Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
GASTRIC RETENTIVE GABAPENTIN DOSAGE FORMS AND METHODS FOR
USING SAME
FIELD OF THE INVENTION
[0001] The invention relates to gastric retentive gabapentin dosage forms and
methods of using
them to reduce or eliminate gabapentin-induced side effects. For example, the
dosage forms of the
invention can be used to reduce or eliminate the side effects associated with
treatment of non-
nociceptive pain states.
BACKGROUND OF THE INVENTION
[0002] Gabapentin (1-(aminomethyl)cyclohexane acetic acid) is a 3-substituted
7-aminobutyric
acid ("GABA") analog that was approved in the United States on December 30
1993 as
NEURONTIN (Pfizer Inc., New York, N.Y.), an immediate irelease dosage form of
gabapentin for
use as adjunctive therapy in the treatment of partial seizures in children and
adults, and was
subsequently approved for treatment of post-herpetic neuralgia ("PHN") in
adults. In addition to
seizures and PHN, gabapentin has also been used in the treatment of
neuropathic pain, restless legs
syndrome, essential tremor, bipolar disorder, migraine headaches, and the
symptoms associated with
menopause, hormonal imbalances, and chemotherapy. Magnus et al., EPILEPSIA
40:S66-S72 (1999);
U.S. Patent No. 6,310,098 to Guttuso. Gabapentin is currently available as
immediate release
NEURONTIN in 100 mg, 300 mg, and 400 mg hard shell capsules; 600 mg and 800
mg film-coated
tablets; and in a liquid formulation having 250 mg/5mL. The recommended dosage
for gabapentin is
a total daily dose of 900 mg to 1800 mg t.i.d. (i.e., three times daily).
[0003] In humans, gabapentin is absorbed throughout the small intestine with
diminished
absorption in the colon. The absorption of immediate release gabapentin occurs
relatively slowly with
the peak plasma concentration occurring approximately 2 to 3 hours after
dosing. The oral
bioavailability of gabapentin is dose-dependent, with approximately 60%
bioavailability for a dose in
the range of 300-400 mg, but with only 3 5 !o bioavailability for a dose of
1600 mg. Bourgeois,
EPILEPSIA36 (Suppl. 5):S1-S7 (1995); Gram, EPILEPSIA 37 (Suppl. 6):S12-S16
(1996); DRUGS OF
ToDAY31:613-9:975-82 (1995); NEUROLOGY 44(Suppl. 5): S17-S32 (2003). The
decrease in
bioavailability with increasing dose of the immediate release tablet has been
attributed to partially
carrier-mediated absorption. Stewart, et al., PHARMACEUTICAL RESEARCH
10(2):276-281 (1993).
[0004) Food has only a small effect on the rate and extent of immediate
release gabapentin
absorption. Further, only 3% of circulating gabapentin is bound to plasma
proteins. Gabapentin is
1
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
not appreciably metabolized in humans, does not induce hepatic enzymes, and is
eliminated
unchanged by renal excretion with a half-life of 5-7 hours regardless of
whether the drug is taken in a
single dose or in multiple doses. (Chadwick; LANCET 343:89-91 (1994); Thomson,
et al., CLIN.
PHARMACOKINET. 23(3):216-230 (1992); and Riva, et al., CLIN. PHARMACOKINET.
31(6):470-493
(1996)).
[0005] Because gabapentin is administered t.i.d., patient compliance with the
conventional
immediate release dosage forms of the drug is an issue. In this respect,
controlled release dosage
forms that would lower the number of daily dosings of gabapentin to once-daily
or twice-daily
dosings would provide a significant advantage over the conventional- immediate
release dosage form;
however, in order for the controlled release dosage form to be effective, the
dosage form must
overcome the poor absorption of the drug in the lower gastrointestinal tract.
[0006] In addition to the compliance and bioavailability issues associated
with the conventional
immediate release dosage form of gabapentin, gabapentin use also suffers from
the adverse side
effects associated with the drug. Side effects reported from gabapentin use
include, most commonly,
somnolence and dizziness, and to a lesser degree, fatigue, ataxia, weight
gain, peripheral edema,
diarrhea, headache, dry mouth, and blurred vision. More recently, gabapentin
use has been associated
with the serious side effect of reversible visual field constriction.
Bekkelund et al., BRIT. MED. J
323:1193 (2006).
[0007] The issue of gabapentin side effects has become increasingly
troublesome since
gabapentin was found to reduce the frequency and severity of hot flashes in
menopausal women. U.S.
Patent No. 6,310,098 to Guttoso. As a result of the prevalence of the
somnolence and dizziness side
effects, many women who have been taking gabapentin for treatment of their
menopausal symptoms
have been forced to discontinue use of the drug.
[0008] In order for gabapentin to gain widespread acceptance for current,
i.e., epilepsy and post-
herpetic neuralgia, and future off-label uses, such as, for example, restless-
leg syndrome, diabetic
neuropathy, back pain, essential tremor, bipolar disorder, migraine
prophylaxis, potentially alcohol
and drug withdrawal, and the symptoms associated with menopause, hormonal
imbalances, and
chemotherapy, there is a need in the art for an improved dosage form of
gabapentin that may be
administered once or twice daily with sufficient absorption of the active
agent to produce the desired
therapeutic effects while increasing the bioavailability of the drug at
elevated doses and reducing the
side effects associated with the drug at the therapeutic dosages.
[0009] Examples of modified release dosage forms, such as controlled-release
dosage forms,
sustained release dosage forms, and extended release dosage forms are known in
the art to which the
invention pertains; however, to the best of the inventor's knowledge,
gabapentin has not been
successfully incorporated into any modified release dosage form because the
pharmacokinetics of the
drug does not facilitate the absorption of the drug in modified release dosage
forms. In fact, the
2
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
innovator noted in the Summary Basis of Approval (NDA 20-235), that a pilot
study (Study #877-
076) of a 600 mg sustained release formulation "indicated unacceptable
sustained release
characteristics," (NDA 20-235) which was attributed to decreasing rate and
extent of absorption as
gabapentin "moves lower in the GI tract." (NDA 20-235). In order to overcome
the shortcomings
inherent in the immediate delivery dosage fonns of gabapentin, a gastric
retentive dosage form of
gabapentin would allow for sustained release of gabapentin while avoiding the
significant loss of
bioavailability of the drug observed with non-gastric retentive controlled
release dosage forms.
[00010] Pain management continues to be a challenge for medical practitioners.
Many pain
medications have unfavorable side effects. In addition, patients can develop
tolerance to pain
medications and require larger doses to reach a previously achieved level of
pain relief.
[00011] Pain is generally classified as either nociceptive pain or non-
nociceptive pain.
Nociceptive pain arises from the stimulation of pain receptors (i.e.,
nociceptive receptors) to heat,
cold, vibration, stretch, and chemical stimulus from damaged cells. Somatic
pain (i.e., muscoskeletal
pain, such as pain specific to skin, muscle, joints, bones, and ligaments) and
visceral pain (i.e., pain
specific to the internal organs and main body cavities) are the two types of
nociceptive pain.
Nociceptive pain is usually time-limited and thus, when the tissue heals, the
pain is resolved. During
periods of pain, nociceptive pain responds well to treatment with opioids.
[00012] Non-nociceptive pain arises from within the peripheral and central
nervous system, where
there are no pain receptors. The pain associated with non-nociceptive pain is
generated from nerve
cell dysfunction. Non-nociceptive pain includes neuropathic pain and
sympathetic pain.
[00013] Neuropathic pain originates in the peripheral nervous system (the
nerves between the
tissue and the spinal cord) or the central nervous system (the nerves between
the spinal cord and the
brain). Neuropathic pain may be caused by nerve degeneration (e.g., by
multiple sclerosis), nerve
pressure (e.g., from a trapped nerve); nerve inflammation (e.g., from a torn
or slipped disc), or nerve
infection (e.g., from shingles or other viral infections). With neuropathic
pain, the injured nerves
become electrically unstable firing of signals in an inappropriate, random,
and disordered fashion.
Neuropathic pain is characterized by nerve malfunctions such as
hypersensitivity to touch, vibrations,
and extreme temperatures and is often described as burning, lancinating, and
shooting pain.
[00014] Sympathetic pain is caused from possible over activity of the
sympathetic system, which
controls blood flow to tissues such as skin and muscle, sweating by the skin,
and the speed and
responsiveness of the peripheral nervous system. Sympathetic pain occurs most
commonly after
fractures and soft tissue injuries of the arms and legs. Sympathetic pain is
characterized by extreme
sensitivity in the skin surrounding the site of injury and peripherally in the
afflicted limb, which may
become so painful that the patient will refuse to use it causing secondary
problems with the limb due
to non-use.
3
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
[00015] Unlike nociceptive pain, non-nociceptive pain is not time limited and
is not easily
treatable. Non-nociceptive pain is generally treated with anti-depressants,
anti-convulsants (i.e., anti-
epileptic drugs), and anti-arrhythmics; however, to date, there is no
effective treatment for non-
nociceptive pain. In commonly owned co-pending U.S. Patent Application Serial
No. 10/280,309
(Publication No. US 2003/0 1 006 1 1 AI), the present inventors disclosed a
gastric-retentive form of
gabapentin and the use of the drug for the treatment of neuropathic pain,
which is a non-nociceptive
pain state.
[0011] , An osmotic dosage form has been described for delivery of gabapentin
prodrugs. U.S.
Patent No. 6,683,112 to Chen et al. describes sustained release formulations
that deliver gabapentin
prodrugs by means of the push-pull osmotic pump system described in U.S. Pat.
No. 4,612,008 to
Wong et al. This system however, is not a gastric retentive dosage fonn and
would be expected to
deliver the drug with poor bioavailability.
[0010] Examples of gastric retentive dosage forms are described in U.S. Patent
No. 4,996,058 to
Sinnreich; U.S. Patent No. 5,232,704 to Franz et al.; U.S. Patent No.
6,120,803 to Wong et al.; and
commonly owned U.S. Patent No. 5,972,389 to Shell et al. and PCT Publication
No. WO 98/055107
to Depomed, Inc. None of these references, however, teach or suggest the use
of the gastric retentive
dosage forms described therein for the administration of gabapentin. To the
best of the inventors'
knowledge, a dosing regimen using a gastric retentive dosage form of
gabapentin for reducing or
eliminating side effects associated with gabapentin treatment has not been
previously described.
Further, to the best of the inventors' knowledge, a dosing regimen using a
gastric retentive dosage
form of gabapentin for pain treatment has not been previously described.
[00111 The present invention overcomes the need in the art for a more
effective gabapentin
dosage form that will increase patient compliance and provide for extended
effective plasma levels so
that patients suffering from a non-nociceptive pain state may be able to more
effectively use
gabapentin for treatment of pain symptoms.
DISCLOSURE OF THE INVENTION
[0012] The present invention overcomes the need in the art for an improved
dosage form of
gabapentin by providing a gastric retentive dosage form of gabapentin that
overcomes the compliance,
bioavailability, and side effect issues associated with the conventional
immediate release dosage
forms of gabapentin.
[0013] The gastric retentive oral dosage form of the present invention has the
advantage of
requiring only once or twice daily dosings; thus improving patient compliance.
Further, if the
decreased bioavailability at all doses of controlled-release gabapentin dosage
forms were due to lack
of colonic absorption, then the gastric retentive gabapentin dosage form of
the present invention has
the additional advantage of keeping the drug in the region of absorption for a
longer period of time;
thus improving the bioavailability of the drug. Moreover, gabapentin has
saturable absorption and
4
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
thus, a gastric retentive dosage form of gabapentin by virtue of its slower
release rate should allow
better absorption at higher doses. This improved bioavailability with
increasing dose allows higher
doses of the drug to be given at once to permit once or twice daily dosing.
This also allows the timing
of the high dose to be in the evening to minimize side effects that may be a
problem during the day,
but not at night; such as somnolence. Furthermore, the slower release rate
allows for a slower rise in
peak plasma levels, which may be associated with side effects.
[00141 In one embodiment of the invention, there is provided a method of
reducing or
eliminating side effects associated with gabapentin use, comprising
administering a therapeutically
effective amount of gabapentin in a gastric retentive dosage form to a patient
in a once-daily dosing
regime within a single 24 hour period.
[0015] In another embodiment ofthe invention, there is provided a method of
reducing or
eliminating side effects associated with gabapentin use, comprising
administering a therapeutically
effective amount of gabapentin in a gastric retentive dosage form to a patient
in a twice-daily dosing
regimen within a single 24 hour period.
[0016] Examples of side effects that may be reduced or eliminated with the
gastric retentive
gabapentin dosage form of the present invention include without limitation,
somnolence, dizziness,
fatigue, ataxia, weight gain, peripheral edema, diarrhea, headache, dry mouth,
blurred vision, and/or
reversible visual field constriction.
[0017] With both the once-daily and twice-daily dosing regimens, upon
administration of the
gastric retentive dosage form to the patient, the patient's blood plasma
exhibits a maximum
concentration (C,,,,,,) of gabapentin that is approximately 15% to
approximately 85% less than C,,,ax for
a comparable dose of immediate release gabapentin. For example the maximum
concentration (C.õa,)
of gabapentin that is approximately 20, 25, 30, 35 %, 40 %, 45%, 50%, 55%,
60%, 65%, 70%, 75%,
80% less than Cma., for a comparable dose of immediate release gabapentin..
The time to Cmjx(Tm-,j is
approximately 1.5 to approximately 5, alternatively, time to CmZ,.(Tm,,x) is
approximately 1.5 to
approximately 3.5, hours longer (equivalent to approximately 40% to
approximately 80% slower)
than Tma,; for a comparable dose of immediate release gabapentin. For example,
the time to Tmax is
approximately 2, 2.5, 3, 3.5, 4, 4.5 hours longer that the Tm.,x for a
comparable dose of immediate
release gabapentin. The bioavailability (AUC) should be at least 70%, 75%,
80%, 85%, 90%, 95%,
96%, 97%, 98% 99% of the AUC, or greater than AUC achieved from a comparable
dose of an
immediate release dosage form and should show greater bioavailability than the
immediate release
dosage forms at higher doses, in particular, at 2400 mg an AUC that is
approximately equal to or
greater than AUC for a comparable dose of immediate release gabapentin. In
some cases the AUC
may be up to 150% or more of the AUC achieved from a comparable dose of an
immediate release
dosage form.
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
[00181 The gastric retentive dosage forms of the present invention may be used
for any disease
state or condition that may be treated with conventional immediate mlease
gabapentin, such as for
example, seizures, PHN (post hepatic neuralgia), diabetic peripheral
neuropathy ("DPN"), pain,
restless-leg syndrome, essential tremor, bipolar disorder, migraine
prophylaxis, and the symptoms
associated with hormonal imbalances and chemotherapy as well as potentially
for alcohol or drug
withdrawal. Within the context of symptoms associated with hormonal
imbalances, such symptoms
include menopausal hot flashes. Within the context of the symptoms associated
with chemotherapy,
such symptoms include chemotherapy-induced hot flashes and nausea.
[0019] For example, gastric retentive dosage forms of gabapentin that may be
administered to a
patient suffering from a pain state in once or twice daily administrations.
The pain states that may be
treated by the gastric retentive dosage forms of the present invention include
non-nociceptive pain
states, such as neuropathic pain or sympathetic pain or pain states that
include a combination of non-
nociceptive pain and nociceptive pain.
[0020] In one embodiment of the invention, there is provided a method of
treating a patient
suffering from a pain state comprising administering to the patient a gastric
retentive dosage form
comprised of a therapeutically effective amount of gabapentin, wherein the
dosage form is
administered to the patient in a once-daily dosing regimen within a single 24-
hour period.
[0021] In another embodiment ofthe invention, there is provided a method of
treating a patient
suffering from a pain state comprising administering to the patient a gastric
retentive dosage form
comprised of a therapeutically effective amount of gabapentin, wherein the
dosage form is
administered to the patient in a twice-daily dosing regimen within a single 24-
hour period.
[0022] In one embodiment, the combination of dosing in a once-daily or twice
daily dosing
regimen with gastric retentive gabapentin allows for a patient to obtain
efficacy in pain or some other
condition for which the gabapentin has been administered due to the unique
combination of the gastric
retentive dosage form, allowing for extended delivery without significant loss
in bioavailability,
dosing once daily with the evening meal or asymmetric dosing with the higher
dose in the evening,
allowing for the potential to ameliorate side effects and thus titrate to a
more efFicacious dose, and the
enhanced bioavailability of the gastric retentive dosage forrn at higher doses
compared to the same
single dose of an immediate release gabapentin dosage form such as NEURONTIN
capsules or
tablets. As noted above, a gastric retentive dosage form allows for extended
delivery without loss in
bioavailability, thus prolonging the plasma concentrations of the drug.
Further, the slower delivery
obtained from the gastric retentive dosage form prevents or ameliorates the
impact of saturation of the
carrier-mediated absorption allowing a higher percent absorption at the higher
dose levels. In
addition, the dosing regimen, either once daily in the evening or twice daily
with the higher dose in
the evening may decrease or ameliorate side effects, allowing the patient to
tolerate a higher dose, and
due to the gastric retentive controlled delivery, is absorbed better than the
equivalent dose of
6
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
immediate release gabapentin if administered as a single dose at the same
dose. Thus, the
combination of gastric retention of gabapentin, the avoidance or amelioration
of impacts of the
carrier-mediated saturation, as well as the once-daily or twice daily
asymmetric dosing, allows for the
subject to obtain efficacy for pain or some other indication for which the
gabapentin is being
administered.
[0023] The invention relates to the use of a medicament comprising a
therapeutically effective
amount of a GABA analog in a gastric retentive dosage form to treat a patient
suffering from a pain
state, wherein said medicament is to be administered to a patient in a once-
daily dosing regimen
within a single 24 hour period.
[0024] The invention also relates to the use of a medicament comprising a
therapeutically
effective amount of a GABA analog in a gastric retentive dosage form to treat
a patient suffering from
a pain state, wherein said medicament is to be administered to a patient in a
twice-daily dosing
regimen within a single 24 hour period.
[0025] Further, the invention relates to the use of a medicament comprising a
therapeutically
effective amount of a GABA analog in a gastric retentive dosage form to reduce
or eliminate side
effects associated with y-aminobutyric acid (GABA) analogs, wherein said
medicament is to be
administered to a patient in a once-daily dosing regimen within a single 24
hour period.
[0026] The invention also relates to the use of a medicament comprising a
therapeutically
effective amount of a GABA analog in a gastric retentive dosage form to reduce
or eliminate side
effects associated with y-aminobutyric acid (GABA) analogs, wherein said
medicament is to be
administered to a patient in a twice-daily dosing regimen within a single 24
hour period.
