Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
Condensed Ring Pyridine Compounds As Subtype-Selective Modulators Of
Sphingosine-1-
Phosphate-2 (SiP2) Receptors
Inventors: Wenkui Ken Fang, Liming Wang, Evelyn G. Corpuz, Ken Chow, Wha-Bin
Im
CROSS REFERENCE TO RELATED APPLICATIONS
The present invention claims priority under 35 U.S.C. 119(e) to United States
Provisional Application No. 61/246,642, filed on Sep 29, 2009 which is
expressly
incorporated herein by reference in its entirety.
BACKGROUND OF THE INVENTION
1. Field of the Invention
The present invention relates to derivatives and/or analogues of sphingosine
which are useful as
anti-fibrotic drugs and are thereby useful for treating ocular, cardiac,
hepatic and pulmonary
fibrosis, proliferative vitreoretinopathy, cicatricial pemphigoid, surgically
induced fibrosis in
cornea, conjunctiva and tenon and for the treatment of eye diseases and
conditions.
2. Summary of the Art
Sphingosine is a compound having the chemical structure shown in the general
formula
described below, in which Y' is hydrogen. It is known that various
sphingolipids, having
sphingosine as a constituent, are widely distributed in the living body
including on the surface of
cell membranes of cells in the nervous system.
H OH NH2
1 1 1
H3C-(CH2)12-C=CH-CH-CH-CH2O-Y1
H
A sphingolipid is one of the lipids having important roles in the living body.
A disease called
lipidosis is caused by accumulation of a specified sphingolipid in the body.
Sphingolipids
present on cell membranes function to regulate cell growth; participate in the
development and
differentiation of cells; function in nerves; are involved in the infection
and malignancy of cells;
etc. Many of the physiological roles of sphingolipids remain to be solved.
Recently the
possibility that ceramide, a derivative of sphingosine, has an important role
in the mechanism of
1
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
cell signal transduction has been indicated, and studies about its effect on
apoptosis and cell
cycle have been reported.
Sphingosine-l-phosphate is an important cellular metabolite, derived from
ceramide that is
synthesized de novo or as part of the sphingomeyeline cycle (in animals
cells). It has also been
found in insects, yeasts and plants.
The enzyme, ceramidase, acts upon ceramides to release sphingosine, which is
phosphorylated
by spingosine kinase, a ubiquitous enzyme in the cytosol and endoplasmic
reticulum, to form
sphingosine-l-phosphate. The reverse reaction can occur also by the action of
sphingosine
phosphatases, and the enzymes act in concert to control the cellular
concentrations of the
metabolite, which concentrations are always low. In plasma, such concentration
can reach 0.2 to
0.9 M, and the metabolite is found in association with the lipoproteins,
especially the HDL. It
should also be noted that sphingosine-l-phosphate formation is an essential
step in the
catabolism of sphingoid bases.
Like its precursors, sphingosine-l-phosphate is a potent messenger molecule
that perhaps
uniquely operates both intra- and inter-cellularly, but with very different
functions from
ceramides and sphingosine. The balance between these various sphingolipid
metabolites may be
important for health. For example, within the cell, sphingosine-l-phosphate
promotes cellular
division (mitosis) as opposed to cell death (apoptosis), which it inhibits.
Intracellularly, it also
functions to regulate calcium mobilization and cell growth in response to a
variety of
extracellular stimuli. Current opinion appears to suggest that the balance
between sphingosine-
1-phosphate and ceramide and/or spingosine levels in cells is critical for
their viability. In
common with the lysophospholipids, especially lysophosphatidic acid, with
which it has some
structural similarities, sphingosine-l-phosphate exerts many of its extra-
cellular effects through
interaction with five specific G protein-coupled receptors on cell surfaces.
These are important
for the growth of new blood vessels, vascular maturation, cardiac development
and immunity,
and for directed cell movement.
Sphingosine-1 phosphate is stored in relatively high concentrations in human
platelets, which
lack the enzymes responsible for its catabolism, and it is released into the
blood stream upon
activation of physiological stimuli, such as growth factors, cytokines, and
receptor agonists and
antigens. It may also have a critical role in platelet aggregation and
thrombosis and could
aggravate cardiovascular disease. On the other hand the relatively high
concentration of the
2
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
metabolite in high-density lipoproteins (HDL) may have beneficial implications
for
atherogenesis. For example, there are recent suggestions that sphingosine-l-
phosphate, together
with other lysolipids such as sphingosylphosphorylcholine and lysosulfatide,
are responsible for
the beneficial clinical effects of HDL by stimulating the production of the
potent antiatherogenic
signaling molecule nitric oxide by the vascular endothelium. In addition, like
lysophosphatidic
acid, it is a marker for certain types of cancer, and there is evidence that
its role in cell division
or proliferation may have an influence on the development of cancers. These
are currently topics
that are attracting great interest amongst medical researchers, and the
potential for therapeutic
intervention in sphingosine-l-phosphate metabolism is under active
investigation.
Fungi and plants have sphingolipids and the major sphingosine contained in
these organisms has
the formula described below. It is known that these lipids have important
roles in the cell growth
of fungi and plants, but details of the roles remain to be solved.
OH OH NH2
1
H3C - (CH2)12 - CH2 - CH - CH - CH - CH2OH
Recently it has been known that derivatives of sphingolipids and their related
compounds exhibit
a variety of biological activities through inhibition or stimulation of the
metabolism pathways.
These compounds include inhibitors of protein kinase C, inducers of apoptosis,
immuno-
suppressive compounds, antifungal compounds, and the like. Substances having
these biological
activities are expected to be useful compounds for various diseases.
Derivatives of sphingosine have been prepared in various patents. For example,
see U.S. Patents
4,952,683; 5,110,987; 6,235,912 B1 and 6,239,297 B1.
Also, compounds which are similar to certain spingosine derivatives, but which
are not reported
as being ligands for the spingosine receptors are reported in various patents
and published patent
applications. See for example, U.S. Patents 5,294,722; 5,102,901; 5,403,851
and 5,580,878.
U.S. Patent Application Publication No. U.S. 2003/0125371 A2.
3
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
SUMMARY OF THE INVENTION
The present invention provides compounds that are able to regulate the
functions of
sphingolipid, and pharmaceutical compositions comprising said compounds.
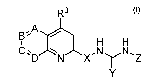
In one aspect of the present invention there are disclosed compounds, having
sphingosine-l-
phosphate receptor agonist and or antagonist biological activity, represented
by the formula I:
R3
/
B'1A H H
C. D \NIX'NyN,Z
Y
wherein:
wherein A is a direct bond or (CR) and B, C and D are independently selected
from the group
consisting of (CR) and N, wherein R is H or alkyl, e.g.lower alkyl; provided
however, not all, of
B, C and D are N and, when A is a direct bond, D is (CR) ;
R3 is selected from the group consisting of alkyl, e.g. lower alkyl:
X is selected from the group consisting of 0, NR4 and CR4R5, wherein R4 and R5
are
independently selected from the group consisting of H and alkyl, e.g. lower
alkyl;
Y is selected from the group consisting of 0 or S; and
Z is a substituted aryl ring and pharmaceutically acceptable salts thereof.
In a first aspect of the present invention, the left most ring is a six
membered ring, i.e. the
compounds of this invention are benzo or pyrido pyridinyl compounds. The
compounds
included in this first aspect of the invention may be represented by the
general formula II,
below:
R1 R3
H H
R2 D N X,NyN,Z
Y
wherein R1 and R2 are independently selected from the group consisting of H
and alkyl, e.g.
lower alkyl, and may include from 1 to 10 carbons, and may be cyclic or
branched chain alkyl
radicals having from 3 to 10 carbons; methoxy, hydroxyl, halogen, nitrile,
trifluoromethyl and
carboxy;
4
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
R3 is selected from the group consisting of alkyl, e.g. lower alkyl, and may
include from 1 to 10
carbons, and may be cyclic or branched chain alkyl radicals having from 3 to
10 carbons;
methoxy, hydroxyl, halogen, nitrile, trifluoromethyl and carboxy;
D is CR or N;
X is 0, NR4 or CR4R5, where R4, R5 are independently selected from the group
consisting of H
and alkyl, e.g. lower alkyl and may have from 1 to 10 carbons, and may be
cyclic or branched
chain alkyl having 3 to 10 carbons, methoxy, hydroxyl, F, Br, I, nitrile,
trifluoromethyl and
carboxy;
Y is 0 or S, preferably 0;
Z is a substituted aryl ring, e.g. a carbocyclic or heterocyclic aryl ring,
having the following
structure:
R6
E
~~/.i
R7
wherein R6 and R7 are independently selected from the group consisting of
alkyl and may
include from 1 to 10 carbons, and may be cyclic or branched chain alkyl having
3 to 10 carbons,
alkyloxy, preferably lower alkyloxy, e.g.ethyloxy, isopropyloxy, n-butyloxy;
hydroxyl, halogen,
preferably chloro; nitrile, trifluoromethyl, and carboxy; and
E is N or CR, preferably N
In a second aspect, of the present invention, the compounds of this invention
the left most ring is
a five membered ring, i.e. the compounds are 1-H pyrazolo[4,3-b] pyridinyl
compounds. The
compounds included in this second aspect of the invention may be represented
by the general
formula III:
R1 R3
N ~ I . N U Z
R2 N X, Y Y
N,
5
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
wherein R', Rand R3 are independently selected from the group consisting of H
and alkyl, e.g.
lower alkyl, and may include from 1 to 10 carbons, and may be cyclic or
branched chain alkyl
radicals having from 3 to 10 carbons; methoxy, hydroxyl, halogen, nitrile,
trifluoromethyl and
carboxy;
R3 is selected from the group consisting of alkyl, e.g. lower alkyl, and may
include from 1 to 10
carbons, and may be cyclic or branched chain alkyl radicals having from 3 to
10 carbons;
methoxy, hydroxyl, halogen, nitrile, trifluoromethyl and carboxy;
D is CR or N;
X is 0, NR4, or CR4R5, where R4, R5 are independently selected from the group
consisting of H
and alkyl, e.g. lower alkyl and may have from 1 to 10 carbons, and may be
cyclic or branched
chain alkyl having 3 to 10 carbons, methoxy, hydroxyl, F, Br, I, nitrile,
trifluoromethyl and
carboxy;
Y is 0 or S, preferably 0;
Z is a substituted aryl ring, e.g. a carbocyclic or heterocyclic aryl ring,
having the following
structure:
R6
E
~~/.i
R7
wherein R6 and R7 are independently selected from the group consisting of
alkyl and may
include from 1 to 10 carbons, and may be cyclic or branched chain alkyl having
3 to 10 carbons,
alkyloxy, preferably lower alkyloxy, e.g.ethyloxy, isopropyloxy, n-butyloxy;
hydroxyl, halogen,
preferably chloro; nitrile, trifluoromethyl, and carboxy; and
E is N or CR, preferably N;
Preferably, Z is a disubstituted aryl, more preferably an o,o- substituted
pyridinyl.
In another aspect of this invention, there is disclosed a method of treating
or preventing a disease
or condition selected from the group consisting of fibrotic conditions e.g.;
ocular, cardiac,
hepatic and pulmonary fibrosis, proliferative vitreoretinopathy, cicatricial
pemphigoid,
surgically induced fibrosis in cornea, conjunctiva and tenon which comprises
administering to a
patient in need thereof a compound represented by the formula I, II, or III,
above.
6
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
DETAILED DESCRIPTION OF THE INVENTION
Novel compounds having this general structure were synthesized and tested for
sphingosine 2-
phosphate receptor activity using the FLIPR assay. Cells expressing the
receptor of interest
(SIP1, SIP2 or SIPS) and a G-protein (Gqi5 or G16) are loaded with fluo-4, a
calcium sensitive
dye. After removal of excess dye by washing, the cells are placed in the FLIPR
TETRA
instrument. Baseline fluorescence readings are taken prior to addition the
compound to be
tested. Agonists will trigger the receptor to interact with the G-protein,
leading to an increase in
intracellular calcium. The increase in intracellular calcium causes an
increase in the
fluorescence of the cells, due to the presence of fluo-4. This fluorescence
increase is recorded
by the FLIPR TETRA. After the calcium transient signal has decreased towards
baseline, the
standard agonist sphingosine 1-phosphate is added. If the test compound is an
antagonist, an
initial calcium signal will not be generated and the antagonist will prevent
the generation of a
calcium signal from sphingosine 1-phosphate. The level of fluorescence is
compared to that of
sphingosine 1-phosphate, and the EC50 or IC50 of the compound determined by
curve fitting.
The compounds in this invention are useful for the treatment of mammals,
including humans, for
diseases or conditions selected from the group consisting of ocular, cardiac,
hepatic and
pulmonary fibrosis, proliferative vitreoretinopathy, cicatricial pemphigoid,
surgically induced
fibrosis in cornea, conjunctiva and tenon
Specific Examples of the compounds of formula I include the compounds of Table
1, below.
