Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02734191 2011-02-11
WO 2010/018474 PCT/IB2009/053131
1
NOUVEAUX DERIVES DE GLUCOPYRANOSE, LEUR PREPARATION ET LEURS
APPLICATIONS BIOLOGIQUES.
La présente invention concerne de nouveaux dérivés de glucopyranose, en
particulier un nouvel ester, le 3,5-di-tertiobutyl-4-hydroxybenzoate de
3,4,5-
trihydroxy-6-méthoxy tétrahydropyran-2-yl méthyl et les dérivés dudit ester.
L'invention concerne également leur procédé de préparation, ainsi que les
applications biologiques dudit composé et de ses dérivés, en particulier pour
le
traitement des infections virales et rétrovirales.
Le nouveau dérivé de l'invention présente une formule développée proche du
composé sucré D-glucopyranose précédemment décrit dans FR 2 887249 au nom
du Demandeur, et dont l'utilisation s'avère particulièrement avantageuse
contre des
infections par des virus à enveloppe, et notamment le virus de l'herpès, du
sida, de la
grippe, des hépatites B et C.
Les travaux poursuivis dans ce domaine ont conduit l'inventeur au nouvel ester
objet
de l'invention et à ses dérivés qui, de manière inattendue, permettent
d'améliorer
encore les résultats obtenus avec le composé sucré D-glucopyranose, comme une
plus grande solubilité, une plus grande stabilité, une synthèse parfaitement
reproductible, une plus grande activité biologique à des concentrations plus
faibles
que celles de l'art antérieur.
L'invention a donc pour but de fournir de nouveaux composés actifs contre les
virus
à enveloppe.
Elle vise également l'utilisation desdits composés pour l'élaboration de
compositions
pharmaceutiques antivirales de grand intérêt.
La présente invention a plus particulièrement pour objet le composé 3,5-di-
tertiobutyl-4-hydroxybenzoate de 3,4,5-trihydroxy-6-méthoxy tétrahydropyran-2-
yl
méthyl défini par la formule (1) suivante :
CA 02734191 2011-02-11
WO 2010/018474 PCT/IB2009/053131
2
OH
HO,, 0 a (I)
OH
Le composé de l'invention peut également se présenter sous la forme d'un
dérivé
comme par exemple sous la forme d'un sel ou d'un acide dudit ester (I).
Le composé (I) de l'invention ainsi que ledit ou lesdits dérivés pourront être
présentés dans une composition comprenant au moins un excipient
pharmaceutiquement acceptable.
L'invention a également pour objet une composition comprenant au moins un
composé tel que défini précédemment en association avec un composé
biologiquement actif.
A titre d'exemple de composés biologiquement actif on pourra citer :
- des composés d'origine végétale,
- l'acide 3,5-di-tertiobutyl-4-hydroxybenzo'ique,
- le 3,5-di-tertiobutyl-4-hydroxybenzoate d'octaoxyéthylèneglycol,
- des dérivés non tissés d'origine synthétique,
- des dérivés proches du cyclopentanophénanthrène,
- des virus statiques antiviraux et antirétroviraux.
L'invention a encore pour objet une composition pharmaceutique renfermant une
quantité thérapeutiquement efficace d'au moins un composé ou d'au moins une
composition tels que définis ci-dessus.
La quantité de composé ou de composition pourra varier selon les applications
envisagées, l'âge et le poids du malade.
La composition pharmaceutique de l'invention pourra se présenter sous une
forme
appropriée pour une administration par voie orale, injectable ou parentérale.
CA 02734191 2011-02-11
WO 2010/018474 PCT/IB2009/053131
3
A titre d'exemple on pourra citer les comprimés, dragées, solutions ou
suspensions
buvables ou injectables, émulsions, suppositoires ... .
Ainsi la présente invention couvre également l'utilisation d'un composé ou
d'une
composition tels que définis ci-dessus, pour la préparation d'un médicament
destiné
au traitement et/ou à la prévention des infections par des virus à enveloppe.
L'invention a encore pour objet un procédé de préparation du nouveau ester 3,5-
di-
tertiobutyl-4-hydroxybenzoate de 3,4,5-trihydroxy-6-méthoxy tétrahydropyran-2-
yl
méthyl de formule (I).
Ce procédé comprend les étapes préalables de formation du chlorure de l'acide
3,5-
di-tertiobutyl-4-hydroxybenzoïque de formule (III) d'une part et du [6-méthoxy-
3,4,5-
tris-(4-méthoxy-benzyloxy)-tétrahydropyran-2-yl] méthanol de formule (V)
d'autre
part, puis une réaction de couplage entre les deux composés (III) et (V) ainsi
obtenus
pour obtenir l'ester protégé (II), qui après déprotection conduit au composé
glucopyranose (I) de l'invention.
