Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
= CA 02818071 2014-02-26
Modified cyclodextrin ring compounds having exactly two hydroxyl
moieties substituted with an amino acid for therapeutics delivery
This application is a divisional application of Canadian Patent Application
2,497,792 filed on September 4, 2003.
Background of the Invention
=
5 Drug delivery of some small molecule therapeutic agents, sucn as
,camptothecin, has been problematic due to their poor pharmacological
profiles.
These therapeutic agents often have low aqueous solubility, their bioactive
forms
exist in equilibrium with an inactive form, or high systemic concentrations of
the
agents lead to toxic side-effects. Some approaches to circumvent the problem
of
10 their delivery have been to conjugate the agent directly to a water-
soluble polymer
such as hydroxypropyl methacrylate (1-IPMA), polyethyleneglycol, and poly-L-
. glutarnic acid. In some cases, such conjugates have been successful in
solubilizing
or stabilizing the bioactive forin of the therapeutic agent, or achieving a
sustained
release formulation which circumvents complications associated with high
systemic
concentrations of the agent.
Another approach to the drug delivery problem has been to form host/guest
. inclusion complexes between the therapeutic agent and cyclodextrins or
derivatives
thereof. Cyclodextrins (a, p, y) and their oxidized forms have unique physico-
chemical properties such as good water solubility, low toxicity and low immune
20 response. To date, most of the drug delivery studies with cyclodextrins
have
focused on their ability to form supra-molecular complexes, wherein
cyclodextrins
form host/guest inclusion complexes with therapeutic molecules and thus alter
the
physical, chemical, and/or biological properties of these guest molecules.
U.S. Patent 5,276,088 describes a method for synthesizing cyclodextrin-
25 containing polymers by either reacting polyvinyl alcohol or cellulose or
derivatives
thereof with cyclodextrin derivatives, or by copolymerization of a
cyclodextrin
derivative with vinyl acetate or methyl methacrylate.
U.S. Patent No. 5,855,900 describes a biodegradable cyclodextrin-
containing polymer. The patent discloses a supramolecular-structured
biodegradable
30 polymeric assembly comprising a Plurality of drug-modified a, p, y-
cyclodextrins
CA 02818071 2013-06-06
and a linear polymeric chain threading through the structural cavity of the
cyclodextrins.
There is an ongoing need for new approaches to the delivery of small
therapeutic agents that have poor pharmacological profiles such as
camptothecin,
paclitaxel, doxorubicin, and cyclosporine A.
Summary of the Invention
The present invention relates to novel compositions of polymer conjugates,
defined as polymeric materials covalently coupled to therapeutic/bioactive
agents, as
carriers for therapeutics delivery. In one aspect, the present invention
provides
water-soluble, biocompatible polymer conjugates comprising a water-soluble,
biocompatible polymer covalently attached to bioactive moieties through
attachments that are cleaved under biological conditions to-release the
bioactive
moieties. In certain such embodiments, the polymer comprises cyclic moieties
alternating with linker moieties that connect the cyclic structures, e.g.,
into linear or
branched polymers, preferably linear polymers. The polymer may be a
polycation,
polyanion, or non-ionic polymer. The bioactive agent, which may be a
therapeutic
agent, a diagnostic agent, or an adjuvant, preferably makes up at least 5%,
10%,
15%, 20%, 25%, 30%, or even 35% by weight of the conjugate. In certain
embodiments, the rate of drug release is dependent primarily upon the rate of
hydrolysis. In certain other embodiments, the rate of drug release is
dependent
primarily on enzymatic cleavage.
The present invention provides cyclodextrin-containing polymeric
compounds for use in drug delivery of these therapeutic agents. The invention
also
provides compounds for use in controlled drug delivery which are capable of
releasing a therapeutic agent in a targeted, predictable, and controlled rate.
Accordingly, one aspect of the present invention is a polymer conjugate
comprising cyclodextrin moieties, a therapeutic agent, and an optional ligand
targeting agent. The polymer may be linear or branched, and may be formed via
2
CA 02818071 2013-06-06
polycondensation of cyclodextrin-containing monomers, copolymerization between
one or more cyclodextrin-containing monomers and one or more comonomers which
do not contain cyclodextrin moieties. Furthermore, the present invention also
contemplates cyclodextrin-containing polymers formed by grafting cyclodextrin
moieties to an already formed polymer. The cyclodextrin moieties contemplated
by
the present invention include, but are not limited to, a, i3, and y
cyclodextrins and
oxidized forms thereof. Depending on the drug/polymer ratio desired, the
therapeutic agent may be attached to a monomer via an optional linker prior to
the
polymerization step, or may be subsequently grafted onto the polymer via an
optional linker. Likewise, the targeting ligand may be attached to a monomer
via an
optional linker prior the polymerization step, or may be subsequently grafted
onto
the polymer via an optional linker,-or may be attached to the polymer as an
inclusion
complex or host-guest interactions.
To illustrate further, one embodiment of the invention is a polymeric
compound represented by Formula 1:
( CD)
I m
L1
) =
al b
L3
T) v
_ W (I)
wherein
P represents a linear or branched polymer chain;
CD represents a cyclic moiety such as a cyclodextrin moiety;
1
CA 02818071 2013-06-06
LI, L2 and L3, independently for each occurrence, may be absent or represent
a linker group; f .
D, independently for each occurrence, represents a therapeutic agent or a
prodrug thereof;
T, independently for each occurrence, represents a targeting ligand or
precursor thereof;
a, m, and v, independently for each occurrence, represent integers in the
range of 1 to 10 (preferably 1 to 8, 1 to 5, or even 1 to 3);
b represents an integer in the range of 1 to about 30,000 (preferably <25,000,
<20,000, <15,000, <10,000, <5,000, <1,000, <500, <100, <50, <25, <10, or even
<5); and
n and w, independently for each occurrence, represents an integer in the
range of 0 to about 30,000 (preferably <25,000, <20,000, <15,000, <10,000,
<5,000,
<1,000, <500, <100, <50, <25, <10, or even <5),
wherein either the polymer chain comprises cyclodextrin moieties or n is at
least I. = .
Another embodiment of the present invention is a compound represented by
Formula II:
_
r _
1 T I
I m =
L7
- i ¨13
=
_________________ L6 P ___________ L8
I[ 1
L10 - - L9
1 I
(CD)in_ (D ) m
n
=
o()
wherein
4
CA 02818071 2013-06-06
P represents a monomer unit of a polymer;
T, independently for each occurrence, represents a targeting ligand or a
precursor thereof;
L6, L7, L8, L9, and L10, independently for each occurrence, may be absent or
represent a linker group;
CD, independently for each occurrence, represents a cyclic moiety such as a
cyclodextrin moiety or a derivative thereof;
D, independently for each occurrence, represents a therapeutic agent or a
prodrug form thereof;
in, independently for each occurrence, represents an integer in the range of 1
to 10 (preferably 1 to 8, I to 5, or even Ito 3);
o represents an integer in the range of 1 to about 30,000 (preferably <25,000,
<20,000, <15,000, <10,000, <5,000, <1,000, <500, <100, <50, <25, <10, or even
<5); and
p, n, and q, independently for each occurrence, represent an integer in the
range of 0 to 10 (preferably 0 to 8, 0 to 5, 0 to 3, or even 0 to about 2),
wherein CD and D are preferably each present at at least 1 location .
(preferably at least 5, 10, 25, 50 or even >100 locations) in the compound.
Another embodiment of the present invention is a compound represented by
Formula III:
T
1 Y
L5
- - Z
____________________ L4 __
CDL7 ______________________________________________________
=
Df (
g L6 -z
( EY) f
- g
5
CA 02818071 2013-06-06
wherein
CD represents a cyclic moiety such as a cyclodextrin moiety, or derivative
thereof;
L4, L5, L6, and L7, independently for each occurrence, may be absent or
represent a linker group;
D and D', independently for each occurrence, represent the same or different
therapeutic agent or prodrugs thereof;
T and T', independently for each occurrence, represent the same or different
targeting ligand or precursor thereof;
f and y, independently for each occurrence, represent an integer in the range
of 1 and 10 (preferably 1 to 8, Ito 5, or even 1 to 3);
g and z, independently for each occurrence, represent an integer in the range
of 0 and 10 (preferably 0 to 8, 0 to 5, 0 to 3, or even 0 to about 2); and
= h represents an integer in the range of.1 and 30,000 (preferably <25,000,
<20,000, <15,000, <10,000, <5,000, <1,000, <500, <100, <50, <25, <10, or even
<5),
wherein at least one occurrence (and preferably at least 5, 10, or even at
least
20, 50, or >100 occurrences) of g represents an integer greater than 0.
Another aspect of the present invention is a method for preparing the
therapeutic cyclodextrin-containing polymeric conjugates described herein.
Another aspect of the present invention is a pharmaceutical composition
comprising a compound or polymer as discussed above.
Another aspect of the present invention is a pharmaceutical dosage form
comprising a polymeric conjugate as described herein.
Another aspect of the present invention is a method for treating a subject
comprising administering a therapeutically effective amount of any of the
polymeric
conjugates described herein.
6
CA 02818071 2013-06-06
Another aspect of the present invention is a method of conducting a
pharmaceutical business comprising manufacturing, licensing, or distributing
kits
containing or relating to any of the polymeric conjugates described herein.
In certain embodiments, these therapeutic polymer conjugates improve drug
stability and/or solubility of the therapeutic agent when used in vivo.
Furthermore,
by selecting from a variety of linker groups, the polymer conjugates present
methods
for controlled release of the therapeutic and/or bioactive agents, or improve
the in
vivo safety and/or therapeutic efficacy of the therapeutic/bioactive agent. In
certain
embodiments, the polymer conjugates are bioerodable or biodegradable.
Brief Description of the Figures
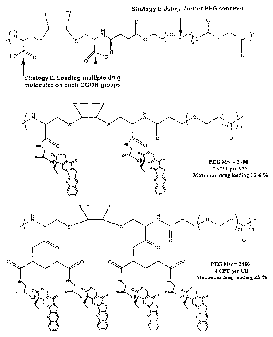
Figure 1 shows strategies for varying polymer conjugates to tune their
characteristics.
Figure 2 demonstrates the effect of peptide tether length on drug release rate
for drug-loaded CD polymer.
I 5 Figure 3 presents the effect that tethering camptothecin has on
enhancing
camptothecin stability, e.g., inhibiting lactone ring-opening.
Figure 4 shows lactone ring opening studies in pH 7.4 KR2PO4 buffer.
Figure 5a and 5b show polymerization control by adjusting polymerization
time.
Figures 6 illustrates CPT release from 11G6 and HGGG6 at 37 C after 24 h
in buffer solutions with pHs ranging from 1.1 to 13.1.
Figure 7 Displays HPLC analysis of degradation of CD-BisCys-SS-Peg3400
Polymer
Figure 8 Shows the tumor growth curve as a function of time for the D5 W,
CPT, irinotecan, LGGGIO at its highest non-toxic dose tested (18 mg CPT/kg),
and
the other three conjugates with high MW polymer (HGGG6, HG6, HGGG10) at
their MTDs in xenograft mice.
Figure 9 presents the median tumor growth curves for HGGG6, HG6 and
HGGG10 in xenograft mice.
Figurel0 presents the medium tumor growth curves for LGGG10 and
HGGG10 each dosed at 9 mg CPT/kg inxenograft mice.
7
CA 02818071 2013-06-06
Figure 11 presents the mean body weight (MBW) losses as a function of
time plotted for D5W, CPT, irinotecan and the three conjugates containing high
MW
polymer at their MTDs in xenograft mice.
Figure 12 shows the correlation of CPT concentration (ng/mg tissue) to
tumor size (in mg) in xenograft mice.
Detailed Description of the Invention
I Overview
The present invention relates to novel compositions of therapeutic
cyclodextrin-containing polymeric compounds designed for drug delivery of
=
therapeutic agents. In certain embodiments, these cyclodextrin-containing
polymers
improve drug stability and/or solubility, and/or reduce toxicity, and/or
improve
efficacy of the small molecule therapeutic when used in vivo. In certain
embodiments, the polymers can be used for delivery of therapeutics such as
camptothecin, taxol, doxorubicin, and arnphotericin. Furthermore, by selecting
from
a variety of linker groups, and/or targeting ligands, the rate of drug release
from the
polymers can be attenuated for controlled delivery. The invention also relates
to
methods of treating subjects with the therapeutic compositions described
herein.
The invention further relates to methods for conducting a pharmaceutical
business
comprising manufacturing, licensing, or distributing kits containing or
relating to the
polymeric compounds described herein.
More generally, the present invention provides water-soluble, biocompatible
polymer conjugates comprising a water-soluble, biocompatible polymer
covalently
attached to bioactive moieties through attachments that are cleaved under
biological
conditions to release the bioactive moieties. In certain such embodiments, the
polymer comprises cyclic moieties alternating with linker moieties that
connect the
cyclic structures, e.g., into linear or branched polymers, preferably linear
polymers.
The cyclic moieties may be any suitable cyclic structures, such as
cyclodextrins,
crown ethers (e.g., 18-crown-6, 15-crown-5, 12-crown-4, etc.), cyclic
oligopeptides
(e.g., comprising from 5 to 10 amino acid residues), cryptands or cryptates
(e.g.,
cryptand [2.2.2], cryptand-2,1,1, and complexes thereof), calixarenes, or
cavitancls,
8
CA 02818071 2013-06-06
= or any combination thereof. Preferably, the cyclic structure is (or is
modified to be)
water-soluble. In certain embodiments, e.g., where a linear polymer is
desired, the
cyclic structure is selected such that under polymerization conditions,
exactly two
moieties of each cyclic structure are reactive with the linker moieties, such
that the
resulting polymer comprises (or consists essentially of) an alternating series
of
cyclic moieties and linker moieties, such as at least four of each type of
moiety.
