Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
WO 2011/067607 PCT/GB2010/052020
TREATMENT OF INFECTIOUS DISEASES
The present invention relates to tonabersat and its analogues and compositions
comprising tonabersat or its analogues for use in the treatment of infectious
disorders
more particularly conditions associated with or resulting from infectious
disease or
infection with an infective agent.
International patent application WO 95/34545 discloses a series of specific
named
compounds, including tonabersat, otherwise known as cis-6-acetyl-4-(S)-(3-
chloro-4-
fluorobenzoylamino)-3,4-dihydro-2,2-dimethyl-2H-1-benzopyran-3-(S)-ol, which
is a
member of the class of drugs called neuronal gap junction blockers, and which
is
currently being investigated for a range of conditions including migraine,
epilepsy,
depression and other neurological conditions.
US Patent No.5948811 (incorporated herein by way of reference) describes a
class of
compounds ('the analogues of formula I') which may be used for the prophylaxis
and
treatment of disorders within the central and peripheral nervous system,
including
migraine, psychosis, epilepsy and other neurological conditions.
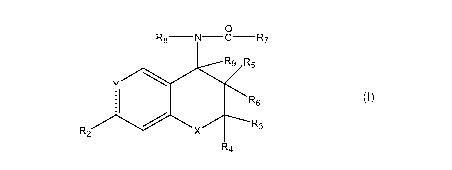
0
Rg N C R7
R9 R5
R6 (I )
R2 X R3
R4
Y is C-R1;
R, is acetyl;
R2 is hydrogen, C3_8 cycloalkyl, C1_6 alkyl optionally interrupted by oxygen
or substituted
by hydroxy, C1_6 alkoxy or substituted aminocarbonyl, C1_6 alkylcarbonyl, C1_6
alkoxycarbonyl, C1_6 alkylcarbonyloxy, C1_6 alkoxy, nitro, cyano, halo,
trifluoromethyl, or
CF3 S; or a group CF3 --A--, where A is --CF2 --,
1
WO 2011/067607 PCT/GB2010/052020
--CO--, --CH2 --, CH(OH), SO2, SO, CH2 --0, or CONH; or a group CF2 H--A'--
where A'
is oxygen, sulphur, SO, SO2, CF2 or CFH; trifluoromethoxy, C1_6
alkylsulphinyl, perfluoro
C2.6 alkylsulphonyl, C1_6 alkylsulphonyl, C1_6 alkoxysulphinyl, C1_6
alkoxysulphonyl, aryl,
heteroaryl, arylcarbonyl, heteroarylcarbonyl, phosphono, arylcarbonyloxy,
heteroarylcarbonyloxy, arylsulphinyl, heteroarylsulphinyl, arylsulphonyl, or
heteroarylsulphonyl in which any aromatic moiety is optionally substituted,
C1_6
alkylcarbonylamino, C1_6 alkoxycarbonylamino, C1_6 alkyl-thiocarbonyl, C1_6
alkoxy-
thiocarbonyl, C1_6 alkyl-thiocarbonyloxy, 1-mercapto C2_7 alkyl, formyl, or
aminosulphinyl,
aminosulphonyl or aminocarbonyl, in which any amino moiety is optionally
substituted
by one or two C1_6 alkyl groups, or C1_6 alkylsulphinylamino, C1_6
alkylsulphonylamino, C,_
6 alkoxysulphinylamino or C1_6 alkoxysulphonylamino, or ethylenyl terminally
substituted
by C1_6 alkylcarbonyl, nitro or cyano, or --C(C1_6 alkyl)NOH or --C(C1_6
alkyl)NNH2 ; or
amino optionally substituted by one or two C1_6 alkyl or by C2_7 alkanoyl; one
of R3 and
R4 is hydrogen or C1_4 alkyl and the other is C1_4 alkyl, CF3 or CH2 Xa is
fluoro, chloro,
bromo, iodo, Ci14 alkoxy, hydroxy, C1_4 alkylcarbonyloxy, --S--C1.4 alkyl,
nitro, amino
optionally substituted by one or two C1_4 alkyl groups, cyano or C1_4
alkoxycarbonyl; or
R3 and R4 together are C2.5 polymethylene optionally substituted by C1.4
alkyl;
R5 is C1.6 alkylcarbonyloxy, benzoyloxy, ON02, benzyloxy, phenyloxy or C1.6
alkoxy and
R6 and R9 are hydrogen or R5 is hydroxy and R6 is hydrogen or C1_2 alkyl and
R9 is
hydrogen;
R7 is heteroaryl or phenyl, both of which are optionally substituted one or
more times
independently with a group or atom selected from chloro, fluoro, bromo, iodo,
nitro,
amino optionally substituted once or twice by C1_4 alkyl, cyano, azido, C1.4
alkoxy,
trifluoromethoxy and trifluoromethyl;
R8 is hydrogen, C1.6 alkyl, OR,, or NHCOR10 wherein R11 is hydrogen, C1.6
alkyl, formyl,
C1_6 alkanoyl, aroyl or aryl-C1.6 alkyl and R10 is hydrogen, C1_6 alkyl, C1_6
alkoxy, mono or
di C<sub>1-6</sub> alkyl amino, amino, amino-C<sub>1-6</sub> alkyl, hydroxy-C1.6 alkyl,
halo-C1.6
alkyl, C1_6 acyloxy-C1.6 alkyl, C1_6 alkoxycarbonyl-C1.6 -alkyl, aryl or
heteroaryl; the R8 --
N--CO--R7 group being cis to the R5 group;
and X is oxygen or NR12 where R12 is hydrogen or C1_6 alkyl.