[0027] For example, the GABA analog is gabapentin. In other examples, the GABA
arnalog is
pregablin.
[0028] In one embodiment, the therapeutically effective amount of gabapentin
is to be
administered to the patient in a total daily dose ranging from approximately
300 mg to approximately
9600 mg.
[0029] In one embodiment, individual dosage units of the gastric retentive
dosage form are
approximately 100 mg to approximately 1800 mg of gastric retentive gabapentin.
[0030] In some embodiments, the dosage form is to be administered to the
patient in an evening
dose in fed mode.
[0031] For example, the dosage form is a tablet or capsule.
[0032] In one embodiment, the medicament is to be administered to a patient
suffering from
menopause-related hot flashes.
[0033] In one embodiment, the medicament is to be administered to a patient
suffering from side
effects such as somnolence, dizziness, fatigue, ataxia, weight gain,
peripheral edema, diarrhea,
headache, dry mouth, blurred vision, and reversible visual field constriction.
7
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
[0034] For example, the side effects are somnolence and/or dizziness.
[0035] In one embodiment, the medicament is to be administered to a patient
suffering from a
pain state that is a mixture of non-nociceptive pain and nociceptive pain.
100361 In one embodiment, the pain state is a non-nociceptive pain. For
example, the non-
nociceptive pain is neuropathic pain.
[0037] The neuropathic pain is, for example, diabetic neuropathy, HIV sensory
neuropathy, post-
herptic neuralgia, post-thoracotomy pain, trigeminal neuralgia, radiculopathy,
neuropathic pain
associated with chemotherapy, reflex sympathetic dystrophy, back pain,
peripheral neuropathy,
entrapment neuropathy, phantom limb pain, or complex regional pain syndrome.
[0038] In one embodiment, the medicament is to be administered to a patient
suffering from a
pain state that is a mixture neuropathic pain and sympathetic pain. For
example, the pain state is a
migraine headache. For example, the non-nociceptive pain is selected from pain
associated with post-
menopausal symptoms or pain associated with chronic pelvic pain syndrome.
10039] In one embodiment, the medicament is to be administered to a patient
suffering from
chemotherapy-related hot flashes and/or nausea.
[0040] In one embodiment, upon administration, bioavailability (AUC) of the
GABA analogue at
2400 mg is approximately 70% to approximately 130% of the AUC for a comparable
total daily dose
of immediate release gabapentin.
[0041] In another embodiment, upon administration, bioavailability (AUC) of
the GABA
analogue at 2400 mg is approximately 80% to approximately 150% of the AUC for
a comparable total
daily dose of immediate release gabapentin.
[00421 In another embodiment, upon administration, bioavailability (AUC) of
the GABA
analogue at 2400 mg is approximately 100% to approximately 150% of the AUC for
a comparable
total daily dose of immediate release gabapentin.
[0043] In another embodiment, upon administration, bioavailability (AUC) of
the GABA
analogue at 1800 mg is approximately 70% to approximately 130% of the AUC for
a comparable total
daily dose of immediate release gabapentin. [0044] In another embodiment, upon
administration, bioavailability (AUC) of the GABA
analogue at 1800 mg is approximately 80% to approximately 150% of the AUC for
a comparable total
daily dose of immediate release gabapentin.
[0045] In another embodiment, upon administration, bioavailability (AUC) of
the GABA
analogue at 1800 mg is approximately 100% to approximately 150% of the AUC for
a comparable
total daily dose of immediate release gabapentin.
[0046] In one embodiment, the medicament, upon administration to a patient as
a single dose,
results in blood plasma levels of the patient that exhibit a maximum
concentration (Cm~) of the
8
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
C~ABA analogue that is approximately 15% to approximately 85% less than
C,,,a,, for a comparable
single dose of immediate release gabapentin.
[0047] In another embodiment, the medicament, upon administration to a patient
as a single
dose, results in blood plasma levels of the patient that exhibit a maximum
concentration (Cm~) of the
GABA analogue that is approximately 35% to approximately 85% less than Cm~ for
a comparable
single dose of immediate release gabapentin.
[0048] In another embodiment, the medicament, upon administration to a patient
as a single
dose, results in blood plasma levels of the patient that exhibit a maximum
concentration (Cm~.) of the
GABA analogue that is approximately 30% to approximately 50% less than Cma,,~
for a comparable
single dose of immediate release gabapentin.
[0049] ' In another embodiment, the medicament, upon administration to a
patient as a single
dose, results in time to C,,,a., (Tm,,,) of gabapentin that is approximately
1.5 to approximately 5 hours
longer than Tmax for a comparable single dose of immediate release gabapentin.
[0050] In another embodiment, the medicament, upon administration to a patient
as a single
. (Tm.) of gabapentin that is approximately 1.5 to approximately 3.5 hours
dose, results in time to C,,,a,
longer than Tmax for a comparable single dose of immediate release gabapentin.
[0051] In one embodiment, the medicament further comprises an additional
active agent selected
from anticonvulsants, tricyclic antidepressants, opioids, and secondary
analgesics.
[0052] For example, the anticonvulsants can be carbamazepine, phenytoin, or
lamotrigine.
[0053] For example, the tricyclic antidepressants are selected from
amitriptyline, imipramine,
clomipramine, and desipramine.
[0054] For example, the opioids are selected from oxycodone and tramadol.
[0055] For example, the secondary analgesic is a non-steroidal anti-
inflammatory drug.
[0056] In one embodiment, the dosage form is administered to the patient in
fed mode in a
morning dose and an evening dose.
[0057] In one embodiment, the morning dose is less than the evening dose.
[0058] For example, the morning dose is equal to or less than about one-half
of the evening dose.
For example, the morning dose is equal to or less than about one-third of the
evening dose. For
example, the morning dose is equal to or less than about one-quarter of the
evening dose.
[0059] The invention relates to the use of a medicament comprising a
therapeutically effective
amount of a GABA analog in a gastric retentive dosage form to reduce or
eliminate side effects
associated with y-aminobutyric acid (GABA) analogs, wherein said medicament is
to be administered
to a patient in a once-daily dosing regimen within a single 24 hour period or
in a twice daily dosing
regimen in which the dose in the morning and dose in the evening are not
equal, also within a single
24 hour period, and wherein said medicament upon administration to a patient
as a single dose results
in blood plasma levels of the patient that exhibit a maximum concentration
(C,,,,,.,) of gabapentin that is
9
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
approximately 15% to approximately 85% less than Cm,_, for a comparable dose
of immediate release
gabapentin also administered as a single dose, and wherein upon
administration, bioavailability (AUC
from 0 to 24 hours at steady-state ) of the gabapentin at 2400 mg is
approximately 80% to
approximately 150 lo of the AUC for a comparable total daily dose of immediate
release gabapentin.
[0060] In one embodiment, the medicament, upon administration to a patient as
a single dose,
results in blood plasma levels of the patient that exhibit a maximum
concentration (Cm,,,) of
gabapentin that is approximately 30% to approximately 50% less than Cm~.' for
a comparable dose of
immediate release gabapentin also administered as a single dose.
[0061] The invention also relates to the use of a medicament comprising a
therapeutically
effective amount of a GABA analog in a gastric retentive dosage form to reduce
or eliminate side
effects associated with y-aminobutyric acid (GABA) analogs, wherein the
medicament is to be
administered to a patient in a once-daily dosing regimen within a single 24
hour period or in a twice
daily dosing regimen in which the dose in the morning and dose in the evening
are not equal, also
within a single 24 hour period, and wherein said medicament upon
administration to a patient as a
single dose results in blood plasma levels of the patient that exhibit a
maximum concentration (Cm,,.,)
of gabapentin that is approximately 15% to approximately 85 l0 less than Cmj"
for a comparable dose
of immediate release gabapentin also administered as a single dose, and
wherein upon administration,
bioavailability (AUC from 0 to 24 hours at steady-state ) of the gabapentin at
1800 mg is
approximately 80% to approximately 150% of the AUC for a comparable total
daily dose of
immediate release gabapentin.
[0062] In one embodiment, the medicament, upon administration to a patient as
a single dose,
results in blood plasma levels of the patient that exhibit a maximum
concentration (Cm~) of
gabapentin that is approximately 30% to approximately 50% less than Cm2,., for
a comparable dose of
immediate release gabapentin also administered as a single dose.
[0063] The invention also relates to the use of a medicament comprising a
therapeutically
effective amount of a GABA analog in a gastric retentive dosage form to treat
a patient suffering from
a pain state, wherein said medicament is to be administered to a patient in a
once-daily dosing
regimen within a single 24 hour period or in a twice daily dosing regimen in
which the dose in the
morning and dose in the evening are not equal, also within a single 24 hour
period, and wherein said
medicament upon administration to a patient as a single dose results in blood
plasma levels of the
patient that exhibit a maximum concentration (Cm,,,) of gabapentin that is
approximately 15% to
approximately 85% less than C.,,, for a comparable dose of immediate release
gabapentin also
administered as a single dose, and wherein upon administration,
bioavailability (AUC from 0 to 24
hours at steady-state ) of the gabapentin at 2400 mg is approximately 70% to
approximately 150% of
the AUC for a comparable total daily dose of immediate release gabapentin.
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
[0064] For example, the medicament, upon administration to a patient as a
single dose, results in
blood plasma levels of the patient that exhibit a maximum concentration
(Cm;,,) of gabapentin that is
approximately 30% to approximately 50% less than Cmax for a comparable dose of
immediate release
gabapentin also administered as a single dose.
[0065] The invention also relates to the use of a medicament comprising a
therapeutically
effective amount of a GABA analog in a gastric retentive dosage form to treat
a patient suffering from
a pain state, wherein said medicament is to be administered to a patient in a
once-daily dosing
regimen within a single 24 hour period or in a twice daily dosing regimen in
which the dose in the
morning and dose in the evening are not equal, also within a single 24 hour
period, and wherein said
medicament upon administration to a patient as a single dose results in blood
plasma levels of the
patient that exhibit a maximum concentration (Cm,,,) of gabapentin that is
approximately 15% to
approximately 85% less than Cm,,, for a comparable dose of immediate release
gabapentin also
administered as a single dose, and wherein upon administration,
bioavailability (AUC from 0 to 24
hours at steady-state ) of the gabapentin at 1800 mg is approximately 70% to
approximately 150% of
the AUC for a comparable total daily dose of immediate release gabapentin.
[00661 For example, the medicament, upon administration to a patient as a
single dose results in
blood plasma levels of the patient that exhibit a maximum concentration
(Cm,,.) of gabapentin that is
approximately 30% to approximately 50% less than Cm. for a comparable dose of
immediate release
gabapentin also administered as a single dose.
[0067] In one embodiment, upon administration, bioavailability (AUC) of the
GABA analogue at
1800 mg is approximately 70% to approximately 130% of the AUC for a comparable
total daily dose
of immediate release gabapentin.
[0068] In another embodiment, upon administration, bioavailability (AUC) of
the GABA
analogue at 1800 mg is approximately 80% to 150% of the AUC for a comparable
total daily dose of
immediate release gabapentin.
[00691 In one embodiment, upon administration, bioavailability (AUC) as
measured over 24
hours of the GABA analogue at 1800 mg is approximately 80% to approximately
150% of the ACJC
for a comparable total daily dose of immediate release gabapentin.
BRIEF DESCRIPTION OF THE IJ'RAWINGS
[0070] FIG. 1 illustrates the dissolution profiles for three gastric retentive
gabapentin
formulations.
[0071] FIG. 2 illustrates the average plasma profile of three gastric
retentive formulations and the
immediate release gabapentin capsule dosage form sold under the trade name
NEURONTIN 0.
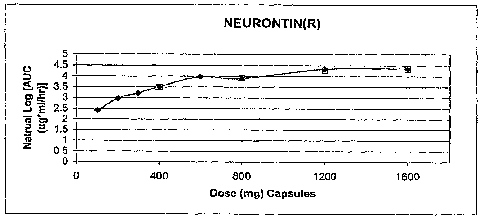
[0072] FIGS. 3 and 5 illustrate the in vivo AUC versus dose for immediate
release
NEURONTIN .
11
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
[0073] FIGS. 4 and 6 illustrate the in vivo AUC versus dose for the gastric
retentive gabapentin
dosage form of the present invention.
DETAILED DESCRIPTION OF THE INVENTION
[0074] DEFINITIONS
[0075] Before describing the present invention in detail, it is to be
understood that the
terminology used herein is for the purpose of describing particular
embodiments only, and is not
intended to be limiting.
[0076] It must be noted that as used in this specification and the appended
claims, the singular
forms "a," "an" and "the" include plural referents unless the context ctearly
dictates otherwise; thus,
for example, reference to "an active agent" or "a pharmacologica.lly active
agent" includes a single
active agent as well a two or more different active agents in combination,
reference to "a polymer"
includes mixtures of two or more polymers as well as a single polymer, and the
like.
[0077] In describing and claiming the present invention, the following
terminology will be used
in accordance with the definitions set out below.
[0078] The terms "effective amount" or a 'therapeutically effective amount"
refer to the amount
of drug or pharmacologically active agent to provide the desired effect
without toxic effects.
100791 The terms "drug," "active agent," and "pharmacologically active agent"
are used
interchangeably herein to refer to any chemical compound, complex or
composition that is suitable for
oral administration and that has a beneficial biological effect, preferably a
therapeutic effect in the
treatment or prevention of a disease or abnormal physiological condition. The
terms also encompass
pharmaceutically acceptable, pharmacologically active derivatives of those
active agents specifically
mentioned herein, including, but not limited to, salts, esters, amides,
prodrugs, active metabolites,
analogs, and the like. When the terms "active agent," "pharmacologically
active agent," and "drug"
are used, then, or when a particular active agent is specifically identified,
it is to be understood that
applicants intend to include the active agent per se as well as
pharmaceutically acceptable,
pharmacologically active salts, esters, amides, prodrugs, metabolites,
analogs, etc.
[0080] The term "dosage form" refers to the physical formulation of the drug
for administration
to the patient. Dosage forms include without limitation, tablets, capsules,
caplets, liquids, syrups,
lotions, lozenges, aerosols, patches, enemas, oils, ointments, pastes, powders
for reconstitution,
sachets, solutions, sponges, and wipes. Within the context of the present
invention, the gabapentin
formulation will generally be administered to patients in the form of tablets
or capsules, although a
liquid formulation is also contemplated under the invention.
[0081] The term "dosage unit" refers to a single unit of the dosage form that
is to be administered
to the patient. The dosage unit will be typically formulated to include an
amount of drug sufficient to
achieve a therapeutic effect with a single administration of the dosage unit
although where the size of
the dosage form is at issue, more than one dosage unit may be necessary to
achieve the desired
12
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
therapeutic effect. For example, a single dosage unit of a drug is typically,
one tablet, one capsule, or
one tablespoon of liquid. More than one dosage unit may be necessary to
administer sufficient drug
to achieve a therapeutic effect where the amount of drug causes physical
constraints on the size of the
dosage form. For example, within the context of the gastric retentive
gabapentin dosage form of the
present invention, where the therapeutic effective amount of gabapentin is
1800 mg, the patient would
be required to take multiple dosage units of gabapentin because a single
dosage unit of 1800 mg of
gabapentin would be too large for a patient to swallow without discomfort. In
such a situation, the
patient would take three 600 mg tablets or capsules or two 900 mg tablets or
capsules of the
gabapentin in order to achieve the 1800 mg therapeutic dose. It is to be
understood that the dosage
units of the gastric retentive gabapentin of the present invention is not
restricted to any particular size
dosage unit (such as the 600 and 900 mg tablets or capsules discussed above)
and that any dosage unit
of a size that would not be restrictive for comfortable ingestion is
contemplated under the present
invention. As an alternative to administering a plurality of 300-900 mg
tablets or capsules, a large
dose of gabapentin could be prepared in a single large dosage unit that is cut
in half at the time of
administration. Thus, with the 1800 mg therapeutic dose, a tablet of 1800 mg
could be prepared that
could be cut in half or in thirds in order to make the 1800 mg dosage unit
more easily ingested.
[0082] "Total daily dose" is the total amount of drug administered to the
patient in one 24 hour
period, regardless of whether the protocol calls for a once-daily, twice-
daily, or thrice-daily
administration of the drug. Thus, the total amount of drug is summed for a
given 24 hour period to
determine how much total drug the patient is to be administered in a given
day. For gabapentin, the
maximum daily total dose deemed reasonable is about 9600 mg with the most
common daily doses of
gabapentin being in the range of 1800 mg to 2400 mg daily; however, it is to
be understood that the
amount of gabapentin to be administered to a particular patient will vary due
to the extent of the
patient's symptoms requiring treatment, the patient's tolerance for gabapentin
or drugs in general, the
size of the patient, and various other factors that one of ordinary skill in
the art must take into
consideration.
[0083] The term "asymmetric dose" refers to the administration of more than
one unequal doses
of a particular drug in a 24 hour period. For example, two asymmetric doses of
a particular drug are
administered in a 24 hour period. Asymmetric doses are typically administered
as a small dose in the
morning and a proportionally larger dose in the evening. Within the context of
the present invention,
a morning dose of the gastric retentive gabapentin of the present invention
may be about one-half,
one-third, or one-fourth the evening dose. Exemplary asymmetrical doses of the
gastric retentive
gabapentin of the present invention may be 600 mg in the morning and 1200 mg
in the evening (1800
total daily dose), 800 mg in the morning and 1500 mg in the evening (2300
total daily dose), 1000 mg
in the morning and 2400 mg in the evening (3400 total daily dose), 800 mg in
the morning and 3600
mg in the evening (4400 total daily dose), or 600 mg in the morning and 6000
mg in the evening
13
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
(6600 total daily dose). While an asymmetric dosing regimen for gabapentin
will generally be
administered with the smaller dose in the morning and the larger dose in the
evening, there may be
situations where the morning dose may need to exceed the evening dose for
reasons based on the
needs of the patient, the degree of the patient's symptoms, and other factors
determined by the
patient's physician. By contrast, the term "symmetric dose" refers to the
administration of two equal
doses in a 24 hour period, such as for example, 300 mg of a given drug in the
morning and 300 mg in
the evening.
[0084] "Titration" is the process of ramping up the total daily amount of drug
administered to the
patient. "Titration" allows the patient's body to get used to the higher dose,
and ensures that the
patient is prepared for subsequent higher doses of the drug through a
succession of daily doses that are
of increasing amount. For example, with gabapentin, where the maintenance dose
is 1500 mg, the
titration protocol might be 300 mg the first day, 600 mg the second say, 900
mg the third day, 1200
mg the fourth day, and 1500 mg the fifth day. In this way, a titration
schedule of 5 days can serve to
adjust the patient to a maintenance dose of 1500 mg.