Suitable difunctionalized cyclic moieties include many that are commercially
available and/or amenable to preparation using published protocols. In certain
embodiments, conjugates are soluble in water to a concentration of at least
0.1 g/mL,
preferably at least 0.25 g/mL.
The polymer may be a polycation, polyanion, or non-ionic polymer. A
polycationic or polyanionic polymer has at least one site that bears a
positive or
negative charge, respectively. In certain such embodiments, at least one of
the linker
moiety and the cyclic moiety comprises such a charged site, so that every
occurrence
of that moiety includes a charged site.
The bioactive agent, which may be a therapeutic agent, a diagnostic agent, or
an adjuvant (such as a radiosensitizer, or a compound that lacks significant
activity
administered alone but that potentiates the activity of another therapeutic
agent),
preferably makes up at least 5%, 10%, 15%, 20%, 25%, 30%, or even 35% by
weight of the conjugate. In preferred embodiments, administration of the
polymer to
a patient results in release of the bioactive agent over a period of at least
6 hours,
preferably at least 12 or 18 hours. For example, the agent may be released
over a
period of time ranging from 6 hours to a month, 6 hours to two weeks, 6 hours
to 3
days, etc. In certain embodiments, the rate of drug release is dependent
primarily
upon the rate of hydrolysis (as opposed to enzymatic cleavage), e.g., the rate
of
release changes by less than a factor of 5, preferably less than a factor of
2, in the
presence of hydrolytic enzymes. In other embodiments, the rate of drug release
may
be dependent primarily on the rate of enzymatic cleavage.
Polymeric conjugates of the present invention may be useful to improve
solubility and/or stability of a bioactive/therapeutic agent, reduce drug-drug
interactions, reduce interactions with blood elements including plasma
proteins,
9
CA 02818071 2013-06-06
reduce or eliminate immunogenicity, protect the agent from metabolism,
modulate
drug-release kinetics, improve circulation time, improve drug half-life (e.g.,
in the
serum, or in selected tissues, such as tumors), attenuate toxicity, improve
efficacy,
normalize drug metabolism across subjects of different species, ethnicities,
and/or
races, and/or provide for targeted delivery into specific cells or tissues.
Poorly
soluble and/or toxic compounds may benefit particularly from incorporation
into
polymeric compounds of the invention.
II. Definitions
(a) General Terms
An 'adjuvant', as the term is used herein, is a compound that has little or no
therapeutic value on its own, but increases the effectiveness of a therapeutic
agent.
Exemplary adjuvants include radiosensitizers, transfection-enhancing agents
(such
as chloroquine and analogs thereof), ehemotactic agents and chemoattractants,
peptides that modulate cell adhesion and/or cell mobility, cell permeabilizing
agents,
inhibitors of multidrug resistance and/or efflux pumps, etc.
The term "agonist", as used herein, is meant to refer to an agent that mimics
.
or up-regulates (e.g., potentiates or supplements) the bioactivity of a
protein of
interest, or an agent that facilitates or promotes (e.g., potentiates or
supplements) an
interaction among polypeptides or between a polypeptide and another molecule
(e.g., a steroid, hormone, nucleic acids, small molecules etc.). An agonist
can be a
wild-type protein or derivative thereof having at least one bioactivity of the
wild-
type protein. An agonist can also be a small molecule that up-regulates the
expression of a gene or which increases at least one bioactivity of a protein.
An
agonist can also be a protein or small molecule which increases the
interaction of a
polypeptide of interest with another molecule, e.g., a target peptide or
nucleic acid.
"Antagonist" as used herein is meant to refer to an agent that down-regulates
(e.g., suppresses or inhibits) the bioactivity of a protein of interest, or an
agent that
inhibits/suppresses or reduces (e.g., destabilizes or decreases) interaction
among
polypeptides or other molecules (e.g., steroids, hormones, nucleic acids,
etc.). An
antagonist can also be a compound that down-regulates the expression of a gene
of
CA 02818071 2013-06-06
interest or which reduces the amount of the wild-type protein present. An
antagonist
can also be a protein or small molecule which decreases or inhibits the
interaction of
a polypeptide of interest with another molecule, e.g., a target peptide or
nucleic acid.
The terms "biocompatible polymer" and "biocompatibility" when used in
relation to polymers are art-recognized. For example, biocompatible polymers
include polymers that are neither themselves toxic to the host (e.g., an
animal or
human), nor degrade (if the polymer degrades) at a rate that produces
monomeric or
oligorneric subunits or other byproducts at toxic concentrations in the host.
In certain
embodiments of the present invention, biodegradation generally involves
degradation of the polymer in an organism, e.g., into its monomeric subunits,
which
may be known to be effectively non-toxic. Intermediate oligomeric products
resulting from such degradation may have different toxicological properties,
however, or biodegradation may involve oxidation or other biochemical
reactions
that generate molecules other than monomeric subunits of the polymer.
Consequently, in certain embodiments, toxicology of a biodegradable polymer
intended for in vivo use, such as implantation or injection into a patient,
may be
=
determined after one or more toxicity analyses. It is not necessary that any
subject
composition have a purity of 100% to be deemed biocompatible. Hence, a subject
composition may comprise 99%, 98%, 97%, 96%, 95%, 90% 85%, 80%, 75% or
even less of biocompatible polymers, e.g., including polymers and other
materials
and excipients described herein, and still be biocompatible.
To determine whether a polymer or other material is biocompatible, it may
be necessary to conduct a toxicity analysis. Such assays are well known in the
art.
One example of such an assay may be performed with live carcinoma cells, such
as
GT3TKB tumor cells, in the following manner: the sample is degraded in 1 M
NaOH at 37 C until complete degradation is observed: The solution is then
neutralized with I M HC1. About 200 p.L of various concentrations of the
degraded
sample products are placed in 96-well tissue culture plates and seeded with
human
gastric carcinoma cells (GT3TKI3) at 104/well density. The degraded sample
products are incubated with the GT3TKB cells for 48 hours. The results of the
assay
may be plotted as % relative growth vs. concentration of degraded sample in
the
11
CA 02818071 2013-06-06
tissue-culture well. In addition, polymers and formulations of the present
invention
may also be evaluated by well-known in vivo tests, such as subcutaneous
implantations in rats to confirm that they do not cause significant levels of
irritation
or inflammation at the subcutaneous implantation sites.
The term "biodegradable" is art-recognized, and includes polymers,
compositions and formulations, such as those described herein, that are
intended to
degrade during use. Biodegradable polymers typically differ from non-
biodegradable polymers in that the former may be degraded during use. In
certain
embodiments, such use involves in vivo use, such as in vivo therapy, and in
other
certain embodiments, such use involves in vitro use. In general, degradation =
attributable to biodegradability involves the degradation of a biodegradable
polymer
into its component subunits, or digestion, e.g., by a biochemical process, of
the
polymer into smaller, non-polymeric subunits. In certain embodiments, two
different
types of biodegradation may generally be identified. For example, one type of
biodegradation may involve cleavage of bonds (whether covalent or otherwise)
in
the polymer backbone. In such biodegradation, monomers and oligomers typically
result, and even more typically, such biodegradation occurs by cleavage of a
bond
connecting one or more of subunits of a polymer. In contrast, another type of
biodegradation may involve cleavage of a bond (whether covalent or otherwise)
internal to sidechain or that connects a side chain to the polymer backbone.
For
example, a therapeutic agent or other chemical moiety attached as a side chain
to the
polymer backbone may be released by biodegradation. In certain embodiments,
one
or the other or both general types of biodegradation may occur during use of a
polymer.
As used herein, the term "biodegradation" encompasses both general types of
biodegradation. The degradation rate of a biodegradable polymer often depends
in
part on a variety of factors, including the chemical identity of the linkage
responsible for any degradation, the molecular weight, crystallinity,
biostability, and
degree of cross-linking of such polymer, the physical characteristics (e.g.,
shape and
size) of an implant, and the mode and location of administration. For example,
the
greater the molecular weight, the higher the degree of crystallinity, and/or
the
12
CA 02818071 2013-06-06
greater the biostability, the biodegradation of any biodegradable polymer is
usually
slower. The term "biodegradable" is intended to cover materials and processes
also
termed "bioerodible".
In certain embodiments wherein the biodegradable polymer also has a
therapeutic agent or other material associated with it, the biodegradation
rate of such
polymer may be characterized by a release rate of such materials. In such
circumstances, the biodegradation rate may depend on not only the chemical
identity
and physical characteristics of the polymer, but also on the identity of
material(s)
incorporated therein. Degradation of the subject compositions includes not
only the
cleavage of intrarnolecular bonds, e.g., by oxidation and/or hydrolysis, but
also the
disruption of intermolecular bonds, such as dissociation of host/guest
complexes by
competitive complex formation with foreign inclusion hosts.
In certain embodiments, polymeric formulations of the present invention
biodegrade within a period that is acceptable in the desired application. In
certain
embodiments, such as in vivo therapy, such degradation occurs in a period
usually
less than about five years, one year, six months, three months, one month,
fifteen
days, five days, three days, or even one day on exposure to a physiological
solution
with a pH between 6 and 8 having a temperature of between 25 and 37 C. In
other
embodiments, the polymer degrades in a period of between about one hour and
several weeks, depending on the desired application.
As used herein the term "bioerodable" refers to polymers which deliver -
sustained effective amounts of therapeutic agent to target tissue over desired
extended periods of time. Thus, a polymer according to the invention in the
biological environment of host tissue and the like, in one aspect, is
subjected to
hydrolytic enzymes and oxidative species under, and in proportion to, the
host's
inflammatory response. This results in release of the therapeutic agent via
the
breaking of the covalent linked bonds. Thus, in certain embodiments, the
materials
of the invention utilize the mammal's own wound-healing repair process in
being
degraded thereby, as hereinbefore described.
13
CA 02818071 2013-06-06
The biodegradable polymers polylactic acid, polyglycolic acid, and
polylactic-glycolic acid copolymer (PLGA), have been investigated extensively
for
nanoparticle formulation. These polymers are polyesters that, upon
implantation in
the body, undergo simple hydrolysis. The products of such hydrolysis are
=
biologically compatible and metabolizable moieties (e.g., lactic acid and
glycolic
acid), which are eventually removed from the body by the citric acid cycle.
Polymer
biodegradation products are formed at a very slow rate, and hence do not
affect
normal cell function. Several implant studies with these polymers have proven
safe
in drug delivery applications, used in the form of matrices, microspheres,
bone
implant materials, surgical sutures, and also in contraceptive applications
for long-
term effects. These polymers are also used as graft materials for artificial
organs,
and recently as basement membranes in tissue engineering investigations.
Nature
Med. 824-826 (1996). Thus, these polymers have been time-tested in various
applications and proven safe for human use. Most importantly, these polymers
are
FDA-approved for human use.
When polymers are used for delivery of pharmacologically active agents in
vivo, it is essential that the polymers themselves be nontoxic and that they
degrade
into non-toxic degradation products as the polymer is eroded by the body
fluids.
Many synthetic biodegradable polymers, however, yield oligom.ers and monomers
upon erosion in vivo that adversely interact with the surrounding tissue. D.
F.
Williams, J. Mater. Sci. 1233 (1982). To minimize the toxicity of the intact
polymer
carrier and its degradation products, polymers have been designed based on
naturally occurring metabolites. Probably the most extensively studied
examples of
such polymers are the polyesters derived from lactic or glycolic acid and
polyamides
derived from amino acids.
A number of bioerodable ot biodegradable polymers are known and used for
controlled release of pharmaceuticals. Such polymers are described in, for
example,
U.S. Pat. No. 4,291,013; U.S. Pat. No. 4,347,234; U.S. Pat. No. 4,525,495;
U.S. Pat No. 4,570,629; U.S. Pat. No. 4,572,832; U.S. Pat. No. 4,587,268;
'U.S. Pat. No. 4,638,045; U.S. Pat. No. 4,675,381; U.S. Pat. No. 4,745,160;
and
U.S. Pat. No. 5,219,980.
14
CA 02818071 2013-06-06
A biohydrolyzable bond (e.g., ester, amide, carbonate, carbamates, or imide)
refers to a bond that is cleaved (e.g., an ester is cleaved to form a hydroxyl
and a
carboxylic acid) under physiological conditions. Physiological conditions
include
the acidic and basic environments of the digestive tract (e.g., stomach,
intestines,
etc.), acidic environment of a tumor, enzymatic cleavage, metabolism, and
other
biological processes, and preferably refer to physiological conditions in a
vertebrate,
such as a mammal.
As used herein the terms "comonomer A precursor", "linker", "linker
group", and "linker moiety" refer to any straight chain or branched, symmetric
or
asymmetric compound which upon reaction with a cyclodextrin monomer precursor
or other suitable cyclic moiety links two such moieties together. In certain
embodiments, a comonomer A precursor is a compound containing at least two
functional groups through which reaction and thus linkage of the cyclodextrin
monomers can be achieved. Examples of functional groups, which may be the same
or different, terminal or internal, of each comonomer A precursor include, but
are
not limited, to amino, acid, imidazole, hydroxyl, thin, acyl halide, or
groups and derivatives thereof. In preferred embodiments, the two functional
groups
are the same and are located at termini of the coinonomer. In certain
embodiments,
a comonomer A precursor contains one or more pendant groups with at least one
functional group through which reaction and thus linkage of therapeutic agent
or
targeting ligand can be achieved, or branched polymerization can be achieved.
Examples of functional groups, which may be the same or different, terminal or
internal, of each comonomer A precursor pendant group include, but are not
limited,
to amino, acid, imidazole, hydroxyl, thiol, acyl halide, ethylene, and ethyne
groups
and derivatives thereof. In certain embodiments, the pendant group is a
(un)substituted branched, cyclic or straight chain Cl-Cl 0 (preferably C I-C6)
alkyl,
or arylalkyl optionally containing one or more heteroatoms, e.g., N, 0, S,
within the
chain or ring.
