Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
1
2-ALCOXY-3 4 5-TRIHYDROXY-ALKYLAMIDE-BENZAZEPINE LEUR
PREPARATION, COMPOSITIONS LES CONTENANT ET UTILISATION
La présente invention concerne notamment des 2-alcoxy-3,4,5-trihydroxy-
alkylamide-benzazépines, leur préparation, des compositions les contenant,
et leur utilisation comme médicament.
Plus particulièrement, et selon un premier aspect, l'invention concerne des 2-
alcoxy-3,4,5-trihydroxy-alkylamide-benzazépines utiles comme agents
anticancéreux.
Des 2-méthoxy-3,4,5-trihydroxy-alkylamides ont été décrits dans US
6239127, US 20010044433 A1, WO 01/85697, WO 00/29382, US 4831135,
EP 687673, et US 2002128474 A1. Ces documents divulguent
essentiellement des analogues et dérivés de bengamide, un produit naturel
isolé d'une éponge marine, Jaspis coriacea.
Ces mêmes produits ont été décrits dans la littérature : J. Org. Chem. (1986),
51(23), 4494-7; J. Org. Chem. (2001), 66(5), 1733-41 ; J. Med. Chem. 2001,
44, 3692-9.
Le problème que se propose de résoudre la présente invention est d'obtenir
de nouveaux produits présentant une activité anticancéreuse. En plus du
maintien d'une activité anticancéreuse, certains de ces nouveaux produits
peuvent également présenter des propriétés avantageuses en relation avec
leur activité pharmacologique, telle que leur pharmacocinétique,
biodisponibilité, solubilité, stabilité, toxicité, absorption ou métabolisme.
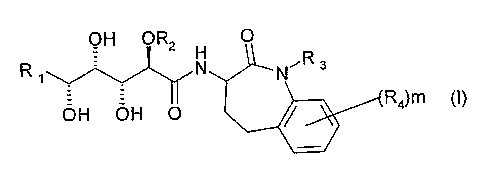
La présente invention a pour objet les produits répondant à la formule
générale (I) suivante :
OH OR2 O
H
R N NR3
OH OH O R4)m (I)
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
2
dans laquelle :
a) R1 est indépendamment sélectionné dans le groupe constitué par (C1-
C12)alkyle, (C2-C12)alcényle, (C2-C12)alcynyle, cycloalkyle,
hétérocyclyle, aryle, hétéroaryle, cycloalkyl(C1-C12)alkyle, cycloalkyl(C2-
C12)alcényle, cycloalkyl(C2-C12)alcynyle, hétérocyclyl(C1-C12)alkyle,
hétérocyclyl(C2-C12)alcényle, hétérocyclyl(C2-C12)alcynyle, aryl(C1-
C12)alkyle, aryl(C2-C12)alcényle, aryl(C2-C12)alcynyle, hétéroaryl(C1-
C12)alkyle, hétéroaryl(C2-C12)alcényle, hétéroaryl(C2-C12)alcynyle, le
groupe aryle de chaque R1 étant éventuellement substitué par un ou
plusieurs halogènes ;
b) R2 est sélectionné dans le groupe constitué par (C1-C6)alkyle, aryl(C1-
C6)alkyle, hétéroaryl(C1-C6)alkyle, aryle, hétéroaryle, (C1-
C6)alkylthio(C1-C6)alkyle, di(C1-C6)alkylamino(C1-C6)alkyle, aryloxy(C1-
C6)alkyle, (C1-C6)alkoxy(C1-C6)aikyle ;
c) R3 est sélectionné dans le groupe constitué par H, COO(R5), CONH(R5),
CO(R5), O(R5), R5;
d) R4 est indépendamment sélectionné dans le groupe constitué par F, CI,
Br, N(R5)2, NO2, CN, COO(R5), CON(R5)2, NHCO(R5), NHCOO(R5),
OCONH(R5), O(R5), R5 ou bien, deux substituants R4, liés à 2 carbones
adjacents du phényle, forment ensemble un cycle choisi parmi cycloalkyle,
hétérocyclyle, aryle ou hétéroaryle, éventuellement substitué par un ou
plusieurs R4;
e) m a pour valeur 0, 1, 2, 3, ou 4;
f) R5 est indépendamment choisi parmi doublet d'électrons non liant, H, (C1-
C12)alkyle, (C2-C12)alcényle, (C2-C12)alcynyle, halogéno(C1-C12)alkyle,
aryl(C1-C12)alkyle, hétéroaryl(C1-C12)alkyle, hétéroarylaryl(C1-
C12)alkyle, aryle, hétéroaryle, cycloalkyle, dans lequel chaque R5 est
éventuellement substitué par au moins un substituant choisi parmi OH,
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
3
halogène, (C1-C4)alkyle, (C1-C4)alkoxy, aryl(C1-C4)alkyle, aryle,
hétéroaryl(C1-C4)alkyl, hétéroaryle, -N(CH3)2, -NH2, CONH2, ~N O ~-N N-Rz =N/~
_ I ,-N
\~
chacun des Rz est indépendamment sélectionné dans le groupe constitué
par H, COO(R5), CONH(R5), CON(R5)2, CO(R5), R5, dans lequel chaque
R5 est indépendamment choisi parmi (C1-C4)alkyle, halogéno(C1-
C4)alkyle, aryl(C1-C4)alkyle, hétéroaryl(C1-C4)alkyle, dans lequel chaque
R5 est éventuellement substitué par un substituant choisi parmi OH,
halogène, (C1-C4)alkyle, (C1-C4)alkoxy, aryl(C1-C4)alkyle, aryle,
hétéroaryl(C1-C4)alkyle, hétéroaryle ;
à la condition que lorsque R1 est (E) -CH=CH-C(CH3)3, R2 est méthyle et R3
est H, alors m est différent de 0.
La présente invention a pour objet les produits de formule générale (I) ci-
dessus dans laquelle R1 est indépendamment sélectionné dans le groupe
constitué par (C1-C12)alkyle, (C2-C12)alcényle, (C2-C12)alcynyle,
cycloalkyle, hétérocyclyle, aryle, hétéroaryle, aryl(C1-C12)alkyle, aryl(C2-
C12)alcényle, aryl(C2-C12)alcynyle, hétéroaryl(C1-C12)alkyle, hétéroaryl(C2-
C12)alcényle, hétéroaryl(C2-C12)alcynyle.
La présente invention a pour objet les produits de formule générale (I) ci-
dessus dans laquelle R2 est sélectionné dans le groupe constitué par (C1-
C6)alkyle, aryl(Cl-C6)alkyle, hétéroaryl(Cl-C6)alkyle, aryle, hétéroaryle, (C2-
C12)alcénylaryle, (C2-C12)alcénylhétéroaryle, (C1-C6)alkylthio(C1-C6)alkyle,
di(C1-C6)alkylamino(C1-C6)alkyle, aryloxy(C1-C6)alkyle.
Selon l'invention, R1 est préférentiellement choisi parmi -C(R6)=C(R7)(R$)
dans lequel R6, R7, et R8 sont indépendamment sélectionnés parmi H, (Cl-
C6)alkyle, cycloalkyle, hétérocyclyle, aryle, hétéroaryle.
Plus préférentiellement, R1 est choisi parmi (E) -CH=CH-CH(CH3)(C2H5), (E) -
CH=CH-CH(CH3)2, (E) -CH=CH-C(CH3)3, ou encore parmi (E) -C(CH3)=CH-
CH(CH3)(C2H5), (E) -C(CH3)=CH-CH(CH3)2, et (E) -C(CH3)=CH-C(CH3)3.
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
4
Plus préférentiellement, R1 est choisi parmi (E) -CH=CH-C5H9 .
Selon l'invention, R2 est préférentiellement méthyle.
Parmi les objets de la présente invention, un premier groupe est caractérisé
en ce que R3 est indépendamment choisi parmi : un groupe méthyle ou un
groupe (3,5-difluoro-phényl)-méthyle. Un second groupe est caractérisé en ce
que R3 est H.
Parmi les objets de la présente invention, un troisième groupe est caractérisé
en ce que R4 est indépendamment choisi parmi : F, CI, Br, phényle,
pyridinyle. Un quatrième groupe est caractérisé en ce que m est 0.
De manière préférée, l'invention concerne les produits exemplifiés dans le
tableau 1.
Selon un autre aspect, l'invention concerne les procédés de préparation des
produits de formule générale (I) ou (I'). Les produits de formule générale
(I')
sont des précurseurs, éventuellement actifs, des produits de formule générale
(I). Les produits de formule générale (I) sont obtenus à partir des produits
de
formule générale (I') par des procédés décrits ou par une ou plusieurs
réactions classiques pour l'homme du métier telle que par exemple une
cyclopropanation, une oxydation ou une séparation chirale.
Les produits de formule générale (I) ou (I') peuvent être obtenus par
hydrolyse d'un produit de formule générale (II) :
OH ORZ O R
R~ N 3
N
(R4)m
ôu ô o
dans lequel R1i R2, R3, R4 et m
sont tels que définis précédemment,.
Les produits de formule générale (II) peuvent être obtenus par réaction d'un
produit de formule générale (III) :
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
0
iRs
H2N N (R4)m
~ (III)
dans lequel R3, R4 et m sont tels que définis
précédemment, avec un produit de formule générale (IV) :
0
0
R OR 2
(IV)
o~ô
dans lequel R1, R2 sont tels que définis
précédemment.
5 Les produits de formule générale (I) ou (I') peuvent également être obtenus
par réaction d'un produit de formule générale (III) tel que défini
précédemment, avec un produit de formule générale (V) :
0
0
R OR2
(V)
ôH ôH dans lequel R1, R2 sont tels que définis
,
précédemment.
Les produits de formule générale (V) peuvent être obtenus par hydrolyse d'un
produit de formule générale (IV) :
0
0
R OR 2
OKO
dans lequel R1, R2 sont tels que définis
précédemment. Des produits de formule générale (V) pour lesquels R1
représente -CH=CH-R'1 peuvent encore être obtenus par hydrolyse d'un
produit de formule générale (VII) :
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
6
0
1I o
OR2
(Vil)
oKô
, dans lequel R2 est tel que défini précédemment, afin
d'obtenir un produit de formule générale (VI) :
O
I o
OR2
(VI)
àH ôH , dans lequel R2 est tel que défini précédemment, qui
subit une métathèse afin d'obtenir un produit de formule générale (V)
O
R OR2
H O" pour lesquels R1 représente -CH=CH-R'i et R'1
représente un groupe (C1-C6)alkyle, cycloalkyle, hétérocyclyle, aryle ou
hétéroaryle.
Les produits de formule générale (VII) peuvent être obtenus par double
déshydratation d'un produit de formule générale (VIII) :
0
OH
pRz
HO , , (VIII)
0 ô
K , dans lequel R2 est tel que défini précédemment.
Les produits de formule générale (I') et (ll) tels que définis précédemment, à
l'exception de ceux pour lesquéls R1 est (E) -CH=CH-C(CH3)3, R2 est méthyle
et R3 est H et m est 0, sont un objet de la présente invention.