Ainsi le procédé de préparation du composé de formule (I) de l'invention est
plus
particulièrement caractérisé en ce qu'il comprend la réaction du chlorure de
3,5-di-
tertiobutyl-4-hydroxybenzoyl de formule (III) :
0H (Il1)
C!
0
avec le [6-méthoxy-3,4,5-tris-(4-méthoxy-benzyloxy)-tétrahydropyran-2-yl]
méthanol
de formule (V) :
CA 02734191 2011-02-11
WO 2010/018474 PCT/IB2009/053131
4
0H
,,,.. 0
0 õ0~ (V)
pour obtenir le composé estérifié 3,5-di-tertiobutyl-4-hydroxybenzoate de
3,4,5-tris-
(4-méthoxy-benzyloxy)-6-méthoxy tétrahydropyran-2-yl méthyl de formule (II)
:
OH
0 Y
0 (II)
qui après déprotection conduit au composé estérifié (I).
Selon un mode de réalisation avantageux du procédé de l'invention, la
préparation
du chlorure d'acide (111) est effectuée à partir de l'acide 3,5-di-tertiobutyl-
4-
hydroxybenzoïque de formule (IV) :
OH (IV)
0
CA 02734191 2011-02-11
WO 2010/018474 PCT/IB2009/053131
La synthèse de l'acide 3,5-di-tertiobutyl-4-hydroxybenzoïque de formule (IV),
de
même que celle de ses halogénures, notamment les bromure et chlorure, a été
décrite dans EP 0 269 981.
5 Cet acide (IV) a été proposé pour préparer des médicaments antiviraux
destinés au
traitement des maladies liées à une infection d'un individu par des virus tels
que ceux
de l'herpès ou du sida.
Selon un autre mode de réalisation avantageux du procédé de l'invention, la
préparation du composé (V) comprend plus particulièrement les étapes suivantes
:
- réaction du 3,4,5-trihydroxy-6-méthoxy-tétrahydropyran--2-yI méthanol de
formule
(VIII)
OH
H o,,..o
(VIII)
Ho 0
0H
avec du p-anisaldéhyde diméthyl acétal de formule (lx) :
,0
i
01, (lx)
pour obtenir le 6-méthoxy-2-(4-méthoxy-phényl)-hexahydro-pyrano [3,2-D] [1,3]
dioxine-7,8-diol de formule (VII) :
.o.~I
(Vil)
H O
OH
CA 02734191 2011-02-11
WO 2010/018474 PCT/IB2009/053131
6
- puis réaction du composé (VII) ainsi obtenu avec du chlorure de p-
méthoxybenzyle
de formule (X)
GI (X)
pour obtenir le 6-méthoxy-7,8-bis-(4-méthoxy-benzyloxy)-2-(4-méthoxy-phényl)-
hexahydro-pyrano [3,2-D] [1,3] dioxine de formule (VI) :
~~
0,,.. (VI)
"0
- et enfin réaction du composé (VI) ainsi obtenu avec du cyanoborohydrure de
sodium (NaBH3CN) et du chlorotriméthylsilane ((CH3)3SiCI) pour obtenir le
composé
de formule (V).
Le composé (I) de la présente invention présente plusieurs avantages notamment
par rapport au composé du brevet FR 2 887 249 de structure assez proche :
- une meilleure solubilité dans l'eau qui facilite l'élaboration de
préparation galénique
plus adaptée pour un médicament,
- une activité virucide à de plus faibles concentrations.
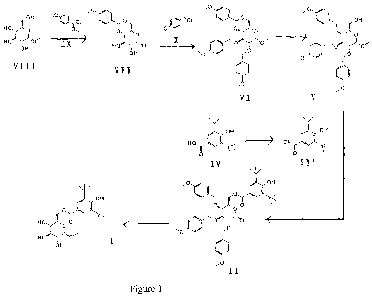
La figure 1 représente le schéma de synthèse du nouvel ester (!).
L'exemple suivant illustre l'invention, il ne la limite en aucune façon.
CA 02734191 2011-02-11
WO 2010/018474 PCT/IB2009/053131
7
EXEMPLE 1 : SYNTHESE DU 3 5-DI-TERTIOBUTYL-4-HYDROXYBENZOATE DE
3 4 5-TRIHYDROXY-6-METHOXY TETRAHYDROPYRAN-2-YL METHYL DE
FORMULE (1) (voir figure 1).
1) SYNTHESE DU CHLORURE DE 3,5-DI-TERTIOBUTYL-4-HYDROXYBENZOYL
DE FORMULE (111).
L'acide 3,5-di-tertiobutyl-4-hydroxybenzoïque (IV) acheté dans le commerce est
activé sous forme de chlorure d'acide par l'action de 5 équivalents de
chlorure de
thionyle (SOC12) au reflux du toluène pendant une nuit à 110 C. Le milieu
réactionnel
est évaporé et le composé chloré (111) est obtenu avec un rendement de 95%.