[0085] "Weaning," which is also referred to as "tapering," is the process of
reducing the daily
total dose a patient is receiving from the maintenance dose to a lesser dose.
"Weaning" occurs when a
patient is experiencing fewer of the symptoms requiring treatment or the
treating physician would like
to test whether the patient can reduce a maintenance dose. Weaning is
effectively the opposite of
titration, and occurs by successively reducing a daily maintenance dose to a
lower level. Weaning can
occur down to 0 mg of drug, depending on whether the patient is in fact ready
to completely stop
taking the medication.
[0086] "Maintenance" is the dosage amount that the patient needs to reach and
maintain a
desired level of relief from the symptoms under treatment. The maintenance
dose is generally a daily
dosage amount, such as, for example 1200 mg, 1500 mg, 1800mg, or 2400 mg. The
maintenance
dose is generally titrated to and maintained for a designated period of time.
As discussed above,
maintenance doses may also be diminished by weaning. As is known by those of
ordinary skill in the
art, maintenance doses should be set to minimize any side effects of the drug.
[0087] The term "controlled release" is intended to refer to any dosage form
in which release of
the drug is not immediate, i.e., with a "controlled release" formulation, oral
administration does not
result in immediate release of.the drug into an absorption pool. The term is
used interchangeably with
"nonimmediate release" as defined in Remington: The Science and Practice of
Pharmacy, 20'h edition
(Lippincott Williams & Wilkins, 2000). Examples of controlled release dosage
forms include
"delayed release," "sustained or extended release," and "modified release"
dosage forms. As
discussed therein, immediate and non-immediate release can be defined
kinetically by reference to the
following equation:
14
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
Dosage kr Absorption ka r. Target ke
-s- ----- ----~
Form drug Pool absorption Area elimination
release
The "absorption pool" represents a solution of the drug administered at a
particular absorption site,
and kr, ka and ke are first-order rate constants for (1) release of the drug
from the formulation, (2)
absorption, and (3) elimination, respectively. For immediate release dosage
forms, the rate constant
for drug release k, is far greater than the absorption rate constant ka. For
controlled release
formulations, the opposite is true, i.e., k, ka, such that the rate of
release of drug from the dosage
form is the rate-limiting step in the delivery of the drug to the target area.
It should be noted that this
simplified model uses a single first order rate constant for release and
absorption, and that the
controlled release kinetics with any particular dosage form may be much more
complicated. In
general, however, the term "controlled release" as used herein includes any
nonimmediate release
formulation.
[00881 "Delayed release" dosage forms are a category of modified release
dosage forms in which
the release of the drug is delayed after oral administration for a finite
period of time after which
release of the drug is unhindered. Delayed release dosage forms are frequently
used to protect an
acid-labile drug from the low pH of the stomach or where appropriate to target
the GI tract for local
effect while minimizing systemic exposure. Enteric coating is frequently used
to manufacture
delayed release dosage forms.
[0089] The terms "sustained release," and "extended release" are used
interchangeably herein to
refer to a dosage form that provides for gradual release of a drug over an
extended period of time.
With extended release dosage forms, the rate of release of the drug from the
dosage form is reduced in
order to maintain therapeutic activity of the drug for a longer period of time
or to reduce any toxic
effects associated with a particular dosing of the drug. Extended release
dosage forms have the
advantage of providing patients with a dosing regimen that allows for less
frequent dosing, thus
enhancing compliance. Extended release dosage forms can also reduce peak-
related side effects
associated with some drugs and can maintain therapeutic concentrations
throughout the dosing period
thus avoiding periods of insufficient therapeutic plasma concentrations
between doses.
[0090] The term "modified release" refers to a dosage form that includes both
delayed and
extended release drug products. The manufacture of delayed, extended, and
modified release dosage
forms are known to ordinary skill in the art and include the formulation of
the dosage forms with
excipients or combinations of excipients necessary to produce the desired
active agent release profile
for the dosage form.
[0091] The "gastric retentive" oral dosage forms described herein are a type
of extended release
dosage form. Gastric retentive dosage forms are beneficial for the delivery of
drugs with reduced
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
absorption in the lower GI tract or for local treatment of diseases of the
stomach or upper GI tract.
For example, in certain embodiments of gastric retentive oral dosage forms of
the present invention,
the dosage form swells in the gastric cavity and is retained in the gastric
cavity of a patient in the fed
med so that the drug may be released for heightened therapeutic effect. See,
Hou et al., Crit. Rev.
Ther. Drug Carrier Syst. 20(6):459-497 (2003).
[0092] The term "AUC" (i.e., "area under the curve," "area under the
concentration curve," or
"area under the concentration-time curve") is a pharmacokinetic term used to
refer a method of
measurement of bioavailability or extent of absorption of a drug based on a
plot of an individual or
pool of individual's blood plasma concentrations sampled at frequent
intervals; the AUC is directly
proportional to the total amount of unaltered drug in the patient's blood
plasma. For example, a linear
curve for a plot of the AUC versus dose (f.e., straight ascending line)
indicates that the drug is being
released slowly into the blood stream and is providing a steady amount of drug
to the patient; if the
AUC versus dose is a linear relationship this generally represents optimal
delivery of the drug into the
patient's blood stream. By contrast, a non-linear AUC versus dose curve
indicates rapid release of
drug such that some of the drug is not absorbed, or the drug is metabolized
before entering the blood
stream. Within the context of the present invention, FIGS. 3-6 show the
difference between the AUC
as a function of dose for immediate release gabapentin (NEURONTIN ) (FIGS. 3
and 5) versus the
AUC as a function of dose for the gastric retentive gabapentin of the present
invention (FIGS. 4 and
6). The data from Table 6 and FIG. 2 indicates that gastric retentive
gabapentin has an AUC that is
approximately equivalent to comparable doses of immediate release gabapentin
while the graphs set
forth at FIGS. 3-6 indicate that gastric retentive gabapentin may actually
have greater bioavailability
than comparable doses of immediate release gabapentin.
[0093] The term "Cmax" (i.e., "maximum concentration") is a pharmacokinetic
term used to
indicate the peak concentration of a particular drug in the blood plasma of a
patient. Within the
context of the present invention, for immediate release formulations of
gabapentin, the Cm. is
generally higher than the Cma,, of gastric retentive gabapentin because the
latter releases the drug more
slowly than the former and thus, the gastric retentive gabapentin at the same
dose does not achieve a
peak concentration as high as the comparable dose of immediate release
gabapentin. As shown in
FIG. 2, and Table 6 of Example 4, the Cma,, of gastric retentive gabapentin
(600 mg) is approximately
30% to approximately 50% less than the Cma., for a comparable dose of
immediate release gabapentin.
[0094] The term "Tm." (i.e., "time of maximum concentration" or "time of Ca,")
is a
pharmacokinetic term used to indicate the time at which the Cm.. is observed
during the time course of
a drug administration. Within the context of the present invention, Tm. is
also longer for gastric
retentive gabapentin when compared to immediate release gabapentin. As shown
in FIG. 2 and Table
6 of Example 4, the Tma, for gastric retentive gabapentin (600 mg) is
approximately 1.5 to
approximately 3.5 hours longer (equivalent to approximately 40% to
approximately 80% slower) than
16
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
the T.,,,,, for a comparable dose of immediate release gabapentin. As would be
expected, a dosage
form that would include an immediate release as well as a gastric retentive
component would have a
Tm,., that is higher than the Tma,, for an immediate release dosage form, but
lower than the Tm,,, for a
purely gastric retentive dosage form.
[00951 The term "half-life" is a pharmacokinetic term used to indicate the
length of time
necessary to eliminate 50% of the remaining amount of drug present in the
body.
[0096] By "pharmaceutically acceptable," such as in the recitation of a
"pharmaceutically
acceptable carrier," or a"pharmaceutically acceptable acid addition salt," is
meant a material that is
not biologically or otherwise undesirable, i.e., the material may be
incorporated into a pharmaceutical
composition administered to a patient without causing any undesirable
biological effects or interacting
in a deleterious manner with any of the other components of the composition in
which it is contained.
The term "pharmacologically active" (or simply "active ) as in a
"pharmacologically active"
derivative, refers to a derivative having the same type of pharmacological
activity as the parent
compound and approximately equivalent in degree. When the term
"pharmaceutically acceptable" is
used to refer to a derivative (e.g., a salt) of an active agent, it is to be
understood that the compound is
pharmacologically active as well. When the term, "pharmaceutically acceptable"
is used to refer to an
excipient, it implies that the excipient has met the required standards of
toxicological and
manufacturing testing or that it is on the Inactive Ingredient Guide prepared
by the FDA, or
comparable agency.
[0097] The term "soluble" as used herein refers to a drug having an aqueous
solubility (measured
in water at 20 C) greater than 10%, preferably greater than 20%, by weight.
The terms "slightly
soluble" and "sparingly soluble" refer to a drug having an aqueous solubility
(measured at 20 C) in
the range of 2% to 10% by weight, while drugs having an aqueous solubility in
the range of 0.001 1o to
less than 2% by weight are referred to as "substantially insoluble."
[0098] The terms "hydrophilic" and "hydrophobic" are generally defined in
terms of a partition
coefficient P; which is the ratio of the equilibrium concentration of a
compound in an organic phase to
that in an.aqueous phase. A hydrophilic compound has a P value less than 1.0,
typically less than
about 0.5, where P is the partition coefficient of the compound between
octanol and water, while
hydrophobic compounds will generally have a P greater than about 1.0,
typically greater than about
5Ø The polymeric carriers herein are hydrophilic, and thus compatible with
aqueous fluids such as
those present in the human body.
[0099] The term "polymer" as used herein refers to a molecule containing a
plurality of
covalently attached monomer units, and includes branched, dendrimeric, and
star polymers as well as
linear polymers. The term also includes both homopolymers and copolymers,
e.g., random
copolymers, block copolymers and graft copolymers, as well as uncrosslinked
polymers and slightly
17
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
to moderately to substantially crosslinked polymers, as well as two or more
interpenetrating cross-
linked networks.
[00100] The term "vesicle" as used herein refers to a small (e.g., 0.01 to 1.0
mm), usually
spherical structure that may contain or be composed of either lipoidal or
aqueous material, or both.
Suitable vesicles include, but are not limited to, liposomes, nanoparticles,
and microspheres composed
of amino acids. While vesicles are usually membrane-bound, they need not
necessarily be membrane
bound and within the context of the present invention, the term "vesicle"
includes both membrane-
bound and non-membrane-bound structures.
1001011 The tenns "swellable" and "bioerodible" (or simply "erodible") are
used to refer to the
polymers used in the present dosage forms, with "swellable" polymers being
those that are capable of
absorbing water and physically swelling as a result, with the extent to which
a polymer can swell
being determined by the molecular weight or degree of crosslinking (for
crosslinked polymers), and
"bioerodible" or "erodible" polymers referring to polymers that slowly
dissolve and/or gradually
hydrolyze in an aqueous fluid, and/or that physically disentangle or undergo
chemical degradation of
the chains themselves, as a result of movement within the stomach or GI tract.
[00102] The in vivo "release rate' and in vivo "release profile" refer to the
time it takes for the
orally administered dosage form, or the active agent-containing layer of a
bilayer or multilayer tablet
(administered when the stomach is in the fed mode) or the content of the
active ingredient to be
reduced to 0-10%, preferably 0-5%, of its original size or level, as may be
observed visually using
NMR shift reagents or paramagnetic species, radio-opaque species or markers,
or radiolabels, or
determined mathematically, such as deconvolution, upon its plasma
concentration profiles.
[00103] The term "fed mode," as used herein, refers to a state which is
typically induced in a
patient by the presence of food in the stomach, the food-giving rise to two
signals, one that is said to
stem from stomach distension and the other a chemical signal based on food in
the stomach. It has
been determined that once the fed mode has been induced, larger particles are
retained in the stomach
for a longer period of time than smaller particles; thus, the fed mode is
typically induced in a patient
by the presence of food in the stomach.
[00104] In the normal digestive process, the passage of matter through the
stomach is delayed by a
physiological condition that is variously referred to as the digestive mode,
the postprandial mode, or
the "fed mode." Between fed modes, the stomach is in the interdigestive or
"fasting" mode. The
difference between the two modes lies in the pattern of gastroduodenal motor
activity.
[00105] In the fasting mode, the stomach exhibits a cyclic activity called the
"interdigestive
migrating motor complex" or 'IMMC". This activity occurs in four phases:
[00106] Phase I, which lasts 45 to 60 minutes, is the most quiescent, with the
stomach
experiencing few or no contractions;
18
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
[00107] Phase II, characterized by sweeping contractions occurring in an
irregular intermittent
pattern and gradually increasing in magnitude;
[00108] Phase III, consisting of intense bursts of peristaltic waves in both
the stomach and the
small bowel, lasting for about 5 to 15 minutes; and
[00109] Phase IV is a transition period of decreasing activity which lasts
until the next cycle
begins.
[00110] The total cycle time for all four phases is approximately 90 minutes.
The greatest activity
occurs in Phase III, when powerful peristaltic waves sweep the swallowed
saliva, gastric secretions,
food particles, and particulate debris, out of the stomach and into the small
intestine and colon. Phase
III thus serves as an intestinal housekeeper, preparing the upper tract for
the next meal and preventing
bacterial overgrowth.
[00111] The fed mode is initiated by nutritive materials entering the stomach
upon the ingestion of
food. Initiation is accompanied by a rapid and profound change in the motor
pattern of the upper GI
tract, over a period of 30 seconds to one minute. The change is observed
almost simultaneously at all
sites along the G.I. tract and occurs before the stomach contents have reached
the distal small
intestine. Once the fed mode is established, the stomach generates 3-4
continuous and regular
contractions per minute, similar to those of the fasting mode but with about
half the amplitude. The
pylorus is partially open, causing a sieving effect in which liquids and small
particles flow
continuously from the stomach into the intestine while indigestible particles
greater in size than the
pyloric opening are retropelled and retained in the stomach. This sieving
effect thus causes the
stomach to retain particles exceeding about 1 cm in size for approximately 4
to 6 hours.
[00112] Within the context of the present invention, the term "pain" as used
herein refers to a pain
state experienced by a human individual or a mammal (also referred to as
a"patient" herein) that
includes a non-nociceptive pain, i.e., a neuropathic pain, a sympathetic pain,
or both. As used herein,
the term "pain" is also intended to include a mixed pain syndrome that
includes a nociceptive pain
state in addition to a non-nociceptive pain state. Examples of neuropathic
pain include, without
limitation, diabetic neuropathy, HIV sensory neuropathy, post-herptic (or post-
shingles) neuralgia,
post-thoracotomy pain, trigeminal neuralgia, radiculopathy, neuropathic pain
associated with
chemotherapy, reflex sympathetic dystrophy or causalgia also kn'own as nerve
damage (for example
carpal tunnel syndrome), back pain, peripheral neuropathy (known as widespread
nerve damage
experienced in the limbs and regions extending from the central nervous
system), entrapment
neuropathy (e.g., carpel tunnel syndrome), phantom limb pain, and complex
regional pain syndrome.
As previously noted, sympathetic pain occurs most commonly after fractures and
soft tissue injuries
of the arms and legs. Because pain is difficult to define and characterize, it
is to be understood that a
patient being treated for a particular non-nociceptive pain state, such as the
neuropathic pain condition
of neuralgia, may also be experiencing a sympathetic pain condition or a
nociceptive pain condition.
19
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
In this respect, the term "pain" as used herein is used to include a mixed
syndrome pain that includes
mixed syndrome non-nociceptive pain (i.e., pain that includes both neuropathic
and sympathetic pain)
or a nociceptive pain that accompanies a non-nociceptive pain state. Examples
of mixed syndrome
non-nociceptive pain are pain associated with post-menopausal symptoms, or
pain associated with
chronic pelvic pain syndrome. A migraine headache is considered to be one
example of a mixed
syndrome pain state that is a mixture of neuropathic and somatic (i.e.,
nociceptive) pain.
[00113] With respect to pain, the terms "treating" and "treatment" as used
herein refer to
reduction in severity and/or frequency of pain symptoms, elimination of pain
symptoms and/or the
underlying cause for pain symptoms and the improvement or remediation of
damage caused by the
pain symptoms. With respect to other conditions or diseases, the terms
"treating" and "treatment"
includes the following actions inhibiting the disease, i.e., arresting its
development; or relieving the
disease, i.e., causing regression of the disease.
[00114] Prevention includes delaying or inhibiting the occurrence of pain
symptoms and/or their
underlying cause, e.g., delaying or inhibiting the disease from occurring in a
subject which may be
predisposed to the disease, but has not yet been diagnosed as having it.
[00115] ACTIVE AGENTS
[00116] Active agents that may be used with the dosage forms of the present
invention include the
GABA analog gabapentin and the related GABA analog pregabalin (S)-(3-
aminomehtyl)-5-
methylhexoanoic acid) or (S)-isobutyl GABA. In addition to gabapentin and
pregabalin, it is
contemplated that other GABA analogs may be used as active agents in the
manufacture of the dosage
forms and the methods described herein. Examples of other GABA analogs that
may be used within the
context of the present invention include, without limitation, cis-(1-S, 3R)-(1-
aminomethyl)-3-
methylcyclohexane)acetic acid; 1a, 3a, 5a-(1-aminomethyl)-(3,5-
dimethylcyclohexane)acetic acid;
(9-(aminomethyi)bicyclo[3.3.1]non-9-yl)acetic acid; and (7-
(aminomethyl)bicycle[2.2.1]hept-7-
yl)acetic acid. Bryans et al. J MED CHEM 41:1838-1845 (1998); Bryans et al.,
MED RES REv 19:149-
177 (1999).
[00117] Within the context of the present invention, the GABA analogs are
preferably used in free
amphoteric form, including zwitterionic form; however, pharmaceutically
acceptable salt forms,
hydrates or solvates that retain the biological effectiveness and properties
of gabapentin, pregabalin,
and/or other GABA analogs are not biologically or otherwise undesirable, can
also be used and may
show superior bioavailability. In this respect, as used herein, the terms
"gabapentin" and "pregabalin"
and "GABA analogs" are intended to include the agent itself, as well as its
pharmaceutically
acceptable salts.
