Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
1
METHODS FOR CONTROLLING WATER AND PARTICULATE PRODUCTION
IN SUBTERRANEAN WELLS
CROSS REFERENCE TO RELATED APPLICATIONS
[0001] This application is a continuation-in-part of U.S. Application Serial
No,
11/545,136 filed on October 10, 2006 which is a continuation-in-part of U.S.
Application
Serial No, 11/183,208 filed on July 15, 2005.
BACKGROUND
[0002] The present invention relates to the stabilization of subterranean
formations.
More particularly, the present invention relates to methods for stabilizing
unconsolidated
portions of a subterranean formation and controlling the production of water
from those
portions.
[0003] Hydrocarbon wells are often located in subterranean formations that
contain
unconsolidated particulates that may migrate out of the subterranean formation
with the oil,
gas, water, and/or other fluids produced by the wells. The presence of
particulates, such as
formation sand and even loose proppant, in produced fluids is undesirable in
that the
particulates may abrade pumping and other producing equipment and reduce the
fluid
production capabilities of the producing zones. Unconsolidated portions of a
subterranean
formation include those that contain loose particulates and those wherein the
bonded
particulates have insufficient bond strength to withstand the forces created
by the production
of fluids through the formation.
[0004] One method of controlling particulates in such unconsolidated portions
has
been to produce fluids from the formations at low flow rates, so that the near
well stability of
sand bridges and the like may be substantially preserved. The collapse of such
sand bridges,
however, may occur due to unintentionally high production rates and/or
pressure cycling as
may occur from repeated shut-ins and start ups of a well. The frequency of
pressure cycling
is critical to the longevity of the near well formation, especially during the
depletion stage of
the well when the pore pressure of the formation has already been
significantly reduced.
[0005] Another method of controlling particulates in unconsolidated formations
involves placing a filtration bed containing gravel near the well bore to
present a physical
barrier to the transport of unconsolidated formation fines with the production
of
hydrocarbons. Typically, such "gravel-packing operations" involve the pumping
and
placement of a quantity of a desired particulate into the unconsolidated
formation in an area
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
2
adjacent to a well bore. One common type of gravel-packing operation involves
placing a
gravel-pack screen in the well bore and packing the surrounding annulus
between the screen
and the well bore with gravel of a specific size designed to prevent the
passage of formation
sand. The gravel-pack screen is generally a filter assembly used to retain the
gravel placed
during the gravel-pack operation. A wide range of sizes and screen
configurations are
available to suit the characteristics of the gravel-pack sand used. Similarly,
a wide range of
sizes of gravel is available to suit the characteristics of the unconsolidated
particulates in the
subterranean formation. The resulting structure presents a barrier to
migrating sand from the
formation while still permitting fluid flow. When installing the gravel pack,
the gravel is
carried to the formation in the form of a slurry by mixing the gravel with a
viscous treatment
fluid. Once the gravel is placed in the well bore, the viscosity of the
treatment fluid is
reduced, and it is returned to the surface.
[0006] Gravel packs act, inter alia, to stabilize the formation while causing
minimal
impairment to well productivity. The gravel, inter alia, acts to prevent
formation particulates
from occluding the screen or migrating with the produced fluids, and the
screen, inter alia,
acts to prevent the gravel from entering the production tubing. Such packs may
be time
consuming and expensive to install. Due to the time and expense needed, it is
sometimes
desirable to place a screen without the gravel. Even in circumstances in which
it is practical
to place a screen without gravel, it is often difficult to determine an
appropriate screen size to
use as formation sands tend to have a wide distribution of grain sizes. When
small quantities
of sand are allowed to flow through a screen, formation erosion becomes a
significant
concern. As a result, the placement of gravel as well as the screen is often
necessary to
assure that the formation sands are controlled. Expandable sand screens have
been developed
and implemented in recent years. As part of the installation, an expandable
sand screen may
be expanded against the well bore, cased hole, or open hole for sand control
purposes without
the need for gravel packing. However, screen erosion and screen plugging are
the main
disadvantages of expandable screens.
[0007] Another method used to control particulates in unconsolidated
formations
involves consolidating unconsolidated subterranean producing zones into
stable, permeable
masses by applying a resin followed by a spacer fluid, a catalyst, and an
after-flush fluid.
Such resin application may be problematic when, for example, an insufficient
amount of
spacer fluid is used between the application of the resin and the application
of the external
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
3
catalyst. The resin may come into contact with the external catalyst in the
well bore itself
rather than in the unconsolidated subterranean producing zone. When resin is
contacted with
an external catalyst an exothermic reaction occurs that may result in rapid
polymerization,
potentially damaging the formation by plugging pore channels, halting pumping
when the
well bore is plugged with solid material, or resulting in a downhole explosion
as a result of
the heat of polymerization. Also, using these conventional processes to treat
long intervals of
unconsolidated regions is not practical due to the difficulty in determining
if the entire
interval has been successfully treated with both the resin and the external
catalyst. Further,
conventional consolidation techniques have often resulted in limited or
inadequate
penetration distances of consolidating agent into formations.
[0008] Often, unconsolidated formation sands migrate out of the formation when
water is produced from the formation. This migration of formation sands is
due, in part, to
the fact that most natural cementation between formation sand grains
disintegrates when in
contact with an aqueous moving phase. The production of water from a
subterranean
producing zone is disadvantageous due to its effect on mobilizing formation
sands, and
because water production constitutes a major expense in the recovery of
hydrocarbons from
subterranean formations, especially in light of the energy expended in
producing, separating,
and disposing of the water.
SUMMARY
[0009] The present invention relates to the stabilization of subterranean
formations.
More particularly, the present invention relates to methods for stabilizing
unconsolidated
portions of a subterranean formation and controlling the production of water
from those
portions.
[0010] Some embodiments of the present invention provide methods of treating a
portion of a subterranean formation that comprises unconsolidated formation
particulates, the
method comprising: introducing a fluid comprising a relative permeability
modifier into at
least a portion of the subterranean formation; and then, introducing a fluid
comprising a
consolidating agent into at least a portion of the subterranean formation so
as to at least
partially consolidate the unconsolidated formation particulates.
[0011 ] Other embodiments of the present invention provide methods of treating
a
portion of a subterranean formation that comprises unconsolidated formation
particulates, the
method comprising introducing a treatment fluid comprising a relative
permeability modifier
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
4
and a consolidating agent into at least a portion of the subterranean
formation so as to at least
partially consolidate the unconsolidated formation particulates.
[0012] Still embodiments of the present invention provide methods of
stabilizing
unconsolidated subterranean formation particulates and reducing the
permeability of water
comprising: providing a portion of a subterranean formation that comprises
unconsolidated
formation particulates; introducing a fluid comprising a relative permeability
modifier into at
least a portion of the subterranean formation so as to at least partially
reduce the permeability
of that portion of the subterranean formation to water; and, introducing a
fluid comprising a
consolidating agent into at least a portion of the subterranean formation so
as to at least
partially consolidate the unconsolidated formation particulates.
[0013] The features and advantages of the present invention will be apparent
to those
skilled in the art. While numerous changes may be made by those skilled in the
art, such
changes are within the spirit of the invention.
BRIEF DESCRIPTION OF THE DRAWINGS
[0014] These drawings illustrate certain aspects of some of the embodiments of
the
present invention and should not be used to limit or define the invention.
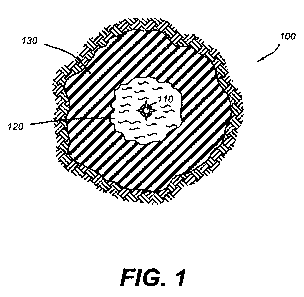
[0015] Figure 1 shows a cross-sectional view of a subterranean formation
penetrated
by a well bore after treatment with a consolidating agent and a relative
permeability modifier
fluid, in which the consolidating agent has been introduced at a rate and
pressure below the
fracture pressure of the subterranean formation.
[0016] Figure 2A shows a cross-sectional view of a subterranean formation
penetrated
by a well bore after treatment with a consolidating agent, followed by
treatment with a
relative permeability modifier fluid which has been introduced at a rate and
pressure
sufficient to create or enhance at least one fracture in the subterranean
formation.
[0017] Figure 2B shows a cross-sectional view of the subterranean formation of
Figure 2A wherein a fracturing fluid comprising proppant particulates has been
used to
extend further the fracture into the formation.
DESCRIPTION OF PREFERRED EMBODIMENTS
[0018] The present invention relates to the stabilization of subterranean
formations.
More particularly, the present invention relates to methods for stabilizing
unconsolidated
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
portions of a subterranean formation and controlling the production of water
from those
portions.
1. Methods of the Present Invention
[0019] One embodiment of the present invention describes a method of
stabilizing an
unconsolidated subterranean formation that is penetrated by a well bore
comprising
introducing a fluid comprising a consolidating agent into at least a portion
of the subterranean
formation so as to transform a portion of the subterranean formation
surrounding the well
bore into a consolidated region; and introducing a relative permeability
modifier fluid into the
subterranean formation through the well bore so as to penetrate at least a
portion of the
consolidated region. The relative permeability modifier fluid, in some
embodiments, may
penetrate beyond the consolidated region.
[0020] Another embodiment of the present invention described a method that
comprises introducing a relative permeability modifier fluid into at least a
portion of a
subterranean formation to form a treated portion of the subterranean
formation; and
introducing a fluid comprising a consolidating agent into the treated portion
of the
subterranean formation so as to transform at least a section of the treated
portion of the
subterranean formation into a consolidated region. In some embodiments, the
subterranean
formation may have been previously stimulated via hydraulically fracturing or
any other
known stimulation method.
[0021 ] Another embodiment of the present invention described a method that
comprises introducing a fluid that comprises a relative permeability modifier
fluid and a
consolidating agent into at least a portion of a subterranean formation so as
to transform at
least a section of the subterranean formation into a treated, consolidated
region. In some
embodiments, the subterranean formation may have been previously stimulated
via
hydraulically fracturing or any other known stimulation method.
[0022] As an example of one embodiment of the methods of the present
invention,
Figure 1 shows a cross-sectional view of subterranean formation 100 penetrated
by well bore
110. First portion 120 of subterranean formation 100 has been treated with a
consolidating
agent to consolidate first portion 120 and form a consolidated region. Prior
to the
consolidation of first portion 120, an after-flush fluid may optionally be
introduced into
subterranean formation 100 to restore the permeability of first portion 120
after introduction
of the consolidating agent. Further, after introduction of an after-flush
fluid, well bore 110
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
6
may optionally be shut-in for a period of time to allow for consolidation of
first portion 120.
Second portion 130 of the subterranean formation 100 may be treated by a
relative
permeability modifier fluid introduced into subterranean formation through
well bore 110 so
as to penetrate first portion 120.
