Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02709982 2015-04-20
PRODUCTION OF AVIATION FUEL FROM BIORENEWABLE FEEDSTOCKS
STATEMENT REGARDING FEDERALLY SPONSORED
RESEARCH OR DEVELOPMENT
BACKGROUND OF THE INVENTION
[0002] This invention relates to a process for producing hydrocarbons
useful as fuel, such
In as aviation fuel, from renewable feedstocks with the glycerides and free
fatty acids found in
materials such as plant oils, fish oils, animal fats, and greases. The process
involves
hydrogenation, decarboxylation, decarbonylation, and/or hydrodeoxygenation,
hydroisomerization, and selective cracking in two or more steps. The selective
cracking step
optimally provides one cracking event per molecule. A reforming step may be
optionally
15 employed to generate hydrogen used in the hydrogenation, deoxygenation,
hydroisomerization, and selective hydrocracking steps.
[00031 As the demand for fuel such as aviation fuel increases worldwide
there is
increasing interest in sources other than petroleum crude oil for producing
the fuel. One such
source is what has been termed renewable sources. These renewable sources
include, but are
20 not limited to, plant oils such as corn, rapeseed, canola, soybean and
algal oils, animal fats
such as tallow, fish oils and various waste streams such as yellow and brown
greases and
sewage sludge. The common feature of these sources is that they are composed
of glycerides
and Free Fatty Acids (FFA). Both of these classes of compounds contain
aliphatic carbon
chains generally having from 8 to 24 carbon atoms. The aliphatic carbon chains
in the
25 glycerides or FFAs can be fully saturated, or mono-, di- or poly-
unsaturated.
[0004] There are reports disclosing the production of hydrocarbons from
oils. For
example, US 4,300,009 discloses the use of crystalline aluminosilicate
zeolites to convert
plant oils such as corn oil to hydrocarbons such as gasoline and chemicals
such as para-
xylene. US 4,992,605 discloses the production of hydrocarbon products in the
diesel boiling
-1-
CA 02709982 2010-06-17
WO 2009/120242
PCT/US2008/085624
range by hydroprocessing vegetable oils such as canola or sunflower oil.
Finally, US
2004/0230085 Al discloses a process for treating a hydrocarbon component of
biological
origin by hydrodeoxygenation followed by isomerization.
[00051 Applicants have developed a process which comprises two or more
steps to
hydrogenate, deoxygenate, isomerize and selectively crack a renewable
feedstock, in order to
generate a fuel such as aviation fuel. Simply deoxygenating the renewable
feedstock typically
results in strait chain paraffins having chain-lengths similar to, or slightly
shorter than, the
fatty acid composition of the feedstock. With many feedstocks, this approach
results in a fuel
meeting the general specification for a diesel fuel, but not for an aviation
fuel. The selective
lo cracking step reduces the chain length of some paraffins to maximize the
selectivity to
aviation fuel range paraffins while minimizing light products. The selective
cracking may
occur before, after, or concurrent with the isomerization. An optional
reforming step may be
included to generate the hydrogen needed in the deoxygenation and the
isomerization steps. In
one embodiment, a recycle from the effluent of the deoxygenation reaction zone
back to the
deoxygenation zone is employed. The volume ratio of recycle hydrocarbon to
feedstock
ranges from 2:1 to 8:1 and provides a mechanism to increase the hydrogen
solubility and
more uniformly distribute the heat of reaction in the deoxygenation reaction
mixture. As a
result of the recycle, some embodiments may have a lower operating pressure.
SUMMARY OF THE INVENTION
[0006] The process is for producing a hydrocarbon fraction useful as fuel
or a fuel
blending component from a renewable feedstock and the process comprises
treating the
renewable feedstock in a reaction zone by hydrogenating and deoxygenating the
feedstock at
reaction conditions to provide a reaction product comprising mostly n-
paraffins, isomerizing
the n-paraffins to improve cold-flow properties, and selectively cracking the
paraffins to
provide paraffins useful as fuel or a fuel blending component. The selective
cracking may
occur before, after, or concurrent with the isomerization. The selective
cracking is a process
step that preferentially cracks Cl-C6 fragments off the end of the long chain
n-paraffins to
increase the selectivity to the desired carbon number range paraffins
significantly in excess of
a non-selective statistical cracking process. In one embodiment, a portion of
the n-paraffins
generated in the deoxygenation step is recycled to the reaction zone with a
volume ratio of
-2-
CA 02709982 2010-06-17
WO 2009/120242
PCT/US2008/085624
recycle to feedstock in the range of 2:1 to 8:1 in order to increase the
solubility of hydrogen in
deoxygenation reaction mixture. An optional reforming step may be included in
order to
produce hydrogen needed in the hydrogenation, deoxygenation,
hydroisomerization, and
selective hydrocracking steps.
BRIEF DESCRIPTION OF THE DRAWINGS
[0007] FIG. 1 is a general flow scheme diagram of the invention where
isomerization
occurs before selective cracking.
[0008] FIG. 2 is a general flow scheme diagram of the invention where
selective cracking
occurs before the isomerization.
[0009] FIG. 3 is a general flow scheme diagram of the invention where
isomerization
occurs concurrently with the selective cracking.
DETAILED DESCRIPTION OF THE INVENTION
[0010] As stated, the present invention relates to a process for
producing a hydrocarbon
stream useful as fuel or a fuel blending component from renewable feedstocks
originating
from plants or animals other than petroleum derived feedstocks. The term
renewable
feedstock is meant to include feedstocks other than those obtained directly
from petroleum
crude oil. Another term that has been used to describe this class of
feedstocks is biorenewable
fats and oils. The renewable feedstocks that can be used in the present
invention include any
of those which comprise glycerides and free fatty acids (FFA). Most of the
glycerides will be
triglycerides, but monoglycerides and diglycerides may be present and
processed as well.
Examples of these renewable feedstocks include, but are not limited to, canola
oil, corn oil,
soy oils, rapeseed oil, soybean oil, colza oil, tall oil, sunflower oil,
hempseed oil, olive oil,
linseed oil, coconut oil, castor oil, peanut oil, palm oil, mustard oil,
cottonseed oil, tallow,
yellow and brown greases, lard, train oil, fats in milk, fich nil, algal oil,
sewage sludge,
cuphea oil, camelina oil, jatropha oil, curcas oil, babassu oil, palm kernel
oil, and the like.