Upon copolymerization of a comonomer A precursor with a cyclodextrin
monomer precursor, two cyclodextrin monomers may be linked together by joining
the primary hydroxyl side of one cyclodextrin monomer with the primary
hydroxyl
CA 02818071 2013-06-06
side of another cyclodextrin monomer, by joining the secondary hydroxyl side
of
one cyclodextrin Monomer with the secondary hydroxyl side of another
cyclodextrin
monomer, or by joining the primary hydroxyl side of one cyclodextrin monomer
with the secondary hydroxyl side of another cyclodextrin monomer. Accordingly,
combinations of such linkages may exist in the final copolymer. Both the
comonomer A precursor and the comonomer A of the final copolymer may be
neutral, cationic (e.g., by containing protonated groups such as, for example,
quaternary ammonium groups), or anionic (e.g., by containing deprotonated
groups,
such as, for example, sulfate, phosphate, borinate or carboxylate). The charge
of
comonomer A of the copolymer may be adjusted by 'adjusting pH conditions.
Examples of suitable comonomer A precursors include, but are not limited to
succinimide (e.g., dithiobis(succinimidyl propionate) DSP, and dissucinimidyl
suberate (DSS)), glutamates, and aspartates).
The cyclodextrin-containing polymers of the present invention may be linear,
branched or grafted. As used herein, the term "linear cyclodextrin-containing
polymer" refers to a polymer comprising (a, or y) cyclodextrin molecules, or
derivatives thereof which are inserted within a polymer chain. As used herein,
the
term "grafted cyclodextrin-containing polymer" refers to a polymer comprising
(a, p, or y) cyclodextrin molecules, or derivatives thereof which are pendant
off of
the polymer chain. The term "graft polymer" as used .herein refers to a
polymer
molecule which has additional moieties attached as pendent groups along a
polymer
backbone. The term "graft polymerization" denotes a polymerization in which a
side chain is grafted onto a polymer chain, which side chain consists of one
or
several other monomers. The properties of the graft copolymer obtained such
as, for
example, solubility, melting point, water absorption, wettability, mechanical
properties, adsorption behavior, etc., deviate more or less sharply from those
of the
initial polymer as a function of the type and amount of the grafted monomers.
The
term "grafting ratio", as used herein, means the weight percent of the amount
of the
monomers grafted based on the weight of the polymer. As used herein, a
branched
cyclodextrin-containing polymer refers to a polymer backbone with a plurality
of
branch points, wherein each branch point is a starting point of yet another
strand of
the polymer backbone, and each section of polymer backbone may have a
plurality
16
CA 02818071 2013-06-06
of (a, (3, or y) cyclodextrin molecules, or derivatives thereof, inserted into
or grafted
onto the chain.
The term "cyclodextrin moiety" refers to (a, f3, or y) cyclodextrin molecules
or derivatives thereof, which may be in their oxidized or reduced forms.
Cyclodextrin moieties may comprise optional linkers. Optional therapeutic
agents
and/or targeting ligands may be further linked to these moieties via an
optional
linker. The linkage may be covalent (optionally via biohydrolyzable bonds,
e.g.,
esters, amides, carbamates, and carbonates) or may be a host-guest complex
between
the cyclodextrin derivative and the therapeuticagent and/or targeting ligand
or the
oPtional linkers of each. Cyclodextrin moieties may further include one or
more
carbohydrate moieties, preferably simple carbohydrate moieties such as
galactose,
attached to the cyclic core, either directly (i.e., via a carbohydrate
linkage) or
through a linker group.
The term "ED50" means the dose of a drug that produces 50% of its
maximum response or effect.
An 'effective amount' of a subject compound, with respect to the subject
method of treatment, refers to an amount of the therapeutic in a preparation
which,
when applied as part of a desired dosage regimen provides a benefit according
to
clinically acceptable standards for the treatment or prophylaxis of a
particular
disorder.
The term "healthcare providers" refers to individuals or organizations that
provide healthcare services to a person, community, etc. Examples of
"healthcare
providers" include doctors, hospitals, continuing care retirement communities,
skilled nursing facilities, subacute care facilities, clinics, multispecialty
clinics,
freestanding ambulatory centers, home health agencies, and HMO's.
"Instruction(s)" as used herein means documents describing relevant
materials or methodologies pertaining to a kit. These materials may include
any
combination of the following: background information, list of components and
their
availability information (purchase information, etc.), brief or detailed
protocols for
17
CA 02818071 2013-06-06
using the kit, trouble-shooting, references, technical support, and any other
related
documents. Instructions can be supplied with the kit or as a separate member
component, either as a paper form or an electronic form which may be supplied
on
computer readable memory device or downloaded from an internet website, or as
recorded presentation. Instructions can comprise one or multiple documents,
and are
meant to include future updates.
"Kit" as used herein means a collection of at least two components
constituting the kit. Together, the components constitute a functional unit
for a
given purpose. Individual member components may be physically packaged
together or separately. For example, a kit comprising an instruction for using
the kit
may or may not physically include the instruction with other individual member
components. Instead, the instruction can be supplied as a separate member
component, either in a paper form Or an electronic form which may be supplied
on
computer readable memory device or downloaded. from an internet webs ite, or
as
recorded presentation.
The term "LD50" means the dose of a drug that is lethal in 50% of test
subjects.
A "patient" or "subject" to be treated by the subject method can mean either
a human or non-human subject.
The "polymerizations" of the present invention include radical, anionic, and
cationic mechanisms, as well as reactions of bifunctional molecules (analogous
to
the formation of nylon, e.g., reacting molecules each of which bears two or
more
different reactive moieties that react with each other (but, preferably, are
disfavored
from reacting intramolecularly by steric, conformational, or other
constraints), or
reacting two or more different compounds, each compound bearing two or more
reactive moieties that react only with reactive moieties of different
compounds (i.e.,
intermolecularly)), as well as metal-catalyzed polymerizations such as olefin
metathesis, and other polymerization reactions known to those of skill in the
art.
18
CA 02818071 2013-06-06
The term "prophylactic or therapeutic" treatment is art-recognized and
includes administration to the host of one or more of the subject
compositions. If it
is administered prior to clinical manifestation of the unwanted condition
(e.g.,
disease or other unwanted state of the host animal) then the treatment is
prophylactic, i.e., it protects the host against developing the unwanted
condition,
whereas if it is administered after manifestation of the unwanted condition,
the
treatment is therapeutic, (i.e., it is intended to diminish, ameliorate, or
stabilize the
existing unwanted condition or side effects thereof).
The term "preventing" is art-recognized, and when used in relation to a
condition, such as a local recurrence (e.g., pain), a disease such as cancer,
a
syndrome complex such as heart failure or any other medical condition, is well
understood in the art, and includes administration of a composition which
reduces
the frequency of, or delays the onset of, symptoms of a medical condition in a
subject relative to a subject which does not receive the composition. Thus,
prevention of cancer includes, for example, reducing the number of detectable
cancerous growths in a population of patients receiving a prophylactic
treatment
relative to an untreated control population, and/or delaying the appearance of
detectable cancerous growths in a treated population versus an untreated
control
population, e.g., by a statistically and/or clinically significant amount.
Prevention of
an infection includes, for example, reducing the number of diagnoses of the
infection in a treated population versus an untreated control population,
and/or
delaying the onset of symptoms of the infection in a treated population versus
an
untreated control population. Prevention of pain includes, for example,
reducing the
frequency of, or alternatively delaying, pain sensations experienced by
subjects in a
treated population versus an untreated control population.
As used herein, the terms "therapeutic agent" include any synthetic or
naturally occurring biologically active compound or composition of matter
which,
when administered to an organism (human or nonhuman animal), induces a desired
pharmacologic, immunogenic, and/or physiologic effect by local and/or systemic
action. The term therefore encomPasses those compounds or chemicals
traditionally
regarded as drugs, vaccines, and biopharmaceuticals including molecules such
as
19
CA 02818071 2013-06-06
proteins, peptides, hormones, nucleic acids, gene constructs and the like.
More
particularly, the term "therapeutic agent" includes compounds or Compositions
for
use in all of the major therapeutic areas including, but not limited to,
adjuvants; anti-
infectives such as antibiotics and antiviral agents; analgesics and analgesic
combinations, anorexics, anti-inflammatory agents, anti-epileptics, local and
general
. anesthetics, hypnotics, sedatives, antipsychotic agents, neuroleptic agents,
antidepressants, anxiolytics, antagonists, neuron blocking agents,
anticholinergic and
cholinomimetic agents, antimuscarinic and muscarinic agents, antiadrenergics,
antiarrhythmics, antihypertensive agents, hormones, and nutrients,
antiarthrities,
antiasthmatic agents, anticonvulsants, antihistamines, antinauseants,
antineoplastics,
antipruritics, antipyretics; antispasmodics, cardiovascular preparations
(including
calcium channel blockers, beta-blockers, beta-agonists and antiarrythmics),
antihypertensives, diuretics, vasodilators; central nervous system stimulants;
cough
and cold preparations; decongestants; diagnostics; hormones; bone growth
stimulants and bone resorption inhibitors; immunosuppressives; muscle
relaxants;
psychostimulants; sedatives; tranquilizers; proteins, peptides, and fragments
thereof
(whether naturally occurring, chemically synthesized or recombinantly
produced);
and nucleic acid molecules (polymeric forms of two or more nucleotides, either
ribonucleotides (RNA) or deoxyribonucleotides (DNA) including both double- and
single-stranded molecules, gene constructs, expression vectors, antisense
molecules
and the like), small molecules (e.g., doxorubicin) and other biologically
active
macromolecules such as, for example, proteins and enzymes. The agent may be a
biologically active agent used in medical, including veterinary, applications
and in
agriculture, such as with plants, as well as other areas. The term therapeutic
agent
also includes without limitation, medicaments; vitamins; mineral supplements;
substances used for the treatment, prevention, diagnosis, cure or mitigation
of
disease or illness; or substances which affect the structure or function of
the body; or
pro-drugs, which become biologically active or more active after they have
been
placed in a predetermined physiological environment.
As used herein the term "low aqueous solubility" refers to water insoluble
compounds having poor solubility in water, that is <5 mg/m1 at physiological
pH
(6.5-7.4). Preferably, their water solubility is <1 mg/ml, more preferably
<0.1
CA 02818071 2013-06-06
mg/mi. It is desirable that the drug is stable in water as a dispersion;
otherwise a
lyophilized or spray-dried solid form may be desirable.
Examples of some preferred water-insoluble drugs include
iinmunosuppressive agents such as cyclosporins including cyclosporine
(cyclosporin
A), immunoactive agents, antiviral and antifungal agents, antineoplastic
agents,
analgesic and anti-inflammatory agents, antibiotics, anti-epileptics,
anesthetics,
hypnotics, sedatives, antipsychotic agents, neuroleptic agents,
antidepressants,
anxiolytics, anticonvulsant agents, antagonists, neuron blocking agents,
anticholinergic and cholinomimetic agents, antimuscarinic and muscarinic
agents, .
antiadrenergic and antiarrhythmics, antihypertensive agents, hormones, and
nutrients. A detailed description of these and other suitable drugs may be
found in
Remington's Pharmaceutical Sciences, 18th edition, 1990, Mack Publishing Co.
Philadelphia, Pa
The term "therapeutic index" refers to the therapeutic index of a drug defined
as LD50/ED50.
A "therapeutically effective amount" of a compound, with respect to a
method of treatment, refers to an amount of the compound(s) in a preparation
which,
when administered as part of a desired dosage regimen (to a mammal, preferably
a
human) alleviates a symptom, ameliorates a condition, or slows the onset of
disease
conditions according to clinically acceptable standards for the disorder or
condition
to be treated or the cosmetic purpose, e.g.,a.t a reasonable benefit/risk
ratio
applicable to any medical treatment.
A "therapeutically effective daily dosage" of a compound, with respect to a
method of treatment, refers to an amount of the compound(s) in a Preparation
which,
when administered as part of a desired daily dosage regimen (to a mammal,
preferably a human) alleviates a symptom, ameliorates a condition, or slows
the
onset of disease conditions according to clinically acceptable standards for
the
disorder or condition to be treated or the cosmetic purpose, e.g., at a
reasonable
benefit/risk ratio applicable to any medical treatment.
(1)) Chemical Terms
21
CA 02818071 2013-06-06
An aliphatic chain comprises the classes of alkyl, alkenyl and alkynyl
defined below. A straight aliphatic chain is limited to unbranched carbon
chain
radicals. As used herein, the term "aliphatic group" refers to a straight
chain,
branched-chain, or cyclic aliphatic hydrocarbon group and includes saturated
and
unsaturated aliphatic groups, such as an alkyl group, an alkenyl group, and an
allcynyl group.
Alkyl refers to a fully saturated branched or unbranched carbon chain radical
having the number of carbon atoms specified, or up to 30 carbon atoms if no
specification is made. For example, alkyl of 1 to 8 carbon atoms refers to
radicals
such as methyl, ethyl, propyl, butyl, pentyl, hexyl, heptyl, and octyl, and
those
radicals which are positional isomers of these radicals. Alkyl of 10 to 30
carbon
atoms includes decyl, undecyl, dodecyl, tridecyl, tetradecyl, pentadecyl,
hexadecyl,
heptadecyl, octadecyl, nonadecyl, eicosyl, heneicosyl, docosyl, tricosyl and
tetracosyl. In preferred embodiments, a straight chain or branched chain alkyl
has
30 or fewer carbon atoms in its backbone (e.g., C1-C30 for straight chains, C3-
C30
for branched chains), and more preferably 20 or fewer. Likewise, preferred
cycloalkyls have from 3-10 carbon atoms in their ring structure, and more
preferably
have 5, 6 or 7 carbons in the ring structure.