7
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
TABLE 1
Example Structure Example Structure
Number Number
N N
1 N~ N N,N N qr CI 2 NN N N q CI
H O O I ~N
Cl CI
H
N\ H IN N,N N N
H H
3 " o iN 4 N'
N N,N N
ci H O qN
Cl
N
N
N~ N N q CI 6 NX 'H H
N CI
N
O I/ N H 0 I ~N
Cl
Cl
N H H N H H
7 N\ N N'N N CF3 8 N\ N N ' N O,,
H O I/ H O qN
CF3 Cl
N
9 N N H Cl 10 N &,, N N O~
N O IO jq~
N
Cl Cl
8
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
Example Structure Example Structure
Number Number
11 NN ,N H 12 NN &N- N N
N H y IC y
O N O N
Cl Cl
N N H 13 NX N N H 14 N~ N N N
O N O N
Cl Cl
N N
15 N~ N NN~N k O 16 N~ N N,N~N
H O I ~N H O I ~N
CI CI
N N
17 N~ I N N \ per/ 18 N\ N N'N~N \ 0,/
N
0 I f N H O I ~N
CI
N \ , / \
19 N H H 0 20 I N N CI
N N N N N' \
N H' H O I/
Oi
0
Cl
CI
9
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
21 N N N"Nu N \ CI 22 N N-N N \ Cl
H IIO I/ H Y I/
CI CI
24 \ N N"N~N \ CI
23 \ I i "N N Cl
N H YO H O I iN
Cl CI
25 9?N)- "N N \ CI 2N"N N \ CI
H O I/ N H O I ~N
CI CI
#N-- 27 N N \ CI
H"
O /
CI
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
Some compounds within the scope of the invention may be prepared as depicted
in the
procedures described below.
The invention is further illustrated by the following examples which are
illustrative of a specific
mode of practicing the invention and are not intended as limiting the scope of
the claims.
Unless otherwise indicated, the following terms as used throughout this
specification have the
following meanings:
DCM refers to dichloromethane
THE refers to tetrahydrofuran
EtOAc refers to ethylacetate
"Me" refers to methyl.
"Ph" refers to phenyl.
"Pharmaceutically acceptable salt" refers to those salts which retain the
biological effectiveness
and properties of the free bases and which are obtained by reaction with
inorganic acids such as
hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric
acid, methanesulfonic
acid, ethanesulfonic acid, p-toluenesulfonic acid, salicylic acid and the
like.
"Alkyl" refers to a straight-chain, branched or cyclic saturated aliphatic
hydrocarbon. Preferably,
the alkyl group has 1 to 12 carbons. More preferably, it is a lower alkyl of
from 1 to 7 carbons,
most preferably 1 to 4 carbons. Typical alkyl groups include methyl, ethyl,
propyl, isopropyl,
butyl, isobutyl, tertiary butyl, pentyl, hexyl and the like. The alkyl group
may be optionally
substituted with one or more substituents selected from the group consisting
of hydroxyl, cyan,
alkoxy, =0, =S, NO2, halogen, dimethyl amino and SH.
"Alkoxy" refers to an "O-alkyl" group.
"Aryl" refers to an aromatic group which has at least one ring having a
conjugated pi electron
system and includes carbocyclic aryl, heterocyclic aryl and biaryl groups. The
aryl group may be
optionally substituted with one or more substituents selected from the group
consisting of
halogen, trihalomethyl, hydroxyl, SH, OH, NO2, amine, thioether, cyan, alkoxy,
alkyl, and
amino.
11
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
"Alkaryl" refers to an alkyl that is covalently joined to an aryl group.
Preferably, the alkyl is a
lower alkyl.
"Carbocyclic" refers to cyclic saturated or unsaturated aliphatic hydrocarbon
and aryl
hydrocarbon groups wherein the ring atoms are exclusively carbons, and
comprises from 6 to 20
carbon atoms, including said ring atoms.
"Carbocyclic aryl" refers to an aryl group wherein the ring atoms are carbon.
"Heterocyclic" refers to cyclic groups wherein the ring atoms comprise carbon
atoms and at
least one oxygen, nitrogen, and/or sulfur atom and may be saturated,
unsaturated, i.e. have one
or more double bonds, or aryl, and comprises up to 20 carbon atoms and from 1
to 5 of the
above heteroatoms.
"Heterocyclic aryl" refers to an aryl group having from 1 to 3 heteroatoms as
ring atoms, the
remainder of the ring atoms being carbon. Heteroatoms include oxygen, sulfur,
and nitrogen.
"Hydrocarbyl" refers to a hydrocarbon radical having only carbon and hydrogen
atoms.
Preferably, the hydrocarbyl radical has from 1 to 20 carbon atoms, more
preferably from 1 to 12
carbon atoms and most preferably from 1 to 7 carbon atoms.
"Substituted hydrocarbyl" refers to a hydrocarbyl radical wherein one or more,
but not all, of the
hydrogen and/or the carbon atoms are replaced by a halogen, nitrogen, oxygen,
sulfur or
phosphorus atom or a radical including a halogen, nitrogen, oxygen, sulfur or
phosphorus atom,
e.g. fluoro, chloro, cyano, nitro, hydroxyl, phosphate, thiol, amide, ester,
thioamide, thiol ester,
amine, thioether, sulfonyl, etc.
"Amide" refers to --C(O)--NH--R', wherein R' is alkyl, aryl, alkylaryl or
hydrogen.
"Ester" refers to --C(O)--O--R', wherein R' is alkyl, aryl or alkylaryl.
"Thioamide" refers to --C(S)--NH--R', wherein R' is alkyl, aryl, alkylaryl or
hydrogen.
"Thiol ester" refers to --C(O)--S--R', wherein R' is alkyl, aryl, alkylaryl or
hydrogen.
"Amine" refers to a --N(R")R"' group, wherein R" and R"' are independently
selected from the
group consisting of alkyl, aryl, and alkylaryl.
"Thioether" refers to --S--R", wherein R" is alkyl, aryl, or alkylaryl.
12
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
"Sulfonyl" refers to --S(O)2 --R"", where R"" is aryl, C(CN)=C-aryl, CH2 CN,
alkyaryl,
sulfonamide, NH-alkyl, NH-alkylaryl, or NH-aryl.
Also, alternatively the substituent on the phenyl moiety may be referred to as
an o, m or p
substituent or a 2, 3 or 4 substituent, respectively. (Obviously, the 5
substituent is also a meta
substituent and the 6 substituent is an ortho substituent.)
The above compounds are evaluated for S1P2 activity according to the above
assay: The results
are reported in TABLE 2, below.
Proton nuclear magnetic resonance (1H NMR) and carbon nuclear magnetic
resonance (13C
NMR) spectra were recorded on a Varian 300 or 500 MHz spectrometer in
deuterated solvent.
Chemical shifts were reported as 6 (delta) values in parts per million (ppm)
relative to
tetramethylsilane (TMS) as an internal standard (0.00 ppm) and multiplicities
were reported as s,
singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad. Data
were reported in the
following format: chemical shift (multiplicity, coupling constant(s) J in
hertz (Hz), integrated
intensity).
General Procedure A for the Synthesis of Substituted 4-Isocyanatopyridine
Intermediates:
R1
HOOC ~R, OCN
/1 1. (C0CI)2/THF/DCM
N 2. NaN3/Acetone N
R2 3.Benzene/reflux R2
Commercially available properly substituted isonicotinic acid (1.87 mmol) was
dissolved in
THF: DCM (4:l Oml) at 0 C. Oxalyl chloride (2.OM in DCM, 1.87m1, 3.75mmol) was
added
followed by 2 drops of DMF. The resulting solution was stirred at 0 C for 30
min and
concentrated to dryness. The solid was dissolved in l Oml of acetone and
cooled to 0 C. NaN3
(195 mg, 3.00 mmol) in 1 mL of water was added. After stirring at 0 C for
30min, the reaction
mixture was concentrated again to dryness and re-dissolved in benzene which
was washed
quickly with ice cold water and dried over Na2SO4. The drying agent Na2SO4 was
filtered out,
and the filtrate was stirred at 110 C then cooled to 50 C to generate the
title compound in situ.
This crude isocyanate was used in the subsequent transformation without
further purification.
13
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
Syntheses of 5-hydrazinyl-7-isopropyl-1,3-dimethyl-1H-pyrazolo[4,3-b]pyridine
and (7-
isopropyl-1,3-dimethyl-1H-pyrazolo [4,3-b] pyridin-5-yl)methanamine
intermediates:
The title intermediate compounds were prepared from commercially available 3-
fluoropyridine
according to the following scheme.
OH
!-I
F 1. LDA/THF/-70 C F red-P/57%Hlaq F I\ 1 N f N /nBuLi/ether
2. Acetone 0C
N -50% N - 50% N 2. CH3CHO /-43%
F F
N
Mn02/Tol McNHNH2 NN MCPBA/CHCI3 N
reflux N H0~\ OH N 70 C /83% X N
N
OH 70 /0 O 1350C/84%
O
POCK N + N N \ Cl NH2NH2/EtOH N N
N'
80 C/45% N Cl N-1151 115 C \ I N NHNH2
( 4:5)
N
TMS-CN/TEA N EtOH/c. HCI/THF N
N N CH3CN/110 C N CN H2/Pd/C/80% N'
NH2
N
80%
0
2-(3-Fluoro pyridin-4 yl) propan-2-ol: 3-Fluoropyridine (11.0 g, 113.0 mmol)
in 100mL of
THE was treated with LDA (1.5M, 100.0 mL, 150.0 mmol) at -78 C for 50 min
under argon.
Acetone (28mL) was added and the resulting reaction mixture was stirred at -78
C to -40 C for
50 min. The reaction was then quenched with aqueous NH4C1 and extracted with
ether. The
combined organic layers were washed with H2O and brine, then dried over Na2SO4
and
concentrated under vacuum. Purification by MPLC (80g column, 0 to 40% ethyl
acetate in
hexane as eluant) afforded the title compound. Spectroscopic data: 'H-NMR (300
MHz,
CDC13): 6 ppm 1.65 (s, 6 H), 7.27 (dd, J=5.57 Hz, 1 H), 8.36-8.39 (m, 2 H).
14
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
3-Fluoro-4-isopropyl-pyridine: 2-(3-Fluoro-pyridin-4-yl)-propan-2-ol (from the
previous step,
6.40 g, 41.3 mmol) was dissolved in 50 mL of 57% HI aqueous solution. Red
phosphorus (4.48
g, 144 .0 mmol) was added and the mixture was stirred at 140 C for 5 hours.
The reaction
mixture was cooled to room temperature, neutralized with 5N aq NaOH and
extracted with
ether. The combined organic layers were washed with H2O and brine, then dried
over Na2SO4
and concentrated under vacuum. The title compound was obtained by
distillation. Spectroscopic
data: 'H-NMR (300 MHz, CDC13): 6 ppm 1.27 (d, J=7.04 Hz, 6 H), 3.25 (hept,
J=7.04 Hz, 1 H),
7.21 (dd, J=5.44 Hz, 1 H), 8.33-8.36 (m, 2 H).
1-(3-Fluoro-4-isopropyl pyridin-2 yl)-ethanol: 1, 4-diazabicyclo [2, 2, 2]
octane (6.16 g, 55.0
mmol) was dissolved in ether (100mL) at -40 C. n-BuLi (2.5M in hexane, 57.50
mL, 23.0
mmol) was added and the resulting reaction mixture was stirred at -20 C for 1
hour. The
reaction solution was then cooled to -60 C and 3-fluoro-4-isopropyl-pyridine
(obtained from the
previous step, 7.00 g, 50.0 mmol) was introduced. The stirring was continued
for 1 hour and
acetaldehyde (3 mL) was added. The solution was then allowed to warm up to -30
C in 1 hour
and quenched with aqueous NH4C1. The aqueous layer was extracted with ether.
The combined
extract was dried over Na2SO4 and concentrated. Purification by MPLC (80g
column, 0 to 15%
ethyl acetate in hexane as eluant) afforded the title compound. Spectroscopic
data: 'H-NMR
(300 MHz, CDC13): 6 ppm 1.27 (d, J=7.04 Hz, 3 H), 1.29 (d, J=7.04 Hz, 3 H),
1.50 (d, J=6.44
Hz, 3 H), 3.28 (hept, J=7.04 Hz, 1 H), 5.13 (q, J=6.44 Hz, 3 H), 7.18 (dd,
J=5.41 Hz, 1 H), 8.29
(d, J=5.27 Hz, 1 H).