Additional examples of renewable feedstocks include non-edible vegetable oils
from the group
comprising Jatropha curcas (Ratanjoy, Wild Castor, Jangli Erandi), Madhuca
indica (Mohuwa),
Pongamia pinnata (Karanji Honge), and Azadiracta indicia (Neem). The
glycerides and 1+ As
of the typical vegetable oil or animal fat or oil contain aliphatic
hydrocarbon chains in their
-3-
CA 02709982 2010-06-17
WO 2009/120242
PCT/US2008/085624
structure which have 8 to 24 carbon atoms with a majority of the oils
containing high
concentrations of fatty acids with 16 and 18 carbon atoms. Mixtures or co-
feeds of renewable
feedstocks and petroleum derived hydrocarbons may also be used as the
feedstock. Other
non-oxygenated feedstock components which may be used, especially as a co-feed
component
in combination with the above listed feedstocks, include liquids derived from
gasification of
coal, biomass, or natural gas followed by a downstream liquefaction step such
as Fischer-
Tropsch technology; liquids derived from depolymerization, thermal or
chemical, of waste
plastics such as polypropylene, high density polyethylene, and low density
polyethylene; and
other synthetic oils generated as byproducts from petrochemical and chemical
processes.
Mixtures of the above feedstocks may also be used as co-feed components. One
advantage of
using a co-feed component is transformation of what may have been considered
to be a waste
product from a petroleum based process into a valuable co-feed component to
the current
process.
[0011] The fuel composition generated in the present invention is
suitable for, or as a
blending component for, uses such as an aviation fuel. Depending upon the
application, various
additives may be combined with the fuel composition generated in order to meet
required
specifications for different specific fuels. In particular, the fuel
composition generated herein
complies with, is a blending component for, or may be combined with one or
more additives to
meet at least one of: ASTM D 1655 Specification for Aviation Turbine Fuels
Defense Stan 91--
91 Turbine Fuel, Aviation Kerosene Type, Jet A-1 NATO code F-35, F-34, F-37
Aviation Fuel
Quality Requirements for Jointly Operated Systems (Joint Checklist) A
combination of ASTM
and Def Stan requirements GOST 10227 Jet Fuel Specifications (Russia) Canadian
CAN/CGSB-3.22 Aviation Turbine Fuel, Wide Cut Type Canadian CAN/CGSB-3.23
Aviation
Turbine Fuel, Kerosene Type MIL-DTL-83133, JP-8, MIL-DTL-5624, JP-4, JP-5 QAV-
1
(Brazil) Especifcacao de Querosene de Aviacao No. 3 Jet Fuel (Chinese)
according to GB6537
DCSEA 134A (France) Carbureacteur Pour Turbomachines D'Aviation, Type Kerosene
Aviation Turbine Fuels of other countries, meeting the general grade
requirements for Jet A, Jet
A-1, Jet B, and TS-1 fuels as described in the IATA Guidance Material for
Aviation Turbine
Fuel Specifications. The aviation fuel is generally termed "jet fuel" herein
and the term "jet
fuel" is meant to encompass aviation fuel meeting the specifications above as
well as to
encompass aviation fuel used as a blending component of an aviation fuel
meeting the
-4-
CA 02709982 2015-01-05
specifications above. Additives may be added to the jet fuel in order to meet
particular
specifications. One particular type of jet fuel is JP-8 which is a military
grade type of highly
refined kerosene based jet propellant specified by the United States
Government. The fuel is
defined by Military Specification MIL-DTL-83133. The jet fuel product is very
similar to
isoparaffinic kerosene or iPK, also known as a synthetic jet fuel.
[0012] Renewable feedstocks that can be used in the present invention
may contain a
variety of impurities. For example, tall oil is a by product of the wood
processing industry and
tall oil contains esters and rosin acids in addition to FFAs. Rosin acids are
cyclic carboxylic
acids. The bio-renewable feedstocks may also contain contaminants such as
alkali metals, e.g.
sodium and potassium, phosphorous as well as solids, water and detergents. An
optional first
step is to remove as much of these contaminants as possible. One possible
pretreatment step
involves contacting the renewable feedstock with an ion-exchange resin in a
pretreatment
zone at pretreatment conditions. The ion-exchange resin is an acidic ion
exchange resin such
as Amberlystm1-15 and can be used as a bed in a reactor through which the
feedstock is
flowed through, either upflow or downflow. Another technique involves
contacting the
renewable feedstock with a bleaching earth, such as bentonite clay, in a
pretreatment zone.
[0013] Another possible means for removing contaminants is a mild acid
wash. This is
carried out by contacting the feedstock with an aqueous solution mixed with an
acid such as
sulfuric, nitric, phosphoric, or hydrochloric acid in a reactor. The acid and
feedstock can be
contacted either in a batch or continuous process. Contacting is done with a
dilute acid
solution usually at ambient temperature and atmospheric pressure. If the
contacting is done in
a continuous manner, it is usually done in a counter current manner. Yet
another possible
means of removing metal contaminants from the feedstock is through the use of
guard beds
which are well known in the art. These can include alumina guard beds either
with or without
demetallation catalysts such as nickel or cobalt. Filtration and solvent
extraction techniques
are other choices which may be employed. Hydroprocessing such as that
described in US Patent No.
7,638,040, is another pretreatment technique which may be employed.
[0014] The renewable feedstock is flowed to a reaction zone comprising
one or more
catalyst beds in one or more reactors. The term feedstock is meant to include
feedstocks that
have not been treated to remove contaminants as well as those feedstocks
purified in a
-5-
CA 02709982 2010-06-17
WO 2009/120242
PCT/US2008/085624
pretreatment zone. In the reaction zone, the renewable feedstock is contacted
with a
hydrogenation or hydrotreating catalyst in the presence of hydrogen at
hydrogenation
conditions to hydrogenate the olefinic or unsaturated portions of the n-
paraffinic chains.
Hydrogenation or hydrotreating catalysts are any of those well known in the
art such as nickel
or nickel/molybdenum dispersed on a high surface area support. Other
hydrogenation
catalysts include one or more noble metal catalytic elements dispersed on a
high surface area
support. Non-limiting examples of noble metals include Pt and/or Pd dispersed
on gamma-
alumina. Hydrogenation conditions include a temperature of 200 C to 300 C or
to 450 C and
a pressure of 1379 kPa absolute (200 psia) to 10,342 kPa absolute (1500 psia),
or to 4826 kPa
absolute (700 psia). Other operating conditions for the hydrogenation zone are
well known in
the art.