Moreover, the term "alkyl" (or "lower alkyl") as used throughout the
specification, examples, and claims is intended to include both "unsubstituted
alkyls" and "substituted alkyls", the latter of which refers to alkyl moieties
having
substituents replacing a hydrogen on one or more carbons of the hydrocarbon
backbone. Such substituents can include, for example, a halogen, a hydroxyl, a
carbonyl (such as a carboxyl, an alkoxycarbonyl, a formyl, or an acyl), a
thiocarbonyt (such as a thioester, a thioacetate, or a thioformate), an
alkoxyl, a
phosphoryl, a phosphate, a phosphonate, a phosphinate, an amino, an amido, an
arnidine, a cyano, a nitro, a sulfhydryl, an allcylthio, a sulfate, a
suIfonate, a
sulfamoyl, a sulfonamido, a sulfonyl, a heterocyclyl, an aralkyl, or an
aromatic or
heteroarornatic moiety. It will be understood by those skilled in the art that
the
moieties substituted on the hydrocarbon chain can themselves be substituted,
if
appropriate. For instance, the substituents of a substituted alkyl may include
substituted and unsubstituted forms of amino, azido, imino, amido, phosphoryl
22
CA 02818071 2013-06-06
(including phosphonate and phosphinate), sulfonyl (including sulfate,
sulfonamido,
sulfamoyl and sulfonate), and silyl groups, as well as ethers, allcylthios,
carbonyls
(including ketones, aldehydes, carboxylates, and esters), -CF3, -CN and the
like.
Exemplary substituted allcyls are described below. Cycloalkyls can be further
substituted with alkyls, alkenyls, alkoxyls, alkylthios, aminoalkyls, carbonyl-
substituted alkyls, -CF3, -CN, and the like.
Unless the number of carbons is otherwise specified, "lower alkyl", as used
herein, means an alkyl group, as defined above, but having from one to ten
carbons,
more preferably from one to six carbon atoms in its backbone structure such as
methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, and tert-
butyl.
Likewise, "lower alkenyl" and "lower alkynyl" have similar chain lengths.
Throughout the application, preferred alkyl groups are lower alkyls. In
preferred
embodiments, a substituent designated herein as alkyl is a lower alkyl.
The term "alkylthio" refers to an alkyl group, as defined above, having a
sulfur
radical attached thereto. In preferred embodiments, the "allcylthio" moiety is
represented by one of -(S)-alkyl, -(S)-alkenyl, -(S)-alkynyl, and -(S)-(CH2)m-
R1,
wherein m and R1 are defined below. Representative alkylthio groups include
methylthio, ethylthio, and the like.
Alkenyl refers to any branched or unbranched unsaturated carbon chain
radical having the number of carbon atoms specified, or. up to 26 carbon atoms
if no
limitation on the number of carbon atoms is specified; and having 1 or more
double
bonds in the radical. Alkenyl of 6 to 26 carbon atoms is exemplified by
hexenyl,
heptenyl, octenyl, nonenyl, decenyl, undecenyl, dodenyl, tridecenyl,
tetradecenyl,
pentadecenyl, hexadecenyl, heptadecenyl, octadecenyl, nonadecenyl, eicosenyl,
heneicosoenyl, docosenyl, tricosenyl and tetracosenyl, in their various
isomeric
forms, where the unsaturated bond(s) can be located anywhere in the radical
and can
have either the (Z) or the (E) configuration about the double bond(s).
Alkynyl refers to hydrocarbyl radicals of the scope of alkenyl, but having 1
or more triple bonds in the radical.
The terms "alkoxyl" or "alkoxy" as used herein refers to an alkyl group, as
defined below, having an oxygen radical attached thereto. Representative
alkoxyl
23
CA 02818071 2013-06-06
groups include methoxy, ethoxy, propoxy, tert-butoxy and the like. An "ether"
is
two hydrocarbons covalently linked by an oxygen. Accordingly, the substituent
of
=
an alkyl that renders that alkyl an ether is or resembles an alkoxyl, such as
can be
represented by one of -0-alkyl, -0-alkenyl, -0-alkynyl, -0-(CH2)m-R1, where m
and RI are described below.
The terms "amine" and "amino" are art-recognized and refer to both
unsubstituted and substituted amines, e.g., a moiety that can be represented
by the
general formulae:
Rg
f-N or
R3 R3
wherein R3, R5 and R6 each independently represent a hydrogen, an alkyl, an
alkenyl, -(CH2)m-R1, or R3 and R5 taken together with the N atom to which they
are attached complete a heterocycle having from 4 to 8 atoms in the ring
structure;
RI represents an alkenyl, aryl, cycloallcyl, a cycloalkenyl, a heterocycly1 or
a
polycyclyl; and m is zero or an integer in the range of Ito 8. In preferred
embodiments, only one of R3 or R5 can be a carbonyl, e.g., R3, R5 and the
nitrogen
together do not form an imide. In even more preferred embodiments, R3 and R5
(and optionally R6) each independently represent a hydrogen, an alkyl, an
alkenyl,
or -(CH2)m-R1. Thus, the term "alkylamine" as used herein means an amine
group,
as defined above, having a substituted or unsubstituted alkyl attached
thereto, i.e., at
least one of R3 and R5 is an alkyl group. In certain embodiments, an amino
group
or an alkylamine is basic, meaning it has a pKa > 7.00. The protonated forms
of
these functional groups have pKas relative to water above 7.00.
The term "carbonyl" is art-recognized and includes such moieties as can be
represented by the general formula:
0 0
X R7 or scsi XJL Rg
24
CA 02818071 2013-06-06
wherein X is a bond or represents an oxygen or a sulfur, and R7 represents a
hydrogen, an alkyl, an alkenyl, -(CH2)m-R1 or a pharmaceutically acceptable
salt,
R8 represents a hydrogen, an alkyl, an alkenyl or -(C112)m-R1, where in and R1
are
as defined above. Where X is an oxygen and R7 or R8 is not hydrogen, the
formula
represents an "ester". Where X is an oxygen, and R7 is as defined above, the
moiety
is referred to herein as a carboxyl group, and particularly when R7 is a
hydrogen, the
formula represents a "carboxylic acid". Where X is an oxygen, and R8 is
hydrogen,
the formula represents a "formate". In general, where the oxygen atom of the
above
formula is replaced by sulfur, the formula represents a "thiocarbonyl" group.
Where
X is a sulfur and R7 or R8 is not hydrogen, the formula represents a
"thioester"
group. Where X is a sulfur and R. is hydrogen, the formula represents a
"thiocarboxylic acid" group. Where X is a sulfur and R8 is hydrogen, the
formula
represents a "thioformate" group. On the other hand, where X is a bond, and R7
is
not hydrogen, the above formula represents a "ketone" group. Where X is a
bond,
and R7 is hydrogen, the above formula represents an "aldehyde" group.
The term "derivatized" refers to chemically modifying molecules. The
chemical modifications may be artificial such as formation of drugs, natural
such as
formation of metabolites. The skilled artisan would readily recognize the
variety of
ways molecules may be modified, such as oxidations, reductions,
electrophilic/nucleophilic substitutions, alkylations, ester/amide formations
and the
like. For example, cyclodextrins of the present invention may be chemically
modified by amination, tosylation, or iodination prior to covalently attaching
them
to the polymeric matrix. Likewise, therapeutic agents may be chemically
modified
by preparing prodrugs (e.g., glycine-carnptothecin).
95 The terms "heterOcycly1" or "heterocyclic group" refer to 3- to 10-
membered
ring structures, more preferably 3- to 7-membered rings, whose ring structures
include one to four heteroatoms. Heterocycles can also be polycycles.
Heterocyclyl
groups include, for example, thiophene, thianthrene, furan, pyran,
isobenzofuran,
chromene, xanthene, phenoxathiin, pyrrole, imidazole, pyrazole, isothiazole,
isoxazole, pyridine, pyrazine, pyrimidine, pyridazine, indolizine, isoindole,
indole,
indazole, purine, quinolizine, isoquinoline, quinoline, phthalazine,
naphthyridine,
CA 02818071 2013-06-06
quinoxaline, quinazoline, cinnolinc, pteridine, carbazole, carboline,
phenanthridine,
acridine, pyrimidine, phenanthro line, phenazine, phenarsazine, phenothiazine,
furazan, phenoxazine, pyrrolidine, oxolane, thiolane, oxazole, piperidine,
piperazine,
tnorpholine, lactones, lactarns such as azetidinones and pyrrolidinones,
sultarns,
sultones, and the like. The heterocyclic ring can be substituted at one or
more
positions with such substituents as described above, as for example, halogen,
alkyl,
aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, amino, nitro, sulfhydryl,
hnino,
amido, phosphate, phosphonate, phosphinate, carbonyl, carboxyl, silyl,
sulfamoyl,
sulfinyl, ether, allcylthio, sulfonyl, ketone, aldehyde, ester, a
heterocyclyl, an
aromatic or heteroaromatic moiety, -CF3, -CN, or the like.
As used herein, the term "substituted" is contemplated to include all
permissible substituents of organic compounds. In a broad aspect, the
permissible
substituents include acyclic and cyclic, branched and unbranched, carbocyclic
and
heterocyclic, aromatic and nonaromatic substituents_ of organic compounds.
Illustrative substituents include, for example, those described herein above.
The
permissible substituents can be one or more and the same or different for
appropriate
organic compounds. For purposes of this invention, the heteroatoms such as
nitrogen may have hydrogen substituents and/or any permissible substituents of
organic compounds described herein which satisfy the valences of the
heteroatoms.
This invention is not intended to be limited in any manner by the permissible
substituents of organic compounds.
The term "hydrocarbyl" refers to a monovalent hydrocarbon radical
comprised of carbon chains or rings of up to 26 carbon atoms to which hydrogen
atoms are attached. The term includes alkyl, cycloalkyl, alkenyl, alkynyl and
aryl
groups, groups which have a mixture of saturated and unsaturated bonds,
carbocyclic rings and includes combinations of such groups. It may refer to
straight
chain, branched-chain, cyclic structures or combinations thereof.
The term "hydrocarbylene" refers to a divalent hydrocarbyl radical.
Representative examples include alkylene, phenylene, or cyclohexylene.
Preferably,
the hydrocarbylene chain is fully saturated and/or has a chain of 1-10 carbon
atoms.
26
CA 02818071 2013-06-06
As used herein, the term "nitro" means -NO2; the term "halogen" designates
-F, -Cl, -Br or :I; the term "sulfhydryl" means -SH; the term "hydroxyl" means
-014;
and the term "sulfonyl" means -S02-.
It will be understood that "substitution" or "substituted with" includes the
implicit proviso that such substitution is in accordance with permitted
valence of the
substituted atom and the substituent, and that the substitution results in a
stable
compound, e.g., which does not spontaneously undergo transformation such as by
rearrangement, cyclization, elimination, etc.
Analogous substitutions can be made to alkenyl and alkynyl groups to
produce, for example, aminoalkenyls, aminoalkynyls, amidoalkenyls,
amidoallcynyls, iminoalkenyls, iminoalkynyls, thioalkenyls, thioalkynyls,
carbonyl-
substituted alkenyls or alkynyls.
As used herein, the definition Of each expression, e.g., alkyl, m, n, etc.,
when
it occurs more than once in any structure, is intended to be independent of
its
definition elsewhere in the same structure.
The terms triflyl, tosyl, mesyl, and nonafiylare art-recognized and refer to
trifluoromethanesulfonyl, p-toluenesulfonyl, methanesulfonyl, and
nonafluorobutanesulfonyl groups, respectively. The terms triflate, tosylate,
mesylate, and nonaflate are art-recognized and refer to
trifluoromethanesulfonate
ester, p-toluenesulfonate ester, methanesulfonate ester, and
nonafluorobutanesulfonate ester functional groups and molecules that contain
said
groups, resPectively.
The abbreviations Me, Et, Ph, Ms represent methyl, ethyl, phenyl, and
= methanesulfonyl, respectively. A more comprehensive list of the
abbreviations
utilized by organic chemists of ordinary skill in the art appears in the first
issue of
each volume of the Journal of Organic Chemistry; this list is typically
presented in a
table entitled Standard List of Abbreviations.
27
CA 02818071 2013-06-06
Certain compounds of the present invention may exist in particular geometric
or stereoisomeric forms. The present invention contemplates all such
compounds,
including cis- and trans-isomers, (R)- and (S)-enantiomers, diastereomers, (d)-
isomers, (I)-isomers, the racemic mixtures thereof, and other mixtures
thereof, as
falling within the scope of the invention. Additional asymmetric carbon atoms
may
. be present in a substituent such as an alkyl group. All such isomers, as
well as
mixtures thereof, are intended to be included in this invention.
If, for instance, a particulafenantiomer of a compound of the present
invention is desired, it may be prepared by asymmetric synthesis, or by
derivatization with a chiral auxiliary, where the resulting diastereomeric
mixture is
separated and the auxiliary group cleaved to provide the pure desired
enantiomers.
Alternatively, where the molecule contains a basic functional group, such as
amino,
or an acidic functional group, such as carboxyl, diastereomeric salts may be
formed
with an appropriate optically active acid or base, folloWed by resolution of
the
diastereomers thus formed by fractional crystallization or chromatographic
means
well known in the art, and subsequent recovery of the pure enantiomers.
Contemplated equivalents of the compounds described above include
compounds which otherwise correspond thereto, and which have the same general
properties thereof, wherein one or more simple variations of substituents are
made
which do not adversely affect the efficacy of the compound. In general, the
compounds of the present invention may be prepared by the methods illustrated
in
the general reaction schemes as, for example, described below, or by
modifications
thereof, using readily available starting materials, reagents and conventional
synthesis procedures_ In these reactions, it is also possible to malce use of
variants
which are in themselves known, but are not mentioned here.
