Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02711726 2010-07-07
WO 2009/098135 PCT/EP2009/050839
-1-
NOVEL IMIDAZOLINYLMETHYL ARYL SULFONAMIDES
This invention relates to an imidazolinylmethyl aryl sulfonamide which is an
alpha- lA adren-
ergic partial agonist, associated pharmaceutical compositions, and methods for
use as a thera-
peutic agent.
Alpha-1 adrenergic receptors (interchangeably named alpha-1 adrenoceptors) are
G-protein
coupled transmembrane receptors that mediate various actions of the
sympathetic nervous
system through the binding of the catecholamines, epinephrine and
norepinephrine (NE).
Currently, several subtypes of the alpha-1 adrenergic receptors are known to
exist for which the
genes have been cloned: alpha- lA (previously known as alpha-1C), alpha-1B and
alpha-1D. The
existence of a low affinity alpha-1 adrenoceptor for prazosin named alpha-1L,
in human prostate
has been determined. However, the gene for the alpha-1L adrenergic receptor
subtype has yet to
be cloned.
The alpha-1 adrenoceptor plays a part in the sympathetic maintenance of smooth
muscle tone
and alpha-1 adrenergic agonists are known to increase muscle tone in the lower
urinary tract
necessary for urine storage and urine emptying thus making adrenergic
receptors important
targets for drug development in urinary dysfunction (Testa, Eur.J.Pharmacol.,
1993, 249, 307-
315. Pharmacological studies resulting in the subdivision of alpha-1
adrenergic receptors have
let to the suggestion that development of subtype-selective compounds may
allow improved
treatment with a lower incidence of side effects, and Tanaguchi et al.,
Eur.J.Pharmacol, 1996,
318, 117-122, have reported that compounds with selectivity for the alpha-IA
receptor and to a
lessen extent to the alpha-1L receptor over the alpha-1B and alpha-1D subtypes
have selectivity
for urethral over vascular tissue.
Urinary incontinence is a condition defined as the involuntary loss of urine
to such an extent as
to become a hygienic or social concern to the patient. Stress urinary
incontinence (SUI) occurs
when the internal sphincter does not close completely. The primary symptom is
minor leakage
from activities, such as coughing, sneezing, laughing, running, lifting, or
even standing, that
apply pressure to a full bladder. Leakage stops when the activity stops. SUI
is most common in
CA 02711726 2010-07-07
WO 2009/098135 PCT/EP2009/050839
-2-
women between the ages of 25 and 50, and many regularly exercising women have
some degree
of SUI.
The methods presently available to treat SUI include physiotherapy and
surgery. Treatment with
pharmaceuticals is limited to the use of non-selective adrenergic agonists.
Only a limited
number of pharmaceutical agents have been employed, with varying success, to
treat stress
incontinence.
Phenylpropanolamine, pseudoephrine and midodrine are considered first-line
therapy for mild to
moderate stress incontinence (Wein, supra; Lundberg (editor), JAMA 1989,
261(18):2685-2690).
These agents are believed to work both by direct activation of alpha-1
adrenoceptors and in-
directly by displacement of endogenous norepinephrine from sympathetic neurons
following
uptake into the nerve terminal (Andersson and Sjogren, Progress in
Neurobiology, 1982, 71-89).
Activation of alpha-1 adrenoceptors located on the smooth muscle cells of the
proximal urethra
and bladder neck (Sourander, Gerontology 1990, 36:19-26; Wein, supra) evokes
contraction and
an increase in urethral closure pressure.
The utility of phenylpropanolamine, pseudoephrine, and midodrine is limited by
a lack of
selectivity among the alpha-1 adrenoceptor subtypes and by the indirect action
of these agents
(i.e. activation of alpha-1, alpha-2, and beta-adrenoceptors in the central
nervous system and
periphery). As a result, any desired therapeutic effect of these agents may be
accompanied by
undesirable side effects such as an increase in blood pressure. The increase
in blood pressure is
dose-dependent and therefore limits the ability to achieve therapeutically
effective circulating
concentrations of these agents (Andersson and Sjogren, supra). Furthermore, in
some patients
these agents produce insomnia, anxiety and dizziness as a result of their
central nervous system
stimulant actions (Andersson and Sjogren, supra, Wein, supra).
Certain alpha-IA/1L agonists are known to be useful in treating various
disease states including
urinary incontinence, nasal congestion, sexual dysfunction such as ejaculation
disorders and
priapism, and CNS disorders such as depression, anxiety, dementia, senility,
Alzheimer's,
deficiencies in attentiveness and cognition, and eating disorders such as
obesity, bulimia, and
anorexia. See e.g. US Patent Nos. 5,952,362, 6,756,395, 6,852,726, and
6,979,696 which
disclose a variety of 2-imidazolinylmethyl aryl and heteroaryl derivatives as
alpha-iA/L agonists.
Full agonists of the alpha IA/1L adrenoceptor subtype, while potentially
effective at treating
urinary incontinence, can be limited by undesirable cardiovascular and central
nervous system
CA 02711726 2010-07-07
WO 2009/098135 PCT/EP2009/050839
-3-
side effects. Selective alpha lA/1L receptor modulators with reduced intrinsic
efficacy (i.e.,
"partial agonists") can reduce such side effects while maintaining the
contractile effects on
urethral smooth muscle needed for treating incontinence.
Due to side effects and /or limited efficacy associated with the current
available medicaments,
there is an unmet medical need for useful compounds. A compound having the
desired alpha-IA
adrenergic partial agonist profile is desirable.
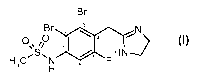
In one aspect, the application provides a compound of formula I:
Br
Br z C N
H C_S_N F HN
s H
or a pharmaceutically acceptable salt or prodrug thereof.
The compound of Formula I, N-[2,3-Dibromo-4-(4,5-dihydro-lH-imidazol-2-
ylmethyl)-5-
fluoro-phenyl]-methanesulfonamide (nomenclature used in this Application is
based on
AUTONOMTM v.4.0), has been found to exhibit unexpectedly enhanced selectivity,
for enhance-
ment of intraurethral pressure (IUP) over blood pressure (MAP), as a partial
agonist of alpha- lA
adrenoceptors. The combination of the dbromo substituents on the 2- and 3-
positions with the
fluoro substituent at the 5-position of the phenyl ring provide unexpected
advantages over the
general class of imidazolinylmethyl aryl sulfonamides in that it has both a
favorable intrinsic
activity, or efficacy, as a partial agonist, which is ideally between 0.35 to
0.60, of 0.38 and an
affinity, or pEC50 value, of 6.7. As full agonist activity is undesirable due
to hypertension
related side effects, the combination of high affinity and partial agonist
behavior is critical for
optimization of urethral activity benefits associated with effective
modulation of alpha-lA
adrenoceptors coupled with minimization of diastolic blood pressure related
side effects.
