,
the hydroxyl groups interact much more strongly with water, resulting in
increased
solubility compared to (3-cyclodextrin. Hydroxypropyl-(3-cyclodextrin is
generally more
soluble than unmodified (3-cyclodextrin. For hydroxypropyl-(3-cyclodextrin
having a
degree of substitution of 7.6, the solubility in aqueous solution is
360g/100m1.
Hydroxypropyl-(3-cyclodextrin is also soluble in aqueous ethanol (225g/100ml
for a 95%
ethanol solution). In preferred formulations, the solubility of the complex
with
zosuquidar is not generally exceeded. Complexes of zosuquidar and
hydroxypropyl-(3-
cyclodextrins exhibit increased solubility and stability when compared to
corresponding
complexes of zosuquidar and unmodified (3-cyclodextrins.
Strong acids, such as hydrochloric acids, can hydrolyze hydroxypropyl-P-
cyclodextrin. The rate of hydrolysis is dependent upon the temperature and
concentration
of the acid. The higher the temperature or concentration of the acid, the more
rapid is the
rate of hydrolysis. Weak acids, such as organic acids, do not hydrolyze
hydroxypropyl-(3-
cyclodextrin, and hydroxypropyl-p-cyclodextrin is stable in bases.
Hydroxypropyl-(3-
cyclodextrin is not hydrolyzed by (3-amylase or glucoamylase, but (3-
cyclodextrin can be
hydrolyzed by some a-amylases. Hydroxypropyl-(3-cyclodextrin generally
exhibits good
stability under physiological conditions when employed in formulations for
intravenous
use.
Zosuquidar - Sulfobutylcyclodextrin Formulations
While the preferred embodiments generally refer to zosuquidar -
sulfobutylcyclodextrin formulations, it is understood that other suitable
cyclodextrins,
such as hydroxypropyl-(3-cyclodextrins, can be used instead of
sulfobutylcyclodextrin to
solubilize zosuquidar. Alternatively, a mixture of two or more different
cyclodextrins can
be used.
-14-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
Use of a sulfobutylcyclodextrin formulation (lyophilized) allows an 800 mg
dose
of zosuquidar to be contained in one (50 inL vial) or two vials (20 or 30 mL
vial) versus
three 100 mL vials for a zosuquidar formulation without cyclodextrin,
resulting in greater
manufacturing efficiency.
The relative amounts of zosuquidar and the cyclodextrin, e.g.,
sulfobutylcyclodextrin, can be adjusted, depending upon the particular
formulation and
the specific cyclodextrin employed. However, a molar ratio of zosuquidar to
modified
cyclodextrin of from about 1:1 or less to about 1:10 or more is generally
preferred,
preferably from about 1:5.0 or 1:5.5 to about 1:8.0, 1:8.5, 1:9.0, or 1:9.5,
and more
preferably from about 1:5.7, 1:5.8, 1:5.9, 1:6.0, 1:6.1, 1:6.2, 1:6.3, 1:6.4,
1:6.5, 1:6.6,
1:6.7 to about 1:6.8, 1:6.9, 1:7.0, 1:7.1, 1:7.2, 1:7.3, or 1.7:4.
The zosuquidar - modified cyclodextrin formulation can by supplied as a powder
and reconstituted. Alternatively, it can be provided in the form of an aqueous
liquid,
which can optionally be freeze dried or lyophilized. In general, the
zosuquidar - modified
cyclodextrin formulations are prepared by dissolving the cyclodextrin in water
and adding
the zosuquidar to the aqueous modified cyclodextrin solution. Excipients, if
any are
desired may be added with or subsequent to adding the active compound. The
resulting
solution can be sterilized using any of the known methods appropriate to
preserving the
active compound. Alternatively, the components can be sterilized by any of the
known
methods appropriate to preserving zosuquidar prior to mixing in water and can
be mixed
using sterile equipment and techniques. The solution can be lyophilized in
sterile
containers and capped. Prior to use, the lyophilized composition of matter can
be
reconstituted using sterile water for injection, deionized sterilized water,
5% dextrose
solution, or other appropriate diluent.
Contemplated routes of administration include topical, oral, subcutaneous,
parenteral, intradermal, intramuscular, intraperitoneal, and intravenous.
However, it is
particularly preferred to administer the zosuquidar - modified cyclodextrin in
intravenous
form.
The intravenous forms containing zosuquidar - modified cyclodextrin are
preferably isotonic with the blood or other body fluid of the patient. The
isotonioity of the
compositions can be attained using sodium tartrate, propylene glycol, sodium
chloride, or
other inorganic or organic solutes. Buffering agents can be employed, such as
acetic acid
and salts, citric acid and salts, boric acid and salts, and phosphoric acid
and salts.
-15-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
Parenteral vehicles include, Ringer's dextrose, lactated Ringer's, or fixed
oils.
Intravenous vehicles can include fluid and nutrient replenishers, electrolyte
replenishers
(such as those based on Ringer's dextrose), and the like. A particularly
preferred vehicle
is dextrose solution, e.g., 5% dextrose. Various excipients can be employed,
depending
upon the route of administration and the preparation desired. Standard texts,
such as
"Remington: The Science and Practice of Pharmacy", Lippincott Williams &
Wilkins;
20th edition (June 1, 2003) and "Remington's Pharmaceutical Sciences," Mack
Pub. Co.;
18th and 19th editions (December 1985, and June 1990, respectively) include
information
regarding such excipients, which can include additional complexing agents,
metal ions,
polymeric compounds such as polyacetic acid, polyglycolic acid, hydrogels,
dextran, and
the like, liposomes, microemulsions, micelles, unilamellar or multilamellar
vesicles,
erythrocyte ghosts or spheroblasts. Suitable lipids for liposomal formulation
include,
without limitation, monoglycerides, diglycerides, sulfatides, lysolecithin,
phospholipids,
saponin, bile acids, and the like. The presence of such additional components
can
influence the physical state, solubility, stability, rate of in vivo release,
and rate of in vivo
clearance, and are thus chosen according to the intended application, such
that the
characteristics of the carrier are tailored to the selected route of
administration.
A pharmaceutically acceptable preservative can be employed to increase the
shelf
life of the pharmaceutical compositions. Benzyl alcohol can be suitable,
although a
variety of preservatives including, for exainple, parabens, thimerosal,
chlorobutanol, or
benzalkoniuin chloride can also be employed. A suitable concentration of the
preservative is typically from about 0.02% to about 2% based on the total
weight of the
composition, although larger or smaller amounts can be desirable depending
upon the
agent selected.