1-(3-Fluoro-4-isopropyl pyridin-2 yl)-ethanone: 1-(3-Fluoro-4-isopropyl-
pyridin-2-yl)-ethanol
(prepared in the previous step, 3.00 g, 16.39 mmol) was dissolved in 60 mL of
anhydrous
toluene. Mn02 (4.05 g, 46.55 mmol) was added and the resulting mixture was
stirred at 140 C
overnight. The mixture was then cooled to room temperature and filtered
thought a pad of
celite. The filtrate was concentrated and purified via MPLC (80g, 0 to 20%
ethyl acetate in
hexane) to afford the title compound. Spectroscopic data: 'H-NMR (300 MHz,
CDC13): 6 ppm
1.28 (d, J=6.73 Hz, 6 H), 2.70 (s, 3 H), 3.35 (hept, J=6.73 Hz, 1 H), 7.38
(dd, J=5.13 Hz, 1 H),
8.39 (d, J=4.69 Hz, 1 H).
7-Isopropyl-1, 3-dimethyl-IH pyrazolo [4, 3-b] pyridine: 1-(3-Fluoro-4-
isopropyl-pyridin-2-yl)-
ethanone (prepared in the previous step, 2.50 g, 15.15 mmol) and methyl
hydrazine (1.04 g,
23.00 mmol) were combined in 8 mL of glycol. The reaction solution was stirred
at 140 C for 2
hours. After cooling, it was quenched with water, extracted with DCM, then
washed with water,
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
brine and concentrated. Column chromatography using 15 to 35% ethyl acetate in
hexane as
eluant afforded the title compound as a pale yellow solid. Spectroscopic data:
'H-NMR (300
MHz, CDC13): 6 ppm 1.39 (d, J=6.73 Hz, 6 H), 2.64 (s, 3 H), 3.65 (hept, J=6.73
Hz, 1 H), 4.23
(s, 3 H), 7.14 (d, J=4.69 Hz, 1 H), 8.44 (d, J=4.69 Hz 1 H).
7-Isopropyl-1, 3-dimethyl-IH-pyrazolo [4, 3-b] pyridine 4-oxide: To 7-
isopropyl- 1, 3-dimethyl-
1H-pyrazolo [4, 3-b] pyridine (prepared in the previous step, 2.50 g, 13.22
mmol) in 100mL of
chloroform was added 77% MCPBA (5.91 g, 26.44 mmol) at 0 C. The resulting
solution was
stirred at 90 C for 3 hours. After cooling, it was neutralized with aq NaHCO3
and extracted
with DCM thoroughly. The combined organic phases were then washed with water,
brine and
concentrated. Column chromatography using 5% 7N NH3 in MeOH /DCM as eluant
afforded
the title compound as a white solid. Spectroscopic data: 'H-NMR (300 MHz,
CDC13): 6 ppm
1.37 (d, J=6.73 Hz, 6 H), 2.83 (s, 3 H), 3.60 (hept, J=6.73 Hz, 1 H), 4.21 (s,
3 H), 6.78 (d,
J=6.30 Hz, 1 H), 8.01 (d, J=6.30 Hz 1 H).
5-Chloro-7-isopropyl-1, 3-dimethyl-IH pyrazolo [4, 3-b] pyridine: To a
solution of 7-isopropyl-
1, 3-dimethyl-1H-pyrazolo [4, 3-b] pyridine 4-oxide (obtained from the
previous step, 2.26 g,
11.00 mmol) in 30mL of toluene was added POC13 (3.37 g, 22.00 mmol). The
resulting reaction
mixture was stirred at 90 C for 2 hours and then concentrated. DCM was added.
The organic
layer was washed with water and brine, then dried over Na2SO4 and
concentrated. Column
chromatography with DCM separated two isomers and afforded the title compound
as a white
solid. Spectroscopic data: 'H-NMR (300 MHz, CDC13): 6 ppm 1.39 (d, J=6.73 Hz,
6 H), 2.58
(s, 3 H), 3.62 (hept, J=6.73 Hz, 1 H), 4.21 (s, 3 H), 7.11 (s, 1 H).
(7-Isopropyl-1, 3-dimethyl-IHpyrazolo [4, 3-b] pyridin-5-yl)-hydrazine: 5-
Chloro-7-isopropyl-
1,3-dimethyl-lH-pyrazolo[4,3-b]pyridine (obtained from the previous step, 620
mg, 2.68 mmol)
and 6 mL of hydrazine monohydrate were combined in 4mL of EtOH. The reaction
solution was
stirred at 120 C for 3 days. The reaction solution was then concentrated under
reduced pressure.
MeOH was added to the residue and concentrated again. Repetition of this
procure several
times until some solid formed. The solid was collected by filtration as the
title compound.
Spectroscopic data: 'H-NMR (300 MHz, CDC13): 6 ppm 1.34 (d, J=6.73 Hz, 6 H),
2.52 (s, 3 H),
3.56 (hept, J=6.73 Hz, 1 H), 4.15 (s, 3 H), 6.59 (s, 1 H).
7-Isopropyl-1, 3-dimethyl-IH pyrazolo [4, 3-b] pyridine-5-carbonitrile: To a
solution of 7-
isopropyl-1,3-dimethyl-lH-pyrazolo[4,3-b]pyridine N-oxide (obtained from the
previous step,
16
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
1.06 g, 5.17 mmol) and TEA (1.08 mL, 7.73 mmol) in 20mL CH3CN was added TMS
cyanide
(1.37 g, 10.34 mmol). The resulting solution was stirred at reflux for 18
hours. Another 0.5 mL
of TMS cyanide was added and the reaction continued for another 7 hours. The
solvent was
removed under reduced pressure and the residue was loaded to silica gel column
and eluted with
20 to 30% ethyl acetate to afford the desired title compound as a solid.
Spectroscopic data: 'H-
NMR (300 MHz, CDC13): 6 ppm 1.40 (d, J=6.74 Hz, 6 H), 2.64 (s, 3 H), 3.69
(hept, J=6.74 Hz,
1 H), 4.26 (s, 3 H), 7.46 (s, 1 H).
C-(7-Isopropyl-1, 3-dimethyl-IH-pyrazolo [4, 3-b] pyridin-5-yl)-methylamine:
7-Isopropyl-1, 3-dimethyl-lH-pyrazolo [4, 3-b] pyridine-5-carbonitrile (from
the previous step,
91.05 g, 5.15 mmol) was dissolved in EtOH:THF:conc.HC1(25 mL:10 mL:1.5 mL).
400 mg of
10% Pd/C was added and the reaction mixture was stirred under hydrogen balloon
overnight.
The catalyst was filtered out and the filtrate was concentrated under reduced
pressure to afford
the title compound as a white solid. Spectroscopic data: 'H-NMR (300 MHz,
CD3OD): 6 ppm
1.41 (d, J=6.74 Hz, 6 H), 2.56 (s, 3 H), 3.77 (hept, J=6.74 Hz, 1 H), 4.02 (s,
2 H), 4.21 (s, 3 H),
7.36 (s, 1 H).
General Procedure B for the Synthesis of Substituted Ureas:
R H H
OCN 6 Ring,X N NR6
Tl
Ring, X,NH2 + ~E THF/RT 0 i~E
R7 R7
A properly substituted amine or hydrazine prepared above and a properly
substituted isocyanate
(either commercial or prepared above, 1.1 eq) were dissolved in 8 mL of THF.
The reaction
mixture was stirred at room temperature for 4 hours and the solvent was
removed under reduced
pressure. The title compound was isolated by column chromatography using 30 to
75% ethyl
acetate in hexane, further purified by recrystallization from MeOH.
Example 1
N
N A-- N H
N N'
N CI
H 0 IqI
CI
17
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
Synthesis of N-(3,5-dichlorophenyl)-2-(7-isopropyl-1,3-dimethyl-1H-pyrazolo
[4,3-
b] pyridin-5-yl)hydrazinecarboxamide:
The title compound was generated from (7-isopropyl-1, 3-dimethyl-1H-pyrazolo
[4, 3-b]
pyridin-5-yl)-hydrazine prepared above and commercially available 3, 5-
dichlorophenyl
isocyanate according to General procedure B described above.
N-(3, 5-Dichlorophenyl)-2-(7-isopropyl-1, 3-dimethyl-JH-pyrazolo[4, 3-
b]pyridin-5-
yl)hydrazinecarboxamide: The title compound was obtained from3, 5-
dichlorophenyl
isocyanate (commercially available, 113 mg, 0.60 mmol), 5-hydrazinyl-7-
isopropyl-1,3-
dimethyl-lH-pyrazolo[4,3-b]pyridine (prepared above, 110 mg, 0.50 mmol)
according to the
protocols as outlined in general procedure B above. Spectroscopic Data: 1H NMR
(300 MHz,
CD3OD) 6 ppm 1.38 (d, J=6.73 Hz, 6 H), 2.44 (s, 3 H), 3.70 (hept, J=6.73 Hz, 1
H), 4.14 (s, 3
H), 6.85 (s, 1 H), 7.02 (dd, J=1.76 Hz, 1 H), 7.55 (d, J=1.76 Hz, 2 H).
Example 2
N
N'\ Nu N ~ CI
N II
O I ~N
CI
Synthesis of 1-(2,6-dichloropyridin-4-yl)-3-((7-isopropyl-1,3-dimethyl-1H-
pyrazolo [4,3-
b] pyridin-5-yl)methyl)urea:
The title compound was generated from (7-isopropyl-1,3-dimethyl-lH-
pyrazolo[4,3-b]pyridin-
5-yl)methanamine prepared above and commercially available 2,6-dichloro-4-
isocyanatopyridine according to General procedure B described above.
1-(2, 6-Dichloropyridin-4 yl)-3-((7-isopropyl-1, 3-dimethyl-JH-pyrazolo[4, 3-
b]pyridin-5-
yl)methyl)urea: The title compound was obtained from 2,6-dichloro-4-
isocyanatopyridine
(commercially available, 180 mg, 0.95 mmol), (7-isopropyl-1,3-dimethyl-lH-
pyrazolo[4,3-
b]pyridin-5-yl)methanamine (prepared above, 200 mg, 0,92 mmol) according to
the protocols as
outlined in general procedure B above. Spectroscopic Data: 1H NMR (300 MHz,
CD3OD) 6
18
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
ppm 1.40 (d, J=6.73 Hz, 6 H), 2.57 (s, 3 H), 3.74 (hept, J=6.73 Hz, 1 H), 4.22
(s, 3 H), 4.61 (s, 2
H), 7.33 (s, 1 H), 7.48 (s, 2 H).
Example 3
N
N'~ H H
N N"NN
H \/ O N
CI
Synthesis of N-(2-butyl-6-chloropyridin-4-yl)-2-(7-isopropyl-1,3-dimethyl-1H-
pyrazolo [4,3-
b] pyridin-5-yl)hydrazinecarboxamide:
The title compound was generated from (7-isopropyl-1, 3-dimethyl-1H-pyrazolo
[4, 3-b]
pyridin-5-yl)-hydrazine prepared above and 2-butyl-6-chloro-4-isocyanato-
pyridine according to
General procedure B described above. Intermediate 2-butyl-6-chloro-4-
isocyanato-pyridine was
prepared according to general procedure A and used in situ without further
purification.
2-Butyl-6-chloro-isonicotinic acid: The titled compound was prepared from
ethyl 2, 4-
dioxooctanate according to the literature reported method in reference 1. The
spectroscopic data
match those reported in the reference and also reported here: 'H-NMR (300 MHz,
CD3OD): 6
ppm 0.96 (t, J=7.33 Hz, 3 H), 1.33-1.43 (m, 2 H), 1.66-1.76 (m, 2 H), 2.82 (t,
J=7.62 Hz, 2 H),
7.70 (s, 1 H).
2-Butyl-6-chloro-4-isocyanato-pyridine: The title compound was prepared from
butyl-6-chloro-
isonicotinic acid (prepared above, 400 mg, 1.87 mmol), oxalyl chloride (2.0 M
in DCM, 1.87
mL, 3.75 mmol) according to general procedure A described above. The crude
title compound
was used in the next step without further purification.
N-(2-Butyl-6-chloropyridin-4 yl)-2-(7-isopropyl-1, 3-dimethyl-JH-pyrazolo[4, 3-
b]pyridin-5-
yl)hydrazinecarboxamide: The title compound was obtained from 2-butyl-6-chloro-
4-
isocyanato-pyridine (prepared above), 5-hydrazinyl-7-isopropyl-1,3-dimethyl-lH-
pyrazolo[4,3-
b]pyridine (prepared above, 200 mg, 0.92 mmol) according to the protocols as
outlined in
general procedure B above. Spectroscopic Data: 1H NMR (300 MHz, CD3OD) 6 ppm
0.91 (t,
J=7.32 Hz, 3 H), 126 (m, 2 H), 1.35 (d, J=6.73 Hz, 6 H), 1.61 (m, 2 H), 2.43
(s, 3 H), 2.61 (t,
19
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
J=7.68 Hz, 2 H), 3.65 (hept, J=6.73 Hz, 1 H), 4.12 (s, 3 H), 6.82 (s, 1 H),
7.30 (s, 1 H), 7.55 (s,
1 H).
Example 4
N
N'\ N N' N N
Fi O ~ N
CI
Synthesis of N-(2-chloro-6-ethoxypyridin-4-yl)-2-(7-isopropyl-1,3-dimethyl-1H-
pyrazolo [4,3-b] pyridin-5-yl)hydrazinecarboxamide:
The title compound was generated from (7-isopropyl-1, 3-dimethyl-lH-pyrazolo
[4, 3-b]
pyridin-5-yl)-hydrazine prepared above and 2-chloro-6-ethoxy-4-
isocyanatopyridine according
to General procedure B described above. Intermediate 2-chloro-6-ethoxy-4-
isocyanatopyridine
was prepared according to general procedure A and used in situ without further
purification.
