Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02730516 2011-01-11
WO 2010/007248 PCT/FR2009/000855
-1-
NOUVEAUX DERIVES TRICYCLIQUES,
LEUR PROCEDE DE PREPARATION
ET LES COMPOSITIONS PHARMACEUTIQUES QUI LES CONTIENNENT
La présente invention concerne de nouveaux dérivés tricycliques, leur procédé
de
préparation et les compositions pharmaceutiques qui les contiennent.
Les composés de la présente invention sont nouveaux et présentent des
caractéristiques
pharmacologiques très intéressantes dans le domaine de l'apoptose et de la
cancérologie.
L'apoptose, ou mort cellulaire programmée, est un processus physiologique
crucial pour le
développement embryonnaire et le maintien de l'homéostasie tissulaire.
La mort cellulaire de type apoptotique fait intervenir des changements
morphologiques, tels
que la condensation du noyau, la fragmentation de l'ADN, ainsi que des
phénomènes
biochimiques, tels que l'activation des caspases qui vont dégrader des
composants
structuraux clés de la cellule.pour induire son désassemblage et sa mort. La
régulation du
processus d'apoptose est complexe et implique l'activation ou la répression de
plusieurs
voies de signalisation intracellulaire (Cory S. et al., Nature Review Cancer,
2002, 2, 647-
656).
Des dérégulations de l'apoptose sont impliquées dans certaines pathologies.
Une apoptose
accrue est liée aux maladies neurodégénératives telles que la maladie de
Parkinson, la
maladie d'Alzheimer et l'ischémie. Inversement, des déficiences dans
l'exécution de
l'apoptose jouent un rôle important dans le développement des cancers et leur
chimiorésistance, des maladies auto-immunes, des maladies inflammatoires et
des infections
virales. Ainsi, l'échappement à l'apoptose fait partie des signatures
phénotypiques du cancer
(Hanahan D. et al., Cell 2000, 100, 57-70).
Les composés de la présente invention outre leur nouveauté, présentent des
propriétés pro-
apoptotiques permettant de les utiliser dans les pathologies impliquant un
défaut d'apoptose,
comme par exemple dans. le traitement du cancer.
CA 02730516 2011-01-11
WO 2010/007248 PCT/FR2009/000855
-2-
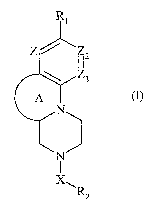
La présente invention concerne plus particulièrement les composés de formule
(I) :
R
Zl/ I 2
Z3
A m
N
N
R2
dans laquelle :
= A représente un cycle aromatique ou non contenant 5, 6 ou 7 chaînons, et
pouvant
contenir 1 ou 2 hétéroatomes choisis parmi oxygène, soufre et azote, ce
dernier
pouvant être substitué par un groupement alkyle (Cl-C6) linéaire ou ramifié,
étant entendu que le cycle A ainsi défini ne peut contenir 2 atomes de soufre
ni 2
atomes d'oxygène et que l'un des chaînons peut représenter un groupement C=O,
= Z1, Z2 et Z3 représentent indépendamment les uns des autres un groupement CH
ou
un atome d'azote, sachant qu'au moins un de ces trois groupes est un atome
d'azote,
e R2 représente un groupement aryle ou hétéroaryle,
= X représente une chaîne alkylène linéaire ou ramifiée contenant de 1 à 6
atomes de
carbone, dont un ou deux des atomes de carbone peut(vent) être remplacé(s) par
un
atome d'oxygène, un groupement cycloalkylène, un groupement arylène, un
groupement hétéroarylène ou un groupement S02,
Ri représente un groupement de formule (II) :
CA 02730516 2011-01-11
WO 2010/007248 PCT/FR2009/000855
-3-
R4
NH \
Yi X502 RS
R3
dans laquelle :
- Y représente un groupement C=O ou CH2,
- R4 représente un atome d'hydrogène et dans ce cas R5 représente un atome
d'hydrogène ou un groupement NR6R'6 ou -CH2 NR6R'6 dans lesquels R6 et R'6,
identiques ou différents, représentent chacun indépendamment un atome
d'hydrogène ou un groupement alkyle (Cl-C6) linéaire ou ramifié substitué par
un ou
plusieurs groupements aryle, hétéroaryle, aryloxy, hétéroaryloxy, arylthio,
hétéroarylthio, hétérocycloalkyle, ou NR9R'9 dans lequel :
* R9 et R'9, identiques ou différents, sont choisis parmi hydrogène, alkyle
(C1-
C6) linéaire ou ramifié, alkoxy (Cl-C6) linéaire ou ramifié, aryle et
hétéroaryle,
ou bien R9 et R'9 forment un groupement cyclique ou bicyclique, saturé ou
non, éventuellement substitué par un hétéroatome choisi parmi oxygène, azote
ou
soufre, étant entendu que un ou plusieurs des chaînons peut (peuvent)
représenter un
groupement C=O ou peut (peuvent) être substitué(s) comme le prévoit la
définition
de l'hétérocycle ci-dessous,
ou R4 et R5 forment avec les deux atomes de carbone qui les portent, un cycle
aromatique ou non contenant 5 ou 6 chaînons dont un atome d'azote en para du
groupement S02, et pouvant contenir en plus de l'atome d'azote un autre atome
d'azote
et/ou un groupement S02, le cycle ainsi défini étant substitué par un
groupement R6 tel
que défini précédemment,
- R3 représente un atome d'halogène ou un groupement NO2, R7, S02-R8, alkyle
(C1-
C6) linéaire ou ramifié, ou alkoxy (C1-C6) linéaire ou ramifié, où. R7 peut
prendre
toutes les valeurs de R6 tel que défini précédemment,
- R8 représente un groupement amino ou un groupement alkyle (Cl-C6) linéaire
ou
ramifié éventuellement substitué par un ou plusieurs atomes d'halogène,
étant entendu que :
- par "aryle", on entend un groupement phényle, naphtyle ou biphényle,
CA 02730516 2011-01-11
WO 2010/007248 PCT/FR2009/000855
-4-
- par "hétéroaryle", on entend tout groupement mono ou bi-cyclique, possédant
au
moins une partie aromatique, et contenant 5 à 10 chaînons et pouvant contenir
1 à 3
hétéroatomes choisis parmi oxygène, soufre ou azote comme les groupements
furane, thiophène, pyrrole, imidazoline, pyridine, quinoléine, isoquinoléine,
chromane, indole, benzothiophène, benzofurane, 1,3-benzodioxole et 2,3-dihydro-
1,4-benzodioxine,
- par "hétérocycloalkyle", on entend tout groupement non aromatique mono ou bi-
cyclique contenant 4 à 10 chaînons et pouvant contenir 1 à 3 hétéroatomes
choisis
parmi oxygène, soufre ou azote,
- par "cycloalkyle", on entend tout groupement non aromatique mono ou bi-
cyclique
contenant 4 à 10 chaînons,
les groupements aryle, hétéroaryle, hétérocycloalkyle et cycloalkyle ainsi
définis pouvant
être substitués par 1 à 3 groupements choisis parmi alkyle (Cl-C6) linéaire ou
ramifié
éventuellement substitué par un groupement hydroxy ou amino, alkoxy (Cl-C6)
linéaire ou
ramifié, hydroxy, carboxy, formyle, nitro, cyano, amino, polyhalogénoalkyle
(C1-C6)
linéaire ou ramifié, alkyloxycarbonyle, ou atomes d'halogène,
- par "arylène", "hétéroarylène"et "cycloalkylène", on entend un groupement
aryle,
hétéroaryle et cycloalkyle respectivement tels que définis précédemment,
insérés à la
place d'un atome de carbone de la chaîne alkylène,
leurs énantiomères et diastéréoisomères, ainsi que leurs sels d'addition à un
acide ou à une
base pharmaceutiquement acceptable.
