Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
DIAGNOSIS OF SEPSIS
CROSS-REFERENCE TO RELATED APPLICATIONS
This application claims priority to Provisional Patent Application No.
60/751,197,
filed December 15, 2005 which is incorporated herein, by reference, in its
entirety. This
application also claims priority to Provisional Patent Application No.
60/819,983, filed July
10, 2006 which is incorporated herein, by reference, in its entirety.
1. FIELD OF THE INVENTION
The present invention relates to methods and compositions for diagnosing or
predicting sepsis and/or its stages of progression in a subject. The present
invention also
relates to methods and compositions for diagnosing systemic inflammatory
response
syndrome in a subject.
2. BACKGROUND OF THE INVENTION
Early detection of a disease condition typically allows for a more effective
therapeutic treatment,with a correspondingly more favorable clinical outcome.
In many
cases, however, early detection of disease symptoms is problematic due to the
complexity of
the disease; hence, a disease may become relatively advanced before diagnosis
is possible.
Systemic inflammatory conditions represent one such class of diseases. These
conditions,
particularly sepsis, typically, but not always, result from an interaction
between a
pathogenic microorganism and the host's defense system that triggers an
excessive and
dysregulated-inflammatory response in the host. The complexity of the host's
response
during the systemic inflammatory response has complicated efforts towards
understanding
disease pathogenesis (reviewed in Healy, 2002, Annul. Pharmacother. 36:648-
54). An
incomplete understanding of the disease pathogenesis, in turn, contributes to
the difficulty
in finding useful diagnostic biomarkers. Early and reliable diagnosis is
imperative,
however, because of the remarkably rapid progression of sepsis into a life-
threatening
condition.
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
The development of sepsis in a subject follows a well-described course,
progressing
from systemic inflammatory response syndrome ("SIRS")-negative, to SIRS-
positive, and
then to sepsis, which may then progress to severe sepsis, septic shock,
multiple organ
dysfunction ("MOD"), and ultimately death. Sepsis may also arise in an
infected subject
when the subject subsequently develops SIRS. "Sepsis" is commonly defined as
the
systemic host response to infection with SIRS plus a documented infection.
"Severe sepsis"
is associated with MOD, hypotension, disseminated intravascular coagulation
("DIC") or
hypoperfiusion abnormalities, including lactic acidosis, oliguria, and changes
in mental
status. "Septic shock" is commonly defined as sepsis-induced hypotension that
is resistant
to fluid resuscitation with the additional presence of hypoperfusion
abnormalities.
Documenting the presence of the pathogenic microorganisms that are clinically
significant to sepsis has proven difficult. Causative microorganisms typically
are detected
by culturing a subject's blood, sputum, urine, wound secretion, in-dwelling
line catheter
surfaces, etc. Causative microorganisms, however, may reside only in certain
body
microenvironments such that the particular material that is cultured may not
contain the
contaminating microorganisms. Detection may be complicated further by low
numbers of
microorganisms at the site of infection. Low numbers of pathogens in blood
present a
particular problem for diagnosing sepsis by culturing blood. In one study, for
example,
positive culture results were obtained in only 17% of subjects presenting
clinical
manifestations of sepsis (Rangel-Frausto et al., 1995, JAMA 273:117-123).
Diagnosis can
be further complicated by contamination of samples by non-pathogenic
microorganisms.
For example, only 12.4% of detected microorganisms were clinically significant
in a study
of 707 subjects with septicemia (Weinstein et al., 1997, Clinical Infectious
Diseases 24:584-
602).
The difficulty in early diagnosis of sepsis is reflected by the high morbidity
and
mortality associated with the disease. Sepsis currently is the tenth leading
cause of death in
the United States and is especially prevalent among hospitalized patients in
non-coronary
intensive care units (ICUs), where it is the most common cause of death. The
overall rate of
mortality is as high as 35 percent, with an estimated 750,000 cases per year
occurring in the
United States alone. The annual cost to treat sepsis in the United States
alone is on the
order of billions of dollars.
A need, therefore, exists for a method of diagnosing sepsis, using techniques
that
have satisfactory specificity and sensitivity performance, sufficiently early
to allow
effective intervention and prevention.
-2-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
3. SUMMARY OF THE INVENTION
The present invention relates to methods and compositions for diagnosing
sepsis,
including the onset of sepsis, in a test subject. The present invention also
relates to methods
and compositions for predicting sepsis in a test subject.
The present invention fiirther relates to methods and compositions for
diagnosing or
predicting stages of sepsis progression in a test subject. The present
invention still further
relates to methods and compositions for diagnosing systemic inflammatory
response
syndrome (SIRS) in a test subject.
In one aspect, the present invention provides a method of predicting the
development of sepsis in a test subject at risk for developing sepsis. This
method comprises
evaluating whether a plurality of features in a biomarker profile of the test
subject satisfies a
value set, wherein satisfying the value set means that the test subject will
develop sepsis
with a likelihood that is determined by the accuracy of the decision rule to
which the
plurality of features are applied in order to determine whether they satisfy
the value set. In
some embodiments, the accuracy of the decision rule is at least 60 percent, at
least 70
percent, at least 80 percent, or at least 90 percent. Therefore,
correspondingly, the
likelihood that the test subject will develop sepsis when the plurality of
features satisfies the
value set is at least 60 percent, at least 70 percent, at least 80 percent, or
at least 90 percent.
Yet another aspect of the invention comprises methods for diagnosing sepsis in
a test
subject. These methods comprise evaluating whether a plurality of features in
a biomarker
profile of the test subject satisfies a value set, wherein satisfying the
value set predicts that
the test subject has sepsis with a likelihood that is determined by the
accuracy of the
decision rule to which the plurality of features are applied in order to
determine whether
they satisfy the value set. When the plurality of biomarkers comprises
complement
component C3 and complement component C4, the plurality of biomarkers
comprises three
or more biomarkers. In some embodiments, the accuracy of the decision rule is
at least 60
percent, at least 70 percent, at least 80 percent, or at least 90 percent.
Therefore,
correspondingly, the likelihood that the test subject has sepsis when the
plurality of features
satisfies the value set is at least 60 percent, at least 70 percent, at least
80 percent, or at least
90 percent.
In a particular embodiment, the biomarker profile comprises at least two
features,
each feature representing a feature of a corresponding biomarker listed in
column three or
four of Table 1. In one embodiment, the biomarker profile comprises at least
two different
biomarkers listed in column three or four of Table 1. In such an embodiment,
the biomarker
-3-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
profile can comprise a respective corresponding feature for the at least two
biomarkers.
Generally, the at least two biomarkers are derived from at least two different
genes. When
the plurality of biomarkers comprises complement component C3 and complement
component C4, the plurality of biomarkers comprises three or more biomarkers.
In the case where a biomarker in the at least two different biomarkers is
listed in
column three of Table 1, the biomarker can be, for example, a transcript made
by the listed
gene, a complement thereof, or a discriminating fragment or complement
thereof, or a
eDNA thereof, or a discriminating fragment of the eDNA, or a discriminating
amplified
nucleic acid molecule corresponding to all or a portion of the transcript or
its complement,
or a protein encoded by the gene, or a discriminating fragment of the protein,
or an
indication of any of the above. Further still, the biomarker can be, for
example, a protein
listed in column four of Table 1 or a discriminating fragment of the protein,
or an indication
of any of the above. Here, a discriminating molecule or fragment is a molecule
or fragment
that, when detected, indicates presence or abundance of the above-identified
transcript,
cDNA, amplified nucleic acid, or protein. In accordance with this embodiment,
the
biomarker profiles of the present invention can be obtained using any standard
assay known
to those skilled in the art, or in an assay described herein, to detect a
biomarker. Such
assays are capable, for example, of detecting the products of expression
(e.g., nucleic acids
and/or proteins) of a particular gene or allele of a gene of interest (e.g., a
gene disclosed in
Table 1). In one embodiment, such an assay utilizes a nucleic acid microarray.
In some
embodiments, the biomarker profile comprises at least 2, 3, 4, 5, 6, 7, 8, 9,
10, 11, 12, 13,
14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 30, 35, 40, 45, or 50
different biomarkers from
Table 1.
In a particular embodiment, the biomarker profile comprises at least two
features,
each feature representing a feature of a corresponding biomarker listed in
column three or
four of Table 4. In one embodiment, the biomarker profile comprises at least
two different
biomarkers listed in column three or four of Table 4. In such an embodiment,
the biomarker
profile can comprise a respective corresponding feature for the at least two
biomarkers.
Generally, the at least two biomarkers are derived from at least two different
genes. In the
case where a biomarker in the at least two different biomarkers is listed in
column three of
Table 4, the biomarker can be, for example, a transcript made by the listed
gene, a
complement thereof, or a discriminating fragment or complement thereof, or a
cDNA
thereof, or a discriminating fragment of the cDNA, or a discriminating
amplified nucleic
acid molecule corresponding to all or a portion of the transcript or its
complement, or a
-4-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
protein encoded by the gene, or a discriminating fragment of the protein, or
an indication of
any of the above. Further still, the biomarker can be, for example, a protein
listed in column
four of Table 4 or a discriminating fragment of the protein, or an indication
of any of the
above. Here, a discriminating molecule or fragment is a molecule or fragment
that, when
detected, indicates presence or abundance of the above-identified transcript,
cDNA,
amplified nucleic acid, or protein. In accordance with this embodiment, the
biomarker
profiles of the present invention can be obtained using any standard assay
known to those
skilled in the art, or in an assay described herein, to detect a biomarker.
Such assays are
capable, for example, of detecting the products of expression (e.g., nucleic
acids and/or
proteins) of a particular gene or allele of a gene of interest (e.g., a gene
disclosed in Table
4). In one embodiment, such an assay utilizes a nucleic acid microarray. In
some
embodiments, the biomarker profile comprises at least 2, 3, 4, 5, 6, 7, 8, 9,
or 10 different
biomarkers from Table 4. In some embodiments, the biomarker profile comprises
SERPINC 1, APOA2, and CRP. In some embodiments, the biomarker profile
comprises at
least one of SERPINCI, APOA2, and CRP, and, additionally, at least 1, 2, 3, 4,
5, 6 or 7
other additional biomarkers in Table 4. In some embodiments, the biomarker
profile
comprises at least one of SERPINCI, APOA2, and CRP and, additionally, at least
1, 2, 3, 4,
5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 or more additional
biomarkers from
any combination of Tables 1, 4, 5, 6, and 7. In some embodiments, the
biomarker profile
comprises at least one of SERPINC1, APOA2, and CRP, and, additionally, at
least 1, 2, 3,
4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 or more
additional biomarkers from
any one of Tables 1, 4, 5, 6, and 7. In some embodiments, each of the
biomarkers in the
profile is a protein. In some embodiments, each of the biomarkers in the
profile is a nucleic
acid. In some embodiments, some of the biomarkers in the profile are nucleic
acids and
some of the biomarkers are proteins.
In a particular embodiment, the biomarker profile comprises at least two
features,
each feature representing a feature of a corresponding biomarker listed in
column three or
four of Table 5. In one embodiment, the biomarker profile comprises at least
two different
biomarkers listed in column three or four of Table 5. In such an embodiment,
the biomarker
profile can comprise a respective corresponding feature for the at least two
biomarkers.
Generally, the at least two biomarkers are derived from at least two different
genes. In the
case where a biomarker in the at least two different biomarkers is listed in
column three of
Table 5, the biomarker can be, for example, a transcript made by the listed
gene, a
complement thereof, or a discriminating fragment or complement thereof, or a
cDNA
-5-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
thereof, or a discriminating fragment of the cDNA, or a discriminating
amplified nucleic
acid molecule corresponding to all or a portion of the transcript or its
complement, or a
protein encoded by the gene, or a discriminating fragment of the protein, or
an indication of
any of the above. Further still, the biomarker can be, for example, a protein
listed in column
four of Table 5 or a discriminating fragment of the protein, or an indication
of any of the
above. Here, a discriminating molecule or fragment is a molecule or fragment
that, when
detected, indicates presence or abundance of the above-identified transcript,
cDNA,
amplified nucleic acid, or protein. In accordance with this embodiment, the
biomarker
profiles of the present invention can be obtained using any standard assay
known to those
skilled in the art, or in an assay described herein, to detect a biomarker.
Such assays are
capable, for example, of detecting the products of expression (e.g., nucleic
acids and/or
proteins) of a particular gene or allele of a gene of interest (e.g., a gene
disclosed in Table
5). In one embodiment, such an assay utilizes a nucleic acid microarray. In
some
embodiments, the biomarker profile comprises at least 2, 3, 4, 5, 6, 7, 8, 9,
10, 11, 12, 13,
14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 30, 35, 40, 45, or 50
different biomarkers from
Table 5.
In a particular embodiment, the biomarker profile comprises at least two
features,
each feature representing a feature of a corresponding biomarker listed in
column three or
four of Table 6. In one embodiment, the biomarker profile comprises at least
two different
biomarkers listed in column three or four of Table 6. In such an embodiment,
the biomarker
profile can comprise a respective corresponding feature for the at least two
biomarkers.
Generally, the at least two biomarkers are derived from at least two different
genes. In the
case where a biomarker in the at least two different biomarkers is listed in
column three of
Table 6, the biomarker can be, for example, a transcript made by the listed
gene, a
complement thereof, or a discriminating fragment or complement thereof, or a
cDNA
thereof, or a discriminating fragment of the cDNA, or a discriminating
amplified nucleic
acid molecule corresponding to all or a portion of the transcript or its
complement, or a
protein encoded by the gene, or a discriminating fragment of the protein, or
an indication of
any of the above. Further still, the biomarker can be, for example, a protein
listed in column
four of Table 6 or a discriminating fragment of the protein, or an indication
of any of the
above. Here, a discriminating molecule or fragment is a molecule or fragment
that, when
detected, indicates presence or abundance of the above-identified transcript,
cDNA,
amplified nucleic acid, or protein. In accordance with this embodiment, the
biomarker
profiles of the present invention can be obtained using any standard assay
known to those
-6-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
skilled in the art, or in an assay described herein, to detect a biomarker.
Such assays are
capable, for example, of detecting the products of expression (e.g., nucleic
acids and/or
proteins) of a particular gene or allele of a gene of interest (e.g., a gene
disclosed in Table
6). In one embodiment, such an assay utilizes a nucleic acid microarray. In
some
embodiments, the biomarker profile comprises at least 2, 3, 4, 5, 6, 7, 8, 9,
10, 11, 12, 13,
14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, or 25 different biomarkers from
Table 6.
In a particular embodiment, the biomarker profile comprises at least two
features,
each feature representing a feature of a corresponding biomarker listed in
column three or
four of Table 7. In one embodiment, the biomarker profile comprises at least
two different
biomarkers listed in column three or four of Table 7. In such an embodiment,
the biomarker
profile can comprise a respective corresponding feature for the at least two
biomarkers.
Generally, the at least two biomarkers are derived from at least two different
genes. In the
case where a biomarker in the at least two different biomarkers is listed in
column three of
Table 7, the biomarker can be, for example, a transcript made by the listed
gene, a
complement thereof, or a discriminating fragment or complement thereof, or a
cDNA
thereof, or a discriminating fragment of the cDNA, or a discriminating
amplified nucleic
acid molecule corresponding to all or a portion of the transcript or its
complement, or a
protein encoded by the gene, or a discriminating fragment of the protein, or
an indication of
any of the above. Further still, the biomarker can be, for example, a protein
listed in column
four of Table 7 or a discriminating fragment of the protein, or an indication
of any of the
above. Here, a discriminating molecule or fragment is a molecule or fragment
that, when
detected, indicates presence or abundance of the above-identified transcript,
cDNA,
amplified nucleic acid, or protein. In accordance with this embodiment, the
biomarker
profiles of the present invention can be obtained using any standard assay
known to those
skilled in the art, or in an assay described herein, to 'detect a biomarker.
Such assays are
capable, for example, of detecting the products of expression (e.g., nucleic
acids and/or
proteins) of a particular gene or allele of a gene of interest (e.g., a gene
disclosed in Table
7). In one embodiment, such an assay utilizes a nucleic acid microarray. In
some
embodiments, the biomarker profile comprises at least 2, 3, 4, 5, 6, 7, 8, 9,
10, 11, 12, 13,
14, 15, 16, 17, 18, 19, 20, 21, 22, 23, or 24 different biomarkers from Table
7.
In a particular embodiment, the biomarker profile comprises at least two
features,
each feature representing a feature of a corresponding biomarker listed in
column three or
four of Table 8. In one embodiment, the biomarker profile comprises at least
two different
biomarkers listed in column three or four of Table 8. In such an embodiment,
the biomarker
-7-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
profile can comprise a respective corresponding feature for the at least two
biomarkers.
Generally, the at least two biomarkers are derived from at least two different
genes.
In the case where a biomarker in the at least two different biomarkers is
listed in
column three of Table 8, the biomarker can be, for example, a transcript made
by the listed
gene, a complement thereof, or a discriminating fragment or complement
thereof, or a
cDNA thereof, or a discriminating fragment of the cDNA, or a discriminating
amplified
nucleic acid molecule corresponding to all or a portion of the transcript or
its complement,
or a protein encoded by the gene, or a discriminating fragment of the protein,
or an
indication of any of the above. Further still, the biomarker can be, for
example, a protein
listed in column four of Table 8 or a discriminating fragment of the protein,
or an indication
of any of the above. Here, a discriminating molecule or fragment is a molecule
or fragment
that, when detected, indicates presence or abundance of the above-identified
transcript,
cDNA, amplified nucleic acid, or protein. In accordance with this embodiment,
the
biomarker profiles of the present invention can be obtained using any standard
assay known
to those skilled in the art, or in an assay described herein, to detect a
biomarker. Such
assays are capable, for example, of detecting the products of expression
(e.g., nucleic acids
and/or proteins) of a particular gene or allele of a gene of interest (e.g., a
gene disclosed in
Table 8). In one embodiment, such an assay utilizes a nucleic acid microarray.
In some
embodiments, the biomarker profile comprises at least 2, 3, 4, 5, 6, 7, 8, 9,
10, I 1, 12, 13,
14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 30, 35, 40, 45, or 50
different biomarkers from
Table 8.
In the case where a biomarker in the at least two different biomarkers is
listed in
column three of Table 9, the biomarker can be, for example, a transcript made
by the listed
gene, a complement thereof, or a discriminating fragment or complement
thereof, or a
cDNA thereof, or a discriminating fragment of the cDNA, or a discriminating
amplified
nucleic acid molecule corresponding to all or a portion of the transcript or
its complement,
or a protein encoded by the gene, or a discriminating fragment of the protein,
or an
indication of any of the above. Further still, the biomarker can be, for
example, a protein
listed in column four of Table 9 or a discriminating fragment of the protein,
or an indication
of any of the above. Here, a discriminating molecule or fragment is a molecule
or fragment
that, when detected, indicates presence or abundance of the above-identified
transcript,
cDNA, amplified nucleic acid, or protein. In accordance with this embodiment,
the
biomarker profiles of the present invention can be obtained using any standard
assay known
to those skilled in the art, or in an assay described herein, to detect a
biomarker. Such
-8-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
assays are capable, for example, of detecting the products of expression
(e.g., nucleic acids
and/or proteins) of a particular gene or allele of a gene of interest (e.g., a
gene disclosed in
Table 9). In one embodiment, such an assay utilizes a nucleic acid microarray.
In some
embodiments, the biomarker profile comprises at least 2, 3, 4, 5, 6, 7, 8, 9,
10, 11, 12, 13,
14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 30, 35, 40, 45, or 50
different biomarkers from
Table 9.
In a particular embodiment, the biomarker profile comprises at least two
features,
each feature representing a feature of a corresponding biomarker listed in
column three or
four of any combination of Tables 1, 4, 5, 6, 7, 8, and 9. In one embodiment,
the biomarker
profile comprises at least two different biomarkers listed in column three or
four of any
combination of Tables 1, 4, 5, 6, 7, 8, and 9. In such an embodiment, the
biomarker profile
can comprise a respective corresponding feature for the at least two
biomarkers. Generally,
the at least two biomarkers are derived from at least two different genes. In
the case where
a biomarker in the at least two different biomarkers is listed in column three
of any
combination of Tables 1, 4, 5, 6, 7, 8, and 9, the biomarker can be, for
example, a transcript
made by the listed gene, a complement thereof, or a discriminating fragment or
complement
thereof, or a cDNA thereof, or a discriminating fragment of the cDNA, or a
discriminating
amplified nucleic acid molecule corresponding to all or a portion of the
transcript or its
complement, or a protein encoded by the gene, or a discriminating fragment of
the protein,
or an indication of any of the above. Further still, the biomarker can be, for
example, a
protein listed in column four of any combination of Tables 1, 4, 5, 6, 7, 8,
and 9 or a
discriminating fragment of the protein, or an indication of any of the above.
Here, a
discriminating molecule or fragment is a molecule or fragment that, when
detected,
indicates presence or abundance of the above-identified transcript, cDNA,
amplified nucleic
acid, or protein. In accordance with this embodiment, the biomarker profiles
of the present
invention can be obtained using any standard assay known to those skilled in
the art, or in
an assay described herein, to detect a biomarker. Such assays are capable, for
example, of
detecting the products of expression (e.g., nucleic acids and/or proteins) of
a particular gene
or allele of a gene of interest (e.g., a gene disclosed in any combination of
Tables 1, 4, 5, 6,
7, 8, and 9). In one embodiment, such an assay utilizes a nucleic acid
microarray. In some
embodiments, the biomarker profile comprises at least 2, 3, 4, 5, 6, 7, 8, 9,
10, 11, 12, 13,
14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 30, 35, 40, 45, or 50
different biomarkers from
any combination of Tables 1, 4, 5, 6, 7, 8, and 9. In some embodiments, the
biomarker
profile comprises the biomarkers CRP, APOA2, and SERPINCI described in Table 4
-9-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
below. In some embodiments these three biomarkers are proteins. In some
embodiments,
these three biomarkers are nucleic acids. In some embodiments, these three
biomarkers are
any combination of proteins and nucleic acids. In some embodiments, the
biomarker profile
comprise at least one of the biomarkers CRP, APOA2, and SERPINCI, and,
additionally, 1,
2, 3, 4, 5, 6, or 7 biomarkers from those set forth in Table 4. In some
embodiments, the
biomarker profile comprises at least one of the biomarkers CRP, APOA2, and
SERPINCI,
and, additionally, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17,
18, 19, 20 or more
additional biomarkers from those listed in columns 3 and/or 4 of any one of
Tables 1, 4, 5,
6, 7, 8, and 9. In some embodiments, the biomarker profile comprises at least
one of the
biomarkers CRP, APOA2, and SERPINC 1, and, additionally, 1, 2, 3, 4, 5, 6, 7,
8, 9, 10, 11,
12, 13, 14, 15, 16, 17, 18, 19, 20 or more additional biomarkers from those
listed in
columns 3 and/or 4 of any combination of Tables 1, 4, 5, 6, 7, 8, and 9.
In a particular embodiment, the biomarker profile comprises at least four
features,
each feature representing a feature of a corresponding biomarker listed in
column three or
four of any combination of Tables 1, 4, 5, 6, 7, 8, and 9. In one embodiment,
the biomarker
profile comprises at least four different biomarkers listed in column three or
four of any
combination of Tables 1, 4, 5, 6, 7, 8, and 9. In such an embodiment, the
biomarker profile
can comprise a respective corresponding feature for the at least four
biomarkers. Generally,
the at least four biomarkers are derived from at least four different genes.
In the case where
a biomarker in the at least four different biomarkers is listed in column
three of any
combination of Tables 1, 4, 5, 6, 7, 8, and 9 the biomarker can be, for
example, a transcript
made by the listed gene, a complement thereof, or a discriminating fragment or
complement
thereof, or a eDNA thereof, or a discriminating fragment of the cDNA, or a
discriminating
amplified nucleic acid molecule corresponding to all or a portion of the
transcript or its
complement, or a protein encoded by the gene, or a discriminating fragment of
the protein,
or an indication of any of the above. Further still, the biomarker can be, for
example, a
protein listed in column four of any combination of Tables 1, 4, 5, 6, 7, 8,
and 9 or a
discriminating fragment of the protein, or an indication of any of the above.
Here, a
discriminating molecule or fragment is a molecule or fragment that, when
detected,
indicates presence or abundance of the above-identified transcript, cDNA,
amplified nucleic
acid, or protein. In accordance with this embodiment, the biomarker profiles
of the present
invention can be obtained using any standard assay known to those skilled in
the art, or in
an assay described herein, to detect a biomarker. Such assays are capable, for
example, of
detecting the products of expression (e.g., nucleic acids and/or proteins) of
a particular gene
-10-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
or allele of a gene of interest (e.g., a gene disclosed in any combination of
Tables 1, 4, 5, 6,
7, 8, and 9). In one embodiment, such an assay utilizes a nucleic acid
microarray. In some
embodiments, the biomarker profile comprises at least 4, 5, 6, 7, 8, 9, 10,
11, 12, 13, 14, 15,
16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 30, 35, 40, 45, or 50 different
biomarkers from any
combination of Tables 1, 4, 5, 6, 7, 8, and 9.
Although the methods of the present invention are particularly useful for
detecting
or predicting the onset of sepsis in SIRS subjects, one of skill in the art
will understand that
the present methods may be used for any subject including, but not limited to,
subjects
suspected of having SIRS or of being at any stage of sepsis. For example, a
biological
sample can be taken from a subject, and a profile of biomarkers in the sample
can be
evaluated in light of biomarker profiles obtained from several different types
of training
populations. Representative training populations variously include, for
example,
populations that include subjects who are SIRS-negative, populations that
include subjects
who are SIRS-positive, and/or populations that include subjects at a
particular stage of
sepsis. Evaluation of the biomarker profile in light of each of these
different training
populations can be used to determine whether the test subject is SIRS-
negative,
SIRS-positive, is likely to become septic, or has a particular stage of
sepsis. Based on the
diagnosis resulting from the methods of the present invention, an appropriate
treatment
regimen can then be initiated.
In particular embodiments, the invention also provides kits that are useful in
diagnosing or predicting the development of sepsis or SIRS in a subject (see
Section 5.3,
infra). Some such kits of the present invention comprise at least 2, 3, 4, 5,
6, 7, 8, 9, 10, 11,
12, 13, 14, 15, 16, 17, 18, 19, 20, 25, 30, 35, 40, 45, 50, 54, 5, 60, 65, 70,
75, 80, 85, 90, 95,
96 or more biomarkers and/or reagents used to detect presence or abundance of
such
biomarkers. In some embodiments, each of these biomarkers is from Table I. In
some
embodiments, each of these biomarkers is from Table 4. In some of these
embodiments
three of the biomarkers in the kit are CRP, APOA2, and SERPINCI. In some
embodiments, the biomarkers in the kit are at least one of SERPINCI, APOA2,
and CRP,
and, additionally, at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15,
16, 17, 18, 19, 20 or
more additional biomarkers from any combination of Tables 1, 4, 5, 6, 7, 8,
and 9. In some
embodiments, the biomarkers in the kit are at least one of SERPINCI, APOA2,
and CRP,
and, additionally, at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15,
16, 17, 18, 19, 20 or
more additional biomarkers from any one of Tables 1, 4, 5, 6, 7, 8, and 9. In
some
embodiments, each of these biomarkers is from Table 5. In some embodiments,
each of
-11-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
these biomarkers is from Table 6. In some embodiments, each of these
biomarkers is from
Table 7. In some embodiments, each of these biomarkers is from any combination
of
Tables l, 4, 5, 6, 7, 8, and 9. In another embodiment, the kits of the present
invention
comprise at least 2, but as many as one hundred or more biomarkers and/or
reagents used to
detect the presence or abundance of such biomarkers.
In a specific embodiment, the kits of the present invention comprise at least
2, 3, 4,
5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 25, 30, 35, 40, 45,
50, 54, 5, 60, 65,
70, 75, 80, 85, 90, 95, 96, 100 or 200 or more reagents that specifically bind
the biomarkers
of the present invention. For example, such kits can comprise nucleic acid
molecules and/or
antibody molecules that specifically bind to biomarkers of the present
invention.
Specific exemplary biomarkers that are useful in the present invention are set
forth
in Tables 1, 4, 5, 6, 7, 8, and 9 of Section 6. The biomarkers of the kit of
the present
invention can be used to generate biomarker profiles according to the present
invention.
Examples of types of biomarkers and/or reagents within such kits include, but
are not
limited to, proteins and fragments thereof, peptides, polypeptides,
antibodies, proteoglycans,
glycoproteins, lipoproteins, carbohydrates, lipids, nucleic acids (e.g., mRNA,
DNA, cDNA,
siRNA), organic and inorganic chemicals, and natural and synthetic polymers or
a
discriminating molecule or fragment thereof.
Still another aspect of the present invention comprises computers and computer
readable media for evaluating whether a test subject is likely to develop
sepsis or SIRS. For
instance, one embodiment of the present invention provides a computer program
product for
use in conjunction with a computer system. The computer prograrn product
comprises a
computer readable storage medium and a computer program mechanism embedded
therein.
The computer program mechanism comprises instructions for evaluating whether a
plurality
of features in a biomarker profile of a test subject at risk for developing
sepsis satisfies a
first value set. Satisfaction of the first value set predicts that the test
subject is likely to
develop sepsis. In some embodiments, the features are measurable aspects of a
plurality of
biomarkers comprising at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15,
16, 17, 18, 19, or
20 biomarkers listed in any one of Tables 1, 4, 5, 6, 7, 8, and 9. When the
plurality of
biomarkers comprises complement component C3 and complement component C4, the
plurality of biomarkers comprises three or more biomarkers. In some
embodiments, the
features are measurable,aspects of a plurality of biomarkers comprising at
least 2, 3, 4, 5, 6,
7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, or 20 biomarkers listed in
any combination of
Tables 1, 4, 5, 6, 7, 8, and 9. In some embodiments, the computer program
product further
-12-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
comprises instructions for evaluating whether the plurality of features in the
biomarker
profile of the test subject satisfies a second value set. Satisfaction of the
second value set
predicts that the test subject is not likely to develop sepsis. In some
embodiments, the
biomarker profile has between 3 and 50 biomarkers listed in one of Tables 1,
4, 5, 6, 7, 8,
and 9, between 3 and 40 biomarkers listed in one of Tables 1, 4, 5, 6, 7, 8,
and 9, at least
four biomarkers listed in one of Tables 1, 4, 5, 6, 7, 8, and 9, or at least
six biomarkers listed
in one of Tables 1, 4, 5, 6, and 7. In some embodiments, the biomarker profile
has at least
1, 2, 3, 4, 5, 6, 7, 8, 9 or 10 biomarkers from columns 3 and/or 4 of Table 4.
In some
embodiments, the biomarker profile comprises CRP, APOA2, and SERPINCI. In some
embodiments, the biomarker profile comprises at least one of SERPINC 1, APOA2,
and
CRP, and, additionally, at least 1, 2, 3, 4, 5, 6 or 7 other additional
biomarkers in Table 4.
In some embodiments, the biomarker profile comprises at least one of SERPINC1,
APOA2,
and CRP, and, additionally, at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12,
13, 14, 15, 16, 17, 18,
19, 20 or more additional biomarkers from any combination of Tables 1, 4, 5,
6, 7, 8, and 9.
In some embodiments, the biomarker profile comprises at leastone of SERPINCI,
APOA2,
and CRP, and, additionally, at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12,
13, 14, 15, 16, 17, 18,
19, 20 or more additional biomarkers from any one of Tables 1, 4, 5, 6, 7, 8,
and 9.
Another computer embodiment of the present invention comprises a central
processing unit and a memory coupled to the central processing unit. The
memory stores
instructions for evaluating whether a plurality of features in a biomarker
profile of a test
subject at risk for developing sepsis satisfies a first value set.
Satisfaction of the first value
set predicts that the test subject is likely to develop sepsis. The features
are measurable
aspects of a plurality of biomarkers. In some embodiments, this plurality of
biomarkers
comprises at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18,
19, or 20
biomarkers from one of Tables 1, 4, 5, 6, 7, 8, and 9. In some embodiments,
this plurality
of biomarkers comprises at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14,
15, 16, 17, 18, 19,
or 20 biomarkers from any combination of Tables 1, 4, 5, 6, 7, 8, and 9. In
some
embodiments, the memory further stores instructions for evaluating whether the
plurality of
features in the biomarker profile of the test subject satisfies a second value
set, wherein
satisfying the second value set predicts that the test subject is not likely
to develop sepsis.
In some embodiments, the biomarker profile consists of between 3 and 50
biomarkers listed
in one of Tables 1, 4, 5, 6, and 7, between 3 and 40 biomarkers listed in one
of Tables 1, 4,
5, 6, 7, 8, and 9, at least four biomarkers listed in one of 1, 4, 5, 6, 7, 8,
and 9, or at least
eight biomarkers listed in one of Tables 1, 4, 5, 6, 7, 8, and 9. In some
embodiments, when
-13-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
the plurality of biomarkers comprises complement component C3 and complement
component C4, the plurality of biomarkers comprises three or more bioma'rkers.
Another computer embodiment in accordance with the present invention comprises
a
computer system for determining whether a subject is likely to develop sepsis.
The
computer system comprises a central processing unit and a memory, coupled to
the central
processing unit. The memory stores instructions for obtaining a biomarker
profile of a test
subject. The biomarker profile comprises a plurality of features. In some
embodiments, the
plurality of features comprises at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12,
13, 14, 15, 16, 17, 18,
19, or 20 biomarkers listed in any one of Tables 1, 4, 5, 6, 7, 8, and 9. In
some
embodiments, the plurality of features comprises at least 2, 3, 4, 5, 6, 7, 8,
9, 10, 11, 12, 13,
14, 15, 16, 17, 18, 19, or 20 biomarkers listed in any combination of Tables
1, 4, 5, 6, 7, 8,
and 9. When the plurality of features comprises features for complement
component C3
and complement component C4, the plurality of features comprises features for
three or
more biomarkers. The memory further comprises instructions for transmitting
the
biomarker profile to a remote computer. The remote computer includes
instructions for
evaluating whether the plurality of features in the biomarker profile of the
test subject
satisfies a first value set. Satisfaction of the first value set predicts that
the test subject is
likely to develop sepsis. The memory further comprises instructions for
receiving a
determination, from the remote computer, as to whether the plurality of
features in the
biomarker profile of the test subject satisfies the first value set. The
memory also comprises
instructions for reporting whether the plurality of features in the biomarker
profile of the
test subject satisfies the first value set. In some embodiments, the remote
computer further
comprises instructions for evaluating whether the plurality of features in the
biomarker
profile of the test subject satisfies a second value set. Satisfaction of the
second value set
predicts that the test subject is not likely to develop sepsis. In such
embodiments, the
memory further comprises instructions for receiving a determination, from the
remote
computer, as to whether the plurality of features in the biomarker profile of
the test subject
satisfies the second set as well as instructions for reporting whether the
plurality of features
in the biomarker profile of the test subject satisfies the second value set.
In some
embodiments, the plurality of biomarkers comprises CRP, APOA2, and SERPINCI.
In
some embodiments, the biomarker profile comprises at least one of SERPINC 1,
APOA2,
and CRP and, additionally, at least 1, 2, 3, 4, 5, 6 or.7 other additional
biomarkers in Table
4. In some embodiments, the biomarker profile comprises at least one of
SERPINC 1,
APOA2, and CRP, and, additionally, at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11,
12, 13, 14, 15,
-14-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
16, 17, 18, 19, 20 or more additional biomarkers from any combination of
Tables 1, 4, 5, 6,
7, 8, and 9. In some embodiments, the biomarker profile comprises at least one
of
SERPINC 1, APOA2, and CRP, and, additionally, at least 1, 2, 3, 4, 5, 6, 7, 8,
9, 10, 11, 12,
13, 14, 15, 16, 17, 18, 19, 20 or more additional biomarkers from any one of
Tables 1, 4, 5,
6, 7, 8, and 9. In some embodiments, the plurality of biomarkers comprises at
least two
biomarkers from Table 4. In some embodimepts, when the plurality of biomarkers
comprises complement component C3 and complement component C4, the plurality
of
biomarkers comprises three or more biomarkers.
Still another embodiment of the present invention comprises a digital signal
embodied on a carrier wave comprising a respective value for each of a
plurality of features
in a biomarker profile. The features are measurable aspects of a plurality of
biomarkers. In
some embodiments, the plurality of biomarkers comprises at least 2, 3, 4, 5,
6, 7, 8, 9, 10,
11, 12, 13, 14, 15, 16, 17, 18, 19, or 20 biomarkers listed in any one of
Tables 1, 4, 5, 6, 7,
8, and 9. In some embodiments, the plurality of biomarkers comprises at least
2, 3, 4, 5, 6,
7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, or 20 biomarkers listed in
any combination of
Tables 1, 4, 5, 6, 7, 8, and 9. In some embodiments, the plurality of
biomarkers comprises
at least 2, 3, 4, 5, 6, 7, 8, 9 or 10 biomarkers from Table 4. In some
embodiments, the
plurality of biomarkers comprises CRP, APOA2, and SERPINCI. In some
embodiments,
the plurality of biomarkers comprises at least one of SERPINCi, APOA2, and
CRP, and,
additionally, at least 1, 2, 3, 4, 5, 6 or 7 other additional biomarkers in
Table 4. In some
embodiments, the plurality of biomarkers comprises at least one of SERPINC 1,
APOA2,
and CRP, and, additionally, at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12,
13, 14, 15, 16, 17, 18,
19, 20 or more additional biomarkers from any combination of Tables 1, 4, 5,
6, 7, 8, and 9.
In some embodiments, when the plurality of biomarkers comprises complement
component
C3 and complement component C4, the plurality of biomarkers comprises three or
more
biomarkers.
Still another aspect of the present invention provides a digital signal
embodied on a
carrier wave comprising a determination as to whether a plurality of features
in a biomarker
profile of a test subject satisfies a value set. The features are measurable
aspects of a
plurality of biomarkers. In some embodiments, this plurality of biomarkers
comprises at
least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, or 20
biomarkers listed in
any one of Tables 1, 4, 5, 6, 7, 8, and 9. In some embodiments, this plurality
of biomarkers
comprises at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18,
19, or 20
biomarkers listed in any combination of Tables 1, 4, 5, 6, 7, 8, and 9.-
Satisfying the value
-15-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
set predicts that the test subject is likely to develop sepsis. In some
embodiments, the
plurality of biomarkers comprises CRP, APOA2, and SERPCI. In some embodiments,
the
plurality of biomarkers comprises at least one of SERPINCI, APOA2, and CRP,
and,
additionally, at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16,
17, 18, 19, 20 or more
additional biomarkers from any combination of Tables 1, 4, 5, 6, 7, 8, and 9.
In some
embodiments, the plurality of biomarkers comprises at least one of SERPINCI,
APOA2,
and CRP, and, additionally, at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12,
13, 14, 15, 16, 17, 18,
19, 20 or more additional biomarkers from any one of Tables 1, 4, 5, 6, 7, 8,
and 9. In some
embodiments, the plurality of biomarkers comprises at least 2, 3, 4, 5, 6, 7,
8, 9, or 10
biomarkers listed in column 3 or 4 of Table 4. In some embodiments, when the
plurality of
biomarkers comprises complement component C3 and complement component C4, the
plurality of biomarkers comprises three or more biomarkers.
Still another embodiment provides a digital signal embodied on a carrier wave
comprising a determination as to whether a plurality of features in a
biomarker profile of a
test subject satisfies a value set. The features are measurable aspects of a
plurality of
biomarkers. In some embodiments, the plurality of biomarkers comprises at
least 2, 3, 4, 5,
6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 or 20 biomarkers listed in
any one of Tables
1, 4, 5, 6, 7, 8, and 9. The plurality of biomarkers comprises at least 2, 3,
4, 5, 6, 7, 8, 9, 10,
11, 12, 13, 14, 15, 16, 17, 18, 19 or 20 biomarkers listed in any combination
of Tables 1, 4,
5, 6, 7, 8, and 9. Satisfaction of the value set predicts that the test
subject is not likely to
develop sepsis. In some embodiments, the plurality of biomarkers comprises
CRP, APOA2,
or SERPINCI. In some embodiments, the plurality of biomarkers comprises at
least one of
SERPINCI, APOA2, and CRP and, additionally, at least 1, 2, 3, 4, 5, 6 or 7
other additional
biomarkers in Table 4. In some embodiments, the plurality of biomarkers
comprises at least
one of SER.PINC1, APOA2, and CRP and, additionally, at least 1, 2, 3, 4, 5, 6,
7, 8, 9, 10,
11, 12, 13, 14, 15, 16, 17, 18, 19, 20 or more additional biomarkers from any
combination
of Tables 1, 4, 5, 6, 7, 8, and 9. In some embodiments, the plurality of
biomarkers
comprises at least one of SERPINCI, APOA2, and CRP and, additionally, at least
1, 2, 3, 4,
5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 or more additional
biomarkers from
any one of Tables 1, 4, 5, 6, 7, 8, and 9. In some embodiments, when the
plurality of
biomarkers comprises complement component C3 and complement component C4, the
plurality of biomarkers comprises three or more biomarkers.
Still another embodiment of the present invention provides a graphical user
interface
for determining whether a subject is likely to develop sepsis. The graphical
user interface
-16
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
comprises a display field for a displaying a result encoded in a digital
signal embodied.on a
carrier wave received from a remote computer. The features are measurable
aspects of a
plurality of biomarkers. In some embodiments, the plurality of biomarkers
comprises at
least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 or 20
biomarkers listed in any
one of Tables 1, 4, 5, 6, 7, 8, and 9. In some embodiments, the plurality of
biomarkers
comprises at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18,
19 or 20 biomarkers
listed in any combination of Tables 1, 4, 5, 6, 7, 8, and 9. The result has a
first value when a
plurality of features in a biomarker profile of a test subject satisfies a
first value set. The
result has a second value when a plurality of features in a biomarker profile
of a test subject
satisfies a second value set. In some embodiments, the plurality of biomarkers
comprises
CRP, APOA2, or SERPINC I. In some embodiments, the plurality of biomarkers
comprises
at least 2, 3, 4, 5, 6, 7, 8, 9 or 10 biomarkers from Table 4. In some
embodiments, the
plurality of biomarkers comprises at least one of SERPINCI, APOA2, and CRP,
and,
additionally, at least 1, 2, 3, 4, 5, 6 or 7 other additional biomarkers in
Table 4. In some
embodiments, the plurality of biomarkers comprises at least one of SERPINC 1,
APOA2,
and CRP, and, additionally, at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12,
13, 14, 15, 16, 17, 18,
19, 20 or more additional biomarkers from any combination of Tables 1, 4, 5,
6, 7, 8, and 9.
In some embodiments, the plurality of biomarkers comprises at least one of
SERPINCI,
APOA2, and CRP, and, additionally, at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11,
12, 13, 14, 15,
16, 17, 18, 19, 20 or more additional biomarkers from any one of Tables 1, 4,
5, 6, 7, 8, and
9. In some embodiments, when the plurality of biomarkers comprises complement
component C3 and complement component C4, the plurality of biomarkers
comprises three
or more biomarkers.
Yet another aspect of the present invention provides a computer system for
determining whether a subject is likely to develop sepsis. The computer system
comprises a
central processing unit and a memory, coupled to the central processing unit.
The memory
stores instructions for obtaining a biomarker profile of a test subject. The
biomarker profile
comprises a plurality of features. The features are measurable aspects of a
plurality of
biomarkers. In some embodiments, the plurality of biomarkers comprise at least
2, 3, 4, 5,
6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 or 20 biomarkers listed in
any one of Tables
1, 4, 5, 6, 7, 8, and 9. In some embodiments, the plurality of biomarkers
comprise at least 2,
3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 or 20 biomarkers
listed in any
combination of Tables 1, 4, 5, 6, 7, 8, and 9. The memory further stores
instructions for
evaluating whether the plurality of features in the biomarker profile of the
test subject
-17-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
satisfies a first value set. Satisfying the first value set predicts that the
test subject is likely
to develop sepsis. The memory also stores instructions for reporting whether
the plurality
of features in the biomarker profile of the test subject satisfies the first
value set. In some
embodiments, the plurality of biomarkers comprises CRP, APOA2, and SERPINCI.
In
some embodiments, the plurality of biomarkers comprises at least one of
SERPINC 1,
APOA2, and CRP, and, additionally, at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11,
12, 13, 14, 15,
16, 17, 18, 19, 20 or more additional biomarkers from any combination of
Tables 1, 4, 5, 6,
7, 8, and 9. In some embodiments, the plurality of biomarkers comprises at
least one of
SERPINC1, APOA2, and CRP, and, additionally, at least 1, 2, 3, 4, 5, 6, 7, 8,
9, 10, 11, 12,
13, 14, 15, 16, 17, 18, 19, 20 or more additional biomarkers from any one of
Tables 1, 4, 5,
6, 7, 8, and 9. In some embodiments, the plurality of biomarkers comprises at
least 2, 3, 4,
5, 6, 7, 8, 9 or 10 biomarkers from Table 4. In some embodiments, when the
plurality of
biomarkers comprises complement component C3 and complement component C4, the
plurality of biomarkers comprises three or more biomarkers.
Each of the methods, computer program products, and computers disclosed herein
optionally further comprise a step of, or instructions for, outputting a
result- (for example, to
a monitor, to a user, to computer readable media, e.g., storage media or to a
remote
computer).
4. BRIEF DESCRIPTION OF THE FIGURES
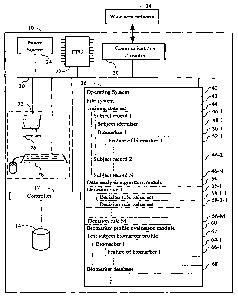
FIG. 1 shows a computer system in accordance with the present invention.
FIG. 2 illustrates the involvement of classical and alternative complement
cascades
in differentiating sepsis from SIRS patients in terms of proteins identified
in the present
invention.
FIG. 3 illustrates the involvement of Intrinsic Prothrombin Activation pathway
in
differentiating Sepsis from SIRS patients using proteins identified in the
present invention.
-18-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
5. DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENTS
The present invention allows for the rapid and accurate diagnosis or
prediction of
sepsis by evaluating biomarker features in biomarker profiles. These biomarker
profiles can
be constructed from one or more biological samples of subjects at a single
time point
("snapshot"), or multiple such time points, during the course of time the
subject is at risk for
developing sepsis. Advantageously, sepsis can be diagnosed or predicted prior
to the onset
of conventional clinical sepsis symptoms, thereby allowing for more effective
therapeutic
intervention.
5.1 DEFINITIONS
"Systemic inflammatory response syndrome," or "SIRS," refers to a clinical
response that is triggered by infectious or noninfectious conditions such as
localized or
generalized infection, trauma, thermal injury, or sterile inflammatory
processes (e.g., acute
pancreatitis). SIRS is considered to be present when a subject is (i)
undergoing a response
to any of the foregoing infectious or noninfectious conditions, and (ii) is
exhibiting some of
the following clinical findings:
fever (body temperature greater than 38.3 C);
hypothermia (body temperature less than 36 C);
heart rate (HR) greater than 90 beats/minute or >2 standard deviations above
the
normal value for age;
tachypnea;
altered mental status;
significant edema or positive fluid balance (> 20 mL/kg over 24 hours);
hyperglycemia (plasma glucose > 120 mg/dL or 7.7 mmol/L) in the absence of
diabetes;
leukocytosis (white cell blood count > 12,000 L"');
leucopenia (white cell blood count < 4,000 L"');
normal white cell blood count > 10% immature forms;
plasma C-reactive protein > 2 standard deviations above the normal value;
plasma procalcitonin > 2 standard deviations above the normal value;
arterial hypotension (SBP < 90 mm Hg, MAP < 80, or an SBP decrease > 40 mm Hg
in adults or < two standard deviations below normal for age);
cardiac index > 3.5 L= min' = M723;
-19-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
arterial hypoxemia (PaO2/FIOZ < 300);
acute oliguria (urine output < 0.5 mL * kg 'o hr"' or 45 mmol/L for at least
two
hours);
creatinine increase > 0.5 mg/dL;
coagulation abnormalities (INR > 1.5 or aPTT > 60 seconds);
Ileus (absent bowel sounds);
Thrombocytopenia (platelet count < 100,000 L"1);
Hyperbilirubinemia (plasma total bilirubin > 4 mg/dL or 70 mmol/L);
Hyperlactatemia (> I mmol/L); and
Decreased capillary refill or mottling.
These symptoms of SIRS represent a consensus definition of SIRS that can be
modified or supplanted by other definitions in the future. The present
definition is used to
clarify current clinical practice and does not represent a critical aspect of
the invention. For
more information on standards used to define SIRs see, for example, American
College of
Chest Physicians/Society of Critical Care Medicine Consensus Conference:
Definitions for
Sepsis and Organ Failure and Guidelines for the Use of Innovative Therapies in
Sepsis,
1992, Crit. Care. llled. 20, 864-874; Levy et al., 2003, "2001
SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference," Crit.
Care
Med. 31, 1250-1256; and Carrigan et al., 2004, "Toward Resolving the
Challenges of
Sepsis Diagnosis," 1301-1314, each of which is incorporated by reference
herein in its
entirety.
A subject with SIRS has a clinical presentation that is classified as SIRS, as
defined
above, but is not clinically deemed to be septic. Methods for determining
which subjects
are at risk of developing sepsis are well known to those in the art. Such
subjects include,
for example, those in an ICU and those who have otherwise suffered from a
physiological
trauma, such as a burn, surgery or other insult. A hallmark of SIRS is the
creation of a
proinflammatory state that can be marked by tachycardia, tachypnea or
hyperpnea,
hypotension, hypoperfusion, oliguria, leukocytosis or leukopenia, pyrexia or
hypothermia
and the need for volume infusion. SIRS characteristically does not include a
documented
source of infection (e.g., bacteremia).
"Sepsis" refers to a state in which a subject has both (i) SIRS and (ii) a
documented
or suspected infection (e.g., a subsequent laboratory confirmation of a
clinically significant
infection such as a positive culture for an organism). Thus, sepsis refers to
the systemic
inflammatory response to an infection (see, e.g., American College of Chest
Physicians
-20-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Society of Critical Care Medicine, Chest, 1997, 101:1644-1655, the entire
contents of which
are herein incorporated by reference). As used here, the term "infection"
means a
pathological process induced by a microorganism. Such an infection can be
caused by
pathogenic gram-negative and gram-positive bacteria, anaerobic bacteria,
fungi, yeast, or
polymicrobial organisms. Examplary non-limiting sites of such infections are
respiratory
tract infactions, genitourinary infections, and intraabdoiminal infections. As
used herein,
"sepsis" includes all stages of sepsis including, but not limited to, the
onset of sepsis, severe
sepsis, septic shock and multiple organ dysfunction ("MOD") associated with
the end stages
of sepsis.
The "onset of sepsis" refers to an early stage of sepsis, e.g., prior to a
stage when
conventional clinical manifestations are sufficient to support a clinical
suspicion of sepsis.
Because the methods of the present invention are used to detect sepsis prior
to a time that
sepsis would be suspected using conventional techniques, the subject's disease
status at
early sepsis can only be confirmed retrospectively, when the manifestation of
sepsis is more
clinically obvious. The exact mechanism by which a subject becomes septic is
not a critical
aspect of the invention. The methods of the present invention can detect the
onset of sepsis
independent of the origin of the infectious process.
"Severe sepsis" refers to sepsis associated with organ dysfunction,
hypoperfusion
abnormalities, or sepsis-induced hypotension. Hypoperfusion abnormalities
include, but are
not limited to, lactic acidosis, oliguria, or an acute alteration in mental
status.
"Septic shock" in adults refers to a state of acute circulatory failure
characterized by
persistent arterial hypotension unexplained by other causes. Hypotension is
defined by a
systolic arterial pressure below 90 mm Hg (or, in children, <2sD below normal
for their
age), a MAP < 60, or a reduction in systolic blood pressure of > 40 mm Hg from
baseline,
despite adequate volume resuscitation, in the absence of other causes for
hypotension.
Children and neontates maintain higher vascular tone than adults. Therefore,
the shock state
occurs long before hypertension in children. Septic shock in pediatric
patients is defined as
a tachychardia (may be absent in the hypothermic patient) with sings of
decreased perfusion
including decreased peripheral pulses compared with central pulses, altered
alertness, flash
capillary refill or capillary refill > 2 seconds, mottled or cool extremities,
or decreased urine
output. Hypotension is a sign of late and decompensated shock in children.
See, for
example, Levy et al., 2003, "2001 SCCM/ESICM/ ACCP/ATS/SIS International
Sepsis
Definitions Conference," Crit. Care Med. 31, 1250-1256; and Carrigan et al.,
2004,
"Toward Resolving the Challenges of Sepsis Diagnosis," 1301-1314; and
Carcillo, 2002,
-21-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
"Clinical Practice Paramaters for Hemodynamic Support of Pediatric and
Neonatal Patients
in Septic Shock," Crit. Care Med. 30, pp. 1-13, each of which is hereby
incorporated by
reference in its entirety.
A "converter" or "converter subject" refers to a SIRS-positive subject who
progresses to clinical suspicion of sepsis during the period the subject is
monitored,
typically during an ICU stay.
A "non-converter" or "non-converter subject" refers to a SIRS-positive subject
who
does not progress to clinical suspicion of sepsis during the period the
subject is monitored,
typically during an ICU stay.
A "biomarker" is virtually any detectable compound, such as a protein, a
peptide, a
proteoglycan, a glycoprotein, a lipoprotein, a carbohydrate, a lipid, a
nucleic acid (e.g.,
DNA, such as cDNA or amplified DNA, or RNA, such as mRNA), an organic or
inorganic
chemical, a natural or synthetic polymer, a small molecule (e.g., a
metabolite), or a
discriminating molecule or discriminating fragment of any of the foregoing,
that is present
in or derived from a biological sample. "Derived from" as used in this context
refers to a
compound that, when detected, is indicative of a particular molecule being
present in the
biological sample. For example, detection of a particular cDNA can be
indicative of the
presence of a particular RNA transcript in the biological sample. As another
example,
detection of binding to a particular antibody can be indicative of the
presence or absence of
a particular antigen (e.g., protein) in the biological sample. Here, a
discriminating molecule
or fragment is a molecule or fragment that, when detected, indicates presence
or abundance
of an above-identified compound.
A biomarker can, for example, be isolated from the biological sample, directly
measured in the biological sample, or detected in or determined to be in the
biological
sample. A biomarker can, for example, be functional, partially functional, or
non-
functional. In one embodiment of the present invention, a biomarker is
isolated and used,
for example, to raise a specifically-binding antibody that can facilitate
biomarker detection
in a variety of diagnostic assays. Any immunoassay may use any antibodies,
antibody
fragment or derivative thereof capable of binding the biomarker molecules
(e.g., Fab,
F(ab')2, Fv, or scFv fragments). Such immunoassays are well-known in the art.
In addition,
if the biomarker is a protein or fragment thereof, it can be sequenced and its
encoding gene
can be cloned using well-established techniques. When a specific biomarker is
listed
herein, for example, as part of a biomarker profile, kit, or otherwise, the
biomarker can be,
for example, the precursor of the listed biomarker, the fully processed
version of the listed
-22-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
biomarker, a splice variant of the biomarker, a fragment thereof, an antibody
thereof, or a
discriminating molecule thereof. For instance, reference to CRP herein is, for
example, a
reference to C-reactive protein, C-reactive protein precursor, a fragment
thereof, an
antibody thereof, a nucleic acid encoding all or a fragment thereof, a
discriminating
molecule thereof, or any other type of biomarker for CRP. Reference to APOA2
herein is,
for example, is a reference to apolipoprotein A-II, apolipoprotein A-II
precursor, a fragment
thereof, an antibody thereof, a nucleic acid encoding all or a fragment
thereof, a
discriminating molecule thereof, or any other type of biomarker for APOA2.
Reference to
SERPINCI herein is, for example, is a reference to serine (or cysteine)
proteinase inhibitor
(or any of its synonyms including, but not limited to, clade C, antithrombin
member 1,
antithrombin-III precursor, ATIII, etc.), a fragment thereof, an antibody
thereof, a nucleic
acid encoding all or a fragment thereof, a discriminating molecule thereof, or
any other type
of biomarker for SERPINC.
As used herein, the term "a species of a biomarker" refers to any
discriminating
portion or discriminating fragment of a biomarker described herein, such as a
splice variant
of a particular gene described herein (e.g., a gene listed in Table 1, 4, 5,
6, 7, 8 and/or 9,
below). Here, a discriminating portion or discriminating fragment is a portion
or fragment
of a molecule that, when detected, indicates presence or abundance of the
above-identified
transcript, cDNA, amplified nucleic acid, or protein.
As used herein, the terms "protein", "peptide", and "polypeptide" are, unless
otherwise indicated, interchangeable.
A' biomarker profile" comprises a plurality of one or more types of biomarkers
(e.g., an mRNA molecule, a cDNA molecule, a protein and/or a carbohydrate,
etc.), or an
indication thereof, together with a feature, such as a measurable aspect
(e.g., abundance) of
the biomarkers. A biomarker profile comprises at least two such biomarkers or
indications
thereof, where the biomarkers can be in the same or different classes, such
as, for example,
a nucleic acid and a carbohydrate. A biomarker profile may also comprise at
least 3, 4, 5, 6,
7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 30,
35, 40, 45, 50, 54, 5,
60, 65, 70, 75, 80, 85, 90, 95, or 100 or more biomarkers or indications
thereof. In one
embodiment, a biomarker profile comprises hundreds, or even thousands, of
biomarkers or
indications thereof. A biomarker profile can further comprise one or more
controls or
internal standards. In one embodiment, the biomarker profile comprises at
least one
biomarker, or indication thereof, that serves as an internal standard. In
another
embodiment, a biomarker profile comprises an indication of one or more types
of
-23-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
biomarkers. The term "indication" as used herein in this context merely refers
to a situation
where the biomarker profile contains symbols, data, abbreviations or other
similar indicia
for a biomarker, rather than the biomarker molecular entity itself. In some
embodiments,
the biomarker profile comprises a nominal indication of the quantity of a
transcript of a
gene from one any one of Tables 1, 4, 5, 6, 7, 8, and 9. Still another
exemplary biomarker
profile of the present invention comprises a microarray to which a physical
quantity of a
gene transcript from one of Tables 1, 4, 5, 6, 7, 8, and 9 is taken. In this
last exemplary
biomarker profile, at least twenty percent, forty percent, or more than forty
percent of the
probes spots are based on sequences in one of Tables 1, 4, 5, 6, 7, 8, and 9.
In another
exemplary biomarker profile, at least twenty percent, forty percent, or more
than forty
percent of the probes spots are based on sequences in probesets for biomarkers
listed in any
one of Tables 1, 4, 5, 6, 7, 8, and 9. In some embodiments, when the
biormarker profile
comprises the biomarkers component C3 and complement component C4, the
biomarker
profile comprises three or more biomarkers.
In typical embodiments, each biomarker in a biomarker profile includes a
corresponding "feature." A "feature", as used herein, refers to a measurable
aspect of a
biomarker. A feature can include, for example, the presence or absence of
biomarkers in
the biological sample from the subject as illustrated in exemplary biomarker
profile 1:
Exemplary biomarker profile 1.
Biomarker Feature
Presence in sample
transcript of gene A Present
transcript of gene B Absent
In exemplary biomarker profile 1, the feature value for the transcript of gene
A is
"presence" and the feature value for the transcript of gene B is "absence."
A feature can include, for example, the abundance of a biomarker in the
biological
sample from a subject as illustrated in exemplary biomarker profile 2:
Exemplary biomarker profile 2.
Biomarker Feature
Abundance in sample in
-24-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
relative units
transcript of gene A 300
transcript of gene B 400
In exemplary biomarker profile 2, the feature value for the transcript of gene
A is
300 units and the feature value for the transcript of gene B is 400 units.
A feature can also be a ratio of two or more measurable aspects of a biomarker
as
illustrated in exemplary biomarker profile 3:
Exemplary biomarker profile 3.
Biomarker Feature
Ratio of abundance of
transcript of gene A/ transcript of
gene Y
transcript of gene A
transcript of gene B 300/400
In exemplary biomarker profile 3, the feature value for the transcript of gene
A and
the feature value for the transcript of gene B is 0.75 (300/400).
A feature may also be the difference between a measurable aspect of the
corresponding biomarker that is taken from two samples, where the two samples
are
collected from a subject at two different time points. For example, consider
the case where
the biomarker is a transcript of a gene A and the "measurable aspect" is
abundance of the
transcript, in samples obtained from a test subject as determined by, e.g., RT-
PCR or
microarray analysis. In this example, the abundance of the transcript of gene
A is measured
in a first sample as well as a second sample. The first sample is taken from
the test subject a
number of hours before the second sample. To compute the feature for gene A,
the
abundance of the transcript of gene A in one sample is subtracted from the
abundance of the
transcript of gene A in the second sample. A feature can also be an indication
as to whether
an abundance of a biomarker is increasing in biological samples obtained from
a subject
over time and/or an indication as to whether an abundance of a biomarker is
decreasing in
biological samples obtained from a subject over time.
In some embodiments, there is a one-to-one correspondence between features and
biomarkers in a biomarker profile as illustrated in exemplary biomarker
profile 1, above.
-25-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
In some embodiments, the relationship between features and biomarkers in a
biomarker
profile of the present invention is more complex, as illustrated in Exemplary
biomarker
profile 3, above.
Those of skill in the art will appreciate that other methods of computation of
a
feature can be devised and all such methods are within the scope of the
present invention.
For example, a feature can represent the average of an abundance of a
biomarker across
biological samples collected from a subject at two or more time points.
Furthermore, a
feature can be the difference or ratio of the abundance of two or more
biomarkers from a
biological sample obtained from a subject in a single time point. A biomarker
profile may
also comprise at least three, four, five, 10, 20, 30 or more features. In one
embodiment, a
biomarker profile comprises hundreds, or even thousands, of features.
In some embodiments, features of biomarkers are measured using microarrays.
The
construction of microarrays and the techniques used to process microarrays in
order to
obtain abundance data is well known, and is described, for example, by
Draghici, 2003,
Data Analysis Tools for DNA Microarrays, Chapman & Hall/CRC, and international
publication number WO 03/061564, each of which is hereby incorporated by
reference in its
entirety. A microarray comprises a plurality of probes. In some instances,
each probe
recognizes, e.g., binds to, a different biomarker. In some instances, two or
more different
probes on a microarray recognize, e.g., bind to, the same biomarker. Thus,
typically, the
relationship between probe spots on the microarray and a subject biomarker is
a two to one
correspondence, a three to one correspondence, or some other form of
correspondence.
However, it can be the case that there is a unique one-to-one correspondence
between
probes on a microarray and biomarkers.
A "phenotypic change" is a detectable change in a parameter associated with a
given
state of the subject. For instance, a phenotypic change can include an
increase or decrease
of a biomarker in a bodily fluid, where the change is associated with SIRS,
sepsis, the onset
of sepsis or with a particular stage in the progression of sepsis. A
phenotypic change can
further include a change in a detectable aspect of a given state of the
subject that is not a
change in a measurable aspect of a biomarker. For example, a change in
phenotype can
include a detectable change in body temperature, respiration rate, pulse,
blood pressure, or
other physiological parameter. Such changes can be determined via clinical
observation and
measurement using conventional techniques that are well-known to the skilled
artisan.