2-Chloro-6-ethoxy-isonicotinic acid: The titled compound was prepared from 2.6-
dicloropyridine-4-carboxylic acid according to the reported method in
reference 2. The
spectroscopic data match those reported in the reference and also listed here
1H-NMR (300
MHz, CD3OD): ppm 1.38 (t, J=7.04 Hz, 3 H), 4.36 (q, J=7.04 Hz, 2 H), 7.17 (d,
J=1.18 Hz, 1
H), 7.39 (d, J=1.18 Hz, 1 H).
2-Chloro-6-ethoxy-4-isocyanato-pyridine: The title compound was prepared from
2-chloro-6-
ethoxy-isonicotinic acid (prepared above), oxalyl chloride (2.0 M in DCM)
according to general
procedure A described above. The crude title compound was used in the next
step without
further purification.
N-(2-Chloro-6-ethoxypyridin-4 yl)-2-(7-isopropyl-1,3-dimethyl-JHpyrazolo[4,3-
b]pyridin-5-
yl)hydrazinecarboxamide: The title compound was obtained from 2-chloro-6-
ethoxy-4-
isocyanato-pyridine (prepared above), 5-hydrazinyl-7-isopropyl-1,3-dimethyl-lH-
pyrazolo[4,3-
b]pyridine (prepared above, 180 mg, 0.82 mmol) according to the protocols as
outlined in
general procedure B above. Spectroscopic Data: 1H NMR (300 MHz, CD3OD) 6 ppm
1.33 (t,
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
J=7.04 Hz, 3 H), 1.37 (d, J=6.73 Hz, 6 H), 2.44 (s, 3 H), 3.67 (hept, J=6.73
Hz, 1 H), 4.14 (s, 3
H), 4.24 (q, J=7.04 Hz, 2 H), 6.83 (s, 1 H), 6.92 (s, 1 H), 7.10 (s, 1 H).
Example 5
N
N'\ I i N N
N ~ CI
O I /
CI
Synthesis of 1-(3,5-dichlorophenyl)-3-((1,3,7-trimethyl-1H-pyrazolo[4,3-
b]pyridin-5-
yl)methyl)urea:
The title compound was generated from (1,3,7-trimethyl-lH-pyrazolo[4,3-
b]pyridin-5-
yl)methanamine prepared using the procedure described above for (7-isopropyl-
1,3-dimethyl-
1H-pyrazolo[4,3-b]pyridin-5-yl)methanamine and commercially available 3, 5-
dichlorophenyl
isocyanate according to General procedure B described above.
1-(3,5-Dichlorophenyl)-3-((1,3,7-trimethyl- 1H JH-pyrazoIo[4,3-b]pyridin-5-yI)
methyl) urea: The
title compound was obtained from3, 5-dichlorophenyl isocyanate (commercially
available, 170
mg, 0.90 mmol), (1,3,7-trimethyl-lH-pyrazolo[4,3-b]pyridin-5-yl)methanamine
(prepared
above, 160 mg, 0,84 mmol) according to the protocols as outlined in general
procedure B above.
Spectroscopic Data: 1H NMR (300 MHz, CD3OD) 6 ppm 2.47 (s, 3 H), 2.74 (s, 3
H), 4.14 (s, 3
H), 4.47 (d, J=4.69 Hz, 2 H), 7.01 (dd, J=2.07 Hz, 1 H), 7.10 (bs, 1 H), 7.21
(s, 1 H), 7.42 (d,
J=2.07 Hz, 2 H), 9.45(s, 1 H).
Example 6
N
N'\ I N N,NUN CI
H I I
O N
CI
Synthesis of N-(2,6-dichloropyridin-4-yl)-2-(7-isopropyl-1,3-dimethyl-1H-
pyrazolo [4,3-
b] pyridin-5-yl)hydrazinecarboxamide:
21
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
The title compound was generated from (7-isopropyl-1, 3-dimethyl-1H-pyrazolo
[4, 3-b]
pyridin-5-yl)-hydrazine prepared above and commercially available 2,6-dichloro-
4-
isocyanatopyridine according to General procedure B described above.
N-(2, 6-Dichloropyridin-4 yl)-2-(7-isopropyl-1, 3-dimethyl-JH-pyrazolo[4, 3-
b]pyridin-5-
yl)hydrazinecarboxamide: The title compound was obtained from 3, 5-
dichlorophenyl
isocyanate (commercially available, 124 mg, 0.66 mmol), 5-hydrazinyl-7-
isopropyl-1,3-
dimethyl-lH-pyrazolo[4,3-b]pyridine (prepared above, 120 mg, 0.55 mmol)
according to the
protocols as outlined in general procedure B above. Spectroscopic Data: 1H NMR
(300 MHz,
CD3OD) 6 ppm 1.37 (d, J=6.73 Hz, 6 H), 2.44 (s, 3 H), 3.68 (hept, J=6.73 Hz, 1
H), 4.14 (s, 3
H), 6.82 (s, 1 H), 7.63 (s, 2 H).
Example 7
N
N'\ N N ' N CF3
H 11:
CF3
Synthesis of N-(3,5-bis(trifluoromethyl)phenyl)-2-(7-isopropyl-1,3-dimethyl-1H-
pyrazolo[4,3-b]pyridin-5-yl)hydrazinecarboxamide:
The title compound was generated from 5-hydrazinyl-7-isopropyl-1,3-dimethyl-lH-
pyrazolo[4,3-b]pyridine prepared above and commercially available 4-isocyanato-
2,6-
bis(trifluoromethyl)pyridine according to General procedure B described above.
N-(3, 5-Bis (trif luoromethyl)phenyl)-2-(7-isopropyl-1, 3-dimethyl-JH-
pyrazolo[4, 3-b]pyridin-5-
yl)hydrazinecarboxamide: The title compound was obtained from 4-isocyanato-2,6-
bis(trifluoromethyl)pyridine (commercially available, 117 mg, 0.44 mmol), 5-
hydrazinyl-7-
isopropyl-1,3-dimethyl-lH-pyrazolo[4,3-b]pyridine (prepared above, 80 mg, 0.36
mmol)
according to the protocols as outlined in general procedure B above.
Spectroscopic Data: 1H
NMR (300 MHz, CD3OD) 6 ppm 1.38 (d, J=6.73Hz, 6 H), 2.44 (s, 3 H), 3.68 (hept,
J=6.73Hz, 1
H), 4.14 (s, 3 H), 6.86 (s, 1 H), 7.53 (s, 1 H), 8.19 (s, 2 H).
22
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
Example 8
N
N' N N. N N 0111
H I I
O IZN
CI
Synthesis of N-(3-chloro-5-methoxypyridin-4-yl)-2-(7-isopropyl-1,3-dimethyl-1H-
pyrazolo [4,3-b] pyridin-5-yl)hydrazinecarboxamide:
The title compound was generated from (7-isopropyl-1, 3-dimethyl-1H-pyrazolo
[4, 3-b]
pyridin-5-yl)-hydrazine prepared above and 2-chloro-6-methoxy-4-
isocyanatopyridine according
to General procedure B described above. Intermediate 2-chloro-6-methoxy-4-
isocyanatopyridine was prepared according to general procedure A and used in
situ without
further purification.
N-(3-Chloro-5-methoxypyridin-4 yl)-2-(7-isopropyl-1,3-dimethyl-JH pyrazolo[4,3-
b]pyridin-5-
yl)hydrazinecarboxamide: The title compound was obtained from 2-chloro-6-
methoxy-4-
isocyanato-pyridine (prepared above according to general procedure A, crude),
5-hydrazinyl-7-
isopropyl-1,3-dimethyl-lH-pyrazolo[4,3-b]pyridine (prepared above, 140 mg,
0.64 mmol)
according to the protocols as outlined in general procedure B above.
Spectroscopic Data: 1H
NMR (300 MHz, CD3OD) 6 ppm 1.37 (d, J=6.8OHz, 6 H), 2.44 (s, 3 H), 3.69 (hept,
J=6.8OHz, 1
H), 3.38 (s, 3 H), 4.14 (s, 3 H), 6.83 (s, 1 H), 6.95 (s, 1 H), 7.01 (s, 1 H).
Example 9
\
N
N N N CI
&N-
0 I /
CI
Synthesis of 1-(2,6-dichlorophenyl)-3-((7-isopropyl-1,3-dimethyl-1H-pyrazolo
[4,3-
b]pyridin-5-yl)methyl)urea:
23
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
The title compound was generated from (7-isopropyl-1,3-dimethyl-lH-
pyrazolo[4,3-b]pyridin-
5-yl)methanamine prepared above and commercially available 1,3-dichloro-5-
isocyanatobenzene according to General procedure B described above.
1-(3, 5-Dichlorophenyl)-3-((7-isopropyl-1, 3-dimethyl-JH-pyrazolo[4, 3-
b]pyridin-5-
yl)methyl)urea: The title compound was obtained from 1,3-dichloro-5-
isocyanatobenzene
(commercially available, 188 mg, 1.0 mmol), (7-isopropyl-1,3-dimethyl-lH-
pyrazolo[4,3-
b]pyridin-5-yl)methanamine (prepared above, 200 mg, 0,92 mmol) according to
the protocols as
outlined in general procedure B above. Spectroscopic Data: 1H NMR (300 MHz,
CD3OD) 6
ppm 1.40 (d, J=6.73Hz, 6 H), 2.57 (s, 3 H), 3.76 (hept, J=6.73Hz, 1 H), 4.21
(s, 3 H), 4.59 (s, 2
H), 6.99 (dd, J=1.76Hz, 1 H), 7.34 (s, 1 H), 7.43 (d, J=1.76Hz, 2 H).
Example 10
N
N &-- N H N \ O\
O I ~N
CI
Synthesis of 1-(2-chloro-6-methoxypyridin-4-yl)-3-((7-isopropyl-1,3-dimethyl-
1H-
pyrazolo [4,3-b] pyridin-5-yl)methyl)urea:
The title compound was generated from (7-isopropyl-1,3-dimethyl-lH-
pyrazolo[4,3-b]pyridin-
5-yl)methanamine prepared above and 2-chloro-4-isocyanato-6-methoxypyridine
(prepared
according to general procedure A described above) according to General
procedure B described
above.
1-(2-Chloro-6-methoxypyridin-4 yl)-3-((7-isopropyl-1,3-dimethyl-JHpyrazolo[4,3-
b]pyridin-5-
yl)methyl)urea: The title compound was obtained from 2-chloro-4-isocyanato-6-
methoxypyridine (prepared above, crude, 1.0 mmol), (7-isopropyl-1,3-dimethyl-
lH-
pyrazolo[4,3-b]pyridin-5-yl)methanamine (prepared above, 200 mg, 0,92 mmol)
according to
the protocols as outlined in general procedure B above. Spectroscopic Data: 1H
NMR (300
MHz, CD3OD) 6 ppm 1.40 (d, J=6.73 Hz, 6 H), 2.57 (s, 3 H), 3.76 (hept,
J=6.73Hz, 1 H), 4.21
(s, 3 H), 4.59 (s, 2 H), 6.82 (d, J=1.76 Hz, 1 H), 7.06 (d, J=1.76 Hz, 1 H),
7.33 (s, 1 H).
24
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
Example 11
N
N'~ ,NUN
N -, II
O N
H
CI
Synthesis of N-(2-chloro-6-propylpyridin-4-yl)-2-(7-isopropyl-1,3-dimethyl-1H-
pyrazolo [4,3-b] pyridin-5-yl)hydrazinecarboxamide:
The title compound was generated from (7-isopropyl-1, 3-dimethyl-lH-pyrazolo
[4, 3-b]
pyridin-5-yl)-hydrazine prepared above and 2-chloro-4-isocyanato-6-
propylpyridine according
to General procedure B described above. Intermediate 2-chloro-4-isocyanato-6-
propylpyridine
was prepared according to general procedure A and used in situ without further
purification.