[0023] As another example of one embodiment of the methods of the present
invention, Figure 2A shows a cross-sectional view of subterranean formation
200 penetrated
by well bore 210. First portion 220 of subterranean formation 200 has been
treated with a
consolidating agent to consolidate first portion 220 and form a consolidated
region. A
relative permeability modifier fluid has been introduced at a rate and
pressure sufficient to
create or enhance fracture 240 in subterranean formation 200. The relative
permeability
modifier fluid may flow into and treat second portion 230. Referring now to
Figure 2B, a
fracturing fluid has been introduced at a rate and pressure sufficient to
extend fracture 240 in
subterranean formation 200. Fracture 240 may be packed with proppant to keep
fracture 240
open. In this way, the relative permeability modifier fluid may treat regions
that are beyond
first portion 220 that have been consolidated using the consolidating agent
and extend into
second portion 230.
[0024] The term, "unconsolidated subterranean formation," as used herein,
refers to
both unconsolidated and weakly consolidated formations. The term,
"consolidating agent,"
as used herein, refers to any agent that may consolidate a portion of the
subterranean
formation, which may, at least in part, stabilize particulates such that loose
or weakly
consolidated particulates are prevented from shifting or migrating once the
consolidation
treatment is complete. The term, "relative permeability modifier fluid," as
used herein refers
to any fluid, which may, among other things, treat a portion of the
subterranean formation so
as to reduce the permeability of the treated portion to water without
substantially reducing the
formation permeability as to hydrocarbons.
[0025] Optionally, other embodiments may include the use of a preflush fluid
and/or
an after-flush fluid. Additional embodiments may include introducing a
fracturing fluid to
create or enhance fractures in the subterranean formation. The term, "create
or enhance," as
used herein also includes the action of extending previously created, or
natural, fractures. In
still other embodiments portions of the subterranean formation may have been
previously
hydraulically fractured to stimulate production through that portion. Further,
the well bore
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
7
may be shut in for a period of time after introduction of the consolidating
agent to allow for
consolidation of the portion of the subterranean formation.
[0026] The term, "preflush fluid," as used herein refers to any fluid that may
be
suitable for preparing the subterranean formation for the later placement of
the consolidating
agent by, among other things, removing oil and/or debris from the pore spaces
within the
formation matrix of the unconsolidated portion. The term, "after-flush fluid,"
as used herein
refers to any fluid that may, among other things, restore the permeability of
the treated
portion of the subterranean formation by displacing at least a portion of the
consolidating
agent from the pore channels of the subterranean formation and forcing the
displaced portion
of the consolidating agent further into the subterranean formation where it
may have
negligible impact on subsequent hydrocarbon production.
[0027] In embodiments of the present invention wherein an optional preflush
fluid is
desired, it may be placed into the subterranean formation either before the
placement of the
fluid comprising a consolidating agent or before the placement of the relative
permeability
modifier fluid. In some preferred embodiments, the preflush fluid is placed
directly before
the fluid comprising a consolidating agent. The preflush fluid acts, inter
alia, to prepare the
subterranean formation for the later placement of the consolidating agent
and/or relative
permeability modifier. Typically, injection of a preflush fluid may occur
prior to
consolidating a portion of a subterranean formation. Injecting a volume of a
preflush fluid
into an unconsolidated portion of a subterranean formation may, among other
things, help to
remove oil and/or debris from the pore spaces within the formation matrix of
the subterranean
formation portion. Generally, the volume of the preflush fluid placed into the
formation is
between 0.1 times to 50 times the volume of the fluid comprising the
consolidating agent
and/or relative permeability modifier fluid. Preflush fluids suitable for use
with the present
invention are described in more detail below.
[0028] Introducing a volume of consolidating agent into the unconsolidated
portion
may among other things, transform a portion of the subterranean formation into
a
consolidated region. Consolidating the region surrounding the well bore may be
advantageous in preventing well bore sloughing, formation sand production, and
the
migration of fines.
[0029] In certain embodiments, the consolidation of a portion of a
subterranean
formation may result in diminishing the permeability of that portion. In
certain
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
8
embodiments, fracturing a portion of the formation may be required to
reconnect the well
bore with portions of the formation (e.g., the reservoir formation) outside
the consolidated
region, as discussed in more detail below. In other embodiments, typically
when no
fracturing step is used, an after-flush fluid may be used to restore
permeability to the portion
of the subterranean formation.
[0030] In certain embodiments of the present invention, after the placement of
the
consolidating agent and/or relative permeability modifier into the
subterranean formation, an
optional after-flush fluid may be placed into the subterranean formation,
inter alia, to restore
the permeability of the treated portion. of the subterranean formation. When
used, the after-
flush fluid is preferably placed into the subterranean formation while the
consolidating agent
is still in a flowing state. For example, an after-flush fluid may be placed
into the formation
prior to a shut-in period. Among other things, the after-flush fluid acts to
displace at least a
portion of the consolidating agent from the pore channels of the subterranean
formation and
to force the displaced portion of the consolidating agent further into the
subterranean
formation where it may have negligible impact on subsequent hydrocarbon
production.
Generally, the after-flush fluid may be any fluid that does not adversely
react with the other
components used in accordance with this invention or with the subterranean
formation. For
example, the after-flush may be an aqueous-based brine, a hydrocarbon fluid
(such as
kerosene, diesel,. or crude oil), or a gas (such as nitrogen or carbon
dioxide). In some
preferred embodiments, the after-flush fluid is a brine. The after-flush fluid
may be placed
into the formation at a matrix flow rate such that a sufficient portion of the
consolidating
agent may be displaced from the pore channels to restore the formation to a
desired
permeability. Generally, a substantial amount of the consolidating agent,
however, should
not be displaced therein. For example, sufficient amounts of the consolidating
agent should
remain in the treated portion to provide effective stabilization of the
unconsolidated portions
of the subterranean formation therein.
[0031 ] Generally, the volume of after-flush fluid placed in the subterranean
formation
ranges from about 0.1 times to about 50 times the volume of the fluid
comprising the
consolidating agent. In some embodiments of the present invention, the volume
of after-flush
fluid placed in the subterranean formation ranges from about 0.1 times to
about 5 times the
volume of the consolidating agent.
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
9
[0032] In another embodiment of the present invention, no after-flush fluid is
placed
into the subterranean formation after placement of a consolidating agent into
the subterranean
formation. Where no after-flush fluid is used, the permeability of the
subterranean formation
may be significantly reduced, because the consolidating agent may remain in
the pore spaces
therein and may convert into a consolidated substance. While a significant
reduction in the
permeability may occur, the unconsolidated portions of the formation may be
stabilized due,
inter alia, to the consolidating agent remaining in the pore spaces of the
formation. In
embodiments in which no after-flush fluid is used, a portion of the formation
may be
fractured so as to reconnect the well bore with portions of the formation
outside the
consolidated region. In embodiments wherein no after-flush fluid is placed
after the
placement of the fluid comprising a consolidating agent, it may be desirable
to perform a
stimulation operation, such as hydrajetting or mini-frac operation, to create
one or more
conduits through the consolidated portion of the subterranean formation.
However,
stimulations operations are not required. Retained permeability of a treated
formation is
function of, among other things, the volume and concentration of the
consolidating agent, and
the volume of any after-flush fluid. For example, in embodiments wherein a
relatively low
amount of consolidating agent is used in the fluid comprising the
consolidating agent, the
treated portion of the subterranean formation may show relatively low
consolidation strength
is obtained and high retained permeability, even without applying after-flush
fluid. In other
embodiments, wherein a relatively high amount of consolidating agent is used
in the fluid
comprising the consolidating agent, the retained permeability may be low, in
which case a
stimulation operation, or use of a higher volume of after-flush fluid may be
useful to restore
formation permeability and for the production of hydrocarbons.
[0033] According to the methods of the present invention, after placement of
the
consolidating agent, the subterranean formation may be shut in for a period of
time to allow
the consolidating agent to consolidate at least a portion of the subterranean
formation. The
shutting-in of the well bore for a period of time may, inter alia, stabilize
unconsolidated
portions of the subterranean formation, for example, by enhancing the coating
and curing of
the resin between formation particulates. Additionally, if a relative
permeability modifier is
placed in the subterranean formation after the placement of the consolidating
agent rather
than before, the shutting in of the well bore may also minimize the washing
away of the
consolidating agent during later placement of a relative permeability
modifier.
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
[0034] Typically, the shut-in period of the well bore occurs after placement
of the
consolidating agent. In embodiments using an after-flush fluid, the shut-in
period preferably
occurs after the use of the after-flush fluid. In embodiments in which a
fracturing step is
performed subsequent to introducing the consolidating agent into the
subterranean formation,
preferably, no shut-in period is used.
[0035] The necessary shut-in time period is dependent, among other things, on
the
composition of the consolidating agent used and the temperature of the
formation. Generally,
the chosen period of time will be between about 0.5 hours and about 72 hours
or longer.
Determining the proper period of time to shut in the formation is within the
ability of one
skilled in the art with the benefit of this disclosure.
[0036] Generally, the relative permeability modifier fluid should reduce the
permeability of the treated portion to water without substantially reducing
the hydrocarbon
permeability. In some embodiments wherein the relative permeability modifier
fluid is
introduced into the formation after the consolidating agent, the relative
permeability modifier
fluid may displace excess portions of the consolidating agent into the
formation and at least
partially restore the permeability to hydrocarbons in that treated portion.
Relative
permeability modifier fluids may be introduced into the subterranean formation
through the
well bore. For example, in some embodiments, the relative permeability
modifier fluids may
penetrate through the consolidated region and into portion of the subterranean
formation
(e.g., unconsolidated portions) that are adjacent to the consolidated region.
[0037] In certain embodiments, a relative permeability modifier fluid is
introduced
into a portion of a subterranean formation after a consolidating agent has
been placed into at
least a portion of that portion of a subterranean formation. In such
embodiments, the relative
permeability modifier fluid may be introduced into the subterranean formation
either before
or after an after-flush fluid has been placed into the portion of the
subterranean formation. In
certain embodiments, the relative permeability modifier fluids may be
introduced into the
subterranean formation at a rate and pressure sufficient to create or enhance
at least one
fracture in a portion of the subterranean formation. In such embodiments, it
may be desirable
that the fracture or fractures extend from a consolidated region of the
subterranean formation
into an unconsolidated region of the subterranean formation. In such
embodiments, the
relative permeability modifier fluid may leak off into the unconsolidated
portion of the
formation along the fracture; this affecting regions of the formation beyond
the consolidated
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
11
region of the formation. In certain embodiments, following the placement of,
at least, a
consolidating agent and a relative permeability modifier fluid an after-flush
fluid may be used
to displace at least a portion of the relative permeability fluid further into
the formation.