Parmi les acides pharmaceutiquement acceptables, on peut citer à titre non
limitatif les
acides chlorhydrique, bromhydrique, sulfurique, phosphonique, acétique,
trifluoroacétique,
lactique, pyruvique, inalonique, succinique, glutarique, fumarique, tartrique,
maléique,
citrique, ascorbique, oxalique, méthane sulfonique, camphorique, etc...
Parmi les bases pharmaceutiquement acceptables, on peut citer à titre non
limitatif
l'hydroxyde de sodium, l'hydroxyde de potassium, la triéthylamine, la
tertbutylamine, etc...
CA 02730516 2011-01-11
WO 2010/007248 PCT/FR2009/000855
-5-
Y représente avantageusement un groupement C=O.
Le groupement R3 préféré est NO2.
Les groupements X-R2 préférés sont les groupements ([1,1'-biphényl]-2-
yl)méthyl
éventuellement substitués par un ou plusieurs atomes d'halogène.
R4 représente de façon préférentielle un atome d'hydrogène.
Le groupement R6 préféré est le groupement 1-(N,N-diméthylamino)-4-
(phénylsulfanyl)-
butan-3-yle.
R'6 représente avantageusement un atome d'hydrogène.
Encore plus particulièrement, l'invention concerne les composés de formule (I)
qui sont :
= le N-({2-[(4'-chloro[1,1'-biphényl]-2-yl)méthyl]-l,2,3,4,10,10a-
hexahydropyrido
[4',3':4,5]pyrrolo[1,2-a]pyrazin-8-yl} carbonyl)-4-({(1R)-3-(diméthylamino)-1-
[(phénylsulfanyl)méthyl]propyl } amino)-3 -nitrobenzènesulfonamide,
= le N-({8-[(4'-chloro[1,1'-biphényl]-2-yl)méthyl]-6,7,8,9,9a,10-
hexahydropyrido
[2',3' :4, 5 ] pyrrolo [ 1,2-a] pyrazin-2-yl } carb onyl)-4- ({ (1 R)-3 -
(diméthylamino) -1-
[(phénylsulfanyl)méthyl]propyl}amino)-3-nitrobenzènesulfonamide,
= le N-({7-[(4'-cl3loro[1,1'-biphényl]-2-yl)méthyl]-5,5a,6,7,8,9-
hexahydropyrido
[3',2':4,5]pyrrolo[1,2-a]pyrazin-3-yl} carbonyl)-4-({(1R)-3-(diméthylamino)-1-
[(phénylsulfanyl)méthyl]propyl} amino)-3 -nitrobenzènesulfonamide,
= le N-({3-[(4'-chloro[1,1'-biphényl]-2-yl)méthyl]-2,3,4,4a,5,6-hexahydro-lH-
pyrazino[1,2-a] [1,5]naphthyridin-8-yl} carbonyl)-4-({(1R)-3-(diméthylamino)-1-
[(phénylsulfanyl)méthyl]propyl} amino)-3 -nitrobenzènesulfonamide,
= le N-({8-[(4'-chloro[1,1'-biphényl]-2-yl)méthyl]-6,6a,7,8,9,10-hexahydro-5H-
pyrazino[1,2-a] [1,7]naphthyridin-3-yl} carbonyl)-4-({(1R)-3-(diméthylamino)-1-
[(phénylsulfanyl)méthyl]propyl} amino)-3 -nitrobenzènesulfonamide,
= le N-({8-[(4'-chloro[1,1'-biphényl]-2-yl)méthyl]-6,6a,7,8,9,10-hexahydro-5H-
CA 02730516 2011-01-11
WO 2010/007248 PCT/FR2009/000855
-6-
pyrazino [ 1,2-a] [ 1, 8]naphthyridin-3 -yl} carbonyl)-4-({ (1 R)-3 -
(diméthylamino)-1-
[(phénylsulfanyl)méthyl]propyl } amino)-3 -nitrobenzènesulfonamide.
Les énantiomères, diastéréoisomères ainsi que les sels d'addition à un acide
ou à une base
pharmaceutiquement acceptable des composés préférés de l'invention font partie
intégrante
de l'invention.