[0015] The hydrogenation and hydrotreating catalysts enumerated above
are also capable
of catalyzing decarboxylation, decarbonylation, and/or hydrodeoxygenation of
the feedstock
to remove oxygen. Decarboxylation, decarbonylation, and hydrodeoxygenation are
herein
collectively referred to as deoxygenation reactions. Decarboxylation and
decarbonylation
conditions pressures including a relatively low pressure of 1724 kPa absolute
(250 psia) to
10,342 kPa absolute (1500 psia), with embodiments in the range of 3447 kPa
(500 psia) to
6895 kPa (1000 psia) or below 4826 kPaa (700 psia); a temperature of 200 C to
460 C with
embodiments in the range of 288 C to 345 C; and a liquid hourly space velocity
of 0.25 to 4
hfiwith embodiments in the range of 1 to 4 hr-I. Since hydrogenation is an
exothermic
reaction, as the feedstock flows through the catalyst bed the temperature
increases and
decarboxylation, decarbonylation, and hydrodeoxygenation will begin to occur.
Although the
hydrogenation reaction is exothermic, some feedstocks may be highly saturated
and not
generate enough heat internally. Therefore, some embodiments may require
external heat
input. Thus, it is envisioned and is within the scope of this invention that
all the reactions
occur simultaneously in one reactor or in one bed. Alternatively, the
conditions can be
controlled such that hydrogenation primarily occurs in one bed and
decarboxylation,
decarbonylation, and/or hydrodeoxygenation occurs in a second or additional
bed(s). If only
one bed is used, it may be operated so that hydrogenation occurs primarily at
the front of the
bed, while decarboxylation, decarbonylation and hydrodeoxygenation occurs
mainly in the
middle and bottom of the bed. Finally, desired hydrogenation can be carried
out in one
-6-
CA 02709982 2010-06-17
WO 2009/120242
PCT/US2008/085624
reactor, while decarboxylation, decarbonylation, and/or hydrodeoxygenation can
be carried
out in a separate reactor. However, the order of the reactions is not critical
to the success of
the process.
[0016] Hydrogen is a reactant in the reactions above, and to be
effective, a sufficient
quantity of hydrogen must be in solution to most effectively take part in the
catalytic reaction.
If hydrogen is not available at the reaction site of the catalyst, the coke
forms on the catalyst
and deactivates the catalyst. To solve this kind of problem, the pressure in a
reaction zone is
often raised to insure enough hydrogen is available to avoid coking reactions
on the catalyst.
However, higher pressure operations are more costly to build and to operate as
compared to
their lower pressure counterparts. An advantage of one embodiment of the
present invention
is that the operating pressure is in the range of 1379 kPa absolute (200 psia)
to 4826 kPa
absolute (700 psia) which is lower than traditionally used in a deoxygenation
zone. In another
embodiment, the operating pressure is in the range of 2413 kPa absolute (350
psia) to 4481
kPa absolute (650 psia), and in yet another embodiment operating pressure is
in the range of
2758 kPa absolute (400 psia) to 4137 kPa absolute (600 psia). Furthermore,
with the increase
hydrogen in solution, the rate of reaction is increased resulting in a greater
amount of
throughput of material through the reactor in a given period of time. The
lower operating
pressures of this embodiment provide an additional advantage in increasing the
decarboxylation reaction while reducing the hydrodeoxygenation reaction. The
result is a
reduction in the amount of hydrogen required to remove oxygen from the
feedstock
component and produce a finished product. Hydrogen can be a costly component
of the feed
and reduction of the hydrogen requirements is beneficial from an economic
standpoint.
[0017] In one embodiment of the invention the desired amount of
hydrogen is kept in
solution at lower pressures by employing a large recycle of hydrocarbon. Other
exothermic
processes have employed hydrocarbon recycle in order to control the
temperature in the
reaction zones. However, the range of recycle to feedstock ratios that may be
used herein is
set based on the need to control the level of hydrogen in the liquid phase and
therefore reduce
the deactivation rate. The amount of recycle is determined not on temperature
control
requirements, but instead, based upon hydrogen solubility requirements.
Hydrogen has a
greater solubility in the hydrocarbon product than it does in the feedstock.
By utilizing a large
hydrocarbon recycle the solubility of hydrogen in the liquid phase in the
reaction zone is
-7-
= CA 02709982 2015-01-05
greatly increased and higher pressures are not needed to increase the amount
of hydrogen in
solution and avoid catalyst deactivation at low pressures. In one embodiment
of the invention,
the volume ratio of hydrocarbon recycle to feedstock is from 2:1 to 8:1. In
another
embodiment the ratio is in the range of 3:1 to 6:1 and in yet another
embodiment the ratio is
in the range of 4:1 to 5:1. The ranges of suitable volume ratios of
hydrocarbon recycle to
feedstock are described in US Patent No. 8,003,836. Suitable ranges for
hydrogen solubility were
shown to begin at a recycle to feed ratio of 2:1. From recycle to feed ratios
of 2:1 through 6:1 the
simulation of US Patent No. 8,003,836 showed that the hydrogen solubility
remained high. Thus, the
specific ranges of vol/vol ratios of recycle to feed for this embodiment is
determined based on achieving
a suitable hydrogen solubility in the deoxygenation reaction zone.
[0018] In
another embodiment, instead of recycling hydrocarbon, one or more of the co-
feed components discussed above may be used to provide the solubility of
hydrogen and
temperature control. Depending upon the relative costs of the hydrocarbon and
the co-feed
component, one embodiment may be more economic than the other. It is important
to note
that the recycle or co-feed is optional and the process does not require
recycle or co-feed.
Complete deoxygenation and hydrogenation may be achieved without recycle or co-
feed
components. In still another embodiment, the process may be conducted with
continuous
catalyst regeneration in order to counteract the catalyst deactivation effects
of the lower
amounts of hydrogen in solution or the higher operating conditions.
[0019] The
reaction product from the deoxygenation reactions in the deoxygenation zone
will comprise a liquid portion and a gaseous portion. The liquid portion
comprises a
hydrocarbon fraction comprising n-paraffins and having a large concentration
of paraffins in
the 15 to 18 carbon number range. Different feedstocks will have different
distributions of
paraffins. A portion of this hydrocarbon fraction, after separation from the
gaseous portion,
may be used as the hydrocarbon recycle described above. Although this
hydrocarbon fraction
is useful as a diesel fuel or diesel fuel blending component, additional
fuels, such as aviation
fuels or aviation fuel blending components which typically have a
concentration of paraffins
in the range of 9 to 15 carbon atoms, may be produced with additional
processing. Also,
because the hydrocarbon fraction comprises essentially all n-paraffins, it
will have poor cold
flow properties. Aviation fuel and blending components must have better cold
flow properties
-8-
CA 02709982 2015-01-05
and so the reaction product is further reacted under isomerization conditions
to isomerize at
least a portion of the n-paraffins to branched paraffins.