2
WO 2011/067607 PCT/GB2010/052020
Infectious diseases are clinically evident diseases resulting from the
presence of
pathogenic agents, including pathogenic viruses, pathogenic bacteria, fungi,
protozoa,
multicellular parasites and aberrant proteins known as prions. A prion is an
infectious
agent which transmits a mis-folded protein. Prion associated diseases include
Creutzfeldt-Jakob disease (CJD), Bovine Spongiform Encephalopathy (BSE)
Scrapie
Disorder, Gerstmann-Straussler-Scheinker syndrome (GSS), Fatal familial
insomnia
(sF1) and Kuru. Collectively these diseases are known as Transmissible
Spongiform
Encephalophathies (TSEs). An infectious disease is not synonymous with an
infection,
and may not cause important clinical symptoms or impair host function.
However,
infectious diseases show a diverse range of symptoms which vary depending on
the
particular infective agent and result in a wide variety of disorders that may
result directly
or indirectly from the presence of, or damage caused by an infectious agent.
An infectious disorder, as opposed to an infectious disease, is a disorder
resulting
directly from the presence of an infectious agent such as prion protein which
can lead to
CJD, or Human Immunodeficiency Virus which can result in the development of
HIV
neuralgia, or Streptococcus pneumoniae which can result in Meningitis, or is a
disorder
resulting indirectly from a current or previous infection or infectious
disease, or is a
disorder which results from damage caused by an infectious agent such as
varicella
zoster virus which can result in the development of postherpetic neruralgia.
Infectious
disorders are often a result of an inflammatory response elicited by the
presence of an
infectious agent.
There are many treatments available for infectious diseases including anti-
viral, anti-
bacterial, anti-fungal treatments. These treatments attack the infectious
agent and in
doing so eventually and hopefully eradicate or control the infectious disease.
However
there exists a critical need for safe, alternative and improved methods and
compositions
for the treatment of infectious disorders.
In a first aspect, the present invention provides for tonabersat, or an
analogue of
formula I,
3
WO 2011/067607 PCT/GB2010/052020
0
Rg N C R7
R9 R5
R6 (I )
R2 X R3
R4
Y is C-R1;
R1 is acetyl;
R2 is hydrogen, C3_8 cycloalkyl, C1.6 alkyl optionally interrupted by oxygen
or substituted
by hydroxy, C1.6 alkoxy or substituted aminocarbonyl, C1.6 alkylcarbonyl, C1.6
alkoxycarbonyl, C1.6 alkylcarbonyloxy, C1.6 alkoxy, nitro, cyano, halo,
trifluoromethyl, or
CF3 S; or a group CF3 --A--, where A is --CF2 --, --CO--, --CH2 --, CH(OH),
SO2, SO, CH2
--0, or CONH; or a group CF2 H--A'-- where A' is oxygen, sulphur, SO, SO2, CF2
or
CFH; trifluoromethoxy, C1.6 alkylsulphinyl, perfluoro C2.6 alkylsulphonyl,
C1.6
alkylsulphonyl, C1.6 alkoxysulphinyl, C1.6 alkoxysulphonyl, aryl, heteroaryl,
arylcarbonyl,
heteroarylcarbonyl, phosphono, arylcarbonyloxy, heteroarylcarbonyloxy,
arylsulphinyl,
heteroarylsulphinyl, arylsulphonyl, or heteroarylsulphonyl in which any
aromatic moiety
is optionally substituted, C1.6 alkylcarbonylamino, C1.6 alkoxycarbonylamino,
C1.6 alkyl-
thiocarbonyl, C1.6 alkoxy-thiocarbonyl, C1.6 alkyl-thiocarbonyloxy, 1-mercapto
C2_7 alkyl,
formyl, or aminosulphinyl, aminosulphonyl or aminocarbonyl, in which any amino
moiety
is optionally substituted by one or two C1.6 alkyl groups, or C1.6
alkylsulphinylamino, C1.6
alkylsulphonylamino, C1.6 alkoxysulphinylamino or C1.6 alkoxysulphonylamino,
or