N-(2-Chloro-6 propylpyridin-4 yl)-2-(7-isopropyl-1,3-dimethyl-JH pyrazolo[4,3-
b]pyridin-5-
yl)hydrazinecarboxamide: The title compound was obtained from 2-chloro-4-
isocyanato-6-
propylpyridine (prepared above), 5-hydrazinyl-7-isopropyl-1,3-dimethyl-lH-
pyrazolo[4,3-
b]pyridine (prepared above, 200 mg, 0.92 mmol) according to the protocols as
outlined in
general procedure B above. Spectroscopic Data: 1H NMR (300 MHz, CD3OD) 6 ppm
0.94 (t,
J=7.32 Hz, 3 H), 1.37 (d, J=6.73 Hz, 6 H), 1.69 (m, 2 H), 2.44 (s, 3 H), 2.61
(t, J=7.68 Hz, 2 H),
3.68 (hept, J=6.73 Hz, 1 H), 4.14 (s, 3 H), 6.84 (s, 1 H), 7.37 (s, 1 H), 7.57
(s, 1 H).
Example 12
N
N '
O I i
CI
Synthesis of 1-(2-chloro-6-propylpyridin-4-yl)-3-((7-isopropyl-1,3-dimethyl-1H-
pyrazolo [4,3-b] pyridin-5-yl)methyl)urea:
The title compound was generated from (7-isopropyl-1,3-dimethyl-lH-
pyrazolo[4,3-b]pyridin-
5-yl)methanamine prepared above and 2-chloro-4-isocyanato-6-propylpyridine
according to
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
General procedure B described above. Intermediate 2-chloro-4-isocyanato-6-
propylpyridine
was prepared according to general procedure A and used in situ without further
purification.
1-(2-Chloro-6 propylpyridin-4 yl)-3-((7-isopropyl-1,3-dimethyl-JH-pyrazolo[4,3-
b]pyridin-5-
yl)methyl)urea: The title compound was obtained from 2-chloro-4-isocyanato-6-
propylpyridine
(prepared above), (7-isopropyl-1,3-dimethyl-lH-pyrazolo[4,3-b]pyridin-5-
yl)methanamine
(prepared above, 200 mg, 0.92 mmol) according to the protocols as outlined in
general
procedure B above. Spectroscopic Data: 1H NMR (300 MHz, CD3OD) 6 ppm 0.94 (t,
J=7.32
Hz, 3 H), 1.37 (d, J=6.73 Hz, 6 H), 1.69 (m, 2 H), 2.44 (s, 3 H), 2.61 (t,
J=7.68 Hz, 2 H), 3.68
(hept, J=6.73 Hz, 1 H), 4.14 (s, 3 H), 6.84 (s, 1 H), 7.37 (s, 1 H), 7.57 (s,
1 H).
Example 13
N
N I ~ H H N
CI
ynt
hesis of 1-(2-chloro-6-ethoxypyridin-4-yl)-3-((7-isopropyl-1,3-dimethyl-1H-
S
pyrazolo [4,3-b] pyridin-5-yl)methyl)urea:
The title compound was generated from (7-isopropyl-1,3-dimethyl-lH-
pyrazolo[4,3-b]pyridin-
5-yl)methanamine prepared above and 2-chloro-6-ethoxy-4-isocyanatopyridine
according to
General procedure B described above. Intermediate 2-chloro-6-ethoxy-4-
isocyanatopyridine
was prepared according to general procedure A and used in situ without further
purification.
1-(2-Chloro-6-ethoxypyridin-4 yl)-3-((7-isopropyl-1, 3-dimethyl-JH-pyrazolo[4,
3-b]pyridin-5-
yl)methyl)urea: The title compound was obtained from 2-chloro-6-ethoxy-4-
isocyanatopyridine
(prepared above), (7-isopropyl-1,3-dimethyl-lH-pyrazolo[4,3-b]pyridin-5-
yl)methanamine
(prepared above, 200 mg, 0.92 mmol) according to the protocols as outlined in
general
procedure B above. Spectroscopic Data: 1H NMR (300 MHz, CD3OD) 6 ppm 1.34 (t,
J=7.04Hz, 3 H), 1.39 (d, J=6.73 Hz, 6 H), 2.57 (s, 3 H), 3.76 (hept, J=6.73
Hz, 1 H), 4.21 (s, 3
H), 4.25 (q, J=7.04 Hz, 2 H), 4.59 (s, 2 H), 6.78 (d, J=1.46 Hz, 1 H), 7.04
(d, J=1.46 Hz, 1 H),
7.33 (s, 1 H).
26
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
Example 14
N
N'\ &-- N H N O~/\
O IZN
CI
Synthesis of 1-(2-chloro-6-propoxypyridin-4-yl)-3-((7-isopropyl-1,3-dimethyl-
1H-
pyrazolo [4,3-b] pyridin-5-yl)methyl)urea:
The title compound was generated from (7-isopropyl-1,3-dimethyl-lH-
pyrazolo[4,3-b]pyridin-
5-yl)methanamine prepared above and 2-chloro-4-isocyanato-6-propoxypyridine
according to
General procedure B described above. Intermediate 2-chloro-4-isocyanato-6-
propoxypyridine
was prepared according to general procedure A and used in situ without further
purification.
1-(2-Chloro-6 propoxypyridin-4 yl)-3-((7-isopropyl-1,3-dimethyl-JH-
pyrazolo[4,3-b]pyridin-5-
yl)methyl)urea: The title compound was obtained from 2-chloro-4-isocyanato-6-
propoxypyridine (prepared above), (7-isopropyl-1,3-dimethyl-lH-pyrazolo[4,3-
b]pyridin-5-
yl)methanamine (prepared above, 180 mg, 0,82 mmol) according to the protocols
as outlined in
general procedure B above. Spectroscopic Data: 1H NMR (300 MHz, CD3OD) 6 ppm
1.00 (t,
J=7.32Hz, 3H), 1.40 (d, J=6.73 Hz, 6 H), 1.75 (m, 2 H), 2.57 (s, 3 H), 3.76
(hept, J=6.73 Hz, 1
H), 4.15 (t, J=6.60 Hz, 2 H), 4.21 (s, 3 H), 4.59 (s, 2 H), 6.78 (d, J=1.46
Hz, 1 H), 7.04 (d,
J=1.46 Hz, 1 H), 7.34 (s, 1 H).
Example 15
N
N' N N,NuN
" O~/\
I I
O I N
CI
Synthesis of N-(2-chloro-6-propoxypyridin-4-yl)-2-(7-isopropyl-1,3-dimethyl-1H-
pyrazolo[4,3-b]pyridin-5-yl)hydrazinecarboxamide:
The title compound was generated from 5-hydrazinyl-7-isopropyl-1,3-dimethyl-lH-
pyrazolo[4,3-b]pyridine prepared above and 2-chloro-4-isocyanato-6-
propoxypyridine according
27
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
to General procedure B described above. Intermediate 2-chloro-4-isocyanato-6-
propoxypyridine
was prepared according to general procedure A and used in situ without further
purification.
N-(3-Chloro-5 propoxyphenyl)-2-(7-isopropyl-1,3-dimethyl-JH pyrazolo[4,3-
b]pyridin-5-
yl)hydrazinecarboxamide: The title compound was obtained from 2-chloro-4-
isocyanato-6-
propoxypyridine (prepared above according to general procedure A, crude), 5-
hydrazinyl-7-
isopropyl-1,3-dimethyl-lH-pyrazolo[4,3-b]pyridine (prepared above, 160 mg,
0,73 mmol)
according to the protocols as outlined in general procedure B above.
Spectroscopic Data: 1H
NMR (300 MHz, CD3OD) 6 ppm 1.00 (t, J=7.34 Hz, 3 H), 1.38 (d, J=6.73 Hz, 6 H),
1.74 (m, 2
H), 2.44 (s, 3 H), 3.67 (hept, J=6.73 Hz, 1 H), 4.12-4.17 (m, 5 H), 6.83 (s, 1
H), 6.94 (s, 1 H),
7.19 (s, 1 H).
Example 16
N
N'\ N N' N N
H I I
O I N
CI
Synthesis of N-(2-butoxy-6-chloropyridin-4-yl)-2-(7-isopropyl-1,3-dimethyl-1H-
pyrazolo [4,3-b] pyridin-5-yl)hydrazinecarboxamide:
The title compound was generated from 5-hydrazinyl-7-isopropyl-1,3-dimethyl-lH-
pyrazolo[4,3-b]pyridine prepared above and 2-butoxy-6-chloro-4-
isocyanatopyridine according
to General procedure B described above. Intermediate 2-butoxy-6-chloro-4-
isocyanatopyridine
was prepared according to general procedure A and used in situ without further
purification.
N-(2-Butoxy-6-chloropyridin-4 yl)-2-(7-isopropyl-1,3-dimethyl-JHpyrazolo[4,3-
b]pyridin-5-
yl)hydrazinecarboxamide: The title compound was obtained from 2-butoxy-6-
chloro-4-
isocyanatopyridine (prepared above according to general procedure A, crude), 5-
hydrazinyl-7-
isopropyl-1,3-dimethyl-lH-pyrazolo[4,3-b]pyridine (prepared above, 160 mg,
0,73 mmol)
according to the protocols as outlined in general procedure B above.
Spectroscopic Data: 1H
NMR (300 MHz, CD3OD) 6 ppm 0.97 (t, J=7.34 Hz, 3 H), 1.38 (d, J=6.73 Hz, 6 H),
1.46 (m, 2
H), 1.71 (m, 2 H), 2.44 (s, 3 H), 3.68 (hept, J=6.73 Hz, 1 H), 4.14 (s, 3 H),
4.20 (t, J=6.44 Hz, 2
H), 6.83 (s, 1 H), 6.93 (s, 1 H), 7.19 (s, 1 H).
28
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
Example 17
\
N
N,\ I Nu H N II
O I N
CI
Synthesis of 1-(2-butoxy-6-chloropyridin-4-yl)-3-((7-isopropyl-1,3-dimethyl-1H-
pyrazolo [4,3-b] pyridin-5-yl)methyl)urea:
The title compound was generated from (7-isopropyl-1,3-dimethyl-lH-
pyrazolo[4,3-b]pyridin-
5-yl)methanamine prepared above and 2-butoxy-6-chloro-4-isocyanatopyridine
according to
General procedure B described above. Intermediate 2-butoxy-6-chloro-4-
isocyanatopyridine
was prepared according to general procedure A and used in situ without further
purification.
1-(2-Butoxy-6-chloropyridin-4 yl)-3-((7-isopropyl-1,3-dimethyl-IH-pyrazolo[4,3-
b]pyridin-5-
yl)methyl)urea: The title compound was obtained from 2-chloro-4-isocyanato-6-
propoxypyridine (prepared above), (7-isopropyl-1,3-dimethyl-lH-pyrazolo[4,3-
b]pyridin-5-
yl)methanamine (prepared above, 170 mg, 0,78 mmol) according to the protocols
as outlined in
general procedure B above. Spectroscopic Data: 1H NMR (300 MHz, CD3OD) 6 ppm
0.97 (t,
J=7.32 Hz, 3 H), 1.40 (d, J=6.73 Hz, 6 H), 1.50 (m, 2 H), 1.72 (m, 2 H), 2.57
(s, 3 H), 3.76
(hept, J=6.73 Hz, 1 H), 4.20 (t, J=6.44 Hz, 2 H), 4.21 (s, 3 H), 4.59 (s, 2
H), 6.80 (d, J=1.46 Hz,
1 H), 7.04 (d, J=1.46 Hz, 1 H), 7.34 (s, 1 H).
Example 18
N
N'\ I N N' N N 0
H Y
0 11::':"Nr
Synthesis of N-(2-ethoxypyridin-4-yl)-2-(7-isopropyl-1,3-dimethyl-1H-pyrazolo
[4,3-
b]pyridin-5-yl)hydrazinecarboxamide:
The title compound was generated from 5-hydrazinyl-7-isopropyl-1,3-dimethyl-lH-
pyrazolo[4,3-b]pyridine prepared above and 2-ethoxy-4-isocyanatopyridine
according to
29
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
General procedure B described above. Intermediate 2-ethoxy-4-
isocyanatopyridine was
prepared according to general procedure A and used in situ without further
purification.
N-(2-Ethoxypyridin-4 yl)-2-(7-isopropyl-1,3-dimethyl-JH-pyrazolo[4,3-b]pyridin-
5-
yl)hydrazinecarboxamide: The title compound was obtained from 2-ethoxy-4-
isocyanatopyridine (prepared above according to general procedure A, crude), 5-
hydrazinyl-7-
isopropyl-1,3-dimethyl-lH-pyrazolo[4,3-b]pyridine (prepared above, 160 mg,
0,73 mmol)
according to the protocols as outlined in general procedure B above.
Spectroscopic Data: 1H
NMR (300 MHz, CD3OD) 6 ppm 1.34 (t, J=7.04 Hz, 3 H), 1.38 (d, J=6.73 Hz, 6 H),
2.44 (s, 3
H), 3.69 (hept, J=6.73 Hz, 1 H), 4.14 (s, 3 H), 4.24 (q, J=7.04 Hz, 2 H), 6.84
(s, 1 H), 7.04-7.07
(m, 2 H), 7.86 (d, J=6.00 Hz,, 1 H).