[0038] In those embodiments in which a fracture is initiated through the use
of a
relative permeability modifier fluid, the fracture may be extended and packed
using any
suitable fracturing methodology known to one skilled in the art with the
benefit of this
disclosure. For example, a fracture may be extended using a crosslinked gelled
fracturing
fluid to further extend the fracture into the formation followed by a
crosslinked gelled fluid
containing proppant, or a viscoelastic surfactant fluid containing proppant.
The proppant
may be coated with a curable resin or consolidating agent to form a hard,
permeable solid
mass in the fracture or fractures, among other things, to prevent proppant
flow back during
production from the well. The proppant also may be blended with fibrous
particulates that
may act to form a stable network with the proppant and/or to control proppant
flow back.
[0039] In certain embodiments a relative permeability modifier fluid may be
placed
into a portion of a subterranean formation before a consolidating agent is
placed into the
subterranean formation. In addition, in some embodiments, the relative
permeability
modifier fluid is placed into the subterranean formation at a rate and
pressure sufficient to
create or an enhance at least one fracture in that portion of the subterranean
formation. In
such embodiments, the relative permeability modifier fluid may be used as a
component in a
fracturing fluid. It may be desirable to place the relative permeability
modifier fluid before
placing a fluid comprising a consolidating agent in situations wherein, if
placed first, the
consolidating agent might coat the subterranean formation surfaces, preventing
the relative
permeability modifier from optimally performing its function. Generally, the
placement of a
relative permeability modifier will not act to prevent a consolidating agent
from performing
its function. Some embodiments may also involve the placement of a relative
permeability
modifier fluid before a stimulation treatment, such as fracturing, frac-
packing, or
hydrajetting, followed by the placement of a fluid comprising a consolidating
agent. Placing
a relative permeability modifier before performing a stimulation operation may
act to
increase the fluid efficiency of the fracturing fluid by improving the fluid
loss characteristics
and in reducing the amount of water inflow into the propped fracture.
Moreover, if the
relative permeability modifier is placed before a fluid comprising a
consolidating agent, the
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
12
placement of the consolidating agent may act to displace the relative
permeability modifier
further into the subterranean formation. and further away from the well bore.
[0040] In certain embodiments a relative permeability modifier fluid may be
placed
into a portion of a subterranean formation substantially simultaneously along
with a
consolidating agent. In some such embodiments, a treatment fluid comprising a
relative
permeability modifier and a consolidating agent may be placed into a portion
of a
subterranean formation as a pre-pad to a hydraulic fracturing or frac-pack
completion. Such
a pre-pad frac-pack placement may, among other things, allow the pre-pad fluid
to enter the
regions of a subterranean formation surrounding fracture faces to, among other
things,
mitigate the production of water from the portion of the subterranean
formation and to control
particulate migration. As used herein, the term "frac-pack completion" refers
to subterranean
operations wherein fracturing and gravel packing are preformed in a single
operation. In
other embodiments, a treatment fluid comprising a relative permeability
modifier and a
consolidating agent may be placed into a portion of a subterranean formation
as a pre-pad to a
gravel pack completion. In embodiments wherein a fluid comprising both a
relative
permeability modifier and a consolidating agent is used, are those in which a
water-soluble
consolidating agent is chosen that does not contain significant quantities of
anionic
substances. In still other embodiments where it is desirable to use an oil-
soluble
consolidating agent, the fluid comprising both a relative permeability
modifier and a
consolidating agent may be formed into a stable emulsion. Such a pre-pad
gravel pack
placement may, among other things, allow the pre-pad fluid to enter the
regions of a
subterranean formation surrounding a gravel pack to, among other things,
mitigate the
production of water from the portion of the subterranean formation and to
control particulate
migration. In other embodiments, a treatment fluid comprising a relative
permeability
modifier and a consolidating agent may be placed into a portion of a
subterranean formation
as a remedial treatment. Such remedial treatment placement may be used in
portions of well
bores that are experiencing undesirable production of water and/or
particulates to, among
other things, mitigate the production of water from the portion of the
subterranean formation
and to control particulate migration. In other embodiments, a treatment fluid
comprising a
relative permeability modifier and a consolidating agent may be placed into a
portion of a
subterranean formation as an after treatment following an acidizing treatment
to that portion
of a subterranean formation. Such remedial post-acidizing placement may be
used mitigate
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
13
the production of water from the portion of the subterranean formation and to
control
particulate migration.
[0041] The methods described herein may be performed repeatedly as desired. In
those instances in which steps are repeated, it may be desirable, for example,
to recommence
the steps described herein starting first with the lowest zones of the
formation and moving up
to higher zones.
II. Fluids Useful in the Methods of the Present Invention
A. Exemplary Preflush Fluids
[0042] Preflush fluids suitable for use with the present invention may
comprise a
brine, a mutual solvent, a surfactant, or any mixture thereof.
[0043] The preflush fluid of the present invention may include any fluid that
does not
adversely interact with the other components used in accordance with this
invention or with
the subterranean formation. For example, the preflush fluid may be an aqueous-
based fluid
or a hydrocarbon-based fluid. In certain embodiments of the present invention,
the preflush
fluid may comprise an aqueous fluid and a surfactant. The aqueous-fluid
component may be
fresh water, salt water, brine, or seawater, or any other aqueous-based fluid
that does not
adversely react with the other components used in accordance with this
invention or with the
subterranean formation. Any surfactant compatible with the later-used
consolidating agent
and relative permeability modifier and capable of aiding the consolidating
agent in flowing to
the contact points between adjacent particulates in the formation may be used
in the present
invention. Such surfactants include, but are not limited to, ethoxylated nonyl
phenol
phosphate esters, mixtures of one or more cationic surfactants, one or more
non-ionic
surfactants, and an alkyl phosphonate surfactant. Suitable mixtures of one or
more cationic
and nonionic surfactants are described in U.S. Patent No. 6,311,773, the
relevant disclosure
of which is incorporated herein by reference. A C12 - C22 alkyl phosphonate
surfactant is
preferred. The surfactant or surfactants used are included in the preflush
fluid in an amount
sufficient to prepare the subterranean formation to receive a treatment of a
consolidating
agent. In some embodiments of the present invention, the surfactant is present
in the preflush
fluid in an amount in the range of from about 0.1% to about 3% by weight of
the aqueous
fluid.
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
14
B. Exemplary Consolidating Agents
[0044] Suitable consolidating agents include any suitable composition for
consolidating a portion of the subterranean formation to stabilize
unconsolidated particulates
therein. Examples of suitable consolidating agents include resins, tackifying
agents, and
gelable liquid compositions.
1. Exemplary Resins
[0045] Resins suitable for use in the consolidation fluids of the present
invention
include any suitable resin that is capable of forming a hardened, consolidated
mass. Many
such resins are commonly used in subterranean consolidation operations, and
some suitable
resins include two component epoxy based resins, novolak resins, polyepoxide
resins,
phenol-aldehyde resins, urea-aldehyde resins, urethane resins, phenolic
resins, furan resins,
furan/furfuryl alcohol resins, phenolic/latex resins, phenol formaldehyde
resins, polyester
resins and hybrids and copolymers thereof, polyurethane resins and hybrids and
copolymers
thereof, acrylate resins, and mixtures thereof. Some suitable resins, such as
epoxy resins,
may be cured with an internal catalyst or activator so that when pumped
downhole, they may
be cured using only time and temperature. Other suitable resins, such as furan
resins
generally require a time-delayed catalyst or an external catalyst to help
activate the
polymerization of the resins if the cure temperature is low (i.e., less than
250 F) but will cure
under the effect of time and temperature if the formation temperature is above
about 250 F,
preferably above about 300 F. It is within the ability of one skilled in the
art, with the benefit
of this disclosure, to select a suitable resin for use in embodiments of the
present invention
and to determine whether a catalyst is required to trigger curing.
[0046] Selection of a suitable resin may be affected by the temperature of the
subterranean formation to which the fluid will be introduced. By way of
example, for
subterranean formations having a bottom hole static temperature ("BHST")
ranging from
about 60 F to about 250 F, two-component epoxy-based resins comprising a
hardenable resin
component and a hardening agent component containing specific hardening agents
may be
preferred. For subterranean formations having a BHST ranging from about 300 F
to about
600 F, a furan-based resin may be preferred. For subterranean formations
having a BHST
ranging from about 200 F to about 400 F, either a phenolic-based resin or a
one-component
HT epoxy-based resin may be suitable. For subterranean formations having a
BHST of at
least about 175 F, a phenol/phenol formaldehyde/furfuryl alcohol resin may
also be suitable.
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
[0047] Any solvent that is compatible with the chosen resin and achieves the
desired
viscosity effect is suitable for use in the present invention. Some preferred
solvents are those
having high flash points (e.g., about 125 F) because of, among other things,
environmental
and safety concerns; such solvents include butyl lactate, butylglycidyl ether,
dipropylene
glycol methyl ether, dipropylene glycol dimethyl ether, dimethyl formamide,
diethyleneglycol methyl ether, ethyleneglycol butyl ether, diethyleneglycol
butyl ether,
propylene carbonate, methanol, butyl alcohol, d'limonene, fatty acid methyl
esters, and
combinations thereof. Other preferred solvents include aqueous dissolvable
solvents such as,
methanol, isopropanol, butanol, glycol ether solvents, and combinations
thereof. Suitable
glycol ether solvents include, but are not limited to, diethylene glycol
methyl ether,
dipropylene glycol methyl ether, 2-butoxy ethanol, ethers of a C2 to C6
dihydric alkanol
containing at least one C1 to C6 alkyl group, mono ethers of dihydric
alkanols,
methoxypropanol, butoxyethanol, hexoxyethanol, and isomers thereof. Selection
of an
appropriate solvent is dependent on the resin chosen and is within the ability
of one skilled in
the art with the benefit of this disclosure. In general, the amount of solvent
needed is based
on the final desired viscosity of the fluid comprising the resin consolidating
agent.
2. Exemplary Tackifying Agents
[0048] Tackifying agents suitable for use in the methods of the present
invention
exhibit a sticky character and, thus, impart a degree of consolidation to
unconsolidated
particulates in the subterranean formation. As used herein, a "tackifying
agent" refers to a
composition having a nature such that it is (or may be activated to become)
somewhat sticky
to the touch. Examples of suitable tackifying agents suitable for use in the
present invention
include non-aqueous tackifying agents; aqueous tackifying agents; and silyl-
modified
polyamides.