The zosuquidar - modified cyclodextrin complex can be provided to an
administering physician or. other health care professional in the form of a
kit. The kit is a
package which houses one or more containers which contain zosuquidar complexed
with
a modified cyclodextrin, such as sulfobutylcyclodextrin or hydroxypropyl-(3-
cyclodextrin,
in a suitable form and instructions for reconstituting and/or administering
the
pharmaceutical composition to a subject. The kit can optionally also contain
one or more
additional therapeutic agents, e.g., mylotarg, daunorubicin, cytarabine,
and/or other
chemotherapeutic agents. The kit can optionally contain one or more diagnostic
tools and
instructions for use. For example, a kit containing a single composition
comprising a
-16-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
complex of zosuquidar and sulfobutylcyclodextrin or hydroxypropyl-(3-
cyclodextrin in
combination with one or more additional therapeutic agents can be provided, or
separate
pharmaceutical compositions containing a coinplex of zosuquidar -
sulfobutylcyclodextrin
and additional therapeutic agents can be provided. The kit can also contain
separate doses
of zosuquidar - sulfobutylcyclodextrin complex for serial or sequential
administration.
The kit can contain suitable delivery devices, e.g., syringes and the like,
along with
instructions for administrating the complex and any other therapeutic agent.
The kit can
optionally contain instructions for storage, reconstitution (if applicable),
and
administration of any or all therapeutic agents included. The kits can include
a plurality
of containers reflecting the number of administrations to be given to a
subject. In a
preferred embodiment, a kit for the treatment of aleukemia or solid tumor is
provided. In
a particularly preferred embodiment, a kit for the treatment of acute
myelogenous
leukemia is provided that includes a zosuquidar - sulfobutylcyclodextrin
complex and
mylotarg (for relapsed patients) or daunorubicin and cytarabine (for newly-
diagnosed
patients) and instructions for administering each. In another particularly
preferred
embodiment, a kit for the treatment of acute myelogenous leukemia is provided
that
includes a zosuquidar - sulfobutylcyclodextrin complex and one or more
diagnostics or
instructions for conducting one or more diagnostics for determining P-gp
expression
and/or efflux pump activity. The kit can also include instructions, an assay,
or a
diagnostic for determining if a patient has acute myelogenous leukemia. The
kit can
contain suitable delivery devices, e.g., syringes, inhalation devices, and the
like, along
with instructions for administrating zosuquidar and/or other therapeutic
agent. The kit
can optionally contain instructions for storage, reconstitution (if
applicable, e.g., for a
lyophilized form reconstituted for intravenous administration), and
administration of any
or all therapeutic agents included. The kits can include a plurality of
containers reflecting
the number of administrations to be given to a subject.
Contemplated amounts of solubilized zosuquidar for intravenous administration
are from about 400 mg/day of zosuquidar or less to about 1,600 mg/day
zosuquidar or
more, preferably from about 500 or 600 mg/day to about 800, 900, 1000, 1100,
1200,
1300, 1400, or 1500 mg/day, and most preferably 700 mg/day. The duration of
the
injection of the zosuquidar - modified cyclodextrin complex can be adjusted
depending
upon various factors, and can comprise a single injection administered over
the course of
a few seconds or less to 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15,
16, 17, 18, 19, 20,
-17-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
21, 22, 23, 24, 26, 28, 30, 32, 34, 36, 40, 44, 48, 54, 60, 66, 72, 78, 84,
90, or 96 hours or
more of continuous intravenous administration.
Chemotherapeutic Regimens Utilizing Zosuguidar - Sulfobutylcyclodextrin
Formulations
The zosuquidar - sulfobutylcyclodextrin complex formulations of preferred
formulations are useful therapeutic agents for treating multidrug resistance
in patients
treated for malignancies, solid tumors, and leukemias. The formulations are
useful for
treatment of cancers that express P-gp, e.g., many solid tumors, bladder
cancer, pancreatic
cancer, liver cancer, myeloma, carcinomas (e.g., breast cancer and ovarian
cancer),
sarcomas, and hematologic malignancies (e.g., acute myelogenous leukemia,
acute
lymphoblastic leukemia, chronic myeloid leukemia, plasma cell dyscrasias,
lymphoma,
myelodysplasia). The zosuquidar - sulfobutylcyclodextrin formulations are
suitable for
use in conjunction with suitable chemotherapeutic agents used to treat
malignancies
wherein multidrug resistance is of concern. However, the formulations are
particularly
suited for use in treating acute myelogenous leukemia. In preferred
embodiments,
relapsed patients are treated with mylotarg in combination with zosuquidar -
sulfobutylcyclodextrin complex formulations. Newly-diagnosed patients can be
treated
with daunorubicin and cytarabine , in combination with zosuquidar -
sulfobutylcyclodextrin complex formulations. Other chemotherapeutic agents can
also be
used in combination with the zosuquidar - sulfobutylcyclodextrin complex
formulations
of preferred embodiments, e.g., anthracyclines (e.g., doxorubicin,
daunorubicin,
epirubicin, idarubicin, mitoxantrone), vincas (e.g., vincristine, vinblastine,
vinorelbine,
vindesine), Topoisomerase-II (e.g., etoposide, teniposide), taxanes (e.g.,
paclitaxel,
docetaxel), and others (e.g., Gleevec, Mylotarg, dactinomycin, mithramycin).
Chemotherapeutic Regimens Utilizing Zosuguidar and M ly otar~
In preferred embodiments, a P-gp expression or efflux pump activity diagnostic
is
conducted to provide information in treating AML patients or patients with
metastatic
breast cancer with a zosuquidar - cyclodextrin complex (e.g., zosuquidar -
sulfobutylcyclodextrin or zosuquidar - hydroxypropyl cyclodextrin) in
combination with
Mylotarg. If the results of the P-gp expression or efflux pump activity
diagnostic
indicates positive P-gp expression or efflux pump activity, then treatment
with a
zosuquidar - cyclodextrin complex in combination with Mylotarg is initiated.
If the
results of the P-gp expression or efflux pump activity diagnostic indicate
negative P-gp
expression or efflux pump activity, then zosuquidar is expected not to yield
an
-18-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
improvement in clinical outcome and another treatment option not involving
administration of a P-gp efflux inhibitor is selected. In relapsed AML
patients, it is
generally considered acceptable clinical practice to wait for P-gp expression
or efflux
pump activity test results before initiating a treatment. However, in certain
embodiments
it can be desirable to initiate treatment before receiving test results, and
then reevaluate
the desirability of continuing treatment, depending upon the test results.
Most preferably,
P-gp expression or efflux pump activity of a sample both in the presence and
absence of
the P-gp efflux inhibitor is compared, whereby the P-gp efflux that is
inhibitable by the P-
gp efflux inhibitor can be determined. However, in certain embodiments wherein
P-gp
expression or function status correlates with expectation of clinical success,
it can be
useful to determine P-gp expression or efflux pump activity at any point in
time.
Mylotarg was approved in May 2000 for relapsed CD33-positive AML patients
over the age of 60. Mylotarg from Wyeth and Celltech is based on antibody-
targeted
chemotherapy. Mylotarg's highly specific antibody recognizes a cell-surface
molecule,
CD33, which is abundant on AML cells (>90%) but absent from normal blood stem
cells,
the seeds from which normal blood and immune cells originate. The antibody is
linked to
calicheamicin, a potent chemotherapy agent. The antibody selectively targets
leukemic
blast cells and delivers calicheamicin to them. The chemical structure of
Mylotarg is
provided below.