Example 19
N O
N H H
N N"N\/N
H 0 0
CI
Synthesis of N-(5-chloro-2,4-dimethoxyphenyl)-2-(7-isopropyl-1,3-dimethyl-1H-
pyrazolo [4,3-b] pyridin-5-yl)hydrazinecarboxamide:
The title compound was generated from 5-hydrazinyl-7-isopropyl-1,3-dimethyl-lH-
pyrazolo[4,3-b]pyridine prepared above and commercially available 1-chloro-5-
isocyanato-2,4-
dimethoxybenzene according to General procedure B described above.
N-(5-Chloro-2, 4-dimethylphenyl)-2-(7-isopropyl-1, 3-dimethyl-JH-pyrazolo[4, 3-
b]pyridin-5-
yl)hydrazinecarboxamide: The title compound was obtained from 1-chloro-5-
isocyanato-2,4-
dimethoxybenzene and 5-hydrazinyl-7-isopropyl-1,3-dimethyl-lH-pyrazolo[4,3-
b]pyridine
(prepared above, 160 mg, 0,73 mmol) according to the protocols as outlined in
general
procedure B above. Spectroscopic Data: 1H NMR (300 MHz, CD3OD) 6 ppm 1.37 (d,
J=6.73
Hz, 6 H), 2.45 (s, 3 H), 3.68 (hept, J=6.73 Hz, 1 H), 3.82 (s, 3 H), 3.85 (s,
3 H), 4.14 (s, 3 H),
6.70 (s, 1 H), 6.86 (s, 1 H), 8.01 (s, 1 H).
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
Example 20
,N N c CI
N N H O
Cl
Synthesis of N-(3,5-dichlorophenyl)-2-(4-methyl-1,8-naphthyridin-2-
yl)hydrazinecarboxamide
The title compound was generated from N-(pyridin-2-yl)pivalamide according to
the chemistry
described in the following scheme. The intermediates were separated and
characterized.
Scheme 1
0 OH 0
1.) n-BuLi, THF, -78 C LDA, THF, -78 C 3N HCI
a~l 1 1 ~~ - -
N NHPiv N 2.) 0 NHPiv 0 N NHPiv 160 C, 5 min
AN'OMe 0 microwave
NCO
NH2NH2 Cl "'dCl
POCI3
I ft' ' ' NN CI
f ftN'
N H 0 toluene N CI 140 C N N NHNH2 CH2Cl2 N N H Y
I\
0
120 C /
Cl
N-(3-Acetylpyridin-2-yl)pivalamide: To a solution of N-(pyridin-2-
yl)pivalamide (2.0 g, 11.22
mmoles) in THF at -78 C was added 9.4 mL (2.1 eq) of n-BuLi. The resulting
mixture was
stirred at 0 C for 3 hours. The reaction mixture was then cooled to -78 C
after which N-
methoxy-N-methylacetamide (1.2 g, 1.1 eq) was added as a solution in THF. The
resulting
mixture was stirred at room temperature for 2 hours. It was then quenched into
ice-H20. The
resulting mixture was extracted with CH2Cl2 (3X 20 mL) and the combined
organic extracts
were washed with H2O (2x 20 mL), brine (lx 20 mL), then dried over MgS04.
Concentration
and purification by MPLC gave N-(3-acetylpyridin-2-yl)pivalamide (1.62 g,
65%).
Spectroscopic data: 1H NMR (300 MHz, CDC13) 6 ppm 1.36 (s, 9 H) 2.65 (s, 3 H)
7.09 (dd,
31
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
J=7.76, 4.83 Hz, 1 H) 8.17 (dd, J=7.76, 1.90 Hz, 1 H) 8.64 (dd, J=4.69, 2.05
Hz, 1 H) 11.49 (br.
s., 1 H).
tent-Butyl 3-hydroxy-3-(2 pivalamidopyridin-3 yl)butanoate: To a solution of
9.5 mL (2.1 eq) of
LDA in 15 mL THE at -78 C was added 2.0 mL of t-butyl acetate (2.1 eq.). The
resulting
mixture was stirred at -78 C for 30 minutes after which a solution of N-(3-
acetylpyridin-2-
yl)pivalamide (1.52 g, 6.90 mmol) in THE was added in a dropwise fashion. The
resulting
reaction mixture was then stirred at -78 C for 30 minutes and was brought up
to room
temperature. The reaction mixture was then quenched with saturated NH4C1. The
resulting
mixture was extracted with EtOAc (3X 20 mL) and the combined organic extracts
were washed
with H2O (2x 20 mL), brine (lx 20 mL), dried over MgSO4 and concentrated to
give the title
compound (1.40 g, 61%). Spectroscopic data: 1H NMR (300 MHz, CDC13) 6 ppm 1.33
(s, 9 H)
1.44 (s, 9 H) 1.58 (s, 3 H) 2.65 (d, J=16.70 Hz, 1 H) 3.06 (d, J=16.70 Hz, 1
H) 5.54 (s, 1 H) 6.96
(dd, J=7.76, 4.83 Hz, 1 H) 7.43 (dd, J=7.91, 1.76 Hz, 1 H) 8.45 (dd, J=4.98,
1.76 Hz, 1 H) 10.29
(s, 1 H).
4-Methyl-1,8-naphthyridin-2(JH)-one: A solution of 1.3 g of tent-butyl 3-
hydroxy-3-(2-
pivalamidopyridin-3-yl)butanoate (3.9 mmol) in 2 mL 3N HC1 was microwaved at
160 C for 5
minutes. The resulting mixture was washed with Et20 (2 x 10 mL). The aqueous
layer was
basified using saturated K2CO3. The precipitate formed was filtered and washed
with water to
give 440 mg (71%) of the desired title compound. Spectroscopic data: 1H NMR
(300 MHz,
DMSO-d6) 6 ppm 2.40 (d, J=1.17 Hz, 3 H) 6.43 (s, 1 H) 7.24 (dd, J=7.91, 4.69
Hz, 1 H) 8.12
(dd, J=7.91, 1.76 Hz, 1 H) 8.49 (dd, J=4.69, 1.76 Hz, 1 H) 11.92 (br. s., 1
H).
2-Chloro-4-methyl-1,8-naphthyridine: To a solution of 4-methyl-1,8-
naphthyridin-2(1H)-one
(440 mg, 2.75 mmol) in toluene was added 302 uL of POC13 (1.2 eq). The
resulting mixture was
refluxed at 120 C for 3 hours. After cooling to room temperature the reaction
mixture was
quenched into ice-H20 and extracted with EtOAc (3X 10 mL). The combined
organic extracts
was washed with H2O (2x 15 mL), brine (lx 20 mL), and then dried over MgSO4.
Concentration
and purification by MPLC gave the desired 2-chloro-4-methyl-1,8-naphthyridine
(330 mg,
67%). Spectroscopic data: 1H NMR (300 MHz, CDC13) 6 ppm 2.72 (s, 3 H) 7.35 (d,
J=1.17 Hz,
1 H) 7.53 (dd, J=8.20, 4.40 Hz, 1 H) 8.36 (dd, J=8.35, 1.90 Hz, 1 H) 9.10 (dd,
1 H).
2-Hydrazinyl-4-methyl-1,8-naphthyridine: A solution of 2-chloro-4-methyl-1,8-
naphthyridine
(330 mg, 1.8 mmol) in NH2NH2 was refluxed at 130 C for 2 hours. The reaction
mixture was
32
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
cooled to room temperature, then excess NH2NH2 was removed on the rotary
evaporator. The
residue was taken up in 20 mL of CH2C12. The mixture was washed with saturated
NaHCO3 (3
x 15 mL), brine (lx 20 mL), and then dried over MgSO4 and concentrated. The
residue was
washed with hexane to give 2-hydrazinyl-4-methyl-1,8-naphthyridine.
Spectroscopic data: 1H
NMR (300 MHz, CDC13) 6 ppm 2.56 (s, 3 H) 6.66 (s, 1 H) 7.22 (dd, J=7.91, 4.40
Hz, 1 H) 8.11
(dd, J=8.06, 1.90 Hz, 1 H) 8.82 (dd, 1 H).
N-(3,5-Dichlorophenyl)-2-(4-methyl-l,8-naphthyridin-2 yl)hydrazinecarboxamide:
To a
solution of 2-hydrazinyl-4-methyl-1,8-naphthyridine prepared above (84 mg,
0.482 mmol) in
CH2C12 was added 3,5-dichlorophenyl isocyanate (91 mg, 1.0 eq). The resulting
mixture was
stirred at room temperature overnight. The resulting precipitate was filtered
off and washed
with EtOAc to give the desired title compound. Spectroscopic data: 1H NMR (300
MHz,
DMSO-d6) 6 ppm 2.51 (s, 3 H) 6.78 (s, 1 H) 7.05 (t, J=1.90 Hz, 1 H) 7.23 (dd,
J=7.91, 4.40 Hz,
1 H) 7.61 (br. s., 2 H) 8.22 (dd, J=8.06, 1.90 Hz, 1 H) 8.57 (br. s., 1 H)
8.68 (dd, J=4.40, 1.76
Hz, 1 H) 8.98 (s, 1 H) 9.22 (br. s., 1 H).
Example 21
,N N q CI
N N H Y
O
CI
Synthesis of N-(3,5-dichlorophenyl)-2-(4-isopropyl-1,8-naphthyridin-2-
yl)hydrazinecarboxamide
The title compound was generated from N-(3-isobutyrylpyridin-2-yl)pivalamide
according to the
chemistry described in scheme 1 described above. The intermediates were
separated and
characterized.
N-(3-Isobutyrylpyridin-2yl)pivalamide: A solution of N-(pyridin-2-
yl)pivalamide (3.20 g, 18.0
mmol), n-BuLi (15.00 mL, 2.1 eq.) and N-methoxy-N-methylisobutyramide (2.60 g,
1.1 eq)
were reacted as outlined in Scheme 1 to give 3.10 g (69%) of N-(3-
isobutyrylpyridin-2-
yl)pivalamide. Spectroscopic data: 1H NMR (300 MHz, CDC13) 6 ppm 1.21 (d,
J=7.03 Hz, 6 H)
1.35 (s, 9 H) 3.55 (dt, J=13.70, 6.78 Hz, 1 H) 7.08 (dd, J=7.91, 4.69 Hz, 1 H)
8.19 (dd, J=8.20,
2.05 Hz, 1 H) 8.63 (dd, J=4.83, 1.90 Hz, 1 H) 11.51 (br. s., 1 H).
33
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
tent-Butyl 3-hydroxy-4-methyl-3-(2 pivalamidopyridin-3 yl)pentanoate: A
solution of N-(3-
isobutyrylpyridin-2-yl)pivalamide (3.10 g, 12.5 mmol), LDA (1.5 M, 17.50 mL,
2.1 eq) and t-
butyl acetate (3.5 mL, 2.1 eq) were reacted as outlined in Scheme 1 to give
tent-butyl 3-hydroxy-
4-methyl-3-(2-pivalamidopyridin-3-yl)pentanoate which was used in the next
step without
further purification.
4-Isopropyl-1,8-naphthyridin-2(]H)-one: A solution of tent-butyl 3-hydroxy-4-
methyl-3-(2-
pivalamidopyridin-3-yl)pentanoate (3.78 g, 10.4 mmol) in 12.0 mL 3N HC1 were
reacted as
outlined in Scheme 1 to give the desired 4-isopropyl- 1,8-naphthyridin-2(l H)-
one (800 mg,
41%). Spectroscopic data: 1H NMR (300 MHz, CDC13) 6 ppm 1.34 (d, J=7.03 Hz, 6
H) 3.34
(quin, J=6.81 Hz, 1 H) 6.66 (s, 1 H) 7.23 (dd, J=12.31, 4.10 Hz, 2 H) 8.09
(dd, J=8.06, 1.61 Hz,
1 H) 8.71 (dd, J=4.69, 1.47 Hz, 1 H) 11.98 (br. s., 1 H).
2-Chloro-4-isopropyl-1,8-naphthyridine: A solution of 4-isopropyl-1,8-
naphthyridin-2(1H)-one
(800 mg, 4.3 mmol) and 475 L of POC13 in toluene were reacted as outlined in
Scheme 1 to
give the desired 2-chloro-4-isopropyl-1,8-naphthyridine (690 mg, 79%).
Spectroscopic data: 1H
NMR (300 MHz, CDC13) 6 ppm 1.41 (d, J=6.74 Hz, 6 H) 3.68 (dq, J=7.03, 6.84 Hz,
1 H) 7.38
(s, 1 H) 7.52 (dd, J=8.35, 4.25 Hz, 1 H) 8.44 (dd, J=8.50, 2.05 Hz, 1 H) 9.09
(dd, 1 H).