L'invention s'étend également au procédé de préparation des composés de
formule (I)
caractérisé en ce que l'on utilise comme produit de départ le composé de
formule (III) :
CY '1_1 -_ ci (III)
dans laquelle Y est tel que défini dans la formule (I) et Cy représente un
système tricyclique
fusionné de formule (IV) :
a
Z1~Z2
Z3
A (IV)
N
1
R2
dans laquelle A, X, Zl, Z2, Z3 et R2 sont tels que définis dans la formule
(I), le groupement
-Y-CI étant rattaché en position a du système tricyclique ainsi défini,
composé de formule (III) sur lequel on condense en milieu basique en présence
ou non d'un
agent de couplage, le composé de formule (V) :
~ Cl
H2N~ I (V)
SO2 R3
dans laquelle R3 est tel que défini dans la formule (I),
CA 02730516 2011-01-11
WO 2010/007248 PCT/FR2009/000855
-7-
pour obtenir le composé de formule (VI) :
C1
I
Y S0 )
Cy~ ~
2 R3
dans laquelle Cy, Y et R3 sont tels que définis précédemment,
sur lequel on condense le composé de formule HNR6R'6 dans laquelle R6 et R'6
sont tels que
définis dans la formule (I) pour conduire au composé de formule (I/a), cas
particulier des
composés de formule (I) :
\ NR6R'6
Cy~ NH (1/a)
Y S02 R3
dans laquelle Cy, Y, R3, R6 et R'6 sont tels que définis précédemment,
qui peut être purifié selon une technique classique de séparation, que l'on
transforme, si on
le souhaite, en ses sels d'addition à un acide ou à une base
pharmaceutiquement acceptable
et dont on sépare éventuellement les isomères selon une technique classique de
séparation.
Les composés de formules (III) et (V) sont soit commerciaux, soit accessibles
à l'homme du
métier par des réactions chimiques classiques et décrites dans la littérature.
Une variante avantageuse concerne le procédé de préparation des composés de
formule (I)
caractérisé en ce que l'on utilise comme produit de départ le composé de
formule (III') :
C Y\OH (III')
dans laquelle Y est tel que défini dans la formule (I) et Cy représente un
système tricyclique
fusionné de formule (IV) :
CA 02730516 2011-01-11
WO 2010/007248 PCT/FR2009/000855
-s-
a
Z~\ 12
Z3
A (IV)
N
1
X11~RZ
dans laquelle A, X, Zl, Z2, Z3 et R2 sont tels que définis dans la formule
(I), le groupement
-Y-OH étant rattaché en position a du système tricyclique ainsi défini,
composé de formule (III') sur lequel on condense en milieu basique en présence
d'un agent
de couplage, le composé de formule (VII) :
R4
Rs
H2N-1, I (Vil)
SO2 R3
dans laquelle R3, R4 et R5 sont tels que définis dans la formule (I),
pour conduire au composé de formule (I) qui peut être purifié selon une
technique classique
de séparation, que l'on transforme, si on le souhaite, en ses sels d'addition
à un acide ou à
une base pharmaceutiquement acceptable et dont on sépare éventuellement les
isomères
selon une technique classique de séparation.
Les composés de formule (III') et (VII) sont soit commerciaux, soit
accessibles à l'homme
du métier par des réactions chimiques classiques et décrites dans la
littérature.
L'étude pharmacologique des dérivés de l'invention a montré qu'ils possédaient
des
propriétés pro-apoptotiques. La capacité à réactiver le processus apoptotique
dans les
cellules cancéreuses représente un intérêt thérapeutique majeur dans le
traitement des
CA 02730516 2011-01-11
WO 2010/007248 PCT/FR2009/000855
-9-
cancers.
Plus particulièrement, les composés selon l'invention seront utiles dans le
traitement des
cancers chimio ou radiorésistants, ainsi que dans les hémopathies malignes et
le cancer du
poumon à petites cellules.
Parmi les traitements des cancers envisagés on peut citer, sans s'y limiter,
les cancers de la
vessie, du cerveau, du sein, de l'utérus, les leucémies lymphoïdes chroniques,
les cancers du
colon, de l'eesophage, du foie, les leucémies lymphoblastiques, les lymphomes
folliculaires,
les mélanomes, les hémopathies malignes, les myélomes, les cancers de
l'ovaire, les cancers
du poumon non à petites cellules, les cancers de la prostate et les cancers du
poumon à
petites cellules.
La présente invention a également pour objet les compositions pharmaceutiques
contenant
au moins un composé de formule (I) seul ou en combinaison avec un ou plusieurs
excipients
pharmaceutiquement acceptables.
Parmi les compositions pharmaceutiques selon l'invention, on pourra citer,
plus
particulièrement celles qui conviennent pour l'administration orale,
parentérale, nasale, per
ou transcutanée, rectale, perlinguale, oculaire ou respiratoire et notamment
les comprimés
simples ou dragéifiés, les comprimés sublinguaux, les sachets, les paquets,
les gélules, les
glossettes, les tablettes, les suppositoires, les crèmes, les pommades, les
gels dermiques, et
les ampoules buvables ou injectables.
La posologie varie selon le sexe, l'âge et le poids du patient, la voie
d'administration, la
nature de l'indication thérapeutique, ou des traitements éventuellement
associés et
s'échelonne entre 0,01 mg et 1 g par 24 heures en une ou plusieurs prises.
Par ailleurs, la présente invention concerne également l'association d'un
composé de
formule (I) avec un agent anticancéreux choisi parmi les agents génotoxiques,
les poisons
mitotiques, les anti-métabolites, les inhibiteurs du protéasome, ou les
inhibiteurs de kinase,
ainsi que l'utilisation de ce type d'association pour la fabrication de
médicaments utiles
dans le traitement du cancer.
CA 02730516 2011-01-11
WO 2010/007248 PCT/FR2009/000855
-10-
Les composés de l'invention peuvent également être utilisés en association
avec une
radiothérapie dans le traitement du cancer.
Les Préparations et Exemples suivants illustrent l'invention et ne la limitent
en aucune
façon.
Préparation 1 :. Acide 2-[(4'-chloro[1,1'-biphényl]-2-yl)méthyl]-
1,2,3,4,10,10a-
hexahydropyrido [4',3' :4,5] pyrrolo [1,2-a] pyrazine-8-carboxylique,
tris(trifluoroacétate)
Stade A : 2-Méthoxy-4-méthyl-5-nitropyridine
A 45 mL d'une solution de méthanolate de sodium dans le méthanol (30 % en
masse) dans
30 mL de méthanol est ajoutée par portions en 45 minutes la 2-chloro-4-méthyl-
5-
nitropyridine (25 g). Le milieu réactionnel ainsi obtenu est agité pendant 4
heures puis
1 heure à reflux avant d'être versé sur 1 L d'eau glacé. Le milieu hétérogène
est agité
pendant une nuit, puis filtré pour conduire après séchage au produit du titre
sous la forme
d'un solide marron.