For purposes of this invention, the chemical elements are identified in
accordance with the Periodic Table of the Elements, CAS version, Handbook of
Chemistry and Physics, 67th Ed., 1986-87, inside cover. Also for purposes of
this
invention, the term "hydrocarbon" is contemplated to include all permissible
compounds having at least one hydrogen and one carbon atom. In a broad aspect,
the permissible hydrocarbons include acyclic and cyclic, branched and
unbranched,
CA 02818071 2013-06-06
carbocyclic and heterocyclic, aromatic and nonarornatic organic compounds
which.
can be substituted or unsubstituted.
III. Exemplary Applications of Method and Compositions
(a) Exemplmy Compositions
The present invention includes polymer conjugates, such as cyclodextrin-
containing polymer conjugates, wherein one or more therapeutic/bioactive
agents
are covalently attached. In certain embodiments, the therapeutic agent is a
small
molecule, a macromolecule, an antibody, a peptide, a protein, an enzyme, a
nucleic
acid, or a polymer that has therapeutic function. The polymers include linear
or
branched cyclodextrin-containing polymers and polymers grafted with
cyclodextrin.
Exemplary cyclodextrin-containing polymers that may be modified as described
herein are taught in U.S. Patent No. 6,509,323, published U.S. application No.
20020151523, and U.S. patent application serial No.s 60/417373, and 10/372723.
These polymers are useful as carriers for small molecule therapeutic delivery,
and
may improve drug stability and solubility when used in vivo.
Accordingly, one embodiment of present invention is a polymeric compound
represented by Formula I:
cD)
I m
Li
1 _fl
{ L2 ( D)
al b
L3
(T) v
w (I)
29
CA 02818071 2013-06-06
wherein
P represents a linear or branched polymer chain;
CD represents a cyclic moiety such as a cyclodextrin moiety;
L, 1.;2 and 1,3, independently for each occurrence, may be absent or represent
a linker group;
D, independently for each occurrence, represents a therapeutic agent or a
prodrag thereof;
T, independently for each occurrence, represents a targeting ligand or
precursor thereof;
a, in, and v, independently for each occurrence, represent integers in the
range of Ito 10 (preferably 1 to 8, Ito 5, or even 1 to 3);
n and w, independently for each occurrence, represent an integer in the range
of 0 to about 30,000 (preferably <25,000, <20,000, <15,000, <10,000, <5,000,
<1,000, <500, <100, <50, <25, <10, or even <5); and
b represents an integer in the range of 1 to about 30,000 (preferably <25,000,
<20,000, <15,000, <10,000, <5,000; <1,000, <500, <100, <50, <25, <10, or even
<5),
wherein either P comprises cyclodextrin moieties or n is at least 1_
In certain embodiments, P contains a plurality of cyclodextrin
moieties within the polymer chain as opposed to the cyclodextrin moieties
being
grafted on to pendant groups off of the polymeric chain. Thus in certain
embodiments, the polymer chain of formula I further comprises n' units of U,
wherein n' represents an integer in the range of 1 to about 30,000 (preferably
<25,000, <20,000, <15,000, <10,000, <5,000, <1,000, <500, <100, <50, <25, <10,
or even <5); and U is represented by the general formula:
CA 02818071 2013-06-06
T
I Y
L5
L
___________________ L4 ___________ CD _______ L7 _____
Df
-g
L6 - 7) -z
D')f
g
wherein
CD represents a cyclic moiety, such as a cyclodextrin moiety, or derivative
thereof;
L4, L5, L6, and L7, independently for each occurrence, may be absent or
represent a linker group;
D and D', independently for each occurrence, represent the same or different
therapeutic agent or prodrug forms thereof;
T and T', independently for each occurrence, represent the same or different
targeting ligand or precursor thereof;
f and y, independently for each occurrence, represent an integer in the range
of 1 and 10; and
g and z, independently for each occurrence, represent an integer in the range
of 0 and 10.
In preferred embodiments, L4 and L7 represent linker groups.
In certain embodiments, the polymer may be selected from polysaccharides,
and other non-protein biocornpatible polymers, and combinations thereof, that
contain at least one terminal hydroxyl group, such as polyvinylpyrrollidone,
poly(oxyethylene)glycol (PEG), polysuccinic anhydride, polysebacic acid, PEG
phosphate, polyglutarnate, polyethylenimine, maleic anhydride divinylether
(DIVMA), cellulose, pullulans, inulin, polyvinyl alcohol (PVA), N-(2-
.
3 1
CA 02818071 2013-06-06
hydroxypropyl)methacrylamide (HPMA), dextran and hydroxyethyl starch (HES),
and have optional pendant groups for grafting therapeutic agents, targeting
ligands
and/or cyclodextrin moieties. In certain embodiments, the polymer may be
biodegradable such as poly(lactic acid), poly(glycolic acid), poly(allcyl 2-
cyanoacrylates), polyanhydrides, and polyorthoesters, or bioerodible such as
polYlactide-glycolide copolymers, and derivatives thereof; non-peptide
polyaminoacids, polyiminocarbonates, poly alpha-amino acids, polyalkyl-cyano-
acrylate, polyphosphazenes or acyloxymethyl poly aspartate and polyglutamate
copolymers and mixtures thereof. -
Another embodiment of the invention is a polymeric compound represented
by Formula II:
[TI
I m
L7
- -
_________________ L6P __________________________ L8 _____
I _
100 L9
/
( (I) rn
m- n -
¨ 0
wherein
P represents a monomer unit of a polymer;
T, independently for each occurrence, represents a targeting ligand or a
precursor thereof;
L6, 1,7, L3, L9, and L10, independently for each occurrence, may be absent or
represent a linker group;
32
CA 02818071 2013-06-06
CD, independently for each occurrence, represents a cyclodextrin moiety or a
derivative thereof;
D, independently for each occurrence, represents a therapeutic agent or a
prodrug form thereof;
m, independently for each occurrence, represents an integer in the range of 1
to 10 (preferably 1 to 8, 1 to 5, or even 1 to 3);
o represents an integer in the range of 1 to about 30,000 (preferably <25,000,
<20,000, <15,000, <10,000, <5,000, <1,000, <500, <100, <50, <25, <10, or even
<5); and
p, n, and q, independently for each occurrence, represent an integer in the
range of 0 to 10 (preferably 0 to 8, 0 to 5, 0 to 3, or even 0 to about 2),
wherein CD and D are preferably each present at at least 1 location
(preferably at least 5, 10, 25, or even 50 or 100 locations) in the compound.
Another embodiment of the invention is a compound represented by Formula
T
Y
L5
- I -z
CD ________________________________________ L7 ____
D (r)
g
L6
1
¨ 11
wherein
CD represents a cyclic moiety, such as a cyclodextrin moiety, or derivative
thereof;
L4, L5, L6, and L7, independently for each occurrence, may be absent or
represent a linker group;
33 =
CA 02818071 2013-06-06
D and D', independently for each occurrence, represent the same or different
therapeutic agent or pro drugs thereof;
T and T', independently for each occurrence, represent the same or different
targeting ligand or precursor thereof;
f and y, independently for each occurrence, represent an integer in the range
of 1 and 10 (preferably 1 to 8, I to 5, or even I to 3);
g and z, independently for each occurrence, represent an integer in the range
of 0 and 10 (preferably 0 to 8, 0 to 5, 0 to 3, or even 0 to about 2); and
h represents an integer in the range of 1 and 30,000 (preferably <25,000,
<20,000, <15,000, <10,000, <5,000, <1,000, <500, <100, <50, <25, <10, or even
<5),
wherein at least one occurrence (and preferably at least 5, 10, or even at
least
20, 50, or 100 occurrences) of g represents an integer greater than 0.
In preferred embodiments, L4 and L7 represent linker groups.
In certain embodiments, the underlying polymers are linear cyclodextrin-
containing polymers, e.g., the polymer backbone includes cyclodextrin
moieties. For
example, the polymer may be a water-soluble, linear cyclodextrin polymer
produced
by providing at least one cyclodextrin derivative modified to bear one
reactive site at
each of exactly two positions, and reacting the cyclodextrin derivative with a
linker
having exactly two reactive moieties capable of forming a covalent bond with
the
reactive sites under polymerization conditions that promote reaction of the
reactive
sites with the reactive moieties to form covalent bonds between the linker and
the
cyclodextrin derivative, whereby a linear polymer comprising alternating units
of
cyclodextrin derivatives and linkers is produced. Alternatively the polymer
may be a
water-soluble, linear cyclodextrin polymer having a linear polymer backbone,
which
polymer comprises a plurality of substituted or unsubstituted cyclodextrin
moieties
and linker moieties in the linear polymer backbone, wherein each of the
cyclodextrin
moieties, other than a cyclodextrin moiety at the terminus of a polymer chain,
is
attached to two of said linker moieties, each linker moiety covalently linking
two
cyclodextrin moieties. In yet another embodiment, the polymer is a water-
soluble,
linear cyclodextrin polymer comprising a plurality of cyclodextrin moieties
= 34
CA 02818071 2013-06-06
covalently linked together by a plurality of linker moieties, wherein each
cyclodextrin moiety, other than a cyclodextrin moiety at the terminus of a
polymer
chain, is attached to two linker moieties to form a linear cyclodextrin
polymer.
The linker group(s) may be an allcylene chain, a polyethylene glycol (PEG)
chain, polysuccinic anhydride, poly-L-glutamic acid, poly(ethyleneimine), an
oligosaccharide, an amino acid chain, or any other suitable linkage. In
certain
embodiments, the linker group itself can be stable under physiological
conditions,
such as an alkylene chain, or it can be cleavable under physiological
conditions,
such as by an enzyme (e.g., the linkage contains a peptide sequence that is a
substrate for a peptidase), or by hydrolysis (e.g., the linkage contains a
hydrolyzable
group, such as an ester or thioester). The linker groups can be biologically
inactive,
such as a PEG, polyglycolic acid, or polylactic acid chain, or can be
biologically
active, such as an oligo- or polypeptide that, when cleaved from the moieties,
binds
a receptor, deactivates an enzyme, etc.- Various oligomeric linker groups that
are
biologically compatible and/or bioerodible are known in the art, and the
selection of
the linkage may influence the ultimate properties of the material, such as
whether it
is durable when implanted, whether it gradually deforms or shrinks after
implantation, or whether it gradually degrades and is absorbed by the body.
The
linker group may be attached to the moieties by any suitable bond or
functional
group, including carbon-carbon bonds, esters, ethers, amides, amines,
carbonates,
carbainates, sulfonamides, etc.
In certain embodiments, the linker group(s) of the present invention represent
a hydrocarbylene group wherein one or more methylene groups is optionally
replaced by a group Y (provided that none of the Y groups are adjacent to each
other), wherein each Y, independently for each occurrence, is selected from,
substituted or urisubstituted aryl, heteroaryI, cycloalkyl, heterocycloalkyl,
or -0-,
C(X) (wherein X is NR, 0 or S), -0C(0)-, -C(=0)0, -NRICO-,
-C(0)N12.1-, -S(0)õ- (wherein n is 0, 1, or 2), -0C(0)-NRI, -NR1-C(0)-NR1-,
-NRi-C(NIZI)-NRI-, and -B(OR1)-; and RI, independently for each occurrence,
represents H or a lower alkyl.
CA 02818071 2013-06-06
In certain embodiments, the linker group represents a derivatized or non-
derivatized amino acid. In certain embodiments, linker groups with one or more
terminal carboxyl groups may be conjugated to the polymer. In certain
embodiments, one or more of these terminal carboxyl groups may be capped by
covalently attaching them to a therapeutic agent, a targeting moiety, or a
cyclodextrin moiety via an (thio)ester or amide bond. In still other
embodiments,
linker groups with one or more terminal hydroxyl, thiol, or amino groups may
be
incorporated into the polymer. In preferred embodiments, one or more of these
terminal hydroxyl groups may be capped by covalently attaching them to a
therapeutic agent, a targeting moiety, or a cyclodextrin moiety via an
(thio)ester,
amide, carbonate, carbamate, thiocarbonate, or thiocarbamate bond. In certain
embodiments, these (thio)ester, amide, (thio)carbonate or (thio)carbamates
bonds
may be biohydrolyzable, i.e., capable of being hydrolyzed under biological
conditions.
In certain embodiments, the polymers as described above have
polydispersities less than about 3, or even less than about 2.
In certain embodiments, the therapeutic agent is a small molecule, a peptide,
a protein, or a polymer that has therapeutic function. In certain embodiments,
the
agent is an anti-cancer (such as camptothecin or related derivatives), anti-
fungal,
anti-bacterial, anti-mycotic, or anti-viral therapeutic. In certain
embodiments, the
agent is a receptor agonist. In certain embodiments, the agent is a receptor
antagonist. In certain embodiments, the therapeutic agent is a protease
inhibitor.
Furthermore, a polymer of the present invention may contain one kind of
therapeutic
agent, or may contain more than one kind of therapeutic agent. For instance,
two or
more different cancer drugs, or a cancer drug and an immunosuppressant, or an
antibiotic and an anti-inflammatory agent may be grafted on to the polymer via
optional linkers. By selecting different linkers for different drugs, the
release of
each drug may be attenuated to achieve maximal dosage and efficacy.
One embodiment of the present invention provides an improved delivery of
certain hydrophobic small molecule therapeutics by covalently conjugating them
to
cyclodextrin containing polymers. Such conjugation improves the aqueous
36
CA 02818071 2013-06-06
solubility and hence the bioavailability of the therapeutic agents.
Accordingly, in
one embodiment of the invention, the therapeutic agent is a hydrophobic
compound
with a log P >0.4, >0.6, >0.8, >1, >2, >3, >4, or even >5. In other
embodiments, a
hydrophobic therapeutic agent, such as camptothecin, may be conjugated to
another
compound, such as an amino acid, prior to covalently attaching the conjugate
on to
the polymer. Examples of amino acid derivatized eamptothecin molecules are
illustrated in Scheme V.
The polymer conjugates of the present invention preferably have molecular
weights in the range of 10,000 to 500,000; 30,000 to 200,000; or even 70,000
to
150,000 amu.