As used herein, the term "complementary," in the context of a nucleic acid
sequence
(e.g., a nucleotide sequence encoding a gene described herein), refers to the
chemical
-26-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
affinity between specific nitrogenous bases as a result of their hydrogen
bonding properties.
For example, guanine (G) forms a hydrogen bond with only cytosine (C), while
adenine
forms a hydrogen bond only with thynzine (T) in the case of DNA, and uracil
(U) in the case
of RNA. These reactions are described as base pairing, and the paired bases (G
with C, or
A with T/U) are said to be complementary. Thus, two nucleic acid sequences may
be
complementary if their nitrogenous bases are able to form hydrogen bonds. Such
sequences
are referred to as "complements" of each other. Such complement sequences can
be
naturally occurring, or, they can be chemically synthesized by any method
known to those
skilled in the art, as for example, in the case of antisense nucleic acid
molecules which are
complementary to the sense strand of a DNA molecule or an RNA molecule (e.g.,
an
mRNA transcript). See, e.g., Lewin, 2002, Genes VII. Oxford University Press
Inc., New
York, New York, which is hereby incorporated by reference herein in its
entirety.
As used herein, "conventional techniques" in the context of diagnosing or
predicting
sepsis or SIRS are those techniques that classify a subject based on
phenotypic changes
without obtaining a biomarker profile according to the present invention.
A "decision rule" is a method used to evaluate biomarker profiles. Such
decision
rules can take on one or more forms that are known in the art, as exemplified
in Hastie et
al., 2001, The Elements ofStatistical Learning, Springer-Verlag, New York,
which is
hereby incorporated by reference in its entirety. A decision rule may be used
to act on a
data set of features to, inter alia, predict the onset of sepsis, to determine
the progression of
sepsis, or to diagnose sepsis. Exemplary decision rules that can be used in
some
embodiments of the present invention are described in fixrther detail in
Section 5.5, below.
"Predicting the development of sepsis" is the determination as to whether a
subject
will develop sepsis. Any such prediction is limited by the accuracy of the
means used to
make this determination. The present invention provides a method, e.g., by
utilizing a
decision rule(s), for making this determination with an accuracy that is 60%
or greater. As
used herein, the terms "predicting the development of sepsis" and "predicting
sepsis" are
interchangeable. In some embodiments, the act of predicting the development of
sepsis
(predicting sepsis) is accomplished by evaluating one or more biomarker
profiles from a
subject using a decision rule that is indicative of the development of sepsis
and, as a result
of this evaluation, receiving a result from the decision rule that indicates
that the subject
will become septic. Such an evaluation of one or more biomarker profiles from
a test
subject using a decision rule uses some or all the features in the one or more
biomarker
profiles to obtain such a result.
-27-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
The terms "obtain" and "obtaining," as used herein, mean "to come into
possession
of," or "coming into possession of," respectively. This can be done, for
example, by
retrieving data from a data store in a computer system. This can also be done,
for example,
by direct measurement.
As used herein, the term "specifically," and analogous terms, in the context
of an
antibody, refers to peptides, polypeptides, and antibodies or fragments
thereof that
specifically bind to an antigen or a fragment and do not specifically bind to
other antigens
or other fragments. A peptide or polypeptide that specifically binds to an
antigen may bind
to other peptides or polypeptides with lower affinity, as determined by
standard
experimental techniques, for example, by any immunoassay well-known to those
skilled in
the art. Such immunoassays include, but are not limited to, radioimmunoassays
(RIAs) and
enzyme-linked immunosorbent assays (ELISAs). Antibodies or fragments that
specifically
bind to an antigen may be cross-reactive with related antigens. Preferably,
antibodies or
fragments thereof that specifically bind to an antigen do not cross-react with
other antigens.
See, e.g., Paul, ed., 2003, Fundamental Immunology, 5th ed., Raven Press, New
York at
pages 69-105, which is incorporated by reference herein, for a discussion
regarding antigen-
antibody interactions, specificity and cross-reactivity, and methods for
determining all of
the above.
As used herein, a"subject" is an animal, preferably a mammal, more preferably
a
non-human primate, and most preferably a human. The terms "subject"
"individual" and
"patient" are used interchangeably herein.
As used herein, a "test subject," typically, is any subject that is not in a
training
population used to construct a decision rule. A test subject can optionally be
suspected of
either having sepsis at risk of developing sepsis.
As used herein, a "tissue type," is a type of tissue. A tissue is an
association of cells
of a multicellular organism, with a common embryoloical origin or pathway and
similar
structure and function. Often, cells of a tissue are contiguous at cell
membranes but
occasionally the tissue may be fluid (e.g., blood). Cells of a tissue may be
all of one type (a
simple tissue, e.g., squamous epithelium, plant parentchyma) or of more than
one type (a
mixed tissue, e.g., connective tissue).
As used herein, a "training population" is a set of samples from a population
of
subjects used to construct a decision rule, using a data analysis algorithm,
for evaluation of
the biomarker profiles of subjects at risk for developing sepsis. In a
preferred embodiment,
-28-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
a training population includes samples from subjects that are converters and
subjects that
are nonconverters.
As used herein, a "data analysis algorithm" is an algorithm used to construct
a
decision rule using biomarker profiles of subjects in a training population.
Representative
data analysis algorithms are described in Section 5.5. A' decision rule" is
the final product
of a data analysis algorithm, and is characterized by one or more value sets,
where each of
these value sets is indicative of an aspect of SIRS, the onset of sepsis,
sepsis, or a prediction
that a subject will acquire sepsis. In one specific example, a value set
represents a
prediction that a subject will develop sepsis. In another example, a value set
represents a
prediction that a subject will not develop sepsis.
As used herein, a "validation population" is a set of samples from a
population of
subjects used to determine the accuracy of a decision rule. In a preferred
embodiment, a
validation population includes samples from subjects that are converters and
subjects that
are nonconverters. In a preferred embodiment, a validation population does not
include
subjects that are part of the training population used to train the decision
rule for which an
accuracy measurement is sought.
As used herein, a "value set" is a combination of values, or ranges of values
for
features in a biomarker profile. The nature of this value set and the values
therein is
dependent upon the type of features present in the biomarker profile and the
data analysis
algorithm used to construct the decision rule that dictates the value set. To
illustrate,
reconsider exemplary biomarker profile 2:
Exemplary biomarker profile 2.
Biomarker Feature
Abundance in sample in
relative units
transcript of gene A 300
transcript of gene B 400
In this example, the biomarker profile of each member of a training population
is
obtained. Each such biomarker profile includes a measured feature, here
abundance, for the
transcript of gene A, and a measured feature, here abundance, for the
transcript of gene B.
These feature values, here abundance values, are used by a data analysis
algorithm to
construct a decision rule. In this example, the data analysis algorithm is a
decision tree,
-29-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
described in Section 5.5.1 and the final product of this data analysis
algorithm, the decision
rule, is a decision tree. An exemplary decision tree is illustrated in Figure.
1. The decision
rule defines value sets. One such value set is predictive of the onset of
sepsis. A subject
whose biomarker feature values satisfy this value set is likely to become
septic. An
exemplary value set of this class is exemplary value set 1:
Exemplary value set 1.
Biomarker Value set component
(Abundance in sample in
relative units)
transcript of gene A < 400
transcript of gene B < 600
Another such value set is predictive of a septic-free state. A subject whose
biomarker feature values satisfy this value set is not likely to become
septic. An exemplary
value set of this class is exemplary value set 2:
Exemplary value set 2.
Biomarker Value set component
(Abundance in sample in
relative units)
transcript of gene A > 400
transcript of gene B > 600
In the case where the data analysis algorithm is a neural network analysis and
the
final product of this neural network analysis is an appropriately weighted
neural network,
one value set is those ranges of biomarker profile feature values that will
cause the weighted
neural network to indicate that onset of sepsis is likely. Another value set
is those ranges of
biomarker profile feature values that will cause the weighted neural network
to indicate that
onset of sepsis is not likely.
As used herein, the term "probe spot" in the context of a microarray refers to
a single
stranded DNA molecule (e.g., a single stranded cDNA molecule or synthetic DNA
oligomer), referred to herein as a "probe," that is used to determine the
abundance of a
particular nucleic acid in a sample. For example, a probe spot can be used to
determine the
-30-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
level of mRNA in a biological sample (e.g., a collection of cells) from a test
subject. In a
specific embodiment, a typical microarray comprises multiple probe spots that
are placed
onto a glass slide (or other substrate) in known locations on a grid. The
nucleic acid for
each probe spot is a single stranded contiguous portion of the sequence of a
gene or gene of
interest (e.g., a 10-mer, 11-mer, 12-mer, 13-mer, 14-mer, 15-mer, 16-mer, 17-
mer, 18-mer,
19-mer, 20-mer, 21-mer, 22-mer, 23-mer, 24-mer, 25-mer or larger) and is a
probe for the
mRNA encoded by the particular gene or gene of interest. Each probe spot is
characterized
by a single nucleic acid sequence, and is hybridized under conditions that
cause it to
hybridize only to its complementary DNA strand or mRNA molecule. As such,
there can be
many probe spots on a substrate, and each can represent a unique gene or
sequence of
interest. In addition, two or more probe spots can represent the same gene
sequence. In
some embodiments, a labeled nucleic sample is hybridized to a probe spot, and
the amount
of labeled nucleic acid specifically hybridized to a probe spot can be
quantified to determine
the levels of that specific nucleic acid (e.g., mRNA transcript of a
particular gene) in a
particular biological sample. Probes, probe spots, and microarrays, generally,
are described
in Draghici, 2003, Data Analysis Tools for DNA Microarrays, Chapman &
Hall/CRC,
Chapter, 2, which is hereby incorporated by reference in its entirety.
As used herein, the term "annotation data" refers to any type of data that
describes a
property of a biomarker. Annotation data includes, but is not limited to,
biological pathway
membership, enzymatic class (e.g., phosphodiesterase, kinase,
metalloproteinase, etc.),
protein domain information, enzymatic substrate information, enzymatic
reaction
information, protein interaction data, disease association, cellular
localization, tissue type
localization, and cell type localization.
As used herein, the term "T.12' refers to the last time blood was obtained
from a
subject before the subject is clinically diagnosed with sepsis. Since, in some
embodiments
of the present invention, blood is collected from subjects each 24 hour
period, T.12
references the average time period prior to the onset of sepsis for a pool of
patients, with
some patients turning septic prior to the twelve hour mark and some patients
turning septic
after the twelve hour mark. However, across a pool of subjects, the average
time period for
this last blood sample is the twelve hour mark, hence the name "T.12."
5.2 METHODS FOR SCREENING SUBJECTS
The present invention allows for accurate, rapid prediction and/or diagnosis
of sepsis
through detection of 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17,
18, 19, 20 or more
-31-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
features of a biomarker profile of a test individual suspected of or at risk
for developing
sepsis in each of one or more biological samples from a test subject. In one
embodiment,
only a single biological sample taken at a single point in time from the test
subject is needed
to construct a biomarker profile that is used to make this prediction or
diagnosis of sepsis.
In another embodiment, multiple biological samples taken at different points
in time from
the test subject are used to construct a biomarker profile that is used to
make this prediction
or diagnosis of sepsis.
In specific embodiments of the invention, subjects at risk for developing
sepsis or
SIRS are screened using the methods of the present invention. In accordance
with these
embodiments, the methods of the present invention can be employed to screen,
for example,
subjects admitted to an ICU and/or those who have experienced some sort of
trauma (such
as, e.g., surgery, vehicular accident, gunshot wound, etc.).
In specific embodiments, a biological sample such as, for example, blood, is
taken
upon admission. In some embodiments, a biological sample is blood, plasma,
serum, saliva,
sputum, urine, cerebral spinal fluid, cells, a cellular extract, a tissue
specimen, a tissue
biopsy, or a stool specimen. In some embodiments a biological sample is whole
blood and
this whole blood is used to obtain measurements for a biomarker profile. In
some
embodiments a biological sample is some component of whole blood. For example,
in
some embodiments some portion of the mixture of proteins, nucleic acid, and/or
other
molecules (e.g., metabolites) within a cellular fraction or within a liquid
(e.g., plasma or
serum fraction) of the blood is resolved as a biomarker profile. This can be
accomplished
by measuring features of the biomarkers in the biomarker profile. In some
embodiments,
the biological sample is whole blood but the biomarker profile is resolved
from biomarkers
in a specific cell type that is isolated from the whole blood. In some
embodiments, the
biological sample is whole blood but the biomarker profile is resolved from
biomarkers
expressed or otherwise found in monocytes that are isolated from the whole
blood. In some
embodiments, the biological sample is whole blood but the biomarker profile is
resolved
from biomarkers expressed or otherwise found in red blood cells that are
isolated from the
whole blood. In some embodiments, the biological sample is whole blood but the
biomarker profile is resolved from biomarkers expressed or otherwise found in
platelets that
are isolated from the whole blood. In some embodiments, the biological sample
is whole
blood but the biomarker profile is resolved from biomarkers expressed or
otherwise found
in neutriphils that are isolated from the whole blood. In some embodiments,
the biological
sample is whole blood but the biomarker profile is resolved from biomarkers
expressed or
-32-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
otherwise found in eosinophils that are isolated from the whole blood. In some
embodiments, the biological sample is whole blood but the biomarker profile is
resolved
from biomarkers expressed or otherwise found in basophils that are isolated
from the whole
blood. In some embodiments, the biological sample is whole blood but the
biomarker
profile is resolved from biomarkers expressed or otherwise found in
lymphocytes that are
isolated from the whole blood. In some embodiments, the biological sample is
whole blood
but the biomarker profile is resolved from biomarkers expressed or otherwise
found in
monocytes that are isolated from the whole blood. In some embodiments, the
biological
sample is whole blood but the biomarker profile is resolved from one, two,
three, four, five,
six, or seven cell types from the group of cells types consisting of red blood
cells, platelets,
neutrophils, eosinophils, basophils, lymphocytes, and monocytes.
In some embodiments, a biomarker profile comprises a plurality of one or more
types of biomarkers (e.g., an mRNA molecule, a cDNA molecule, a protein and/or
a
carbohydrate, etc.), or an indication thereof, together with features, such as
a measurable
aspect (e.g., abundance) of the biomarkers. A biomarker profile can comprise
at least two
such biomarkers or indications thereof, where the biomarkers can be in the
same or different
classes, such as, for example, a nucleic acid and a carbohydrate. In some
embodiments, a
biomarker profile comprises at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13,
14, 15, 16, 17, 18,
19, 20, 25, 30, 35, 40, 45, 50, 54, 5, 60, 65, 70, 75, 80, 85, 90, 95, 96, or
100 or more
biomarkers or indications thereof. In one embodiment, a biomarker profile
comprises
hundreds, or even thousands, of biomarkers or indications thereof. In some
embodiments, a
biomarker profile comprises at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13,
14, 15, 16, 17, 18,
19, 20, 25, 30, 35, 40, 45, 50, or more biomarkers or indications thereof. In
one example, in
some embodiments, a biomarker profile comprises at least 2, 3, 4, 5, 6, 7, 8,
9, 10, 11, 12,
13, 14, 15, 16, 17, 18, 19, 20 or more biomarkers selected from Table 1 or
indications
thereof. In another example, in some embodiments, a biomarker profile
comprises at least
2, 3, 4, 5, 6, 7, 8, 9 or more biomarkers selected from Table 4 or indications
thereof. In
another example, in some embodiments, a biomarker profile comprises at least
CRP,
APOA2, and SERPINCI, or indications thereof. In some embodiments, the
biomarker
profile comprises at least one of SERPINCI, APOA2, and CRP, and, additionally,
at least 1,
2, 3, 4, 5, 6 or 7 other additional biomarkers in Table 4. In some
embodiments, the
biomarker profile comprises at least one of SERPINCI, APOA2, and CRP, and,
additionally, at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16,
17, 18, 19, 20 or more
additional biomarkers from any combination of Tables 1, 4, 5, 6, 7, 8, and 9.
In some
- 33 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
embodiments, the biomarker profile comprises at least one of SERPINCI, APOA2,
and
CRP, and, additionally, at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13,
14, 15, 16, 17, 18, 19,
20 or more additional biomarkers from any one of Tables 1, 4, 5, 6, 7, 8, and
9. In another
example, in some embodiments, a biomarker profile comprises at least 2, 3, 4,
5, 6, 7, 8, 9,
10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 or more biomarkers selected from
Table 5 or
indications thereof. In yet another example, in some embodiments, a biomarker
profile
comprises at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18,
19, 20 or more
biomarkers selected from Table 6 or indications thereof. In one example, in
some
embodiments, a biomarker profile comprises at least 2, 3, 4, 5, 6, 7, 8, 9,
10, 11, 12, 13, 14,
15, 16, 17, 18, 19, 20 or more biomarkers selected from Table 7 or indications
thereof. In
one example, in some embodiments, a biomarker profile comprises at least 2, 3,
4, 5, 6, 7, 8,
9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 or more biomarkers selected from
Table 8 or
indications thereof. In one example, in some embodiments, a biomarker profile
comprises
at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 or
more biomarkers
selected from Table 8 or indications thereof. In some embodiments, when the
biomarker
profile comprises complement component C3 and complement component C4, the
biomarker profile comprises three or more biomarkers. In typical embodiments,
each
biomarker in the biomarker profile is represented by a feature. In other
words, there is a
correspondence between biomarkers and features. In some embodiments, the
correspondence between biomarkers and features is 1:1, meaning that for each
single
biomarker there is a corresponding single feature. In some embodiments, there
is more than
one feature for each biomarker. In some embodiments the number of features
corresponding to one biomarker in the biomarker profile is different than then
number of
features corresponding to another biomarker in the biomarker profile. As such,
in some
embodiments, a biomarker profile can include at least 2, 3, 4, 5, 6, 7, 8, 9,
10, 11, 12, 13, 14,
15, 16, 17, 18, 19, 20, 25, 30, 35, 40, 45, 50, 54, 5, 60, 65, 70, 75, 80, 85,
90, 95, 96, 100 or
more features, provided that there a r e a t least 2, 3, 4, 5, 6, 7, 8, 9, 10,
11, 12, 13, 14, 15, 16,
17, 18, 19, 20, 25, 30, 35, 40, 45, 50, 54, 5, 60, 65, 70, 75, 80, 85, 90, 95,
96, 100 or more
biomarkers in the biomarker profile. In some embodiments, a biomarker profile
can include
at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20,
25, 30, 35, 40, 45, 50,
or more features.
Regardless of embodiment, the aforementioned features can be determined
through
the use of any reproducible measurement technique or combination of
measurement
techniques. Such techniques include those that are well known in the art
including any
-34-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
technique described herein or, for example, any technique disclosed below.
Typically, such
techniques are used to measure feature values using a biological sample taken
from a
subject at a single point in time or multiple samples taken at multiple points
in time. In one
embodiment, an exemplary technique to obtain a biomarker profile from a sample
taken
from a subject is a cDNA microarray. In another embodiment, an exemplary
technique to
obtain a biomarker profile from a sample taken from a subject is a protein-
based assay or
other form of protein-based technique such as described in the BD Cytometric
Bead Array
(CBA) Human Inflammation Kit Instruction Manual (BD Biosciences) or the bead
assay
described in United States Patent Number 5,981,180, each of which is
incorporated herein
by reference in its entirety, and in particular for their teachings of various
methods of assay
protein concentrations in biological samples. In still another embodiment, the
biomarker
profile is mixed, meaning that it comprises some biomarkers that are nucleic
acids, or
indications thereof, and some biomarkers that are proteins, or indications
thereof. In such
embodiments, both protein based and nucleic acid based techniques are used to
obtain a
biomarker profile from one or more samples taken from a subject. In other
words, the
feature values for the features associated with the biomarkers in the
biomarker profile that
are nucleic acids are obtained by nucleic acid based measurement techniques
(e.g., a nucleic
acid microarray) and the feature values for the features associated with the
biomarkers in
the biomarker profile that are proteins are obtained by protein based
measurement
techniques. In some embodiments biomarker profiles can be obtained using a
kit, such as a
kit described in Section 5.3 below.
In specific embodiments, a subject is screened using the methods and
compositions
of the invention as frequently as necessary (e.g., during their stay in the
ICU) to diagnose or
predict sepsis or SIRS in a subject. In a preferred embodiment, subjects are
screened soon
after they arrive in the ICU or other medical establishment. In some
embodiments, subjects
are screened daily after they arrive in the ICU or other medical
establishment. In some
embodiments, subjects screened every i to 4 hours, 1 to 8 hours, 8 to 12
hours, 12 to 16
hours, or 16 to 24 hours after they arrive in the ICU or other medical
establishment.
5.3 KITS
The invention also provides kits that are useful in diagnosing or predicting
the
development of sepsis or diagnosing SIRS in a subject. In some embodiments,
the kits of
the present invention comprise at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12,
13, 14, 15, 16, 17, 18,
19, 20, 25, 30, 35, 40, 45, 50, 54, 5, 60, 65, 70, 75, 80, 85, 90, 95, 96,
100, 105, 110, 115,
- 35 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
120, 125, 130, 135, 140, 145, 150 or more biomarkers. In other embodiments,
the kits of
the present invention comprise at least 2, but as many as several hundred or
more
biomarkers. In a specific embodiment, the kits of the present invention
comprise at least 2,
3, 4, 5, 6, 7, 8, 9, 10, 1 1 , 12, 13, 14, 15, 16, 17, 18, 19, 20, 25, 30, 35,
40, 45, 50, 54, 5, 60,
65, 70, 75, 80, 85, 90, 95, 96, 100, 105, 110, 115, 120, 125, 130, 135, 140,
145, 150 or more
reagents that specifically bind the biomarkers of the present invention.
Specific biomarkers
that are useful in the present invention are set forth in Section 5.6 as well
as Tables 1, 4, 5,
6, 7, 8, and 9 of Section 6. The biomarkers of the kit can be used to generate
biomarker
profiles according to the present invention. Examples of classes of compounds
of the kit
include, but are not limited to, proteins and fragments thereof, peptides,
proteoglycans,
glycoproteins, lipoproteins, carbohydrates, lipids, nucleic acids (e.g., DNA,
such as cDNA
or amplified DNA, or RNA, such as mRNA), organic or inorganic chemicals,
natural or
synthetic polymers, small molecules (e.g., metabolites), or discriminating
molecules or
discriminating fragments of any of the foregoing. Here, a discriminating
molecule or
fragment is a molecule or fragment that, when detected, indicates presence or
abundance of
a molecule of interest (e.g., a cDNA, amplified nucleic acid molecule, or
protein). In a
specific embodiment, a biomarker is of a particular size, (e.g., at least 10,
15, 20, 25, 30, 35,
40, 45, 50, 54, 5, 60, 65, 70, 75, 80, 85, 90, 95, 100, 105, 110, 115, 120,
125, 130, 135, 140,
145, 150, 155, 160, 165, 170, 175, 180, 185, 190, 195 or 200 Da or greater).
The
biomarker(s) may be part of an array, or the biomarker(s) may be packaged
separately
and/or individually. The kit may also comprise at least one internal standard
to be used in
generating the biomarker profiles of the present invention. Likewise, the
internal standard
or standards can be any of the classes of compounds described above.
In one embodiment, the invention provides kits comprising probes and/or
primers
that may or may not be immobilized at an addressable position on a substrate,
such as
found, for example, in a microarray. In a particular embodiment, the invention
provides
such a microarray.
The kits of the present invention may also contain reagents that can be used
to detect
biomarkers contained in the biological samples from which the biomarker
profiles are
generated. In a specific embodiment, the invention provides a kit for
predicting the
development of sepsis in a test subject comprises a plurality of antibodies
that specifically
bind a plurality of biomarkers listed in any one of Tables 1, 4, 5, 6, 7, 8,
and 9. In another
specific embodiment, the invention provides a kit for predicting the
development of sepsis
in a test subject comprises a plurality of antibodies that specifically bind a
plurality of
-36-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
biomarkers listed in any combination of Tables 1, 4, 5, 6, 7, 8, and 9. In
accordance with
this embodiment, the kit may comprise a set of antibodies or functional
fragments or
derivatives thereof (e.g., Fab, F(ab')2, Fv, or scFv fragments) that
specifically bind at least 2,
3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23,
24, 25, or more of the
protein-based biomarkers set forth in any one of Tables 1, 4, 5, 6, 7, 8, and
9. In some
embodiments, when the kit comprises antibodies to complement component 0 and
complement component C4, the kit comprises an antibody to at least one other
biomarker in
any one of Table 1, 4, 5, 6, 7, 8, and 9. In accordance with this embodiment,
the kit may
include antibodies, fragments or derivatives thereof (e.g., Fab, F(ab')2, Fv,
or scFv
fragments) that are specific for the biomarkers of the present invention. In
one
embodiment, the antibodies may be detectably labeled. In one embodiment, the
kit
comprises antibodies to any combination of the proteins set forth in Table 4.
In one
embodiment, the kit comprises antibodies to CRP, APOA2, and SERPINCI. In some
embodiments, the biomarker profile comprises antibodies to at least one of
SERPINC 1,
APOA2, and CRP, and, additionally, antibodies to at least 1, 2, 3, 4, 5, 6 or
7 other
additional biomarkers in Table 4. In some embodiments, the biomarker profile
comprises
antibodies to at least one of SERPINCI, APOA2, and CRP, and, additionally,
antibodies to
at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20
or more additional
biomarkers from any combination of Tables 1, 4, 5, 6, 7, 8, and 9. In some
embodiments,
the biomarker profile comprises antibodies to at least one of SERPINCI, APOA2,
and CRP,
and, additionally, antibodies to at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11,
12, 13, 14, 15, 16, 17,
18, 19, 20 or more additional biomarkers from any one of Tables 1, 4, 5, 6, 7,
8, and 9.
In other embodiments of the invention, a kit may comprise a specific biomarker
binding component, such as an aptamer. If the biomarkers comprise a nucleic
acid, the kit
may provide an oligonucleotide probe that is capable of forming a duplex with
the
biomarker or with a complementary strand of a biomarker. The oligonucleotide
probe may
be detectably labeled.
The kits of the present invention may also include reagents such as buffers,
or other
reagents that can be used in constructing the biomarker profile. Prevention of
the action of
microorganisms can be ensured by the inclusion of various antibacterial and
antifungal
agents, for example, paraben, chlorobutanol, phenol sorbic acid, and the like.
It may also be
desirable to include isotonic agents such as sugars, sodium chloride, and the
like.
Some kits of the present invention comprise a microarray. In one embodiment
this
microarray comprises a plurality of probe spots, wherein at least twenty
percent of the probe
-37-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
spots in the plurality of probe spots correspond to biomarkers in any one of
Tables 1, 4, 5, 6,
7, 8, and 9. In some embodiments, at least forty percent, or at least sixty
percent, or at least
eighty percent of the probe spots in the plurality of probe spots correspond
to biomarkers in
any one of Tables 1, 4, 5, 6, 7, 8, and 9. In one embodiment this microarray
comprises a
plurality of probe spots, wherein at least twenty percent of the probe spots
in the plurality of
probe spots correspond to biomarkers in any combination of Tables 1, 4, 5, 6,
7, 8, and 9.
In some embodiments, at least forty percent, or at least sixty percent, or at
least eighty
percent of the probe spots in the plurality of probe spots correspond to
biomarkers in any
combination of Tables 1, 4, 5, 6, 7, 8, and 9. In some embodiments, when the
plurality of
probe spots contain a spot the corresponds to complement component C3 and
complement
component C4, the plurality of probe spots comprises a probe spot for at least
one other
biomarker in any of Tables 1, 4, 5, 6, 7, 8, and 9. In some embodiments, the
microarray
consists of between about three and about one hundred probe spots on a
substrate. In some
embodiments, the microarray consists of between about three and about one
hundred probe
spots on a substrate. As used in this context, the term "about" means within
five percent of
the stated value, within ten percent of the stated value, or within twenty-
five percent of the
stated value.
Some kits of the invention may further comprise a computer program product for
use in conjunction with a computer system. In such kits, the computer program
product
comprises a computer readable storage medium and a computer program mechanism
embedded therein. The computer program mechanism comprises instructions for
evaluating
whether a plurality of features in a biomarker profile of a test subject at
risk for developing
sepsis satisfies a first value set. Satisfying the first value set predicts
that the test subject is
likely to develop sepsis. In one embodiment, the plurality of features
corresponds to
biomarkers listed in any one of Tables 1, 4, 5, 6, 7, 8, and 9. In one
embodiment, the
plurality of features corresponds to biomarkers listed in any combination of
Tables 1, 4, 5,
6, 7, 8, and 9: In one embodiment, the plurality of features comprises
features for CRP,
APOA2, and SERPINC1. In some embodiments, the plurality of features comprises
features for at least one of SERPINCI, APOA2, and CRP, and, additionally,
features for at
least 1, 2, 3, 4, 5, 6 or 7 other additional biomarkers in Table 4. In some
embodiments, the
plurality of features comprises features for at least one of SERPINCl, APOA2,
and CRP,
and, additionally, features for at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11,
12, 13, 14, 15, 16, 17,
18, 19, 20 or more additional biomarkers from any combination of Tables 1, 4,
5, 6, 7, 8,
and 9. In some embodiments, the plurality of features comprises features for
at least one of
-38-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
SERPINC1, APOA2, and CRP, and, additionally, features for at least 1, 2, 3, 4,
5, 6, 7, 8, 9,
10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 or more additional biomarkers from
any one of
Tables 1, 4, 5, 6, 7, 8, and 9. In some embodiments, where the plurality of
features
comprises features for component C3 and complement component C4, the plurality
of
features comprises a feature for at least one other biomarker in any of Tables
1, 4, 5, 6, 7, 8,
and 9. In some kits, the computer program product further comprises
instructions for
evaluating whether the plurality of features in the biomarker profile of the
test subject
satisfies a second value set. Satisfying the second value set predicts that
the test subject is
not likely to develop sepsis.
Some kits of the present invention comprise a computer having a central
processing
unit and a memory coupled to the central processing unit. The memory stores
instructions
for evaluating whether a plurality of features in a biomarker profile of a
test subject at risk
for developing sepsis satisfies a first value set. Satisfying the first value
set predicts that the
test subject is likely to develop sepsis. In one embodiment, the plurality of
features
corresponds to biomarkers listed in any one of Tables 1, 4, 5, 6, 7, 8, and 9.
In one
embodiment, when the plurality of features includes a feature for complement
component
C3 and complement component C4, the plurality of features includes a feature
for at least
one other biomarker in any of Tables 1, 4, 5, 6, 7, 8, and 9. In one
embodiment, the
plurality of features corresponds to biomarkers listed in any combination of
Tables 1, 4, 5,
6, 7, 8, and 9.
Fig. 1 details an exemplary system that supports the functionality described
above.
The system is preferably a computer system 10 having:
= a central processing unit 22;
= a main non-volatile storage unit 14, for example, a hard disk drive, for
storing software and data, the storage unit 14 controlled by storage
controller
12;
= a system memory 36, preferably high speed random-access memory (RAM),
for storing system control programs, data, and application programs,
comprising programs and data loaded from non-volatile storage unit 14;
system memory 36 may also include read-only memory (ROM);
= a user interface 32, comprising one or more input devices (e.g., keyboard
28)
and a display 26 or other output device;
= a network interface card 20 for connecting to any wired or wireless
communication network 34 (e.g., a wide area network such as the Internet);
-39-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
= an internal bus 30 for interconnecting the aforementioned elements of the
system; and
= a power source 24 to power the aforementioned elements.
= Operation of computer 10 is controlled primarily by operating system 40,
which is executed by central processing unit 22. Operating system 40 can be
stored in system memory 36. In addition to operating system 40, in a typical
implementation system memory 36 includes:
= file system 42 for controlling access to the various files and data
structures
used by the present invention;
= a training data set 44 for use in construction one or more decision rules in
accordance with the present invention;
= a data analysis algorithm module 54 for processing training data and
constructing decision rules;
= one or more decision rules 56;
= a biomarker profile evaluation module 60 for determining whether a plurality
of features in a biomarker profile of a test subject satisfies a first value
set or
a second value set;
= a test subject biomarker profile 62 comprising biomarkers 64 and, for each
such biomarkers, features 66; and
= a database 68 of select biomarkers of the present invention (e.g., Tables 1,
4,
5, 6, 7, 8 and/or 9).
Training data set 46 comprises data for a plurality of subjects 46. For each
subject
46 there is a subject identifier 48 and a plurality of biomarkers 50. For each
biomarker 50,
there is at least one feature 52. Although not shown in Figure 1, for each
feature 52, there is
at least one feature value. For each decision rule 56 constructed using data
analysis
algorithms, there is at least one decision rule value set 58.
As illustrated in Figure 1, computer 10 comprises software program modules and
data structures. The data structures stored in computer 10 include training
data set 44,
decision rules 56, test subject biomarker profile 62, and biomarker database
68. Each of
these data structures can comprise any form of data storage system including,
but not
limited to, a flat ASCII or binary file, an Excel spreadsheet, a relational
database (SQL), or
an on-line analytical processing (OLAP) database (MDX and/or variants
thereof). In some
-40-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
specific embodiments, such data structures are each in the form of one or more
databases
that include hierarchical structure (e.g., a star schema). In some
embodiments, such data
structures are each in the form of databases that do not have explicit
hierarchy (e.g.,
dimension tables that are not hierarchically arranged).
In some embodiments, each of the data structures stored or accessible to
system 10
are single data structures. In other embodiments, such data structures in fact
comprise a
plurality of data structures (e.g., databases, files, archives) that may or
may not all be hosted
by the same computer 10. For example, in some embodiments, training data set
44
comprises a plurality of Excel spreadsheets that are stored either on computer
10 and/or on
computers that are addressable by computer 10 across wide area network 34. In
another
example, training data set 44 comprises a database that is either stored on
computer 10 or is
distributed across one or more computers that are addressable by computer 10
across wide
area network 34.
It will be appreciated that many of the modules and data structures
illustrated in
Figure 1 can be located on one or more remote computers. For example, some
embodiments of the present application are web service-type implementations.
In such
embodiments, biomarker profile evaluation module 60 and/or other modules can
reside on a
client computer that is in communication with computer 10 via network 34. In
some
embodiments, for example, biomarker profile evaluation module 60 can be an
interactive
web page.
In some embodiments, training data set 44, decision rules 56, and/or biomarker
database 68 illustrated in Figure 1 are on a single computer (computer 10) and
in other
embodiments one or more of such data structures and modules are hosted by one
or more
remote computers (not shown). Any arrangement of the data structures and
software
modules illustrated in Figure 1 on one or more computers is within the scope
of the present
invention so long as these data structures and software modules are
addressable with respect
to each other across network 34 or by other electronic means. Thus, the
present invention
fully encompasses a broad array of computer systems.
Still another kit of the present invention comprises computers and computer
readable
media for evaluating whether a test subject is likely to develop sepsis or
SIRS. For
instance, one embodiment of the present invention provides a computer program
product for
use in conjunction with a computer system. The computer program product
comprises a
computer readable storage medium and a computer program mechanism embedded
therein.
The computer program mechanism comprises instructions for evaluating whether a
plurality
-41-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
of features in a biomarker profile of a test subject at risk for developing
sepsis satisfies a
first value set. Satisfaction of the first value set predicts that the test
subject is likely to
develop sepsis. In some embodiments, this plurality of features is measurable
aspects of a
plurality of biomarkers. The plurality of biomarkers can comprise at least 2,
3, 4, 5, 6, 7, 8,
9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, or 20 biomarkers listed in any one
of Tables 1, 4, 5,
6, 7, 8, and 9. In certain embodiments, the plurality of biomarkers comprises
at least 2, 3, 4,
5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, or 20 biomarkers listed
in any
combination of Tables 1, 4, 5, 6, 7, 8, and 9. In some embodiments, the
plurality of
biomarkers comprises CRP, APOA2, and SER.PINCI. In some embodiments, the
plurality
of biomarkers comprises at least one of SERPINC1, APOA2, and CRP, and,
additionally, at
least 1, 2, 3, 4, 5, 6 or 7 other additional biomarkers in Table 4. In some
embodiments, the
plurality of biomarkers comprises at least one of SERPINC 1, APOA2, and CRP,
and,
additionally, at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16,
17, 18, 19, 20 or more
additional biomarkers from any combination of Tables 1, 4, 5, 6, 7, 8, and 9.
In some
embodiments, the plurality of biomarkers comprises at least one of SERPINCI,
APOA2,
and CRP, and, additionally, at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12,
13, 14, 15, 16, 17, 18,
19, 20 or more additional biomarkers from any one of Tables 1, 4, 5, 6, 7, 8,
and 9. In some
embodiments, when the plurality of biomarkers comprises complement component
C3 and
complement component C4, the plurality of biomarkers comprises at least one
other
biomarker from any one of Tables 1, 4, 5, 6, 7, 8, and 9. In some embodiments,
the
computer program product further comprises instructions for evaluating whether
the
plurality of features in the biomarker profile of the test subject satisfies a
second value set.
Satisfaction of the second value set predicts that the test subject is not
likely to develop
sepsis. In some embodiments, the biomarker profile has between 3 and 50
biomarkers listed
in any one of Tables 1, 4, 5, 6, 7, 8, and 9, between 3 and 40 biomarkers
listed in any one of
Tables 1, 4, 5, 6, 7, 8, and 9, at least four biomarkers listed in any one of
Tables 1, 4, 5, 6, 7,
8, and 9, or at least eight biomarkers listed in any one of Tables 1, 4, 5, 6,
7, 8, and 9. In
some embodiments, the biomarker profile has between 3 and 50 biomarkers listed
in any
combination of Tables 1, 4, 5, 6, 7, 8, and 9 between 3 and 40 biomarkers
listed in any
combination of Tables 1, 4, 5, 6, 7, 8, and 9 at least four biomarkers listed
in any
combination of Tables 1, 4, 5, 6, 7, 8, and 9 or at least eight biomarkers
listed in any
combination of Tables 1, 4, 5, 6, 7, 8, and 9.
Another kit of the present invention comprises a central processing unit and a
memory coupled to the central processing unit. The memory stores instructions
for
- 42 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
evaluating whether a plurality of features in a biomarker profile of a
test.subject at risk for
developing sepsis satisfies a first value set. Satisfaction of the first value
set predicts that
the test subject is likely to develop sepsis. The plurality of features is
measurable aspects of
a plurality of biomarkers. In some embodiments, this plurality of biomarkers
comprises at
least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, or 20
biomarkers from any
one of Tables 1, 4, 5, 6, 7, 8, and 9. In some embodiments, this plurality of
biomarkers
comprises at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18,
19, or 20
biomarkers from any combination of Tables 1, 4, 5, 6, 7, 8, and 9. In some
embodiments,
this plurality of biomarkers comprises CRP, APOA2, and SEitPINCl . In some
embodiments, the plurality of biomarkers comprises at least one of SERPINCI,
APOA2,
and CRP, and, additionally, at least 1, 2, 3, 4, 5, 6 or 7 other additional
biomarkers in Table
4. In some embodiments, the plurality of biomarkers comprises at least one of
SERPINCI,
APOA2, and CRP, and, additionally, at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11,
12, 13, 14, 15,
16, 17, 18, 19, 20 or more additional biomarkers from any combination of
Tables 1, 4, 5, 6,
7, 8, and 9. In some embodiments, the plurality of biomarkers comprises at
least one of
SERPINC1, APOA2, and CRP, and, additionally, at least 1, 2, 3, 4, 5, 6, 7, 8,
9, 10, 11, 12,
13, 14, 15, 16, 17, 18, 19, 20 or more additional biomarkers from any one of
Tables 1, 4, 5,
6, 7, 8, and 9. In some embodiments, when the plurality of biomarkers
comprises
complement component C3 and complement component C4, the plurality of
biomarkers
comprises at least one other biomarker from any one of Tables 1, 4, 5, 6, 7,
8, and 9. In
some embodiments, the memory further stores instructions for evaluating
whether the
plurality of features in the biomarker profile of the test subject satisfies a
second value set.
Satisfaction of the second value set predicts that the test subject is not
likely to develop
sepsis. In some embodiments, the biomarker profile consists of between 3 and
50
biomarkers listed in any one of Tables 1, 4, 5, 6, 7, 8, and 9, between 3 and
40 biomarkers
listed in any one of Tables 1, 4, 5, 6, 7, 8, and 9 at least four biomarkers
listed in any one of
Tables 1, 4, 5, 6, 7, 8, and 9 or at least eight biomarkers listed in any one
of Tables 1, 4, 5,
6, 7, 8 and 9 (for example, all found in Table 1, all found in Table 4, all
found in Table 5, all
found in Table 6, or all found in Table 7, or all found in Table 8). In some
embodiments,
the biomarker profile consists of between 3 and 50 biomarkers listed in any
combination of
Tables 1, 4, 5, 6, 7, 8, and 9 between 3 and 40 biomarkers listed in any
combination of
Tables 1, 4, 5, 6, 7, 8, and 9 at least 3, 4, 5, 6, 7, 8, 9, or 10 biomarkers
listed in any
combination of Tables 1, 4, 5, 6, 7, 8 and 9 (for example, all found in Tables
1 or 4, all
found in Table 4 or 5, all found in Tables 1, 5, 7, 8 and 9).
-43-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Another kit in accordance with the present invention comprises a computer
system
for determining whether a subject is likely to develop sepsis. The computer
system
comprises a central processing unit and a memory, coupled to the central
processing unit.
The memory stores instructions for obtaining a biomarker profile of a test
subject. The
biomarker profile comprises a plurality of features. Each feature in the
plurality of features
is a measurable aspect of a corresponding biomarker in a plurality of
biomarkers. In some
embodiments, the plurality of biomarkers comprises at least 2, 3, 4, 5, 6, 7,
8, 9, 10, 11, 12,
13, 14, 15, 16, 17, 18, 19, 20 or biomarkers listed in any combination of
Tables 1, 4, 5, 6, 7,
8, or 9. In some embodiments, the plurality of biomarkers comprises at least
2, 3, 4, 5, 6, 7,
8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 or biomarkers listed in any
one of Tables 1, 4,
5, 6, 7, 8, or 9. In some embodiments, the plurality of biomarkers comprises
CRP, APOA2,
and SERPINCI. In some embodiments, the biomarker profile comprises at least
one of
SERPINCI, APOA2, and CRP, and, additionally, at least 1, 2, 3, 4, 5, 6 or 7
other
additional biomarkers in Table 4. In some embodiments, the biomarker profile
comprises at
least one of SERPINCI, APOA2, and CRP, and, additionally, at least 1, 2, 3, 4,
5, 6, 7, 8, 9,
10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 or more additional biomarkers from
any one of
Tables 1, 4, 5, 6, 7, 8, and 9. In some embodiments, when the plurality of
biomarkers
comprises complement component C3 and complement component C4, the plurality
of
biomarkers comprises at least one other biomarker from any one of Tables 1, 4,
5, 6, 7, 8,
and 9. The memory further comprises instructions for transmitting the
biomarker profile to
a remote computer. The remote computer includes instructions for evaluating
whether the
plurality of features in the biomarker profile of the test subject satisfies a
first value set.
Satisfaction of the first value set predicts that the test subject is likely
to develop sepsis.
The memory further comprises instructions for receiving a determination, from
the remote
computer, as to whether the plurality of features in the biomarker profile of
the test subject
satisfies the first value set. The memory also comprises instructions for
reporting whether
the plurality of features in the biomarker profile of the test subject
satisfies the first value
set. In some embodiments, the plurality of biomarkers comprises at least 2, 3,
4, 5, 6, 7, 8,
9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, or 20 biomarkers listed in any one
of Tables 1, 4, 5,
6, 7, 8, and 9. In some embodiments, the plurality of biomarkers comprises at
least 2, 3, 4,
5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, or 20 biomarkers listed
in any
combination of Tables 1, 4, 5, 6, 7, 8, and 9. In some embodiments, the remote
computer
further comprises instructions for evaluating whether the plurality of
features in the
biomarker profile of the test subject satisfies a second value set.
Satisfaction of the second
-44-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
value set predicts that the test subject is not likely to develop sepsis. In
such embodiments,
the memory further comprises instructions for receiving a determination, from
the remote
computer, as to whether the plurality of features in the biomarker profile of
the test subject
satisfies the second set as well as instructions for reporting whether the
plurality of features
in the biomarker profile of the test subject satisfies the second value set.
Still another aspect of the present invention comprises a digital signal
embodied on a
carrier wave comprising a respective value for each of a plurality of features
in a biomarker
profile. The plurality of features is measurable aspects of a plurality of
biomarkers. In
some embodiments, the plurality of biomarkers comprises at least 2, 3, 4, 5,
6, 7, 8, 9, 10,
11, 12, 13, 14, 15, 16, 17, 18, 19, 20 or biomarkers listed in any one of
Tables 1, 4, 5, 6, 7,
8, and 9. In some embodiments, the plurality of biomarkers comprises at least
2, 3, 4, 5, 6,
7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 or biomarkers listed in
any combination of
Tables 1, 4, 5, 6, 7, 8, and 9. In some embodiments, the plurality of
biomarkers comprises
at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20,
21, 22, 23, 24, or 25
biomarkers from Table 1. In some embodiments, the plurality of biomarkers
comprises at
least 2, 3, 4, 5, 6, 7, 8, 9, 10 biomarkers from Table 4. In some embodiments,
the plurality
of biomarkers comprises at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14,
15, 16, 17, 18, 19,
20, 21, 22, 23, 24, or 25 biomarkers from Table 5. In some embodiments, the
plurality of
biomarkers comprises at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15,
16, 17, 18, 19, 20,
21, 22, 23, 24, or 25 biomarkers from Table 6. In some embodiments, the
plurality of
biomarkers comprises at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15,
16, 17, 18, 19, 20,
21, 22, 23, 24, or 25 biomarkers from Table 7. In some embodiments, the
plurality of
biomarkers comprises at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15
biomarkers from
Table 8. In some embodiments, the plurality of biomarkers comprises at least
2, 3, 4, 5, 6,
7, 8, 9, 10, 11, 12, 13, 14, 15 biomarkers from Table 9. In some embodiments,
the plurality
of biomarkers comprises CRP, APOA2, and SERPINC 1. In some embodiments, the
plurality of biomarkers comprises at least one of SERPINCI, APOA2, and CRP,
and,
additionally, at least 1, 2, 3, 4, 5, 6 or 7 other additional biomarkers in
Table 4. In some
embodiments, the plurality of biomarkers comprises at least one of SERPINCI,
APOA2,
and CRP, and, additionally, at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12,
13, 14, 15, 16, 17, 18,
19, 20 or more additional biomarkers from any combination of Tables 1, 4, 5,
6, 7, 8, and 9.
In some embodiments, the plurality of biomarkers comprises at least one of
SERPINCI,
APOA2, and CRP, and, additionally, at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11,
12, 13, 14, 15,
16, 17, 18, 19, 20 or more additional biomarkers from any one of Tables 1, 4,
5, 6, 7, 8, and
- 45 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
9. In some embodiments, when the plurality of biomarkers comprises complement
component C3 and complement component C4, the plurality of biomarkers
comprises at
least one other biomarker from any one of Tables 1, 4, 5, 6, 7, 8, and 9.
Still another aspect of the present invention provides a digital signal
embodied on a
carrier wave comprising a determination as to whether a plurality of features
in a biomarker
profile of a test subject satisfies a value set. The plurality of features is
measurable aspects
of a plurality of biomarkers. In some embodiments, the plurality of biomarkers
comprises
at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 or
biomarkers listed in
any one of Tables 1, 4, 5, 6, 7, 8, and 9. In some embodiments, the plurality
of biomarkers
comprises at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18,
19, 20 or
biomarkers listed in any combination of Tables 1, 4, 5, 6, 7, 8, and 9. In
some
embodiments, the plurality of biomarkers comprises at least 2, 3, 4, 5, 6, 7,
8, 9, 10, 11, 12,
13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, or 25 biomarkers from Table 1.
In some
embodiments, the plurality of biomarkers comprises at least 2, 3, 4, 5, 6, 7,
8, 9, or 10
biomarkers from Table 4. In some embodiments, the plurality of biomarkers
comprises at
least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21,
22, 23, 24, or 25
biomarkers from Table 5. In some embodiments, the plurality of biomarkers
comprises at
least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21,
22, 23, 24, or 25
biomarkers from Table 6. In some embodiments, the plurality of biomarkers
comprises at
least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21,
22, 23, 24, or 25
biomarkers from Table 7. In some embodiments, the plurality of biomarkers
comprises at
least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 biomarkers from Table 8.
In some
embodiments, the plurality of biomarkers comprises at least 2, 3, 4, 5, 6, 7,
8, 9, 10, 11, 12,
13, 14, 15 biomarkers from Table 9. In some embodiments, the plurality of
biomarkers
comprises CRP, APOA2, and SERPINCI . In some embodiments, the plurality of
biomarkers comprises at least one of SERPINCI, APOA2, and CRP, and,
additionally, at
least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 or
more additional
biomarkers from any combination of Tables 1, 4, 5, 6, 7, 8, and 9. In some
embodiments,
the plurality of biomarkers comprises at least one of SER.PINC1, APOA2, and
CRP and,
additionally, at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16,
17, 18, 19, 20 or more
additional biomarkers from any one of Tables 1, 4, 5, 6, 7, 8, and 9. In some
embodiments,
when the plurality of biomarkers comprises complement component C3 and
complement
component C4, the plurality of biomarkers comprises at least one other
biomarker from any
one of Tables 1, 4, 5, 6, 7, 8, and 9.
-46-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Still another embodiment provides a digital signal embodied on a carrier wave
comprising a determination as to whether a plurality of features in a
biomarker profile of a
test subject satisfies a value set. The plurality of features is measurable
aspects of a
plurality of biomarkers. In some embodiments, the plurality of biomarkers
comprises at
least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 or
biomarkers listed in
any one of Tables 1, 4, 5, 6, 7, 8, and 9. In some embodiments, the plurality
of biomarkers
comprises at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18,
19, 20 or
biomarkers listed in any combination of Tables 1, 4, 5, 6, 7, 8, and 9. In
some
embodiments, the plurality of biomarkers comprises at least 2, 3, 4, 5, 6, 7,
8, 9, 10, 11, 12,
13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, or 25 biomarkers from Table 1.
In some
embodiments, the plurality of biomarkers comprises at least 2, 3, 4, 5, 6, 7,
8, 9, 10
biomarkers from Table 4. In some embodiments, the plurality of biomarkers
comprises at
least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21,
22, 23, 24, or 25
biomarkers from Table 5. In some embodiments, the plurality of biomarkers
comprises at
least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21,
22, 23, 24, or 25
biomarkers from Table 6. In some embodiments, the plurality of biomarkers
comprises at
least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21,
22, 23, 24, or 25
biomarkers from Table 7. In some embodiments, the plurality of biomarkers
comprises at
least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 biomarkers from Table 8.
In some
embodiments, the plurality of biomarkers comprises at least 2, 3, 4, 5, 6, 7,
8, 9, 10, 11, 12,
13, 14, 15 biomarkers from Table 9. In some embodiments, the plurality of
biomarkers
comprises CRP, APOA2, and SERPINC 1. In some embodiments, the plurality of
biomarkers comprises at least one of SERPINCI, APOA2, and CRP, and,
additionally, at
least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 or
more additional
biomarkers from any combination of Tables 1, 4, 5, 6, 7, 8, and 9. In some
embodiments,
the plurality of biomarkers comprises at least one of SERPINCI, APOA2, and
CRP, and,
additionally, at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16,
17, 18, 19, 20 or more
additional biomarkers from any one of Tables 1, 4, 5, 6, 7, 8, and 9. In some
embodiments,
when the plurality of biomarkers comprises complement component C3 and
complement
component C4, the plurality of biomarkers comprises at least one other
biomarker from any
one of Tables 1, 4, 5, 6, 7, 8, and 9.
Still another embodiment of the present invention provides a graphical user
interface
for determining whether a subject is likely to develop sepsis. The graphical
user interface
comprises a display field for a displaying a result encoded in a digital
signal embodied on a
-47-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
carrier wave received from a remote computer. The plurality of features is
measurable
aspects of a plurality of biomarkers. In some embodiments, the plurality of
biomarkers
comprises at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18,
19, 20 or
biomarkers listed in any one of Tables 1, 4, 5, 6, 7, 8, and 9. In some
embodiments, the
plurality of biomarkers comprises at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12,
13, 14, 15, 16, 17,
18, 19, 20 or biomarkers listed in any combination of Tables 1, 4, 5, 6, 7, 8,
and 9. In some
embodiments, the plurality of biomarkers comprises at least 2, 3, 4, 5, 6, 7,
8, 9, 10, 11, 12,
13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, or 25 biomarkers from Table 1.
In some
embodiments, the plurality of biomarkers comprises at least 2, 3, 4, 5, 6, 7,
8, 9, 10
biomarkers from Table 4. In some embodiments, the plurality of biomarkers
comprises at
least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11; 12, 13, 14, 15, 16, 17, 18, 19, 20, 21,
22, 23, 24, or 25
biomarkers from Table 5. In some embodiments, the plurality of biomarkers
comprises at
least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21,
22, 23, 24, or 25
biomarkers from Table 6. In some embodiments, the plurality of biomarkers
comprises at
least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21,
22, 23, 24, or 25
biomarkers from Table 7. In some embodiments, the plurality of biomarkers
comprises at
least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13,14, 15 biomarkers from Table 8.
In some
embodiments, the plurality of biomarkers comprises at least 2, 3, 4, 5, 6, 7,
8, 9, 10, 11, 12,
13, 14, 15 biomarkers from Table 9. In some embodiments, the plurality of
biomarkers
comprises CRP, APOA2, and SERPINC 1. In some embodiments, the plurality of
biomarkers comprises at least one of SERPINC1, APOA2, and CRP, and,
additionally, at
least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 or
more additional
biomarkers from any combination of Tables 1, 4, 5, 6, 7, 8, and 9. In some
embodiments,
the plurality of biomarkers comprises at least one of SERPINCI, APOA2, and CRP
and,
additionally, at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16,
17, 18, 19, 20 or more
additional biomarkers from any one of Tables 1, 4, 5, 6, 7, 8, and 9. In some
embodiments,
when the plurality of biomarkers comprises complement component C3 and
complement
component C4, the plurality of biomarkers comprises at least one other
biomarker from any
one of Tables 1, 4, 5, 6, 7, 8, and 9.
Yet another kit of the present invention provides a computer system for
determining
whether a subject is likely to develop sepsis. The computer system comprises a
central
processing unit and a memory, coupled to the central processing unit. The
memory stores
instructions for obtaining a biomarker profile of a test subject. The
biomarker profile
comprises a plurality of features. The plurality of features is measurable
aspects of a
-48-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
plurality of biomarkers. In some embodiments, the plurality of biomarkers
comprises at
least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 or
biomarkers listed in
any one of Tables 1, 4, 5, 6, 7, 8, and 9. In some embodiments, the plurality
of biomarkers
comprises at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18,
19, 20 or
biomarkers listed in any combination of Tables 1, 4, 5, 6, 7, 8, and 9. The
memory further
stores instructions for evaluating whether the plurality of features in the
biomarker profile of
the test subject satisfies a first value set. Satisfying the first value set
predicts that the test
subject is likely to develop sepsis. The memory also stores instructions for
reporting
whether the plurality of features in the biomarker profile of the test subject
satisfies the first
value set. In some embodiments, the plurality of biomarkers comprises at least
2, 3, 4, 5, 6,
7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, or 25
biomarkers from
Table 1. In some embodiments, the plurality of biomarkers comprises at least
2, 3, 4, 5, 6,
7, 8, 9, 10 biomarkers from Table 4. In some embodiments, the plurality of
biomarkers
comprises at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18,
19, 20, 21, 22, 23,
24, or 25 biomarkers from Table 5. In some embodiments, the plurality of
biomarkers
comprises at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18,
19, 20, 21, 22, 23,
24, or 25 biomarkers from Table 6. In some embodiments, the plurality of
biomarkers
comprises at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18,
19, 20, 21, 22, 23,
24, or 25 biomarkers from Table 7. In some embodiments, the plurality of
biomarkers
comprises at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 biomarkers
from Table 8. In
some embodiments, the plurality of biomarkers comprises at least 2, 3, 4, 5,
6, 7, 8, 9, 10,
11, 12, 13, 14, 15 biomarkers from Table 9. In some embodiments, the plurality
of
biomarkers comprises CRP, APOA2, and SERPINC 1. In some embodiments, the
plurality
of biomarkers comprises at least one of SERPINC 1, APOA2, and CRP, and,
additionally, at
least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 or
more additional
biomarkers from any combination of Tables 1, 4, 5, 6, 7, 8, and 9. In some
embodiments,
the plurality of biomarkers comprises at least one of SERPINCI, APOA2, and
CRP, and,
additionally, at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16,
17, 18, 19, 20 or more
additional biomarkers from any one of Tables 1, 4, 5, 6, 7, 8, and 9. In some
embodiments,
when the plurality of biomarkers comprises complement component C3 and
complement
component C4, the plurality of biomarkers comprises at least one other
biomarker from any
one of Tables 1, 4, 5, 6, 7, 8, and 9.
- 49 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
5.4 GENERATION OF BIOMARKER PROFILES
According to one ernbodiment, the methods of the present invention comprise
generating a biomarker profile from a biological sample taken from a subject.
The
biological sample may be, for example, whole blood, plasma, serum, red blood
cells,
platelets, neutrophils, eosinophils, basophils, lymphocytes, monocytes,
saliva, sputum,
urine, cerebral spinal fluid, cells, a cellular extract, a tissue sample, a
tissue biopsy, a stool
sample or any sample that may be obtained from a subject using techniques well
known to
those of skill in the art. In a specific embodiment, a biomarker profile is
determined using
samples collected from a subject at one or more separate time points. In
another specific
embodiment, a biomarker profile is generated using samples obtained from a
subject at
separate time points. In one example, these samples are obtained from the
subject either
once or, alternatively, on a daily basis, or more frequently, e.g., every 4,
6, 8 or 12 hours. In
some embodiments, these samples are collected from the subject on multiple
different time
points, but on an irregular time basis. In a specific embodiment, a biomarker
profile is
determined using samples collected from a single tissue type. In another
specific
embodiment, a biomarker profile is determined using samples collected from at
least 2, 3, 4,
4, 5, 6 or 7 different tissue types.
5.4.1 Methods of detecting nucleic acid biomarkers
In specific embodiments of the invention, biomarkers in a biomarker profile
are
nucleic acids. Such biomarkers and corresponding features of the biomarker
profile may be
generated, for example, by detecting the expression product (e.g., a
polynucleotide or
polypeptide) of one or more genes described herein (e.g., a gene listed in
Tables 1, 4, 5, 6,
7, 8, and/or 9). In a specific embodiment, the biomarkers and corresponding
features in a
biomarker profile are obtained by detecting and/or analyzing one or more
nucleic acids
expressed from a gene disclosed herein (e.g., a gene listed in Tables 1, 4, 5,
6, 7, 8 and/or 9)
using any method well known to those skilled in the art including, but by no
means limited
to, hybridization, microarray analysis, RT-PCR, nuclease protection assays and
Northern
blot analysis.
In certain embodiments, nucleic acids detected and/or analyzed by the methods
and
compositions of the invention include RNA molecules such as, for example,
expressed
RNA molecules which include messenger RNA (mRNA) molecules, mRNA spliced
variants as well as regulatory RNA, cRNA molecules (e.g., RNA molecules
prepared from
-50-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
cDNA molecules that are transcribed in vitro) and discriminating fragments
thereof.
Nucleic acids detected and/or analyzed by the methods and compositions of the
present
invention can also include, for example, DNA molecules such as genomic DNA
molecules,
cDNA molecules, and discriminating fragments thereof (e.g., oligonucleotides,
ESTs, STSs,
etc.).
The nucleic acid molecules detected and/or analyzed by the methods and
compositions of the invention may be naturally occurring nucleic acid
molecules such as
genomic or extragenomic DNA molecules isolated from a sample, or RNA
molecules, such
as mRNA molecules, present in, isolated from or derived from a biological
sample. The
sample of nucleic acids detected and/or analyzed by the methods and
compositions of the
invention comprise, e.g., molecules of DNA, RNA, or copolymers of DNA and RNA.
Generally, these nucleic acids correspond to particular genes or alleles of
genes, or to
particular gene transcripts (e.g., to particular mRNA sequences expressed in
specific cell
types or to particular cDNA sequences derived from such mRl'+TA sequences).
The nucleic
acids detected and/or analyzed by the methods and compositions of the
invention may
correspond to different exons of the same gene, e.g., so that different splice
variants of that
gene may be detected and/or analyzed.
In specific embodiments, the nucleic acids are prepared in vitro from nucleic
acids
present in, or isolated or partially isolated from biological a sample. For
example, in one
embodiment, RNA is extracted from a sample (e.g., total cellular RNA, poly(A)+
messenger RNA, fraction thereof) and messenger RNA is purified from the total
extracted
RNA. Methods for preparing total and poly(A)+ RNA are well known in the art,
and are
described generally, e.g., in Sambrook et al., 2001, Molecular Cloning: A
Laboratory
Manual. 3a edition, Cold Spring Harbor Laboratory Press (Cold Spring Harbor,
New York),
which is incorporated by reference herein in its entirety. In one embodiment,
RNA is
extracted from a sample using guanidinium thiocyanate lysis followed by CsCI
centrifugation and an oligo dT purification (Chirgwin et al., 1979,
Biochemistry
18:5294-5299). In another embodiment, RNA is extracted from a sample using
guanidinium thiocyanate lysis followed by purification on RNeasy columns
(Qiagen,
Valencia, California). eDNA is then synthesized from the purified mRNA using,
e.g.,
oligo-dT or random primers. In specific embodiments, the target nucleic acids
are cRNA
prepared from purified messenger RNA extracted from a sample. As used herein,
cRNA is
defined here as RNA complementary to the source RNA. The extracted RNAs are
amplified using a process in which doubled-stranded cDNAs are synthesized from
the
-51-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
RNAs using a primer linked to an RNA polymerase promoter in a direction
capable of
directing transcription of anti-sense RNA. Anti-sense RNAs or eRNAs are then
transcribed
from the second strand of the double-stranded cDNAs using an RNA polymerase
(see, e.g.,
U.S. Patent Nos. 5,891,636, 5,716,785; 5,545,522 and 6,132,997, which are
hereby
incorporated by reference). Both oligo-dT primers (U.S. Patent Nos. 5,545,522
and
6,132,997, hereby incorporated by reference herein) or random primers that
contain an RNA
polymerase promoter or complement thereof can be used. In some embodiments the
target
nucleic acids are short and/or fragmented nucleic acid molecules which are
representative of
the original nucleic acid population of the sample.
In one embodiment, nucleic acids of the invention can be detectably labeled.
For
example, cDNA can be labeled directly, e.g., with nucleotide analogs, or
indirectly, e.g., by
making a second, labeled cDNA strand using the first strand as a template.
