Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02735654 2011-02-28
WO 2010/028206 PCT/US2009/055976
LIQUID FUEL COMPOSITIONS BASED ON CATALYTICALLY DEOXYGENATED AND CONDENSATED
OXYGENATED CARBONHYDRATES
Field of the Invention
The present invention relates to liquid fuel
compositions comprising a component derived from a water-
soluble oxygenated hydrocarbon.
Background of the Invention
Significant amount of attention has been placed on
developing new technologies for providing energy from
resources other than fossil fuels. Biomass is a resource
that shows promise as a fossil fuel alternative. As
opposed to fossil fuel, biomass is also renewable.
One type of biomass is plant biomass. Plant biomass
is the most abundant source of carbohydrate in the world
due to the lignocellulosic materials composing the cell
walls in higher plants. Plant cell walls are divided into
two sections, primary cell walls and secondary cell
walls. The primary cell wall provides structure for
expanding cells and is composed of three major
polysaccharides (cellulose, pectin, and hemicellulose)
and one group of glycoproteins. The secondary cell wall,
which is produced after the cell has finished growing,
also contains polysaccharides and is strengthened through
polymeric lignin covalently cross-linked to
hemicellulose. Hemicellulose and pectin are typically
found in abundance, but cellulose is the predominant
polysaccharide and the most abundant source of
carbohydrates.
Most transportation vehicles, whether boats, trains,
planes and automobiles, require high power density
provided by internal combustion and/or propulsion
engines. These engines require clean burning fuels which
1
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
are generally in liquid form or, to a lesser extent,
compressed gases. Liquid fuels are more portable due to
their high energy density and their ability to be pumped,
which makes handling easier.
Currently, biomass provides the only renewable
alternative for liquid transportation fuels.
Unfortunately, the progress in developing new
technologies for producing liquid biofuels has been slow
in developing, especially for liquid fuel products that
fit within the current infrastructure. Although a variety
of fuels can be produced from biomass resources, such as
ethanol, methanol, biodiesel, Fischer-Tropsch diesel and
kerosene, and gaseous fuels, such as hydrogen and
methane, these fuels can require either new distribution
technologies and/or combustion technologies appropriate
for their characteristics. The production of these fuels
also tends to be expensive.
Ethanol, for example, is made by converting the
carbohydrate from biomass into sugar, which is then
converted into ethanol in a fermentation process. Ethanol
is the most widely used biofuel today with current
capacity of 4.3 billion gallons per year based on starch
crops, such as corn. Ethanol, however, has very
substantial disadvantages with respect its energy value
as a fuel relative to the amount of energy needed to
produce it. Ethanol produced by fermentation contains
large amounts of water, typically comprising only about 5
percent of ethanol by volume in the water/alcohol
fermentation product. The removal of this water is highly
energy-consuming, and often requires the use of natural
gas as a heat source. Ethanol also has less energy
content than gasoline, which means that it takes more
fuel to go the same distance. Ethanol is very corrosive
2
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
to fuel systems and cannot be transported in petroleum
pipelines. As a result, ethanol is transported over-the-
road in tank trucks, which increases its overall cost and
energy consumption. When considering the total energy
consumed by farm equipment, cultivation, planting,
fertilizers, pesticides, herbicides, petroleum-based
fungicides, irrigation systems, harvesting,
transportation to processing plants, fermentation,
distillation, drying, transport to fuel terminals and
retail pumps, and lower ethanol fuel energy content, the
net energy content value added and delivered to consumers
is very small.
Biodiesel is another potential energy source.
Biodiesel can be made from vegetable oil, animal fats,
waste vegetable oils, microalgae oils or recycled
restaurant greases, and is produced through a process in
which organically derived oils are combined with alcohol
(ethanol or methanol) in the presence of a catalyst to
form ethyl or methyl ester. The biomass-derived ethyl or
methyl esters can then be blended with conventional
diesel fuel or used as a neat fuel (100% biodiesel).
Biodiesel is also expensive to manufacture, and poses
various issues in its use and combustion. For example,
special handling may be required to avoid gelling in cold
temperatures.
Biomass can also be gasified to produce a synthesis
gas composed primarily of hydrogen and carbon monoxide,
also called syngas or biosyngas. Syngas produced today is
used directly to generate heat and power, but several
types of biofuels may be derived from syngas. Hydrogen
can be recovered from syngas, or it can be catalytically
converted to methanol. The gas can also be run through a
biological reactor to produce ethanol or converted using
3
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
Fischer-Tropsch catalyst into a liquid stream with
properties similar to diesel fuel, called Fischer-Tropsch
diesel. These processes, however, tend to be expensive.
There exists a need for liquid fuel compositions
which contain a component that is derivable from biomass
and that is capable of use in the current infrastructure,
namely the same distribution system and the same engines
without the need for special modifications. There also
exists a need for liquid fuel compositions which contain
a component that is derivable from biomass that do not
depend on microorganisms, enzymes or other expensive and
delicate manufacturing processes.
Summary of the Invention
The present invention provides a liquid fuel
composition comprising a distillation fraction of a
component having at least one C4+ compound derived from a
water-soluble oxygenated hydrocarbon prepared by a method
comprising:
providing water and a water-soluble oxygenated
hydrocarbon comprising a C1+01+ hydrocarbon in an aqueous
liquid phase and/or a vapor phase;
providing H2;
catalytically reacting in the liquid and/or vapor
phase the oxygenated hydrocarbon with the H2 in the
presence of a deoxygenation catalyst at a deoxygenation
temperature and deoxygenation pressure to produce an
oxygenate comprising a C1+01_3 hydrocarbon in a reaction
stream; and
catalytically reacting in the liquid and/or vapor
phase the oxygenate in the presence of a condensation
catalyst at a condensation temperature and condensation
pressure to produce the C4+ compound,
4
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
wherein the C4+ compound comprises a member selected
from the group consisting of C4+ alcohol, C4+ ketone, C4+
alkane, C4+ alkene, C5+ cycloalkane, C5+ cycloalkene, aryl,
fused aryl, and a mixture thereof;
wherein the liquid fuel composition is selected from:
a gasoline composition having an initial boiling
point in the range of from 15 C to 70 C (IP123), a
final boiling point of at most 230 C (IP123), a RON in
the range of from 85 to 110 (ASTM D2699) and a MON in the
range of from 75 to 100 (ASTM D2700);
a diesel fuel composition having an initial boiling
point in the range of from 130 C to 230 C (IP123), a
final boiling point of at most 410 C (IP123) and a
cetane number in the range of from 35 to 120 (ASTM D613);
and
a kerosene composition having an initial boiling
point in the range of from 80 to 150 C, a final boiling
point in the range of from 200 to 320 C and a viscosity
at -20 C in the range of from 0.8 to 10 mm2/s (ASTM
D445).
The present invention also provides a gasoline
composition comprising a component having at least one
C4+ compound derivable from a water-soluble oxygenated
hydrocarbon having a final boiling point in the range of
from 150 to 220 C, a density at 15 C in the range of
from 700 to 890 kg/m3, a sulphur content of at most
5 mg/kg, an oxygen content of at most 3.5 %wt., a RON in
the range of from 80 to 110, and a MON in the range of
from 70 to 100, wherein said gasoline composition has an
initial boiling point in the range of from 15 C to 70 C
(IP123), a final boiling point of at most 220 C (IP123),
a RON in the range of from 85 to 110 (ASTM D2699) and a
MON in the range of from 75 to 100 (ASTM D2700).
5
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
The present invention also provides a diesel fuel
composition comprising a component having at least one
C4+ compound derivable from a water-soluble oxygenated
hydrocarbon having t a T95 in the range of from 220 to
380 C, a flash point in the range of from 30 to 70 C, a
density at 15 C in the range of from 700 to 900 kg/m3, a
sulphur content of at most 5 mg/kg, an oxygen content of
at most 10 %wt., and a viscosity at 40 C in the range of
from 0.5 to 6 cSt, wherein said diesel fuel composition
has an initial boiling point in the range of from 130 C
to 230 C (IP123), a final boiling point of at most
410 C (IP123) and a cetane number in the range of from
35 to 120 (ASTM D613).
The present invention also provides a kerosene
composition comprising a component having at least one
C4+ compound derivable from a water-soluble oxygenated
hydrocarbon having an initial boiling point in the range
of from 120 to 215 C, a final boiling point in the range
of from 220 to 320 C, a density at 15 C in the range of
from 700 to 890 kg/m3, a sulphur content of at most
0.1 %wt., a total aromatics content of at most 30 %vol.,
a freeze point of -40 C or lower, a smoke point of at
least 18 mm, a viscosity at -20 C in the range of from 1
to 10 cSt, and a specific energy content in the range of
from 40 to 47 MJ/kg, wherein said kerosene composition
has an initial boiling point in the range of from 80 to
150 C, a final boiling point in the range of from 200 to
320 C and a viscosity at -20 C in the range of from 0.8
to 10 mm2/s (ASTM D445).
The present invention also provides a method for
preparing a liquid fuel composition according to the
present invention, comprising admixing:
6
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
(a) a distillation fraction of a component having at
least one C4+ compound derived from a water-soluble
oxygenated hydrocarbon prepared by a method
comprising:
providing water and a water-soluble oxygenated
hydrocarbon comprising a C1+01+ hydrocarbon in an
aqueous liquid phase and/or a vapor phase;
providing H2;
catalytically reacting in the liquid and/or
vapor phase the oxygenated hydrocarbon with the H2
in the presence of a deoxygenation catalyst at a
deoxygenation temperature and deoxygenation pressure
to produce an oxygenate comprising a C1+01-3
hydrocarbon in a reaction stream; and
catalytically reacting in the liquid and/or
vapor phase the oxygenate in the presence of a
condensation catalyst at a condensation temperature
and condensation pressure to produce the C4+
compound,
wherein the C4+ compound comprises a member
selected from the group consisting of C4+ alcohol,
C4+ ketone, C4+ alkane, C4+ alkene, C5+ cycloalkane,
C5+ cycloalkene, aryl, fused aryl, and a mixture
thereof, with
(b) at least one fuel component.
Brief Description of the Drawings
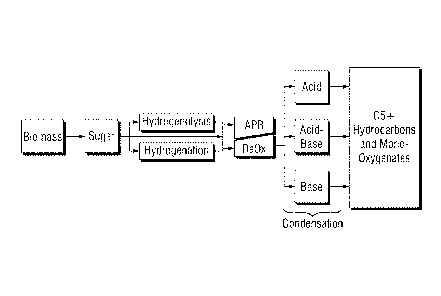
Figure 1 is a flow diagram illustrating various
production pathways associated with the present
invention.
Figure 2 illustrates potential chemical routes that
allow carbohydrates, such as sugars, to be converted to
non-oxygenated hydrocarbons.
7
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
Figure 3 is an illustration of various reaction
pathways involved in the deoxygenation of sorbitol to
oxygenates and APR hydrogen.
Figure 4 is an illustration of the thermodynamic
equilibriums along the reaction pathway for converting
acetone to 2-methyl pentane at 100 C and 400 C.
Figure 5 is a graph illustrating the equilibrium
constants associated with the intermediate reaction
products and the overall conversion for the reaction of 2
moles of acetone with 3 moles of hydrogen to form 1 mole
of 2-methylpentane and 2 moles of water.
Figure 6 is a flow diagram illustrating a reactor
system configured to allow for the recycle of hydrogen,
oxygenates and oxygenated hydrocarbons.
Figure 7 is a flow diagram illustrating a reactor
system configured to allow for the use of air or an oil
as a temperature control element.
Figure 8 a flow diagram illustrating a reactor
system for the present invention.
Figure 9 is a flow diagram illustrating a reactor
system utilizing two reactors.
Figure 10 is a flow diagram illustrating a reactor
system utilizing two feedstock lines.
Figure 11 is an illustration of a reactor useful in
practicing the present invention.
Figure 12 is a graph illustrating the carbon
distribution of mono-oxygenates produced from glycerol.
Figure 13 is a graph illustrating the axial
temperature profile for a reactor when used to produce
compounds from a feedstock of oxygenated hydrocarbons.
Figure 14 is a graph illustrating the percentage of
feed carbon exiting as oxygenates from the conversion of
8
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
an oxygenate feed stream to C5+ compounds as a function
of time.
Figure 15 is a graph illustrating the percentage of
feed carbon exiting as C5+ hydrocarbons from the
conversion of an oxygenate feed stream as a function of
time.
Figure 16 is a graph illustrating the percentage of
feed carbon exiting as C5+ aromatic hydrocarbons from the
conversion of an oxygenate feed stream as a function of
time.
Figure 17 is a graph showing the total weight
percentage of paraffin and aromatic compounds derived
from the conversion of a feed stream of sucrose and
xylose.
Figure 18 is a graph illustrating the heating value
of C5+ hydrocarbons derived from the production of
gasoline from sorbitol, as a percentage of the heating
value of the feed.
Figure 19 is a graph illustrating the percentage of
carbon recovered as aromatic hydrocarbons from the
production of gasoline from sorbitol, shown as a
percentage of the carbon present in the feed.
Detailed Description of the Invention
The liquid fuel compositions of the present
invention comprise a component having at least one C4+
compound derived from a water-soluble oxygenated
hydrocarbon. Preferably, the water-soluble oxygenated
hydrocarbon is derived from biomass.
Typically, the process of preparing the component
having at least one C4+ compound derived from a water-
soluble oxygenated hydrocarbon produces hydrocarbons,
ketones and alcohols from biomass-derived oxygenated
hydrocarbons, such as sugars, sugar alcohols,
9
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
cellulosics, lignocelluloses, hemicelluloses, saccharides
and the like.
The component derived from a water-soluble
oxygenated hydrocarbon comprises C4+ alkanes, C4+ alkenes,
C5+ cycloalkanes, C5+ cycloalkenes, aryls, fused aryls, C4+
alcohols, C4+ ketones, and mixtures thereof (collectively
referred to herein as "C4+ compounds"). The C4+
hydrocarbons typically have from 4 to 30 carbon atoms and
may be branched or straight chained alkanes or alkenes,
or unsubstituted, mono-substituted or multi-substituted
aromatics (aryls) or cycloalkanes. The C4+ alcohols and
C4+ ketones may be cyclic, branched or straight chained,
and have from 4 to 30 carbon atoms.
Lighter fractions, primarily C4-C9, may be separated
for gasoline use. Moderate fractions, such as C7-C14, may
be separated for kerosene, for example for use in jet
fuel, while heavier fractions, i.e., C12-C24, may be
separated for diesel fuel use. The heaviest fractions may
be used as lubricants or cracked to produce additional
gasoline and/or diesel fractions. The C4+ compounds
derived from water-soluble oxygenated hydrocarbons may
also find use as industrial chemicals, such as xylene,
whether as an intermediate or an end product.
Process of Preparing the Component Derived from a Water-
Soluble Oxygenated Hydrocarbon
The general process of preparing the component
derived from a water-soluble oxygenated hydrocarbon is
illustrated in Figure 1. A feedstock solution containing
a water-soluble oxygenated hydrocarbon having one or more
carbon atoms is reacted with hydrogen over a
deoxygenation catalyst to produce oxygenates, and then
the oxygenates are reacted over a condensation catalyst
under conditions of temperature and pressure effective to
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
cause a condensation reaction that produces the C4+
compounds. The hydrogen may originate from any source,
but is preferably derived in situ or in parallel from
biomass using aqueous phase reforming. The hydrogen and
oxygenated hydrocarbons may also be supplemented with
recycled hydrogen and oxygenated hydrocarbons derived
from the process. The oxygenated hydrocarbon may be a
monosaccharide, disaccharide, polysaccharide, cellulose,
hemicellulose, lignin, sugar, sugar alcohol or other
polyhydric alcohols, or may be derived from the
hydrogenation of a sugar, furfural, carboxylic acid,
ketone, or furan, or the hydrogenolysis of a sugar, sugar
alcohol, polysaccharide, monosaccharide, disaccharide or
polyhydric alcohol.
One unique aspect about the process of preparing the
component derived from a water-soluble oxygenated
hydrocarbon in the present invention is that the C4+
compounds are derived from biomass components using
catalytic processes instead of microorganisms, enzymes,
high temperature gasification or transesterification
methods. The process of preparing the component derived
from a water-soluble oxygenated hydrocarbon in the
present invention can also generate hydrogen in situ to
avoid reliance on external hydrogen sources, such as
hydrogen generated from the steam reforming of natural
gas, or the electrolysis or thermolysis of water. The
process of preparing the component derived from a water-
soluble oxygenated hydrocarbon in the present invention
also generates water, which may be recycled and used in
upstream processes or returned to the environment. The
process of preparing the component derived from a water-
soluble oxygenated hydrocarbon in the present invention
is also able to generate non-condensable fuel gases for
11
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
purposes of providing a heat source within the reactor
system or for external processes.
Carbohydrates are the most widely distributed,
naturally occurring organic compounds on Earth.
Carbohydrates are produced during photosynthesis, a
process in which the energy from the sun is converted
into chemical energy by combining carbon dioxide with
water to form carbohydrates and oxygen:
Szail
H.,0
The energy from sunlight is stored through this
process as chemical energy in the form of carbohydrates
in plants. The carbohydrates, especially when in a sugar
form, are highly reactive compounds that are readily
oxidized by living material to generate energy, carbon
dioxide and water. Plant materials store these
carbohydrates either as sugars, starches, polymeric
cellulose, and/or hemi-cellulose.
The presence of oxygen in the molecular structure of
carbohydrates contributes to the reactivity of sugars in
biological systems. Ethanol fermentation technology takes
advantage of this highly reactive nature by forming
ethanol at ambient temperatures. The fermentation
technology essentially de-functionalizes the highly
reactive sugar to generate a partially oxidized
hydrocarbon, ethanol. Ethanol, however, has very
substantial disadvantages with respect its energy value
as highlighted above.
Figure 2 shows potential chemical routes that allow
carbohydrates, such as sugars, to be converted to non-
oxygenated hydrocarbons. Water soluble carbohydrates are
known to react with hydrogen over catalyst(s) to generate
polyhydric alcohols, either by hydrogenation or
hydrogenolysis. The hydrogen has historically been
12
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
generated externally, i.e., from natural gas or by other
processes, but can now be generated in situ or in
parallel according to the present invention through the
aqueous-phase reforming of the polyhydric alcohol.
The aqueous-phase reforming (APR) of the polyhydric
alcohol proceeds through the formation of an aldehyde
(shown in Figure 2) where the aldehyde reacts over a
catalyst with water to form hydrogen, carbon dioxide, and
a smaller polyhydric alcohol. The polyhydric alcohol can
further react with hydrogen over a catalyst through a
series of deoxygenation reactions to form either alcohol,
ketone, or aldehydes species that can undergo
condensation reactions to form either larger carbon
number straight chain compounds, branched chain
compounds, or cyclic compounds. The condensation
reactions can be either acid catalyzed, base catalyzed,
or both acid and base catalyzed. The resulting compounds
may be hydrocarbons or hydrocarbons containing oxygen,
the oxygen of which can be removed through the reaction
with hydrogen over a catalyst. The resulting condensed
products include C4+ alcohols, C4+ ketones, C4+ alkanes,
C4+ alkenes, C5+ cycloalkanes, C5+ cycloalkenes, aryls,
fused aryls, and mixtures thereof. The mixtures can be
fractionated and blended to produce the appropriate
mixtures of molecules typically used in gasoline,
kerosene (for example as jet fuel), or diesel fuels.
The de-functionalization begins by reacting the
glucose with hydrogen in either a hydrogenation reaction
or hydrogenolysis reaction to convert the cyclic sugar
molecule to its corresponding linear alcohol, sorbitol,
or lower polyhydric alcohols, such as glycerol, propylene
glycol, ethylene glycol, xylitol, among others. As
indicated above, the hydrogen may be from any source, but
13
CA 02735654 2016-04-28
is preferably hydrogen generated in situ by aqueous phase
reforming or excess hydrogen recycled from the reactor
system.
During the aqueous phase reforming process, the
carbohydrate first undergoes dehydrogenation to provide
adsorbed intermediates, prior to cleavage of C-C or C-0
bonds. Subsequent cleavage of C-C bonds leads to the
formation of CO and H2, with the CO then reacting with
water to form CO2 and H2 by the water-gas shift reaction.
Various APR methods and techniques are described in U.S.
Patent Nos. 6,699,457; 6,964,757 and 6,964,758; and U.S.
Patent Application No. 11,234,727 (all to Cortright et
al., and entitled "Low-Temperature Hydrogen Production
from Oxygenated Hydrocarbons"); and U.S. Patent No.
6,953,873 (to Cortright et al., and entitled "Low
Temperature Hydrocarbon Production from Oxygenated
Hydrocarbons"); and WO 2007/075476 A2 (to Cortright et
al., and entitled "Catalyst and Methods for Reforming
Oxygenated Compounds").
The term "aqueous phase reforming"
and "APR" shall generically denote the reforming of
oxygenated hydrocarbons and water to yield hydrogen and
carbon dioxide, regardless of whether the reactions takes
place in the gaseous phase or in the condensed liquid
phase. "APR H2" shall generically refer to the hydrogen
produced by the APR process.
The resulting oxygenated hydrocarbon, namely the
sorbitol or glycerol, propylene glycol, ethylene glycol,
xylitol, etc., are further defunctionalized through
deoxygenation reactions to form oxygenates, such as
alcohols, ketones, aldehydes, furans, diols, triols,
hydroxy carboxylic acids, and carboxylic acids for use in
later condensation reactions. Figure 3 illustrates
14
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
various reaction pathways involved in the deoxygenation
of sorbitol to oxygenates and APR hydrogen. In general,
without being limited to any particular theory, it is
believed that the deoxygenation reactions involves a
combination of various different reaction pathways,
including without limitation: hydrodeoxygenation,
consecutive dehydration-hydrogenation, hydrogenolysis,
hydrogenation and dehydration reactions, resulting in the
removal of oxygen from the oxygenated hydrocarbon to
arrive at a hydrocarbon molecule having the general
formula C1+01_3.
The oxygenates produced are then converted into C4+
compounds by condensation. Without being limited to any
specific theories, it is believed that the acid
condensation reactions generally consist of a series of
steps involving: (a) the dehydration of oxygenates to
olefins; (b) oligomerization of the olefins; (c) cracking
reactions; (d) cyclization of larger olefins to form
aromatics; (e) paraffin isomerization; and (f) hydrogen-
transfer reactions to form paraffins. Basic condensation
reactions are believed to generally consist of a series
of steps involving: (1) aldol condensation to form a p-
hydroxyketone or p-hydroxyaldehyde; (2) dehydration of
the p-hydroxyketone or p-hydroxyaldehyde to form a
conjugated enone; (3) hydrogenation of the conjugated
enone to form a ketone or aldehyde, which may participate
in further condensation reactions or conversion to an
alcohol or hydrocarbon; and (4) hydrogenation of
carbonyls to alcohols, or vice-versa. Acid-base
condensation reactions are believed to generally involve
any of the previous acidic and/or basic reactions steps.
In certain embodiments, the condensation reactions
occur at typical condensation temperatures and pressures.
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
However, in various embodiments, it may also be more
favorable to conduct the condensation reactions at
temperature and/or pressure conditions that are elevated
as compared to typical condensation processes. Generally,
conducting condensation reactions under elevated
conditions results in unfavorable thermodynamics that
limit the extent of conversion to condensation products.
The present invention has revealed that conducting the
reaction with the condensation catalysts and at the
temperatures and pressures described below overcomes
these limitations and unexpectedly promotes an immediate
conversion of the condensation products to hydrocarbons,
ketones and alcohols. The conversion, in turn, removes
the condensation products from the reaction, thereby
overcoming the thermodynamic limitations of the system to
allow additional condensation reactions to occur.
Elevated temperature and/or pressure conditions also
avoid excessive conversion of the oxygenates directly to
their corresponding hydrocarbons. The process also has
the added benefit of allowing for the condensation
reactions, deoxygenation reactions and APR reactions to
occur in a single reactor and under steady-state
equilibrium.
For any given reaction, the free energy change is
indicative of the favorability of the forward reaction.
The more negative the free energy change, the more
favorable the reaction. As a result, reactions associated
with a highly negative change in free energy are
generally favorable and have the potential to exhibit
high conversions to reaction products. Conversely,
reactions associated with positive changes in free energy
are not favorable and are inherently limited in the
extent to which reactants are converted to products. As
16
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
an illustration, Figure 4 shows the free energy changes
associated with steps along the reaction pathway for
converting acetone and hydrogen to a C6 hydrocarbon (2-
methylpentane) and water at 100 C and 400 C. The known
free energy levels of the stable intermediates derived
along this pathway are shown with a solid line. The first
step in the reaction pathway is the aldol condensation of
two molecules of acetone to form one molecule of
diacetone alcohol. The reaction at the lower temperature
(100 C) has a free energy change of -53 KJ/mole and is
thermodynamically favored, while the reaction at the
higher temperature (400 C) is less favorable due to a
free energy change of -10 KJ/mole. The implication is
that the maximum conversion of pure acetone to diacetone
alcohol for this step decreases as the temperature is
increased (greater than 99% theoretical maximal
conversion at 100 C at atmospheric pressure, to only 15%
at 400 C at atmospheric pressure). Accordingly, the
thermodynamic equilibrium limitation imposes an absolute
limit to the amount of diacetone alcohol that may be
produced under given conditions and in the absence of
other reactions. This is further illustrated in Figure 5,
which provides the equilibrium constants associated with
the intermediate reaction products and the overall
conversion for the reaction of 2 moles of acetone with
3 moles of hydrogen to form 1 mole of 2-methylpentane and
2 moles of water. It can be seen that the equilibrium
constant for the conversion of acetone to diacetone
alcohol decreases with increasing temperature.
The present invention obviates this issue by
immediately converting the condensation product to a
compound that provides a more favorable reaction
environment. In the case above, by removing the diacetone
17
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
alcohol from the reaction mixture through a dehydration
reaction that forms mesityl oxide, additional diacetone
alcohol can be formed. In particular, the combination of
a condensation and dehydration step to provide mesityl
oxide and water from acetone provides a slightly more
favorable reaction environment. As illustrated in Figure
5, the conversion of acetone to mesityl oxide and water
is slightly more favorable at the higher temperatures.
The total reaction system pressure also has a
beneficial effect on the maximal theoretical extent to
which reactant may form a product. Considering the
condensation reaction example above, the conversion of
acetone to diacetone alcohol is limited to 15% at 400 C
at atmospheric pressure with pure acetone feed. By
increasing the system pressure to 600 psi gauge pressure,
the equilibrium conversion shifts so that up to 76%
conversion may be achieved at the same temperature. For
reactions exhibiting a net decrease in the number of
moles of product as compared to the moles of reactant, an
increase in system pressure (with all other conditions
held constant) will act to increase the equilibrium
product conversion. For the overall conversion of ketones
to hydrocarbons, there is typically a net decrease in the
moles of product compared to the moles of reactant, thus
higher reaction pressures would lead to higher potential
equilibrium conversions.
The process of preparing the component derived from
a water-soluble oxygenated hydrocarbon in the present
invention strikes a balance with the above thermodynamic
limitations by operating with condensation catalysts and
at temperature and pressure conditions that offset any
reduction in the production of condensation products with
an increase in the conversion to other downstream
18
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
products. The kinetics of the entire system is also more
favorable such that products may be produced continuously
and at a more desirable rate. In terms of scaled-up
production, after start-up, the reactor systems may be
process controlled, and the reactions could proceed at
steady-state equilibrium.
Oxygenates
The C.4+ compounds are derived from oxygenates. As
used herein in relation to the process of preparing the
component derived from a water-soluble oxygenated
hydrocarbon, "oxygenates" generically refers to
hydrocarbon compounds having 1 or more carbon atoms and
between 1 and 3 oxygen atoms (referred to herein as CI+01_
3 hydrocarbons), such as alcohols, ketones, aldehydes,
furans, hydroxy carboxylic acids, carboxylic acids, diols
and triols. Preferably, the oxygenates have from 1 to 6
carbon atoms, or 2 to 6 carbon atoms, or 3 to 6 carbon
atoms. Alcohols may include, without limitation, primary,
secondary, linear, branched or cyclic C1+ alcohols, such
as methanol, ethanol, n-propyl alcohol, isopropyl
alcohol, butyl alcohol, isobutyl alcohol, butanol,
pentanol, cyclopentanol, hexanol, cyclohexanol, 2-methyl-
cyclopentanonol, heptanol, octanol, nonanol, decanol,
undecanol, dodecanol, and isomers thereof. The ketones
may include, without limitation, hydroxyketones, cyclic
ketones, diketones, acetone, propanone, 2-oxopropanal,
butanone, butane-2,3-dione, 3-hydroxybutan-2-one,
pentanone, cyclopentanone, pentane-2,3-dione, pentane-
2,4-dione, hexanone, cyclohexanone, 2-methyl-
cyclopentanone, heptanone, octanone, nonanone, decanone,
undecanone, dodecanone, methylglyoxal, butanedione,
pentanedione, diketohexane, and isomers thereof. The
aldehydes may include, without limitation,
19
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
hydroxyaldehydes, acetaldehyde, propionaldehyde,
butyraldehyde, pentanal, hexanal, heptanal, octanal,
nonal, decanal, undecanal, dodecanal, and isomers
thereof. The carboxylic acids may include, without
limitation, formic acid, acetic acid, propionic acid,
butanoic acid, pentanoic acid, hexanoic acid, heptanoic
acid, isomers and derivatives thereof, including
hydroxylated derivatives, such as 2-hydroxybutanoic acid
and lactic acid. The diols may include, without
limitation, ethylene glycol, propylene glycol, 1,3-
propanediol, butanediol, pentanediol, hexanediol,
heptanediol, octanediol, nonanediol, decanediol,
undecanediol, dodecanediol, and isomers thereof. The
triols may include, without limitation, glycerol, 1,1,1
tris(hydroxymethyl)-ethane (trimethylolethane),
trimethylolpropane, hexanetriol, and isomers thereof.