2-Hydrazinyl-4-isopropyl-1,8-naphthyridine: A solution of chloro-4-isopropyl-
1,8-
naphthyridine (690 mg, 3.4 mmol) in NH2NH2 was reacted as outlined in Scheme 1
above to
give the desired title compound (620 mg, 92%). Spectroscopic data: 1H NMR (300
MHz,
CDC13) 6 ppm 1.35 (d, J=6.74 Hz, 6 H) 3.45 - 3.59 (m, 1 H) 6.70 (br. s., 1 H)
7.21 (dd, J=8.06,
4.54 Hz, 1 H) 8.19 (d, J=7.62 Hz, 1 H) 8.79 (br. s., 1 H).
N-(3,5-Dichlorophenyl)-2-(4-isopropyl-1,8-naphthyridin-
2yl)hydrazinecarboxamide: A 100 mg
(0.5 mmol) sample of 2-hydrazinyl-4-isopropyl-1,8-naphthyridine and 94 mg (1.0
eq) of 3,5-
dichlorophenyl isocyanate were reacted as outlined in Scheme 1 above to give
the desired title
compound. Spectroscopic data: 1H NMR (300 MHz, DMSO-d6) 6 ppm 1.23 (d, J=6.74
Hz, 6 H)
3.56 (d, J=6.74 Hz, 1 H) 6.82 (s, 1 H) 7.04 (t, J=1.76 Hz, 1 H) 7.23 (dd,
J=8.20, 4.40 Hz, 1 H)
7.59 (br. s., 2 H) 8.31 (d, J=1.47 Hz, 1 H) 8.54 (br. s., 1 H) 8.66 (d, J=1.17
Hz, 1 H) 8.99 (br. s.,
1 H) 9.25 (br. s., 1 H).
34
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
Example 22
N N' qrCl
NYN
H 0 Cl
Synthesis of N-(3,5-dichlorophenyl)-2-(4-isopropyl-5,8-dimethylquinolin-2-
yl)hydrazinecarboxamide
The title compound was generated from 2,5-dimethylaniline according to the
chemistry
described in the following scheme (Scheme 2). The intermediates were separated
and
characterized.
Scheme 2
OEt
0 0 0 0 H2SO4 POCI3
NHZ 160 C H 100 C N OH toluene N Cl
120 C
NCO
NH2NH2 \ \ CIJ CI /
H N
Y CI
140 C N NHNHZ CH3CN \ N '
II
H
0
Cl
N-(2,5-Dimethylphenyl)-4-methyl-3-oxopentanamide: A mixture of 2,5-
dimethylaniline (7.70 g,
63.54 mmol) and ethyl 4-methyl-3-oxopentanoate (10.00 g, 1.0 eq) were refluxed
at 160 C
overnight. After the reaction mixture was cooled to room temperature, it was
triturated with
hexane. The resulting precipitate was filtered and dried under high vacuum to
yield the desired
N-(2,5-dimethylphenyl)-4-methyl-3-oxopentanamide. Spectroscopic data: 1H NMR
(300 MHz,
CDC13) 6 ppm 1.18 (d, J=7.03 Hz, 6 H) 2.30 (d, J=7.62 Hz, 6 H) 2.66 - 2.85 (m,
1 H) 3.64 (s, 2
H) 6.87 (d, J=8.50 Hz, 1 H) 7.06 (d, J=7.62 Hz, 1 H) 7.77 (s, 1 H) 9.13 (br.
s., 1 H).
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
4-Isopropyl-5, 8-dimethylquinolin-2-ol: A sample of N-(2,5-dimethylphenyl)-4-
methyl-3-
oxopentanamide and 10.0 mL of H2SO4 was reacted at 100 C for 1 hour. After
the reaction was
cooled to room temperature it was quenched into ice-H20. The resulting mixture
was extracted
with CH2C12 (3 x 20 mL). The combined organic extracts were washed with
saturated NaHCO3
(3 x 15 mL), brine (lx 20 mL), dried over MgSO4 and concentrated. The residue
was
recrystallized in EtOAc to give 1.30 g of the desired 4-isopropyl-5,8-
dimethylquinolin-2-ol.
Spectroscopic data: 1H NMR (300 MHz, CDC13) 6 ppm 1.29 (d, J=6.45 Hz, 6 H)
2.41 (s, 3 H)
2.77 (s, 3 H) 3.84 (quin, J=6.59 Hz, 1 H) 6.67 (s, 1 H) 6.93 (d, J=7.62 Hz, 1
H) 7.19 (d, J=7.62
Hz, 1 H) 8.81 (br. s., 1 H).
2-Chloro-4-isopropyl-5,8-dimethylquinoline: A solution of 4-isopropyl-5,8-
dimethylquinolin-2-
ol (1.30 g, 6.05 mmol), 700 L (1.2 eq) of POC13 in toluene were reacted as
outlined in Scheme
1 above to yield 1.00 g of 2-chloro-4-isopropyl-5,8-dimethylquinoline (71%).
Spectroscopic
data: 1H NMR (300 MHz, CDC13) 6 ppm 1.34 (d, J=6.74 Hz, 6 H) 2.71 (s, 3 H)
2.87 (s, 3 H)
4.12 (quin, J=6.74 Hz, 1 H) 7.21 (d, J=7.33 Hz, 1 H) 7.32 (s, 1 H) 7.40 (d,
J=7.33 Hz, 1 H).
2-Hydrazinyl-4-isopropyl-5,8-dimethylquinoline: A solution of 2-chloro-4-
isopropyl-5,8-
dimethylquinoline (1.00 g, 4.3 mmol) in NH2NH2 was reacted as outlined in
Scheme 1 above to
give the title compound. Spectroscopic data: 1H NMR (300 MHz, CDC13) 6 ppm
1.30 (d,
J=6.74 Hz, 6 H) 2.64 (s, 3 H) 2.82 (s, 3 H) 3.95 - 4.10 (m, 1 H) 6.72 (s, 1 H)
6.98 (d, J=7.33 Hz,
1 H) 7.30 (d, J=7.33 Hz, 1 H).
N-(3,5-Dichlorophenyl)-2-(4-isopropyl-5,8-dimethylquinolin-2
yl)hydrazinecarboxamide: A
solution of 2-hydrazinyl-4-isopropyl-5,8-dimethylquinoline (100 mg, 0.44 mmol)
and 82 mg
(1.0 eq) of 3,5-dichlorophenyl isocyanate was reacted as outlined in Scheme 1
above to give the
desired title compound. Spectroscopic data: 1H NMR (300 MHz, DMSO-d6) 6 ppm
1.23 (d,
J=6.74 Hz, 6 H) 2.42 (s, 3 H) 2.74 (s, 3 H) 3.88 - 4.06 (m, 1 H) 6.89 (s, 1 H)
6.94 (d, J=7.33 Hz,
1 H) 7.07 (t, J=1.90 Hz, 1 H) 7.22 (d, J=7.03 Hz, 1 H) 7.65 (br. s., 2 H) 8.27
(br. s., 1 H) 8.58 (s,
1 H) 9.24 (br. s., 1 H).
36
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
Example 23
/ I \
N N.N~N \ CI
H 0 /
CI
Synthesis of N-(3,5-dichlorophenyl)-2-(4-isopropylquinolin-2-
yl)hydrazinecarboxamide
The title compound was generated from aniline according to the chemistry
described in the
Scheme 1 and Scheme 2. The intermediates were separated and characterized.
4-Methyl-3-oxo-N-phenylpentanamide: Aniline (5.80 g, 62.3 mmol) and 10.0 g
(1.0 eq.) of ethyl
isobutyryl acetate were reacted as outlined in Scheme 2 described above to
give 6.70 g (52%) of
the desired title compound. Spectroscopic data: 1H NMR (300 MHz, CDC13) 6 ppm
1.17 (d,
J=7.03 Hz, 6 H) 2.64 - 2.83 (m, 1 H) 3.61 (s, 2 H) 7.11 (t, J=7.33 Hz, 1 H)
7.32 (t, J=7.91 Hz, 2
H) 7.51 - 7.59 (m, 2 H) 9.20 (br. s., 1 H).
4-Isopropylquinolin-2-ol: A solution of 4-methyl-3-oxo-N-phenylpentanamide
(3.70 g, 18.03
mmol) in 15 mL H2SO4 was reacted as outlined in Scheme 2 above to give 1.95 g
(57%) of the
desired title compound. Spectroscopic data: 1H NMR (300 MHz, CDC13) 6 ppm 1.36
(d, J=6.74
Hz, 6 H) 3.45 (quin, J=6.81 Hz, 1 H) 6.67 (s, 1 H) 7.15 - 7.32 (m, 1 H) 7.40 -
7.61 (m, 2 H) 7.79
(d, J=8.50 Hz, 1 H) 12.28 (br. s., 1 H).
2-Chloro-4-isopropylquinoline: 4-Isopropylquinolin-2-ol (1.95 g, 10.43 mmol)
and 1.14 mL
(1.2 eq) of POC13 in toluene were reacted as outlined in Scheme 1 above to
yield 1.8 g (84%) of
the desired title compound. Spectroscopic data: 1H NMR (300 MHz, CDC13) 6 ppm
1.40 (d,
J=6.74 Hz, 6 H) 3.71 (quin, J=6.81 Hz, 1 H) 7.28 (s, 1 H) 7.48 - 7.63 (m, 1 H)
7.65 - 7.78 (m, 1
H)7.98-8.10(m,2H).
2-Hydrazinyl-4-isopropylquinoline: A solution of 2-chloro-4-isopropylquinoline
(1.80 g, 8.8
mmol) in NH2NH2 was reacted as outlined in Scheme 1 above to give the desired
title
compound. Spectroscopic data: 1H NMR (300 MHz, CDC13) 6 ppm 1.41 (d, J=6.74
Hz, 6 H)
3.51-3.76(m,1H)7.20-7.35(m,1H)7.44-7.60 (m, 2 H) 7.61 - 7.79 (m,1H)7.84-8.06
(m, 1 H).
37
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
N-(3,5-Dichlorophenyl)-2-(4-isopropylquinolin-2 yl)hydrazinecarboxamide: A
solution of 2-
hydrazinyl-4-isopropylquinoline (97 mg, 0.48 mmol) and 91 mg (1.0 eq) of 3,5-
dichlorophenyl
isocyanate was reacted as outlined in Scheme 1 above to give the desired title
compound.
Spectroscopic data: 1H NMR (300 MHz, DMSO-d6) 6 ppm 1.29 (d, J=6.74 Hz, 6 H)
3.53 - 3.71
(m, 1 H) 6.83 (s, 1 H) 7.09 (t, J=1.90 Hz, 1 H) 7.28 (td, J=7.47, 1.46 Hz, 1
H) 7.45 - 7.62 (m, 2
H) 7.69 (br. s., 2 H) 7.94 (d, J=7.62 Hz, 1 H) 8.50 (br. s., 1 H) 8.70 (s, 1
H) 9.23 (br. s., 1 H).
Example 24
N N"NYN CI
I N
O
CI
Synthesis of N-(2,6-dichloropyridin-4-yl)-2-(4,8-dimethylquinolin-2-
yl)hydrazinecarboxamide
The title compound was generated from 2-chloro-4,8-dimethylquinoline according
to the
chemistry described in Scheme 1 and Scheme 2. The intermediates were separated
and
characterized.
2-Hydrazinyl-4,8-dimethylquinoline: A solution of 2-chloro-4,8-
dimethylquinoline (1.00 g, 5.2
mmoles) in NH2NH2 was reacted as outlined in Scheme 1 above to give the
desired title
compound which was used in the next step without further purification.
N-(2,6-Dichloropyridin-4yl)-2-(4,8-dimethylquinolin-2yl)hydrazinecarboxamide:
A solution of
2-hydrazinyl-4,8-dimethylquinoline (110 mg, 0.60 mmol) and 111 mg (1.0 eq) of
2,6-dichloro-
4-isocyanatopyridine was reacted as outlined in Scheme 1 above to give the
desired title
compound. Spectroscopic data: 1H NMR (300 MHz, DMSO-d6) 6 ppm 2.50 (s, 3 H)
2.56 (s, 3
H) 6.79 (s, 1 H) 7.18 (t, J=7.62 Hz, 1 H) 7.41 (d, J=7.03 Hz, 1 H) 7.70 (d,
J=8.20 Hz, 2 H) 8.72
(s, 2 H).
38
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
Example 25
N/\ CI
9?N)--- N
H" Y
O
CI
Synthesis of N-(3,5-dichlorophenyl)-2-(4,8-dimethylquinolin-2-
yl)hydrazinecarboxamide
The title compound was generated from 2-hydrazinyl-4,8-dimethylquinoline
according to the
chemistry described in Scheme 1. The intermediate 2-hydrazinyl-4,8-
dimethylquinoline was
prepared according to Scheme 1 and it's characterization was presented earlier
(see above).