Alternatively,
the double-stranded cDNA can be transcribed into cRNA and labeled.
In some embodiments the detectable label is a fluorescent label, e.g., by
incorporation of nucleotide analogs. Other labels suitable for use in the
present invention
include, but are not limited to, biotin, imminobiotin, antigens, cofactors,
dinitrophenol,
lipoic acid, olefinic compounds, detectable polypeptides, electron rich
molecules, enzymes
capable of generating a detectable signal by action upon a substrate, and
radioactive
isotopes. Suitable radioactive isotopes include 32P, 35S, IaC, "N and "SI.
Fluorescent
molecules suitable for the present invention include, but are not limited to,
fluorescein and
its derivatives, rhodamine and its derivatives, Texas red, 5 carboxy-
fluorescein ("FMA"),
6-carboxy-4',5'-dichloro-2',7'-dimethoxyfluorescein, succinimidyl ester
("JOE"),
6-carboxytetramethylrhodamine ("TAMRA"), 6Ncarboxy-X-rhodamine ("ROX"), HEX,
TET, IRD40, and IRD41. Fluorescent molecules that are suitable for the
invention further
include, but are not limited to: cyamine dyes, including by not limited to
Cy3, Cy3.5 and
Cy5; BODIPY dyes including but not limited to BODIPY-FL, BODIPY-TR,
BODIPY-TMR, BODIPY-630/650, BODIPY-650/670; and ALEXA dyes, including but not
limited to ALEXA-488, ALEXA-532, ALEXA-546, ALEXA-568, and ALEXA-594; as
well as other fluorescent dyes which will be known to those who are skilled in
the art.
Electron-rich indicator molecules suitable for the present invention include,
but are not
limited to, ferritin, hemocyanin, and colloidal gold. Alternatively, in some
embodiments the
target nucleic acids may be labeled by specifically complexing a first group
to the nucleic
acid. A second group, covalently linked to an indicator molecules and which
has an affinity
for the first group, can be used to indirectly detect the target nucleic acid.
In such an
- 52 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
embodiment, compounds suitable for use as a first group include, but are not
limited to,
biotin and iminobiotin. Compounds suitable for use as a second group include,
but are not
limited to, avidin and streptavidin.
5.4.1.1 Nucleic acid arrays
In certain embodiments of the invention, nucleic acid arrays are employed to
generate features of biomarkers in a biomarker profile by detecting the
expression of any
one or more of the genes described herein (e.g., a gene listed in Tables 1, 4,
5, 6, 7, 8 and/or
9). In one embodiment of the invention, a microarray, such as a cDNA
microarray, is used
to determine feature values of biomarkers in a biomarker profile. The
diagnostic use of
eDNA arrays is well known in the art. (See, e.g., Zou et. al., 2002, Oncogene
21:4855-
4862; as well as Draghici, 2003, Data Analysis Tools for DNA Microarrays,
Chapman &
Hall/CRC, each of which is hereby incorporated by reference herein in its
entirety).
In certain embodiments, the feature values for biomarkers in a biomarker
profile are
obtained by hybridizing to the array detectably labeled nucleic acids
representing or
corresponding to the nucleic acid sequences in mRNA transcripts present in a
biological
sample (e.g., fluorescently labeled cDNA synthesized from the sample) to a
microarray
comprising one or more probe spots.
Nucleic acid arrays, for example, microarrays, can be made in a number of
ways, of
which several are described herein below. Preferably, the arrays are
reproducible, allowing
multiple copies of a given array to be produced and results from said
microarrays compared
with each other. Preferably, the arrays are made from materials that are
stable under
binding (e.g., nucleic acid hybridization) conditions. Those skilled in the
art will know of
suitable supports, substrates or carriers for hybridizing test probes to probe
spots on an
array, or will be able to ascertain the same by use of routine
experimentation.
Arrays, for example, microarrays, used can include one or more test probes. In
some embodiments each such test probe comprises a nucleic acid sequence that
is
complementary to a subsequence of RNA or DNA to be detected. Each probe
typically has
a different nucleic acid sequence, and the position of each probe on the solid
surface of the
array is usually known or can be determined. Arrays useful in accordance with
the
invention can include, for example, oligonucleotide microarrays, cDNA based
arrays, SNP
arrays, spliced variant arrays and any other array able to provide a
qualitative, quantitative
or semi-quantitative measurement of expression of a gene described herein
(e.g., a gene
listed in Tables 1, 4, 5, 6, 7, 8 and/or 9). Some types of microarrays are
addressable arrays.
-53-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
More specifically, some microarrays are positionally addressable arrays. In
some
embodiments, each probe of the array is located at a known, predetermined
position on the
solid support so that the identity (e.g., the sequence) of each probe can be
determined from
its position on the array (e.g., on the support or surface). In some
embodiments, the arrays
are ordered arrays. Microarrays are generally described in Draghici, 2003,
Data Analysis
Tools for DNA Microarrays, Chapman & Hall/CRC, which is hereby incorporated by
reference herein in its entirety.
In some embodiments of the present invention, an expressed transcript (e.g., a
transcript of a gene described herein) is represented in the nucleic acid
arrays. In such
embodiments, a set of binding sites can include probes with different nucleic
acids that are
complementary to different sequence segments of the expressed transcript.
Exemplary
nucleic acids that fall within this class can be of length of 15 to 200 bases,
20 to 100 bases,
25 to 50 bases, 40 to 60 bases or some other range of bases. Each probe
sequence can also
comprise one or more linker sequences in addition to the sequence that is
complementary to
its target sequence. As used herein, a linker sequence is a sequence between
the sequence
that is complementary to its target sequence and the surface of support. For
example, the
nucleic acid arrays of the invention can comprise one probe specific to each
target gene or
exon. However, if desired, the nucleic acid arrays can contain at least 2, 5,
10, 100, or 1000
or more probes specific to some expressed transcript (e.g., a transcript of a
gene described ,
herein, e.g., in Tables 1, 4, 5, 6, 7, 8 and/or 9). For example, the array may
contain probes
tiled across the sequence of the longest mRNA isqform of a gene.
It will be appreciated that when cDNA complementary to the RNA of a cell, for
example, a cell in a biological sample, is made and hybridized to a microarray
under
suitable hybridization conditions, the level of hybridization to the site in
the array
corresponding to a gene described herein (e.g., a gene listed in Tables 1, 4,
5, 6, 7, 8 and/or
9) will reflect the prevalence in the cell of mRNA or mRNAs transcribed from
that gene.
Alternatively, in instances where multiple isoforms or alternate splice
variants produced by
particular genes are to be distinguished, detectably labeled (e.g., with a
fluorophore) cDNA
complementary to the total cellular mRNA can be hybridized to a microarray,
and the site
on the array corresponding to an exon of the gene that is not transcribed or
is removed
during RNA splicing in the cell will have little or no signal (e.g.,
fluorescent signal), and a
site corresponding to an exon of a gene for which the encoded mRNA expressing
the exon
is prevalent will have a relatively strong signal. The relative abundance of
different
-54-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
mRNAs produced from the same gene by alternative splicing is then determined
by the
signal strength pattern across the whole set of exons monitored for the gene.
In one embodiment, hybridization levels at different hybridization times are
measured separately on different, identical microarrays. For each such
measurement, at
hybridization time when hybridization level is measured, the microarray is
washed briefly,
preferably in room temperature in an aqueous solution of high to moderate salt
concentration (e.g., 0.5 to 3 M salt concentration) under conditions which
retain all bound
or hybridized nucleic acids while removing all unbound nucleic acids. The
detectable label
on the remaining, hybridized nucleic acid molecules on each probe is then
measured by a
method which is appropriate to the particular labeling method used. The
resulting
hybridization levels are then combined to form a hybridization curve. In
another
embodiment, hybridization levels are measured in real time using a single
microarray. In
this embodiment, the microarray is allowed to hybridize to the sample without
interruption
and the microarray is interrogated at each hybridization time in a non-
invasive manner. In
still another embodiment, one can use one array, hybridize for a short time,
wash and
measure the hybridization level, put back to the same sample, hybridize for
another period
of time, wash and measure again to get the hybridization time curve.
In some embodiments, nucleic acid hybridization and wash conditions are chosen
so
that the nucleic acid biomarkers to be analyzed specifically bind or
specifically hybridize to
the complementary nucleic acid sequences of the array, typically to a specific
array site,
where its complementary DNA is located.
Arrays containing double-stranded probe DNA situated thereon can be subjected
to
denaturing conditions to render the DNA single-stranded prior to contacting
with the target
nucleic acid molecules. Arrays containing single-stranded probe DNA (e.g.,
synthetic
oligodeoxyribonucleic acids) may need to be denatured prior to contacting with
the target
nucleic acid molecules, e.g., to remove hairpins or dimers which form due to
self
complementary sequences.
Optimal hybridization conditions will depend on the length (e.g., oligomer
versus
polynucleotide greater than 200 bases) and type (e.g., RNA, or DNA) of probe
and target
nucleic acids. General parameters for specific (i.e., stringent) hybridization
conditions for
nucleic acids are described in Sambrook et al., (supra), and in Ausubel et
al., 1988, Current
Protocols in Molecular Biology, Greene Publishing and Wiley-Interscience, New
York.
When the cDNA microarrays of Shena et al. are used, typical hybridization
conditions are
hybridization in 5 X SSC plus 0.2% SDS at 65 C for four hours, followed by
washes at
-55-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
25 C in low stringency wash buffer (1 X SSC plus 0.2% SDS), followed by 10
minutes at
25 C in higher stringency wash buffer (0.1 X SSC plus 0.2% SDS) (Shena et al.,
1996,
Proc. Natl. Acad. Sci. U.S.A. 93:10614). Useful hybridization conditions are
also provided
in, e.g., Tijessen, 1993, Hybridization With Nucleic Acid Probes, Elsevier
Science
Publishers B.V.; Kricka,1992, Nonisotopic DNA Probe Techniques, Academic
Press, San
Diego, CA; and Zou et. al., 2002, Oncogene 21:4855-4862; and Draghici, Data
Anal,ysis
Tools for DNA Microanalysis, 2003, CRC Press LLC, Boca Raton, Florida, pp. 342-
343,
which are hereby incorporated by reference herein in their entirety.
In a specific embodiment, a microarray can be used to sort out RT-PCR products
that have been generated by the methods described, for example, below in
Section 5.4.1.2.
5.4.1.2 RT-PCR
In certain embodiments, to determine the feature values of biomarkers in a
biomarker profile of the invention, the level of expression of one or more of
the genes
described herein (e.g., a gene listed in Tables 1, 4, 5, 6, 7, 8 and/or 9) is
measured by
amplifying RNA from a sample using reverse transcription (RT) in combination
with the
polymerase chain reaction (PCR). In accordance with this embodiment, the
reverse
transcription may be quantitative or semi-quantitative. The RT-PCR methods
taught herein
may be used in conjunction with the microarray methods described above, for
example, in
Section 5.4.1.1. For example, a bulk PCR reaction may be performed, the PCR
products
may be resolved and used as probe spots on a microarray.
Total RNA, or mRNA from a sample is used as a template and a primer specific
to
the transcribed portion of the gene(s) is used to initiate reverse
transcription. Methods of
reverse transcribing RNA into cDNA are well known and described in Sambrook et
al.,
2001, supra. Primer design can be accomplished based on known nucleotide
sequences that
have been published or available from any publicly available sequence database
such as
GenBank. For example, primers may be designed for any of the genes described
herein
(see, e.g., Table 1, 4, 5, 6, 7, 8 and/or 9, which provides the GenBank
accession numbers of
the nucleotide and amino acid sequences of the genes described herein).
Further, primer
design may be accomplished by utilizing commercially available software (e.g.,
Primer
Designer 1.0, Scientific Software etc.). The product of the reverse
transcription is
subsequently used as a template for PCR.
PCR provides a method for rapidly amplifying a particular nucleic acid
sequence by
using multiple cycles of DNA replication catalyzed by a thermostable, DNA-
dependent
-56-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
DNA polymerase to amplify the target sequence of interest. PCR requires the
presence of a
nucleic acid to be amplified, two single-stranded oligonucleotide primers
flanking the
sequence to be amplified, a DNA polymerase, deoxyribonucleoside triphosphates,
a buffer
and salts. The method of PCR is well known in the art. PCR is performed, for
example, as
described in Mullis and Faloona, 1987, Methods Enzymol. 155:335, which is
hereby
incorporated by reference herein in its entirety.
PCR can be performed using template DNA or cDNA (at least 1 fg; more usefully,
1-
1000 ng) and at least 25 pmol of oligonucleotide primers. A typical reaction
mixture
includes: 2 l of DNA, 25 pmol of oligonucleotide primer, 2.5 l of 10 M PCR
buffer I
(Perkin-Elmer, Foster City, California), 0.4 l of 1.25 M dNTP, 0.15 1 (or
2.5 units) of Taq
DNA polymerase (Perkin Elmer, Foster City, California) and deionized water to
a total
volume of 25 l. Mineral oil is overlaid and the PCR is performed using a
programmable
thermal cycler.
The length and temperature of each step of a PCR cycle, as well as the number
of
cycles, are adjusted according to the stringency requirements in effect.
Annealing
temperature and timing are determined both by the efficiency with which a
primer is
expected to anneal to a template and the degree of mismatch that is to be
tolerated. The
ability to optimize the stringency of primer annealing conditions is well
within the
knowledge of one of moderate skill in the art. An annealing temperature of
between 30 C
and 72 C is used. Initial denaturation of the template molecules,normally
occurs at between
92 C and 99 C for 4 minutes, followed by 20-40 cycles consisting of
denaturation (94-99 C
for 15 seconds to 1 minute), annealing (temperature determined as discussed
above; 1-2
minutes), and extension (72 C for 1 minute). The final extension step is
generally carried
out for 4 minutes at 72 C, and may be followed by an indefinite (0-24 hour)
step at 4 C.
Quantitative RT-PCR ("QRT-PCR"), which is quantitative in nature, can also be
performed to provide a quantitative measure of gene expression levels. In QRT-
PCR
reverse transcription and PCR can be performed in two steps, or reverse
transcription
combined with PCR can be performed concurrently. One of these techniques, for
which
there are commercially available kits such as Taqman (Perkin Elmer, Foster
City, CA) or as
provided by Applied Biosystems (Foster City, CA) is performed with a
transcript-specific
antisense probe. This probe is specific for the PCR product (e.g: a nucleic
acid fragment
derived from a gene) and is prepared with a quencher and fluorescent reporter
probe
complexed to the 5' end of the oligonucleotide. Different fluorescent markers
are attached
to different reporters, allowing for measurement of two products in one
reaction. When Taq
-57-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
DNA polymerase is activated, it cleaves off the fluorescent reporters of the
probe bound to
the template by virtue of its 5'-to-3' exonuclease activity. In the absence of
the quenchers,
the reporters now fluoresce. The color change in the reporters is proportional
to the amount
of each specific product and is measured by a fluorometer; therefore, the
amount of each
color is measured and the PCR product is quantified. The PCR reactions are
performed in
96-well plates so that samples derived from many individuals are processed and
measured
simultaneously. The Taqman system has the additional advantage of not
requiring gel
electrophoresis and allows for quantification when used with a standard curve.
A second technique useful for detecting PCR products quantitatively without is
to
use an intercolating dye such as the commercially available QuantiTect SYBR
Green PCR
(Qiagen, Valencia California). RT-PCR is performed using SYBR green as a
fluorescent
label which is incorporated into the PCR product during the PCR stage and
produces a
flourescense proportional to the amount of PCR product.
Both Taqman and QuantiTect SYBR systems can be used subsequent to reverse
transcription of RNA. Reverse transcription can either be performed in the
same reaction
mixture as the PCR step (one-step protocol) or reverse transcription can be
performed first
prior to amplification utilizing PCR (two-step protocol).
Additionally, other systems to quantitatively measure mRNA expression products
are known including Molecular BeaconsO which uses a probe having a fluorescent
molecule and a quencher molecule, the probe capable of forming a hairpin
structure such
that when in the hairpin form, the fluorescence molecule is quenched, and when
hybridized
the fluorescence increases giving a quantitative measurement of gene
expression.
Additional techniques to quantitatively measure RNA expression include, but
are not
limited to, polymerase chain reaction, ligase chain reaction, Qbeta replicase
(see, e.g.,
International Application No. PCT/US87/00880, which is hereby incorporated by
reference
herein in its entirety), isothermal amplification method (see, e.g., Walker el
al., 1992, PNAS
89:382-396, which is hereby incorporated by reference herein in its entirety),
strand
displacement amplification (SDA), repair chain reaction, Asymmetric
Quantitative PCR
(see, e.g., U.S. Publication No. US 2003/30134307A1, herein incorporated by
reference)
and the multiplex microsphere bead assay described in Fuja et al., 2004,
Journal of
Biotechnology 108:193-205, herein incorporated by reference.
The level of expression of one or more of the genes described herein (e.g.,
the genes
listed in Tables 1, 4, 5, 6, 7, 8 and/or 9) can, for example, be measured by
amplifying RNA
from a sample using amplification (NASBA). See, e.g., Kwoh et al.,1989, PNAS
USA
-58-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
86:1173; International Publication No. WO 88/10315; and U.S. Patent No.
6,329,179, each
of which is hereby incorporated by reference. In NASBA, the nucleic acids may
be
prepared for amplification using conventional methods, e.g., phenol/chloroform
extraction,
heat denaturation, treatment with lysis buffer and minispin columns for
isolation of DNA
and RNA or guanidinium chloride extraction of RNA. These amplification
techniques
involve annealing a primer that has target specific sequences. Following
polymerization,
DNA/RNA hybrids are digested with RNase H while double stranded DNA molecules
are
heat denatured again. In either case the single stranded DNA is made fully
double stranded
by addition of second target specific primer, followed by polymerization. The
double-
stranded DNA molecules are then multiply transcribed by a polymerase such as
T7 or SP6.
In an isothermal cyclic reaction, the RNA's are reverse transcribed into
double stranded
DNA, and transcribed once with a polymerase such as T7 or SP6. The resulting
products,
whether trun.cated or complete, indicate target specific sequences.
Several techniques may be used to separate amplification products. For
example,
amplification products may be separated by agarose, agarose-acrylamide or
polyacrylamide
gel electrophoresis using conventional methods. See Sambrook et al., 2001.
Several
techniques for detecting PCR products quantitatively without electrophoresis
may also be
used according to the invention (see, e.g., PCR Protocols, A Guide to Methods
and
Applications, Innis et al., 1990, Academic Press, Inc. N.Y., which is hereby
incorporated by
reference). For example, chromatographic techniques may be employed to effect
separation. There are many kinds of chromatography which may be used in the
present
invention: adsorption, partition, ion-exchange and molecular sieve, HPLC, and
many
specialized techniques for using them including column, paper, thin-layer and
gas
chromatography (Freifelder, Physical Biochemistry Applications to Biochemistry
and
Molecular Biology, 2nd ed., Wm. Freeman and Co., New York, N.Y., 1982, which
is
hereby incorporated by reference).
Another example of a separation methodology is to covalently label the
oligonucleotide primers used in a PCR reaction with various types of small
molecule
ligands. In one such separation, a different ligand is present on each
oligonucleotide. A
molecule, perhaps an antibody or avidin if the ligand is biotin, that
specifically binds to one
of the ligands is used to coat the surface of a plate such as a 96 well ELISA
plate. Upon
application of the PCR reactions to the surface of such a prepared plate, the
PCR products
are bound with specificity to the surface. After washing the plate to remove
unbound
reagents, a solution containing a second molecule that binds to the first
ligand is added. This
-59-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
second molecule is linked to some kind of reporter system. The second molecule
only binds
to the plate if a PCR product has been produced whereby both oligonucleotide
primers are
incorporated into the final PCR products. The amount of the PCR product is
then detected
and quantified in a commercial plate reader much as ELISA reactions are
detected and
quantified. An ELISA-like system such as the one described here has been
developed by
Raggio Italgene (under the C-Track tradename).
Amplification products should be visualized in order to confirm amplification
of the
nucleic acid sequences of interest, i.e., nucleic acid sequences of one or
more of the genes
described herein (e.g., a gene listed in Tables 1, 4, 5, 6, 7, 8 and/or 9).
One typical
visualization method involves staining of a gel with ethidium bromide and
visualization
under UV light. Alternatively, if the amplification products are integrally
labeled with
radio- or fluorometrically-labeled nucleotides, the amplification products may
then be
exposed to x-ray film or visualized under the appropriate stimulating spectra,
following
separation.
In one embodiment, visualization is achieved indirectly. Following separation
of
amplification products, a labeled, nucleic acid probe is brought into contact
with the
amplified nucleic acid sequence of interest, i.e., nucleic acid sequences of
one or more of
the genes described herein (e.g., a gene listed in Tables 1, 4, 5, 6, 7, 8
and/or 9). The probe
preferably is conjugated to a chromophore but may be radiolabeled. In another
embodiment, the probe is conjugated to a binding partner, such as an antibody
or biotin,
where the other member of the binding pair carries a detectable moiety.
In another embodiment, detection is by Southern blotting and hybridization
with a
labeled probe. The techniques involved in Southern blotting are well known to
those of
skill in the art and may be found in many standard books on molecular
protocols. See
Sambrook et al., 2001. Briefly, amplification products are separated by gel
electrophoresis.
The gel is then contacted with a membrane, such as nitrocellulose, permitting
transfer of the
nucleic acid and non-covalent binding. Subsequently, the membrane is incubated
with a
chromophore-conjugated probe that is capable of hybridizing with a target
amplification
product. Detection is by exposure of the membrane to x-ray film or ion-
emitting detection
devices. One example of the foregoing is described in U.S. Pat. No. 5,279,721,
incorporated by reference herein, which discloses an apparatus and method for
the
automated electrophoresis and transfer of nucleic acids. The apparatus permits
electrophoresis and blotting without external manipulation of the gel and is
ideally suited to
carrying out methods according to the present invention.
-60-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
5.4.1.3 Nuclease protection assays
In particular embodiments, feature values for biomarkers in a biomarker
profile can
be obtained by performing nuclease protection assays (including both
ribonuclease
protection assays and S 1 nuclease assays) to detect and quantify specific
mRNAs (e.g.,
mRNAs of a gene described in Tables 1, 4, 5, 6, 7, 8 and/or 9). Such assays
are described
in, for example, Sambrook et al., 2001, supra. In nuclease protection assays,
an antisense
probe (labeled with, e.g., radiolabeled or nonisotopic) hybridizes in solution
to an RNA
sample. Following hybridization, single-stranded, unhybridized probe and RNA
are
degraded by nucleases. An acrylamide gel is used to separate the remaining
protected
fragments. Typically, solution hybridization is more efficient than membrane-
based
hybridization, and it can accommodate up to 100 g of sample RNA, compared
with the
20-30 g maximum of blot hybridizations.
The ribonuclease protection assay, which is the most common type of nuclease
protection assay, requires the use of RNA probes. Oligonucleotides and other
single-
stranded DNA probes can only be used in assays containing S1 nuclease. The
single-
stranded, antisense probe must typically be completely homologous to target
RNA to
prevent cleavage of the probe:target hybrid by nuclease.
5.4.1.4 Northern blot assays
Any hybridization technique known to those of skill in the art can be used to
generate feature values for biomarkers in a biomarker profile. In other
particular
embodiments, feature values for biomarkers in a biomarker profile can be
obtained by
Northern blot analysis (to detect and quantify specific RNA molecules (e.g.,
RNAs of a
gene described in Tables 1, 4, 5, 6, 7, 8 and/or 9). A standard Northern blot
assay can be
used to ascertain an RNA transcript size, identify alternatively spliced RNA
transcripts, and
the relative amounts of one or more genes described herein (in particular,
mRNA) in a
sample, in accordance with conventional Northern hybridization techniques
known to those
persons of ordinary skill in the art. In Northern blots, RNA samples are first
separated by
size via electrophoresis in an agarose gel under denaturing conditions. The
RNA is then
transferred to a membrane, crosslinked and hybridized with a labeled probe.
Nonisotopic or
high specific activity radiolabeled probes can be used including random-
primed, nick-
translated, or PCR-generated DNA probes, in vitro transcribed RNA probes, and
oligonucleotides. Additionally, sequences with only partial homology (e.g.,
cDNA from a
-61-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
different species or genomic DNA fragments that might contain an exon) may be
used as
probes. The labeled probe, e.g., a radiolabelled cDNA, either containing the
full-length,
single stranded DNA or a fragment of that DNA sequence may be at least 20, at
least 30, at
least 50, or at least 100 consecutive nucleotides in length. The probe can be
labeled by any
of the many different methods known to those skilled in this art. The labels
most
commonly employed for these studies are radioactive elements, enzymes,
chemicals that
fluoresce when exposed to ultraviolet light, and others. A number of
fluorescent materials
are known and can be utilized as labels. These include, but are not limited
to, fluorescein,
rhodamine, auramine, Texas Red, AMCA blue and Lucifer Yellow. The radioactive
label
can be detected by any of the currently available counting procedures. Non-
limiting
examples of isotopes include 3H, 14C' 32P, 35S, 36C,I, 51Cr, 57C0, 58C'O,
59Fe, 90Y, 125I11311, and
186Re. Enzyme labels are likewise useful, and can be detected by any of the
presently
utilized colorimetric, spectrophotometric, fluorospectrophotometric,
amperometric or
gasometric techniques. The enzyme is conjugated to the selected particle by
reaction with
bridging molecules such as carbodiimides, diisocyanates, glutaraldehyde and
the like. Any
enzymes known to one of skill in the art can be utilized. Examples of such
enzymes
include, but are not limited to, peroxidase, beta-D-galactosidase, urease,
glucose oxidase
plus peroxidase and alkaline phosphatase. U.S. Patent Nos. 3,654,090,
3,850,752, and
4,016,043 are referred to by way of example for their disclosure of alternate
labeling
material and methods.
5.4.2 Methods of detecting proteins
In specific embodiments of the invention, feature values of biomarkers in a
biomarker profile can be obtained by detecting proteins, for example, by
detecting the
expression product (e.g., a nucleic acid or protein) of one or more genes
described herein
(e.g., a gene listed in Tables 1, 4, 5, 6, 7, 8 and/or 9), or post-
translationally modified, or
otherwise modified, or processed forms of such proteins. In a specific
embodiment, a
biomarker profile is generated by detecting and/or analyzing one or more
proteins and/or
discriminating fragments thereof expressed from a gene disclosed herein (e.g.,
a gene listed
in Tables 1, 4, 5, 6, 7, 8 and/or 9) using any method known to those skilled
in the art for
detecting proteins including, but not limited to protein nlicroarray analysis,
immunohistochemistry and mass spectrometry.
Standard techniques may be utilized for determining the amount of the protein
or
proteins of interest (e.g., proteins expressed from genes listed in Tables 1,
4, 5, 6, 7, 8
-62-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
and/or 9) present in a sample. For example, standard techniques can be
employed using,
e.g., immunoassays such as, for example Western blot, immunoprecipitation
followed by
sodium dodecyl sulfate polyacrylamide gel electrophoresis, (SDS-PAGE),
immunocytochemistry, and the like to determine the amount of protein or
proteins of
interest present in a sample. One exemplary agent for detecting a protein of
interest is an
antibody capable of specifically binding to a protein of interest, preferably
an antibody
detectably labeled, either directly or indirectly.
For such detection methods, if desired a protein from the sample to be
analyzed can
easily be isolated using techniques which are well known to those of skill in
the art. Protein
isolation methods can, for example, be such as those described in Harlow and
Lane, 1988,
Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory Press (Cold
Spring
Harbor, New York), which is incorporated by reference herein in its entirety.
In certain embodiments, methods of detection of the protein or proteins of
interest
involve their detection via interaction with a protein-specific antibody. For
example,
antibodies directed to a protein of interest (e.g., a protein expressed from a
gene described
herein, e.g., a protein listed in Tables 1, 4, 5, 6, 7, 8 and/or 9).
Antibodies can be generated
utilizing standard techniques well known to those of skill in the art. In
specific
embodiments, antibodies can be polyclonal, or more preferably, monoclonal. An
intact
antibody, or an antibody fragment (e.g., scFv, Fab or F(ab')2) can, for
example, be used.
For example, antibodies, or fragments of antibodies, specific for a protein of
interest
can be used to quantitatively or qualitatively detect the presence of a
protein. This can be
accomplished, for example, by immunofluorescence techniques. Antibodies (or
fragments
thereof) can, additionally, be employed histologically, as in
immunofluorescence or
inununoelectron microscopy, for in situ detection of a protein of interest. In
situ detection
can be accomplished by removing a biological sample (e.g., a biopsy specimen)
from a
patient, and applying thereto a labeled antibody that is directed to a protein
of interest (e.g.,
a protein expressed from a gene in Tables 1, 4, 5, 6, 7, 8 and/or 9). The
antibody (or
fragment) is preferably applied by overlaying the antibody (or fragment) onto
a biological
sample. Through the use of such a procedure, it is possible to determine not
only the
presence of the protein of interest, but also its distribution, in a
particular sample. A wide
variety of well-known histological methods (such as staining procedures) can
be utilized to
achieve such in situ detection.
Immunoassays for a protein of interest typically comprise incubating a
biological
sample of a detectably labeled antibody capable of identifying a protein of
interest, and
- 63 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
detecting the bound antibody by any of a number of techniques well-known in
the art. As
discussed in more detail, below, the term "labeled" can refer to direct
labeling of the
antibody via, e.g., coupling (i.e., physically linking) a detectable substance
to the antibody,
and can also refer to indirect labeling of the antibody by reactivity with
another reagent that
is directly labeled. Examples of indirect labeling include detection of a
primary antibody
using a fluorescently labeled secondary antibody.
The biological sample can be brought in contact with and immobilized onto a
solid
phase support or carrier such as nitrocellulose, or other solid support which
is capable of
immobilizing cells, cell particles or soluble proteins. The support can then
be washed with
suitable buffers followed by treatment with the detectably labeled fingerprint
gene-specific
antibody. The solid phase support can then be washed with the buffer a second
time to
remove unbound antibody. The amount of bound label on solid support can then
be
detected by conventional methods.
By "solid phase support or carrier" is intended any support capable of binding
an
antigen or an antibody. Well-known supports or carriers include glass,
polystyrene,
polypropylene, polyethylene, dextran, nylon, amylases, natural and modified
celluloses,
polyacrylamides and magnetite. The nature of the carrier can be either soluble
to some
extent or insoluble for the purposes of the present invention. The support
material can have
virtually any possible structural configuration so long as the coupled
molecule is capable of
binding to an antigen or antibody. Thus, the support configuration can be
spherical, as in a
bead, or cylindrical, as in the inside surface of a test tube, or the external
surface of a rod.
Alternatively, the surface can be flat such as a sheet, test strip, etc.
Preferred supports
include polystyrene beads. Those skilled in the art will know many other
suitable carriers
for binding antibody or antigen, or will be able to ascertain the same by use
of routine
experimentation.
One of the ways in which an antibody specific for a protein of interest can be
detectably labeled is by linking the same to an enzyme and use in an enzyme
immunoassay
(EIA) (Voller, 1978, "The Enzyme Linked Immunosorbent Assay (ELISA),"
Diagnostic
Horizons 2:1-7, Microbiological Associates Quarterly Publication,
Walkersville, MD;
Voller et al., 1978, J. Clin. Pathol. 31:507-520; Butler, J.E., 1981, Meth.
Enzymol.
73:482-523; Maggio, E. (ed.), 1980, Enzyme Immunoassay, CRC Press, Boca Raton,
FL;
Ishikawa, E. et al., (eds.), 1981, Enzyme Immunoassay, Kgaku Shoin, Tokyo,
each of which
is hereby incorporated by reference in its entirety). The enzyme which is
bound to the
antibody will react with an appropriate substrate, preferably a chromogenic
substrate, in
-64-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
such a manner as to produce a chemical moiety which can be detected, for
example, by
spectrophotometric, fluorimetric or by visual means. Enzymes which can be used
to
detectably label the antibody include, but are not limited to, malate
dehydrogenase,
staphylococcal nuclease, delta-5-steroid isomerase, yeast alcohol
dehydrogenase, alpha-
glycerophosphate, dehydrogenase, triose phosphate isomerase, horseradish
peroxidase,
alkaline phosphatase, asparaginase, glucose oxidase, beta-galactosidase,
ribonuclease,
urease, catalase, glucose-6-phosphate dehydrogenase, glucoamylase and
acetylcholinesterase. The detection can be accomplished by colorimetric
methods which
employ a chromogenic substrate for the enzyme. Detection can also be
accomplished by
visual comparison of the extent of enzymatic reaction of a substrate in
comparison with
similarly prepared standards.
Detection can also be accomplished using any of a variety of other
immunoassays.
For example, by radioactively labeling the antibodies or antibody fragments,
it is possible to
detect a protein of interest through the use of a radioimmunoassay (RIA) (see,
for example,
Weintraub, 1986, Principles of Radioimmunoassays, Seventh Training Course on
Radioligand Assay Techniques, The Endocrine Society, March, which is hereby
incorporated by reference herein). The radioactive isotope (e.g., 1251, 1311,
35S or 3H) can be
detected by such means as the use of a gamma counter or a scintillation
counter or by
autoradiography.
It is also possible to label the antibody with a fluorescent compound. When
the
fluorescently labeled antibody is exposed to light of the proper wavelength,
its presence can
then be detected due to fluorescence. Among the most commonly used fluorescent
labeling
compounds are fluorescein isothiocyanate, rhodamine, phycoerythrin,
phycocyanin,
allophycocyanin, o-phthaldehyde and fluorescamine.
The antibody can also be detectably labeled using fluorescence emitting metals
such
as 152,Eu, or others of the lanthanide series. These metals can be attached to
the antibody
using such metal chelating groups as diethylenetriaminepentacetic acid (DTPA)
or
ethylenediaminetetraacetic acid (EDTA).
The antibody also can be detectably labeled by coupling it to a
chemiluminescent
compound. The presence of the chemiluminescent-tagged antibody is then
determined by
detecting the presence of luminescence that arises during the course of a
chemical reaction.
Examples of particularly useful chemiluminescent labeling compounds are
luminol,
isoluminol, theromatic acridinium ester, imidazole, acridinium salt and
oxalate ester.
-65-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Likewise, a bioluminescent compound can be used to label the antibody of the
present invention. Bioluminescence is a type of chemiluminescence found in
biological
systems in, which a catalytic protein increases the efficiency of the
chemiluminescent
reaction. The presence of a bioluminescent protein is determined by detecting
the presence
of luminescence. Important bioluminescent compounds for purposes of labeling
are
luciferin, luciferase and aequorin.
In another embodiment, specific binding molecules other than antibodies, such
as
aptamers, may be used to bind the biomarkers. In yet another embodiment, the
biomarker
profile may comprise a measurable aspect of an infectious agent (e.g.,
lipopolysaccharides
or viral proteins) or a component thereof.
In some embodiments, a protein chip assay (e.g., The ProteinChip Biomarker
System, Ciphergen, Fremont, California) is used to measure feature values for
the
biomarkers in the biomarker profile. See also, for example, Lin, 2004, Modern
Pathology,
1-9; Li, 2004, Journal of Urology 171, 1782-1787; Wadsworth, 2004, Clinical
Cancer
Research, 10, 1625-1632; Prieto, 2003, Journal of Liquid Chromatography &
Related
Technologies 26, 2315-2328; Coombes, 2003, Clinical Chemistry 49, 1615-1623;
Mian,
2003, Proteomics 3, 1725-1737; Lehre et al., 2003, BJU International 92, 223-
225; and
Diamond, 2003, Journal of the American Society for Mass Spectrometry 14, 760-
765, each
of which is hereby incorporated by reference herein in its entirety.
In some embodiments, a bead assay is used to measure feature values for the
biomarkers in the biomarker profile. One such bead assay is the Becton
Dickinson
Cytometric Bead Array (CBA). CBA employs a series of particles with discrete
fluorescence intensities to simultaneously detect multiple soluble analytes.
CBA is
combined with flow cytometry to create a multiplexed assay. The Becton
Dickinson CBA
system, as embodied for example in the Becton Dickinson Human Inflammation
Kit, uses
the sensitivity of amplified fluorescence detection by flow cytometry to
measure soluble
analytes in a particle-based immunoassay. Each bead in a CBA provides a
capture surface
for a specific protein and is analogous to an individually coated well in an
ELISA plate.
The BD CBA capture bead mixture is in suspension to allow for the detection of
multiple
analytes in a small volume sample.
In some embodiments the multiplex analysis method described in U.S. Pat. No.
5,981,180 ("the '180 patent"), hereby incorporated by reference herein in its
entirety, and in
particular for its teachings of the general methodology, bead technology,
system hardware
and antibody detection, is used to measure feature values for the biomarkers
in a biomarker
-66-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
profile. For this analysis, a matrix of microparticles is synthesized, where
the matrix
consists of different sets of microparticles. Each set of microparticles can
have thousands of
molecules of a distinct antibody capture reagent immobilized on the
microparticle surface
and can be color coded by incorporation of varying amounts of two fluorescent
dyes. The
ratio of the two fluorescent dyes provides a distinct emission spectrum for
each set of
microparticles, allowing the identification of a microparticle a set following
the pooling of
the various sets of microparticles. See also United States Patent Nos.
6,268,222 and
6,599,331, also hereby incorporated by reference herein in their entireties,
and in particular
for their teachings of various methods of labeling microparticles for
multiplex analysis.
5.4.3 Use of other methods of detection
In some embodiments, a separation method may be used determine feature values
for biomarkers in a biomarker profile, such that only a subset of biomarkers
within the
sample is analyzed. For example, the biomarkers that are analyzed in a sample
may be
mRNA species from a cellular extract which has been fractionated to obtain
only the nucleic
acid biomarkers within the sample, or the biomarkers may be from a fraction of
the total
complement of proteins within the sample, which have been fractionated by
chromatographic techniques.
Feature values for biomarkers in a biomarker profile can also, for example, be
generated by the use of one or more of the following methods described below.
For
example, methods may include nuclear magnetic resonance (NMR) spectroscopy, a
mass
spectrometry method, such as electrospray ionization mass spectrometry (ESI-
MS), ESI-
MS/MS, ESI-MS/(MS)' (n is an integer greater than zero), matrix-assisted laser
desorption
ionization time-of-flight mass spectrometry (MALDI-TOF-MS), surface-enhanced
laser
desorption/ionization time-of-flight mass spectrometry (SELDI-TOF-MS),
desorption/ionization on silicon (DIOS), secondary ion mass spectrometry
(SIMS),
quadrupole time-of-flight (Q-TOF), atmospheric pressure chemical ionization
mass
spectrometry (APCI-MS), APCI-MS/MS, APCI-(MS) , atmospheric pressure
photoionization mass spectrometry (APPI-MS), APPI-MS/MS, and APPI-(MS)". Other
mass spectrometry methods may include, inter alia, quadrupole, Fourier
transform mass
spectrometry (FTMS) and ion trap. Other suitable methods may include chemical
extraction partitioning, column chromatography, ion exchange chromatography,
hydrophobic (reverse phase) liquid chromatography, isoelectric focusing, one-
dimensional
polyacrylamide gel electrophoresis (PAGE), two-dimensional polyacrylamide gel
-67-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
electrophoresis (2D-PAGE) or other chromatography, such as thin-layer, gas= or
liquid
chromatography, or any combination thereof. In one embodiment, the biological
sample
may be fractionated prior to application of the separation method.
In one embodiment, laser desorption/ionization time-of-flight mass
spectrometry is
used to create determine feature values in a biomarker profile where the
biomarkers are
proteins or protein fragments that have been ionized and vaporized off an
immobilizing
support by incident laser radiation and the feature values are the presence or
absence of
peaks representing these fragments in the mass spectra profile. A variety of
laser
desorption/ionization techniques are known in the art (see, e.g., Guttman el
al., 2001, Anal.
Chem. 73:1252-62 and Wei et al., 1999, Nature 399:243-246, each of which is
hereby
incorporated herein by reference in its entirety).
Laser desorption/ionization time-of-flight mass spectrometry allows the
generation
of large amounts of information in a relatively short period of time. A
biological sample is
applied to one of several varieties of a support that binds all of the
biomarkers, or a subset
thereof, in the sample. Cell lysates or samples are directly applied to these
surfaces in
volumes as small as 0.5 L, with or without prior purification or
fractionation. The lysates
or sample can be concentrated or diluted prior to application onto the support
surface. Laser
desorption/ionization is then used to generate mass spectra of the sample, or
samples, in as
little as three hours.
5.5 DATA ANALYSIS ALGORITHMS
Biomarkers whose corresponding feature values are capable of discriminating
between converters and nonconverters are identified in the present invention.
The identity
of these biomarkers and their corresponding features (e.g., expression levels)
can be used to
develop a decision rule, or plurality of decision rules, that discriminate
between converters
and nonconverters. Data analysis algorithms can be used to construct a number
of decision
rules. Each such data analysis algorithm uses features (e.g., expression
values) of a subset
of the biomarkers identified in the present invention across a training
population that
includes converters and nonconverters. Typically, a SIRS subject is considered
a
nonconverter when the subject does not develop sepsis in a defined time period
(e.g.,
observation period). This defined time period can be, for example, twelve
hours, twenty
four hours, forty-eight hours, a day, a week, a month, or longer. Specific
data analysis
algorithms for building a decision rule, or plurality of decision rules, that
discriminate
between subjects that develop sepsis and subjects that do not develop sepsis
during a
-68-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
defined period will be described in the subsections below. Once a decision
rule has been
built using these exemplary data analysis algorithms or other techniques known
in the art,
the decision rule can be used to classify a test subject into one of the two
or more
phenotypic classes (e.g., a converter or a nonconverter). This is accomplished
by applying
the decision rule to a biomarker profile obtained from the test subject. Such
decision rules,
therefore, have enormous value as diagnostic indicators.
The present invention provides, in one aspect, for the evaluation of a
biomarker
profile from a test subject to biomarker profiles obtained firom a training
population. In
some embodiments, each biomarker profile obtained from subjects in the
training
population, as well as the test subject, comprises a feature for each of a
plurality of different
biomarkers. In some embodiments, this comparison is accomplished by (i)
developing a
decision rule using the biomarker profiles from the training population and
(ii) applying the
decision rule to the biomarker profile from the test subject. As such, the
decision rules
applied in some embodiments of the present invention are used to determine
whether a test
subject having SIRS will or will not likely acquire sepsis.
In some embodiments of the present invention, when the results of the
application of
a decision rule indicate that the subject will likely acquire sepsis, the
subject is diagnosed as
a "sepsis" subject. If the results of an application of a decision rule
indicate that the subject
will not acquire sepsis, the subject is diagnosed as a "SIRS" subject. Thus,
in some
embodiments, the result in the above-described binary decision situation has
four possible
outcomes:
(i) truly septic, where the decision rule indicates that the subject will
acquire sepsis
and the subject does in fact acquire sepsis during the definite time period
(true positive, TP);
(ii) falsely septic, where the decision rule indicates that the subject will
acquire
sepsis and the subject, in fact, does not acquire sepsis during.the definite
time period (false
positive, FP);
(iii) truly SIRS, where the decision rule indicates that the subject will not
acquire
sepsis and the subject, in fact, does not acquire sepsis during the definite
time period (true
negative, TN); or
(iv) falsely SIRS, where the decision rule indicates that the subject will not
acquire
sepsis and the subject, in fact, does acquire sepsis during the definite time
period (false
negative, FN).
It will be appreciated that other definitions for TP, FP, TN, FN can be made.
For
example, TP could have been defined as instances where the decision rule
indicates that the
-69-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
subject will not acquire sepsis and the subject, in fact, does not acquire
sepsis during the
definite time period. While all such alternative definitions are within the
scope of the
present invention, for ease of understanding the present invention, the
definitions for TP,
FP, TN, and FN given by definitions (i) through (iv) above will be used
herein, unless
otherwise stated.
As will be appreciated by those of skill in the art, a number of quantitative
criteria
can be used to communicate the performance of the comparisons made between a
test
biomarker profile and reference biomarker profiles (e.g., the application of a
decision rule to
the biomarker profile from a test subject). These include positive predicted
value (PPV),
negative predicted value (NPV), specificity, sensitivity, accuracy, and
certainty. In
addition, other constructs such a receiver operator curves (ROC) can be used
to evaluate
decision rule performance. As used herein:
PPV = TP
TP+FP
NPV = TN
TN+FN
TN
specificity =
TN +FP
TP
sensitivity =
TP + FN
accuracy = certainty = TP +TN
N
Here, N is the number of samples compared (e.g., the number of test samples
for
which a determination of sepsis or SIRS is sought). For example, consider the
case in
which there are ten subjects for which SIRS/sepsis classification is sought.
Biomarker
profiles are constructed for each of the ten test subjects. Then, each of the
biomarker
profiles is evaluated by applying a decision rule, where the decision rule was
developed
based upon biomarker profiles obtained from a training population. In this
example, N,
from the above equations, is equal to 10. Typically, N is a number of samples,
where each
sample was collected from a different member of a population. This population
can, in fact,
be of two different types. In one type, the population comprises subjects
whose samples
and phenotypic data (e.g., feature values of biomarkers and an indication of
whether or not
the subject acquired sepsis) was used to construct or refine a decision rule.
Such a
population is referred to herein as a training population. In the other type,
the population
-70-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
comprises subjects that were not used to construct the decision rule. Such a
population is
referred to herein as a validation population. Unless otherwise stated, the
population
represented by N is either exclusively a training population or exclusively a
validation
population, as opposed to a mixture of the two population types. It will be
appreciated that
scores such as accuracy will be higher (closer to unity) when they are based
on a training
population as opposed to a validation population. Nevertheless, unless
otherwise explicitly
stated herein, all criteria used to assess the performance of a decision rule
(or other forms of
evaluation of a biomarker profile from a test subject) including certainty
(accuracy) refer to
criteria that were measured by applying the decision rule corresponding to the
criteria to
either a training population or a validation population. Furthermore, the
definitions for
PPV, NPV, specificity, sensitivity, and accuracy defined above can also be
found in
Draghici, Data Analysis Tools for DNA Microanalysis, 2003, CRC Press LLC, Boca
Raton,
Florida, pp. 342-343, which is hereby incorporated by reference herein in its
entirety.
In some embodiments the training population comprises nonconverters and
converters. In some embodiments, biomarker profiles are constructed from this
population
using biological samples collected from the training population at some time
period prior to
the onset of sepsis by the converters of the population. As such, for the
converters of the
training population, a biological sample can be collected two weeks before,
one week
before, four days before, three days before, one day before, or any other time
period before
the converters became septic. In practice, such collections are obtained by
collecting a
biological sample at regular time intervals after admittance into the hospital
with a SIRS
diagnosis. For example, in one approach, subjects who have been diagnosed with
SIRS in a
hospital are used as a training population. Once admitted to the hospital with
SIRS, the
biological samples are collected from the subjects at selected times (e.g.,
hourly, every eight
hours, every twelve hours, daily, etc.). A portion of the subjects acquire
sepsis and a
portion of the subjects do not acquire sepsis. For the subjects that acquire
sepsis, the
biological sample taken from the subjects just prior to the onset of sepsis
are termed the T.12
biological samples. All other biological samples from the subjects are
retroactively indexed
relative to these biological samples. For instance, when a biological sample
has been taken
from a subject on a daily basis, the biological sample taken the day before
the T_ia sample is
referred to as the T_36 biological sample. Time points for biological samples
for a
nonconverter in the training population are identified by "time-matching" the
nonconverter
subject with a converter subject. To illustrate, consider the case in which a
subject in the
training population became clinically-defined as septic on his sixth day of
enrollment. For
-71-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
the sake of illustration, for this subject, T_36 is day four of the study, and
the T_36 biological
sample is the biological sample that was obtained on day four of the study.
Likewise, T_36
for the matched nonconverter subject is deemed to be day four of the study on
this paired
nonconverter subject.
In some embodiments, N is more than one, more than five, more than ten, more
than
twenty, between ten and 100, more than 100, or less than 1000 subjects. A
decision rule (or
other forms of comparison) can have at least about 99% certainty, or even
more, in some
embodiments, against a training population or a validation population. In
other
embodiments, the certainty is at least about 97%, at least about 95%, at least
about 90%, at
least about 85%, at least about 80%, at least about 75%, or at least about
70%, at least about
65%, or at least about 60%, against a training population or a validation
population (and
therefore against a single subject that is not part of a training population
such as a clinical
patient). The useful degree of certainty may vary, depending on the particular
method of
the present invention. As used herein, "certainty" means "accuracy." In one
embodiment,
the sensitivity andlor specificity is at is at least about 97%, at least about
95%, at least about
90%, at least about 85%, at least about 80%, at least about 75%, or at least
about 70%
against a training population or a validation population. In some embodiments,
such
decision rules are used to predict the development of sepsis with the stated
accuracy. In
some embodiments, such decision rules are used to diagnoses sepsis with the
stated
accuracy. In some embodiments, such decision rules are used to determine a
stage of sepsis
with the stated accuracy.
The number of features that may be used by a decision rule to classify a test
subject
with adequate certainty is two or more. In some embodiments, it is three or
more, four or
more, ten or more, or between 10 and 200. Depending on the degree of certainty
sought,
however, the number of features used in a decision rule can be more or less,
but in all cases
is at least two. In one embodiment, the number of features that may be used by
a decision
rule to classify a test subject is optimized to allow a classification of a
test subject with high
certainty.
Relevant data analysis algorithms for developing a decision rule include, but
are not
limited to, discriminant analysis including linear, logistic, and more
flexible discrimination
techniques (see, e.g., Gnanadesikan, 1977, Methods for Statistical Data
Analysis of
Multivariate Observations, New York: Wiley 1977, which is hereby incorporated
by
reference herein in its entirety); tree-based algorithms such as
classification and regression
trees (CART) and variants (see, e.g., Breiman, 1984, Classifzcation and
Regression Trees,
-72-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Belmont, California: Wadsworth International Group, which is hereby
incorporated by
reference herein in its entirety, as well as Section 5.1.3, below);
generalized additive models
(see, e.g., Tibshirani , 1990, Generalized Additrve Models, London: Chapman
and Hall,
which is hereby incorporated by reference herein in its entirety); and neural
networks (see,
e.g., Neal, 1996, Bayesian Learningfor Neural Networks, New York: Springer-
Verlag; and
Insua, 1998, Feedforward neural networks for nonparametric regression In:
Practical
Nonparametric and Semiparametrie Bayesian Statistics, pp. 181-194, New York:
Springer,
which is hereby incorporated by reference herein in its entirety).
In one embodiment, comparison of a test subject's biomarker profile to a
biomarker
profiles obtained from a training population is performed, and comprises
applying a
decision rule. The decision rule is constructed using a data analysis
algorithm, such as a
computer pattern recognition algorithm. Other suitable data analysis
algorithms for
constructing decision rules include, but are not limited to, logistic
regression or a
nonparametric algorithm that detects differences in the distribution of
feature values (e.g., a
Wilcoxon Signed Rank Test (unadjusted and adjusted)). The decision rule can be
based
upon two, three, four, five, 10, 20 or more features, corresponding to
measured observables
from one, two, three, four, five, 10, 20 or more biomarkers. In one
embodiment, the
decision rule is based on hundreds of features or more. Decision rules may
also be built
using a classification tree algorithm. For example, each biomarker profile
from a training
population can comprise at least three features, where the features are
predictors in a
classification tree algorithm. The decision rule predicts membership within a
population (or
class) with an accuracy of at least about at least about 70%, of at least
about 75%, of at least
about 80%, of at least about 85%, of at least about 90%, of at least about
95%, of at least
about 97%, of at least about 98%, of at least about 99%, or about 100%.
Suitable data analysis algorithms are known in the art, some of which are
reviewed
in Hastie et al., supra. In a specific embodiment, a data analysis algorithm
of the invention
comprises Classification and Regression Tree (CART), Multiple Additive
Regression Tree
(MART), Prediction Analysis for Microarrays (PAM) or Random Forest analysis.
Such
algorithms classify complex spectra from biological materials, such as a blood
sample, to
distinguish subjects as normal or as possessing biomarker expression levels
characteristic of
a particular disease state. In other embodiments, a data analysis algorithm of
the invention
comprises ANOVA and nonparametric equivalents, linear discriminant analysis,
logistic
regression analysis, nearest neighbor classifier analysis, neural networks,
principal
component analysis, quadratic discriminant analysis, regression classifiers
and support
-73-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
vector machines. While such algorithms may be used to construct a decision
rule and/or
increase the speed and efficiency of the application of the decision rule and
to avoid
investigator bias, one of ordinary skill in the art will realize that computer-
based algorithms
are not required to cany out the methods of the present invention.
Decision rules can be used to evaluate biomarker profiles, regardless of the
method
that was used to generate the biomarker profile. For example, suitable
decision rules that
can be used to evaluate biomarker profiles generated using gas chromatography,
as
discussed in Harper, "Pyrolysis and GC in Polymer Analysis," Dekker, New York
(1985).
Further, Wagner et al., 2002, Anal. Chem. 74:1824-1835 disclose a decision
rule that
improves the ability to classify subjects based on spectra obtained by static
time-of-flight
secondary ion mass spectrometry (TOF-SIMS). Additionally, Bright et al., 2002,
J.
Microbiol. Methods 48:127-38, hereby incorporated by reference herein in its
entirety,
disclose a method of distinguishing between bacterial strains with high
certainty (79-89%
correct classification rates) by analysis of MALDI-TOF-MS spectra. Dalluge,
2000,
Fresenius J. Anal. Chem. 366:701-711, hereby incorporated by reference herein
in its
entirety, discusses the use of MALDI-TOF-MS and liquid chromatography-
electrospray
ionization mass spectrometry (LC/ESI-MS) to classify profiles of biomarkers in
complex
biological samples.
5.5.1 Decision Trees
One type of decision rule that can be constructed using the feature values of
the
biomarkers identified in the present invention is a decision tree. Here, the
"data analysis
algorithm" is any technique that can build the decision tree, whereas the
final "decision
tree" is the decision rule. A decision tree is constructed using a training
population and
specific data analysis algorithms. Decision trees are described generally by
Duda, 2001,
Pattern Classification, John Wiley & Sons, Inc., New York. pp. 395-396, which
is hereby
incorporated by reference herein. Tree-based methods partition the feature
space into a set
of rectangles, and then fit a model (like a constant) in each one.
The training population data includes the features (e.g., expression values,
or some
other observable) for the biomarkers of the present invention across a
training set
population. One specific algorithm that can be used to construct a decision
tree is a
classification and regression tree (CART). Other specific decision tree
algorithms include,
but are not limited to, ID3, C4.5, MART, and Random Forests. CART, ID3, and
C4.5 are
described in Duda, 2001, Pattern Classffrcation, John Wiley & Sons, Inc., New
York. pp.
-74-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
396-408 and pp. 411-412, which is hereby incorporated by reference. CART,
MART, and
C4.5 are described in Hastie et al., 2001, The Elements of Statistical
Learning, Springer-
Verlag, New York, Chapter 9, which is hereby incorporated by reference in its
entirety.
Random Forests are described in Breiman, 1999, "Random Forests - Random
Features,"
Technical Report 567, Statistics Department, U.C.Berkeley, September 1999,
which is
hereby incorporated by reference in its entirety.
In some embodiments of the present invention, decision trees are used to
classify
subjects using features for combinations of biomarkers of the present
invention. Decision
tree algorithms belong to the class of supervised learning algorithms. The aim
of a decision
tree is to induce a classifier (a tree) from real-world example data. This
tree can be used to
classify unseen examples that have not been used to derive the decision tree.
As such, a
decision tree is derived from training data. Exemplary training data contains
data for a
plurality of subjects (the training population). For each respective subject
there is a
plurality of features the class of the respective subject (e.g., sepsis /
SIRS). In one
embodiment of the present invention, the training data is expression data for
a combination
of biomarkers across the training population.
The following algorithm describes an exemplary decision tree derivation:
Tree(Examples,Class,Features)
Create a root node
If all Examples have the same Class value, give the root this label
Else if Features is empty label the root according to the most common value
Else begin
Calculate the information gain for each Feature
Select the Feature A with highest information gain and make this the root
Feature
For each possible value, v, of this Feature
Add a new branch below the root, corresponding to A= v
Let Examples(v) be those examples with A = v
-75-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
If Examples(v) is empty, make the new branch a leaf node labeled with the most
common value among Examples
Else let the new branch be the tree created by Tree(Examples(v),Class,Features
-
{A})
end
A more detailed description of the calculation of information gain is shown in
the
following. If the possible classes v; of the examples have probabilities P(v;)
then the
information content I of the actual answer is given by:
I(P071);...SP07 -,)) _ P(v;)log , P(v;)
s=1
The I- value shows how much information we need in order to be able to
describe
the outcome of a classification for the specific dataset used. Supposing that
the dataset
contains p positive (e.g. will develop sepsis) and n negative (e.g. will not
develop sepsis)
examples (e.g. subjects), the information contained in a correct answer is:
I( p , n ) = - p log2 ~' - . log, rz
p+n p+n p+n p+rx p+n p+n
where loga is the logarithm using base two. By testing single features the
amount of
information needed to make a correct classification can be reduced. The
remainder for a
specific feature A (e.g. representing a specific biomarker) shows how much the
information
that is needed can be reduced.
''
_ I Pi n;
Re inaifzder=(A) + n;
p + n p;+n,pf+n;
"v" is the number of unique attribute values for feature A in a certain
dataset, "i" is a
certain attribute value, "pi" is the number of examples for feature A where
the classification
is positive (e.g. will develop sepsis), "ni" is the number of examples for
feature A where the
classification is negative (e.g. will not develop sepsis).
The information gain of a specific feature A is calculated as the difference
between the information content for the classes and the remainder of feature
A:
T1
Gaan(14) = I( p p tz _ p+n) - Re ~Airlder~(~4)
-76-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
The information gain is used to evaluate how important the different features
are for
the classification (how well they split up the examples), and the feature with
the highest
information.
In general there are a number of different decision tree algorithms, many of
which
are described in Duda, Pattern Classification, Second Edition, 2001, John
Wiley & Sons,
Inc. Decision tree algorithms often require consideration of feature
processing, impurity
measure, stopping criterion, and pruning. Specific decision tree algorithms
include, but are
not limited to classification and regression trees (CART), multivariate
decision trees, ID3,
and C4.5.
In one approach, when a decision tree is used, the gene expression data for a
select
combination of genes described in the present invention across a training
population is
standardized to have mean zero and unit variance. The members of the training
population
are randomly divided into a training set and a test set. For example, in one
embodiment,
two thirds of the members of the training population are placed in the
training set and one
third of the members of the training population are placed in the test set.
The expression
values for a select combination of biomarkers described in the present
invention is used to
construct the decision tree. Then, the ability for the decision tree to
correctly classify
members in the test set is determined. In some embodiments, this computation
is performed
several times for a given combination of biomarkers. In each computational
iteration, the
members of the training population are randomly assigned to the training set
and the test set.
Then, the quality of the combination of biomarkers is taken as the average of
each such
iteration of the decision tree computation.
In addition to univariate decision trees in which each split is based on a
feature value
for a corresponding biomarker, among the set of biomarkers of the present
invention, or the
relative feature values of two such biomarkers, multivariate decision trees
can be
implemented as a decision rule. In such multivariate decision trees, some or
all of the
decisions actually comprise a linear combination of feature values for a
plurality of
biomarkers of the present invention. Such a linear combination can be trained
using known
techniques such as gradient descent on a classification or by the use of a sum-
squared-error
criterion. To illustrate such a decision tree, consider the expression:
0.04xt+0.16x2<500
-77-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Here, xl and X2 refer to two different features for two different biomarkers
from
among the biomarkers of the present invention. To poll the decision rule, the
values of
features xi and X2 are obtained from the measurements obtained from the
unclassified
subject. These values are then inserted into the equation. If a value of less
than 500 is
computed, then a first branch in the decision tree is taken. Otherwise, a
second branch in
the decision tree is taken. Multivariate decision trees are described in Duda,
2001, Pattern
Classifrcation, John Wiley & Sons, Inc., New York, pp. 408-409, which is
hereby
incorporated by reference.
Another approach that can be used in the present invention is multivariate
adaptive
regression splines (MARS). MARS is an adaptive procedure for regression, and
is well
suited for the high-dimensional problems addressed by the present invention.
MARS can be
viewed as a generalization of stepwise linear regression or a modification of
the CART
method to improve the performance of CART in the regression setting. MARS is
described
in Hastie et al., 2001, The Elements ofStatistical Learning, Springer-Verlag,
New York, pp.
283-295, which is hereby incorporated by reference in its entirety.
5.5.2 Predictive analysis of microarrays (PAM)
One approach to developing a decision rule using feature values of biomarkers
of the
present invention is the nearest centroid classifier. Such a technique
computes, for each
class (sepsis and SIRS), a centroid given by the average feature levels of the
biomarkers in
the class, and then assigns new samples to the class whose centroid is
nearest. This
approach is similar to k-means clustering except clusters are replaced by
known classes.
This algorithm can be sensitive to noise when a large number of biomarkers are
used. One
enhancement to the technique uses shrinkage: for each biomarker, differences
between class
centroids are set to zero if they are deemed likely to be due to chance. This
approach is
implemented in the Prediction Analysis of Microarray, or PAM. See, for
example,
Tibshirani et al., 2002, Proceedings of the National Academy of Science USA
99; 6567
6572, which is hereby incorporated by reference in its entirety. Shrinkage is
controlled by a
threshold below which differences are considered noise. Biomarkers that show
no
difference above the noise level are removed. A threshold can be chosen by
cross-
validation. As the threshold is decreased, more biomarkers are included and
estimated
classification errors decrease, until they reach a bottom and start climbing
again as a result
of noise biomarkers - a phenomenon known as overfitting.
-78-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
5.5.3 Bagging, boosting, and the random subspace method
Bagging, boosting, the random subspace method, and additive trees are data
analysis
algorithms known as combining techniques that can be used to improve weak
decision
rules. These techniques are designed for, and usually applied to, decision
trees, such as the
decision trees described in Section 5.5.1, above. In addition, such techniques
can also be
useful in decision rules developed using other types of data analysis
algorithms such as
linear discriminant analysis.
In bagging, one samples the training set, generating random independent
bootstrap
replicates, constructs the decision rule on each of these, and aggregates them
by a simple
majority vote in the final decision rule. See, for example, Breiman, 1996,
Machine
Learning 24, 123-140; and Efron & Tibshirani, An Introduction to Boostrap,
Chapman &
Hall, New York, 1993, which is hereby incorporated by reference in its
entirety.
In boosting, decision rules are constructed on weighted versions of the
training set,
which are dependent on previous classification results. Initially, all
features under
consideration have equal weights, and the first decision rule is constructed
on this data set.
Then, weights are changed according to the performance of the decision rule.
Erroneously
classified features get larger weights, and the next decision rule is boosted
on the
reweighted training set. In this way, a sequence of training sets and decision
rules is
obtained, which is then combined by simple majority voting or by weighted
majority voting
in the final decision rule. See, for example, Freund & Schapire, "Experiments
with a new
boosting algorithm," Proceedings 13th International Conference on Machine
Learning,
1996, 148-156, which is hereby incorporated by reference in its entirety.