-19-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
n, average loading of
'n- P 6 7.6 calichearrcicin derivative
on antibody, is 2 to 3
moles/mole
HN
H~ CH~
O~~'~~' '::?~= C3 C
Nf-iN-kA Ht7.~ 0
C~{3 S !
3 ~H~ O ~H3 p ~ ~H p
~ S CH3 ~~#3
t~ t~H3 C~H HN H
~ M1~13 H~J O
CN~~~
~tC3 CH2CH~I'r~~~
H3 ~ ~ ~
C~t~ If CCHs
0 -J n
There is a growing body of evidence to suggest that the calicheamicin
component
of Mylotarg is also an MDR substrate and subject to the P-gp efflux pump. In
several
studies, the cytotoxic effect of Mylotarg has been shown to be inversely
correlated with
the amount of P-gp present. Two MDR modulators, valspodar and the quinolone
derivative MS-209, have both been shown to reverse the resistance to Mylotarg
in P-gp
expressing CD33(+) leukemia cells and clinical studies are underway in
combination with
cyclosporine.
The combination of zosuquidar, a highly specific and safe P-gp efflux
inhibitor,
complexed with cyclodextrin, in combination with Mylotarg or another
calicheamicin-
antibody conjugate is effective for treatment of relapsed AML. The effective
dose of the
zosuquidar - cyclodextrin complex and the timing of administration of
zosuquidar and
Mylotarg are critical to achieving improved complete remission rates and
enhanced
leukemia free and overall survival rates in the relapsed AML patient
population. While
the methods and formulations of preferred embodiments are especially preferred
for
treatment of relapsed AML patients, the methods and formulations can be
adapted to
other drugs and indications. For example, P-gp efflux inhibitors other than
Mylotarg can
be administered according to the disclosed dosing regimens, or slightly
modified dosing
-20-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
regimens. Likewise, the formulations and dosing regimens employing a
zosuquidar -
cyclodextrin complex and Mylotarg can be employed in treating AML patients
other than
relapsed AML patients, or for other types of leulcemia or other cancers that
express P-gp,
e.g., many solid tumors, lymphomas, bladder cancer, pancreatic cancer, ovarian
cancer,
liver cancer, myeloma, lymphocytic leulcemia, breast cancer, and sarcoma.
The duration of the injection of a zosuquidar - cyclodextrin complex and/or
Mylotarg can be adjusted depending upon various factors, and can comprise a
single
injection administered over the course of a few seconds or less to 1, 2, 3, 4,
5, 6, 7, 8, 9,
10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 26, 28, 30, 32,
34, 36, 40, 44, 48,
54, 60, 66, 72, 78, 84, 90, or 96 hours or more of continuous intravenous
administration.
A zosuquidar - cyclodextrin complex and a therapeutic agent that is a
substrate for
P-gp efflux can be administered to patients suffering from AML prior to
confirmation of
P-gp expression or function, or to AML patients other than relapse AML
patients.
However, such therapy is preferably administered to relapsed AML patients. The
administration route, amount administered, and frequency of administration can
vary
depending on the age of the patient, status as relapsed or newly diagnosed AML
patient,
and severity of the condition.
Contemplated amounts of Mylotarg for intravenous administration to treat
relapsed AML are from about 10 mg/day or less to about 1000 mg/day or more
administered on one, two, or more separate days. The dosage is preferably
administered
intravenously at a rate of about 1 mg/m2 or less to about 10 mg/m2 or more
continuously
over the course of about 2, 3, or 4 hours to about 5, 6, 7, 8, 9, 10, 11, 12,
13, 14, 15, 16,
17, 18, 19, 20, 21, 22, 23, or 24 hours, more preferably over the course of
about 2 hours to
about 6 hours; however, administration at a rate of 5 mg/m2, 7 mg/m2, or 9
mg/m2 over
about 2 hours is particularly preferred. Preferably, doses of Mylotarg are
administered on
Day 1 and Day 15 of the treatment regimen. However, in certain embodiments,
the
second dose can be administered on Day 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 16,
17, 18, 19,
20, 21, or 22, or another day of the treatment regimen. Other dosing regimens
include
administering three doses total over a week.
Contemplated amounts of zosuquidar (in the form of a cyclodextrin complex) for
intravenous administration to treat relapsed AML are from about 400 mg/day or
less to
about 1,600 mg/day or more, preferably from about 500, 600, or 700 mg/day to
about 900,
1000, 1100, 1200, 1300, 1400, or 1500 mg/day, and most preferably from about
500
-21-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
mg/day to about 800 mg/day. It is generally preferred to start the infusion of
the
zosuquidar - cyclodextrin complex from about 2 hours or less to about 6 hours
or more
prior to the administration of Mylotarg. In the course of a treatment regimen,
the
zosuquidar - cyclodextrin complex is preferably administered on two, three, or
four
separate days. The dosage is preferably administered intravenously
continuously over the
course of about 6 to 90 hours, more preferably over the course of 12, 18, 24,
30, 36, or 42
hours to about 54, 60, 66, 72, 78, or 84 hours, most preferably over about 24
hours, 48
hours, or 72 hours, depending upon the treatment regimen. Preferably the
zosuquidar -
cyclodextrin complex is administered on Day 1 of the treatment regimen. In
certain
embodiments, additional zosuquidar - cyclodextrin complex is administered on
Day 2, on
Days 2 and 3, or on Days 2, 15, and 16. However, in certain embodiments, one,
two, or
three or more additional doses can be administered on other days of the
treatment
regimen.
Table 1 provides various dosing regimes that can be used in treating relapsed
AML.
Table 1.
Dose Mylotarg Zosuquidar
Level (complexed with cyclodextrin)
-1 * 5 mg/m2 IV over 4 hr Day 1 and 15 800 mg/day continuous IV over 24 hr,
Day1and15
1 5 mg/m2 IV over 4 hr Day 1 and 15 800 mg/day continuous IV over 48 hr
Day 1&2 and 15&16
2 7 mg/m2 IV over 4 hr Day 1 and 15 800 mg/day continuous IV over 48 hr
Day 1&2 and 15&16
3 9 mg/m2 IV over 4 hr Day 1 and 15 800 mg/day continuous IV over 48 hr
Day 1&2 and 15&16
4 9 mg/m2 IV over 4 hr Day 1 and 15 800 mg/day continuous IV over 72 hr
Day 1-3 and 15-17
* Only if level 1 has a dose limiting toxicity (DLT).
Tables 2 and 3 provide alternative dosing regimes that can be used in treating
relapsed AML.
-22-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
Table 2.