Furthermore, the compound of Formula I, in comparison to analogue compounds,
exhibits im-
proved durability of IUP response over time which is necessary for effective
treatment of in-
continence.
In one embodiment, the application provides the compound of formula I, wherein
the pharma-
ceutically acceptable salt is hydrochloride.
CA 02711726 2010-07-07
WO 2009/098135 PCT/EP2009/050839
-4-
In one embodiment, the application provides a composition comprising the
compound of formula
1 and further comprising a pharmaceutically acceptable carrier.
In one embodiment, the application provides the above composition, wherein the
composition is
suitable for administration to a subject having a disease state which is
alleviated by treatment
with an alpha-lA receptor partial agonist.
In one embodiment, the application provides a method for preventing,
alleviating, or treating a
disorder modulated by alpha-IA adrenoceptors, said method comprising
administering to a
subject in need thereof an effective amount of the compound of formula 1.
In one embodiment, the application provides the above method, wherein the
disorder is selected
from urge incontinence, stress incontinence, overflow incontinence, and
functional incontinence.
In one embodiment, the application provides a method for preventing,
alleviating, or treating a
disorder modulated by alpha-IA adrenoceptors, wherein the disorder is stress
incontinence.
In one embodiment, the application provides a method for preventing,
alleviating, or treating a
disorder modulated by alpha-IA adrenoceptors, wherein the disorder is urge
incontinence.
In one embodiment, the application provides a method for preventing,
alleviating, or treating a
disorder modulated by alpha-IA adrenoceptors, wherein the disorder is overflow
incontinence.
In one embodiment, the application provides a method for preventing,
alleviating, or treating a
disorder modulated by alpha-IA adrenoceptors, wherein the disorder is
functional incontinence.
In one embodiment, the application provides a method for preventing,
alleviating, or treating a
disorder modulated by alpha-IA adrenoceptors, said method comprising
administering to a
subject in need thereof an effective amount of the compound of formula 1 in
combination with a
second modulator of alpha-IA adrenoceptors.
In one embodiment, the application provides a method of treating or preventing
a disease state
characterized by urinary incontinence comprising administering to a subject in
need thereof an
effective amount of a compound of formula 1.
IN THE DRAWINGS
Figure I a. Effect of vehicle on IUP, MAP and HR in Conscious Pig Model
CA 02711726 2010-07-07
WO 2009/098135 PCT/EP2009/050839
-5-
Figure lb. Formula I in Conscious Pig Model
Figure lc. Analogue Compound in Conscious Pig Model
Unless otherwise stated, the following terms used in this Application,
including the specification
and claims, have the definitions given below. It must be noted that, as used
in the specification
and the appended claims, the singular forms "a", "an," and "the" include
plural referents unless
the context clearly dictates otherwise.
All patents and publications identified herein are incorporated herein by
reference in their
entirety.
As used herein, "IUP" means intraurethral pressure and is measured as the 2
minute mean from
the first peak of the urethral response.
As used herein, "MAP" means mean arterial blood pressure and is measured as
the average
blood pressure during the 2 minute section where IUP is measured.
As used herein, "durability of IUP response over time" means the slope of the
IUP response in
mmHg/min and is calculated immediately after the 2 minute IUP response for 5
minutes (2-7
minutes post the first peak) for the top 3 doses.
"Aryl" means the monovalent cyclic aromatic hydrocarbon radical consisting of
one or more
fused rings in which at least one ring is aromatic in nature, which can
optionally be substituted
with hydroxy, cyan, lower alkyl, lower alkoxy, alkylthio, halo, haloalkyl,
hydroxyalkyl, nitro,
alkoxycarbonyl, amino, alkylamino, dialkylamino, amino carbonyl,
carbonylamino, aminosulf-
onyl, sulfonylamino, nitro, and/or alkylsulphonyl, unless otherwise indicated.
Examples of aryl
radicals include, but are not limited to, phenyl, naphthyl, biphenyl, indanyl,
anthraquinolyl, and
the like.
"Arylsulfonyl" means a radical -S(O)2R where R is an aryl group as defined
herein.
"2-Imidazolinylmethyl", "imidazolin-2-ylmethyl", "imidazolinylmethyl", and 4,5-
dihydro-lH-
imidazol-2-ylmethyl", which may be used interchangeably, mean the moiety
designated by the
structure
HN~
CA 02711726 2010-07-07
WO 2009/098135 PCT/EP2009/050839
-6-
It is to be understood that the double bond in 2-imidazoline and 2-
imidazolinylmethyl may
assume other resonance forms. The terms 2-imidazoline 2-imidazolinylmethyl
include all such
resonance forms.
"Isomerism" means compounds that have identical molecular formulae but that
differ in the
nature or the sequence of bonding of their atoms or in the arrangement of
their atoms in space.
Isomers that differ in the arrangement of their atoms in space are termed
"stereoisomers".
Stereoisomers that are not mirror images of one another are termed
"diastereoisomers", and
stereoisomers that are non-superimposable mirror images are termed
"enantiomers", or some-
times optical isomers. A carbon atom bonded to four nonidentical substituents
is termed a
"chiral center".
"Chiral compound" means a compound with one or more chiral center. It has two
enantiomeric
forms of opposite chirality and may exist either as an individual enantiomer
or as a mixture of
enantiomers. A mixture containing equal amounts of individual enantiomeric
forms of opposite
chirality is termed a "racemic mixture". A compound that has more than one
chiral center has 2"-
1 enantiomeric pairs, where n is the number of chiral centers. Compounds with
more than one
chiral center may exist as either an individual diastereomer or as a mixture
of diastereomers,
termed a "diastereomeric mixture". When chiral centers are present, the
stereoisomers may be
characterized by the absolute configuration (R or S ) of the chiral centers.
Absolute configure-
tion refers to the arrangement in space of the substituents attached to a
chiral center. The sub-
stituents attached to a chiral center under consideration are ranked in
accordance with the
Sequence Rule of Cahn, Ingold and Prelog. (Cahn et al. Angew. Chem. Inter.,
1966, Edit., 5,
385; errata 511; Cahn et al. Angew. Chem., 1966, 78, 413; Cahn and Ingold, J.