[00118] Pharmaceutically acceptable salts may be amphoteric and may be present
in the form of
internal salts. In this respect, the gabapentin, pregabalin, and other GABA
analogs described herein
may form acid addition salts and salts with bases. Exemplary acids that can be
used to form such salts
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
include, by way of example and not limitation, mineral acids such as
hydrochloric, hydrobromic,
sulfuric or phosphoric acid or organic acids such as organic sulfonic acids
and organic carboxylic
acids. Salts formed with inorganic bases include, for example, the sodium,
potassium, lithium,
ammonium, calcium, and magnesium salts. Salts derived from organic bases
include, for example,
the salts of primary, secondary and tertiary amines, substituted amines
including naturally-occurring
substituted amines, and cyclic amines, including isopropylamine,
trimethylamine, diethylamine,
triethylamine, tripropylamine, ethanolamine, 2-dimethyl aminoethanol,
tromethamine, lysine,
arginine, histidine, caffeine, procaine, hydrabamine, choline, betaine,
ethylenediamine, glucosamine,
N-alkylglucamines, theobromine, purines, piperazine, piperidine, N-
ethylpiperidine, fumarate,
maleate, succinate, acetate and oxalate. The invention also contemplates
administering one or more
additional therapeutic agents with the gabapentin treatment. The selection of
these additional
therapeutic agents will depend upon the specific disease state or condition
that is being treated.
[00119] Additional therapeutic agents that can be used with the gastric
retentive gabapentin of the
present invention to treat any of the various pain states described above,
include anticonvulsants,
tricyclic antidepressants, opioids, and secondary analgesics. Examples of
suitable anticonvulsants
include carbamazepine, phenytoin, and lamotrigine. Examples of suitable
tricyclic antidepressants
include amitriptyline, imipramine, clomipramine, and desipramine. Examples of
suitable opioids
include oxycodone and tramadol.
[00120] For those embodiments of the invention where the gabapentin gastric
retentive dosage
form is administered for prophylactic treatment of migraine headaches, such
additional therapeutic
agents can be selected from the group consisting of tricyclic antidepressants
(amitriptyline, doxepin,
imipramine, maprotiline, protriptyline, desipramine), SSRI (fluoxetine),
triptine (sumatriptan, etc.),
and ergotamine.
[00121] Where the additional therapeutic agent is a secondary analgesic, any
analgesic that would
complement the treatment protocol of the gastric retentive gabapentin of the
present invention can be
administered with gabapentin, either at the same time or at different times in
order to treat the pain
condition at hand. The secondary analgesic would typically be administered at
least once in a 24-hour
period, and can be any analgesic effective for treatment of pain. One type of
analgesic that may be
used in conjunction with the gastric retentive gabapentin of the present
invention is non-steroidal anti-
inflammatory drugs ("NSAIDs").
[00122] DOSING REGIMENS AND PHARMACOKINETICS
[00123] Generally, the frequency of administration of a particular dosage form
is determined to
provide the most effective results in an efficient manner without overdosing
and varies according to
the characteristics of the particular drug, including both its pharmacological
characteristics and its
physical characteristics, such as solubility. In some embodiments, the
characteristics of the swellable
matrix, such as its permeability, and the relative amounts of the drug and
polymer will also affect
21
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
frequency of administration. In most cases, the dosage form is prepared such
that effective results are
achieved with administration once every eight hours, once every twelve hours,
or once every twenty-
four hours. As previously discussed, due to the physical constraints placed on
a tablet or capsule that
is to be swallowed by a patient, most dosage forms can only support a limited
amount of drug within a
single dosage unit.
[00124] In one embodiment, the present invention relates to a method of
treating a pain state, by
administering a therapeutically effective amount of gabapentin, or a
pharmaceutically acceptable salt
thereof, to a patient in need of such treatment in a gastric retentive dosage
form that is administered to
the patient in a once-daily or twice-daily dosing regimen. The method of the
present invention is
useful for treating numerous pain states that are currently being treated with
conventional immediate
release formulations of gabapentin and include, by way of illustration and not
limitation, pain states
exhibiting neuropathic pain, sympathetic pain, a mixture of neuropathic pain
and sympathetic pain, or
a mixture of neuropathic pain or sympathetic pain with nociceptive pain.
[00125] Within the context of the present invention, the gastric retentive
gabapentin of the present
invention has the advantage of improving patient compliance with gabapentin
administration
protocols because the drug is administered in a once-daily or twice=daily
dosing regimen, rather than
the multiple dosing administrations necessary for the immediate release dosage
forms of gabapentin.
For one embodiment of the present invention, when administered in the fed
mode, the gastric retentive
gabapentin dosage forms are retained for a period of time in the stomach and
the release of the drug is
extended beyond the release time of an immediate mlease dosage form. One
embodiment of the
invention relates to a method of administering a therapeutically effective
amount of gabapentin to a
patient in need thereof, comprising administering gabapentin or a
phannaceutically acceptable salt
thereof, in a gastric retentive dosage form once in the morning or evening in
a once a day daily dosing
regimen. Another embodiment comprises administering a therapeutically
effective amount of
gabapentin to a patient in need thereof, comprising administering gabapentin
or a pharmaceutically
acceptable salt thereof, in a gastric retentive dosage fonn twice a day, for
example once in the
morning and once in the evening in a twice a day daily dosing regimen.
[00126] In addition to the foregoing, one embodiment ofthe gastric retentive
dosage forms of the
present invention can lower the maximum plasma concentration of the drug in a
patient's blood when
compared to a similar does of immediate release (IR) gabapentin thereby
reducing any side effects
from the drug. The following discussion references gabapentin for purposes of
illustration and not for
purposes of limiting the dosage forms of the present invention solely to
gabapentin; in this respect,
pregabalin and other GABA analogs are expected to behave similarly to
gabapentin.
[00127] Since gabapentin is absorbed high in the GI (gastrointestinal) tract
by means of a
saturable transport mechanism, a gastric retentive dosage form is particularly
beneficial for delivery of
gabapentin since the dosage form can keep the drug in the region of absorption
for an extended period
22
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
of time and consequently improve bioavailability of the drug. By virtue of the
slower release rate of
the gastric retentive gabapentin of the present invention, saturation of the
carrier-mediated transport of
conventional dosages is avoided. The gastric retentive dosage form of the
present invention is
particularly beneficial for delivery of gabapentin due to its prolonged
transit in the upper GI tract,
which allows the drug to be absorbed adequately in the preferred region of
absorption. Further, as
shown in FIG. 2, the gastric retentive gabapentin dosage forms of the present
invention increase the
T,,,a,, for the drug and lower the Cma,, for the drug, which may result in
reduced incidence and/or
severity of the central nervous system (CNS) side effects of the drug, such as
somnolence, dizziness,
fatigue, ataxia, weight gain, peripheral edema, diarrhea, headache, dry mouth,
blurred vision, and/or
reversible visual field constriction.
[00128] In one embodiment of the present invention, the gastric retentive
gabapentin is
administered in a once-daily dosing regimen with a total daily dose of
gabapentin ranging from about
300 mg/day to about 9600 mg/day, depending on the disease state or condition
that is being treated.
[00129] In another embodiment of the invention, the gastric retentive
gabapentin is administered
in the morning and evening in a twice a day daily dosing regimen with a total
daily dose of gabapentin
ranging from about 300 mg/day to about 9600 mg/day.
[00130] Where the total daily dose of gabapentin is 1000 mg or greater, the
patient is preferably
titrated up to the maximum maintenance dose that the patient is capable of
tolerating. Titration is
preferable with both the once-daily and twice-daily dosing regimens.
[00131] . With the twice-daily dosing regimen, the two dosings may be
administered in a
symmetric or asymmetric dosing regimen. With a symmetric dosing regimen, the
morning dose is the
same as the evening dose. Thus, a symmetric dosing regimen may consist of 300
mg of gabapentin in
the morning and 300 mg of gabapentin in the evening for a total daily dose of
600 mg of gabapentin
for a sirigle 24 hour period. With gabapentin, symmetric dosing regimens are
best used where lower
dosages of gabapentin are being used for treating a particular disease state
or condition. With the
twice-daily dosing regimen in an asymmetric dosing regimen, the morning and
evening doses will not
be the same. Where high doses of gabapentin are being used to treat a
particular disease state or
condition, asymmetric dosing regimens are preferred. Examples of asymmetric
dosing regimens can
be, for example, 300 mg in the morning and 1200 mg in the evening for.a total
daily dose of 1500
mg/day; 600 mg in the morning and 3600 mg in the evening for a total daily
dose of 4200 mg/day; or
900 mg in the morning and 6000 mg in the evening for a total daily dose of
6900 mg/day. Differential
dosing levels may also be used for a dosing regimen, such as for example,
where different dosages are
administered for the morning and evening doses during the asymmetric treatment
course. Under this
scenario, a patient may have a decreasing morning dose and an increasing
evening dose, or an
increasing morning dose and a decreasing evening dose during a particular part
of the dosing regimen.
23
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
[00132] Individual dosage units for both the once-daily and twice-daily dosing
regimens will
generally contain from about 100 mg to about 1800 mg of gabapentin per dosage
unit. Presently, any
dosing regimen for the gastric retentive gabapentin of the present invention
must take into
consideration both the amount of the gabapentin in a single dosage unit and
the number of tablets or
capsules that can be consumed together to reach the desired daily dose and/or
maintenance dose. For
example, for a dosing regimen comprised of a once-daily dosing of 1800 mg of
gastric retentive
gabapentin, three 600 mg tablets or a 1200 mg tablet or capsule may be taken
together with a 600 mg
tablet at the evening meal. If the patient finds that the 1200 mg tablet is
too large, then the patient
may take two 600 mg tablets or three 400 mg tablets. For a dosing regimen
comprised of a twice-
daily dosing of 1800 mg of gastric retentive gabapentin, 600 mg may be taken
with a morning meal
and 1200 mg (in one or multiple dosage units) may be taken with an evening
meal.
[00133] For all modes of administration, for the embodiment of the present
invention for which
the gastric retentive dosage is a swellable matrix which relies upon dosing in
the fed mode to increase
the gastric residence time, the gastric retentive gabapentin dosage forms are
preferably administered
in the fed mode, i.e:, with orjust after consumption of a meal. Because two of
the side effects of
gabapentin are somnolence and dizziness, it is preferable, when possible, for
the patient to take the
once-daily dose or the larger of the twice-daily doses with an evening meal.
In this way, the patient
may avoid the side effects by sleeping through the side effects, thus
permitting better compliance and
optimization of the dosing regimen. When administered in the evening fed mode,
one embodiment of
the gastric retentive gabapentin of the present invention will provide the
patient with continued relief
from the particular disease state or condition under treatment through the
night and into the next day.
The gastric retentive gabapentin dosage form of the present invention is able
to provide relief from the
disease state or condition under treatment for an extended period of time
because the dosage form
allows for both extended release of the gabapentin and the superior absorption
of the drug in the GI
tract.
[00134] As previously discussed, with both the once-daily and twice-daily
dosing regimens
described herein, the total daily dose of gabapentin may be titrated up to a
maximum amount per day,
also called the maintenance dose. The length of time for the titration process
will vary with the
individual patients, but will generally range from approximately two days to
approximately two
weeks. Likewise, where a patient is nearing the completion of a therapeutic
course of gabapentin, the
patient may be weaned off the maintenance dose over a period of days or weeks
so that the patient's
body has a chance to adjust to the reduction of medication slowly.
[00135] That the gastric retentive gabapentin dosage form of the present
invention may be
administered in once-daily or twice-daily dosing regimens as described herein
is particularly
surprising and unexpected when compared to the behavior of immediate release
gabapentin.
Specifically, immediate release gabapentin is not well -absorbed in the colon
and has such a short
24
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
half-life that it must be administered at least three times a day in order to
achieve a desired level of
pain relief. Non gastric-retentive extended release gabapentin formulations
may deliver a significant
fraction of the drug to the lower GI tract, and therefore the drug is not well
absorbed, thereby
decreasing bioavailability of the drug when compared to the bioavailability of
an equivalent
immediate release dose. Thus, gastric retentive dosage forms of gabapentin
allow for extended
delivery of gabapentin without significant loss of bioavailability. However,
the slower absorption of
the gastric retentive gabapentin of the present invention allows for
administration of the drug in a
once or twice daily dosing regimen with improved pain relief and without
exacerbation of the
incidence of adverse side effects. FIG. 2 and Table 6 of Example 4 provide
evidence of the slower
release of gabapentin as compared to immediate release gabapentin into the
bloodstream through Cm,,,
measurements. The data in Tables 6 indicates that gastric retentive gabapentin
has a C,,,,,, that is
approximately 30% to approximately 50% less than the Cma, of a comparable dose
of immediate
release gabapentin.
[00136] In addition to a decrease in Cm,,,, the gastric retentive dosage forms
of the present
invention also show increased Tma., (FIG. 2 and Table 6 of Example 4)
providing further evidence for
the longer lasting effects of the gastric retentive gabapentin of the present
invention when compared
to immediate release gabapentin. The data in Table 6 indicates that 600 mg of
gastric retentive
gabapentin has a Tma,, that is approximately 1.5 to approximately 3.5 hours
longer (equivalent to
approximately 40% to approximately 80% slower) than the Tmax of a comparable
dose of immediate
release gabapentin.
[00137] A further surprising and unexpected feature of the gastric retentive
gabapentin dosage
forms of the present invention is that the gastric retentive dosage forms
enable greater bioavailability
of the higher doses of gabapentin when compared to a comparable dose of
immediate release
gabapentin. Specifically, according to the Summary Basis of Approval for
NEUROTNIN (New
Drug Application ("NDA") 20-235), the normalized AUC(soo-,ns) of 600-mg, 800-
mg, 1200mg and
1600-mg in four subjects were 53%, 37 fa, 38% and 29%, respectively where the
AUC was obtained
at steady-state. For one embodiment of the present invention, the AUCo{,
(ng*hr/ml) (geometric
mean of 19 subjects) values for escalating doses of 600-mg, 1200-mg, 1800-mg
and 2400-mg
administered as single-doses immediately after a meal to healthy volunteers
were 35698, 63209,
90894, and 108572, respectively (rounded to the nearest whole number). Because
the AUCo.W for a
single dose is equivalent to the steady-state AUC as measured between dosing
intervals, the AUC
values in both cases represent the AUCo-., and comparisons can be made.
Therefore, if a ratio of the
AUC to that obtained at 600-mg dose, and normalized for the dose administered
is made, the ratios for
the NEUROTNIN capsules are 1.0, 0.698, 0.717 and 0.547 for the 600-mg, 800-
mg, 1200mg and
1600-mg respectively compared to ratios for one embodiment of the present
invention of 1.0, 0.882,
0.843 and 0.753 for the 600-mg, 1200-mg, 1800-mg and 2400-mg doses,
respectively. Thus, the
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
fraction of the dose that is absorbed decreases by about 50% when the dose is
increased from 600-mg
to 1600 mg for NEUROTNIN capsules. In contrast, for one embodiment of the
present invention,
the fraction of the dose that is absorbed decreases by about 25% when the dose
is increased from 600
mg to 2400 mg, and only 15% at 1800 mg, which is still above the 1600 mg dose
of immediate
release NEURONTIN which demonstrated a loss of almost 50% compared to the 600-
mg dose of
immediate release NEURONTIN . FIGS. 3-6 provide evidence of the enhanced
bioavailability of
the gastric retentive gabapentin of the present invention as compared to
immediate release gabapentin
through AUC measurements. In addition, Example 12 demonstrates comparable AUC
over 24 hours
for the twice -daily gabapentin as compared to three times daily NEURONTIN at
the same total
daily dose of 1800 mg.
[00138] Further, as gabapentin is known to exhibit saturable absorption (i.e.,
where a drug is
absorbed only to the amount of saturation), it is also surprising and
unexpected that the gastric
retentive gabapentin dosage forms of the present invention are capable of
being administered in doses
that are two to six times the doses administered for immediate release
gabapentin while retaining
sufficient bioavailability to attain the therapeutic effects of the
conventional immediate release
gabapentin dosage forms. As shown in FIGS. 4 and 6, the slower absorption of
the gastric retentive
gabapentin of the present invention permits nearly linear absorption over the
range of 600 mg to
2400mg of gastric retentive gabapentin administered at one dosing. FIGS. 3 and
5 show that
immediate release gabapentin (i.e., NEURONTIN ) is saturated at 1200 mg and
thus, is unavailable
to deliver additional drug to the patient at dosages above 1200 mg (although
not all 1200 mg is
absorbed for either case). The saturable absorption mechanism may also explain
the higher loss of
bioavailability observed in the NDA for the NEURONTINOD when compared to the
present invention.
[00139] A particularly beneficial advantage of the once or twice daily dosing
regimens for the
gastric retentive gabapentin of the present invention is that when the once-
daily dosing is
administered in the evening, or when the larger of the twice-daily dosing is
administered in the
evening, the patient is able to experience the therapeutic effects of
gabapentin throughout the night.
As a result of the linear absorption of the drug in the gastric retentive
dosage forms of the present
invention, the gabapentin is released continuously throughout the night thus
providing continuous
therapy. Tables 9-12 of Example 10 shows data obtained from a clinical trial
conducted with the
gastric retentive gabapentin dosage form of the present invention where the
gabapentin was
administered in once daily and twice daily dosing regimens. In addition,
Example 12 demonstrates
data obtained from a clinical trial conducted with the gastric retentive
gabapentin dosage form of the
present, invention where the gabapentin was administered in once daily and
twice daily dosing
regimens.
[00140] Another beneficial advantage of the gastric retentive gabapentin of
the present invention
is that when it is administered at a sufficiently high dose with an evening
meal, the somnolence and/or
26
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
dizziness, typically associated with higher dosages of gabapentin administered
in other ways, are
ameliorated with nighttime sleep. Another advantage of administering a high
evening dose of gastric
retentive gabapentin is that the dosage form will allow for continued therapy
upon waking and
potentially throughout the next day until the next evening administration.
Where appropriate and if
necessary, a small morning dosing (e.g., 300 mg) may be administered to
supplement the larger
evening dosages. There is also an advantage of the reduced gastric motility at
night time over that in
the day time (induced by the fed mode) which has to be maintained for an
extended period of time
(with meals).
[001411 Another advantage of the present invention is that it may allow
patients who are unable to
reach an effective therapeutic dose of gabapentin to be effectively treated
with gabapentin. For those
patients who are unable to reach an effective dose of gabapentin due to
inability to tolerate side
effects, the combination of extended release with a lower maximum
concentration (Cmax) compared
to the equivalent IR dose if both are administered as single doses, as well as
the dosing regimen in
which all or a larger fraction of the dose, is administered after the evening
meal, may lower the
incidence of side effects sufficiently to allow the patient to reach a
therapeutically effective dose.
Moreover, some patients may not be effectively treated with gabapentin because
they experience no
increase in efficacy with increased dose. The present invention will allow for
a larger percentage of
the administered dose to be absorbed at higher doses when compared to
immediate release
gabapentin, and thus may allow some patients to obtain efficacious treatment
who found immediate
release gabapentin insufficient. Thus, the combination of dosing regimen,
increased bioavailability
(when compared to immediate release gabapentin) and the extended release of
gabapentin over time
may allow for reduced incidence of side effects and as a result, improved
efficacy as more patients
may be able to reach an efficacious dose.