Furans and furfurals include, without limitation, furan,
tetrahydrofuran, dihydrofuran, 2-furan methanol, 2-
methyl-tetrahydrofuran, 2,5-dimethyl-tetrahydrofuran, 2-
methyl furan, 2-ethyl-tetrahydrofuran, 2-ethyl furan,
hydroxylmethylfurfural, 3-hydroxytetrahydrofuran,
tetrahydro-3-furanol, 2,5-dimethyl furan, 5-
hydroxymethy1-2(5H)-furanone, dihydro-5-(hydroxymethyl)-
2(3H)-furanone, tetrahydro-2-furoic acid, dihydro-5-
(hydroxymethyl)-2(3H)-furanone, tetrahydrofurfuryl
alcohol, 1-(2-furyl)ethanol,
hydroxymethyltetrahydrofurfural, and isomers thereof.
The oxygenates may originate from any source, but
are preferably derived from biomass. As used herein, the
term "biomass" refers to, without limitation, organic
materials produced by plants (such as leaves, roots,
seeds and stalks), and microbial and animal metabolic
wastes. Common sources of biomass include: (1)
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
agricultural wastes, such as corn stalks, straw, seed
hulls, sugarcane leavings, bagasse, nutshells, and manure
from cattle, poultry, and hogs; (2) wood materials, such
as wood or bark, sawdust, timber slash, and mill scrap;
(3) municipal waste, such as waste paper and yard
clippings; and (4) energy crops, such as poplars,
willows, switch grass, alfalfa, prairie bluestream, corn,
soybean, and the like. The term also refers to the
primary building blocks of the above, namely,
saccharides, lignin, cellulosics, hemicellulose and
starches, among others.
Oxygenates from biomass may be produced by any known
method. Such methods include fermentation technologies
using enzymes or microorganisms, Fischer-Tropsch
reactions to produce C2-10 alpha alcohols, and pyrolysis
technologies to produce alcohols from oil, among others.
In one embodiment, the oxygenates are produced using
catalytic reforming technologies, such as the BioFormingTM
technology developed by Virent Energy Systems, Inc.
(Madison, Wisconsin).
Oxygenated Hydrocarbons
In one embodiment, the oxygenates are derived from
the catalytic reforming of oxygenated hydrocarbons. The
oxygenated hydrocarbons may be any water-soluble
oxygenated hydrocarbon having one or more carbon atoms
and at least one oxygen atom (referred to herein as C1+01+
hydrocarbons). Preferably, the oxygenated hydrocarbon has
2 to 12 carbon atoms (C1_1201_11 hydrocarbon), and more
preferably 2 to 6 carbon atoms (C1_601-6 hydrocarbon). The
oxygenated hydrocarbon may also have an oxygen-to-carbon
ratio ranging from 0.5:1 to 1.5:1, including ratios of
0.75:1.0, 1.0:1.0, 1.25:1.0, 1.5:1.0, and other ratios
between. In one example, the oxygenated hydrocarbon has
21
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
an oxygen-to-carbon ratio of 1:1. Nonlimiting examples of
preferred water-soluble oxygenated hydrocarbons include
monosaccharides, disaccharides, polysaccharides, sugar,
sugar alcohols, alditols, ethanediol, ethanedione, acetic
acid, propanol, propanediol, propionic acid, glycerol,
glyceraldehyde, dihydroxyacetone, lactic acid, pyruvic
acid, malonic acid, butanediols, butanoic acid,
aldotetroses, tautaric acid, aldopentoses, aldohexoses,
ketotetroses, ketopentoses, ketohexoses, alditols,
hemicelluloses, cellulosic derivatives, lignocellulosic
derivatives, starches, polyols and the like. Preferably,
the oxygenated hydrocarbon includes sugar, sugar
alcohols, saccharides and other polyhydric alcohols. More
preferably, the oxygenated hydrocarbon is a sugar, such
as glucose, fructose, sucrose, maltose, lactose, mannose
or xylose, or a sugar alcohol, such as arabitol,
erythritol, glycerol, isomalt, lactitol, malitol,
mannitol, sorbitol, xylitol, ribitol, or glycol.
Oxygenated hydrocarbons shall also refer to and
include alcohols derived by hydrogenation or
hydrogenolysis of any of the foregoing. In certain
embodiments, it may be preferable to convert the starting
oxygenated hydrocarbon to another oxygenated hydrocarbon
form that can be more readily converted to the desired
oxygenates (e.g., primary, secondary, tertiary or
polyhydric alcohols). For instance, some sugars may not
convert as efficiently to oxygenates as compared to their
corresponding sugar alcohol derivatives. It may therefore
be desirable to convert the starting material, such as a
sugar, furfural, carboxylic acid, ketone, or furan, into
its corresponding alcohol derivative, such as by
hydrogenation, or to smaller alcohol molecules, such as
by hydrogenolysis.
22
CA 02735654 2016-04-28
Various processes are known for hydrogenating
sugars, furfurals, carboxylic acids, ketones, and furans
to their corresponding alcohol form, including those
disclosed by: B.S. Kwak et al. (W02006/093364A1 and WO
2005/021475A1), involving the preparation of sugar
alditols from monosaccharides by hydrogenation over a
ruthenium catalyst; and Elliot et al. (U.S. Patent Nos.
6,253,797 and 6,570,043), disclosing the use of a nickel
and rhenium free ruthenium catalyst on a more than 75%
rutile titania support to convert sugars to sugar
alcohols. Other
suitable ruthenium catalysts are described by Arndt et
al. in published U.S. patent application 2006/0009661
(filed December 3, 2003), and Arena in U.S. Patent Nos.
4,380,679 (filed April 12, 1982), 4,380,680 (filed May
21, 1982), 4,503,274 (filed August 8, 1983), 4,382,150
(filed January 19, 1982), and 4,487,980 (filed April 29,
1983). The
hydrogenation catalyst generally includes Cu, Re, Ni, Fe,
Co, Ru, Pd, Rh, Pt, Os, Ir, and alloys or combinations
thereof, either alone or with promoters such as W, Mo,
Au, Ag, Cr, Zn, Mn, Sn, B, P, Bi, and alloys or
combinations thereof. The hydrogenation catalyst may also
include any one of the supports further described below,
and depending on the desired functionality of the
catalyst. Other effective hydrogenation catalyst
materials include either supported nickel or ruthenium
modified with rhenium. In general, the hydrogenation
reaction is carried out at hydrogenation temperatures of
between about 80 C to 250 C, and hydrogenation pressures
in the range of about 100 psig to 2000 psig. The hydrogen
used in the reaction may include in situ generated H2r
external H2r recycled H2, or a combination thereof.
23
CA 02735654 2016-04-28
The hydrogenation catalyst may also include a
supported Group VIII metal catalyst and a metal sponge
material, such as a sponge nickel catalyst. Activated
sponge nickel catalysts (e.g., Raney nickel) are a well-
known class of materials effective for various
hydrogenation reactions. One type of sponge nickel
catalyst is the type A7063 catalyst available from
Activated Metals and Chemicals, Inc., Sevierville, Tenn.
The type A7063 catalyst is a molybdenum promoted
catalyst, typically containing approximately 1.5%
molybdenum and 85% nickel. The use of the sponge nickel
catalyst with a feedstock comprising xylose and dextrose
is described by M. L. Cunningham et al. in U.S.
6,498,248, filed September 9, 1999.
The use of a Raney nickel catalyst with
hydrolyzed cornstarch is also described in U.S.
4,694,113, filed June 4, 1986.
The preparation of suitable Raney nickel
hydrogenation catalysts is described by A. Yoshino et al.
in published U.S. patent application 2004/0143024, filed
November 7, 2003. The
Raney nickel catalyst may be prepared by treating an
alloy of approximately equal amounts by weight of nickel
and aluminum with an aqueous alkali solution, e.g.,
containing about 25 wt. % of sodium hydroxide. The
aluminum is selectively dissolved by the aqueous alkali
solution leaving particles having a sponge construction
and composed predominantly of nickel with a minor amount
of aluminum. Promoter metals, such as molybdenum or
chromium, may be also included in the initial alloy in an
amount such that about 1-2 wt. % remains in the sponge
nickel catalyst.
24
CA 02735654 2016-04-28
In another embodiment, the hydrogenation catalyst is
prepared by impregnating a suitable support material with
a solution of ruthenium (III) nitrosylnitrate, ruthenium
(III) nitrosylnitrate, or ruthenium (III) chloride in
water to form a solid that is then dried for 13 hours at
120 C in a rotary ball oven (residual water content is
less than 1% by weight). The solid is then reduced at
atmospheric pressure in a hydrogen stream at 300 C
(uncalcined) or 400 C (calcined) in the rotary ball
furnace for 4 hours. After cooling and rendering inert
with nitrogen, the catalyst may then be passivated by
passing over 5% by volume of oxygen in nitrogen for a
period of 120 minutes.
In yet another embodiment, the hydrogenation
reaction is performed using a catalyst comprising a
nickel-rhenium catalyst or a tungsten-modified nickel
catalyst. One example of a suitable hydrogenation
catalyst is the carbon-supported nickel-rhenium catalyst
composition disclosed by Werpy et al. in U.S. 7,038,094,
filed September 30, 2003.
In other embodiments, it may also be desirable to
convert the starting oxygenated hydrocarbon, such as a
sugar, sugar alcohol or other polyhydric alcohol, to a
smaller molecule that can be more readily converted to
the desired oxygenates, such as by hydrogenolysis. Such
smaller molecules may include primary, secondary,
tertiary or polyhydric alcohols having less carbon atoms
than the originating oxygenated hydrocarbon. Various
processes are known for such hydrogenolysis reactions,
including those disclosed by: Werpy et al. in U.S. Patent
Nos. 6,479,713 (filed October 23, 2001), 6,677,385 (filed
August 6, 2002), 6,6841,085 (filed October 23, 2001) and
CA 02735654 2016-04-28
7,083,094 (filed September 30, 2003),
and describing the hydrogenolysis of
and 6 carbon sugars and sugar alcohols to propylene
glycol, ethylene glycol and glycerol using a rhenium-
5 containing multi-metallic catalyst. Other systems include
those described by Arena in U.S. Patent No. 4,401,823
(filed May 18, 1981) directed to the use of a
carbonaceous pyropolymer catalyst containing transition
metals (such as chromium, molybdenum, tungsten, rhenium,
manganese, copper, cadmium) or Group VIII metals (such as
iron, cobalt, nickel, platinum, palladium, rhodium,
ruthenium, iridium and osmium) to produce alcohols,
acids, ketones, and ethers from polyhydroxylated
compounds, such as sugars and sugar alcohols, and U.S.
Patent No. 4,496,780 (filed June 22, 1983) directed to
the use of a catalyst system having a Group VIII noble
metal on a solid support with an alkaline earth metal
oxide to produce glycerol, ethylene glycol and 1, 2-
propanediol from carbohydrates.
Another system includes that described by
Dubeck et al. in U.S. Patent No. 4,476,331 (filed
September 6, 1983) directed to the use of a sulfide-
modified ruthenium catalyst to produce ethylene glycol
and propylene glycol from larger polyhydric alcohols,
such as sorbitol.
Other systems include those described by Saxena et al.,
"Effect of Catalyst Constituents on
(Ni,MoandCu)/Kieselguhr-Catalyzed Sucrose
Hydrogenolysis," Ind. Eng. Chem. Res. 44, 1466-1473
(2005), describing the use of Ni, W, and Cu on a
kieselguhr support.
In one embodiment, the hydrogenolysis catalyst
includes Cr, Mo, W, Re, Mn, Cu, Cd, Fe, Co, Ni, Pt, Pd,
26
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
Rh, Ru, Ir, or Os, and alloys or combinations thereof,
either alone or with promoters such as Au, Ag, Cr, Zn,
Mn, Sn, Bi, B, 0 and alloys or combinations thereof.
Other effective hydrogenolysis catalyst materials may
include the above metals combined with an alkaline earth
metal oxide or adhered to catalytically active support,
such as kieselguhr, or any one of the supports further
described below.
The process conditions for carrying out the
hydrogenolysis reaction will vary depending on the type
of feedstock and desired products. In general, the
hydrogenolysis reaction is conducted at temperatures of
at least 110 C, or between 110 C and 300 C, or between
170 C and 240 C. The reaction should also be conducted
under basic conditions, preferably at a pH of about 8 to
about 13, or at a pH of about 10 to about 12. The
reaction should also be conducted at pressures of between
about 10 psig and 2400 psig, or between about 250 psig
and 2000 psig, or between about 700 psig and 1600 psig.
The hydrogen used in the reaction may include in
situ generated H2, external H2, recycled H2, or a
combination thereof.
Production of Oxygenates
The oxygenates are prepared by reacting an aqueous
feedstock solution containing water and the water soluble
oxygenated hydrocarbons with hydrogen over a catalytic
material to produce the desired oxygenates. Preferably,
the hydrogen is generated in situ using aqueous phase
reforming (in situ generated H2 or APR H2), or a
combination of APR H2, external H2 or recycled H2, or just
simply external H2 or recycled H2. The term "external H2"
refers to hydrogen that does not originate from the
feedstock solution, but is added to the reactor system
27
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
from an external source. The term "recycled H2" refers to
unconsumed hydrogen that originates from the feedstock
solution, and which is collected and then recycled back
into the reactor system for further use. External H2 and
recycled H2 may also be referred to collectively or
individually as "supplemental H2." In general,
supplemental H2 may be added for purposes of
supplementing the APR hydrogen, or to substitute the
inclusion of an APR hydrogen production step, or to
increase the reaction pressure within the system, or to
increase the molar ratio of hydrogen to carbon and/or
oxygen in order to enhance the production yield of
certain reaction product types, such as ketones and
alcohols.
In processes utilizing APR H2, the oxygenates are
prepared by catalytically reacting a portion of the
aqueous feedstock solution containing water and the water
soluble oxygenated hydrocarbons in the presence of an APR
catalyst at a reforming temperature and reforming
pressure to produce the APR H2, and catalytically
reacting the APR H2 (and recycled H2 and/or external H2)
with a portion of the feedstock solution in the presence
of a deoxygenation catalyst at a deoxygenation
temperature and deoxygenation pressure to produce the
desired oxygenates. In systems utilizing recycled H2 or
external H2 as a hydrogen source, the oxygenates are
simply prepared by catalytically reacting the recycled H2
and/or external H2 with the feedstock solution in the
presence of the deoxygenation catalyst at the
deoxygenation temperatures and pressures. In each of the
above, the oxygenates may also include recycled
oxygenates (recycled C1+01_2 hydrocarbons). Unless
otherwise indicated, any discussions of APR catalysts and
28
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
deoxygenation catalysts are non-limiting examples of
suitable catalytic materials.
The deoxygenation catalyst is preferably a
heterogeneous catalyst having one or more materials
capable of catalyzing a reaction between hydrogen and the
oxygenated hydrocarbon to remove one or more of the
oxygen atoms from the oxygenated hydrocarbon to produce
alcohols, ketones, aldehydes, furans, carboxylic acids,
hydroxy carboxylic acids, diols and triols. In general,
the materials will be adhered to a support and may
include, without limitation, Cu, Re, Fe, Ru, Ir, Co, Rh,
Pt, Pd, Ni, W, Os, Mo, Ag, Au, alloys and combinations
thereof. The deoxygenation catalyst may include these
elements alone or in combination with one or more Mn, Cr,
Mo, W, V, Nb, Ta, Ti, Zr, Y, La, Sc, Zn, Cd, Ag, Au, Sn,
Ge, P, Al, Ga, In, Tl, and combinations thereof. In one
embodiment, the deoxygenation catalyst includes Pt, Ru,
Cu, Re, Co, Fe, Ni, W or Mo. In yet another embodiment,
the deoxygenation catalyst includes Fe or Re and at least
one transition metal selected from Ir, Ni, Pd, P, Rh, and
Ru. In another embodiment, the catalyst includes Fe, Re
and at least Cu or one Group VIIIB transition metal. The
support may be any one of the supports further described
below, including a nitride, carbon, silica, alumina,
zirconia, titania, vanadia, ceria, zinc oxide, chromia,
boron nitride, heteropolyacids, kieselguhr,
hydroxyapatite, and mixtures thereof. The deoxygenation
catalyst may also be atomically identical to the APR
catalyst or the condensation catalyst.
The deoxygenation catalyst may also be a bi-
functional catalyst. For example, acidic supports (e.g.,
supports having low isoelectric points) are able to
catalyze dehydration reactions of oxygenated compounds,
29
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
followed by hydrogenation reactions on metallic catalyst
sites in the presence of H2, again leading to carbon
atoms that are not bonded to oxygen atoms. The bi-
functional dehydration/ hydrogenation pathway consumes H2
and leads to the subsequent formation of various polyols,
diols, ketones, aldehydes, alcohols and cyclic ethers,
such as furans and pyrans. Catalyst examples include
tungstated zirconia, titania zirconia, sulfated zirconia,
acidic alumina, silica-alumina, zeolites and
heteropolyacid supports. Heteropolyacids are a class of
solid-phase acids exemplified by such species as H3+PM012-
xVx040 1 H 4S iW 12 40 1 H3PW 12040 f and H6P2W1B062. Heteropolyacids
are solid-phase acids having a well-defined local
structure, the most common of which is the tungsten-based
Keggin structure.
Loading of the first element (i.e., Cu, Re, Fe, Ru,
Ir, Co, Rh, Pt, Pd, Ni, W, Os, Mo, Ag, Au, alloys and
combinations thereof) is in the range of 0.25 wt% to 25
wt% on carbon, with weight percentages of 0.10% and 0.05%
increments between, such as 1.00%, 1.10%, 1.15%, 2.00%,
2.50%, 5.00%, 10.00%, 12.50%, 15.00% and 20.00%. The
preferred atomic ratio of the second element (i.e., Mn,
Cr, Mo, W, V, Nb, Ta, Ti, Zr, Y, La, Sc, Zn, Cd, Ag, Au,
Sn, Ge, P, Al, Ga, In, Tl, and combinations thereof) is
in the range of 0.25-to-1 to 10-to-1, including any
ratios between, such as 0.50, 1.00, 2.50, 5.00, and 7.50-
to-1. If the catalyst is adhered to a support, the
combination of the catalyst and the support is from 0.25
wt% to 10 wt% of the primary element.
To produce oxygenates, the oxygenated hydrocarbon is
combined with water to provide an aqueous feedstock
solution having a concentration effective for causing the
formation of the desired reaction products. The water-to-
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
carbon ratio on a molar basis is preferably from about
0.5:1 to about 100:1, including ratios such as 1:1, 2:1,
3:1, 4:1, 5:1, 6:1, 7:1, 8:1, 9:1, 10:1, 15:1, 25:1, 50:1
75:1, 100:1, and any ratios there-between. The feedstock
solution may also be characterized as a solution having
at least 1.0 weight percent (wt%) of the total solution
as an oxygenated hydrocarbon. For instance, the solution
may include one or more oxygenated hydrocarbons, with the
total concentration of the oxygenated hydrocarbons in the
solution being at least about 1%, 5%, 10%, 20%, 30%, 40%,
50%, 60%, 70%, 80% or greater by weight, including any
percentages between, and depending on the oxygenated
hydrocarbons used. In one embodiment, the feedstock
solution includes at least about 10%, 20%, 30%, 40%, 50%,
or 60% of a sugar, such as glucose, fructose, sucrose or
xylose, or a sugar alcohol, such as sorbitol, mannitol,
glycerol or xylitol, by weight. Water-to-carbon ratios
and percentages outside of the above stated ranges are
also included. Preferably the balance of the feedstock
solution is water. In some embodiments, the feedstock
solution consists essentially of water, one or more
oxygenated hydrocarbons and, optionally, one or more
feedstock modifiers described herein, such as alkali or
hydroxides of alkali or alkali earth salts or acids. The
feedstock solution may also include recycled oxygenated
hydrocarbons recycled from the reactor system. The
feedstock solution may also contain negligible amounts of
hydrogen, preferably less than about 1.5 mole of hydrogen
per mole of feedstock. In the preferred embodiments,
hydrogen is not added to the feedstock solution.
The feedstock solution is reacted with hydrogen in
the presence of the deoxygenation catalyst at
deoxygenation temperature and pressure conditions, and
31
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
weight hourly space velocity, effective to produce the
desired oxygenates. The specific oxygenates produced will
depend on various factors, including the feedstock
solution, reaction temperature, reaction pressure, water
concentration, hydrogen concentration, the reactivity of
the catalyst, and the flow rate of the feedstock solution
as it affects the space velocity (the mass/volume of
reactant per unit of catalyst per unit of time), gas
hourly space velocity (GHSV), and weight hourly space
velocity (WHSV). For example, an increase in flow rate,
and thereby a reduction of feedstock exposure to the
catalysts over time, will limit the extent of the
reactions which may occur, thereby causing increased
yield for higher level diols and triols, with a reduction
in ketone and alcohol yields.
The deoxygenation temperature and pressure are
preferably selected to maintain at least a portion of the
feedstock in the liquid phase at the reactor inlet. It is
recognized, however, that temperature and pressure
conditions may also be selected to more favorably produce
the desired products in the vapor-phase. In general, the
reaction should be conducted at process conditions
wherein the thermodynamics of the proposed reaction are
favorable. For instance, the minimum pressure required to
maintain a portion of the feedstock in the liquid phase
will likely vary with the reaction temperature. As
temperatures increase, higher pressures will generally be
required to maintain the feedstock in the liquid phase,
if desired. Pressures above that required to maintain the
feedstock in the liquid phase (i.e., vapor-phase) are
also suitable operating conditions.
In condensed phase liquid reactions, the pressure
within the reactor must be sufficient to maintain the
32
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
reactants in the condensed liquid phase at the reactor
inlet. For liquid phase reactions, the reaction
temperature may be from about 80 C to 300 C, and the
reaction pressure from about 72 psig to 1300 psig. In one
embodiment, the reaction temperature is between about
120 C and 300 C, or between about 200 C and 280 C, or
between about 220 C and 260 C, and the reaction pressure
is preferably between about 72 and 1200 psig, or between
about 145 and 1200 psig, or between about 200 and 725
psig, or between about 365 and 700 psig, or between about
600 and 650 psig.
For vapor phase reactions, the reaction should be
carried out at a temperature where the vapor pressure of
the oxygenated hydrocarbon is at least about 0.1 atm (and
preferably a good deal higher), and the thermodynamics of
the reaction are favorable. This temperature will vary
depending upon the specific oxygenated hydrocarbon
compound used, but is generally in the range of from
about 100 C to 600 C for vapor phase reactions.
Preferably, the reaction temperature is between about
120 C and about 300 C, or between about 200 C and about
280 C, or between about 220 C and about 260 C.
In another embodiment, the deoxygenation temperature
is between about 100 C and 400 C, or between about 120 C
and 300 C, or between about 200 C and 280 C, and the
reaction pressure is preferably between about 72 and 1300
psig, or between about 72 and 1200 psig, or between about
200 and 725 psig, or between about 365 and 700 psig.
A condensed liquid phase method may also be
performed using a modifier that increases the activity
and/or stability of the catalyst system. It is preferred
that the water and the oxygenated hydrocarbon are reacted
at a suitable pH of from about 1.0 to about 10.0,
33
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
including pH values in increments of 0.1 and 0.05
between, and more preferably at a pH of from about 4.0 to
about 10Ø Generally, the modifier is added to the
feedstock solution in an amount ranging from about 0.1%
to about 10% by weight as compared to the total weight of
the catalyst system used, although amounts outside this
range are included within the present invention.
In general, the reaction should be conducted under
conditions where the residence time of the feedstock
solution over the catalyst is appropriate to generate the
desired products. For example, the WHSV for the reaction
may be at least about 0.1 gram of oxygenated hydrocarbon
per gram of catalyst per hour, and more preferably the
WHSV is about 0.1 to 40.0 g/g hr, including a WHSV of
about 0.25, 0.5, 0.75, 1.0, 1.0, 1.1, 1.2, 1.3, 1.4, 1.5,
1.6, 1.7, 1.8, 1.9, 2.0, 2.1, 2.2, 2.3, 2.4, 2.5, 2.6,
2.7, 2.8, 2.9, 3.0, 3.1, 3.2, 3.3, 3.4, 3.5, 3.6, 3.7,
3.8, 3.9, 4.0, 4.1, 4.2, 4.3, 4.4, 4.5, 4.6, 4.7, 4.8,
4.9, 5.0, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 20, 25, 30,
35, 40 g/g hr.
The hydrogen used in the deoxygenation reaction is
preferably in-situ generated H2, but may also be external
or recycled H2. When present, the amount of external H2
is preferably provided sparingly. Most preferably, the
amount of external H2 is provided in amounts that provide
less than one hydrogen atom per oxygen atom in all of the
oxygenated hydrocarbons in the feedstock stream prior to
contacting the deoxygenation catalyst. For example, the
molar ratio between the external H2 and the total water-
soluble oxygenated hydrocarbons in the feedstock solution
is preferably selected to provide no more than one
hydrogen atom per oxygen atom in the oxygenated
hydrocarbon. The molar ratio of the oxygenated
34
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
hydrocarbons in the feedstock to the external H2
introduced to the feedstock is also preferably not more
than 1:1, or more preferably up to 2:1, 3:1, 5:1, 10:1,
20:1 or greater (including 4:1, 6:1, 7:1, 8:1, 9:1, 11:1,
12:1, 13:1, 14:1, 15:1, 16:1, 17:1, 18:1 and 19:1). The
amount (moles) of external H2 introduced to the feedstock
is between 0 - 100%, 0 - 95%, 0 - 90%, 0 - 85%, 0 - 80%,
0 - 75%, 0 - 70%, 0 - 65%, 0 - 60%, 0 - 55%, 0 - 50%, 0 -
45%, 0 - 40%, 0 - 35%, 0 - 30%, 0 - 25%, 0 - 20%, 0 -
15%, 0 - 10%, 0 - 5%, 0 - 2%, or 0 - 1% of the total
number of moles of the oxygenated hydrocarbon(s) in the
feedstock, including all intervals between. When the
feedstock solution, or any portion thereof, is reacted
with APR hydrogen and external H2, the molar ratio of APR
hydrogen to external H2 is at least 1:20; 1:15, 1:10,
1:5; 1:3, 1:2, 1:1, 2:1, 3:1, 5:1, 10:1, 15:1, 20:1, and
ratios between (including 4:1, 6:1, 7:1, 8:1, 9:1, 11:1,
12:1, 13:1, 14:1, 15:1, 16:1, 17:1, 18:1 and 19:1, and
vice-versa). Preferably, the oxygenated hydrocarbon is
reacted with H2 in the presence of an insignificantly
effective amount of external H2.
The amount of external H2 (or supplemental H2) added
may be calculated by considering the concentration of the
oxygenated hydrocarbons in the feedstock solution.
Preferably, the amount of external H2 added should
provide a molar ratio of oxygen atoms in the oxygenated
hydrocarbons to moles of hydrogen atoms (i.e., 2 oxygen
atoms per molecule of H2 gas) of less than or equal to
1Ø For example, where the feedstock is an aqueous
solution consisting of glycerol (3 oxygen atoms), the
amount of supplemental H2 added to the feedstock is
preferably not more than about 1.5 moles of H2 per mole
of glycerol (C3H803), and preferably not more than about
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
1.25, 1.0, 0.75, 0.50 or 0.25. In general, the amount of
supplemental H2 added is less than 0.75-times, and more
preferably not more than 0.67, 0.50, 0.33, 0.30, 0.25,
0.20, 0.15, 0.10, 0.05, 0.01-times the amount of total H2
(APR H2 and external H2) that would provide a 1:1 atomic
ratio of oxygen to hydrogen atoms.
The amount of APR H2 within a reactor may be
identified or detected by any suitable method. APR H2 may
be determined based on the composition of the product
stream as a function of the composition of the feedstock
stream, the catalyst composition(s) and the reaction
conditions, independent of the actual reaction mechanism
occurring within the feedstock stream. The amount of APR
H2 may be calculated based on the catalyst, reaction
conditions (e.g., flow rate, temperature, pressure, etc.)
and the contents of the feedstock and the reaction
products. For example, the feedstock may be contacted
with the APR catalyst (e.g., platinum) to generate APR H2
in situ and a first reaction product stream in the
absence of a deoxygenation catalyst. The feedstock may
also be contacted with both the APR catalyst and the
deoxygenation catalyst to produce a second reaction
product stream. By comparing the composition of the first
reaction product stream and the second reaction product
stream at comparable reaction conditions, one may
identify the presence of APR H2 and calculate the amount
of APR H2 produced. For example, an increase in the
amount of oxygenated compounds with greater degrees of
hydrogenation in the reaction product compared to the
feedstock components may indicate the presence of APR H2.
In-situ Hydrogen Production
One advantage of the process of preparing the
component derived from a water-soluble oxygenated
36
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
hydrocarbon in the present invention is that it allows
for the production and use of in-situ generated H2. The
APR H2 is produced from the feedstock under aqueous phase
reforming conditions using an aqueous phase reforming
catalyst (APR catalyst). The APR catalyst is preferably a
heterogeneous catalyst capable of catalyzing the reaction
of water and oxygenated hydrocarbons to form H2 under the
conditions described below. In one embodiment, the APR
catalyst includes a support and at least one Group VIIIB
metal, Fe, Ru, Os, Ir, Co, Rh, Pt, Pd, Ni, alloys and
combinations thereof. The APR catalyst may also include
at least one additional material from Group VIIIB, Group
VIIB, Group VIB, Group VB, Group IVB, Group IIB, Group
IB, Group IVA or Group VA metals, such as Cu, B, Mn, Re,
Cr, Mo, Bi, W, V, Nb, Ta, Ti, Zr, Y, La, Sc, Zn, Cd, Ag,
Au, Sn, Ge, P, Al, Ga, In, Tl, alloys and combinations
thereof. The preferred Group VIIB metal includes Re, Mn,
or combinations thereof. The preferred Group VIB metal
includes Cr, Mo, W, or a combination thereof. The
preferred Group VIIIB metals include Pt, Rh, Ru, Pd, Ni,
or combinations thereof. The supports may include any one
of the catalyst supports described below, depending on
the desired activity of the catalyst system.
The APR catalyst may also be atomically identical to
the deoxygenation catalyst or the condensation catalyst.
For instance, the APR and deoxygenation catalyst may
include Pt alloyed or admixed with Ni, Ru, Cu, Fe, Rh,
Re, alloys and combinations thereof. The APR catalyst and
deoxygenation catalyst may also include Ru alloyed or
admixed with Ge, Bi, B, Ni, Sn, Cu, Fe, Rh, Pt, alloys
and combinations thereof. The APR catalyst may also
include Ni alloyed or admixed with Sn, Ge, Bi, B, Cu, Re,
Ru, Fe, alloys and combinations thereof.