In certain embodiments, the cyclodextrin moieties make up at least about
2%, 5% or 10% by weight, up to 20%, 30%, 50% or even 80% of the cyclodextrin-
modified polymer by weight. In certain embodiments, the therapeutic agents, or
targeting ligands make up at least about 1%, 5%, 10% or 15%, 20%, 25%, 30% or
even 35% of the cyclodextrin-modified polymer by weight. Number-average
molecular weight (Me) may also vary widely, but generally fall in the range of
about
1,000 to about 500,000 daltons, preferably from about 5000 to about 200,000
daltons and, even more preferably, from about 10,000 to about 100,000. Most
preferably, Mn varies between about 12,000 and 65,000 daltons_ In certain
other
embodiments, Mn varies between about 3000 and 150,000 daltons. Within a given
sample of a subject polymer, a wide range of molecular weights may be present.
For
example, molecules within the sample may have molecular weights that differ by
a
factor of 2, 5, 10, 20, 50, 100, or more, or that differ from the average
molecular
weight by a factor of 2, 5, 10, 20, 50, 100, or more. Exemplary cyclodextrin
moieties include cyclic structures consisting essentially of from 7 to 9
saccharide
moieties, such as cyclodextrin and .oxidized cyclodextrin. A cyclodextrin
moiety
optionally comprises a linker moiety that forms a covalent linkage between the
cyclic structure and the polymer backbone, preferably having from 1 to 20
atoms in
the chain, such as allcyl chains, including dicarboxylic acid derivatives
(such as
glutaric acid derivatives, succinic acid derivatives, and the like), and
heteroalkyl
chains, such as oligoethylene glycol chains.
37
CA 02818071 2013-06-06
Cyclodextrins are cyclic polysaccharides containing naturally occurring D-
(+)-glucopyranose units in an a-(1,4) linkage. The most common cyclodextrins
are
alpha ((a)-cyclodextrins, beta (13)-cyclodextrins and gamma (7)-cyclodextrins
which
contain, respectively six, seven, or eight glucopyranose units. Structurally,
the
cyclic nature of a cyclodextrin forms a torus or donut-like shape having an
inner
apolar or hydrophobic cavity, the secondary hydroxyl groups situated on one
side of
the cyclodextrin torus and the primary hydroxyl groups situated on the other.
Thus,
using (13)-cyclodextrin as an example, a cyclodextrin is often represented
schematically as follows.
HO 0
__________________________ NOH
OH H
OHO OH
H01.7
0 secondary hydroxyl
OHO
OH HO
0
OH
0
HO
primary hydroxyl
OH
OH
HO =
0 .
0
0
OH
0 _______________ \:)H
0 HO
OH
OH
0
The side on which the secondary hydroxyl groups are located has a wider
diameter than the side on which the primary hydroxyl groups are located. The
present invention contemplates covalent linkages to cyclodextrin moieties on
the
primary and/or secondary hydroxyl groups. The hydrophobic nature of the
cyclodextrin inner cavity allows for host-guest inclusion complexes of a
variety of
compounds, e.g., adamantane. (Comprehensive Supramolecular Chemistry, Volume
38
CA 02818071 2013-06-06
3, J.L. Atwood et al., eds., Pergamon Press (1996); T. Cserhati, Analytical
Biochemistry, 225:328-332(1995); Husain et al., Applied Spectroscopy, 46:652-
658
(1992); FR 2 665 169). Additional methods for modifying polymers are disclosed
in
Suh, J. and Noh, Y., Bioorg. Med. Chem. Lett. 1993, 8, 1327-1330.
In certain embodiments, the present invention contemplates linear, water-
soluble, cyclodextrin-containing polymer, wherein a plurality of bioactive
moieties
are covalently attached to the polymer through attachments that are cleaved
under
biological conditions to release the bioactive moieties, wherein
administration of the
polymer to a patient results in release of the bioactive agent over a period
of at least
2,3, 5, 6, 8, 10, 15, 20, 24, 36, 48 or even 72 hours.
In certain embodiments, the present invention contemplates attenuating the
rate of release of the therapeutic agent by introducing various linking groups
between the therapeutic agent and/or targeting ligand and the polymer. Thus,
in
certain embodiments, the polymeric therapeutics of the present invention are
compositions for controlled delivery of therapeutic agents. One skilled in the
art
would also recognize that by labeling the therapeutic agent and/or targeting
ligand
with radionuclei, or by forming complexes of NIVIR active nuclei, e.g.,
technetium,
gadolinium, or dysprosium, the polymers of the present invention can achieve a
dual
diagnostic/therapeutic utility.
=
= 20 In other embodiments, the polymeric compounds stabilize
the bioactive form
of a therapeutic agent which exists in equilibrium between an active and
inactive
form. For instance, conjugating the therapeutic agent to the polymers of the
present
invention may shift the equilibrium between two tautomeric forms of the agent
to
the bioactive tautomer. In other embodiment, the polymeric compounds may
attenuate the equilibrium between lactonic and acid forms of a therapeutic
agent.
One method to determine molecular weight is by gel permeation
chromatography ("GPC"), e.g., mixed bed columns, CH2Cl2 solvent, light
scattering
detector, and off-line dn/dc. Other methods are known in the art.
39
CA 02818071 2013-06-06
In other embodiments, the polymer conjugate of the invention may be a
flexible or flowable material. When the polymer used is itself flowable, the
polymer
composition of the invention, even when viscous, need not include a
biocompatible
solvent to be flowable, although trace or residual amounts of biocompatible
solvents
may still be present. =
While it is possible that the biodegradable polymer or the biologically active
agent may be dissolved in a small quantity of a solvent that is non-toxic to
more
efficiently produce an amorphous, monolithic distribution or a fine dispersion
of the
biologically active agent in the flexible or flowable composition, it is an
advantage
of the invention that, in a preferred embodiment, no solvent is needed to form
a
flowable composition. Moreover, the use of solvents is preferably avoided
because,
once a polymer composition containing solvent is placed totally or partially
within
the body, the solvent dissipates or diffuses away from the polymer and must be
processed and eliminated by the body, placing an extra burden on the body's
clearance ability at a time when the illness (and/or other treatments for the
illness)
may have already deleteriously affected it.
However, when a solvent is used to facilitate mixing or to maintain the
flowability of the polymer conjugate of the invention, it should be non-toxic,
otherwise biocompatible, and should be used in relatively small amounts.
Solvents
that are toxic should not be used in any material to be placed even partially
within a
living body. Such a solvent also must not cause substantial tissue irritation
or
necrosis at the site of administration.
Examples of suitable biocompatible solvents, when used, include N-methyl-
2-pyrrolidone, 2-pyiTolidone, ethanol, propylene glycol, acetone, methyl
acetate,
ethyl acetate, methyl ethyl ketone, dimethylformamide, dimethylsulfoxide,
tetrahydrofuran, caprolactam, oleic acid, or 1-dodecylazacylcoheptanone.
Preferred
solvents include N-methylpyrrolidone, 2-pyrrolidone, dimethylsulfoxide, and
acetone because of their solvating ability and their biocompatibility.
In certain embodiments, the subject polymer conjugates are soluble in one or
more common organic solvents for ease- of fabrication and processing. Common
CA 02818071 2013-06-06
organic solvents include such solvents as chloroform, dichloromethane,
dichloroethane, 2-butanone, butyl acetate, ethyl butyrate, acetone, ethyl
acetate,
dimethylacetamide, N-methylpyrrolidone, dimethylformamide, and
dimethylsulfoxide.
One aspect of the present invention contemplates attaching a hydrophobic
therapeutic agent such as (S)-20-camptothecin to linear or branched
cyclodextrin-
containing polymers for better delivery of the drug. (S)-20-carnptothecin
(CPT), an
alkaloid isolated from Carnptitheca accuminata in the late 1950's, was found
to
exhibit anticancer activity by inhibiting the action of topoisornerase I
during the S-
phase of the cell cycle. Its application in human cancer treatment, however,
is
limited due to several factors, especially its undesirable interactions with
human
serum albumin, instability of the bioactive lactone form, and poor aqueous
solubility. In order to circuMvent this problem, many CPT analogs have been
developed to improve lactone stability-and aqueous solubility. Topotecan and
irinotecan are analogs of CPT that have already been approved by FDA for human
cancer treatment. The present invention discloses various types of linear,
branched,
or grafted cyclodextrin.-containing polymers wherein (8)-20-camptothecin is
covalently bound to the polymer. In certain embodiments, the drug is
covalently
linked via a biohydrolyzable bond selected from an ester, amide, carbamates,
or
carbonate.
An exemplary synthetic scheme for covalently bonding a derivatized CD to
20(S)-camptothecin is shown in Scheme I.
Scheme I
0
COCl2 CD-NH,
N N N
0 0 0
HO 0 0 .
\ \
0
() CPT-CD
a \ NH \
CD
41
CA 02818071 2013-06-06
Without intending to limit the scope of the invention, a general strategy for
synthesizing linear, branched or grafted cyclodextrin-containing polymers (CD
Polymer) for loading a therapeutic agent such as camptothecin, and an optional
targeting ligand is shown in Scheme II.
Scheme II
Exemplary cyclodextrin monomers
for linear, branch or graft
cyclodextrin polymers
g0
1I. Therapeutic agent (D)
-)NH2 with optional linker (L)
0E0
k
Tosyl
CD Polymer CD Polymer
Multifunctional
1
polyrnerizable ____________________________________ in
monomers
Optional targeting
ligand (1), with
optional linker (LI
To illustrate further, without intending to be limiting, comonomer A
precursors, cyclodextrin moieties, therapeutic agents, and/or targeting
ligands may
be assembled as shown in Schemes Ila-lib. Note that in schemes Lla-b, in any
given
reaction there may be more than one comonomer A precursor, cyclodextrin
moiety,
therapeutic agent or targeting ligand that is of the same type or different.
Furthermore, prior to polymerization, one or more comonomer A precursor,
cyclodextrin moiety, therapeutic agent or targeting ligand may be covalently
linked
with each other in one or more separate step.
Scheme ha: General scheme for graft polymers. The comonomer A
precursor, cyclodextrin moiety, therapeutic agent and targeting ligand are as
defined
above. Furthermore, one skilled in the art may choose from a variety of
reactive
groups, e.g., hydroxyls, carboxyls, halides, arnines, and activated ethenes,
ethynes,
or aromatic groups in order achieve polymerization. For further examples of
42
CA 02818071 2013-06-06
reactive groups are disclosed in Advanced Organic Chemistry: Reactions,
Mechanisms, and Structure, 5th Edition, 2000.
I r ____
Reactivei __ _ i Comonomer A ____1 Reactive
Group
Group
Pendant
. Group(s)
Reactive
Group
+
_____________________________________________ =
Reactive ¨ Optional ¨ Cyclodextrin
Group Linker Moiety
+ OPTIONALLY
Reactive , Optional Therapeutic
Group Linker Agent
AND/OR
Reactive Optional . , Targeting
Group Linker Ligand =
_
' o
Polymerization
_ ¨
_________________________ . Comonomer A
. precursor
. .
_ __
Targeting __________ Optional Pendant ¨ Optional ¨ Targeting
Ligand Linker Group(s) Linker Ligand
Optional
Linker
¨ ¨
B
Cyclodextrin
Moiety
.
Scheme lib: General scheme of preparing linear cyclodextrin-containing
polymers. One skilled in the art would recognize that by choosing a comonomer
A
precursor that has multiple reactive groups polymer branching can be achieved.
43
CA 02818071 2013-06-06
Reactive ______________________ Comonomer A
Group precursor
ReactiveReactive
____________________________ Cyclodextrin __
Group Group
Moiety
+ OPTIONALLY
Reactive Optional Therapeutic
Group Linker Agent
AND/OR
Reactive . Optional Targeting
Group Linker Ligand
Polymerization
Optional _______________ Cyclodextrin __ Comonomer A _____________
Linker Moiety precursor
¨n
Wherein R is a therapeutic agent
and/or targeting ligand which may
be absent or present.
Examples for different ways of synthesizing linear cyclodextrin-CPT
polymers are shown in Schemes
Scheme III
44
CA 02818071 2013-06-06
MN" ti2N COON =
-..s.-Ths, 111111W C001-1
Vi
...õ.\\ 0
_I . 0 1 n
0 ¨ S S-= 0
11 11 C 0 01 1
o o
o
=-.1...---71
(W)anL______).- lrAW 1-1
N S
H
R 0
wherein
W represents an optional linking group; and
R represents W-CPT or a
Scheme IV
OBz OBz OBz OBz
y_Zi, 1,0 0:_-_
---...-0 ----.. NHz _
1-10,1c.N 01-1 H2ts!-------).--- Ny
H " 0 H H 0 0
0
OH OH
W¨CPT
Y
H --
2
--------).-
H...HN,,....Ø--....õ.-0-õ..,,-.Ny
o 0
W-CPT W-CPT
wherein
W represents an optional linking group
1)riNi N
H Hi" .
0 0
Scheme V
- =
CA 02818071 2013-06-06
Oi.OH Or),OH
o o
tso-r"...tr't
=-6
____________________________ )
0 1OH
0 0
0y0CPT
CPT or Gly-CPT, H
o ty00pr LocpT
0 0
0
0 OH
.5\
0
N 0
0 N-r- HOji'l
NH2 0 _kW \
0
OH
CPT, )1,..1
W 0
W-CPT
0
tyW
wherein CPT0
W represents an optional linking oup, e.g., glycyt residue
=
Scheme VI
46
CA 02818071 2013-06-06
0
ON
CPT
HN
0
HO OH
EDC
NHS
0 0
0
N
CPT
HN=
n
Scheme VII
Bac
0
ie Boc
OH H
0
0 Etc .NH
0 NH
Soc-
0 Gly-CPT
0
HO...k.õThrGly-CPT HN 0 0
HC
0 NH
Gly-CPT
Scheme VIII
47
CA 02818071 2013-06-06
OCPT
Y=7,
H2N NH2
0 Lys-Gly-CPT
H
in
0
HN 0
Oy
OCPT
Examples for grafting cyclodextrins on to side-chains of CPT-containing
polymers subunits are shown in Schemes Each subunit
may repeat any
number of times, and one subunit may occur with substantially the same
frequency,
more often, or less often than another subunit, such that both subunits may be
present in approximately the same amount, or in differing amounts, which may
differ slightly or be highly disparate, e.g., one subunit is present nearly to
the
= 10 exclusion of the other.