2) SYNTHESE DU [6-METHOXY-3,4,5-TRIS-(4-METHOXY-BENZYLOXY)-
TETRAHYDROPYRAN-2-YL1 METHANOL DE FORMULE (V).
a) Etape 1 : Synthèse du 6-méthoxy-2-(4-méthoxy-phényl)-hexahydro-pyrano [3,2-
D]
[1,3] dioxine-7,8-diol de formule (VII).
La synthèse débute par la protection de l'alcool primaire (VIII) (le 3,4,5-
trihydroxy-6-
méthoxy-tétrahydropyran-2-yl méthanol) sous forme d'un acétal (VII) (le 6-
méthoxy-
2-(4-méthoxy-phényl)-hexahydro-pyrano [3,2-D] [1,3] dioxine-7,8-diol) par
action du
du p-anisaldéhyde diméthyl acétal (I11) en présence d'acide camphrosulfonique
dans
le DMF (diméthylformamide).
La réaction est conduite à température ambiante sous vide industriel (pression
= 40
mbar) (évaporation du méthanol formé) pendant 2 heures, pour que l'on observe
un
avancement significatif de la formation du produit désiré, de telle sorte
qu'il ne reste
que des traces de produit de départ.
Après un traitement aqueux en milieu basique, les traces de produit de départ
sont
éliminées par un lavage au cyclohexane, cela permet d'isoler le composé (VII)
sous
forme d'un solide blanc avec des rendements compris entre 60 et 80%.
b) Etape 2: Synthèse du 6-méthoxy-7,8-bis-(4-méthoxy-benzyloxy)-2-(4-méthoxy-
phényl)-hexahydro-pyrano [3,2-D] [1,3] dioxine de formule (VI).
CA 02734191 2011-02-11
WO 2010/018474 PCT/IB2009/053131
8
La protection des deux fonctions alcools secondaires libres du composé (VII)
est
effectuée par l'action de l'hydrure de sodium et du chlorure de p-
méthoxybenzyle (X)
dans un mélange DMF/THF (80/20).
L'addition de l'hydrure de sodium se fait à 0 C, puis après ajout du dérivé
chloré, un
chauffage à 90 C maintenu pendant 1 heure conduit à la formation totale du
produit
protégé (VI).
Un lavage au cyclohexane conduit à l'obtention d'un produit pur sous forme
d'un
solide blanc et avec un rendement de 86%.
c) Etape 3: Synthèse du [6-méthoxy-3,4,5-tris-(4-méthoxy-benzyloxy)-
tétrahydropyran-2-ylj méthanol de formule (V).
L'ouverture sélective de l'acétal (VI) préalablement formé est réalisée par le
cyanoborohydrure de sodium et le chlorotriméthylsilane en présence de tamis
moléculaire 3Å dans l'acétonitrile. La réaction est effectuée en 1 heure à -20
C, et
après traitement basique suivi d'une purification sur gel de silice, le
produit ouvert (V)
est isolé sous forme d'un solide blanc avec un rendement de 60%.
3) OBTENTION DU 3 5-DI-TERTIOBUTYL-4-HYDROXYBENZOATE DE 3,4-5
TRIHYDROXY-6-METHOXY TETRAHYDROPYRAN-2-YL METHYL (I)
Le couplage entre le chlorure d'acide (III) prélablement préparé et le composé
ouvert
(V) est réalisé en présence de 1,5 équivalents de triéthylamine (Et3N) dans le
dichlorométhane (CH2CI2) à température ambiante en 2 heures. Le produit de
couplage (II) (le 3,5-di-tertiobutyl-4-hydroxybenzoate de 3,4,5-tris-(4-
méthoxy-
benzyloxy)-6-méthoxy tétrahydropyran-2-yl méthyl) est isolé par
chromatographie sur
gel de silice sous forme d'une huile jaune avec un rendement de 70%.
Les protections p-méthoxybenzyl des trois positions alcools secondaires du
composé
(ll) sont clivées par l'acide trifluoroacétique (12 équivalents) dans le
dichlorométhane
(CH2CIz).
Au bout d'une nuit à température ambiante, la totalité du composé (II) est
convertie
en composé libre (I). Il est isolé par un passage rapide sur silice pour
fournir un
solide blanc avec un rendement de 60%.
CA 02734191 2011-02-11
WO 2010/018474 PCT/IB2009/053131
9
On obtient ainsi 10,5 g de 3,5-di-tertiobutyl-4-hydroxybenzoate de 3,4,5-
trihydroxy-6-
méthoxy tétrahydropyran-2-yl méthyl, avec une pureté HPLC de 99,3% et un
rendement global de synthèse de 15%.