Les produits de formule générale (III) pour lesquels R3 est H, méthyle ou
(3,5-difluorophényl)-méthyle et R4 est un atome de brome, phényle ou bien m
a pour valeur 0, à l'exception de ceux pour lesquels R3 est H et m a pour
valeur 0, sont un objet de la présente invention.
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
7
Les produits de formule générale (IV) et (V) pour lesquels R2 est méthyle et
R1 est -(E) -CH=CH-C5H9 sont un objet de la présente invention.
Les produits de formule générale (VI) pour lesquels R2 est méthyle sont un
objet de la présente invention. Les produits de formule générale (VII) pour
lesquels R2 est méthyle sont un objet de la présente invention.
Les produits selon la présente invention peuvent exister à l'état de bases, de
sels d'additions avec des acides, de solvats, d'hydrates ou de prodrogues.
Les produits selon l'invention peuvent être sous forme non chirale, ou
racémique, ou enrichie en un stéréo-isomère, ou enrichie en un énantiomère ;
et peuvent éventuellement être salifiés. Les produits pour lesquels le carbone
lié à l'amine exocyclique est de configuration (S) sont préférés.
Un produit conforme à l'invention pourra être utilisé pour la fabrication d'un
médicament utile pour prévenir ou traiter un état pathologique, en particulier
un cancer.
Les produits de la présente invention peuvent également être utilisés pour la
fabrication d'un médicament utile pour prévenir ou traiter un état
pathologique
dans lequel une néovascularisation ou angiogenèse se fait de façon
inappropriée c'est-à-dire dans les cancers en général et dans des cancers
particuliers tels que le sarcome de Kaposi ou l'hémoangiome infantile, mais
aussi dans l'arthrite rhumatoïde, l'osthéoarthrite et/ou ses douleurs
associées, les maladies inflammatoires de l'intestin telle que la recto-colite
hémorragique ou la maladie de Crohn's, les pathologies de I'ceil telle que la
dégénérescence maculaire liée à l'âge, les rétinopathies diabétiques,
l'inflammation chronique, le psoriasis.
L'angiogenèse est un processus de génération de nouveaux vaisseaux
capillaires à partir des vaisseaux pré-existants. L'angiogenèse tumorale
(formation de néovaisseaux sanguins), indispensable à la croissance
tumorale, est également un des facteurs essentiels de la dissémination
métastatique (Oncogene. 2003 May 19;22(20):3172-9 ; Nat Med. 1995
Jan;1(1):27-31.).
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
8
La présente invention concerne aussi les compositions thérapeutiques
contenant un composé selon l'invention, en association avec un excipient
pharmaceutiquement acceptable selon le mode d'administration choisi. La
composition pharmaceutique peut se présenter sous forme solide, liquide ou
de liposomes.
Parmi les compositions solides on peut citer les poudres, les gélules, les
comprimés. Parmi les formes orales on peut aussi inclure les formes solides
protégées vis-à-vis du milieu acide de l'estomac. Les supports utilisés pour
les formes solides sont constitués notamment de supports minéraux comme
les phosphates, les carbonates ou de supports organiques comme le lactose,
les celluloses, l'amidon ou les polymères. Les formes liquides sont
constituées de solutions de suspensions ou de dispersions. Elles contiennent
comme support dispersif soit l'eau, soit un solvant organique (éthanol, NMP
ou autres) ou de mélanges d'agents tensioactifs et de solvants ou d'agents
complexants et de solvants.
Les formes liquides seront de préférence injectables et, de ce fait, auront
une
formulation acceptable pour une telle utilisation.
Des voies d'administration par injection acceptables incluent les voies
intraveineuse, intra-péritonéale, intramusculaire, et sous cutanée, là voie
intraveineuse étant préférée.
La dose administrée des composés de l'invention sera adaptée par le
praticien en fonction de la voie d'administration du patient et de l'état de
ce
dernier.
Les composés de la présente invention peuvent être administrés seuls ou en
mélange avec d'autres anticancéreux. Parmi les associations possibles on
peut citer :
= les agents alkylants et notamment le cyclophosphamide, le melphalan,
l'ifosfamide, le chlorambucil, le busulfan, le thiotepa, la prednimustine, la
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
9
carmustine, la lomustine, la semustine, la steptozotocine, la decarbazine, la
témozolomide, la procarbazine et l'hexaméthylmélamine
= les dérivés du platine comme notamment le cisplatine, le carbopiatine ou
l'oxaliplatine
= les agents antibiotiques comme notamment la bléomycine, la mitomycine,
la dactinomycine
= les agents antimicrotubules comme notamment la vinblastine, la
vincristine, la vindésine, la vinorelbine, les taxoides (paclitaxel et
docétaxel)
= les anthracyclines comme notamment la doxorubicine, la daunorubicine,
l'idarubicine, l'épirubicine, la mitoxantrone, la losoxantrone
= les topoisomérases des groùpes I et Il telles que l'étoposide, le
teniposide,
l'amsacrine, l'irinotecan, le topotecan et le tomudex
= les fluoropyrimidines telles que le 5-fluorouracile, I'UFT, la floxuridine
= les analogues de cytidine telles que la 5-azacytidine, la cytarabine, la
gemcitabine, la 6-mercaptopurine, la 6-thioguanine
= les analogues d'adénosine telles que la pentostatine, la cytarabine o,u le
phosphate de fludarabine
= le méthotrexate et l'acide folinique
= les enzymes et composés divers tels que la L-asparaginase, l'hydroxyurée,
l'acide trans-rétinoique, la suramine, la dexrazoxane, l'amifostine,
l'herceptin
ainsi que les hormones oestrogéniques, androgéniques
= les agents antivasculaires tels que les dérivés de la combretastatine, par
exemple la CA4P, des chalcones ou de la colchicine, par exemple le ZD6126,
et leurs prodrogues.
= Les inhibiteurs de kinases tels que l'ertonilib ou l'imatinib.
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
= Les agents biothérapeutiques comme les anticorps tels que le rituximab, le
bevacizumab, le cetuximab, le trastuzumab ou l'alemtuzumab.
= Les inhibiteurs du protéasome tel que le bortesomib.
Il est également possible d'associer aux composés de la présente invention
5 un traitement par des radiations. Ces traitements peuvent être administrés
simultanément, séparément, séquentiellement. Le traitement sera adapté par
le praticien en fonction du malade à traiter.
Définitions
Le terme halogène fait référence à un élément choisi parmi F, CI, Br, et
I.
10 Le terme alkyle fait référence à un substituant hydrocarboné saturé,
linéaire ou ramifié, ayant de 1 à 12 atomes de carbone. Les substituants
méthyle, éthyle, propyle, 1 -méthyléthyl, butyle, 1 -méthylpropyl,
2-méthylpropyle, 1,1-diméthyléthyle, pentyle, 1-méthylbutyle, 2-méthylbutyle,
3-méthylbutyle, 1,1-diméthylpropyle, 1,2-diméthylpropyle, 2,2-diméthyl-
propyle, 1-éthylpropyle, hexyle, 1-méthylpentyle, 2-méthylpentyle, 1-éthyl-
butyle, 2-éthylbutyle, 3,3-diméthylbutyle, heptyle, 1-éthylpentyle, octyle,
nonyle, décyle, undécyle, et dodécyle sont des exemples de substituant
alkyle.
Le terme alcényle fait référence à un substituant hydrocarboné linéaire ou
ramifié ayant une ou plusieurs insaturations, ayant de 2 à 12 atomes de
carbone. Les substituants éthylènyle, 1 -méthyléthylènyle, prop-1 -ènyle, prop-
2-ènyle, Z-1-méthylprop-1-ènyle, E-1-méthylprop-1-ènyle, Z-1,2-diméthyl-
prop-1-ènyle, E-1,2-diméthylprop-1-ényle, but-1,3-diényle, 1 -méthylidènyl-
prop-2-ènyle, Z-2-méthylbut-1,3-diényle, E-2-méthylbut-1,3-diényle, 2-méthyl-
1-méthylidènylprop-2-ènyle, undéc-1-ènyle et undéc-10-ènyle sont des
exemples de substituant alkylène.
Le terme alcynyle fait référence à un substituant hydrocarboné linéaire ou
ramifié ayant au moins deux insaturations portées par une paire d'atomes de
carbone vicinaux, ayant de 2 à 12 atomes de carbone. Les substituants
éthynyle; prop-1-ynyle; prop-2-ynyle; et but-1-ynyle sont des exemples de
substituant alkynyle.
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
11
Le terme aryle fait référence à un substituant aromatique mono- ou
polycyclique ayant de 6 à 14 atomes de carbone. Les substituants phényle,
napht-1-yle ; napht-2-yle ; anthracen-9-yl ; 1,2,3,4-tétrahydronapht-5-yle ;
et
1,2,3,4-tétrahydronapht-6-yle sont des exemples de substituant aryle.
Le terme hétéroaryle fait référence à un substituant hétéroaromatique
mono- ou polycyclique ayant de 1 à 13 atomes de carbone et de 1 à 4
hétéroatomes. Les substituants pyrroi-1-yle ; pyrrol2-yle ; pyrrol3-yle ;
furyle ;
thienyle ; imidazolyle ; oxazolyle ; thiazolyle ; isoxazolyle ; isothiazolyle
;
1,2,4-triazolyle ; oxadiazolyle ; thiadiazolyle ; tétrazolyle ; pyridyle ;
pyrimidyle ; pyrazinyle ; 1,3,5-triazinyle ; indolyle ; benzo[b]furyle ;
benzo[b]thiényle ; indazolyle ; benzimidazolyle ; azaindolyle ; quinoiéyle ;
isoquinoléyle ; carbazolyle ; et acridyle sont des exemples de substituant
hétéroaryle.
Le terme hétéroatome fait référence ici à un atome au moins divalent,
différent du carbone. N; O; S; et Se sont des exemples d'hétéroatome.
Le terme cycloalkyle fait référence à un substituant hydrocarboné cyclique
saturé ou partiellement insaturé ayant de 3 à 12 atomes de carbone. Les
substituants cyclopropyle; cyclobutyle; cyclopentyle; cyclopentènyle;
cyclopentadiényle; cyclohexyle; cyclohexènyle; cycloheptyle;
bicyclo[2.2.1 ]heptyle ; cyclooctyle; bicyclo[2.2.2]octyle ; adamantyle ; et
perhydronapthyle sont des exemples de substituant cycloalkyle.
Le terme hétérocyclyle fait référence à un substituant hydrocarboné
cyclique saturé ou partiellement insaturé ayant de 1 à 13 atomes de carbone
et de 1 à 4 hétéroatomes. De préférence, le substituant hydrocarboné
cyclique saturé ou partiellement insaturé sera monocyclique et comportera 4
ou 5 atomes de carbone et 1 à 3 hétéroatomes.