ethylenyl terminally substituted by C1.6 alkylcarbonyl, nitro or cyano, or --
C(C1.6
alkyl)NOH or --C(C1.6 alkyl)NNH2 ; or amino optionally substituted by one or
two C1.6
alkyl or by C2_7 alkanoyl; one of R3 and R4 is hydrogen or C1.4 alkyl and the
other is C1.4
alkyl, CF3 or CH2 Xa is fluoro, chloro, bromo, iodo, C1.4 alkoxy, hydroxy, C1-
4
alkylcarbonyloxy, --S--C1.4 alkyl, nitro, amino optionally substituted by one
or two C1.4
alkyl groups, cyano or C1.4 alkoxycarbonyl; or R3 and R4 together are C2.5
polymethylene
optionally substituted by C1.4 alkyl;
R5 is C1.6 alkylcarbonyloxy, benzoyloxy, ON02, benzyloxy, phenyloxy or C1.6
alkoxy and
4
WO 2011/067607 PCT/GB2010/052020
R6 and R9 are hydrogen or R5 is hydroxy and R6 is hydrogen or C1_2 alkyl and
R9 is
hydrogen;
R7 is heteroaryl or phenyl, both of which are optionally substituted one or
more times
independently with a group or atom selected from chloro, fluoro, bromo, iodo,
nitro,
amino optionally substituted once or twice by C1_4 alkyl, cyano, azido, C1.4
alkoxy,
trifluoromethoxy and trifluoromethyl;
R8 is hydrogen, C1.6 alkyl, OR,, or NHCOR10 wherein R11 is hydrogen, C1.6
alkyl, formyl,
C1_6 alkanoyl, aroyl or aryl-C1.6 alkyl and R10 is hydrogen, C1_6 alkyl, C1_6
alkoxy, mono or
di C<sub>1-6</sub> alkyl amino, amino, amino-C<sub>1-6</sub> alkyl, hydroxy-C1.6 alkyl,
halo-C1.6
alkyl, C1_6 acyloxy-C1.6 alkyl, C1_6 alkoxycarbonyl-C1.6 -alkyl, aryl or
heteroaryl; the R8 --
N--CO--R7 group being cis to the R5 group;
and X is oxygen or NR12 where R12 is hydrogen or C1_6 alkyl or a
pharmaceutically
acceptable composition thereof, for use in the treatment of infectious
disorders.
A preferred analogue of formula 1 is the compound carabersat or (trans-(+)-6-
acetyl-4-
(S)-(4-fluorobenzoylamino)-3,4-dihydro-2,2-dimethyl-2H-l-benzo[b]pyran-3R
ol,hemihydrate.
For therapeutic administration according to the present invention, tonabersat
or an
analogue of formula I, is most preferably employed in the form of its free
base, but may
also be used in the form of a pharmaceutically acceptable salt, preferably the
hydrochloride salt. Alternative salts with pharmaceutically acceptable acids
may also be
utilised in prophylactic and/or therapeutic administration, for example salts
derived from
acids including, but not limited to, hydrobromic acid, phosphoric acid, acetic
acid,
fumaric acid, maleic acid, salicylic acid, citric acid, oxalic acid, lactic
acid, malic acid,
methanesulphonic acid and p-toluene sulphonic acid.
All references to tonabersat or an analogue of formula I, herein includes all
pharmaceutically acceptable salts, and all solvates thereof.
Also included within the scope of the present invention are polymorphs,
solvates and
radiolabelled derivatives of tonabersat or an analogue of formula I, and
pharmaceutically acceptable compositions thereof. References to tonabersat or
an
analogue of formula I, include such polymorphs, solvates and radiolabelled
derivatives
thereof.
WO 2011/067607 PCT/GB2010/052020
For prophylactic and/or therapeutic administration according to the invention,
tonabersat or an analogue of formula I, may be administered in pure form, but
will
preferably be formulated into any suitable pharmaceutically acceptable and
effective
composition which provides effective levels of the active ingredient in the
body.
Accordingly, the present invention provides a method for the treatment of
infectious
disorders, comprising administering to a patient in need thereof a
pharmaceutically
effective amount of tonabersat or an analogue of formula I, or a
pharmaceutically
acceptable composition thereof.