[0049] One type of tackifying agent suitable for use in the present invention
is a non-
aqueous tackifying agent. An example of a suitable tackifying agent may
comprise
polyamides that are liquids or in solution at the temperature of the
subterranean formation
such that they are, by themselves, non-hardening when introduced into the
subterranean
formation. A particularly preferred product is a condensation reaction product
comprised of
commercially available polyacids and a polyamine. Such commercial products
include
compounds such as mixtures of C36 dibasic acids containing some trimer and
higher
oligomers and also small amounts of monomer acids that are reacted with
polyamines. Other
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
16
polyacids include trimer acids, synthetic acids produced from fatty acids,
maleic anhydride,
acrylic acid, and the like. Such acid compounds are commercially available
from companies
such as Witco Corporation, Union Camp, Chemtall, and Emery Industries. The
reaction
products are available from, for example, Champion Technologies, Inc. and
Witco
Corporation. Additional compounds which may be used as non-aqueous tackifying
compounds include liquids and solutions of, for example, polyesters,
polycarbonates and
polycarbamates, natural resins such as shellac and the like. Other suitable
non-aqueous
tackifying agents are described in U.S. Patent Numbers 5,853,048 and
5,833,000, the entire
disclosures of which are herein incorporated by reference.
[0050] Non-aqueous tackifying agents suitable for use in the present invention
may be
either used such that they form a non-hardening coating, or they may be
combined with a
multifunctional material capable of reacting with the non-aqueous tackifying
agent to form a
hardened coating. A "hardened coating," as used herein, means that the
reaction of the
tackifying compound with the multifunctional material will result in a
substantially non-
flowable reaction product that exhibits a higher compressive strength in a
consolidated
agglomerate than the tackifying compound alone with the particulates. In this
instance, the
non-aqueous tackifying agent may function similarly to a hardenable resin.
Multifunctional
materials suitable for use in the present invention include, but are not
limited to, aldehydes
such as formaldehyde, dialdehydes such as glutaraldehyde, hemiacetals or
aldehyde releasing
compounds, diacid halides, dihalides such as dichlorides and dibromides,
polyacid
anhydrides such as citric acid, epoxides, furfuraldehyde, glutaraldehyde or
aldehyde
condensates and the like, and combinations thereof. In some embodiments of the
present
invention, the multifunctional material may be mixed with the tackifying
compound in an
amount of from about 0.01 to about 50 percent by weight of the tackifying
compound to
effect formation of the reaction product. In some preferable embodiments, the
compound is
present in an amount of from about 0.5 to about 1 percent by weight of the
tackifying
compound. Suitable multifunctional materials are described in U.S. Patent
Number
5,839,510, the entire disclosure of which is herein incorporated by reference.
[0051 ] Solvents suitable for use with the non-aqueous tackifying agents of
the present
invention include any solvent that is compatible with the non-aqueous
tackifying agent and
achieves the desired viscosity effect. The solvents that can be used in the
present invention
preferably include those having high flash points (most preferably above about
125 F).
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
17
Examples of solvents suitable for use in the present invention include, but
are not limited to,
butylglycidyl ether, dipropylene glycol methyl ether, butyl bottom alcohol,
dipropylene
glycol dimethyl ether, diethyleneglycol methyl ether, ethyleneglycol butyl
ether, methanol,
butyl alcohol, isopropyl alcohol, diethyleneglycol butyl ether, propylene
carbonate,
d'limonene, 2-butoxy ethanol, butyl acetate, furfuryl acetate, butyl lactate,
dimethyl
sulfoxide, dimethyl formamide, fatty acid methyl esters, and combinations
thereof. It is
within the ability of one skilled in the art, with the benefit of this
disclosure, to determine
whether a solvent is needed to achieve a viscosity suitable to the
subterranean conditions and,
if so, how much. In general, the amount of solvent needed is based on the
final desired
viscosity of the fluid comprising the non-aqueous tackifier consolidating
agent. In some
embodiments, the amount of solvent needed is based on a target viscosity of
less than about
20 cP, in still other embodiments, the target viscosity may be less than about
5 cP.
[0052] Aqueous tackifier agents suitable for use in the present invention are
preferably not significantly tacky when placed onto a particulate, but are
capable of being
"activated" (that is, destabilized, coalesced, and/or reacted) to transform
the compound into a
sticky, tackifying compound at a desirable time. Such activation may occur
before, during, or
after the aqueous tackifier agent is placed in the subterranean formation. In
some
embodiments, a pretreatment may be first contacted with the surface of a
particulate to
prepare it to be coated with an aqueous tackifier agent. Suitable aqueous
tackifying agents
are generally charged polymers that comprise compounds that, when in an
aqueous solvent or
solution, will form a non-hardening coating (by itself or with an activator)
and, when placed
on a particulate, will increase the continuous critical resuspension velocity
of the particulate
when contacted by a stream of water. The aqueous tackifier agent may enhance
the grain-to-
grain contact between the individual particulates within the formation (be
they proppant
particulates, formation fines, or other particulates), helping bring about the
consolidation of
the particulates into a cohesive, flexible, and permeable mass.
[0053] Examples of aqueous tackifier agents suitable for use in the present
invention
include, but are not limited to, acrylic acid polymers, acrylic acid ester
polymers, acrylic acid
derivative polymers, acrylic acid homopolymers, acrylic acid ester
homopolymers (such as
poly(methyl acrylate), poly (butyl acrylate), and poly(2-ethylhexyl
acrylate)), acrylic acid
ester co-polymers, methacrylic acid derivative polymers, methacrylic acid
homopolymers,
methacrylic acid ester homopolymers (such as poly(methyl methacrylate),
poly(butyl
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
18
methacrylate), and poly(2-ethylhexyl methacryate)), acrylamido-methyl-propane
sulfonate
polymers, acrylamido-methyl-propane sulfonate derivative polymers, acrylamido-
methyl-
propane sulfonate co-polymers, and acrylic acid/acrylamido-methyl-propane
sulfonate co-
polymers, and combinations thereof. Methods of determining suitable aqueous
tackifier
agents and additional disclosure on aqueous tackifier agents can be found in
U.S. Patent
Application Number 10/864,061, filed June 9, 2004, and U.S. Patent 7,131,491
issued
November 7, 2006, the entire disclosures of which are hereby incorporated by
reference. In
some embodiments, the concentration of aqueous tackifier agent is from about
0.01 to 10%
wt./vol. of the fluid comprising a consolidating agent. In other embodiments,
the
concentration of aqueous tackifier agent is from about 0.1 to 2% wt. vol. of
the fluid
comprising a consolidating agent.
[0054] Silyl-modified polyamide compounds suitable for use in the tackifying
agents
in the methods of the present invention may be described as substantially self-
hardening
compositions that are capable of at least partially adhering to particulates
in the unhardened
state, and that are further capable of self-hardening themselves to a
substantially non-tacky
state to which individual particulates such as formation fines will not adhere
to, for example,
in formation or proppant pack pore throats. Such silyl-modified polyamides may
be based,
for example, on the reaction product of a silating compound with a polyamide
or a mixture of
polyamides. The polyamide or mixture of polyamides may be one or more
polyamide
intermediate compounds obtained, for example, from the reaction of a polyacid
(e.g., diacid
or higher) with a polyamine (e.g., diamine or higher) to form a polyarriide
polymer with the
elimination of water. Other suitable silyl-modified polyamides and methods of
making such
compounds are described in U.S. Patent Number 6,439,309, the entire disclosure
of which is
herein incorporated by reference. In some embodiments, the concentration of
silyl-modified
polyamide is from about 0.01 to 10% wt./vol. of the fluid comprising a
consolidating agent.
In other embodiments, the concentration of silyl-modified polyamide is from
about 1 to 3%
wt. vol. of the fluid comprising a consolidating agent.
3. Exemplary Gelable Liquid Compositions
[0055] The gelable liquid composition may be any gelable liquid composition
capable
of converting into a gelled substance capable of substantially plugging the
permeability of the
formation while allowing the formation to remain flexible. That is, the gelled
substance
should negatively impact the ability of the formation to produce desirable
fluids such as
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
19
hydrocarbons. As discussed above, the permeability of the formation may be
restored
through use of an after-flush fluid or by fracturing through the consolidated
region. As
referred to herein, the term "flexible" refers to a state wherein the treated
formation is
relatively malleable and elastic and able to withstand substantial pressure
cycling without
substantial breakdown of the formation. Thus, the resultant gelled substance
should be a
semi-solid, immovable, gel-like substance, which, among other things,
stabilizes the treated
portion of the formation while allowing the formation to absorb the stresses
created during
pressure cycling. As a result, the gelled substance may aid in preventing
breakdown of the
formation both by stabilizing and by adding flexibility to the formation
sands. Examples of
suitable gelable liquid compositions include, but are not limited to, resin
compositions that
cure to form flexible gels, gelable aqueous silicate compositions,
crosslinkable aqueous
polymer compositions, and polymerizable organic monomer compositions.
[0056] Certain embodiments of the gelable liquid compositions of the present
invention comprise curable resin compositions. Curable resin compositions are
well known
to those skilled in the art and have been used to consolidate portions of
unconsolidated
formations and to consolidate proppant materials into hard, permeable masses.
While the
curable resin compositions used in accordance with the present invention may
be similar to
those previously used to consolidate sand and proppant into hard, permeable
masses, they are
distinct in that resins suitable for use with the present invention do not
cure into hard,
permeable masses; rather they cure into flexible, gelled substances. That is,
suitable curable
resin compositions form resilient gelled substances between the particulates
of the treated
zone of the unconsolidated formation and thus allow that portion of the
formation to remain
flexible and to resist breakdown. It is not necessary or desirable for the
cured resin
composition to solidify and harden to provide high consolidation strength to
the treated
portion of the formation. On the contrary, upon being cured, the curable resin
compositions
useful in accordance with this invention form semi-solid, immovable, gelled
substances.
[0057] Generally, the curable resin compositions useful in accordance with
this
invention may comprise a curable resin, a diluent, and a resin curing agent.
When certain
resin curing agents, such as polyamides, are used in the curable resin
compositions, the
compositions form the semi-solid, immovable, gelled substances described
above. Where the
resin curing agent used may cause the organic resin compositions to form hard,
brittle
material rather than a desired gelled substance, the curable resin
compositions may further
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
comprise one or more "flexibilizer additives" (described in more detail below)
to provide
flexibility to the cured compositions.
[0058] Examples of curable resins that can be used in the curable resin
compositions
of the present invention include, but are not limited to, organic resins such
as polyepoxide
resins (e.g., bisphenol A-epichlorihydrin resins), polyester resins, urea-
aldehyde resins, furan
resins, urethane resins, and mixtures thereof. Of these, polyepoxide resins
are preferred. One
of skill in the art will be able to determine a desired amount of curable
resin to be included in
the fluid through, for example, porosity fill calculation determinations based
on estimated
depth of coverage for the volume and quantity of resin needed for the
particular formation.