37
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
Preferred loading of the primary Group VIIIB metal
is in the range of 0.25 wt% to 25 wt% on carbon, with
weight percentages of 0.10% and 0.05% increments between,
such as 1.00%, 1.10%, 1.15%, 2.00%, 2.50%, 5.00%, 10.00%,
12.50%, 15.00% and 20.00%. The preferred atomic ratio of
the second material is in the range of 0.25-to-1 to 10-
to-1, including ratios between, such as 0.50, 1.00, 2.50,
5.00, and 7.50-to-1.
A preferred catalyst composition is further achieved
by the addition of oxides of Group IIIB, and associated
rare earth oxides. In such event, the preferred
components would be oxides of either lanthanum or cerium.
The preferred atomic ratio of the Group IIIB compounds to
the primary Group VIIIB metal is in the range of 0.25-to-
1 to 10-to-1, including ratios between, such as 0.50,
1.00, 2.50, 5.00, and 7.50-to-1.
Another preferred catalyst composition is one
containing platinum and rhenium. The preferred atomic
ratio of Pt to Re is in the range of 0.25-to-1 to 10-to-
1, including ratios there-between, such as 0.50, 1.00,
2.50, 5.00, and 7.00-to-1. The preferred loading of the
Pt is in the range of 0.25 wt% to 5.0 wt%, with weight
percentages of 0.10% and 0.05% between, such as .35%,
.45%, .75%, 1.10%, 1.15%, 2.00%, 2.50%, 3.0%, and 4.0%.
Preferably, the APR catalyst and the deoxygenation
catalyst are of the same atomic formulation. The
catalysts may also be of different formulations. In such
event, the preferred atomic ratio of the APR catalyst to
the deoxygenation catalyst is in the range of 5:1 to 1:5,
such as, without limitation, 4.5:1, 4.0:1,3.5:1, 3.0:1,
2.5:1, 2.0:1, 1.5:1, 1:1, 1:1.5, 1:2.0, 1:2.5, 1:3.0,
1:3.5, 1:4.0, 1:4.5, and any amounts between.
38
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
Similar to the deoxygenation reactions, the
temperature and pressure conditions are preferably
selected to maintain at least a portion of the feedstock
in the liquid phase at the reactor inlet. The reforming
temperature and pressure conditions may also be selected
to more favorably produce the desired products in the
vapor-phase. In general, the APR reaction should be
conducted at a temperature where the thermodynamics are
favorable. For instance, the minimum pressure required to
maintain a portion of the feedstock in the liquid phase
will vary with the reaction temperature. As temperatures
increase, higher pressures will generally be required to
maintain the feedstock in the liquid phase. Any pressure
above that required to maintain the feedstock in the
liquid phase (i.e., vapor-phase) is also a suitable
operating pressure. For vapor phase reactions, the
reaction should be conducted at a reforming temperature
where the vapor pressure of the oxygenated hydrocarbon
compound is at least about 0.1 atm (and preferably a good
deal higher), and the thermodynamics of the reaction are
favorable. The temperature will vary depending upon the
specific oxygenated hydrocarbon compound used, but is
generally in the range of from about 100 C to 450 C, or
from about 100 C to 300 C, for reactions taking place in
the vapor phase. For liquid phase reactions, the reaction
temperature may be from about 80 C to 400 C, and the
reaction pressure from about 72 psig to 1300 psig.
In one embodiment, the reaction temperature is
between about 100 C and 400 C, or between about 120 C and
300 C, or between about 200 C and 280 C, or between about
150 C and 270 C. The reaction pressure is preferably
between about 72 and 1300 psig, or between about 72 and
1200 psig, or between about 145 and 1200 psig, or between
39
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
about 200 and 725 psig, or between about 365 and 700
psig, or between about 600 and 650 psig.
A condensed liquid phase method may also be
performed using a modifier that increases the activity
and/or stability of the APR catalyst system. It is
preferred that the water and the oxygenated hydrocarbon
are reacted at a suitable pH of from about 1.0 to 10.0,
or at a pH of from about 4.0 to 10.0, including pH value
increments of 0.1 and 0.05 between. Generally, the
modifier is added to the feedstock solution in an amount
ranging from about 0.1% to about 10% by weight as
compared to the total weight of the catalyst system used,
although amounts outside this range are included within
the present invention.
Alkali or alkali earth salts may also be added to
the feedstock solution to optimize the proportion of
hydrogen in the reaction products. Examples of suitable
water-soluble salts include one or more selected from the
group consisting of an alkali or an alkali earth metal
hydroxide, carbonate, nitrate, or chloride salt. For
example, adding alkali (basic) salts to provide a pH of
about pH 4.0 to about pH 10.0 can improve hydrogen
selectivity of reforming reactions.
The addition of acidic compounds may also provide
increased selectivity to the desired reaction products in
the hydrogenation reactions described below. It is
preferred that the water-soluble acid is selected from
the group consisting of nitrate, phosphate, sulfate,
chloride salts, and mixtures thereof. If an acidic
modifier is used, it is preferred that it be present in
an amount sufficient to lower the pH of the aqueous feed
stream to a value between about pH 1.0 and about pH 4Ø
Lowering the pH of a feed stream in this manner may
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
increase the proportion of oxygenates in the final
reaction products.
In general, the reaction should be conducted under
conditions where the residence time of the feedstock
solution over the APR catalyst is appropriate to generate
an amount of APR hydrogen sufficient to react with a
second portion of the feedstock solution over the
deoxygenation catalyst to provide the desired oxygenates.
For example, the WHSV for the reaction may be at least
about 0.1 gram of oxygenated hydrocarbon per gram of APR
catalyst, and preferably between about 1.0 to 40.0 grams
of oxygenated hydrocarbon per gram of APR catalyst, and
more preferably between about 0.5 to 8.0 grams of
oxygenated hydrocarbon per gram of APR catalyst. In terms
of scaled-up production, after start-up, the APR reactor
system should be process controlled so that the reactions
proceed at steady-state equilibrium.
Condensation Step
The oxygenates produced are then converted into C.4+
compounds by condensation. Without being limited to any
specific theories, it is believed that the acid
condensation reactions generally consist of a series of
steps involving: (a) the dehydration of oxygenates to
olefins; (b) oligomerization of the olefins; (c) cracking
reactions; (d) cyclization of larger olefins to form
aromatics; (e) paraffin isomerization; and (f) hydrogen-
transfer reactions to form paraffins. Basic condensation
reactions are believed to generally consist of a series
of steps involving: (1) aldol condensation to form a p-
hydroxyketone or p-hydroxyaldehyde; (2) dehydration of
the p-hydroxyketone or p-hydroxyaldehyde to form a
conjugated enone; (3) hydrogenation of the conjugated
enone to form a ketone or aldehyde, which may participate
41
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
in further condensation reactions or conversion to an
alcohol or hydrocarbon; and (4) hydrogenation of
carbonyls to alcohols, or vice-versa. Acid-base
condensation reactions are believed to generally involve
any of the previous acidic and/or basic reactions steps.
Production of the C.4+ compounds occurs by
condensation of the oxygenates in the presence of a
condensation catalyst. The condensation catalyst will
generally be a catalyst capable of forming longer chain
compounds by linking two oxygen containing species
through a new carbon-carbon bond, and converting the
resulting compound to a hydrocarbon, alcohol or ketone,
such as an acid catalyst, basic catalyst or a multi-
functional catalyst having both acid and base
functionality. The condensation catalyst may include,
without limitation, carbides, nitrides, zirconia,
alumina, silica, aluminosilicates, phosphates, zeolites,
titanium oxides, zinc oxides, vanadium oxides, lanthanum
oxides, yttrium oxides, scandium oxides, magnesium
oxides, cerium oxides, barium oxides, calcium oxides,
hydroxides, heteropolyacids, inorganic acids, acid
modified resins, base modified resins, and combinations
thereof. The condensation catalyst may include the above
alone or in combination with a modifier, such as Ce, La,
Y, Sc, P, B, Bi, Li, Na, K, Rb, Cs, Mg, Ca, Sr, Ba, and
combinations thereof. The condensation catalyst may also
include a metal, such as Cu, Ag, Au, Pt, Ni, Fe, Co, Ru,
Zn, Cd, Ga, In, Rh, Pd, Ir, Re, Mn, Cr, Mo, W, Sn, Os,
alloys and combinations thereof, to provide a metal
functionality. The condensation catalyst may also be
atomically identical to the APR catalyst and/or the
deoxygenation catalyst.
42
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
The condensation catalyst may be self-supporting
(i.e., the catalyst does not need another material to
serve as a support), or may require a separate support
suitable for suspending the catalyst in the reactant
stream. One particularly beneficial support is silica,
especially silica having a high surface area (greater
than 100 square meters per gram), obtained by sol-gel
synthesis, precipitation or fuming. In other embodiments,
particularly when the condensation catalyst is a powder,
the catalyst system may include a binder to assist in
forming the catalyst into a desirable catalyst shape.
Applicable forming processes include extrusion,
pelletization, oil dropping, or other known processes.
Zinc oxide, alumina, and a peptizing agent may also be
mixed together and extruded to produce a formed material.
After drying, this material is calcined at a temperature
appropriate for formation of the catalytically active
phase, which usually requires temperatures in excess of
450 C. Other catalyst supports may include those
described in further detail below.
Acid Catalysts
The acid condensation reaction is performed using
acidic catalysts. The acid catalysts may include, without
limitation, aluminosilicates (zeolites), silica-alumina
phosphates (SAPO), aluminum phosphates (ALPO), amorphous
silica alumina, zirconia, sulfated zirconia, tungstated
zirconia, tungsten carbide, molybdenum carbide, titania,
acidic alumina, phosphated alumina, phosphated silica,
sulfated carbons, phosphated carbons, acidic resins,
heteropolyacids, inorganic acids, and combinations
thereof. In one embodiment, the catalyst may also include
a modifier, such as Ce, Y, Sc, La, P, B, Bi, Li, Na, K,
Rb, Cs, Mg, Ca, Sr, Ba, and combinations thereof. The
43
CA 02735654 2016-04-28
catalyst may also be modified by the addition of a metal,
such as Cu, Ag, Au, Pt, Ni, Fe, Co, Ru, Zn, Cd, Ga, In,
Rh, Pd, Ir, Re, Mn, Cr, Mo, W, Sn, Os, alloys and
combinations thereof, to provide metal functionality,
and/or sulfides and oxide of Ti, Zr, V, Nb, Ta, Mo, Cr,
W, Mn, Re, Al, Ga, In, Fe, Co, Ir, Ni, Si, Cu, Zn, Sn,
Cd, P, and combinations thereof. Gallium has also been
found to be particularly useful as a promoter for the
present process. The acid catalyst may be homogenous,
self-supporting or adhered to any one of the supports
further described below, including supports containing
carbon, silica, alumina, zirconia, titania, vanadia,
ceria, nitride, boron nitride, heteropolyacids, alloys
and mixtures thereof.
Ga, In, Zn, Fe, Mo, Ag, Au, Ni, P, Sc, Y, Ta, and
lanthanides may also be exchanged onto zeolites to
provide a zeolite catalyst having activity. The term
"zeolite" as used herein refers not only to microporous
crystalline aluminosilicate but also for microporous
crystalline metal-containing aluminosilicate structures,
such as galloaluminosilicates and gallosilicates. Metal
functionality may be provided by metals such as Cu, Ag,
Au, Pt, Ni, Fe, Co, Ru, Zn, Cd, Ga, In, Rh, Pd, Ir, Re,
Mn, Cr, Mo, W, Sn, Os, alloys and combinations thereof.
Examples of suitable zeolite catalysts include ZSM-
5, ZSM-11, ZSM-12, ZSM-22, ZSM-23, ZSM-35 and ZSM-48.
Zeolite ZSM-5, and the conventional preparation thereof,
is described in U.S. Pat. Nos. 3,702,886; Re. 29,948
(highly siliceous ZSM-5); 4,100,262 and 4,139,600.
Zeolite ZSM-11, and the
conventional preparation thereof, is described in U.S.
Pat. No.3,709,979.
Zeolite ZSM-12, and the conventional
44
CA 02735654 2016-04-28
preparation thereof, is described in U.S. Pat.
No.3,832,449. Zeolite
ZSM-23, and the conventional preparation thereof, is
described in U.S. Pat. No. 4,076,842.
Zeolite ZSM-35, and the conventional
preparation thereof, is described in U.S. Pat. No.
4,016,245. Another
preparation of ZSM-35 is described in U.S. Pat. No.
4,107,195.
ZSM-48, and the conventional preparation
thereof, is taught by U.S. Pat. No.4,375,573.
Other examples of
zeolite catalysts are described in US Patent 5,019,663
and US Patent 7,022,888.
As described in U.S. Patent 7,022,888, the acid
catalyst may be a bifunctional pentasil zeolite catalyst
including at least one metallic element from the group of
Cu, Ag, Au, Pt, Ni, Fe, Co, Ru, Zn, Cd, Ga, In, Rh, Pd,
Ir, Re, Mn, Cr, Mo, W, Sn, Os, alloys and combinations
thereof, or a modifier from the group of Ga, In, Zn, Fe,
Mo, Au, Ag, Y, Sc, Ni, P, Ta, lanthanides, and
combinations thereof. The zeolite preferably has a strong
acidic and dehydrogenation sites, and may be used with
reactant streams containing and an oxygenated hydrocarbon
at a temperature of below 500 C. The bifunctional
pentasil zeolite may have ZSM-5, ZSM-8 or ZSM-11 type
crystal structure consisting of a large number of 5-
membered oxygen-rings, i.e., pentasil rings. The zeolite
with ZSM-5 type structure is a particularly preferred
catalyst. The bifunctional pentasil zeolite catalyst is
preferably Ga and/or In-modified ZSM-5 type zeolites such
as Ga and/or In-impregnated H-ZOM-5, Ga and/or In-
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
exchanged H-ZSM-5, H-gallosilicate of ZSM-5 type
structure and H-galloaluminosilicate of ZSM-5 type
structure. The bifunctional ZSM-5 type pentasil zeolite
may contain tetrahedral aluminum and/or gallium present
in the zeolite framework or lattice and octahedral
gallium or indium. The octahedral sites are preferably
not present in the zeolite framework but are present in
the zeolite channels in a close vicinity of the zeolitic
protonic acid sites, which are attributed to the presence
of tetrahedral aluminum and gallium in the zeolite. The
tetrahedral or framework Al and/or Ga is believed to be
responsible for the acid function of zeolite and
octahedral or non-framework Ga and/or In is believed to
be responsible for the dehydrogenation function of the
zeolite.
In one embodiment, the condensation catalyst may be
a H-galloaluminosilicate of ZSM-5 type bifunctional
pentasil zeolite having framework (tetrahedral) Si/A1 and
Si/Ga mole ratio of about 10-100 and 15-150,
respectively, and non-framework (octahedral) Ga of about
0.5-5.0 wt. %. When these pentasil H-galloaluminosilicate
zeolites are used as a condensation catalyst, the density
of strong acid sites can be controlled by the framework
Al/Si mole ratio: the higher the Al/Si ratio, the higher
the density of strong acid sites. The highly dispersed
non-framework gallium oxide species can be obtained by
the degalliation of the zeolite by its pre-treatment with
H2 and steam. The zeolite containing strong acid sites
with high density and also highly dispersed non-framework
gallium oxide species in close proximity of the zeolite
acid site is preferred. The catalyst may optionally
contain any binder such as alumina, silica or clay
material. The catalyst can be used in the form of
46
CA 02735654 2016-04-28
pellets, extrudates and particles of different shapes and
sizes.
The acidic catalysts may include one or more zeolite
structures comprising cage-like structures of silica-
alumina. Zeolites are crystalline microporous materials
with well-defined pore structure. Zeolites contain active
sites, usually acid sites, which can be generated in the
zeolite framework. The strength and concentration of the
active sites can be tailored for particular applications.
Examples of suitable zeolites for condensing secondary
alcohols and alkanes may comprise aluminosilicates
optionally modified with cations, such as Ga, In, Zn, Mo,
and mixtures of such cations, as described, for example,
in U.S. Patent No. 3,702,886.
As recognized in the art, the
structure of the particular zeolite or zeolites may be
altered to provide different amounts of various
hydrocarbon species in the product mixture. Depending on
the structure of the zeolite catalyst, the product
mixture may contain various amounts of aromatic and
cyclic hydrocarbons.
Alternatively, solid acid catalysts such as alumina
modified with phosphates, chloride, silica, and other
acidic oxides could be used in practicing the present
invention. Also, either sulfated zirconia or tungstated
zirconia may provide the necessary acidity. Re and Pt/Re
catalysts are also useful for promoting condensation of
oxygenates to C51. hydrocarbons and/or C5i. mono-oxygenates.
The Re is sufficiently acidic to promote acid-catalyzed
condensation. Acidity may also be added to activated
carbon by the addition of either sulfates or phosphates.
47
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
Base Catalysts
The base condensation reaction is performed using a
base catalyst. The base catalyst includes at least Li,
Na, K, Cs, B, Rb, Mg, Ca, Sr, Si, Ba, Al, Zn, Ce, La, Y,
Sc, Y, Zr, Ti, hydrotalcite, zinc-aluminate, phosphate,
base-treated aluminosilicate zeolite, a basic resin,
basic nitride, alloys or combinations thereof. The base
catalyst may also include an oxide of Ti, Zr, V, Nb, Ta,
Mo, Cr, W, Mn, Re, Al, Ga, In, Co, Ni, Si, Cu, Zn, Sn,
Cd, Mg, P, Fe, and combinations thereof. In one
embodiment, the condensation catalyst further includes a
metal, such as Cu, Ag, Au, Pt, Ni, Fe, Co, Ru, Zn, Cd,
Ga, In, Rh, Pd, Ir, Re, Mn, Cr, Mo, W, Sn, Os, alloys and
combinations thereof. Preferred Group IA materials
include Li, Na, K, Cs and Rb. Preferred Group IIA
materials include Mg, Ca, Sr and Ba. Preferred Group IIB
materials include Zn and Cd. Preferred Group IIIB
materials include Y and La. Basic resins include resins
that exhibit basic functionality, such as Amberlyst. The
base catalyst may be self-supporting or adhered to any
one of the supports further described below, including
supports containing carbon, silica, alumina, zirconia,
titania, vanadia, ceria, nitride, boron nitride,
heteropolyacids, alloys and mixtures thereof.
The base catalyst may also include zeolites and
other microporous supports that contain Group IA
compounds, such as Li, Na, K, Cs and Rb. Preferably, the
Group IA material is present in an amount greater than
that required to neutralize the acidic nature of the
support. These materials may be used in any combination,
and also in combination with alumina or silica. A metal
function may also be provided by the addition of group
VIIIB metals, or Cu, Ga, In, Zn or Sn.
48
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
In one embodiment, the condensation catalyst is
derived from the combination of MgO and A1203 to form a
hydrotalcite material. Another preferred material
contains ZnO and A1203 in the form of a zinc aluminate
spinel. Yet another preferred material is a combination
of ZnO, A1203, and CuO. Each of these materials may also
contain an additional metal function provided by a Group
VIIIB metal, such as Pd or Pt. In one embodiment, the
base catalyst is a metal oxide containing Cu, Ni, Zn, V,
Zr, or mixtures thereof. In another embodiment, the base
catalyst is a zinc aluminate metal containing Pt, Pd Cu,
Ni, or mixtures thereof.
Preferred loading of the primary metal is in the
range of 0.10 wt% to 25 wt%, with weight percentages of
0.10% and 0.05% increments between, such as 1.00%, 1.10%,
1.15%, 2.00%, 2.50%, 5.00%, 10.00%, 12.50%, 15.00% and
20.00%. The preferred atomic ratio of the second metal,
if any, is in the range of 0.25-to-1 to 10-to-1,
including ratios there between, such as 0.50, 1.00, 2.50,
5.00, and 7.50-to-1.
Acid-Base Catalysts
The acid-base condensation reaction is performed
using a multi-functional catalyst having both acid and
base functionality. The acid-base catalyst may include
hydrotalcite, zinc-aluminate, phosphate, Li, Na, K, Cs,
B, Rb, Mg, Si, Ca, Sr, Ba, Al, Ce, La, Sc, Y, Zr, Ti, Zn,
Cr, and combinations thereof. In further embodiments, the
acid-base catalyst may also include one or more oxides
from the group of Ti, Zr, V, Nb, Ta, Mo, Cr, W, Mn, Re,
Al, Ga, In, Fe, Co, Ir, Ni, Si, Cu, Zn, Sn, Cd, P, and
combinations thereof. The acid-base catalyst may also
include a metal functionality provided by Cu, Ag, Au, Pt,
Ni, Fe, Co, Ru, Zn, Cd, Ga, In, Rh, Pd, Ir, Re, Mn, Cr,
49
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
Mo, W, Sn, Os, alloys or combinations thereof. In one
embodiment, the catalyst further includes Zn, Cd or
phosphate. In one embodiment, the condensation catalyst
is a metal oxide containing Pd, Pt, Cu or Ni, and even
more preferably an aluminate or zirconium metal oxide
containing Mg and Cu, Pt, Pd or Ni. The acid-base
catalyst may also include a hydroxyapatite (HAP) combined
with any one or more of the above metals. The acid-base
catalyst may be self-supporting or adhered to any one of
the supports further described below, including supports
containing carbon, silica, alumina, zirconia, titania,
vanadia, ceria, nitride, boron nitride, heteropolyacids,
alloys and mixtures thereof.
The condensation catalyst may also include zeolites
and other microporous supports that contain Group IA
compounds, such as Li, NA, K, Cs and Rb. Preferably, the
Group IA material is present in an amount less than that
required to neutralize the acidic nature of the support.
A metal function may also be provided by the addition of
group VIIIB metals, or Cu, Ga, In, Zn or Sn.
In one embodiment, the condensation catalyst is
derived from the combination of MgO and A1203 to form a
hydrotalcite material. Another preferred material
contains a combination of MgO and Zr02, or a combination
of ZnO and A1203. Each of these materials may also
contain an additional metal function provided by copper
or a Group VIIIB metal, such as Ni, Pd, Pt, or
combinations of the foregoing.
If a Group IIB, VIB, VIIB, VIIIB, IIA or IVA metal
is included, the loading of the metal is in the range of
0.10 wt% to 10 wt%, with weight percentages of 0.10% and
0.05% increments between, such as 1.00%, 1.10%, 1.15%,
2.00%, 2.50%, 5.00% and 7.50%, etc. If a second metal is
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
included, the preferred atomic ratio of the second metal
is in the range of 0.25-to-1 to 5-to-1, including ratios
there between, such as 0.50, 1.00, 2.50 and 5.00-to-1.
Condensation Reactions
The specific C.4+ compounds produced will depend on
various factors, including, without limitation, the type
of oxygenates in the reactant stream, condensation
temperature, condensation pressure, the reactivity of the
catalyst, and the flow rate of the reactant stream as it
affects the space velocity, GHSV and WHSV. Preferably,
the reactant stream is contacted with the condensation
catalyst at a WHSV that is appropriate to produce the
desired hydrocarbon products. The WHSV is preferably at
least about 0.1 grams of oxygenate in the reactant stream
per hour, more preferably the WHSV is between about 0.1
to 40.0 g/g hr, including a WHSV of about 1, 2, 3, 4, 5,
6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 20, 25, 30, 35 g/g
hr, and increments between.
In general, the condensation reaction should be
carried out at a temperature at which the thermodynamics
of the proposed reaction are favorable. For condensed
phase liquid reactions, the pressure within the reactor
must be sufficient to maintain at least a portion of the
reactants in the condensed liquid phase at the reactor
inlet. For vapor phase reactions, the reaction should be
carried out at a temperature where the vapor pressure of
the oxygenates is at least about 0.1 atm (and preferably
a good deal higher), and the thermodynamics of the
reaction are favorable. The condensation temperature will
vary depending upon the specific oxygenate used, but is
generally in the range of from about 80 C to 500 C for
reactions taking place in the vapor phase, and more
preferably from about 125 C to 450 C. For liquid phase
51
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
reactions, the condensation temperature may be from about
80 C to 500 C, and the condensation pressure from about
0 psig to 1200 psig. Preferably, the condensation
temperature is between about 125 C and 300 C, or between
about 125 C and 250 C, or between about 250 C and 425 C.
The reaction pressure is preferably at least about 0.1
atm, or between about 0 and 1200 psig, or between about
0 and 1000 psig, or between about 0 and 700 psig.
Varying the factors above, as well as others, will
generally result in a modification to the specific
composition and yields of the C4+ compounds. For example,
varying the temperature and/or pressure of the reactor
system, or the particular catalyst formulations, may
result in the production of C4+ alcohols and/or ketones
instead of C4+ hydrocarbons. The C4+ hydrocarbon product
may also contain a variety of olefins, and alkanes of
various sizes (typically branched alkanes). Depending
upon the condensation catalyst used, the hydrocarbon
product may also include aromatic and cyclic hydrocarbon
compounds. The C4+ hydrocarbon product may also contain
undesirably high levels of olefins, which may lead to
coking or deposits in combustion engines, or other
undesirable hydrocarbon products. In such event, the
hydrocarbon molecules produced may be optionally
hydrogenated to reduce the ketones to alcohols and
hydrocarbons, while the alcohols and unsaturated
hydrocarbon may be reduced to alkanes, thereby forming a
more desirable hydrocarbon product having low levels of
olefins, aromatics or alcohols.
The finishing step will generally be a hydrogenation
reaction that removes the remaining carbonyl group or
hydroxyl group. In such event, any one of the
hydrogenation catalysts described above may be used. Such
52
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
catalysts may include any one or more of the following
metals, Cu, Ni, Fe, Co, Ru, Pd, Rh, Pt, Ir, Os, alloys or
combinations thereof, alone or with promoters such as Au,
Ag, Cr, Zn, Mn, Sn, Cu, Bi, and alloys thereof, may be
used in various loadings ranging from about 0.01 to about
20 wt% on a support as described above.
In general, the finishing step is carried out at
finishing temperatures of between about 80 C to 250 C,
and finishing pressures in the range of about 100 psig to
2000 psig. The finishing step may be conducted in the
vapor phase or liquid phase, and may use in situ
generated H2, external H2, recycled H2, or combinations
thereof, as necessary.
Other factors, such as the concentration of water or
undesired oxygenates, may also effect the composition and
yields of the C4+ compounds, as well as the activity and
stability of the condensation catalyst. In such event,
the process may include a dewatering step that removes a
portion of the water prior to condensation, or a
separation unit for removal of the undesired oxygenates.
For instance, a separator unit, such as a phase
separator, extractor, purifier or distillation column,
may be installed prior to the condensation step so as to
remove a portion of the water from the reactant stream
containing the oxygenates. A separation unit may also be
installed to remove specific oxygenates to allow for the
production of a desired product stream containing
hydrocarbons within a particular carbon range, or for use
as end products or in other systems or processes.
C4+ Compounds
The practice of the process of preparing the
component derived from a water-soluble oxygenated
hydrocarbon in the present invention results in the
53
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
production of C4+ alkanes, C4+ alkenes, C5+ cycloalkanes,
C5+ cycloalkenes, aryls, fused aryls, C4+ alcohols, C4+
ketones, and mixtures thereof. The C4+ alkanes and C4+
alkenes have from 4 to 30 carbon atoms (C4-30 alkanes and
C4-30 alkenes) and may be branched or straight chained
alkanes or alkenes. The C4+ alkanes and C4+ alkenes may
also include fractions of C4-9, C7-14, C12-24 alkanes and
alkenes, respectively, with the C4-9 fraction directed to
gasoline, the C7_14 fraction directed to kerosene (e.g.
jet fuels), and the C12-24 fraction directed to diesel fuel
and other industrial applications. Examples of various
C4+ alkanes and C4+ alkenes include, without limitation,
butane, butane, pentane, pentene, 2-methylbutane, hexane,
hexane, 2-methylpentane, 3-methylpentane, 2,2-
dimethylbutane, 2,3-dimethylbutane, heptane, heptene,
octane, octene, 2,2,4,-trimethylpentane, 2,3-dimethyl
hexane, 2,3,4-trimethylpentane, 2,3-dimethylpentane,
nonane, nonene, decane, decene, undecane, undecene,
dodecane, dodecene, tridecane, tridecene, tetradecane,
tetradecene, pentadecane, pentadecene, hexadecane,
hexadecane, heptyldecane, heptyldecene, octyldecane,
octyldecene, nonyldecane, nonyldecene, eicosane,
eicosene, uneicosane, uneicosene, doeicosane, doeicosene,
trieicosane, trieicosene, tetraeicosane, tetraeicosene,
and isomers thereof.
The C5+ cycloalkanes and C5+ cycloalkenes have from 5
to 30 carbon atoms and may be unsubstituted, mono-
substituted or multi-substituted. In the case of mono-
substituted and multi-substituted compounds, the
substituted group may include a branched C3+ alkyl, a
straight chain C1+ alkyl, a branched C3+ alkylene, a
straight chain C1+ alkylene, a straight chain C2+
alkylene, a phenyl or a combination thereof. In one
54
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
embodiment, at least one of the substituted groups
include a branched C3-12 alkyl, a straight chain C1-12
alkyl, a branched C3-12 alkylene, a straight chain C1-12
alkylene, a straight chain C2-12 alkylene, a phenyl or a
combination thereof. In yet another embodiment, at least
one of the substituted groups include a branched C3-4
alkyl, a straight chain C1_4 alkyl, a branched C3-4
alkylene, straight chain C1_4 alkylene, straight chain C2-4
alkylene, a phenyl or a combination thereof. Examples of
desirable C5+ cycloalkanes and C5+ cycloalkenes include,
without limitation, cyclopentane, cyclopentene,
cyclohexane, cyclohexene, methyl-cyclopentane, methyl-
cyclopentene, ethyl-cyclopentane, ethyl-cyclopentene,
ethyl-cyclohexane, ethyl-cyclohexene, and isomers
thereof.