Concernant le phényle fusionné, lorsque m a pour valeur zéro on entend qu'il
s'agit d'un phényle non substitué (ou substitué par 4 atomes d'hydrogène), et
lorsque m a pour valeur 1, 2, 3 ou 4, on entend qu'l, 2, 3 ou 4 atomes
d'hydrogènes sont substitués par un substituant R4.
Les avantages de l'invention seront plus particulièrement illustrés par les
exemples suivants :
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
12
Abréviations :
Ac acétate ; Bn benzyle ; C degré Celsius ; cat. catalyseur ;'CCM
chromatographie sur couche mince ; CCP chromatographie sur colonne
préparative ; cm centimètre ; S déplacement chimique ; d doublet ; dd doublet
de doublets ; DMF diméthylformamide ; DMSO-d6 diméthylsulfoxyde
deutéré ; dt doublet de triplets ; éq. équivalent ; ES+/- electrospray (modes
positif / négatif) ; Et éthyle ; g gramme ; h heure ; Hz hertz ; IC50
constante
d'inhibition à 50% d'activité ; iPr isopropyle ; j. jour ; J constante de
couplage ; LCMS chromatographie liquide couplée à une spectrométrie de
masse; m multiplet ; Me méthyle ; mg milligramme ; MHz mégahertz ; mL
millilitre ; pL microlitre ; mm millimètre ; pm micromètre ; mmol millimole ;
mn minute ; N mol.L"1; PF point de fusion ; Ph phényle ; ppm parties par
million ; q quadruplet ; Rdt rendement; Rf rapport frontal ; RMN 1H
résonance magnétique nucléaire du proton ; s singulet ; sl singulet large ; t
triplet ; TA température ambiante ; tBu tertiobutyle ; TFA acide
trifluoroacétique ; THF tétrahydrofuranne ; tR temps de rétention ; U.V.
ultraviolet ; V volt.
Ex1: N-[1-(3,5-difluoro-benzyl)-2-oxo-2,3,4,5-tetrahydro-1 H-1-benzazépin-3-
yl]- ((E)-(2R,3R,4S,5R)-3,4,5-trihydroxy-2-méthoxy-8,8-diméthyl-non-6-
énamide
F
oH ô 0
N
OH OH O F
Ex1
Etape 1: Préparation du (2-oxo-2,3,4,5-tetrahydro-lH-1-benzazépin-3-yl)-
carbamate de tert-butyle (2)
o N 0
HzN 0 N N
1~
2
Dans un ballon de 250 mL contenant 2,0 g de 1 (11,4 mmol) (3-amino-
1,3,4,5,-tetrahydo-2H-1-benzazépin-2-one, commercial chez Interchim), 1,15
mL de TEA (11,4 mmol) et 50 mL de CHC13, on additionne à 0 C une solution
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
13
de 2,48 g de tBoc2O (11,4 mmol) dans le 50 mL de CHCI3. On agite le milieu
pendant 2h à 0 C sous argon. On lave le milieu rationnel avec 10 mL de HCI
(1 N) et 100 mL d'eau. La phase organique est séchée sur MgSO4, filtrée et
évaporée à sec, on obtient 3,75 g d'un solide crème qui est trituré dans 20
mL d'éther isopropylique. Après essorage, on obtient 2,3 g de 2(solide blanc
cassé).
RMN 'H (400 MHz, DMSO-d6), S(ppm): 1,33 (s, 9H) ; 2,06 (m, 1 H) ; 2,18 (m,
1 H) ; de 2,60 à 2,70 (m, 2H) ; 3,87 (m, 1 H) ; 6,92 (d, J = 8,0 Hz, 1 H) ;
7,00
(d, J = 7,5 Hz, 1 H) ; 7,11 (t large, J =7,5 Hz, 1 H) ; de 7,21 à 7,29 (m, 2H)
;
9,68 (s, 1 H) .
Etape 2: Préparation du [1-(3,5-difluoro-benzyl)-2-oxo-2,3,4,5-tetrahydro-1 H-
1-benzazépin-3-yl]-carbamate de tert-butyle (3)
F
O O
O N N H N ~~
~ i ~ ~-- 0" F
O
2 3
Dans un ballon de 100 mL, sous agitation et atmosphère d'argon, contenant
20 mL de THF et 0,5 g de 2(1,81 mmol), on introduit à TA 72 mg d'hydrure
de sodium en suspension à 60% dans l'huile (1,81 mmol). On agite le milieu
pendant 1 h, puis ajoute 0,75 g (3,62 mmol) de bromure de 3,5-
difluorobenzyle. On laisse le milieu sous agitation à TA pendant une nuit. On
ajoute 50 mL d'AcOEt, lave la phase organique avec 50 mL d'eau. La phase
organique est séchée sur MgSO4, filtrée puis évaporée à sec. On obtient 1 g
d'un solide blanc. Après trituration avec 20 mL d'éther isopropylique et
essorage, on obtient 0,62 g de produit 3(solide blanc).
RMN 1H (400 MHz, DMSO-ds), b(ppm): 1,35 (s, 9H) ; 2,10 (m, 2H) ; 2,52 (m
partiellement masqué, 1 H) ; 2,61 (m, 1 H) ; 3,94 (m, 1 H) ; 4,92 (d, J = 16,0
Hz, 1 H) ; 5,10 (d, J= 16,0 Hz, 1 H) ; 6,96 (m, 2H) ; 7,09 (tt, J = 2,5 et 9,5
Hz,
1 H) ; de 7,17 à 7,22 (m, 3H) ; de 7,25 à 7,34 (m, 2H).
Etape 3: Préparation du chlorhydrate de 3-amino-l-(3,5-difluoro-benzyl)-
1,3,4,5-tetrahydro-1-benzazépin-2-one (4)
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
14
F F
0 0
O
N N HzN N
I F
C I
HCI
3 4
On reprend 0,62 g de 3(1,54 mmol) dans un ballon de 25 mL et on ajoute 10
mL d'une solution d'acide chlorhydrique dans le dioxane (4M). On agite le
milieu pendant 5h à TA sous argon. Après évaporation du solvant, on obtient
0,46 g d'amine 4 sous forme de chlorhydrate qui est utilisé directement pour
l'étape suivante.
RMN ' H(300 MHz, DMSO-d6), â(ppm): 2,16 (m, 1 H) ; 2,43 (m, 1 H) ; 2,57 (m,
1 H) ; 2,70 (m, 1 H) ; 3,76 (dd, J = 7,5 et 11,5 Hz, 1 H) ; 5,04 (d, J= 15,5
Hz,
1 H) ; 5,12 (d, J = 15,5 Hz, 1 H) ; 6,99 (m, 2H) ; 7,14 (tt, J = 2,5 et 9,5
Hz, 1 H)
de 7,22 à 7,40 (m, 4H) ; 8,31 (s large, 3H) .
Etape 4: Préparation de N-[1-(3,5-difluoro-benzyl)-2-oxo-2,3,4,5-tetrahydro-
1 H-1-benzazépin-3-yl]-2-[(4R,5S,6R)-6-((E)-3,3-diméthyl-but-1-ényl)-5-
hydroxy-2,2-diméthyl-1,3-dioxinan-4-yl]-2-méthoxy-acétamide (6)
F
F
HCI C -- ~HO oo
H
o~ + HZN N N N
o F
oô F
4 6
On introduit successivement dans un tube Wheaton, sous agitation et sous
atmosphère d'argon, 100 mg de 5(352 pmol) (qui peut être préparé selon les
modes opératoire décrites dans Org. Process Res. Dev. 2003, 7(6), 856-865),
239 mg de 4 (0,70 mmol), 146 mg de 2-éthylhexanoate de sodium (0,77
mmol) dans 2 mL de THF. On maintient l'agitation à TA pendant 24 h. On
ajoute 3 mL d'acétate d'éthyle au milieu réactionnel. On lave successivement
avec 3 mL d'une solution de HCI (1 N), puis 3 mL d'une solution saturée de
NaHCO3 et 3 mL d'eau. La phase organique est séchée sur sulfate de
magnésium anhydre, filtrée puis évaporée à sec. On obtient 450 mg d'une
huile incolore, qui est chromatographié sur une cartouche de silice (20g,
éluant CH2CI2/MeOH- en gradient MeOH 1 à 10%). On recueille 122 mg de
produit attendu 6.
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
RMN 1H (400 MHz, DMSO-d6), S(ppm): (un mélange 50% - 50% d'isomères)
0,98 (s, 4,5H) ; 0,99 (s, 4,5H) ; 1,20 (s, 1,5H) ; 1,21 (s, 1,5H) ; 1,26 (s,
1,5H)
1,27 (s, 1,5H) ; 2,15 (m, 2H) ; 2,53 (m partiellement masqué, 1H) ; 2,63 (m,
1 H) ; 3,23 (s, 1,5H) ; 3,25 (m masqué, 1 H) ; 3,26 (s, 1,5H) ; 3,75 (d, J =
8,5
5 Hz, 0,5H) ; 3,80 (d, J = 8,5 Hz, 0,5H) ; 3,91 (d large, J 8,5 Hz, 0,5H) ;
3,96
(d large, J = 8,5 Hz, 0,5H) ; de 4,20 à 4,42 (m, 3H) ; de 4,95 à 5,11 (m, 2H)
;
5,44 (dd dédoublé, J = 7,0 et 16,0 Hz, 1 H) ; 5,67 (d dédoublé, J = 16,0 Hz,
1 H) ; 6,94 (m, 2H) ; 7,10 (m, 1 H) ; de 7,18 à 7,37 (m, 4H) ; 8,26 (d, J =
8,0
Hz, 0,5H) ; 8,35 (d, J = 7,0 Hz, 0,5H).
10 Etape 5: Préparation du N-[1-(3,5-difluoro-benzyl)-2-oxo-2,3,4,5-tetrahydro-
1 H-1 -benzazépin-3-yl]- ((E)-(2R,3R,4S,5R)-3,4,5-trihydroxy-2-méthoxy-8,8-
diméthyl-non-6-énamide (Ex1)
F
/
OH 0 0 I OH O 0
N m N
ôô o F = F
OH OH O
g Ex1
Dans un ballon de 10 mL, on mélange 92 mg de 6(157 pmol) dans 0,8 mL de
15 THF et 1,57 mL d'acide chlorhydrique 1 N (1,57 mmol), sous agitation et
sous
argon. L'agitation est maintenue 5 h à TA. Il se forme un précipité, qui est
essoré sur un verre fritté. Après lavage avec 0,5 ml THF puis 2 mL d'éther
isopropylique et séchage sous vide, on obtient 70 mg de Ex1 (solide blanc).
ES : m/z = 569 MNa+.