Stade B : 3-(2-Méthoxy-5-nitro-4-pyridyl)-2-oxopropanoate d'éthyle
A 30 mL d'éthanol sont ajoutés le tert-butylate de potassium (15.14 g), le
diéthyléther
(300 mL) et le diéthyloxalate (18.43 mL). Après un temps de contact de 15
minutes est
ajouté le composé du Stade A (22.69 g), puis le milieu réactionnel ainsi
obtenu est agité
pendant 4 heures à reflux et une nuit à température ambiante. Le précipité
rouge obtenu est
filtré et rincé au diéthyléther, puis dilué dans 500 mL d'eau. De l'acide
acétique glacial est
ajouté jusqu'à obtenir un pH de 4, puis le milieu réactionnel est agité à
température
ambiante pendant 2 heures, puis filtré pour coduire au produit du titre sous
la forme d'un
solide beige.
CA 02730516 2011-01-11
WO 2010/007248 PCT/FR2009/000855
-11-
Stade C : 5-Méthoxy-lH-pyrrolo[2,3-c]pyridine-2-carboxylate d'éthyle
A une solution hétérogène du composé du Stade B (27.84 g) dans 350 mL
d'éthanol est
ajouté le palladium sur charbon (5.9 g, 20 % en masse). Le mélange réactionnel
est
hydrogéné pendant 24 heures, puis filtré et concentré. Le résidu obtenu est
repris dans un
mélange éther / eau, puis le produit est extrait à l'éther. Les phases
organiques sont lavées
par une solution aqueuse saturée en carbonate de potassium, séchées sur
sulfate de
magnésium, filtrées et concentrées pour conduire au produit du titre sous la
forme d'un
solide brun.
Stade D : 1-Acétyl-5-méthoxy-2,3-dihydro-1H-pyrrolo[2,3-c]pyridine-2-
carboxylate
d'éthyle
A une solution de log du composé du Stade C dans 60 mL de dichlorométhane sont
additionnés la triéthylamine (7.57 mL), la 4-diméthylaminopyridine (550 mg),
et goutte à
goutte l'anhydride acétique (5.48 mL). Le mélange réactionnel est agité à
température
ambiante pendant 24 heures, puis dilué dans l'eau. Le produit est extrait au
dichlorométhane, puis les phases organiques sont lavées, à l'eau et à l'aide
d'une solution
aqueuse saturée en chlorure de sodium. Elles sont ensuite séchées sur sulfate
de magnésium,
filtrées et concentrées.
A une solution du ainsi résidu obtenu dans 200 m L d'éthanol est ajouté le
chlorure de
palladium (II) (798 mg,). Puis, le milieu réactionnel est hydrogéné pendant 24
heures. Après
filtration et concentration, le résidu est purifié par chromatographie flash
sur gel de silice
(heptane /acétate d'éthyle) pour conduire au produit du titre sous la forme
d'un solide blanc
cassé.
Stade E : 5-Méthoxy-2,3-dihydro-1H-pyrrolo[2,3-c]pyridine-2-carboxylate
d'éthyle
Une solution de l'azaindoline protégée du stade précédent (17.9 g) dans 75 mL
d'éthanol
chlorhydrique 1N est mise à reflux pendant 8 heures. Après retour à
température ambiante,
le milieu réactionnel est neutralisé avec une solution aqueuse saturée en
hydrogénocarbonate de sodium, puis le produit est extrait à l'acétate
d'éthyle. La phase
CA 02730516 2011-01-11
WO 2010/007248 PCT/FR2009/000855
-12-
organique est concentrée et le résidu est directement utilisé dans l'étape
suivante.
Stade F : 8-Méthoxy-2,3,10,10a-tétrahydropyrido[4',3':4,5]pyrrolo[1,2-
a]pyrazine-
1,4-dione
A une solution d'acide benzyloxycarbonylaminoacétique (17 g) dans 300 mL de
tétrahydrofurane est ajouté par portions à 0 C du PC15 (15g). Le mélange
réactionnel est
agité à cette même température pendant 2 heures, puis y est additionné goutte
à goutte une
solution du composé du Stade E dans 100 mL de tétrahydrofurane et 50 mL de
pyridine.
Après retour à température ambiante, le milieu réactionnel est agité pendant
12 heures. Le
mélange réactionnel est hydrolysé à 0 C, goutte à goutte, puis extrait à
l'acétate d'éthyle.
Les phases organiques sont lavées par une solution aqueuse saturée
d'hydrogénocarbonate
de sodium et une solution aqueuse saturée de chlorure de sodium, séchées sur
sulfate de
magnésium, filtrées et concentrées. Après filtration sur silice, le solide
obtenu est mis en
solution dans 100 mL de tétrahydrofurane et 350 mL de méthanol. Le formiate
d'ammonium (4.8 g) est ensuite additionné par portions, et le milieu
réactionnel est mis à
reflux pendant 10 heures. Après filtration et rinçage au tétrahydrofurane
chaud et au
diméthylformamide chaud,. le filtrat est concentré et repris dans le méthanol
froid pour
conduire au produit du titre sous la forme d'un solide blanc.
Stade G : 2-Eenzyl-8-méthoxy-2,3,10,10a-tétrahydropyrido[4',3':4,5]pyrrolo[1,2-
a]
pyrazine-1,4-dione
A une solution hétérogène d'hydrure de sodium (1.82 g) dans 50 mL de
diméthylformamide
est additionnée goutte à goutte à 0 C une solution du composé du stade F (7.09
g) dans
450 mL de diméthylformamide pendant une durée de 2 heures. Après un retour à
température ambiante pendant 4 heures, le bromure de benzyle (4.54 mL) est
ajouté en 25
minutes. Le milieu réactionnel est agité pendant 17 heures, puis est
concentré. Le résidu
obtenu est repris dans un mélange d'acétate d'éthyle et d'eau, et le produit
est extrait à
l'acétate d'éthyle. Les phases organiques sont lavées par une solution aqueuse
saturée de
chlorure de lithium, séchées sur sulfate de magnésium, filtrées et
concentrées. Après
CA 02730516 2011-01-11
WO 2010/007248 PCT/FR2009/000855
-13-
purification par chromatographie flash sur gel de silice (heptane /acétate
d'éthyle), le
produit du titre est obtenu sous la forme d'un solide blanc cassé.