In some embodiments, the concentration of curable resin is from about 0. 1 to
25% wt./vol. of
the fluid comprising a consolidating agent. In other embodiments, the
concentration of
curable resin is from about 1 to 5% wt. vol. of the fluid comprising a
consolidating agent.
[0059] Any diluent that is compatible with the curable resin and achieves the
desired
viscosity effect is suitable for use in the present invention. Examples of
diluents that may be
used in the curable resin compositions of the present invention include, but
are not limited to,
phenols; foimaldehydes; furfuryl alcohols; furfurals; alcohols; ethers such as
butyl glycidyl
ether and cresyl glycidyl etheiphenyl glycidyl ether; and mixtures thereof. In
some
embodiments of the present invention, the diluent comprises butyl lactate. The
diluent may
be used to reduce the viscosity of the curable resin composition to from about
3 to about
3,000 centipoises ("cP") at 80 F. Among other things, the diluent acts to
provide flexibility
to the cured composition. The diluent may be included in the curable resin
composition in an
amount sufficient to provide the desired viscosity effect. Generally, the
diluent used is
included in the curable resin composition in amount in the range of from about
5% to about
75% by weight of the curable resin.
[0060] Generally, any resin curing agent that may be used to cure an organic
resin is
suitable for use in the present invention. When the resin curing agent chosen
is an amide or a
polyamide, generally no flexibilizer additive will be required because, inter
alia, such curing
agents cause the curable resin composition to convert into a semi-solid,
immovable, gelled
substance. Other suitable resin curing agents (such as an amine, a polyamine,
methylene
dianiline, and other curing agents known in the art) will tend to cure into a
hard, brittle
material and will thus benefit from the addition of a flexibilizer additive.
Generally, the resin
curing agent used is included in the curable resin composition, whether a
flexibilizer additive
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
21
is included or not, in an amount in the range of from about 5% to about 75% by
weight of the
curable resin. In some embodiments of the present invention, the resin curing
agent used is
included in the curable resin composition in an amount in the range of from
about 20% to
about 75% by weight of the curable resin.
[0061] As noted above, flexibilizer additives may be used, inter alia, to
provide
flexibility to the gelled substances formed from the curable resin
compositions. Flexibilizer
additives should be used where the resin curing agent chosen would cause the
organic resin
composition to cure into a hard and brittle material - not the desired gelled
substances
described herein. For example, flexibilizer additives may be used where the
resin curing
agent chosen is not an amide or polyamide. Examples of suitable flexibilizer
additives
include, but are not limited to, an organic ester, an oxygenated organic
solvent, an aromatic
solvent, and combinations thereof. Of these, ethers, such as dibutyl
phthalate, are preferred.
Where used, the flexibilizer additive may be included in the curable resin
composition in an
amount in the range of from about 5% to about 80% by weight of the curable
resin. In some
embodiments of the present invention, the flexibilizer additive may be
included in the curable
resin composition in an amount in the range of from about 20% to about 45% by
weight of
the curable resin.
[0062] In other embodiments, the gelable liquid compositions of the present
invention
may comprise a gelable aqueous silicate composition. Generally, the gelable
aqueous silicate
compositions that are useful in accordance with the present invention
generally comprise an
aqueous alkali metal silicate solution and a temperature activated catalyst
for gelling the
aqueous alkali metal silicate solution.
[0063] The aqueous alkali metal silicate solution component of the gelable
aqueous
silicate compositions generally comprises an aqueous liquid and an alkali
metal silicate. The
aqueous liquid component of the aqueous alkali metal silicate solution
generally may be fresh
water, salt water (e.g., water containing one or more salts dissolved
therein), brine (e.g.,
saturated salt water), seawater, or any other aqueous liquid that does not
adversely react with
the other components used in accordance with this invention or with the
subterranean
formation. Examples of suitable alkali metal silicates include, but are not
limited to, one or
more of sodium silicate, potassium silicate, lithium silicate, rubidium
silicate, or cesium
silicate. Of these, sodium silicate is preferred. While sodium silicate exists
in many forms,
the sodium silicate used in the aqueous alkali metal silicate solution
preferably has a Na2O-
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
22
to-SiO2 weight ratio in the range of from about 1:2 to about 1:4. Most
preferably, the sodium
silicate used has a Na2O-to-SiO2 weight ratio in the range of about 1:3.2.
Generally, the
alkali metal silicate is present in the aqueous consolidating fluid in an
amount in the range of
from about 0.1% to about 42% by weight of the aqueous consolidating fluid.
Typically, the
alkali metal silicate is present in the aqueous consolidating fluid in an
amount in the range of
from about 3% to about 8% by weight of the aqueous consolidating fluid.
[0064] The temperature activated catalyst component of the gelable aqueous
silicate
gelable liquid compositions is used, inter alia, to convert the gelable
aqueous silicate
compositions into the desired semi-solid, immovable, gelled substance
described above.
Selection of a temperature activated catalyst is related, at least in part, to
the temperature of
the subterranean formation to which the gelable aqueous silicate composition
will be
introduced. The temperature activated catalysts which can be used in the
gelable aqueous
silicate compositions of the present invention include, but are not limited
to, ammonium
sulfate, which is most suitable in. the range of from about 60 F to about 240
F; sodium acid
pyrophosphate, which is most suitable in the range of from about 60 F to about
240 F; citric
acid, which is most suitable in the range of from about 60 F to about 120 F;
and ethyl
acetate, which is most suitable in the range of from about 60 F to about 120
F. Generally,
the temperature activated catalyst is present in the aqueous consolidating
fluid in the range of
from about 0.1% to about 5% by weight of the aqueous consolidating fluid.
[0065] In other embodiments, the gelable liquid composition consolidating
fluids of
the present invention may comprise crosslinkable aqueous polymer compositions.
Generally,
suitable crosslinkable aqueous polymer compositions may comprise an aqueous
solvent, a
crosslinkable polymer, and a crosslinking agent.
[0066] The aqueous solvent may be any aqueous solvent in which the
crosslinkable
composition and the crosslinking agent may be dissolved, mixed, suspended, or
dispersed
therein to facilitate gel formation. For example, the aqueous solvent used may
be fresh water,
salt water, brine, seawater, or any other aqueous liquid that does not
adversely react with the
other components used in accordance with this invention or with the
subterranean formation.
[0067] Examples of crosslinkable polymers that can be used in the
crosslinkable
aqueous polymer compositions include, but are not limited to, carboxylate-
containing
polymers and acrylamide-containing polymers. Preferred acrylamide-containing
polymers
include polyacrylamide, partially hydrolyzed polyacrylamide, copolymers of
acrylamide and
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
23
acrylate, and carboxylate-containing terpolymers and tetrapolymers of
acrylate. Additional
examples of suitable crosslinkable polymers include hydratable polymers
comprising
polysaccharides and derivatives thereof and that contain one or more of the
monosaccharide
units galactose, mannose, glucoside, glucose, xylose, arabinose, fructose,
glucuronic acid, or
pyranosyl sulfate. Suitable natural hydratable polymers include, but are not
limited to, guar
gum, locust bean gum, tara, konjak, tamarind, starch, cellulose, karaya,
xanthan, tragacanth,
and carrageenan, and derivatives of all of the above. Suitable hydratable
synthetic polymers
and copolymers that may be used in the crosslinkable aqueous polymer
compositions include,
but are not limited to, polyacrylates, polymethacrylates, polyacrylamides,
maleic anhydride,
methylvinyl ether polymers, polyvinyl alcohols, and polyvinylpyrrolidone. The
crosslinkable
polymer used should be included in the crosslinkable aqueous polymer
composition in an
amount sufficient to form the desired gelled substance in the subterranean
formation. In
some embodiments of the present invention, the crosslinkable polymer is
included in the
crosslinkable aqueous polymer composition in an amount in the range of from
about 1% to
about 30% by weight of the aqueous solvent. In another embodiment of the
present
invention, the crosslinkable polymer is included in the crosslinkable aqueous
polymer
composition in an amount in the range of from about 1% to about 20% by weight
of the
aqueous solvent.
[0068] The crosslinkable aqueous polymer compositions of the present invention
may
further comprise a crosslinking agent for crosslinking the crosslinkable
polymers to form the
desired gelled substance. In some embodiments, the crosslinking agent may be a
molecule or
complex containing a reactive transition metal cation. A most preferred
crosslinking agent
comprises trivalent chromium cations complexed or bonded to anions, atomic
oxygen, or
water. Examples of suitable crosslinking agents include, but are not limited
to, compounds or
complexes containing chromic acetate and/or chromic chloride. Other suitable
transition
metal cations include chromium VI within a redox system, aluminum III, iron
II, iron III, and
zirconium IV.
[0069] The crosslinking agent should be present in the crosslinkable aqueous
polymer
compositions of the present invention in an amount sufficient to provide,
inter alia, the
desired degree of crosslinking. In some embodiments of the present invention,
the
crosslinking agent is present in the crosslinkable aqueous polymer
compositions of the
present invention in an amount in the range of from 0.01% to about 5% by
weight of the
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
24
crosslinkable aqueous polymer composition. The exact type and amount of
crosslinking
agent or agents used depends upon the specific crosslinkable polymer to be
crosslinked,
formation temperature conditions, and other factors known to those individuals
skilled in the
art.
[0070] Optionally, the crosslinkable aqueous polymer compositions may further
comprise a crosslinking delaying agent, such as a polysaccharide crosslinking
delaying agents
derived from guar, guar derivatives, or cellulose derivatives. The
crosslinking delaying agent
may be included in the crosslinkable aqueous polymer compositions, inter alia,
to delay
crosslinking of the crosslinkable aqueous polymer compositions until desired.
One of
ordinary skill in the art, with the benefit of this disclosure, will know the
appropriate amount
of the crosslinking delaying agent to include in the crosslinkable aqueous
polymer
compositions for a desired application.
[0071] In other embodiments, the gelled liquid compositions of the present
invention
may comprise polymerizable organic monomer compositions. Generally, suitable
polymerizable organic monomer compositions may comprise an aqueous-base fluid,
a water-
soluble polymerizable organic monomer, an oxygen scavenger, and a primary
initiator.
[0072] The aqueous-base fluid component of the polymerizable organic monomer
composition generally may be fresh water, salt water, brine, seawater, or any
other aqueous
liquid that does not adversely react with the other components used in
accordance with this
invention or with the subterranean formation.