Chem. Soc.
(London), 1951, 612; Cahn et al., Experientia, 1956, 12, 81; Cahn, J.,
Chem.Educ., 1964, 41,
116).
"Tautomers" refers to compounds whose structures differ markedly in
arrangement of atoms, but
which exist in easy and rapid equilibrium. It should also be understood that
when compounds
have tautomeric forms, all tautomeric forms are intended to be within the
scope of the invention,
and the naming of the compounds does not exclude any tautomer form.
"Pharmaceutically acceptable" means that which is useful in preparing a
pharmaceutical compo-
sition that is generally safe, non-toxic, and neither biologically nor
otherwise undesirable and
includes that which is acceptable for veterinary as well as human
pharmaceutical use.
CA 02711726 2010-07-07
WO 2009/098135 PCT/EP2009/050839
-7-
"Pharmaceutically acceptable salts" of a compound means salts that are
pharmaceutically
acceptable, as defined herein, and that possess the desired pharmacological
activity of the parent
compound. Such salts include:
(1) acid addition salts formed with inorganic acids such as hydrochloric acid,
hydrobromic
acid, sulfuric acid, nitric acid, phosphoric acid, and the like; or formed
with organic acids such as
acetic acid, benzenesulfonic acid, benzoic, camphorsulfonic acid, citric acid,
ethanesulfonic acid,
fumaric acid, glucoheptonic acid, gluconic acid, glutamic acid, glycolic acid,
hydroxynaphtoic
acid, 2-hydroxyethanesulfonic acid, lactic acid, maleic acid, malic acid,
malonic acid, mandelic
acid, methanesulfonic acid, muconic acid, 2-naphthalenesulfonic acid,
propionic acid, salicylic
acid, succinic acid, tartaric acid, p-toluenesulfonic acid, trimethylacetic
acid, and the like; or
(2) salts formed when an acidic proton present in the parent compound either
is replaced by a
metal ion, e.g., an alkali metal ion, an alkaline earth ion, or an aluminum
ion; or coordinates with
an organic or inorganic base. Acceptable organic bases include diethanolamine,
ethanolamine,
N-methylglucamine, triethanolamine, tromethamine, and the like. Acceptable
inorganic bases
include aluminum hydroxide, calcium hydroxide, potassium hydroxide, sodium
carbonate and
sodium hydroxide.
It should be understood that all references to pharmaceutically acceptable
salts include solvent
addition forms (solvates) or crystal forms (polymorphs) as defined herein, of
the same acid
addition salt.
The preferred pharmaceutically acceptable salts are the salts formed from
acetic acid, hydro-
chloric acid, sulphuric acid, methanesulfonic acid, maleic acid, phosphoric
acid, tartaric acid,
citric acid, sodium, potassium, calcium, zinc, and magnesium.
"Solvates" means solvent additions forms that contain either stoichiometric or
non stoichiometric
amounts of solvent. Some compounds have a tendency to trap a fixed molar ratio
of solvent
molecules in the crystalline solid state, thus forming a solvate. If the
solvent is water the solvate
formed is a hydrate, when the solvent is alcohol, the solvate formed is an
alcoholate. Hydrates
are formed by the combination of one or more molecules of water with one of
the substances in
which the water retains its molecular state as H20, such combination being
able to form one or
more hydrate.
"Subject" means mammals and non-mammals. Mammals means any member of the
Mammalia
class including, but not limited to, humans; non-human primates such as
chimpanzees and other
CA 02711726 2010-07-07
WO 2009/098135 PCT/EP2009/050839
-8-
apes and monkey species; farm animals such as cattle, horses, sheep, goats,
and swine; domestic
animals such as rabbits, dogs, and cats; laboratory animals including rodents,
such as rats, mice,
and guinea pigs; and the like. Examples of non-mammals include, but are not
limited to, birds,
and the like. The term "subject" does not denote a particular age or sex.
"Therapeutically effective amount" means an amount of a compound that, when
administered to
a subject for treating a disease state, is sufficient to effect such treatment
for the disease state.
The "therapeutically effective amount" will vary depending on the compound,
disease state being
treated, the severity or the disease treated, the age and relative health of
the subject, the route and
form of administration, the judgement of the attending medical or veterinary
practitioner, and
other factors.
"Pharmacological effect" as used herein encompasses effects produced in the
subject that achieve
the intended purpose of a therapy. For example, a pharmacological effect would
be one that
results in the prevention, alleviation or reduction of urinary incontinence in
a treated subject.
"Disease state" means any disease, condition, symptom, or indication.
"Treating" or "treatment" of a disease state includes:
(1) preventing the disease state, i.e. causing the clinical symptoms of the
disease state not to
develop in a subject that may be exposed to or predisposed to the disease
state, but does not yet
experience or display symptoms of the disease state;
(2) inhibiting the disease state, i.e., arresting the development of the
disease state or its
clinical symptoms; or
(3) relieving the disease state, i.e., causing temporary or permanent
regression of the disease
state or its clinical symptoms.
"ai-adrenergic receptors", "a1A-adrenergic receptors" (previously known as
"air-adrenergic re-
ceptors"), "aiL-adrenergic receptors", or "a1A/iL-adrenergic receptors", which
may be used inter-
changeably with "ai-adrenoceptors", "aiA-adrenoceptors" (previously known as
"aic-adrenocep-
tors receptors"), "aiL-adrenoceptors" or "a1A/1L-adrenoceptors" respectively,
refers to a molecule
conforming to the seven membrane-spanning G-protein receptors, which under
physiologic con-
ditions mediate various actions, e.g., in the central and/or peripheral
sympathetic nervous system
through the binding of the catecholamines, epinephrine and norepinephrine.
CA 02711726 2010-07-07
WO 2009/098135 PCT/EP2009/050839
-9-
"Agonist" or "full agonist" means a molecule, such as a compound, a drug, an
enzyme activator,
or a hormone, that enhances the activity of another molecule or receptor site.
"Partial agonist" means activates a receptor, but only produces a partial
physiological response
compared to a full agonist.
"Urinary Incontinence" is a condition characterized by the involuntary loss of
urine, which is
objectively demonstrable. It is both a social and hygienic problem. Stated
simply, incontinence
results from the failure of the bladder and/or the urethra to work properly,
or when the coordina-
tion of their functions is defective. While the prevalence of incontinence is
two-fold higher in
females, with the greatest incidence in postmenopausal women, it also affects
males.
Urinary incontinence can be classified into four basic types: urge, stress,
overflow and functional,
and as used herein the term "urinary incontinence" encompasses all four types.