Stade H : 2-Benzyl-8-méthoxy-1,2,3,4,10,10a-
hexahydropyrido[4',3':4,5]pyrrolo[1,2-a]
pyrazine
A une solution du composé su Stade G (7.72 g) dans 225 mL de tétrahydrofurane
et 225 mL
de toluène est additionnée goutte à goutte à 0 C en 50 minutes une solution
molaire
d'hydrure d'aluminium lithium dans le tétrahydrofurane (75 mL). Le milieu
réactionnel est
mis à reflux pendant 6 heures. Après retour à température ambiante, il est
hydrolysé goutte à
goutte à 0 C par un ajout respectif de 10 mL d'eau, 10 mL d'une solution
aqueuse de soude
à 15 % et 30 mL d'eau. Après retour à température ambiante, le mélange est
agité
vigoureusement pendant 6 heures, puis filtré. Le filtrat est concentré et
repris dans un
mélange acétate d'éthyle et une solution aqueuse saturée d'hydrogénocarbonate
de sodium.
Le produit est ensuite extrait à l'acétate d'éthyle. Après concentration des
phases
organiques, le résidu est purifié par chromatographie flash sur gel de silice
(dichlorométhane /méthanol) pour conduire au produit du titre sous la forme
d'une huile
brune.
Stade I : 2-Benzyl-1,2,3,4,10,10a-hexahydropyrido[4',3':4,5]pyrrolo[1,2-
a]pyrazin-8-
yl trifluoromethanesulfonate
Une solution du composé du Stade H (2.6 g) dans une solution à 33 % d'acide
bromhydrique dans l'acide acétique est portée à reflux pendant 3 heures. Après
retour à
température ambiante, une solution aqueuse d'hydrogénocarbonate de sodium est
additionnée, goutte à goutte, à 0 C jusqu'à un pH de S. Le produit est extrait
au
dichlorométhane puis les phases organiques sont réunies, séchées sur sulfate
de magnésium,
filtrées et concentrées. A une solution du résidu ainsi obtenu dans 70 mL de
pyridine est
additionné, goutte à goutte, à 0 C en 50 minutes l'anhydride triflique (13
mL). Le mileu
réactionnel est remonté à température ambiante en 16 heures puis concentré. Le
résidu est
repris dans un mélange d'acétate d'éthyle et d'eau. Le produit est extrait à
l'acétate
d'éthyle, puis les phases organiques sont lavées par une solution aqueuse
CA 02730516 2011-01-11
WO 2010/007248 PCT/FR2009/000855
-14
d'hydrogénocarbonate de sodium, séchées sur sulfate de magnésium, filtrées et
concentrées.
Le résidu est purifié par chromatographie flash sur gel de silice (heptane
/acétate d'éthyle)
pour conduire au produit du titre sous la forme d'une huile verte.
Stade J : 2-Benzyl-1,2,3,4,10,10a-hexahydropyrido[4',3':4,5]pyrrolo[1,2-
a]pyrazine-8-
carboxylate de méthyle
A une solution du composé triflate du stade précédent (2.3 g) dans 58 mL d'un
mélange
diméthylsufoxyde / méthanol (3/2) sont additionnés la triéthylamine (1.8 mL),
le 1,1'-
bis(diphénylphosphino)ferrocène (643 mg) et le diacétate de palladium (II)
(130.2 mg). Un
bullage d'argon pendant 15 min, puis de monoxyde de carbone, est appliqué au
mélange
réactionnel. Le ballon est ensuite mis sous pression de monoxyde de carbone.
Le mélange
réactionnel est porté à 65 C, puis agité pendant 18 heures. Après
concentration, le résidu est
repris dans un mélange acétate d'éthyle / eau. Le produit est extrait à
l'acétate d'éthyle puis
les phases organiques sont lavées par une solution aqueuse saturée en
hydrogénocarbonate
de sodium, séchées sur sulfate de magnésium, filtrées et concentrées. Après
lyophilisation
dans le dioxane, le résidu est purifié par chromatographie flash sur gel de
silice
(dichlorométhane / méthanol) pour conduire au produit du titre.
Stade K : 1,2,3,4,10,10a-Hexahydropyrido[4',3':4,5]pyrrolo[1,2-a]pyrazine-8-
carboxylate de méthyle, bischlorhydrate
A une solution du composé du stade précédent (1.49 g) dans 35 mL d'une
solution de
méthanol chlorhydrique 1N est ajouté l'hydroxyde de palladium (II) sur charbon
(300 mg,
20 % en masse). Le mélange réactionnel est hydrogéné pendant 16 heures puis
filtré et
concentré pour conduire au produit du titre sous la forme d'un solide brun.
Stade L: 2-[(4'-Chloro [1,1'-biphényl]-2-yl)méthyl]-1,2,3,4,10,10a-
hexahydropyrido
[4',3':4,5]pyrrolo[1,2-a]pyrazine-8-carboxylate de méthyle
A une solution du composé du Stade K (1.016 g) dans 15 mL de diméthylformamide
sont
additionnés la triéthylamine (1.84 mL), le 4'-chloro-2-chlorométhylbiphényle
(787 mg) et
CA 02730516 2011-01-11
WO 2010/007248 PCT/FR2009/000855
-15-
l'iodure de sodium (50 mg). Le mélange réactionnel est agité à 50 C pendant 3
heures,
concentré, puis repris dans un mélange d'acétate d'éthyle et d'une solution
aqueuse saturée
en hydrogénocarbonate de sodium. Après extraction à l'acétate d'éthyle, les
phases
organiques sont lavées par une solution saturée en chlorure de lithium,
séchées sur sulfate
de magnésium, filtrées et concentrées. Le résidu est purifié par
chromatographie flash sur
gel de silice (heptane / acétate d'éthyle) pour conduire au produit du titre
sous la forme d'un
solide blanc cassé.
Stade M: Acide 2-[(4'-chloro[1,1'-biphényl]-2-yl)méthyl]-1,2,3,4,10,10a-
hexahydro-
pyrido [4',3' :4,5] pyrrolo [1,2-a] pyrazine-8-carboxylique,
tris(trifluoroacétate)
A une suspension du composé ester du stade précédent (1.085 g) dans un mélange
dioxane /
eau (4/1) est additionnée la lithine (525 mg). Le mélange réactionnel est
agité à température
ambiante pendant 24 heures puis concentré et repris dans 20 mL d'un mélange
d'eau et
d'une solution aqueuse d'acide chlorhydrique 1N (1/1). Le milieu réactionnel
est agité
pendant 2 heures à température ambiante puis concentré. Le résidu est purifié
sur une
cartouche Oasis (eau / acétonitrile / acide trifluoroacétique) et lyophilisé
dans un mélange
d'acétonitrile et d'eau pour conduire au produit du titre.