RMN'H (400 MHz, DMSO-d6), â(ppm): (un mélange 50% - 50% d'isomères)
0,94 (s, 4,5H) ; 0,95 (s, 4,5H) ; 2,18 (m, 2H) ; 2,53 (m partiellement masqué,
1 H) ; 2,63 (m, 1 H) ; 3,22 (s, 1,5H) ; 3,24 (s, 1,5H) ; de 3,26 à 3,33 (m
masqué, 1 H) ; 3,52 (m, 1 H) ; 3,68 (d, J = 8,0 Hz, 1 H) ; 3,94 (m, 1 H) ; de
4,18
à 4,36 (m, 3H) ; 4,54 (d large, J = 4,5 Hz, 1 H) ; de 4,98 à 5,10 (m, 2H) ;
5,28
(dd dédoublé, J 7,0 et 16,0 Hz, 1 H) ; 5,61 (d, J = 16,0 Hz, 1 H) ; 6,95 (m,
2H) ; 7,10 (tt, J 2,5 et 10,0 Hz, 1 H) ; de 7,17 à 7,36 (m, 4H) ; 8,16 (d, J
8,0 Hz, 0,5H) ; 8,24 (d, J = 7,0 Hz, 0,5H).
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
16
Ex2: N-[1-méthyl-2-oxo-2,3,4,5-tetrahydro-1 H-1-benzazépin-3-yl]-((E)-
(2R,3 R,4S,5R)-3,4,5-trihyd roxy-2-méthoxy-8,8-diméthyl-non-6-énamide
oH o~
_
oH oH o tJ / \
Ex2
Etape 1: Préparation du [1-méthyl-2-oxo-2,3,4,5-tetrahydro-1 H-1-benzazépin-
3-yl]-carbamate de tert-butyle (7)
o O
e
O` 'N N
/x\ l'OI{ N OyN ~ ~
O
2 7 ~
Dans un ballon de 100 mL, sous agitation et atmosphère d'argon, contenant
50 mL de THF et 1,2 g de 2 (4,34 mmol), on introduit à TA 174 mg d'hydrure
de sodium en suspension à 60% dans l'huile (4,34 mmol). On agite le milieu
pendant 1 h, et puis ajoute 1,23 g (8,69 mmol) d'iodure de méthyle. On laisse
le milieu sous agitation pendant une nuit, on ajoute 50 mL d'AcOEt, lave la
phase organique avec 100 mL d'eau. La phase organique est séchée sur
MgSO4, filtrée puis évaporée à sec. On obtient 1,3 g d'un solide blanc. Après
chromatographie sur une cartouche de silice (40 g) (éluant CH2CI2/MeOH- en
gradient MeOH 1 à 10%). On obtient 0,66 g de produit 7 (solide blanc).
RMN'H (400 MHz, DMSO-d6), 8(ppm): 1,33 (s, 9H) ; 2,05 (m, 2H) ; 2,62 (m,
2H) ; 3,26 (s, 3H) ; 3,86 (m, 1 H) ; 7,00 (d, J= 8,5 Hz, 1 H) ; 7,21 (m, 1 H)
; 7,28
(m, 1 H) ; 7,36 (m, 2H).
Etape 2: Préparation du chlorhydrate de 3-amino-1-méthyl-1,3,4,5-tetrahydro-
1-benzazépin-2-one (8)
0 0
b o~N N HZN N
HCI
o 7 $
On reprend 0,66 g de 7 (2,27 mmol) dans un ballon de 50 mL et on ajoute 15
mL d'une solution d'acide chlorhydrique dans le dioxane (4M). On agite
pendant 5h à TA sous argon. Après évaporation du solvant, on obtient 0,53 g
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
17
d'amine 8 sous forme de chlorhydrate qui est utilisé directement pour l'étape
suivante.
RMN 1H (300 MHz, DMSO-d6), 8(ppm):: 2,12 (m, 1H) ; 2,45 (m partiellement
masqué, 1 H) ; de 2,68 à 2,79 (m, 2H) ; 3,34 (s, 3H) ; 3,59 (dd, J = 7,5 et
11,5
Hz, 1 H) ; 7,25 (m, 1 H) ; 7,35 (d large, J = 7,5 Hz, 1 H) ; 7,40 (m, 2H) ;
8,38 (s
large, 3H).
Etape 3: Préparation du (3R,4R,5S)-4-Hydroxy-5-((E)-(R)-1-hydroxy-4,4-
diméthyl-pent-2-ényl)-3-méthoxy-dihydro-furan-2-one (9)
: .
H ôH
9
Dans un ballon de 250 mL contenant 40 mL d'eau et 3,6 g de 3 en
suspension, on ajoute 17 mL de TFA dans 10 mL d'eau. On agite le milieu
pendant 1,5 h à TA. On dilue le milieu avec 290 mL d'eau, congèle puis
lyophilise. On obtient 4 g d'une huile qui est cristallisé dans 20 mL d'éther
isopropylique à TA. Après essorage, lavage à l'éther isopropylique, et
séchage sous vide à 40 C, on obtient 2,46 g de produit attendu 7 (cristaux
blanc).
PF: 123 C.
IC : m/z = 262 MNH4+
RMN'H (400 MHz, DMSO-d6), â(ppm): 1,00 (s, 9H) ; 3,41 (s, 3H) ; 3,93 (dd, J
= 2,5 et 9,0 Hz, 1 H) ; de 4,22 à 4,31 (m, 3H) ; 5,19 (d, J = 5,0 Hz, 1 H) ;
5,42
(dd, J= 5,0 et 16,0 Hz, 1 H) ; 5,43 (d, J= 4,5 Hz, 1 H) ; 5,87 (d, J= 16,0 Hz,
1H).
IR (KBr): 3239; 2964; 2914; 1701; 1499; 1312; 1253; 1047 & 751 cm-1.
Etape 4: Préparation du N-[1-méthyl-2-oxo-2,3,4,5-tetrahydro-1 H-1-
benzazépin-3-yl]-((E)-(2R,3R,4S,5R)-3,4,5-trihydroxy-2-méthoxy-8,8-
diméthyl-non-6-énamide (Ex2)
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
18
H4
N
~I + HCI / \ _ = tJ
OH oH
OH ôH
g Ex2
On introduit successivement dans un tube Wheaton, sous agitation et sous
atmosphère d'argon, 36 mg de 9(147 pmoi), 66,8 mg de 8(0,30 mmol), 61
mg de 2-éthylhexanoate de sodium (0,37 mmol) dans 1,0 mL de THF. On
maintient l'agitation à TA pendant 24 h. On évapore directement le milieu
réactionnel à sec. Le brut est chromatographié sur une cartouche de silice
(5g, éluant CH2CI2/MeOH - en gradient MeOH 1 à 10%). On recueille 39 mg
de produit attendu Ex2 .
ES : m/z = 457 M Na+ ; m/z = 435 M H+.
RMN 1H (400 MHz, DMSO-d6), S(ppm): (un mélange 50% - 50% d'isomères)
0,95 (s, 4,5H) ; 0,96 (s, 4,5H) ; 2,06 (m, 1 H) ; 2,21 (m, 1 H) ; de 2,60 à
2,75
(m, 2H) ; 3,21 (s, 3H) ; de 3,25 à 3,35 (m masqué, 1 H) ; 3,29 (s large, 3H) ;
3,51 (m large, 1 H) ; 3,66 (d, J = 7,5 Hz, 0,5H) ; 3,67 (d, J= 7,5 Hz, 0,5H) ;
3,93 (m, 1 H) ; 4,21 (m, 1 H) ; de 4,28 à 4,60 (m large, 3H) ; 5,28 (dd, J =
7,0
et 16,0 Hz, 0,5H) ; 5,30 (dd, J = 7,0 et 16,0 Hz, 0,5H) ; 5,61 (d, J = 16,0
Hz,
0,5H) ; 5,63 (d, J = 16,0 Hz, 0,5H) ; 7,22 (m, 1 H) ; 7,31 (m, 1 H) ; de 7,37
(m,
2H) ; 8,03 (d, J = 8,0 Hz, 0,5H) ; 8,05 (d, J = 7,0 Hz, 0,5H).
'Ex3: N-(7-bromo-2-oxo-2,3,4,5-tetrahydro-1 H-1-benzazépin-3-yl)-(E)-
(2R,3R,4S,5R)-3,4,5-trihydroxy-2-méthoxy-8,8-diméthyl-non-6-énamide et
Ex3a & Ex3b
I CH' N.. O N I O_H o~ H O I oH Of O
N
OH OH O oH oH O OH OH O
Ex3 Ex3a Ex3b
Br Br Br
Etape 1: Préparation du (7-bromo-2-oxo-2,3,4,5-tetrahydro-1 H-1 -benzazépin-
3-yl)-carbamate de tert-butyle (10)
o ~
N
ON N ON
O \ '~C\ O
2 10 Br
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
19
Dans un ballon de 100 mL, sous agitation et atmosphère d'argon, contenant
40 mL d'AcOEt et 0,5 g de 2 (1,8 mmol); on introduit à TA 472 mg de N-
bromosuccinimide (2,65 mmol). On laisse le milieu sous agitation pendant
une nuit, lave avec 40 mL de HCI (1 N), 40 mL d'eau saturée en NaHCO3 et
40 mL d'eau. La phase organique est séchée sur MgSO4, filtrée puis
évaporée à sec. On obtient 1,3 g d'une huile, qui cristallise dans 10 mL
d'éther isopropylique. Après essorage, on obtient 450 mg de produit 10
(solide blanc).
RMN 'H (400 MHz, DMSO-d6), â(ppm): 1,34 (s, 9H) ; 2,05 (m, 1 H) ; 2,21 (m,
1 H) ; de 2,58 à 2,72 (m, 2H) ; 3,86 (m, 1 H) ; 6,95 (d, J = 8,5 Hz, 1 H) ;
6,98 (d,
J= 8,5 Hz, 1 H) ; 7,44 (dd, J = 2,0 et 8,5 Hz, 1 H) ; 7,50 (d, J = 2,0 Hz, 1
H) ;
9,7 (s, 1 H).
Etape 2: Préparation du chlorhydrate de 3-amino-7-bromo-1,3,4,5-tetrahydro-
1-benzazépin-2-one (11)
0 0
H H
o"r N N HZN N
p ' HCI
10 Br 11 Br
On reprend 0,45 g de 10 (1,27 mmol) dans un ballon de 25 mL et on ajoute
10 mL d'une solution d'acide chlorhydrique dans le dioxane (4M). On agite le
milieu pendant 5 h à TA sous argon. Un précipité s'est formé, qui est filtré
sur
un verre fritté, lavé avec 5 mL de dioxane puis 5 mL d'éther isopropylique. On
obtient 0,38 g d'amine 11 sous forme de chlorhydrate.
RMN iH (400 MHz, DMSO-d6), â(ppm): 2,12 (m, 1 H) ; 2,51 (m partiellement
masqué, 1 H) ; de 2,65 à 2,84 (m, 2H) ; 3,72 (dd, J = 8,0 et 11,5 Hz, 1 H) ;
6,99
(d, J = 8,5 Hz, 1 H) ; 7,48 (dd, J = 2,5 et 8,5 Hz, 1 H) ; 7,58 (d, J = 2,5
Hz, 1 H) ;
8,25 (s, 3H) ; 10,35 (s, 1 H) .