[00142] As previously noted, the patient may be titrated up to the maintenance
dose (i.e., the
highest maximum dose allowable or preferred for a patient). Titration may
occur over a period of
days or weeks, depending on the patient's therapeutic needs, the magnitude of
the maintenance dose,
and the patient's apparent tolerance for gabapentin. Titration will generally
be determined by the
administrating practitioner.
[00143] Likewise the patient may be weaned from the high maintenance dose down
to a zero dose
in a gradual process that allows the patient's body to adjust to reduced
medication and to determine
whether the gabapentin therapy is sufficient at the lower dose.
[00144] When the administration of an additional therapeutic agent in addition
to the gabapentin is
desired, the additional active agent may be administered at the same time or
at a different time than
gabapentin. For purposes of facilitating patient compliance, administration of
any of the
aforementioned additional agents at the same time is preferred.
27
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
[00145] DOSAGE Foxms
[00146] There are several drug delivery systems that are suitable for use in
delivering gabapentin
in the method of the invention as they are particularly tailored to be gastric-
retentive dosages, such as
the swellable bilayer described in U.S. Pat. No. 5,232,704 to Franz et al.;
the multilayer tablet with a
band described in U.S. Pat. No. 6,120,803 to Wong et al.; the membrane sac and
gas generating agent
described in U.S. Pat. No. 4,996,058 to Sinnreich; the swellable, hydrophilic
polymer system
described in U.S. Pat. No. 5,972,389 to Shell et al. and WO 98/55107 to Shell
et al.; all of which are
incorporated herein by reference.
[00147] Of particular interest are gastric retentive dosage forms that contain
hydrophilic polymers
that swell to a size such that the dosage form is retained in the fed mode.
For example, the gastric
retentive dosage form can contain polymers with a high swelling capacity such
as polyethylene oxide,
hydroxyethylcellulose, and hydroxypropylmethylcellulose. The polymers are
preferably of a
moderate to high molecular weight (4 x 103 to greater that 107) to enhance
swelling and provide
control of the release of gabapentin. In one embodiment ofthe invention, a
hydroxypropylmethylcellulose polymer of such molecular weight is utilized so
that the viscosity of a
1% aqueous solution is about 4000 cps to greater than 100,000 cps. An example
of suitable
polyethylene oxide polymers are those having molecular weights (viscosity
average) on the order of
2-7 million. A typical dosage form should swell to approximately 115% of its
original volume within
one hour after administration, and at a later time should swell to a volume
that is 130% or more of the
original volume. Fillers, binders, lubricants and other additives may also be
included in the gastric
retentive dosage form, such as are well known to those of skill in the art.
[00148] The gastric retentive dosage forms of the present invention provide
for a drug delivery
profile such that gabapentin, both on an in vivo and in vitro basis, is
delivered for at least 5 hours and
more typically over a time period of about 8-10 hours. In order to provide for
sustained delivery, it is
preferable that at least 40 wt % of gabapentin is retained in the dosage form
after 1 hour, i.e., no more
than 60 wt % of the drug is administered in the first hour. In addition, it
may be desired to utilize a
dosage form that provides for substantially all of the gabapentin to be
delivered over the intended
duration, which is typically about 6-12 hours, where substantially all is
taken to mean at least about 85
(generally the art teaches that substantially all is 80 wt %) of the
gabapentin is administered.
[00149] The gastric retentive gabapentin dosage forms of the present invention
can be made by
techniques that are well established in the art, including wet granulation,
fluid-bed granulation, dry
granulation, direct compression, and so forth.
[00150] In one embodiment of the invention, the gabapentin dosage forms
contain one or more
hydrophilic polymers that swell unrestrained dimensionally to a size such that
the dosage form is
retained in the fed mode. The polymers are preferably of a moderate to high
molecular weight (4 x
103 to greater that 107) to enhance swelling and provide control of the
release of gabapentin. A typical
28
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
dosage form should swell to approximately 115% of its original volume within
one hour after
administration, and at a later time should swell to a volume that is 130% or
more of the original
volume. The molecular weight of the polymer may be selected based upon
viscosity of the polymer
in solution. For example, the polymer may be selected such that a 1% aqueous
solution has a
viscosity in a range of 4000 cps (centipoise) to greater than 100,000 cps.
Examples of such polymers
include, by way of illustration and not limitation, polymers with a high
swelling capacity such as
polyethylene oxide ("PEO"), hydroxyethylcellulose, and
hydroxypropylmethylcellulose ("HPMC"
also known as hypromellose). Examples of suitable PEO polymers are those
having molecular
weights (viscosity average) on the order of 2-7 million.
[00151] In another embodiment of the present invention, there is provided a
gastric retentive
swellable, sustained-release dosage form having a matrix comprised of PEO and
HPMC that releases
gabapentin to the upper GI tract. The dosage form may be a single-layer or a
bilayer tablet. Where
the dosage form is a bilayer tablet, one layer is the active agent containing
layer that releases the
gabapentin while the other layer is a swelling or floating layer. Further
details on the formulation of
gastric retentive swellable, sustained-release may be found in commonly owned
U.S. Patent No.
6,723,340 Gusler et al., which is incorporated herein by reference. Examples
1, 2, and 3 describe the
formulation of gastric retentive swellable gabapentin dosage forms made by dry
granulation with PEO
and I-IPMC.
[00152] A typical dosage form would provide for a drug delivery profile such
that gabapentin both
on an in vivo and in vitro basis is delivered for at least 5 hours, and
typically over a time period of
about 8-10 hours. In order to provide for sustained delivery, it is preferable
that at least 40 wt % of
gabapentin is retained in the dosage form after 1 hour, i.e., no more than 60
wt % of the drug is
administered in the first hour. In addition, it may be desired to utilize a
dosage form that provides for
substantially all of the gabapentin to be delivered over the intended
duration, which is typically about
6-12 hours, where substantially all is taken to mean at least about 85
(generally the art teaches that
substantially all is 80) wt % of the gabapentin is administered.
[001531 In one embodiment of the invention, the gastric retentive dosage form
of gabapentin is a
capsule dosage form that allows for the extended release of gabapentin in the
stomach and comprises:
(a) at least one component that expands on contact with gastric juice and
contains an agent capable of
releasing carbon dioxide or nitrogen, and gabapentin or a pharmaceutically
acceptable salt thereof; (b)
at least one hydrophilic membrane in the form of a sachet which contains
component (a), expands by
inflation, floats on the aqueous phase in the stomach and is permeable to
gastric juice and; (c) capsule
dosage form which contains components (a) and (b) and which disintegrates
without delay in the
stomach under the action of gastric juice. Component (a) may also contain a
pharmaceutically
acceptable hydrophilic swelling agent such as lower alkyl ethers of cellulose,
starches, water-soluble
aliphatic or cyclic poly-N-vinylamides, polyvinyl alcohols, polyacrylates,
polymethacrylates,
29
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
polyethylene glycols and mixtures thereof, as well as other materials used in
the manufacture of
pharmaceutical dosage forms. Further details regarding an example of this type
of dosage form can
be found in U.S. Pat. No. 4,996,058 to Sinnreich.
[00154] In another embodiment of the invention, the gastric retentive dosage
form of gabapentin
is an extended release oral drug dosage form for releasing gabapentin into the
stomach, duodenum
and small intestine of a patient, and comprises: a single or a plurality of
solid particles consisting of
gabapentin or a pharmaceutically acceptable salt thereof dispersed within a
polymer that (i) swells
unrestrained dimensionally by imbibing water from gastric fluid to increase
the size of the particles to
promote gastric retention in the stomach of the patient in which the fed mode
has been induced; (ii)
gradually the gabapentin diffuses or the polymer erodes over a time period of
hours, where the
diffusion or erosion commences upon contact with the gastric fluid; and (iii)
releases gabapentin to
the stomach, duodenum and small intestine of the patient, as a result of the
diffusion or polymeric
erosion at a rate corresponding to the time period. Exemplary polymers include
polyethylene oxides,
alkyl substituted cellulose materials and combinations thereof, for example,
high molecular weight
polyethylene oxides and high molecular weight or viscosity
hydroxypropylmethylcellulose materials.
Further details regarding an example of this type of dosage form can be found
in U.S. Pat. No.
5,972,389 to Shell et al. and WO 9855107 to Shell et al..
[00155] In yet another embodiment, a bi-layer tablet releases gabapentin to
the upper GI tract
from an active cotitaining layer, while the other layer is a swelling or
floating layer. Details of this
dosage may be found in U.S. Pat. No. 5,232,704 to Franz et al. This dosage
form may be surrounded
by a band of insoluble material as described in U.S. Pat. No. 6,120,803 to
Wong et al.
[00156] Another embodiment of the invention uses a gastric retentive
swellable, sustained-release
tablet having a matrix comprised of poly(ethylene oxide) and
hydroxypropylmethylcellulose. This
dosage form is illustrated in Example I and further details may be found in
U.S. Patent Application
Publication No. 20030104053 to Gusler et al.
[00157] In a further embodiment of the present invention, there is provided a
dosage form that is
formulated to have a large enough size so as to provide for prolonged transit
in the upper GI tract.
Such tablets would contain at least 800 mg of gabapentin, typically 800-1200
mg. Materials and
techniques useful for manufacturing these large-sized dosage forms are
described in REMINGTON:
THE SCIENCE AND PRACTICE OF PHARMACY, 20'h edition (Lippincott Williams &
Wilkins, 2000) and
Ansel et al., PHARMACEUTICAL DOSAGE FORMS AND DRUG DELIVERY SYSTEMs, 6d' Ed.
(Media, PA:
Williams & Wilkins, 1995). It is preferred that these large size dosage forms
are either coated with a
membrane or equipped with an osmotic pump system. Suitable membranes include
polymer coatings,
such as cellulose, cellulose acetate, cellulose acetate butyrate, and ethyl
cellulose. Osmotic pumps are
described in U.S. Patent No. 3,845,770 to Theeuwes et al. and U.S. Patent No.
3,977,404 to
Theeuwes.
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
[00158] Numerous materials useful for manufacturing these large-sized dosage
forms are
described in Remington: The Science and Practice of Pharmacy, 20'h edition
(Lippincott Williams &
Wilkins, 2000) and Ansel et al., Pharmaceutical Dosage Forms and Drug Delivery
Systems, 6'h Ed.
(Media, PA: Williams & Wilkins, 1995). Along with gabapentin, the core may
contain
pharmaceutically acceptable additives or excipients to facilitate
manufacturing. These include binders
(e.g., ethyl cellulose, gelatin, gums, polyethylene glycol,
polyvinylpyrrolidone, polyvinylalcohol,
starch, sugars, waxes), coloring agents, diluents (e.g., calcium sulfate,
cellulose, dicalcium phosphate,
kaolin, lactose, mannitol, microcrystalline cellulose, sodium chloride,
sorbitol, starch, sucrose),
flavoring agents, glidants (e_g., colloidal silicon dioxide, talc), and
lubricants (e.g., calcium stearate,
glyceryl behenate, hydrogenated vegetable oils, magnesium stearate,
polyethylene glycol, sodium
stearyl fumarate, stearic acid; stearyl behenate, talc). The core may also
contain pharmaceutically
acceptable additives or excipients that serve to provide desirable physical
characteristics to the dosage
form. These include sweeteners, polymers, waxes, and solubility-retarding
materials. These dosage
forms can be made by techniques that are well established in the art,
including wet granulation, fluid-
bed granulation, dry granulation, direct compression, and so forth.
[00159] In another embodiment of the present invention, there is provided a
film coated dosage
form or a capsule dosage form that allows for the controlled, sustained,
and/or extended release of
gabapentin in the stomach. In this embodiment, the dosage form may have a drug-
containing core
surrounded by a controlled release film coating that provides for controlled,
sustained, or extended
drug release, i.e., continuous diffusion of drug from the core into the upper
GI tract.
[00160] In certain embodiments, the dosage form may have a drug-containing
core surrounded by
a swellable coating. See also, US patent application 20030104062, published
June 5, 2003
(application serial number 10/213,823) as one potential embodiment of a
similar concept.
[00161] The controlled release film coating can also be applied by techniques
that are well
established in the art, for example, by dissolving the material in an
appropriate solvent such as
acetone or methylene chloride and is then applying the coating to the dosage
form core by molding,
air spraying, dipping or brushing a solvent-based solution of the material
onto the core. Materials
suitable for use in controlled release film coatings include by way of
illustration, and not limitation,
mixtures of waxes such as beeswax and carnuba wax, shellac, and zein,
celluloses such as ethyl
cellulose, acrylic resins, cellulose acetates including diacetates and
triacetates and other cellulose
esters, and silicone elastomers. Additional examples are set forth below.
[00162] Of particular interest are controlled release film coating materials
that can form a
semipermeable membrane or coating, which can be porous or non-porous, and
which are permeable to
external fluid, and substantially impermeableto the unsolubilized drug
contained within the core.
Typically, external fluids are aqueous fluids or biological fluids in the
environment of use, such as the
upper GI tract. External fluid passes through the semipermeable membrane into
the core, where it
31
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
solubilizes the drug. The solubilized drug then moves from the core through
the membrane into the
GI tract.
[00163] After application of the controlled release film coating to the core,
a drying step is
required and then a suitable exit means for the gabapentin must be formed
through the semipermeable
membrane. Depending on the properties of the gabapentin and other ingredients
within the internal
compartment and the desired release rate for the dosage form, one or more
orifices for gabapentin
delivery can be formed through the membrane by mechanical drilling, laser
drilling, or the like. The
orifice(s) may range in size from a single large orifice containing
substantially an entire surface of the
dosage form to one or more small orifices selectively located on the surface
of the semipermeable
membrane. One specific embodiment of a semiperrneable membrane-coated core,is
the elementary
osmotic pump. The membrane is provided with one or more delivery orifices,
e.g., pierced with a
laser to create one or more delivery orifices. Fluid passing through the
membrane into the core
generates an osmotic pressure that serves to "pump" the solubilized drug
through the delivery
orifice(s). See, e.g., U.S. Patent No. 3,845,770 to Theeuwes et al. and U.S.
Patent No. 3,977,404 to
Theeuwes.
[00164] The materials used in forming the semipermeable membrane can be
substantially
insoluble in the external fluid or they can erode after a predetermined period
of time with erosion
taking place at the end of the gabapentin release period. Suitable materials
include, by way of
illustration and not limitation: acetaldehyde dimethyl acetate and
acetaldehyde dimethylcellulose
acetate; agar acetate; alkylene oxide and alkyl glycidyl ether copolymers;
amylose triacetate; beta
glucan acetate and beta glucan triacetate; cellulosic materials, which include
cellulose esters (e.g.,
mono-, di- and tricellulose acetates, cellulose acetate butyl sulfonate,
cellulose acetate butyrate,
cellulose acetate chloroacetate, cellulose acetate dimethylaminoacetate,
cellulose acetate ethyl
carbamate, cellulose acetate ethyl carbonate, cellulose acetate ethyl oxalate,
cellulose acetate laurate,
cellulose acetate methyl carbamate, cellulose acetate methyl sulfonate,
cellulose acetate octate,
cellulose acetate phthalate, cellulose acetate propionate, cellulose acetate
succinate, cellulose acetate
p-toluene sulfonate, cellulose acetate valerate, cellulose propionate,
cellulose propionate succinate,
dimethyl cellulose acetate, mono-, di- and tricellulose acrylates, mono-, di-
and tricellulose
alkanylates, mono, di and tricellulose aroylates, cellulose triacylates such
as cellulose trilaurate,
cellulose tripalmitate, cellulose trisuccinate and cellulose trivalerate, and
cellulose diacylates such as
cellulose dicaprylate, cellulose dioctanoate, cellulose dipalmatate, cellulose
dipentanlate and cellulose
disuccinate), cellulose ethers (e.g., ethyl cellulose, hydroxyethylcellulose,
hydroxypropylcellulose,
and methylcellulose), cellulose ester-ether polymers, mono-, di- and
tricellulose acrylates, mono-, di-
and tricellulose alkenylates; hydroxylated ethylene-vinyl acetate; perm-
selective aromatic nitrogen
containing polymeric membranes that exhibit water permeability and essentially
no solute
permeability; polyamides; polyalkylene oxides such as crosslinked and non-
crosslinked polyethylene
32
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
oxide; polyether and polyamide copolymers; polyglycolic acid and polylactic
acid and derivatives
thereof; polymeric epoxides; poly(methacrylate) copolymer salts such as
poly(ammonium
methacrylate) copolymer, poly(ammonium methacrylate) copolymer,
poly(aminoalkyl methacrylate)
copolymer, and (ethyl acrylate)-(methyl methacrylabe)-[(trimethylammonium)-
ethylmethacrylate]
(1:2:0.2) copolymer; cross-linked poly(sodium styrene sulfonate); crosslinked
polystyrenes;
polyurethanes; polyvinyl alcohol; crosslinked poly(vinylbenzyltrimethyl
ammonium chloride);
polyvinyl chloride; poly(vinylmethyl ether) copolymers; polyvinylpyrrolidone;
propylcarbamate;
sulfonated polystyrenes; polyvinylacetate; copolymers of vinyl acetate and
vinylpyrrolidone;
triacetate of locust gum bean; and so forth; and combinations thereof.
[00165] Preferred materials for use in forming the semipermeable membrane
include by way of
illustration and not limitation, cellulose esters; cellulose ethers;
polyvinylpyrrolidone; polyvinyl
alcohol; polyalkylene oxides; and combinations thereof.
[00166] The semipermeable membrane may also include one or more plasticizers,
including:
acetylated monoglycerides; dibutyl phthalate, diethyl phthalate; isopropyl
phthalate, dimethyl
phthalate, and dactyl phthalate; dibutyl sebacate and dimethyl sebacate;
esters such as acetyl triethyl
citrate, acetyl tributyl citrate, citrate ester, dibutyl sebacate, tetraethyl
acetate, triethyl citrate and other
citrate esters; fatty acids such as stearic acid; glyceryl behenate; glycols
such as 1,2-butylene glycol,
2,3-butylene glycol, diethylene glycol, ethylene glycol, propylene glycol,
tetraethylene glycol,
triethylene glycol and polyalkylene glycols such as polyethylene glycol; oils
such as castor oil and
fractionated coconut oil; glycerin; glycerol and glycerol monostearate;
triacetin; and so forth; and
combinations thereof. Preferred plasticizers include by way of illustration
and not limitation, esters
and fatty acids.
[00167] An example of a core/coating system that can be used with gabapentin
to provide for a
gastric retentive dosage form is the delayed release tablet described in U.S.