Urge incontinence (detrusor instability) is the involuntary loss of urine
associated with a strong
urge to void. This type of incontinence is the result of either an overactive
or hypersensitive
detrusor muscle. The patient with detrusor overactivity experiences
inappropriate detrusor
contractions and increases in intravesical pressure during bladder filling.
Detrusor instability
resulting from a hypersensitive detrusor (detrusor hyperreflexia) is most
often associated with a
neurological disorder.
Genuine stress incontinence (outlet incompetence) is the involuntary loss of
urine occurring
when increases in intra-abdominal pressure cause a rise in intravesical
pressure which exceeds
the resistance offered by urethral closure mechanisms. Stress incontinent
episodes can result
from normal activities such as laughing, coughing, sneezing, exercise, or, in
severe stress in-
continent patients, standing or walking. Physiologically, stress incontinence
is often character-
rized by a descensus of the bladder neck and funneling of the bladder outlet.
This type of in-
continence is most common in multiparous women, as pregnancy and vaginal
delivery can cause
loss of the vesicourethral angle and damage to the external sphincter.
Hormonal changes
associated with menopause may exacerbate this condition.
Overflow incontinence is an involuntary loss of urine resulting from a weak
detrusor or from the
failure of the detrusor to transmit appropriate signals (sensory) when the
bladder is full. Over-
flow incontinent episodes are characterized by frequent or continuous
dribbling of urine and
incomplete or unsuccessful voiding.
CA 02711726 2010-07-07
WO 2009/098135 PCT/EP2009/050839
-10-
Functional incontinence, in contrast to the types of incontinence described
above, is not defined
by an underlying physiological dysfunction in the bladder or urethra. This
type of incontinence
includes the involuntary loss of urine resulting from such factors as
decreased mobility, medica-
tions (e.g., diuretics, muscarinic agents, or alpha-1 adrenoceptor
antagonists), or psychiatric
problems such as depression or cognitive impairment.
"A method of treating or preventing incontinence" refers to the prevention of
or relief from the
symptoms of incontinence including involuntary voiding of feces or urine, and
dribbling or
leakage of feces or urine which may be due to one or more causes including,
but not limited to,
pathology altering sphincter control, loss of cognitive function,
overdistention of the bladder,
hyper- reflexia and/or involuntary urethral relaxation, weakness of the
muscles associated with
the bladder, or neurologic abnormalities.
In general, the nomenclature used in this Application is based on AUTONOMTM
v.4.0, a
Beilstein Institute computerized system for the generation of IUPAC systematic
nomenclature.
Chemical structures shown herein were prepared using ISIS version 2.4. Any
open valency
appearing on a carbon, oxygen, sulfur or nitrogen atom in the structures
herein indicates the
presence of a hydrogen atom. Whenever a chiral carbon is present in a chemical
structure, it is
intended that all stereoisomers associated with that chiral carbon are
encompassed by the
structure. Whenever a chemical structure shown herein can exist in a different
tautomeric form,
it is intended that the structure encompasses such different tautomeric forms.
EXAMPLES
The following preparations and examples are given to enable those skilled in
the art to more
clearly understand and to practice the present invention. They should not be
considered as
limiting the scope of the invention, but merely as being illustrative and
representative thereof.
The compound of the present invention may be made by the methods depicted in
the illustrative
synthetic reaction schemes shown and described below.
The starting materials and reagents used in preparing Formula I generally are
either available
from commercial suppliers, such as Aldrich Chemical Co., or are prepared by
methods known to
those skilled in the art following procedures set forth in standard
references. Where necessary,
conventional protecting group techniques were used as described by Greene et
al., Protecting
Groups in Organic Synthesis, 3rd Ed., Wiley Interscience, 1999. The following
synthetic re-
CA 02711726 2010-07-07
WO 2009/098135 PCT/EP2009/050839
-11-
action schemes are merely illustrative of some methods by which the compound
of the present
invention may be synthesized, and various modifications to these synthetic
reaction schemes
may be made and will be suggested to one skilled in the art having referred to
the disclosure
contained in this Application.
The starting materials and the intermediates of the synthetic reaction schemes
may be isolated
and purified if desired using conventional techniques, including but not
limited to filtration,
distillation, crystallization, chromatography, and the like. Such materials
may be characterized
using conventional means, including physical constants and spectral data.
Unless specified to the contrary, the reactions described herein preferably
take place at atom-
spheric pressure over a temperature range from about -78 C to about 150 C,
more preferably
from about 0 C to about 125 C, and most preferably and conveniently at about
room (or
ambient) temperature (RT), e.g., about 20 C.
Synthetic Scheme
F F N
F DBDMH BNC,,C02tBu Br F
SOa NaOH / McCN Br
NO2 NO2 NO2
N N
MSCI /
SnCI2- 2H20 Br F Et3N Br L F EDA / CS2
EtOH
Br Br
NH2 N(S02Me)2
NH /-NH HCI
N N
Br F HCI MeOH Br F
I I
Br Br
NHSO2Me NHSO2Me
A. Preparation of 2,3-Dibromo-4,5-difluoro-l-nitro-benzene (1)
F F
F DBDMH Br F
H2SO4 Br
NO2 NO2
CA 02711726 2010-07-07
WO 2009/098135 PCT/EP2009/050839
-12-
1,2-Difluoro-4-nitro-benzene (16g) was dissolved in sulfuric acid (IOOmL). To
this solution was
added 1,3-dibromo-5,5-dimethyl-imidazolidine-2,4-dione (30.2g). As the
reaction mixture began
to warm, it was cooled in ice and wrapped in aluminum foil. Stirring was
continued overnight.
The reaction mixture was poured onto a mixture of ice and ethyl acetate and
the layers were
separated. The water layer was extracted once with ethyl acetate and the
combined organic ex-
tracts were washed with sodium bisulfite solution, dried, and solvent removed
in vacuo. The
resulting orange semisolid (33.61g) was filtered through a thick plug of
silica gel in dichloro-
methane to remove a large amount of colored impurity. The resulting light
yellow oil (32.6g) ran
as a single spot on tlc (1:9 ethyl acetate - hexane) but nmr analysis revealed
it to be a mixture of
the title compound mixed with other components. No further purification was
carried out at this
point.