Dose Mylotarg Zosuquidar
Level (complexed with cyclodextrin)
-1 * 5 mg/m2 IV over 6-24 hr Day 1 and 500-700 mg/day continuous IV over 24
15 hr Day l and 15
1 5 mg/m2 IV over 6-24 hr Day 1 and 500-700 mg/day continuous IV over 48
15 hrDayl&2and15&16
2 7 mg/m2 IV over 6-24 hr Day 1 and 500-700 mg/day continuous IV over 48
15 hr Day 1&2 and 15&16
3 9 mg/m2 IV over 6-24 hr Day 1 and 500-700 mg/day continuous IV over 48
15 hr Day 1&2 and 15&16
4 9 mg/m2 IV over 6-24 hr Day 1 and 500-700 mg/day continuous IV over 72
15 lir Day 1-3 and 15-17
* Only if level 1 has a dose limiting toxicity (DLT).
A clinical study was conducted to determine the efficacy of Mylotarg in the
treatment of relapsed AML. It was determined that the rate of complete
remission (CR +
CRp) for P-gp negative patients treated with Mylotarg was 64% (N=36). In
contrast, the
rate of coinplete remission for P-gp positive patients was only 9% (N=22).
This indicates
that P-gp efflux plays an important role in survival rates for relapsed AML,
and further
indicates that inhibition of P-gp efflux, e.g., by also administering
zosuquidar or another
P-gp efflux inhibitor, has the potential to significantly improve response
rates in P-gp
positive patients. The diagnostic and assay methods described herein are
therefore useful
in treating relapsed AML. Likewise, a diagnostic or assay to determine P-gp
expression
or function or efflux pump activity can be useful in devising treatment
regimens for other
cancers, such as metastatic breast cancer, that also exhibit P-gp expression.
Chemotherapeutic Regimens Utilizing Zosuguidar, Daunorubicin, and Cytarabine
In preferred embodiments, a P-gp expression or efflux pump activity diagnostic
is
conducted to provide information in treating newly diagnosed AML patients with
a
zosuquidar - cyclodextrin complex (e.g., zosuquidar - sulfobutylcyclodextrin
or
zosuquidar - hydroxypropyl cyclodextrin) in combination with daunorubicin and
cytarabine. In newly diagnosed AML patients, it is generally not considered
acceptable
clinical practice to wait for P-gp expression or efflux pump activity test
results before
initiating a treatment. Accordingly, treatment is initiated immediately after
diagnosis.
-23-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
When test results become available, the desirability of continuing treatment
can be
evaluated, depending upon the test results. Typically, when the results of the
P-gp
expression or efflux pump activity diagnostic indicate negative P-gp
expression, then
treatment with a P-gp efflux inhibitor is discontinued because administration
of the drug
is not expected to contribute to an improved clinical outcome. Preferably, P-
gp
expression or function or efflux pump activity is determined both in the
presence and the
absence the P-gp efflux inhibitor to determine the P-gp expression that is
inhibitable by
the P-gp efflux inhibitor.
Daunorubicin is an antibiotic chemotherapy treatment that is widely used to
treat
acute myeloid leukemia and acute lymphocytic leukemia. It is produced by the
bacteria
Streptomyces coeruleorubidis and was approved by the FDA as a first line
therapy
treatment for leukemia in 1998. Daunorubicin is typically administered
intravenously. It
is marketed under the brand names Cerubidine, DaunoXome, and Liposomal
daunorubicin. Daunorubicin has the following structure:
0 H
~QOCH3
OH
ome 0 OH 0 H-CHy-CH-CH-yi 4-CFl,"'H'Ct
Cytarabine is a deoxycytidine analogue, cytosine arabinoside (ara-C), which is
metabolically activated to the triphosphate nucleotide (ara-CTP), which acts
as a
competitive inhibitor of DNA polymerase and produces S phase-specific
cytotoxicity. It
is used as an antineoplastic, generally as part of a combination chemotherapy
regimen, in
the treatment of acute lymphocytic and acute myelogenous leukemia, the blast
phase of
chronic myelogenous leukemia, erythroleukemia, and non-Hodgkin's lymphoma. It
is
typically administered intravenously and subcutaneously, and for the
prophylaxis and
treatment of meningeal leulcemia, administered intrathecally. Cytarabine has
the
following structure:
-24-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
0 N
7HOCH ry OH
T he combination of a zosuquidar - cyclodextrin complex, the antibiotic
chemotherapeutic daunorubicin, and the antineoplastic cytarabine, is effective
for
treatment of newly diagnosed AML. The effective dose of the zosuquidar -
cyclodextrin
complex and the timing of administration of the zosuquidar - cyclodextrin
complex,
daunorubicin, and cytarabine are critical to achieving improved complete
remission rates
and enhanced leukemia free survival rates in the newly diagnosed AML patient
population. While the methods and formulations of preferred embodiments are
especially
preferred for treatment of newly diagnosed AML patients, the methods and
formulations
can be adapted to other drugs and indications. For example, chemotherapeutics
other than
daunorubicin and cytarabine can be administered according to the disclosed
dosing
regimens, or slightly modified dosing regimens. Likewise, the formulations and
dosing
regimens employing a zosuquidar - cyclodextrin complex, daunorubicin, and
cytarabine
can be employed in treating AML patients other than newly diagnosed AML
patients, or
for treating other types of leukemia or other cancers that exhibit P-gp
expression.
Zosuquidar - cyclodextrin complex, daunorubicin, and cytarabine can be
formulated as described above for zosuquidar - cyclodextrin complex and
Mylotarg, and
can be included in kits, also as describe4 above.
The zosuquidar - cyclodextrin complex, daunorubicin, and/or cytarabine can be
administered to patients suffering from AML prior to confirmation of the P-gp
expression
or function, or to AML patients other than newly diagnosed AML patients (e.g.,
relapsed
AML patients). However, therapy is preferably administered to newly diagnosed
AML
patients. The administration route, amount administered, and frequency of
administration
can vary depending on the age of the patient, status as relapsed or newly
diagnosed AML
patient, and severity of the condition.
Contemplated amounts of zosuquidar (in the form of a cyclodextrin complex) for
intravenous administration to treat newly diagnosed AML are from about 400
mg/day or
-25-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
less to about 1,600 mg/day or more, prelerably iTom anoui :)vv, ovv, or /vv
mgiaay io
about 900, 1000, 1100, 1200, 1300, 1400, or 1500 mg/day, and most preferably
700
mg/day. In the course of a treatment regimen, the zosuquidar - cyclodextrin
complex is
preferably administered on two, three, or four separate days. The dosage is
preferably
administered in intravenously continuously over the course of about 6 to about
90 hours,
more preferably over the course of about 12, 18, 24, 30, 36, or 42 hours to
about 54, 60,
66, 72, 78, or 84 hours, most preferably over about 24 hours, 48 hours, or 72
hours,
depending upon the treatment regimen. Preferably the zosuquidar - cyclodextrin
complex
is administered on Day 1 of the treatment regimen. In certain embodiments,
additional
zosuquidar - cyclodextrin complex is administered on Day 2, on Days 2 and 3,
or on Days
2, 15, and 16. However, in certain embodiments, one, two, or three or more
additional
doses can be administered on other days of the treatment regimen.