Aryls will generally consist of an aromatic
hydrocarbon in either an unsubstituted (phenyl), mono-
substituted or multi-substituted form. In the case of
mono-substituted and multi-substituted compounds, the
substituted group may include a branched C3+ alkyl, a
straight chain C1+ alkyl, a branched C34_ alkylene, a
straight chain C2+ alkylene, a phenyl or a combination
thereof. In one embodiment, at least one of the
substituted groups include a branched C3-12 alkyl, a
straight chain C1-12 alkyl, a branched C3-12 alkylene, a
straight chain C2-12 alkylene, a phenyl or a combination
thereof. In yet another embodiment, at least one of the
substituted groups include a branched C3-4 alkyl, a
straight chain C1_4 alkyl, a branched 03-4 alkylene,
straight chain 02-4 alkylene, a phenyl or a combination
thereof. Examples of various aryls include, without
limitation, benzene, toluene, xylene (dimethylbenzene),
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
ethyl benzene, para xylene, meta xylene, ortho xylene, C9
aromatics.
Fused aryls will generally consist of bicyclic and
polycyclic aromatic hydrocarbons, in either an
unsubstituted, mono-substituted or multi-substituted
form. In the case of mono-substituted and multi-
substituted compounds, the substituted group may include
a branched C3+ alkyl, a straight chain Cl+ alkyl, a
branched C3+ alkylene, a straight chain C2+ alkylene, a
phenyl or a combination thereof. In another embodiment,
at least one of the substituted groups include a branched
C3-4 alkyl, a straight chain C1_4 alkyl, a branched C3-4
alkylene, straight chain C2-4 alkylene, a phenyl or a
combination thereof. Examples of various fused aryls
include, without limitation, naphthalene, anthracene,
tetrahydronaphthalene, and decahydronaphthalene, indane,
indene, and isomers thereof.
The C4+alcohols may also be cyclic, branched or
straight chained, and have from 4 to 30 carbon atoms. In
general, the C4+alcohols may be a compound according to
the formula R'-OH, wherein Rl is a member selected from
the group consisting of a branched C4+ alkyl, straight
chain C4+ alkyl, a branched C4+ alkylene, a straight chain
C4+ alkylene, a substituted C5+ cycloalkane, an
unsubstituted C5+ cycloalkane, a substituted C5+
cycloalkene, an unsubstituted C5+ cycloalkene, an aryl, a
phenyl and combinations thereof. Examples of desirable
C4+ alcohols include, without limitation, butanol,
pentanol, hexanol, heptanol, octanol, nonanol, decanol,
undecanol, dodecanol, tridecanol, tetradecanol,
pentadecanol, hexadecanol, heptyldecanol, octyldecanol,
nonyldecanol, eicosanol, uneicosanol, doeicosanol,
trieicosanol, tetraeicosanol, and isomers thereof.
56
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
The C.4+ ketones may also be cyclic, branched or
straight chained, and have from 4 to 30 carbon atoms. In
general, the C.4+ ketone may be a compound according to
the formula
\R3
,C=0
/
R4
wherein R3 and R4 are independently a member selected
from the group consisting of a branched C3+ alkyl, a
straight chain C1+ alkyl, a branched C3+ alkylene, a
straight chain C2+ alkylene, a substituted C5+
cycloalkane, an unsubstituted C5+ cycloalkane, a
substituted C5+ cycloalkene, an unsubstituted C5+
cycloalkene, an aryl, a phenyl and a combination thereof.
Examples of desirable C4+ ketones include, without
limitation, butanone, pentanone, hexanone, heptanone,
octanone, nonanone, decanone, undecanone, dodecanone,
tridecanone, tetradecanone, pentadecanone, hexadecanone,
heptyldecanone, octyldecanone, nonyldecanone, eicosanone,
uneicosanone, doeicosanone, trieicosanone,
tetraeicosanone, and isomers thereof.
The lighter fractions of the above, primarily C4-c9,
may be separated for gasoline use. Moderate fractions,
such as C7-C14, may be separated for kerosene use (e.g.
jet fuel), while heavier fractions, i.e., C12-C24, may be
separated for diesel fuel use. The heaviest fractions may
be used as lubricants or cracked to produce additional
gasoline and/or diesel fractions.
Catalyst Supports
In various embodiments above, the catalyst systems
include a support suitable for suspending the catalyst in
the feedstock solution. The support should be one that
provides a stable platform for the chosen catalyst and
57
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
the reaction conditions. The support may take any form
which is stable at the chosen reaction conditions to
function at the desired levels, and specifically stable
in aqueous feedstock solutions. Such supports include,
without limitation, carbon, silica, silica-alumina,
alumina, zirconia, titania, ceria, vanadia, nitride,
boron nitride, heteropolyacids, hydroxyapatite, zinc
oxide, chromia, and mixtures thereof. Nanoporous supports
such as zeolites, carbon nanotubes, or carbon fullerene
may also be used.
One particularly preferred catalyst support is
carbon, especially carbon supports having relatively high
surface areas (greater than 100 square meters per gram).
Such carbons include activated carbon (granulated,
powdered, or pelletized), activated carbon cloth, felts,
or fibers, carbon nanotubes or nanohorns, carbon
fullerene, high surface area carbon honeycombs, carbon
foams (reticulated carbon foams), and carbon blocks. The
carbon may be produced via either chemical or steam
activation of peat, wood, lignite, coal, coconut shells,
olive pits, and oil based carbon. Another preferred
support is granulated activated carbon produced from
coconuts. In one embodiment, the APR and deoxygenation
catalyst system consists of Pt on carbon, with the Pt
being further alloyed or admixed with Ni, Ru, Cu, Fe, Rh,
Re, alloys and combinations thereof.
Another preferred catalyst support is zirconia. The
zirconia may be produced via precipitation of zirconium
hydroxide from zirconium salts, through sol-gel
processing, or any other method. The zirconia is
preferably present in a crystalline form achieved through
calcination of the precursor material at temperatures
exceeding 400 C and may include both tetragonal and
58
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
monoclinic crystalline phases. A modifying agent may be
added to improve the textural or catalytic properties of
the zirconia. Such modifying agents include, without
limitation, sulfate, tungstenate, phosphate, titania,
silica, and oxides of Group IIIB metals, especially Ce,
La, or Y. In one embodiment, the APR and deoxygenation
catalyst consists of Pt on a primarily tetragonal phase
silica modified zirconia, with the Pt being further
alloyed or admixed with Ni, Ru, Cu, Fe, Rh, Re, alloys
and combinations thereof.
Yet another preferred catalyst support is titania.
The titania may be produced via precipitation from
titanium salts, through sol-gel processing, or any other
method. The titania is preferably present in a
crystalline form and may include both anatase and rutile
crystalline phases. A modifying agent may be added to
improve the textural or catalytic properties of the
titania. Such modifying agents include, without
limitation, sulfate, silica, and oxides of Group IIIB
metals, especially Ce, La, or Y. In one embodiment, the
APR and oxygenate forming catalyst system consists of Ru
on a primarily rutile phase titania, with the Ru being
further alloyed or admixed with Ge, Bi, B, Ni, Sn, Cu,
Fe, Re, Rh, Pt, alloys and combinations thereof.
Another preferred catalyst support is silica. The
silica may be optionally combined with alumina to form a
silica-alumina material. In one embodiment, the APR
catalyst system is Pt on silica-alumina or silica, with
the Pt being further alloyed or admixed with Ni, Ru, Cu,
Fe, Rh, Re, alloys and combinations thereof. In another
embodiment, the APR catalyst system is Ni on silica-
alumina or silica, with the nickel being further alloyed
59
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
or admixed with Sn, Ge, Bi, Bu, Cu, Re, Ru, Fe, alloys
and combinations thereof.
The support may also be treated or modified to
enhance its properties. For example, the support may be
treated, as by surface-modification, to modify surface
moieties, such as hydrogen and hydroxyl. Surface hydrogen
and hydroxyl groups can cause local pH variations that
affect catalytic efficiency. The support may also be
modified, for example, by treating it with sulfates,
phosphates, tungstenates, silanes, lanthanides, alkali
compounds or alkali earth compounds. For carbon supports,
the carbon may be pretreated with steam, oxygen (from
air), inorganic acids or hydrogen peroxide to provide
more surface oxygen sites. The preferred pretreatment
would be to use either oxygen or hydrogen peroxide. The
pretreated carbon may also be modified by the addition of
oxides of Group IVB and Group VB. It is preferred to use
oxides of Ti, V, Zr and mixtures thereof.
The catalyst systems, whether alone or mixed
together, may be prepared using conventional methods
known to those in the art. Such methods include incipient
wetting, evaporative impregnation, chemical vapor
deposition, wash-coating, magnetron sputtering
techniques, and the like. The method chosen to fabricate
the catalyst is not particularly critical to the function
of the invention, with the proviso that different
catalysts will yield different results, depending upon
considerations such as overall surface area, porosity,
etc.
Supplemental Materials
Supplemental materials and compositions
("supplements") may be added to the feedstock solution at
various stages of the process in order to enhance the
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
reaction or to drive it to the production of the desired
reaction products. Supplements may include, without
limitation, acids, salts and additional hydrogen or
feedstock. Such supplements may be added directly to the
feedstock stream prior to or contiguous with contacting
the relevant catalyst, or directly to the reaction bed
for the appropriate reactions.
In one embodiment, the supplement may include an
additional feedstock solution for providing additional
oxygenated hydrocarbons for oxygenate formation. The
feedstock may include any one or more oxygenated
hydrocarbons listed above, including any one or more
sugar alcohols, glucose, polyols, glycerol or
saccharides. For instance, the supplemental material may
include glycerol. In this embodiment, crude glycerol is
used to initiate the reaction and to produce hydrogen so
as to avoid polluting the deoxygenation catalyst with
contaminants from the crude glycerol. Purified glycerol
is then added to the feedstock solution prior to or at
the same time the original feedstock solution is placed
in contact with the deoxygenation catalyst to increase
the oxygenated hydrocarbons available for processing. It
is anticipated that the opposite may be employed with the
crude glycerol serving as the supplement depending on the
characteristics of the APR catalyst and deoxygenation
catalyst.
In another embodiment, the supplement may include
additional oxygenates for the condensation reaction. The
oxygenates may include any one or more oxygenates listed
above. For instance, the supplemental material may
include a propyl alcohol. In this embodiment, the propyl
alcohol may be produced in a parallel system from a
glycerol feedstock and then combined with oxygenates
61
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
produced by the processing of a sorbitol feedstock in
order to provide a reactant stream most effective to
produce a product containing a combination of C6-12
hydrocarbons.
In yet another embodiment, the supplemental material
may include recycled oxygenates and/or oxygenated
hydrocarbons not fully reacted during the production
process. The oxygenates and oxygenated hydrocarbons may
include any one or more of oxygenates and oxygenated
hydrocarbons listed above.
In still yet another embodiment, the supplemental
material may include acids and salts added to the
process. The addition of acidic compounds may provide
increased selectivity to the desired oxygenates and,
ultimately, C.4+ compounds. Water-soluble acids may
include, without limitation, nitrate, phosphate, sulfate,
chloride salts, and mixtures thereof. If an optional
acidic modifier is used, it is preferred that it be
present in an amount sufficient to lower the pH of the
aqueous feed stream to a value between about pH 1.0 and
about pH 4Ø Lowering the pH of a feed stream during
oxygenate formation in this manner may increase the
proportion of diols, polyols, ketones or alcohols for
further condensation.
Reactor System
The reactions described herein may be carried out in
any reactor of suitable design, including continuous-
flow, batch, semi-batch or multi-system reactors, without
limitation as to design, size, geometry, flow rates, etc.
The reactor system may also use a fluidized catalytic bed
system, a swing bed system, fixed bed system, a moving
bed system, or a combination of the above. Preferably,
62
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
the present invention is practiced utilizing a
continuous-flow system at steady-state equilibrium.
In a continuous flow system, the reactor system
includes at least a reforming bed adapted to receive an
aqueous feedstock solution to produce hydrogen, a
deoxygenation bed adapted to produce oxygenates from the
hydrogen and a portion of the feedstock solution, and a
condensation bed to produce C4+ compounds from the
oxygenates. The reforming bed is configured to contact
the aqueous feedstock solution in a vapor phase or liquid
phase with the APR catalyst to provide hydrogen in a
reactant stream. The deoxygenation bed is configured to
receive the reactant stream for contact with the
deoxygenation catalyst and production of the desired
oxygenates. The condensation bed is configured to receive
the reactant stream for contact with the condensation
catalyst and production of the desired C4+ compounds. For
systems not involving an APR hydrogen production step,
the reforming bed may be removed. For systems not
involving a hydrogen or oxygenate production step, the
reforming and deoxygenation beds may be removed. Because
the APR catalyst, deoxygenation catalyst and condensation
catalyst may also be atomically identical, the catalysts
may exist as the same bed. For systems with a
hydrogenation or hydrogenolysis step, an additional
reaction bed may be included prior to the deoxygenation
and/or reforming bed. For systems with a finishing step,
an additional reaction bed for conducting the finishing
process may be included after the condensation bed.
In systems producing both hydrogen and oxygenates,
the condensation bed may be positioned within the same
reactor vessel along with the reforming bed or in a
second reactor vessel in communication with a first
63
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
reactor vessel having the reforming bed. The condensation
bed may be within the same reactor vessel along with the
reforming or deoxygenation bed or in a separate reactor
vessel in communication with the reactor vessel having
the deoxygenation bed. Each reactor vessel preferably
includes an outlet adapted to remove the product stream
from the reactor vessel. In systems including a
hydrogenation step or hydrogenolysis step, the
hydrogenation or hydrogenolysis reaction bed may be
within the same reactor vessel along with the reforming
or deoxygenation bed or in a separate reactor vessel in
communication with the reactor vessel having the
reforming bed and/or deoxygenation bed. For systems with
a finishing step, the finishing reaction bed may be
within the same reactor vessel along with the
condensation bed or in a separate reactor vessel in
communication with the reactor vessel having the
condensation bed.
The reactor system may also include additional
outlets to allow for the removal of portions of the
reactant stream to further advance or direct the reaction
to the desired reaction products, and to allow for the
collection and recycling of reaction byproducts for use
in other portions of the system. The reactor system may
also include additional inlets to allow for the
introduction of supplemental materials to further advance
or direct the reaction to the desired reaction products,
and to allow for the recycling of reaction byproducts for
use in the reforming process. For example, the system may
be designed such that excess hydrogen is produced over
the APR catalyst, with a portion of the excess hydrogen
removed and reintroduced downstream in the process to
supplement the reaction of the oxygenates over the
64
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
condensation catalyst or the finishing of the
condensation product to arrive at the desired C4+
compounds. Alternatively, the system may be designed such
that excess hydrogen is produced over the APR catalyst,
with a portion of the excess hydrogen removed and used in
other upstream processes, such as feedstock pretreatment
processes and hydrogenation or hydrogenolysis reactions.
The reactor system may also include elements which
allow for the separation of the reactant stream into
different components which may find use in different
reaction schemes or to simply promote the desired
reactions. For instance, a separator unit, such as a
phase separator, extractor, purifier or distillation
column, may be installed prior to the condensation step
to remove water from the reactant stream for purposes of
advancing the condensation reaction to favor the
production of hydrocarbons. A separation unit may also be
installed to remove specific oxygenates to allow for the
production of a desired product stream containing
hydrocarbons within a particular carbon range, or for use
as end products or in other systems or processes.
In one embodiment, the reaction system is configured
such that the flow direction of the aqueous feedstock
solution is established to ensure maximal interaction
with the in-situ generated H2. The reactor may be
designed so that the reactant stream flows horizontally,
vertical or diagonally to the gravitational plane so as
to maximize the efficiency of the system. In systems
where the reactant stream flows vertically or diagonally
to the gravitational plan, the stream may flow either
against gravity (up-flow system), with gravity (down-flow
system), or a combination of both. In one preferred
embodiment, the APR and/or deoxygenation reactor vessel
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
is designed as an up-flow system while the condensation
reactor vessel is designed as a down-flow system. In this
embodiment, the feedstock solution first contacts a
reforming bed containing the APR catalyst to produce in-
situ generated H2. Due to the configuration of the
reactor, the APR H2 is then able to, under certain
conditions, percolate through a second reaction bed
containing the deoxygenation catalyst at a rate greater
than or equal to the feedstock solution to maximize the
interaction of the feedstock solution with the H2 and
deoxygenation catalyst. The resulting reactant stream is
then feed into the condensation reactor in a down-flow
configuration for processing.
If the APR catalyst and deoxygenation catalyst are
within a single chamber, the APR catalyst and
deoxygenation catalyst may be placed in a stacked
configuration to allow the feedstock solution to first
contact the APR catalyst and then the deoxygenation
catalyst, or a series of deoxygenation catalysts
depending on the desired reaction products. The reaction
beds for the APR catalyst and deoxygenation catalyst, or
catalysts, may also be placed side-by-side dependent upon
the particular flow mechanism employed. In either case,
the feedstock solution may be introduced into the
reaction vessel through one or more inlets, and then
directed across the catalysts for processing. In another
embodiment, the feedstock solution is directed across the
APR catalyst to produce APR H2, and then both the APR H2
and the remaining feedstock solution are directed across
the deoxygenation catalyst, or catalysts, to produce the
desired oxygenates. In a parallel configuration, the
feedstock solution may be separated to direct a first
portion of the feedstock solution to the reforming bed
66
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
where APR H2 is produced, and a second portion to a
deoxygenation bed where the desired oxygenates are
produced using the in situ generated APR H2.
Alternatively, the reactor may be configured to
accommodate the use of two separate feedstock solutions,
with the first feedstock solution directed to the APR
reactor vessel and the second feedstock solution directed
to the deoxygenation reactor vessel. In a sequential
configuration, the reactor may be designed so that the
feedstock solution flows through the APR reactor vessel
and into the deoxygenation reactor vessel. In embodiments
employing a combined APR/deoxygenation catalyst, the
generation of APR H2 and oxygenates occurs
simultaneously. In either of these systems, because the
APR H2 is produced in-situ, the pressure is provided by a
pumping mechanism that also drives the feedstock solution
through the reactor chambers.
Figure 6 is a process diagram illustrating one
potential reactor system useful in practicing the process
of preparing the component derived from a water-soluble
oxygenated hydrocarbon in the present invention. A feed
stream of oxygenated hydrocarbons 1 (with or without
water) is mixed with a stream of recycled water and
recycled oxygenates at 2 to provide an aqueous feedstock
solution 3. The feedstock solution 3 is then hydrogenated
in a pretreatment step 4 to provide a feedstock solution
5 that is more readily converted to the desired
oxygenates. The H2 for the hydrogenation step may derive
from an external source 22 or hydrogen recycled from the
system as illustrated in steps 13 - 21 below. The
feedstock solution 5 is reacted in a reactor vessel 8
that contains an APR catalyst and a deoxygenation
catalyst to produce product stream 7 containing water,
67
CA 02735654 2016-04-28
=
H2, carbon dioxide, hydrocarbons and oxygenates. Water in
product stream 7 is then removed at 8 to provide a
product stream 10 containing oxygenates, hydrogen, carbon
dioxide and hydrocarbons. Water from dewatering step 8 is
then recycled at 9 and 15 for mixing with the stream of
oxygenated hydrocarbons at 2. Product stream 10 is then
passed through reactor vessel 11, which includes a
condensation catalyst to produce product stream 12
containing C4+ compounds, water, H2 and carbon dioxide.
Product stream 12 is then passed through a three-phase
separator 13 to separate the non-condensable gases 16
(i.e., hydrogen, carbon dioxide, methane, ethane, and
propane) from the hydrocarbon product stream 14
containing C41- compounds and water 15. Water 15 from the
separator can be either recycled or exported from the
system. The non-condensable gas stream 16 can be passed
through a separation unit 17 to provide a purified H2
stream 19 and a raffinate stream 18 containing carbon
dioxide, methane, ethane, propane, and some hydrogen. The
purified H219 may then be either exported from the
system at 20 or passed through a recycle compressor 21 to
provide recycled hydrogen stream 23.
In another preferred reactor system, illustrated in
Figure 7, a first reactor system is provided for
converting the desired feedstock solution to C4+
compounds. The feedstock solution is stored in tankinand
then passed through feed linelminto charge pumpin.
Charge pumplo3increases the pressure of the feedstock
solution to the desired reaction pressure, e.g., 600 psi,
and then discharges the solution through linelo4into an
electric preheaterlosthat heats the feed to the desired
inlet temperature. The heated solutionio6is then passed
into the process side of a reactor having essentially a
68
CA 02735654 2016-04-28
tube-within-tube configuration (tubelo7within tube 108)
Depending on the pressure of the reactor and the
temperatures at which the several stages are operated,
the reactant stream flowing through the reactor tube 107
=
will generally be maintained substantially in the liquid
phase throughout, but may vaporize due to the heat of the
condensation of the distal portion 107b such that most of
the product exiting the outlet end of the reactor through
line 115 is in vapor form.
The stages and stage regions of the reactor tube 10
include an APR/deoxygenation catalyst (combined) and a
condensation catalyst, each packed in successive
catalytic beas (i.e., one on top of another). In this
example, reactor tubelo7contains an APR/deoxygenation
catalyst in the proximal portion 107a of reactor tubelcrand
a condensation catalyst at the distal portion 107b. The
catalyst system is supported at the bottom with small
mesh stainless steel spheres setting on a stainless steel
frit. Stainless steel spheres are also place on top of
the catalyst bed. To facilitate separation of spent
catalyst for recycling or regeneration, the catalyst beds
are separated by means of a porous material, such as
glass wool. The reactor may also be physically separated
in separate tubes with conduits connecting the tubes to
permit continuous flow. Such an arrangement may permit
better thermal management, allowing optimization of
temperature according to the requirements of the
reactions in the several reactor stages.
The APR reaction is typically endothermic, while the
condensation reaction is typically highly exothermic.
Preferably, the reactor system permits the heat generated
in the condensation reaction to be used to heat the APR
and deoxygenation reactions. An advantage of conducting
69
CA 02735654 2016-04-28
both of these reactions together is that heat is
immediately transferred from the exothermic condensation
reaction to the endothermic reforming/deoxygenation
reactions.
The process tubelo7is preferably formed from a heat-
conducting material configured to transfer heat from the
distalportion 107b to the proximal portion 107a. In
addition, the process tube may be heated with hot oil or
hot air flowing through an annular space between process
tubelo7and outer tubeicm. The hot air may be generated by
heating ambient air from a blower in with an electrical
heater 112 and sent to the reactor through line in. Hot
oil may also be used and generated by a heater and pump
(not shown) and sent to the reactor through line 113 as
well. The flow configuration for this system is such that
the hot air (or oil) in tubelo8flows countercurrent to
the process fluid in tube 107. Accordingly, the reactor
tubelo7is preferably warmer at the bottom than at the
top.
Alternatively, the process tubelo7may be separated
into two separate tubes or regions to facilitate the
optimization of reaction conditions separately for the
APR and deoxygenation reactions, and for the condensation
reaction. For example, the separation of spent catalyst
for regeneration may be simplified in this manner. In a
two-region second stage in a vertical reactor, heat
generated by condensation in the lower region may be
permitted to move by convection to the upper region for
use in the reformation reaction. The second region may
also be configured to provide a continuous or step-wise
gradient of mixed reformation and condensation catalysts,
with more reformation catalyst at the upper end and more
condensation catalyst at the lower end.
CA 02735654 2016-04-28
The effluent 115 from reactor tubevrincludes gaseous
products (such as hydrogen, CO and CO2) as well as
aqueous and organic liquid products. The effluent is
cooled to ambient temperature using a water cooled tube
in a tube condenser 116. Effluent 117 from the condenser116
is then directed to a three-phase separator to separate
the product phases: the non-condensable gas 118 (upper
phase), a lower density organic-liquid phase 119 (middle
phase) and a higher-density aqueous-liquid phase 120
(lower phase). The system pressure is maintained by
controlling the flow of non-condensable gas through line
121. The liquid level is maintained by controlling the
flow of the aqueous-phase components through line 122. The
organic-liquid phase is then skimmed off the top of the
aqueous phase through line 122.
The aqueous phase 120 is withdrawn through line 123.
If the aqueous phase 120 contains significant levels of
residual oxygenates (i.e., products of incomplete
reformation), the aqueous phase 120 may be conducted
through line 123 back to feed source1o6where it is used
for feedstock directed back into the reactor. In this
way, the carbon content and energy value of the
intermediate processes are recovered.
The middle phase 119 contains C5+ compounds.
Typically, this phase contains hydrocarbons and mono-
oxygenates ranging primarily from C4 to C30. Lighter
fractions, primarily C4-C9, may be separated for gasoline
use. The moderate fraction, i.e., C7-C14, may be separated
for use as kerosene (e.g. in jet fuel). Heavier
fractions, i.e., C12-C24, may be separated for diesel use.
The heaviest fractions may be used as lubricants or
cracked to produce additional gasoline and/or diesel
fractions.
71
CA 02735654 2016-04-28
The vapor phase 118 contains hydrogen and other APR
reaction products, such as carbon monoxide, carbon
dioxide, methane, ethane, propane, butane, pentane,
and/or hexane gas. Part of this gas is purged from the
system to prevent the build-up of light hydrocarbons and
CO2 in the system through line 122. The gases may also be
used as a fuel source for purposes of providing heat to
the reactor system. In terms of scaled-up production,
after start-up, the reactor systems could be process
controlled, and the reactions would proceed at steady-
state equilibrium.
Component Having at Least One C4+ Compound
The component having at least one C4+ compound
derivable from a water-soluble oxygenated hydrocarbon by
the process as described above is preferably separated
into various distillation fractions by any known means
before it is used in the liquid fuel compositions of the
present invention. Preferably, the component having at
least one C4+ compound derived from the process as
described above is separated into more than one
distillation fraction, wherein at least one of the
distillation fractions is a lighter, moderate or heavier
fraction described herein below.
As described above, the lighter fractions, primarily
C4-C9, may be separated for gasoline use. The moderate
fractions, e.g. C7-C14, may be separated for use as
kerosene, e.g. for jet fuel use. Heavier fractions, e.g.
C12-C24, may be separated for diesel fuel use. The
heaviest fractions may be used as lubricants or may be
cracked to produce additional fractions for use in
gasoline, kerosene and/or diesel fractions.
Because the component having at least one C4+
compound derivable from a water-soluble oxygenated
72
CA 02735654 2016-04-28
hydrocarbon is conveniently derived from biomass, the age
of the component, or fractions thereof, is less than 100
years old, preferably less than 40 years old, more
preferably less than 20 years old, as calculated from the
carbon 114 concentration of the component.
Lighter Fractions
The lighter fractions of the component having at
least one C41- compound derivable from a water-soluble
oxygenated hydrocarbon preferably has one or more of the
following properties (LF-i to LF-vi):
(LF-i) a final boiling point in the range of from 150
to 220 C, more preferably in the range of from
160 to 210 C;
(LF-ii) a density at 15 C in the range of from 700 to
890 kg/m3, more preferably in the range of from
720 to 800 kg/m3;
(LF-iii) a sulphur content of at most 5 mg/kg, more
preferably at most 1 mg/kg;
(IF-iv) an oxygen content of at most 3.5 %wt., more
preferably at most 3.0 %wt., typically at most
2.7 %wt.;
(LF-v) a RON in the range of from 80 to 110, more
preferably in the range of from 90 to 100;
(LF-vi) a MON in the range of from 70 to 100, more
preferably in the range of from 80 to 90.
Conveniently, the lighter fractions of the component
having at least one C4, compound derivable from a water-
soluble oxygenated hydrocarbon has properties which
accord with each of the properties detailed in LF-i to
LF-vi above, more conveniently with each of the preferred
values for each of the properties detailed in LF-i to LF-
-
vi above.
73
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
Moderate Fractions
The moderate fractions of the component having at
least one C4+ compound derivable from a water-soluble
oxygenated hydrocarbon preferably has one or more of the
following properties (MF-i to MF-vix):
(MF-i) an initial boiling point in the range of
from 120 to 215 C, more preferably in the
range of from 130 to 205 C;
(MF-ii) a final boiling point in the range of from
220 to 320 C, more preferably in the range
of from 230 to 320 C;
(MF-iii) a density at 15 C in the range of from 700
to 890 kg/m3, more preferably in the range
of from 730 to 840 kg/m3;
(MF-iv) a sulphur content of at most 0.1 %wt., more
preferably at most 0.01 %wt.;
(MF-v) a total aromatics content of at most
30 %vol., more preferably at most 25 %vol.,
even more preferably at most 20 %vol., most
preferably at most 15 %vol.;
(MF-vi) a freeze point of -40 C or lower, more
preferably at least -47 C or lower;
(MF-vii) a smoke point of at least 18 mm, more
preferably at least 19 mm, even more
preferably at least 25 mm;
(MF-viii) a viscosity at -20 C in the range of from
1 to 10 cSt, more preferably in the range of
from 2 to 8 cSt.
(MF-vix) a specific energy content in the range of
from 40 to 47 MJ/kg, more preferably in the
range of from 42 to 46 MJ/kg.
Conveniently, the moderate fractions of the
component having at least one C4+ compound derivable from
74
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
a water-soluble oxygenated hydrocarbon has properties
which accord with each of the properties detailed in MF-i
to MF-vix above, more conveniently with each of the
preferred values for each of the properties detailed in
MF-i to MF-vix above.
Heavier Fractions
The heavier fractions of the component having at
least one C4+ compound derivable from a water-soluble
oxygenated hydrocarbon preferably has one or more of the
following properties (HF-i to HF-vi):
(HF-i) a T95 in the range of from 220 to 380 C, more
preferably in the range of from 260 to 360 C;
(HF-ii) a flash point in the range of from 30 to 70 C,
more preferably in the range of from 33 to
60 C;
(HF-iii) a density at 15 C in the range of from 700 to
900 kg/m3, more preferably in the range of from
750 to 850 kg/m3;
(HF-iv) a sulphur content of at most 5 mg/kg, more
preferably at most 1 mg/kg;
(HF-v) an oxygen content of at most 10 %wt., more
preferably at most 8 %wt.;
(HF-vi) a viscosity at 40 C in the range of from 0.5 to
6 cSt, more preferably in the range of from 1 to
5 cSt.
Conveniently, the heavier fractions of the component
having at least one C4+ compound derivable from a water-
soluble oxygenated hydrocarbon has properties which
accord with each of the properties detailed in HF-i to
HF-vi above, more conveniently with each of the preferred
values for each of the properties detailed in HF-i to HF-
vi above.