Patent No. 6,350,471 to
Seth, which comprises a drug/excipient core and a coating of a water-
insoluble, water-permeable ftlm-
forming polymer such as ethyl cellulose, a plasticizer such as stearic acid,
and a water-soluble
polymer such as polyvinylpyrrolidone or hydroxypropylcellulose.
[00168] Another suitable core/coating system has a polyvinyl alcohol coating,
which is either a
water-soluble polyvinyl alcohol blended with a water insoluble polyvinyl
alcohol, or a polyvinyl
alcohol that has been crosslinked with a material such as boric acid or sodium
borate. Such a coating
may also include one or more plasticizers.
[00169] Examples 5 to 8 illustrate the formulation of the gastric retentive
core/coating dosage
forms described above made by wet granulation.
[00170] In addition to the active agent and the polymers, the gastric
retentive gabapentin dosage
forms of the present invention may also include additional excipients, which
are known to those of
33
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
skill in the art to which the invention pertains; such excipients may include,
for example, binders,
lubricants, diluents, fillers, glidants, and other additives.
[00171] Examples of binders that may be used to formulate the dosage forins of
the present
invention include by way of illustration and not limitation, HPMC,
hydroxypropylcellulose ("HPC"),
methylcellulose ("MC"), microcrystalline cellulose ("MCC"), ethyl cellulose,
polyethylene glycol
("PEG"), polyvinylpyrrolidone ("PVP" also known as povidone), vinylpyrolidone-
vinyl acetate
copolymer (also known as copovidone), polyvinylalcohol ("PVA"), gelatin,
starch, and gums.
[00172] Examples of lubricants that may be used to formulate the dosage forms
of the present
invention include by way of illustration and not limitation, magnesium
stearate, calcium stearate,
glyceryl behenate, hydrogenated vegetable oils, PED, sodium stearyl fumarate,
stearic acid, stearyl
behenate, and talc. Examples 1, 2, and 3 describe gastric retentive gabapentin
dosage forms
formulated using a dry blend process (Examples I and 2) or standard
granulation (Example 3) with
magnesium stearate as a lubricant.
[00173] Examples of diluents that may be used to fon-nulate the dosage forms
of the present
invention include by way of illustration and not limitation, calcium sulfate,
cellulose, dicalcium
phosphate, kaolin, lactose, mannitol, MCC, sodium chloride, sorbitol, starch,
and sucrose.
100174] Fillers may include dicalcium phosphate ("DCP"), MCC, spray-dried
lactose, maltose,
starch, sugars, sugar alcohols, and waxes; glidants may include colloidal
silicon dioxide and talc; and
other additives may include coloring agents, flavoring agents, sweeteners, and
solubility retarding
agents.
[001751 For those embodiments of the invention that include further
administering additional
therapeutic agents simultaneously with gabapentin, these agents can either be
administered in the
gastric retentive dosage form that includes gabapentin or can be administered
in a dosage form that is
separate from gabapentin; such dosages can be any suitable formulation as are
well known in the art.
Where appropriate, the additional therapeutic agent may be contained in a
vesicle within the dosage
form or as one layer of a bilayer or multilayer dosage form.
[00176] For those additional agents where controlled release is desirable, the
agent may be
incorporated in the gabapentin gastric retentive dosage form or be
administered in a separate gastric
retentive or other controlled release formulation dosage form. For those
additional agents where
immediate release is desirable, the agent may be incorporated in a coating
around the gabapentin
gastric retentive dosage form or in a separate layer of a bilayer tablet, the
agent may be simply
enclosed in the capsule of the aforementioned gabapentin gastric retentive
capsule dosage form, or the
agent may be administered in a separate immediate release dosage form.
[00177] Typically, dosage forms contain the additional agent (i.e., another
analgesic or
antineuralgic or anticonvulsant agent) in combination with one or more
pharmaceutically acceptable
ingredients. The carrier may be in the form of a solid, semi-solid, or liquid
diluent, or a capsule.
34
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
Usually the amount of active agent is about 0.1-95 wt %, more typically about
1-50 wt %. Actual
methods of preparing such dosage forms are known, or will be apparent, to
those skilled in this art
(see, e.g., REMINGTON: THE SCIENCE AND PRACTICE OF PHARMACY, 20', edition
(Lippincott
Williams & Wilkins, 2000)). The dosage form to be administered will, in any
event, contain a
quantity of the additional therapeutic agents in an amount effective to
alleviate the symptoms of the
subject being treated.
[00178] In the preparation of pharmaceutical formulations containing the
additional therapeutic
agent in the form of dosage units for oral administration the agent may be
mixed with solid, powdered
ingredients, such as lactose, microcrystalline cellulose, maltodextrin,
saccharose, sorbitol, mannitol,
starch, amylopectin, cellulose derivatives, gelatin, or another suitable
ingredient, as well as with
disintegrating agents and lubricating agents such as magnesium stearate,
calcium stearate, sodium
stearyl fumarate, and polyethylene glycol waxes. The mixture is then processed
into granules or
pressed into tablets such as chewable and oral disintegrating tablets.
[00179] Soft gelatin capsules may be prepared by mixing the active agent and
vegetable oil, fat, or
other suitable vehicle. Hard gelatin capsules may contain granules of the
active agent, alone or in
combination with solid powdered ingredients such as lactose, saccharose,
sorbitol, mannitol, potato
starch, cornstarch, amylopectin, cellulose derivatives, or gelatin.
[00180] Liquid preparations for oral administration may be prepared in the
form of syrups or
suspensions, e.g. solutions or suspensions containing about 0.2-20 wt % of the
active agent and the
remainder consisting of sugar or sugar alcohols and a mixture of ethanol,
water, glycerol, propylene
glycol, and PEG. The liquid preparations may contain coloring agents,
flavoring agents, sweeteners
such as saccharin, and carboxymethyl cellulose or other thickening agents.
Liquid preparations for
oral administration may also be prepared in the form of a dry powder to be
reconstituted with a
suitable solvent prior to use.
[00181] When the method of the invention includes administering another agent,
such as
secondary analgesics, anticonvulsant agents, antidepressants, or opioids, the
additional agent may be
obtained from a commercial source in a variety of dosage forms (e.g., tablets,
capsules, oral
suspensions, and syrups). The additional agent may be administered as a
separate dosage form or the
gastric retentive gabapentin dosage form of the present invention may be
designed to include the
additional agent. Additional analgesic agents may be selected from among the
many available
NSAIDs on the market. Examples of suitable commercially available anti-
convulsants include
TEGRETOL (carbamazepine; Novartis, Summit, N.J), DILANTIN (Pfizer Inc., New
York,
N.Y.), and LAMICTAL (lamotrigine (G1axoSmithKline, Philadelphia, PA).
Suitable
antidepressants include the tricyclic antidepressants LIMBITROLO
(amitriptyline; Hoffmann-
LaRoche, Nutley, N.J.), TOFRANIL (imipramine; Tyco Healthcare, Mansfiled,
MA),
ANAFRANILTM (clomipramine; Tyco Healthcare, Mansfield, MA), and NORPRAIVIIN
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
(desipramine; Sanofi-Aventis, Bridgewater, N.J.). Examples of suitable
commercially available
opioids include PERCOCET (oxycodone; Dupont Merck Pharmaceuticals,
Wilmington, DE),
ULTRACET (tramadol; Johnson & Johnson, New Brunswick, N.J.), and CLONOPINT"'
(clonazepam; Hoffmann-LaRoche, Nutley, N.J.).
1001821 UTILITY
[00183] As previously discussed, the gastric retentive dosage forms of the
present invention
reduce the side effects that have been reported with conventional immediate
release gabapentin
dosage forms. The dosage forms of the present invention are comprised of a
therapeutically effective
amount of gabapentin, pregabalin, another GABA analog, or a pharmaceutically
acceptable salt
thereof, to a patient in need of such treatment in a gastric retentive dosage
form that is administered to
the patient in a once-daily or twice-daily dosing regimen. Examples of
conditions that may benefit
from the gastric retentive dosage forms of the present invention include all
conditions that are
treatable with immediate release gabapentin, such as for example, seizures,
PHN, neuropathic pain,
restless legs syndrome, essential tremor, bipolar disorder, migraine
headaches, and the symptoms
associated with hormonal imbalances and chemotherapy. Within the context of
hormonal imbalances,
the gastric retentive dosage forms may be used to reduce or eliminate the
severity of menopausal hot
flashes and within the context of chemotherapy, the gastric retentive dosage
forms may be used to
reduce or eliminate the severity of chemotherapy-related nausea and hot
flashes. .
[00184] All patent applications, patents, publications, and other published
documents mentioned
or referred to in this specification are incorporated herein by reference in
their entireties, to the same
extent as if each individual patent application, patent, publication, and
other published document was
specifically and individually indicated to be incorporated by reference.
[00185] The general methods of the invention are best understood with
reference to the following
examples which are intended to enable those skilled in the art to more clearly
understand and to
practice the present invention. The following examples are not intended, nor
are they to be construed,
as limiting the scope of the invention, but are merely intended to be
illustrative and representative of
the invention.
EXPERIMENTAL
1001861 The practice of the present invention will use, unless otherwise
indicated, conventional
techniques of pharmaceutical formulation, medicinal chemistry and the like,
which are within the skill
of the art. Such techniques are explained fully in the literature. Preparation
of various types of
pharmaceutical formulations are described, for example, in REMINGTON: THE
SCIENCE AND PRACTICE
OF PHARMACY, 20'h edition (Lippincott Williams & Wilkins, 2000) and Ansel et
al.,
PHARMACEUTICAL DOSAGE FORMS AND DRUG DELIVERY SYSTEMS, 6a' Ed. (Media, PA:
Williams &
Wilkins, 1995).
36
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
[00187] In all human clinical trials described in the examples, all
investigators involved in the
studies conducted the clinical trials in accordance with the International
Conference on Harmonization
Harmonized Tripartite Guideline for Good Clinical Practice, the Guidelines of
the Declaration of
Helsinki, Finland, 1964 and its subsequent amendments up through at least 1996
(Tokyo, Japan, 1975;
Venice, Italy, 1983; Hong Kong, 1989; Republic of South Africa, 1996; and
Scotland 2000), and
either United States IND regulations (21. C.F.R. 50, 54 and 56) and all
other national, state and
local laws if conducted within the United States or applicable national, sate,
and local laws of the
pertinent regulatory authorities if conducted outside the United States. All
patients provided written
Informed Consent before any study-related procedures were undertaken.
[00188] Gabapentin was obtained from a variety of sources, including Plantex
U.S.A. (Englewood
Cliffs, N.J.). METHOCEL brand hydroxypropyl methylcellulose (also known as
hypromellose),
and SENTRY POLYOX brand polyethylene oxide were obtained from Dow Chemical
(Midland,
Mich.). METHOCEL E5, premium is a USP type 2910 hydroxypropyl methylcellulose
with
number average molecular weight of on the order of 6000-8000 and a viscosity
of 5 cps as a 2%
aqueous solution at 20 C. METHOCEL K4M and METHOCELO K15M are USP type 2208
hydroxypropyl methylcellulose with viscosities of 4000 cps and 15,000 cps,
respectively, as a 2%
aqueous solution at 20 C, and number average molecular weights on the order of
80,000 and 100,000,
respectively. SENTRY POLYOX WSR 301, NF FP, SENTRY POLYOX WSR Coagulant,
NF FP and SENTRY POLYOX WSR 303, NF FP have viscosity-average molecular
weights of
approximately 4,000,000, 5,000,000 and 7,000,000, respectively. AVICEL PH-101,
NF is
microcrystalline cellulose supplied by FMC Corporation (Philadelphia, Pa.).
Magnesium stearate, NF
was supplied by Spectrum Quality Products (New Brunswick, N.J.) or an
alternative supplier.
EXAMPLE 1
[00189] Gastric retentive gabapentin tablets were manufactured using a dry
blend process, and
hand made on a Carver Auto C Press (Fred Carver, Inc., Indiana). The dry blend
process consisted of
blending all of the ingredients in a plastic bag, and compressing into a 1000
mg tablet (600 mg
gabapentin dose) using a 0.7086" x 0.3937" Mod Oval die (Natoli Engineering,
St. Charles, MO).
The parameters for the operation of the Carver Auto C Press were as follows:
40001bs force, 0-
second dwell time (the setting on the Carver Press), and 100% pump speed. The
formulation for the
tablets is set froth in Table 1:
37
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
TABLE 1
SAMPLE FORMULATION COMPOSITION wt%)
NO. GABAPENTIN PEO METHOCEL MAGNESIUM
COAGULANT K100M STEARATE
1 60.0 39.0 0.0 1
2 60.0 24.3 14.7 1
3 60.0 0.0 39.0 1
[00190] The dissolution was determined in USP apparatus 1 (40 mesh baskets),
100 rpm, in
deionized water. Samples, 5 ml at each time-point, were taken without media
replacement at 1, 4, and
8 hours. The resulting cumulative dissolution profile, based upon a
theoretical percent active added to
the formulations, is set forth in Table 2:
TABLE 2
TIME THEORETICAL wt% OF ACTIVE RELEASED
(HOURS) SAMPLE 1 SAMPLE 2 SAMPLE 3
1 15.4 14.8 18.6
4 39.4 37.4 43.3
8 61.7 57.8 64.7
EXAMPLE 2
[00191] Gastric retentive gabapentin tablets were manufactured using a dry
blend process, and
hand made on a Carver Auto C Press (Fred Carver, Inc., Indiana). The dry blend
process consisted of
blending all of the ingredients in a plastic bag, and compressing into a 600
mg tablet (300 mg .
gabapentin) using a 0.6299" x 0.3937" Mod Oval die (Natoli Engineering, St.
Charles, MO). The
parameters for the operation of the Carver'Auto C' Press were as follows: -
2000-2500 lbs. force, 0-
second dwell time (the setting on the Carver Press), and 100% pump speed. The
formulation for the
tablets is set forth in Table 3:
TABLE 3
SAMPLE FORMULATION COMPOSITION wt%
NO. ACTIVE PEO METHOCEL MAGNESIUM
COAGULANT K15M STEARATE
4 50.0 24.5 24.50 1
[00192] The dissolution was determined in USP apparatus 1 (40 mesh baskets),
100 rpm, in
deionized water. Samples, 5 ml at each time-point, were taken without media
replacement at 1, 2, 4,
6, 8 and 10 hours. The resulting cumulative dissolution profile, based upon a
theoretical percent
active added to the formulation is set forth in Table 4:
38
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
TABLE 4
TIME THEORETICAL wt% OF
(HOURS) ACTIVE RELEASED SAMPLE 4
1 20.6
2 32.4
4 49.7
6 63.1
8 74.0
82.6
ExAMPLE 3
[00193] Three gastric retentive gabapentin formulations were manufactured
utilizing a standard
granulation technique. The formulations manufactured are shown Table 5.
TABLE 5
GR GABAPENTIN FORMULATIONS
GABAPENTIN GR6, 300-MG GABAPENTIN GR8, 300-MG GABAPENTIN GR8, 600-MG
(GR6, 300-MG) (GR8, 300-MG) (GR8, 600-MG)
44.765,'o Gabapentin 44.76% Gabapentin 61.11 % Gabapentin
16.46% METHOCEL 21.99% METHOCEL 7.59% METHOCEL
K4M, premium K15M, premium K15M, premium
21.99 .'o SENTRY 21.99% SENTRY 27.09% SENTRY
POLYOX WSR 303, NF FP POLYOX WSR Coagulant, pOLYOX WSR 303, NF FP
NF FP
12.98% AVICELO 7.49% AVICEL 0.00% AVICEL
PH-101,NF PH-101, NF PH-101, NF
2.75% METHOCEL 2.75% METHOCEL 3.22% METHOCEL
E5, premium E5, premium E5, premium
1.00% Magnesium Stearate, 1.00% Magnesium Stearate, NF 1.00% Magnesium
Stearate, NF
NF
670-mg 670-mg 982-mg
0.3937" X 0.6299" 0.3937" X 0.6299" 0.4062" X 0.75"
Mod Oval Mod Oval Mod Cap
[00194] The dissolution profiles, as determined by USP Apparatus I(100 rpm) in
modified
simulated gastric fluid, for three prototypes formulations are shown in FIG.
1.
EXAMPLE 4
[001951 The pharmacokinetic profiles of the three gastric retentive ("GR")
formulations described
in Example 3, administered as a 600-mg dose, were compared to NEURONTIN
immediate release
300-mg capsule in a randomized four-way cross-over experiment involving 15
healthy volunteers.
Each subject was administered treatment of 600-mg gabapentin as one of the
three formulations (1 x
39
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
600-mg tablet or 2 x 300-mg tablet) or NEURONTIN capsules (2 x 300-mg) within
5 minutes of
completing a high fat breakfast (FDA breakfast). Plasma samples were taken up
to 48 hours post-
dose. FIG. 2 illustrates the average plasma profile for the four treatments
administered, and the
pharmacokinetic data are shown in table 6.
TABLE 6
GABAPENTIN PLASMA DATA - AVERAGE FOR 15 SUBJECTS
DOSING AUC,nF Cmax (11g1ml) *Tma,, (hours)
ml *hr
NEURONTIN , Mean 46.65 4.72 3.93
300-mg % CV 19.0 20.2 15.1
2 x capsules
GR6, 300-mg Mean 44.43 2.97 6.63
2 x tablets fo CV 34.9 29.7 45.1
GR8, 3 00-mg Mean 41.84 3.10 5.63
2 x tablets % CV 34.4 26.2 34.9
GR8, 600-mg Mean 48.01 3.13 7.13
1 x tablet % CV 26.8 18.7 42.2
Geometric Mean and Geometric % CV (coefficient of variation) are reported here
*Arithmetic mean
AUCiõf = area under the concentration-time curve from time zero to infinity.
[001961 As demonstrated in Table 6 and FIG. 2, the gastric retentive (GR)
formulations of the
present invention demonstrated sustained release with a lower maximum plasma
concentration (Cm.)
and a larger value for the time of the maximum concentration (Tm~,,) compared
to the immediate
release (IR) capsules without any significant loss in the bioavailability of
the gabapentin as measured
by the plasma AUCiõf. The Cm,,,, for the GR dosage forms were approximately
30% to approximately
40% lower (rounded to the nearest 5%) than the Cm,,., for the IR dosage form.
The Tm,,, for each of the
three GR dosage ranged from 1.5 to 3.5 hours longer than the Tm,,,, for the IR
dosage form, which
indicates that Tm,,, for the gastric retentive dosage form of the present
invention is approximately 40%
to approximately 80% slower (rounded to the nearest 5%) than Tm,,., for
immediate release dosages
forms.