Etape 3: Préparation du (R)-N-(7-bromo-2-oxo-2,3,4,5-tetrahydro-1 H-1-
benzazépin-3-yl)-2-[(4R,5S,6R)-6-((E)-3,3-diméthyl-but-1-ényl)-5-hydroxy-2,2-
diméthyl-1,3-dioxinan-4-yl]-2-méthoxy-acétamide (12)
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
HCI O
I H oH ô 0
o~+HzN
0 0
K 0 x
5 qq Br 12 Br
On introduit successivement dans un tube Wheaton, sous agitation et sous
atmosphère d'argon, 100 mg de 5(352 pmol), 205 mg de 11 (0,70 mmol),
146 mg de 2-éthylhexanoate de sodium (0,77 mmol) dans 2 mL de THF. On
5 maintient l'agitation à TA pendant 24 h. On évapore à sec puis purifie par
chromatographie sur une cartouche de silice (20g) avec un éluant
CH2CI2/MeOH (en gradient MeOH 1 à 10%). On recueille 160 mg de produit
attendu 12.
RMN ' H(300 MHz, DMSO-d6), 5(ppm): (un mélange 70% - 30% d'isomères)
10 0,99 (s, 9H) ; 1,23 (s, 0,9H) ; 1,25 (s, 2,1 H) ; 1,28 (s, 0,9H) ; 1,29 (s,
2,1 H) ;'
2,08 (m, 1 H) ; 2,27 (m, 1 H) ; de 2,65 à 2,76 (m, 2H) ; de 3,21 à 3,34 (m
masqué, 1 H) ; 3,24 (s, 2,1 H) ; 3,26 (s, 0,9H) ; 3,73 (d, J 8,5 Hz, 0,7H) ;
3,78
(d, J = 8,5 Hz, 0,3H) ; 3,90 (d large, J = 8,5 Hz, 0,3H) ; 3,96 (d large, J=
8,5
Hz, 0,7H) ; de 4,12 à 4,43 (m, 3H) ; 5,43 (dd, J 7,0 et 16,0 Hz, 0,3H) ; 5,44
15 (dd, J = 7,0 et 16,0 Hz, 0,7H) ; 5,68 (d large, J 16,0 Hz, 1 H) ; 6,97 (d,
J =
8,5 Hz, 1 H) ; 7,46 (dd, J = 2,5 et 8,5 Hz, 1 H) ; 7,54 (d, J = 2,5 Hz, 1 H) ;
8,08
(d, J = 8,0 Hz, 0,7H) ; 8,15 (d, J = 7,5 Hz, 0,3H) ; 9,85 (s, 0,3H) ; 9,89 (s,
0,7H).
Etape 4: Préparation du N-(7-bromo-2-oxo-2,3,4,5-tetrahydro-1 H-1 -
20 benzazépin-3-yl)-(E)-(2R,3R,4S,5R)-3,4,5-trihydroxy-2-méthoxy-8,8-diméthyl-
non-6-énamide (Ex3) et Ex3a & Ex3b
i OH O O OH O
0/K\ ` /0 0 OH OH 0
12 Br Ex3
Br
Dans un ballon de 25 mL, on introduit -150 mg de 12 (258 pmol) dans 1,4 mL
de THF et 2,78 mL d'acide chlorhydrique 1 N, sous agitation et sous argon.
L'agitation est maintenue 4,5 h à TA. On neutralise le milieu avec NaOH (1 N)
jusqu'à pH 7, et extrait avec 2 fois 3 mL d'AcOEt. Les phases organiques
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
21
réunies sont séchées sur MgSO4, filtrées et évaporées à sec. Le brut est
chromatographié sur une cartouche de silice (10 g) en éluant avec
CH2CI2/MeOH (en gradient MeOH 1 à 10%). On recueille 65 mg de produit
attendu Ex3. 40 mg d'Ex3 est séparé par HPLC chiral préparative (Chirapad
AD-H 5 pm 250 x 4.6 mm, débit 1 mI/min, phase mobile : Heptane 50%-EtOH
50%, TEA 0,1 %), pour donner 9 mg d'Ex3a (tr = 7,48 min) et 20 mg d'Ex3b
(tr = 9,71 min).
ES : m/z = 497 (M-H)".
Ex3: RMN 'H (400 MHz, DMSO-d6), â(ppm): (un mélange 70% - 30%
d'isomères) 0,95 (s, 6,3 H) ; 0,96 (s, 2,7H) ; 2,09 (m, 1 H) ; 2,31 (m, 1 H) ;
de
2,65 à 2,73 (m, 2H) ; de 3,19 à 3,33 (m masqué, 1 H) ; 3,22 (s, 2,1 H) ; 3,29
(s,
0,9H) ; 3,52 (m, 1 H) ; 3,66 (d, J = 8,0 Hz, 0,3H) ; 3,68 (d, J = 7,5 Hz,
0,7H) ;
3,94 (m, 1 H) ; de 4,16 à 4,41 (m, 3H) ; 4,55 (d, J = 4,5 Hz, 1 H) ; 5,28 (dd,
J
7,0 et 16,0 Hz, 0,3H) ; 5,30 (dd, J = 7,0 et 16,0 Hz, 0,7H) ; 5,61 (d J= 16,0
Hz, 0,3H) ; 5,63 (d, J 16,0 Hz, 0,7H) ; 6,97 (d, J = 8,5 Hz, 1 H) ; 7,46 (dd,
J=
2,5 et 8,5 Hz, 1 H) ; 7,54 (d, J = 2,5 Hz, 1 H) ; 8,04 (d, J = 7,5 Hz, 1 H) ;
9,91 (s,
0,3H) ; 9,95 (s, 0,7H) .
Ex3a: RMN 1H (400 MHz, DMSO-d6), b(ppm): 0,96 (s, 9H) ; 2,07 (m, 1H) ;
2,30 (m, 1 H) ; 2,70 (m, 2H) ; 3,22 (s, 3H) ; de 3,27 à 3,35 (m masqué, 1 H) ;
3,51 (m, 1 H) ; 3,67 (d, J = 7,5 Hz, 1 H) ; 3,91 (m, 1 H) ; 4,19 (m, 1 H) ;
4,29 (m,
2H) ; 4,54 (d, J = 4,5 Hz, 1 H) ; 5,29 (dd, J = 7,0 et 16,0 Hz, 1 H) ; 5,62
(d, J =
16,0 Hz, 1 H) ; 6,97 (d, J = 8,5 Hz, 1 H) ; 7,45 (dd, J = 2,5 et 8,5 Hz, 1 H)
; 7,54
(d, J = 2,5 Hz, 1 H) ; 8,02 (d, J = 7,5 Hz, 1 H) ; 9,90 (s, 1 H).
Ex4: N-(7-phenyi-2-oxo-2,3,4,5-tetrahydro-1 H-1-benzazépin-3-yl)-(E)-
(2R,3R,4S,5R)-3,4,5-trihydroxy-2-méthoxy-8,8-diméthyl-non-6-énamide et
Ex4a & Ex4b
_
OH O F{ O I OH O O H I OH O
OH OH O I\ OH OH O OH OH O
Ex4 Ex4a Ex4b
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
22
Etape 1: Préparation du (7-phenyl-2-oxo-2,3,4,5-tetrahydro-1 H-1 -benzazépin-
3-yl)-carbamate de tert-butyle (13)
0 0
Oy N N Oir N N
O O
Br 13
Dans un ballon de 100 mL, sous agitation et atmosphère d'argon, contenant
5 22 mL d'eau, 6,5 mL de dioxane et 1,08 g de 10 (3,04 mmol), on introduit 371
mg d'acide phenylboronique (3,04 mmol), 62 mg de chlorure de 1,1'-
bis(diphenylphosphino)ferrocene palladium (C35H30CI4FeP2Pd, PM 816,65,
0,025 mmol) et 3,96 g de carbonate de Césium (12,16 mmol). On chauffe à
100 C le milieu sous agitation pendant 1 h. On filtre le milieu sur célite,
lave
10 avec 10 mL de dioxane, 10 mL de CH2CI2 et 10 mL de MeOH. Le filtrat est
concentré sous vide. On ajoute ensuite 25 ml d'AcOEt et lave avec 2 fois 25
mL d'eau. La phase organique est séchée sur MgSO4, filtrée et évaporé à
sec. Le brut est chromatographié sur une cartouche de silice (50 g) en éluant
avec CH2CI2/MeOH (en gradient MeOH : 1 à 10%). On recueille 350 mg de
produit attendu 13 (solide blanc).
RMN 1H(300 MHz, DMSO-d6), â(ppm): 1,34 (s, 9H) ; 2,09 (m, 1 H) ; 2,25 (m,
1 H) ; de 2,63 à 2,79 (m, 2H) ; 3,94 (m, 1 H) ; 6,97 (d, J = 8,5 Hz, 1 H) ;
7,09
(d, J = 8,5 Hz, 1 H) ; 7,34 (t, J =7,5 Hz, 1 H) ; 7,46 (t, J = 7,5 Hz, 2H) ;
7,56 (dd,
J = 2,0 et 8,5 Hz, 1 H) ; 7,58 (s large, 1 H) ; 7,66 (d, J =7,5 Hz, 2H) ; 9,77
(s,
1H).
Etape 2: Préparation du chlorhydrate de 3-amino-7-phenyl-1,3,4,5-tetrahydro-
1-benzazapin-2-one (14)
0 0
H H
O T'( ` /N N _HZN
Jx HCI
/ \
0
~ ~
13 / ~ 14
On reprend 0,35 g de 13 (0,99 mmol) dans un ballon de 20 mL et on ajoute 5
mL d'une solution d'acide chlorhydrique dans le dioxane (4M). On agite le
milieu pendant 5h à TA sous argon. Il se forme un précipité que l'on essore et
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
23
lave avec 5 mL de dioxane puis 5 mL d'éther isopropylique. Après séchage,
on obtient 0,23 g d'amine 14 sous forme de chlorhydrate.
RMN 'H (400 MHz, DMSO-d6), 8(ppm):: 2,18 (m, 1 H) ; 2,55 (m partiellement
masqué, 1 H) ; de 2,74 à 2,90 (m, 2H) ; 3,76 (dd, J 8,5 et 12,0 Hz, 1 H) ;
7,12 (d, J 8,0 Hz, 1 H) ; 7,36 (t, J = 7,5 Hz, 1 H) ; 7,47 (t, J = 7,5 Hz, 2H)
;
7,60 (dd, J 2,0 et 8,0 Hz, 1 H) ; de 7,64 à 7,69 (m, 3H) ; 8,19 (s large, 3H)
;
10,35 (s, 1 H) .