In certain instances, the polymers are random copolymers, in which the
different subunits and/or other monomeric units are distributed randomly
throughout
the polymer chain. Thus, where the formula Xõ,-Yii-Zo appears, wherein X, Y
and Z
are polymer subunits, these subunits may be randomly interspersed throughout
the
polymer backbone. In part, the term "random" is intended to refer to the
situation in
which the particular distribution or incorporation of monomeric units in a
polymer
that has more than one type of monomeric units is not directed or controlled
directly
by the synthetic protocol, but instead results from features inherent to the
polymer
48
CA 02818071 2013-06-06
system, such as the reactivity, amounts of subunits and other characteristics
of the
synthetic reaction or other methods of manufacture, processing, or treatment.
Scheme IX
0 cn, 0 r
) ______ \
A Boioso_ , 11 1 ii
0 0 > p-0-0,i_cHr_0* --C12--).¨
V I
o NP I n n
--% ---H H CI
.517 0 CH, 0 CH,
0 TH3
CPT ¨ __
H2 ( 11 11 11
).- N NaOH
)-- ---). P 0¨CH¨CH? 0 ) ( P 0¨CH¨CH2 0 ) ( P 0¨CH¨CH?-0--)¨
In I m I o
NH OCPT CI
,LI
=
Scheme X
=
________ o 0 ___ 0
5
n rn o
A1BN /
\
o
0
0
\
0 0 HN _____________ IP¨ 0 0
or ATRP \
(0 HN
CPT CPT
)
O o
\ \
Scheme XI '
49
CA 02818071 2013-06-06
,
,
.,,
µ
-s( n
H2N
Y-7
H20 n ,
) ),---
0 0 0 0 0 0 0
0 0 0 0' -0 0- HNJ vi 0'
is-2 o
, st,
*
-
Scheme XII
= 0
NH
0 0
0
H2N . N õ,..¨ 1
/ `,,NH2 0
H 0
H m
0
//o-
HNN
HOo/ o o
0 ______________________________________________ '', 0
0111.= z
N
-..õ.
/ \
N
0
The present invention further contemplates CD-polymers synthesized using
CD-biscysteine monomer and a di-NHS ester such as PEG-DiSPA or PEG-BTC as
shown in Schemes XIII-XIV.
-
CA 02818071 2013-06-06
Scheme XIII
õ...V¨Z o
/ 0
0
s s DMSO
4- ________________________________________________________________ 4.--
N N DEA
-,
HO o
---0 OH
6 MWpEc= 3400 0
H H EDC/NHS
),
0 0
0
. :,00
HC, 0 o 0 0 N
OH NE12
. 95-98% 1 \ PI
Mn 55,700; Mw 99,500; Mw/Mn= L74 m., Gly-CPT
iSr
H
. tAir
m n
0
,,,,,(3, 0
litrCt
j.....,0 0 0 ojs,...0 0
0
0 ,... 0 . ..,..
* *
Scheme XIV
.L
o 0
HO 0 0 o Ho-r'''--"--n*--OH ). 0--(/
OH*
n 0
DMAP 0 0
,
0 0 0 0
NHS
---).¨
I
DCC u 0 N V10---0 0 OH
_____________________________________________________________________ ).
0 0
0 dr
0 0
H H
N..----õ,._ ..i7... .....õ.õN 04,--O,y....õ...-- Gly-CPT
..õ.....õ,,,---õ,, 0
HO 0 0 OH
0 0
0
=
CPT-Gly 0 0 Gly-CPT
\=---..¨ n
Degradable ester bond linkage
51
CA 02818071 2013-06-06
In certain embodiments, the present invention discloses several strategies to
increase drug loading as shown in Figure 1.
(b) Targeting Ligand
As mentioned above, one aspect of the present invention contemplates
attaching a therapeutic agent to the polymer conjugates described herein.
In certain embodiments, the polymer conjugate further comprises a targeting
ligand. Thus in certain embodiments, a receptor, cell, and/or tissue-targeting
ligand,
or a precursor thereof is coupled to a polymer conjugate. As used herein the
term
"targeting ligand" refers to any material or substance which may promote
targeting
of receptors, cells, and/or tissues in vivo or in vitro with the compositions
of the
present invention. The targeting ligand may be synthetic, semi-synthetic, or
naturally-occurring. Materials or substances which may serve as targeting
ligands
include, for example, proteins, including antibodies, antibody fragments,
hormones,
hormone analogues, glycoproteins and lectins, peptides, polypeptides, amino
acids,
sugars, saccharides, including monosaccharides and polysaccharides,
carbohydrates,
small molecules, vitamins, steroids, steroid analogs, hormones, cofactors,
bioactive
agents, and genetic material, including nucleosides, nucleotides, nucleotide
acid
constructs and polynucleotides. As used herein, the term "precursor" to a
targeting
ligand refers to any material or substance which may be converted to a
targeting
ligand. Such conversion may involve, for example, anchoring a precursor to a
targeting ligand. Exemplary targeting precursor moieties include maleimide
groups,
disulfide groups, such as ortho-pyridyl disulfide, vinylsulfone groups, azide
groups,
and a-iodo acetyl groups. The attachment of the targeting ligand or precursor
thereof
to the polymer may be accomplished in various ways including but not limited
to
chelation, covalent attachment, or formation of host-guest complexes. In
certain
embodiments, an optional linker group may be present between the targeting
ligand
or precursor thereof and the polymer, wherein the linker group is attached to
the
polymer via chelation, covalent attachment or form host guest complexes. For
example, the one terminal end of a linker group may be attached to the
targeting
52
CA 02818071 2013-06-06
= ligand while the other may be attached to an adamantane group, or other
such
hydrophobic moiety, which forms a host guest complex with a cyclodextrin
moiety.
Thus the targeting ligand may be attached to a grafted cyclodextrin moiety, to
a
cyclodextrin moiety within the polymeric chain, or to the polymeric chain
itself.
The number of targeting ligands per polymeric chain may vary according to
various
factors including but not limited to the identity of the therapeutic agent,
nature of the
disease, type of polymer chain. Structures of possible linker groups are the
same as
linker groups defined elsewhere in this application.
(c) Pharmaceutical Compositions. Formulations and Dosages
In part, a biocompatible polymer composition of the present invention
includes a biocompatible and optionally biodegradable polymer, such as one
having
the recurring monomeric units shown in one of the foregoing formulas,
optionally
including any other biocompatible and optionally biodegradable polymer
mentioned
above or known in the art. In certain embodiments, the compositions are non-
pyrogenic, e.g., do not trigger elevation of a patient's body temperature by
more
than a clinically acceptable amount.
The subject compositions may contain a "drug", "therapeutic agent,"
"medicament," or "bioactive substance," which are biologically,
physiologically, or
pharmacologically active substances that act locally or systemically in the
human or
animal body. For example, a subject composition may include any of the other
compounds discussed above.
Various forms of the medicaments or biologically active materials may be
used which are capable of being released from the polymer matrix into adjacent
tissues or fluids. They may be hydrophobic molecules, neutral molecules, polar
molecules, or molecular complexes capable of hydrogen bonding. They may be in
the form of ethers, esters, amides and the like, including prodrugs which are
biologiCally activated when injected into the human or animal body, e.g., by
cleavage of an ester or amide. A therapeutic agent in a subject composition
may
vary widely with the purpose for the composition.
53
CA 02818071 2013-06-06
Plasticizers and stabilizing agents known in the art may be incorporated in
polymers of the present invention. In certain embodiments, additives such as
plasticizers and stabilizing agents are selected for their biocompatibility.
In certain
embodiments, the additives are lung surfactants, such as 1,2-
dipalmitoylphosphatidycholine (DPPC) and 1,-a-phosphatidylcholine (PC).
A composition of this invention may further contain one or more adjuvant
substances, such as fillers, thickening agents or the like. In other
embodiments,
materials that serve as adjuvants may be associated with the polymer matrix.
Such
additional materials may affect the characteristics of the polymer matrix that
results.
For example, fillers, such as bovine serum albumin (BSA) or mouse serum
albumin (MSA), may be associated with the polymer matrix. In certain
embodiments, the amount of filler may range from about 0.1 to about 50% or
more
by weight of the polymer matrix, or about 2.5, 5, 10, 25, or 40 percent.
Incorporation of such fillers may affect the biodegradation of the polymeric
material
and/or the sustained release rate of any encapsulated substance. Other fillers
known
to those of skill in the art, such as carbohydrates, sugars, starches,
saccharides,
celluloses and polysaccharides, including mannitose and sucrose, may be used
in
certain embodiments of the present invention.
In other embodiments, spheronization enhancers facilitate the production of
subject polymeric matrices that are generally spherical in shape. Substances
such as
zein, microcrystalline cellulose or microcrystalline cellulose co-processed
with
sodium carboxymethyl cellulose may confer plasticity to the subject
compositions as
well as implant strength and integrity. In particular embodiments, during
spheronization, extrudates that are rigid, but not plastic, result in the
formation of
dumbbell shaped implants and/or a high proportion of fines, and extrudates
that are
plastic, but not rigid, tend to agglomerate and form excessively large
implants. In
such embodiments, a balance between rigidity and plasticity is desirable. The
percent of spheronization enhancer in a formulation typically range from 10 to
90%
(w/w).
54
CA 02818071 2013-06-06
In certain embodiments, a subject composition includes an excipient. A
particular exeipient may be selected based on its melting point, solubility in
a
selected solvent (e.g., a solvent that dissolves the polymer and/or the
therapeutic
agent), and the resulting characteristics of the microparticles.
= Excipients may comprise a few percent, about 5%, 10%, 15%, 20%, 25%,
30%, 40%, 50%, or higher percentage of the subject compositions.
Buffers, acids and bases may be incorporated in the subject compositions to
adjust their pH. Agents to increase the diffusion distance of agents released
from the
polymer matrix may also be included.
Disintegrants are substances that, in the presence of liquid, promote the
disruption of the subject compositions. Disintegrants are most often used in
implants, in which the function of the disintegrant is to countei-act or
neutralize the
effect of any binding materials used in the subject formulation. In general,
the
mechanism of disintegration involves moisture absorption and swelling by an
insoluble material.
Examples of disintegyants include croscarmellose sodium and crospovidone
which, in certain embodiments, may be incorporated into the polymeric matrices
in
the range of about 1-20% of total matrix weight. In other cases, soluble
fillers such
as sugars (mannitol and lactose) may also be added to facilitate
disintegration of
implants.
Other materials may be used to advantage or to control the desired release
rate of a .therapeutic agent for a Particular treatment protocol. For example,
if the
sustained release is too slow for a particular application, a pore-forming
agent may
be added to generate additional pores in the matrix. Any biocompatible water-
soluble material may be used as the pore-forming agent. They may be capable of
dissolving, diffusing or dispersing out of the formed polymer system whereupon
pores and microporous channels are generated in the system. The amount of pore-
forming agent (and size of dispersed particles of such pore-forming agent, if
CA 02818071 2013-06-06
appropriate) within the composition should affect the size and number of the
pores
in the polymer system.
Pore-forming agents include any pharmaceutically acceptable organic or
inorganic substance that is substantially miscible in water and body fluids
and will
dissipate from the forming and formed matrix into aqueous medium or body
fluids
or water-immiscible substances that rapidly degrade to water-soluble
substances.
Suitable pore-forming agents include, for example, sugars such as sucrose
and dextrose, salts such as sodium chloride and sodium carbonate, and polymers
such as hydroxylpropylcellulose, carboxymethylcellulose, polyethylene glycol,
and
PVP. The size and extent of the pores may be varied over a wide range by
changing
the molecular weight and percentage of pore-forming agent incorporated into
the
polymer system.
The charge, lipophilicity or -hydrophilicity of any subject polymeric matrix
may be modified by attaching in some fashion an appropriate compound to the
surface of the matrix. For example, surfactants may be used to enhance
wettability
of poorly soluble or hydrophobic compositions. Examples of suitable
surfactants
include dextran, polysorbates and sodium lauryl sulfate. In general,
surfactants are
used in low concentrations, generally less than about 5%.
Binders are adhesive materials that may be incorporated in polymeric
formulations to bind and maintain matrix integrity. Binders may be added as
dry
powder or as solution. Sugars and natural and synthetic polymers may act as
binders.
Materials added specifically as binders are generally included in the range of
about 0.5%-15% w/w of the matrix formulation.. Certain materials, such as
microcrystalline cellulose, also used as a spheronization enhancer, also have
additional binding properties.
Various coatings may be applied to modify the properties of the matrices.
56
CA 02818071 2013-06-06
Three exemplary types of coatings are seal, gloss and enteric coatings. Other
types of coatings having various dissolution or erosion properties may be used
to
further modify subject matrices behavior, and such coatings are readily known
to
one of ordinary skill in the art.
The seal coat may prevent excess moisture uptake by the matrices during the
application of aqueous based enteric coatings. The gloss coat generally
improves
the handling of the finished matrices. Water-soluble materials such as
hydroxypropylcellulose may be used to seal coat and gloss coat implants. The
seal
coat and gloss coat are generally sprayed onto the matrices until an increase
in
weight between about 0.5% and about 5%, often about 1% for a seal coat and
about
3% for a gloss coat, has been obtained.