[0073] A variety of monomers are suitable for use as the water-soluble
polymerizable
organic monomers in the present invention. Examples of suitable monomers
include, but are
not limited to, acrylic acid, methacrylic acid, acrylamide, methacrylamide, 2-
methacrylamido-2-methylpropane sulfonic acid, 2-dimethylacrylamide, vinyl
sulfonic acid,
N,N-dimethylaminoethylmethacrylate, 2-triethylammoniumethylmethacrylate
chloride, N,N-
dimethyl-aminopropylmethacryl-amide, methacrylamidepropyltriethylammonium
chloride,
N-vinyl pyrrolidone, vinyl-phosphonic acid, and methacryloyloxyethyl
trimethylammonium
sulfate, and mixtures thereof. Preferably, the water-soluble polymerizable
organic monomer
should be self crosslinking. Examples of suitable monomers which are self
crosslinking
include, but are not limited to, hydroxyethylacrylate, hydroxymethylacrylate,
hydroxyethylmethacrylate, N-hydroxymethylacrylamide, N-hydroxymethyl-
methacrylamide,
polyethylene glycol acrylate, polyethylene glycol methacrylate, polypropylene
gylcol
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
acrylate, polypropylene glycol methacrylate, and mixtures thereof. Of these,
hydroxyethylacrylate is preferred. An example of a particularly preferable
monomer is
hydroxyethylcellulose-vinyl phosphoric acid.
[0074] The water-soluble polymerizable organic monomer (or monomers where a
mixture thereof is used) should be included in the polymerizable organic
monomer
composition in an amount sufficient to form the desired gelled substance after
placement of
the polymerizable organic monomer composition into the subterranean formation.
In some
embodiments of the present invention, the water-soluble polymerizable organic
monomer(s)
are included in the polymerizable organic monomer composition in an amount in
the range of
from about 1% to about 30% by weight of the aqueous-base fluid. In another
embodiment of
the present invention, the water-soluble polymerizable organic monomer(s) are
included in
the polymerizable organic monomer composition in an amount in the range of
from about I%
to about 20% by weight of the aqueous-base fluid.
[0075] The presence of oxygen in the polymerizable organic monomer composition
may inhibit the polymerization process of the water-soluble polymerizable
organic monomer
or monomers. Therefore, an oxygen scavenger, such as stannous chloride, may be
included
in the polymerizable monomer composition. In order to improve the solubility
of stannous
chloride so that it may be readily combined with the polymerizable organic
monomer
composition on the fly, the stannous chloride may be pre-dissolved in a
hydrochloric acid
solution. For example, the stannous chloride may be dissolved in a 0.1 % by
weight aqueous
hydrochloric acid solution in an amount of about 10% by weight of the
resulting solution.
The resulting stannous chloride-hydrochloric acid solution may be included in
the
polymerizable organic monomer composition in an amount in the range of from
about 0.1%
to about 10% by weight of the polymerizable organic monomer composition.
Generally, the
stannous chloride may be included in the polymerizable organic monomer
composition of the
present invention in an amount in the range of from about 0.005% to about 0.1
% by weight of
the polymerizable organic monomer composition.
[0076] The primary initiator is used, inter alia, to initiate polymerization
of the water-
soluble polymerizable organic monomer(s) used in the present invention. Any
compound or
compounds which form free radicals in aqueous solution may be used as the
primary initiator.
The free radicals act, inter alia, to initiate polymerization of the water-
soluble polymerizable
organic monomer(s) present in the polymerizable organic monomer composition.
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
26
Compounds suitable for use as the primary initiator include, but are not
limited to, alkali
metal persulfates; peroxides; oxidation-reduction systems employing reducing
agents, such as
sulfites in combination with oxidizers; and azo polymerization initiators.
Preferred azo
polymerization initiators include 2,2'-azobis(2-imidazole-2-hydroxyethyl)
propane, 2,2'-
azobis(2-aminopropane), 4,4'-azobis(4-cyanovaleric acid), and 2,2'-azobis(2-
methyl-N-(2-
hydroxyethyl) propionamide. Generally, the primary initiator should be present
in the
polymerizable organic monomer composition in an amount sufficient to initiate
polymerization of the water-soluble polymerizable organic monomer(s). In
certain
embodiments of the present invention, the primary initiator is present in the
polymerizable
organic monomer composition in an amount in the range of from about 0.1% to
about 5% by
weight of the water-soluble polymerizable organic monomer(s).
[0077] Optionally, the polymerizable organic monomer compositions further may
comprise a secondary initiator. A secondary initiator may be used, for
example, where the
immature aqueous gel is placed into a subterranean formation that is
relatively cool as
compared to the surface mixing, such as when placed below the mud line in
offshore
operations. The secondary initiator may be any suitable water-soluble compound
or
compounds that may react with the primary initiator to provide free radicals
at a lower
temperature. An example of a suitable secondary initiator is triethanolamine.
In some
embodiments of the present invention, the secondary initiator is present in
the polymerizable
organic monomer composition in an amount in the range of from about 0.1% to
about 5% by
weight of the water-soluble polymerizable organic monomer(s).
[0078] Optionally, the polymerizable organic monomer compositions of the
present
invention further may comprise a crosslinking agent for crosslinking the
polymerizable
organic monomer compositions in the desired gelled substance. In some
embodiments, the
crosslinking agent is a molecule or complex containing a reactive transition
metal cation. A
most preferred crosslinking agent comprises trivalent chromium cations
complexed or
bonded to anions, atomic oxygen, or water. Examples of suitable crosslinking
agents include,
but are not limited to, compounds or complexes containing chromic acetate
and/or chromic
chloride. Other suitable transition metal cations include chromium VI within a
redox system,
aluminum III, iron II, iron III, and zirconium IV. Generally, the crosslinking
agent may be
present in polymerizable organic monomer compositions in an amount in the
range of from
0.01% to about 5% by weight of the polymerizable organic monomer composition.
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
27
C. Relative Permeability Modifier Fluids
[0079] The relative permeability modifier fluids of the present invention may
comprise an aqueous fluid and a relative permeability modifier. As used
herein, "relative
permeability modifier" refers to any material capable of reducing the
permeability of a
subterranean formation to aqueous fluids without substantially reducing the
permeability of
the subterranean formation to hydrocarbons. A variety of additional additives
suitable for use
in subterranean operations also may be included in the relative permeability
modifier fluids
as desired. The aqueous fluid of the relative permeability modifier fluids of
the present
invention may include freshwater, saltwater, brine (e.g., saturated or
unsaturated saltwater),
or seawater. Generally, the aqueous fluid may be from any source, provided
that it does not
contain components that may adversely affect other components in the treatment
fluid.
[0080] The relative permeability modifiers useful in the present invention may
be any
relative permeability modifier that is suitable for use in subterranean
operations. After
introducing the relative permeability modifier fluid into a portion of the
subterranean
formation, the relative permeability modifier preferably attaches to surfaces
within the
porosity of the subterranean formation, so as to reduce the permeability of
the portion of the
subterranean formation to aqueous fluids without substantially changing its
permeability to
hydrocarbons. Examples of suitable relative permeability modifiers include
water-soluble
polymers with or without hydrophobic or hydrophilic modification. As used
herein, "water-
soluble" refers to at least 0.01 weight percent soluble in distilled water. A
water-soluble
polymer with hydrophobic modification is referred to herein as a
"hydrophobically modified
polymer." As used herein, the term "hydrophobic modification," or
"hydrophobically
modified," refers to the incorporation into the hydrophilic polymer structure
of hydrophobic
groups, wherein the alkyl chain length is from about 4 to about 22 carbons. A
water-soluble
polymer with hydrophilic modification is referred to herein as a
"hydrophilically modified
polymer." As used herein, the term "hydrophilic modification," or
"hydrophilically
modified," refers to the incorporation into the hydrophilic polymer structure
of hydrophilic
groups, such as to introduce branching or to increase the degree of branching
in the
hydrophilic polymer. Combinations of hydrophobically modified polymers,
hydrophilically
modified polymers, and water-soluble polymers without hydrophobic or
hydrophilic
modification may be included in the relative modifier fluids of the present
invention.
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
28
[0081 ] The hydrophobically modified polymers useful in the present invention
typically have molecular weights in the range of from about 100,000 to about
10,000,000.
While these hydrophobically modified polymers have hydrophobic groups
incorporated into
the hydrophilic polymer structure, they should remain water-soluble. In some
embodiments,
a mole ratio of a hydrophilic monomer to the hydrophobic compound in the
hydrophobically
modified polymer is in the range of from about 99.98:0.02 to about 90:10,
wherein the
hydrophilic monomer is a calculated amount present in the hydrophilic polymer.
In certain
embodiments, the hydrophobically modified polymers may comprise a polymer
backbone,
the polymer backbone comprising polar heteroatoms. Generally, the polar
heteroatoms
present within the polymer backbone of the hydrophobically modified polymers
include, but
are not limited to, oxygen, nitrogen, sulfur, or phosphorous.
[0082] The hydrophobically modified polymers may be synthesized using any
suitable method. In one example, the hydrophobically modified polymers may be
a reaction
product of a hydrophilic polymer and a hydrophobic compound. In another
example, the
hydrophobically modified polymers may be prepared from a polymerization
reaction
comprising a hydrophilic monomer and a hydrophobically modified hydrophilic
monomer.
Those of ordinary skill in the art, with the benefit of this disclosure, will
be able to determine
other suitable methods for the synthesis of suitable hydrophobically modified
polymers.
[0083] In certain embodiments, suitable hydrophobically modified polymers may
be
synthesized by the hydrophobic modification of a hydrophilic polymer. The
hydrophilic
polymers suitable for forming hydrophobically modified polymers of the present
invention
should be capable of reacting with hydrophobic compounds. Suitable hydrophilic
polymers
include, homo-, co-, or terpolymers such as, but not limited to,
polyacrylamides,
polyvinylamines, poly(vinylamines/vinyl alcohols), alkyl acrylate polymers in
general, and
derivatives thereof. Additional examples of alkyl acrylate polymers include,
but are not
limited to, polydimethylaminoethyl methacrylate, polydimethylaminopropyl
methacrylamide,
poly(acrylamide/dimethylaminoethyl methacrylate), poly(methacrylic
acid/dimethylaminoethyl methacrylate), poly(2-acrylamido-2-methyl propane
sulfonic
acid/dimethylaminoethyl methacrylate), poly(acrylamide/dimethylaminopropyl
methacrylamide), poly (acrylic acid/dimethylaminopropyl methacrylamide), and
poly(methacrylic acid/di.methylaminopropyl methacrylamide). In certain
embodiments, the
hydrophilic polymers comprise a polymer backbone and reactive amino groups in
the
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
29
polymer backbone or as pendant groups, the reactive amino groups capable of
reacting with
hydrophobic compounds. In some embodiments, the hydrophilic polymers comprise
dialkyl
amino pendant groups. In some embodiments, the hydrophilic polymers comprise a
dimethyl
amino pendant group and a monomer comprising dimethylaminoethyl methacrylate
or
dimethylaminopropyl methacrylamide. In certain embodiments of the present
invention, the
hydrophilic polymers comprise a polymer backbone, the polymer backbone
comprising polar
heteroatoms, wherein the polar heteroatoms present within the polymer backbone
of the
hydrophilic polymers include, but are not limited to, oxygen, nitrogen,
sulfur, or
phosphorous. Suitable hydrophilic polymers that comprise polar heteroatoms
within the
polymer backbone include homo-, co-, or terpolymers, such as, but not limited
to, celluloses,
chitosans, polyamides, polyetheramines, polyethyleneimines,
polyhydroxyetheramines,
polylysines, polysulfones, gums, starches, and derivatives thereof. In one
embodiment, the
starch is a cationic starch. A suitable cationic starch may be formed by
reacting a starch,
such as corn, maize, waxy maize, potato, tapioca, and the like, with the
reaction product of
epichlorohydrin and trialkylamine.