Etape 3: Préparation du (R)-N-(7-phenyl-2-oxo-2,3,4,5-tetrahydro-1 H-1-
benzazépin-3-yl)-2-[(4R,5S,6R)-6-((E)-3,3-diméthyl-but-1-ényl)-5-hydroxy-2,2-
diméthyl-1,3-dioxinan-4-yl]-2-méthoxy-acétamide (15)
H
HzN N oH ô 0
HCI ~~ N
._ O` 'X\ O O
74 ~ ~ J 15
On introduit successivement dans un tube Wheaton, sous agitation et sous
atmosphère d'argon, 100 mg de 5(352 pmol), 203 mg de 14 (0,70 mmol),
146 mg de 2-éthylhexanoate de sodium (0,77 mmol) dans 2 mL de THF. On
maintient l'agitation à TA pendant 24 h. On évapore à sec puis purifie par
chromatographie sur une cartouche de silice (20g) en éluant avec
CH2CI2/MeOH (en gradient MeOH ; 1 à 10%). On recueille 150 mg de produit
attendu 15.
RMN 1H (300 MHz, DMSO-d6), 8(ppm): (un mélange 60% - 40% d'isomères):
0,99 (s, 9H) ; 1,23 (s, 1,2H) ; 1,25 (s, 1,8H) ; 1,28 (s, 1,2H) ; 1,30 (s,
1,8H)
2,11 (m, 1 H) ; 2,32 (m, 1 H) ; 2,78 (m, 2H) ; de 3,22 à 3,35 (m masqué, 1 H)
;
3,25 (s, 1,8H) ; 3,27 (s, 1,2H) ; 3,74 (d, J = 8,5 Hz, 0,6H) ; 3,77 (d, J =
8,5 Hz,
0,4H) ; 3,91 (d large, J = 8,5 Hz, 0,4H) ; 3,96 (d large, J = 8,5 Hz, 0,6H) ;
de
4,20 à 4,44 (m, 3H) ; 5,44 (dd, J = 7,0 et 16,0 Hz, 1 H) ; 5,68 (d, J = 16,0
Hz,
1 H) ; 7,10 (d, J = 8,5 Hz, 1 H) ; 7,35 (t, J = 7,5 Hz, 1 H) ; 7,46 (t, J =
7,5 Hz,
2H) ; 7,57 (d large, J = 8,5 Hz, 1 H) ; 7,62 (s large, 1 H) ; 7,67 (d, J = 7,5
Hz,
2H) ; 8,09 (d, J = 7,5 Hz, 0,6H) ; 8,15 (d, J = 7,5 Hz, 0,4H) ; 9,87 (s, 0,4H)
;
9,92 (s, 0,6H).
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
24
Etape 4: Préparation du N-(7-phenyl-2-oxo-2,3,4,5-tetrahydro-1 H-1-
benzazépin-3-yl)-(E)-(2R,3R,4S,5R)-3,4,5-trihydroxy-2-méthoxy-8,8-diméthyl-
non-6-énamide (Ex4) et Ex4a & Ex4b
~ O
OH 0 O
N N
OH O N H \HAHO
O\ O O ~ \ / \
sx\ 15 / \ Ex4
Dans un ballon de 20mL, on introduit 150 mg de 15 (280 pmol) dans 1,4 mL
de THF et 2,78 mL d'acide chlorhydrique 1 N, sous agitation et sous argon.
L'agitation est maintenue 4 h à TA. On neutralise le milieu avec NaOH (1 N)
jusqu'à pH 7, et extrait avec 2 fois 5 mL d'AcOEt. Les phases organiques
réunies sont séchées sur MgSO4, filtrées et évaporées à sec. Le brut est
chromatographié sur une cartouche de silice (10 g) avec un éluant
CH2CI2/MeOH (en gradient MeOH 1 à 109ïo/). On recueille 85 mg de produit
attendu Ex4. 58 mg d'Ex4 est séparé par HPLC chiral préparative (Chirapad
AD-H 5 pm 250 x 4.6 mm, débit 1 mI/min, phase mobile : Heptane 70%-EtOH
30%, TEA 0,1%), pour donner 16 mg d'Ex4a (tr = 13,42 min) et 40 mg
d'Ex4b (tr = 18,96 min).
ES : 497 MH+
Ex4: RMN 1H (400 MHz, DMSO-d6), 8(ppm): (un mélange 70% - 30%
d'isomères) 0,95 (s, 2,7H) ; 0,99 (s, 6,3H) ; 2,11 (m, 1 H) ; 2,35 (m, 1 H) ;
2,79
(m, 2H) ; 3,23 (s, 3H) ; de 3,25 à 3,35 (m masqué, 1 H) ; 3,53 (m, 1 H) ; 3,68
(d, J= 8,5 Hz, 0,3H) ; 3,70 (d, J = 8,5 Hz, 0,7H) ; 3,95 (m, 1 H) ; de 4,22 à
4,33 (m, 2,3H) ; 4,41 (d, J = 6,0 Hz, 0,7H) ; 4,55 (m, 1 H) ; 5,30 (m, 1 H) ;
5,61
(d, J = 16,0 Hz, 0,3H) ; 5,63 (d, J = 16,0 Hz, 0,7H) ; 7,10 (d, J = 8,5 Hz, 1
H) ;
7,35 (t, J = 7,5 Hz, 1 H) ; 7,46 (t, J 8,0 Hz, 2H) ; 7,57 (dd, J = 2,5et 8,5
Hz,
1 H) ; 762 (s large, 1 H) ; 7,67 (d, J 8,0 Hz, 2H) ; 8,04 (d, J = 7,5 Hz, 1 H)
;
9,93 (s, 03H) ; 9,99 (s, 0,.7H) .
Ex4a : RMN iH (400 MHz, DMSO-d6), â(ppm): 0,95 (s, 9H) ; 2,10 (m, 1H) ;
2,37 (m, 1 H) ; 2,79 (m, 2H) ; 3,24 (s, 3H) ; de 3,27 à 3,33 (m masqué, 1 H) ;
3,51 (m, 1 H) ; 3,68 (d, J = 7,5 Hz, 1 H) ; 3,93 (m, 1 H) ; de 4,22 à 4,32 (m,
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
3H) ; 4,54 (d, J = 4,5 Hz, 1 H) ; 5,29 (dd, J = 7,0 et 16,0 Hz, 1 H) ; 5,61
(d, J =
16,0 Hz, 1 H) ; 7,10 (d, J = 8,0 Hz, 1 H) ; 7,35 (t, J = 7,5 Hz, 1 H) ; 7,46
(t, J =
7,5 Hz, 2H) ; 7,56 (dd, J = 2,0 et 8,0 Hz, 1 H) ; 7,62 (d, J = 2,0 Hz, 1 H) ;
7,67
(m, 2H) ; 8,02 (d, J = 7,5 Hz, 1 H) ; 9,93 (s, 1 H) .
5 Ex4a: (2R,3R,4S,5R,6E)-3,4,5-trihydroxy-2-méthoxy-8,8-diméthyl-N-[(3S)-2-
oxo-7-phényl-2,3,4,5-tétrahydro-1 f-I-1-benzazépin-3-yl]non-6-énamide
OH ô O
N,
OH OH O
Ex4a
Etape 1: Préparation du [(3S)-2-oxo-2,3,4,5-tétrahydro-1 H-1-benzazépin-3-
yl]carbamate de tert-butyle (2a)
0
N 0
H N
C~r N",,,
o
1a 2a
Dans un ballon de 2 L contenant 20,0 g de 1 a(113,5 mmol) ((s)-3-amino-
1,3,4,5,-tetrahydo-2H-1-benzazépin-2-one, préparé selon les modes
opératoires décrites dans J. Org. Chem. 1997, 62, 8271), et 500 mL de
CHCI3, on additionne à 0 C, 16 mL de TEA (113,5 mmol), une solution de
24,77g de tBoc2O (113,5 mmol) dans le 500 mL de CHCI3. On agite le milieu
pendant 1 nuit. On lave le milieu rationnel avec 500 mL de HCI (0.5N) et 500
mL d'eau. La phase organique est séchée sur MgSO4, filtrée et évaporée à
sec, on délite les solides avec 200 mL d'éther isopropylique. Après essorage,
on obtient 30.02 g de 2a (solide blanc cassé).
[a]o : -229,0 +/- 2,5 (c=1,852 mg/0,5 mL MeOH)
RMN ' H(400 MHz, DMSO-d6), 8(ppm): 1,33 (s, 9H) ; de 1,98 à 2,25 (m, 2H) ;
de 2,59 à 2,71 (m, 2H) ; 3,89 (m, 1 H) ; 6,90 (d, J = 8,0 Hz, 1 H) ; 7,00 (d,
J
7,5 Hz, 1 H) ; 7,11 (d, J = 7,5 Hz, 1 H) ; de 7,20 à 7,30 (m, 2H) ; 9,68 (s, 1
H).
Etape 2: Préparation du [(3S)-7-bromo-2-oxo-2,3,4,5-tétrahydro-1 H-1-
benzazépin-3-yl]carbarnate de tert-butyle (10a)
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
26
H o p
N
pyN,,,,. N / \ \ ~1- p, r N,,,,.
O
2a 10a Br
Dans un tricol de 2 L, sous agitation et atmosphère d'argon, contenant 1,5 L
d'AcOEt et 8,47 g de 2a (103,0 mmol), on introduit à TA 23,5 g de N-
bromosuccinimide (136 mmol) en 4 fois pendant 3 jours. On laisse le milieu
sous agitation une nuit supplémentaire, lave avec 500 mL de HCI (0,5N), 500
mL d'eau saturée en NaHCO3 et 500 mL d'eau. La phase organique est
séchée sur MgSO4, filtrée puis évaporée à sec. On obtient 38 g de meringue
marron. On purifie par chromatographie sur une cartouche de silice (600g) en
éluant avec Heptane/AcOEt (70/30). On recueille 20,46 g de produit attendu
10a (solide blanc).
RMN iH (400 MHz, DMSO-d6), S(ppm): 1,34 (s, 9H) ; 2,05 (m, 1 H) ; 2,20 (m,
1 H) ; de 2,56 à 2,73 (m, 2H) ; 3,87 (m, 1 H) ; 6,97 (m, 2H) ; 7,45 (dd, J=
2,0 et
8,0 Hz, 1 H) ; 7,51 (d, J= 2,0 Hz, 1 H) ; 9,75 (s, 1 H).
Etape 3: Préparation du [(3S)-2-oxo-7-phényl-2,3,4,5-tétrahydro-1H-1-
benzazépin-3-yl]carbamate de tert-butyle (13a)
~
H H
pN,,, . N OYN,., .
N / \
p O
~
~ ~
10a Br 13a
Dans un ballon de 500 mL, sous agitation et atmosphère d'argon, contenant
142 mL d'eau, 42 mL de dioxane et 7,0 g de 10a (19,71 mmol), on introduit
2,643 g d'acide phenylboronique (21,68 mmol), 402 mg de chlorure de 1,1'-
bis(diphenylphosphino)ferrocene palladium (C35H30C14FeP2Pd, PM 816,65,
0,493 mmôi) et 25,69 g de carbonate de césium (78,84 mmoi). On chauffe à
100 C le milieu sous agitation pendant 4 h. On concentre sous vide = le
dioxane, ajoute 100 mL d'eau distillée, puis extrait par 2x200 mL d'AcOEt. La
phase organique est séchée sur MgSO4, filtrée et évaporé à sec. On obtient
7 g d'un solide marron qui est cristallisé dans 55 mL de toluène à chaud.