Contemplated amounts of daunorubicin for intravenous administration to treat
newly diagnosed AML are from about 10 mg/m2/day or less to about 100 mg/m2/day
or
more administered at initiation of zosuquidar - cyclodextrin complex infusion
or up to
about 1, 2, 3, 4, 5, or 6 or more hours after initiation of zosuquidar -
cyclodextrin
complex infusion. The dosage is preferably administered intravenously at a
rate of about
mg/m2/day or less to about 90 mg/m2/day or more, preferably about 30, 35, or
40
mg/m2/day or less to about 50, 55, 60, 65, 70, 75, 80, or 85 mg/m2/day, and
most
20 preferably about 45 mg/m2/day continuously over the course of about 2 or
2.5 days to
about 3.5 or 4 days, preferably about 3 days.
Contemplated amounts of cytarabine for intravenous administration to treat
newly
diagnosed AML patients are from about 10 mg/day or less to about 3,000 mg/day
or more
administered at initiation of zosuquidar - cyclodextrin complex infusion or
after initiation
25 of zosuquidar - cyclodextrin complex infusion. The dosage is preferably
administered
intravenously at a rate of about 50 mg/m2/day or less to about 200 mg/m2/day
or more,
preferably 60, 70, 80, or 90 mg/m2/day or less to about 110, 120, 130, 140,
150, 160, 170,
180, or 190 mg/m2/day, and most preferably about 100 mg/m2/day continuously
over the
course of about 1, 2, 3, 4, 5, or 6 days up to about 8, 9, or 10 days or more,
preferably over
about 7 days.
A particularly preferred dosing regimen for newly diagnosed AML includes
continuous intravenous administration of 550 mg of zosuquidar (as a
cyclodextrin
complex) over 6 hours (3 days), continuous intravenous administration of
cytarabine at a
-26-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
rate of 100 mg/m2/day (7 days), and intravenous administration of daunorubicin
at a dose
of 45 mg/ma/day (3 days), wherein infusion of daunorubicin is started 1 hour
after
initiation of zosuquidar infusion. Another particularly preferred dosing
regimen includes
continuous intravenous administration (preferably about 1 to 24 hours in
duration, more
preferably about 6 to 24 hours in duration, most preferably about 24 hours in
duration) of
500 to 700 mg/day of zosuquidar (3 days), continuous intravenous
administration of
cytarabine at a rate of 100 mg/mZ/day (7 days), and intravenous administration
of
daunorubicin at a dose of 45 mg/m2/day (3 days), wherein infusion of
daunorubicin is
started 1 to 4 hours after initiation of zosuquidar - cyclodextrin complex
infusion. While
in the above described embodiments infusion of daunorubicin is started after a
specified
time period has lapsed after initiation of zosuquidar - cyclodextrin complex
infusion, in
other embodiments other start times can be preferred, e.g., immediately after
or during
initiation of zosuquidar - cyclodextrin complex infusion up to about 1, 2, 3,
4, 5, 6, 7, 8,
9, 10, 11, 12, or more hours after initiation of zosuquidar - cyclodextrin
complex
infusion.
Experiments
Dissolution testing was conducted to determine fill voluines required to
achieve an
800 mg dose of zosuquidar at various concentrations of sulfobutylcyclodextrin
(CAPTISOL , (3-cyclodextrin derivative sodium salt, CyDex, Inc., Lenexa, KS).
CAPTISOL is a polyanionic B-cyclodextrin derivative with a sodium sulfonate
salt
separated from the lipophilic cavity by a butyl ether spacer group, or
sulfobutylether.
Upon intravenous administration, CAPTISOL exhibits limited plasma protein
binding
and distributes to intracellular fluid. IV doses of 14C-labeled CAPTISOL
administered to
rats, mice, dogs, rabbits and humans were rapidly and completely cleared
intact from the
circulation. Excretion is primarily in urine, with clearance approximating the
glomerular
filtration rate.
As demonstrated in the data of Table 3, acceptable fill volumes for 800 mg
zosuquidar can be achieved for solutions containing from about 8 mg/mL to
about 50
mg/mL zosuquidar, and from about 5 wt. % (based on solvent, i.e., water,
weight) to
about 30 wt. % (based on solvent, i.e., water, weight) of
sulfobutylcyclodextrin. Larger or
smaller amounts of zosuquidar can be filled into vials by varying fill volume.
-27-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
Table 3.
Dissolution Summary~800 mgZosuquidar
Zosuquidar Sulfobutyl Twee Glycine *Fill *Fill
Sample mg/mL Cyclodextrin n 80 mg/mL pH Dissolution Volume Volume
(solvent wt.) 1 Vial 2 Vials
1 50.00 30% 1.33 6 min 5 sec 16.0 mL 8.0 mL
2 41.67 25% 1.37 5 min 5 sec 19.2 mL 9.6 mL
3 33.33 20% 1.47 1 min 50 sec 24.0 mL 12.0 mL
4 33.33 20% 5 1.49 0 min 55 sec 24.0 mL 12.0 mL
33.33 20% 20 2,19 1 min 5 sec 24.0 mL 12.0 mL
6 33.33 20% 0.1% 3.09 1 min 55 sec 24,0 mL 12.0 mL
7 33.33 20% 0.25% 1.56 2 min 10 sec 24,0 mL 12.0 mL
8 25.00 15% 1.54 0 min 55 sec 32.0 mL 16.0 mL
9 16.67 10% 1.69 < 30 sec 48.0 mL 24.0 mL
8.33 5% 1.94 < 30 sec 96.0 mL 48.0 mL
*Based on an 800 mg dose
5
Data were also obtained demonstrating the feasibility of achieving acceptable
fill
volume for 900 mg zosuquidar (Table 4)'.
Table 4.
10 Dissolution Summary - 900 mg Zosu uq idar
Zosuquidar Sulfobutyl **Fill **Fill
Sample pH Dissolution Volume Volume
mg/mL cyclodextrin 1 Vial 2 Vials
la 65.2 m/mL 30% 1.19 7.5 minutes 13.8 mL 6.9 mL
4a 43.0 mg/mL 20% 1.30 2.5 minutes 20.9 mL 10.5 mL
The data suggest that the solubility of the formulations is pH sensitive. When
formulated at 50 mg/mL zosuquidar and 30 % sulfobutylcyclodextrin, the normal
pH is
approximately 1.3. Titration experiments showed that when the pH of this
solution was
increased to around 3.5, active ingredient precipitated out of solution. Based
on this
observation, material was formulated and the pH adjusted to 3.0 with NaOH. The
samples were then freeze-dried. The active ingredients exhibited satisfactory
solubility in
solution at this pH. However, when diluted into IV administration fluids
(normal saline
or 5% dextrose (900 mg - 500mL)) there was a significant decrease in
solubility. The
normal saline solution became hazy immediately. The 5% dextrose solution
became
turbid over the course of 1 hour. The pH of both solutions was determined and
found to
be 3.97 and 4.05 for saline and dextrose, respectively. Phosphoric acid was
added to each
formulation until the pH was measured at less than or equal to 2, and the
haziness
-28-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
disappeared. The data suggests that the common ion effect plays a small role
in
precipitation; however, pH appears to be a major force.