B. Preparation of (2,3-Dibromo-6-fluoro-4-nitro-phenyl)-acetonitrile (2)
F N
BrI F NCB.CO2t-Bu Br F
Br NaOH / MeCN Br
N O2 N O2
32g of a mixture containing 2,3-dibromo-4,5-difluoro-l-nitro -benzene was
dissolved in aceto-
nitrile (500mL) and t-butyl cyanoacetate (19g) was added. Sodium hydroxide
(15.6g in small
pearls) was added and the mixture was vigorously stirred overnight. The
solvent was removed in
vacuo and the residue was partitioned between ethyl acetate and dilute
hydrochloric acid. The
layers were separated and the organic layer was washed with saturated sodium
chloride solution.
The solution was dried and solvent removed in vacuo to afford 51g of an oil.
The crude material
was partially purified on a silica gel column to remove more polar material.
The major less polar
component was heated in vacuo with stirring at 150 C for about an hour after
which time, the
bubbling had stopped. The crude material was purified by chromatography on
silica gel (hexane
- ethyl acetate) to afford 24g of an oil which contained the title compound as
a major component.
No further purification was attempted at this point.
C. Preparation of (4-Amino-2,3-dibromo-6-fluoro-phenyl)-acetonitrile (3)
CA 02711726 2010-07-07
WO 2009/098135 PCT/EP2009/050839
-13-
NI~rl N
Br F SnC12- 2H20 Br F
Br ( EtOH
Br
NO2 NH2
Twenty-four grams of a mixture containing (2,3-dibromo-6-fluoro-4-nitro-
phenyl)-acetonitrile
was dissolved in ethanol (500mL) and tin (II) chloride dihydrate (85g) was
added. The mixture
was stirred and heated at 750 C for 5h. lOg more tin chloride was added and
the mixture was
heated at reflux for 30 additional minutes. The solution was cooled and most
of the ethanol was
removed in vacuo. The reaction mixture was diluted to a volume of 600mL with
ethyl acetate
and lOOmL water was added. With stirring, sodium bicarbonate (75g) was slowly
added. The
ethyl acetate layer was separated and the aqueous layer extracted a second
time with ethyl
acetate. The combined organic extracts were dried and the solvent removed in
vacuo to afford
22.8g of crude product. The title compound (3.8g) was obtained from the crude
mixture by
column chromatography (hexane - ethyl acetate; then hexane - methylene
chloride mixtures).
D. Preparation of (4-(bis-methylsulfonyl)amino--2,3-dibromo-6-fluoro-phenyl)-
acetonitrile
(4)
N N
Br F MsCI / Et3N Br F
Br Br
NH2 N(S02Me)2
(4-Amino-2,3-dibromo-6-fluoro-phenyl)-acetonitrile (3.61 g) was dissolved in
dichloromethane
(125 ml) and triethyl amine (5.OmL) was added. The solution was cooled in ice
and a solution of
methanesulfonyl chloride (2.30 mL) in dichloromethane (25 mL) was added
dropwise with
stirring under nitrogen. The ice bath was removed and the reaction mixture was
allowed to reach
room temperature and stirred overnight. The reaction mixture was diluted with
water and the
organic layer separated, dried and evaporated to dryness. The crude residue
was purified by
column chromatography. Elution with ethyl acetate - hexane mixtures afforded
2.7g of the title
compound.
E. Preparation of N-[2,3-Dibromo-4-(4,5-dihydro-lH-imidazol-2-ylmethyl)-5-
fluoro-
phenyl]-methanesulfonamide hydrochloride (hydrochloride salt of Formula I)
CA 02711726 2010-07-07
WO 2009/098135 PCT/EP2009/050839
-14-
NH HCI
N N
Br F 1. EDA / CS2 Br F
2. HCI
Br; Br
N(SO2Me)2 NHSO2Me
(4-(bis-methylsulfonyl)amino-2,3-dibromo-6-fluoro-phenyl)-acetonitrile (1.91g)
was dissolved
in anhydrous ethylene diamine (20mL). Following the initial exotherm, 1 drop
of carbon di-
sulfide was added. The clear solution was flushed with argon and sealed with a
Teflon-lined cap.
The reaction vessel was placed in an oil bath preheated to 130 C and stirred
at that temperature
for 2h. The crude reaction mixture was concentrated and purified on silica
gel, eluting first with
ethyl acetate followed by a mixture of dichloromethane-methanol-ammonium
hydroxide.
Further purification by preparative tlc afforded the pure free base which was
converted to its
hydrochloride salt (0.276 g) by treatment with HCl in ether (MP 182.3 - 183.5;
MS [M+H]+
428/430/432).
The compound of the present invention have selective alpha-lA adrenergic
selective activity and
as such are expected to be useful in the treatment of various disease states,
such as urinary in-
continence; nasal congestion; sexual dysfunction, such as ejaculation
disorders and priapism;
CNS disorders such as depression, anxiety, dementia, senility, Alzheimer's,
deficiencies in
attentiveness and cognition, and eating disorders such as obesity, bulimia,
and anorexia.
Urinary incontinence (UI) is a condition defined as the involuntary loss of
urine to such an extent
as to become a hygienic or social concern to the patient. Involuntary loss of
urine occurs when
pressure inside the bladder exceeds retentive pressure of the urethral
sphincters (intraurethral
pressure). Four major types of urinary incontinence have been defined based on
symptoms,
signs and condition: stress, urge, overflow and functional incontinence.
Stress urinary incontinence (SUI) is the involuntary loss of urine during
coughing, sneezing,
laughing, or other physical activities. The present methods to treat SUI
include physiotherapy
and surgery. Treatment with pharmaceutical agents is limited to the use of non
selective-
adrenergic agonists like phenylproanolamine and midodrine. The rationale for
the use of
adrenergic agonists for the treatment of SUI is based on physiological data
indicating an
abundant noradrenergic input to smooth muscle of the urethra.
CA 02711726 2010-07-07
WO 2009/098135 PCT/EP2009/050839
-15-
Urge incontinence (detrusor instability) is the involuntary loss of urine
associated with a strong
urge to void. This type of incontinence is the result of either an overactive
or hypersensitive
detrusor muscle. The patient with detrusor overactivity experiences
inappropriate detrusor
contractions and increases in intravesical pressure during bladder filling.
Detrusor instability
resulting from a hypersensitive detrusor (detrusor hyperreflexia) is most
often associated with a
neurological disorder.
Overflow incontinence is an involuntary loss of urine resulting from a weak
detrusor or from the
failure of the detrusor to transmit appropriate signals (sensory) when the
bladder is full. Over-
flow incontinent episodes are characterized by frequent or continuous
dribbling of urine and in-
complete or unsuccessful voiding.