To illustrate boosting, consider the case where there are two phenotypes
exhibited
by the population under study, phenotype 1 (e.g., acquiring sepsis during a
defined time
periond), and phenotype 2 (e.g., SIRS only, meaning that the subject does
acquire sepsis
within a defined time period). Given a vector of predictor biomarkers (e.g., a
vector of
features that represent such biomarkers) from the training set data, a
decision rule G(X)
produces a prediction taking one of the type values in the two value set: {
phenotype 1,
phenotype 2}. The error rate on the training sample is
_ N
err = 1I I( y; # G(x, ))
N ;o,
where N is the number of subjects in the training set (the sum total of the
subjects
that have either phenotype 1 or phenotype 2). For example, if there are 49
organisms that
acquire sepsis and 72 organisms that remain in the SIRS state, N is 121. A
weak decision
-79-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
rule is one whose error rate is only slightly better than random guessing. In
the boosting
algorithm, the weak decision rule is repeatedly applied to modified versions
of the data,
thereby producing a sequence of weak decision rules Gm(x), m, = 1, 2, ..., M.
The
predictions from all of the decision rules in this sequence are then combined
through a
weighted majority vote to produce the final decision rule:
G(x) = sign am Gm (x)
m=1
Here a,, a2, ..., aM are computed by the boosting algorithm and their purpose
is to
weigh the contribution of each respective decision rule Gm(x). Their effect is
to give higher
influence to the more accurate decision rules in the sequence.
The data modifications at each boosting step consist of applying weights wi,
w2, ...,
wõ to each of the training observations (xi, y;), i = 1, 2, ..., N. Initially
all the weights are set
to w; = 1/N, so that the first step simply trains the decision rule on the
data in the usual
manner. For each successive iteration m= 2, 3, ..., M the observation weights
are
individually modified and the decision rule is reapplied to the weighted
observations. At
step m, those observations that were misclassified by the decision rule Gm-
1(x) induced at
the previous step have their weights increased, whereas the weights are
decreased for those
that were classified correctly. Thus as iterations proceed, observations that
are difficult to
correctly classify receive ever-increasing influence. Each successive decision
rule is
thereby forced to concentrate on those training observations that are missed
by previous
ones in the sequence.
The exemplary boosting algorithm is summarized as follows:
1. Initialize the observation weights wi = 1/N, i= 1, 2, ..., N.
2.Form=1toM:
(a) Fit a decision rule Gm(x) to the training set using weights w;.
(b) Compute
N,x'rI(yr ~Gm(x;))
err,,, =
Z=1 wi
(c) Compute am=log((1-errm)/errm).
(d) Set w; < w; = exp[am - I(yi # Gm (x; ))], i= 1, 2,..., N.
3. Output G(x) = sign ~m , amGm (x),
-90-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
In one embodiment in accordance with this algorithm, each object is, in fact,
a
factor. Furthermore, in the algorithm, the current decision rule G,õ(x) is
induced on the
weighted observations at line 2a. The resulting weighted error rate is
computed at line 2b.
Line 2c calculates the weight am given to Gm(x) in producing the final
classifier G(x) (line
3). The individual weights of each of the observations are updated for the
next iteration at
line 2d. Observations misclassified by Gm(x) have their weights scaled by a
factor exp(am),
increasing their relative influence for inducing the next classifier G,,,+1(x)
in the sequence.
In some embodiments, modifications of the Freund and Schapire, 1997, Journal
of
Computer and System Sciences 55, pp. 119-139, boosting methods are used. See,
for
example, Hasti et al., The Elements of Statistical Learning, 2001, Springer,
New York,
Chapter 10, which is hereby incorporated by reference in its entirety. For
example, in some
embodiments, feature preselection is performed using a technique such as the
nonparametric
scoring methods of Park et al., 2002, Pac. Symp. Biocomput. 6, 52-63, which is
hereby
incorporated by reference in its entirety. Feature preselection is a form of
dimensionality
reduction in which the genes that discriminate between classifications the
best are selected
for use in the classifier. Then, the LogitBoost procedure introduced by
Friedman et al.,
2000, Ann Stat 28, 337-407 is used rather than the boosting procedure of
Freund and
Schapire. In some embodiments, the boosting and other classification methods
of Ben-Dor
et al., 2000, Journal of Computational Biology 7, 559-583, hereby incorporated
by reference
in its entirety, are used in the present invention. In some embodiments, the
boosting and
other classification methods of Freund and Schapire, 1997, Journal of Computer
and System
Sciences 55, 119-139, hereby incorporated by reference in its entirety, are
used.
In the random subspace method, decision rules are constructed in random
subspaces
of the data feature space. These decision rules are usually combined by simple
majority
voting in the final decision rule. See, for example, Ho, "The Random subspace
method for
constructing decision forests," IEEE Trans Pattern Analysis and Machine
Intelligence,
1998; 20(8): 832 844, which is hereby incorporated by reference in its
entirety.
5.5.4 Multiple additive regression trees
Multiple additive regression trees (MART) represents another way to construct
a
decision rule that can be used in the present invention. A generic algorithm
for MART is:
1. Initialize fo(x) = arg miny L(y, y).
2.Form=ltoM:
(a) For I = 1,2, , N compute
-81-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
aL(y; I .l~(xr ))
r~m a.f(xr)
jt=tM-i
(b) Fit a regression tree to the targets r;m giving terminal regions Rj,,,, j
1,2, , Jm=
(c) For j= 1, 2, , Jm compute
yjm =argmin ZL(y,,fm-,(x;)+Y)=
r xJeRj~
(d) Update fm(x) =.fm-1(x) +E J', Y;,,, I(x E Rj,,, )
3. Ouput f(x) =fM (x).
Specific algorithms are obtained by inserting different loss criteria
L(y,f(x)). The
first line of the algorithm initializes to the optimal constant model, which
is just a single
terminal node tree. The components of the negative gradient computed in line
2(a) are
referred to as generalized pseudo residuals, r. Gradients for commonly used
loss functions
are summarized in Table 10.2, of Hastie et al., 2001, The Elements of
Statistical Learning,
Springer-Verlag, New York, p. 321, which is hereby incorporated by reference.
The
algorithm for classification is similar and is described in Hastie et al.,
Chapter 10, which is
hereby incorporated by reference in its entirety. Tuning parameters associated
with the
MART procedure are the number of iterations M and the sizes of each of the
constituent
trees Jm, m= 1, 2, , M.
5.5.5 Decision rules derived by regression
In some embodiments, a decision rule used to classify subjects is built using
regression. In such embodiments, the decision rule can be characterized as a
regression
classifier, preferably a logistic regression classifier. Such a regression
classifier includes a
coefficient for each of the biomarkers (e.g., a feature for each such
biomarker) used to
construct the classifier. In such embodiments, the coefficients for the
regression classifier
are computed using, for example, a maximum likelihood approach. In such a
computation,
the features for the biomarkers (e.g., RT-PCR, microarray data) is used. In
particular
embodiments, molecular marker data from only two trait subgroups is used
(e.g., trait
subgroup a: will acquire sepsis in a defined time period and trait subgroup b:
will not
acquire sepsis in a defined time period) and the dependent variable is absence
or presence of
a particular trait in the subjects for which biomarker data is available.
-82-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
In another specific embodiment, the training population comprises a plurality
of trait
subgroups (e.g., three or more trait subgroups, four or more specific trait
subgroups, etc.).
These multiple trait subgroups can correspond to discrete stages in the
phenotypic
progression from healthy, to SIRS, to sepsis, to more advanced stages of
sepsis in a training
population. In this specific embodiment, a generalization of the logistic
regression model
that handles multicategory responses can be used to develop a decision that
discriminates
between the various trait subgroups found in the training population. For
example,
measured data for selected molecular markers can be applied to any of the
multi-category
logit models described in Agresti, An Introduction to Categorical Data
Analysis, 1996, John
Wiley & Sons, Inc., New York, Chapter 8, hereby incorporated by reference in
its entirety,
in order to develop a classifier capable of discriminating between any of a
plurality of trait
subgroups represented in a training population.
5.5.6 Neural networks
In some embodiments, the feature data measured for select biomarkers of the
present
invention (e.g., RT-PCR data, mass spectrometry data, microarray data) can be
used to train
a neural network. A neural network is a two-stage regression or classification
decision rule.
A neural network has a layered structure that includes a layer of input units
(and the bias)
connected by a layer of weights to a layer of output units. For regression,
the layer of
output units typically includes just one output unit. However, neural networks
can handle
multiple quantitative responses in a seamless fashion.
In multilayer neural networks, there are input units (input layer), hidden
units
(hidden layer), and output units (output layer). There is, furthermore, a
single bias unit that
is connected to each unit other than the input units. Neural networks are
described in Duda
et al., 2001, Pattern Classification, Second Edition, John Wiley & Sons, Inc.,
New York;
and Hastie et al., 2001, The Elements of Statistical Learning, Springer-
Verlag, New York,
each of which is hereby incorporated by reference in its entirety. Neural
networks are also
described in Draghici, 2003, Data Analysis Tools for DNA Microarrays, Chapman
&
Hall/CRC; and Mount, 2001, Bioinformatics: sequence and genome analysis, Cold
Spring
Harbor Laboratory Press, Cold Spring Harbor, New York, each of which is hereby
incorporated by reference in its entirety. What is disclosed below is some
exemplary forms
of neural networks.
The basic approach to the use of neural networks is to start with an untrained
network, present a training pattern to the input layer, and to pass signals
through the net and
-83-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
determine the output at the output layer. These outputs are then compared to
the target
values; any difference corresponds to an error. This error or criterion
function is some
scalar function of the weights and is minimized when the network outputs match
the desired
outputs. Thus, the weights are adjusted to reduce this measure of error. For
regression, this
error can be sum-of-squared errors. For classification, this error can be
either squared error
or cross-entropy (deviation). See, e.g., Hastie et al., 2001, The Elements
ofStatistical
Learning, Springer-Verlag, New York, which is hereby incorporated by reference
in its
entirety.
Three commonly used training protocols are stochastic, batch, and on-line. In
stochastic training, patterns are chosen randomly from the training set and
the network
weights are updated for each pattern presentation. Multilayer nonlinear
networks trained by
gradient descent methods such as stochastic back-propagation perform a maximum-
likelihood estimation of the weight values in the classifier defined by the
network topology.
In batch training, all patterns are presented to the network before learning
takes place.
Typically, in batch training, several passes are made through the training
data. In online
training, each pattern is presented once and only once to the net.
In some embodiments, consideration is given to starting values for weights. If
the
weights are near zero, then the operative part of the sigmoid commonly used in
the hidden
layer of a neural network (see, e.g., Hastie et al., 2001, The Elements
ofStatistical Learning,
Springer-Verlag, New York, hereby incorporated by reference) is roughly
linear, and hence
the neural network collapses into an approximately linear classifier. In some
embodiments,
starting values for weights are chosen to be random values near zero. Hence
the classifier
starts out nearly linear, and becomes nonlinear as the weights increase.
Individual units
localize to directions and introduce nonlinearities where needed. Use of exact
zero weights
leads to zero derivatives and perfect symmetry, and the algorithm never moves.
Alternatively, starting with large weights often leads to poor solutions.
Since the scaling of inputs determines the effective scaling of weights in the
bottom
layer, it can have a large effect on the quality of the final solution. Thus,
in some
embodiments, at the outset all expression values are standardized to have mean
zero and a
standard deviation of one. This ensures all inputs are treated equally in the
regularization
process, and allows one to choose a meaningful range for the random starting
weights.
With standardization inputs, it is typical to take random uniform weights over
the range
[-0.7, +0.7].
-84-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
A recurrent problem in the use of three-layer networks is the optimal number
of
hidden units to use in the network. The number of inputs and outputs of a
three-layer
network are determined by the problem to be solved. In the present invention,
the number
of inputs for a given neural network will equal the number of biomarkers
selected from the
training population. The number of output for the neural network will
typically be just one.
However, in some embodiments more than one output is used so that more than
just two
states can be defined by the network. For example, a multi-output neural
network can be
used to discriminate between, healthy phenotypes, various stages of SIRS,
and/or various
stages of sepsis. If too many hidden units are used in a neural network, the
network will
have too many degrees of freedom and is trained too long, there is a danger
that the network
will overfit the data. If there are too few hidden units, the training set
cannot be learned.
Generally speaking, however, it is better to have too many hidden units than
too few. With
too few hidden units, the classifier might not have enough flexibility to
capture the
nonlinearities in the date; with too many hidden units, the extra weight can
be shrunk
towards zero if appropriate regularization or pruning, as described below, is
used. In typical
embodiments, the number of hidden units is somewhere in the range of 5 to 100,
with the
number increasing with the number of inputs and number of training cases.
One general approach to determining the number of hidden units to use is to
apply a
regularization approach. In the regularization approach, a new criterion
function is
constructed that depends not only on the classical training error, but also on
classifier
complexity. Specifically, the new criterion function penalizes highly complex
classifiers;
searching for the minimum in this criterion is to balance error on the
training set with error
on the training set plus a regularization term, which expresses constraints or
desirable
properties of solutions:
J = Jpac + AJ,eg.
The parameter.Z is adjusted to impose the regularization more or less
strongly. In
other words, larger values for A will tend to shrink weights towards zero:
typically cross-
validation with a validation set is used to estimate A. This validation set
can be obtained by
setting aside a random subset of the training population. Other forms of
penalty have been
proposed, for example the weight elimination penalty (see, e.g., Hastie et
al., 2001, The
Elements of Statistical Learning, Springer-Verlag, New York, hereby
incorporated by
reference).
Another approach to determine the number of hidden units to use is to
eliminate -
prune - weights that are least needed. In one approach, the weights with the
smallest
- 85 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
magnitude are eliminated (set to zero). Such magnitude-based pruning can work,
but is
nonoptimal; sometimes weights with small magnitudes are important for learning
and
training data. In some embodiments, rather than using a magnitude-based
pruning
approach, Wald statistics are computed. The fundamental idea in Wald
Statistics is that
they can be used to estimate the importance of a hidden unit (weight) in a
classifier. Then,
hidden units having the least importance are eliminated (by setting their
input and output
weights to zero). Two algorithms in this regard are the Optimal Brain Damage
(OBD) and
the Optimal Brain Surgeon (OBS) algorithms that use second-order approximation
to
predict how the training error depends upon a weight, and eliminate the weight
that leads to
the smallest increase in training error.
Optimal Brain Damage and Optimal Brain Surgeon share the same basic approach
of training a network to local minimum error at weight w, and then pruning a
weight that
leads to the smallest increase in the training error. The predicted functional
increase in the
error for a change in full weight vector Sw is:
t Z
~u -C ~~ - ~ + 2 ~t ~Z ~ + o(l~'ll3 }
z
where 2 is the Hessian matrix. The first term vanishes at a local minimum in
error; third and higher order terms are ignored. The general solution for
minimizing this
function given the constraint of deleting one weight is:
z
8w- _'~'' H' u~ and L~ _?- '''9
lH _' J99 2 LH ' ~J9
Here, uq is the unit vector along the qth direction in weight space and Lq is
approximation to the saliency of the weight q - the increase in training error
if weight q is
pruned and the other weights updated Sw. These equations require the inverse
of H. One
method to calculate this inverse matrix is to start with a small value, Ho' =
a"jI, where a is
a small parameter - effectively a weight constant. Next the matrix is updated
with each
pattern according to
H_l H- Hm'Xm+1Xm+1Hml E n. I
m+l m n q
+Xm+lHnr Xm+t
am
-86-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
where the subscripts correspond to the pattern being presented and a,,,
decreases with
m. After the full training set has been presented, the inverse Hessian matrix
is given by H-1
= H. In algorithmic form, the Optimal Brain Surgeon method is:
begin initialize nH, w, 0
train a reasonably large network to minimum error
do compute H"' by Eqn. 1
q* 4-- arg min wq /(2[H-' Iq )(saliency Lq)
n
w
w F- w- H 9H-'eg (saliency Lq)
~
until J(w) > 0
return w
end
The Optimal Brain Damage method is computationally simpler because the
calculation of the inverse Hessian matrix in line 3 is particularly simple for
a diagonal
matrix. The above algorithm terminates when the error is greater than a
criterion initialized
to be 0. Another approach is to change line 6 to terminate when the change in
J(w) due to
elimination of a weight is greater than some criterion value. In some
embodiments, the
back-propagation neural network See, for example Abdi, 1994, "A neural network
primer," J. Biol System. 2, 247-283, hereby incorporated by reference in its
entirety.
5.5.7 Clustering
In some embodiments, features for select biomarkers of the present invention
are
used to cluster a training set. For example, consider the case in which ten
features
(corresponding to ten biomarkers) described in the present invention is used.
Each member
m of the training population will have feature values (e.g. expression values)
for each of the
ten biomarkers. Such values from a member m in the training population define
the vector:
XIm X2m Xbm x4m XSm X6m X7m X8m X9m x10m
where Xim is the expression level of the i'h biomarker in organism m. If there
are m
organisms in the training set, selection of i biomarkers will define m
vectors. Note that the
methods of the present invention do not require that each the expression value
of every
single biomarker used in the vectors be represented in every single vector m.
In other
words, data from a subject in which one of the ith biomarkers is not found can
still be used
-87-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
for clustering. In such instances, the missing expression value is assigned
either a "zero" or
some other normalized value. In some embodiments, prior to clustering, the
feature values
are normalized to have a mean value of zero and unit variance.
Those members of the training population that exhibit similar expression
patterns
across the training group will tend to cluster together. A particular
combination of genes of
the present invention is considered to be a good classifier in this aspect of
the invention
when the vectors cluster into the trait groups found in the training
population. For instance,
if the training population includes class a: subjects that do not develop
sepsis, and class b:
subjects that develop sepsis, an ideal clustering classifier will cluster the
population into two
groups, with one cluster group uniquely representing class a and the other
cluster group
uniquely representing class b.
Clustering is described on pages 211-256 of Duda and Hart, Pattern
C'lassification
and Scene Analysis, 1973, John Wiley & Sons, Inc., New York, (hereinafter
"Duda 1973 )
which is hereby incorporated by reference in its entirety. As described in
Section 6.7 of
Duda 1973, the clustering problem is described as one of finding natural
groupings in a
dataset. To identify natural groupings, two issues are addressed. First, a way
to measure
similarity (or dissimilarity) between two samples is determined. This metric
(similarity
measure) is used to ensure that the samples in one cluster are more like one
another than
they are to samples in other clusters. Second, a mechanism for partitioning
the data into
clusters using the similarity measure is determined.
Similarity measures are discussed in Section 6.7 of Duda 1973, where it is
stated
that one way to begin a clustering investigation is to define a distance
function and to
compute the matrix of distances between all pairs of samples in a dataset. If
distance is a
good measure of similarity, then the distance between samples in the same
cluster will be
significantly less than the distance between samples in different clusters.
However, as
stated on page 215 of Duda 1973, clustering does not require the use of a
distance metric.
For example, a nonmetric similarity function s(x, x') can be used to compare
two vectors x
and x'. Conventionally, s(x, x') is a symmetric function whose value is large
when x and x'
are somehow "similar". An example of a nonmetric similarity function s(x, x')
is provided
on page 216 of Duda 1973.
Once a method for measuring "similarity" or "dissimilarity" between points in
a
dataset has been selected, clustering requires a criterion function that
measures the
clustering quality of any partition of the data. Partitions of the data set
that extremize the
-88-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
criterion function are used to cluster the data. See page 217 of Duda 1973.
Criterion
functions are discussed in Section 6.8 of Duda 1973.
More recently, Duda et al., Pattern Classification, 2 d edition, John Wiley &
Sons,
Inc. New York, has been published. Pages 537-563 describe clustering in
detail. More
information on clustering techniques can be found in Kaufman and Rousseeuw,
1990,
Finding Groups in Data: An Introduction to Cluster Analysis, Wiley, New York,
NY;
F,veritt, 1993, Cluster analysis (3d ed.), Wiley, New York, NY; and Backer,
1995,
Computer Assisted Reasoning in Cluster Analysis, Prentice Hall, Upper Saddle
River, New
Jersey. Particular exemplary clustering techniques that can be used in the
present invention
include, but are not limited to, hierarchical clustering (agglomerative
clustering using
nearest-neighbor algorithm, farthest-neighbor algorithm, the average linkage
algorithm, the
centroid algorithm, or the sum-of-squares algorithm), k-means clustering,
fuzzy k-means
clustering algorithm, and Jarvis-Patrick clustering.
5.5.8 Principal component analysis
Principal component analysis (PCA) has been proposed to analyze gene
expression
data. More generally, PCA can be used to analyze feature value data of
biomarkers of the
present invention in order to construct a decision rule that discriminates
converters from
nonconverters. Principal component analysis is a classical technique to reduce
the
dimensionality of a data set by transforming the data to a new set of variable
(principal
components) that summarize the features of the data. See, for example,
Jolliffe, 1986,
Principal Component Analysis, Springer, New York, which is hereby incorporated
by
reference. Principal component analysis is also described in Draghici, 2003,
Data Analysis
7'ools for DNA Microarrays, Chapman & Hall/CRC, which is hereby incorporated
by
reference. What follows is non-limiting examples of principal components
analysis.
Principal components (PCs) are uncorrelated and are ordered such that the k'h
PC has
the kth largest variance among PCs. The k'h PC can be interpreted as the
direction that
maximizes the variation of the projections of the data points such that it is
orthogonal to the
first k - 1 PCs. The first few PCs capture most of the variation in the data
set. In contrast,
the last few PCs are often assumed to capture only the residual 'noise' in the
data.
PCA can also be used to create a classifier in accordance with the present
invention.
In such an approach, vectors for the select biomarkers of the present
invention can be
constructed in the same manner described for clustering above. In fact, the
set of vectors,
-89-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
where each vector represents the feature values (e.g., abundance values) for
the select genes
from a particular member of the training population, can be viewed as a
matrix. In some
embodiments, this matrix is represented in a Free-Wilson method of qualitative
binary
description of monomers (Kubinyi, 1990, 3D QSAR in drug design theory methods
and
applications, Pergamon Press, Oxford, pp 589-638), and distributed in a
maximally
compressed space using PCA so that the first principal component (PC) captures
the largest
amount of variance information possible, the second principal component (PC)
captures the
second largest amount of all variance information, and so forth until all
variance
information in the matrix has been considered.
Then, each of the vectors (where each vector represents a member of the
training
population) is plotted. Many different types of plots are possible. In some
embodiments, a
one-dimensional plot is made. In this one-dimensional plot, the value for the
first principal
component from each of the members of the training population is plotted. In
this form of
plot, the expectation is that members of a first subgroup (e.g: those subjects
that do not
develop sepsis in a determined time period) will cluster in one range of first
principal
component values and members of a second subgroup (e.g., those subjects that
develop
sepsis in a determined time period) will cluster in a second range of first
principal
component values.
In one ideal example, the training population comprises two subgroups:
"sepsis" and
"SIRS." The first principal component is computed using the molecular marker
expression
values for the select biomarkers of the present invention across the entire
training
population data set. Then, each member of the training set is plotted as a
function of the
value for the first principal component. In this ideal example, those members
of the training
population in which the first principal component is positive are the
"responders" and those
members of the training population in which the first principal component is
negative are
"subjects with sepsis."
In some embodiments, the members of the training population are plotted
against
more than one principal component. For example, in some embodiments, the
members of
the training population are plotted on a two-dimensional plot in which the
first dimension is
the first principal component and the second dimension is the second principal
component.
In such a two-dimensional plot, the expectation is that members of each
subgroup
represented in the training population will cluster into discrete groups. For
example, a first
cluster of members in the two-dimensional plot will represent subjects that
develop sepsis in
-90-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
a given time period and a second cluster of members in the two-dimensional
plot will
represent subjects that do not develop sepsis in a given time period.
5.5.9 Nearest neighbor analysis
Nearest neighbor classifiers are memory-based and require no classifier to be
fit.
Given a query point xo, the k training points x(,), r, , k closest in distance
to xo are
identified and then the point xo is classified using the k nearest neighbors.
Ties can be
broken at random. In some embodiments, Euclidean distance in feature space is
used to
determine distance as:
d(+) - IIxUa - x. 11 Typically, when the nearest neighbor algorithm is used,
the expression data used to
compute the linear discriminant is standardized to have mean zero and variance
1. In the
present invention, the members of the training population are randomly divided
into a
training set and a test set. For example, in one embodiment, two thirds of the
members of
the training population are placed in the training set and one third of the
members of the
training population are placed in the test set. A select combination of
biomarkers of the
present invention represents the feature space into which members of the test
set are plotted.
Next, the ability of the training set to correctly characterize the members of
the test set is
computed. In some embodiments, nearest neighbor computation is performed
several times
for a given combination of biomarkers of the present invention. In each
iteration of the
computation, the members of the training population are randomly assigned to
the training
set and the test set. Then, the quality of the combination of biomarkers is
taken as the
average of each such iteration of the nearest neighbor computation.
The nearest neighbor rule can be refined to deal with issues of unequal class
priors,
differential misclassification costs, and feature selection. Many of these
refinements
involve some form of weighted voting for the neighbors. For more information
on nearest
neighbor analysis, see Duda, Pattern Classffication, Second Edition, 2001,
John Wiley &
Sons, Inc; and Hastie, 2001, The Elements of Statistical Learning, Springer,
New York,
each of which is hereby incorporated by reference in its entirety.
5.5.10 Linear discriminant analysis
Linear discriminant analysis (LDA) attempts to classify a subject into one of
two
categories based on certain object properties. In other words, LDA tests
whether object
-91-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
attributes measured in an experiment predict categorization of the objects.
LDA typically
requires continuous independent variables and a dichotomous categorical
dependent
variable. In the present invention, the feature values for the select
combinations of
biomarkers of the present invention across a subset of the training population
serve as the
requisite continuous independent variables. The trait subgroup classification
of each of the
members of the training population serves as the dichotomous categorical
dependent
variable.
LDA seeks the linear combination of variables that maximizes the ratio of
between-
group variance and within-group variance by using the grouping information.
Implicitly,
the linear weights used by LDA depend on how the feature values of a molecular
marker
across the training set separates in the two groups (e.g., a group a that
develops sepsis
during a defined time period and a group b that does not develop sepsis during
a defined
time period) and how these feature values correlate with the feature values of
other
biomarkers. In some embodiments, LDA is applied to the data matrix of the N
members in
the training sample by K biomarkers in a combination of biomarkers described
in the
present invention. Then, the linear discriminant of each member of the
training population
is plotted. Ideally, those members of the training population representing a
first subgroup
(e.g. those subjects that develop sepsis in a defined time period) will
cluster into one range
of linear discriminant values (e.g., negative) and those member of the
training population
representing a second subgroup (e.g. those subjects that will not develop
sepsis in a defined
time period) will cluster into a second range of linear discriminant values
(e.g., positive).
The LDA is considered more successful when the separation between the clusters
of
discriminant values is larger. For more information on linear discriminant
analysis, see
Duda, Pattern C7assification, Second Edition, 2001, John Wiley & Sons, Inc;
and Hastie,
2001, The Elements ofStatistical Learning, Springer, New York; and Venables &
Ripley,
1997, Modern Applied Statistics with s-plus, Springer, New York, each of which
is hereby
incorporated by reference in its entirety.
5.5.11 Quadratic discriminant analysis
Quadratic discriminant analysis (QDA) takes the same input parameters and
returns
the same results as LDA. QDA uses quadratic equations, rather than linear
equations, to
produce results. LDA and QDA are interchangeable, and which to use is a matter
of
-92-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
preference and/or availability of software to support the analysis. Logistic
regression takes
the same input parameters and returns the same results as LDA and QDA.
5.5.12 Support vector machines
In some embodiments of the present invention, support vector machines (SVMs)
are
used to classify subjects using feature values of the genes described in the
present invention.
SVMs are a relatively new type of learning algorithm. See, for example,
Cristianini and
Shawe-Taylor, 2000, An Introduction to Support Vector Machines, Cambridge
University
Press, Cambridge; Boser et al., 1992, "A training algorithm for optimal margin
classifiers,"
in Proceedings of the Sth Annual ACM Workshop on Computational Learning
Theory, ACM
Press, Pittsburgh, PA, pp. 142-152; Vapnik, 1998, Statistical Learning Theory,
Wiley, New
York; Mount, 2001, Bioinformatics: sequence and genome analysis, Cold Spring
Harbor
Laboratory Press, Cold Spring Harbor, New York, Duda, Pattern Classification,
Second
Edition, 2001, John Wiley & Sons, Inc.; and Hastie, 2001, The Elements
ofStatistical
Learning, Springer, New York; and Furey et al., 2000, Bioinformatics 16, 906-
914, each of
which is hereby incorporated by reference in its entirety. When used for
classification,
SVMs separate a given set of binary labeled data training data with a hyper-
plane that is
maximally distance from them. For cases in which no linear separation is
possible, SVMs
can work in combination with the technique of 'kernels', which automatically
realizes a
non-linear mapping to a feature space. The hyper-plane found by the SVM in
feature space
corresponds to a non-linear decision boundary in the input space.
In one,approach, when a SVM is used, the feature data is standardized to have
mean
zero and unit variance and the members of a training population are randomly
divided into a
training set and a test set. For example, in one embodiment, two thirds of the
members of
the training population are placed in the training set and one third of the
members of the
training population are placed in the test set. The expression values for a
combination of
genes described in the present invention is used to train the SVM. Then the
ability for the
trained SVM to correctly classify members in the test set is determined. In
some
embodiments, this computation is performed several times for a given
combination of
molecular markers. In each iteration of the computation, the members of the
training
population are randomly assigned to the training set and the test set. Then,
the quality of the
combination of biomarkers is taken as the average of each such iteration of
the SVM
computation.
- 93 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
5.5.13 Evolutionary methods
Inspired by the process of biological evolution, evolutionary methods of
decision
rule design employ a stochastic search for an decision rule. In broad
overview, such
methods create several decision rules - a population - from a combination of
biomarkers
described in the present invention. Each decision rule varies somewhat from
the other.
Next, the decision rules are scored on feature data across the training
population. In
keeping with the analogy with biological evolution, the resulting (scalar)
score is sometimes
called the fitness. The decision rules are ranked according to their score and
the best
decision rules are retained (some portion of the total population of decision
rules). Again,
in keeping with biological terminology, this is called survival of the
fittest. The decision
rules are stochastically altered in the next generation - the children or
offspring. Some
offspring decision rules will have higher scores than their parent in the
previous generation,
some will have lower scores. The overall process is then repeated for the
subsequent
generation: the decision rules are scored and the best ones are retained,
randomly altered to
give yet another generation, and so on. In part, because of the ranking, each
generation has,
on average, a slightly higher score than the previous one. The process is
halted when the
single best decision rule in a generation has a score that exceeds a desired
criterion value.
More information on evolutionary methods is found in, for example, Duda,
Pattern
Classifzcatian, Second Edition, 2001, John Wiley & Sons, Inc.
5.5.14 Other data analysis algorithms
The data analysis algorithms described above are merely examples of the types
of
methods that can be used to construct a decision rule for discriminating
converters from
nonconverters. Moreover, combinations of the techniques described above can be
used.
Some combinations, such as the use of the combination of decision trees and
boosting, have
been described. However, many other combinations are possible. In addition, in
other
techniques in the art such as Projection Pursuit and Weighted Voting can be
used to
construct decision rules.
-94-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
5.6 BIOMARKERS
In specific embodiments, the present invention provides biomarkers that are
useful
in diagnosing or predicting sepsis and/or its stages of progression in a
subject. While the
methods of the present invention may use an unbiased approach to identifying
predictive
biomarkers, it will be clear to the artisan that specific groups of biomarkers
associated with
physiological responses or with various signaling pathways may be the subject
of particular
attention. This is particularly the case where biomarkers from a biological
sample are
contacted with an array that can be used to measure the amount of various
biomarkers
through direct and specific interaction with the biomarkers (e.g., an antibody
array or a
nucleic acid array). In this case, the choice of the components of the array
may be based on
a suggestion that a particular pathway is relevant to the determination of the
status of sepsis
or SIRS in a subject. The indication that a particular biomarker has a feature
that is
predictive or diagnostic of sepsis or SIRS may give rise to an expectation
that other
biomarkers that are physiologically regulated in a concerted fashion likewise
may provide a
predictive or diagnostic feature. The artisan will appreciate, however, that
such an
expectation may not be realized because of the complexity of biological
systems. For
example, if the amount of a specific mRNA biomarker were a predictive feature,
a
concerted change in mRNA expression of another biomarker might not be
measurable, if
the expression of the other biomarker was regulated at a post-translational
level. Further,
the mRNA expression level of a biomarker may be affected by multiple
converging
pathways that may or may not be involved in a physiological response to
sepsis.
Biomarkers can be obtained from any biological sample, which can be, by way of
example and not of limitation, whole blood, plasma, saliva, serum, red blood
cells, platelets,
neutrophils, eosinophils, basophils, lymphocytes, monocytes, urine, cerebral
spinal fluid,
sputum, stool, cells and cellular extracts, or other biological fluid sample,
tissue sample or
tissue biopsy from a host or subject. The precise biological sample that is
taken from the
subject may vary, but the sampling preferably is minimally invasive and is
easily performed
by conventional techniques.
Measurement of a phenotypic change may be carried out by any conventional
technique. Measurement of body temperature, respiration rate, pulse, blood
pressure, or
other physiological parameters can be achieved via clinical observation and
measurement.
Measurements of biomarker molecules may include, for example, measurements
that
indicate the presence, concentration, expression level, or any other value
associated with a
biomarker molecule. The form of detection of biomarker molecules typically
depends on
-95-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
the method used to form a profile of these biomarkers from a biological
sample. See
Section 5.4, above, and Tables 1, 4, 5, 6, 7, 8 and/or 9, below.
In a particular embodiment, the biomarker profile comprises at least 2, 3, 4,
5, 6, 7,
8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, or 20 different biomarkers
listed in any one of
Tables 1, 4, 5, 6, 7, 8 and 9, below. In another particular embodiment, the
biomarker profile
comprises at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18,
19, or 20 different
biomarkers listed in any combination of Tables 1, 4, 5, 6, 7, 8, and 9 below.
In still another
particular embodiment, the biomarker profile comprises at least 2, 3, 4, 5, 6,
7, 8, 9, or 10
different biomarkers listed in 4 below. In still another particular
embodiment, the
biomarker profile comprises at least CRP, APOA2, and SERPINCI. In some
embodiments,
the biomarker profile comprises at least one of SERPINCI, APOA2, and CRP, and,
additionally, at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16,
17, 18, 19, 20 or more
additional biomarkers from any combination of Tables 1, 4, 5, 6, 7, 8, and 9.
In some
embodiments, the biomarker profile comprises at least one of SERPINCI, APOA2,
and
CRP, and, additionally, at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13,
14, 15, 16, 17, 18, 19,
20 or more additional biomarkers from any one of Tables 1, 4, 5, 6, 7, 8, and
9. The
biomarker profile further comprises a respective corresponding feature for
each of the
biomarkers in the profile. Such biomarkers can be, for example, mRNA
transcripts, cDNA
or some other nucleic acid, for example amplified nucleic acid, or proteins.
Generally, the
biomarkers in a biomarker profile are derived from at least two different
genes. In the case
where a biomarker in the biomarker profile is listed in Tables 1, 4, 5, 6, 7,
8 and/or 9, the
biomarker can be, for example, a transcript made by the listed gene, a
complement thereof,
or a discriminating fragment or complement thereof, or a cDNA thereof, or a
discriminating
fragment of the cDNA, or a discriminating amplified nucleic acid molecule
corresponding
to all or a portion of the transcript or its complement, or a protein encoded
by the gene, or a
discriminating fragment of the protein, or an indication of any of the above.
Further still,
the biomarker can be, for example, a protein of a gene listed in Tables 1, 4,
5, 6, 7, 8 and/or
9, or a discriminating fragment of the protein, or an indication of any of.the
above. Here, a
discriminating molecule or fragment is a molecule or fragment that, when
detected,
indicates presence or abundance of the above-identified transcript, cDNA,
amplified nucleic
acid, or protein. In accordance with this embodiment, the biomarker profiles
of the present
invention can be obtained using any standard assay known to those skilled in
the art, or in
an assay described herein, to detect a biomarker. Such assays are capable, for
example, of
detecting the products of expression (e.g., nucleic acids and/or proteins) of
a particular gene
-96-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
or allele of a gene of interest (e.g., a gene disclosed in Tables 1, 4, 5, 6,
7, 8 and/or 9). In
one embodiment, such an assay utilizes a nucleic acid microarray.
In some embodiments the biomarker profile has between 2 and 100 biomarkers
listed in Table 1. In some embodiments, the biomarker profile has between 3
and 50
biomarkers listed in Table 1. In some embodiments, the biomarker profile has
between 4
and 25 biomarkers listed in Table 1. In some embodiments, the biomarker
profile has at
least 3 biomarkers listed in Table 1. In some embodiments, the biomarker
profile has at
least 4 biomarkers listed in Table 1. In some embodiments, the biomarker
profile has at
least 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 25, 30, 35, 40,
45, 50, 54, 5, 60, 65,
70, 75, 80, 85, 90, 95, 96, or 100 biomarkers listed in Table 1. In some
embodiments, each
such biomarker is a nucleic acid. In some embodiments, each such biomarker is
a protein.
In some embodiments, some of the biomarkers in the biomarker profile are
nucleic acids
and some of the biomarkers in the biomarker profile are proteins.
In some embodiments the biomarker profile has between 2 and 10 biomarkers
listed
in Table 4. In some embodiments, the biomarker profile has between 3 and 8
biomarkers
listed in Table 4. In some embodiments, the biomarker profile has at least 2,
3, 4, 5, 6, 7, 8,
9, or 10 biomarkers listed in Table 4. In some embodiments, each such
biomarker is a
nucleic acid. In some embodiments, each such biomarker is a protein. In some
embodiments, some of the biomarkers in the biomarker profile are nucleic acids
and some
of the biomarkers in the biomarker profile are proteins. In some embodiments,
the
biomarker profile comprises at least CRP, APOA2, and SERPINC1. In some
embodiments,
the biomarker profile comprises at least one of SERPINCI, APOA2, and CRP, and,
additionally, at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16,
17, 18, 19, 20 or more
additional biomarkers from any combination of Tables 1, 4, 5, 6, 7, 8, and 9.
In some
embodiments, the biomarker profile comprises at least one of SERPINCI, APOA2,
and
CRP, and, additionally, at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13,
14, 15, 16, 17, 18, 19,
20 or more additional biomarkers from any one of Tables 1, 4, 5, 6, 7, 8, and
9.
In some embodiments, some of the biomarkers in the biomarker profile are
nucleic
acids and some of the biomarkers in the biomarker profile are proteins. In
some
embodiments the biomarker profile has between 2 and 100 biomarkers listed in
Table 5. In
some embodiments, the biomarker profile has between 3 and 50 biomarkers listed
in Table
5. In some embodiments, the biomarker profile has between 4 and 25 biomarkers
listed in
Table 5. In some embodiments, the biomarker profile has at least 3 biomarkers
listed in
Table 5. In some embodiments, the biornarker profile has at least 4 biomarkers
listed in
-97-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Table 5. In some embodiments, the biomarker profile has at least 6, 7, 8, 9,
10, 11, 12, 13,
14, 15, 16, 17, 18, 19, 20, 25, 30, 35, 40, 45, 50, 54, 5, 60, 65, 70, 75, 80,
85, 90 biomarkers
listed in Table 5. In some embodiments, each such biomarker is a nucleic acid.
In some
embodiments, each such biomarker is a protein. In some embodiments, some of
the
biomarkers in the biomarker profile are nucleic acids and some of the
biomarkers in the
biomarker profile are proteins.
In some embodiments the biomarker profile has between 2 and 30 biomarkers
listed
in Table 6. In some embodiments, the biomarker profile has between 3 and 50
biomarkers
listed in Table 6. In some embodiments, the biomarker profile has between 4
and 25
biomarkers listed in Table 6. In some embodiments, the biomarker profile has
at least 3
biomarkers listed in Table 6. In some embodiments, the biomarker profile has
at least 4
biomarkers listed in Table 6. In some embodiments, the biomarker profile has
at least 6, 7,
8, 9, 10, 11, 12,13, 14, 15, 16, 17, 18, 19, 20, 25, 30, 35, 40, 45, 50, 54,
5, 60, 65, 70, 75,
80, 85, 90 biomarkers listed in Table 6. In some embodiments, each such
biomarker is a
nucleic acid. In some embodiments, each such biomarker is a protein. In some
embodiments, some of the biomarkers in the biomarker profile are nucleic acids
and some
of the biomarkers in the biomarker profile are proteins.
In some embodiments the biomarker profile has between 2 and 20 biomarkers
listed
in Table 7. In some embodiments, the biomarker profile has between 3 and 25
biomarkers
listed in Table 7. In some embodiments, the biomarker profile has between 4
and 25
biomarkers listed in Table 7. In some embodiments, the biomarker profile has
at least 3
biomarkers listed in Table 7. In some embodiments, the biomarker profile has
at least 4
biomarkers listed in Table 7. In some embodiments, the biomarker profile has
at least 6, 7,
8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 or 20 biomarkers listed in Table
7. In some
embodiments, each such biomarker is a nucleic acid. In some embodiments, each
such
biomarker is a protein. In some embodiments, some of the biomarkers in the
biomarker
profile are nucleic acids and some of the biomarkers in the biomarker profile
are proteins.
In some embodiments the biomarker profile has between 2 and 20 biomarkers
listed
in Table 8. In some embodiments, the biomarker profile has between 3 and 25
biomarkers
listed in Table 8. In some embodiments, the biomarker profile has between 4
and 25
biomarkers listed in Table 8. In some embodiments, the biomarker profile has
at least 3
biomarkers listed in Table 8. In some embodiments, the biomarker profile has
at least 4
biomarkers listed in Table 8. In some embodiments, the biomarker profile has
at least 6, 7,
8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 or 20 biomarkers listed in Table
8. In some
-98-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
embodiments, each such biomarker is a nucleic acid. In some embodiments, each
such
biomarker is a protein. In some embodiments, some of the biomarkers in the
biomarker
profile are nucleic acids and some of the biomarkers in the biomarker profile
are proteins.
In some embodiments the biomarker profile has between 2 and 20 biomarkers
listed
in Table 9. In some embodiments, the biomarker profile has between 3 and 25
biomarkers
listed in Table 9. In some embodiments, the biomarker profile has between 4
and 25
biomarkers listed in Table 9. In some embodiments, the biomarker profile has
at least 3
biomarkers listed in Table 9. In some embodiments, the biomarker profile has
at least 4
biomarkers listed in Table 9. In some embodiments, the biomarker profile has
at least 6, 7,
8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 or 20 biomarkers listed in Table
9. In some
embodiments, each such biomarker is a nucleic acid. In some embodiments, each
such
biomarker is a protein. In some embodiments, some of the biomarkers in the
biomarker
profile are nucleic acids and some of the biomarkers in the biomarker profile
are proteins.
Biomarkers are listed in Tables 1, 4, 5, 6, 7, 8, and 9 by their gene symbol
and gene
name for reference purposes. However, the present invention encompasses, inter
alia, both
the nucleic acid product from and protein product form, as well as
discriminatory fragments
thereof, of such genes. A more detailed description of the biomarkers listed
in Tables 1, 4,
5, 6, 7, 8, and 9 is provided in Section 5.6.1, below.
5.6.1 Isolation of Useful Biomarkers
The accession numbers in this section refer to Natioanl Center for
Biotechnology
Information (NCBI) accession numbers, through the NCBI portal Entrez, to the
NCBI
nucleotide database and the NCBI protein sequence database. The NCBI
nucleotide
database is a collection of sequences from several sources, including GenBank
, the EST
database, the GSS database, HomoloGene, the HTG database, the SNPs database,
RefSeq
(Release 17), UniSTS, UniGene, and the PDB. GenBank is the NIH genetic
sequence
database, an annotated collection of all publicly available DNA sequences
(Nucleic Acids
Research 2005 January 13;33(Database Issue):D34-D36). There are approximately
59,750,386,305 bases in 54,584,635 sequence records in the traditional GenBank
divisions
and 63,183,065,091 bases in 12,465,546 sequence records in the WGS division as
of
February 2006. The EST database is a collection of expressed sequence tags, or
short,
single-pass sequence reads from mRNA (cDNA). The GSS database is a database of
genome survey sequences, or short, single-pass genomic sequences. HomoloGene
is a gene
- 99 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
homology tool that compares nucleotide sequences between pairs of organisms in
order to
identify putative orthologs. The HTG database is a collection of high-
throughput genome
sequences from large-scale genome sequencing centers, including unfinished and
finished
sequences. The SNPs database is a central repository for both single-base
nucleotide
substitutions and short deletion and insertion polymorphisms. The RefSeq
database is a
database of non-redundant reference sequences standards, including genomic DNA
contigs,
mRNAs, and proteins for known genes. For more information on RefSeq, see NCBI
Reference Sequence (RefSeq): a curated non-redundant sequence database of
genomes,
transcripts and proteins Pruitt et al., 2005, Nucleic Acids Res 33: D501=D504.
The STS
database is a database of sequence tagged sites, or short sequences that are
operationally
unique in the genome. The UniSTS database is a unified, non-redundant view of
sequence
tagged sites (STSs). The UniGene database is a collection of ESTs and full-
length mRNA
sequences organized into clusters, each representing a unique known or
putative human
gene annotated with mapping and expression information and cross-references to
other
sources. The NCBI protein database has been compiled from a variety of
sources, including
SwissProt, Protein Information Resource (PIR), PRF, Protein Data Bank (PDB)
(sequences
from solved structures), and translations from annotated coding regions in
GenBank and
RefSeq.
The nucleotide sequence of C4B, (identified by accession no. K02403) is
disclosed
in, e.g., Belt et al., 1984, "The structural basis of the multiple forms of
human complement
component C4," published in Cell 36, 907-914, and the amino acid sequence of
C4B
(identified by accession no. AAB67980) is disclosed in, e.g., Xie et al.,
2003, "Analysis of
the gene-dense major histocompatibility complex class III region and its
comparison to
mouse" published in Genome Res. 13, 2621-2635, each of which is incorporated
by
reference herein in its entirety.
The nucleotide sequence of SERPINA3 (identified by accession no. NM 001085) is
disclosed in, e.g., Furiya, Y. et al., 2005, "Alpha-l-antichymotrypsin gene
polymorphism
and susceptibility to multiple system atrophy (MSA)," published in Brain Res.
Mol. Brain
Res. 138 (2), 178-181, and the amino acid sequence of SERPINA3 (identified by
accession
no. NP001076) is disclosed in, e.g., Furiya, Y. et al., 2005, "Alpha-l-
antichymotrypsin
gene polymorphism and susceptibility to multiple system atrophy (MSA),"
published in
Brain Res. Mol. Brain Res. 138 (2), 178-181, each of which is incorporated by
reference
herein in its entirety.
- 100 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
The nucleotide sequence of ACTB (identified by accession no. NM_001101) is
disclosed in, e.g., Dahlen, A. et al., 2004, "Molecular genetic
characterization of the
genomic ACTB-GLI fusion in pericytoma with t(7; 12)," published in Biochem.
Biophys.
Res. Commun. 325 (4), 1318-1323, and the amino acid sequence of ACTB
(identified by
accession no. AAS79319) is disclosed in Livingston, RJ et al., and is
unpublished and each
of which is incorporated by reference herein in its entirety.
The nucleotide sequence of AFM (identified by accession no. NM 001133) is
disclosed in, e.g., Jerkovic, L. et al., 2005, "Afamin is a novel human
vitamin E-binding
glycoprotein characterization and in vitro expression," published in J.
Proteome Res. 4 (3),
889-899, and the amino acid sequence of AFM (identified by accession no.
AAA21612) is
disclosed in, e.g., Lichenstein, H.S. et al., 1994, "Afamin is a new member of
the albumin,
alpha-fetoprotein, and vitamin D-binding protein gene family," published in J.
Biol. Chem.
269 (27), 18149-18154, each of which is incorporated by reference herein in
its entirety.
The nucleotide sequence of AGT (identified by accession no. NM_000029) is
disclosed in, e.g., Rasmussen-Torvik, L.J. et a1., 2005, "A population
association study of
angiotensinogen polymorphisms and haplotypes with left ventricular
phenotypes,"
published in Hypertension 46 (6), 1294-1299, and the amino acid sequence of
AGT
(identified by accession no. AAR03501) is disclosed in, e.g., Crawford, D.C.
et al., 2004,
"Haplotype diversity across 100 candidate genes for inflammation, lipid
metabolism, and
blood pressure regulation in two populations," published in Am. J. Hum. Genet.
74 (4), 610-
622, each of which is incorporated by reference herein in its entirety.
The nucleotide sequence of AHSG (identified by accession no. NM_001622) is
disclosed in, e.g., Matsushima, K. et al., 1982, "Purification and
physicochemical
characterization of human alpha2-HS-glycoprotein," published in Biochim.
Biophys. Acta
701 (2), 200-205; Keeley, F.W. et a1.,1985, "Identification and quantitation
of alpha 2-HS-
glycoprotein in the mineralized matrix of calcified plaques of atherosclerotic
human aorta"
published in Atherosclerosis 55 (1), 63-69; Yoshioka, Y. et al., 1986, "The
complete amino
acid sequence of the A-chain of human plasma alpha 2HS-glycoprotein" published
in J.
Biol. Chem. 261 (4), 1665-1676 and the amino acid sequence of AHSG (identified
by
accession no. NP 001613) is disclosed in, e.g., Matsushima, K. et al., 1982,
"Purification
and physicochemical characterization of human alpha 2-HS-glycoprotein,"
published in
Biochim. Biophys. Acta 701 (2), 200-205 (1982); Keeley, F.W. et a1.,1985,
"Identification
and quantitation of alpha 2-HS-glycoprotein in the mineralized matrix of
calcified plaques
of atherosclerotic human aorta" published in Atherosclerosis 55 (1), 63-69;
Yoshioka,Y. et
- 101 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
al., 1986, "The complete amino acid sequence of the A-chain of human plasma
alpha 2HS-
glycoprotein" published in J. Biol. Chem. 261 (4), 1665-1676, each of which is
incorporated
by reference herein in its entirety.
The nucleotide sequence of AMBP (identified by accession no. NM 001633) is
disclosed in, e.g., Ekstrom, B. et al.., 1977, "Human alphal-microglobulin.
Purification
procedure, chemical and physiochemical properties," published in J. Biol.
Chem. 252 (22),
8048-8057; Grubb, A.O. et al., 1983, "Isolation of human complex-forming
glycoprotein,
heterogeneous in charge (protein HC), and its IgA complex from plasma.
Physiochemical
and immunochemical properties, normal plasma concentration" published in J.
Biol. Chem.
258 (23), 14698-14707; Bourguignon, J. et al., 1985, "Human inter-alpha-
trypsin-inhibitor:
characterization and partial nucleotide sequencing of a light chain-encoding
cDNA"
published in Biochem. Biophys. Res. Commun. 131 (3), 1146-1153 and the amino
acid
sequence of AMBP (identified by accession no. NP 001624) is disclosed in,
e.g., Ekstrom,
B. et al., 1977, "Human alphal-microglobulin. Purification procedure, chemical
and
physiochemical properties," published in J. Biol. Chem. 252 (22), 8048-8057;
Grubb, A.O.
et al., 1983, "Isolation of human complex-forming glycoprotein, heterogeneous
in charge
(protein HC), and its IgA complex from plasma. Physiochemical and
immunochemical
properties, normal plasma concentration" published in J. Biol. Chem. 258 (23),
14698-
14707; Bourguignon, J. et al., 1985, "Human inter-alpha-trypsin-inhibitor:
characterization
and partial nucleotide sequencing of a light chain-encoding cDNA" published in
Biochem.
Biophys. Res. Conunun. 131 (3), 1146-1153, each of which is incorporated by
reference
herein in its entirety.
The nucleotide sequence of APOF (identified by accession no. NM 001638) is
disclosed in, e.g., Olofsson, S.O. et al., 1978, "Isolation and partial
characterization of a
new acidic apolipoprotein (apolipoprotein F) from high density lipoproteins of
human
plasma," published in Biochemistry 17 (6), 1032-1036; Koren, E. et al.,
"Isolation and
characterization of simple and complex lipoproteins containing apolipoprotein
F from
human plasma," published in Biochemistry 21 (21), 5347-535 1; Day, J.R. et
al., 1994,
"Purification and molecular cloning of human apolipoprotein F," published in
Biochem.
Biophys. Res. Commun. 203 (2), 1146-1151, and the amino acid sequence of APOF
(identified by accession no. AAA65642) is disclosed in, e.g., Day, J.R. et
al., 1994,
"Purification and molecular cloning of human apolipoprotein F," published in
Biochem.
Biophys. Res. Commun. 203 (2), 1146-1151, each of which is incorporated by
reference
herein in its entirety.
-102-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
The nucleotide sequence of APOA1 (identified by accession no. NM 000039) is
disclosed in, e.g., Breslow et al., 1987, "Isolation and characterization of
cDNA clones for
human apolipoprotein A-I," published in Proc. Natl. Acad. Sci. U.S.A. 79 (22),
6861-6865;
Karathanasis et al., 1983, "An inherited polymorphism in the human
apolipoprotein A-I
gene locus related to the development of atherosclerosis," published in Nature
301 (5902),
718-720; Law et al., 1983, "cDNA cloning of human apoA-I: amino acid sequence
of
preproapoA-I," published in Biochem. Biophys. Res. Commun. 112 (1), and the
amino acid
sequence of APOAI (identified by accession no. AAD34604) is disclosed in,
e.g., Hamidi
et al., 1999, "A novel apolipoprotein A-1 variant, Arg173Pro, associated with
cardiac and
cutaneous amyloidosis," Biochem. Biophys. Res. Commun. 257, 584-588, each of
which is
incorporated by reference herein in its entirety.
The nucleotide sequence of APOAI precursor (identified by accession no.
NM 000039) is disclosed in, e.g., Breslow, J.L. et al., 1987, "Isolation and
characterization
of cDNA clones for human apolipoprotein A-I," published in Proc. Natl. Acad.
Sci. U.S.A.
79 (22), 6861-6865; Karathanasis, S.K. et al., 1983, "An inherited
polymorphism in the
human apolipoprotein A-I gene locus related to the development of
atherosclerosis,"
published in Nature 301 (5902), 718-720; Law, S.W. et al., 1983, "cDNA cloning
of human
apoA-I: amino acid sequence of preproapoA-I," published in Biochem. Biophys.
Res.
Commun. 112 (1), and the amino acid sequence of APOA1 precursor (identified by
accession no. P02647) is disclosed in, e.g., Shoulders, 1983, "Gene structure
of human
apolipoprotein Al," Nucleic Acids Research 1, 1983, 2827-2837, each of which
is
incorporated by reference herein in its entirety.
The nucleotide sequence of APOA2 (identified by accession no. NM_001643) is
disclosed in, e.g., Brewer, H.B. Jr. et al., 1972, "Amino acid sequence of
human apoLp-
Gln-II (apoA-II), an apolipoprotein isolated from the high-density lipoprotein
complex,"
published in Proc. Natl. Acad. Sci. U.S.A. 69 (5), 1304-1308; Servillo, L. et
al., 1981,
"Evaluation of the mixed interaction between apolipoproteins A-II and C-I
equilibrium
sedimentation," published in Biophys. Chem. 13 (1), 29-38; Koren, E. et al.,
1982,
"Isolation and characterization of simple and complex lipoproteins containing
apolipoprotein F from human plasma," published in Biochemistry 21 (21), 5347-
535 1), and
the amino acid sequence of APOA2 (identified by accession no. AAA51701) is
disclosed in,
e.g., Chan, L. et al., 1987, "Molecular cloning and sequence analysis of human
apolipoprotein A-II cDNA," published in Meth. Enzymol. 128, 745-752, each of
which is
incorporated by reference herein in its entirety.
=103-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
The nucleotide sequence of apoliprotein AII precursor (identified by accession
no.
X00955) is disclosed in, e.g., Brewer et al., 1972, "Amino acid sequence of
human apoLp-
Gln-II (ApoA-II), an apoliprotein isolated from the high-density lipoprotein
complex," Proc.
Natl. Acad. Sci. U.S.A. 69, 1304-1308, and the amino acid sequence of
apolipoprotein All
precursor (identified by accession no. P02652) is disclosed in, e.g., Knott et
al., 1985, "The
human apolipoprotein All gene structural organization and sites of
expression," Nucleic
Acids Research 13, 6387-6398, each of which is incorporated by reference
herein in its
entirety.
The nucleotide sequence of APOA4 (identified by accession no. NM 000482) is
disclosed in, e.g., Karathanasis, S.K., 1985, "Apolipoprotein multigene
family: tandem
organization of human apolipoprotein Al, CIII, and AIV genes," published in
Proc. Natl.
Acad. Sci. U.S.A. 82 (19), 6374-6378; Elshourbagy, N.A. et al., 1986, "The
nucleotide and
derived amino acid sequence of human apolipoprotein A-IV mRNA and the close
linkage of
its gene to the genes of apolipoproteins A-I and C-Ill," published in J. Biol.
Chem. 261 (5),
1998-2002; Karathanasis, S.K. et al., 1986, "Structure, evolution, and tissue-
specific
synthesis of human apolipoprotein AIV," published in Biochemistry 25 (13),
3962-3970,
and the amino acid sequence of APOA4 (identified by accession no. AAS68228) is
disclosed in, e.g., Fullerton et al., 2004, "The effects of scale: variation
in the
APOA1/C3/A4/A5 gene cluster," Hum. Genet. 115 (1), 36-56 (2004) each of which
is
incorporated by reference herein in its entirety.
The nucleotide sequence of APOB (identified by accession no. NM_000384) is
disclosed in, e.g., Mahley, R.W. et al., 1984, "Plasma lipoproteins:
apolipoprotein structure
and function," published in J. Lipid Res. 25 (12), 1277-1294; Lusis, A.J. et
al., 1985,
"Cloning and expression of apolipoprotein B, the major protein of low and very
low density
lipoproteins," published in Proc. Natl. Acad. Sci. U.S.A. 82 (14), 4597-4601;
Deeb, S.S. et
al., 1985, "A partial eDNA clone for human apolipoprotein B," published in
Proc. Natl.
Acad. Sci. U.S.A. 82 (15), 4983-4986, and the amino acid sequence of APOB
(identified by
accession no. AAP72970) is disclosed in, Yang et al., 1986, "The complete cDNA
and
amino acid sequence of human apolipoprotein B-100," J. Biol. Chem. 261, 12918-
12921,
each of which is incorporated by reference herein in its entirety.
The nucleotide sequence of APOC 1(identified by accession no. NIvI 001645) is
disclosed in, e.g., Servillo, L. et al., 1981, "Evaluation of the mixed
interaction between
apolipoproteins A-II and C-I equilibrium sedimentation," published in Biophys.
Chem. 13
(1), 29-38; Curry, M.D. et al., 1981, "Quantitative determination of
apolipoproteins C-I and
-104-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
C-II in human plasma by separate electroimmunoassays," published in Clin.
Chem. 27 (4),
543-548; Knott, T.J. et al., 1984, "Characterisation of mRNAs encoding the
precursor for
human apolipoprotein CI," published in Nucleic Acids Res. 12 (9), 3909-3915,
and the
amino acid sequence of APOC1 (identified by accession no. AAQ91813) is
disclosed in,
Jackson et al., 1974, "The primary structure of apolipoprotein-serine," J.
Biol. Chem.
249:5308-5313, each of which is incorporated by reference herein in its
entirety.
The nucleotide sequence of APOC3 (identified by accession no. BC027977) is
disclosed in, e.g., Strausberg, R.L.et al., 2002, "Generation and initial
analysis of more than
15,000 full-length human and mouse cDNA sequences," published in Proc. Natl.
Acad. Sci.
U.S.A. 99 (26), 16899-16903, and the amino acid sequence of APOC3 (identified
by
accession no. AAB59372) is disclosed in, e.g., Maeda, H. et al., 1987,
"Molecular cloning
of a human apoC-III variant: Thr 74----Ala 74 mutation prevents 0-
glycosylation,"
published in J. Lipid Res. 28 (12), 1405-1409, each of which is incorporated
by reference
herein in its entirety.
The nucleotide sequence of APOE (identified by accession no. NM 000041) is
disclosed in, e.g., Utermann, G. et al., 1979, "Polymorphism of apolipoprotein
E. III. Effect
of a single polymorphic gene locus on plasma lipid levels in man," published
in Clin. Genet.
15 (1), 63-72; Rail, S.C. Jr. et al., 1982, "Structural basis for receptor
binding heterogeneity
of apolipoprotein E from type III hyperlipoproteinemic subjects," published in
Proc. Natl.
Acad. Sci. U.S.A. 79 (15), 4696-4700; Breslow, J.L. et al., 1982,
"Identification and DNA
sequence of a human apolipoprotein E cDNA clone," published in J. Biol. Chem.
257 (24),
14639-14641 , and the amino acid sequence of APOE (identified by accession no.
AAB59397) is disclosed in, e.g., Emi, M. et al., 1988, "Genotyping and
sequence analysis
of apolipoprotein E isoforms," published in Genomics 3 (4), 373-379; Das, H.K.
et al.,
1985, "Isolation, characterization, and mapping to chromosome 19 of the human
apolipoprotein E gene," published in J. Biol. Chem. 260 (10), 6240-6247, each
of which is
incorporated by reference herein in its entirety.
The nucleotide sequence of APOH (identified by accession no. NM_000042) is
disclosed in, e.g, Lee,.N.S. et al., 1983, "beta 2-Glycoprotein I. Molecular
properties of an
unusual apolipoprotein, apolipoprotein H," published in J. Biol. Chem. 258
(8), 4765-4770;
Lozier et al., 1984, "Complete amino acid sequence of human plasma beta 2-
glycoprotein
I," published in Proc. Natl. Acad. Sci. U.S.A. 81 (12), 3640-3644; Henry et
al., 1988,
"Inhibition of the activation of Hageman factor (factor XII) by beta 2-
glycoprotein I,"
published in J. Lab. Clin. Med. 111 (5), 519-523, and the amino acid sequence
of APOH
-105-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
(identified by accession no. CAA40977) is disclosed in, e.g. Mehdi et al.,
1991, "Nucleotide
sequence and expression of the human gene encoding apolipoprotein H (beta 2-
glycoprotein
I)," published in Gene 108 (2), 293-298, each of which is incorporated by
reference herein
in its entirety.
The nucleotide sequence of SERPINCI (identified by accession no. NM_000488) is
disclosed in, e.g., Bjork et aL, 1981, "The site in human antithrombin for
functional
proteolytic cleavage by human thrombin," FEBS Lett. 126 (2), 257-260; Lijnen,
H.R. et al.,
1983, "Heparin binding properties of human histidine-rich glycoprotein.
Mechanism and
role in the neutralization of heparin in plasma," published in J. Biol. Chem.