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
Liquid Fuel Composition
The amount of the component having at least one C.4+
compound derivable from a water-soluble oxygenated
hydrocarbon present in the liquid fuel composition of the
present invention is at least 0.1 %vol., based on the
overall volume of the liquid fuel composition. More
preferably, the amount of the component derived from a
water-soluble oxygenated hydrocarbon present in the
liquid fuel composition of the present invention
additionally accords with one or more of the parameters
(i) to (xx) listed below:
(i) at least 0.5 %vol.
(ii) at least 1 %vol
(iii) at least 1.5 %vol
(iv) at least 2 %vol
(v) at least 2.5 %vol
(vi) at least 3 %vol
(vii) at least 3.5 %vol
(viii) at least 4 %vol
(ix) at least 4.5 %vol
(x) at least 5 %vol
(xi) at most 99.5 %vol.
(xii) at most 99 %vol.
(xiii) at most 98 %vol.
(xiv) at most 97 %vol.
(xv) at most 96 %vol.
(xvi) at most 95 %vol.
(xvii) at most 90 %vol.
(xviii) at most 85 %vol.
(xix) at most 80 %vol.
(xx) at most 75 %vol.
Conveniently, the amount of the component having at
least one C.4+ compound derivable from a water-soluble
76
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
oxygenated hydrocarbon present in the liquid fuel
composition of the present invention accords with one
parameter selected from (i) to (x) above, and one
parameter selected from (xi) to (xx) above.
Conveniently, for gasoline compositions according to
the present invention, the amount of the component having
at least one C.4+ compound derivable from a water-soluble
oxygenated hydrocarbon present in the gasoline
composition will be in the range of from 0.1 to 60 %vol,
0.5 to 55 %vol or 1 to 50 %vol.
Conveniently, for diesel fuel compositions according
to the present invention, the amount of the component
having at least one C.4+ compound derivable from a water-
soluble oxygenated hydrocarbon present in the diesel fuel
composition will be in the range of from 0.1 to 60 %vol,
0.5 to 55 %vol or 1 to 50 %vol.
Conveniently, for kerosene compositions according to
the present invention, the amount of the component having
at least one C.4+ compound derivable from a water-soluble
oxygenated hydrocarbon present in the kerosene
composition will be in the range of from 0.1 to 90 %vol,
0.5 to 85 %vol or 1 to 80 %vol, such as in the range of
from 0.1 to 60 %vol, 0.5 to 55 %vol or 1 to 50 %vol.
The liquid fuel composition comprising of the
present invention is typically selected from a gasoline,
kerosene or diesel fuel composition.
If the liquid fuel composition is a gasoline
composition, then the gasoline composition has an initial
boiling point in the range of from 15 C to 70 C
(IP123), a final boiling point of at most 230 C (IP123),
a RON in the range of from 85 to 110 (ASTM D2699) and a
MON in the range of from 75 to 100 (ASTM D2700).
77
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
If the liquid fuel composition is a kerosene
composition, then the kerosene composition has an initial
boiling point in the range of from 110 to 150 C, a final
boiling point in the range of from 200 to 320 C and a
viscosity at -20 C in the range of from 0.8 to 10 mm2/s
(ASTM D445).
If the liquid fuel composition is a diesel fuel
composition, then the diesel fuel composition has an
initial boiling point in the range of from 130 C to
230 C (IP123), a final boiling point of at most 410 C
(IP123) and a cetane number in the range of from 35 to
120 (ASTM D613).
Preferably, the liquid fuel composition of the
present invention additionally comprises one or more fuel
additive.
Gasoline Compositions
The gasoline composition according to the present
invention typically comprise mixtures of hydrocarbons
boiling in the range from 15 to 230 C, more typically in
the range of from 25 to 230 C (EN-ISO 3405). The initial
boiling point of the gasoline compositions according to
the present invention are in the range of from 15 to
70 C (IP123), preferably in the range of from 20 to
60 C, more preferably in the range of from 25 to 50 C.
The final boiling point of the gasoline compositions
according to the present invention is at most 230 C,
preferably at most 220 CC, more preferably at most
210 C. The optimal ranges and distillation curves
typically varying according to climate and season of the
year.
In addition to the component having at least one C4+
compound derivable from a water-soluble oxygenated
hydrocarbon, the hydrocarbons in the gasoline composition
78
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
may be derived by any means known in the art,
conveniently the hydrocarbons may be derived in any known
manner from straight-run gasoline, synthetically-produced
aromatic hydrocarbon mixtures, thermally or catalytically
cracked hydrocarbons, hydro-cracked petroleum fractions,
catalytically reformed hydrocarbons or mixtures of these.
The research octane number (RON) of the gasoline
compositions according to the present invention is in the
range of from 85 to 110 (ASTM D2699). Preferably, the RON
of the gasoline composition will be at least 90, for
instance in the range of from 90 to 110, more preferably
at least 91, for instance in the range of from 91 to 105,
even more preferably at least 92, for instance in the
range of from 92 to 103, even more preferably at least
93, for instance in the range of from 93 to 102, and most
preferably at least 94, for instance in the range of from
94 to 100.
The motor octane number (MON) of the gasoline
compositions according to the present invention is in the
range of from 75 to 100 (ASTM D2699). Preferably, the MON
of the gasoline composition will be at least 80, for
instance in the range of from 80 to 100, more preferably
at least 81, for instance in the range of from 81 to 95,
even more preferably at least 82, for instance in the
range of from 82 to 93, even more preferably at least 83,
for instance in the range of from 83 to 92, and most
preferably at least 84, for instance in the range of from
84 to 90.
Typically, gasoline compositions comprise a mixture
of components selected from one or more of the following
groups; saturated hydrocarbons, olefinic hydrocarbons,
aromatic hydrocarbons, and oxygenated hydrocarbons.
Conveniently, the gasoline composition may comprise a
79
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
mixture of saturated hydrocarbons, olefinic hydrocarbons,
aromatic hydrocarbons, and, optionally, oxygenated
hydrocarbons.
Typically, the olefinic hydrocarbon content of the
gasoline composition is in the range of from 0 to 40
percent by volume based on the gasoline (ASTM D1319);
preferably, the olefinic hydrocarbon content of the
gasoline composition is in the range of from 0 to 30
percent by volume based on the gasoline composition, more
preferably, the olefinic hydrocarbon content of the
gasoline composition is in the range of from 0 to 20
percent by volume based on the gasoline composition.
Typically, the aromatic hydrocarbon content of the
gasoline composition is in the range of from 0 to 70
percent by volume based on the gasoline (ASTM D1319), for
instance the aromatic hydrocarbon content of the gasoline
composition is in the range of from 10 to 60 percent by
volume based on the gasoline composition; preferably, the
aromatic hydrocarbon content of the gasoline composition
is in the range of from 0 to 50 percent by volume based
on the gasoline composition, for instance the aromatic
hydrocarbon content of the gasoline composition is in the
range of from 10 to 50 percent by volume based on the
gasoline composition.
The benzene content of the gasoline composition is
at most 10 percent by volume, more preferably at most 5
percent by volume, especially at most 1 percent by volume
based on the gasoline composition.
The gasoline composition preferably has a low or
ultra low sulphur content, for instance at most 1000 ppmw
(parts per million by weight), preferably no more than
500 ppmw, more preferably no more than 100, even more
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
preferably no more than 50 and most preferably no more
than even 10 ppmw.
The gasoline composition also preferably has a low
total lead content, such as at most 0.005 g/l, most
preferably being lead free - having no lead compounds
added thereto (i.e. unleaded).
When the gasoline composition comprises oxygenated
hydrocarbons, at least a portion of non-oxygenated
hydrocarbons will be substituted for oxygenated
hydrocarbons. The oxygen content of the gasoline may be
up to 30 percent by weight (EN 1601) based on the
gasoline composition. For example, the oxygen content of
the gasoline may be up to 25 percent by weight,
preferably up to 10 percent by weight. Conveniently, the
oxygenate concentration will have a minimum concentration
selected from any one of 0, 0.2, 0.4, 0.6, 0.8, 1.0, and
1.2 percent by weight, and a maximum concentration
selected from any one of 5, 4.5, 4.0, 3.5, 3.0, and 2.7
percent by weight.
Examples of oxygenated hydrocarbons, other than the
oxygenated hydrocarbons that may be present in the
component having at least one C.4+ compound derivable from
a water-soluble oxygenated hydrocarbon, that may be
incorporated into the gasoline include alcohols, ethers,
esters, ketones, aldehydes, carboxylic acids and their
derivatives, and oxygen containing heterocyclic
compounds. Preferably, the oxygenated hydrocarbons that
may be incorporated into the gasoline are selected from
alcohols (such as methanol, ethanol, propanol, iso-
propanol, butanol, tert-butanol and iso-butanol), ethers
(preferably ethers containing 5 or more carbon atoms per
molecule, e.g., methyl tert-butyl ether) and esters
(preferably esters containing 5 or more carbon atoms per
81
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
molecule); a particularly preferred oxygenated
hydrocarbon is ethanol.
When oxygenated hydrocarbons are present in the
gasoline composition, the amount of oxygenated
hydrocarbons in the gasoline composition may vary over a
wide range. For example, gasolines comprising a major
proportion of oxygenated hydrocarbons are currently
commercially available in countries such as Brazil and
U.S.A, e.g. E85, as well as gasolines comprising a minor
proportion of oxygenated hydrocarbons, e.g. E10 and E5.
Therefore, the amount of oxygenated hydrocarbons present
in the gasoline composition is preferably selected from
one of the following amounts: up to 85 percent by volume;
up to 65 percent by volume; up to 30 percent by volume;
up to 20 percent by volume; up to 15 percent by volume;
and, up to 10 percent by volume, depending upon the
desired final formulation of the gasoline. Conveniently,
the gasoline composition may contain at least 0.5, 1.0 or
2.0 percent by volume oxygenated hydrocarbons.
Examples of suitable gasoline compositions include
gasolines which have an olefinic hydrocarbon content of
from 0 to 20 percent by volume (ASTM D1319), an oxygen
content of from 0 to 5 percent by weight (EN 1601), an
aromatic hydrocarbon content of from 0 to 50 percent by
volume (ASTM D1319) and a benzene content of at most 1
percent by volume.
Whilst not critical to the present invention, the
gasoline compositions of the present invention may
conveniently additionally include one or more fuel
additive. The concentration and nature of the fuel
additive(s) that may be included in the gasoline
composition of the present invention is not critical.
Non-limiting examples of suitable types of fuel additives
82
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
that can be included in the gasoline composition of the
present invention include anti-oxidants, corrosion
inhibitors, detergents, dehazers, antiknock additives,
metal deactivators, valve-seat recession protectant
compounds, dyes, friction modifiers, carrier fluids,
diluents and markers. Examples of suitable such additives
are described generally in US Patent No.5,855,629.
Conveniently, the fuel additives can be blended with
one or more diluents or carrier fluids, to form an
additive concentrate, the additive concentrate can then
be admixed with the gasoline composition of the present
invention.
The (active matter) concentration of any additives
present in the gasoline composition of the present
invention is preferably up to 1 percent by weight, more
preferably in the range from 5 to 1000 ppmw,
advantageously in the range of from 75 to 300 ppmw, such
as from 95 to 150 ppmw.
Alternatively, the gasoline composition of the
present invention may be an aviation gasoline. If the
gasoline composition is an aviation gasoline then,
depending upon the grade of the aviation gasoline, the
Lean Mixture Motor Octane Number will be at least 80
(ASTM D2700) and the Rich Mixture Octane Number will be
at least 87 (ASTM D 909), or the Lean Mixture Motor
Octane Number will be at least 99.5 (ASTM D2700) and the
Performance Number will be at least 130 (ASTM D 909).
Furthermore, if the gasoline composition is an aviation
gasoline then the Reid Vapour Pressure at 37.8 C will be
in the range of from 38.0 to 49.0 kPa (ASTM D323), the
final boiling point will be at most 170 C (ASTM D 86),
and the tetraethyl lead content will be at most
0.85 gPb/1.
83
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
Kerosene Fuel Compositions
The kerosene fuel compositions of the present
invention have use in aviation engines, such as jet
engines or aero diesel engines, but also in any other
suitable power or lighting source.
In addition to the component having at least one C4+
compound derivable from a water-soluble oxygenated
hydrocarbon, the kerosene fuel composition may comprise a
mixture of two or more different fuel components, and/or
be additivated as described below.
The kerosene fuel compositions will typically have
boiling points within the range of 80 to 320 C,
preferably in the range of 110 to 320 C, more preferably
in the range of from 130 to 300 C, depending on grade
and use. They will typically have a density from 775 to
845 kg/m3, preferably from 780 to 830 kg/m3, at 15 C
(e.g. ASTM D4502 or IP 365). They will typically have an
initial boiling point in the range 80 to 150 C,
preferably in the range 110 to 150 C, and a final
boiling point in the range 200 to 320 C. Their kinematic
viscosity at -20 C (ASTM D445) is typically in the range
of from 0.8 to 10 mm2/s, preferably from 1.2 to
8.0 mm2/s.
It may be desirable for the kerosene fuel
composition to contain a Fischer-Tropsch derived fuel, if
the kerosene fuel composition does contain a Fischer-
Tropsch derived fuel, then it will conveniently contain
5%v or greater, preferably 10%v or greater, or more
preferably 25%v or greater, of a Fischer-Tropsch derived
fuel.
The Fischer-Tropsch derived fuel should be suitable
for use as a kerosene fuel. Its components (or the
majority, for instance 95%w or greater, thereof) should
84
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
therefore have boiling points within the range given
above, i.e. from 110 to 320 C, preferably from 130 to
300 C. It will suitably have a 90%v/v distillation
temperature (190) of from 180 to 250 C, preferably 180 to
230 C.
By "Fischer-Tropsch derived" is meant that the fuel
is, or derives from, a synthesis product of a Fischer-
Tropsch condensation process. The Fischer-Tropsch
reaction converts carbon monoxide and hydrogen into
longer chain, usually paraffinic, hydrocarbons:
n(CO + 2H2) = (-CH2-)n + nH20 + heat,
in the presence of an appropriate catalyst and typically
at elevated temperatures (e.g. 125 to 300 C, preferably
175 to 250 C) and/or pressures (e.g. 500 to 10000 kPa,
preferably 1200 to 5000 kPa). Hydrogen:carbon monoxide
ratios other than 2:1 may be used if desired.
The carbon monoxide and hydrogen may themselves be
derived from organic or inorganic, natural or synthetic
sources, typically either from natural gas or from
organically derived methane.
A kerosene product may be obtained directly from
this reaction, or indirectly for instance by
fractionation of a Fischer-Tropsch synthesis product or
from a hydrotreated Fischer-Tropsch synthesis product.
Hydrotreatment can involve hydrocracking to adjust the
boiling range (see, e.g. GB-B-2077289 and EP-A-0147873)
and/or hydroisomerisation which can improve base fuel
cold flow properties by increasing the proportion of
branched paraffins. EP-A-0583836 describes a two-step
hydrotreatment process in which a Fischer-Tropsch
synthesis product is firstly subjected to hydroconversion
under conditions such that it undergoes substantially no
isomerisation or hydrocracking (this hydrogenates the
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
olefinic and oxygen-containing components), and then at
least part of the resultant product is hydroconverted
under conditions such that hydrocracking and
isomerisation occur to yield a substantially paraffinic
hydrocarbon fuel. The desired kerosene fraction(s) may
subsequently be isolated for instance by distillation.
Other post-synthesis treatments, such as
polymerisation, alkylation, distillation, cracking-
decarboxylation, isomerisation and hydroreforming, may be
used to modify the properties of Fischer-Tropsch
condensation products, as described for example in US-A-
4125566 and US-A-4478955.
Typical catalysts for the Fischer-Tropsch synthesis
of paraffinic hydrocarbons comprise, as the catalytically
active component, a metal from Group VIII of the periodic
table, in particular ruthenium, iron, cobalt or nickel.
Suitable such catalysts are described for example in EP-
A-0583836 (pages 3 and 4).
An example of a Fischer-Tropsch based process is the
SMDS (Shell Middle Distillate Synthesis) described in
"The Shell Middle Distillate Synthesis Process", van der
Burgt et al (paper delivered at the 5th Synfuels
Worldwide Symposium, Washington DC, November 1985; see
also the November 1989 publication of the same title from
Shell International Petroleum Company Ltd., London, UK).
This process (also sometimes referred to as the ShellTM
"Gas-to-Liquids" or "GTL" technology) produces middle
distillate range products by conversion of a natural gas
(primarily methane) derived synthesis gas into a heavy
long-chain hydrocarbon (paraffin) wax which can then be
hydroconverted and fractionated to produce liquid
transport fuels such as kerosene fuel compositions. A
version of the SMDS process, using a fixed-bed reactor
86
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
for the catalytic conversion step, is currently in use in
Bintulu, Malaysia and its products have been blended with
petroleum derived gas oils in commercially available
automotive fuels.
Gas oils and kerosenes prepared by the SMDS process
are commercially available from the Royal Dutch/Shell
Group of Companies.
Suitably, in accordance with the present invention,
the Fischer-Tropsch derived kerosene fuel will consist of
at least 90%w, preferably at least 95%w, more preferably
at least 98%w, even more preferably at least 99%w, most
preferably at least 99.8%w, of paraffinic components,
typically normal and iso-paraffins. The weight ratio of
normal to iso-paraffins will preferably be in the ranges
indicated above. The actual value for this ratio will be
determined, in part, by the hydroconversion process used
to prepare the kerosene from the Fischer-Tropsch
synthesis product. Some cyclic paraffins may also be
present.
By virtue of the Fischer-Tropsch process, a Fischer-
Tropsch derived kerosene has essentially no, or
undetectable levels of, sulphur and nitrogen. Compounds
containing these heteroatoms tend to act as poisons for
Fischer-Tropsch catalysts and are therefore removed from
the synthesis gas feed. Further, the process as usually
operated produces no or virtually no aromatic components.
The aromatics content of a Fischer-Tropsch kerosene, as
determined by ASTM D4629, will typically be below 5%w,
preferably below 2%w, more preferably below 1%w and most
preferably below 0.2%w.
The Fischer-Tropsch derived kerosene which may be
used in the kerosene fuel compositions of the present
invention will typically have a density from 730 to 770
87
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
kg/m3 at 15 C; a kinematic viscosity from 1.2 to 6,
preferably from 2 to 5, more preferably from 2 to 3.5,
mm2/s at -20 C; and a sulphur content of 20 ppmw (parts
per million by weight) or less, preferably of 5 ppmw or
less.
Preferably it is a product prepared by a Fischer-
Tropsch methane condensation reaction using a
hydrogen/carbon monoxide ratio of less than 2.5,
preferably less than 1.75, more preferably from 0.4 to
1.5, and ideally using a cobalt containing catalyst.
Suitably it will have been obtained from a hydrocracked
Fischer-Tropsch synthesis product (for instance as
described in GB-B-2077289 and/or EP-A-0147873), or more
preferably a product from a two-stage hydroconversion
process such as that described in EP-A-0583836 (see
above). In the latter case, preferred features of the
hydroconversion process may be as disclosed at pages 4 to
6, and in the examples, of EP-A-0583836.
The kerosene fuel composition of the present
invention preferably contains no more than 3000 ppmw
sulphur, more preferably no more than 2000 ppmw, or no
more than 1000 ppmw, or no more than 500 ppmw sulphur.
The kerosene fuel composition or the components
thereof may be additivated (additive-containing) or
unadditivated (additive-free). If additivated, e.g. at
the refinery or in later stages of fuel distribution, it
will contain minor amounts of one or more additives
selected for example from anti-static agents (e.g.
STADISTm 450 (ex. Octel)), antioxidants (e.g. substituted
tertiary butyl phenols), metal deactivator additives
(e.g. N,N'-disalicylidene 1,2-propanediamine), fuel
system icing inhibitor additives (e.g. diethylene glycol
monomethyl ether), corrosion inhibitor/lubricity improver
88
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
additives (e.g. APOLLOTM PRI 19 (ex. Apollo), DCI 4A (ex.
Octel), NALCOTM 5403 (ex. Nalco)), or thermal stability
improving additives (e.g. APA 1O1TM, (ex. Shell)) that are
approved in international civil and/or military jet fuel
specifications.
Unless otherwise stated, the (active matter)
concentration of each such additional component in the
additivated kerosene fuel composition is at levels
required or allowed in international jet fuel
specifications.
In the above, amounts (concentrations, %v, ppmw,
wt%) of components are of active matter, i.e. exclusive
of volatile solvents/diluent materials, unless otherwise
stipulated in the relevant specification.
The kerosene fuel composition of the present
invention is particularly applicable where the kerosene
fuel composition is used or intended to be used in a jet
engine.
Diesel Fuel Compositions
The diesel fuel composition according to the present
invention typically comprise mixtures of hydrocarbons
boiling in the range from 130 to 410 C, more typically
in the range of from 150 to 400 C. The initial boiling
point of the diesel fuel compositions according to the
present invention are in the range of from 130 to 230 C
(IP123), preferably in the range of from 140 to 220 C,
more preferably in the range of from 150 to 210 C. The
final boiling point of the diesel fuel compositions
according to the present invention is at most 410 C,
preferably at most 405 C, more preferably at most
400 C.
In addition to the component having at least one C4+
compound derivable from a water-soluble oxygenated
89
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
hydrocarbon, the diesel fuel composition may comprise a
mixture of two or more different diesel fuel components,
and/or be additivated as described below.
Such diesel fuel compositions will contain one or
more base fuels which may typically comprise liquid
hydrocarbon middle distillate gas oil(s), for instance
petroleum derived gas oils. Such fuels will typically
have boiling points within the range described above,
depending on grade and use. They will typically have a
density from 750 to 1000 kg/m3, preferably from 780 to
860 kg/m3, at 15 C (e.g. ASTM D4502 or IP 365) and a
cetane number (ASTM D613) of from 35 to 120, more
preferably from 40 to 85. They will typically have an
initial boiling point in the range described above and a
final boiling point of at most 410 C, preferably at most
405 C, more preferably at most 400 C, most preferably
in the range 290 to 400 C. Their kinematic viscosity at
40 C (ASTM D445) might suitably be from 1.2 to 4.5 mm2/s.
An example of a petroleum derived gas oil is a
Swedish Class 1 base fuel, which will have a density from
800 to 820 kg/m3 at 15 C (SS-EN ISO 3675, SS-EN ISO
12185), a T95 of 320 C or less (SS-EN ISO 3405) and a
kinematic viscosity at 40 C (SS-EN ISO 3104) from 1.4 to
4.0 mm2/s, as defined by the Swedish national
specification EC1.
Optionally, non-mineral oil based fuels, such as
biofuels (other than the component having at least one
C.4+ compound derivable from a water-soluble oxygenated
hydrocarbon) or Fischer-Tropsch derived fuels, may also
form or be present in the diesel fuel. Such Fischer-
Tropsch fuels may for example be derived from natural
gas, natural gas liquids, petroleum or shale oil,
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
petroleum or shale oil processing residues, coal or
biomass.
The amount of Fischer-Tropsch derived fuel used in
the diesel fuel composition of the present invention may
be from 0% to the remaining portion of the diesel fuel
composition (i.e. the portion of the diesel fuel
composition that is not the component having at least one
C.4+ compound derivable from a water-soluble oxygenated
hydrocarbon), preferably from 5% to the remaining portion
of the diesel fuel composition, more preferably from 5%
to 75%v of the diesel fuel composition. It may be
desirable for such a diesel fuel composition to contain
10%v or greater, more preferably 20%v or greater, still
more preferably 30%v or greater, of the Fischer-Tropsch
derived fuel. It is particularly preferred for such
diesel fuels to contain 30 to 75%v, and particularly 30
or 70%v, of the Fischer-Tropsch derived fuel. The balance
of the diesel fuel is made up of the component having at
least one C.4+ compound derivable from a water-soluble
oxygenated hydrocarbon and optionally one or more other
diesel fuel components.
Such a Fischer-Tropsch derived fuel component is any
fraction of the middle distillate fuel range, which can
be isolated from the (optionally hydrocracked) Fischer-
Tropsch synthesis product. Typical fractions will boil in
the naphtha, kerosene or gas oil range. Preferably, a
Fischer-Tropsch product boiling in the kerosene or gas
oil range is used because these products are easier to
handle in for example domestic environments. Such
products will suitably comprise a fraction larger than
90 wt% which boils between 160 and 400 C, preferably to
about 370 C. Examples of Fischer-Tropsch derived kerosene
and gas oils are described in EP-A-0583836, WO-A-
91
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
97/14768, WO-A-97/14769, WO-A-00/11116, WO-A-00/11117,
WO-A-01/83406, WO-A-01/83648, WO-A-01/83647, WO-A-
01/83641, WO-A-00/20535, WO-A-00/20534, EP-A-1101813, US-
A-5766274, US-A-5378348, US-A-5888376 and US-A-6204426.
The Fischer-Tropsch product will suitably contain
more than 80 wt% and more suitably more than 95 wt% iso
and normal paraffins and less than 1 wt% aromatics, the
balance being naphthenics compounds. The content of
sulphur and nitrogen will be very low and normally below
the detection limits for such compounds. For this reason
the sulphur content of a diesel fuel composition
containing a Fischer-Tropsch product may be very low.
The diesel fuel composition preferably contains no
more than 5000ppmw sulphur, more preferably no more than
500ppmw, or no more than 350ppmw, or no more than
150ppmw, or no more than 100ppmw, or no more than 7Oppmw,
or no more than 5Oppmw, or no more than 3Oppmw, or no
more than 2Oppmw, or most preferably no more than 15ppmw
sulphur.
The diesel fuel typically also includes one or more
fuel additive.
The diesel base fuel may itself be additivated
(additive-containing) or unadditivated (additive-free).
If additivated, e.g. at the refinery, it will contain
minor amounts of one or more additives selected for
example from anti-static agents, pipeline drag reducers,
flow improvers (e.g. ethylene/vinyl acetate copolymers or
acrylate/maleic anhydride copolymers), lubricity
additives, antioxidants and wax anti-settling agents.
Detergent-containing diesel fuel additives are known
and commercially available. Such additives may be added
to diesel fuels at levels intended to reduce, remove, or
slow the build up of engine deposits.
92
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
Examples of detergents suitable for use in diesel
fuel additives for the present purpose include polyolefin
substituted succinimides or succinamides of polyamines,
for instance polyisobutylene succinimides or
polyisobutylene amine succinamides, aliphatic amines,
Mannich bases or amines and polyolefin (e.g.
polyisobutylene) maleic anhydrides. Succinimide
dispersant additives are described for example in GB-A-
960493, EP-A-0147240, EP-A-0482253, EP-A-0613938, EP-A-
0557516 and WO-A-98/42808. Particularly preferred are
polyolefin substituted succinimides such as
polyisobutylene succinimides.
The diesel fuel additive mixture may contain other
components in addition to the detergent. Examples are
lubricity enhancers; dehazers, e.g. alkoxylated phenol
formaldehyde polymers; anti-foaming agents (e.g.
polyether-modified polysiloxanes); ignition improvers
(cetane improvers) (e.g. 2-ethylhexyl nitrate (EHN),
cyclohexyl nitrate, di-tert-butyl peroxide and those
disclosed in US-A-4208190 at column 2, line 27 to column
3, line 21); anti-rust agents (e.g. a propane-1,2-diol
semi-ester of tetrapropenyl succinic acid, or polyhydric
alcohol esters of a succinic acid derivative, the
succinic acid derivative having on at least one of its
alpha-carbon atoms an unsubstituted or substituted
aliphatic hydrocarbon group containing from 20 to 500
carbon atoms, e.g. the pentaerythritol diester of
polyisobutylene-substituted succinic acid); corrosion
inhibitors; reodorants; anti-wear additives; anti-
oxidants (e.g. phenolics such as 2,6-di-tert-butylphenol,
or phenylenediamines such as N,N'-di-sec-butyl-p-
phenylenediamine); metal deactivators; combustion
93
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
improvers; static dissipator additives; cold flow
improvers; and wax anti-settling agents.
The diesel fuel additive mixture may contain a
lubricity enhancer, especially when the diesel fuel
composition has a low (e.g. 500 ppmw or less) sulphur
content. In the additivated diesel fuel composition, the
lubricity enhancer is conveniently present at a
concentration of less than 1000 ppmw, preferably between
50 and 1000 ppmw, more preferably between 70 and 1000
ppmw. Suitable commercially available lubricity enhancers
include ester- and acid-based additives. Other lubricity
enhancers are described in the patent literature, in
particular in connection with their use in low sulphur
content diesel fuels, for example in:
- the paper by Danping Wei and H.A. Spikes, "The
Lubricity of Diesel Fuels", Wear, III (1986) 217-235;
- WO-A-95/33805 - cold flow improvers to enhance
lubricity of low sulphur fuels;
- WO-A-94/17160 - certain esters of a carboxylic
acid and an alcohol wherein the acid has from 2 to 50
carbon atoms and the alcohol has 1 or more carbon atoms,
particularly glycerol monooleate and di-isodecyl adipate,
as fuel additives for wear reduction in a diesel engine
injection system;
- US-A-5490864 - certain dithiophosphoric diester-
dialcohols as anti-wear lubricity additives for low
sulphur diesel fuels; and
- WO-A-98/01516 - certain alkyl aromatic compounds
having at least one carboxyl group attached to their
aromatic nuclei, to confer anti-wear lubricity effects
particularly in low sulphur diesel fuels.
It may also be preferred for the diesel fuel
composition to contain an anti-foaming agent, more
94
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
preferably in combination with an anti-rust agent and/or
a corrosion inhibitor and/or a lubricity enhancing
additive.
Unless otherwise stated, the (active matter)
concentration of each such additive component in the
additivated diesel fuel composition is preferably up to
10000 ppmw, more preferably in the range from 0.1 to 1000
ppmw, advantageously from 0.1 to 300 ppmw, such as from
0.1 to 150 ppmw.
The (active matter) concentration of any dehazer in
the diesel fuel composition will preferably be in the
range from 0.1 to 20 ppmw, more preferably from 1 to 15
ppmw, still more preferably from 1 to 10 ppmw,
advantageously from 1 to 5 ppmw. The (active matter)
concentration of any ignition improver present will
preferably be 2600 ppmw or less, more preferably 2000
ppmw or less, conveniently from 300 to 1500 ppmw. The
(active matter) concentration of any detergent in the
diesel fuel composition will preferably be in the range
from 5 to 1500 ppmw, more preferably from 10 to 750 ppmw,
most preferably from 20 to 500 ppmw.