Solubility studies were conducted using zosuquidar and various co-solvents and
complexing agents. Solutions, as described in Table 5, were formulated and 3
mL
aliquots were placed in 6 mL scintillation vials. An excess of zosuquidar was
added to
each solution and the vials were capped. Samples were placed on a Burrell
Model 75
shaker and shaken on high speed. The samples were watched over the course of
several
hours and additional zosuquidar was added where needed. The samples were
shaken for
approximately 20 hours. The samples were removed fiom the shaker and visually
observed. Table 6 provides the visual data results.
Table 5.
Formulations for Solubili Testing
Sample # Formulation Sample # Formulation
1 Purified Water 8 5% Sulfobu lc clodextrin
2 5% Ethanol 9 10% Sulfobu lc clodextrin
3 10% Ethanol 10 20% Sulfobu lc clodextrin
4 15% Ethanol 11 40% Sulfobu lc clodextrin
5 20% Propylene Gl co15% Ethanol 12 20% Sulfobu lc clodextrin pH
6 20% Sulfobu lc clodextrin 5% Ethanol 13 20% Sulfobu lc clodextrin H 7
7 2.5% Sulfobu lc clodextrin 14 20% Sulfobu lc clodextrin H 9
Table 6.
Formulations for Solubili Testing
Sample # Visual Results
1* Thin, milky yellow suspension
2* Very viscous (gelled), millcy yellow suspension. Some a re ated solids were
present
3* Very viscous (gelled), milky yellow suspension. Some a re ated solids were
present
4 Thin, milk yellow suspension
5* Thin, milky yellow, peariescent suspension
6 Thin, clear, yellow solution containing undissdved, a re ated solids
7 Thin, clear, yellow solution containing undissolved, a re ated solids
8 Thin, clear, yellow solution containing undissolved, a re ated solids
9 Thin, clear, yellow solution containing undissolved, a re ated solids
10 Thin, clear, yellow solution containing undissolved, a re ated solids
11 Sli htl viscous, clear, yellow solution containing undissolved, a re ated
solids
12* Thin, mil yellow suspension
13 Thin, clear, yellow solution containing undissolved, a re ated solids
14 Thin, clear, yellow solution containing undissolved, a re ated solids
*Samples 1, 2, 3, 5, and 12 could not be filtered - undissolved solids finely
suspended
The samples were filtered through 0.45 m syringe filters to remove the
undissolved solids. Samples 1, 2, 3, 5, and 12 could not be filtered because
the
-29-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
undissolved solids were so finely suspended that the filter was easily
blocked. These
samples were centrifuged at 4500 rpm in an attempt to separate the solids;
however, only
samples 5 and 12 could be separated. Sample 11, although clear, was too
viscous to pass
through the 0.45 m membrane and so was instead filtered using a 5 m
membrane. The
potency of each sample (if able to be separated) was determined using an HPLC
potency
assay. The results (reported as the free base) are listed in Table 7.
Table 7.
Formulations for Solubility Testin~
Sample # Potency (Free base), mg/mL
15% Ethanol 13.7
20% Propylene Glycol 5% Ethanol 21.8
20% Sulfobu lc clodextrin 5% Ethanol 30.9
2.5% Sulfobu lc clodextrin 5.5
5% Sulfobu Ic clodextrin 3.9
10% Sulfobu lc clodextrin 9.7
20% Sulfobu lc clodextrin (pH 6.4) 32.0
40% Sulfobu lc clodextrin 87.1
20% Sulfobu lc clodextrin pH 5 34.2
20% Sulfobu lc clodextrin pH 7 29.5
20% Sulfobu lc clodextrin pH 9 30.6
The data demonstrate that the solubility of zosuquidar is significantly
increased
when CAPTISOL ((3-cyclodextrin derivative sodium salt) is incorporated into
the
formulation. The graph in Figure 1 illustrates the increase of zosuquidar
solution
concentration as a function of sulfobutylcyclodextrin concentration.
A study was conducted to determine the optimum ratio of zosuquidar to
CAPTISOL . Additionally, the use of glycine and Polysorbate 80 additions were
investigated as a means of decreasing dissolution time. The different
formulations tested
are listed in Table 8. After formulation, the clear solutions were separated
from the hazy
solutions and 2 mL aliquots were placed into 5 mL x 13 mm vials and were
freeze-dried.
After lyophilization, the vials were removed and a visual description was
recorded. All
calces were yellow, slightly shrunken, and showed no signs of collapse.
Samples were
reconstituted with 2 mL of purified water, the dissolution time was recorded,
and a visual
description of the reconstituted solution was recorded. Solutions containing a
haze or
insoluble material were shalcen for at least five minutes before being listed
as N/A.
-30-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
Table 8.
Zosuquidar % % Glycine Solution Dissolution Reconstituted
Sample m /mL CAPTIS Tween
g OL 80 mg/mL Description Time Description
1 10.8 5 0 0 very sl. haze n/a n/a
2 10.8 5 0.25 very sl. haze n/a n/a
3 10.8 5 5 sl. haze n/a n/a
4 10.8 5 10 hazy n/a n/a
10.8 5 20 hazy n/a n/a
6 21.5 10 0 0 Clear 45 secs. Insoluble matter
in via]
7 21.5 10 0.25 Clear 35 secs Small bead of
insoluble matter
8 21.5 10 5 hazy n/a n/a
9 21.5 10 10 sl. haze n/a n/a
21.5 10 20 sl. haze n/a n/a
11 32.3 15 0 0 Clear 3.3 min. Some Insoluble
matter in via]
12 32.3 15 0.25 Clear 3.2 min. Small bead of
insoluble matter
13 32.3 15 5 Clear 2.7 min, Some Insoluble
matter in vial
14 32.3 15 10 very sl. haze n/a n/a
32.3 15 20 sl. haze n/a n/a
16 43.0 20 0 0 Clear 3.2 min. Some Insoluble
matter in vial
17 43.0 20 0.25 Clear 4.3 min Some Insoluble
matter in vial
18 43.0 20 5 Clear 3.3 min. No insoluble
matter observed
19 43.0 20 10 Clear 2 min Some Insoluble
matter in vial
43.0 20 20 very sl. haze n/a n/a
Based on the results from Table 8, while some of the samples with higher
5 concentrations do go completely into solution when formulating, most of them
do not
reconstitute in an acceptable amount of time and or go completely back into
solution.
Neither Tween 80 nor glycine had a significant impact on the dissolution time
or the
reconstitution solubility. The only sample that did reconstitute to a complete
solution was
sample 18; however, the reconstitution time was relatively high considering
this was a 2
10 mL fill and reconstitution times would likely increase as the sample volume
increased.