The present invention also provides for tonabersat, or an analogue of formula
I, or a
pharmaceutically acceptable composition thereof, for use in the manufacture of
a
medicament for the treatment of infectious disorders.
By infectious disorders we mean:
i) disorders resulting directly from the presence of pathogenic microbial
agents, such
as viruses, bacteria, fungi, protozoa or prions. Such disorders include
bacterial
disorders such as Meningitis from Streptococcus pneumoniae. Such disorders
also
include prion related disorders such as CJD, BSE, Scrapie Disorder, Gerstmann-
Straussler-Scheinker syndrome (GSS), Fatal familial insomnia (sF1) and Kuru.
Collectively these diseases are known as Transmissible Spongiform
Encephalophaties (TSEs).
ii) disorders resulting indirectly from a current or previous infection or
infectious
disease, such as chronic fatigue syndrome, peptic ulcer.
iii) disorders resulting from damage caused by an infectious agent such as one
of the
herpes viruses including varicella zoster virus, herpes simplex, Epstein-Barr
virus
and cytomegalovirus which can result in the development of postherpetic
neruralgia, HIV which causes extensive damage to the nervous system and
results
in a severe form of neuropathic pain called HIV neuralgia. Diphtheria and
leprosy
are bacterial diseases characterized by extensive peripheral nerve damage, and
Lyme disease which can cause a wide range of neuropathic disorders.
All treatments may be acute or prophylactic. For acute treatment, a rapid
onset of
action is preferred, and therefore, drugs that reach maximum plasma
concentrations
shortly after administration would be most beneficial. Accordingly,
compositions
6
WO 2011/067607 PCT/GB2010/052020
providing rapid drug-release and/or dissolution are preferred.
Tonabersat or an analogue of formula I, may be delivered alone, but will
generally be
delivered in the form of a pharmaceutically acceptable composition thereof,
which
comprises tonabersat or an analogue of formula I, and one or more
pharmaceutically
acceptable diluents or carriers selected with regard to the intended route of
administration.
Treatment with tonabersat or an analogue of formula I, or a pharmaceutically
acceptable
composition thereof, may be conducted at a unit dose of between 1 to 1000 mg,
suitably
1 to 500 mg, for example an amount in the range of from 2 to 400 mg such as 2,
5, 10,
20, 30, 40, 50, 80, 100, 200, 300 and 400 mg of the active compound.
Unit doses will normally be administered once or more than once per day, for
example
1, 2, 3, 4, 5 or 6 times a day, more usually 1 to 4 times a day, such that the
total daily
dose is normally in the range, for a 70 kg adult of 1 to 1000 mg, for example
1 to 500
mg, that is in the range of approximately 0.01 to 15 mg/kg/day, more usually
0.1 to 6
mg/kg/day, for example 1 to 6 mg/kg/day.
Preferably, the tonabersat or an analogue of formula I, or a pharmaceutically
acceptable
salt thereof, is administered to the patient at dose ranges of approximately
0.01 to 15
mg/kg/day, more usually 0.1 to 6 mg/kg/day, for example 1 to 6 mg/kg/day.
It is preferred that tonabersat or an analogue of formula I, is administered
in the form of
a pharmaceutical composition, such as a composition for oral, including sub-
lingual,
intranasal, rectal, topical, parenteral (especially intravenous), ocular or
aural
administration.
Pharmaceutical compositions suitable for the delivery of tonabersat or an
analogue of
formula I, and methods for their preparation will be readily apparent to those
skilled in
the art. Such compositions and methods for their preparation may be found, for
example, in Remington's Pharmaceutical Sciences, 19th Edition (Mack Publishing
Company, 1995).
Compositions suitable for oral administration include solid formulations such
as tablets,
capsules containing particulates, liquids, or powders, lozenges (including
liquid-filled),
chews, multi- and nano-particulates, gels, solid solution, liposome, films,
ovules, sprays
and liquid formulations. Liquid formulations include suspensions, solutions,
syrups and
7
WO 2011/067607 PCT/GB2010/052020
elixirs. Liquid formulations may also be prepared by the reconstitution of a
solid, for
example, from a sachet.
Orally administrable compositions may be in the form of oral solid
compositions, such as
tablets, capsules, pastilles, pellets, pills, lozenges powders and granules.
The
composition may be in solid form which melts on contact with the tongue of the
patient,
for example in the form of disintegrating tablets sold under the trade name
ZYDIS .
Shaped oral compositions are preferred, since they are more convenient for
general
use.
Solid forms for oral administration are usually presented in a unit dose, and
contain
conventional additives such as adjuvants, binding agents, diluents,
disintegrants,
dispersing agents, excipients, fillers, tabletting agents, lubricants,
colorants, flavourings,
desiccants, humectants, and wetting agents.