Préparation 2 Acide 8-[(4'-chloro[1,1'-biphényl]-2-y1)méthyl]-6,7,8,9,9a,10-
hexahydro
pyrido [2',3' :4,5] pyrrolo [1,2-a] pyrazine-2-carboxylique,
trifluoroactétate
On procède comme dans les stades A à M de la Préparation 1 en remplaçant au
Stade A la
2-chloro-4-méthyl-5-nitropyridine par la 6-chloro-2-méthyl-3-nitropyridine.
Préparation 3 : Acide 7-[(4'-chloro[1,1'-biphényl]-2-yl)méthyl]-5,5a,6,7,8,9-
hexahydro
pyrido [3',2' :4,5] pyrrolo [1,2-a] pyrazine-3-carboxylique,
trifluoroactétate
CA 02730516 2011-01-11
WO 2010/007248 PCT/FR2009/000855
-16-
Stade A : 3-Méthyl-2-pyridylcarbamate de tert-butyle
A une solution de di-tert-butyldicarbonate (161.5 g) dans 400 mL d'hexane est
ajoutée en 2
heures une solution de 3-méthyl-2-aminopyridine (50 g) dans 50 mL d'acétate
d'éthyle. Le
milieu réactionnel est porté à reflux pendant 1 heure, puis dilué dans 550 mL
d'hexane et
agité à température ambiante pendant 2 heures. Le précipité est filtré pour
conduire au
produit du titre sous la forme d'un solide blanc.
Stade B : 3-{2-[(tert-Butoxycarbonyl)amino]-3-pyridyl}-2-oxopropanoate
d'éthyle
A une solution du composé du stade précédent (10 g) dans 200 mL de
tétrahydrofurane est
ajouté à une température inférieure à 5 C le n-butyllithium. Le mélange
réactionnel est
ensuite agité 1 heure et 15 minutes à 0 C avant d'être canulé à une
température inférieure à
-3 C sur une solution de l'oxalate de diéthyle (14 mL) dans 50 mL de
tétrahydrofurane.
Stade C : 1H-Pyrrolo[2,3-b]pyridine-2-carboxylate d'éthyle
Le milieu réactionnel du stade précédent est agité à température ambiante
pendant 2 heures
et 15 minutes puis versé doucement sur 30 mL d'une solution d'acide
chlorhydrique 5N à
une température n'excédant pas 10 C. La solution obtenue est mise à reflux
pendant 2
heures. Après retour à température ambiante, elle est neutralisée jusqu'à pH 3
par une
solution de soude 5N. Le produit est extrait à l'éther puis les phases
organiques sont lavées
avec une solution saturée en carbonate de potassium, séchées sur sulfate de
magnésium,
filtrées et concentrées. Le résidu est repris dans le méthanol pour conduire
au produit du
titre sous la forme d'un solide beige.
Stade D : 1-Acétyl-2,3-dihydro-lH-pyrrolo[2,3-b]pyridine-2-carboxylate
d'éthyle
La procédure décrite au Stade D de la Préparation 1 est appliquée à
l'azaindole obtenu au
stade précédent. Le produit du titre est finalement sous la forme d'un solide
vert clair.
CA 02730516 2011-01-11
WO 2010/007248 PCT/FR2009/000855
-17-
Stade E : 5,5a,7,8-Tétrahydropyrido[3',2':4,5]pyrrolo[1,2-a]pyrazine-6,9-dione
La procédure décrite au Stade E de la Préparation 1 est appliquée à
l'azaindoline obtenue au
stade précédent. Le produit du titre est finalement sous la forme d'un solide
blanc.
Stade F : 7-Benzyl-5,5a,7,8-tétrahydropyrido[3',2':4,5]pyrrolo[1,2-a]pyrazine-
6,9-
dione
On applique au composé du stade précédent la procédure décrite au stade F de
la
Préparation 1. Le produit du titre est finalement sous la forme d'un solide
jaune pâle.
Stade G : 7-Benzyl-5,5a,6,7,8,9-hexahydropyrido[3',2':4,5]pyrrolo[1,2-
alpyrazine
On applique au composé du stade précédent la procédure décrite au stade G de
la
Préparation 1. Le produit du titre est finalement sous la forme d'une huile
brune.
Stade H : 7-Benzyl-3-bromo-5,5a,6,7,8,9-hexahydropyrido[3',2':4,5]pyrrolo[1,2-
a]pyrazine
A une solution du composé du Stade G dans 50 mL de diméthylformamide est
additionnée à
0 C, goutte à goutte, une solution de N-bromosuccinimide (785 mg) dans 30 mL
de
diméthylformamide pendant 45 minutes. Le mélange réactionnel est agité à cette
même
température pendant 2.5 heures avant d'être hydrolysé. Le produit est ensuite
extrait à
l'acétate d'éthyle. Les phases organiques sont lavées avec un solution saturée
en chlorure de
lithium, séchées sur sulfate de magnésium, filtrées et concentrées. Le résidu
est purifié par
chromatographie flash sur gel de silice (heptane /acétate d'éthyle) pour
obtenir le produit du
titre sous la forme d'une huile brune.
Stade I : 7-Benzyl-5,5a,6,7,8,9-hexahydropyrido[3',2':4,5]pyrrolo[1,2-
a]pyrazine-3-
carboxylate de méthyle
CA 02730516 2011-01-11
WO 2010/007248 PCT/FR2009/000855
-18-
On applique au composé du stade précédent la procédure décrite au Stade J de
la
Préparation 1. Le produit du titre est finalement sous la forme d'une huile
brune.
Stade J : 5,5a,6,7,8,9-Hexahydropyrido[3',2':4,5]pyrrolo[1,2-a]pyrazine-3-
carboxylate
de méthyle, bischlorhydrate
On applique au composé du stade précédent la procédure décrite au Stade K de
la
Préparation 1. Le produit du titre est finalement sous la forme d'un solide
brun.
Stade K : 7-[(4'-Chloro[1,1'-biphényl]-2-yl)méthyl]-5,5a,6,7,8,9-
hexahydropyrido
[3',2':4,5]pyrrolo[1,2-a]pyrazine-3-carboxylate de méthyle
On applique au composé du stade précédent la procédure décrite au Stade L de
la
Préparation 1. Le produit du titre est finalement sous la forme d'un solide
blanc.