[0020] Catalysts
and conditions for isomerization are well known in the art. See for
example US 2004/0230085 Al. The same catalyst may be employed for both the
isomerization and
the selective cracking, or two or more different catalysts may be employed.
Isomerization can be
carried out in a separate bed of the same reaction zone, i.e. same reactor,
described above or the
isomerization can be carried out in a separate reactor. Therefore, the product
of the deoxygenation
reaction zone is contacted with an isomerization catalyst in the presence of
hydrogen at isomerization
conditions to isomerize at least a portion of the normal paraffins to branched
paraffins. The
isomerization catalyst may be the same catalyst as the selective cracking
catalyst, or it may be a
different catalyst. Due to the presence of hydrogen, this reaction may also be
called
hydroisomerization. Only minimal branching is required, enough to overcome
cold-flow problems of
the normal paraffins.
[0021] Overall, the
isomerization of the paraffinic product can be accomplished in any
manner known in the art or by using any suitable catalyst known in the art.
Many of the
isomerization catalysts are also suitable selective cracking catalysts,
although some may require
different conditions than would be employed for isomerization alone. Catalysts
having small or
medium sized pores, which are therefore shape selective, are favorable for
catalyzing both the
isomerization reaction and the selective cracking. In general, suitable
isomerization catalysts
comprise a metal of Group VIII (IUPAC 8-10) of the Periodic Table and a
support material.
Suitable Group VIII metals include platinum and palladium, each of which may
be used alone
or in combination. The support material may be amorphous or crystalline.
Suitable support
materials include amorphous alumina, amorphous silica-alumina, ferrierite,
ALPO-31, SAPO-
11, SAPO-31, SAPO-37, SAPO-41, SM-3, MgAPS0-31, FU-9, NU-10, NU-23, ZSM-I2,
ZSM-22, ZSM-23, ZSM-35, ZSM-48, ZSM-50, ZSM-57, MeAP0-11, MeAP0-31, MeAPO-
41, MeAPS0-11, MeAPS0-31, MeAPS0-41, MeAPS0-46, ELAP0-11, ELAPO-31, ELAPO-
41, ELAPSO-11, ELAPSO-31, ELAPSO-41, laumontite, cancrinite, offretite,
hydrogen form of
stillbite, magnesium or calcium form of mordenite, and magnesium or calcium
form of
partheite, each of which may be used alone or in combination. ALPO-31 is
described in US
4,310,440. SAPO-11, SAPO-31, SAPO-37, and SAPO-41 are described in US
4,440,871. SM-3
-9-
CA 02709982 2015-01-05
is described in US 4,943,424; US 5,087,347; US 5,158,665; and US 5,208,005.
MgAPSO is a
MeAPSO, which is an acronym for a metal aluminumsilicophosphate molecular
sieve, where
the metal Me is magnesium (Mg). Suitable MeAPS0-31 catalysts include MgAPS0-
31.
MeAPSOs are described in US 4,793,984, and MgAPSOs are described in US
4,758,419.
MgAPS0-31 is a preferred MgAPSO, where 31 means a MgAPSO having structure type
31.
Many natural zeolites, such as ferrierite, that have an initially reduced pore
size can be
converted to forms suitable for olefin skeletal isomerization by removing
associated alkali metal
or alkaline earth metal by ammonium ion exchange and calcination to produce
the substantially
hydrogen form, as taught in US 4,795,623 and US 4,924,027. Further catalysts
and conditions
for skeletal isomerization are disclosed in US 5,510,306, US 5,082,956, and US
5,741,759.
[0022] The isomerization catalyst may also comprise a modifier selected
from the group
consisting of lanthanum, cerium, praseodymium, neodymium, samarium,
gadolinium, terbium,
and mixtures thereof, as described in US 5,716,897 and US 5,851,949. Other
suitable support
materials include ZSM-22, ZSM-23, and ZSM-35, which are described for use in
dewaxing in
US 5,246,566 and in the article entitled "New molecular sieve process for lube
dewaxing by
wax isomerization," written by S. J. Miller, in Microporous Materials 2 (1994)
439-449.
Additional suitable catalyst modifiers or supports are disclosed in US
4,310,440; US 4,440,871; US
4,793,984; US 4,758,419; US 4,943,424; US 5,087,347; US 5,158,665; US
5,208,005; US 5,246,566;
US 5,716,897; and US 5,851,949.
[0023] US 5,444,032 and US 5,608,134 teach a suitable bifunctional catalyst
which is
constituted by an amorphous silica-alumina gel and one or more metals
belonging to Group
VIIIA, and is effective in the hydroisomerization of long-chain normal
paraffins containing
more than 15 carbon atoms. US 5,981,419 and 5,968,344 teach a suitable
bifunctional catalyst
which comprises: (a) a porous crystalline material isostructural with beta-
zeolite selected from
boro-silicate (BOR-B) and boro-alumino-silicate (Al-BOR-B) in which the molar
Si02:A1203
ratio is higher than 300:1; (b) one or more metal(s) belonging to Group VIIIA,
selected from
platinum and palladium, in an amount comprised within the range of from 0.05
to 5% by
weight. Article V. Calemma et al., App. Catal. A: Gen., 190 (2000), 207
teaches yet another
suitable catalyst.
[00241 isomerization zone conditions include a temperature of about 150 C
to about 360 C
and a pressure of about 1724 kPa absolute (250 psia) to about 4826 kPa
absolute (700 psia). In
-10-
CA 02709982 2010-06-17
WO 2009/120242
PCT/US2008/085624
another embodiment the isomerization conditions include a temperature of 300 C
to 360 C and
a pressure of 3102 kPa absolute (450 psia) to 3792 kPa absolute (550 psia).
[0025] The product of the hydrogenation, deoxygenation, and
isomerization steps
contains paraffinic hydrocarbons suitable for use as diesel fuel or as a
blending component for
diesel fuel, but further processing results in paraffinic hydrocarbons meeting
the
specifications for other fuels or as blending components for other fuels. As
illustrative of this
concept, a concentration of paraffins formed from renewable feedstocks
typically have 15 to
18 carbon atoms, but additional paraffins may be formed to provide a range of
from 8 to 24
carbon atoms. A portion of the normal paraffins are isomerized to branched
paraffins, but the
to carbon number range of paraffins does not alter with only isomerization.