Pills, pellets and tablets may be coated according to well known methods in
the art.
Oral solid formulations also include conventional sustained release
formulations, such
as tablets or granules having an enteric coating.
Suitable fillers include cellulose, mannitol, lactose and other similar
agents. Suitable
disintegrants include starch, polyvinylpyrrolidone and starch derivatives such
as sodium
starch glycollate. Suitable lubricants include, for example, magnesium
stearate. Suitable
pharmaceutically acceptable wetting agents include sodium lauryl sulphate.
Solid oral compositions are prepared by admixture, and may be prepared by
conventional methods of blending, filling, tabletting or the like. Repeated
blending
operations may be used to distribute the active agent throughout those
compositions
employing large quantities of fillers. Such operations are, of course,
conventional in the
art.
They may also be in the form of oral fluid preparations, including liquid
preparations,
such as aqueous or oily blends, mixtures, suspensions, solutions, emulsions,
syrups,
tinctures and elixirs, and gel preparations.
They may also be presented as dilutable fluid concentrates or dry product
reconstitutable powders for dilution or reconstitution with water or other
suitable vehicle
before use.
8
WO 2011/067607 PCT/GB2010/052020
Oral fluid preparations, including gels and liquid preparations may contain
conventional
additives such as: suspending agents, for example sorbitol, syrup, methyl
cellulose,
gelatin, hydroxyethylcelIulose, carboxymethyl cellulose, aluminium stearate
gel or
hydrogenated edible fats; emulsifying agents, for example lecithin, sorbitan
monooleate,
or acacia; non-aqueous vehicles (which may include edible oils), for example,
almond
oil, fractionated coconut oil, oily esters such as esters of glycerine,
propylene glycol, or
ethyl alcohol; preservatives, for example methyl or propyl p-hydroxybenzoate
or sorbic
acid; and, if desired, conventional flavouring or colouring agents.
Compositions for oral administration may be formulated to be immediate and/or
modified release. Modified release formulations include delayed-, sustained-,
pulsed-,
controlled-, targeted and programmed release.
Compositions suitable for parenteral administration include injectable and
infusible
aqueous or oily blends, mixtures, suspensions, solutions, emulsions and low-
viscosity
gel preparations. Compositions for parenteral administration may be formulated
to be
immediate and/or modified release. Modified release formulations include
delayed-,
sustained-, pulsed-, controlled-, targeted and programmed release.
The parenteral compositions for use in the invention may be prepared as long
acting
depot preparations. Such formulations may be administered by intramuscular
injection.
Thus, for example, the composition of the invention may be formulated with
suitable
polymeric or hydrophobic materials (for example as an emulsion in a
pharmaceutically
acceptable oil) or ion exchange resins, or as sparingly soluble derivatives,
for example,
as a sparingly soluble salt.
Advantageously, adjuvants such as a local anaesthetic, preservatives and
buffering
agents are also dissolved, emulsified or suspended in the vehicle.
Such compositions are prepared by admixture of the compound and a solvent or
vehicle. The compound, depending on the vehicle and the concentration, can be
emulsified, suspended or dissolved. Parenteral compositions are normally
prepared by
with the compound and a vehicle which is sterile, and/or the composition is
sterilised,
before filling into a suitable vial or ampoule and sealing.
To enhance the stability, the composition may also be presented as a dry
product
9
WO 2011/067607 PCT/GB2010/052020
reconstitutable powder for reconstitution with water or other suitable vehicle
before use.
A fluid composition can be frozen after filling into the vial and freeze-dried
under
vacuum.
Tonabersat or an analogue of formula I, may also be administered topically to
the skin
or mucosa, that is, dermally or transdermally. Typical formulations for this
purpose
include gels, hydrogels, lotions, solutions, creams, ointments, dusting
powders,
dressings, foams, films, skin patches, wafers, implants, sponges, fibres,
bandages and
microemulsions. Liposomes may also be used. Typical carriers include alcohol,
water,
mineral oil, liquid petrolatum, white petrolatum, glycerin, polyethylene
glycol and
propylene glycol. Penetration enhancers may be incorporated - see, for
example, J
Pharm Sci, 88 (10), 955-958, by Finnin and Morgan (October 1999).
Other means of topical administration include delivery by electroporation,
iontophoresis,
phonophoresis, sonophoresis and microneedle or needle-free (e.g. PowderjectTM
BiojectTM, etc.) injection.
Formulations for topical administration may be formulated to be immediate
and/or
modified release. Modified release formulations include delayed-, sustained-,
pulsed-,
controlled-, targeted and programmed release.