258, 3803-3808;
Chandra et al., 1983, "Isolation and sequence characterization of a cDNA clone
of human
antithrombin III," published in Proc. Natl. Acad. Sci. U.S.A. 80, 1845-1848,
and the amino
acid sequence of SERPINCI (identified by accession no. CAI14923) is disclosed
in, e.g.,
Sehra, 2005, Direct Submission, Wellcome Trust Sanger Institute, Hinxton,
Cambridgeshire, each of which is incorporated by reference herein in its
entirety.
The nucleotide sequence of antithrombin-III precursor (ATIII) (identified by
accession no. X68793) is disclosed in Bock et al. 1988, "Antithrombin III
Utah: proline-407
to leucine mutation in a highly conserved region near the inhibitor reactive
site,"
Biochemistry 27, 6171-6178 and the amino acid sequence of antithrombin-III
precursor is
disclosed in Bock, 1982, "Cloning and expression of the cDNA for human
antithrombin
III," Nucleic Acids Res 10, 8113-8125 each of which is incorporated by
reference herein in
its entirety.
The nucleotide sequence of AZGP 1(identified by accession no. NM_001185) is
disclosed in, e.g., Burgi et al., 1981, "Preparation and properties of Zn-
alpha 2-glycoprotein
of normal human plasma," published in J. Biol. Chem. 236, 1066-1074; Shibata
et al., 1982,
"Nephritogenic glycoprotein. IX. Plasma Zn-alpha2-glycoprotein as a second
source of
nephritogenic glycoprotein in urine," published in Nephron 31 (2), 170-176;
Ueyama, H. et
al., 1991, "Cloning and nucleotide sequence of a human Zn-alpha 2-glycoprotein
cDNA and
chromosomal assignment of its gene," published in Biochem. Biophys. Res.
Commun. 177
(2), 696-703, and the amino acid sequence of AZGP1 (identified by accession
no.
NP 001176) is disclosed in, e.g., Burgi, W. et al., 1981, "Preparation and
properties of Zn-
alpha 2-glycoprotein of normal human plasma," published in J. Biol. Chem. 236,
1066-
1074; Shibata el al., 1982, "Nephritogenic glycoprotein. IX. Plasma Zn-alpha2-
glycoprotein
as a second source of nephritogenic glycoprotein in urine," published in
Nephron 31 (2),
170-176; Ueyama, H. et al., 1991, "Cloning and nucleotide sequence of a human
Zn-alpha
- 106 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
2-glycoprotein cDNA and chromosomal assignment of its gene," published in
Biochem.
Biophys. Res. Commun. 177, 696-703, each of which is incorporated by reference
herein in
its entirety.
The nucleotide sequence of BF (identified by accession no. NM_001710) is
disclosed in, e.g., Arnason, A. et al., 1977, "Very close linkage between HLA-
B and BF
inferred from allelic association" published in Nature 268 (5620), 527-528;
Woods, et al.,
1982, "Isolation of cDNA clones for the human complement protein factor B, a
class III
major histocompatibility complex gene product" published in Proc. Natl. Acad.
Sci. U.S.A.
79 (18), 5661-5665; Campbell, R.D et al., 1983, "Molecular cloning and
characterization of
the gene coding for human complement protein factor B," published in Proc.
Natl. Acad.
Sci. U.S.A. 80 (14), 4464-4468.
The nucleotide sequence of SERPINGI (identified by accession no. NM_000062) is
disclosed in, e.g., Chesne, S. et al., 1982, "Fluid-phase interaction of C1
inhibitor (C1 Inh)
and the subcomponents C 1 r and C 1 s of the first component of complement, Cl
published
in Biochem. J. 201 (1), 61-70; Brower, M.S. et al., 1982, "Proteolytic
cleavage and
inactivation of alpha 2-plasmin inhibitor and C1 inactivator by human
polymorphonuclear
leukocyte elastase" published in J. Biol. Chem. 257 (16), 9849-9854; Nilsson,
T. et al.,
1983, "Structural and circular-dichroism studies on the interaction between
human CI-
esterase inhibitor and Cls" published in Biochem. J. 213 (3), 617-624 (1983),
and the
amino acid sequence of SERPINGI (identified by accession no. AAW69393) is
disclosed
in, e.g., Stoppa-Lyonnet, 1990, "Clusters of intragenic Alu repeats predispose
the human
Cl inhibitor locus to deleterious rearrangements," Proc. Natl. Acad. Sci. USA
87:1551-
1555, each of which is incorporated by reference herein in its entirety.
The nucleotide sequence of plasma protease C 1 inhibitor precursor (identified
by
accession no. AB209826) is disclosed in, e.g., Carter, 1988, "Genomic and cDNA
cloning
of the human CI inhibitor. Intron-exon junctions and comparison to other
serpins," Eur. J.
Biochem. 173: 163-169, and the amino acid sequence of plasma protease Cl
inhibitor
precursor (identified by accession no. P05155) is disclosed in, e.g., Dunbar
and Fothergill,
1988, Eur. J. Biochm 173, 163-169, each of which is incorporated by reference
herein in its
entirety.
The nucleotide sequence of C 1 QB (identified by accession no. NM 000491) is
disclosed in, e.g., Reid, K.B. et al., 1978, "Amino acid sequence of the N-
terminal 108
amino acid residues of the B chain of subcomponent C 1 q of the first
component of human
complement" published in Biochem. J. 173 (3), 863-868; Reid, K.B., 1979,
"Complete
- 107 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
amino acid sequences of the three collagen-like regions present in
subcomponent C 1 q of the
first component of human complement" published in Biochem. J. 179 (2), 367-
371; Reid,
K.B. et al., 1982, "Completion of the amino acid sequences of the A and B
chains of
subcomponent C 1 q of the first component of human complement" published in
Biochem. J.
203 (3), 559-569, and the amino acid sequence of C1QB (identified by accession
no.
NP_000482) is disclosed in, Reid, K.B. et al., 1978, "Amino acid sequence of
the N-
terminal 108 amino acid residues of the B chain of subcomponent C 1 q of the
first
component of human complement" published in Biochem. J. 173 (3), 863-868;
Reid, K.B.,
1979, "Complete amino acid sequences of the three collagen-like regions
present in
subcomponent C 1 q of the first component of human complement" published in
Biochem. J.
179 (2), 367-371; Reid, K.B. et al., 1982, "Completion of the amino acid
sequences of the A
and B chains of subcomponent C 1 q of the first component of human complement"
published in Biochem. J. 203 (3), 559-569, each of which is incorporated by
reference
herein in its entirety.
The nucleotide sequence of C1 S(identified by accession no. NM 201442) is
disclosed in, e.g., Nilsson, T. et al., 1983, "Structural and circular-
dichroism studies on the
interaction between human C1-esterase inhibitor and Cls" published in Biochem.
J. 213(3),
617-624; Bock, S.C. et aL, 1986, "Human Cl inhibitor: primary structure, cDNA
cloning,
and chromosomal localization" published in Biochemistry 25 (15), 4292-4301
(1986);
Mackinnon, C.M., 1987, "Molecular cloning of cDNA for human complement
component
Cls. The complete amino acid sequence" published in Eur. J. Biochem. 169 (3),
547-553,
and the amino acid sequence of CIS (identified by accession no. AAW69393) is
disclosed
in, e.g, Nilsson, T. et al., 1983, "Structural and circular-dichroism studies
on the interaction
between human C 1-esterase inhibitor and C1 s" published in Biochem. J.
213(3), 617-624;
Bock, S.C. et al., 1986, "Human Cl inhibitor: primary structure, eDNA cloning,
and
chromosomal localization" published in Biochemistry 25 (15), 4292-4301;
Mackinnon,C.M.
1987, "Molecular cloning of cDNA for human complement component C I s. The
complete
amino acid sequence" published in Eur. J. Biochem. 169 (3), 547-553, each of
which is
incorporated by reference herein in its entirety.
The nucleotide sequence of C2 (identified by accession no. NM 000063) is
disclosed in, e.g., Lutsenko, S.M. et al., 1976, "Circulating blood volume and
regional
hemodynamics in acute gastrointestinal hemorrhage" published in J. Biol. Chem.
2, 38-41;
Bentley, D.R. et al., 1984, "Isolation of cDNA clones for human complement
component
-108-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
C2 published in Proc. Natl. Acad. Sci. U.S.A. 81 (4), 1212-1215; Bentley,
D.R., 1985,
"DNA polymorphism of the C2 locus" published in Immunogenetics 22 (4), 377-
390.
The nucleotide sequence of C3 (identified by accession no. NM 000064) is
disclosed in, e.g., Alper, C.A. et al., 1970, "Studies in vivo and in vitro on
an abnormality in
the metabolism ofC3 in a patient with increased susceptibility to infection"
published in J.
Clin. Invest. 49 (11), 1975-1985; Renwick, A.G. et al., 1978, "The fate of
saccharin
impurities: the excretion and metabolism of [3-14C]Benz[d]-isothiazoline-l,l-
dioxide (BIT)
in man and rat" published in Xeriobiotica 8 (8), 475-486; Bischof, P. et al.,
1984,
"Pregnancy-associated plasma protein A (PAPP-A) specifically inhibits the
third component
of human complement (C3)" published in Placenta 5 (1), 1-7, and the amino acid
sequence
of C3 (identified by accession no. AAR89906) is disclosed in, e.g., Hugli,
1975," Human
anaphylatoxin (C3a) from the third component of complment," J. Biol. Chem.
250: 8293-
8301; Oxvig et al., 1995, "Idnentification of angiotensinogen and complement
C3dg as
novel proteins binding the proform of eosinophil major basic protein in human
pregnancy
serum and plasma," J. Biol. Chem. 270: 13645013651; and Thomas et al., 1982,
"Third
compoment of human complement: localization of the internal thiolester bond,"
Proc. Natl.
Acad. Sci. USA 79: 1054-1058, each of which is incorporated by reference
herein in its
entirety.
The nucleotide sequence of C4BPA (identified by accession no. NM 001017367) is
disclosed in, e.g., Hillarp, A. et al., 1988, "Novel subunit in C4b-binding
protein required
for protein S binding" published in J. Biol. Chem. 263 (25), 12759-12764
(1988); Hillarp.
A., 1990, "T Cloning of eDNA coding for the beta chain of human complement
component
C4b-binding protein: sequence homology with the alpha chain" published in
Proc. Natl.
Acad. Sci. U.S.A. 87 (3), 1183-1187; Andersson, A. et al., 1990, "Genes for
C4b-binding
protein alpha- and beta-chains (C4BPA and C4BPB) are located on chromosome 1,
band
1 q32, in humans and on chromosome 13 in rats" published in Somat. Cell Mol.
Genet. 16
(5), 493-500 which is incorporated by reference herein in its entirety.
The nucleotide sequence of C5 (identified by accession no. NM_001736) is
disclosed in, e.g., Gerard, N.P. et aL, 1991, "The chemotactic receptor for
human C5a
anaphylatoxin" published in Nature 349 (6310), 614-617; Boulay, F., et al.,
1991, "T
Expression cloning of a receptor for C5a anaphylatoxin on differentiated HL-60
cells "
published in Biochemistry 30 (12), 2993-2999; Bao, L., et al., 1992, "Mapping
of genes for
the human C5a receptor (C5AR), human FMLP receptor (FPR), and two FMLP
receptor
homologue orphan receptors(FPRH1, FPRH2) to chromosome 19 published in
Genomics
- 109 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
13 (2), 437-440, and the amino acid sequence of C5 (identified by accession
no.
NP 001726) is disclosed in, e.g., Tack, B.F. et al., 1979, "Fifth component of
human
complement: purification from plasma and polypeptide chain structure"
published in
Biochemistry 18 (8), 1490-1497; Lundwall, A.B. et al., 1985, "Isolation and
sequence
analysis of a cDNA clone encoding the fifth complement component" published in
J. Biol.
Chem. 260 (4), 2108-2112; Wetsel, R.A. et aL, 1988, "Molecular analysis of
human
complement component C5: localization of the structural gene to chromosome 9
published
in Biochemistry 27 (5), 1474-1482, each of which is incorporated by reference
herein in its
entirety.
The nucleotide sequence of C8A (identified by accession no. NM_000562) is
disclosed in, e.g., Matthews, 1980, "Recurrent meningococcal infections
associated with a
functional deficiency of the C8 component of human complement" published in
Clin. Exp.
Immunol. 39 (1), 53-59; Stewart, J.L. et al., 1985, "Analysis of the specific
association of
the eighth and ninth components of human complement: identification of a
direct role for
the alpha subunit of C8 Biochemistry 24 (17), 4598-4602; Rao, A.G., 1987,
"Complementary DNA and derived amino acid sequence of the alpha subunit of
human
complement protein C8: evidence for the existence of a separate alpha subunit
messenger
RNA" published in Biochemistry 26 (12), 3556-3564, and the amino acid sequence
of C8A
(identified by accession no. CA119172) is disclosed in, e.g., Steckel, 1980,
"The eight
component of human complment," J. Biol. Chem. 255:11997-12005, each of which
is
incorporated by reference herein in its entirety.
The nucleotide sequence of C8G (identified by accession no. NM 000606) is
disclosed in, e.g., Ng, S.C. et al., 1987, "The eighth component of human
complement:
evidence that it is an oligomeric serum protein assembled from products of
three different
genes," published in Biochemistry 26 (17), 5229-5233; Haefliger, J.A. et al.,
1987,
"Structural homology of human complement component C8 gamma and plasma protein
HC:
identity of the cysteine bond pattern," published in Biochem. Biophys. Res.
Commun. 149
(2), 750-754; Kaufman, K.M. et al., 1994, "Genomic structure of the human
complement
protein C8 gamma: homology to the lipocalin gene family," published in
Biochemistry 33
(17), 5162-5166, and the amino acid sequence of C8G (identified by accession
no.
CAI19172) is disclosed in, e.g., Schreck et al., 2000, "Human complement
protein C8
gamma," Biochim. Biophys. Acta 1482: 199-208; Ortlund et al., "Crystal
structure of
human complement protein C8 gamma," Biochemistry 41:7030-7037, each of which
is
incorporated by reference herein in its entirety.
-110-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
The nucleotide sequence of C9 (identified by accession no. NM 001737) is
disclosed in, e.g., DiScipio , R.G. et al., 1984, "Nucleotide sequence of cDNA
and derived
amino acid sequence of human complement component C9," published in Proc.
Natl. Acad.
Sci. U.S.A. 81 (23), 7298-7302; Stanley, K.K. et al., 1985, "The sequence and
topology of
human complement component C9," published in EMBO J. 4 (2), 375-3 82; Stewart,
J.L. et
al., 1985, "Analysis of the specific association of the eighth and ninth
components of human
complement: identification of a direct role for the alpha subunit of C8,"
published in
Biochemistry 24 (17), 4598-4602, and the amino acid sequence of C9 (identified
by
accession no. NP_001728) is disclosed in, e.g., DiScipio, R.G. et al., 1984,
"Nucleotide
sequence of cDNA and derived amino acid sequence of human complement component
C9," published in Proc. Natl. Acad. Sci. U.S.A. 81 (23), 7298-7302; Stanley,
K.K. et al.,
1985, "The sequence and topology of human complement component C9," published
in
EMBO J. 4 (2), 375-382; Stewart, J.L. et al., 1985, "Analysis of the specific
association of
the eighth and ninth components of human complement: identification of a
direct role for
the alpha subunit of C8," published in Biochemistry 24 (17), 4598-4602, each
of which is
incorporated by reference herein in its entirety.
The nucleotide sequence of SERPINA6 (identified by accession no. NM_001756) is
disclosed in, e.g., Rosner, W. et al., 1976, "Identification of corticosteroid-
binding globulin
in human milk: measurement with a filter disk assay," published in J. Clin.
Endocrinol.
Metab. 42 (6), 1064-1073; Agrimonti, F. et al., 1982, "Circadian and
circaseptan
rhythmicities in corticosteroid-binding globulin (CBG) binding activity of
human milk,"
published in J. Chromatogr. 9 (3), 281-290; Hammond, G.L. et al., 1986,
"Identification and
measurement of sex hormone binding globulin (SHBG) and corticosteroid binding
globulin
(CBG) in human saliva," published in Acta Endocrinol. 112 (4), 603-608, and
the amino
acid sequence of SERPINA6 (identified by accession nos. NP001002236, NP000286)
is
disclosed in, e.g., Kurachi, K. et al., 1981, "Cloning and sequence of cDNA
coding for
alpha 1-antitrypsin," published in Proc. Natl. Acad. Sci. U.S.A. 78 (11), 6826-
6830;
Lobermann, H. et al., 1982, "Interaction of human alpha 1-proteinase inhibitor
with
chymotrypsinogen A and crystallization of a proteolytically modified alpha 1-
proteinase
inhibitor," published in Hoppe-Seyler's Z. Physiol. Chem. 363 (11), 1377-1388;
Bollen, A.
et al., 1983, "Cloning and expression in Escherichia coli of full-length
complementary DNA
coding for human alpha 1-antitrypsin," published in DNA 2 (4), 255-264, each
of which is
incorporated by reference herein in its entirety.
- 111 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
The nucleotide sequence of CD14 (identified by accession no. NM000591) is
disclosed in, e.g., Goyert, S.M. et al., 1988, "The CD14 monocyte
differentiation antigen
maps to a region encoding growth factors and receptors," published in Science
239 (4839),
497-500; Ferrero, E. et aL, 1988, "Nucleotide sequence of the gene encoding
the monocyte
differentiation antigen, CD 14," published in Nucleic Acids Res. 16 (9), 4173;
Simmons,
D.L. et aL, 1989, "Monocyte antigen CD14 is a phospholipid anchored membrane
protein,"
published in Blood 73 (1), 284-289 which is incorporated by reference herein
in its entirety.
The nucleotide sequence of CLU (identified by accession no. NM_203339) is
disclosed in, e.g., Murphy, B.F. et al., 1988, "SP-40,40, a newly identified
normal human
serum protein found in the SC5b-9 complex of complement and in the immune
deposits in
glomerulonephritis," published in J. Clin. Invest. 81 (6), 1858-1864;
Yokoyama, M. et al.,
1988, "Isolation and characterization of sulfated glycoprotein from human
pancreatic juice,"
published in Biochim. Biophys. Acta 967 (1), 34-42; Kirszbaum, L. et al.,
1989, "Molecular
cloning and characterization of the novel, human complement-associated
protein, SP-40,40:
a link between the complement and reproductive systems," published in EMBO J.
8 (3),
711-718, and the amino acid sequence of CLU (identified by accession no.
AAP88927) is
disclosed in, e.g., James et al., 1991, "Characterization of a human high
density lipoprotein-
associated protein," Arterioscler. Thromb. 11:645-652; de Silva et al., 1990,
"Purification
and characterization of apolipoprotein J," J. Biol. Chem. 265: 14292-14297,
each of which
is incorporated by reference herein in its entirety.
The nucleotide sequence of CP (identified by accession no. NM 000096) is
disclosed in, e.g., Kingston, I.B. et al., 1977, "Chemical evidence that
proteolytic cleavage
causes the heterogeneity present in human ceruloplasmin preparations,"
published in Proc.
Nati. Acad. Sci. U.S.A. 74 (12), 5377-5381; Polosatov, M.V. et al., 1979,
"Interaction of
synthetic human big gastrin with blood proteins of man and animals," published
in Proc.
Natl. Acad. Sci. U.S.A. 26 (2), 154-159; Rask, L. et al., 1983, "Subcellular
localization in
normal and vitamin A-deficient rat liver of vitamin A serum transport
proteins, albumin,
ceruloplasmin and class I major histocompatibility antigens," published in
Exp. Cell Res.
143 (1), 91-102, and the amino acid sequence of CP (identified by accession
no.
NP_000087) is disclosed in, e.g., Kingston, I.B. et al., 1977, "Chemical
evidence that
proteolytic cleavage causes the heterogeneity present in human ceruloplasmin
preparations," published in Proc. Natl. Acad. Sci. U.S.A. 74 (12), 5377-5381;
Polosatov,
M.V. et al., 1979, "Interaction of synthetic human big gastrin with blood
proteins of man
and animals," published in Proc. Natl. Acad. Sci. U.S.A. 26 (2), 154-159;
Rask, L. et al.,
- 112 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
1983, "Subcellular localization in normal and vitamin A-deficient rat liver of
vitamin A
serum transport proteins, albumin, ceruloplasmin and class I major
histocompatibility
antigens," published in Exp. Cell Res. 143 (1), 91-102, each of which is
incorporated by
reference herein in its entirety.
The nucleotide sequence of CRP (identified by accession no. NM 000567) is
disclosed in, e.g., Osmand, A.P. et al., 1977, "Characterization of C-reactive
protein and the
complement subcomponent Clt as homologous proteins displaying cyclic
pentameric
symmetry (pentraxins)," published in Proc. Natl. Acad. Sci. U.S.A. 74 (2), 739-
743;
Oliveira,E.B. et al., 1979, "Primary structure of human C-reactive protein,"
published in J.
Biol. Chem. 254 (2), 489-502; Whitehead, A.S. et al., 1983, "Isolation of
human C-reactive
protein complementary DNA and localization of the gene to chromosome 1,"
published in
Science 221 (4605), 69-7 1, and the amino acid sequence of CRP (identified by
accession
no. NP 000558) is disclosed in, e.g., Osmand, A.P. et al., 1977,
"Characterization of C-
reactive protein and the complement subcomponent Cit as homologous proteins
displaying
cyclic pentameric symmetry (pentraxins)," published in Proc. Natl. Acad. Sci.
U.S.A. 74
(2), 739-743; Oliveira,E.B. et aL, 1979, "Primary structure of human C-
reactive protein,"
published in J. Biol. Chem. 254 (2), 489-502; Whitehead,A.S. et al., 1983,
"Isolation of
human C-reactive protein complementary DNA and localization of the gene to
chromosome
1," published in Science 221 (4605), 69-71, each of which is incorporated by
reference
herein in its entirety.
The nucleotide sequence and the amino acid sequence of C-reactive protein
precursor (respectively identified by accession nos. M11880 and AAB59526) is
disclosed
in, e.g., Who et al., 1985, "Characterization of genomic and complementary DNA
sequence
of human C-reactive poretin, comparison with the complementary DNA sequence of
serum
amyloid P component," J. Biol. Chem. 260, 13384-13388 which is incorporated by
reference herein in its entirety.
The nucleotide sequence of F2 (identified by accession no. NM 000506) is
disclosed in, e.g., Bergmann et al., 1982, "Receptor-bound thrombin is not
internalized
through coated pits in mouse embryo cells," published in J. Cell. Biochem. 20
(3), 247-258;
Degen et aL, 1983, "Characterization of the complementary deoxyribonucleic
acid and gene
coding for human prothrombin," published in Biochemistry 22 (9), 2087-2097;
Wicki, A.N.
et al., 1985, "Structure and function of platelet membrane glycoproteins lb
and V. Effects of
leukocyte elastase and other proteases on platelets response to von Willebrand
factor and
thrombin," published in Eur. J. Biochem. 153 (1), 1-11, and the amino acid
sequence of F2
-113-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
(identified by accession no. AAL77436) is disclosed in Walz et al., 1972,
"Amino Acid
Sequence of human prothrombin fragments 1 and 2," Proc. Nati. Acad. Sci.
U.S.A.
74:1969-1972; Butkowski et al., 1977, "Primary structure of human prethrombin
2 and
alpha-thrombin," J. Biol. Chem. 252: 4942-4957, each of which is incorporated
by reference
herein in its entirety.
The nucleotide sequence of F9 (identified by accession no. NM_000133) is
disclosed in, e.g., Davie et al., 1975, "Basic mechanisms in blood
coagulation," published in
Annu. Rev. Biochem. 44, 799-829; Gentry,P.A., 1977, "Interaction of heparin
with canine
coagulation proteins: in vivo and in vitro studies," published in Can. J.
Comp. Med. 41 (4),
396-403; Choo, K.H. et al., 1982, "Molecular cloning of the gene for human
anti-
haemophilic factor IX," published in Nature 299 (5879), 178-180, and the amino
acid
sequence of F9 (identified by accession no. NP_000124) is disclosed in, e.g.,
Scherer
Davie,E.W. et al., 1975, "Basic mechanisms in blood coagulation," published in
Annu. Rev.
Biochem. 44, 799-829; Gentry, P.A., 1977, "Interaction of heparin with canine
coagulation
proteins: in vivo and in vitro studies," published in Can. J. Comp. Med. 41
(4), 396-403;
Choo, K.H. et al., 1982, "Molecular cloning of the gene for human anti-
haemophilic factor
IX," published in Nature 299 (5879), 178-180, each of which is incorporated by
reference
herein in its entirety.
The nucleotide sequence of FGA (identified by accession no. BC070246) is
disclosed in, e.g., Strausberg et al., 2002, "Generation and initial analysis
of more than
15,000 full-length human and mouse cDNA sequences," published in Proc. Natl.
Acad. Sci.
U.S.A. 99 (26), 16899-16903, and the amino acid sequence of FGA (identified by
accession
no. BAC55116) is disclosed in, e.g., Hamasaki,N., 2002, Direct Submission,
Naotaka
Hamasaki, Kyushu University Hospital, Department of clinical chemistry and
laboratory; 3-
1-1 maidasi, Higasi-ku Fukuokasi, Fukuoka 812-8582, Japan, Watanabe,K. et al.,
unpublished, "Identification of simultaneous mutation of fibrinogen alpha;
chain and protain
C genes in a Japanese kindred," each of which is incorporated by reference
herein in its
entirety.
The nucleotide sequence of FGB (identified by accession no. NM_005141) is
disclosed in, e.g., Tarakhovskii et al., 1979, "Temperature-dependent changes
in the profile
of the sarcoplasmic reticulum membrane hydrophobic zones," published in
Biokhimiia 44
(5), 897-902; Weinger,R.S. et al., 1980, "Fibrinogen Houston: a dysfibrinogen
exhibiting
defective fibrin monomer aggregation and alpha-chain cross-linkages,"
published in Am. J.
Hematol. 9 (3), 237-248; Chung, D.W. et al., 1983, "Characterization of
complementary
- 114 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
deoxyribonucleic acid and genomic deoxyribonucleic acid for the beta chain of
human
fibrinogen," published in Biochemistry 22 (13), 3244-3250, and the amino acid
sequence of
FGB (identified by accession no. AAA18024) is disclosed in, e.g., Chung, D.W.
et al.,
1991, "Nucleotide sequences of the three genes coding for human fibrinogen",
Fibrinogen,
Thrombosis, Coagulation and Fibrinolysis: 39-48; Plenum Press, New York, each
of which
is incorporated by reference herein in its entirety.
The nucleotide sequence of FGG (identified by accession no. NM_000509) is
disclosed in, e.g., Olaisen, B. et al., 1982, "Fibrinogen gamma chain locus is
on
chromosome 4 in man," published in Hum. Genet. 61 (1), 24-26, Hawiger,J. et
al., 1982,
"gamma and alpha chains of human fibrinogen possess sites reactive with human
platelet
receptors," published in Proc. Nati. Acad. Sci. U.S.A. 79 (6), 2068-2071;
Chung, D.W. et
a1.,1983, "Characterization of a complementary deoxyribonucleic acid coding
for the
gamma chain of human fibrinogen," published in Biochemistry 22 (13), 3250-
3256, and the
amino acid sequence of FGG (identified by accession no. AAB59531) is disclosed
in, e.g.,
Rixon, M. W. et aL, 1985, "Nucleotide sequence of the gene for the gamma chain
of human
fibrinogen," published in Biochemistry 24 (8), 2077-2086, each of which is
incorporated by
reference herein in its entirety.
The nucleotide sequence of FLNA (identified by accession no. NM_001456) is
disclosed in, e.g., Wallach, D. et al., 1978, "Cyclic AMP-dependent
phosphorylation of the
actin-binding protein filamin," published in Proc. Natl. Acad. Sci. U.S.A. 9,
371-379;
Gorlin, J.B. et aL, 1990, "Human endothelial actin-binding protein (ABP-280,
nonmuscle
filamin): a molecular leaf spring," published in J. Cell Biol. 111 (3), 1089-
1105;
Maestrini, E. et al., 1990, "Probes for CpG islands on the distal long arm of
the human X
chromosome are clustered in Xq24 and Xq28," published in Genomics 8 (4), 664-
670, and
the amino acid sequence of FLNA (identified by accession no. CA143227) is
disclosed in,
e.g., Heath, P., 2002, Direct Submission, Wellcome Trust Sanger Institute,
Hinxton,
Cambridgeshire, CB 10 1 SA, UK, each of which is incorporated by reference
herein in its
entirety.
The nucleotide sequence of FN1 (identified by accession no. BT006856) is
disclosed
in, e.g., Kalnine et al., 2003, Direct Submission, BD Biosciences Clontech,
1020 East
Meadow Circle, Palo Alto, California 94303, USA; Kalnine,N. et ar,
unpublished,
"Cloning of human full-length CDSs in BD CreatorTM System Donor vector," and
the,
amino acid sequence of FN1 (identified by accession no. BAD52437) is disclosed
in, e.g.,
Kato, 2004, Direct Submission, Seishi Kato, National Rehabilitation Center for
Persons
-115-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
with Disabilities, Research Institute, Department of Rehabilitation
Engineering; 4-1 Namiki,
Tokorozawa, Saitama 359-8555, Japan; Kato, 2004, "Human full-length cDNA
starting
with the capped site sequence," Published only in database, each of which is
incorporated
by reference herein in its entirety.
The nucleotide sequence of GC (identified by accession no. NM 000583) is
disclosed in, e.g., Mikkelsen, M. et al., 1977, "Possible localization of Gc-
System on
chromosome 4. Loss of long arm 4 material associated with father-child
incompatibility
within the Ge-System," published in Hum. Hered. 27 (2), 105-107; Constans, J.
et al., 1981,
"Binding of the apo and holo forms of the serum vitamin D-binding protein to
human
lymphocyte cytoplasm and membrane by indirect immunofluorescence," published
in
Immunol. Lett. 3 (3), 159-162; Wooten, M.W. et al., 1985, "Identification of a
major
endogenous substrate for phospholipid/Ca2+-dependent kinase in pancreatic
acini as
Gc(vitamin D-binding protein)," published in FEBS Lett. 191 (1), 97-101, and
the amino
acid sequence of GC (identified by accession no. NP_000574) is disclosed in,
e.g.,
Mikkelsen, M. et al., 1977, "Possible localization of Gc-System on chromosome
4. Loss of
long arm 4 material associated with father-child incompatibility within the Gc-
System,"
published in Hum. Hered. 27 (2), 105-107; Constans, J. et al., 1981, "Binding
of the apo
and holo forms of the serum vitamin D-binding protein to human lymphocyte
cytoplasm
and membrane by indirect immunofluorescence," published in Immunol. Lett. 3
(3), 159-
162; Wooten, M.W. et al., 1985, "Identification of a major endogenous
substrate for
phospholipid/Ca2+-dependent kinase in pancreatic acini as Gc(vitamin D-binding
protein),"
published in FEBS Lett. 191 (1), 97-101, each of which is incorporated by
reference herein
in its entirety.
The nucleotide sequence of GSN (identified by accession no. BC026033) is
disclosed in, e.g., Strausberg et al., 2002, "Generation and initial analysis
of more than
15,000 full-length human and mouse cDNA sequences," published in Proc. Nat1.
Acad. Sci.
U.S.A. 99 (26), 16899-16903 which is incorporated by reference herein in its
entirety.
The nucleotide sequence of HBB (identified by accession no. NM000518) is
disclosed in, e.g., Marotta, C.A. et al., 1976, "Nucleotide sequence analysis
of coding and
noncoding regions of human beta-globin mRNA," published in Prog. Nucleic Acid
Res.
Mol. Biol. 19, 165-175; Proudfoot,N.J., 1977, "Complete 3 noncoding region
sequences of
rabbit and human beta-globin messenger RNAs, published in Cell 10 (4), 559-
570; Marotta,
C.A. et al., 1977, "Human beta-globin messenger RNA. III. Nucleotide sequences
derived
from complementary DNA," published in J. Biol. Chem. 252 (14), 5040-5053, and
the
-116-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
amino acid sequence of HBB (identified by accession no. AAD19696) is disclosed
in, e.g.,
Braunitzer et al., 1961, "The constitution of normal adult human haemoglobin,"
Hoppe-Seyler's Z. Physiol. Chem. 325:283-286, each of which is incorporated by
reference
herein in its entirety.
The nucleotide sequence of SERPIND 1(identified by accession no. NM_000185) is
disclosed in, e.g., Ragg, H., 1986, "A new member of the plasma protease
inhibitor gene
family," published in Nucleic Acids Res. 14 (2), 1073-1088; Inhorn,R.C. et
al., 1986,
"Isolation and characterization of a-partial cDNA clone for heparin cofactor
II1," published
in Biochem. Biophys. Res. Commun. 137 (1), 431-436; Hortin, G. et al., 1986,
"Identification of two sites of sulfation of human heparin cofactor II,"
published in J. Biol.
Chem. 261 (34), 15827-15830, and the amino acid sequence of SERPINDI
(identified by
accession no. CAG30459) is disclosed in, e.g., Collins et al., 2004, "A genome
annotation-driven approach to cloning the human ORFeome," published in Genome
Biol. 5
(10), R84, each of which is incorporated by reference herein in its entirety.
The nucleotide sequence of HP (identified by accession no. BC107587) is
disclosed
in, e.g. Strausberg et al., 2002, "Generation and initial analysis of more
than 15,000 full-
length human and mouse cDNA sequences," published in Proc. Natl. Acad. Sci.
U.S.A. 99
(26), 16899-16903, and the amino acid sequence of HP (identified by accession
no.
NP_005134) is disclosed in, e.g., Kazim, A.L. et al., 1980, "Haemoglobin
binding with
haptoglobin. Unequivocal demonstration that the beta-chains of human
haemoglobin bind to
haptoglobin, published in Biochem. J. 185 (1), 285-287; Eaton et al., 1982,
"Haptoglobin: a
natural bacteriostat," published in Science 215 (4533), 691-693; Costanzo et
al., Sequence
of hurnan haptoglobin cDNA: evidence that the alpha and beta subunits are
coded by the
same mRNA," published in Nucleic Acids Res. 11 (17), 5811-5819, each of which
is
incorporated by reference herein in its entirety.
The nucleotide sequence of HPX (identified by accession no. NM 000613) is
disclosed in, e.g., Morgan, W.T. et al., 1978, "Interaction of rabbit
hemopexin with
bilirubin," published in Biochim. Biophys. Acta 532 (1), 57-64; Takahashi,N.
et al., 1984,
"Structure of human hemopexin: 0-glycosyl and N-glycosyl sites and unusual
clustering of
tryptophan residues," published in Proc. Natl. Acad. Sci. U.S.A. 81 (7), 2021-
2025;
Frantikova, V. et al., .Amino acid sequence of the N-terminal region of human
hemopexin,"
published in FEBS Lett. 178 (2), 213-216, and the amino acid sequence of HPX
(identified
by accession no. NP_000604) is disclosed in, e.g., Morgan, W.T. et al., 1978,
"Interaction
of rabbit hemopexin with bilirubin," published in Biochim. Biophys. Acta 532
(1), 57-64;
-117-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Takahashi, N. et al., 1984, "Structure of human hemopexin: 0-glycosyl and N-
glycosyl
sites and unusual clustering of tryptophan residues," published in Proc. Natl.
Acad. Sci.
U.S.A. 81 (7), 2021-2025; Frantikova,V. et al., Amino acid sequence of the N-
terminal
region of human hemopexin," published in FEBS Lett. 178 (2), 213-216, each of
which is
incorporated by reference herein in its entirety.
The nucleotide sequence of HRG (identified by accession no. NM_000412) is
disclosed in, e.g., Heimburger, N. et al., 1972, "Human serum proteins with
high affinity to
carboxymethylcellulose. II. Physico-chemical and immunological
characterization of a
histidine-rich 3,8S- 2 -glycoportein (CM-protein I)," published in Hoppe-
Seyler's Z.
Physiol. Chem. 353 (7), 1133-1140; Silverstein, R.L. et al., 1984, "Complex
formation of
platelet thrombospondin with plasminogen. Modulation of activation by tissue
activator,"
published in J. Clin. Invest. 74 (5), 1625-1633; Leung, L.L., 1986,
"Interaction of histidine-
rich glycoprotein with fibrinogen and fibrin," published in J. Clin. Invest.
77 (4), 1305-
1311, and the amino acid sequence of HRG (identified by accession no.
NP_000403) is
disclosed in, e.g., Heimburger,N. et al., 1972, "Human serum proteins with
high affinity to
carboxymethylcellulose. II. Physico-chemical and immunological
characterization of a
histidine-rich 3,8S- 2 -glycoportein (CM-protein I)," published in Hoppe-
Seyler's Z.
Physiol. Chem. 353 (7), 1133-1140; Silverstein, R.L. et al., 1984, "Complex
formation of
platelet thrombospondin with plasminogen. Modulation of activation by tissue
activator,"
published in J. Clin. Invest. 74 (5), 1625-1633; Leung, L.L., 1986,
"Interaction of histidine-
rich glycoprotein with fibrinogen and fibrin," published in J. Clin. Invest.
77 (4), 1305-
1311, each of which is incorporated by reference herein in its entirety.
The nucleotide sequence of IF (identified by accession no. NM 000204) is
disclosed
in, e.g., Catterall, C.F. et al., 1987, "Characterization of primary amino
acid sequence of
human complement control protein factor I from an analysis of cDNA clones,"
published in
Biochem. J. 242 (3), 849-856; Goldberger,G. et al., 1987, "Human complement
factor I:
analysis of cDNA-derived primary structure and assignment of its gene to
chromosome 4
published in J. Biol. Chem. 262 (21), 10065-10071; Shiang, R. et al., 1989,
"Mapping of the
human complement factor I gene to 4q25," published in Genomics 4 (1), 82-86,
and the
amino acid sequence of IF (identified by accession no. NP_000195) is disclosed
in, e.g.,
Catterall, C.F. et al., 1987, "Characterization of primary amino acid sequence
of human
complement control protein factor I from an analysis of cDNA clones,"
published in
Biochem. J. 242 (3), 849-856; Goldberger, G. et al., 1987, "Human complement
factor I:
analysis of eDNA-derived primary structure and assignment of its gene to
chromosome 4
-118-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
published in J. Biol. Chem. 262 (21), 10065 10071; Shiang,R. et al., 1989,
"Mapping of the
human complement factor I gene to 4q25," published in Genomics 4 (1), 82-86,
each of
which is incorporated by reference herein in its entirety.
The nucleotide sequence of IGFALS (identified by accession no. NM_004970) is
disclosed in, e.g., Baxter, R.C. et al., 1989, "High molecular weight insulin-
like growth
factor binding protein complex. Purification and properties of the acid-labile
subunit from
human serum," published I J. Biol. Chem. 264 (20), 11843-11848; Leong, S.R. et
al., 1992,
"Structure and functional expression of the acid-labile subunit of the insulin-
like growth
factor-binding protein complex," published in Mol. Endocrinol. 6 (6), 870-876;
Dai, J. et
al., 1992, "Molecular cloning of the acid-labile subunit of the rat insulin-
like growth factor
binding protein complex," published in Biochem. Biophys. Res. Commun. 188 (1),
304-
309, and the amino acid sequence of IGFALS (identified by accession no.
NP004691) is
disclosed in, e.g., Kubisch, C. et al., 1999, "KCNQ4, a novel potassium
channel expressed
in sensory outer hair cells, is mutated in dominant deafness," published in
Cell 96 (3), 437-
446; Selyanko, A.A. et al., 2000, "Inhibition of KCNQ1-4 potassium channels
expressed in
mammalian cells via Ml muscarinic acetylcholine receptors," published in J.
Physiol.
(Lond.) 522 PT 3, 349-355; Sogaard, R. et al., 2001, "KCNQ4 channels expressed
in
mammalian cells: functional characteristics and pharmacology," published in
Am. J.
Physiol., Cell Physiol. 280 (4), C859-C866, each of which is incorporated by
reference
herein in its entirety.
The nucleotide sequence of ITGA1 (identified by accession no. NM_181501) is
disclosed in, e.g., Takada et al., 1987, "The very late antigen family of
heterodimers is part
of a superfamily of molecules involved in adhesion and embryogenesis,"
published in Proc.
Natl. Acad. Sci. U.S.A. 84 (10), 3239-3243; MacDonald, T.T. et al., 1990,
"Increased
expression of laminin/collagen receptor (VLA-1) on epithelium of inflamed
human
intestine," published in J. Clin. Pathol. 43 (4), 313-315; Tawil,N.J. et al.,
1990, "Alpha I
beta 1 integrin heterodimer functions as a dual laminin/collagen receptor in
neural cells,"
published in Biochemistry 29 (27), 6540-6544, and the amino acid sequence of
ITGA1
(identified by accession no. NP_852478) is disclosed in, e.g., Scherer
Takada,Y. et al.,
1987, "The very late antigen family of heterodimers is part of a superfamily
of molecules
involved in adhesion and embryogenesis," published in Proc. Natl. Acad. Sci.
U.S.A. 84
(10), 3239-3243; MacDonald, T.T. et al., 1990, "Increased expression of
laminin/collagen
receptor (VLA-1) on epithelium of inflamed human intestine," published in J.
Clin. Pathol.
43 (4), 313-315; Tawil, N.J. et al., 1990, "Alpha 1 beta 1 integrin
heterodimer functions as a
- 119 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
dual laminin/collagen receptor in neural cells," published in Biochemistry 29
(27), 6540-
6544, each of which is incorporated by reference herein in its entirety.
The nucleotide sequence of ITIH1 (identified by accession no. BC109115) is
disclosed in, e.g., NIH MGC Project, 2005, Direct Submission, National
Institutes of
Health, Mammalian Gene Collection (MGC), Bethesda, MD 20892-2590, USA, and the
amino acid sequence of ITIH1 (identified by accession nos. NP_002206,
NP_032432) is
disclosed in, e.g., Salier, J.P. et al., 1987, "Isolation and characterization
of cDNAs
encoding the heavy chain of human inter-alpha-trypsin inhibitor (I alpha TI):
unambiguous
evidence for multipolypeptide chain structure of I alpha TI," published in
Proc. Natl. Acad.
Sci. U.S.A. 84 (23), 8272-8276; Diarra-Mehrpour, M. et al., 1989, "Human
plasma inter-
alpha-trypsin inhibitor is encoded by four genes on three chromosomes,"
published in Eur.
J. Biochem. 179 (1), 147-154; Gebhard, W. et al., 1989, "Two out of the three
kinds of
subunits of inter-alpha-trypsin inhibitor are structurally related," published
in Eur. J.
Biochem. 181 (3), 571-576, each of which is incorporated by reference herein
in its entirety.
The nucleotide sequence of ITIH2 (identified by accession no. NM 002216) is
disclosed in, e.g., Salier, J.P. et al., 1987, "Isolation and characterization
of cDNAs
encoding the heavy chain of human inter-alpha-trypsin inhibitor (I alpha TI):
unambiguous
evidence for multipolypeptide chain structure of I alpha TI," published in
Proc. Natl. Acad.
Sci. U.S.A. 84 (23), 8272-8276; Gebhard, W. et al., 1988, "Complementary DNA
and
derived amino acid sequence of the precursor of one of the three protein
components of the
inter-alpha-trypsin inhibitor complex," published in FEBS Lett. 229 (1), 63-
67; Salier, J.P.
et aL, 1988, "Human inter-alpha-trypsin inhibitor. Isolation and
characterization of heavy
(H) chain cDNA clones coding for a 383 amino-acid sequence of the H chain,"
published in
Biol. Chem. Hoppe-Seyler 369 SUPPL, 15-18, and the amino acid sequence of
ITIH2
(identified by accession no. NP 002207) is disclosed in, e.g., Salier, J.P. et
al., 1987,
"Isolation and characterization of cDNAs encoding the heavy chain of human
inter-alpha-
trypsin inhibitor (I alpha TI): unambiguous evidence for multipolypeptide
chain structure of
I alpha TI," published in Proc. Natl. Acad. Sci. U.S.A. 84 (23), 8272-8276;
Gebhard, W. et
al., 1988, "Complementary DNA and derived amino acid sequence of the precursor
of one
of the three protein components of the inter-alpha-trypsin inhibitor complex,"
published in
FEBS Lett. 229 (1), 63-67; Salier, J.P. et al., 1988, "Human inter-alpha-
trypsin inhibitor.
Isolation and characterization of heavy (H) chain eDNA clones coding for a 383
amino-acid
sequence of the H chain," published in Biol. Chem. Hoppe-Seyler 369 SUPPL, 15-
18, each
of which is incorporated by reference herein in its entirety.
-120-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
The nucleotide sequence of ITIH4 (identified by accession no. NM_002218) is
disclosed in, e.g., Tobe, T. et aL, 1995, "Mapping of human inter-alpha-
trypsin inhibitor
family heavy chain-related protein gene (ITIHL 1) to human chromosome 3p21-->p
14,"
published in Cytogenet. Cell Genet. 71 (3), 296-298; Saguchi, K. et al., 1995,
"Cloning and
characterization of cDNA for inter-alpha-trypsin inhibitor family heavy chain-
related
protein (IHRP), a novel human plasma glycoprotein," published in J. Biochem.
117 (1), 14-
18; Nishimura, H. et al., 1995, "cDNA and deduced amino acid sequence of human
PK-
120, a plasma kallikrein-sensitive glycoprotein," published in FEBS Lett. 357
(2), 207-211,
and the amino acid sequence of ITIH4 (identified by accession no. NP 002209)
is disclosed
in, e.g., Tobe, T. et al., 1995, "Mapping of human inter-alpha-trypsin
inhibitor family heavy
chain-related protein gene (ITIHLI) to human chromosome 3p2l-->pl4," published
in
Cytogenet. Cell Genet. 71 (3), 296-298; Saguchi et al., 1995, "Cloning and
characterization
of cDNA for inter-alpha-trypsin inhibitor family heavy chain-related protein
(IHRP), a
novel human plasma glycoprotein," published in J. Biochem. 117 (1), 14-18;
Nishimura, H.
et al., 1995, "cDNA and deduced amino acid sequence of human PK-120, a plasma
kallikrein-sensitive glycoprotein," published in FEBS Lett. 357 (2), 207-211,
each of which
is incorporated by reference herein in its entirety.
The nucleotide sequence of KLKB1 (identified by accession no. NM_000892) is
disclosed in, e.g., Aznar, J.A. et al., 1978, "Fletcher factor deficiency:
report of a new
family," published in J. Biol. Chem. 21 (2), 94-98; Thompson, R.E. etal.,
"Studies of
binding of prekallikrein and Factor XI to high molecular weight kininogen and
its light
chain," published in Proc. Natl. Acad. Sci. U.S.A. 76 (10), 4862-4866; Chung,
D.W., et al.,
"Human plasma prekallikrein, a zymogen to a serine protease that contains four
tandem
repeats," published in Biochemistry 25 (9), 2410-2417, and the amino acid
sequence of
KLKB1 (identified by accession no. NP_000883) is disclosed in, e.g., Aznar,
J.A. et al.,
1978, "Fletcher factor deficiency: report of a new family," published in J.
Biol. Chem. 21
(2), 94-98; Thompson,R.E. et al., "Studies of binding of prekallikrein and
Factor XI to high
molecular weight kininogen and its light chain," published in Proc. Natl.
Acad. Sci. U.S.A.
76 (10), 4862-4866; Chung,D.W., et al., "Human plasma prekallikrein, a zymogen
to a
serine protease that contains four tandem repeats," published in Biochemistry
25 (9), 2410-
2417, each of which is incorporated by reference herein in its entirety.
The nucleotide sequence of KNGI (identified by accession no. NM 000893) is
disclosed in, e.g., Colman, R.W. et al., 1975, "Williams trait. Human
kininogen deficiency
with diminished levels of plasminogen proactivator and prekallikrein
associated with
- 121 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
abnormalities of the Hageman factor-dependent pathways," published in J. Clin.
Invest. 56
(6), 1650-1662; Thompson, R.E. et aL, 1979, "Studies of binding of
prekallikrein and
Factor XI to high molecular weight kininogen and its light chain," published
in Proc. Natl.
Acad. Sci. U.S.A. 76 (10), 4862-4866; Kerbiriou, D.M. et aL, 1979, "Human high
molecular weight kininogen. Studies of structure-function relationships and of
proteolysis of
the molecule occurring during contact activation of plasma," published in J.
Biol. Chem.
254 (23), 12020-12027, and the amino acid sequence of KNG 1(identified by
accession no.
NP000884) is disclosed in, e.g., Colman, R.W. et al., 1975, "Williams trait.
Human
kininogen deficiency with diminished levels of plasminogen proactivator and
prekallikrein
associated with abnormalities of the Hageman factor-dependent pathways,"
published in J.
Clin. Invest. 56 (6), 1650-1662; Thompson, R.E. et al., 1979, "Studies of
binding of
prekallikrein and Factor XI to high molecular weight kininogen and its light
chain,"
published in Proc. Natl. Acad. Sci. U.S.A. 76 (10), 4862-4866; Kerbiriou, D.M.
et aL, 1979,
"Human high molecular weight kininogen. Studies of structure-function
relationships and of
proteolysis of the molecule occurring during contact activation of plasma,"
published in J.
Biol. Chem. 254 (23), 12020-12027, each of which is incorporated by reference
herein in its
entirety.
The nucleotide sequence of KRT1 (identified by accession no. BC063697) is
disclosed in, e.g., Strausberg et aL, 2002, "Generation and initial analysis
of more than
15,000 full-length human and mouse cDNA sequences," published in Proc. Natl.
Acad. Sci.
U.S.A. 99 (26), 16899-16903, and the amino acid sequence of KRT1 (identified
by
accession no. NP_000412) is disclosed in, e.g., Darmon, M.Y. et aL, 1987,
"Sequence of a
cDNA encoding human keratin No 10 selected according to structural homologies
of
keratins and their tissue-specific expression," published in Mol. Biol. Rep.
12 (4), 277-283;
Zhou, X.M. et aL, 1988, "The complete sequence of the human intermediate
filament chain
keratin 10. Subdomainal divisions and model for folding of end domain
sequences,"
published in J. Biol. Chem. 263 (30), 15584-15589, each of which is
incorporated by
reference herein in its entirety.
The nucleotide sequence of LGALS3BP (identified by accession nos. NM005567,
BC015761, BC002403, BC002998) is disclosed in, e.g., Rosenberg, I. et aL,
1991, "Mac-2-
binding glycoproteins. Putative ligands for a cytosolic beta-galactoside
lectin," published in
J. Biol. Chem. 266 (28), 18731-18736; Koths, K. etal., 1993, "Cloning and
characterization
of a human Mac-2-binding protein, a new member of the superfamily defined by
the
macrophage scavenger receptor cysteine-rich domain," published in J. Biol.
Chem. 268
- 122 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
(19), 14245-14249; Ullrich,A. et al., 1994, "The secreted tumor-associated
antigen 90K is a
potent immune stimulator," published in J. Biol. Chem. 269 (28), 18401-18407;
Strausberg
et al., 2002, "Generation and initial analysis of more than 15,000 full-length-
human and
mouse cDNA sequences," published in Proc. Natl. Acad. Sci. U.S.A. 99 (26),
16899-16903;
and the amino acid sequence of LGALS3BP (identified by accession no.
NP_005558) is
disclosed in, e.g., Rosenberg, I. et al., 1991, "Mac-2-binding glycoproteins.
Putative ligands
for a cytosolic beta-galactoside lectin," published in J. Biol. Chem. 266
(28), 18731-18736;
Koths, K. et al., 1993, "Cloning and characterization of a human Mac-2-binding
protein, a
new member of the superfamily defined by the macrophage scavenger receptor
cysteine-
rich domain," published in J. Biol. Chem. 268 (19), 14245-14249; Ullrich, A.
et al., 1994,
"The secreted tumor-associated antigen 90K is a potent immune stimulator,"
published in J.
Biol. Chem. 269 (28), 18401-18407, each of which is incorporated by reference
herein in its
entirety.
The nucleotide sequence of LPA (identified by accession no. NM 005577) is
disclosed in, e.g., McLean, J.W. et al., 1987, "cDNA sequence of human
apolipoprotein(a)
is homologous to plasminogen," published in Nature 330 (6144), 132-137; Frank,
S.L. et
al., 1998, "The apolipoprotein(a) gene resides on human chromosome 6q26-27, in
close
proximity to the homologous gene for plasminogen," published in Hum. Genet. 79
(4),
352-356; Salonen, E.M. et al., 1989, "Lipoprotein(a) binds to fibronectin and
has serine
proteinase activity capable of cleaving it," published in EMBO J. 8 (13), 4035-
4040, and the
amino acid sequence of LPA (identified by accession no. NP005568) is disclosed
in, e.g.,
McLean, J.W. et al., 1987, "cDNA sequence of human apolipoprotein(a) is
homologous to
plasminogen," published in Nature 330 (6144), 132-137; Frank,S.L. et al.,
1998, "The
apolipoprotein(a) gene resides on human chromosome 6q26-27, in close proximity
to the
homologous gene for plasminogen," published in Hum. Genet. 79 (4), 352-356;
Salonen,
E.M. et al., 1989, "Lipoprotein(a) binds to fibronectin and has serine
proteinase activity
capable of cleaving it," published in EMBO J. 8(13), 4035-4040, each of which
is
incorporated by reference herein in its entirety.
The nucleotide sequence of MLL (identified by accession no. NM_005934) is
disclosed in, e.g., Tkachuk, D.C. et al., 1992, "Involvement of a homolog of
Drosophila
trithorax by 11 q23 chromosomal translocations in acute leukemias," published
in Cell 71
(4), 691-700; Yamamoto, K. et al., 1993, "Two distinct portions of LTG19/ENL
at 19p13
are involved in t(11;19) leukemia," published in Oncogene 8 (10), 2617-2625;
Rubnitz, J.E.
et al., 1994, "ENL, the gene fused with HRX in t(11;19) leukemias, encodes a
nuclear
-123-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
protein with transcriptional activation potential in lymphoid and myeloid
cells," published
in Blood 84 (6), 1747-1752, and the amino acid sequence of MLL (identified by
accession
no. NP005924) is disclosed in, e.g., Ziemin-van der Poel, S. et al., 1991,
"Identification of
a gene, MLL, that spans the breakpoint in 11 q23 translocations associated
with human
leukemias," published in Proc. Natl. Acad. Sci. U.S.A. 88 (23), 10735-10739;
Djabali, M. et
al., 1992, "A trithorax-like gene is interrupted by chromosome 11 q23
translocations in acute
leukaemias," published in Nat. Genet. 2 (2), 113-118; Tkachuk, D.C. et al.,
1992,
"Involvement of a homolog of Drosophila trithorax by 11 q23 chromosomal
translocations
in acute leukemias," published in Cell 71 (4), 691 700, each of which is
incorporated by
reference herein in its entirety.
The nucleotide sequence of MRC1 (identified by accession no. NM002438) is
disclosed in, e.g., Taylor, M.E. et al., 1990, "Primary structure of the
mannose receptor
contains multiple motifs resembling carbohydrate-recognition domains,"
published in J.
Biol. Chem. 265 (21), 12156-12162; Ezekowitz, R.A. et al., 1990, Molecular
characterization of the human macrophage mannose receptor: demonstration of
multiple
carbohydrate recognition-like domains and phagocytosis of yeasts in Cos-1
cells," published
in J. Exp. Med. 172 (6), 1785-1794; Taylor, M.E. et al., 1992, "Contribution
to ligand
binding by multiple carbohydrate-recognition domains in the macrophage mannose
reoeptor," published in J. Biol. Chem. 267 (3), 1719-1726, and the amino acid
sequence of
MRC1 (identified by accession no. NP_002429) is disclosed in, e.g., Taylor,
M.E. et al.,
1990, "Primary structure of the mannose receptor contains multiple motifs
resembling
carbohydrate-recognition domains," published in J. Biol. Chem. 265 (21), 12156-
12162;
Ezekowitz, R.A. et al., 1990, "Molecular characterization of the human
macrophage
mannose receptor: demonstration of multiple carbohydrate recognition-like
domains and
phagocytosis of yeasts in Cos-1 cells," published in J. Exp. Med. 172 (6),
1785-1794;
Taylor, M.E. et al., 1992, "Contribution to ligand binding by multiple
carbohydrate-
recognition domains in the macrophage mannose receptor," published in J. Biol.
Chem. 267
(3), 1719-1726, each of which is incorporated by reference herein in its
entirety.
The nucleotide sequence of MYL2 (identified by accession no. NM_000432) is
disclosed in, e.g., Dalla Libera, L. et al., 1989, "Isolation and nucleotide
sequence of the
cDNA encoding human ventricular myosin light chain 2," published in Nucleic
Acids Res.
17 (6), 2360; Macera, M.J. et al., "Localization of the gene coding for
ventricular myosin
regulatory light chain (MYL2) to human chromosome 12q23-q24.3," published in
Genomics 13 (3), 829-831; Wadgaonkar, R. et al., 1993, "Interaction of a
conserved peptide
-124-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
domain in recombinant human ventricular myosin light chain-2 with myosin heavy
chain,"
published in Cell. Mol. Biol. Res. 39 (1), 13-26, and the amino acid sequence
of MYL2
(identified by accession no. AAH31006) is disclosed in, e.g., Strausberg,
R.L., "Generation
and initial analysis of more than 15,000 full-length human and mouse cDNA
sequences,"
Proc. Natl. Acad. Sci. U.S.A. 99 (26), 16899-16903 (2002), each of which is
incorporated
by reference herein in its entirety.
The nucleotide sequence of MYO6 (identified by accession no. NM004999) is
disclosed in, e.g., Bement, W.M. et al., 1994, "Identification and overlapping
expression of
multiple unconventional myosin genes in vertebrate cell types," published in
Proc. Natl.
Acad. Sci. U.S.A. 91 (14), 6549-6553; Avraham, K.B. et al., 1995, "The mouse
Snell's
waltzer deafness gene encodes an unconventional myosin required for structural
integrity of
inner ear hair cells," published in Nat. Genet. 11 (4), 3 69-375; Avraham,
K.B. et al., 1997,
"Characterization of unconventional MY06, the human homologue of the gene
responsible
for deafness in Snell's waltzer mice," published in Hum. Mol. Genet. 6 (8),
1225-1231, and
the amino acid sequence of KCTD7 (identified by accession no. NP_004990) is
disclosed
in, e.g., Bement, W.M. et al., 1994, "Identification and overlapping
expression of multiple
unconventional myosin genes in vertebrate cell types," published in Proc.
Natl. Acad. Sci.
U.S.A. 91 (14), 6549-6553; Avraham, K.B. et al., 1995, "The mouse Snell's
waltzer
deafness gene encodes an unconventional myosin required for structural
integrity of inner
ear hair cells," published in Nat. Genet. 11 (4), 369-375; Avraham, K.B. et
al., 1997,
"Characterization of unconventional MYO6, the human homologue of the gene
responsible
for deafness in Snell's waltzer mice," published in Hum. Mol. Genet. 6 (8),
1225-1231, each
of which is incorporated by reference herein in its entirety.
The nucleotide sequence of ORM1 (identified by accession no. NM 000607) is
disclosed in, e.g., Schmid, K. et al., 1974, "The disulfide bonds of aiphal-
acid
glycoprotein," published in Biochemistry 13 (13), 2694-2697; Mbuyi, J.M. et
al., 1982,
"Plasma proteins in human cortical bone: enrichment of alpha 2 HS-
glycoprotein, alpha 1
acid-glycoprotein, and IgE," published in Calcif. Tissue Int. 34 (3), 229-231;
Dente,L. et al.,
1985, "Structure of the human alpha 1-acid glycoprotein gene: sequence
homology with
other human acute phase protein genes," published in Nucleic Acids Res. 13
(11),
3941-3952, and the amino acid sequence of ORMI (identified by accession no.
CAI16859)
is disclosed in, e.g., Schmid et al., 1973, "Structure of alpha 1-acid
glycoprotein"
Biochemistry 12:2711-2724, each of which is incorporated by reference herein
in its
entirety.
-125-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
The nucleotide sequence of SERPINF 1(identified by accession nos. NM 002615,
BC013984) is disclosed in, e.g., Steele, F.R. et al., 1993, "Pigment
epithelium-derived
factor: neurotrophic activity and identification as a member of the serine
protease inhibitor
gene family," published in Proc. Natl. Acad. Sci. U.S.A. 90 (4), 1526-1530;
Pignolo, R.J. et
al., 1993, "Senescent WI-38 cells fail to express EPC-1, a gene induced in
young cells upon
entry into the GO state," published in J. Biol. Chem. 268 (12), 8949-8957;
Becerra, S.P. et
al., 1993, "Overexpression of fetal human pigment epithelium-derived factor in
Escherichia
coli. A functionally active neurotrophic factor," published in J. Biol. Chem.
268 (31),
23148-23156, and the amino acid sequence of SERPINFI (identified by accession
no.
AAH13984) is disclosed in, e.g., Petersen et al., 2003, "Pigment-epithelium-
derived factor
occurs at a physiologically relevant concentration in human blood:
purification and
characterization," Biochem J. 374: 199-206, each of which is incorporated by
reference
herein in its entirety.
The nucleotide sequence of SERPINAl (identified by accession nos. BC015642,
NM 000295) is disclosed in, e.g., NIH MGC Project, 2001, Direct Submission,
National
Institutes of Health, Mammalian Gene Collection (MGC), Bethesda, MD 20892-
2590,
USA; ; Strausberg,R.L. et al., 2002, "Generation and initial analysis of more
than 15,000
full-length human and mouse cDNA sequences," published in Proc. Natl. Acad.
Sci. U.S.A.
99 (26), 16899-16903; Kurachi, K. et al., 1981, "Cloning and sequence of cDNA
coding for
alpha 1-antitrypsin," published in Proc. Natl. Acad. Sci. U.S.A. 78 (11), 6826-
6830;
Lobermann, H. et al., 1982, "Interaction of human alpha 1-proteinase inhibitor
with
chymotrypsinogen A and crystallization of a proteolytically modified alpha 1-
proteinase
inhibitor," published in Hoppe-Seyler's Z. Physiol. Chem. 363 (11), 1377-1388;
Bollen, A.
et al., 1983, "Cloning and expression in Escherichia coli of full-length
complementary DNA
coding for human alpha 1-antitrypsin," published in DNA 2 (4), 255-264, and
the amino
acid sequence of SERPINA1 (identified by accession nos. NP 001002235,
NP_000286) is
disclosed in, e.g., Kurachi, K. et al., 1981, "Cloning and sequence of eDNA
coding for
alpha 1-antitrypsin," published in Proc. Natl. Acad. Sci. U.S.A. 78 (11), 6826-
6830;
Lobermann,H. et al., 1982, "Interaction of human alpha 1-proteinase inhibitor
with
chymotrypsinogen A and crystallization of a proteolytically modified alpha I -
proteinase
inhibitor," published in Hoppe-Seyler's Z. Physiol. Chem. 363 (11), 1377-1388;
Bollen, A.
et al., 1983, "Cloning and expression in Escherichia coli of full-length
complementary DNA
coding for human alpha 1-antitrypsin," published in DNA 2 (4), 255-264, each
of which is
incorporated by reference herein in its entirety.