The 9 to 24 carbon
number range is a desired paraffin carbon number range for diesel fuel, which
is a valuable
fuel itself. Aviation fuel, however, generally comprises paraffins having
boiling points from
150 C to 300 C which is lower than that of diesel fuel. To convert the diesel
range fuel to a
fuel useful for aviation, the larger chain-length paraffins are cracked.
Typical cracking
processes are likely to crack the paraffins too much and generate a large
quantity of undesired
low molecular weight molecules which have much lower economic value. In the
present
invention, the paraffins generated from the renewable feedstock are
selectively cracked in
order to control the degree of cracking and maximize the amount of product
formed in the
desired carbon number range. The selective cracking is controlled through
catalyst choice and
reaction conditions in an attempt to restrict the degree of cracking
occurring. Ideally, each
paraffin molecule would experience only a single cracking event and ideally
that single
cracking event would result in at least one paraffin in the C9 to C15 carbon
number range.
[0026] However, fuel specifications are typically not based upon carbon
number ranges.
Instead, the specifications for different types of fuels are often expressed
through acceptable
ranges of chemical and physical requirements of the fuel. For example,
aviation turbine fuels,
a kerosene type fuel including JP-8, are specified by MIL-DTL-83133, JP-4, a
blend of
gasoline, kerosene and light distillates, is specified by MIL-DTL-5624 and JP-
5 a kerosene
type fuel with low volatility and high flash point is also specified by MIL-
DTL-5624, with the
written specification of each being periodically revised. Often a distillation
range from 10
percent recovered to a final boiling point is used as a key parameter defining
different types of
fuels. The distillations ranges are typically measured by ASTM Test Method D
86 or D2887.
-11-
CA 02709982 2010-06-17
WO 2009/120242
PCT/US2008/085624
Therefore, blending of different components in order to meet the specification
is quite
common. While the product of the present invention may meet fuel
specifications, it is
expected that some blending of the product with other blending components may
be required
to meet the desired set of fuel specifications. In other words, the product of
this invention is a
composition which may be used with other components to form a fuel meeting at
least one of
the specifications for aviation fuel such as JP-8. The desired product is a
highly paraffinic
distillate fuel component having a paraffin content of at least 75% by volume.
[0027] The selective cracking step and the isomerization step may be
either co-current or
sequential. The cracking may be conducted first to minimize the over-cracking
of the highly
branched hydrocarbons resulting from the isomerization. The selective cracking
may proceed
through several different routes. The catalysts for the selective cracking
process typically
comprise at least a cracking component and a non cracking component.
Compositing the
catalyst with active and non active cracking components may positively affect
the particle
strength, cost, porosity, and performance. The non cracking components are
usually referred to
as the support. However, some traditional support materials such as silica-
alumina may make
some contribution to the cracking capability of the catalyst. One example of a
suitable catalyst is
a composite of zeolite beta and alumina or silica alumina. Other inorganic
refractory materials
which may be used as a support in addition to silica-alumina and alumina
include for example
silica, zirconia, titania, boria, and zirconia-alumina. These support
materials may be used alone
or in any combination. Another example is a catalyst based on zeolite Y, or
one having
primarily amorphous cracking components.
[0028] The catalyst of the subject process can be foimulated using
industry standard
techniques. It is may be manufactured in the form of a cylindrical extrudate
having a diameter of
from 0.8 to 3.2 mm (1/32 in to about 1/8 in). The catalyst can be made in any
other desired form
such as a sphere or pellet. The extrudate may be in forms other than a
cylinder such as the form
of a well-known trilobe or other shape which has advantages in terms or
reduced diffusional
distance or pressure drop.
[0029] A
non-selective catalyst may be utilized under conditions optimized to result in
selective cracking, where primary cracking is accomplished with minimal
secondary cracking.
Furthermore, a non-selective catalyst may be modified to weaken the acidity of
the catalyst in
order to minimize undesired cracking.
-12-
CA 02709982 2010-06-17
WO 2009/120242
PCT/US2008/085624
[0030]
One class of suitable selective cracking catalysts are the shape-selective
catalysts.
Highly isomerized paraffins are more readily cracked as compared to straight
chain or mono-
substituted paraffins since they can crack through stabilized carbenium-ion
intermediates.
Unfortunately, this leads to the tendency for these molecules to over crack
and form lighter
molecules outside the preferred aviation fuel range. Highly isomerized
paraffins are also more
likely to crack than the other paraffins and can be prevented from entering
the pore structures of
some molecular sieves. A shape-selective catalyst would prevent the majority
of highly
isomerized molecules from entering the pore structure and cracking leaving
only straight-chain
or slightly isomerized paraffins to crack in the catalyst pores. Furthermore,
by selective small to
medium size pore molecular sieves, the smaller pore size will prevent easy
diffusion of the long
chain paraffin deep into the pore system. The end of a long chain paraffin
enters the pore
channel of the catalyst and encounters a dehydrogenation active site, such as
platinum, resulting
in an olefin. Protonation of the olefins yields a carbenium ion which
rearranges by methyl shift
to form a carbenium ion with a single methyl branch, then via n-elimination,
the hydrocarbon
cracks at the site of the methyl branch yielding two olefins, one short chain
and one long chain.
In this way, beta scission cracking, the primary mechanism for bronsted acids,
will therefore
occur close to the pore mouth of the catalyst. Since diffusion is limited,
cracking will be
primarily at the ends of the paraffins. Examples of suitable catalysts for
this route include ZSM-
5, ZSM-23, ZSM-11, ZSM-22 and ferrierite. Further suitable catalysts are
described in Arroyo,
J. A. M.; Martens, G. G.; Froment, G. F.; Mann, G. B.; Jacobs, P. A.; martens,
J. A., Applied
Catalysis, A: General, 2000, 192(1) 9-22; Souverijins, W.; martins, J. A.;
Froment, G. F.;
Jacobs, P. A., Journal of Catalysis, 1998, 174(2) 177-184; Huang, W.; Li, D.;
Kang, X; Shi, Y.;
Nie, H. Studies in Surface Science and Catalysis, 2004, 154(c) 2353-2358;
Claude, M. C.;
Martens J. A. Journal of Catalysis, 2000, 190(1), 39-48; Sastre, G.; Chica,
A.; Corma, A.,
Journal of Catalysis, 2000, 195(2), 227-236.