Compositions for use in surgical wounds may be prepared as long acting depot
preparations. Such formulations may be administered by implantation (for
example
subcutaneously or intramuscularly) or by intramuscular injection. Thus, for
example, the
composition of the invention may be formulated with suitable polymeric or
hydrophobic
materials (for example as an emulsion in an acceptable oil) or ion exchange
resins, or
as sparingly soluble derivatives, for example, as a sparingly soluble salt.
It is more preferred that tonabersat or an analogue of formula (I) is
administered in the
form of a unit-dose composition for administration into a temporary or
permanent human
or other animal bodily orifice, such as the trachea, nostril, nasal passage,
rectum, udder
duct, urethra or vagina, or a surgical would, e.g. an incision, or any device
inserted in
such a temporary or permanent orifice, such as a catheter, trochar, cannula,
endotracheal or other endoscopic tube or an ostomy tube, e.g. a tracheostomy
or
colostomy tube. Intranasal administration is greatly preferred
Intranasally mucosal administrable compositions may be in the form of
intranasal
mucosal solid compositions, such as powders and granules. They may also be in
the
WO 2011/067607 PCT/GB2010/052020
form of intranasal mucosal fluid preparations, including liquid preparations,
such as
aqueous or oily blends, mixtures, suspensions, solutions, emulsions and
elixirs, and gel
preparations. They may also be presented as dilutable fluid concentrates or
dry product
reconstitutable powders for dilution or reconstitution with water or other
suitable vehicle
before use.
Tonabersat or an analogue of formula I, may be administered intranasally or by
inhalation, typically in the form of a dry powder from a dry powder inhaler
(either alone,
as a mixture, for example, in a dry blend with lactose, or as a mixed
component particle,
for example, mixed with phospholipids, such as phosphatidylcholine) from a dry
powder
inhaler or as an aerosol spray from a pressurised container, pump, spray,
atomiser
(preferably an atomiser using electrohydrodynamics to produce a fine mist), or
nebuliser, with or without the use of a suitable propellant, such as 1,1,1,2-
tetrafluoroethane or 1,1,1,2,3,3,3-heptafluoropropane. For intranasal use, the
powder
may comprise a bioadhesive agent, for example, chitosan or cyclodextrin.
Solid forms for intranasal mucosal administration are usually presented in a
unit dose,
and contain conventional additives, such as adjuvants, diluents, dispersing
agents,
excipients, colorants, desiccants, humectants, and wetting agents.
Powders and granules may be coated according to well known methods in the art.
Intranasal mucosal solid formulations also include conventional sustained
release
formulations, such as powders or granules having a resistant coating.
Suitable excipients include cellulose, mannitol, lactose, chitosan, pectin,
cellulose
derivatives such as hydroxypropylmethylcelIulose, methyl cellulose,
hydroxyethylcelIulose, carboxymethyl cellulose, polyoxamers, such as
poly(ethylene
oxides), gelatin, polyvinylpyrrolidone and starch. Suitable pharmaceutically
acceptable
wetting agents include sodium lauryl sulphate.
Solid intranasal mucosal compositions are prepared by admixture, and may be
prepared
by conventional methods of blending or the like. Repeated blending operations
may be
used to distribute the active agent throughout those compositions employing
large
quantities of excipients. Such operations are, of course, conventional in the
art.
Intranasal mucosal fluid preparations, including gels and liquid preparations
may contain
conventional additives such as suspending agents, for example sorbitol, methyl
11
WO 2011/067607 PCT/GB2010/052020
cellulose, gelatin, hydroxyethylcelIulose, carboxymethyl cellulose, aluminium
stearate
gel or hydrogenated edible fats, emulsifying agents, for example lecithin,
sorbitan
monooleate, or acacia; non-aqueous vehicles (which may include edible oils),
for
example, almond oil, fractionated coconut oil, oily esters such as esters of
glycerine,
propylene glycol, or ethyl alcohol; preservatives, for example methyl or
propyl p-
hydroxybenzoate or sorbic acid, and if desired conventional colouring agents.
The pressurised container, pump, spray, atomizer, or nebuliser contains a
solution or
suspension of the compound(s) of the invention comprising, for example,
ethanol,
aqueous ethanol, or a suitable alternative agent for dispersing, solubilising,
or extending
release of the active, a propellant(s) as solvent and an optional surfactant,
such as
sorbitan trioleate, oleic acid, or an oligolactic acid.
Prior to use in a dry powder or suspension formulation, the drug product is
micronised to
a size suitable for delivery by inhalation (typically less than 5 microns).
This may be
achieved by any appropriate comminuting method, such as spiral jet milling,
fluid bed jet
milling, supercritical fluid processing to form nanoparticles, high pressure
homogenisation, or spray drying.