In the case of a diesel fuel composition, for
example, the fuel additive mixture will typically contain
a detergent, optionally together with other components as
described above, and a diesel fuel-compatible diluent,
which may be a mineral oil, a solvent such as those sold
by Shell companies under the trade mark "SHELLSOL", a
polar solvent such as an ester and, in particular, an
alcohol, e.g. hexanol, 2-ethylhexanol, decanol,
isotridecanol and alcohol mixtures such as those sold by
Shell companies under the trade mark "LINEVOL",
especially LINEVOL 79 alcohol which is a mixture of C7_9
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
primary alcohols, or a C12_14 alcohol mixture which is
commercially available.
The total content of the additives in the diesel
fuel composition may be suitably between 0 and 10000 ppmw
and preferably below 5000 ppmw.
In the above, amounts (concentrations, % vol, ppmw,
% wt) of components are of active matter, i.e. exclusive
of volatile solvents/diluent materials.
Process of Preparing a Liquid Fuel Composition
The liquid fuel composition of the present invention
is produced by admixing:
(a) a component derivable from a water-soluble
oxygenated hydrocarbon, with
(b) at least one fuel component.
By the term "fuel component" it is meant a component
used in the preparation of a liquid fuel composition, or
a liquid fuel composition per se, which is not a
component derived from a water soluble oxygenated
hydrocarbon. Examples of "fuel components" include fuel
components that are currently used in the preparation of
gasoline, kerosene and/or diesel fuel, such as petroleum
derived product streams, Fischer-Tropsch derived product
streams, oxygenates and biofuel components.
Typically, the petroleum derived product streams are
product streams produced at an oil refinery, also
referred to herein as refinery streams. Non-limiting
examples of such refinery streams include:
= "C4," (light boiling fraction typically comprising
greater than 80 %vol paraffinic and olefinic C4
compounds)
= Straight Run Tops (SR Tops) or Naphtha (light
boiling fraction of C4 - C7 hydrocarbons (final
boiling point below about 140 C). These
96
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
hydrocarbons are mainly paraffins and have a low
octane rating. The stream is low in aromatics and
has some naphthenes.)
= Isomerate (light boiling fraction (final boiling
point below about 110 C) obtained by isomerising C5
and C6 paraffins in SR Tops/Naphtha to produce
higher octane isomers)
= Light, full-range (FR) or heavy Platformate (or
light, full-range (FR) or heavy Reformate) (fraction
obtained by catalytic reforming of hydrotreated
naphtha to produce a high octane, high aromatic, low
olefin stream)
= Alkylate (including aviation alkylate)(fraction
produced by the alkylation of isobutane with C4 and
03 olefins to produce high-octane branched chain
paraffins)
= Light or heavy catalytic cracked gasoline (LCCG or
HCCG) (fraction produced by the treatment of heavier
streams in a fluidised catalytic cracker to produce
lighter hydrocarbons, including olefins)
= Straight run kerosene (fraction containing high
levels of paraffinic hydrocarbons, typically having
a distillation temperature in the range of from
about 150 C, +/- 10 C, to about 260 C, +/- about
20 C)
= Sweetened kerosene (straight run kerosene that has
been treated to reduce mercaptan levels and also
reduce total acid content)
= Hydrotreated or hydrofined kerosene (mildly
hydroprocessed straight run kerosene that has
reduced mercaptan, sulphur, olefins, acid and metals
levels)
97
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
= Severely hydroprocessed kerosene (such as deeply
hydrogenated kerosene, hydrodesulphurised kerosene,
hydrocracked kerosene (straight run kerosene that
has been subjected to a more severe hydroprocessing
than hydrotreated kerosene, typically leading to
lower sulphur and nitrogen levels))
= Straight run distillates boiling in the diesel fuel
range
= Hydrocracked gas oil (AGO)
= Light cracked and hydrotreated oils from the coker
= Catalytically cracked gas oil (VGO)
= Thermally and steam cracked gas oils
The oxygenates and biofuel components are any such
component which is suitable for use in a liquid fuel
composition, such as those described hereinbefore.
Process of Preparing a Gasoline Composition
In order to prepare a gasoline composition of the
present invention, a component derivable from a water-
soluble oxygenated hydrocarbon, as described above, in
particular the lighter fraction of a component derived
from a water-soluble oxygenated hydrocarbon, is combined
with at least one fuel component.
Typical fuel components which may be admixed with
the component derivable from a water-soluble oxygenated
hydrocarbon to prepare a gasoline composition according
to the present invention include:
= Gasoline compositions per se;
= Refinery streams, such as C48, SR Tops, Isomerate,
Light Platformate, FR Platformate, Heavy
Platformate, Alkylate, LCCG and HCCG;
= Oxygenates, such as alcohols and ethers. Preferred
oxygenate components include methanol, ethanol,
98
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
propanol, butanol, methyl tertiary-butyl ether
(MTBE), and ethyl tertiary-butyl ether (ETBE); and
= Biofuel components such as those described
hereinbefore.
By the term "gasoline composition" when used in
reference to a fuel component, it is meant a composition
as defined in the gasoline composition section above.
Typically, a gasoline composition of the present
invention is prepared by admixing:
(a) the lighter fraction of a component derived from a
water-soluble oxygenated hydrocarbon as described
above, with
(b) at least one fuel component selected from a refinery
stream.
Preferably, a gasoline composition of the present
invention is prepared by admixing:
(a) the lighter fraction of a component derived from a
water-soluble oxygenated hydrocarbon as described
above, with
(b) at least one fuel component selected from the
refinery streams C4õ SR Tops, Isomerate, Light
Platformate, FR Platformate, Heavy Platformate,
Alkylate, LCCG and HCCG; and optionally an oxygenate
selected from ethanol, MTBE and ETBE.
More preferably, a gasoline composition of the
present invention is prepared by admixing:
(a) the lighter fraction of a component derived from a
water-soluble oxygenated hydrocarbon as described
above, with
(b) at least one fuel component selected from the
refinery streams C4õ SR Tops, Heavy Platformate,
Alkylate and LCCG; and optionally ethanol.
99
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
Depending upon the amount of the lighter fraction of
a component derived from a water-soluble oxygenated
hydrocarbon that is used, the amount of the fuel
components used will be varied to prepare a gasoline
composition having the desired properties.
For instance, a gasoline composition of the present
invention may be prepared by admixing:
(a) at least 0.1 %vol based on the overall fuel
composition of the lighter fraction of a component
derived from a water-soluble oxygenated hydrocarbon
as described above, with
(b) at least one fuel component selected from the
refinery streams in the following amounts, 1 to
%vol C48, 3 to 25 %vol SR Tops, 0 to 50 %vol Heavy
15 Platformate, 5 to 20 %vol Alkylate, and 10 to
35 %vol LCCG, based on the overall fuel composition;
and optionally up to 85 % ethanol, based on the
overall fuel composition.
In the preparation of the gasoline compositions
according to the present invention, it may be desirable
to reduce the relative amount of any SR Tops and/or Heavy
Platformate in the gasoline composition according to the
present invention with increasing amounts of the lighter
fraction of a component derived from a water-soluble
oxygenated hydrocarbon as described above. Therefore, a
gasoline composition of the present invention may be
prepared by substituting at least a portion of any SR
Tops and/or Heavy Platformate used in the preparation of
a gasoline with the lighter fraction of a component
derived from a water-soluble oxygenated hydrocarbon as
described above.
Alternatively, a gasoline composition of the present
invention is prepared by admixing:
100
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
(a) the lighter fraction of a component derived from a
water-soluble oxygenated hydrocarbon as described
above, with
(b) a gasoline composition.
If the gasoline composition being prepared is an
aviation gasoline, then no oxygenates would be used in
the preparation of such an aviation gasoline according to
the present invention.
Process of Preparing a Kerosene Composition
In order to prepare a kerosene composition of the
present invention, a component derivable from a water-
soluble oxygenated hydrocarbon, as described above, in
particular the moderate fraction of a component derived
from a water-soluble oxygenated hydrocarbon, is combined
with at least one fuel component.
Typical fuel components which may be admixed with
the component derivable from a water-soluble oxygenated
hydrocarbon to prepare a kerosene composition according
to the present invention include refinery streams, such
as straight run kerosene, sweetened kerosene,
hydrotreated or hydrofined kerosene, and severely
hydroprocessed kerosene.
Typically, a kerosene composition of the present
invention is prepared by admixing:
(a) the moderate fraction of a component derived from a
water-soluble oxygenated hydrocarbon as described
above, with
(b) at least one fuel component selected from the
refinery streams straight run kerosene, sweetened
kerosene, hydrotreated or hydrofined kerosene, and
severely hydroprocessed kerosene.
101
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
Process of Preparing a Diesel Fuel Composition
In order to prepare a diesel fuel composition of the
present invention, a component derivable from a water-
soluble oxygenated hydrocarbon, as described above, in
particular the heavier fraction of a component derived
from a water-soluble oxygenated hydrocarbon, is combined
with at least one fuel component.
Typical fuel components which may be admixed with
the component derivable from a water-soluble oxygenated
hydrocarbon to prepare a diesel fuel composition
according to the present invention include:
. Diesel fuel compositions per se;
. Refinery streams, such as straight run distillates
boiling in the diesel fuel range, hydrocracked gas
oil (AGO), light cracked and hydrotreated oils from
the coker, catalytically cracked gas oil (VGO),
thermally and steam cracked gas oils;
= Biofuel components such as those described
hereinbefore.
By the term "diesel fuel composition" when used in
reference to a fuel component, it is meant a composition
as defined in the diesel fuel composition section above.
Typically, a diesel fuel composition of the present
invention is prepared by admixing:
(a) the heavier fraction of a component derived from a
water-soluble oxygenated hydrocarbon as described
above, with
(b) at least one fuel component selected from a refinery
stream.
Preferably, a diesel fuel composition of the present
invention is prepared by admixing:
102
CA 02735654 2016-04-28
(a) the heavier fraction of a component derived from a
water-soluble oxygenated hydrocarbon as described
above, with
(b) at least one fuel component selected from the
refinery streams straight run distillates boiling in
the diesel fuel range, hydrocracked gas oil (AGO),
light cracked and hydrotreated oils from the coker,
catalytically cracked gas oil (VGO), thermally and
steam cracked gas oils; and optionally a bifuel
component.
The present invention further provides a method of
operating an internal combustion engine, jet engine, or a
boiler, which method involves introducing into a
combustion chamber of the engine or boiler, a liquid fuel
composition according to the present invention.
The present invention will be further understood
from the following examples.
Examples
Exemplary Reactor Systems
Example 1
Figure 8 shows a process diagram illustrating one
reactor system useful in practicing the present
invention. A feedstock tankauacts as a reservoir for
holding the feedstock solutions. The feedstock solution
is delivered from the feedstock tankauto feed pump2o3
through feed line 202, where it is then passed through
discharge line2o4to preheater2o5. The preheater2o5may be a
heat exchanger heated by an electrical resistance heater,
or any other heat exchanger known in the art. The
preheated feed is then passed through line206and, in some
cases, combined with hydrogen2o7before entering reactor2o9
through line 208. One illustration of a potential reactor 209
1()3
CA 02735654 2016-08-31
is set forth in Figure 11 and more fully described in
Example 4 below.
The temperature of the walls of reactor 209is
maintained by block heaters, 210a, 210b, 210c, and 210d, in
this case, electrical resistance heaters. Upon exiting
the reactor 209, reaction products enter the reactor outlet
line 211 and are cooled to near ambient temperature in
reactor product cooler 212, resulting in a potential three
phase product stream. From reactor product cooler 212, the
reaction products proceed through line 213 to pressure
regulating valve 214, which is used to control the
pressure at the reactor outlet if required.
After valve 214, the products enter a phase separator
216 through line 215 where it segregates into three
separate phases: (1) non-condensable gas components 217
containing predominately hydrogen, carbon dioxide,
methane, ethane, and propane; (2) an organic liquid
fraction 218 containing both hydrocarbons and C.
alcohols, ketones and carboxylic acids; and (3) an
aqueous layer 219 containing mostly water and water
soluble oxygenated compounds, such as ethanol,
isopropancl, acetone, propanol and acetic acid. The non-
condensable gas fraction 217 may be routed through the gas
product line 220 to pressure reducing valve 221. The
pressure of separator 216 is maintained by pressure
reducing valve 221. In an alternate mode of operation, the
separator 216 may be maintained at a pressure nearly the
same as the reactor outlet by opening or eliminating
valve 214. In the alternate mode of operation, the reactor
outlet pressure is then controlled by action of pressure
reducing valve 221. Gas flow rate and composition are
measured upon exiting the system through line 222.
104
CA 02735654 2016-08-31
The organic liquid fraction 218exits the separator
through line 223 before entering organic draw-off valve
224. The level of organic phase within the separator is
controlled by adjustment of valve 224. The flow rate and
composition of the organic fraction are determined after
the organic fraction exit the system through line 225. The
aqueous liquid fraction 219 exits the separator through
line 226 before entering separator bottoms draw-off valve
227. The level of aqueous phase within the separator is
controlled by adjustment of valve 227.
The flow rate and composition of the aqueous
fraction may be determined after the aqueous fraction
exits the system through line 228. In an alternate mode of
operation, both the organic liquid fraction 218 and the
aqueous liquid fraction 219 exit the system through the
bottom draw-off valve 227 of the separator and line 228
before being separated in a decanter for measurement of
the individual phase compositions and flow rates.
In all cases, the alternate modes of operation do
not affect the catalytic processes being investigated.
The alternate modes of operation may be employed as
deemed prudent to achieve optimal control of the process,
depending on the relative flow rates of the gaseous phase
217, organic liquid phase 218, and aqueous phase 219.
Prior to initiating a flow of feed to the reactors,
unless otherwise noted, catalysts were reduced in a
stream of flowing hydrogen at 400 C, regardless of
whether a reduction was completed prior to loading the
catalyst into the reactors.
Example 2
Figure 9 shows a process diagram illustrating
another reactor system useful for practicing the present
invention. This reactor configuration contains two
105
CA 02735654 2016-08-31
separate reactors with the capability of operating both
reactors in series or operating only the first reactor.
In addition, this configuration allows the catalyst in
the second reactor to be taken off line and regenerated
in situ. After regeneration, the second reactor may be
returned to service without impacting the first reactor
operation.
The reactor is similar to the reactor of Example 1,
except that the reaction products from reactor product
cooler 312 could be routed into the second reactor through
line 314 or routed to bypass the second reactor by passing
into line 344. When utilizing the second reactor, flow
would proceed from line 314 to pressure regulating valve
315. Pressure regulating valve 315 may be used to control
the pressure at the outlet of the first reactor. From
pressure regulating valve 315 the flow proceeds to the
second reactor inlet isolation valve 317 and into line 318.
From line 318 the flow continues to line 319 and into the
second reactor preheater 320. In the illustrated
embodiment, preheater 320 is a heat exchanger heated by an
electrical resistance heater.
The preheated feed is then passed through line 319
into the second reactor 322, which is more fully described
in Example 4. The temperature of the wall of reactor 32
is maintained by block heaters, 323a, 323b, 323c, and 323d,
in this case, electrical resistance heaters. Upon exiting
the reactor, the reaction products enter the second
reactor outlet line 324 and are then cooled in second
reactor product cooler 325. From second reactor product
cooler 326 the process flow may be routed through lines 326
and 327 to second reactor outlet isolation valve 328, into
lines 328 followed by 330 and then into the product
separator 331.
106
CA 02735654 2016-10-13
When operation of the second reactor is desired,
valve and valve 326 are open while the second reactor
bypass valve 345 is closed to prevent the flow from
bypassing the second reactor. When operation of only the
first reactor is desired, or when the second reactor is
being regenerated, valve 317 and valve 325j are closed while
valve 345 is open. When the second reactor is bypassed,
the first reactor product flows directly from line 313
into line 344, through bypass valve 345, into line 346 and
on to line 330. In either case, whether the second reactor
is in operation or bypassed, the flow would proceed from
line 330 into the product separator.
In phase separator 331, reaction products are
separated into a gaseous fraction 332, an organic fraction
3Y?, and an aqueous fraction 334 as described above in
Example 1. The gaseous fraction 332 is routed through the
gas product line 335 to pressure reducing valve 336. The
pressure of separator 331 is maintained by pressure
reducing valve 336. When the second reactor 322 is in
service, the pressure at the second reactor 322 outlet is
controlled by action of pressure reducing valve 336. When
the second reactor 322 is bypassed, the pressure at the
outlet of the first reactor ]!()1=,is controlled by action of
pressure reducing valve 336.
Gas flow rate and composition are measured upon
exiting the system through line 337. The organic liquid
fraction 133 exits the separator through line 336 before
entering organic draw-off valve 339. The level of organic
phase within the separator is controlled by adjustment of
valve 339. The flow rate and composition of the organic
fraction are determined after the organic fraction exits
the system through line 340. The aqueous liquid fraction
334 exits the separator through line 341 before entering
107
CA 02735654 2016-04-28
separator bottoms draw-off valve 342. The level of aqueous
phase within the separator is controlled by adjustment of
valve 342. The flow rate and composition of the aqueous
fraction are determined after the aqueous fraction exits
the system through line 343. In an alternate mode of
operation, both the organic liquid fraction 333 and the
aqueous liquid fraction 334 exit the system through the
separator bottoms draw-off valve 342 and line 343 before
being separated in a decanter for measurement of the
individual phase compositions and flow rates. In all
cases, the alternate modes of operation do not affect the
catalytic processes being investigated. The alternate
modes of operation are employed as deemed prudent to
achieve optimal control of the process, depending on the
relative flow rates of the gaseous phase 335, organic
liquid phase 333, and aqueous phase 334.
Example 3
Figure 10 shows a process diagram illustrating a
dual feed pump reactor system useful for practicing the
present invention. A dual feed pump system is used when
the desired mix of feed components would not exist in a
single liquid phase. For example, when a mix of 50% by
weight 2-pentanol and 50% by weight water is the desired
feed, two feed pumps are used, one to deliver 2-pentanol
and the other to deliver water. A similar system may also
be used to mix feedstock derived from two separate
sources, such as a virgin feedstock and an oxygenated
hydrocarbon feedstock derived from an effluent stream of
the reactor system itself.
First feedstock tank4olacts as a reservoir for a
first feedstock solution, while second feedstock tank44o
acts as a reservoir for a second feedstock solution. A
first feed is delivered from first feedstock tank4olto
108
CA 02735654 2016-08-31
first feed pump4o3through first feed line 402. The first
feed is then passed through the first feed pump discharge
line4o4to combined feed line444. The second feed is
delivered from the second feedstock tank 440 to second
feed pump 442 through second feed line 441. The second feed
is then passed through second feed pump discharge line 443
to combined feed line 444. From combined feed line 444 the
combined feed passes into preheater4os.All other elements
are as set forth in Example 1, except that the aqueous
phase 19 may be recycled to feedstock tank 440 for further
processing or used in other processes.
Example 4
Figure 11 shows a schematic illustration of one type
of reactor which may be employed in reactor systems as
described in Examples 1, 2 and 3. Reactor tubesoiis
composed of 316 stainless steel with either an inside
diameter of 8.5 mms or an inside diameter of 21.2 mm,
depending on the experiment. Inlet linesozis provided to
allow feedstock or intermediate product, such as
oxygenates, to enter the reactor. Outlet line5o3is
provided to remove product from the reactor. Inlet frit
504, composed of stainless steel, acts to secure the beds
of preheat media and catalyst in place. Preheat media 505,
consisting of stainless steel beads, acts as a zone to
allow transfer of heat from the reactor walls so that the
feed is at the desired temperature upon entering the
catalvstso7. A stainless steel screen may be placed
between preheat mediasosand catalyst5o7to prevent the
materials from mixing. Catalyst5o7may be supported in
position by a second stainless steel frit sos.
A thermowellso9 may be installed in some cases to
allow measurement of the temperatures within catalyst 507
and preheating zone sos. Control of temperature at the
109
CA 02735654 2016-04-28
reactor inlet is accomplished by the use of an external
preheater prior to the feed entering the reactor through
line 502, and may be further adjusted by control of the
heat transfer that occurs in the preheat media. In some
cases, the preheat media is not required to achieve the
desired temperature profile. Control of the reactor wall
temperature is achieved by the use of external heaters in
contact with the outer wall of the reactor. Independently
controlled heating zones may be used to control the
temperature of the reactor wall as desired.
Example 5 - Analysis Techniques
Product streams from the examples described below
were analyzed as follows. The organic liquid phase was
collected and analyzed using either gas chromatograph
with mass spectrometry detection or flame ionization
detection. Component separation was achieved using a
column with a bonded 100% dimethyl polysiloxane
stationary phase. Relative concentrations of individual
components were estimated via peak integration and
dividing by the sum of the peak areas for an entire
chromatogram. Compounds were identified by comparison to
standard retention times and/or comparison of mass
spectra to a compiled mass spectral database. Gas phase
compositions were determined by gas chromatography with a
thermal conductivity detector and flame ionization or
mass spectrometry detectors for other gas phase
components. The aqueous fraction was analyzed by gas
chromatography with and without a derivatization of the
organic components of the fraction using a flame
ionization detector. Product yields are represented by
the feed carbon present in each product fraction. The
weight hourly space velocity (WHSV) was defined as the
weight of feed introduced into the system per weight of
110
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
catalyst per hour, and based on the weight of the
oxygenated hydrocarbon feed only, excluding water present
in the feed.
Production of Oxygenates
Example 6 - Hydrogenation Catalyst
A hydrogenation catalyst was prepared by adding an
aqueous solution of dissolved ruthenium nitrosyl nitrate
to a carbon catalyst support (UU Carbon, Calgon, with
particle sizes restricted to those that were maintained
on a 120 mesh screen after passing through an 60 mesh
screen) to a target loading of 2.5% ruthenium. Water was
added in excess of the pore volume and evaporated off
under vacuum until the catalyst was free flowing. The
catalyst was then dried overnight at 100 C in a vacuum
oven.
Example 7 - APR/Deoxygenation Catalyst
A combined APR and deoxygenation catalyst was
prepared by dissolving hexachloroplatinic acid and
perrhenic acid in water and then adding the mixture to a
monoclinic zirconia catalyst support (NorPro Saint-
Gobain, Product code SZ31164, with particle sizes
restricted to those that were maintained on a 60 mesh
screen after passing through an 18 mesh screen) using an
incipient wetness technique to target a platinum loading
of 1.8% and a rhenium loading of 6.3% on the catalyst
after subsequent decomposition of the metal precursors.
The preparation was dried overnight in a vacuum oven and
subsequently calcined in a stream of flowing air at
400 C.
Example 8 - Conversion of Sucrose to Oxygenates
The catalyst systems referenced in Examples 6 and 7
were investigated for the conversion of sucrose to an
intermediate product containing oxygenates using the
111
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
reactor system described in Example 1. The study was
conducted using a 21.2 mm internal diameter stainless
steel tube reactor shown in Example 4, with an analysis
completed as described in Example 5.
31 grams of hydrogenation catalyst from Example 6
and 76 grams of APR catalyst from Example 7 were loaded
into the reactor, with the hydrogenation catalyst on top
of the APR catalyst, separated by a stainless steel
screen. External hydrogen was combined with the feed
prior to the feed entering the reactor. Heaters external
to the reactor, shown in Figure 8 as 10a, 10b, 10c, 10d,
were maintained at the following reactor wall
temperatures; 10a - 125 C, 10b - 200 C, 10c - 265 C, 10d
- 265 C, resulting in reactor bed temperatures of
approximately -110-150 C for hydrogenation, and 150-265 C
for the APR/Deoxygenation catalyst. The ranges indicate
the approximate reactor wall temperatures at the inlet
and outlet of each catalyst bed, respectively. Results
from the experiment across 39 hours of operation are
shown in Table 1. The WHSV is based on the weight of the
APR/Deoxygenation catalyst. Total mono-oxygenates
includes alcohols, ketones, tetrahydrofurans and cyclic
mono-oxygenates. Cyclic mono-oxygenates includes
compounds in which the ring does not include oxygen, such
as cyclopentanone and cyclohexanone. The fraction of feed
carbon contained within unknown components in the aqueous
phase was determined as the difference of carbon
accounted for by known, measured components and the total
organic carbon.
112
Table 1
0
w
o
1-,
Conversion of Sucrose to Oxygenates Across a Hydrogenation and APR Catalyst
o
Hours on Stream 5 16
27 39
w
m
WHSV Wtfeed/ (Wtcatalyst hr) 1.8 1.8
1.7 1.5 w
o
c.,
Added Hydrogen mo1H2/molfeed 3.4 3.4
3.6 4.0
Organic Phase Yield % of feed carbon 27 25
20 22
Breakdown of Reactor Outlet Composition
Carbon Dioxide % of feed carbon 19.4 21.2
18.1 17.7
Paraffins % of feed carbon 14.1 13.5
9.2 10.8
Mono-oxygenates % of feed carbon 31.5 30.6
27.5 30.8
Alcohols % of feed carbon 11.1 11.8
11.2 11.6 n
Ketones % of feed carbon 8.2 7.0
7.1 9.0 0
KJ
Tetrahydrofurans % of feed carbon 10.6 10.7
8.1 8.6 --3
w
Cyclic Mono-oxygenates % of feed carbon 1.6 1.1
1.1 1.5 m
m
m
Unknown Aqueous Species % of feed carbon 21.2 27.8
28.3 32.0 a,
I.)
0
H
H
I
0
KJ
I
KJ
CO
.0
n
,-i
cp
w
=
=
-:,--
u,
u,
-.1
c.,
113
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
Example 9 - APR/Deoxygenation Catalyst
A catalyst was prepared as described in Example 7,
except that the catalyst support was a tetragonal
zirconia (NorPro Saint-Gobain, Product code SZ61152) with
particle sizes restricted to those that were maintained
on a 60 mesh screen after passing through an 18 mesh
screen.
Example 10 - APR/Deoxygenation Catalyst
Hexachloroplatinic acid and perrhenic acid dissolved
in water were added to a monoclinic zirconia catalyst
support (NorPro Saint-Gobain, Product code 5Z61164, with
particle sizes restricted to those that were maintained
on a 60 mesh screen after passing through an 18 mesh
screen) using an incipient wetness technique to target a
platinum loading of 1.9% and a rhenium loading of 1.8% on
the catalyst after subsequent decomposition of the metal
precursors. The preparation was dried overnight in a
vacuum oven and subsequently calcined in a stream of
flowing air at 400 C.
Example 11 - APR/Deoxygenation Catalyst
A catalyst was prepared as described in Example 7
except that the support was a hydrogen peroxide
functionalized activated carbon. The support was first
prepared by adding activated carbon (Calgon UU 60x120
mesh carbon) slowly to a 30% hydrogen peroxide solution,
with the mixture then left overnight. The aqueous phase
was decanted and the carbon was washed three times with
deionized water, and then dried under vacuum at 100 C. A
solution of hexachloroplatinic acid and perrhenic acid in
water was then added to the support using an incipient
wetness technique to target a platinum loading of 1.8%
and a rhenium loading of 6.3% after subsequent
114
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
decomposition of the metal precursors. The preparation
was dried overnight in a vacuum oven at 100 C.
Example 12 - Conversion of Sorbitol and Glycerol
The catalyst systems referenced in Example 9,
Example 10, and Example 11, were investigated for the
conversion of sorbitol or glycerol to an intermediate
product containing oxygenates using the reactor
configuration described in Example 1, with an analysis
completed as described in Example 5. The study was
conducted using the 8.5 mm internal diameter stainless
steel tube reactor shown in Example 4. In all cases, the
reactor pressure was maintained at 625 psig. Reactor
inlet and outlet temperatures, shown in Table 2 were
controlled using heaters external to the reactor as shown
in Figure 8 as 10a, 10b, 10c, 10d. Results of these
experiments are shown in Table 2.
Table 2 shows the impact of catalyst composition,
feedstock composition, and operating conditions on the
conversion performance. Figure 12 shows the carbon number
distribution of the mono-oxygenates produced in
Experiment D and Experiment E. The primary difference
between these two experiments was the reaction
temperature. For Experiment D, mono-oxygenates containing
three or fewer carbon atoms predominated while for
Experiment E, a significant fraction of the mono-
oxygenates contained four or more carbon atoms,
indicating that condensation reactions were occurring
within the same reaction zone as the hydrogen generation
and deoxygenation reactions. The WHSV is based on the
weight of the APR/Deoxygenation catalyst. The net
hydrogen produced is the hydrogen present at the reactor
outlet as H2, which does not include hydrogen produced
and consumed in situ. Total mono-oxygenates include
115
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
alcohols, ketones, tetrahydrofurans and cyclic mono-
oxygenates. Cyclic mono-oxygenates include compounds in
which the ring does not include oxygen, such as
cyclopentanone and cyclohexanone. The fraction of feed
carbon contained within unknown components in the aqueous
phase was determined as the difference of carbon
accounted for by known, measured components and the total
organic carbon.
116
Table 2
0
w
o
1-,
Conversion of Polyols to Oxygenates Across a APR/Deoxygenation Catalyst
o
Experiment A B
C D E 'a
w
m
50% 50%
65% 50% 50% w
o
c.,
Feed Sorbitol Sorbitol Sorbitol
Glycerol Glycerol
Catalyst Composition Example No. 11 9
10 10 10
Wtfeed/ (Wtcatalyst
WHSV hr) 2.1 1.8
1.7 1.5 1.5
Catalyst Inlet Temp. C 241 240
240 260 310
Catalyst Outlet
Temperature C 240 241
321 260 350 n
Net Hydrogen Produced mo1H2/molfeed 0.6 0.9
0.7 1.2 0.7 0
I.)
Organic Phase Yield % of feed carbon 17 24
38 0 38 --3
w
Breakdown of Reactor Outlet Composition
m
m
m
Carbon Dioxide % of feed carbon 32.4 34.0
23.5 31.3 16.0 a,
Paraffins % of feed carbon 37.4 25.3
7.8 6.6 7.4 "
0
H
Total Mono-oxygenates % of feed carbon 33.9 32.9
40.0 45.9 41.0 H
I
Alcohols % of feed carbon 6.3 8.5
2.6 40.6 4.6 0
I.)
1
Ketones % of feed carbon 23.5 16.9
15.2 5.2 24.1 I.)
0
Tetrahydrofurans % of feed carbon 4.1 7.2
10.7 0.1 2.7
Cyclic Mono-oxygenates % of feed carbon 0.0 0.4
11.6 0.0 9.7
Unknown Aqueous Species % of feed carbon 1.2 7.8
15.8 30.4 10.7
Iv
n
,-i
cp
t..)