Functional incontinence, in contrast to the types of incontinence described
above, is not defined
by an underlying physiological dysfunction in the bladder or urethra. This
type of incontinence
includes the involuntary loss of urine resulting from such factors as
decreased mobility, medica-
tions (e.g., diuretics, muscarinic agents, or alpha-1 adrenoceptor
antagonists), or psychiatric
problems such as depression or cognitive impairment.
The compound of this invention are also particularly useful for the treatment
of nasal congestion
associated with allergies, colds, and other nasal disorders, as well as the
sequelae of congestion
of the mucous membranes (e.g., sinusitis and otitis media). with less or no
undesired side effects.
These and other therapeutic uses are described, e.g., in Goodman & Gilman's,
The
Pharmacological Basis of Therapeutics, ninth edition, McGraw-Hill, New York,
1996, Chapter
26:601-616; and Coleman, Pharmacological Reviews, 1994, 46:205-229.
General Strategy for Testing Alpha- lA adrenoceptor Partial Agonists:
In general, IUP is the intraurethral pressure and is measured as the 2 minute
mean from the first
peak of the urethral response (Figure 1). MAP is the mean arterial blood
pressure and is
measured as the average blood pressure during the 2 minute section where IUP
is measured. The
durability is the slope of the IUP response in mmHg/min and is calculated
immediately after the
2 minute IUP response for 5 minutes (2-7 minutes post the first peak) for the
top 3 doses.
CONSCIOUS PIG Model
CA 02711726 2010-07-07
WO 2009/098135 PCT/EP2009/050839
-16-
Sling Training: Female Yucatan Micro-Swine were trained to stay in a sling for
up to 4 hours.
Swine were exposed to progressively longer sling durations per IACUC sling-
training guideline.
Swine were chosen for surgical instrumentation only if they exhibited
acceptable tolerance for
sling exposure.
Surgical Instrumentation: Female Yucatan Micro-swine were instrumented with a
telemetry
device with both pressure and ECG monitoring capabilities (Data Sciences
International St Paul
MN). In addition, a Bardport low profile titanium VAP was placed
subcutanteously for blood
sampling. All devices were implanted by the surgical veterinarian at Roche
Palo Alto. Briefly,
the telemetry probe body was placed subcutaneously in the cervical region. The
intra-arterial
pressure catheter was advanced via the superficial cervical artery into the
subclavian artery for
blood pressure measurement. The ECG leads were placed intra-muscularly: one in
the inter-
costal muscle between the T8-T10 regions on the left side, and the other in
the intercostal
muscles between the T1-T3 regions on the right side. The VAP was placed
subcutaneously in
the neck region, with the catheter advanced into the jugular vein. Swine were
allowed to fully
recover from the surgery (typically 10 days).
Pre-Experiment: On the day of the study, the swine were anesthetized to effect
with Iso-
flurane/02 in the animal colony. A catheter was placed into an ear vein and
the swine sedated
with approximately 2mg/kg, po bolus ofpropofol as isoflurane/02 was
discontinued. The swine
was then transported to the study room and placed on intravenous propofol
infusion
(-12mg/kg/hr, iv). The vulva and surrounding area were aseptically prepared
and a sterile 8-Fr
4-sensor solid state pressure transducer (Unisensor, Roterdam, Switzerland)
was inserted through
the external urethral meatus into the bladder. Placement of the catheter was
confirmed via
urethral profilometry (the third most distal transducer was placed at the high
pressure zone of the
urethra). The catheter was anchored into place by sutures placed in the skin
surrounding the
vulva and attached to tape fixed to the catheter. After aseptic preparation of
the VAP site a huber
needle with tubing assembly was placed in the VAP for serial blood sampling.
Propofol infusion
was ceased and the swine was allowed to wake up.
Experiment: After the swine was fully awake and stable (usually approximately
1 hour post
wake up), baseline blood pressure, IUP, and heart rate parameters were
determined. Formula I
or vehicle (0.9 % saline) was infused via the ear vein at 1 ml/min via an
infusion pump for two
hours. Blood samples were taken at 5, 15, 30, 45, 60, 75, 90, 105, and 120
minutes post infusion
initialization (Figure 2a). The pig was offered food and water during the
experiment.
CA 02711726 2010-07-07
WO 2009/098135 PCT/EP2009/050839
-17-
Data Generating and Analysis Systems: Cardiovascular readings were generated
by the
TL11M3-D70-PCP (Data Sciences International, St. Paul, MN) telemetry device
and associated
hardware. The device, when activated, transmits its signal to a receiver which
forwards this
signal to the data exchange matrix. The data exchange matrix then sends its
signal stream to the
Data-Quest ART Gold version 4.0 which processes and generates cardiovascular
data. IUP was
monitored by a solid state catheter connected to a TA6000 Polygraph (Data
Sciences
International, St. Paul, MN). An analogue signal from the TA6000 was sent to
either a Gould
Acquisition Interface or Power Lab data acquisition system and this data was
processed by either
the Ponema software version 3.2 (Data Sciences International, St. Paul, MN) or
Power Lab Chart
version 5Ø2 (ADlnstruments, Colorado Springs, CO).
Measurements: Baseline included a two-minute sampling period just prior to
infusion for IUP,
MAP, and HR. Post-dose time points included two-minute sampling periods at
approximately 5,
15, 30, 45, 60, 75, 90, 105, and 120 minutes post-infusion initialization for
IUP, MAP, and HR.
Typically, post-dose time points were sampled for two minutes leading up to
the blood sample
(e.g. 58-60 min post dose). If pig activity caused aberrant data points
(typically a marked
increase in HR) the two minute sampling period was moved either a few minutes
before or after
active period. Data was considered invalid for a sampling period and not
reported if the
following was observed: 1) Persistent activity during a specific time point
which lead to a
sustained increase in HR and/ or 2) swine defecation which led to a
substantial change in IUP.
Change from baseline was calculated as post-dose value- pre-dose value.
The compound of Formula I has been found to exhibit unexpectedly enhanced
selectivity, for
enhancement of intraurethral pressure (IUP) over blood pressure (MAP), as a
partial agonist of
alpha-IA adrenoceptors. The combination of the dibromo substituents on the 2-
and 3-positions
with the fluoro substituent at the 5-position of the phenyl ring provide
unexpected advantages
over the general class of imidazolinylmethyl aryl sulfonamides in that it has
both a favorable
intrinsic activity, or efficacy, as a partial agonist, which is ideally
between 0.35 to 0.60, of 0.38
and an affinity, or pEC50 value, of 6.7. As full agonist activity is
undesirable due to hyper-
tension related side effects, the combination of high affinity and partial
agonist behavior is
critical for optimization of urethral activity benefits associated with
effective modulation of
alpha-lA adrenoceptors coupled with minimization of diastolic blood pressure
related side
effects. Furthermore, the compound of Formula I, in comparison to analogue
compounds, ex-
CA 02711726 2010-07-07
WO 2009/098135 PCT/EP2009/050839
-18-
hibits improved durability of IUP response over time which is necessary for
effective treatment
of incontinence.