[0084] The hydrophobic compounds that are capable of reacting with the
hydrophilic
polymers of the present invention include, but are not limited to, alkyl
halides, sulfonates,
sulfates, organic acids, and organic acid derivatives. Examples of suitable
organic acids and
derivatives thereof include, but are not limited to, octenyl succinic acid;
dodecenyl succinic
acid; and anhydrides, esters, imides, and amides of octenyl succinic acid or
dodecenyl
succinic acid. In certain embodiments, the hydrophobic compounds may have an
alkyl chain
length of from about 4 to about 22 carbons. In another embodiment, the
hydrophobic
compounds may have an alkyl chain length of from about 7 to about 22 carbons.
In another
embodiment, the hydrophobic compounds may have an alkyl chain length of from
about 12
to about 18 carbons. For example, where the hydrophobic compound is an alkyl
halide, the
reaction between the hydrophobic compound and hydrophilic polymer may result
in the
quaternization of at least some of the hydrophilic polymer amino groups with
an alkyl halide,
wherein the alkyl chain length is from about 4 to about 22 carbons.
[0085] As previously mentioned, in certain embodiments, suitable
hydrophobically
modified polymers also may be prepared from a polymerization reaction
comprising a
hydrophilic monomer and a hydrophobically modified hydrophilic monomer.
Examples of
suitable methods of their preparation are described in U.S. Patent Number
6,476,169, the
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
entire disclosure of which is incorporated herein by reference. The
hydrophobically modified
polymers synthesized from the polymerization reactions may have estimated
molecular
weights in the range of from about 100,000 to about 10,000,000 and mole ratios
of the
hydrophilic monomer(s) to the hydrophobically modified hydrophilic monomer(s)
in the
range of from about 99.98:0.02 to about 90:10.
[0086] A variety of hydrophilic monomers may be used to form the
hydrophobically
modified polymers useful in the present invention. Examples of suitable
hydrophilic
monomers include, but are not limited to acrylamide, 2-acrylamido-2-methyl
propane
sulfonic acid, N,N-dimethylacrylamide, vinyl pyrrolidone, dimethylaminoethyl
methacrylate,
acrylic acid, dimethylaminopropylmethacrylamide, vinyl amine, vinyl acetate,
trimethylammoniumethyl methacrylate chloride, methacrylamide, hydroxyethyl
acrylate,
vinyl sulfonic acid, vinyl phosphonic acid, methacrylic acid, vinyl
caprolactam, N-
vinylformamide, N,N-diallylacetamide, dimethyldiallyl ammonium halide,
itaconic acid,
styrene sulfonic acid, methacrylamidoethyltrimethyl ammonium halide,
quaternary salt
derivatives of acrylamide, and quaternary salt derivatives of acrylic acid.
[0087] A variety of hydrophobically modified hydrophilic monomers also may be
used to form the hydrophobically modified polymers useful in the present
invention.
Examples of suitable hydrophobically modified hydrophilic monomers include,
but are not
limited to, alkyl acrylates, alkyl methacrylates, alkyl acrylamides, alkyl
methacrylamides
alkyl dimethylammoniumethyl methacrylate halides, and alkyl
dimethylammoniumpropyl
methacrylamide halides, wherein the alkyl groups have from about 4 to about 22
carbon
atoms. In another embodiment, the alkyl groups have from about 7 to about 22
carbons. In
another embodiment, the alkyl groups have from about 12 to about 18 carbons.
In certain
embodiments, the hydrophobically modified hydrophilic monomer comprises
octadecyldimethylammoniumethyl methacrylate bromide,
hexadecyldimethylammoniumethyl
methacrylate bromide, hexadecyldimethylammoniumpropyl methacrylamide bromide,
2-
ethylhexyl methacrylate, or hexadecyl methacrylamide.
[0088] Suitable hydrophobically modified polymers that may be formed from the
above-described reactions include, but are not limited to,
acrylamide/octadecyldimethylammoniumethyl methacrylate bromide copolymer,
dimethylaminoethyl methacrylate/vinyl
pyrrolidone/hexadecyldimethylammoniumethyl
methacrylate bromide terpolymer, and acrylamide/2-acrylamido-2-methyl propane
sulfonic
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
31
acid/2-ethylhexyl methacrylate terpolymer. Another suitable hydrophobically
modified
polymer formed from the above-described reaction is an amino
methacrylate/alkyl amino
methacrylate copolymer. A suitable dimethlyaminoethyl methacrylate/alkyl-
dimethylammoniumethyl methacrylate copolymer is a dimethylaminoethyl
methacrylate/hexadecyl-dimethylammoniumethyl methacrylate copolymer. As
previously
discussed, these copolymers may be formed by reactions with a variety of alkyl
halides. For
example, in some embodiments, the hydrophobically modified polymer may be a
dimethylaminoethyl methacrylate/hexadecyl-dimethylammoniumethyl methacrylate
bromide
copolymer.
[0089] In another embodiment of the present invention, the relative
permeability
modifier fluid of the present invention may comprise a water-soluble
hydrophilically
modified polymer. The hydrophilically modified polymers of the present
invention typically
have molecular weights in the range of from about 100,000 to about 10,000,000.
In certain
embodiments, the hydrophilically modified polymers comprise a polymer
backbone, the
polymer backbone comprising polar heteroatoms. Generally, the polar
heteroatoms present
within the polymer backbone of the hydrophilically modified polymers include,
but are not
limited to, oxygen, nitrogen, sulfur, or phosphorous.
[0090] The hydrophilically modified polymers may be synthesized using any
suitable
method. In one example, the hydrophilically modified polymers may be a
reaction product of
a hydrophilic polymer and a hydrophilic compound. Those of ordinary skill in
the art, with
the benefit of this disclosure, will be able to determine other suitable
methods for the
preparation of suitable hydrophilically modified polymers.
[0091] In certain embodiments, suitable hydrophilically modified polymers may
be
formed by additional hydrophilic modification, for example, to introduce
branching or to
increase the degree of branching, of a hydrophilic polymer. The hydrophilic
polymers
suitable for forming the hydrophilically modified polymers used in the present
invention
should be capable of reacting with hydrophilic compounds. In certain
embodiments, suitable
hydrophilic polymers include, homo-, co-, or terpolymers, such as, but not
limited to,
polyacrylamides, polyvinylamines, poly(vinylamines/vinyl alcohols), and alkyl
acrylate
polymers in general. Additional examples of alkyl acrylate polymers include,
but are not
limited to, polydimethylaminoethyl methacrylate, polydimethylaminopropyl
methacrylamide,
poly(acrylamide/dimethylaminoethyl methacrylate), poly(methacrylic
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
32
acid/dimethylaminoethyl methacrylate), poly(2-acrylamido-2-methyl propane
sulfonic
acid/dimethylaminoethyl methacrylate), poly(acrylamide/dimethylaminopropyl
methacrylamide), poly (acrylic acid/dimethylaminopropyl methacrylamide), and
poly(methacrylic acid/dimethylaminopropyl methacrylamide). In certain
embodiments, the
hydrophilic polymers comprise a polymer backbone and reactive amino groups in
the
polymer backbone or as pendant groups, the reactive amino groups capable of
reacting with
hydrophilic compounds. In some embodiments, the hydrophilic polymers comprise
dialkyl
amino pendant groups. In some embodiments, the hydrophilic polymers comprise a
dimethyl
amino pendant group and at least one monomer comprising dimethylaminoethyl
methacrylate
or dimethylaminopropyl methacrylamide. In other embodiments, the hydrophilic
polymers
comprise a polymer backbone comprising polar heteroatoms, wherein the polar
heteroatoms
present within the polymer backbone of the hydrophilic polymers include, but
are not limited
to, oxygen, nitrogen, sulfur, or phosphorous. Suitable hydrophilic polymers
that comprise
polar heteroatoms within the polymer backbone include homo-, co-, or
terpolymers, such as,
but not limited to, celluloses, chitosans, polyamides, polyetheramines,
polyethyleneimines,
polyhydroxyetheramines, polylysines, polysulfones, gums, starches, and
derivatives thereof.
In one embodiment, the starch is a cationic starch. A suitable cationic starch
may be formed
by reacting a starch, such as corn, maize, waxy maize, potato, tapioca, and
the like, with the
reaction product of epichlorohydrin and trialkylamine.
[0092] The hydrophilic compounds suitable for reaction with the hydrophilic
polymers include polyethers that comprise halogens, sulfonates, sulfates,
organic acids, and
organic acid derivatives. Examples of suitable polyethers include, but are not
limited to,
polyethylene oxides, polypropylene oxides, and polybutylene oxides, and
copolymers,
terpolymers, and mixtures thereof. In some embodiments, the polyether
comprises an
epichlorohydrin-terminated polyethylene oxide methyl ether.
[0093] The hydrophilically modified polymers formed from the reaction of a
hydrophilic polymer with a hydrophilic compound may have estimated molecular
weights in
the range of from about 100,000 to about 10,000,000 and may have weight ratios
of the
hydrophilic polymers to the polyethers in the range of from about 1:1 to about
10:1. Suitable
hydrophilically modified polymers having molecular weights and weight ratios
in the ranges
set forth above include, but are not limited to, the reaction product of
polydimethylaminoethyl methacrylate and epichlorohydrin-terminated
polyethyleneoxide
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
33
methyl ether; the reaction product of polydimethylaminopropyl methacrylamide
and
epichlorohydrin-terminated polyethyleneoxide methyl ether; and the reaction
product of
poly(acrylamide/dimethylaminopropyl methacrylamide) and epichlorohydrin-
terminated
polyethyleneoxide methyl ether. In some embodiments, the hydrophilically
modified
polymer comprises the reaction product of a polydimethylaminoethyl
methacrylate and
epichlorohydrin-terminated polyethyleneoxide methyl ether having a weight
ratio of
polydimethylaminoethyl methacrylate to epichlorohydrin-terminated
polyethyleneoxide
methyl ether of about 3:1.