[0031] In one embodiment, the selective cracking catalyst also contains
a metallic
hydrogenolysis component. The hydrogenolysis component is provided as one or
more base
metals uniformly distributed in the catalyst particle. Noble metals such as
platinum and
palladium could be applied, or the composition of the metal hydrogenolysis
component may be,
for example, nickel, iridium, rhenium, rhodium, or mixtures thereof. The
hydrogenolysis
function preferentially cleaves Cl to C6 fragments from the end of the
paraffin molecule. Two
-13-
CA 02709982 2010-06-17
WO 2009/120242
PCT/US2008/085624
classes of catalysts are suitable for this approach. A first class is a
catalyst having a
hydrogenolysis metal with a mechanistic preference to crack the ends of the
paraffin molecules.
See, for example, Carter, J. L.; Cusumano, J. A.; Sinfelt, J. H. Journal of
Catalysis, 20, 223-229
(1971) and Huang, Y. J.; Fung, S. C.; Gates, W. E.; McVicker, G. B. journal of
Catalysis 118,
192-202 (1989). The second class of catalysts include those where the
hydrogenolysis function
is located in the pore moth of a small to medium pore molecular sieve that
prevent facile
diffusion of the ling chain paraffin molecule into the pores system. Also,
since olefins are easy
to protonate, and therefore crack, as compared to paraffins, the
dehydrogenation function
component may be minimized on the external surface of the catalyst to maintain
the
selectivity of the cracking. Examples of suitable catalysts for this
hydrogenolysis route of
selective cracking include silicalite, ferrierite, ZSM-22, ZSM-23 and small to
medium pore
molecular sieves.
[0032] Another suitable type of catalysts include molecular sieves with
strong pore
acidity, which when used a higher operating temperatures promote Haag Dessau
cracking; a
type of acid-catalyst cracking that does not require isomerization or a
bifunctional catalyst as
described in Weitkamp et al. Agnew. Chem. hit. ed. 2001, 40, No. 7, 1244. The
intermediate
is a carbonium ion formed after prontonation of a carbon-carbon or carbon-
hydrogen bond.
The catalyst does not need a significant dehydrogenation function since the
olefin is not
necessary. Residence time on these strong acid sites would need to be
minimized to prevent
extensive cracking by techniques such as reducing the acid site density or
operating at a
higher space velocity. An example of a suitable catalyst for this approach is
ZSM-5.
[0033] The selective cracking is operated at a range of conditions that
provide product in
the targeted carbon number range. Therefore, the operating conditions in many
instances are
refinery or processing unit specific. They may be dictated in large part by
the construction and
limitations of the existing selective cracking unit, which normally cannot be
changed without
significant expense, the composition of the feed and the desired products. The
inlet temperature
of the catalyst bed should be in the range of from 232 C to 454 C (450 F to
850 F), and the
inlet pressure should be above 1379 kPa gauge to 13,790 kPa gauge (200 to
2,000 psig). The
feed stream is admixed with sufficient hydrogen to provide hydrogen
circulation rate of 168 to
1684 n.1/1 (1000 to 10000 SCF/barrel, hereafter SCFB) and passed into one or
more reactors
containing fixed beds of the catalyst. The hydrogen will be primarily derived
from a recycle gas
-14-
CA 02709982 2010-06-17
WO 2009/120242
PCT/US2008/085624
stream which may pass through purification facilities for the removal of acid
gases. The
hydrogen rich gas admixed with the feed and in one embodiment any recycle
hydrocarbons will
contain at least 90 mol percent hydrogen. The feed rate in terms of liquid
hourly space velocity
(L.H.S.V.) will normally be within the broad range of 0.3 to about 5 hr-I,
with a L.H.S.V. below
1.2 being used in one embodiment.
[0034] The two reactions types, isomerization and selective cracking
may be carried out
together using the same catalyst, or separately using the same or different
catalysts. hi the
situation where the isomerization and selective cracking catalysts are the
same, the acidity of
the catalyst is selected to be great enough to perform both the isomerization
and the selective
cracking. In this embodiment, both isomerization and selective cracking occur
concurrently.
Examples of catalysts suitable for both reaction types include, but are not
limited to, zeolite
Y, amorphous silica alumina, MOR, SAPO-11 and SM3. An example of combined
isomerization and selective cracking conditions include a temperature of 150 C
to 360 C or
150 C to 375 C and a pressure of 1724 kPa absolute (250 psia) to 4826 kPa
absolute (700
psia). In another embodiment the combined isomerization and selective cracking
conditions
include a temperature of 300 C to 360 C and a pressure of 3102 kPa absolute
(450 psia) to
3792 kPa absolute (550 psia).
[0035] On the other hand, when the isomerization and selective
cracking are conducted in
separate reaction zones, the catalysts for the two reaction types need not be
the same. Any of
the above catalysts may be employed. The selective cracking may be done before
or after the
isomerization step. Specific examples of isomerization catalysts include those
having moderate
acidity, enough for isomerization but weak enough to prevent significant
cracking, include
platinum modified MAPSO-31, platinum modified MAPSO-SM3, platinum modified
SAPO-
11, and platinum modified and acid washed UZM-15. The prevention of
significant cracking is
important since the desired product range is C9 to C15 and significant
uncontrolled cracking
may result in a large amount of C8 and lower carbon atoms paraffins being
produced. The
selective cracking catalyst may have a higher acidity than the isomerization
catalyst, and
specific examples include ZSM-5, Y zeolite, and MOR.
[0036] Optionally the process may employ a steam reforming zone in order
to provide
hydrogen to the hydrogenation/deoxygenation zone, isomerization zone, and/or
selective
cracking zone. The steam reforming process is a well known chemical process
for producing
-15-
CA 02709982 2010-06-17
WO 2009/120242
PCT/US2008/085624
hydrogen, and is the most common method of producing hydrogen or hydrogen and
carbon
oxide mixtures. A hydrocarbon and steam mixture is catalytically reacted at
high temperature to
form hydrogen, and the carbon oxides: carbon monoxide and carbon dioxide.
Since the
refoi __ ming reaction is strongly endothermic, heat must be supplied to the
reactant mixture, such
as by heating the tubes in a furnace or reformer. A specific type of steam
reforniing is
autothermal reforming, also called catalytic partial oxidation. This process
differs from catalytic
steam reforming in that the heat is supplied by the partial internal
combustion of the feedstock
with oxygen or air, and not supplied from an external source. In general, the
amount of
reforming achieved depends on the temperature of the gas leaving the catalyst;
exit temperatures
in the range of 700 C to 950 C are typical for conventional hydrocarbon
reforming. Pressures
may range up to 4000 kPa absolute. Steam reforming catalysts are well known
and conventional
catalysts are suitable for use in the present invention.