Après avoir laissé reposer pendant 1 nuit à TA, on essore les aiguilles, et
lave
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
27
avec 5 mL de toluène, et 5 mL d'éther isopropylique. On obtient après
séchage sous vide 5,63 g de produit attendu 13a.
PF= 198,6 + /- 1 C
[a]D : -157,0 +/- 2,1 (c=2,332 mg/0.5 mL MeOH)
MS : m/z = 353 [M+H]+
RMN 1 H (400 MHz, DMSO-d6), b(ppm): 1,34 (s, 9H) ; 2,09 (m, 1 H) ; 2,25 (m,
1 H) ; de 2,65 à 2,80 (m, 2H) ; 3,94 (m, 1 H) ; 6,95 (d, J = 8,0 Hz, 1 H) ;
7,09
(d,J=8,5Hz, 1 H) ; 7,35 (t, J = 7,5 Hz, 1H)7,46(t,J=7,5Hz,2H)7,54
(dd, J = 2,0 et 8,5 Hz, 1 H) ; 7,59 (d, J = 2,0 Hz, 1 H) ; 7,67 (d, J = 7,5
Hz,
2H) ; 9,76 (s, 1 H).
Etape 3 Préparation du (3S)-3-amino-7-phényl-1,3,4,5-tétrahydro-2H-1-
benzazépin-2-one chlorhydrate (14a)
0 0
H H
NN HZNõ,,. N
yx\ p HCI
13a 14a
On reprend 5,63 g de 13a (15,97 mmol) dans un ballon de 250 mL et on
ajoute 140 mL d'une solution d'acide chlorhydrique dans le dioxane (4M). On
agite le milieu pendant 4h à TA sous argon. Il se forme un précipité que l'on
essore et lave avec 5 mL de dioxane puis 5 mL d'éther isopropylique. Après
séchage, on obtient 5,7 g d'amine 14a sous forme de chlorhydrate.
PF=198,6 + /- 1 C
[a]p :-292,0 +/- 3,2 (c=1,894 mg/0.5 mL MeOH)
RMN'H (400 MHz, DMSO-d6), b(ppm): 2,19 (m, 1 H) ; 2,57 (m, 1 H) ; de 2,72 à
2,91 (m, 2H) ; 3,78 (m, 1 H) ; 7,13 (d, J 8,5 Hz, 1 H) ; 7,38 (t, J = 7,5 Hz,
1 H) ; 7,48 (t, J = 7,5 Hz, 2H) ; 7,60 (dd, J 2,0 et 8,5 Hz, 1 H) ; 7,69 (m,
3H) ;
8,29 (s large, 3H) ; 10,35 (s, 1 H).
Etape 4: Préparation du (2R,3R,4S,5R,6E)-3,4,5-trihydroxy-2-méthoxy-8,8-
diméthyl-N-[(3S)-2-oxo-7-phényl-2,3,4,5-tétrahydro-1 H-1-benzazépin-3-
yl]non-6-énamide (Ex4a)
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
28
0
O
H,N,, O_H O O
I O "=.
o~+ HCI
oH OH o
OH ôH
14a Ex4a
On introduit successivement dans un ballon de 150 mL, sous agitation et
sous atmosphère d'argon, 2,3 g de 9(9,42 mmol), 2,72 g de 14a (9,42
mmol), 3,63 g de 2-éthylhexanoate de sodium (21,84 mmoi) dans 50,0 mL de
THF. On maintient l'agitation à TA pendant 48 h. On évapore directement le
milieu réactionnel à sec. Le brut est chromatographié sur une cartouche de
silice (300g, éluant AcOEt), on obtient 3,7 g d'un solide blanc qui est
repurifié
sur une cartouche de silice (240g, éluant CH2CI2/MeOH - en gradient MeOH :
1 à 5%). On recueille 3,23 g de produit attendu Ex4a (solide blanc).
[a]p -54,6 +/- 0,9 (c=2,989 mg/0.5 mL MeOH)
MS : m/z = 497 [M+H]+
RMN 'H (400 MHz, DMSO-d6), â(ppm): 0,95 (s, 9H) ; 2,11 (m, 1 H) ; 2,38 (m,
1 H) ; 2,79 (m, 2H) ; 3,24 (s, 3H) ; 3,30 (m masqué, 1 H) ; 3,52 (m, 1 H) ;
3,69 (d,
J = 8,0 Hz, 1 H) ; 3,93 (m, 1 H) ; de 4,20 à 4,32 (m, 3H) ; 4,53 (d, J = 5,5
Hz,
1H);5,30(dd,J=7,5et16,OHz, 1H);5,62(d,J16,OHz, 1 H) ; 7,10 (d, J =
8,5 Hz, 1 H) ; 7,35 (t, J= 7,5 Hz, 1 H) ; 7,45 (t, J 7,5 Hz, 2H) ; 7,57 (dd,
J=
2,0 et 8,5 Hz, 1 H) ; 7,62 (d, J= 2,0 Hz, 1 H) ; 7,68 (d, J = 7,5 Hz, 2H) ;
8,02 (d,
J = 8,0 Hz, 1 H) ; 9,92 (s, 1 H).
Ex5: (2R,3R,4S,5R,6E)-7-cyclopentyl-3,4,5-trihydroxy-2-méthoxy-N-[(3S)-2-
oxo-7-phényl-2,3,4,5-tétrahydro-1 H-1 -benzazép in-3-yl]hept-6-én amide
OH OO 0
I = q,
OH OH o
Ex5
Etape 1: Préparation du (4R,4aS,7R,7aR)-7-méthoxy-2,2-diméthyl-4-
vinyitétrahydro-6H-furo[3,2-dj[1,3]dioxin-6-one (17)
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
29
HO O O
0 p ~ p
HO
O0 O~O
16 17
Dans un ballon de 4000 mL, équipé d'une agitation mécanique, on charge
sous azote 178,2 g de PPh3 (0,679 moI), 84,1 g d'imidazole (1,235 mol), et
2430 mL de THF anhydre, On additionne avec précaution 156,8 g d'lode
bisublimé (0,618 mol) en maintenant la température du mélange réactionnel à
30 C. On porte ce milieu au reflux (66 C) durant 1 h, puis on ajoute
progressivement 81 g de 16 (0,309 mol) (qui peut être préparé selon les
modes opératoires décrits dans Org. Process Res. Dev. 2003, 7(6), 856-865)
à 66 C +/- 2 C. Le milieu homogène ainsi obtenu est chauffé au reflux
pendant 3 h. On laisse revenir à 20 C +/- 5 C, puis on coule 1000 mL d'une
solution de NaHCO3 à 10% (effervescence, athermique) (pH 8,0-8,5). Ensuite
on additionne 185,5 g de Na2S2O3 jusqu'à la décoloration quasi totale
(apparition d'un précipité minéral). Après agitation à 20 C +/- 5 C pendant 30
minutes, on filtre et rince avec du THF le solide. Le filtrat THF/H20 est
concentré partiellement à l'évaporateur rotatif à une température inférieure à
35 C. Le concentrat aqueux est saturé avec NaCI et extrait avec 1500ml de
CH2CI2. La phase organique est séchée sur MgSO4, filtrée et évaporée à sec.
Le résidu est repris dans 2000 mL d'un mélange H20/Acétone (75/25), les
insolubles sont filtrés, et rincés avec le mélange H20/Acétone (75/25). Les
filtrats sont concentrés à l'évaporateur rotatif à 50 C et 20 mbars, et
filtrés de
nouveau sur un verre fritté (porosité N 4). La phase aqueuse est saturée
avec NaCI, est extraite 3 fois avec CH2CI2 (1000 mL, 500 mL, et 250 mL). Les
phases organiques sont réunies, séchées sur MgSO4, filtrées et évaporées à
sec pour donner 60 g de produit brut, qui est dissout dans 250 mL de CH2CI2.
On ajoute ensuite dans la solution 30 g de silice. Après agitation pendant 15
min, on filtre la silice, rince 2 fois avec de CH2CI2 (250 mL et 100 mL). Le
filtrat est concentré à sec et séché sous 1 mbar à 20 C pour donner 54,8 g de
produit attendu 17 (solide blanc)
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
RMN 1H (300 MHz, DMSO-d6), S(ppm): 5.85 (m, 1 H) ; 5.35 (d, 1 H) ; 5.25 (d,
1 H) ; 4.80 (m, 1 H) ; 4.69 (m, 1 H) ; 4.43 (d, 1 H) ; 4.22 (m, 1 H) ; 3.40
(s, 3
H) ; 1.49 (s, 3 H) ; 1.30 (s, 3 H).
Etape 2: Préparation du (3R,4R,5S)-4-hydroxy-5-[(1 R)-1-hydroxyprop-2-én-1-
5 yl]-3-méthoxydihydrofuran-2(3H)-one (18)
o
0 0
o ~~,~'=
O O
~ OH OH
17 18
Dans un ballon de 100 mL contenant 1,0 g de 17 (4,38 mmol), 10 mL d'eau et
14 mL de THF, on ajoute à 0 C goutte à goutte 10 mL de TFA. On laisse le
milieu revenir à TA et agite pendant 1 nuit. On concentre ensuite le milieu à
10 pression réduite à TA et ajoute 50 mL d'eau, congèle et lyophilise. Le
lyophilisat est empâté dans de l'heptane en présence d'un minimum de
méthanol, après évaporation des solvants, on obtient 778 mg de 18 attendu
(solide blanc).
MS : m/z = 211 [M+Na]+, 189 [M+H]+
15 RMN 1H (400 MHz, DMSO-d6), S(ppm): 3,42 (s, 3H) ; 3,98 (dd, J = 2,5 et 9,0
Hz, 1 H) ; de 4,25 à 4,34 (m, 3H) ; 5,22 (dm, J = 10,5 Hz, 1 H) ; 5,29 (d, J =
5,0
Hz, 1 H) ; 5,44 (d partiellement masqué, J= 16,5 Hz, 1 H); 5,46 (m, 1 H) ;
5,97
(m, 1 H).
Etape 3: Préparation du (3R,4R,5S)-5-[(1 R,2E)-3-cyclopentyl-1 -hydroxyprop-
20 2-én-1-yl]-4-hydroxy-3-méthoxydihydrofuran-2(3"-one (19)
0 0
O O I
OH OH OH OH
18 19
Dans un vial de 5 mL, introduire 100 mg (0,53 mmol) de 18, 4 mL de CH2CI2,
726 L (5,3 mmol) de vinylcyclopentane puis 90,2 mg (106 mol) de
catalyseur de Grubbs de seconde génération. Chauffer la solution 10 min à
25 60 C aû micro-ondes. Le solvant est ensuite évaporé à sec sous pression
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
31
réduite puis le résidu est purifié sur colonne de silice Biotage 12-M (éluant
:
Hept/AcOEt 40/60). Le produit 19 (76,3 mg, Rf = 0.35 dans les conditions
d'élution utilisées) est obtenu sous forme d'un solide marron.