[0094] In another embodiment of the present invention, the water-soluble
relative
permeability modifiers comprise a water-soluble polymer without hydrophobic or
hydrophilic
modification. Examples of suitable water-soluble polymers include, but are not
limited to,
homo-, co-, and terpolymers of acrylamide, 2-acrylamido-2-methyl propane
sulfonic acid,
N,N-dimethylacrylamide, vinyl pyrrolidone, dimethylaminoethyl methacrylate,
acrylic acid,
dimethylaminopropylmethacrylamide, vinyl amine, vinyl acetate,
trimethylammoniumethyl
methacrylate chloride, methacrylamide, hydroxyethyl acrylate, vinyl sulfonic
acid, vinyl
phosphonic acid, methacrylic acid, vinyl caprolactam, N-vinylformamide, N,N-
diallylacetamide, dimethyldiallyl ammonium halide, itaconic acid, styrene
sulfonic acid,
methacrylamidoethyltrimethyl ammonium halide, quaternary salt derivatives of
acrylamide
and quaternary salt derivatives of acrylic acid.
[0095] Sufficient concentrations of a suitable relative permeability modifier
may be
present in the treatment fluids of the present invention to provide the
desired degree of
diversion. The amount of the relative permeability modifier to include in the
treatment fluid
depends on a number of factors including, the composition of the fluid to be
diverted and the
porosity of the formation. In some embodiments, a relative permeability
modifier may be
present in a treatment fluid of the present invention in an amount in the
range of from about
0.02% to about 10% by weight of the composition. In some embodiments, a
relative
permeability modifier may be present in an amount in the range of from about
0.05% to about
1.0% by weight of the composition. In certain embodiments of the present
invention, the
relative permeability modifier may be provided in a concentrated aqueous
solution prior to its
combination with the other components necessary to form a treatment fluid of
the present
invention.
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
34
[0096] Additional additives may be included in the treatment fluids of the
present
invention as deemed appropriate for a particular application by one skilled in
the art, with the
benefit of this disclosure. Examples of such additives include, but are not
limited to, acids,
weighting agents, surfactants, scale inhibitors, antifoaming agents,
bactericides, salts,
foaming agents, fluid loss control additives, viscosifying agents, gel
breakers, clay stabilizers,
and combinations thereof.
[0097] Therefore, the present invention is well adapted to attain the ends and
advantages mentioned as well as those that are inherent therein. While
numerous changes
may be made by those skilled in the art, such changes are encompassed within
the spirit of
this invention as defined by the appended claims. The terms in the claims have
their plain,
ordinary meaning unless otherwise explicitly and clearly defined by the
patentee.
[0098] Water control treatments are often considered as the last resort to
provide
solutions to the problems affecting the production and operation of the wells.
Instead of
treating subterranean formations with water and sand production problems
separately, the
present invention allows for water and sand control treatments to be performed
simultaneously or one directly after the other. Although experimental testing
showed that
relative permeability modifiers may be applied either before or after that of
a consolidation
treatment, it may be more convenient and cost effective to treat the interval
with RPM
solution before the consolidating agent such that the action of placing the
consolidating agent
displaces the relative permeability modifier deeper into the formation.
[0099] To facilitate a better understanding of the present invention, the
following
examples of certain aspects of some embodiments are given. In no way should
the following
examples be read to limit, or define, the scope of the invention.
EXAMPLES
[00100] Preparation of Sand Packs. Both synthetic sand packs and Brown
sandstone
outcrop material were used in the study. Sand-pack samples were prepared from
a sand
mixture composed of 88% (wt/wt) 70/170-mesh sand, 10% silica flour, and 2%
smectite.
The sand mixture was blended using a kitchen mixer to help ensure homogeneity
of the sand
pack. After being well blended, each sand mixture was hand packed into the
Hassler sleeve
and assembled in a consolidation chamber. The Hassler sleeve had a diameter of
2.54 cm and
a length of 13.3 cm. Each sand column was packed in the Hassler sleeve in the
following
order of materials: 10 g of 70/170-mesh sand, 20 g of resieved 20/40-mesh
Ottawa sand, 175
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
g of sand mixture (described above), and 10 g of resieved 20/40-mesh Ottawa
sand. Screen
pieces with 60-mesh size were installed on top and at the bottom of the sand
pack. The
chamber was equipped with a heated jacket to simulate downhole temperatures
during curing.
A piston displacement ISCO pump was used to inject all fluids with a
backpressure of 50 psi
applied during injection and an annular confining pressure of 2,500 psi placed
on the sand
pack to ensure no fluid bypassed the pack. The entire system was maintained at
170 F during
treatment.
[00101] Relative Permeability Modifier. The selected relative permeability
modifier
was a hydrophobically modified, water-soluble polymer. A relative permeability
modifier
solution with concentration of 2,000 ppm was prepared in 2% KCl that was then
adjusted
with an acid buffer to obtain a pH of 6Ø
[00102] Consolidating Agent. The selected consolidating agent was an epoxy-
based
resin system capable of acting as a consolidating agent at temperatures
between 170-325 F.
An epoxy based resin with a volume of 30 mL, (i.e., 15 mL of hardenable resin
component
and 15 mL of internal activator component) was prepared to form a single
mixture just before
the consolidation treatment. This resin mixture has a viscosity of about 15 cP
at 72 F.
[00103] Test 1 - Consolidation Treatment of Sand Pack without RPM Treatment.
This test was performed to determine the consolidation performance of the
consolidating
agent on the sand pack. All injections were maintained at 5 mL/min pump rate.
Initial
permeability values of kerosene and API brine for the sand pack were first
established in the
production direction. The consolidation treatment was then followed in the
injection
direction. The consolidation treatment typically involved injecting a preflush
fluid, the resin
mixture described above having a viscosity of about 15 cP at 72 F, and an
after -flush fluid.
Once the after-flush treatment was completed, the treated sand pack was then
shut in for 48
hours at 170 F to allow complete curing of the epoxy-based resin consolidating
agent. After
curing, the regained permeability of the sand pack was determined with
kerosene in the
production direction. The consolidated sand pack was then cut into cores of
desired length
and unconfined compressive strengths (UCS) of the cores were determined.
[00104] Test 2 - Relative Permeability Modifier Followed by a Consolidating
Agent.
This test was performed to determine the compatibility of a relative
permeability modifier
treatment with that of consolidating agent treatment in a sand pack. Following
measurements
of initial permeability of kerosene and API brine through the sand pack, as
described above, a
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
36
relative permeability modifier treatment was performed on the sand pack. The
relative
permeability modifier treatment typically consists of injecting an API brine,
followed by a
relative permeability modifier solution, followed by an after-flush brine
displacement,
followed by treatment with the resin mixture described above having a
viscosity of about 15
cP at 72 F. The treated sand pack was then allowed to cure at 170 F for 48
hours. Both
kerosene and API brine were used in determining the regained permeability with
respect to
kerosene and brine. The UCS, consolidation strength values were then obtained
for the
consolidated sand pack.
[00105] Test 3 - Relative Permeability Modifier Followed by a Consolidating
Agent.
This test was performed to determine if consolidation of a sand pack could be
achieved when
a very low viscosity fluid comprising a consolidating agent is used following
the placement
of a relative permeability modifier. Similar to the procedure described in
Test 2, above, a
sand pack was first treated with a relative permeability modifier solution and
followed by
placement of the resin mixture described above having a viscosity of about 15
cP at 72 F.
[00106] Test 4 - Treatments of Consolidating Agent and Relative Permeability
Modifier on Sequential Set Up of Sand Pack and Brown Sandstone Core. This test
was
performed to examine the performance of a relative permeability modifier
treatment in a sand
pack and in a Brown sandstone core after the placement of a consolidating
agent. A flow
chamber which had a Hassler sleeve containing a sand pack, as described above,
was set up
in front of the flow chamber with Hassler sleeve containing the Brown
sandstone core. The
Hassler sleeve containing sandstone core was equipped with multitaps where
pressure
transducers were installed to allow measurements of differential pressures
between various
interval lengths of the cores. The Brown sandstone had a 6-in. length and a 1-
in. diameter.
Initial values of permeability of kerosene and API brine were obtained for the
sand pack and
sandstone core in the production direction. A consolidation treatment was then
performed on
the sand pack in the injection direction. The sandstone core was never exposed
to
consolidating agent. Following the consolidation treatment, a relative
permeability modifier
treatment was applied also in the injection direction sequentially through
both the sand pack
and the sandstone. The treated sand pack and sandstone core were then shut in
for 48 hours
at 170 F. After the shut-in period, kerosene and API brine were used in
regained
CA 02710449 2010-06-21
WO 2009/087349 PCT/GB2008/004151
37
[00107] permeability measurements by injecting in the production direction.
The
UCS, consolidation strength, values were then obtained for the consolidated
sand pack.
[00108] Results. Table 1, below, provides summary results of regained
permeability
of kerosene, the amount of water shutoff, and consolidation strengths (i.e.,
UCS) obtained for
the treated sand packs in these tests.
Test I Test 2 Test 3 Test 4
treatment Resin only Relative Relative Sand Pack: Sandstone:
type Permeability Permeability Relative Relative
Modifier Modifier Permeability Permeability
followed by followed by Modifier Modifier only
Resin Low followed by
Viscosity Resin
Resin
% regained 74 84 98 92 80
permeability
to kerosene
% water ----- 38 65 62 75
shutoff
UCS (psi) 990 1010 <10 1270 ------
[00109] Therefore, the present invention is well adapted to attain the ends
and
advantages mentioned as well as those that are inherent therein. The
particular embodiments
disclosed above are illustrative only, as the present invention may be
modified and practiced
in different but equivalent manners apparent to those skilled in the art
having the benefit of
the teachings herein. Furthermore, no limitations are intended to the details
of construction or
design herein shown, other than as described in the claims below. It is
therefore evident that
the particular illustrative embodiments disclosed above may be altered or
modified and all
such variations are considered within the scope and spirit of the present
invention. In
particular, every range of values (of the form, "from about a to about b," or,
equivalently,
"from approximately a to b," or, equivalently, "from approximately a-b")
disclosed herein is
to be understood as referring to the power set (the set of all subsets) of the
respective range of
values, and set forth every range encompassed within the broader range of
values. Also, the
terms in the claims have their plain, ordinary meaning unless otherwise
explicitly and clearly
defined by the patentee