- 126 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
The nucleotide sequence of SERPINA4 (identified by accession no. NM 006215) is
disclosed in, e.g., Wang, M.Y. et aL, 1989, "Human kallistatin, a new tissue
kallikrein-
binding protein: purification and characterization," published in Adv. Exp.
Med. Biol.
247B, 1-8; Zhou, G.X. et al., 1992, "Kallistatin: a novel human tissue
kallikrein inhibitor.
Purification, characterization, and reactive center sequence," published in J.
Biol. Chem.
267 (36), 25873-25880; Chai, K.X. et al., 1993, "Kallistatin: a novel human
serine
proteinase inhibitor. Molecular cloning, tissue distribution, and expression
in Escherichia
coli," published in J. Biol. Chem. 268 (32), 24498-24505, and the amino acid
sequence of
SERPINA4 (identified by accession no. NP006206) is disclosed in, e.g., Wang,
M.Y. et
al., 1989, "Human kallistatin, a new tissue kallikrein-binding protein:
purification and
characterization," published in Adv. Exp. Med. Biol. 247B, 1-8; Zhou,G.X. et
al., 1992,
"Kallistatin: a novel human tissue kallikrein inhibitor. Purification,
characterization, and
reactive center sequence," published in J. Biol. Chem. 267 (36), 25873-25880;
Chai, K.X. et
al., 1993, "Kallistatin: a novel human serine proteinase inhibitor. Molecular
cloning, tissue
distribution, and expression in Escherichia coli," published in J. Biol. Chem.
268 (32),
24498-24505, each of which is incorporated by reference herein in its
entirety.
The nucleotide sequence of SERPINF2 (identified by accession no. BC031592) is
disclosed in, e.g., NIH MGC Project, 2002, Direct Submission, National
Institutes of
Health, Mammalian Gene Collection (MGC), Bethesda, MD 20892-2590, USA;
Strausberg,
R.L. et al., 2002, "Generation and initial analysis of more than 15,000 full-
length human
and mouse eDNA sequences," published in Proc. Natl. Acad. Sci. U.S.A. 99 (26),
16899-
16903, and the amino acid sequence of SERPINF2 (identified by accession no.
NP_000925)
is disclosed in, e.g., Wiman, B. et al., 1979, "On the mechanism of the
reaction between
human alpha 2-antiplasmin and plasmin," published in J. Biol. Chem. 254 (18),
9291-9297;
Yoshioka, A. et al., 1982, "Congenital deficiency of alpha 2-plasmin inhibitor
in three
sisters," published in Haemostasis 11 (3), 176-184; Brower, M.S. et al_, 1982,
"Proteolytic
cleavage and inactivation of alpha 2-plasmin inhibitor and Cl inactivator by
human
polymorphonuclear leukocyte elastase," published in J. Biol. Chem. 257 (16),
9849-9854,
each of which is incorporated by reference herein in its entirety.
The nucleotide sequence of PROS1 (identified by accession no. NM 000313) is
disclosed in, e.g., Dahlback, B. et al., 1981, "High molecular weight complex
in human
plasma between vitamin K-dependent protein S and complement component C4b-
binding
protein," published in Proc. Natl. Acad. Sci. U.S.A. 78 (4), 2512-2516; Comp,
P.C. et al.,
1984, "Recurrent venous thromboembolism in patients with a partial deficiency
of protein
- 127 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
S," published in N. Engi. J. Med. 311 (24), 1525-1528; Lundwall,A. et al.,
1986, "Isolation
and sequence of the cDNA for human protein S, a regulator of blood
coagulation,"
published in Proc. Natl. Acad. Sci. U.S.A. 83 (18), 6716-6720, and the amino
acid sequence
of PROS1 (identified by accession no. NP 000304) is disclosed in, e.g.,
Dahlback, B. et al.,
1981, "High molecular weight complex in human plasma between vitamin K-
dependent
protein S and complement component C4b-binding protein," published in Proc.
Natl. Acad.
Sci. U.S.A. 78 (4), 2512-2516; Comp, P.C. et al., 1984, "Recurrent venous
thromboembolism in patients with a partial deficiency of protein S," published
in N. Engl. J.
Med. 311 (24), 1525-1528; Lundwall, A. et al., 1986, "Isolation and sequence
of the cDNA
for human protein S, a regulator of blood coagulation," published in Proc.
Natl. Acad. Sci.
U.S.A. 83 (18), 6716-6720, each of which is incorporated by reference herein
in its entirety.
The nucleotide sequence of QSCN6 (identified by accession no. NM002826) is
disclosed in, e.g. Coppock, D.L. et al., 1993, "Preferential gene expression
in quiescent
human lung fibroblasts" published in Cell Growth Differ. 4(6), 483-493 (1993);
Hoober,
K.L., et al., 1999, "Homology between egg white sulfl-iydryl oxidase and
quiescin Q6
defines a new class of flavin-linked sulfhydryl oxidases" published in "J.
Biol. Chem. 274
(45), 31759-31762 (1999); Coppock, D. et al., 2000, "Regulation of the
quiescence-induced
genes: quiescin Q6, decorin, and ribosomal protein S29 published in Biochem.
Biophys.
Res. Commun. 269 (2), 604-610 (2000) and the amino acid sequence of QSCN6
(identified
by accession no. AAQ89300) is disclosed in, e.g., Clark, H.F. et al., 2003,
"The Secreted
Protein Discovery Initiative (SPDI), a Large-ScaleEffort to Identify Novel
Human Secreted
and Transmembrane Proteins: A Bioinformatics Assessment" published in Genome
Res. 13
(10), 2265-2270 (2003), each of which is incorporated by reference herein in
its entirety.
The nucleotide sequence of RGS4 (identified by accession no. NM_005613) is
disclosed in, e.g., Druey, K.M. et al., 1996, "Inhibition of G-protein-
mediated MAP kinase
activation by a new mammalian gene family," published in Nature 379 (6567),
742-746;
Berman, D.M. et al., 1996, "GAIP and RGS4 are GTPase-activating proteins for
the Gi
subfamily of G protein alpha subunits," published in Ce1186 (3), 445-452;
Heximer, S.P. et
al., 1997, "RGS2/GOS8 is a selective inhibitor of Gqalpha function," published
in Proc.
Nati. Acad. Sci. U.S.A. 94 (26), 14389-14393, and the amino acid sequence of
RGS4
(identified by accession no. NP 005604) is disclosed in, e.g. Druey, K.M. et
al., 1996,
"Inhibition of G-protein-mediated MAP kinase activation by a new mammalian
gene
family," published in Nature 379 (6567), 742-746; Berman, D.M. et al., 1996,
"GAIP and
RGS4 are GTPase-activating proteins for the Gi subfamily of G protein alpha
subunits,"
- 128 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
published in Ce1186 (3), 445-452; Heximer, S.P. et aL, 1997, "RGS2/GOS8 is a
selective
inhibitor of Gqatpha function," published in Proc. Nati. Acad. Sci. U.S.A. 94
(26), 143 89-
14393, each of which is incorporated by reference herein in its entirety.
The nucleotide sequence of SAA1 (identified by accession no. BC105796) is
disclosed in, e.g., NIH MGC Project, 2005, Direct Submission, National
Institutes of
Health, Mammalian Gene Collection (MGC), Bethesda, Maryland; Strausberg et
al., 2002,
"Generation and initial analysis of more than 15,000 full-length human and
mouse cDNA
sequences," published in Proc. Natl. Acad. Sci. U.S.A. 99, 16899-16903, and
the amino acid
sequence of SAA1 (identified by accession nos. AAA64799, AAA3 0968) is
disclosed in,
e.g., Kluve-Beckerman, B. et al., 1998, "Human serum amyloid A. Three hepatic
mRNAs
and the corresponding proteins in one person," published in J. Clin. Invest.
82 (5),
1670-1675; Marhaug, G. et al., 1990, "Mink serum amyloid A protein. Expression
and
primary structure based on cDNA sequences," published in J. Biol. Chem. 265,
10049-
10054, each of which is hereby incorporated by reference herein in its
entirety.
The nucleotide sequence of SAA4 (identified by accession no. NM 006512) is
disclosed in, e.g., Bausserman, L.L. et al.,1983, "Interaction of the serum
amyloid A
proteins with phospholipid," published in J. Biol. Chem. 258 (17), 10681-
10688;
Whitehead, A.S. et al., 1992, "Identification of novel members of the serum
amyloid A
protein superfamily as constitutive apolipoproteins of high density
lipoprotein," published
in J. Biol. Chem. 267 (6), 3862-3867; Watson, G. et al., 1992, "Analysis of
the genomic and
derived protein structure of a novel human serum amyloid A gene, SAA4,"
published in
Scand. J. Immunol. 36 (5), 703-712, and the amino acid sequence of SAA4
(identified by
accession no. NP 006503) is disclosed in, e.g., Bausserman, L.L. et al.,1983,
"Interaction of
the serum amyloid A proteins with phospholipid," published in J. Biol. Chem.
258 (17),
10681-10688; Whitehead, A.S. et al., 1992, "Identification of novel members of
the serum
amyloid A protein superfamily as constitutive apolipoproteins of high density
lipoprotein,"
published in J. Biol. Chem. 267 (6), 3862-3867; Watson, G. et al., 1992,
"Analysis of the
genomic and derived protein structure of a novel human serum amyloid A gene,
SAA4,"
published in Scand. J. Immunol. 36 (5), 703-712; and Kang et al., 1987, "The
precursor of
Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor,
Nature 325,
733-736, each of which is incorporated by reference herein in its entirety.
The nucleotide sequence of serum amyloid A-4 protein precursor (identified by
accession no. M81349) is disclosed in, e.g., Whitehead et al., 1992,
"Identification of novel
members of the serum amyloid A protein superfamily as constitutive
apolipoproteins of
-129-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
high density lipoprotein and the amino acid sequence of SAA4," and the amino
acid
sequence of serum amyloid A-4 protein precursor (identified by accession no.
P02375) is
disclosed in Sipe, 1985, "Human serum amyloid A (SAA): biosynthesis and
postsynthetic
processing of preSAA and structural variants defined by complementary DNA,"
Biochemistry 24, 2931-2936, each of which is incorporated by reference herein
in its
entirety.
The nucleotide sequence of SERPINA7 (identified by accession no. NM000354) is
disclosed in, e.g., Flink, I.L. et al., 1986, "Complete amino acid sequence of
human
thyroxine-binding globulin deduced from cloned DNA: close homology to the
serine
antiproteases," published in Proc. Natl. Acad. Sci. U.S.A. 83 (20), 7708-7712;
Takeda, K. et
al., 1989, "Sequence of the variant thyroxine-binding globulin of Australian
aborigines.
Only one of two amino acid replacements is responsible for its altered
properties,"
published in J. Clin. Invest. 83 (4), 1344-1348; Mori, Y. et aL, 1990,
"Replacement of
Leu227 by Pro in thyroxine-binding globulin (TBG) is associated with complete
TBG
deficiency in three of eight families with this inherited defect," published
in J. Clin.
Endocrinol. Metab. 70 (3), 804-809, and the amino acid sequence of SERPINA7
(identified
by accession no. CAB06092) is disclosed in, e.g., Cheng, "Partial amino acid
sequence of
human thyroxine binding globulin," Biochem. Biophys. Res. Commun. 79: 1212-
1218, each
of which is incorporated by reference herein in its entirety.
The nucleotide sequence of TF (identified by accession no. NM_001063) is
disclosed in, e.g., Enns, C.A. et al., 1981, "Physical characterization of the
transferrin
receptor in human placentae," published in J. Biol. Chem. 256 (19), 9820-9823;
Sass-Kuhn,
S.P. et al., 1984, "Human granulocyte/pollen-binding protein. Recognition and
identification as transferrin," published in J. Clin. Invest. 73 (1), 202-210;
Uzan, G. et al.,
1984, "Molecular cloning and sequence analysis of cDNA for human transferrin,"
published
in Biochem. Biophys. Res. Commun. 119 (1), 273-281, and the amino acid
sequence of TF
(identified by accession no. NP_001054) is disclosed in, e.g., Enns, C.A. et
al., 1981,
"Physical characterization of the transferrin receptor in human placentae,"
published in J.
Biol. Chem. 256 (19), 9820-9823; Sass-Kuhn, S.P. et al., 1984, "Human
granulocyte/pollen-binding protein. Recognition and identification as
transferrin," published
in J. Clin. Invest. 73 (1), 202-210; Uzan, G. et al., 1984, "Molecular cloning
and sequence
analysis of eDNA for human transferrin," published in Biochem. Biophys. Res.
Commun.
119 (1), 273-28 1, each of which is incorporated by reference herein in its
entirety.
- 130 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
The nucleotide sequence of TFRC (identified by accession no. NM 003234) is
disclosed in, e.g., Enns, C.A. et al., 1981, "Physical characterization of the
transferrin
receptor in human placentae," published in J. Biol. Chem. 256 (19), 9820-9823;
Omary,
M.B. et aL, 1981, "Biosynthesis of the human transferrin receptor in cultured
cells,"
published in J. Biol. Chem. 256 (24), 12888-12892; Miller, Y.E. et al., 1983,
"Chromosome
3q (22-ter) encodes the human transferrin receptor," published in Am. J. Hum.
Genet. 35
(4), 573-583, and the amino acid sequence of TFRC (identified by accession no.
NP_003225) is disclosed in, e.g., Enns, C.A. et al., 1981, "Physical
characterization of the
transferrin receptor in human placentae," published in J. Biol. Chem. 256
(19), 9820-9823;
Omary, M.B. et al., 1981, "Biosynthesis of the human transferrin receptor in
cultured cells,"
published in J. Biol. Chem. 256 (24), 12888-12892; Miller,Y.E. et aL, 1983,
"Chromosome
3q (22-ter) encodes the human transferrin receptor," published in Am. J. Hum.
Genet. 35
(4), 573-583, each of which is incorporated by reference herein in its
entirety.
The nucleotide sequence of TTN (identified by accession no. BC013396) is
disclosed in, e.g., Strausberg et al., 2002, "Generation and initial analysis
of more than
15,000 full-length human and mouse cUNA sequences," published in Proc. Natl.
Acad. Sci.
U.S.A. 99 (26), 16899-16903, and the amino acid sequence of TTN (identified by
accession
no. CADI2456) is disclosed in, e.g., Bang et al., 2001, "The complete gene
sequence of
titin, expression of an unusual approximately 700-kDa titin isoform, and its
interaction with
obscurin identify a novel Z-line to I-band linking system," published in Circ.
Res. 89 (11),
1065-1072, each of which is incorporated by reference herein in its entirety.
The nucleotide sequence of TTR (identified by accession no. NM_000371) is
disclosed in, e.g., Fex et al., 1979, "Interaction between prealbumin and
retinol-binding
protein studied by affinity chromatography, gel filtration and two-phase
partition,"
published in Eur. J. Biochem. 99 (2), 353-360; Mita et al., 1984, "Cloning and
sequence
analysis of cDNA for human prealbumin," published in Biochem. Biophys. Res.
Commun.
124 (2), 558-564, and the amino acid sequence of TTR (identified by accession
nos.
AAH05310, AAP35853) is disclosed in, e.g., Kanda et al., "The amino acid
sequence of
human plasma prealbumin," J. Biol. Chem. 249: 6796-6805, each of which is
incorporated
by reference herein in its entirety.
The nucleotide sequence of transthyretin precursor (prealbumin) (TBPA) (TTR)
(ATTR) is disclosed in Gu et al., 1991, "Transthyretin (prealbumin) gene in
human primary
hepatic cancer," Chem. Life Scie Earth Sci. 34, 1312-1318 and the amino acid
sequence is
disclosed in Mita et al., 1984, "Cloning and sequence analysis of cDNA for
human
-131-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
prealbumin," Biophys. Res. Commun. 124, 558-564 each of which is hereby
incorporated
by reference in its entirety.
The nucleotide sequence of UBC (identified by accession no. NM_021009) is
disclosed in, e.g., Wiborg, O. et al., 1985, "The human ubiquitin multigene
family: some
genes contain multiple directly repeated ubiquitin coding sequences,"
published in EMBO J.
4(3), 755-759; Einspanier, R. et al., 1987, "Cloning and sequence analysis of
a cDNA
encoding poly-ubiquitin in human ovarian granulosa cells," published in
Biochem. Biophys.
Res. Commun. 147 (2), 581-587; Baker, R.T. et al., 1989, "Unequal crossover
generates
variation in ubiquitin coding unit number at the human UbC polyubiquitin
locus," published
in Am. J. Hum. Genet. 44 (4), 534-542, and the amino acid sequence of UBC
(identified by
accession no. NP_066289) is disclosed in, e.g., Wiborg, O. et al., 1985, "The
human
ubiquitin multigene family: some genes contain multiple directly repeated
ubiquitin coding
sequences," published in EMBO J. 4 (3), 755-759; Einspanier, R. et al., 1987,
"Cloning and
sequence analysis of a cDNA encoding poly-ubiquitin in human ovarian granulosa
cells,"
published in Biochem. Biophys. Res. Commun. 147 (2), 581-587; Baker, R.T. et
al., 1989,
"Unequal crossover generates variation in ubiquitin coding unit number at the
human UbC
polyubiquitin locus," published in Am. J. Hum. Genet. 44 (4), 534-542, each of
which is
incorporated by reference herein in its entirety.
The nucleotide sequence of VTN (identified by accession no. NM 000638) is
disclosed in, e.g., Suzuki, S. et al., 1984, "Domain structure of vitronectin.
Alignment of
active sites," published in J. Biol: Chem. 259 (24), 15307-15314; Suzuki, S.
et al., 1985,
"Complete amino acid sequence of human vitronectin deduced from cDNA.
Similarity of
cell attachment sites in vitronectin and fibronectin," published in EMBO J. 4
(10), 2519-
2524; Jenne, D. et al., 1985, "Molecular cloning of S-protein, a link between
complement,
coagulation and cell-substrate adhesion," published in EMBO J. 4 (12), 3153-
3157, and the
amino acid sequence of VTN (identified by accession no. P04004) is disclosed
in, e.g.,
Zhou, A. et al., 2003, "How vitronectin binds PAI-1 to modulate fibrinolysis
and cell
migration," published in Nat. Struct. Biol. 10 (7), 541-544; Kamikubo,Y. et
al., 2002,
"Identification of the disulfide bonds in the recombinant somatomedin B domain
of human
vitronectin," published in J. Biol. Chem. 277 (30), 27109-27119; Seger, D. et
al., 1998,
"Phosphorylation of vitronectin by casein kinase II. Identification of the
sites and their
promotion of cell adhesion and spreading," published in J. Biol. Chem. 273
(38), 24805-
24813, each of which is incorporated by reference herein in its entirety
- 132 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
The nucleotide sequence of VWF (identified by accession no. NM 000552) is
disclosed in, e.g., Coller, B.S. et aL, 1983, "Studies with a murine
monoclonal antibody that
abolishes ristocetin-induced binding of von Willebrand factor to platelets:
additional
evidence in support of GPIb as a platelet receptor for von Willebrand factor,"
published in
Blood 61 (1), 99-110; Lynch, D.C. et al., 1985, "Molecular cloning of cDNA for
human
von Willebrand factor: authentication by a new method," published in Cell 41
(1), 49-56;
Ginsburg,D. et al., 1985, "Human von Willebrand factor (vWF): isolation of
complementary DNA(cDNA) clones and chromosomal localization," published in
Science
228 (4706), 1401-1406, and the amino acid sequence of VWF (identified by
accession no.
AAB59458) is disclosed in, e.g., Mancuso, D.J. et al., 1989, "Structure of the
gene for
human von Willebrand factor," published in J. Biol. Chem. 264 (33), 19514-
19527, each of
which is incorporated by reference herein in its entirety.
The nucleotide sequence of ALMS1 (identified by accession no. NM_015120) is
disclosed in, e.g., Collin, G.B. et al., 1999, "Alstrom syndrome: further
evidence for linkage
to human chromosome 2p13," published in Hum. Genet. 105 (5), 474-479; Collin,
G.B. et
al., 2002, "Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory
degeneration in Alstrom syndrome," published in Nat. Genet. 31 (1), 74-78;
Hearn, T. et al.,
Mutation of ALMS1, a large gene with a tandem repeat encoding 47 amino acids,
causes
Aistrom syndrome," published in Nat. Genet. 31 (1), 79-83, and the amino acid
sequence of
ALMS1 (identified by accession no. NP 055935) is disclosed in, e.g., Collin,
G.B. et al.,
1999, "Alstrom syndrome: further evidence for linkage to human chromosome
2pl3,"
published in Hum. Genet. 105 (5), 474-479; Collin, G.B. et al., 2002,
"Mutations in
ALMS 1 cause obesity, type 2 diabetes and neurosensory degeneration in Alstrom
syndrome," published in Nat. Genet. 31 (1), 74-78; Hearn,T. et al., Mutation
of ALMS 1, a
large gene with a tandem repeat encoding 47 amino acids, causes Alstrom
syndrome,"
published in Nat. Genet. 31 (1), 79-83, each of which is incorporated by
reference herein in
its entirety.
The nucleotide sequence of ATRN (identified by accession nos. BC101705,
NM139321) is disclosed in, e.g., Strausberg, R.L. et al., 2002, "Generation
and initial
analysis of more than 15,000 full-length human and mouse cDNA sequences,"
published in
Proc. Natl. Acad. Sci. U.S.A. 99 (26), 16899-16903; Mori,M. et al., 1992,
"Topical timolol
and blood-aqueous barrier permeability to protein in human eyes," published in
Nippon
Ganka Gakkai Zasshi 96 (11), 1418-1422; Duke-Cohan, J.S. et al., 1996, "Serum
high
molecular weight dipeptidyl peptidase IV (CD26) is similar to a novel antigen
DPPT-L
-133-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
released from activated T cells," published in J. Immunol. 156 (5), 1714-1721;
Duke-
Cohan, J.S. et al., 1998, "Attractin (DPPT-L), a member of the CUB family of
cell adhesion
and guidance proteins, is secreted by activated human T lymphocytes and
modulates
immune cell interactions," published in Proc. Natl. Acad. Sci. U.S.A. 95 (19),
11336-11341,
and the amino acid sequence of ATRN (identified by accession no. CAI22615) is
disclosed
in, e.g., Sehra, H., 2005, Direct Submission, Wellcome Trust Sanger Institute,
Hinxton,
Cambridgeshire, CB 10 1 SA, UK, each of which is incorporated by reference
herein in its
entirety.
The nucleotide sequence of APOL1 (identified by accession nos. BC017331,
NM_003661) is disclosed in, e.g., Strausberg et al., 2002, "Generation and
initial analysis
of more than 15,000 full-length human and mouse cDNA sequences," published in
Proc.
Natl. Acad. Sci. U.S.A. 99 (26), 16899-16903; Duchateau, P.N. et al., 1997,
"Apolipoprotein L, a new human high density lipoprotein apolipoprotein
expressed by the
pancreas. Identification, cloning, characterization, and plasma distribution
of apolipoprotein
L," published in Biol. Chem. 272 (41), 25576-25582; Duchateau, P.N. et al.,
2000, "Plasma
apolipoprotein L concentrations correlate with plasma triglycerides and
cholesterol levels in
normolipidemic, hyperlipidemic, and diabetic subjects," published in J. Lipid
Res. 41 (8),
1231-1236; Duchateau, P.N. et al., 2001, "Apolipoprotein L gene family: tissue
specific
expression, splicing, promoter regions; discovery of a new gene," published in
J. Lipid Res.
42 (4), 620-630, and the amino acid sequence of APOLI (identified by accession
no.
AAK20210) is disclosed in, e.g., Page et al., 2001, "The human apolipoprotein
L gene
cluster: identification, classification, and sites of distribution," published
in Genomics 74
(1), 71-78, each of which is incorporated by reference herein in its entirety.
The nucleotide sequence of TRIP11 (identified by accession no. NM_004239) is
disclosed in, e.g., Lee, J.W. et al., 1995, "Two classes of proteins dependent
on either the
presence or absence of thyroid hormone for interaction with the thyroid
hormone receptor,"
published in Mol. Endocrinol. 9 (2), 243-254; Chang,K.H. et al., 1997, "A
thyroid hormone
receptor coactivator negatively regulated by the retinoblastoma protein,"
published in Proc.
Natl. Acad. Sci. U.S.A. 94 (17), 9040-9045; Abe,A. et aL, 1997, "Fusion of the
platelet-
derived growth factor receptor beta to a novel gene CEV14 in acute myelogenous
leukemia
after clonal evolution," published in Blood 90 (11), 4271 4277, and the amino
acid sequence
of TRIP11 (identified by accession no. NP_004230) is disclosed in, e.g., Lee,
J.W. et al.,
1995, "Two classes of proteins dependent on either the presence or absence of
thyroid
hormone for interaction with the thyroid hormone receptor," published in Mol.
Endocrinol.
-134-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
9 (2), 243-254; Chang, K.H. et al., 1997, "A thyroid hormone receptor
coactivator
negatively regulated by the retinoblastoma protein," published in Proc. Natl.
Acad. Sci.
U.S.A. 94 (17), 9040-9045; Abe, A. et al., 1997, "Fusion of the platelet-
derived growth
factor receptor beta to a novel gene CEV 14 in acute myelogenous leukemia
after clonal
evolution," published in Blood 90 (11), 4271-4277, each of which is
incorporated by
reference herein in its entirety.
The nucleotide sequence of PDCD11 (identified by accession no. NM_014976) is
disclosed in, e.g., Lacana, E. et al., 1999, "Regulation of Fas ligand
expression and cell
death by apoptosis-linked gene 4," published in Nat. Med. 5 (5), 542-547;
Sweet, T. et al.,
2003, "Identification of a novel protein from glial cells based on its ability
to interact with
NF-kappaB subunits," published in J. Cell. Biochem. 90 (5), 884-891, and the
amino acid
sequence of PDCD11 (identified by accession no. NP_055791) is disclosed in,
e.g., Lacana,
E. et al., 1999, "Regulation of Fas ligand expression and cell death by
apoptosis-linked gene
4," published in Nat. Med. 5 (5), 542-547; Sweet, T. et aL, 2003,
"Identification of a novel
protein from glial cells based on its ability to interact with NF-kappaB
subunits," published
in J. Cell. Biochem. 90 (5), 884-891, each of which is incorporated by
reference herein in its
entirety.
The nucleotide sequence of KIAA0433 (identified by accession no. AB007893) is
disclosed in, e.g., Ishikawa et al., 1997, "Prediction of the coding sequences
of unidentified
human genes. VIII. 78 new cDNA clones from brain which code for large proteins
in vitro,"
DNA Res. 4 (5), 307-313, and the amino acid sequence of KIAA0433 (identified
by
accession no. BAA24863) is disclosed in, e.g., Kisarazu et aL, 1997,
"Prediction of the
coding sequences of unidentified human genes. VIII. 78 new eDNA clones from
brain
which code for large proteins in vitro," published in DNA Res. 4 (5), 307-313,
each of
which is incorporated by reference herein in its entirety.
The nucleotide sequence of SERPINAIO (identified by accession no. NM 016186)
is disclosed in, e.g., Han, X. et aL, 1998, "Isolation of a protein Z-
dependent plasma
protease inhibitor," published in Proc. Natl. Acad. Sci. U.S.A. 95 (16), 9250-
9255; Han, X.
et al., 1999, "The protein Z-dependent protease inhibitor is a serpin,"
published in
Biochemistry 38 (34), 11073-11078; Yin, Z.F. et aL, 2000, "Prothrombotic
phenotype of
protein Z deficiency," published in Proc. Natl. Acad. Sci. U.S.A. 97 (12),
6734-6738, and
the amino acid sequence of SERPINAI0 (identified by accession no. NP057270) is
disclosed in, e.g., Han, X. et aL, 1998, "Isolation of a protein Z-dependent
plasma protease
inhibitor," published in Proc. Natl. Acad. Sci. U.S.A. 95 (16), 9250-9255;
Han, X. et aL,
- 135 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
1999, "The protein Z-dependent protease inhibitor is a serpin," published in
Biochemistry
38 (34), 11073-11078; Yin, Z.F. et aL, 2000, "Prothrombotic phenotype of
protein Z
deficiency," published in Proc. Nati. Acad. Sci. U.S.A. 97 (12), 6734-6738,
each of which is
incorporated by reference herein in its entirety.
The nucleotide sequence of BCOR (identified by accession no. BC063536) is
disclosed in, e.g., Strausberg et al., 2002, "Generation arid initial analysis
of more than
15,000 full-length human and mouse cDNA sequences," Proc. Natl. Acad. Sci.
U.S.A. 99
(26), 16899-16903, and the amino acid sequence of BCOR (identified by
accession no.
AAG41429) is disclosed in, e.g., Huynh et al., 2000, "BCOR, a novel
corepressor involved
in BCL-6 repression," Genes Dev. 14 (14), 1810-1823, each of which is
incorporated by
reference herein in its entirety.
The nucleotide sequence of C10orfl 8 (identified by accession no. BC001759) is
disclosed in, e.g., Strausberg et al., 2002, "Generation and initial analysis
of more than
15,000 full-length human and mouse cDNA sequences," Proc. Natl. Acad. Sci.
U.S.A. 99
(26), 16899-16903, and the amino acid sequence of C10orfl 8 (identified by
accession no.
CAI13368) is disclosed in, e.g., Wray, P., 2005, Direct Submission, Wellcome
Trust Sanger
Institute, Hinxton, Cambridgeshire, CB 10 1 SA, UK, each of which is
incorporated by
reference herein in its entirety.
The nucleotide sequence of YY1AP1 (identified by accession nos. BC044887,
BC014906) is disclosed in, e.g., Strausberg et al., 2002, "Generation and
initial analysis of
more than 15,000 full-length human and mouse cDNA sequences," published in
Proc. Natl.
Acad. Sci. U.S.A. 99 (26), 16899-16903 and the amino acid sequence of YYlAPI
(identified by accession nos. AAL75971, CAH71646) is disclosed in, e.g., Liang
et al.,
"Cloning and characterization of a novel YYI associated protein," unpublished,
Almeida,
J., 2005, Direct Submission, WelIcome Trust Sanger Institute, Hinxton,
Cambridgeshire,
CB 10 1 SA, UK each of which is incorporated by reference herein in its
entirety.
The nucleotide sequence of FLJ10006 (identified by accession nos. BC110537,
BC1 10536) is disclosed in, e.g., Strausberg et al., 2002, "Generation and
initial analysis of
more than 15,000 full-length human and mouse cDNA sequences," Proc. Natl.
Acad. Sci.
U.S.A. 99 (26), 16899-16903, and the amino acid sequence of FLJ10006
(identified by
accession no. AAH17012) is disclosed in, e.g., Director MGC Project, 2005,
Direct
Submission, National Institutes of Health, Mammalian Gene Collection (MGC),
Cancer
Genomics Office, National Cancer Institute, 31 Center Drive, Room 11A03,
Bethesda, MD
20892-2590,USA; Strausberg et al., 2002, "Generation and initial analysis of
more than
- 136 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
15,000 full-length human and mouse cDNA sequences," Proc. Natl. Acad. Sci.
U.S.A. 99
(26), 16899-16903, each of which is incorporated by reference herein in its
entirety.
The nucleotide sequence of BDP1 (identified by accession no. NM_018429) is
disclosed in, e.g., Schramm, L. et al., 2000, "Different human TFIIIB
activities direct RNA
polymerase III transcription from TATA-containing and TATA-less promoters,"
published
in Genes Dev. 14 (20), 2650-2663; Kelter, A.R. et al., 2000, "The
transcription factor-like
nuclear regulator (TFNR) contains a novel 55-amino-acid motif repeated nine
times and
maps closely to SMN1," published in Genomics 70 (3), 315-326; Weser, S. et
al.,
Transcription factor (TF)-like nuclear regulator, the 250-kDa form of Homo
sapiens
TFIIIB', is an essential component of human TFIIICI activity," published in J.
Biol. Chem.
279 (26), 27022-27029, and the amino acid sequence of BDP1 (identified by
accession no.
AAH32146) is disclosed in, e.g., Strausberg, R.L et al., "Generation and
initial analysis of
more than 15,000 full-length human and mouse cDNA sequences" published in
Proc. Natl.
Acad. Sci. U.S.A. 99 (26), 16899-16903 (2002), each of which is incorporated
by reference
herein in its entirety.
The nucleotide sequence of SMARCADI (identified by accession no. NM 020159)
is disclosed in, e.g., Soininen, R. et al., 1992, "The mouse Enhancer trap
locus 1(Etl-1): a
novel mammalian gene related to Drosophila and yeast transcriptional regulator
genes,"
published in Mech. Dev. 39 (1-2), 111-123; Adra, C.N. et al., 2000, "SMARCADI,
a novel
human helicase family-defining member associated with genetic instability:
cloning,
expression, and mapping to 4q22-q23, a band rich in breakpoints and deletion
mutants
involved in several human diseases," published in Genomics 69 (2), 162-173,
and the amino
acid sequence of SMARCADI (identified by accession no. NP064544) is disclosed
in,
e.g., Soininen, R. et al., 1992, "The mouse Enhancer trap locus 1(Etl-1): a
novel
mammalian gene related to Drosophila and yeast transcriptional regulator
genes," published
in Mech. Dev. 39 (1-2), 111-123; Adra, C.N. et al., 2000, "SMARCAD1, a novel
human
helicase family-defining member associated with genetic instability: cloning,
expression,
and mapping to 4q22-q23, a band rich in breakpoints and deletion mutants
involved in
several human diseases," published in Genomics 69 (2), 162-173, each of which
is
incorporated by reference herein in its entirety.
The nucleotide sequence of MKL2 (identified by accession no. NM_014048) is
disclosed in, e.g., Cen, B. et al., 2003, "Megakaryoblastic leukemia 1, a
potent
transcriptional coactivator for serum response factor (SRF), is required for
serum induction
of SRF target genes," published in Mol. Cell. Biol. 23 (18), 6597-6608;
Selvaraj, A. et al.,
-137-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
2003, "Megakaryoblastic leukemia-1/2, a transcriptional co-activator of serum
response
factor, is required for skeletal myogenic differentiation," published in J.
Biol. Chem. 278
(43), 41977-41987, and the amino acid sequence of MKL2 (identified by
accession no.
AAH47761) is disclosed in, e.g., Strausberg et al., 2002, "Generation and
initial analysis of
more than 15,000 full-length human and mouse cDNA sequences," Proc. Natl.
Acad. Sci.
U.S.A. 99 (26), 16899-16903, each of which is incorporated by reference herein
in its
entirety.
The nucleotide sequence of CHST8 (identified by accession nos. NM 022467,
BC018723) is disclosed in, e.g., Xia et al., 2000, "Molecular cloning and
expression of the
pituitary glycoprotein hormone N-acetylgalactosamine-4-O-sulfotransferase,"
published in
J. Biol. Chem. 275 (49), 38402-38409; Okuda, T. et al., 2000, "Molecular
cloning and
characterization of GaINAc 4-sulfotransferase expressed in human pituitary
gland,"
published in J. Biol. Chem. 275 (51), 40605-40613; Hiraoka, N et al., 2001,
"Molecular
cloning and expression of two distinct human N-acetylgalactosamine 4-0-
sulfotransferases
that transfer sulfate to Ga1NAc beta 1-->4G1cNAc beta 1-->R in both N- and 0-
glycans,"
Glycobiology 11 (6), 495-504; Strausberg et al., 2002, "Generation and initial
analysis of
more than 15,000 full-length human and mouse cDNA sequences," Proc. Natl.
Acad. Sci.
U.S.A. 99 (26), 16899-16903, and the amino acid sequence of CHST8 (identified
by
accession no. NP_071912) is disclosed in, e.g., Xia et al., 2000, "Molecular
cloning and
expression of the pituitary glycoprotein hormone N acetylgalactosamine-4-O-
sulfotransferase, " J. Biol. Chem. 275 (49), 38402-38409; Okuda et al., 2000,
"Molecular
cloning and characterization of Ga1NAc 4-sulfotransferase expressed in human
pituitary
gland," published in J. Biol. Chem. 275 (51), 40605-40613; Hiraoka, N et al.,
2001,
"Molecular cloning and expression of two distinct human N-acetylgalactosamine
4-0-
sulfotransferases that transfer sulfate to GaINAc beta 1-->4G1cNAc beta 1-->R
in both N-
and 0-glycans," published in Glycobiology 11 (6), 495-504, each of which is
incorporated
by reference herein in its entirety.
The nucleotide sequence of MCPH1 (identified by accession nos. NM_024596,
BC030702) is disclosed in, e.g., Jackson, A.P. et al., 1998, "Primary
autosomal recessive
microcephaly (MCPH1) maps to chroinosome 8p22-pter," published in Am. J. Hum.
Genet.
63 (2), 541-546; Jackson, A.P. et al., 2002, "Identification of microcephalin,
a protein
implicated in determining the size of the human brain," published in Am. J.
Hum. Genet. 71
(1), 136-142; Kumar, A. et al., 2002, "Primary microcephaly: microcephalin and
ASPM
determine the size of the human brain," published in J. Biosci. 27 (7), 629
632, and the
-138-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
amino acid sequence of MCPHI (identified by accession no. AAH30702) is
disclosed in,
e.g., Strausberg, R.L. et al., 2002, "Generation and initial analysis of more
than 15,000 full-
length human and mouse eDNA sequences" published in Proc. Natl. Acad. Sci.
U.S.A. 99
(26), 16899-16903 (2002), each of which is incorporated by reference herein in
its entirety.
The nucleotide sequence of MYO18B (identified by accession no. NM032608) is
disclosed in, e.g., Nishioka, M. et al., 2002, "MYO18B, a candidate tumor
suppressor gene
at chromosome 22q12.1, deleted, mutated, and methylated in human lung cancer,"
published in Proc. Natl. Acad. Sci. U.S.A. 99 (19), 12269-12274; Salamon, M.
et al., 2003,
"Human MYOI8B, a novel unconventional myosin heavy chain expressed in striated
muscles moves into the myonuclei upon differentiation," J. Mol. Biol. 326 (1),
137-149;
Yanaihara, N. et al., 2004, "Reduced expression of MYO18B, a candidate tumor-
suppressor
gene on chromosome arm 22q, in ovarian cancer," published in Int. J. Cancer
112 (1), 150-
154, and the amino acid sequence of MYO I 8B (identified by accession no.
NP_115997) is
disclosed in, e.g., Nishioka, M. et al., 2002, "MYO18B, a candidate tumor
suppressor gene
at chromosome 22q 12.1, deleted, mutated, and methylated in human lung
cancer,"
published in Proc. Natl. Acad. Sci. U.S.A. 99 (19), 12269-12274; Salamon, M.
et al., 2003,
"Human MYO18B, a novel unconventional myosin heavy chain expressed in striated
muscles moves into the myonuclei upon differentiation," J. Mol. Biol. 326 (1),
137-149;
Yanaihara,N. et al., 2004, "Reduced expression of MYO 18B, a candidate tumor-
suppressor
gene on chromosome arm 22q, in ovarian cancer," published in Int. J. Cancer
112 (1), 150-
154, each of which is incorporated by reference herein in its entirety.
The nucleotide sequence of MICAL-L1 (identified by accession no. NM_033386) is
disclosed in, e.g., Marzesco, A.M. et al., 2002, "The small GTPase Rabl3
regulates
assembly of functional tight junctions in epithelial cells," published in Mol.
Biol. Cell 13
(6), 1819-1831; Terman, J.R. et al., 2002, "MICALs, a family of conserved
flavoprotein
oxidoreductases, function in plexin-mediated axonal repulsion," published in
Cell 109 (7),
887-900; Collins, J.E. et al., 2004, "A genome annotation driven approach to
cloning the
human ORFeome," published in Genome Biol. 5 (10), R84, and the amino acid
sequence of
MICAL-L1 (identified by accession nos. AAH82243, AAH01090) is disclosed in,
e.g.,
Strausberg et al., 2002, "Generation and initial analysis of more than 15,000
full-length
human and mouse cDNA sequences," published in Proc. Natl. Acad. Sci. U.S.A. 99
(26),
16899-16903, each of which is incorporated by reference herein in its
entirety.
The nucleotide sequence of PGLYRP2 (identified by accession no. NM_052890) is
disclosed in, e.g., Liu, C. et al., 2002, "Peptidoglycan recognition proteins:
a novel family
- 139 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
of four human innate immunity pattern recognition molecules," published in J.
Biol. Chem.
276 (37), 34686-34694; Xu, X.R. et al., 2001, "Insight into hepatocellular
carcinogenesis at
transcriptome level by comparing gene expression profiles of hepatocellular
carcinoma with
those of corresponding noncancerous liver," published in Proc. Nati. Acad.
Sci. U.S.A. 98
(26), 15089-15094; Kibardin, A.V. et al., 2003, "Expression analysis of
proteins encoded
by genes of the tag7/tagL(PGRP-S,L) family in human peripheral blood cells,"
Genetika 39
(2), 244-249, and the amino acid sequence of PGLYRP2 (identified by accession
no.
Q96PD5) is disclosed in, e.g., Zhang, H. et al., 2003, "Identification and
quantification of
N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and
mass
spectrometry," published in Nat. Biotechnol. 21 (6), 660-666; Wang,Z.M. et
al., 2003,
"Human peptidoglycan recognition protein-L is an N-acetylmuramoyl-L-alanine
amidase,"
published in J. Biol. Chem. 278 (49), 49044-49052; Zhang, Z. et al., 2004,
"Signal peptide
prediction based on analysis of experimentally verified cleavage sites,"
published in Protein
Sci. 13 (10), 2819-2824, each of which is incorporated by reference herein in
its entirety.
The nucleotide sequence of LRG1 (identified by accession no. NM 052972) is
disclosed in, e.g., Takahashi, N. et al., 1985, "Periodicity of leucine and
tandem repetition
of a 24-amino acid segment in the primary structure of leucine-rich alpha 2
glycoprotein of
human serum," published in Proc. Natl. Acad. Sci. U.S.A. 82 (7), 1906 1910;
O'Donnell,
L.C. et al., 2002, "Molecular characterization and expression analysis of
leucine-rich
alpha2-glycoprotein, a novel marker of granulocytic differentiation,"
published in J.
Leukoc. Biol. 72 (3), 478-485; Bunkenborg, J. et al., 2004, "Screening for N-
glycosylated
proteins by liquid chromatography mass spectrometry," published in Proteomics
4 (2), 454-
465, and the amino acid sequence of LRG1 (identified by accession no.
AAH70198) is
disclosed in, e.g., Strausberg et al., 2002, "Generation and initial analysis
of more than
15,000 full-length human and mouse cDNA sequences," Proc. Natl. Acad. Sci.
U.S.A. 99
(26), each of which is incorporated by reference herein in its entirety.
The nucleotide sequence of KCTD7 (identified by accession no. NM_153033) is
disclosed in, e.g., Scherer, S.W. et al., 2003, "Human chromosome 7: DNA
sequence and
biology," published in Science 300 (5620), 767-772, and the amino acid
sequence of
KCTD7 (identified by accession no. NP 694578) is disclosed in, e.g., Scherer,
S.W. et al.,
2003, "Human chromosome 7: DNA sequence and biology," published in Science 300
(5620), 767-772, each of which is incorporated by reference herein in its
entirety.
The nucleotide sequence of MGC27165 (identified by accession nos. BC087841 and
BC005951) is disclosed in, e.g., Strausberg et al., 2002, "Generation and
initial analysis of
- 140 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
more than 15,000 full-length human and mouse cDNA sequences," Proc. Natl.
Acad. Sci.
U.S.A. 99 (26), 16899-16903 and the amino acid sequence of MGC27165
(identified by
accession no. AAH87841) is disclosed in, e.g., Strausberg et al., 2002,
"Generation and
initial analysis of more than 15,000 full-length human and mouse eDNA
sequences,"
published in Proc. Natl. Acad. Sci. U.S.A. 99 (26), 16899-16903, each of which
is
incorporated by reference herein in its entirety.
The nucleotide sequence of A1BG (identified by accession no. NM130786) is
disclosed in, e.g., Ishioka, N. et al., 1986, "Amino acid sequence of human
plasma alpha
1 B-glycoprotein: homology to the immunoglobulin supergene family," published
in Proc.
Natl. Acad. Sci. U.S.A. 83 (8), 2363-2367; Gahne, B. et al., 1987, "Genetic
polymorphism
of human plasma alpha 1B-glycoprotein: phenotyping by immunoblotting or by a
simple
method of 2-D electrophoresis," published in Hum. Genet. 76 (2), 111 115;
Eiberg, H. et al.,
1989, "Linkage between alpha 1B-glycoprotein (AlBG) and Lutheran (LU) red
blood group
system: assignment to chromosome 19: new genetic variants of AIBG,' published
in Clin.
Genet. 36 (6), 415-418, and the amino acid sequence of A1BG (identified by
accession no.
NP_570602) is disclosed in, e.g., Ishioka, N. et al., 1986, "Amino acid
sequence of human
plasma alpha IB-glycoprotein: homology to the immunoglobulin supergene
family," .
published in Proc. Natl. Acad. Sci. U.S.A. 83 (8), 2363-2367; Gahne, B. et
al., 1987,
"Genetic polymorphism of human plasma aIpha 1B-glycoprotein: phenotyping by
immunoblotting or by a simple method of 2-D electrophoresis," published in
Hum. Genet.
76 (2), 111-115; Eiberg, H. et al., 1989, "Linkage between alpha 1B-
glycoprotein (A1BG)
and Lutheran (LU) red blood group system: assignment to chromosome 19: new
genetic
variants of A1BG," published in Clin. Genet. 36 (6), 415-418, each of which is
incorporated
by reference herein in its entirety.
The nucleotide sequence of A2M (identified by accession no. NM_000014) is
disclosed in, e.g., Nartikova et al., 1979, "Uniform method for determining
the alpha
1-antitrypsin and alpha 2-macroglobulin activity in human blood serum
(plasma),"
published in Vopr. Med. Khim. 25 (4), 494-499; Gustavsson et al., 1980,
"Interaction
between human pancreatic elastase and plasma protease inhibitors," Hoppe-
Seyler's Z.
Physiol. Chem. 361 (2), 169-176; Murata et al., 1983, "Radioimmunoassay of
human
pancreatic elastase 1. In vitro interaction of human pancreatic elastase I
with serum
protease inhibitors," Enzyme 30 (1), 29-37, and the amino acid sequence of A2M
(identified
by accession no. AAT02228) is disclosed in, e.g., Sottrup-Jensen et al., 1984,
"Primary
-141-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
structure of human alpha 2-macroglobulin," J. Biol. Chem 259:8318-8327, each
of which is
incorporated by reference herein in its entirety.
The nucleotide sequence of ABLIMI (identified by accession no. NM 002313) is
disclosed in, e.g., Adams et al., 1995, "Initial assessment of human gene
diversity and
expression patterns based upon 83 million nucleotides of cDNA sequence,"
Nature 377
(6547 SUPPL), 3-174; Roof et al., 1997, "Molecular characterization of abLIM,
a novel
actin-binding and double zinc finger protein," J. Cell Biol. 138 (3), 575-588;
Kim et al.,
1997, "Limatin (LIMAB 1), an actin-binding LIM protein, maps to mouse
chromosome 19
and human chromosome 10q25, a region frequently deleted in human cancers,"
published in
Genomics 46 (2), 291-293, and the amino acid sequence of ABLIMI (identified by
accession no. CAI10910) is disclosed in, e.g., Tracey, A., 2005, Direct
Submission,
Wellcome Trust Sanger Institute, Hinxton, Cambridgeshire, CB 10 1 SA, UK, each
of which
is incorporated by reference herein in its entirety.
The nucleotide sequence of ACTAI (identified by accession no. NIVI 001100) is
disclosed in, e.g., Gunning, P. et al., 1983, "Isolation and characterization
of full-length
cDNA clones for human alpha-, beta-, and gamma-actin mRNAs: skeletal but not
cytoplasmic actins have an amino-terminal cysteine that is subsequently
removed,"
published in Mol. Cell. Biol. 3 (5), 787-795; Hanauer, A. et al., 1983,
"Isolation and
characterization of cDNA clones for human skeletal muscle alpha actin,"
published in
Nucleic Acids Res. 11 (11), 3503-3516; Kedes, L. et al., 1985, "The human beta-
actin
multigene family," published in Trans. Assoc. Am. Physicians 98, 42-46, and
the amino
acid sequence of ACTA1 (identified by accession no. CAI19052) is disclosed in,
e.g.,
Matthews, N., 2005, Direct Submission, Wellcome Trust Sanger Institute,
Hinxton,
Cambridgeshire, CB 10 1 SA, UK, each of which is incorporated by reference
herein in its
entirety.
The nucleotide sequence of ANK3 (identified by accession no. NM 020987) is
disclosed in, e.g., Kordeli, E. et al., 1995, "AnkyrinG. A new ankyrin gene
with neural-
specific isoforms localized at the axonal initial segment and node of
Ranvier," published in
J. Biol. Chem. 270 (5), 2352-2359; Kapfhamer, D. et al., Chromosomal
localization of the
ankyrinG gene (ANK3/Ank3) to human I0q21 and mouse 10," published in Genomics
27
(1), 189-191; Devarajan,P. et al., 1996, "Identification of a small
cytoplasmic ankyrin
(AnkG119) in the kidney and muscle that binds beta I sigma spectrin and
associates with the
Golgi apparatus," published in J. Cell Biol. 133 (4), 819-830, and the amino
acid sequence
of ANK3 (identified by accession no. CA140519) is disclosed in, e.g.,
Chapman,J., 2005,
- 142 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Direct Submission, Wellcome Trust Sanger Institute, Hinxton, Cambridgeshire,
CB 10 1 SA,
UK, each of which is incorporated by reference herein in its entirety.
The nucleotide sequence of APCS (identified by accession no. BT006750) is
disclosed in, e.g., Mantzouranis et al., 1985, "Human serum amyloid P
component. cDNA
isolation, complete sequence of pre-serum amyloid P component, and
localization of the
gene to chromosome 1," J. Biol. Chem. 260:7752-7756, and the amino acid
sequence of
APCS (identified by accession no. CAH73651) is disclosed in, e.g., Cobley, V.,
2005,
Direct Submission, Wellcome Trust Sanger Institute, Hinxton, Cambridgeshire,
CB 10 1 SA,
UK, each of which is incorporated by reference herein in its entirety.
The nucleotide sequence of serum amyloid P component precursor (identified by
accession no. BC007058) is disclosed in, Strausberg, 2002, "Generation and
Initial analyis
of more than 15,000 full-length human and mouse eDNA sequences," Proc. Natl.
Acad. Sci.
USA 99, 16899-16802 and the amino acid sequence of serum amyloid P component
precursor (identified by accession no. NP001630) is disclosed in, e.g.,
Veerhuis et'al., 2005,
"Activation of human microglia by fibrillar prion protein-related peptides is
enhanced by
amyloid-associated factors SAP and Clq," Neurobiol. Dis. 19, 273-282, each of
which is
incorporated by reference herein in its entirety.
The nucleotide sequence of B2M (identified by accession no. NM 004048) is
disclosed in, e.g., Krangel, M.S. et al., 1979, "Assembly and maturation of
HLA-A and
HLA-B antigens in vivo," published in Cell 18 (4), 979-991; Suggs, S.V. et
al., 1981, "Use
of synthetic oligonucleotides as hybridization probes:isolation of cloned cDNA
sequences
for human beta 2-microglobulin," published in Proc. Natl. Acad. Sci. U.S.A. 78
(11), 6613-
6617; Rosa, F. et al., 1983, "The beta2-microglobulin mRNA in human Daudi
cells has a
mutated initiation codon but is still inducible by interferon," published in
EMBO J. 2 (2),
239-243, and the amino acid sequence of B2M (identified by accession no.
AAA51811) is
disclosed in, e.g., Gussow, D. et al., 1987, "The human beta 2-microglobulin
gene. Primary
structure and definition of the transcriptional unit," published in J.
Immunol. 139 (9), 3132-
3138, each of which is incorporated by reference herein in its entirety.
The nucleotide sequence of C1R (identified by accession no. NM_001733) is
disclosed in, e.g., Lee, S.L. et al., 1978, "Familial deficiency of two
subunits of the first
component of complement. C1r and C1s associated with a lupus erythematosus-
like
disease," published in Arthritis Rheum. 21 (8), 958-967; Leytus, S.P. et al.,
1986,
"Nucleotide sequence of the eDNA coding for human complement Cir," published
in
Biochemistry 25 (17), 4855-4863; Journet, A. et al., 1986, "Cloning and
sequencing of full-
-143-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
length cDNA encoding the precursor of human complement component Clr,"
published in
Biochem. J. 240 (3), 783-787, and the amino acid sequence of C1R (identified
by accession
no. NP_001724) is disclosed in, e.g., Lee, S.L. et al., 1978, "Familial
deficiency of two
subunits of the first component of complement. C 1 r and C 1 s associated with
a lupus
erythematosus-like disease," published in Arthritis Rheum. 21 (8), 958-967;
Leytus, S.P. et
al., 1986, "Nucleotide sequence of the eDNA coding for human complement Clr,"
published in Biochemistry 25 (17), 4855-4863; Joumet, A. et al., 1986,
"Cloning and
sequencing of full-length cDNA encoding the precursor of human complement
component
C 1 r," published in Biochem. J. 240 (3), 783-787, each of which is
incorporated by reference
herein in its entirety.
The nucleotide sequence of C4B (identified by accession no. NM 000592) is
disclosed in, e.g., Teisberg, P. et aL, 1976, "Genetic polymorphism of C4 in
man and
localisation of a structural C4 locus to the HLA gene complex of chromosome
6," published
in Nature 264 (5583), 253-254; Moon, K.E. et al., 1981, "Complete primary
structure of
human C4a anaphylatoxin," published in J. Biol. Chem. 256 (16), 8685-8692;
Mascart-
Lemone, F. et al., 1983, "Genetic deficiency of C4 presenting with recurrent
infections and
a SLE-like disease. Genetic and immunologic studies," published in Am. J. Med.
75 (2),
295-304, and the amino acid sequence of C4B (identified by accession no.
AAR89095) is
disclosed in, e.g., Sayer, D. et al., 2003, Direct Submission, Dept of
Clinical Immunology,
Royal Perth Hospital, Wellington Street, Perth, Westem Australia, Australia;
Sayer, D. et
al., unpublished, "Molecular genetics of complement C4: implications for MHC
evolution
and disease susceptibility gene mapping," each of which is incorporated by
reference herein
in its entirety.
The nucleotide sequence of C6 (identified by accession no. NM_000065) is
disclosed in, e.g., Hetland, G. et al., 1986, "Synthesis of complement
components C5, C6,
C7, C8 and C9 in vitro by human monocytes and assembly of the terminal
complement
complex," published in Scand. J. Immunol. 24 (4), 421-428; Chakravarti, D.N.
et al., 1988,
"Biochemical characterization of the human complement protein C6. Association
with
alpha-thrombin-like enzyme and absence of serine protease activity in
cytolytically active
C6," published in J. Biol. Chem. 263 (34), 18306-18312; Chakravarti, D.N. et
al., 1989,
"Structural homology of complement protein C6 with other channel-forming
proteins of
complement," published in Proc. Natl. Acad. Sci. U.S.A. 86 (8), 2799-2803, and
the amino
acid sequence of C6 (identified by accession no. BAD02322) is disclosed in,
e.g., Soejima
et al., 2005, "Nucleotide sequence analyses of human complement 6 (C6) gene
suggest
- 144 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
balancing selection," Ann. Hum. Genet. 69 (PT 3), 239-252, each of which is
incorporated
by reference herein in its entirety.
The nucleotide sequence of C7 (identified by accession no. NM 000587) is
disclosed in, e.g., DiScipio, R.G. et al., 1988, "The structure of human
complement
component C7 and the C5b-7 complex," published in J. Biol. Chem. 263 (1), 549-
560;
Nurnberger, W. et al., 1989, "Familial deficiency of the seventh component of
complement
associated with recurrent meningococcal infections," published in Eur. J.
Pediatr. 148 (8),
758-760; Coto, E. et al., 1991, "DNA polymorphisms and linkage relationship of
the human
complement component C6, C7, and C9 genes," published in Immunogenetics 33
(3), 184-
187, and the amino acid sequence of C7 (identified by accession no. CAA72407)
is
disclosed in, e.g., Gonzalez, S., 1997, Direct Submission, S. Gonzalez,
Servicio de
Immunologia, Hospital Central Asturias, Julian Claveria s.n., 33006 Oviedo,
Asturias,
SPAIN; Gonzalez, S. et al., 2002, "Cloning and characterization of human
complement
component C7 promoter," each of which is incorporated by reference herein in
its entirety.
The nucleotide sequence of C8B (identified by accession no. NM 000066) is
disclosed in, e.g., Howard, O.M. et al., 1987, "Complementary DNA and derived
amino
acid sequence of the beta subunit of human complement protein C8:
identification of a close
structural and ancestral relationship to the alpha subunit and C9," published
in Biochemistry
26 (12), 3565-3570; Haefliger, J.A. et al., 1987, "Complementary DNA cloning
of
complement C8 beta and its sequence homology to C9," published in Biochemistry
26 (12),
3551-3556; Stewart, J.L. et al., 1987, "Evidence that C5b recognizes and
mediates C8
incorporation into the cytolytic complex of complement," published in J.
Immunol. 139 (6),
1960-1964, and the amino acid sequence of C8B (identified by accession no. CAC
18532) is
disclosed in, e.g., Howden, P., 2005, Direct Submission, Wellcome Trust Sanger
Institute,
Hinxton, Carnbridgeshire, CB 10 I SA, UK, each of which is incorporated by
reference
herein in its entirety.
The nucleotide sequence of CDK5RAP2 (identified by accession no. NM_018249)
is disclosed in, e.g., Ching, Y.P. et al., 2000, "Cloning of three novel
neuronal Cdk5
activator binding proteins," published in Gene 242 (1-2), 285-294; Wang, X. et
al., 2000,
"Identification of a common protein association region in the neuronal Cdk5
activator,"
published in J. Biol. Chem. 275 (41), 3 1 763-3 1 769; Andersen, J.S. et al.,
2003, "Proteomic
characterization of the human centrosome by protein correlation profiling,"
published in
Nature 426 (6966), 570-574, and the amino acid sequence of CDK5RAP2
(identified by
accession no. CA140927) is disclosed in, e.g., Beasley, H., 2005, Direct
Submission,
- 145 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Wellcome Trust Sanger Institute, Hinxton, Cambridgeshire, CB 10 1 SA, UK, each
of which
is incorporated by reference herein in its entirety.
The nucleotide sequence of CHGB (identified by accession no. NM_001819) is
disclosed in, e.g., Benedum, U.M. et al., 1987, "The primary structure of
human
secretogranin I (chromogranin B): comparison with chromogranin A reveals
homologous
terminal domains and a large intervening variable region," published in EMBO
J. 6 (5),
1203-1211; Gill, B.M. et al., 1991, "Chromogranin B: isolation from
pheochromocytoma,
N-terminal sequence, tissue distribution and secretory vesicle processing,"
published in
Regul. Pept. 33 (2), 223-235; Levine, M.A. et al., 1991, "Mapping of the gene
encoding the
alpha subunit of the stimulatory G protein of adenylyl cyclase (GNAS1) to
20q13.2----q13.3
in human by in situ hybridization," published in Genomics 11 (2), 478-479, and
the amino
acid sequence of CHGB (identified by accession no. CAB55272) is disclosed in,
e.g., Pelan,
S., 2005, Direct Submission, Wellcome Trust Sanger Institute, Hinxton,
Cambridgeshire,
CB 10 1 SA, UK, each of which is incorporated by reference herein in its
entirety.
The nucleotide sequence of COMP (identified by accession no. NM 000095) is
disclosed in, e.g., Briggs, M.D. et al., 1993, "Genetic linkage of mild
pseudoachondroplasia
(PSACH) to markers in the pericentromeric region of chromosome 19," published
in
Genomics 18 (3), 656-660; Oehlmann, R. et al., 1994, "Genetic linkage mapping
of
multiple epiphyseal dysplasia to the pericentromeric region of chromosome 19,"
published
in Am. J. Hum. Genet. 54 (1), 3-10; Newton, G. et al., 1994, "Characterization
of human
and mouse cartilage oligomeric matrix protein," published in Genomics 24 (3),
435-439,
and the amino acid sequence of COMP (identified by accession no. AAC83643) is
disclosed
in, e.g., Deere et al., 2001, "Analysis of the promoter region of human
cartilage oligomeric
matrix protein (COMP)," Matrix Biol. 19 (8), 783-792, each of which is
incorporated by
reference herein in its entirety.
The nucleotide sequence of COROIA (identified by accession no. NM 007074) is
disclosed in, e.g., Suzuki, K. et al., 1995, "Molecular cloning of a novel
actin-binding
protein, p57, with a WD repeat and a leucine zipper motif," published in FEBS
Lett. 364
(3), 283-288; Okumura,M., et al., 1998, "Definition of family of coronin-
related proteins
conserved between humans and mice: close genetic linkage between coronin-2 and
CD45-
associated protein," published in DNA Cell Biol. 17 (9), 779-787; Ferrari,G.
et al., 1999, "A
coat protein on phagosomes involved in the intracellular survival of
mycobacteria,"
published in Cell 97 (4), 435-447, and the amino acid sequence of COROIA
(identified by
accession no. NP 009005) is disclosed in, e.g., Suzuki, K. et al., 1995,
"Molecular cloning
-146-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
of a novel actin-binding protein, p57, with a WD repeat and a leucine zipper
motif,"
published in FEBS Lett. 364 (3), 283-288; Okumura, M.et al., 1998, "Definition
of family
of coronin-related proteins conserved between humans and mice: close genetic
linkage
between coronin-2 and CD45-associated protein," published in DNA Cell Biol. 17
(9), 779-
787; Ferrari, G. et al., 1999, "A coat protein on phagosomes involved in the
intracellular
survival of mycobacteria," published in Cell 97 (4), 435-447, each of which is
incorporated
by reference herein in its entirety.
The nucleotide sequence of CPN1 (identified by accession no. NM_001308) is
disclosed in, e.g., Skidgel, R.A. et al., 1988, "Amino acid sequence of the N-
terminus and
selected tryptic peptides of the active subunit of human plasma
carboxypeptidase N:
comparison with other carboxypeptidases," published in Biochem. Biophys. Res.
Commun.
154 (3), 1323-1329; Gebhard, W. et al., 1989, "cDNA cloning of kininase 1,"
published in
Adv. Exp. Med. Biol. 247B, 261-264; Gebhard, W. et al., 1989, "cDNA cloning
and
complete primary structure of the small, active subunit of human
carboxypeptidase N
(kininase 1), " published in Eur. J. Biochem. 178 (3), 603-607, and the amino
acid sequence
of CPN1 (identified by accession no. NP001299) is disclosed in, e.g., Skidgel,
R.A. et al.,
1988, "Amino acid sequence of the N-terminus and selected tryptic peptides of
the active
subunit of human plasma carboxypeptidase N: comparison with other
carboxypeptidases,"
published in Biochem. Biophys. Res. Commun. 154 (3), 1323-1329; Gebhard, W. et
al.,
1989, "cDNA cloning of kininase 1," published in Adv. Exp. Med. Biol. 247B,
261-264;
Gebhard, W. et al., 1989, "cDNA cloning and complete primary structure of the
small,
active subunit of human carboxypeptidase N (kininase 1)," published in Eur. J.