The compound of formula I tested in the conscious pig model exhibited
unexpectedly increased
durability of IUP response over time (Figure lb) compared to an analogue
compound, differing
from the compound of formula I with the absence of the 3-bromo and 5-flouro
substituents and
substitution of a 2-chloro for the 2-bromo on the phenyl ring (Figure 1 c).
Additionally, the
compound of formula I has selectivity for enhancement of intraurethral
pressure (IUP) over
blood pressure (MAP). The compound of formula I also has a correspondingly low
maximum
arterial blood pressure effect. These characteristics, in combination,
contribute to render the
compound of formula I a remarkably superior pharmaceutical candidate over
structurally similar
analogues, both selectively, for enhancement of intraurethral pressure (IUP)
over blood pressure
(MAP), and effectively over time, as an alpha-1 A partial agonist for the
treatment of
incontinence.
The present invention includes pharmaceutical compositions comprising the
compound of the
present invention, or an individual isomer, racemic or non-racemic mixture of
isomers or a
pharmaceutically acceptable salt or solvate thereof, together with at least
one pharmaceutically
acceptable carrier, and optionally other therapeutic and/or prophylactic
ingredients.
In general, the compound of the present invention will be administered in a
therapeutically
effective amount by any of the accepted modes of administration for agents
that serve similar
utilities. Suitable dosage ranges are typically 1-500 mg daily, preferably 1-
100 mg daily, and
most preferably 1-30 mg, depending upon numerous factors such as the severity
of the disease to
be treated, the age and relative health of the subject, the potency of the
compound, the route and
form of administration, the indication towards which the administration is
directed, and the pre-
ferences and experience of the medical practitioner involved. One of ordinary
skill in the art of
treating such diseases will be able, without undue experimentation and in
reliance upon personal
knowledge and the disclosure of this Application, to ascertain a
therapeutically effective amount
of the compound of the present invention for a given disease.
In general, the compound of the present invention will be administered as
pharmaceutical
formulations including those suitable for oral (including buccal and sub-
lingual), rectal, nasal,
topical, pulmonary, vaginal, or parenteral (including intramuscular,
intraarterial, intrathecal,
subcutaneous and intravenous) administration or in a form suitable for
administration by
CA 02711726 2010-07-07
WO 2009/098135 PCT/EP2009/050839
-19-
inhalation or insufflation. The preferred manner of administration is
generally oral using a
convenient daily dosage regimen which can be adjusted according to the degree
of affliction.
The compound of the present invention, together with one or more conventional
adjuvants,
carriers, or diluents, may be placed into the form of pharmaceutical
compositions and unit
dosages. The pharmaceutical compositions and unit dosage forms may be
comprised of con-
ventional ingredients in conventional proportions, with or without additional
active compounds
or principles, and the unit dosage forms may contain any suitable effective
amount of the active
ingredient commensurate with the intended daily dosage range to be employed.
The pharmaceu-
tical compositions may be employed as solids, such as tablets or filled
capsules, semisolids,
powders, sustained release formulations, or liquids such as solutions,
suspensions, emulsions,
elixirs, or filled capsules for oral use; or in the form of suppositories for
rectal or vaginal ad-
ministration; or in the form of sterile injectable solutions for parenteral
use. Formulations con-
taining about one (1) milligram of active ingredient or, more broadly, about
0.01 to about one
hundred (100) milligrams, per tablet, are accordingly suitable representative
unit dosage forms.
The compound of the present invention may be formulated in a wide variety of
oral administra-
tion dosage forms. The pharmaceutical compositions and dosage forms may
comprise the com-
pound the present invention or pharmaceutically acceptable salts thereof as
the active component.
The pharmaceutically acceptable carriers may be either solid or liquid. Solid
form preparations
include powders, tablets, pills, capsules, cachets, suppositories, and
dispersible granules. A solid
carrier may be one or more substances which may also act as diluents,
flavoring agents, solubi-
lizers, lubricants, suspending agents, binders, preservatives, tablet
disintegrating agents, or an
encapsulating material. In powders, the carrier generally is a finely divided
solid which is a mix-
ture with the finely divided active component. In tablets, the active
component generally is
mixed with the carrier having the necessary binding capacity in suitable
proportions and com-
pacted in the shape and size desired. The powders and tablets preferably
contain from about one
(1) to about seventy (70) percent of the active compound. Suitable carriers
include but are not
limited to magnesium carbonate, magnesium stearate, talc, sugar, lactose,
pectin, dextrin, starch,
gelatin, tragacanth, methylcellulose, sodium carboxymethylcellulose, a low
melting wax, cocoa
butter, and the like. The term "preparation" is intended to include the
formulation of the active
compound with encapsulating material as carrier, providing a capsule in which
the active com-
ponent, with or without carriers, is surrounded by a carrier, which is in
association with it.
CA 02711726 2010-07-07
WO 2009/098135 PCT/EP2009/050839
-20-
Similarly, cachets and lozenges are included. Tablets, powders, capsules,
pills, cachets, and
lozenges may be as solid forms suitable for oral administration.
Other forms suitable for oral administration include liquid form preparations
including emulsions,
syrups, elixirs, aqueous solutions, aqueous suspensions, or solid form
preparations which are
intended to be converted shortly before use to liquid form preparations.
Emulsions may be
prepared in solutions, e.g., in aqueous propylene glycol solutions or may
contain emulsifying
agents, e.g., such as lecithin, sorbitan monooleate, or acacia. Aqueous
solutions can be prepared
by dissolving the active component in water and adding suitable colorants,
flavors, stabilizing,
and thickening agents. Aqueous suspensions can be prepared by dispersing the
finely divided
active component in water with viscous material, such as natural or synthetic
gums, resins,
methylcellulose, sodium carboxymethylcellulose, and other well known
suspending agents.
Solid form preparations include solutions, suspensions, and emulsions, and may
contain, in
addition to the active component, colorants, flavors, stabilizers, buffers,
artificial and natural
sweeteners, dispersants, thickeners, solubilizing agents, and the like.