An additional study was conducted to further investigate the ratio of
zosuquidar to
CAPTISOL and the effects on dissolution time and completeness of solution
after
reconstitution. Samples were formulated 2 mL aliquots were placed into 5 mL x
13 mm
vials and were freeze-dried using a conservative cycle. After drying, all of
the samples
15 were inspected, and all vials contained a yellow, slightly shrunlcen plug
with no signs of
-31-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
collapse. Samples were reconstituted with 2 mL of purified water and the
reconstitution
times were recorded. Upon inspection of the reconstituted solutions, all
samples visually
formed a complete solution with no undissolved solids stuck to the sides of
the vial or
floating free in solution. Table 9 lists the different samples tested and the
results. The
theoretical fill volumes of 1 vial per dose and 2 vials per dose based on an
800 mg dose
are included in this table.
Table 9.
Zosuquidar Tween Glycine *Fill *Fill
Sample m/mL CAPTIS L 80 mg/mL' pH Dissolution Volume Volume
mg/mL I Vial 2 Vials
1 50.00 30% 1.33 6 min 5 sec 16.0 mL 8.0 mL
2 41.67 25% 1.37 5 min 5 sec 19.2 mL 9.6 mL
3 33.33 20% 1.47 1 min 50 sec 24.0 mL 12.0 mL
4 33.33 20% 5 1.49 0 min 55 sec 24.0 mL 12.0 mL
5 33.33 20% 20 2.19 1 min 5 sec 24.0 mL 12.0 mL
6 33.33 20% 0.1% 3.09 1 min 55 sec 24.0 mL 12.0 mL
7 33.33 20% 0.25% 1.56 2 min 10 sec 24.0 mL 12.0 mL
8 25.00 15% 1.54 0 min 55 sec 32.0 mL 16.0 mL
** 22.5 15% 1.6 1 min 30 sec --- 12.5 mL
9 16.67 10% 1.69 < 30 sec 48.0 mL 24.0 mL
8.33 5% 1.94 < 30 sec 96.0 mL 48.0 mL
*Based on an 800 mg dose
**Preferred formulation based on a 550 mg dose
The results shown in Table 9 show that based on dissolution time, Tween 80
does
not improve the reconstitution properties of the formulation. In contrast,
Tween 80
appears to slow down the dissolution time. Adding glycine to the formulation
did appear
to offer some benefit in reducing the dissolution time.
Samples were formulated containing 20% CAPTISOL , 33.3 mg/mL of
zosuquidar, and different amounts of glycine. 2 mL aliquots were placed into 5
mL x 13
mm vials and were freeze-dried using a conservative cycle. Half of the samples
were held
aside and freeze-dried using a conservative cycle with an annealing step (hold
at -15 C
for 2 hours prior to re-cooling back to -45 C and freeze-drying). All of the
vials
contained yellow cakes, which were slightly shrunken, and no signs of collapse
were
observed. Samples were reconstituted with 2 mL of purified water, and the
dissolution
time and the description of the solution was recorded. Table 10 contains the
samples
tested and the results.
-32-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
Table 10.
Sample Lyo Cycle Glycine, Dissolution Time Description
mg/mL
1 Normal 0 1 min. 51 secs No insoluble matter present
2 Normal 1 1 min. 53 secs No insoluble matter present
3 Annealed 1 2 min. 28 secs No insoluble matter present
4 Normal 2 1 min. 46 secs No insoluble matter present
Annealed 2 2 min. 18 secs No insoluble matter present
6 Normal 3 1 min. 43 secs No insoluble matter present
7 Annealed 3 2 min. 23 secs No insoluble matter present
8 Normal 4 1 min. 38 secs No insoluble matter present
9 Annealed 4 2 min. 31 secs No insoluble matter present
Normal 5 1 min. 39 secs No insoluble matter present
11 Annealed 5 2 min. 21 sees No insoluble matter present
12* Normal 5 3 min. 28 secs No insoluble matter present
13** Normal 5 5 min.24 secs No insoluble matter present
*12 mL fill in 20 mL vial; **24 mL fill in 50 mL vial
The results in Table ' 10 demonstrate a formulation and process which yield a
5 product that will reconstitute in an acceptable amount of time. However,
Samples 12 and
13, which reflect actual sample fill volumes, show that the total amount of
product in a
vial does affect the dissolution time. Glycine had a very minimal effect on
dissolution
time and that annealing seemed to increase dissolution time.
To further examine the dissolution time, the formulation concentration, vial
size
10 and fill, and glycine content were also examined. Samples were formulated
to contain
150 mg/mL and 25 mg/mL zosuquidar. Glycine was also added to several of the
samples
to determine if there is an effect on the dissolution time. 16 mL aliquots
were filled into
either 20 mL x 20 mm vials or 30 mL x 20 mm vials, and were freeze-dried using
a
conservative cycle. The amount of added glycine, the vial size, the
dissolution times, and
a visual description of the samples after reconstitution are listed in Table
11. Samples
were reconstituted with 16 mL of purified water. All vials contained yellow
calces, which
were slightly shrunken and fractured.
-33-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
Table 11.
Sample Glycine, Vial Size pH Dissolution Description
mg/mL Time
1 0 20 mL 1.57 1 min. 48 sec No insoluble matter
present
2 0 30 mL 1.57 1 min. 52 sec No insoluble matter
present
3 5 20 mL 2.37 1 min. 51 sec No insoluble matter
present
4 5 30 mL 2.37 1 min. 47 sec No insoluble matter
present
20 20 mL 3.19 1 min. 26 sec No insoluble matter
present
6 20 30 mL 3.19 1 inin. 23 sec No insoluble matter
present
These results show that the ratio and total solids amount in the samples
tested
5 produced cakes which dissolve in under 2 minutes, assuming an 800 mg dose
delivered in
2 vials. Added glycine does not appear to affect the dissolution time. Glycine
does
however affect the pH of the formulation, which is problematic because
previous studies
have shown that as the pH increases the solubility of the API decreases.
Because of these
factors, it is desirable not to add glycine to the forinulation.
The 25 mg/mL zosuquidar and 150 mg/mL CAPTISOL formulation exhibits
desirable formulation attributes; however, during lyophilization studies, it
was observed
that there was a small amount of undissolved "crust" stuck to the bottom of
some of the
samples after lyophilization and reconstitution. After watching samples during
the
freezing step in the lyophilization cycle, it was believed that CAPTISOL was
releasing
the zosuquidar as the solution temperature decreased. The unbound zosuquidar
would
then precipitate and sink to the bottom of the vial where it would form a
slowly dissolving
crust. Upon reconstitution, most of the solids within the vial were completely
dissolved
in approximately 1 minute. The crust at the bottom of the vial on the other
hand, would
talce several hours to completely dissolve.
Based on these results, a study was conducted to investigate the effects of
varying
the ratio of zosuquidar to CAPTISOL and the amounts in an attempt to prevent
zosuquidar from being released from the CAPTISOL during freezing. Samples
were
prepared according to the concentrations listed in Table 12. 16 mL aliquots
were filled
into 30 mL x 20 mm tubing vials and lyophilized using a conservative cycle.
The 16 mL
-34-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
fill samples were reconstituted with 20 mL of purified water, and the 8 mL
fill samples
were reconstituted with 10 mL of purified water.