=
=
'a
u,
u,
-4
c7,
117
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
Condensation of Oxygenates Using Basic Catalysts
Example 13
A zinc aluminate catalyst support was prepared by
mixing zinc oxide powder and alumina powder (Dispal 18N4-
80, Sasol North America, Houston, Texas) to a target
ratio of 1.0 moles of ZnO to 1 mole of A1203. Dilute
nitric acid was then added at a level of 1 wt% HNO3 to
alumina. The dough consistency of the mixture was
adjusted with water addition to form a workable dough,
which was then extruded using a laboratory scale
extruder. The extrudates were dried overnight under
vacuum at 100 C, then further dried at 200 C for one hour
under flowing air, and then subsequently calcined at
750 C for 4 hours under flowing air. The resulting
material was then ground and sieved. Material that was
maintained on a 60 mesh screen after passing through an
18 mesh screen was recovered.
Example 14
Hexachloroplatinic acid was added to the calcined
material of Example 13 using an incipient wetness
impregnation technique to achieve a target platinum
loading of 1.0 wt%. The catalyst was dried overnight
under vacuum at 100 C and calcined at 400 C under flowing
air.
Example 15
Palladium nitrate was added to the calcined material
of Example 13 using an incipient wetness impregnation
technique to achieve a target palladium loading of 0.5
wt%. The catalyst was dried overnight under vacuum at
100 C and calcined at 400 C under flowing air.
Example 16
A copper zinc aluminate catalyst was prepared by
mixing zinc oxide, copper (I) oxide, and alumina powder
118
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
(Dispal 18N4-80) at a target ratio of 0.11 moles of CuO
and 0.9 moles of ZnO to one mole of A1203. Dilute nitric
acid was then added at a level of 1 wt% HNO3 to alumina.
The dough consistency of the mixture was adjusted with
water addition to form a workable dough, which was then
extruded using a laboratory scale extruder. The
extrudates were dried overnight under vacuum at 100 C,
then further dried at 200 C for one hour under flowing
air, and then subsequently calcined at 750 C for 4 hours
under flowing air. The resulting material was then ground
and sieved. Material that was maintained on a 60 mesh
screen after passing through an 18 mesh screen was
recovered.
Example 17
A cesium modified silica-alumina catalyst was
prepared by adding cesium carbonate dissolved in water to
Siralox silica-alumina catalyst support (Sasol North
America, Houston, Texas). The target loading of cesium
was 25 wt% based on final catalyst weight. This material
was dried for 24 hours under vacuum at 100 C and calcined
at 500 C for 6 hours under flowing air. After calcining,
platinum was added using an incipient wetness
impregnation technique to achieve a final platinum
loading of 1 wt%. After impregnation, the catalyst was
dried and then calcined at 500 C for 6 hours under
flowing air.
Example 18
A cerium modified silica was prepared by adding
cerium nitrate solution to a silica gel (Davisil grade
636, WR Grace Company) to a final loading of 25 wt% Ce02.
The resulting material was then dried at 120 C for six
hours and further calcined at 550 C for six hours under
flowing air. Palladium nitrate was added to the calcined
119
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
material using an incipient wetness impregnation
technique to achieve a target palladium loading of 0.5
wt%. This material was then dried at 120 C for six hours
and further calcined at 550 C for six hours under flowing
air.
Example 19
The catalyst systems referenced in Examples 14-18
were investigated for the vapor-phase condensation of
various oxygenates. The studies were conducted using 8.5
mm and 21.2 mm internal diameter size stainless steel
tube reactors as described in Example 4 and in the
reactor systems illustrated by Figures 8 and 10. Between
and 18 milliliters of catalyst was loaded into the
smaller reactor, with between 50 and 70 milliliters of
15 catalyst loaded into the larger reactor. In all cases the
catalyst was reduced at 400 C under flowing hydrogen
prior to use.
The organic liquid phase was collected and analyzed
as described in Example 5. Table 3 shows organic product
yields and composition as a function of operating
conditions, feedstock composition, and the added metal
component for the catalysts described in Examples 14 - 18
above. Greater than 100% reported organic phase yields
stem from experimental uncertainty in the measurement of
process stream flow rates or composition. Non-condensed
components are those components that do not require the
formation of new carbon-carbon bonds to be produced from
the given feed. For simplicity, all compounds containing
five or fewer carbon atoms are considered to be non-
condensed components. Total condensation products are
those compounds containing six or more carbon atoms,
which require the formation of new carbon-carbon bonds to
be formed from the given feedstocks.
120
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
Experiments F and G demonstrate that product
selectivity can be affected by the choice of
hydrogenation function, e.g. Pt or Pd. Paraffins were
produced to a larger extent over the catalyst containing
1% platinum compared to the catalyst containing 0.5%
palladium. The later favored the production of mono-
oxygenates, primarily ketones. Experiments H and I
further reinforce this concept. Experiment H shows that
condensed mono-oxygenate components can be obtained at
high yield with isopropyl alcohol as a feed, accounting
for >97% of the organic product and containing >90% of
the overall carbon at the reactor outlet. By increasing
the reaction temperature and using copper to drive the
hydrogenation reactions, the selectivity can be shifted
to obtain a significant yield of olefins (Experiment I).
Experiments J, K and L show that a number of other
heterogeneous catalysts can be used to promote the
condensation of oxygenates followed by hydrogenation of
the initial condensation products. Experiments K and L
show that as the temperature is decreased from 300 C to
250 C, the rate of condensation drops so that the
conversion to condensed products drops from 81 wt% to 18
wt% in the resulting organic phase.
121
Table 3 Vapor Phase Condensation of Oxygenates Over Basic Catalysts
0
w
o
1% Pt / Cs
=
Impregnated
0.5% Pd / 0.5% Pd / -1
m
Siralox
Ce Ce t-.)
=
1% Pt / 0.5% Pd / 0.5% Pd / CuO/ZnO Silica-
Modified Modified c:
Catalyst ZnO/A1203 ZnO/A1203 ZnO/A1203 /A1203 Alumina
Silica Silica
Experiment F G H I J
K L
Feed Feed A Feed A Feed B Feed B Feed A
Feed B Feed B
wtfeed/
WHSV (wtcaf hr) 1 1.5 1.5 2 1.1
1.9 1.9
moliu/
Added Hydrogen molfeed 1 1 0 0 1
0 0 n
Temperature C 375 375 300 375 325
300 250 0
Pressure Psig 600 600 600 625 600
600 600 1.)
-.3
w
Organic Phase % of feed
m
m
Yield carbon 75 99 95 55 107
74 98 m
.1.
Organic Phase Composition Breakdown
1.)
0
C5-
H
H
Hydrocarbons wt% 9.6 7.3 0.0 2.4 1.6
0.0 0.0 1
0
C5- Oxygenates wt% 6.2 20.9 1.9 14.6 75.8
18.5 81.8 "
1
1.)
Total Non-
0
Condensed
Components wt% 15.8 28.2 1.9 16.9 77.4
18.5 81.8
C6+ Paraffins wt% 49.5 18.9 0.3 1.0 0.0
20.1 0.2
C6+ Olefins wt% 4.6 0.0 0.0 15.9 0.0
0.0 0.0
Other C6+
Hydrocarbons wt% 0.0 0.0 0.0 1.1 0.0
0.0 0.0 1-0
C6+ Mono-
n
,-i
oxygenates wt% 30.2 51.8 97.3 64.5 22.6
61.0 18.0
cp
Total Cond.
t-.)
=
Products wt% 84.2 70.7 97.6 82.5 22.6
81.1 18.2 =
Feed A - 49.5% 2-Pentanone, 50.5% 2-Pentanol
-1
un
un
Feed B - 100% Isopropyl Alcohol
--.1
c,
122
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
Condensation of Oxygenates Using Acid-Base Catalysts
Example 20
A hydrotalcite catalyst was prepared from a
commercially available hydrotalcite support (ESM-350, ASM
Catalysts, Baton Rouge, LA) by grinding the material and
passing through graduated screens to achieve particles
sizes larger than 60 mesh and less than 18 mesh. The
material was then calcined in a quartz tube reactor at
450 C for 6 hours under flowing nitrogen.
Example 21
Platinum was added to the hydrotalcite catalyst of
Example 20 using an incipient wetness impregnation
technique to achieve a final target platinum loading of
1 wt%. The platinum containing precursor was
hexachloroplatinic acid, H2PtC16. The impregnated
material was dried overnight under vacuum at 100 C and
subsequently calcined at 400 C for 2 hours under flowing
air.
Example 22
Platinum and tin were added to the hydrotalcite
catalyst of Example 20 using an incipient wetness
impregnation technique to achieve a final target loading
of 1 wt% Pt and 0.2 wt% Sn. The platinum containing
precursor was hexachloroplatinic acid, H2PtC16 while tin
was derived from tin chloride, SnC12*2H20. The
impregnated material was dried overnight under vacuum at
100 C and subsequently calcined at 450 C for 8 hours
under flowing nitrogen.
Example 23
A 5% magnesium oxide catalyst supported on granular
zirconia was prepared using an incipient wetness
impregnation technique to achieve a final target loading
of 5 wt% Mg. Magnesium was added as magnesium nitrate and
123
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
dried overnight under vacuum at 100 C and subsequently
calcined at 450 C for 8 hours under flowing air. An
aqueous palladium nitrate solution was added to the
calcined material to achieve a target palladium loading
of 0.5 wt% using an incipient wetness impregnation
technique. The catalyst was dried a second time and
calcined at 400 C for six hours under flowing air.
Example 24
A zinc aluminate catalyst support was prepared by
mixing zinc oxide powder and alumina powder (Dispal 18N4-
80, Sasol North America, Houston, Texas) to a target
ratio of 0.85 moles of ZnO to 1 mole of A1203. Dilute
nitric acid was added at a level of 1 wt% HNO3 to total
solids. The dough consistency was adjusted with water
addition to form a workable dough suitable for extrusion
and the mixture was extruded using a laboratory scale
extruder. The extrudates were dried overnight under
vacuum at 100 C and subsequently calcined at 750 C for 8
hours under flowing air. The material was then sized to
18 by 60 mesh. An aqueous palladium nitrate solution was
added to the calcined material to achieve a target
palladium loading of 0.5 wt% using an incipient wetness
impregnation technique. This catalyst was then dried a
second time and calcined at 400 C for six hours under
flowing air.
Example 25
The catalyst systems referenced in Examples 21-24
were used to conduct vapor-phase condensation reactions
with various oxygenates. The studies were conducted using
8.5 mm and 21.2 mm internal diameter size stainless steel
tube reactors as described in Example 4 and reactor
systems as illustrated in Examples 1 and 3. Between 15
and 18 milliliters of catalyst was loaded into the
124
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
smaller reactor, with between 50 and 70 milliliters of
catalyst loaded into the larger reactor. In all cases the
catalyst was reduced at 400 C under flowing hydrogen
prior to use.
The organic liquid phase was collected and analyzed
as described in Example 5. Table 4 shows the organic
product yields and composition as a function of operating
conditions, feedstock composition, and the added metal
component for the hydrotalcite catalysts described in
Examples 21 and 22 above. The data from the experiments
show that a primarily hydrocarbon product can be formed
from acetone and isopropyl alcohol in the absence of an
added metal hydrogenation component. In Experiment M, the
organic phase product contained primarily nine carbon
methyl substituted cyclohexenes, categorized as other C6+
hydrocarbons in Table 4. The addition of platinum
(Experiment N) to this catalyst favored the formation of
condensed mono-oxygenate products, mainly ketones and
alcohols, and the formation of some paraffins as a result
of deoxygenation of the ketones and alcohols. The
selectivity was further shifted in favor of condensed
mono-oxygenates by attenuating the platinum with tin and
operating at a higher pressure (Experiment 0).
Experiments P, Q, R and S illustrate the impact of
reaction temperature for the condensation of a mixed feed
containing pentanol and pentanone. As the reaction
temperature was raised from 300 C to 375 C, a gradual
change in product composition became apparent, with the
selectivity to condensed mono-oxygenates decreasing and
the selectivity to condensed paraffins increasing as the
temperature was raised.
Table 5 shows the impact of feedstock components and
reaction temperature on organic product yields and
125
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
composition for the catalysts of Examples 23 and 24.
Experiments T and U compare the condensation of 2-
pentanone and 2-methylterahydrofuran. Overall, the
condensation of 2-pentanone is faster than 2-
methyltetrahydrofuran. Nonetheless, around 30% of the
tetrahydrofuran was converted to condensation products
under these conditions. Experiments 10 and 11 show the
impact of reaction temperature when using a pure
isopropyl alcohol feed. At 300 C (Experiment V), mono-
oxygenated condensation products predominate, while at
400 C (Experiment W) a significant portion of the
products consisted of hydrocarbons. Compared to other
experiments listed in Tables 4and 5, Experiment W is
notable in that the organic product contained a higher
level of olefins. The addition of valeric acid to the
feed (Experiment X) suppressed overall condensation rates
and shifted the selectivity away from paraffins and
towards other hydrocarbons, primarily substituted aryl
compounds.
Greater than 100% reported organic phase yields stem
from experimental uncertainty in the measurement of
process stream flow rates or composition. Non-condensed
components are those components that do not require the
formation of new carbon-carbon bonds to be produced from
the given feed. For simplicity, all compounds containing
five or fewer carbon atoms are considered to be non-
condensed components. Total condensation products are
those compounds containing six or more carbon atoms,
which require the formation of new carbon-carbon bonds to
be formed from the given feedstocks.
126
Table 4 Vapor Phase Condensation of Oxygenates Over Hydrotalcite Catalysts
0
w
o
1% Pt, 1% Pt, 1%
Pt, 1% Pt, 1% Pt,
o
Metal Function None 1% Pt 0.2% Sn 0.2% Sn 0.2%
Sn 0.2% Sn 0.2% Sn -1
m
Experiment M N 0 P 4
R S w
o
Feed Feed
c:
Feed C C Feed C Feed A Feed
A Feed A Feed A
wtfeed/
WHSV wtcat hr 1.0 0.9 0.7 0.7 0.7
0.7 0.7
moliu/
Added Hydrogen MOlfeed 0.5 0 0 1 1
1 1
Temperature C 350 350 350 300 325
350 375
Pressure Psig 100 100 600 600 600
600 600 n
% of
0
I.)
feed
w
Organic Phase Yield carbon 61 95 91 108 104
108 85 m
m
Organic Phase Composition Breakdown
ul
.1.
C5- Hydrocarbons wt% 2.8 3.6 1.0 4.6 7.1
9.4 20.0 1.)
0
C5- Oxygenates wt% 11.9 16.0 5.8 41.9
21.4 13.7 8.8 H
H
I
Total Non-Condensed
0
Components wt% 14.7 19.6 6.8 46.5
28.5 23.1 28.8 1.)
1
C6+ Paraffins wt% 0.0 13.1 7.6 2.2
11.3 28.6 53.0 1.)
0
C6+ Olefins wt% 5.1 1.2 1.0 0.0 0.2
0.0 0.0
Other C6+
Hydrocarbons wt% 72.8 0.0 0.0 0.0 0.0
0.0 0.0
C6+ Mono-oxygenates wt% 5.7 54.3 80.4 51.4
60.1 47.8 18.2
Total Condensation
Products wt% 83.5 68.6 89.0 53.6
71.6 76.5 71.2 IV
n
Feed A - 49.5% 2-Pentanone, 50.5% 2-Pentanol
Feed C - 50% Isopropyl Alcohol, 50% Acetone
cp
w
o
o
-1
vi
vi
--.1
c,
127
Table 5
0
w
o
1-,
o
-1
w
Vapor Phase Condensation of Oxygenates Over Magnesium Impregnated Zirconia
a:
w
and Zinc Aluminate Catalysts
o
c:
0.5% Pd / Zinc 0.5%
Pd / Zinc 0.5% Pd / Zinc
0.5% Pd / 0.5% Pd / Aluminate
Aluminate Aluminate
5% Mg 5% Mg (0.85:1
(0.85:1 (0.85:1
Catalyst Zirconia Zirconia ZnO:A1203)
ZnO:A1203) ZnO:A1203)
Experiment T U V
W X
Feed Feed D Feed E Feed B
Feed B Feed F
wtfeed/
n
WHSV (wtcat hr) 2 2 1
1 1
0
moliu/
1.)
-.3
Added Hydrogen MOlfeed 1 1 0
0 0 W
m
Temperature C 400 400 300
400 400 m
m
.1.
Pressure psig 600 625 600
600 600 1.)
Organic Phase % of feed
0
H
Yield carbon 85 76 104
58 53 H
1
0
Organic Phase Composition Breakdown
1.)
1
C5- Hydrocarbons wt% 7.4 4.0 0.4
2.8 2.0 1.)
0
C5- Oxygenates wt% 21.4 66.5 5.2
6.9 17.3
Total Non-
Condensed
Components wt% 28.8 70.6 5.6
9.7 19.3
C6+ Paraffins wt% 22.1 10.9 3.4
17.1 5.6
C6+ Olefins wt% 0.0 2.8 0.0
23.8 13.6 IV
Other C6+
n
,-i
Hydrocarbons wt% 1.3 0.3 0.0
8.1 19.8
cp
t-4
=
=
-1
un
un
-4
c:
128
Table 5 (continued)
0
w
o
1-,
o
-1
w
Vapor Phase Condensation of Oxygenates Over Magnesium Impregnated Zirconia
m
w
and Zinc Aluminate Catalysts
o
c,
0.5% Pd / Zinc 0.5% Pd
/ Zinc 0.5% Pd / Zinc
0.5% Pd / 0.5% Pd / Aluminate
Aluminate Aluminate
5% Mg 5% Mg (0.85:1 (0.85:1 (0.85:1
Catalyst Zirconia Zirconia ZnO:A1203)
ZnO:A1203) ZnO:A1203)
C6+ Mono-
oxygenates wt% 46.5 14.7 90.8
41.2 38.6
Total Cond.
n
Products wt% 69.9 28.8 94.2
90.1 77.7
0
Feed B - 100% Isopropyl alcohol
I.)
...3
w
m
Feed D - 100% 2-pentanone
m
m
a,
Feed E - 100% 2-methyltetrahydrofuran
I.)
0
H
Feed F - 90% Isopropyl alcohol, 10% Valeric Acid
H
I
0
KJ
I
KJ
CO
Iv
n
1-i
cp
w
=
=
-,i,--
u,
u,
-.1
c,
129
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
Base Condensation of Oxygenates Followed by Deoxygenation
Example 26
A zinc aluminate catalyst support was prepared
similar to that in Example 13 except that the amount of
zinc oxide was reduced to target a ratio of 0.85 moles of
ZnO to 1 mole of A1203.
Example 27
Hexachloroplatinic acid was added to the calcined
material of Example 26 using an incipient wetness
impregnation technique to achieve a target platinum
loading of 1.0 wt%. The catalyst was dried overnight
under vacuum at 100 C and calcined at 400 C under flowing
air.
Example 28
The catalyst systems referenced in Examples 27 and
15 were investigated for the vapor-phase condensation of
various oxygenates and subsequent conversion to
hydrocarbons. The studies were conducted using 21.2 mm
internal diameter size stainless steel tube reactors as
described in Example 4, and reactor systems as
illustrated by Examples 2 and 3. Approximately 100
milliliters of each catalyst was loaded into two separate
reactors. The two reactors were arranged so that the
effluent of the first reactor flowed into the second
reactor. The first reactor contained the catalyst of
Example 15 and the second reactor contained the catalyst
of Example 27. The catalyst was reduced at 400 C under
flowing hydrogen prior to use. In all cases, hydrogen was
combined with the feed prior to entering the reactor.
Products were separated and analyzed as described in
Example 5. Table 6 shows organic product yields and
composition as a function of operating conditions and
feedstock composition obtained from the consecutive
130
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
reactions. Non-condensed components are those components
that do not require the formation of new carbon-carbon
bonds to be produced from the given feed. For simplicity,
all compounds containing five or fewer carbon atoms are
considered to be non-condensed components. Total
condensation products are those compounds containing six
or more carbon atoms, which require the formation of new
carbon-carbon bonds to be formed from the given
feedstocks.
Experiments AA, BB, CC, and DD demonstrate that
various oxygenates can be employed in the consecutive
condensation and deoxygenation reactions to yield a
product containing primarily C6+ alkanes. The products
contain a larger fraction of alkanes and low levels of
oxygenated compounds compared to the results shown in
Table 3. This demonstrates that the use of catalysts with
different functionalities (i.e. a basic+hydrogenation
catalyst in a first reactor followed by
acid+basic+hydrogenation catalyst in the second reactor)
can be more effective for the production of hydrocarbons
from oxygenated compounds than the use of a catalyst that
contains only basic and hydrogenation functionality. In
Experiment EE, the organic product produced in
Experiments AA through DD was recycled through the
reaction system. After this treatment, the final product
contained primarily alkanes with only traces of oxygen
containing components. The hydrocarbons thus produced
would be valuable for use as liquid fuels such as
gasoline, diesel, and jet fuel.
131
Table 6
0
w
=
1-,
Vapor Phase Condensation and Deoxygenation of Oxygenates
=
Experiment AA BB
CC DD EE
w
oo
Feed Feed B Feed G Feed D
Feed H Feed I w
=
c.,
WHSV Wtfeed/ (Wtcat hr) 1.9 2.2
2.1 2.0 2.0
Added Hydrogen mo1H2/molfeed 1.5 1.7
2 2 >2
Reactor 1 Temperature C 300 300
300 300 325
Reactor 2 Temperature C 350 375
375 375 375
Pressure psig 625 625
625 625 625
Organic Phase Yield % of feed carbon 81 76
80 93 87
Product Composition Breakdown
n
C5- Hydrocarbons % of feed carbon 8 11
15 33 15 0
I.)
C5- Oxygenates % of feed carbon 3 2
2 4 0 --3
w
Total Non-Condensed Components % of feed carbon 11 13
18 37 15 m
m
m
C6+ Alkanes % of feed carbon 71 71
65 56 74 a,
C6+ Alkenes % of feed carbon 0 0
0 0 0 I.)
0
H
Other C6+ Hydrocarbons % of feed carbon 0 0
0 0 0 H
I
C6+ Mono-oxygenates % of feed carbon 6 5
3 2 0 0
I.)
1
Total Products (Condensation) % of feed carbon 77 76
68 58 74 I.)
co
Feed B - 100% Isopropyl Alcohol
Feed D - 100% 2-pentanone
Feed G - 50% Isopropyl Alcohol, 50% 2-Pentanone
Feed H - 50% Acetone, 50% 2-Pentanone
Iv
n
Feed I - Organic Phase From AA-DD
cp
w
=
=
vD
vl
vl
vD
--.1
c.,
132
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
Product Fractionation
Example 29
The material of Experiment EE of Example 28 was
collected and subjected to a distillation step. The
distillation was conducted at atmospheric pressure using
a simple, single stage laboratory batch distillation
apparatus. 2.950 liters of liquid product was added to a
heated round bottomed flask which acted at the reboiler
at the beginning of the experiment. The overhead product
was condensed and segregated into separate samples based
on the temperature of the vapor phase in equilibrium with
the boiling liquid, with an analysis of the fractions
completed as described in Example 5. The carbon number
distribution of the product fractions is shown in
Table 7. All fractions contained primarily alkanes.
The fractions recovered with a boiling point less
than 150 C contain alkanes mainly in the C5-10 range and
would be suitable as a gasoline blending component. The
higher boiling point range materials could be potentially
useful for incorporation into distillate fuels, kerosene
and diesel.
Example 30
The distilled product boiling in the range of 150 C
to 250 C was analyzed for suitability as a Jet Fuel by a
commercial testing service (Intertek Testing Services,
Illinois) according to ASTM testing method D1655. The
sample passed all required specifications with the
exception of the flash point and density specifications.
It is probable that the flash point specification could
be met through adoption of improved product distillation,
while the low density may be attributed to the high
levels of alkanes in the sample.
133
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
Table 7
Results from Distillation of the Product of Example 30
Boiling Starting Less 100 to 150 to Greater
Range C Material than 100 150 250 than 250
Volume
Recovered ml 2950 750 750 1300 180
Total
Alkanes wt% 99.8 100.0 100.0 99.4 91.4
Carbon
Number Breakdown by Species Carbon Number
C4- Wt% 0.2 0.4
C5-9 Wt% 52.6 96.0 78.1 13.7
C1o_14 wt% 41.3 3.6 21.9 78.3 29.9
C1.5 wt% 5.7 7.4 61.5
Production of C5+ Compounds from Glycerol Using a Single
Catalytic System
Example 31
A bimetallic catalyst system containing platinum and
rhenium (5 wt% platinum with a molar ratio of Pt:Re of
1:2.5) supported on activated carbon (Calgon UU 60x120
mesh carbon) was prepared using incipient wetness
techniques. Activated carbon was added slowly to a 30%
hydrogen peroxide solution. After addition of the carbon
was completed, the mixture was left overnight. The
aqueous phase was decanted and the carbon was washed
three times with of deionized water, and then dried under
vacuum at 100 C. An aqueous solution, with a volume equal
to incipient wetness volume for the carbon to be
impregnated, 10.4mL, and containing dihydrogen
hexachloroplatinate (IV) hexahydrate (Alfa Aesar, 39.85%
Pt) and perrhemic acid solution (Alfa Aesar, 76.41%
HRe04) was applied drop wise, while stirring, to hydrogen
peroxide functionalized carbon. The wetted carbon was
dried at 100 C under vacuum.
Example 32
104.4 grams of the 1:2.5 Pt/Re catalyst were loaded
into a 63.5 cm long reactor tube as described in Example
134
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
4 and Example 1, except that the temperature profile was
controlled by heat exchange with a hot air stream
provided by a blower and heater as illustrated in Figure
7. The catalyst was reduced with flowing hydrogen at
350 C for two hours before liquid feed was introduced to
the catalyst bed. A 50 wt% glycerol (Colgate Palmolive
USP Grade) containing about 2Oppm sulfate in water
solution was fed downflow across the reactor after being
preheated to 182 C at a weight hourly space velocity of
0.97 grams of glycerol per gram of catalyst per hour. Hot
air was fed upflow through the annular space at 409 C.
The axial temperature profile within the center of the
catalyst bed was measured using a sliding thermocouple as
shown in Example 4, and is illustrated in Figure 13. The
separator pressure was maintained at 600 psig. The
effluent from the reactor was cooled down with a water
cooled condenser and separated in a three-phase
separator. The gas-phase products were analyzed with a
gas chromatograph that allowed the analysis of hydrogen,
carbon dioxide, methane, ethane, propane, butane,
pentane, and hexane. An organic phase was collected,
weighed, and sent to Southwest Research Institute (San
Antonio, Texas) for gasoline analysis. The aqueous-phase
was collected and weighed, and then analyzed using both a
GCMS as well as GC-FID. In this system, there was
complete conversion of the glycerol. Table 8 below shows
the yields of hydrogen as well as the yields of carbon
containing product compounds.
135
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
Table 8
Yields for the Conversion of Glycerol from Example 32
Products
moles of H2/mole of glycerol feed 1.03
%Carbon/Carbon in Feed
CO2 31.79
Methane 7.35
Ethane 7.28
Propane 5.25
Butane 0.56
Pentane 1.40
Hexane 2.05
C7 - C13 Normal 0.87
C4 - C13 Iso 2.87
C6 - C12 Aromatic 3.87
C8 - C11 Naphthalene/Napthenes 1.89
C5 - C10 Olefins 5.67
C4- C6 Oxygenated Compounds in
Organic Phase 1.86
Ethanol in Aqueous Phase 0.39
Acetic Acid in Aqueous Phase 1.33
Acetone in Aqueous Phase 13.19
Propionic Acid in Aqueous Phase 4.69
Propylene Glycol in Aqueous Phase 2.79
1-Propanol in Aqueous Phase 1.71
Isopropyl Alcohol in Aqueous Phase 1.28
C4/C5/C6 in Aqueous Phase 2.20
Production of C5+ Compounds from Sugar Alcohols
Example 33
Experiments were conducted with aqueous solutions of
oxygenated hydrocarbons (e.g., 50 wt. % glycerol/water
mixture or 50 wt% sorbitol/water mixture) introduced in
to the reactor system of Example 1. The feedstock was
further modified by the addition of K2SO4 at various
concentrations (1, 20, or 50 ppm).
Example 34
A total of 10.61 grams of the 1:2.5 Pt/Re catalyst
were loaded into the 8.5 mm stainless steel reactor tube
described in Example 4. The catalyst was reduced with
flowing hydrogen at 350 C for two hours before liquid
feed was introduced to the catalyst bed. A 50 wt%
glycerol solution containing about 1 ppm sulfate in water
136
CA 02735654 2011-02-28
WO 2010/028206 PCT/US2009/055976
solution was fed downflow across the reactor at a WHSV of
1.24 grams of glycerol per gram of catalyst per hour.
Subsequent tests were performed with 20 ppm and 50 ppm
sulfate added as K2SO4. The block heaters were controlled
at 260 C and the separator pressure was maintained at
600 psig.
An organic phase was collected from the separated,
weighed, and analyzed with a GC-MS as described in
Example 5. Table 9 below shows the yields of hydrogen as
well as the yields of carbon containing product compounds
with the different amounts of sulfate added to the
system. In this system, there was complete conversion of
the glycerol. The table shows that a liquid organic phase
was generated with the addition of sulfate greater than
20 ppm.