N-(3,5-Dichlorophenyl)-2-(4,8-dimethylquinolin-2 yl)hydrazinecarboxamide: A
solution of 2-
hydrazinyl-4,8-dimethylquinoline (100 mg, 0.53 mmol) and 100 mg (1.0 eq) of
1,3-dichloro-5-
isocyanatobenzene was reacted as outlined in Scheme 1 above to give the
desired title
compound. Spectroscopic data: 1H NMR (300 MHz, DMSO-d6) 6 ppm 2.51 (s, 3 H)
2.55 (s, 3
H) 6.77 - 6.80 (m, 1 H) 7.09 (t, J=1.76 Hz, 1 H) 7.17 (dd, J=8.20, 7.03 Hz, 1
H) 7.40 (d, J=6.74
Hz, 1 H) 7.65 - 7.72 (m, 3 H) 8.40 (br. s., 1 H) 8.62 (s, 1 H) 9.25 (br. s., 1
H).
Example 26
/ I \
N NNYN \ CI
H O I ~N
CI
Synthesis of N-(2,6-dichloropyridin-4-yl)-2-(4-methylquinolin-2-
yl)hydrazinecarboxamide
The title compound was generated from commercially available 2-chloro-4-
methylquinoline
according to the chemistry described above in Scheme 1. The intermediate 2-
hydrazinyl-4-
methylquinoline was used in the next step without further purification.
2-Hydrazinyl-4-methylquinoline: A solution of 2-chloro-4-methylquinoline (1.00
g, 5.63 mmol)
in NH2NH2 was reacted as outlined in Scheme 1 above to give the title compound
which was
used in the next step without further purification.
39
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
N-(2,6-Dichloropyridin-4 yl)-2-(4-methylquinolin-2 yl)hydrazinecarboxamide: A
solution of 2-
hydrazinyl-4-methylquinoline (100 mg, 0.58 mmol) and 110 mg (1.0 eq) of 2,6-
dichloro-4-
isocyanatopyridine was reacted as outlined in Scheme 1 above to give the
desired title
compound. Spectroscopic data: 1H NMR (300 MHz, DMSO-d6) 6 ppm 2.57 (s, 3 H)
6.80 (s, 1
H) 7.30 (t, J=7.47 Hz, 1 H) 7.50 - 7.64 (m, 2 H) 7.87 (d, J=8.20 Hz, 1 H) 8.78
(s, 1 H) 8.91 (br.
s., 1 H).
Example 27
/ I \ I'll N N,N N \ CI
H O /
CI
Synthesis of N-(3,5-dichlorophenyl)-2-(4,5,8-trimethylquinolin-2-
yl)hydrazinecarboxamide
The title compound was generated from 2,5-dimethylaniline according to the
chemistry
described in Scheme 1 and Scheme 3 below. The intermediates were separated and
characterized.
Scheme 3
Yy OEt
0 0 0 0 H2SO4 \ POCi3
NH2 180 C, 5 min H 140 C, 5 min I / N OH toluene / N CI
120 C
microwave microwave
NCO
NH2NH2 CI Cl 4N' H H
14 N NHNH2 N,NYN Cl
CH3CN H
/
Cl
N-(2,5-Dimethylphenyl)-3-oxobutanamide: A mixture of 2,5-dimethylaniline (1.00
g, 8.25
mmol) and ethyl 3-oxobutanoate (1.0 mL, 1.0 eq) was microwaved at 180 C for 5
minutes.
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
After the reaction mixture was cooled to room temperature, it was triturated
with hexane. The
resulting precipitate was filtered and dried under high vacuum to yield the
desired title
compound. Spectroscopic data: 1H NMR (300 MHz, CDC13) 6 ppm 2.28 (s, 3 H) 2.32
(s, 3 H)
2.34 (s, 3 H) 3.63 (s, 2 H) 6.88 (d, J=7.62 Hz, 1 H) 7.07 (d, J=7.62 Hz, 1 H)
7.75 (s, 1 H) 9.00 -
9.12 (m, 1 H).
4,5,8-Trimethylquinolin-2-ol: A 5.50 g (26.8 mmol) sample of N-(2,5-
dimethylphenyl)-3-
oxobutanamide in 10.0 mL of H2SO4 was microwaved at 140 C for 5 minutes.
After the
reaction was cooled to room temperature it was quenched into ice-H20. The
resulting mixture
was basified with saturated NaHCO3. The resulting mixture was extracted with
CH2C12 (3 x 20
mL) and the combined organic extracts were washed with H20 (l x 15 mL), brine
(lx 20 mL),
dried over MgSO4 and concentrated to give the desired title compound (1.4 g,
28%).
Spectroscopic data: 1H NMR (300 MHz, CDC13) 6 ppm 2.41 (s, 3 H) 2.69 (d,
J=1.17 Hz, 3 H)
2.73 (s, 3 H) 6.48 (s, 1 H) 6.90 (d, J=7.33 Hz, 1 H) 7.19 (d, J=7.62 Hz, 1 H)
8.87 (br. s., 1 H).
2-Chloro-4,5,8-trimethylquinoline: A solution of 4,5,8-trimethylquinolin-2-ol
(2.40 g, 12.8
mmol) and 1.4 mL (1.2 eq) of POC13 in toluene was reacted as outlined in
Scheme 1 above to
yield the desired title compound (2.2 g, 85%). Spectroscopic data: 1H NMR (300
MHz, CDC13)
6 ppm 2.68 (s, 3 H) 2.82 (s, 3 H) 2.85 (s, 3 H) 7.12 (s, 1 H) 7.18 (d, J=7.33
Hz, 1 H) 7.39 (d, 1
H).
2-Hydrazinyl-4,5,8-trimethylquinoline: A solution of 2-chloro-4,5,8-
trimethylquinoline (2.2 g,
10.7 mmoles) in NH2NH2 was reacted as outlined in Scheme 1 above to give the
desired title
compound (830 mg, 38%). Spectroscopic data: 1H NMR (300 MHz, CDC13) 6 ppm 2.62
(s, 3
H) 2.78 (s, 6 H) 4.13 (br. s., 2 H) 5.74 (br. s., 1 H) 6.52 (s, 1 H) 6.95 (d,
J=7.03 Hz, 1 H) 7.28 (d,
1 H).
N-(3,5-Dichlorophenyl)-2-(4,5,8-trimethylquinolin-2 yl)hydrazinecarboxamide: A
solution of 2-
hydrazinyl-4,5,8-trimethylquinoline (100 mg, 0.50 mmol) and 93.53 mg (1.0 eq)
of 3,5-
dichlorophenyl isocyanate was reacted as outlined in Scheme 1 above to give
the desired title
compound. Spectroscopic data: 1H NMR (300 MHz, DMSO-d6) 6 ppm 2.43 (s, 3 H)
2.73 (s, 3
H) 2.77 (s, 3 H) 6.69 (s, 1 H) 6.92 (d, J=7.03 Hz, 1 H) 7.09 (t, J=1.90 Hz, 1
H) 7.23 (d, J=7.62
Hz, 1 H) 7.68 (br. s., 2 H) 8.35 (s, 1 H) 8.53 (s, 1 H) 9.24 (br. s., 1 H).
41
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
TABLE 2
FLIPR
Object ID EC50 (nM) /(% Antagonism)
S1P1 S1P2 S1P3
1 >8300 47 (96) >8300
2.9 (33P S1 P)
2 >8300 118(91) >8300
23.8 (33P S1P)
3 >8300 8 (99) >8300
5.7 (33P S1 P)
4 >8300 1.5 (33P Si P) >8300
>8300 135(82) >8300
6 >8300 11(95) >8300
2.2 (33P S1 P)
7 >8300 196 (98) >8300
8 >8300 12(99) >8300
6.4 (33P S1 P)
9 >8300 149(52) >8300
26.4 (33P S1P)
>8300 58(98) >8300
9.9 (33P S1 P)
11 >8300 9(98) >8300
5 (33P S1P)
12 >8300 24(99) >8300
2.1 (33P S1 P)
13 >8300 0.5 (33P S1 P) >8300
14 >8300 0.5 (33P S1 P) >8300
>8300 0.2 (33P S1 P) >8300
16 >8300 0.1 (33P S1 P) >8300
17 >8300 0.1 (33P S1 P) >8300
18 >8300 266(33P S1P) >8300
19 >8300 40 (33P S1 P) >8300
42
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
FLIPR
Example No. EC50 (nM) /(% Antagonism)
S1P1 S1P2 S1P3
20 >8300 196 (33P S1 P) >8300
21 >8300 81 (33P S1P) >8300
22 >8300 199 (33P S1 P) >8300
23 >8300 188 (33P S1 P) >8300
24 >8300 390 (33P S1 P) >8300
25 >8300 451 (33P S1 P) >8300
26 >8300 196 (33P S1 P) >8300
27 >8300 386 (33P S1 P) >8300
As can be seen from the above results, the pyrazolopyridinyl compounds of this
invention
are preferred over the benzopyridinyl or pyrido pyridinyl compounds. In
particular, the
pyrazolopyridinyl compounds which comprise substituted aryl which is pyridinyl
are especially
preferred. And finally, the pyrazolopyridinyl compounds which comprise
substituted aryl which
is pyridinyl and wherein the substitution pattern is o-halo, o-alkyloxy, more
preferably o-chloro,
o-ethyyloxy, propyloxy or butyloxy are most preferred.
As a result of the above activity of the compounds utilized in the method of
the present
invention, it is clear that such compounds may be used in treating and/or
preventing the
following diseases and conditions of the eye as well as other diseases and
conditions discussed
below. (It should be noted that "treating" means ameliorating and/or
modulating a disease or
disorder that exists in a subject (whether the subject is aware of the disease
or disorder or not) or
delaying the onset of the disease or disorder and "preventing" means
preventing the recurrence,
onset or development of one or more symptoms of a disease or disorder in a
subject by
administering one or more compounds of the invention.)
Ocular diseases; wet and dry age-related macular degeneration, diabetic
retinopathy,
retinopathy of prematurity, geographic atrophy, glaucomatous optic neuropathy
43
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
Systemic vascular barrier related diseases;, various inflammatory diseases,
including acute
lung injury, its prevention, sepsis, tumor metastasis, atherosclerosis,
pulmonary edemas, and
ventilation-induced lung injury
Allergies and other inflammatory diseases; urticaria, bronchial asthma, and
other airway
inflammations including pulmonary emphysema and chronic obstructive pulmonary
diseases
Cardiac protection; Ischemia/reperfusion injury, atherosclerosis
Anti-fibrosis; ocular, cardiac, hepatic and pulmonary fibrosis, proliferative
vitreoretinopathy,
cicatricial pemphigoid, surgically induced fibrosis in cornea, conjunctiva and
tenon
Pains and anti-inflammation; Acute pain, flare-up of chronic pain, musculo-
skeletal pains,
visceral pain, pain associated with diabetic neuropathy, rheumatoid arthritis,
chronic knee and
joint pain, tendonitis, osteoarthritis, bursitis, neuropathic pains
Wound Healing; scar-free healing of wounds from cosmetic skin surgery and
ocular surgery,
GI surgery, general surgery, various mechanical and heat injuries
See, for example, the following articles:
1. Hla, Timothy "Inhibitor of the Receptor Activity of the S1P2 Receptor for
Inhibiting
Pathological Angiogenesis in the eye." PCT Int. Appl. (2008), 54 pp. WO
2008154470
Al
2. Athanasia Skoura, Teresa Sanchez, Kevin Claffey, Suzanne M. Mandala,
Richard L.
Proia, and Timothy Hla "Essential role of sphingosine 1-phosphate receptor 2
in
pathological angiogenesis of the mouse retina." J Clin Invest. 2007 September
4; 117(9):
2506-2516.
3. Serriere-Lanneau V, Teixeira-Clerc F, Li L, et al. The sphingosine 1-
phosphate receptor
S1P2 triggers hepatic wound healing. FASEB J2007 21:2005-13.
44
CA 02775587 2012-03-26
WO 2011/041287 PCT/US2010/050486
The foregoing description details specific methods and compositions that can
be employed to
practice the present invention, and represents the best mode contemplated.
Thus, however
detailed the foregoing may appear in text, it should not be construed as
limiting the overall scope
hereof, rather, the ambit of the present invention was to be governed only by
the lawful
construction of the appended claims. In particular, the present invention
includes, as preferred
novel compounds, having subtype-selective modulating activity of sphingosine-l-
phosphate-2
(SIP2) receptors, compounds selected from the group consisting of 1-(2-chloro-
6-
loweralkyloxypyridin-4-yl)-3-((7-isopropyl-1,3-dimethyl-lH-pyrazolo [4,3-
b]pyridin-5-
yl)methyl)ureas and 1-(2-chloro-6-loweralkyloxypyridin-4-yl)-3-((7-isopropyl-
l,3-dimethyl-lH-
pyrazolo[4,3-b]pyridin-5-yl)imino)ureas.