[00371 Typically, natural gas is the most predominate feedstock to a
steam reforming
process. However, in the present invention, hydrocarbons that are too light
for the desired
product may be generated at any of the reaction zones. For example, in the
deoxygenation zone,
propane is a common by product. Other Cl to C3 paraffins may be present as
well. These lighter
components may be separated from the desired portion of the deoxygenation
effluent and routed
to the steam reforming zone for the generation of hydrogen. Similarly,
paraffins having eight or
less carbon atoms from the effluent of the collective isomerization and
selective cracking steps
may be conducted to the reforming zone. Therefore, the lighter materials from
the
deoxygenation, isomerization and cracking zones are directed, along with
stream, to a reforming
zone. In the reforming zone, the lighter hydrocarbons and steam are
catalytically reacted to form
hydrogen and carbon oxides. The steam reforming product may be recycled to any
of the
reaction zones to provide at least hydrogen to the reaction zone. Optionally,
the hydrogen may
be separated from the carbon oxides generated in the steam reforming reaction,
and the
separated hydrogen may be recycled to any of the reaction zones. Since
hydrogen is an
expensive resource, generating at least a portion of the required hydrogen
from the undesired
products of the reaction zones can decrease the cost of the process. This
feature becomes more
valuable when an external source of hydrogen is not readily available.
[0038] In an alternative embodiment, catalytic reforming may be employed
instead of steam
reforming. In a typical catalytic reforming zone, the reactions include
dehydrogenation,
-16-
CA 02709982 2010-06-17
WO 2009/120242
PCT/US2008/085624
isomerization and hydrocracking. The dehydrogenation reactions typically will
be the
dehydroisomerization of alkylcyclopentanes to aromatics, the dehydrogenation
of paraffins to
olefins, the dehydrogenation of cyclohexanes to aromatics and the
dehydrocyclization of acyclic
paraffins and acyclic olefins to aromatics. The isomerization reactions
included isomerization of
n-paraffins to isoparaffins, the hydroisomerization of olefins to
isoparaffins, and the
isomerization of substituted aromatics. The hydrocracking reactions include
the hydrocracking
of paraffins. The aromatization of the n-paraffins to aromatics is generally
considered to be
highly desirable because of the high octane rating of the resulting aromatic
product. In this
application, the hydrogen generated by the reactions is also a highly desired
product, for it is
recycled to at least the deoxygenation zone. The hydrogen generated is
recycled to any of the
reaction zones, the hydrogenation/deoxygenation zone, the isomerization zone,
and or the
selective cracking zone.
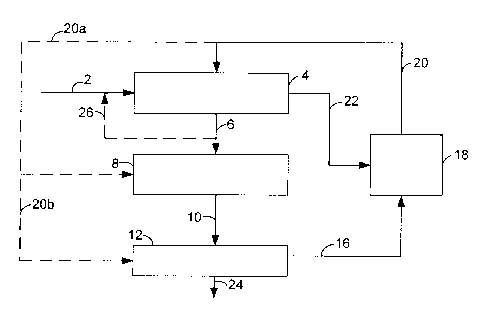
[0039] Three general flow schemes are shown in the figures. FIG. 1
shows the sequence
of reaction zones as a deoxygenation zone followed by an isomerization zone
followed by a
selective cracking zone. In FIG. 2, the order of the isomerization zone and
selective cracking
zone is reversed as compared to FIG. 1. In Fig. 3, the isomerization zone and
the selective
cracking zone are combined into a single combined zone.
[0040] In FIG. 1, renewable feedstock 2 enters deoxygenation reaction
zone 4 along with
recycle hydrogen stream 20 and optional product recycle 26. Contacting the
renewable
feedstock with the deoxygenation catalyst generates deoxygenated product 6
which is directed
to isomerization zone 8. Carbon oxides, possibly hydrogen sulfide, and water
vapor may be
removed from the reaction mixture (not shown). C3 and lighter components may
be separated
and removed in line 22 and conducted to reforming zone 18. Optionally, line 22
may contain
the C3 and light components as well as the carbon oxides, possibly hydrogen
sulfide, and
water vapor, thus eliminating a separation. The deoxygenated liquid product is
passed to the
isomerization reaction zone 8 for conversion of normal paraffins to branched
paraffins.
Branched paraffin effluent 10 of isomerization zone 8 is passed to selective
cracking zone 12
to crack the higher carbon number paraffins and form paraffins in the desired
aviation fuel
range. After selective cracking the desired aviation fuel range of paraffin-
rich product is
collected via line 24 and the C8 and lighter components are separated and
recycled via line 16
to reforming zone 18. Hydrogen generated in reforming zone 18 is recycled via
line 20 to the
-17-
CA 02709982 2015-01-05
deoxygenation zone 4. Optionally, hydrogen generated in reforming zone 18 is
recycled via
line 20a to the isomerization zone 8, and or via line 20b to the selective
cracking zone 12.
Other components may be removed from reforming zone 18 (not shown).
[00411 In FIG. 2, renewable feedstock 2 enters deoxygenation reaction
zone 4 along with
recycle hydrogen stream 20 and optional product recycle 26. Contacting the
renewable
feedstock with the deoxygenation catalyst generates deoxygenated product 6
which is directed
to isomerization zone 8. Carbon oxides, possibly hydrogen sulfide, and water
vapor may be
removed from the reaction mixture (not shown). C3 and lighter components may
be separated
and removed in line 22 and conducted to reforming zone 18. Optionally, line 22
may contain
the C3 and light components as well as the carbon oxides, possibly hydrogen
sulfide, and
water vapor, thus eliminating a separation. The deoxygenated liquid product is
passed to
selective cracking zone 12 to crack the higher carbon number paraffins and
form paraffins in
the desired aviation fuel range. Effluent 14 of the selective cracking zone 12
is passed to the
isomerization reaction zone 8 for conversion of normal paraffins to branched
paraffins. After
isomerization in isomerization zone 8 the desired aviation fuel range of
paraffin-rich product
is collected via line 24 and the C8 and lighter components are separated and
recycled via line
16 to reforming zone 18. Optionally, the liquid portion of the recycle in line
16 may be
separated and sold as a product, added to a gasoline pool, or upgraded by
other refinery
processes (not shown). Hydrogen generated in reforming zone 18 is recycled via
line 20 to the
deoxygenation zone 4. Optionally, hydrogen generated in reforming zone 18 is
recycled via
line 20a to the isomerization zone 8, and or via line 20b to the selective
cracking zone 12.
Other components may be removed from reforming zone 18 (not shown).