EXAMPLE 5
[00197] A gastric retentive tablet containing 900 mg of gabapentin is prepared
by granulation with
90 mg of PVP and 10 mg of magnesium stearate and then tableted as a 1000 mg
tablet on a Carver
press with 4000 lbs force, 0-second dwell time. These tablet cores are then
coated from an alcohol-
water solution that dries with approximately 2% dry coat weight of 10 mg ethyl
cellulose, 7 mg PVP,
and 3 mg stearic acid.
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
EXAMPLE 6
[00198]' A gastric retentive tablet containing 1200 mg of gabapentin is
prepared by granulation
with 120 mg of PVP and 10 mg of magnesium stearate and then tableted as a 1330
mg tablet on a
Carver press with 4000 lbs force, 0-second dwell time. These tablet cores are
then coated from an
alcohol-water solution that dries with approximately 25 mg dry coat weight of
10 mg ethyl cellulose,
mg HPC, and 5 mg glyceryl behenate.
EXAMPLE 7
[00199] A gastric retentive tablet containing 900 mg of gabapentin is prepared
by granulation with
90 mg of PVP and 10 mg of magnesium stearate and then tableted as a 1000 mg
tablet on a Carver
press with 4000 lbs force, 0-second dwell time. These tablet cores are then
coated from an aqueous
solution that dries with approximately 2% dry coat weight of 15 mg PVA, 5 mg
PVP, and 3 mg
stearic acid. The coated tablets are then sprayed with an aqueous solution of
1% sodium borate to
crosslink the PVA and dried.
EXAMPLE 8
[00200] A gastric retentive tablet containing 900 mg of gabapentin is prepared
by granulation with
90 mg of PVP, 250 mg MCC, and 10 mg of magnesium stearate and then tableted as
a 1250 mg tablet
on a Carver press with 4000 lbs force, 0-second dwell time. These tablet cores
are then coated from
an alcohol-water solution that dries with approximately 2% dry coat weight of
10 mg ethyl cellulose,
7 mg PVP, and 3 mg stearic acid.
EXAMPLE 9
[00201] To study the rate and extent of absorption of the gastric retentive
gabapentin dosage
forms of the present invention, a four-arm, non-randomized, open-label, single
dose, fed designed
study was conducted on 24 healthy non-smoking males.
[00202) The objective of the study was to compare the rate and extent of
absorption of gabapentin
following administration of four escalating doses of a test formulation of 600
mg tablets of gastric
retentive gabapentin (Depomed Inc., Menlo Park, CA) administered once daily
under fed condition.
[00203] The subjects of the study were 24 nonsmoking males in the age range of
18-65 years old.
The 24 subjects were separated into four treatment groups of six subjects per
group. The drug
administration protocol was as follows:
[00204] TREATMENT GROUP A - following an overnight fast of at least 10 hours,
one 600 mg
gastric retentive gabapentin tablet with 240 mL of ambient temperature water
was administered 20
minutes after the start of a standardized moderate fat content meal. Treatment
dose was 600 -mg.
41
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
[00205] TREATMENT GROUP B - following an overnight fast of at least 10 hours,
two 600 mg
gastric retentive gabapentin tablets with 240 mL of ambient temperature water
were administered 20
minutes after the start of a standardized moderate fat content meal. Treatment
dose was 1200 mg.
[00206] TREATMENT GROUP C - following an overnight fast of at least 10 hours,
three 600 mg
gastric retentive gabapentin tablets with 240 mL of ambient temperature water
were administered 20
minutes after the start of a standardized moderate fat content meal. Treatment
dose was 1800 mg.
[00207] TREATMENT GROUP D - following an overnight fast of at least 10 hours,
four 600 mg
gastric retentive gabapentin tablets with 240 mL of ambient temperature water
were administered 20
minutes after the start of a standardized moderate fat content meal. Treatment
dose was 2400 mg.
[002081 The meals for all the treatment groups were a 500-600 calorie meal
with moderate fat
(about 40% fat), with approximately 80 calories from protein, 252 calories
from carbohydrates, and
about 207 calories from fats. As noted above, the meals were provided after an
overnight fast of at
least 10 hours. Additional moderate fat meals with beverages were provided for
the subjects at 4.5
and 9.5 hours post-dose and a standardized snack was provided 13.5 hours post-
dose. All meals and
beverages were free of alcohol, grapefruit products, xanthine, and caffeine
and were identical during
the study periods.
[00209] The length of the study was four three-day periods separated by at
least one-week
washout period between treatments. Eighteen blood samples of 4 mL each were
drawn in each three-
day-period according to the following schedule (in hours): 0.0 (pre-dose),
0.5, 1.0, 2.0, 3.0, 4.0, 5.0,
6.0, 7.0, 8.0, 10.0, 12.0, 14.0, 16.0, 24.0, 35.0, and 36.0 hours post-dose.
The total blood volume was
315 mL. Vital signs (blood pressure, temperature, respiration rate, and heart
rate) were measured at
the following time periods: 0.0 (predose), 2.0, 4.0, 8.0, 12.0, and 24.0 hours
post dose. Data obtained
from the study is shown in Tables 7 and 8 and in FIG. 4. Data reported in
table 7 represents the
arithmetic mean.
TABLE 7
DOSE (mg) Cmax FOR GR GABAPENTIN
( /mL
600 2.96
1200 4.93
1800 6.68
2400 7.85
42
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
TABLE 8
DOSE (mg) AUC FOR GR GABAPENTIN
n -h/mL
400 25000
600 36247
800 42000
1200 63765
1500 79000
1600 80000
1800 91147
2000 99000
2400 108677
2500 110000
3000 120000
'Some data is extrapolated or interpolated - 600 mg, 1200 mg, 1800 mg, and
2400 mg are actual data
2 AUC values reported here are is the arithmetic mean AUCo_t where t is 36
hours for 19 subjects
[002101 To determine the rate and extent of absorption of the gastric
retentive gabapentin dosage
forms, the pharmacokinetic measurements obtained for the AUC (in ng-hlmL) for
each treatment
group at 24 hours (Table 8) was plotted against the amount of drug
administered to each group. A
comparison of the data for the GR and IR gabapentin in FIGS. 3 and 4
demonstrate the enhanced
bioavailability of the gastric retentive gabapentin at high doses of the
present invention. As shown in
FIG. 4, patients were administered 600 mg, 1200 mg, 1800 mg, and 2400 mg of
gastric retentive
gabapentin in once-daily dosings (AUC values for the additional dosages of the
GR gabapentin in
Table 8 and FIG. 4 were extrapolated based upon the curve of FIG. 4). FIG. 3
shows the same
experiment conducted with 400 mg, 800 mg, 1200 mg, and 1600 mg of immediate
release gabapentin
. The nearly linear curve of FIG. 4 of AUC up to 24 hours demonstrates, the
gastric retentive
gabapentin dosage forms of the present invention demonstrated continued
bioavailability at doses as
high as 2400 mg. By contrast, the plateau seen in FIG. 3 after 1200 mg ofIR
gabapentin indicates
that the body is incapable of effectively absorbing more drug as the dose
increases above 1200 mg of
IR gabapentin.
[002111 The difference in bioavailability of gastric retentive and immediate
release gabapentin
depicted in FIGS. 4 and 3 is shown more dramatically in FIGS. 6, and 5,
respectively, where the
log(AUC) is plotted against the dosage. The log graphs show the sharp decline
in bioavailability of
immediate release gabapentin with the 1200 mg dosing. By contrast, even at a
dosing of 1800 rng, the
gastric retentive gabapentin of the present invention demonstrates continued
bioavailability of the
drug (FIG. 6).
[002121 Further evidence of enhanced bioavailability of at least one
embodiment of the present
invention, as shown in Figures 4 and 6 is obtained by comparing the data on a
dose-normalized basis.
According to the Summary Basis of Approval for NEUROTI\TIN (New Drug
Application ("NDA")
43
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
20-235), the normalized AUC(600-mg) of 600-mg, 800-mg, 1200mg and 1600-mg in
four subjects were
53%, 37%, 38% and 29%, respectively where the AUC was obtained at steady-
state. For one
embodiment of the present invention, the geometric mean (19 subjects) values
of AUCo_. (ng*hr/ml)
values for escalating doses of 600-mg, 1200-mg, 1800-mg and 2400-mg
administered as single-doses
immediately after a meal to healthy volunteers were 35698, 63209, 90894, and
108572, respectively
(rounded to the nearest whole number). Because the AUCo.m for a single dose is
equivalent to the
steady-state AUC as measured between dosing intervals, the AUC values in both
cases represent the
AUCo_õ and comparisons can be made. Therefore, if a ratio of the AUC to that
obtained at 600-mg
dose, and normalized for the dose administered is made, the ratios for the
NEUROTNIN capsules
are 1.0, 0.698, 0.717 and 0.547 for the 600-mg, 800-mg, 1200mg and 1600-mg
respectively compared
to ratios for one embodiment of the present invention of 1.0, 0.882, 0.843 and
0.753 for the 600-mg,
1200-mg, 1800-mg and 2400-mg doses, respectively. Thus, the fraction of the
dose that is absorbed
decreases by about 50% when the dose is increased from 600-mg to 1600 mg for
NEUROTNIN
capsules. In contrast, for one embodiment of the present invention, the
fraction of the dose that is
absorbed decreases by about 25% when the dose is increased from 600 mg to 2400
mg, and only 15%
at 1800 mg, which is still above the 1600 mg dose of immediate release
NEURONTIN which
demonstrated a loss of almost 50% compared to the 600-mg dose of immediate
release
NEURONTIN .
EXAMPLE 10
[00213] To study the efficacy of once-daily versus twice-daily administration
of gastric retentive
gabapentin, a randomized, double-blind multi-center trial was conducted on 158
patients (consisting
of both males and females older than 18 years of age) with PHN. The aim of the
study was to
determine if the gastric retentive gabapentin dosage forms was successful in
reducing the patients'
mean daily pain scores from the baseline week to end of the efficacy treatment
period (Treatment
Week 4). Secondary efficacy measures included changes from baseline in mean
weekly sleep
interference scores, Short-Form McGill Pain Questionnaire (SF-MPQ), the
Neuropathic Pain Scale
(NPS), Patient Global Impression of Change (PGIC), and Investigator-rated
Clinical Global
Impression of Change (CGIC).
[00214] Patients suffering from PHN were eligible for the study if they had
experienced pain for
at least three months after the healing of an acute herpes zoster skin rash
with a pain intensity of at
least 4 on the 11-point Lickert scale (f.e_, 0-10) at screening. Baseline pain
values for all eligible
patients were determined during a one-week pretreatment period where the
patients were to base their
pain during this week on the 11-point Lickert scale; patients who recorded at
least a 4 on the 11-point
scale together with the completion of at least 4 days of diary entries were
deemed eligible to
participate in the study. All patients were required to undergo 7-day washout
for medications
44
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
prescribed for PHN and a 14-day washout (tapered appropriately) for strong
opiates (i.e., morphine or
fentanyl). Patients were permitted to take acetaminophen or acetaminophen with
hydrocodone (if
required for treatment of pain during the study).
[00215] The 158 patients selected for the study were randomly assigned to
treatment with 1800
mg of gastric retentive gabapentin dosed once daily following the evening meal
(55 patients), or
dosed twice daily with 600 mg in the morning and 1200 mg in the evening (52
patients) against a
placebo (51 patients). The course of the study was five weeks. Patients
randomized to active
treatment were gradually titrated over a two-week period to a total daily dose
of 1500 mg, followed
by an additional two-week period at the 1800 mg/day maintenance dose. All
patients, regardless of
treatment, took the same number of tablets of identical appearance each day to
maintain the study
blind; accordingly, patients assigned to placebo received no drug, but took
the same number of tablets
each day as those patients assigned to active treatments. A one week blinded
tapering period followed
the four-week efficacy treatment period. End of study safety assessments were
completed at the
Week 5 visit.
[00216] The gastric retentive gabapentin dosage units prepared for
administration to the patients
were 300 mg and 600 mg white film coated, modified oval-shaped tablets with a
total mass of 714 mg
and 1020 mg, respectively. In addition to gabapentin, the tablets included the
following inactive
ingredients: polyethylene oxide, hypromellose, magnesium stearate, and
coating, and the 300 mg
dose also included MCC. The placebo tablets were coated to match the
appearance of the active
tablets.
[00217] Patients assigned to placebo randomly took the required number of
placebo tablets each
morning and evening to match the dosing of patients assigned to the two active
treatment groups.
[00218] The results of the study are shown in Tables 9-13. In all tables,
patients who had both
baseline and endpoint values were included in the data analysis. In accordance
with standard
statistical analyses, a lower p-value represents stronger evidence against the
null hypothesis of no
effect in the population being tested (p = 1.00).
[00219] In Tables 9, 10, and 12 the "p-values" (both overall and vs. placebo)
are based on a Type
III sum of squares statistical analysis; the LS mean and SEM values for the
Baseline are estimated
from the ANOVA (ANalysis Of VAriance between groups) model that includes
treatment, center, and
treatment by center interaction factor; and the LS mean and SEM values for the
Endpoint and Change
from Baseline to Endpoint are estimated from the ANCOVA (ANalysis of
COVAriance ) model that
includes, treatment, center, treatment by center interaction factor, and
baseline value as a covariate:
[00220] In Table 10, the "responders" are defined as patients with at least
50% reduction in LOCF
average daily pain score from baseline; the "overall p-value" is based on a
Cochran-Mantel-Haenszel
test for the general association stratified by the baseline pain score
category (less than 8 vs. at least 8);
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
and the "p-value vs. placebo" is based on the Z test for the difference in
proportions between the two
groups (i.e., the treatment groups and the placebo group).
TABLE 9
ANALYSIS OF LOCF AVERAGE DAILY PAIN SCORE
TREATMENT GROUP OVERALL
AVERAGE DAILY GR GABAPENTIN GR GABAPENTIN TREATMENT
PAIN SCORE (1800 mg PM) (1800 mg AM/PM) PLACEBO p-value
n=55 n-52 n=51
Baseline
Mean (SD) 6.56 (1.43) 6.32 (1.27) 6.59 (1.58)
LS Mean (SEM) 6.54 (0.20) 6.28 (0.21) 6.56 (0.21) 0.528
95% CI (6.13, 6.94) (5.87, 6.69) (6.14, 6.97)
p-value (vs. placebo) 0.943 0.315
LOCF Endpoint
Mean (SD) 4.69 (2.20) 4.21 (2.27) 5.32 (2.09)
LS Mean (SEM) 4.56 (0.28) 4.25 (0.29) 5.20 (0.29) 0.042
95% CI (4.00, 5.12) (3.68, 4.82) (4.62, 5.78)
Change from Baseline to LOCF Endpoint
Mean (SD) -1.87 (1.78) -2.11 (2.12) -1.27 (1.93)
LS Mean (SEM) -1.93 (0.28) -2.24 (0.29) -1.29 (0.29) 0.042
95% CI -2.49, -1.37) -2.81, -1.67 -1.86, -0.71)
GR Gabapentin minus Placebo
LS Mean A (SEM) -0.64, (0.37) -0.95 (0.38)
95% CI for A (-1.38, 0.10) (-1.71, -0.20) N/A
p-value (vs. placebo) 0.089 0.014
n = sample size; GR = gastric retentive; LOCF = last observation carried
forward; LS = least squares;
SEM = standard error of LS mean; CI = confidence interval; A = Difference; N/A
= not applicable
[002211 The Overall Treatment p-values in Table 9 show that the group as a
whole experienced a
statistically significant decrease in pain from Baseline to LOCF. While Table
9 shows a relatively
large placebo effect, most-likely due to self-administration ofacetaminophen
(with or without
hydrocodone) during the course of the study, the p-values (vs. placebo) for
the two gabapentin
treatment groups indicate a statistically significant decrease in the pain
experienced by the patients
from Baseline to LOCF (see, p-values (vs. placebo) at Baseline and for GR
Gabapentin minus
Placebo). Between the two treatment groups, patients administered the twice-
daily gastric retentive
gabapentin showed more pain reduction than did the patients on the once-daily
dosing regimen;
however, the difference was not great (see, values for LOCF Endpoint and
Changes from Baseline to
LOCF Endpoint).
[00222] Table 10 shows that 25.5% of the patients following the once-daily
dosing regimen and
28.8% of the patients following the twice-daily dosing regimen experienced a
50% reduction in pain
from Baseline to LOCF and Table 11 outlines the pain reduction from 0%
Decrease to 100%
Decrease for each of the patients in each of the Treatment Groups. Within the
o'nce-daily Treatment
46
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
Group, two patients reported a 90% decrease in pain and within the twice-daily
Treatment Group,
three patients reported a 100% decrease.
TABLE 10
PROPORTION OF RESPONDERS AT ENDPOINT
TREATMENT GROUP OVERALL
AVERAGE DAILY GR GABAPENTIN GR GABAPENTIN TREATMENT
PAIN SCORE (1800 mg PM) (1800 nig AM/PM) PLACEBO p-value
n=55 n=52 n=51
Responders at End oint
Yes 14 (25.5%) 15 (28.8%) 6(11.8%) 0.094
No 41 (74.5%) 37 (71.2%) 41 (88.2%)
GR Gabapentin minus Placebo
A in Yes Responders 13.70% 17.00%
95% CI of AP . (-0.83 l0, 28.23%) {1.84%, 32.16%) N/A
p-value (vs. placebo) 0.072 0.032
n = sample size; GR = Gastric Retentive; A = Difference; AP = Difference in
proportions;
N/A = not applicable
47
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
TABLE 11
PERCENT CHANGE FROM BASELINE TO ENDPOINT IN LOCF AVERAGE DAILY PAIN
SCORE
TREATMENT GROUP
AVERAGE DAILY GR GABAPENTIN GR GABAPENTIN
PAIN SCORE PLACEBO
(1800 mg PM) (1800 mg AM/PM) n=51
n=55 n=52
Percent Change from Baseline to LOCF Endpoint: n(%)
Any Increase 6(10.91 !0) 5(9.72%) 12 (23.53%)
No Change 2(3.64%) 3(5.77%) 5(8.80%)
> 0% Decrease 47 (85.45%) 44 (84.62%) 34 (66.67%)
> 10% Decrease 40 (72.73%) 39 (75.00%) 33 (64.71%)
> 20 fo Decrease 31(56.36%) 31(59.62%) 23 (45.10%)
> 30% Decrease 24(43.64%) 25 (48.08%) 16(31.37%)
> 40% Decrease 18 (32.73 fo) 19 (36.54%) 11 (21.57%)
> 50% Decrease 14 (25.45%) 15 (28.85%) 6(11.76%)
? 60% Decrease 9(16.36%) 11(21.15%) 5(9.80%)
> 70% Decrease 3(5.45%) 10(19.23%) 3(5.88 fo)
> 80% Decrease 3(5.45%) 7 (13.46%) 0(0.00%)
90% Decrease 2(3.64 fo) 4(7.69%) 0(0.00 1n)
= 100% Decrease 0(0.00%) 3(5.77 Jo) 0(0.00%)
n = sample size; LOCF = last observation carried forward
[00223] Table 12 sets forth the LOCF Average Daily Pain Score from Table 9 for
those patients at
least 65 years of age. The data from Table 12 shows statistical differences
from placebo in pain
management between the patients on the once-daily dosing regimen and the twice-
daily dosing
regimen (see, p-value (vs. placebo) for GR Gabapentin minus Placebo) and is
more consistent than for
the complete age group (Table 9).