Capsules (made, for example, from gelatin or hydroxypropylmethylcellulose),
blisters
and cartridges for use in an inhaler or insufflator may be formulated to
contain a powder
mix of the compound of the invention, a suitable powder base such as lactose
or starch
and a performance modifier such as I-leucine, mannitol, or magnesium stearate.
The
lactose may be anhydrous or in the form of the monohydrate, preferably the
latter. Other
suitable excipients include dextran, glucose, maltose, sorbitol, xylitol,
fructose, sucrose
and trehalose.
A suitable solution formulation for use in an atomiser using
electrohydrodynamics to
produce a fine mist may contain from 1 pg to 20mg of the compound of the
invention per
actuation and the actuation volume may vary from 1 pI to 100pl. A typical
formulation
may comprise an analogue of formula I, propylene glycol, sterile water,
ethanol and
sodium chloride. Alternative solvents which may be used instead of propylene
glycol
include glycerol and polyethylene glycol.
Suitable flavours, such as menthol and levomenthol, or sweeteners, such as
saccharin
or saccharin sodium, may be added to those formulations of the invention
intended for
inhaled/intranasal administration.
12
WO 2011/067607 PCT/GB2010/052020
Tonabersat or an analogue of formula I, may also be administered rectally or
vaginally,
for example, in the form of a suppository, pessary, or enema.
Tonabersat or an analogue of formula I, may also be administered directly to
the eye or
ear, typically in the form of drops of a micronised suspension or solution in
isotonic, pH-
adjusted, sterile saline. Other formulations suitable for ocular and aural
administration
include ointments, biodegradable (e.g. absorbable gel sponges, collagen) and
non-
biodegradable (e.g. silicone) implants, wafers, lenses and particulate or
vesicular
systems, such as niosomes or liposomes. A polymer such as crossed-linked
polyacrylic acid, polyvinylalcohol, hyaluronic acid, a cellulosic polymer, for
example,
hydroxypropylmethylcelIulose, hydroxyethylcelIulose, or methyl cellulose, or a
heteropolysaccharide polymer, for example, gelan gum, may be incorporated
together
with a preservative, such as benzalkonium chloride. Such formulations may also
be
delivered by iontophoresis.
The compositions of tonabersat or an analogue of formula I, may also be in the
form of
fast-dispersing dosage forms such as those described in Expert Opinion in
Therapeutic
Patents, 11 (6), 981-986, by Liang and Chen (2001) and Verma RK et.al. Current
Status
of Drug Delivery Technologies and Future Directions, Pharmaceutical Technology
On-
Line, 2001, 25(2), 1-14. Such dosage forms are also known as oral fast-
dissolving,
rapid-dissolve, rapid-melt, mouth-dissolving and fast-disintegrating tablet.
The
composition may be in solid form which melts on contact with the tongue of the
patient,
for example in the form of disintegrating tablets sold under the trade name
ZYDIS (RP
Scherer, UK). Alternatively, the composition may be in the form of the EFVDAS
(effervescent drug absorption system, Elan Corporation), Fast Melt (highly
porous
microfine matrix tablet, Elan Corporation), Flashdose (floss matrix utilising
shearform
technology, (Fuisz Technologies, USA), Flashtab (orodispersible
multiparticulate tablet,
Prographarm, France), Multiflash (fast disintegrating multi-unit,
multiparticulate tablet,
Prographarm), Orasolv (effervescent dispersed microcapsule tablet, Cima Labs
Inc,
USA), Wowtab tablets (Yamanouchi Pharma Technologies, USA), LYOC (freeze dried
fast dispersing tablets, Farmalyoc, France) or Quicksolve (freeze dried fast
dispersing
tablets, Janssen Pharamceutica, USA).
Other suitable formulation technologies may include INDAS (insoluble drug
absorption
system, Elan Corporation), which utilises a stabilised amorphous form of the
drug with
enhanced solubility, NanoCrystal technology (Elan Corporation), which utilises
nanoparticles of the drug, typically having a particle size of less than 400nm
in diameter,
or SoftGel (RP Scherer), which utilises a soft gelatin capsule formulation.
13
WO 2011/067607 PCT/GB2010/052020
The formulation technologies described herein may advantageously provide more
rapid
drug dissolution and absorption. For those compositions that disintegrate in
the oral
cavity, such as beneath the tongue, the rate of absorption may be increased
and first-
pass metabolism effects reduced.
As is common practice, the compositions will usually be accompanied by written
or
printed directions for use in the medical treatment concerned.
The compositions for use in the invention may contain from 0.1% to 99% by
weight,
preferably from 1% - 60% by weight, of the active material, depending on the
method of
administration.