MS : m/z = 279 [M+Na]', 257 [M+H]+
RMN 'H (300 MHz, DMSO-d6), S(ppm): 5.82 (dd, 1 H, J=8 Hz et 16 Hz,), 5.50
(dd, 1 H, J=6 Hz et 16 Hz), 5.39 (d, 1 H, J=4.5 Hz), 5.15 (d, 1 H, J=6 Hz),
4.27
(m, 3H), 3.95 (dd, 1 H, J=3 Hz et 8 Hz), 3.41 (s, 3H), 2.43 (m, 1 H), 1.73 (m,
2H), 1.56 (m, 4H), 1.26 (m, 2H)
Etape 4: Préparation du (2R,3R,4S,5R,6E)-7-cyclopentyl-3,4,5-trihydroxy-2-
méthoxy-N-[(3S)-2-oxo-7-phényl-2,3,4,5-tétrahydro-1 H-1-benzazépin-3-
yl]hept-6-énamide (Ex5)
o
HzN,, H OH o O
I O I -
+ HCI N,,
OH OH O
OH OH
9 14a Ex5
On introduit successivement dans un tube Wheaton, sous agitation et sous
atmosphère d'argon, 100 mg de 19 (0,39 mmol), 112,6 mg de 14a (0,39
mmol), 162 mg de 2-éthylhexanoate de sodium (0,98 mmol) dans 2,0 mL de
THF. On maintient l'agitation à TA pendant 24 h. On évapore directement le
milieu réactionnel à sec. Le brut est chromatographié sur une cartouche de
silice (12g, éluant AcOEt), on obtient 92 mg de produit attendu Ex5 (solide
blanc).
ES: m/z = 531 MNa+; m/z = 509 MH+.
RMN 1H (400 MHz, DMSO-d6), 5(ppm): De 1,13 à 1,73 (m, 8H) ; 2,11 (m,
1 H) ; de 2,29 à 2,45 (m, 2H) ; de 2,70 à 2,85 (m, 2H) ; 3,23 (s, 3H) ; 3,30
(m masqué, 1 H) ; 3,50 (m, 1 H) ; 3,69 (d, J = 8,0 Hz, 1 H) ; 3,92 (m, 1 H) ;
de
4,19 à 4,38 (m, 3H) ; 4,52 (s large, 1 H) ; 5,36 (dd, J = 7,5 et 16,0 Hz, 1 H)
;
5,58 (dd, J = 8,0 et 16,0 Hz, 1 H) ; 7,10 (d, J = 8,5 Hz, 1 H) ; 7,36 (t, J =
7,5
Hz, 1 H) ; 7,46 (t, J = 7,5 Hz, 2H) ; 7,58 (d large, J = 8,5 Hz, 1 H) ; 7,62
(s
large, 1 H) ; 7,69 (d, J = 7,5 Hz, 2H) ; 8,02 (d, J = 8,0 Hz, 1 H) ; 9,92 (s,
1 H).
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
32
Ex6: (2R,3R,4S,5R,6E)-7-cyclopentyl-3,4,5-trihydroxy-2-méthoxy-N-[(35-1-
méthyl-2-oxo-2,3,4,5-tétrahydro-1 H-1 -benzazépi n-3-yl] hept-6-én amide
OH H O /
N,
OH OH 0
Ex5
Etape 1: Préparation du [(3S)-1-méthyl-2-oxo-2,3,4,5-tétrahydro-1 H-1 -
benzazépin-3-yl]carbamate de tert-butyle (7a)
0 0
H N H O
py N,~,,. / -~ C~N .., N
C O
2a 7a
Dans un ballon de 500 mL, sous agitation et atmosphère d'argon, contenant
260 mL de THF et 6,2 g de 2a (22,44 mmol), on introduit à TA 0,898 mg
d'hydrure de sodium en suspension à 60% dans l'huile (22,44 mmol). On
agite le milieu pendant 1 h, et puis ajoute 3,823 g (26,94 mmoi) d'iodure de
méthyle. On laisse le milieu sous agitation pendant une nuit, on ajoute 500
mL d'AcOEt, lave 2 fois la phase organique avec 500 mL d'eau. La phase
organique est séchée sur MgSO4, filtrée puis évaporée à sec. On obtient 6,85
g de produit brut. Après chromatographie sur une cartouche de silice (250 g,
éluant Heptane/AcOEt- en gradient AcOEt : 10 à 50%). On obtient 4,47 g de
produit 7a (solide blanc).
PF= 133,2 +/- 1 C
[(X]o : -204,8 +/- 2,2 (c=2,484 mg/0.5 mL MeOH)
Etape 2: Préparation du (3S)-3-amino-1-méthyl-1,3,4,5-tétrahydro-2H-1-
benzazépin-2-one chlorhydrate (8a)
0 0
H b Oy N,,.,, N 0 HCI
7a 8a
On reprend 4,36 g de 7a (15,01 mmol) dans un ballon de 250 mL et on ajoute
110 mL d'une solution d'acide chlorhydrique dans le dioxane (4M). On agite
pendant 4h à TA sous argon. Après évaporation du solvant, on délite le solide
avec 25 mL d'éther isopropylique, et essore sur frité. On obtient après
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
33
. séchage 3,1 g d'amine 8a sous forme de chlorhydrate qui est utilisé
directement pour l'étape suivante.
PF=221,7+/-1 C
[(X]D : -257,1 +/- 2,8 (c=2,696 mg/0.5 mL MeOH)
Etape 3: Préparation du (2R,3R,4S,5R,6E)-7-cyclopentyl-3,4,5-trihydroxy-2-
méthoxy-N-[(3S)-1-méthyl-2-oxo-2,3,4,5-tétrahydro-1 H-1-benzazépin-3-
yl]hept-6-énamide (Ex6)
HaN.., OH O
HCI
oH dH oH oH o
19 6a Ex6
On introduit successivement dans un tube Wheaton, sous agitation et sous
atmosphère d'argon, 100 mg de 19 (0,39 mmol), 88,42 mg de 8a (0,39
mmoi), 162 mg de 2-éthylhexanoate de sodium (0,98 mmol) dans 2,0 mL de
THF. On maintient l'agitation à TA pendant 24 h. On évapore directement le
milieu réactionnel à sec. Le brut est chromatographié sur une cartouche de
silice (12g, éluant AcOEt), on obtient 60 mg de produit attendu Ex6 (solide
blanc).
ES : m/z = 447 MH+.
RMN 1H (500 MHz, DMSO-d6), S(ppm): 1,21 (m, 2H) ; de 1,41 à 1,73 (m, 6H) ;
2,03 (m, .1 H) ; 2,20 (m, 1 H) ; 2,37 (m, 1 H) ; de 2,60 à 2,75 (m, 2H) ; 3,21
(s, 3H) ; de
3,25 à 3,35 (m masqué, 1 H) ; 3,30 (s, 3H) ; 3,48 (m, 1 H) ; 3,66 (d, J= 8,0
Hz, 1 H) ;
3,91 (m, 1 H) ; 4,20 (m, 1 H) ; 4,29 (m, 2H) ; 4,53 (d, J = 4,0 Hz, 1 H) ;
5,35 (dd, J = 7,5
et 16,0 Hz, 1 H) ; 5,57 (dd, J = 8,5 et 16,0 Hz, 1 H) ; 7,23 (m, 1 H) ; 7,31
(d, J = 7,5 Hz,
1 H) ; 7,39 (m, 2H) ; 8,04 (d, J = 8,0 Hz, 1 H).
Activité biologique des produits préparés:
Au jour du dépôt de la démande, il a été mesuré que le Caco2-TC7 du produit
de l'exemple 4a (Papp single point = 48.10"' cm.sec 1) est meilleur que celui
du produit de l'exemple 22a (Papp single point = 6.10"' cm.sec"') décrit dans
la demande de brevet W02006/056696.
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
34
L'activité antiproliférative des produits des exemples du tableau 1 a été
déterminée par mesure de l'inhibition de la prolifération cellulaire de
cellules
HCT116. Les cellules sont ensemencées dans un milieu de culture cellulaire
à une concentration de 10 000 cellules par puits, dans 0.17 mL de milieu, et
20 pL de produit à tester, à différentes concentrations, et 10 pL de Thymidine
[methyl-14C] (100 pCi/ml - activité spécifique 47.90 mCi/mmol; NEN
Technologies référence NEC568 batch 3550-001) sont ajoutés, puis les
cellules sont incubées à 37 C et 5% de C02.
Milieu utilisé pour la culture de cellules HCT116 : milieu DMEM 2 mM L-
glutamine, 200 UI/ml pénicilline, 200 pg/ml streptomycine and 10% (VN)
Sérum de veau foetal (Life Technologies).
Après 96 heures, l'incorporation de 14C-thymidine est comptée dans un
compteur à scintillation liquide 1450 Microbeta Wallac Trilux. Les résultats R
sont exprimés en cpm (coups par minute) et convertis en pourcentage
d'inhibition de croissance Gl% en faisant premièrement la soustraction de la
moyenne du nombre de cpm des puits sans cellules B et en divisant ensuite
par le nombre de cpm des puits des cellules non traitées C comprenant 20NL
de milieu de dilution du produit contentant 1% d'éthanol. (GI %=(R - B) x
100/C%).
Les valeurs d'IC50 sont calculées à l'aide de l'équation 205 du logiciel XLFit
(IDBS company, UK) par analyse de régréssion non linéaire utilisant
l'algorithme Marquardt (Donald W. MARQUARDT, J.Soc.industry.appl, vol 11,
No. 2, June, 1963).
Les produits du tableau 1 présentent une IC50 sur les cellules HCT116
généralement inférieure à 30pM et de préférence inférieure à 100nM. Par
exemple, le produit de l'exemple 1 a une IC50 de 14 nM et le produit de
l'exemple 3 a une IC50 de 58 nM. Le produit de l'exemple 2 a une IC50 de 15
nM, le produit de l'exemple 3a a une IC50 de 32 nM, le produit de l'exemple 4
a une IC50 de 75 nM et le produit de l'exemple 4a a une IC50 de 14 nM.
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
- Tableau 1 -
Exemple Structure
Ex1 F
I OH ô O
N N
OH OH O \
Ex2
OH O
N N
OH OH O
Ex3
I H 0
N N
OH OH
Br
Ex3a
I OH 0~ O
N, N
OH OH O
Br
Ex4
I OH ô 0
N H
OH OH O ~ ~
~ \
CA 02649336 2008-10-15
WO 2007/135293 PCT/FR2007/000866
36
Ex4a
OH O O
' F~ H
OH OH O
Ex5
I oH r~,., o r~
OH OH o
Ex6
OH ô O
N
OH OH 0