Table 12.
Vial Zosuquidar Free CAPTISOL
Base Concentration Solution Description after Reconstitution
Concentration
1 20 mg/mL 150 mg/mL No crust or residue or free a re ates
2 20 mg/mL 150 mg/mL No crust or residue or free aggregates
3 22.5 mg/mL 150 mg/mL No crust or residue or free aggregates
4 22.5 mg/mL 150 m/mL No crust or residue or free aggregates
25 mg/mL 150 mg/mL Slight crust present at bottom, no free
aggregates
6 25 mg/mL 150 mg/mL Slight crust present at bottom, no free
aggregates
7* 25 mg/mL 150 mg/mL Slight crust present at bottom, no free
aggregates
8* 25 mg/mL 150 mg/mL Slight crust present at bottom, no free
aggregates
9* 25 mg/mL 150 mg/mL Slight crust present at bottom, no free
aggregates
10* 25 mg/mL 150 mg/mL Slight crust present at bottom, no free
aggregates
11 25 mg/mL 175 mg/mL No crust or residue or free aggregates, slow
dissolution
12 25 mg/mL 175 mg/mL No crust or residue or free aggregates, slow
dissolution
13 25 mg/mL 200 mg/mL No crust or residue or free aggregates, slow
dissolution
14 25 mghnL 200 mg/mL No crust or residue or free aggregates, slow
dissolution
25 mg/mL 225 mg/mL No crust or free aggregates, very difficult to
dissolve
16 25 mg/mL 225 mg/mL No crust or free aggregates, very difficult to
dissolve
5 * 8mLfi11ina30mLx20mmvial
Based on these results, the optimal concentration of CAPTISOL and zosuquidar
was 150 mg/mL (15%) and 22.5 mg/mL, respectively.
Three vials of zosuquidar (275 mg/vial zosuquidar, 1850 mg/vial CAPTISOL )
10 were each reconstituted with 15 mL of 5% Dextrose Injection, USP, 500 mL. A
total of
47 mL of sample solution was removed from the vials with a 50 mL syringe and
was
injected into the 500 mL bag of 5% Dextrose Injection. The Zosuquidar/Dextrose
solution was held at room temperature, and samples were removed at 0, 2, 4, 8,
12, 24,
-35-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
and 48 hours. All samples were held at -70 C after being pulled and were
analyzed after
all samples had been collected. Each sample was tested for pH, HPLC
concentration, and
related substances. The data obtained from this study is summarized in Table
13. There
was essentially no change in potency, impurities, and pH for all samples.
Based on these
results, zosuquidar with CAPTISOL is stable at room temperature for 48 hours
when
reconstituted with 5% Dextrose Injection.
Table 13.
Time Point pH Zosuquidar Free Base
Conc. (mg/mL)
t= 0 hours 2.63 1.41
t=2hours 2.62 1.41
t=4hours 2.64 1.41
t= 8 hours 2.63 1.42
t= 12 hours 2.63 1.40
t= 24 hours 2.64 1.40
t= 48 hours 2.64 1.40
Discussion
A drug product comprising 275 mg of zosuquidar trihydrochloride in CAPTISOL
was formulated that exhibited superior solubility characteristics. Use of
CAPTISOL
afforded over a 5-fold increase in water solubility of zosuquidar
trihydrochloride,
enabling lyophilization of a greater quantity of active ingredient in a 30 mL
vial. To
provide a dose of 275 mg/vial, a fill volume of 12.2 mL per 30 mL vial was
employed.
The total CAPTISOL concentration per vial was 1.83 g. This concentration of
CAPTISOL solubilized zosuquidar and provided an acceptable reconstitution
rate for
the vial. Reconstitution of vials with 15 mL of 5% Dextrose Injection provided
a solution
that contained 16.9 mg/mL of zosuquidar.
Use of CAPTISOL achieves a higher drug content per vial, an acceptable
reconstitution time, and an acceptable lyophilized cake compared to other
solubilizers
such as mannitol and glycine, as demonstrated by the data in Table 14.
-36-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
Table 14.
Parameter Zosuquidar at 50 mg/vial Zosuquidar at 275 mg/vial
Bulk Formulation 5 mg/mL zosuquidar 22.5 mg/mL zosuquidar
Concentration 20 mg/mL mannitol 150 mg/mL CAPTISOL
1.5 mg/mL glycine
Fill volume per vial 10 mL 12.7 mL
Vial Content 50 mg zosuquidar 286 mg zosuquidar
200 mg mannitol 1905 mg CAPTISOL
15 mg glycine
Vial size 30 mL Type 1 glass tubing vial 30 mL Type 1 glass tubing
vial
Appearance Light yellow solid calce Light yellow solid cake
Reconstitution time Approx. 1 to 2 min Approx 1 to 1.5 min
Reconstitution 10 mL 17.3 mL
volume
Reconstitution 5 mg/mL 16.5 mg/mL
concentration
Number of vials 11 2
required per 550
mg/day dose
Use of CAPTISOL in combination with zosuquidar provides a stable
formulation, as demonstrated by the real time stability data in Table 15.
Table 15.
Test Initial 1 month
Appearance (solid) Pale light yellow solid cake Light yellow solid cake
Appearance (liquid) Light yellow liquid Light yellow liquid
pH 1.68 1.81
Assay by HPLC 102.7% 102.9%
Total Related substances by HPLC 0.18% 0.32%
Moisture by KF 1.0% 1.0%
All references cited herein, including but not limited to published and
unpublished
applications, patents, and literature references, are incorporated herein by
reference in
their entirety and are hereby made a part of this specification. To the extent
publications
and patents or patent applications incorporated by reference contradict the
disclosure
contained in the specification, the specification is intended to supersede
and/or take
precedence over any such contradictory material.
The term "comprising" as used herein is synonymous with "including,"
"containing," or "characterized by," and is inclusive or open-ended and does
not exclude
additional, unrecited elements or method steps.
-37-
CA 02630087 2008-01-04
WO 2007/008496 PCT/US2006/026031
All numbers expressing quantities of ingredients, reaction conditions, and so
forth
used in the specification are to be understood as being modified in all
instances by the
term "about." Accordingly, unless indicated to the contrary, the numerical
parameters set
forth herein are approximations that may vary depending upon the desired
properties
sought to be obtained. At the very least, and not as an attempt to limit the
application of
the doctrine of equivalents to the scope of any claims in any application
claiming priority
to the present application, each numerical parameter should be construed in
light of the
number of significant digits and ordinary rounding approaches.
The above description discloses several methods and materials of the present
invention. This inveiition is susceptible to modifications in the methods and
materials, as
well as alterations in the fabrication methods and equipment. Such
modifications will
become apparent to those skilled in the art from a consideration of this
disclosure or
practice of the invention disclosed herein. Consequently, it is not intended
that this
invention be limited to the specific embodiments disclosed herein, but that it
cover all
modifications and alternatives coming within the true scope and spirit of the
invention.
-38-