The following are given by way of example only to illustrate and aid
understanding of the
invention:
Studies with tonabersat have employed a number of different formulations
including:
- Direct compression tablets 0.05, 1.0, 10 and 25 mg with tablet core weight
250 mg
- Direct compression tablets 15, 25, 40 and 80 mg with tablet core weight 400
mg
- Direct compression tablets 20 mg with core weight 400 mg
- Nanoparticulate tablets 10, 20 and 40 mg with tablet core weight 400 mg
The direct compression tablets utilise micronized drug substance whilst the
nanoparticulate tablets were direct compression tablets utilising wet bed
milled spray
dried nanoparticulate drug substance. Clinical trials have been conducted
utilising 10,
20, 30, 40, 60 and 80 mg round white uncoated direct compression tablets with
a core
weight of 400mg with the following unit composition (20 mg tablet only
presented; all
other strengths differ only in tonabersat and lactose content):
A representative formulation suitable for use in the present invention is
detailed in
Table 1.
14
WO 2011/067607 PCT/GB2010/052020
Ingredient 20mg Tablet
Quantity (mg)
Tonabersat 20.0
Lactose 330.0
Microcrystalline Cellulose 20.0
Sodium Starch Glycollate, 24.0
Type A
Colloidal Silicon Dioxide 2.0
Magnesium Stearate 4.0
Total Weight 400.0
Table 1
Experimental Methods
Example 1: Investigation of prion neuroinflammation in mice
Aim
To investigate the effect of tonabersat on two models of prion
neuropathogenesis: prion
infected mice using mock-inoculated mice as controls; and Pneumococcus induced
brain inflammation, as a model of inflammation.
Materials and Method
1. C57b16N mice are used as a model of scrapie and BSE strain prion
replication
(intracerebral injection of 106 LD50 prion infectivity). They are compared to
mock-
inoculated similar mice. Acute and chronic injection of Tonabersat is done, at
doses
ranging from 0.5mg/kg to 10 and 20 mg/kg (in saline or phosphate buffers
solution,
with i.p. or p.o. administration). The group is composed of 10 animals for EEG
analysis and 10 animals for behavioural test and mortality. Analyses include,
during
the incubation period (at the first third, the second third and the last
third):
(a) EEG relative power analysis (EEG spectral powers were calculated as the
ratio of
the spectral power obtained per minute in the treatment recording session over
the
spectral power obtained in the vehicle recording treatment), comparing
infected and
non-infected mice. ANOVA procedure determines the significance of the results.
These tests also include the comparison with drugs that modify the EEG signals
or
behaviour of the rodents (imipramine, venlafine, diazepam, amphetamine,
fluoxetine, caffeine, chlorpromazine, buspirone, at pharmacological doses,
i.e.
WO 2011/067607 PCT/GB2010/052020
generally from 1 to 32 mg/kg, in NaCL or PBS), with or without gap junction
modulators (Tonabersat). That helps evaluate the level of relation between gap
junctions and specific pathological neurotransmitter systems, most of which
are
damaged during the infection.
(b) Behavioural tests: forced swim (i.e. Porsolt test or behavioutal despair),
Open Field,
Activity Meter, Tail Suspension Test, Dark Light Box. Tests are also made on
mice
injected with drugs that induce behaviour changes. T test helps determine the
significance of the results.
(c) Mortality and morbidity are evaluated and significance is evaluated using
Kaplan-
Meier estimator.
(d) Histological analyses are made to evaluate the localisation of
inflammation (using
GFAP and Vimentin labellings) and connexins.
2. Meningitis (and therefore neuroinflammation) is triggered in mice using a
pathogenic bacteria, Streptococcus pneumoniae, at 105 CFU (Colony Forming
Unit), in the right cerebral hemisphere. Similar parameters as those
concerning
prion infected mice will be evaluated and compared to antibiotic-treated
infected
mice (the antibiotic will be chosen among ceftriaxone, vancomycin, cefotaxim,
cefdinir at pharmacological doses.
3. Mouse glial and neuronal cell lines infectable by scrapie strain prion
(PrPres
Biochemical quantification) are used for prion susceptibility investigations.
Effects of
Tonabersat given before or after infection (22L prion strain 103 104, 105
LD50, in
triplicate in six well plate format) are investigated. PrPres will be
quantified after 3
passages. The toxicity of Tonabersat in these cellular models is investigated
prior to
study initiation (a dose-ranging study from 1 nM to 100 mM will be done at
this time).
Other gap junction inhibitor will be tested as well (meclofenamic acid,
glycyrrhetinic
acid, fluflenamic acid, after dose-rangind studies).
16