[0042] In FIG. 3, renewable feedstock 2 enters deoxygenation reaction
zone 4 along with
recycle hydrogen stream 20 and optional product recycle 26. Contacting the
renewable
feedstock with the deoxygenation catalyst generates deoxygenated product 6
which is directed
to isomerization zone 8. Carbon oxides, possibly hydrogen sulfide, and water
vapor may be
removed from the reaction mixture (not shown). C3 and lighter components may
be separated
and removed in line 22 and conducted to reforming zone 18. Optionally, line 22
may contain
the C3 and light components as well as the carbon oxides, possibly hydrogen
sulfide, and
water vapor, thus eliminating a separation. The deoxygenated liquid product is
passed to the
combined isomerization and selective cracking zone 15 for both conversion of
normal
-18-
CA 02709982 2015-01-05
paraffins to branched paraffins and selective cracking of the higher carbon
number paraffins
to form paraffins in the desired aviation fuel range. After isomerization and
selective cracking
the desired aviation fuel range of paraffin-rich product is collected via line
24 and the C8 and
lighter components are separated and recycled via line 16 to reforming zone
18. Hydrogen 20c
generated in reforming zone 18 is recycled via line 20 to the deoxygenation
zone 4. Other
components may be removed from reforming zone 18 (not shown).
[0043] The final effluent stream, i.e. the stream obtained after all
reactions have been
carried out, may be processed through one or more separation steps to obtain a
purified
hydrocarbon stream useful as an aviation fuel. Because the final effluent
stream comprises
to -- both a liquid and a gaseous component, the liquid and gaseous components
are separated
using a separator. The separated liquid component comprises the product
hydrocarbon stream
useful as an aviation fuel. Further separations may be performed to remove
naphtha and LPG
from the product hydrocarbon stream. The separated gaseous component comprises
mostly
hydrogen and the carbon dioxide from the decarboxylation reaction. The carbon
dioxide can
-- be removed from the hydrogen by means well known in the art, reaction with
a hot carbonate
solution, pressure swing absorption, etc. Also, absorption with an amine in
processes such as
described in US Patents No. 7,982,077 and 7,982,078, may by employed. If
desired, essentially pure
carbon dioxide can be recovered by regenerating the spent absorption media.
The hydrogen remaining
after the removal of the carbon dioxide may be recycled to the reaction zone
where hydrogenation
-- primarily occurs and/or to any subsequent beds/reactors.
[0044] Finally, a portion of the product hydrocarbon is recycled to the
hydrogenating and
deoxygenating reaction zone. The recycle stream may be taken from the product
hydrocarbon
stream after the hydrogenating and deoxygenating reactor(s) and separation
from gaseous
components, and recycled back to the hydrogenating and deoxygenating
reactor(s). A portion
of a hydrocarbon stream may also be cooled down if necessary and used as cool
quench liquid
between the beds of the deoxygenation reaction zone to further control the
heat of reaction
and provide quench liquid for emergencies. The recycle stream may be
introduced to the inlet
of the deoxygenation reaction zone and/or to any subsequent beds or reactors.
One benefit of
the hydrocarbon recycle is to control the temperature rise across the
individual beds.
However, as discussed above, the amount of hydrocarbon recycle herein is
determined based
-19-
CA 02709982 2015-01-05
upon the desired hydrogen solubility in the reaction zone. Increasing the
hydrogen solubility
in the reaction mixture allows for successful operation at lower pressures,
and thus reduced
cost. Operating with high recycle and maintaining high levels of hydrogen in
the liquid phase
helps dissipate hot spots at the catalyst surface and reduces the formation of
undesirable
heavy components which lead to coking and catalyst deactivation.
[0045] The following example is presented in illustration of this
invention and is not
intended as an undue limitation on the generally broad scope of the invention.
EXAMPLE
[0046] Deoxygenation of refined-bleached-deodorized (RBD) soybean oil over
the
deoxygenation catalyst CAT-DO was accomplished by mixing the RBD soybean oil
with a
2500 ppm S co-feed and flowing the mixture down over the catalyst in a tubular
furnace at
330 C, 3447 kPa gauge (500 psig), LHSV of lh-land an H2/feed ratio of 4000
scf/bbl. The
soybean oil was completely deoxygenated and the double bonds hydrogenated to
produce an n-
iS paraffin mixture having predominantly from 15 to 18 carbon atoms;
deoxygenation products
CO, CO2, H20, and propane; with removal of the sulfur as H2S.
[0047] The n-paraffin product from the deoxygenation stage was fed over
a cracking
catalyst CAT-C1 in a second process step. The n-paraffin mixture having
predominantly from
about 15 to about 18 carbon atoms was delivered down flow over the cracking
catalyst in a
tubular furnace at 280 C, 3447 kPa gauge (500 psig), 0.8 LHSV and an H2/feed
ratio of 2500
scf/bbl. This step produced 50% jet fuel-range paraffins but the product was
not highly
isomerized to meet the required freeze point properties. Therefore, the
product of this stage was
fed over isomerization catalyst CAT-Iso in a similar tubular furnace at 330 C,
3447 kPa gauge
(500 psig), 1 LHSV, and an H2/ feed ratio of 2500 scf/bbl. The product from
this isomerization
step was fractionated and the jet fuel range material (as defined in the
specification for JP-8,
MIL-DTL-83133) was collected. The final yield of jet fuel (normal and
isoparaffins) was 36
wt-% of vegetable oil feed. The properties of final jet fuel produced are
shown in the Table.
CA 02709982 2015-01-05
TABLE
Freeze Flash
aromatic Point, Point, Density,
Sample: added C C g/cc
JP-8 Specifications -47 38 0.775
Soybean oil paraffin 0% -52.6 53 0.759
[0048] In a second iteration of the experiment, the RBD soybean oil feed
was again
deoxygenated over CAT-DO using the same conditions as above. The deoxygenated
paraffin
product was then processed over CAT-C2 at 345C, 3447 kPa gauge (500 psig), 1
LHSV, and an
H2/feed ratio of 2500 scf/bbl. However, this catalyst contained a selective
cracking function
that also produced a much higher iso/normal ratio paraffin product. Therefore,
a separate
isomerization processing step (the third step of the first example) was not
required. After
fractionation the jet fuel yield was 40 wt-% of the vegetable oil feed. The
properties of this
in product also met the freeze and flash point requirements for JP-8 as
defined by MIL-DTL-
83133.
[0049] The scope of the claims should not be limited by the preferred
embodiments set forth in the
examples, but should be given the broadest interpretation consistent with the
description as a whole.
-21-