Biochem.
178 (3), 603-607, each of which is incorporated by reference herein in its
entirety.
The nucleotide sequence of CULl (identified by accession no. NM003592) is
disclosed in, e.g., Kipreos, E.T. et al., 1996, "cul-1 is required for cell
cycle exit in C.
elegans and identifies a novel gene family," published in Cell 85 (6), 829-
839; Lisztwan, J.
et al., 1998, "Association of human CUL-1 and ubiquitin-conjugating enzyme
CDC34 with
the F-box protein p45(SKP2): evidence for evolutionary conservation in the
subunit
composition of the CDC34-SCF pathway," published in EMBO J. 17 (2), 368-383;
Michel,
J.J. et al., 1998, "Human CUL- 1, but not other cullin family members,
selectively interacts
with SKP I to form a complex with SKP2 and cyclin A," published in Cell Growth
Differ. 9
(6), 435-449, and the amino acid sequence of CUL 1(identified by accession no.
NP_003583) is disclosed in, e.g., Kipreos, E.T. et al., 1996, "cul-1 is
required for cell cycle
exit in C. elegans and identifies a novel gene family," published in Cell 85
(6), 829-839;
-147-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Lisztwan, J. et al., 1998, "Association of human CUL-1 and ubiquitin-
conjugating enzyme
CDC34 with the F-box protein p45(SKP2): evidence for evolutionary conservation
in the
subunit composition of the CDC34-SCF pathway," published in EMBO J. 17 (2),
368-383;
Michel,J.J. et al., 1998, "Human CUL-1, but not other cullin family members,
selectively
interacts with SKP1 to form a complex with SKP2 and cyclin A," published in
Cell Growth
Differ. 9 (6), 435-449, each of which is incorporated by reference herein in
its entirety.
The nucleotide sequence of DET1 (identified by accession no. NM_017966) is
disclosed in, e.g., Eastman, S.W. et al., 2005, "Identification of human
VPS37C, a
component of endosomal sorting complex required for transport-I important for
viral
budding," published in J. Biol. Chem. 280 (1), 628-636, and the amino acid
sequence of
DET1 (identified by accession no. NP_060466) is disclosed in, e.g., Wertz,
I.E. et al., 2004,
"Human De-etiolated-1 regulates c-Jun by assembling a CUL4A ubiquitin ligase,"
published in Science 303 (5662), 1371-1374, each of which is incorporated by
reference
herein in its entirety.
The nucleotide sequence ofDSC1 (identified by accession no. BC109161) is
disclosed in, e.g., Strausberg,R.L. et al., 2002, "Generation and initial
analysis of more than
15,000 full-length human and mouse cDNA sequences," published in Proc. Natl.
Acad. Sci.
U.S.A. 99 (26), 16899-16903, and the amino acid sequence of DET1 (identified
by
accession no. NP 060466) is disclosed in, e.g., Wertz, I.E. et al., 2004,
"Human De-
etiolated-1 regulates c-Jun by assembling a CUL4A ubiquitin ligase," published
in Science
303 (5662), 1371-1374, each of which is incorporated by reference herein in
its entirety.
The nucleotide sequence of F13A1 (identified by accession no. NM000129) is
disclosed in, e.g., Takahashi, N. et al., 1986, "Primary structure of blood
coagulation factor
XIIIa (fibrinoligase, transglutaminase) from human placenta," published in
Proc. Natl.
Acad. Sci. U.S.A. 83 (21), 8019-8023; Grundmann, U. et al., 1986,
"Characterization of
cDNA coding for human factor XIIIa," published in Proc. Natl. Acad. Sci.
U.S.A. 83 (21),
8024-8028; Ichinose, A. et al., 1986, "Amino acid sequence of the a subunit of
human
factor XIII," published in Biochemistry 25 (22), 6900-6906, and the amino acid
sequence of
F13A1 (identified by accession no. CAC36886) is disclosed in, e.g., Sehra, H.,
2005, Direct
Submission, Wellcome Trust Sanger Institute, Hinxton, Cambridgeshire, CB 10 1
SA, UK,
each of which is incorporated by reference herein in its entirety.
The nucleotide sequence of F5 (identified by accession no. N1VI 000130) is
disclosed in, e.g., Suzuki, K. et al., 1982, "Thrombin-catalyzed activation of
human
coagulation factor V," published in J. Biol. Chem. 257 (11), 6556-6564; Kane,
W.H. et al.,
- 148 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
1986, "Cloning of a cDNA coding for human factor V, a blood coagulation factor
homologous to factor VIII and ceruloplasmin," published in Proc. Natl. Acad.
Sci. U.S.A.
83 (18), 6800-6804; Jenny, R.J. et al., 1987, "Complete cDNA and derived amino
acid
sequence of human factor V," published in Proc. Natl. Acad. Sci. U.S.A. 84
(14), 4846
4850, and the amino acid sequence of F5 (identified by accession nos.
CA123065,
CAB 16748) is disclosed in, e.g., Bird, C., 2005, Direct Submission, Wellcome
Trust Sanger
Institute, Hinxton, Cambridgeshire, CB 10 1 SA, UK, each of which is
incorporated by
reference herein in its entirety.
The nucleotide sequence of GOLGAI (identified by accession no. NM002077) is
disclosed in, e.g., Griffith, K.J. et al., 1997, "Molecular cloning of a novel
97-kd Golgi
complex autoantigen associated with Sjogren's syndrom," published in Arthritis
Rheum. 40
(9), 1693-1702; Barr, F.A., 1999, "A novel Rab6-interacting domain defines a
family of
Golgi-targeted coiled-coil proteins," published in Curr. Biol. 9 (7), 381-384;
Lu, L. et al.,
2003, "Interaction of Ar11-GTP with GRIP domains recruits autoantigens Golgin-
97 and
Golgin-245/p230 onto the Golgi," published in Mol. Biol. Cell 14 (9), 3767-
3781, and the
amino acid sequence of GOLGAI (identified by accession no. CA139632) is
disclosed in,
e.g., Tracey, A., 2005, Direct Submission, Wellcome Trust Sanger Institute,
Hinxton,
Cambridgeshire, CB 10 1 SA, UK, each of which is incorporated by reference
herein in its
entirety.
The nucleotide sequence of HBA 1(identified by accession no. NM 000558) is
disclosed in, e.g., Kleihauer, E.F. et al., 1968, "Hemoglobin-Bibba or alpha-2-
136Pro-beta
2, an unstable alpha chain abnormal hemoglobin," published in Biochim.
Biophys. Acta 154
(1), 220-222; Boyer, S.H. et al., 1968, "A survey of hemoglobins in the
Republic of Chad
and characterization of hemoglobin Chad:alpha-2-23G1u--Lys-beta-2," published
in Am. J.
Hum. Genet. 20 (6), 570-578; Fujiwara, N. et al., 1971, "Hemoglobin Atago
(alpha2-85Tyr
beta-2) a new abnormal human hemoglobin found in Nagasaki. Biochemical studies
on
hemoglobins and myoglobins. VI," published in Int. J. Protein Res. 3 (1), 3 5-
39, and the
amino acid sequence of HBAI (identified by accession no. AA022464) is
disclosed in, e.g.,
Elam, D. et al., 2002, Direct Submission, Medicine/Hematology-
Oncology/Hemoglobin
DNA Laboratory, Medical College of Georgia, 15 th Street, AC-1000, Augusta,
Georgia
30912, USA, each of which is incorporated by reference herein in its entirety.
The nucleotide sequence of HSPA5 (identified by accession no. NM 005347) is
disclosed in, e.g., Munro, S. et al., 1986, "An Hsp70-like protein in the ER:
identity with
the 78 kd glucose-regulated protein and immunoglobulin heavy chain binding
protein,"
- 149 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
published in Ce1146 (2), 291-300; Pollok, B.A. et al., 1987, "Molecular basis
of the
cell-surface expression of immunoglobulin mu chain without light chain in
human B
lymphocytes," published in Proc. Nati. Acad. Sci. U.S.A. 84 (24), 9199-9203;
Ting, J. et al.,
1988, "Human gene encoding the 78,000-dalton glucose-regulated protein and its
pseudogene: structure, conservation, and regulation," published in DNA 7 (4),
275-286, and
the amino acid sequence of HSPA5 (identified by accession no. NP_005338) is
disclosed in,
e.g., Munro, S. et al., 1986, "An Hsp70-like protein in the ER: identity with
the 78 kd
glucose-regulated protein and immunoglobulin heavy chain binding protein,"
published in
Ce1146 (2), 291-300; Pollok, B.A. et al., 1987, "Molecular basis of the cell-
surface
expression of immunoglobulin mu chain without light chain in human B
lymphocytes,"
published in Proc. Natl. Acad. Sci. U.S.A. 84 (24), 9199-9203; Ting, J. et
al., 1988,
"Human gene encoding the 78,000-dalton glucose-regulated protein and its
pseudogene:
structure, conservation, and regulation," published in DNA 7 (4), 275-286,
each of which is
incorporated by reference herein in its entirety.
The nucleotide sequence of HUNK (identified by accession no. NM 014586) is
disclosed in, e.g., Gardner, H.P. et al., 2002, "Cloning and characterization
of Hunk, a novel
mammalian," published in Genomics 63 (1), 46-59; Korobko,I.V. et al., 2000,
"The
MAK-V protein kinase regulates endocytosis in mouse," published in Mol. Gen.
Genet. 264
(4), 411-418; Korobko, I.V. et al., 2004, "Proteinkinase MAK-V/Hunk as a
possible
diagnostic and prognostic marker of human breast carcinoma," published in
Arkh. Patol. 66
(5), 6-9, and the amino acid sequence of HUNK (identified by accession no.
NP055401) is
disclosed in, e.g., Gardner, H.P. et al., 2002, "Cloning and characterization
of Hunk, a novel
mammalian," published in Genomics 63 (1), 46-59; Korobko, I.V. et al., 2000,
"The
MAK-V protein kinase regulates endocytosis in mouse," published in Mol. Gen.
Genet. 264
(4), 411-418; Korobko, I.V. et al., 2004, "Proteinkinase MAK-V/Hunk as a
possible
dianostic and prognostic marker of human breast carcinoma," published in Arkh.
Patol. 66
(5), 6-9, each of which is incorporated by reference herein in its entirety.
The nucleotide sequence of IGFBP5 (identified by accession no. NM_000599) is
disclosed in, e.g., Kiefer, M.C. et al., 1991, "Molecular cloning of a new
human insulin-like
growth factor binding protein," published in Biochem. Biophys. Res. Commun.
176 (1),
219-225; Ehrenborg, E. et al., 1991, "Structure and localization of the human
insulin-like
growth factor-binding protein 2 gene," published in Biochem. Biophys. Res.
Commun. 176
(3), 1250-1255; Shimasaki, S. et al., 1991, "Identification of five different
insulin-like
growth factor binding proteins (IGFBPs) from adult rat serum and molecular
cloning of a
- 150 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
novel IGFBP-5 in rat and human," published in J. Biol. Chem. 266 (16), 10646-
10653, and
the amino acid sequence of IGFBP5 (identified by accession no. NP_000590) is
disclosed
in, e.g., Kiefer, M.C. et al., 1991, "Molecular cloning of a new human insulin-
like growth
factor binding protein," published in Biochem. Biophys. Res. Commun. 176 (1),
219-225;
Ehrenborg, E. et al., 1991, "Structure and localization of the human insulin-
like growth
factor-binding protein 2 gene," published in Biochem. Biophys. Res. Commun.
176 (3),
1250-1255; Shimasaki, S. et al., 1991, "Identification of five different
insulin-like growth
factor binding proteins (IGFBPs) from adult rat serum and molecular cloning of
a novel
IGFBP-5 in rat and human," published I J. Biol. Chem. 266 (16), 10646-10653,
each of
which is incorporated by reference herein in its entirety.
The nucleotide sequence of IGHG1 (identified by accession no. BC092518) is
disclosed in, e.g., Strausberg, R.L. et al., 2002, "Generation and initial
analysis of more than
15,000 full-length human and mouse cDNA sequences," published in Proc. Natl.
Acad. Sci.
U.S.A. 99 (26), 16899-16903, and the amino acid sequence of IGHG1 (identified
by
accession no. CAC20454) is disclosed in, e.g., McLean et al., 2000, "Human and
murine
immunoglobulin expression vector cassettes," Mol. Immunol. 37 (14), 837-845,
each of
which is incorporated by reference herein in its entirety.
The nucleotide sequence of IGLV4-3 (identified by accession no. BC020236) is
disclosed in, e.g., Strausberg, R.L. et al., 2002, "Generation and initial
analysis of more than
15,000 full-length human and mouse cDNA sequences," published in Proc. Natl.
Acad. Sci.
U.S.A. 99 (26), 16899-16903, and the amino acid sequence of IGLV4-3
(identified by
accession no. AAH20236) is disclosed in, e.g., Strausberg, R.L. et al., 2002,
"Generation
and initial analysis of more than 15,000 full-length human and mouse cDNA
sequences,"
published in Proc. Natl. Acad. Sci. U.S.A. 99 (26), 16899-16903, each of which
is
incorporated by reference herein in its entirety.
The nucleotide sequence of KIF5C (identified by accession no. NM 004984) is
disclosed in, e.g., Niclas, J. et al., "Cloning and localization of a
conventional kinesin motor
expressed exclusively in neurons," Neuron 12 (5), 1059-1072, 1994; and the
amino acid
sequence of KIF5C (identified by accession no. AAH17298) is disclosed in,
e.g.,
Strausberg, R.L. et al., "Generation and initial analysis of more than 15,000
fu11-length
human and mouse cDNA sequences," Proc. Nati. Acad. Sci. U.S.A. 99 (26), 16899-
16903
(2002) each of which is incorporated by reference herein in its entirety.
The nucleotide sequence of KRT10 (identified by accession no. NM 000421) is
disclosed in, e.g., Darmon, M.Y. et al., 1987, "Sequence of a cDNA encoding
human
-151-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
keratin No. 10 selected according to structural homologies of keratins and
their
tissue-specific expression," published in Mol. Biol. Rep. 12 (4), 277-283;
Zhou, X.M. et aL,
1988, "The complete sequence of the human intermediate filament chain keratin
10.
Subdomainal divisions and model for folding of end domain sequences,"
published in J.
Biol. Chem. 263 (30), 15584-15589; Lessin, S.R. et al., 1988, "Chromosomal
mapping of
human keratin genes: evidence of non-linkage," published in J. Invest.
Dermatol. 91 (6),
572-578, and the amino acid sequence of KRT10 (identified by accession no.
NP_000412)
is disclosed in, e.g., Darmon, M.Y. et al., 1987, "Sequence of a cDNA encoding
human
keratin No 10 selected according to structural homologies of keratins and
their
tissue-specific expression," published in Mol. Biol. Rep. 12 (4), 277-283;
Zhou, X.M. et al.,
1988, "The complete sequence of the human intermediate filament chain keratin
10.
Subdomainal divisions and model for folding of end domain sequences,"
published in J.
Biol. Chem. 263 (30), 15584-15589; Lessin, S.R. et al., 1988, "Chromosomal
mapping of
human keratin genes: evidence of non-linkage," published in J. Invest.
Dermatol. 91 (6),
572-578, each of which is incorporated by reference herein in its entirety.
The nucleotide sequence of KRT9 (identified by accession no. NM_000226) is
disclosed in, e.g., Reis, A. et al., 1992, "Mapping of a gene for
epidermolytic palmoplantar
keratoderma to the region of the acidic keratin gene cluster at 17q12-q21,"
published in
Hum. Genet. 90 (1-2), 113-116; Rogaev, E.I. et al., 1993, "Identification of
the genetic
locus for keratosis palmaris et plantaris on chromosome 17 near the RARA and
keratin type
I genes," published in Nat. Genet. 5 (2), 158-162; Langbein, L. et al., 1993,
"Molecular
characterization of the body site-specific human epidermal cytokeratin 9: cDNA
cloning,
amino acid sequence, and tissue specificity of gene expression," published in
Differentiation
55 (1), 57-7 1, and the amino acid sequence of KRT9 (identified by accession
no.
NP_000217) is disclosed in, e.g., Reis, A. et al., 1992, "Mapping of a gene
for
epidermolytic palmoplantar keratoderma to the region of the acidic keratin
gene cluster at
17q12-q21," published in Hum. Genet. 90 (1-2), 113-116; Rogaev, E.I. et al.,
1993,
"Identification of the genetic locus for keratosis palmaris et plantaris on
chromosome 17
near the RARA and keratin type I genes," published in Nat. Genet. 5 (2), 158-
162;
Langbein, L. et al., 1993, "Molecular characterization of the body site-
specific human
epidermal cytokeratin 9: cDNA cloning, amino acid sequence, and tissue
specificity of gene
expression," published in Differentiation 55 (1), 57-71, each of which is
incorporated by
reference herein in its entirety.
- 152 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
The nucleotide sequence of LBP (identified by accession no. AF105067) is
disclosed in, e.g., Long et al., 1998, "Cloning and sequencing of human
lipopolysaccharide-
binding protein gene," Shengwu Huaxue Yu Shengwu Wuli Jinzhan 25, 469-471, and
the
amino acid sequence of LBP (identified by accession no. AAC39547) is disclosed
in, e.g.,
Kirschning et al., 1997, "Similar organization of the lipopolysaccharide-
binding protein
(LBP) and phospholipid transfer protein (PLTP) genes suggests a common gene
family of
lipid-binding proteins," Genomics 46 (3), 416-425, each of which is
incorporated by
reference herein in its entirety.
The nucleotide sequence of LUM (identified by accession no. BC035997) is
disclosed in, e.g., Strausberg et al., 2002, "Generation and initial analysis
of more than
15,000 full-length human and mouse cDNA sequences," Proc. Natl. Acad. Sci.
U.S.A. 99
(26), 16899-16903, and the amino acid sequence of LUM (identified by accession
no.
AAP35353) is disclosed in, e.g., Kalnine, N. et al., 2003, Direct Submission,
BD
Biosciences Clontech, 1020 East Meadow Circle, Palo Alto, California 94303,
USA, each
of which is incorporated by reference herein in its entirety.
The nucleotide sequence of MMP14 (identified by accession no. NM_004995) is
disclosed in, e.g., Sato, H. et al., 1994, "A matrix metalloproteinase
expressed on the
surface of invasive tumour cells," published in Nature 370 (6484), 61-65;
Okada, A. et al.,
1995, "Membrane-type matrix metalloproteinase (MT-MMP) gene is expressed in
stromal
cells of human colon, breast, and head and neck carcinomas," published in
Proc. Natl. Acad.
Sci. U.S.A. 92 (7), 2730-2734; Takino, T. et al., 1995, "Cloning of a human
gene
potentially encoding a novel matrix metalloproteinase having a C-terminal
transmembrane
domain," published in Gene 155 (2), 293-298, and the amino acid sequence of
MMP14
(identified by accession no. AAV40837) is disclosed in, e.g., Livingston, R.J.
et al., 2004,
Direct Submission, Genome Sciences, University of Washington, 1705 NE Pacific,
Seattle,
Washington 98195, USA, each of which is incorporated by reference herein in
its entirety.
The nucleotide sequence of MYH4 (identified by accession no. NM017533) is
disclosed in, e.g., Soussi-Yanicostas et al., 1993, "Five skeletal myosin
heavy chain genes
are organized as a multigene complex in the human genome," Hum. Mol. Genet. 2
(5),
563-569; Sant'ana Pereira et al., 1995, "New method for the accurate
characterization of
single human skeletal muscle fibres demonstrates a relation between mATPase
and MyHC
expression in pure and hybrid fibre types," J. Muscle Res. Cell. Motil. 16
(1), 21-34, and the
amino acid sequence of MYH4 (identified by accession no. NP 060003) is
disclosed in,
e.g., Soussi-Yanicostas et al., 1993, "Five skeletal myosin heavy chain genes
are organized
-153-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
as a multigene complex in the human genome," Hum. Mol. Genet. 2 (5), 563-569;
Sant'ana
Pereira et al., 1995, "New method for the accurate characterization of single
human skeletal
muscle fibres demonstrates a relation between mATPase and MyHC expression in
pure and
hybrid fibre types," J. Muscle Res. Cell. Motil. 16 (1), 21-34; and Weiss et
al., 1999,
"Organization of human and mouse skeletal myosin heavy chain gene clusters is
highly
conserved," Proc. Natl. Acad. Sci. U.S.A. 96 (6), 295 8-2963, each of which is
incorporated
by reference herein in its entirety.
The nucleotide sequence of NEB (identified by accession no. NM_004543 ) is
disclosed in, e.g., Stedman, H. et al., 1988, "Nebulin cDNAs detect a 25-
kilobase transcript
in skeletal muscle and localize to human chromosome 2," published in Genomics
2 (1), 1-7;
Zeviani, M. et al., 1988, "Cloning and expression of human nebulin cDNAs and
assignment
of the gene to chromosome 2q31-q32," published in Genomics 2 (3), 249-256;
Labeit, S. et
al., 1991, "Evidence that nebulin is a protein-ruler in muscle thin
filaments," published in
FEBS Lett. 282 (2), 313-316, and the amino acid sequence of NEB (identified by
accession
no. NP004534) is disclosed in, e.g., Stedman, H. et al., 1988, "Nebulin cDNAs
detect a
25-kilobase transcript in skeletal muscle and localize to human chromosome 2,"
published
in Genomics 2 (1), 1-7; Zeviani, M. et al., 1988, "Cloning and expression of
human nebulin
cDNAs and assignment of the gene to chromosome 2q31-q32," published in
Genomics 2
(3), 249-256; Labeit, S. et al., 1991, "Evidence that nebulin is a protein-
ruler in rnuscle thin
filaments," published in FEBS Lett. 282 (2), 313-316, each of which is
incorporated by
reference herein in its entirety.
The nucleotide sequence of NUCB2 (identified by accession no. NM_005013) is
disclosed in, e.g., Barnikol-Watanabe, S. et al., 1994, "Human protein NEFA, a
novel DNA
binding/EF-hand/leucine zipper protein. Molecular cloning and sequence
analysis of the
cDNA, isolation and characterization of the protein," published in Biol. Chem.
Hoppe-
Seyler 375 (8), 497-512; Kroll, K.A. et al., 1999, "Heterologous
overexpression of human
NEFA and studies on the two EF-hand calcium-binding sites," published in
Biochem.
Biophys. Res. Commun. 260 (1), 1-8; Taniguchi, N. et al., 2000, "The post
mitotic growth
suppressor necdin interacts with a calcium-binding protein (NEFA) in neuronal
cytoplasm,"
published in J. Biol. Chem. 275 (41), 31674-31681, and the amino acid sequence
of
NUCB2 (identified by accession no. NP_005004) is disclosed in, e.g., Barnikol-
Watanabe,
S. et al., 1994, "Human protein NEFA, a novel DNA binding/EF-handlleucine
zipper
protein. Molecular cloning and sequence analysis of the cDNA, isolation and
characterization of the protein," published in Biol. Chem. Hoppe-Seyler 375
(8), 497-512;
-154-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Kroll, K.A. et al., 1999, "Heterologous overexpression of human NEFA and
studies on the
two EF-hand calcium-binding sites," published in Biochem. Biophys. Res.
Commun. 260
(1), 1-8; Taniguchi, N. et al., 2000, "The postmitotic growth suppressor
necdin interacts
with a calcium-binding protein (NEFA) in neuronal cytoplasm," published in J.
Biol. Chem.
275 (41), 31674-31681, each of which is incorporated by reference herein in
its entirety.
The nucleotide sequence of ORM2 (identified by accession no. NM000608) is
disclosed in, e.g., Schmid, K. et al., 1973, "Structure of 1-acid
glycoprotein. The complete
amino acid sequence, multiple amino acid substitutions, and homology with the
immunoglobulins," published in Biochemistry 12 (14), 2711-2724; Schmid, K. el
al., 1974,
"The disulfide bonds of alphal-acid glycoprotein," published in Biochemistry
13 (13),
2694-2697; Dente, L. et al., 1987, "Structure and expression of the genes
coding for human
alpha 1-acid glycoprotein," published in EMBO J. 6 (8), 2289-2296, and the
amino acid
sequence of ORM2 (identified by accession no. NP 000599) is disclosed in,
e.g., Schmid,
K. et al., 1973, "Structure of 1-acid glycoprotein. The complete amino acid
sequence,
multiple amino acid substitutions, and homology with the immunoglobulins,"
published in
Biochemistry 12 (14), 2711-2724; Schmid, K. et al., 1974, "The disulfide bonds
of
alphal-acid glycoprotein," published in Biochemistry 13 (13), 2694-2697;
Dente, L. et al.,
1987, "Structure and expression of the genes coding for human alpha 1-acid
glycoprotein,"
published in EMBO J. 6(8), 2289-2296, each of which is incorporated by
reference herein
in its entirety.
The nucleotide sequence of PF4V 1(identified by accession no. NM 002620) is
disclosed in, e.g., Green, C.J. et al., 1989, "Identification and
characterization of PF4varl, a
human gene variant of platelet factor 4," published in Mol. Cell. Biol. 9(4),
1445-1451;
Eisman, R. et al., 1990, "Structural and functional comparison of the genes
for human
platelet factor 4 and PF4alt," published in Blood 76 (2), 336-344, and the
amino acid
sequence of PF4V 1(identified by accession no. NP_002611) is disclosed in,
e.g., Green, C.
J. et al., 1989, "Identification and characterization of PF4varl, a human gene
variant of
platelet factor 4," published in Mol. Cell. Biol. 9 (4), 1445-145 1; Eisman,
R. et al., 1990,
"Structural and functional comparison of the genes for human platelet factor 4
and PF4alt,"
, published in Blood 76 (2), 336-344, each of which is incorporated by
reference herein in its
entirety.
The nucleotide sequence of PIGR (identified by accession no. NM 002644) is
disclosed in, e.g., Mizoguchi, A. et al., 1982, "Structures of the
carbohydrate moieties of
secretory component purified from human milk," published in J. Biol. Chem. 257
(16),
-155-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
9612-9621; Hood, L. et al., 1985, "T cell antigen receptors and the
immunoglobulin
supergene family," published in Cell 40 (2), 225-229; Davidson, M. K. et al.,
1988,
"Genetic mapping of the human polymeric immunoglobulin receptor gene to
chromosome
region 1 q31----q41," published in Cytogenet. Cell Genet. 48 (2), 107-111, and
the amino
acid sequence of PIGR (identified by accession no. CAC10060) is disclosed in,
e.g.,
Schjerven, 2000, "Mechanism of IL-4-mediated up-regulation of the polymeric Ig
receptor:
role of STAT6 in cell type-specific delayed transcriptional response," J.
Immunol. 165 (7),
3898-3906, each of which is incorporated by reference herein in its entirety.
The nucleotide sequence of PLG (identified by accession no. NM 000301) is
disclosed in, e.g., Robbins, K.C. et al., 1967, "The peptide chains of human
plasmin.
Mechanism of activation of human plasminogen to plasmin," published in J.
Biol. Chem.
242 (10), 2333-2342; Deutsch, D.G. et al. 1970, "Plasminogen: purification
from human
plasma by affinity chromatography," published in Science 170 (962), 1095-1096;
Wiman,
B. et al., 1979, "On the mechanism of the reaction between human alpha 2-
antiplasmin and
plasmin," published in J. Biol. Chem. 254 (18), 9291-9297, and the amino acid
sequence of
PLG (identified by accession no. AAH60513) is disclosed in, e.g., Strausberg
et al., 2002,
"Generation and initial analysis of more than 15,000 full-length human and
mouse cDNA
sequences," Proc. Natl. Acad. Sci. U.S.A. 99 (26), 16899-16903, each of which
is
incorporated by reference herein in its entirety.
The nucleic acid sequence for plasminogen precursor is disclosed in, e.g.,
Forsgren
et al., 1987, FEBS Lett 213, 254-260, and amino acid sequence of plasminogen
precursor
(identified by accession no. P00747) is disclosed in, e.g., Petersen et al.,
"Characterization
of the gene for human plasminogen, a key proenzyme in the fibrinolytic system,
1990, J.
Biol. Chem. 265 (11), 6104-6111; and Forsgren et al., 1987, "Molecular cloning
and
characterization of a full-length cDNA clone for human plasminogen, FEBS Lett.
213 (2),
254-260, each of which is incorporated by reference herein in its entirety.
The nucleotide sequence of PON1 (identified by accession no. NM_000446) is
disclosed in, e.g., Ortigoza-Ferado, J. et al., 1984, "Paraoxon hydrolysis in
human serum
mediated by a genetically variable arylesterase and albumin," published in Am.
J. Hum.
Genet. 36 (2), 295-305; Gan, K.N. et al., 1991, "Purification of human serum
paraoxonase/arylesterase. Evidence for one esterase catalyzing both
activities," published in
Drug Metab. Dispos. 19 (1), 100-106; Hassett, C. et al., 1991,
"Characterization of cDNA
clones encoding rabbit and human serum paraoxonase: the mature protein retains
its signal
sequence," published in Biochemistry 30 (42), 10141-10149, and the amino acid
sequence
-156-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
of PON1 (identified by accession no. NP 000437) is disclosed in, e.g.,
Ortigoza-Ferado, J.
et al., 1984, "Paraoxon hydrolysis in human serum mediated by a genetically
variable
arylesterase and albumin," published in Am. J. Hum. Genet. 36 (2), 295-305;
Gan, K.N. et
al., 1991, "Purification of human serum paraoxonase/arylesterase. Evidence for
one esterase
catalyzing both activities," published in Drug Metab. Dispos. 19 (1), 100-106;
Hassett, C. et
al., 1991, "Characterization of cDNA clones encoding rabbit and human serum
paraoxonase: the mature protein retains its signal sequence," published in
Biochemistry 30
(42), 10141-10149, each of which is incorporated by reference herein in its
entirety.
The nucleotide sequence of PPBP (identified by accession no. NM 002704) is
disclosed in, e.g., Begg, G.S. et al., 1978, "Complete covalent structure of
human
beta-thromboglobulin," published in Biochemistry 17 (9), 1739-1744; Kaplan,
K.L., 1979,
"Platelet alpha-granule proteins: studies on release and subeellular
localization," published
in Blood 53 (4), 604-618; McLaren, K.M. et al., 1982, "Immunological
localisation of
beta-thromboglobulin and platelet factor 4 in human megakaryocytes and
platelets,"
published in J. Clin. Pathol. 35 (11), 1227-1231, and the amino acid sequence
of PPBP
(identified by accession no. CAG33086) is disclosed in, e.g., Ebert, L. et
al., 2004, Direct
Submission, RZPD Deutsches Ressourcenzentrum fuer Genomforschung GmbH, Im
Neuenheimer Feld 580, D-69120 Heidelberg, Germany; Ebert,L. et al.,
unpublished,
"Cloning of human full open reading frames in Gateway(TM) system entry vector
(pDONR201)," each of which is incorporated by reference herein in its
entirety.
The nucleotide sequence of RBP4 (identified by accession no. NM_006744) is
disclosed in, e.g., Rask, L. et al., 1971, "Studies on two physiological forms
of the human
retinol-binding protein differing in vitamin A and arginine content,"
published in J. Biol.
Chem. 246 (21), 6638-6646; Fex, G. et al., 1979, "Retinol-binding protein from
human
urine and its interaction with retinol and prealbumin," published in Eur. J.
Biochem. 94 (1),
307-313; Fex, G. et al., 1979, "Interaction between prealbumin and retinol-
binding protein
studied by affinity chromatography, gel filtration and two-phase partition,"
published in
Eur. J. Biochem. 99 (2), 353-360, and the amino acid sequence of RBP4
(identified by
accession no. CAH72328) is disclosed in, e.g., Brown, A., 2005, Direct
Submission,
Wellcome Trust Sanger Institute, Hinxton, Cambridgeshire, CB 10 1 SA, UK, each
of which
is incorporated by reference herein in its entirety.
The nucleotide sequence of RIMS 1(identified by accession no. NIVI 014989) is
disclosed in, e.g., Kelsell, R.E. et al., 1998, "Localization of a gene
(CORD7) for a
dominant cone-rod dystrophy to chromosome 6q," published in Am. J. Hum. Genet.
63 (1),
- 157 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
274-279; Betz, A. et al., 2001, "Functional interaction of the active zone
proteins Munc13-1
and RIMI in synaptic vesicle priming," published in Neuron 30 (1), 183-196;
Coppola, T. et
al., 2001, "Direct interaction of the Rab3 effector RIM with Ca2+ channels,
SNAP-25, and
synaptotagmin," published in J. Biol. Chem. 276 (35), 32756-32762, and the
amino acid
sequence of RIMS1 (identified by accession no. NP_055804) is disclosed in,
e.g., Kelsell,
R.E. et al., 1998, "Localization of a gene (CORD7) for a dominant cone-rod
dystrophy to
chromosome 6q," published in Am. J. Hum. Genet. 63 (1), 274-279; Betz, A. et
al., 2001,
"Functional interaction of the active zone proteins Munc13-1 and RIM1 in
synaptic vesicle
priming," published in Neuron 30 (1), 183-196; Coppola, T. et al., 2001,
"Direct interaction
of the Rab3 effector RIM with Ca2+ channels, SNAP-25, and synaptotagmin,"
published in
J. Biol. Chem. 276 (35), 32756-32762, each of which is incorporated by
reference herein in
its entirety.
The nucleotide sequence of RNF6 (identified by accession no. NM 005977) is
disclosed in, e.g., Macdonald, D.H. et al., 1999, "Cloning and
characterization of RNF6, a
novel RING finger gene mapping to 13q12 published in Genomics 58 (1), 94-97;
Lopez,
P., et al., 2002, "Gene control in germinal differentiation: RNF6, a
transcription regulatory
protein in the mouse sertoli cell" published in Mol. Cell. Biol. 22 (10), 3488-
3496; Lo, H.S.
et al., 2002, "Identification of somatic mutations of the RNF6 gene in human
esophageal
squamous cell carcinoma" published in Cancer Res. 62 (15), 4191-4193, and the
amino acid
sequence of RNF6 (identified by accession no. CAH73183) is disclosed in, e.g.,
Tromans,
A. et al., 2004, Direct Submission, Wellcome Trust Sanger Institute, Hinxton,
Cambridgeshire, CB 10 1 SA, UK, each of which is incorporated by reference
herein in its
entirety.
The nucleotide sequence of SEMA3D (identified by accession no. N1V1 152754) is
disclosed in, e.g., Scherer, S.W. et al., 2003, "Human chromosome 7: DNA
sequence and
biology" published in Science 300 (5620), 767-772; Clark, H.F., et al., 2003,
"The secreted
protein discovery initiative (SPDI), a large-scale effort to identify novel
human secreted and
transmembrane proteins:a bioinformatics assessment" published in Genome Res.
13 (10),
2265-2270; and the amino acid sequence of SEMA3D (identified by accession no.
EAL24I84) is disclosed in, e.g., Scherer et al., 2003, "Human chromosome 7:
DNA
sequence and biology," Science 300 (5620), 767-772, each of which is
incorporated by
reference herein in its entirety.
The nucleotide sequence of SF3B1 (identified by accession no. N1V1 012433) is
disclosed in, e.g., Wang, C. et al., 1998, "Phosphorylation of spliceosomal
protein SAP 155
-158-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
coupled with splicing catalysis," published in Genes Dev. 12 (10), 1409-1414;
Pauling,
M.H. et al. 2000, "Functional Cusl p is found with Hsh155p in a multiprotein
splicing factor
associated with U2 snRNA" published in Mol. Cell. Biol. 20 (6), 2176-2185;
Will, C.L. et
al., 2001, "A novel U2 and U11/U12 snRNP protein that associates with the pre-
mRNA
branch site" published in EMBO J. 20 (16), 4536-4546, and the amino acid
sequence of
SF3B1 (identified by accession no. NP_006833) is disclosed in, e.g., Gozani,
O. et aL,
1996, "Evidence that sequence-independent binding of highly conserved U2 snRNP
proteins upstream of the branch site is required for assembly of spliceosomal
complex
A"published in Genes Dev. 10 (2), 233-243; Agell,N., et al., 1998, "New
nuclear functions
for calmodulin" published in Cell Calcium 23 (2-3), 115-121; Das,B.K., et al.,
1999,
"Characterization of a protein complex containing spliceosomal proteins SAPs
49, 130, 145,
and 155 published in Mol. Cell. Biol. 19 (10), 6796-6802, each of which is
incorporated by
reference herein in its entirety.
The nucleotide sequence of SPINK 1 (identified by accession no. NIVI 043122)
is
disclosed in, e.g., Huhtala, M.L. et al.; 1982, inhibitor from the urine of a
patient with =
ovarian cancer," published in J. Biol. Chem. 257 (22), 13713-13716;
Yamamoto,T. et al.,
1985, "Molecular cloning and nucleotide sequence of human pancreatic secretory
trypsin
inhibitor (PSTI) eDNA" published in Biochem. Biophys. Res. Commun. 132 (2),
605-612;
Horii,A:, et al., 1987, "Primary structure of human pancreatic secretory
trypsin
inhibitor(PSTI) gene" published in Biochem. Biophys. Res. Commun. 149 (2), 635-
641,
and the amino acid sequence of SPINKI (identified by accession no. NP 003113)
is
disclosed in, e.g., Huhtala, M.L. et al., 1982, inhibitor from the urine of a
patient with
ovarian cancer," published in J. Biol. Chem. 257 (22), 13713-13716; Yamamoto,
T. et al.,
1985, "Molecular cloning and nucleotide sequence of human pancreatic secretory
trypsin
inhibitor (PSTI) cDNA " published in Biochem. Biophys. Res. Commun. 132 (2),
605-612;
Horii, A. et al., 1987, "Primary structure of human pancreatic secretory
trypsin inhibitor
(PSTI) gene" published in Biochem. Biophys. Res. Commun. 149 (2), 635-641,
each of
which is incorporated by reference herein in its entirety.
The nucleotide sequence of SPP1 (identified by accession no. NM_000582) is
disclosed in, e.g., Kiefer, M.C. et al., 1989, "The cDNA and derived amino
acid sequence
for human osteopontin" published in Nucleic Acids Res. 17 (8), 3306; Nemir,
M., et al.,
1989, "Normal rat kidney cells secrete both phosphorylated and
nonphosphorylated forms
of osteopontin showing different physiological properties" published in J.
Biol. Chem. 264
(30), 18202-18208; Fisher, L.W. et al., "Human bone sialoprotein. Deduced
protein
-159-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
sequence and chromosomal localization" published in J. Biol. Chem. 265 (4),
2347-2351,
and the amino acid sequence of SPP1 (identified by accession no. AAH17387) is
disclosed
in, e.g., Strausberg, R.L. et al., 2002, "T Generation and initial analysis of
more than 15,000
full-length human and mouse cDNA sequences published in Proc. Natl. Acad. Sci.
U.S.A.
99 (26), 16899-16903, each of which is incorporated by reference herein in its
entirety.
The nucleotide sequence of SPTB (identified by accession no. NM 001024858) is
disclosed in, e.g., Carlier, M.F. et al., 1984, "Interaction between
microtubule-associated
protein tau and spectrin" published in Biochimie 66 (4), 305-311; Prchal,
J.T., et al.,1987,
"Isolation and characterization of cDNA clones for human erythrocyte beta-
spectrin"
published in Proc. Natl. Acad. Sci. U.S.A. 84 (21), 7468-7472; Winkelmann,
J.C. el al.,
1988, "Molecular cloning of the cDNA for human erythrocyte beta-spectrin"
published in
Blood 72 (1), 328-334, and the amino acid sequence of SPTB (identified by
accession no.
BAD92652) is disclosed in, e.g., Totoki, Y. et al., 2000, Direct Submission,
Osamu Ohara,
Kazusa DNA Research Institute, Department of Human Gene Research; 2-6-7
Kazusa-kamatari, Kisarazu, Chiba, 292-0818, Japan, each of which is
incorporated by
reference herein in its entirety.
The nucleotide sequence of SYNE 1(identified by accession no. NM_182961) is
disclosed in, e.g., Zhang, Q. et al., "Nesprins: a novel family of spectrin-
repeat-containing
proteins that localize to the nuclear membrane in multiple tissues," J. Cell.
Sci. 114 (PT 24),
4485-4498 (2001); Apel, E.D. et al., "Syne-1, a dystrophin- and Klarsicht-
related protein
associated with synaptic nuclei at the neuromuscular junction," J. Biol. Chem.
275 (41),
31986-31995 (2000); Nedivi, E., et al., "A set of genes expressed in response
to light in the
adult cerebral cortex and regulated during development," Proc. Natl. Acad.
Sci. U.S.A. 93
(5), 2048-2053 (1996), and the amino acid sequence of SYNE1 (identified by
accession no.
AAH39121) is disclosed in, e.g., Strausberg, R.L. et al., "Generation and
initial analysis of
more than 15,000 full-length human and mouse cDNA sequences," Proc. Natl.
Acad. Sci.
U.S.A. 99 (26), 16899-16903 (2002), each of which is incorporated by reference
herein in
its entirety.
The nucleotide sequence of TAF4B (identified by accession no. NM_003187) is
disclosed in, e.g., Parada, C.A., et al., "A novel LBP-1-mediated restriction
of HIV-1
transcription at the level of elongation in vitro," J. Biol. Chern. 270 (5),
2274-2283 (1995);
Zhou and Sharp, "Novel mechanism and factor for regulation by HIV-1 Tat," EMBO
J. 14
(2), 321-328 (1995); Ou et al., "Role of flanking E box motifs in human
immunodeficiency
virus type 1 TATA element function," J. Virol. 68 (11), 7188-7199 (1994);
Kashanchi, F. et
- 160 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
al. "Direct interaction of human TFIID with the HIV-1 transactivator tat,"
Nature 367
(6460), 295-299 (1994), each of which is hereby incorporated herein by
reference in its
entirety and the amino acid sequence of TAF4B (identified by accession no. XP
290809) is
predicted by automated computational analysis of the annotated genomic
sequence
(NT 010966) using gene prediction method: GNOMON.
The nucleotide sequence of TBC1D1 (identified by accession no. NM 015173) is
disclosed in, e.g., White, R.A. et al., 2000, "The gene encoding TBCIDI with
homology to
the tre-2/USP6 oncogene, BUB2, and cdcl6 maps to mouse chromosome 5 and human
chromosome 4 published in Cytogenet. Cell Genet. 89 (3-4), 272-275; and the
amino acid
sequence of TBC1D1 (identified by accession no. NP 055988) is disclosed in,
e.g., White,
R.A. et al., 2000, "The gene encoding TBC1D1 with homology to the tre-2/USP6
oncogene, BUB2, and cdc 16 maps to mouse chromosome 5 and human chromosome 4,"
published in Cytogenet. Cell Genet. 89 (3-4), 272-275, each of which is
incorporated by
reference herein in its entirety.
The nucleotide sequence of TLN1 (identified by accession no. NM 006289) is
disclosed in, e.g., Kupfer et al., 1990, "The PMA-induced specific association
of LFA-1 and
talin in intact cloned T helper cells" published in J. Mol. Cell. Immunol. 4
(6), 317-325;
Luna, E. J. et al., 1992, "Cytoskeleton--plasma membrane interactions"
published in
Science 258 (5084), 955-964; Salgia, R. et al., 1995, "Molecular cloning of
human paxillin,
a focal adhesion protein phosphorylated by P210BCR/ABL" published in J. Biol.
Chem.
270 (10), 5039-5047, and the amino acid sequence of TLN1 (identified by
accession no.
NP_006280) is disclosed in, e.g., Kupfer, A. et al., 1990, "The PMA-induced
specific
association of LFA-1 and talin in intact cloned T helper cells" published in
J. Mol. Cell.
Immunol. 4 (6), 317-325; Luna, E.J. et al., 1992, "Cytoskeleton--plasma
membrane
interactions" published in Science 258 (5084), 955-964; Salgia, R. et al.,
1995, "Molecular
cloning of human paxillin, a focal adhesion protein phosphorylated by
P210BCR/ABL"
published in J. Biol. Chem. 270 (10), 5039-5047, each of which is incorporated
by reference
herein in its entirety.
The nucleotide sequence of TMSB4X (identified by accession no. NM_021109) is
disclosed in, e.g., Erickson-Viitanen, S. et al., 1983, "Distribution of
thymosin beta 4 in
vertebrate classes" published in Arch. Biochem. Biophys. 221 (2), 570-576;
Friedman, R.L.
et al., 1984, "Transcriptional and posttranscriptional regulation of
interferon-induced gene
expression in human cells" published in Cell 38 (3), 745-755; Soma, G. et al.,
1985,
"Detection of a countertranscript in promyelocytic leukemia cells HL60 during
early
-161-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
differentiation by TPA" published in Biochem. Biophys. Res. Commun. 132 (1),
100-109,
and the amino acid sequence of TMSB4X (identified by accession no. NP 066932)
is
disclosed in, e.g., Erickson-Viitanen, S. et al., 1983, "Distribution of
thymosin beta 4 in
vertebrate classes" published in Arch. Biochem. Biophys. 221 (2), 570-576;
Friedman, R.L.
et al., 1984, "Transcriptional and posttranscriptional regulation of
interferon-induced gene
expression in human cells" published in Cel138 (3), 745-755; Soma, G. et al.,
1985,
"Detection of a countertranscript in promyelocytic leukemia cells HL60 during
early
differentiation by TPA" published in Biochem. Biophys. Res. Commun. 132 (1),
100-109,
each of which is incorporated by reference herein in its entirety.
The nucleotide sequence of UROC1 (identified by accession no. NM_144639) is
disclosed in, e.g., Yamada, S. et al., 2004, "Expression profiling and
differential screening
between hepatoblastomas and the corresponding normal livers: identification of
high
expression of the PLK1 oncogene as a poor-prognostic indicator of
hepatoblastomas"
published in Oncogene 23 (35), 5901-5911, and the amino acid sequence of UROC1
(identified by accession no. NP_653240) is disclosed in, e.g., Yamada, S. et
al., 2004,
"Expression profiling and differential screening between hepatoblastomas and
the
corresponding normal livers: identification of high expression of the PLK1
oncogene as a
poor-prognostic indicator of hepatoblastomas" published in Oncogene 23 (35),
5901-5911,
each of which is incorporated by reference herein in its entirety.
The nucleotide sequence of ZFHX2 (identified by accession no. NM_033400) is
disclosed in, e.g., Nagase, T.'et al., 2000, "Prediction of the coding
sequences of
unidentified human genes. XIX. The complete sequences of 100 new cDNA clones
from
brain which code for large proteins in vitro" published in DNA Res. 7 (6), 347-
355; and the
amino acid sequence of ZFHX2 (identified by accession no. NP_207646) is
disclosed in,
e.g., Nagase, T. et al., 2000, "Prediction of the coding sequences of
unidentified human
genes. XIX. The complete sequences of 100 new cDNA clones from brain which
code for
large proteins in vitro" published in DNA Res. 7 (6), 347-355, each of which
is incorporated
by reference herein in its entirety.
The nucleotide sequence of ZYX (identified by accession no. NM_003461) is
disclosed in, e.g., Wang, L.F. et al., 1994, "Gene encoding a mammalian
epididymal
protein" published in Biochem. Mol. Biol. Int. 34 (6), 1131-1136; Reinhard, M.
et al., 1995,
"Identification, purification, and characterization of a zyxin-related protein
that binds the
focal adhesion and microfilament protein VASP (vasodilator-stimulated
phosphoprotein),"
Proc. Natl. Acad. Sci. U.S.A. 92 (17), 7956-7960; Hobert, O. et al., 1996,
"SH3
- 162 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
domain-dependent interaction of the proto-oncogene product Vav with the focal
contact
protein zyxin" published in Oncogene 12 (7), 1577-1581, and the amino acid
sequence of
ZYX (identified by accession no. NP 001010972) is disclosed in, e.g., Wang,
L.F. et erl.,
1994, "Gene encoding a mammalian epididymal protein" published in Biochem.
Mol. Biol.
Int. 34 (6), 1131-1136; Reinhard, M. et al., 1995, "Identification,
purification, and
characterization of a zyxin-related protein that binds the focal adhesion and
microfilament
protein VASP (vasodilator-stimulated phosphoprotein)" Proc. Natl. Acad. Sci.
U.S.A. 92
(17), 7956-7960; Hobert, O. et al., 1996, "SH3 domain-dependent interaction of
the proto-
oncogene product Vav with the focal contact protein zyxin" published in
Oncogene 12 (7),
1577-1581, each of which is incorporated by reference herein in its entirety.
5.6.2 Isolation of Useful Biomarkers
The biomarkers of the present invention may, for example, be used to raise
antibodies that bind the biomarker if it is a protein (using methods described
herein, or any
method well known to those of skill in the art), or they may be used to
develop a specific
oligonucleotide probe, if it is a nucleic acid, for example, using a method
described herein,
or any method well known to those of skill in the art. The skilled artisan
will readily
appreciate that useful features can be further characterized to determine the
molecular
structure of the biomarker. Methods for characterizing biomarkers in this
fashion are well-
known in the art and include X-ray crystallography, high-resolution mass
spectrometry,
irzfrared spectrometry, ultraviolet spectrometry and nuclear magnetic
resonance. Methods
for determining the nucleotide sequence of nucleic acid biomarkers, the amino
acid
sequence of polypeptide biomarkers, and the composition and sequence of
carbohydrate
biomarkers also are well-known in the art.
5.7 APPLICATION OF THE PRESENT INVENTION TO SIRS
SUBJECTS
In one embodiment, the presently described methods are used to screen SIRS
subjects who are at risk for developing sepsis. A one or more biological
samples are taken
from a SIRS-positive subject at one or more different time points and used to
construct a
biomarker profile. The biomarker profile is then evaluated to determine
whether the feature
values of the biomarker profile satisfy a first value set associated with a
particular decision
rule. This evaluation classifies the subject as a converter or a nonconverter.
A treatment
-163-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
regimen may then be initiated to forestall or prevent the progression of
sepsis when the
subject is classified as a converter.
5.8 APPLICATION OF THE PRESENT INVENTION TO STAGES
OF SEPSIS
In one embodiment, the presently described methods are used to screen subjects
who
are particularly at risk for developing a certain stage of sepsis. A
biological sample is taken
from a subject and used to construct a biomarker profile. The biomarker
profile is then
evaluated to determine whether the feature values of the biomarker profile
satisfy a first
value set associated with a particular decision rule. This evaluation
classifies the subject as
having or not having a particular stage of sepsis. A treatment regimen may
then be initiated
to treat the specific stage of sepsis. In some embodiments, the stage of
sepsis is for
example, onset of sepsis, severe sepsis, septic shock, or multiple organ
dysfunction.
5.9 EXEMPLARY EMBODIMENTS
In some embodiments of the present invention, a biomarker profile is obtained
using
a biological sample from a test subject, particularly a subject at risk of
developing sepsis,
having sepsis, or suspected of having sepsis. The biomarker profile in such
embodiments is
evaluated. This evaluation can be made, for example, by applying a decision
rule to the test
subject. The decision rule can, for example, be or have been constructed based
upon the
biomarker profiles obtained from subjects in the training population. The
training
population, in one embodiment, includes (a) subjects that had SIRS and were
then
diagnosed as septic during an observation time period as well as (b) subjects
that had SIRS
and were not diagnosed as septic during an observation time period. If the
biomarker
profile from the test subject contains appropriately characteristic features,
then the test
subject is diagnosed as having a more likely chance of becoming septic, as
being afflicted
with sepsis or as being at the particular stage in the progression of sepsis.
Various
populations of subjects including those who are suffering from SIRS (e.g.,
SIRS-positive
subjects) or those who are suffering from an infection but who are not
suffering from SIRS
(e.g., SIRS-negative subjects) can serve as training populations. Accordingly,
the present
invention allows the clinician to distinguish, inter alia, between those
subjects who do not
have SIRS, those who have SIRS but are not likely to develop sepsis within a
given time
- 164 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
frame, those who have SIRS and who are at risk of eventually becoming septic,
and those
who are suffering from a particular stage in the progression of sepsis.
5.10 USE OF ANNOTATION DATA TO IDENTIFY
DISCRIMINATING BIOMARKERS
In some embodiments, data analysis algorithms identify a large set of
biomarkers
whose features discriminate between converters and nonconverters. For example,
in some
embodiments, application of a data analysis algorithm to a training population
results in the
selection of more than 500 biomarkers, more than 1000 biomarkers, or more than
10,000
biomarkers. In some embodiments, further reduction in the number of biomarkers
that are
deemed to be discriminating is desired. Accordingly, in some embodiments,
filtering rules
that are complementary to data analysis algorithms (e.g., the data analysis
algorithms of
Section 5.5) are used to further reduce the list of discriminating biomarkers
identified by the
data analysis algorithms. Specifically, the list of biomarkers identified by
application of one
or more data analysis algorithms to the biomarker profile data measured in a
training
population is further refined by application of annotation data based
filtering rules to the
list. In such embodiments, those biomarkers in the set of biomarkers
identified by the one
or more data analysis algorithms that satisfy the one or more applied
annotation data based
filtering rules remain in the set of discriminating biomarkers. In some
instances, those
biomarkers in the set of biomarkers identified by the one or more data
analysis algorithms
that do not satisfy the one or more applied annotation data based filtering
rules are removed
from the set. In other instances, those biomarkers in the set of biomarkers
identified by the
one or more data analysis algorithms that do not satisfy the one or more
applied annotation
data based filtering rules stay in the set and those that satisfy the one or
more applied
annotation data based filtering rules are removed from the set. In this way,
annotation data
can be used to reduce the number of biomarkers in the set of discriminating
biomarkers
identified by the data analysis algorithms.
Annotation data based filtering rules are rules based upon annotation data.
Annotation data refers to any type of data that describes a property of a
biomarker. An
example of annotation data is the identification of biological pathways to
which a given
biomarker belongs. Another example of annotation data is enzymatic class
(e.g.,
phosphodiesterases, kinases, metalloproteinases, etc.). Still other examples
of annotation
data include, but are not limited to, protein domain information, enzymatic
substrate
-165-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
information, enzymatic reaction information, and protein interaction data. Yet
another
example of annotation data is disease association, in other words, which
disease process a
given biomarker has been linked to or otherwise affects. Another form of
annotation data is
any type of data that associates biomarker expression, other forms of
biomarker abundance,
and/or biomarker activity, with cellular localization, tissue type
localization, and/or cell type
localization.
As the name implies, annotation data is used to construct an annotation data
based
filtering rule. An example of an annotation data based filtering rule is:
Annotation rule 1:
remove all transcription factors from the training set.
Application of this filtering rule to a set of biomarkers will remove all
transcription
factors from the set.
Another type of annotation data based filtering rule is:
Annotation rule 2:
keep all biomarkers that are enriched for annotation X
in a biomarker list.
Application of this filtering rule will only keep those biomarkers in a given
list that
are enriched (overrepresented) for annotation X in the list. To more fully
appreciate this
filtering rule, consider an exemplary biomarker set that has been identified
by application of
a data analysis algorithm (Section 5.5) to biomarker profiles measured using
training
population data measured in accordance with a technique disclosed in Section
5.4. This
exemplary biomarker set has 500 biomarkers. Assume, for in this illustrative
example, that
the full set of biomarkers in a human consists of 25,000 biomarkers. Here, the
25,000
biomarkers is a population and the 500 biomarker set is the sample. As used
here, the term
"population" consists of all possible observable biomarkers. The term
"sarnple" is the data
that is actually considered. Now, for this example, let X = kinases. Suppose
there are 800
known human kinases and fiarther suppose that the set of 500 biomarkers was
randomly
selected with respect to kinases. Under these circumstances, the list of 500
biomarkers
identified by the data analysis algorithms should select about (500 / 25,000)
* 800 = 16
- 166 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
kinases. Since there are, in fact, 50 kinases in the sample, a conclusion can
be reached that
kinases are indeed enriched in the sample relative to the population.
More formally, in this example, a deterrnination can be made as to whether
kinases
are enriched in the set of biomarkers identified by the data analysis
algorithm (the sample)
relative to the population by analysis of the two-way contingency table that
describes the
observed sample and population:
Kinase
Group Yes No Total
Population 800 24,200 25,000
Sample 50 450 500
Following Agresti, 1996, An Introduction to Categorical Data Analysis, John
Wiley
& Sons, New York, which is hereby incorporated by reference in its entirety,
this two-way
contingency table can be analyzed by treating each row as an independent
bionomial
variable. In such instances, the true difference in proportions, termed 7c, -
7c2, compares the
probabilities in the two rows. This difference falls between -1 and +1. It
equals zero when
IC1 =TEz, that is, when the selection of kinases in the sample from the
population is
independent of the kinase annotation. Of the NI = 25,000 biomarkers in the
population, 800
are kinases, a proportion ofpl = 800 / 25,000 = 0.032. Of the N2 = 500
biomarkers in the
sample identified using a data analysis algorithm, 50 are kinases, a
proportionpz of 50/500
= 0.10. The sample difference of proportions is 0.032 - 0.10 =-0.068. In
accordance with
Agresti, when the counts in the two rows are independent binomial samples, the
estimated
standard error ofpl -P2 is:
a'(Pi -Pa)= Pz (1 - P2)
N, N2
where N, and N2 are the samples sizes for the population and the sample
selected by
data analysis algorithm, respectively. The standard error decreases, and hence
the estimate
of 7ci -n2 improves, as the sample sizes increase. A large-sample (100(1 -
a))% confidence
interval for n, - 7t2 is
(Pi + Pa ) -!" Z ~,2/ _ Zo.o2s = 1.96
Thus, for this example, the estimated error is
- 167 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
0.032(1- 0.032) + 0.10(1- 0.10) = 0.013
25,000 500
and a 95% confidence interval for the true difference n, - 7r2 is -0.068
1.96(0.013), or -0.068 0.025. Since the 95% confidence interval contains
only negative
values, the conclusion can be reached that kinases are enriched in the sample
(the biomarker
set produced by the data analysis algorithm) relative to the population of
25,000 biomarkers.
The two-way contingency table in the example above can be analysed using
methods known in the art other than the one disclosed above. For example, the
chi-square
test for independence and/or Fisher's exact test can be used to test the null
hypothesis that
the row and column classification variables of the two-way contingency table
are
independent.
The term "X" in annotation rule 2 can be any form of annotation data. In one
embodiment, "X" is any biological pathway. As such the annotation data based
filtering
rule has the following form.
Annotation rule 3:
Select all biomarkers that are in any biological pathway
that is enriched in the biomarker list.
To determine whether a particular biological pathway is enriched, the number
of
biomarkers in a particular biological pathway in the sample is compared with
the number of
biomarkers that are in the particular biological pathway in the population
using, for
example, the two-way contingency table analysis described above, or other
techniques
known in the art. If the biological pathway is enriched in the sample, then
all biomarkers in
the sample that are also in the biological pathway are retained for further
analysis, in
accordance with the annotation data based filtering rule.
An example of enrichment, in which it was shown that the proportion of kinases
in
the sample was greater than the proportion of kinases in the population across
its entire 95%
confidence interval has been given. In one embodiment, biomarkers having a
given
annotation are considered enriched in the sample relative to the population
when the
proportion of biomarkers having the annotation in the sample is greater than
the proportion
of biomarkers having the annotation in the population across its entire 95%
degree
confidence interval as determined by two-way contingency table analysis. In
another
-168-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
embodiment, biomarkers having a given annotation are considered enriched in
the sample
relative to the population if ap value as determined by the Fisher exact test,
Chi-square test,
or relative algorithms is 0.05 or less, 0.005 or less or 0.0005 or less.
Another form of annotation data based filtering rule has the following form:
Annotation rule 4:
Select all biomarkers that are in biological pathway X.
In an embodiment, a set of biomarkers is determined using a data analysis
algorithm.
Exemplary data analysis algorithms are disclosed in Section 5.5. In addition,
Section 6
describes certain tests that can also serve as data analysis algorithms. These
tests include,
but are not limited to a Wilcoxon test and the like with a statistically
significantp value
(e.g., 0.05 or less, 0.04, or less, etc.), and/or a requirement that a
biomarker exhibit a mean
differential abundance between biological samples obtained from converters and
biological
samples obtained from nonconverters in a training population. Upon application
of the data
analysis algorithm, a set of biomarkers that discriminates between converters
and
nonconverters is determined. Next, an annotation rule, for exarnple annotation
rule 4, is
applied to the set of discriminating biomarkers in order to further reduce the
set of
biomarkers. Those of skill in the art will appreciate that the order in which
these rules are
applied is generally not important. For example, annotation rule 4 can be
applied first and
then certain data analysis algorithms can be applied, or vice versa. In some
embodiments,
biomarkers ultimately deemed as discriminating between converters and
nonconverters
satisfy each of the following criteria: (i) ap value of 0.05 or less (p <
0.05) as determined
from a Wilcoxon adjusted test using static (single time point) data; (ii) a
mean-fold change
of 1.2 or greater between converters and nonconverters across the training set
using static
(single time point data), and (iii) present in a specific biological pathway.
See also, Section
6.7, infra, for a detailed example. In this example, there is no requirement
that members of
the pathway are enriched in the set of biomarkers identified by the data
analysis algorithms.
Furthermore, it is noted that criteria (i) and (ii) are forms of data analysis
algorithms and
criterion (iii) is a annotation data based filtering rule.
In another embodiment, once a list of discriminating biomarkers is identified,
the
biomarkers can then be used to determine the identity of the particular
biological pathways
from which the discriminating biomarkers are implicated. In certain
embodiments,
annotation data-based filtering rules are applied to the list of
discriminating biomarkers
- 169 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
identified by the methods of the present invention (e.g., the methods
described in Sections
5.4, 5.5 and 6). Such annotation data-based filtering rules identify the
particular biological
pathway or pathways that are enriched in the discriminating list of biomarkers
identified by
the data analysis algorithms. In an exemplary embodiment of the invention,
DAVID 2.0
software is used to identify and apply such annotation data-based filtering
rules to the set of
biomarkers identified by the data analysis algorithms in order to identify
pathways that are
enriched in the set. In some embodiments, those biomarkers that are in an
enriched
biological pathway are selected for use as discriminating biomarkers in the
kits of the
present invention.
In some embodiments of the present invention, biomarkers that are in
biological
pathways that are enriched in the biomarker set determined by application of a
data analysis
algorithm to a training population that includes converters and nonconverters
can be used as
filtering step to reduce the number of biomarkers in the set. In one such
approach,
biological samples from subjects in a training population are obtained using,
e.g., any of one
or more of the methods described in Section 5.4, supra, and in Section 6,
infra. In
accordance with this embodiment, a nucleic acid array, such as a cDNA
microarray, may be
employed to generate features of biomarkers in a biomarker profile by
detecting the
expression of any one or more of the genes known to be or suspected to be
involved in the
selected biological pathways. Data derived from the cDNA microarray analysis
may then
be analyzed using any one or more of the analysis algorithms described in
Section 5.5,
supra, to identify biomarkers whose features discriminate between converters
and
nonconverters. Biomarkers whose corresponding feature values are capable of
discriminating, for example, between converters (i.e., SIRS patients who
subsequently
develop sepsis) and non-converters (i.e., SIRS patients who do not
subsequently develop
sepsis) can thus be identified and classified as discriminating biomarkers.