Table 9
Yields of Hydrogen and Carbon Containing Products from
Example 34
K2SO4 loading Sulfate 1 20 50
Block 1 Temperature ( C)
(Figure 8, 10a) 260 260 260
Block 2 Temperature ( C)
(Figure 8, 10b) 260 260 260
Block 3 Temperature ( C)
(Figure 8, 10c) 260 260 260
Block 4 Temperature ( C)
(Figure 8, 10d) 260 260 260
H2 produced/mole of glycerol feed 1.67 1.26 0.72
%Carbon/Carbon in Feed
CO2 48.9% 44.4% 27.4%
CH4 14.5% 12.7% 6.1%
C2H6 18.9% 16.0% 6.0%
C3H8 9.4% 7.4% 4.8%
C4H10 0.6% 0.7% 0.2%
C5H12 1.0% 1.0% 0.3%
C6H14 1.1% 0.7% 0.1%
C6-' Hydrocarbons in Organic Phase 0.0% 0.4% 5.4%
C2 - C6 Oxygenates in Organic Phase 0.0% 1.7% 7.9%
C2 - C6 Oxygenates in Aqueous Phase 6.9% 13.3% 42.6%
137
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
Example 35
A total of 10.61 grams of the 1:2.5 Pt/Re catalyst
were loaded into the 8.5 mm stainless steel reactor tube
described in Example 4 and the reactor system illustrated
in Example 1. The catalyst was reduced with flowing
hydrogen at 350 C for two hours before liquid feed was
introduced to the catalyst bed. A 50 wt% glycerol
solution containing either 1 ppm or 20 ppm sulfate in
water was fed downflow across the reactor at a WHSV of
1.24 grams of glycerol per gram of catalyst per hour. The
block heaters were controlled such that the first 10.1 cm
of the reactor was held at 260 C, the second 10.1 cm of
the reactor was at approximate 306 C, the next 10.1 cm of
the reactor was at approximately 355 C, and the last 10.1
cm of the reactor at 400 C. The separator pressure was
maintained at 600 psig.
The effluent from the reactor was cooled down with a
water cooled condenser, separated in a three-phase
separator, and then analyzed as described in Example 5.
In this system, there was complete conversion of the
glycerol. Table 10 below shows the yields of hydrogen as
well as the yields of carbon containing product
compounds.
138
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
Table 10
Yields of Hydrogen and Carbon Containing Products from
Example 35
K2SO4 loading Sulfate 1 20
Block 1 Temperature ( C) 260 260
Block 2 Temperature ( C) 307 305
Block 3 Temperature ( C) 354 356
Block 4 Temperature ( C) 400 400
H2 produced/mole of glycerol feed 1.01 0.83
%Carbon/Carbon in Feed
CO2 42.8% 41.7%
CH4 15.7% 16.1%
C2H6 15.8% 11.9%
C3H8 19.9% 18.2%
C4B10 1.8% 3.0%
C5E-112 2.3% 3.4%
C6E-114 1.0% 1.7%
C6-' Hydrocarbons in Organic Phase 0.0% 1.1%
C2 - C6 Oxygenates in Organic Phase 0.0% 0.7%
C2 - C6 Oxygenates in Aqueous Phase 0.2% 0.1%
Example 36
A bimetallic catalyst system containing platinum and
rhenium (5 wt% platinum with a molar ratio of Pt:Re of
1:5) supported on activated carbon (Calgon UU 60x120 mesh
carbon) was prepared using an incipient wetness
technique. Activated carbon was added slowly to a 30%
hydrogen peroxide solution. After addition of the carbon
was completed, the mixture was left overnight. The
aqueous phase was decanted and the carbon was washed
three times with deionized water, and then dried under
vacuum at 100 C. An aqueous solution, with a volume equal
to the incipient wetness volume for the carbon to be
impregnated and containing dihydrogen hexachloroplatinate
(IV) hexahydrate (Alfa Aesar, 39.85% Pt) and perrhemic
acid solution (Alfa Aesar, 76.41% HRe04) was applied drop
wise, while stirring, to hydrogen peroxide functionalized
carbon. The wetted carbon was then dried at 100 C under
vacuum.
139
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
Example 37
11.97 grams of the 1:5 Pt/Re catalyst described in
Example 36 were loaded into the 8.5 mm diameter stainless
steel tube as described in Example 4 and the reactor
system illustrated in Example 1. The catalyst was reduced
with flowing hydrogen at 350 C for two hours before
liquid feed was introduced to the catalyst bed. A 57.2
wt% sorbitol solution containing 0 ppm sulfate in water
solution was fed downflow across the reactor at a WHSV of
1.20 grams of sorbitol per gram of catalyst per hour. The
block heaters were controlled such that the first 10.1 cm
of the reactor was held at 260 C, the second 10.1 cm of
the reactor was at 260 C, the next 10.1 cm of the reactor
was at 360 C, and the last 10.1 cm of the reactor at
410 C. The separator pressure was maintained at 600 psig.
The effluent from the reactor was cooled down with a
water cooled condenser and separated in a three-phase
separator. The product fractions were analyzed as
described in Example 5. In addition, the organic phase
was collected, separated, and weighed, with a sample sent
to Southwest Research Institute (San Antonia, Texas) for
gasoline analysis. In this system, there was complete
conversion of the glycerol. Table 11 below shows the
yields of hydrogen as well as the yields of carbon
containing product compounds.
140
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
Table 11
Yields of Hydrogen and Carbon Containing Products from
Example 37
Block 1 Temperature ( C) (Figure 8, 10a) 260
Block 2 Temperature ( C) (Figure 8, 10b) 260
Block 3 Temperature ( C) (Figure 8, 10c) 360
Block 4 Temperature ( C) (Figure 8, 10d) 410
Products
moles of H2/mole of Sorbitol feed 1.36
%Carbon/Carbon in Feed
CO2 44.37
Methane 9.24
Ethane 8.25
Propane 11.74
Butane 6.53
Pentane 5.66
Hexane 3.79
C7 - C13 Normal 0.08
C4 - C13 Isoparaffin 0.99
C6 - C12 Aromatic 2.45
C8 - C11 Naphthalene/Napthenes 0.93
C5 - C10 Olefins 0.45
C4- C6 Oxygenated Compounds in Organic Phase 1.68
Oxygenates in Aqueous Phase 3.83
Conversion of Oxygenates to C5+ Compounds Using Acidic
Catalysts
Example 38
An aqueous 1.0 molar lanthanum nitrate solution was
prepared and added to H-mordenite extrudates (BASF 712A-
5-2641-1) for a target of 3 weight % La on the catalyst
after the subsequent decomposition of the metal
precursor. The La solution was mixed briefly with the
catalyst and then soaked at 80 C for 6 hours. The excess
liquid was then removed and the catalyst rinsed with
deionized water. The catalyst was then dried in a vacuum
oven and calcined in air at 55 0C. Following this, the
catalyst was ground and sieved to restrict the particles
sizes to those that were maintained on a 60 mesh screen
after passing through an 18 mesh screen.
141
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
Example 39
Deionized water was added to H-mordenite extrudates
(BASF 712A-5-2641-1, with particle sizes restricted to
those that were maintained on a 60 mesh screen after
passing through an 18 mesh screen) until extra water
covered the support. An aqueous 0.36 molar nickel nitrate
solution was then added to the wet support to target 1
weight % Ni after decomposition of the metal precursor.
The catalyst was mixed briefly and left to soak for 48
hours. The catalyst was then dried in a vacuum oven and
calcined in air at 400 C.
Example 40
An aqueous 1.0 molar europium chloride solution was
prepared and added to H-Mordenite (BASF 712A-5-2641-1,
with particle sizes restricted to those that were
maintained on a 60 mesh screen after passing through an
18 mesh screen) for a target of 3 weight % Eu on the
catalyst after the subsequent decomposition of the metal
precursors. The Eu solution was mixed briefly with the
catalyst and then soaked at 80 C for 6 hours. The excess
liquid was then removed and the catalyst rinsed with
deionized water. The catalyst was then dried in a vacuum
oven and calcined in air at 550 C. Following this the
catalyst was ground and sieved to restrict the particles
sizes to those that were maintained on a 60 mesh screen
after passing through an 18 mesh screen.
Example 41
H-Beta zeolite extrudates (1.6mm diameter
extrudates) were ground and sieved to restrict the
particle sizes to those that were maintained on a 60 mesh
screen after passing through an 18 mesh screen. An
aqueous gallium nitrate solution was added by incipient
wetness to target 1.2 weight % Ga on the catalyst after
142
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
decomposition of the metal precursor. The catalyst was
then dried in a vacuum oven and calcined in air at 400 C.
Example 42
Phosphoric acid was diluted with deionized water and
added by incipient wetness to a Davicat Si02/A1203 support
(Grace-Davis, with particle sizes restricted to those
that were maintained on a 60 mesh screen after passing
through an 18 mesh screen) to target 5 weight %
phosphorous on the catalyst. The catalyst was then dried
in a vacuum oven overnight and subsequently calcined in a
stream of flowing air at 500 C.
Example 43
An aqueous nickel nitrate solution was added to an
alumina bound ZSM-5 zeolite preparation (Si02:A1203 30:1,
with particle sizes restricted to those that were
maintained on a 60 mesh screen after passing through an
18 mesh screen) using an incipient wetness technique to
target a nickel loading of 1.0 weight %. The preparation
was dried overnight in a vacuum oven and subsequently
calcined in a stream of flowing air at 400 C.
Example 44
An aqueous gallium nitrate solution was added to an
alumina bound ZSM-5 zeolite preparation (5i02:A1203 80:1,
with particle sizes restricted to those that were
maintained on a 60 mesh screen after passing through an
18 mesh screen) using an incipient wetness technique to
target a gallium loading of 1.2 weight %. The preparation
was dried overnight in a vacuum oven and subsequently
calcined in a stream of flowing air at 400 C.
Example 45
Catalyst systems produced using the methods of
Examples 38 to 44 were investigated for the vapor-phase
condensation of various oxygenates at a temperature from
143
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
325 C to 375 C and a total pressure between 200 psig and
625 psig, and with WHSVs ranging from 1.9 to 42.8. In
these investigations, two different size reactors were
used; 15 and 18 milliliters of catalyst were loaded into
a 8.5 mm internal diameter stainless steel tube reactor
or between 50 and 70 milliliters of catalyst were loaded
into a 21.2 mm stainless steel tube reactor (Example 4).
The reaction process flow was as described in Example 1
or Example 3 depending on the feedstock, with an analysis
completed as described in Example 5.
Operating conditions and results from these
experiments are shown in Table 12. Where feed
compositions add up to less than 100%, the balance was
water. As these results show, a variety of oxygenates,
including alcohols and ketones, both 3 carbon and 5
carbon, are substrates which may be converted to C5+
hydrocarbons across a broad range of conditions. Zeolites
are particularly useful in these conversions, as shown by
experiments FF, GG, HH, II, JJ, LL, and MM. Experiments
FF, GG, HH, II, and JJ show that the main products of
alcohol conversion across mordenite and beta zeolites
were olefinic condensation products. The phosphorous
impregnated silica alumina catalyst, experiment KK,
demonstrated a similar product selectivity profile. In
contrast, the ZSM-5 based catalysts, Experiments LL and
MM, produced significant fractions of aromatic and
paraffinic components.
144
Table 12
0
t-4
=
1-,
Vapor Phase Condensation of Oxygenates Over Acid Catalysts =
5%
Ni / -1
t-4
m
Phosphorous
30:1 Ga/ 80:1 t-4
=
La/ Ni/ Eu/ Eu/ Ga/
/ Silica- Si02:A1203 Si02:A1203 c:
Catalyst mordenite mordenite mordenite mordenite Beta
Alumina ZSM-5 ZSM-5
Experiment FF GG HH II JJ
KK LL MM
Feed
Feed Feed J Feed K Feed L Feed K K
Feed M Feed K Feed N
wtfeed/
(wtcat
WHSV hr) 1.9 2.1 2.2 1.9 3.1
2.7 42.8 2.1 n
Reactor
0
Temperature C 325 350 325 375 375
375 375 375 1.)
-.3
w
Pressure psig 625 625 600 600 600
600 200 625 m
m
Reactor Outlet Yield Distribution
m
.1.
wt% of
1.)
0
feed
H
H
C4_ Alkanes carbon 2.9 0.7 3.9 3.6 1.2
1.6 9.6 7.0 1
0
wt% of
1.)
I
feed
1.)
0
C4_ Olefins carbon 19.5 47.7 11.3 32.9 32.5
73.5 10.8 0.5
wt% of
Total C4_ feed
Hydrocarbons carbon 22.3 48.4 15.3
36.5 33.7 75.1 20.5 7.5
wt% of
C5+ feed
IV
Paraffins carbon 6.6 0.8 16.9 3.1 4.3
1.9 29.6 8.5 n
,-i
wt% of
feed
cp
t-4
C5+ Olefins carbon 56.2 46.9 43.1 56.6 52.0
18.4 21.7 0.1 =
=
wt% of
-1
un
feed
un
Naphthenes carbon 0.0 2.5 1.5 5.6 3.2
3.4 2.7 1.0 -4
c:
145
0
Table 12 (continued)
=
1-,
=
-1
Reactor Outlet Yield Distribution
t-.)
m
5%
Ni / t-.)
=
Phosphorous
30:1 Ga/ 80:1 cr
La/ Ni/ Eu/ Eu/ Ga/
/ Silica- Si02:A1203 Si02:A1203
Catalyst mordenite mordenite mordenite mordenite Beta
Alumina ZSM-5 ZSM-5
wt% of
feed
Aromatics carbon 0.0 0.0 1.4 0.0 2.0
0.0 18.0 79.1
wt% of
Other C5_ feed
n
Hydrocarbons carbon 0.8 0.1 5.7 1.5 0.2
0.0 7.1 0.0 0
1.)
wt% of
w
Total C5_ feed
m
m
Hydrocarbons carbon 63.6 50.3 68.6 66.7 61.8 23.7
79.2 88.6 m
.1.
Feed J - 50% 2-pentanol
I.)
0
H
Feed K - 50% isopropyl alcohol
H
I
0
KJ
Feed L - 59% 2-pentanol
1
"
co
Feed M - 90% isopropyl alcohol
Feed N - 89.6% Acetone
Iv
n
1-i
cp
w
=
=
-,i,--
u,
u,
-.1
c,
146
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
Production of C5+ Compounds from Oxygenated Hydrocarbons
Example 46
A catalyst preparation technique identical to that
of Example 44 was followed with the exception that the
alumina bound ZSM-5 material had a Si02:A1203 ratio of
30:1.
Example 47
A catalyst produced using the method of Example 46
was investigated for the vapor-phase condensation of a
mixture of oxygenates at 375 C and 200 psig. In this
investigation, 11.3 grams of catalyst were loaded into a
8.5 mm internal diameter stainless steel tube reactor as
described in Example 4. The reaction process flow was as
described in Example 3. The oxygenate mix included, by
weight, 25% 2-pentanone, 20% 3-pentanone, 20% 2-pentanol,
10% isopropyl alcohol, 10% valeric acid, 5% 2-methyl
tetrahydrofuran. This mixture was added using one pump in
the Example 3 reactor system while the second pump added
water so that the total combined feed contained 60 weight
% water and 40 weight% of mixed oxygenates.
The process was monitored for a period of 128 hours,
with samples periodically removed from the system to
analyze the process performance. Each analysis was
completed as described in Example 5. Figure 15 shows the
fraction of feed carbon that exited the reactor system as
C5+ compounds as a function of time. Figure 16 shows the
fraction of feed carbon that exited the reactor system as
an aromatic hydrocarbon as a function of time. Figure 14
shows the fraction of feed carbon that exited the reactor
system as oxygenates as a function of time.
As Figures 14, 15 and 16 show, the catalyst system
is able to operate for extended periods of time with an
oxygenate mix that contains a mixture of oxygenates,
147
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
including alcohols, ketones, an acid, and a
tetrahydrofuran. Over time the production of C5+
compounds remains relatively stable, while the amount of
aromatic hydrocarbons present in the product drops and
the breakthrough of oxygenated compounds increases
(Figure 14). It is believed that the catalyst
deactivation is primarily due to the accumulation of
carbonaceous deposits limiting the accessibility of the
reactants to the active sites.
Example 48
An aqueous solution of hexachloroplatinic acid and
perrhenic acid was added to a carbon catalyst support
(OLC-AW, Calgon, with particle sizes restricted to those
that were maintained on a 50 mesh screen after passing
through an 120 mesh screen) using an incipient wetness
technique to target a platinum loading of 1.8% and a
rhenium loading of 6.3% on the catalyst after subsequent
decomposition of the metal precursors. The preparation
was dried overnight in a vacuum oven and subsequently
reduced in a stream of flowing hydrogen at 400 C. After
being reduced the catalyst was stored in a nitrogen
atmosphere until ready for use.
Example 49
A catalyst preparation technique identical to that
of Example 44 was followed with the exception that the
alumina bound ZSM-5 material had a Si02:A1203 ratio of
150:1.
Example 50
Hexachloroplatinic acid and perrhenic acid dissolved
in water were added to a monoclinic zirconia catalyst
support (NorPro Saint Gobain, product code 5Z31164, with
particle sizes restricted to those that were maintained
on a 60 mesh screen after passing through an 18 mesh
148
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
screen) using an incipient wetness technique to target a
platinum loading of 1.8% and a rhenium loading of 6.3% on
the catalyst after subsequent decomposition of the metal
precursors. The preparation was dried overnight in a
vacuum oven and subsequently calcined in a stream of
flowing air at 400 C.
Example 51
The same procedure used for preparing the catalyst
of Example 50 was followed with the exception that the
target rhenium loading was 1.8%.
Example 52
An 80:1 Si02:A1203 ratio ZSM-5 zeolite (Zeolyst
International, CBV 8014) was mixed with a 1:1 molar ratio
of ZnO and A1203 powders so that the ZnO and A1203 (Dispal
18N4-80, Sasol North America, Houston, Texas) combined
comprised 30 weight % of the total solids. Dilute nitric
acid was added at a level of 2 weight % HNO3 to the
combined ZnO and A1203. The dough consistency was
adjusted with water addition to form a workable dough
suitable for extrusion and the mixture was extruded using
a laboratory scale extruder. The extrudates were dried
overnight under vacuum at 100 C and subsequently calcined
at 600 C under flowing air.
Example 53
An aqueous solution of gallium nitrate was added to
the material of Example 52, with particle sizes
restricted to those that were maintained on a 60 mesh
screen after passing through an 18 mesh screen, using an
incipient wetness technique to target a gallium loading
of 1.2 weight %. The preparation was dried overnight in a
vacuum oven and subsequently calcined in a stream of
flowing hydrogen at 400 C.
149
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
Example 54
An aqueous solution of nickel nitrate was added to
the material of Example 52, with particle sizes
restricted to those that were maintained on a 60 mesh
screen after passing through an 18 mesh screen, using an
incipient wetness technique to target a nickel loading of
1.0 weight %. The preparation was dried overnight in a
vacuum oven and subsequently calcined in a stream of
flowing hydrogen at 400 C.
Example 55
The catalyst systems referenced in Examples 6, 46,
48, 49, 51, 53, and 54 were investigated for the
conversion of glycerol, sorbitol, sucrose, and xylose to
hydrocarbons using the reactor configuration described in
Example 2. The studies were conducted using two 21.2 mm
internal diameter stainless steel tube reactors shown in
Example 4, with an analysis completed as described in
Example 5. Tungstated zirconia (NorPro-Saint Gobain,
product code SZ61143, with particle sizes restricted to
those that were maintained on a 60 mesh screen after
passing through an 18 mesh screen) was placed on top of
the condensation catalyst installed in the second reactor
to provide for a zone for vaporization of the first
reactor effluent prior to entering the condensation
catalyst.
Table 13 shows the results of these investigations.
For Experiment NN (38% Sucrose + 7% Xylose), a stream of
hydrogen with a targeted flow rate equal to 3 times the
moles of sucrose plus 1.5 times the moles of xylose was
combined with the feed prior to entering the reactor. The
other experiments were conducted without externally
supplied hydrogen. Heaters external to the reactor, shown
in Figure 9 as 10a, 10b, 10c, 10d, 23a, 23b, 23c, and
150
CA 02735654 2014-08-28
23d, were used to maintain the reactor wall temperatures,
as indicated in Table 13. The hydrocarbon products of
these studies, disclosed in Table 13, were grouped into a
C4 _ fraction, which are predominately present in the gas
phase at ambient temperature and pressure, and a C5_,
fraction, which are generally suitable for incorporation
into liquid fuels. The results show that a variety of
sugars and polyhydric alcohols may be readily converted to
C5, hydrocarbons by the processes described here. The
products contained mainly paraffin and aromatic
constituents. The breakdown of paraffins and aromatics
within this sample is shown in Fig. 17.
151
Table 13
0
w
=
1-,
Conversion of Sugars and Polyhydric Alcohols to C5+ Hydrocarbons
=
Experiment NN 00
PP QQ
w
oo
Catalyst Descriptions
w
=
c.,
Hydrogenation Example 6 None
None None
APR/Deoxygenation Example 48 Example 51
Example 51 Example 50
Condensation Example 49 Example 53
Example 46 Example 54
Catalyst Loadings
Hydrogenation grams 10 -
- -
APR/Deoxygenation grams 40 52
60 60
Tungstated Zirconia grams 71 60
-60 58 n
Condensation grams 62 60
60 60 0
I.)
Heater Block Temperature Ranges, Inlet of Catalyst Bed - Outlet of Catalyst
Bed --3
w
Hydrogenation C 100-150 -
- - m
m
m
APR/Deoxygenation C 245-265 250-270
335-365 275-285 a,
Tungstated Zirconia C 250-375 370-370
395-375 395-375 I.)
0
H
Condensation C 375-375 385-385
375-375 375-375 H
I
0
Pressures
I.)
I
First Reactor Outlet psig 625 625
625 625 "
co
2nd Reactor Outlet psig 625 350
250 350
Feed Feed 0 Feed P
Feed P Feed Q
Hydrogen production mol/mol feed -2.85 0.73
0.57 0.50
WHSV Wtfeed/ (Wtcat hr) 1.6 1.9
2.0 2.0
Reactor Outlet Yield Distribution
C.4_ Alkanes wt% of feed carbon 21.2 26.9
8.1 13.0 Iv
n
C4_ Olefins wt% of feed carbon 1.1 1.4
1.3 5.2
Total C4_ Hydrocarbons wt% of feed carbon 22.3 28.3
9.4 18.1 cp
w
C5+ Paraffins wt% of feed carbon 20.0 7.9
9.5 11.3 =
=
vD
C5+ Olefins wt% of feed carbon 0.8 1.9
1.2 7.8
vl
Naphthenes wt% of feed carbon 1.9 1.4
1.6 1.2 vl
vD
--.1
c.,
152
Table 13 (continued)
0
w
o
Experiment NN 00
PP 44
o
Reactor Outlet Outlet Yield Distribution
w
m
Aromatics wt% of feed carbon 25.0 17.8
48.4 22.3 w
o
c.,
Other 05+ Hydrocarbons wt% of feed carbon 0.0 1.1
0.2 3.4
Total 05+ Hydrocarbons wt% of feed carbon 47.7 30.1
61.0 46.1
Feed 0 - 38% Sucrose, 7% Xylose
Feed P - 50% Glycerol
Feed Q - 50% Sorbitol
n
0
1.)
--3
w
in
m
in
a,
1.)
0
H
H
I
0
KJ
I
KJ
CO
.0
n
,-i
cp
w
=
=
-:,--
u,
u,
-.1
c.,
153
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
Example 56
The process described in Example 55 and exemplified
by Experiment QQ in Table 13 was operated for a period of
more than 400 hours. After an initial period of time in
operation, the conversion to aromatic components and the
yield of hydrocarbons dropped, shown in Figures 18 and 19
as Cycle 1. In Figure 18, the heating value of C5+
hydrocarbons present at the outlet of the second reactor,
as a percentage of the heating value of the feed, is
shown. In Figure 19, the carbon present as aromatic
hydrocarbons at the outlet of the second reactor is shown
as a percentage of the carbon present in the feed. After
approximately 120 hours on stream, the second reactor was
bypassed while the first reactor continued operating. An
oxidative regeneration of the catalyst in the second
reactor was then performed. During the regeneration, a
flow of nitrogen and air was initiated so that the target
oxygen concentration at the second reactor inlet was 1
mol %. The second reactor block temperatures were then
raised to 500 C and the flow of nitrogen and oxygen
continued until carbon dioxide was no longer detected at
the second reactor outlet. The oxygen concentration was
then raised to a target level of 5 mol %. This flow was
continued until carbon dioxide was no longer detected at
the second reactor outlet. At this time the oxygen flow
was discontinued while the nitrogen flow continued. The
second reactor block temperatures were then reduced to
400 C while the composition of the gas flowing through
the catalyst bed was changed to hydrogen. The second
reactor block temperatures were then adjusted to those
shown for Experiment QQ in Table 13. The second reactor
was then placed back on line, targeting the conditions
shown for Experiment QQ in Table 13. The second reactor
154
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
was then subjected to multiple cycles of operation and
regeneration, with the results for the period of time in
operation shown in Figures 18 and 19. As these results
show, the regeneration of the condensation catalyst
resulted in a restoration of activity, consistent with
the theory that deposition of carbonaceous materials were
the main cause of a drop in catalyst performance over
time. Furthermore, the results show that the condensation
catalyst may be regenerated multiple times without a
significant loss of performance.
Preparation of Gasoline Compositions
Example 57
A gasoline composition (gasoline composition GC1)
was prepared by admixing a base gasoline with 5 %vol.,
based on the volume of the final gasoline composition, of
the product of the process described in Example 55 and
exemplified by Experiment PP in Table 13.
The base gasoline used and the gasoline composition
prepared are detailed in Table 14.
Example 58
A gasoline composition (gasoline composition GC2)
was prepared by admixing a base gasoline with 10 %vol.,
based on the volume of the final gasoline composition, of
the product of the process described in Example 55 and
exemplified by Experiment 00 in Table 13.
The base gasoline used and the gasoline composition
prepared are detailed in Table 14.
155
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
Table 14
Property Unit Base Gasoline GC1
GC2
RVP kPa 92.9 88.7 85.8
Density Kg/m3 727.5 735.2 736.6
IBP C 26.9 27.6 27.6
FBP C 194.3 206.8 198.2
Residue %v 1 1.2 1.1
Recovery %v 95.9 95.8 958
Loss %v 3.1 3 3.1
T10 C 39.9 40.2 41.2
T20 C 51 51.5 52.8
T30 C 63.4 64.3 65.9
T40 C 76.4 78.4 79.8
T50 C 89.9 92.5 94.2
T60 C 102.6 106 107.4
T70 C 114.8 118.8 120.1
T80 C 129 134 136.1
T90 C 149 154.1 155.6
T95 C 165 170.4 171
E70 %v 35.1 34.1 32.9
E100 %v 57.9 55.3 54
E120 %v 74 70.7 69.8
E150 %v 90 88 87
E180 %v 97.5 96.6 96.7
RON 95.6 96.4 96.3
MON 85.4 85.5 85.1
It can be seen from the information presented in
Table 14 above that the gasoline compositions comprising
the products prepared by the process described in Example
55 have very similar distillation characteristics to the
base gasoline, and advantageously have a higher Road
Octane Number (RON) than the base gasoline. It can also
be seen from the information presented in Table 14 above
that the gasoline compositions comprising the products
prepared by the process described in Example 55 have a
lower Reid Vapour Pressure (RVP) compared to the base
gasoline, which may advantageously be used to decrease
the RVP of a gasoline in order to meet specific
requirements of the gasoline composition, or
156
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
alternatively allow the addition of greater amounts of
gasoline components whose addition to gasoline
compositions may have been limited due to the volatility
of such components.
Example 59
Two separate gasoline compositions were prepared
using sorbitol and sucrose as a feedstock, and then
analyzed for their carbon 14 (14C) concentration as
compared to petroleum-based gasoline.
The first gasoline sample (V-18510) was produced
from a 50% solution of sorbitol in water using a reactor
system similar to that described in Example 55, except
that the system included a second condensation reactor in
a lead/lag configuration. The APR/deoxygenation catalyst
was prepared as described in Example 50, while the
condensation catalyst was prepared as described in
Example 43. Multiple runs using the sorbitol feedstock
where conducted over various temperatures and conditions.
APR/deoxygenation reactions were conducted in a single
reactor over a low to high temperature profile with
temperatures varied from 110 C to 130 C at the inlet and
from 235 C to 250 C at the outlet. Condensation
reactions were conducted in a two reactor system with the
temperature in the lead reactor varied from 345 C to
385 C and the lag reactor varied from 260 C to 385 C.
Pressure conditions were also varied from 620psig to
630psig for the APR/deoxygenation reactions and 95 psig
to 105 psig for the condensation reactions. WHSV was
varied from 0.9 to 1.1 hr-1 for the APR/deoxygenation
catalyst.
A second gasoline sample (V-18512) was prepared from
a collective sample of gasoline produced from sucrose
using a reactor system as illustrated in Figure 6.
157
CA 02735654 2011-02-28
WO 2010/028206
PCT/US2009/055976
Sucrose was fed at various temperatures, pressures and
WHSV over a hydrogenation catalyst containing 2.5%
ruthenium on UU carbon (Calgon, with particle sizes
restricted to those that were maintained on a 120 mesh
screen after passing through a 60 mesh screen), and the
APR/deoxygenation catalyst and condensation catalyst
described above for the sorbitol feed. Conditions for
the hydrogenation reaction varied with temperatures in
the range of 115 C to 140 C, and pressures in the range
of 620 psig to 680 psig. The WHSV, temperature and
pressure conditions for the APR/deoxygenation were as
described for sorbitol. The condensation reaction was
conducted in a single condensation reactor with the WHSV,
temperature and pressure conditions the same as that
described above for the lead reactor.
The product streams from the various runs of
sorbitol were combined into a single sample (V-18510) and
then subjected to a distillation step as described in
Example 29. The product streams from the various runs of
sucrose were also combined into a single sample (V-18512)
and then subjected to distillation as described in
Example 29. Distillation fractions having a boiling
point of less than 210 C were collected for further
blending into final fuel compositions. A sample of each
fraction was also collected for 14C testing.
Carbon 14 testing was performed by Beta Analytical
Inc (Miami, Florida USA) using ASTM-D6866, Test Methods
for Determining the Biobased Content of Natural Range
Materials Using Radiocarbon and Isotope Ratio Mass
Spectrometry Analysis. In addition to 14C testing on
samples V-18510 and V-18512, a biobased determination was
also performed on two additional samples collected from
separate retail gas stations in Madison, Wisconsin. The
158
CA 02735654 2011-02-28
WO 2010/028206 PCT/US2009/055976
first sample (V-RRGWE) was a regular unleaded gasoline
identified as containing up to 10% ethanol, while the
second sample (V-SVPNE) was premium gasoline. The
results of the study are listed in Table 15 below.
Mean Percentage of
Sample No. ASTM-D6866 Method
Biobased Material
V-18510 Method - B 99%
V-18512 Method - B 99%
V-RRGWE Method - B 7%
V-SVPNE Method - B 2%
159