48
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
TABLE 12
ANALYSIS OF LOCF AVERAGE DAILY PAIN SCORE FOR PATIENTS OF AT LEAST 65
YEARS OF AGE
TREATMENT GROUP OVERALL
AVERAGE DAILY GR GABAPENTIN GR GABAPENTIN TREATMENT
PAIN SCORE (1800 mg PM) (1800 mg AM/PM) PLACEBO p-value
n=41 n=38 n=33
Baseline
Mean (SD) 6.46 (1.57) 6.18 (1.58) 6.68 (1.58)
LS Mean (SEM) 6.46 (0.23) 6.18 (0.24) 6.68 (0.26)
0.362
95% CI (6.01,6.92) (5.71,6.65) (6.17,7.18)
p-value (vs. placebo) 0.532 0.158
LOCF Endpoint
Mean (SD) 5.81 (2.21) 4.37 (2.26) 5.89 (2.17)
LS Mean (SEM) 4.79 (0.28) 4.60 (0.29) 5.67 (0.31) 0.033
95% CI (4.23, 5.34) 4.02, 5.18 (5.05, 6.29)
Change from Baseline to LOCF Endpoint
Mean (SD) -1.65 (1.71) -1.80 (2.12) -0.79 (1.42)
LS Mean (SEM) -1.64 (0.28) -1.83 (0.29) -0.76 (0.31) 0.033
95% CI (-2.20, -1.09) (-2.41, -1.25) ( 0,14)
GR Gabapentin minus Placebo
LS Mean A (SEM) -0.88 (0.42) -1.07 (0.43)
95% CI for A (-1.71, -0.05) (-1.92, -0.22) N/A
p-value (vs. placebo) 0.037 0.014
n= sample size; GR = gastric retentive; LOCF last observation carried forward;
LS = least squares;
SEM = standard error of LS mean; CI = confidence interval; A = Difference; N/A
= not applicable
EXAMPLE 11
[00224] To study the efficacy of once-daily and twice-daily administration of
gastric retentive
gabapentin (gabapentin GR) versus placebo, a randomized, double-blind multi-
center placebo
controlled trial was conducted on 147 patients (consisting of both males and
females not less than 18
years of age) with painful diabetic peripheral neuropathy (DPN). The aim of
the study was to
determine if the gastric retentive gabapentin GR dosage forms were successful
in reducing the
patients' mean daily pain scores from the Baseline week to end of the efficacy
treatment period
(Treatment Week 4). Secondary efficacy measures included changes from Baseline
in mean weekly
sleep interference scores, Short-Form McGill Pain Questionnaire (SF-MPQ), the
Neuropathic Pain
Scale (NPS), Patient Global Impression of Change (PGIC), and Investigator-
rated Clinical Global
Impression of Change (CGIC).
[00225] Patients with a diagnosis of type 1 or type 2 diabetes were eligible
for the study if they
had experienced symmetrical painful symptoms in distal extremities for 1-5
years and the symptoms
were attributable to sensorimotor diabetic peripheral neuropathy (DPN).
Patients were also required
49
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
to be on a stable dosing regimen of antidiabetic medication, and have a
hemoglobin Alc of not greater
than 11 % at screening, and a fasting plasma glucose of not greater than 310
mg/dL at screening.
Patients who had previously not responded to treatment for DPN with gabapentin
at doses greater than
or equal to 1200 mg, patients who experienced dose-limiting adverse events
that prevented titration of
gabapentin to an effective dose, and patients who were hypersensitive to
gabapentin were excluded
from the study. Additional exclusion criteria, primarily focused on co-
existing medical conditions,
were also applied to patients. Baseline pain values for all eligible patients
were determined during a
one-week pretreatment period where the patients based their pain on the 11-
point Lickert scale.
Patients with a mean baseline score of at least a 4 on the 11-point scale
together with the completion
of at least 4 days of diary entries were eligible to participate in the study.
All patients were required
to undergo an appropriate washout for medications prescribed for pain
associated with DPN prior to
the Baseline period.
[00226] The 147 patients selected for the study were randomly assigned to
treatment with 3000
mg of gastric retentive gabapentin GR dosed once daily following the evening
meal (46 patients), or
dosed twice daily with 1200 mg in the morning and 1800 mg in the evening (50
patients), against a
placebo (51 patients). The course of the study was five weeks. Patients
randomized to active
treatment were gradually titrated over a two-week period to a total daily dose
of 3000 mg, followed
by an additional two-week period at the 3000 mg/day maintenance dose. All
patients, regardless of
treatment, took the same number of tablets of identical appearance each day to
maintain the study
blind; accordingly, patients assigned to placebo received no drug, but took
the same number of tablets
each day as those patients assigned to active treatments. A one- week blinded
tapering period
followed the four-week effrcacy treatment period. End of study safety
assessments were completed at
the Week 5 visit.
[00227] The gastric retentive gabapentin GR dosage units prepared for
administration to the
patients were 300 mg and 600 mg white film coated, modified oval-shaped
tablets with a total mass of
714 mg and 1020 mg, respectively. In addition to gabapentin, the tablets
included the following
inactive ingredients: polyethylene oxide, hypromellose (hydroxypropyl
methylcellulose), magnesium
stearate, binder, and coating. The placebo tablets were coated tablets
comprised of only excipients,
and were manufactured to have the same appearance as the active product.
[002281 The results of the study are shown in Tables 13-15. In all tables,
patients who had both
baseline and endpoint values were included in the data analysis. In accordance
with standard
statistical analyses, a lower p-value represents stronger evidence against the
null hypothesis of no
effect in the population being tested (p = 1.00)
[00229] In Tables 13, 14 and 15 the "p-values" (both overall and vs. placebo)
are based on a Type
III sum of squares statistical analysis; the LS (least square) mean and SEM
(standard error of the
mean) values for the Baseline are estimated from the ANOVA (ANalysis Of
Variance) model that
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
includes treatment, center, and treatment by center interaction factor; and
the LS mean and SEM
values for the Endpoint and Change from Baseline to Endpoint are estimated
from the ANCOVA
(ANalysis of COVAriance) model that includes, treatment, center, treatment by
center interaction
factor, and baseline value as a covariate.
[00230] In Table 14, the "responders" are defined as patients with at least
50% reduction in BOCF
(baseline observation carried forward) average daily pain score from Baseline;
the "overall p-value" is
based on a Cochran-Mantel-Haenszel test for the general association stratified
by the baseline pain
score category (less than 8 vs. at least 8); and the "p-value vs. placebo" is
based on the Z test for the
difference in proportions between the two groups (i.e., the treatment groups
and the placebo group).
TABLE 13
ANALYSIS OF LOCF AVERAGE DAILY PAIN SCORE
TREATMENT GROUP OVERALL
AVERAGE DAILY GABAPENTIN GR GABAPENTIN GR TREATMENT
PAIN SCORE (3000 mg PM) (3000 mg AM/PM) PLACEBO p-value [1]
n=46 N=50 n=51
Baseline
Mean (SD) 6.71 (1.34) 6.44 (1.51)
LS Mean (SEM) 6.79 (0.23) 6.69 (0.23) 6.74 (1.37)
95% CI 6.97 (0.22) 0.660
p-value (vs. placebo) C6.33, 7.25) (6.24, 7.14} (6.53, 7.41)
2 0.571 0.370
LOCF Endpoint
Mean (SD) 3.84 (1.83) 4.48 (2.27) 5.26 (1.93)
LS Mean (SEM) 3.87 (0.29) 4.65 (0.28) 5.24 (0.28) 0.002
95% CI (3.29,4.44) (4.10, 5.21) (4.69, 5.80)
Change from Baseline to LOCF Endpoint
Mean (SD) -2.87 (1.97) -1.96 (2.04) -1.48 (1.94)
LS Mean (SEM) -2.76 (0.29) -1.98 (0.28) -1.38 (0.28) 0.002
95% CI (-3.34, -2.19) (-2.53, -1.42) (-1.94,-0.83)
Gabapentin GR minus Placebo
LS Mean A (SEM) -1.38 (0.38) -0.59 (0.37)
95% CI for A (-2.13, -0.63) (-1.33, 0.14) N/A
p-value (vs. placebo) < 0.001 0.114
2
n= sample size; GR = gastric retentive; LOCF = last observation carried
forward; LS = least squares;
SEM = standard error of LS mean; CI = confidence interval; A = Difference; N/A
= not applicable
[1] The p-value (overall) for the overall comparison among all treatment
groups is based on type III analysis
[2] The p-value (vs. Placebo) for the pairwise test of difference of the LS
mean change from baseline between
Gabapentin GR and placebo groups is based on the t-test of Type III analysis
from the models described above.
51
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
TABLE 14
PROPORTION OF RESPONDERS AT ENDPOINT BASED ON BOCF
TREATMENT GROUP OVERALL
AVERAGE DAILY GABAPENTIN GR GABAPENTIN GR TREATMENT
PAIN SCORE (3000 mg PM) (3000 mg AM/PM) PLACEBO p-value
n=46 n=50 n=51
Responders at Endpoint
Yes 16 (34.8 %) 13 (26.0 %) 4(7.8 %) 0.005
No 30(65. 2 %) 37 (74.0 %) 47 (92.2%)
Gabapentin GR minus Placebo
Difference in Yes 27.00% 18.20%
Responders (11.39%, 42.61%) (3.99% 32.41 Jo) N/A
95% CI of DP 0.001 0.015
p-value (vs. placebo)
n= sample size; GR = Gastric Retentive; DP = Difference in proportions; CI=
confidence interval
N/A = not applicable; BOCF = Baseline Observation Carried Forward
52
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
TABLE 15
PERCENT CHANGE FROM BASELINE TO ENDPOINT IN BOCF AVERAGEDAILY SLEEP
INTERFERENCE SCORE
TREATMENT GROUP OVERALL
AVERAGE DAILY GABAPENTIN GR GABAPENTIN GR TREATMENT
PAIN SCORE (3000 mg PM) (3000 mg AM/PM) PLACEBO p-value [1]
n=46 n=50 n=51
Baseline
Mean (SD) 6,24 (1.81) 5.61 (2.37)
LS Mean (SEM) 6.30 (0.35) 5.79 (0.34) 6'22 (1'94)
95% Cl 6.38 (0.34) 0.421
p-value (vs. placebo) (5.61, 7.00) (5.11, 6.47) (5.71, 7.04)
2 0.879 0.225
LOCF Endpoint
Mean (SD) 3.16 (2.21) 3.23 (2.73) 4.41 (2.32)
LS Mean (SEM) 2.98 (0.31) 3.49 (0.30) 5.23 (0.30) 0.009
95% CI (2.36, 3.59 2.89, 4.08) (3.64, 4.82)
Change from Baseline to LOCF Endpoint
Mean (SD) -3.08 (2.06) -2.39 (2.21) -1.81 (2.25)
LS Mean (SEM) -3.04 (0.31) -2.53 (0.30) -1.79 (0.30) 0.009
95% CI -3.65, -2.42) (-3.13, -l .94) (-2.38,-1.20)
Gabapentin GR minus Placebo
LS Mean A (SEM) -1,25 (0.41) -0.74 (0.40)
95% CI for 0 (-2,05, -0.45) (-1.53, 0.05) N/A
p-value (vs. piacebo) 0.003 0.066
[2l
n = sample size; GR = gastric retentive; LOCF = last observation carried
forward;
LS = least squares; SEM = standard error of LS mean; Cl = confidence interval;
N/A = not applicable
(1] The p-value (overall) for the overall comparison among all treatment
groups is based on type III analysis
from the models described above
[2] The p-value (vs. Placebo) for the pairwise test of difference of the LS
mean change from baseline between
Gabapentin GR and placebo groups is based on the t-test of Type III analysis
from the models described above.
1002311 The Overall Treatment p-values in Table 13 show that the group as a
whole experienced a
statistically significant decrease in pain from Baseline to the end of
efficacy based upon LOCF (Last
Observation Carried Forward). While Table 13 shows a relatively large placebo
effect, the p-value
(vs. placebo) for the gabapentin treatment group for once-daily evening dosing
indicates a statistically
significant decrease in the pain experienced by the patients from Baseline to
the end of efficacy period
(see, p-values (vs. placebo) at Baseline and for GR Gabapentin minus Placebo).
Between the two
treatment groups, patients administered the twice-daily gastric retentive
gabapentin showed less pain
reduction than did the patients on the once-daily dosing regimen.
[00232] Table 14 shows that 34.6% of the patients following the once-daily
dosing regimen and
26.0% of the patients following the twice-daily dosing regimen experienced a
50% reduction in pain
53
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
from Baseline to the end of efficacy period (based on BOCF analysis). For both
the once-daily dosing
regimen and the twice-daily dosing regimen the difference in responders
defined as those patients
who experienced a 50% reduction in pain from Baseline to the end of efficacy
period was statistically
different from placebo.
[002331 Table 15 sets forth the change in Average Daily Sleep Interference
Score from baseline to
the end of the efficacy period. The data from Table 15 shows statistical
differences from placebo in
the change in sleep interference score from Baseline to the end of efficacy
period for patients on the
once-daily dosing regimen. For the twice-daily dosing regimen, the difference
does not reach
statistical significance.
EXAMPLE 12
[00234] To study the rate and extent of absorption of the gastric retentive
gabapentin (gabapentin
GR) dosage forms of the present invention, a three-arm, randomized open-label,
multiple dose, study
was conducted on healthy non-smoking male and female subjects between the ages
of 18 and 65.
[00235] The objective of the study was to compare the pharmacokinetics of
gabapentin on Day I
and on Day 8 (after 5 days of administration) for 2 different dosing regimes
of gastric retentive
gabapentin GR (Depomed Inc., Menlo Park, CA) administered under fed condition
compared to three
times daily dosing of the reference product, NEURONTIN tablets.
[00236] Twenty-four nonsmoking male or female subjects between the ages of 18-
65 years old
were enrolled. Twenty-one subjects completed the study. The drug
administration protocol was as
follows:
[00237] TREATMENT GROUP A - after an overnight fast of at least 10 hours, one
600 mg
gastric retentive gabapentin GR tablet was administered with 240 mL of a water
20 minutes after the
start of a standardized meal at 8:00 a.m. and the second and third 600 mg
gastric retentive gabapentin
GR tablets (2 tablets) were administered with 240 mL of water 20 minutes after
the start of a
standardized meal at 8:00 p.m. (20:00). The total treatment dose was 1800 mg.
The Day 1 dosing
was followed by two days without drug administration. Then a steady-state
period was initiated in
which this treatment regimen was repeated for five consecutive days (Days 4-
8).
[00238] TREATMENT GROUP B - three 600 mg gastric retentive gabapentin GR
tablets were
administered with 240 mL of water 20 minutes after the start of a standardized
meal at 8:00 p.m.
(20:00). The total treatment dose was 1800 mg. The Day 1 dosing was followed
by two days without
drug administration. Then a steady-state period was initiated in which this
treatment regimen was
repeated for five consecutive days (Days 4-8).
[00239] TREATMENT GROUP C - after an overnight fast of at least 10 hours, one
NEURONTIND tablet containing 600 mg gabapentin was administered with 240 mL of
water at 8:00
a.m., 2:00 p.m. (14:00) and 8:00 p.m. (20:00). All doses were administered 20
minutes after the start
of standardized meals. The total treatment dose was 1800 mg. The Day I dosing
was followed by
54
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
two days without drug administration. Then a steady-state period was initiated
in which this
treatment regimen was repeated for five consecutive days (Days 4-8).
1002401 The length of the study was three periods in which treatment was
administered on day 1
and days 4-8 separated by at least one-week washout period between treatments.
Blood samples were
drawn in each three-day-period up to 36.0 hours post-dose.
[00241] A summary of some of the important steady-state pharmacokinetic
parameters are shown
in Table 16 below. As shown in table 16, at steady-state the gabapentin GR
dosed twice per day (600
mg in the morning, 1200 mg in the evening) shows similar bioavailability to
NEURONTIN
immediate release tablets dosed three times per day (600-mg dose at 8:00 a.m.,
2:00 p.m. and 8:00
p.m.) as evidenced by the ratio of mean AUC "' f of 102%. Also note that the
Gabapentin GR
maximum plasma concentration is about 19% lower and the minimum plasma
concentration is about
18% higher than the maximum and minimum concentrations, respectively, for the
TID dosing of the
NEURONTIN immediate release tablets. The data is an illustration that this
embodiment of the
present invention has provided improved convenience of dosing (twice per day
versus three times per
day) with a smoother plasma profile as evidenced by the lower maximum and
higher minimum
plasma concentrations without any loss of bioavailability.
[00242] Furthennore, the once daily dosing of this embodiment of the present
invention also
compares favorably with the TID dosing of the of the NEURONTIN immediate
release tablets.
Although the minimum concentration is about 50% lower, the maximum plasma
concentration is only
about 16% higher than that observed for NEURONTIN 'immediate release tablets.
There is a
modest loss in bioavailability (ratio of the means of the bioavailability for
gabapentin GR dosed once
daily to the NEURONTIN immediate release tablet is 93%), but a significant
increase in dosing
convenience with a regimen of three times daily dosing compared to once daily
dosing after the
evening meal.
CA 02635466 2008-06-26
WO 2007/079195 PCT/US2006/049511
TABLE 16:
COMPARISON OF DOSING REGIMENS BASED ON DAY 8 DOSING
Parameter 90% C.I. Ratio of Means Intra-Subject CV
Treatment A versus Treatment B
AUCo.Z4 102% to 117% 110% 13%
C,õA: 66% to 75% 70% 12%
Cmiõ 205% to 248 00 225% .18%
Treatment A versus Treatment C
AUCa24 95% to 109% 102% 13%
Cmai 76% to 86 !o 81% 12%
Cmin 107% to 130% 118% 18%
Treatment B versus Treatment C
AUCa24 87% to 100% 93% 13%
CMa= 109% to 123 !0 116% 12%
Cmia 48% to 56% 52% 18%
56