Stade L : Acide 7-[(4'-chloro[1,1'-biphényl]-2-yl)méthyl]-5,5a,6,7,8,9-
hexahydropyrido[3',2':4,5]pyrrolo[1,2-a]pyrazine-3-carboxylique,
trifluoroactétate
On applique au composé du stade précédent la procédure décrite au Stade M de
la
Préparation 1. Le produit du titre est finalement sous la forme d'un solide
blanc.
Préparation 4 :.Acide 3-[(4'-chloro[1,1'-biphényl]-2-yl)méthyl]-2,3,4,4a,5,6-
hexahydro-
1I- pyrazino[1,2-a][1,5]naphthyridine-8-carboxylique, trifluoroacétate
On reprend les opérations décrites aux Stades F à M de la Préparation 1 en
remplaçant au
Stade F le composé du Stade E par le 6-méthoxy-1,2,3,4-
tétrahydro[1,5]naphthyridine-2-
carboxylate de méthyle.
Préparation 5 : Acide 8-[(4'-chloro[1,1'-biphényl]-2-y1)méthyl]-6,6a,7,8,9,10-
hexahydro-SH-pyrazino[1,2-a] [1,7]naphthyridine-3-carboxylique,
trifluoroacétate
On reprend les opérations décrites aux Stades F à M de la Préparation 1 en
remplaçant au
CA 02730516 2011-01-11
WO 2010/007248 PCT/FR2009/000855
-19-
Stade F le composé du Stade E par le 6-méthoxy-1,2,3,4-
tétrahydro[1,7]naphthyridine-2-
carboxylate de méthyle.
Préparation 6 : Acide 8-[(4'-chloro[1,1'-biphényl]-2-yl)méthyl]-6,6a,7,8,9,10-
hexahydro-5H-pyrazino [1,2-a] [1,8] naphthyridine-3-carboxylique,
trifluoroacétate
On reprend les opérations décrites aux Stades F à M de la Préparation 1 en
remplaçant au
Stade F le composé du Stade E par le 6-méthoxy-1,2,3,4-
tétrahydro[1,8]naphthyridine-2-
carboxylate de méthyle.
CA 02730516 2011-01-11
WO 2010/007248 PCT/FR2009/000855
-20-
Exemple 1: N-({2-[(4'-Chloro[1,1'-biphényl]-2-y1)méthyl]-1,2,3,4,10,10a-
hexahydropyrido [4',3':4,5]pyrrolo [1,2-a]pyrazin-8-yl}carbonyl)-4-({(1R)-
3-(diméthylamino)-1-[(phénylsulfanyl)méthyl] propyl}amino)-3-
nitrobenzènesulfonamide, trichlorhydrate
Stade A : N-({2-[(4'-Chloro[1,1'-biphényl]-2-yl)méthyl]-1,2,3,4,10,10a-
hexahydropyrido [4',3' :4,5] pyrrolo [1,2-a]pyrazin-8-yl}carbonyl)-4-({(1R)-3-
(diméthylamino)-1-[(phénylsulfanyl)méthyl]propyl}amino)-3-
nitrobenzènesulfonamide
A une solution du composé de la Préparation 1 (346 mg) dans 10 mL de
dichlorométhane
sont additionnés la diisopropyléthylamine (300 L), le 4-({(1R)-3-
(diméthylamino)-1-
[(phénylsulfanyl)méthyl]propyl}amino)-3-nitrobenzènesulfonamide (191 mg), la N-
éthyl-
N'-3-diméthylaminopropylcarbodiimine (121 mg) et la 4-diméthylaminopyridine
(77 mg,
0.63 mmol, 1.4 éq.). Le mélange réactionnel est agité à température ambiante
pendant 6
jours, puis hydrolysé par une solution aqueuse saturée en chlorure d'ammonium.
Le produit
est extrait à l'acétate d'éthyle. Les phases organiques sont séchées sur
sulfate de
magnésium, filtrées et concentrées. Le résidu est purifié par chromatographie
flash sur gel
de silice (dichlorométhane / méthanol ammoniacale) pour conduire au produit du
titre sous
la forme d'un solide jaune pâle.
Stade B : N-({2-[(4'-Chloro[1,1'-biphényl]-2-yl)méthyl]-1,2,3,4,10,10a-
hexahydropyrido [4',3':4,5]pyrrolo [1,2-a]pyrazin-8-yl}carbonyl)-4-({(1R)-3-
(diméthylamino)-1- [(phénylsulfanyl)méthyl]propyl} amino)-3-
nitrobenzènesulfonamide, trichlorhydrate
Le composé du Stade A est mis en solution dans 4 mL de dichlorométhane, puis
une
solution d'éther chlorhydrique 1N (805 L) est additionnée. Le mélange
réactionnel est
agité à température ambiante pendant 2 heures, puis concentré pour conduire au
produit du
titre sous la forme d'un solide jaune après lyophilisation.
CA 02730516 2011-01-11
WO 2010/007248 PCT/FR2009/000855
-21-
Microanalyse élémentaire :
%C %H %N %S %Cl
Calculé 50,66 5,56 9,85 6,44 12,80
Trouvé 50,43 5,38 10,02 6,37 12,93
Exemple 2 : N-({8-[(4'-Chloro[1,1'-biphényl]-2-yl)méthyl]-6,7,8,9,9a,10-
hexahydro
pyrido [2',3':4,5] pyrrolo [1,2-a]pyrazin-2-yl} carb onyl)-4-({(1R)-3-
(diméthylamino)-1- [(phénylsulfanyl)méthyl] propyl}amino)-3-
nitrobenzènesulfonamide, bischlorhydrate
On procède comme dans les stades A et B de l'Exemple 1 en remplaçant au Stade
A le
composé de la Préparation 1 par le composé de la Préparation 2.
Microanalyse élémentaire :
%C %H %N %S %Cl
Calculé 54,40 5,27 10,57 6,91 12,23
Trouvé 54,73 5,21 10,45 6,45 12,49
Exemple 3 : N-({7-[(4'-Chloro [1,1'-biphényl]-2-yl)méthyl]-5,5a,6,7,8,9-
hexahydropyrido [3',2' :4,5] pyrrolo [1,2-a]pyrazin-3-y1}carbonyl)-4-({(1R)-
3-(diméthylamino)-1-[(phénylsulfanyl)méthyl]propyl}amino)-3-
nitrobenzènesulfonamide, trichlorhydrate
On procède comme dans les stades A et B de l'Exemple 1 en remplaçant au Stade
A le
composé de la Préparation 1 par le composé de la Préparation 3.