Biomarkers that
are in enriched biological pathways can be selected from this set by applying
Annotation
rule 3, from above. Representative biological pathways that could be found
include, for
example, genes involved in the Thl/Th2 cell differentiation pathway). In one
embodiments,
biomarkers ultimately deemed as discriminating between converters and
nonconverters
satisfy each of the following criteria: (i) ap value of 0.05 or less (p <
0.05) as determined
from a Wilcoxon adjusted test; (ii) a mean-fold change of 1.2 or greater
between converters
and nonconverters across the training set, and (iii) present in a biological
pathway that is
enriched in the set of biomarkers derived by application of criteria (i) and
(ii).
- 170 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
In some embodiments of the present invention, annotation data based filtering
rules
are used to identify biological pathways that are enriched in a given
biomarker set. This
biomarker set can be, for example, a set of biomarkers that is identified by
application of a
data analysis algorithm to training data comprising converters and
nonconverters. Then,
biomarkers in these enriched biological pathways are analyzed using any of the
data
analysis algorithms disclosed herein in order to identify biomarkers that
discriminate
between converters and nonconverters. In some instances, some of the
biomarkers analyzed
in the enriched biological pathways were not among the biomarkers in the
original given
biomarker set. In some instances, some of the biomarkers in the enriched
biological
pathways are among the biomarkers in the original given biomarker set. In some
embodiments, a secondary assay is used to collect feature data for biomarkers
that are in
enriched pathways and it is this data that is used to determine whether the
biomarkers in the
enriched biological pathways discriminate between converters and
nonconverters.
In some embodiments, biomarkers in biological pathways of interest are
identified.
In one example, genes involved in the Th1/Th2 cell differentiation pathway are
identified.
Then, these biomarkers are evaluated using the data analysis algorithms
disclosed herein to
determine whether they discriminate between converters and nonconverters.
6. EXAMPLES
SIRS positive subjects admitted to an ICU were recruited for the study.
Subjects
were eighteen years of age or older and gave informed consent to comply with
the study
protocol. Subjects were excluded from the study if they were (i) pregnant,
(ii) taking
antibiotics to treat a suspected infection, (iii) were taking systemic
corticosteroids (total
dosage greater than 100 mg hydrocortisone or equivalent in the past 48 hours
prior to study
entry), (iv) had a spinal cord injury or other illness requiring high-dose
corticosteroid
therapy, (v) pharmacologically immunosuppressed (e.g., azathioprine,
methotrexate,
cyclosporin, tacrolimus, cyclophosphamide, etanercept, anakinra, infliximab,
leuflonamide,
mycophenolic acid, OKT3, pentoxyphylin, etc.), (vi) were an organ transplant
recipient,
(vii) had active or metastatic cancer, (viii) had received chemotherapy or
radiation therapy
within eight weeks prior to enrollment, and/or (ix) had taken investigational
use drugs
within thirty days prior to enrollment.
In the study SIRS criteria were evaluated daily. APACHE II and SOFA scoring
was
performed following ICU admission. APACHE II is a system for rating the
severity of
medical illness. APACHE stands for "Acute Physiology and Chronic Health
Evaluation,"
and is most frequently used to predict in-hospital death for patients in an
intensive care unit.
-171-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
See, for example, Gupta et al., 2004, Indian Journal of Medical Research 119,
273-282,
which is hereby incorporated herein by reference in its entirety. SOFA is a
test to measure
the severity of sepsis. See, for example, Vincent et aL, 1996, Intensive Care
Med. 22, 707-
710, which is hereby incorporated herein by reference in its entirety.
Patients were
monitored daily for up to two weeks for clinical suspicion of sepsis
including, but not
limited to, any of the following signs and symptoms:
= pneumonia: temperature > 38.3 C or <36 C + white blood cell count (WBC) >
12,000/mm3 or < 4,000/mm3 + new-onset of purulent sputum + new or progressive
infiltrate
on chest radiograph (3 out of 4 findings);
= wound infection: temperature > 38.3 C or < 36 C + pain + erythema + purulent
discharge (3 out of 4 findings);
= urinary tract infection: temperature > 38.3 C or WBC > 12,000/mm3 or <
4,000/mm3 + bacteruria and pyuria (>10 WBC/hpf or positive leukocyte esterase)
(all
findings);
= line sepsis: temperature > 38.3 C or < 36 C + erythema / pain /purulence at
catheter exit site (3 out of 4 findings, including fever);
= intra-abdominal abscess: temperature > 38.3 C or < 36 C + WBC > 12,000/mm3
or
<4,000/mm3 + radiographic evidence of fluid collection (2 out of 3 criteria);
= CNS Infection: temperature > 38.3 C or < 36 C + WBC > 12,000/mm3 or
<4,000/mm3 + CSF pleocytosis via LP or Ventricular drainage.
Blood was drawn daily for a minimum of four consecutive days beginning within
24
hours following study entry. Patients were followed and blood samples were
drawn daily
for a maximum of fourteen consecutive days unless clinical suspicion of
infection occurred.
The maximum volume of blood drawn from any one subject did not exceed 210 mL
over
the course of a fourteen day study maximum. Blood draws for the study were
discontinued
if the loss of blood posed risk to the patient as defined by physician's
judgment. Each
patient had two Paxgene (RNA) tubes drawn on each day.
Blood samples were collected in PreAnalytiX PAXgeneTM blood collection tubes
from male and female patients over 18 years of age admitted to a trauma center
or ICU. All
patients were at risk of sepsis based on meeting two of four SIRS criteria.
Blood samples
were drawn each day for a minimum of four and maximum of fourteen days. Thus,
plasma
samples were collected prospectively upon admission to an ICU and divided into
septic
versus SIRS patients retrospectively based on whether they developed
infection. Sepsis
-172-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
samples represent time points prior to the clinical diagnosis of sepsis and
were compared to
time-matched uninfected SIRS patients.
The protein profiling experiment was divided into two separate parts. Part I
examined plasma for differentially expressed proteins between Sepsis and SIRS
pooled
samples using three-dimensional LC fractionation with electrospray ion trap
mass
spectrometry (3D LC-MS/MS). Two rounds (batches) of plasma samples were pooled
at
study time points "DOE (Day of Entry)", T_60 and T-12. An additional round
(batch) also
included a pool from the T.36 time point for a total of three batches each for
Sepsis and
SIRS. Part II of the experiment also examined plasma for differentially
expressed proteins
between Sepsis and SIRS pooled samples. For part II, a single round of LCMS/MS
was run
on a pool from time point T_,Z.
Part I. Plasma samples from 25 SIRS and 25 septic patients (150 L) were used
in
this part of the study. Equal volumes of plasma (50 L) were taken randomly
from
individual patient samples from the same disease state (either sepsis or SIRS)
for the
creation of six pools (three sepsis batches and three SIRS batches), each
containing a total
of 20 individual samples. The goal of these studies was to identify proteins
common to
each pooled dataset that were either up-regulated or down-regulated in the
early stages of
sepsis, ultimately allowing for the identification of protein biomarkers.
Immunodepletion. Plasma samples first were immunodepleted to remove abundant
proteins. The pooled plasma samples were immunodepleted using the Agilent
Multiple
Affinity Removal System (5185-5985, 4.6 mm x 50mm, Agilent Technologies). This
immunodepletion column is based upon affinity purified polyclonal antibody
technology
(Maccarone et al., 2004, Electrophoresis 25, 2402-2412; Bjorhall et al., 2005,
Proteomics 5,
307-317; and Echan et al., 2005, Proteomics 5, 3292-3303, each of which is
hereby
incorporated herein by reference in its entirety), containing six types of
antibodies to
specifically remove six target proteins: albumin, transferrin, haptoglobin,
anti-trypsin, IgG,
and IgA. The six antibodies are oriented on the surface of solid beads and
chemically cross
linked via the FC (Fragment Crystallizable) region resulting in a stable, non-
leaching
product, with the material packed in an HPLC column format.
A 25 L aliquot from each plasma sample was diluted five times with column
loading buffer A (Agilent Technologies, #5185-5987) and followed by
centrifugation at
12,000rpm for ten minutes. The column was attached to a capillary HPLC pump
and
equilibrated with buffer A at a flow rate of 0.5 mL/min for 10 min. The
diluted plasma
sample was then injected onto the column and the column was washed with buffer
A at a
-173-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
flow rate of 0.25 mL/min. The first 1.5 mL flow-through containing low
abundance plasma
proteins was collected. The retained proteins were then removed by elution
using buffer B
(Agilent Technologies # 5185-5988) at a flow rate of 0.5 mL/min for seven
minutes.
Retained proteins were checked by 2D-Gel and no other proteins except these
six binding
proteins have been found (Maccarone et al., 2004, Electrophoresis 25, 2402-
2412; and
Echan et al., 2005, Proteomics 5, 3292-3303). The column was either stored or
re-used.
The protein concentrations of samples before and after immunodepletion were
measured by
Coomassie protein assay reagent kit (Pierce #23200) in order to validate the
protein removal
approach. It was determined that -85% of total protein was removed from the
original
plasma sample after immunodepletion.
Protein Digestion. A 2 mg protein alliquot from the immunodepletion column was
collected for each pooled plasma sample. The proteins were concentrated by
Vacufuge
(eppendorf) to 0.25mL (4mg/mL protein) and denatured in 8M urea (ACROS) for 10
minutes. The proteins were reduced and alkylated using 5mM DL-dithiothreitol
(Sigma) at
37 C for 30 minutes and 10 mM iodoacetamide (Sigma) at 37 C for 30 minutes,
respectively. The samples were then diluted four times to a final
concentration of 2M urea
using 100mM ammonium hydrogen carbonate (Fluka). The proteins were digested
with
trypsin (Promega) at the ratio of 1:50 (w/w) overnight and followed by the
second trypsin
digestion under the same conditions. The protein digestion efficiency was
checked with a
Coomassie protein assay reagent kit.
Three dimensional chromatography and sample loading. Using this approach, the
peptide mixture was pre-fractionated by a first reverse phase column (RP 1)
based on
hydrophobicity and then each fraction was then further fractionated by a SCX
column based
on the peptide ion strengths. The final high resolution separation was
performed on a
second reverse phase (RP2) column by a shallow reverse phase gradient which
was
determined by the first reverse phase (RP1) fractionation gradient.
Approximately 2mg digested protein was harvested from each pooled sample and
0.5mg was then subjected to 3-D LC-MS/MS analysis using an Agilent 1100 LC/MSD
Trap
system coupled directly to an Agilent 1100 nano-pump and a micro-autosampler.
Using an
in-house constructed pressure cell, 5 m Zorbax SB-C18 packing material
(Agilent
Technologies) was packed into a 500 m ID 1/32 OD PEEK tubing (Upchurch
Scientific).
A 10 cm section was cut off to form the first dimension RP column (RP1). A
similar
column (500 m ID, 4 cm length) packed with 5 m PolySulfoethyl (Western
Analytic
Production) packing material was used as the SCX column. A second C18 column
4cm in
-174-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
length and 250pm ID was used as the trap column. A zero dead volume 1 .m
filter
(Upchurch, M548) was attached to the exit of each column for column packing
and
connecting. A fused silica capillary (100 m ID, 360 um OD, 20 cm length)
packed with
m Zorbax SB-C18 packing material (Agilent Technologies) was used as the
analytical
column (RP2). One end of the fused silica tubing was pulled to a sharp tip
with the ID
smaller than 5 m using a Sutter P-2000 laser puller (Sutter Instrument
Company, Novato,
California, USA). The peptide mixtures were loaded onto the first Cl 8 column
(RP1) using
the same in-house pressure cell. To avoid sample carry-over, a new set of the
four columns
was used for each sample. In order to maintain good reproducibility for
quantitation, each
of the above four columns was packed to the exact same length for every 3D
experiment.
The 3-dimensional LC separation consisted of two HPLC pumps, four micro- and
nano-
flow LC columns constructed In-house, together with a switch valve. A I mg
alliquot of
each digested protein sample was loaded onto the first dimension reverse phase
(RP)
column for every analysis. Up to five RP fractions and up to eight strong
cation exchange
(SCX) fractions were eluted sequentially from the loading column to the
analytical column
for high resolution peptide separation. All fractionation and separation
methods were
identical for the samples within the same batch. The runtime for each fraction
was about
2.5 hours and total runtime for each sample was about 3 days. A scan range of
200-2000rn/z
was employed in the positive mode.
Spectra were analyzed using Spectrum Mill MS Proteomics Workbench (version 2.7
software, Agilent Technologies). Over ten million spectra were generated and
peaks
identified using Agilent Technologies SpectrumMill Xtractor. The total spectra
numbers
were normalized across all rounds and entries were removed if they had a
sequence tag
length of 1 or less. Remaining MS/MS spectra were searched against the
National Center
for Biotechnology Information (NCBI) non-redundant protein database (NCBI-nr
human
11/06/2003, 97027 sequences) limited to human taxonomy. The enzyme parameter
was
limited to full tryptic peptides with a maximum miscleavage of 2. All other
search
parameters were set to SpectrumMill's default settings (carbamidomethylation
of cysteines,
+/- 2.5 Dalton tolerance for precursor ions, +/- 0.7 Dalton tolerance for
fragment ions, and a
minimum matched peak intensity of 50%).
The false positive rate was estimated by searching one 3D dataset against a
combined forward-reverse database (NCBI-nr human 11/06/2003, 97027 sequences,
Peng,
2003 J. Proteome Res. 2, 43-50). A total of 4294 spectra and 107 proteins were
- 175 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
auto-validated. Among them, 16 spectra and 12 proteins were from the reverse
database.
Thus the false positive rate of the filtering criteria was 0.75/ 1o spectra,
and 22% protein.
The false positive rate for proteins with a minimum unique peptide of 2 was
0.19/% spectra,
and 2.8% protein. Only proteins with at least 2 unique peptides were selected
for relative
quantitative analysis.
SpectrumMill grouped the proteins with the same set or subsets of unique
peptides
together in order to minimize protein redundancy. The number of identified
proteins
reported in SpectrumMill is the number of identified "protein groups" rather
than the
number of identified protein sequences in the database. Spectrum ounting was
used for
relative protein quantitation. The number of valid MS/MS spectra from each
protein was
normalized to the total MS/MS spectra number of each dataset. Samples were
divided into
two patient groups, SIRS vs. sepsis. The Z-test was used for statistical
analysis. The
Z-scores (A/stdev) of each protein were calculated between those 2 groups and
proteins with
Z-scores above 2 were corn.sidered to be biomarker candidates. The candidate
list was
further filtered by relative standard deviation (<100%), absolute MS/MS
spectrum number
(>=10), unique peptide number (>=2) and manual inspection to remove obvious
false hits
such as keratin.
The Spectrum Mill Workbench output produced 2,810 protein entries across the
three pools. The total spectra numbers were normalized across all pools and
entries with a
distinct sum tag score less than 13. The Genbank Accession numbers for each
hit were
cross refenced with their corresponding Entrez Gene ID using the
gene2accession table
from the National Institute of Health. Only proteins with a gene ID were
included for
further analysis (484 remaining entries). Sepsis to SIRS ratios were
calculated using the
normalized total spectra numbers. Where SIRS > Sepsis, the ratio was
calculated using 1/
(Sepsis/SIRS). If either number was zero, so a ratio cannot be calculated, the
value was
tagged SEPSIS+ or SIRS+ as appropriate. The range of values across time points
was
calculated for each round and protein entries were included if they had a
range > 1.5 or
contained SEPSIS+ or SIRS+ at any time point. This left 151 remaining entries
constituting
103 unique gene IDs identitified in Table 1 below.
-176-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Table 1: Biomarkers that discriminate between Sepsis and Sirs
Gene Symbol Gene Name Gene Accession Protein Accession
Number Number
Column 1 Column 1 Column 3 Column 4
SERPINA3 serine (or cysteine) NM_001085 NP_001076
proteinase inhibitor, clade
A (alpha-1 antiproteinase,
antitrypsin), member 3
ACTB actin, beta NM001101 AAS79319
AFM Afamin NM 001133 AAA21612
AGT angiotensinogen (serine NM_000029 AAR03501
(or cysteine) proteinase
inhibitor, clade A (alpha-1
antiproteinase,
antitrypsin), member 8)
AHSG alpha-2-HS-glycoprotein NM 001622 NP_001613
AMBP alpha-l- NM_00163 3 NP_001624
microglobulin/bikunin
precursor
APOF apolipoprotein F NM_001638 AAA65642
APOAI apolipoprotein A-I NM 000039 AAD34604
APOA2 apolipoprotein A-II NM001643 AAA51701
APOA4 apolipoprotein A-IV NM_000482 AAS68228
APOB apolipoprotein B NM000384 AAP72970
(including Ag(x) antigen)
APOC1 apolipoprotein C-I NM001645 AAQ91813
APOC3 apolipoprotein C-III BC027977 AAB59372
APOE apolipoprotein E NM000041 AAB59397
APOH apolipoprotein H (beta-2- NM000042 CAA40977
glycoprotein I)
SERPINCI serine (or cysteine) NM_000488 CAI19423
proteinase inhibitor, clade X68793 P01008
C (antithrombin), member
1; Antithrombin-III
precursor (ATIII)
AZGPI alpha-2-glycoprotein 1, NM-001185 NP_001176
zinc
-177-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Gene Symbol Gene Name Gene Accession Protein Accession
Number Number
Column 1 Column 1 Column 3 Column 4
BF B-factor, properdin NM001710 CAI17456
SERPINGI serine (or cysteine) NM_000062; AAW69393
proteinase inhibitor, clade BC011171
G (Cl inhibitor), member
1, (angioedema,
hereditary)
C1QB complement component 1, NM_000491 NP_000482
q subcomponent, beta
polypeptide
C1S complement component 1, NM_201442; NP_958850
s subcomponent NM 001734
C2 complement component 2 NM 000063 CAI17451
C3 complement component 3 NM_000064 AAR89906
C4BPA complement component 4 NM_001017367 CAH70782
binding protein, alpha
C5 complement component 5 NM 001736 NP_001726
C8A complement component 8, NM000562 CAI19172
alpha polypeptide
C8G complement component 8, NM 000606 NP 000597
gamma polypeptide
C9 complement component 9 NM 001737 NP_001728
SERPINA6 serine (or cysteine) NM_001756 NP001002236
proteinase inhibitor, clade NP_000286
A (alpha-1 antiproteinase,
antitrypsin), member 6
CD14 CD14 antigen NM_000591 AAP35995
CLU clusterin (complement NM_001831 AAP88927
lysis inhibitor, SP-40,40,
sulfated glycoprotein 2,
testosterone-repressed
prostate message 2,
apolipoprotein J)
CP ceruloplasmin NM 000096 NP000087
(ferroxidase) ~
CRP C-reactive protein NM_000567 NP_000558
CSK c-src tyrosine kinase NM 004383 NP004374
F2 coagulation factor II NM_000506 AAL77436
-178-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Gene Symbol Gene Name Gene Accession Protein Accession
Number Number
Column 1 Column 1 Column 3 Column 4
(thrombin)
F9 coagulation factor IX NM_000133 NP_000124
(plasma thromboplastic
component, Christmas
disease, hemophilia B)
FGA fibrinogen alpha chain BC070246 BAC55116
FGB fibrinogen beta chain NM 005141 AAA18024
FGG fibrinogen gamma chain NM_000509 AAB59531
FLNA filamin A, alpha (actin NM001456 CA143227
binding protein 280)
FN1 fibronectin 1 BT006856 BAD52437
GC group-specific component NM_000583 NP_000574
(vitamin D binding
protein)
GSN gelsolin (amyloidosis, BC026033 CA114413
Finnish type)
HBB hemoglobin, beta NM_000518 AAD19696
SERPINDI serine (or cysteine) NM_000185 CAG30459
proteinase inhibitor, clade
D (heparin cofactor),
member 1
HP Haptoglobin BC107587 NP_005134
HPX Hemopexin NM_000613 NP_000604
HRG histidine-rich glycoprotein NM 000412 NP_000403
IF I factor (complement) NM_000204 NP_000195
IGFALS insulin-like growth factor NM_004970 NP_004961
binding protein, acid labile
subunit
ITGAI integrin, alpha 1 NM 181501 NP 852478
ITIH1 inter-alpha (globulin) BC109115 NP_002206
inhibitor HI NP 032432
ITIH2 inter-alpha (globulin) NM 002216 NP_002207
inhibitor H2 '
ITIH4 inter-alpha (globulin) NM_002218 NP_002209
inhibitor H4 (plasma
Kallikrein-sensitive
glycoprotein)
-179-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Gene Symbol Gene Name Gene Accession Protein Accession
Number Number
Column 1 Column 1 Column 3 Column 4
KLKB1 kallikrein B, plasma NM_000892 NP_000883
(Fletcher factor) I
KNG1 kininogen 1 NM_000893 NP_000884
KRTI keratin 1 (epidermolytic BC063697 NP_000412
hyperkeratosis)
LGALS3BP lectin, galactoside- NM005567 NP_005558
binding, soluble, 3 binding BC015761
protein BC002403
BC002998
LPA lipoprotein, Lp(a) NM 005577 NP 005568
MLL myeloid/lymphoid or NM 005934 NP_005924
mixed-lineage leukemia
(trithorax homolog,
Drosophila)
MRCI mannose receptor, C type NM002438 NP002429
1
MYL2 myosin, light polypeptide NM_000432 AAH31006
2, regulatory, cardiac,
slow
MYO6 Myosin VI NM004999 NP_004990
ORM1 orosomucoid 1 NM 000607 CAI16859
SERPINF 1 serine (or cysteine) NM_002615 AAH 13984
proteinase inhibitor, clade BC013984
F (alpha-2 antiplasmin,
pigment epithelium
derived factor), member 1
SERPINAI serine (or cysteine) BC015642 NP_001002235
proteinase inhibitor, clade NM_000295 NP_000286
A (alpha-1 antiproteinase,
antitrypsin), member 1
SERPINA4 serine (or cysteine) NM 006215 NP006206
proteinase inhibitor, clade
A (alpha-1 antiproteinase,
antitrypsin), member 4
SERPINF2 serine (or cysteine) BC031592 NP_000925
proteinase inhibitor, clade
F (alpha-2 antiplasmin,
pigment epithelium
derived factor), member 2
PROS 1 Protein S (alpha) NM_000313 NP_000304
QSCN6 quiescin Q6 NM002826 AAQ89300
- 180 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Gene Symbol Gene Name Gene Accession Protein Accession
Number Number
Column 1 Column 1 Column 3 Column 4
RGS4 regulator of G-protein NM005613 NP_005604
signalling 4
SAAI serum amyloid Al BC105796 AAA64799
AAA30968
SAA4 serum amyloid A4, NM_006512 NP_006503
constitutive / Serum P05067
Amyloid A-4 protein
precursor (constitutively
expressed serum amyloid
A) (C-SAA)
SERPINA7 serine (or cysteine) NM000354 CAB06092
proteinase inhibitor, clade
A (alpha-1 antiproteinase,
antitrypsin), member 7
TF transferrin NM 001063 NP 001054
TFRC transferrin receptor (p90, NM003234 NP003225
CD71)
TTN titin BC013396 CAD12456
TTR transthyretin (prealbumin, NM_000371 AAH05310
amyloidosis type 1) AAP35853
UBC ubiquitin C NM 021009 NP_066289
VTN vitronectin (serum NM000638 P04004
spreading factor,
somatomedin B,
complement S-protein)
VWF von Willebrand factor NM 000552 AAB59458
ALMS1 Alstrom syndrome 1 NM015120 NP055935
ATRN attractin BC101705 CA122615
NM_139321
APOLI apolipoprotein L, I BC017331 AAK20210
NM 003661
TRIP11 thyroid hormone receptor NM_004239 NP_004230
interactor 11
PDCD 1 i programmed cell death 11 NM 014976 NP_055791
KIAA0433 - AB007893 BAA24863
SERPINA 10 serine (or cysteine) NM_016186 NP_057270
proteinase inhibitor, clade
A (alpha-1 antiproteinase,
antitrypsin), member 10
-181-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Gene Symbol Gene Name Gene Accession Protein Accession
Number Number
Column 1 Column 1 Column 3 Column 4
BCOR BCL6 co-repressor BC063536 AAG41429
C l 0orfl 8 chromosome 10 open BC001759 CA113368
reading frame 18
YY1AP1 YY1 associated protein 1 BC044887 AAL75971
BC014906 CAH71646
FLJ10006 - BC110537 AAH17012
BC110536
BDP1 B double prime 1, subunit NM 018429 AAH32146
of RNA polymerase III ~
transcription initiation
factor IIIB
SMARCAD1 SWI/SNF-related, matrix- NM_020159 NP_064544
associated actin-dependent
regulator of chromatin,
subfamily a, containing
DEAD/H box I
MKL2 MKL/myocardin-like 2 NM 014048 AAH47761
CHST8 carbohydrate (N- NM_022467 NP 071912
acetylgalactosamine 4-0) BC018723
sulfotransferase 8
MCPHI microcephaly, primary NM024596 AAH30702
autosomal recessive 1 BC0_30702
MYO18B myosin XVIIIB NM_032608 NP_l 15997
MICAL-L1 - NM 033386 AAH82243
~ AAH01090
PGLYRP2 peptidoglycan recognition NM_052890 Q96PD5
protein 2
LRG1 leucine-rich alpha-2- NM_052972 AAH70198
glycoprotein 1
KCTD7 potassium channel NM 153033 NP_694578
tetramerisation domain ~
containing 7
MGC27165 - BC087841 AAH87841
BC005951
The 103 proteins (Entrez Gene ID's) were uploaded into DAVID 2.1 (Database for
Annotation, Visualization and Integrated Discovery, Dennis et al., 2003,
Genome Biol.
4:P3). All 103 genes were recognized by DAVID. The canonical pathways
contained in
the data were examined by selecting output from Biocarta and KEGG pathways. As
set
-182-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
forth in Table 2, below, any pathways containing at least two genes from the
list of 103 and
having a probability score (p-value) <_ 0.1 were included. In Table 2, the
"count" is the
number of proteins from that particular pathway that are present in Table 1
and the
"Percent" is the above-described count divided by the total protein number of
proteins in the
given database that are in the pathway. The data indicates participation of
the Complement
and Coagulation systems.
Table 2: Pathways associated with Sepsis and Sirs
Categorgy System Term Count Percent P Value
KEGG-PATHWAY Complement and 25 24 2.25E-33
Coagulation
Cascades
BIOCARTA Intrinsic 8 7 5.81E-09
Prothrombin
Activation
Pathway
BIOCARTA Complement 7 6 1.04E-06
Pathway
BIOCARTA Classical 6 5 2.70E-06
Complement
Pathway
BIOCARTA Alternative 5 4 2.86E-05
Complement
Pathway
BIOCARTA Lectin Induced 5 4 8.36E-05
Complement
Pathway
BIOCARTA Extrinsic 4 3 8.54E-04
Prothrombin
Activation
Pathway
BIOCARTA Acute Myocardial 4 3 1.44E-03
Infaretion
KEGG-PATHWAY Regulation of 8 7 8.59E-03
Actin
Cytoskeleton
KEGG PATHWAY Focal Adhesion 7 6 4.48E-02
KEGG-PATHWAY ECM-Receptor 4 3 7.31 E-02
Interaction
-183-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Additionally the molecular functions and biological process inherent in the
data set
set forth in Table 1 were examined by outputting any "over-represented" gene
ontology
categories. Gene ontology categories over-represented in the data are set
forth in Table 3
below. As in the case of Table 2,""count" is the number of proteins present in
Table 1 from
that particular pathway and "Percent" is the count (as defined here) / total
protein number
from that pathway in the database. Similar to the pathway output, Complement
and
Coagulation activity is highly represented in this data set. The major theme
of the data
present in Table 3 is immune system activity. Additionally, lipid transport
(apolipoproteins)
is a functional process that may prove to be important in distinguishing
Sepsis from SIRS.
Table 3: Gene Ontology Categories Over-represented in Table 1.
Term Count Percent P Value
Acute-Phase Response 18 17 5.07E-28
Response to Pest, Pathogen or Parasite 34 33 5.70E-26
Complement Activation 14 13 9.01E-20
Blood Coagulation 16 15 1.13E-17
Serine-Type Endopeptidase Inhibitor Activity 16 15 1.52E-17
Wound Healing 16 15 2.96E-17
Lipid Transport 12 11 1.76E-13
Humoral Immune Response 14 13 2.56E-11
Immune Response 39 37 4.73E-11
Humoral Defense Mechanism (Sensu Vertebrata) 12 11 1.61E-10
Inflammatory Response 12 11 3.87E-08
The ontologies were filtered to include only those with a level 5 distinction
(most
specific gene ontologies) and greater than 10 percent of the input gene list
(Table 1). Only
ontologies from the Molecular Function or Biological Processes were
incorporated. See the
website at geneontology.org. The canonical pathways identified by DAVID are
shown in
Table 2. Each pathway was over-represented in this data set, implying that
they contained
more proteins from the data than would be expected by chance. The results
indicate a
significant focus on both the complement and coagulation cascades, known to
play a major
part in sepsis.
The complement pathway consists of a complex series of over thirty plasma
proteins
which are part of the immune response, providing a critical defense against
infection.
Figure 2 shows the identified proteins from this study in the complement
cascade.
-184-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Activation of the complement system lyses bacterial cells, forms chemotactic
peptides (C3a
and C5a) that attract immune cells, and increases phagocytotic clearance of
infecting cells.
Additionally, the complement pathway can result in increased permeability of
vascular
walls and inflammation. Most complement proteins exist in plasma as inactive
precursors
that cleave and activate each other in a proteolytic cascade leading
ultimately to the
formation of the membrane attack complex (MAC), which causes lysis of cells.
MAC
formation may be activated by three pathways distinct in the initiation of the
proteolytic
cascade but share most of their components; the classical pathway, alternative
pathway and
membrane attack pathway. Here, the classical and alternative pathways are
discussed. The
classical pathway is activated by the recognition of foreign cells by
antibodies bound to the
surface of the cells. In this data, the proteins Cl S and C2 were unique to
this pathway.
Proteolysis is triggered in the alternative pathway by the spontaneous
activation of C3
convertase from C3. Complement Factor-B (Protein BF, properdin) was found in
the data
presented here and is unique to the alternative activation pathway.
Additionally the proteins
C3, C5, C8 and C9, discovered in the plasma samples in this study, are common
to all
methods of complement activation.
Activation of coagulation is a normal component of the acute inflammatory
response
and disorders of coagulation are common in sepsis. Tissue factor production is
increased
and leads to the activation of both the intrinsic and extrinsic prothrombin
activation
pathways. In this study, the data strongly indicated participation of the
intrinsic
prothrombin activation pathway (Figure 3). Briefly, blood coagulation or
clotting takes
place in three essential phases. First is the activation of a prothrombin
activator complex,
followed by the second stage of prothrombin activation. The third stage is
clot formation as
a result of fibrinogen cleavage by activated thrombin. The prothrombin
activation complex
is formed by two pathways, each of which results in a different form of the
prothrombin
activator. The intrinsic mechanism of prothrombin activator formation begins
with trauma
to the blood or exposure of blood to collagen in a traumatized vessel wall.
While the
extrinsic pathway was identified by DAVID, it appeared to be included because
of the
overlapping proteins PROS, SERPINCI, Thrombin and Fibrinogen. The data also
contained SERPING 1, KNG, KLKB 1 and F9 which are all uniquely involved in the
formation of the prothrombin activator complex specific to the intrinsic
prothrombin
activation pathway. The inclusion of gene ontologies in Table 3 covering both
complement
and coagulation also further support the role of these pathways in
distinguishing sepsis from
SIRS samples. Utilizing these criteria, seven proteins showed a common
increase in the
- 185 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
plasma from sepsis patients in all three batches, while three proteins (where
both the
precursor and the final product are counted as a single biomarker) showed a
common
decrease as illustrated in Table 4.
Table 4: Up-regulated and down-regulated proteins in the plasma of sepsis
patients.
compared to SIRS patients.
Gene Symbol Gene Name Gene Accession Protein Accession
Number Number
Column 1 Column 1 Column 3 Column 4
UPREGULATED
C4B Complement component K02403 AAB67980
C4
CRP C-reactive protein NM_000567 NP_000558
CRP C-reactive protein M11880 AAB59526
precursor
PLG plasminogen NM_000301 AAH60513
PLG plasminogen precursor X05199 P00747
APOA2 apolipoprotein A-II NM 001643 AAA51701
APOA2 apolipoprotein A-II X00955 P02652
precursor
SERPING1 serine (or cysteine) NM_000062; AAW69393
proteinase inhibitor, clade BC011171
G (C 1 inhibitor), member
1, (angioedema,
hereditary)
SERPINGI plasma protease Cl AB209826 P05155
inhibitor precursor
TTR transthyretin (prealbumin, NM_000371 AAH05310
amyloidosis type I) AAP35853
TTR, TBPA, ATTR transthyretin precursor U19780 P02766
(prealbumin) (TBPA)
(TTR)(ATTR)
APCS amyloid P component, BT006750 CAH73651
serum
-186-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Gene Symbol Gene Name 1 Gene Accession Protein Accession
Number Number
Column 1 Column 1 Column 3 Column 4
UPREGULATED
APCS serum amyloid BC007058 NP001630
P-component precursor
DOWNREGULATED
APOA1 apolipoprotein A-I NM 000039 AAD34604
APOA1 apolipoprotein A-I NM_000039 P02647
precursor
SERPINCI serine (or cysteine) NM_000488 CAI19423
proteinase inhibitor, clade X68793 P01008
C (antithrombin), member
1; Antithrombin-III
precursor (ATIII)
SAA4 serum amyloid A4, NIvI_006512 NP_006503
constitutive / Serum P05067
Amyloid A-4 protein
precursor (constitutively
expressed serum amyloid
A) (C-SAA)
SAA4 Serum amyloid A-4 M81349 P02375
protein precursor
The possibility of non-specific protein binding to the immunodepletion column,
which could cause losses of the lower abundance proteins, was investigated by
randomly
analyzing the samples twice without immunodepletion. Even in the absence of
immunodepletion, these ten proteins were still identified as strong biomarker
candidates.
Interrogation of the data using DAVID had shown the complement and coagulation
pathways to be over-represented, suggesting that they could play an important
role in
distinguishing sepsis from SIRS. These findings were supported by some of the
proteins
identified here. Many of the proteins in Table 4 are known to be acute phase
proteins (C-
reactive protein, plasminogen and serum amyloid P), involved in the complement
pathway
(complement component C4), the coagulation pathway (antithrombin) or both
(plasma
protease Cl inhibitor) or lipid transport (apolipoproteins). Altered levels of
several of these
proteins have been reported to correlate with SIRS and sepsis (Mesters, 1996,
Mannucci et
-187-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
al., Blood 88, 881-886 (antithrombin-III); Nakae et al., 1996, Surg Today 26,
225-229
(complement component C3 and complement component C4); Chenaud et al., 2004,
Crit.
Care Med. 32, 632-637 (ApoAl); Roemish et al., 2002, Blood Coagul.
Fibrinolysis 13, 657-
670 (antithrombin III); and Sierra et al., 2004, Intensive Care Med. 30, 2038-
2045 (C-
reactive protein) , each of which is hereby incorporated by reference in its
entirety), as both
the complement and coagulation pathways are known to be activated (Mesters et
al., 1996,
Blood 88, 881-886; Haeney 1998, J. Antimicrob Chemother. 41, Suppl A:41-6;
Wheeler et
aL, 1999, N. Engl J. Med. 340, 207-214; and Aird, 2005, Crit. Care Clin 21,
417-431, each
of which is hereby incorporated by reference).
Sepsis is a complex disease, common in the critically ill, that still has no
truly
effective early diagnosis strategy or treatment. It can strike rapidly, in a
matter of days, and
is associated with substantial morbidity and mortality. Plasma represents a
proven resource
in the quest for understanding the complex interactions of the biochemical
cascades that
lead to disease and, further, in the identification of biomarkers for disease
diagnosis. The
above experimental data provides a unique combination of immunodepletion, 3D
LC
separation and MS/MS analysis to offer some important insights into the
interactions that
surround the onset of sepsis and the potential identification of protein
biomarkers in this
event. This platform allowed for the removal of the highly abundant proteins
and thus the
detection of previously suppressed low abundance proteins. Subsequent analysis
using an
in-house developed high resolution separation and tandem mass analysis enabled
the
detection of -3000 lower abundance plasma proteins and the ultimate
observation of these
ten potential sepsis biomarkers (where both the precursor and the final
product are counted
as a single biomarker), with the down-regulation of seven proteins including
those involved
in lipid transport, as well as the up-regulation of three proteins observed in
plasma from
SIRS patients.
Part H. Methods used in part II vary slightly from that given for Table I
above, in
that only a single set of pooled samples, at the T_12 hour time point was
analyzed.
Procedurally, the difference was that samples were analyzed using LC/MS-MS
(not LC3)
and no immunodepletion of the samples was performed prior to analysis. Thus,
part II of
the experiment also examined plasma for differentially expressed proteins
between Sepsis
and SIRS pooled samples. A single round of LCMS/MS was run on a pool from time
point
T_12. The data was also analyzed using Spectrum Mill Workbench software. The
final
output report contained 142 entries all with a distinct sum tag score > 13.
The data was
normalized to the same scale as used in part I. Each entry was identified
using a Uniprot ID
- 188 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
and was cross references to its appropriate Entrez Gene ID using data from the
International
Protein Index and annotated using data from NCBI. Entries that could be linked
to an
Entrez gene ID were included. Ratio data was calculated as in part I. Since
only a single
time point was examined, it's not possible to calculate a range of ratios over
time. Entries
were included when the ratio > 1.5 or was SEPSIS+ or SIRS+. That left 93
remaining
entries representing 93 unique genes identified in Table 5 below.
Table 5: Biomarkers that discriminate between Sepsis and Sirs
Gene Symbol Gene Name Gene Accession Protein Accession
Number Number
Column 1 Column 2 Column 3 Column 4
A 1 BG alpha-l-B glycoprotein NM 130786 NP 570602
A2M alpha-2-macroglobulin NM_000014 AAT02228
ABLIMI Actin binding LIM NM 002313 CAI10910
protein 1 ~
ACTA1 Actin, alpha 1, skeletal NM_001100 CA119052
muscle
AGT angiotensinogen (serine NM 000029 AAR03501
(or cysteine) proteinase ~
inhibitor, clade A (alpha-
1 antiproteinase,
antitrypsin), member 8)
AHSG alpha-2-HS-glycoprotein NM_001622 NP 001613
ANK3 ankyrin 3, node of NM_020987 CA140519
Ranvier (ankyrin G)
APCS amyloid P component, BT006750 CAH73651
serum
APOA1 apolipoprotein A-I NM 000039 AAD34604
APOA4 apolipoprotein A-IV NM_000482 AAS68228
APOB apolipoprotein B NM_000384 AAP72970
(including Ag(x) antigen)
APOC3 apolipoprotein C-III BC027977 AAB59372
APOLI apolipoprotein L, 1 BC017331 AAK20210
NM_003661
AZGP1 alpha-2-glycoprotein 1, N1VI 001185 NP 001176
-189-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Gene Symbol Gene Name Gene Accession Protein Accession
Number Number
Column 1 Column 2 Column 3 Column 4
zinc
B2M beta-2-microglobulin NM_004048 AAA51811
BF B-factor, properdin NM 001710 CAI17456
C 1 R complement component NM00173 3 NP_001724
1, r subcomponent
C1S complement component NM_201442; NP_958850
1, s subcomponent NM 001734 NP_001725
C2 complement component 2 NM 000063 CAI17451
C4B complement component NM_000592 AAR89095
4B
C5 complement component 5 NM 001736 NP_001726
C6 complement component 6 NM 000065 BAD02322
C7 complement component 7 NM000587 CAA72407
C8A complement component NM000562 CA119172
8, alpha polypeptide
C8B complement component NM_000066 CAC18532
8, beta polypeptide '
CDK5RAP2 CDK5 regulatory subunit NM_018249 CA140927
associated protein 2
CHGB chromogranin B NIVI_001819 CAB55272
(secretogranin 1)
CLU clusterin (complement NM001831 AAP88927
lysis inhibitor, SP-40,40,
sulfated glycoprotein 2,
testo sterone-repre ssed
prostate message 2,
apolipoprotein J)
COMP cartilage oligomeric NM 000095 AAC83643
matrix protein ~
CORO 1 A coronin, actin binding NM_007074 NP_009005
protein, 1 A
CPN1 carboxypeptidase N, NM_001308 NP_001299
polypeptide 1, 50kD
CULI cullin 1 NM 003592 NP 003583
DETI de-etiolated homolog 1 NM_017996 NP_060466
(Arabidopsis)
DSCI desmocollin 1 BC109161 NP 060466
-190-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Gene Symbol Gene Name Gene Accession Protein Accession
Number Number
Column 1 Column 2 Column 3 Column 4
F13A1 coagulation factor XIII, NM000129 CAC36886
Al polypeptide
F2 coagulation factor II NM 000506 AAL77436
(thrombin)
F5 coagulation factor V NM_000130 CA123065
(proaccelerin, labile CAB 16748
factor)
FGB fibrinogen beta chain NM_005141 AAA18024
GOLGAI golgi autoantigen, golgin NM_002077 CA139632
subfamily a, 1
GSN gelsolin (amyloidosis, BC026033 CA114413
Finnish type)
HBA1 hemoglobin, alpha 1 NM 000558 AA022464
HBB hemoglobin, beta NM 000518 AAD19696
HP haptoglobin BC107587 NP005134
HPX hemopexin NM 000613 NP000604
HSPA5 heat shock 70kDa protein NM_005347 NP_005338
(glucose-regulated
protein, 78kDa)
HUNK hormonally upregulated NM 014586 NP_055401
Neu-associated kinase
IGFBP5 insulin-like growth factor NM 000599 NP000590
binding protein 5
IGHG1 immunoglobulin heavy BC092518 CAC20454
constant gamma 1 (G1 m
marker)
IGLV4-3 immunoglobulin lambda BC020236 AAH20236
variable 4-3
KIF5C kinesin family member NM 004984 AAH17298
5C
KNGI kininogen 1 NM 000893 NP 000884
KRT1 keratin 1 (epidermolytic BC063697 NP_000412
hyperkeratosis)
KRT10 keratin 10 (epidermolytic NM000421 NP000412
hyperkeratosis; keratosis
palmaris et plantaris)
KRT9 keratin 9(epidermolytic NM 000226 NP 000217
- 191 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Gene Symbol Gene Name Gene Accession Protein Accession
Number Number
Column 1 Column 2 Column 3 Column 4
palmoplantar
keratoderma)
LBP lipopolysaccharide AF105067 AAC39547
binding protein
LGALS3BP lectin, galactoside- NM_005567 NP_005558
binding, soluble, 3 BC015761
binding protein BC002403
BC002998
LRG1 leucine-rich alpha-2- NM 052972 AAH70198
glycoprotein I
~
LUM lumican BC035997 AAP35353
MMP14 matrix metalloproteinase NM004995 AAV40837
14 (membrane-inserted)
MYH4 myosin, heavy NM_017533 NP_060003
polypeptide 4, skeletal
muscle
NEB nebulin NM 004543 NP 004534
NUCB2 nucleobindin 2 NM 005013 NP 005004
ORM2 orosomucoid 2 NM 000608 NP 000599
PF4V 1 platelet factor 4 variant 1 NM 002620 NP_002611
PIGR polymeric NM_002644 CAC 10060
inununoglobulin receptor
PLG plasminogen NM_000301 AAH60513
PON1 paraoxonase 1 NM_000446 NP 000437
PPBP pro-platelet basic protein NM_002704 CAG33086
(chemokine (C-X-C
motif) ligand 7)
RBP4 retinol binding protein 4, NM006744 CAH72328
plasma
RIMSI regulating synaptic NM_014989 NP_055804
membrane exocytosis I
RNF6 ring finger protein NM_005977 CAH73183
(C3H2C3 type) 6
SAA1 serum amyloid Al BC105796 AAA64799
AAA30968
SEMA3D sema domain, NM_152754 EAL24184
immunoglobulin domain
(Ig), short basic domain,
secreted, sema horin
- 192 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Gene Symbol Gene Name Gene Accession Protein Accession
Number Number
Column 1 Column 2 Column 3 Column 4
3D
SERPINAI serine (or cysteine) BC015642 NP_001002235
proteinase inhibitor, clade NM_000295 NP000286
A (alpha-I antiproteinase,
antitrypsin), member 1
SERPINDI serine (or cysteine) NM_000185 CAG30459
proteinase inhibitor, clade
D (heparin cofactor),
member 1
SERPINF2 serine (or cysteine) BC031592 NP_000925
proteinase inhibitor, clade
F (alpha-2 antiplasmin,
pigment epithelium
derived factor), member 2
SERPINGI serine (or cysteine) NM000062; AAW69393
proteinase inhibitor, clade gC011171
G (C 1 inhibitor), member
1, (angioedema,
hereditary)
SF3B 1 splicing factor 3b, subunit NM_012433 NP_006833
1, 1551eDa
SPINKI serine protease inhibitor, NM_003122 NP_003113
Kazal type I
SPP1 secreted phosphoprotein 1 NM 000582 AAH17387
(osteopontin, bone '
sialoprotein I, early T-
lymphocyte activation 1)
SPTB spectrin, beta, NM_001024858 BAD92652
erythrocytic (includes
spherocytosis, clinical
type I)
SYNE1 spectrin repeat NM_182961 AAH39121
containing, nuclear
envelope I
TAF4B TAF4b RNA polymerase NM_003187 XP290809
II, TATA box binding
protein (TBP)-associated
factor, 105kDa
TBC I D I TBC 1(tre-2/USP6, NM_015173 NP_055988
BUB2, cdc16) domain
family, member 1
-193-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Gene Symbol Gene Name Gene Accession Protein Accession
Number Number
Column 1 Column 2 Column 3 Column 4
TLN1 talin 1 NM 006289 NP 006280
TMSB4X thymosin, beta 4, X- NM_021109 NP_066932
linked
TRIP11 thyroid hormone receptor NM_004239 NP_004230
interactor 11
TTR transthyretin (prealbumin, NM000371 AAH05310
amyloidosis type I) AAP35853
UROC1 urocanase domain NM_144639 NP 653240
containing 1 !
VTN Vitronectin{serum NM_000638 P04004
spreading factor,
somatomedin B,
complement S-protein)
VWF von Willebrand factor NM 000552 AAB59458
ZFHX2 zinc finger homeobox 2 NM 033400 NP_207646.
ZYX Zyxin NM 003461 NP_001010972
An embodiment of the present invention consists of those biomarkers that are
present in both Tables 1 and Table 5, which is listed in Table 6 below.
Table 6: Biomarkers that are present in both tables Tables 1 and 5
Gene Symbol Gene Name Gene Accession Protein
Number Accession
Number
Column 1 Column 2 Column 3 Column 4
AGT angiotensinogen (serine or NM_000029 AAR03501
cysteine) proteinase inhibitor,
clade A (alpha-1 antiproteinase,
antitrypsin), member 8)
AHSG alpha-2-HS-glycoprotein NM 001622 NP 001613
APOA1 apolipoprotein A-I NM000039 AAD34604
APOA4 apolipoprotein A-IV NIv1 000482 AAS68228
APOB apolipoprotein B (including NM 000384 AAP72970
Ag(x) antigen)
-194-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Gene Symbol Gene Name Gene Accession Protein
Number Accession
Number
Column 1 Column 2 Column 3 Column 4
APOC3 apolipoprotein C-III BC027977 AAB59372
AZGPI alpha-2-glycoprotein 1, zinc NM 001185 NP_001176
BF B-factor, properdin NM001710 CAI17456
SERPINGI serine (or cysteine) proteinase NM000062; AAW69393
inhibitor, clade G(C1 inhibitor), BC011171
member 1, (angioedema,
hereditary)
C1S complement component 1, s NM_201442; NP_958850
subcomponent NM 001734 NP 001725
C2 complement component 2 NM_000063 CAI17451
C5 complement component 5 NM 001736 NP_001726
C8A complement component 8, alpha NM_000562 CA119172
polypeptide
CLU clusterin (complement lysis NM_001831 AAP88927
inhibitor, SP-40,40, sulfated
glycoprotein 2, testosterone-
repressed prostate message 2,
apolipoprotein J)
F2 coagulation factor II (thrombin) NM 000506 AAL77436
FGB fibrinogen beta chain NM005141 AAA18024
GSN gelsolin (amyloidosis, Finnish BC026033 CAI14413
type)
HBB hemoglobin, beta NM 000518 AAD19696
SERPIND1 serine (or cysteine) proteinase NM_000185 CAG30459
inhibitor, clade D (heparin
cofactor), member 1
HP haptoglobin BC107587 NP_005134
HPX hemopexin NM 000613 NP 000604
KNGI kininogen 1 NM_000893 NP_000884
KRT1 keratin 1 (epidermolytic BC063697 NP_000412
hyperkeratosis)
LGALS3BP lectin, galactoside-binding, NM_005567 NP_005558
soluble, 3 binding protein BC015761
BC002403
BC002998
SERPINAI serine (or cysteine) proteinase BC015642 NP_001002235
-195-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Gene Symbol Gene Name Gene Accession Protein
Number Accession
Number
Column 1 Column 2 Column 3 Column 4
inhibitor, clade A(alpha-1 NM000295 NP_000286
antiproteinase, antitrypsin),
member 1
SERPINF2 serine (or cysteine) proteinase BC031592 NP_000925
inhibitor, clade F (alpha-2
antiplasmin, pigment epithelium
derived factor), member 2
SAA 1 serum amyloid A 1 BC105796 AAA64799
AAA30968
TTR transthyretin (prealbumin, NM_000371 AAH05310
amyloidosis type I) AAP35853
VTN vitronectin (serum spreading NM_000638 P04004
factor, somatomedin B,
complement S-protein)
VWF von Willebrand factor NM 000552 AAB59458
AP L1 apolipoprotein L, 1 BC017331 AAK20210
NM_003661
TRIP 11 thyroid hormone receptor NM_004239 NP_004230
interactor 11
LRGI leucine-rich alpha-2- NM052972 AAH70198
glycoprotein 1
AGT angiotensinogen (serine (or NM_000029 AAR03501
cysteine) proteinase inhibitor,
clade A (alpha-1 antiproteinase,
antitrypsin), member 8)
Biomarkers from Table 1 or 5 known to be associated with coagulation were
determined and constitute another embodiment of the present invention as set
forth in Table
7.
Table 7: Biomarkers from Table I or 5 known to be associated with coagulation
Gene Symbol Gene Name Gene Accession Gene
Name Protein
Name
Column 1 Column 2 Column 3 Column 4
AGT angiotensinogen (serine (or NM000029 AAR03501
c steine proteinase inhibitor,
- 196 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Gene Symbol Gene Name Gene Accession Gene
Name Protein
Name
Column 1 Column 2 Column 3 Column 4
clade A (alpha-1 antiproteinase,
antitrypsin), member 8)
APCS amyloid P component, serum BT006750 CAH73651
BF B-factor, properdin NM_001710 CAI17456
SERPINGI serine (or cysteine) proteinase NM_000062 AAW69393
inhibitor, clade G(C1 inhibitor), gC011171
member I. (angioedema,
hereditary)
C 1 QB complement component 1, q NM_000491 NP000482
subcomponent, beta polypeptide CA122896
CIR complement component 1, r NM_001733 NP_001724
subcomponent
C1S complement component 1, s NM_201442; NP_958850
subcomponent NM 001734 NP_001725
C2 complement component 2 NM_000063 CAI17451
CA141858
C3 complement component 3 NM 000064 AAR89906
C4BPA complement component 4 NM_001017367 CAH70782
binding protein, alpha
C5 complement component 5 NM_001736 NP_001726
C6 complement component 6 NM_000065 BAD02322
C7 complement component 7 NM 000587 CAA72407
C8A complement component 8, alpha NM_000562 CA119172
polypeptide
C8B complement component 8, beta NM_000066 CAC18532
polypeptide
C8G complement component 8, NM_000606 NP_000597
gamma polypeptide
C9 complement component 9 NM_001737 NP001728
AAH20721
CLU clusterin (complement lysis NM_001831 AAP88927
inhibitor, SP-40,40, sulfated
glycoprotein 2, testosterone-
repressed prostate message 2,
apolipoprotein J)
CPN1 carboxypeptidase N, polypeptide NM_001308 NP_001299
1, 50kD
- 197 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Gene Symbol Gene Name Gene Accession Gene
Name Protein
Name
Column 1 Column 2 Column 3 Column 4
CRP C-reactive protein, pentraxin- NM_000567 NP_000558
related
IF I factor (complement) NM000204 LOOK UP
LATER
IGFBP5 insulin-like growth factor binding NM_000599 NP000590
protein 5
KRT1 keratin 1(epidermolytic BC063697 NP_000412
hyperkeratosis)
PLG Plasminogen NM 000301 AAH60513
C4B - NM 000592 AAR89095
Biomarkers in accordane with another embodiment of the present invention are
set
forth in Table 8.
Table 8: Biomarkers in accordance with another embodiment of the present
invention.
Gene Symbol Gene Name Gene Accession Protein
Number Accession
Number
Column 1 Column 2 Column 3 Column 4
AGT angiotensinogen (serine or NM 000029 AAR03501
cysteine) proteinase inhibitor,
clade A (alpha-1 antiproteinase,
antitrypsin), member 8)
AHSG alpha-2-HS-glycoprotein NM 001.622 NP_001613
APOAI apolipoprotein A-I NM 000039 AAD34604
APOA4 apolipoprotein A-IV Niv! 000482 AAS68228
APOB apolipoprotein B (including NM_000384 AAP72970
Ag(x) antigen)
APOC3 apolipoprotein C-III BC027977 AAB59372
AZGP1 alpha-2-glycoprotein 1, zinc NM 001185 NP_001176
BF B-factor, properdin NM 001710 CA117456
SERPINGI serine (or cysteine) proteinase NM_000062; AAW69393
inhibitor, clade G(C1 inhibitor), BC011171
member 1, an ioedema,
-198-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Gene Symbol Gene Name Gene Accession Protein
Number Accession
Number
Column 1 Column 2 Column 3 Column 4
hereditary)
C1S complement component 1, s NM_201442; NP_958850
subcomponent NM 001734 NP_001725
C2 complement component 2 NM_000063 CAI17451
C8A complement component 8, alpha NM_000562 CA119172
polypeptide
CLU clusterin (complement lysis NIVI_001831 AAP88927
inhibitor, SP-40,40, sulfated
glycoprotein 2, testosterone-
repressed prostate message 2,
apolipoprotein J)
F2 coagulation factor II (thrombin) NM 000506 AAL77436
FGB fibrinogen beta chain NM_005141 AAA18024
GSN gelsolin (amyloidosis, Finnish BC026033 CA114413
type)
HBB hemoglobin, beta NM 000518 AAD19696
SERPINDI serine (or cysteine) proteinase NM000185 CAG30459
inhibitor, clade D (heparin
cofactor), member 1
HP haptoglobin BC107587 NP_005134
HPX hemopexin NM_000613 NP_000604
KNGI kininogen 1 NM000893 NP_000884
KRT1 keratin 1 (epidermolytic BC063697 NP_000412
hyperkeratosis)
LGALS3BP lectin, galactoside-binding, NM_005567 NP_005558
soluble, 3 binding protein BC015761
BC002403
BC002998
SERPINAI serine (or cysteine) proteinase BC015642 NP_001002235
inhibitor, clade A(alpha-1 NM000295 NP_000286
antiproteinase, antitrypsin),
member 1
SERPINF2 serine (or cysteine) proteinase BC031592 NP_000925
inhibitor, clade F (alpha-2
antiplasmin, pigment epithelium
derived factor), member 2
SAA1 serum amyloid Al BC105796 AAA64799
-199-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Gene Symbol Gene Name Gene Accession Protein
Number Accession
Number
Column 1 Column 2 Column 3 Column 4
AAA30968
TTR transthyretin (prealbumin, NM_000371 AAH05310
amyloidosis type I) AAP35853
VTN vitronectin (serum spreading NM_000638 P04004
factor, somatomedin B,
complement S-protein)
VWF von Willebrand factor NM 000552 AAB59458
AP4L1 apolipoprotein L, 1 BC017331 AAK20210
NM_003661
TRIP11 thyroid hormone receptor NM_004239 NP_004230
interactor 11
LRG1 leucine-rich alpha-2- NM_052972 AAH70198
glycoprotein 1
AGT angiotensinogen (serine (or NM_000029 AAR03501
cysteine) proteinase inhibitor,
clade A (alpha-1 antiproteinase,
antitrypsin), member 8)
Biomarkers in accordane with another embodiment of the present invention are
set
forth in Table 9.
Table 9: Biomarkers in accordance with another embodiment of the present
invention.
Gene Symbol Gene Name Gene Accession Protein
Number Accession
Number
Column 1 Column 2 Column 3 Column 4
AGT angiotensinogen (serine or NM_000029 AAR03501
cysteine) proteinase inhibitor,
clade A (alpha-1 antiproteinase,
antitrypsin), member 8)
AHSG alpha-2-HS-glycoprotein NM_001622 NP 001613
APOA4 apolipoprotein A-IV NM 000482 AAS68228
APOB apolipoprotein B (including NM_000384 AAP72970
Ag(x) antigen)
- 200 -
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Gene Symbol Gene Name Gene Accession Protein
Number Accession
Number
Column 1 Column 2 Column 3 Column 4
APOC3 apolipoprotein C-Ill BC027977 AAB59372
AZGPI alpha-2-glycoprotein 1, zinc NIvI 001185 NP 001176
BF B-factor, properdin NM 001710 CA117456
SERPINGI serine (or cysteine) proteinase NIvI_000062; AAW69393
inhibitor, clade G(C 1 inhibitor), BC011171
member 1, (angioedema,
hereditary)
C1S complement component 1, s NM_201442; NP_958850
subcomponent NM 001734 NP_001725
C2 complement component 2 NM 000063 CAI17451
C8A complement component 8, alpha NM_000562 CA119172
polypeptide
CLU clusterin (complement lysis NM_001831 AAP88927
inhibitor, SP-40,40, sulfated
glycoprotein 2, testosterone-
repressed prostate message 2,
apolipoprotein J)
F2 coagulation factor II (thrombin) NM 000506 AAL77436
FGB fibrinogen beta chain NM 005141 AAA18024
GSN gelsolin (amyloidosis, Finnish BC026033 CA114413
type)
HBB hemoglobin, beta NM_000518 AAD 19696
SERPINDI serine (or cysteine) proteinase NM_000185 CAG30459
inhibitor, clade D (heparin
cofactor), member 1
HP haptoglobin BC 107587 NP 0(Y5134
HPX hemopexin NM_000613 NP000604
KNG1 kininogen 1 NM 000893 NP_000884
KRT1 keratin I (epidermolytic BC063697 NP_000412
hyperkeratosis)
LGALS3BP lectin, galactoside-binding, NM_005567 NP_005558
soluble, 3 binding protein BC015761
BC002403
BC002998
SERPINAI serine (or cysteine) proteinase BC015642 NP_001002235
inhibitor, clade A(alpha-1 NM_000295 NP 000286
anti roteinase, antit sin , ~
-201-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
Gene Symbol Gene Name Gene Accession Protein
Number Accession
Number
Column 1 Column 2 Column 3 Column 4
member I
SERPINF2 serine (or cysteine) proteinase BC031592 NP000925
inhibitor, clade F (alpha-2
antiplasmin, pigment epithelium
derived factor), member 2
SAA1 serum amyloid A1 BC105796 AAA64799
AAA30968
TTR transthyretin (prealbumin, NM000371 AAH05310
amyloidosis type I) AAP35853
VTN vitronectin (serum spreading NM_000638 P04004
factor, somatomedin B,
complement S-protein)
APOL1 apolipoprotein L, I BC017331 AAK20210
NM_003661
TRIP 11 thyroid hormone receptor - NM004239 NP004230
interactor 11
LRGI leucine-rich alpha-2- NIv1_052972 AAH70198
glycoprotein 1
AGT angiotensinogen (serine (or NM000029 AAR03501
cysteine) proteinase inhibitor,
clade A (alpha-1 antiproteinase,
antitrypsin), member 8)
7. ALTERNATIVE EMBODIMENTS AND REFERENCES CITED
All references cited herein are incorporated herein by reference in their
entirety and
for all purposes to the same extent as if each individual publication or
patent or patent
application was specifically and individually indicated to be incorporated by
reference in its
entirety for all purposes.
The present invention can be implemented as a computer program product that
comprises a computer program mechanism embedded in a computer readable storage
medium. Further, any of the methods of the present invention that don't
involve a
measuring step can be implemented in one or more computers or computer
systems.
Further still, any of the methods of the present invention that don't involve
a measuring step
can be implemented in one or more computer program products. Some embodiments
of the
-202-
CA 02633291 2008-06-13
WO 2007/078841 PCT/US2006/047737
present invention provide a computer system or a computer program product that
encodes or
has instructions for performing any or all of the methods disclosed herein.
Such
methods/instructions can be stored on a CD-ROM, DVD, magnetic disk storage
product, or
any other computer readable data or program storage product. Such methods can
also be
embedded in permanent storage, such as ROM, one or more programrnable chips,
or one or
more application specific integrated circuits (ASICs). Such permanent storage
can be
localized in a server, 802.11 access point, 802.11 wireless bridge/station,
repeater, router,
mobile phone, or other electronic devices. Such methods encoded in the
computer program
product can also be distributed electronically, via the Internet or otherwise,
by transmission
of a computer data signal (in which the software modules are embedded) either
digitally or
on a carrier wave.
Some embodiments of the present invention provide a computer program product
that contains any or all of the program modules shown in Fig. 1. These program
modules
can be stored on a CD-ROM, DVD, magnetic disk storage product, or any other
computer
readable data or program storage product. The program modules can also be
embedded in
permanent storage, such as ROM, one or more programmable chips, or one or more
application specific integrated circuits (ASICs). Such permanent storage can
be localized in
a server, 802.11 access point, 802.11 wireless bridge/station, repeater,
router, mobile phone,
or other electronic devices. The software modules in the computer program
product can
also be distributed electronically, via the Internet or otherwise, by
transmission of a
computer data signal (in which the software modules are embedded) either
digitally or on a
carrier wave.
Having now fully described the invention with reference to certain
representative
embodiments and details, it will be apparent to one of ordinary skill in the
art that changes
and modifications can be made thereto without departing from the spirit or
scope of the
invention as set forth herein. The specific embodiments described herein are
offered by
way of example only, and the invention is to be limited only by the terms of
the appended
claims, along with the full scope of equivalents to which such claims are
entitled.
-203-