The compound of the present invention may be formulated for parenteral
administration (e.g., by
injection, e.g. bolus injection or continuous infusion) and may be presented
in unit dose form in
ampoules, pre-filled syringes, small volume infusion or in multi-dose
containers with an added
preservative. The composition may take such form as a suspension, solution, or
emulsion in oily
or aqueous vehicles, e.g. a solution in aqueous polyethylene glycol. Examples
of oily or non-
aqueous carriers, diluents, solvents or vehicles include propylene glycol,
polyethylene glycol,
vegetable oils (e.g., olive oil), and injectable organic esters (e.g., ethyl
oleate), and may contain
formulatory agents such as preserving, wetting, emulsifying or suspending,
stabilizing and/or
dispersing agents. Alternatively, the active ingredient may be in powder form,
obtained by a-
septic isolation of sterile solid or by lyophilisation from solution for
constitution before use with
a suitable vehicle, e.g., sterile, pyrogen-free water.
The compound of the present invention may be formulated for topical
administration to the
epidermis as ointments, creams or lotions, or as a transdermal patch.
Ointments and creams may,
e.g., be formulated with an aqueous or oily base with the addition of suitable
thickening and/or
gelling agents. Lotions may be formulated with an aqueous or oily base and
will in general also
containing one or more emulsifying agents, stabilizing agents, dispersing
agents, suspending
agents, thickening agents, or coloring agents. Formulations suitable for
topical administration in
the mouth include lozenges comprising active agents in a flavored base,
usually sucrose and
CA 02711726 2010-07-07
WO 2009/098135 PCT/EP2009/050839
-21-
acacia or tragacanth; pastilles comprising the active ingredient in an inert
base such as gelatin
and glycerin or sucrose and acacia; and mouthwashes comprising the active
ingredient in a
suitable liquid carrier.
The compound of the present invention may be formulated for administration as
suppositories.
A low melting wax, such as a mixture of fatty acid glycerides or cocoa butter
is first melted and
the active component is dispersed homogeneously, e.g., by stirring. The molten
homogeneous
mixture is then poured into convenient sized molds, allowed to cool, and to
solidify.
The compound of the present invention may be formulated for vaginal
administration. Pessaries,
tampons, creams, gels, pastes, foams or sprays containing in addition to the
active ingredient
such carriers as are known in the art to be appropriate.
The compound of the present invention may be formulated for nasal
administration. The solu-
tions or suspensions are applied directly to the nasal cavity by conventional
means, e.g., with a
dropper, pipette or spray. The formulations may be provided in a single or
multidose form. In
the latter case of a dropper or pipette, this may be achieved by the patient
administering an
appropriate, predetermined volume of the solution or suspension. In the case
of a spray, this may
be achieved e.g. by means of a metering atomizing spray pump.
The compound of the present invention may be formulated for aerosol
administration, particular-
ly to the respiratory tract and including intranasal administration. The
compound will generally
have a small particle size e.g. of the order of five (5) microns or less. Such
a particle size may be
obtained by means known in the art, e.g. by micronization. The active
ingredient is provided in a
pressurized pack with a suitable propellant such as a chlorofluorocarbon
(CFC), e.g., dichlorodi-
fluoromethane, trichlorofluoromethane, or dichlorotetrafluoroethane, or carbon
dioxide or other
suitable gas. The aerosol may conveniently also contain a surfactant such as
lecithin. The dose
of drug may be controlled by a metered valve. Alternatively the active
ingredients may be pro-
vided in a form of a dry powder, e.g. a powder mix of the compound in a
suitable powder base
such as lactose, starch, starch derivatives such as hydroxypropylmethyl
cellulose and polyvinyl-
pyrrolidine (PVP). The powder carrier will form a gel in the nasal cavity. The
powder composi-
tion may be presented in unit dose form e.g. in capsules or cartridges of
e.g., gelatin or blister
packs from which the powder may be administered by means of an inhaler.
CA 02711726 2010-07-07
WO 2009/098135 PCT/EP2009/050839
-22-
When desired, formulations can be prepared with enteric coatings adapted for
sustained or con-
trolled release administration of the active ingredient. For example, the
compound of the present
invention can be formulated in transdermal or subcutaneous drug delivery
devices. These
delivery systems are advantageous when sustained release of the compound is
necessary and
when patient compliance with a treatment regimen is crucial. Compounds in
transdermal
delivery systems are frequently attached to an skin-adhesive solid support.
The compound of
interest can also be combined with a penetration enhancer, e.g., Azone (1-
dodecylaza-cyclo-
heptan-2-one). Sustained release delivery systems are inserted subcutaneously
into to the sub-
dermal layer by surgery or injection. The subdermal implants encapsulate the
compound in a
lipid soluble membrane, e.g., silicone rubber, or a biodegradable polymer,
e.g., polyactic acid.
The pharmaceutical preparations are preferably in unit dosage forms. In such
form, the prepara-
tion is subdivided into unit doses containing appropriate quantities of the
active component. The
unit dosage form can be a packaged preparation, the package containing
discrete quantities of
preparation, such as packeted tablets, capsules, and powders in vials or
ampoules. Also, the unit
dosage form can be a capsule, tablet, cachet, or lozenge itself, or it can be
the appropriate
number of any of these in packaged form.
Other suitable pharmaceutical carriers and their formulations are described in
Remington: The
Science and Practice of Pharmacy 1995, edited by Martin, Mack Publishing
Company, 19th
edition, Easton, Pennsylvania. Representative pharmaceutical formulations
containing the
compound of the present invention are described in the Examples.
The following preparations and examples are given to enable those skilled in
the art to more
clearly understand and to practice the present invention. They should not be
considered as
limiting the scope of the invention, but merely as being illustrative and
representative thereof.
Efforts have been made to ensure accuracy with respect to numbers used (e.g.,
amounts,
temperatures, etc.), but some experimental error and deviation should, of
course, be allowed for
as well as due to differences such as, e.g., in calibration, rounding of
numbers, and the like.
While the present invention has been described with reference to the specific
embodiments
thereof, it should be understood by those skilled in the art that various
changes may be made and
equivalents may be substituted without departing from the true spirit and
scope of the invention.
In addition, many modifications may be made to adapt a particular situation,
material, composi-
CA 02711726 2010-07-07
WO 2009/098135 PCT/EP2009/050839
-23-
tion of matter, process, process step or steps, to the objective spirit and
scope of the present
invention. All such modifications are intended to be within the scope of the
claims appended
hereto.