Microanalyse élémentaire :
%C %H %N %S % CI
Calculé 53,50 5,21 10,40 6,80 13,91
Trouvé 53,82 5,24 10,20 6,74 14,13
CA 02730516 2011-01-11
WO 2010/007248 PCT/FR2009/000855
-22-
Exemple 4: N-({3-[(4'-Chloro[1,1'-biphényl]-2-y1)méthyl]-2,3,4,4a,5,6-
hexahydro-lH-
pyrazino[1,2-a] [1,5]naphthyridin-8-yl}carbonyl)-4-({(1R)-3-
(diméthylamino)-1-[(phénylsulfanyl)méthyl] propyl}amino)-3-
nitrobenzènesulfonamide, bischlorhydrate
On procède comme dans les stades A et B de l'Exemple 1 en remplaçant au Stade
A le
composé de la Préparation 1 par le composé de la Préparation 4.
Exemple 55 : N-({8-[(4'-Chloro[1,1'-biphényl]-2-y1)méthyl]-6,6a,7,8,9,10-
hexahydro-
5H-pyrazino[1,2-a] [1,7]naphthyridin-3-yl}carbonyl)-4-({(1R)-3-
(diméthylamino)-1- [(phénylsulfanyl)méthyl]propyl} amino)-3-
nitrobenzènesulfonamide, bischlorhydrate
On procède comme dans les stades A et B de l'Exemple 1 en remplaçant au Stade
A le
composé de la Préparation 1 par le composé de la Préparation 5.
Exemple 6 : N-({8-[(4'-Chloro[1,1'-biphényl]-2-yl)méthyl]-6,6a,7,8,9,10-
hexahydro-5H-
l0 pyrazino[1,2-al[1.,8]naphthyridin-3-yl}carbonyl)-4-({(1R)-3- .
(diméthylamino)-1-[(phénylsulfanyl)méthyl] propyl} amino)-3-
nitrobenzènesulfonamide, bischlorhydrate
On procède comme dans les stades A et B de l'Exemple 1 en remplaçant au Stade
A le
composé de la Préparation 1 par le composé de la Préparation 6.
CA 02730516 2011-01-11
WO 2010/007248 PCT/FR2009/000855
-23 -
ETUDE PHARMACOLOGIQUE
EXEMPLE A : Induction de l'activité caspase in vitro.
Cette étude a porté sur la lignée cellulaire tumorale humaine suivante :
1 carcinome du poumon à petites cellules, H146.
Cette lignée cellulaire est cultivée dans un incubateur à 37 C en présence de
5% de C02.
Plus précisément, les cellules H146 sont cultivées dans du milieu RPMI 1640
complet
contenant 10% de sérum de veau foetal, 2 mM de glutamine, 50 unités/ml de
pénicilline, 50
gg/ml de streptomycine et 10 mM de tampon Hepes, pH = 7,4.
Les cellules sont réparties dans des plaques 6 puits et exposées aux composés
à tester
pendant 6 heures. Elles sont ensuite collectées et lysées et l'activité
caspase est mesurée
dans les lysats cellulaires.
Cette mesure enzymatique est réalisée en dosant l'apparition d'un produit de
clivage
fluorigénique (Pharmacia).
Les résultats montrent que les composés de l'invention sont de puissants
inducteurs
d'apoptose, évaluée en mesurant l'activité caspase 3, dans la lignée tumorale
testée.
A titre d'exemple, les composés des Exemples 1 à 3 montrent respectivement une
activité à
10 M de 15395, 9948 et 17776 RFU (Relative Fluorescence Units).
EXEMPLE B : Cytotoxicité in vitro.
Les études de cytotoxicité ont été réalisées sur la lignée tumorale présentée
dans l'Exemple
A.
Les cellules sont réparties dans des microplaques et exposées aux composés à
tester pendant
48 heures. La viabilité cellulaire est ensuite quantifiée par un essai
colorimétrique, le
Microculture Tetrazolium Assay (Cancer Res., 1987, 47, 93 9-942).
Les résultats sont exprimés en IC50 (concentration en composé qui inhibe à 50
% la viabilité
cellulaire), et montrent que les composés de l'invention sont cytotoxiques.
CA 02730516 2011-01-11
WO 2010/007248 PCT/FR2009/000855
-24-
EXEMPLE C : Induction de l'activité caspase in vivo.
La capacité des composés de l'invention à activer la caspase 3 est évaluée
dans un modèle
de xénogreffe de cellules de carcinome pulmonaire à petites cellules H146.
5.106 cellules H146 sont greffées par voie sous-cutanée dans des souris
immunodéprimées
(souche NOD SCID). 25 à 30 jours après la greffe, les composés à tester sont
injectés par
voie intra-péritonéale dans un mélange tween80/eau. Seize heures après le
traitement, les
masses tumorales sont récupérées, lysées et l'activité caspase 3 est mesurée
dans les lysats
tumoraux.
Les résultats obtenus montrent que les composés de l'invention sont capables
d'induire
l'apoptose dans les cellules tumorales H146 in vivo.
EXEMPLE D : Composition pharmaceutique : Comprimés
1000 comprimés dosés à 5 mg de N-({2-[(4'-chloro[l,l'-biphényl]-2-yl)méthyl]-
1,2,3,4,10,10a-hexahydropyrido[4',3':4,5]pyrrolo [1,2-a]pyrazin-8-yl}
carbonyl)-4-({(1R)-3-
(diméthylamino)-1- [(phénylsulfanyl)méthyl]propyl } amino)-3 -
nitrobenzènesulfonamide,
trichlorhydrate (Exemple 1)
...............................................................................
............ 5 g
Amidon de blé
...............................................................................
.......................... 20 g
Amidon de maïs
...............................................................................
....................... 20 g
Lactose
...............................................................................
..................................:.. 30 g
Stéarate de magnésium
...............................................................................
............. 2 g
Silice
...............................................................................
......................................... 1 g
Hydroxypropylcellulose
...............................................................................
........... 2 g