Enteric coatings consist of polymers which are insoluble in the low pH (less
than 3.0) of the stomach, but are soluble in the elevated pH (greater than
4.0) of the
small intestine. Polymers such as EUDRAGITTm, RohmTech, Inc., Malden, Mass.,
and AQUATERICTm, FMC Corp., Philadelphia, Penn., may be used and are layered
as thin membranes onto the implants from aqueous solution or suspension or by
a
spray drying method. The enteric. coat is generally sprayed to a weight
increase of
about 1% to about 30%, preferably about 10 to about 15% and may contain
coating
adjuvants such as plasticizers, surfactants, separating agents that reduce the
tackiness of the implants during coating, and coating permeability adjusters.
The present compositions may additionally contain one or more optional
additives such as fibrous reinforcement, colorants, perfumes, rubber
modifiers,
modifying agents, etc. In practice, each of these optional additives should be
compatible with the resulting polymer and its intended use. Examples of
suitable
fibrous reinforcement include PGA microfibrils, collagen microfibrils,
cellulosic
microfibrils, and olefinic microfibrils. The amount of each of these optional
additives employed in the composition is an amount necessary to achieve the
desired
effect.
The therapeutic polymer conjugates as described herein can be administered
in various pharmaceutical formulations, depending on the disorder to be
treated and
57
CA 02818071 2013-06-06
the age, condition and body weight of the patient, as is well known in the
art. For
example, where the compounds are to be administered orally, they may be
formulated as tablets, capsules, granules, powders or syrups; or for
parenteral
administration, they may be formulated as injections (intravenous,
intramuscular or
subcutaneous), drop infusion preparations or suppositories. For application by
the
ophthalmic mucous membrane route, they may be formulated as eyedrops or eye
ointments. These formulations can be prepared by conventional means, and, if
desired, the active ingredient may be mixed with any conventional additive,
such as
an excipient, a binder, a disintegrating agent, a lubricant, a corrigent, a
solubilizing
agent, a suspension aid, an emulsifying agent or a coating agent. Although the
dosage will vary depending on the symptoms, age and body weight of the
patient,
the nature and severity of the disorder to be treated or prevented, the route
of -
administration and the form of the drug, in general, a daily dosage of from
0.01 to
2000 mg of the therapeutic agent is recommended for an adult human patient,
and
this may be administered in a single dose or in divided doses.
The precise time of administration and/or amount of therapeutic polymer
conjugate that will yield the most effective results in terms of efficacy of
treatment
in a given patient will depend upon the activity, pharmacokinetics, and
bioavailability of a particular compound, physiological condition of the
patient
(including age, sex, disease type and stage, general physical condition,
responsiveness to a given dosage and type of medication), route of
administration,
etc. However, the above guidelines can be used as the basis for fine-tuning
the
treatment, e.g., determining the optimum time and/or amount of administration,
which will require no more than routine experimentation consisting of
monitoring
=
the subject and adjusting the dosage and/or timing.
The phrase "pharmaceutically acceptable" is employed herein to refer to
those therapeutic polymer conjugates, materials, compositions, and/or dosage
forms
which are, within the scope of sound medical judgment, suitable for use in
contact
with the tissues of human beings and animals without excessive toxicity,
irritation,
allergic response, or other problem or complication, commensurate with a
reasonable benefit/risk ratio.
58
CA 02818071 2013-06-06
The phrase "pharmaceutically acceptable carrier" as used herein means a
pharmaceutically acceptable material, composition or vehicle, such as a liquid
or
solid filler, diluent, excipient, solvent or encapsulating material, involved
in carrying
or transporting the subject chemical from one organ, or portion of the body,
to
another organ, or portion of the body. Each carrier must be "acceptable" in
the
sense of being compatible with the other ingredients of the formulation and
not
injurious to the patient. Some examples of materials which can serve as
pharmaceutically acceptable carriers include: (1) sugars, such as lactose,
glucose and
sucrose; (2) starches, such as corn starch and potato starch; (3) cellulose,
and its
derivatives, such as sodium carboxymethyl cellulose, ethyl cellulose and
cellulose
acetate; (4) powdered tragacanth; (5) malt; (6) gelatin; (7) talc; (8)
excipients, such
as cocoa butter and suppository waxes; (9) oils, such as peanut oil,
cottonseed oil,
safflower oil, sesame oil, olive oil, corn oil and soybean oil; (10) glycols,
such as
propylene glycol; (11) polyols, such as glycerin, sorbitol, mannitol and
polyethylene
glycol; (12) esters, such as ethyl oleate and ethyl laurate; (13) agar; (14)
buffering
agents, such as magnesium hydroxide and aluminum hydroxide; (15) alginic acid;
(16) pyrogen-free water; (17) isotonic saline; (18) Ringer's solution; (19)
ethyl
alcohol; (20) phosphate buffer solutions; and (21) other non-toxic compatible
substances employed in pharmaceutical formulations.
The term "pharmaceutically acceptable salts" refers to the relatively non-
toxic, inorganic and organic acid addition salts of the therapeutic polymer
conjugates. These salts can be prepared in situ during the final isolation and
purification of the therapeutic polymer conjugates, or by separately reacting
a
purified polymer in its free base form with a suitable organic or inorganic
acid, and
isolating the salt thus formed. Representative salts include the hydrobromide,
hydrochloride, sulfate, bisulfate, phosphate, nitrate, acetate, valerate,
oleate,
palmitate, stearate, laurate, benzoate, lactate, phosphate, tosylate, citrate,
maleate,
fumarate, succinate, tartrate, naplithylate, mesylate, glucoheptonate,
lactobionate,
and laurylsulphonate salts and the like. (See, for example, Berge et al.
(1977)
"Pharmaceutical Salts", J. Phann. Sci. 66:1-19)
59
CA 02818071 2013-06-06
-
In other cases, the therapeutic polymer conjugates useful in the methods of
the present invention may contain one or more acidic functional groups and,
thus,
=
are capable of forming pharmaceutically acceptable salts with pharmaceutically
acceptable bases. The term "pharmaceutically acceptable salts" in these
instances
refers to the relatively non-toxic, inorganic and organic base addition salts
of the
polymer(s). These salts can likewise be prepared in situ during the final
isolation
and purification of the polymer(s), or by separately reacting the purified
polymer(s)
in its free acid form with a suitable base, such as the hydroxide, carbonate
or
bicarbonate of a pharmaceutically acceptable metal cation, with ammonia, or
with a
pharmaceutically acceptable organic primary, secondary or tertiary amine.
Representative alkali or alkaline earth salts include the lithium, sodium,
potassium,
calcium, magnesium, and aluminum salts and the like. Representative organic
amines useful for the formation of base addition salts include ethylamine,
diethylamine, ethylefiediamine, ethanolamine, diethanolamine, piperazine and
the
like (see, for example, Berge et al., supra).
Wetting agents, emulsifiers and lubricants, such as sodium lauryl sulfate and
magnesium stearate, as well as coloring agents, release agents, coating
agents,
sweetening, flavoring and perfuming agents, preservatives and antioxidants can
also
be present in the compositions.
=
Examples of pharmaceutically acceptable antioxidants include: (1) water
soluble antioxidants, such as ascorbic acid, cysteine hydrochloride, sodium
bisulfate,
sodium rnetabisulfite, sodium sulfite and the like; (2) oil-soluble
antioxidants, such
as ascorbyl palmitate, butylated hydroxyanisole (BHA), butylated
hydroxytoluene
(BHT), lecithin, propyl gallate, alpha-tocopherol, and the like; and (3) metal
chelating agents, such as citric acid, ethylenediamine tetraacetic acid
(EDTA),
sorbitol, tartaric acid, phosphoric acid, and the like.
Formulations useful in the methods of the present invention include those
suitable for oral, nasal, topical (including ophthalmic, otic, buccal and
sublingual),
rectal, vaginal, aerosol and/or parenteral administration. The formulations
may
conveniently be presented in unit dosage form and may be prepared by any
methods
well known in the art of pharmacy. The amount of active ingredient which can
be
CA 02818071 2013-06-06
_
combined with a carrier material to produce a single dosage form will vary
depending upon the host being treated, the particular mode of administration.
The
amount of active ingredient which can be combined with a carrier material to
produce a single dosage form will generally be that amount of the compound
which
produces a therapeutic effect. Generally, out of one hundred per cent, this
amount
will range from about 1 per cent to about ninety-nine percent of active
ingredient,
preferably from about 5 per cent to about 70 per cent, most preferably from
about 10
per cent to about 30 per cent.
Methods of preparing these formulations or compositions include the step of
bringing into association a therapeutic polymer conjugate(s) with the carrier
and,
optionally, one or more accessory ingredients. In general, the formulations
are
prepared by uniformly and intimately bringing into association a therapeutic
polymer conjugate with liquid carriers, or finely divided solid carriers, or
both, and
then, if necessary, shaping the product:
Formulations suitable for oral administration may be in the form of capsules,
cachets, pills, tablets, gums, lozenges (using a flavored basis, usually
sucrose and
acacia or tragacanth), powders, granules, or as a solution or a suspension in
an
aqueous or non-aqueous liquid, or as an oil-in-water or water-in-oil liquid
emulsion,
or as an elixir or syrup, or as pastilles (using an inert base, such as
gelatin and
glycerin, or sucrose and acacia) and/or as mouthwashes and the like, each
containing
a predetermined amount of a therapeutic polymer conjugate(s) as an active
ingredient. A compound may also be administered as a bolus, electuary or
paste.
A tablet may be made by compression or molding, optionally with one or
more accessory ingredients. Compressed tablets may be prepared using binder
(for
example, gelatin or hydroxypropylmethyl cellulose), lubricant, inert diluent,
preservative, disintegrant (for example, sodium starch glycolate or cross-
linked
sodium carboxymethyl cellulose), surface-active or dispersing agent. Molded
tablets
may be made by molding in a suitable machine a mixture of the powdered peptide
or
peptidomimetic moistened with an inert liquid diluent.
61
CA 02818071 2013-06-06
--
Tablets, and other solid dosage forms, such as dragees, capsules, pills and
granules, may optionally be scored or prepared with coatings and shells, such
as
enteric coatings and other coatings well known, in the pharmaceutical-
formulating
art. They may also be formulated so as to provide slow or controlled release
of the
active ingredient therein using, for example, hydroxypropylmethyl cellulose in
varying proportions to provide the desired release profile, other polymer
matrices,
liposomes and/or microspheres. They may be sterilized by, for example,
filtration
through a bacteria-retaining filter, or by incorporating sterilizing agents in
the form
of sterile solid compositions which can be dissolved in sterile water, or some
other
sterile injectable medium immediately before use. These compositions may also
optionally contain pacifying agents and may be of a composition that they
release
the active ingredient(s) only, or preferentially, in a certain portion of the
gastrointestinal tract, optionally, in a delayed manner. Examples of embedding
compositions which can be used include polymeric substances and waxes. The
active ingredient can also be in micro-encapsulated form, if appropriate, with
one or
more of the above-described excipients.
Liquid dosage forms for oral administration include pharmaceutically
acceptable emulsions, microemulsions, solutions, suspensions, syrups and
elixirs. In
addition to the active ingredient, the liquid dosage forms may contain inert
diluents
commonly used in the art, such as, for example, water or other solvents,
solubilizing
agents and emulsifiers, such as ethyl alcohol, isopropyl alcohol, ethyl
carbonate,
ethyl acetate, benzyl alcohol, benzyl benzoate, propylene glycol, 1,3-butylene
glycol, oils (in particular, cottonseed, groundnut, corn, germ, olive, castor
and
sesame oils), glycerol, tetrahydrofiiryl alcohol, polyethylene glycols and
fatty acid
esters of sorbitan, and mixtures thereof.
Besides inert diluents, the oral compositions can also include adjuvants such
as wetting agents, emulsifying and suspending agents, sweetening, flavoring,
coloring, perfuming and preservative agents.
Suspensions, in addition to the active therapeutic polymer conjugates may
contain suspending agents as, for example, ethoxylated isostearyl alcohols,
62
CA 02818071 2013-06-06
polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose,
aluminum
rnetahydroxide, bentonite, agar-agar and tragacanth, and mixtures thereof.
Formulations for rectal or vaginal administration may be presented as a
suppository, which may be prepared by mixing one or more therapeutic polymer
conjugates with one or more suitable nonirritating excipients or carriers
comprising
for example, cocoa butter, polyethylene glycol, a suppository wax or a
salicylate,
and which is solid at room temperature, but liquid at body temperature and,
therefore, will melt in the rectum or vaginal cavity and release the active
agent.
Formulations which are suitable for vaginal administration also include
pessaries, tampons, creams, gels, pastes, foams or spray formulations
containing
such carriers as are known in the art to be appropriate_
Dosage forms for the topical or transdermal administration of a therapeutic
polymer conjugate(s) include powders, sprays, ointments, pastes, creams,
lotions,
gels, solutions, patches and inhalants. The active component may be mixed
under
sterile conditions with a pharmaceutically acceptable carrier, and with any
preservatives, buffers, or propellants which may be required.
The ointments, pastes, creams and gels may contain, in addition to ligand(s),
excipients, such as animal and vegetable fats, oils, waxes, paraffins, starch,
tragacanth, cellulose derivatives, polyethylene glycols, silicones,
bentonites, silicic
acid, talc and zinc oxide, or mixtures thereof.
Powders and sprays can contain, in addition to a therapeutic polymer
conjugate(s), excipients such as lactose, talc, silicic acid, aluminum
hydroxide,
calcium silicates and polyamide powder, or mixtures of these substances.
Sprays
can additionally contain customary propellants, such as
chlorofluorohydrocarbons
and volatile unsubstituted hydrocarbons, such as butane and propane.
The therapeutic polymer conjugate(s) can be alternatively administered by
aerosol. This is accomplished by preparing an aqueous aerosol, liposomal
preparation or solid particles containing the compound_ A nonaqueous (e.g.,
fluorocarbon propellant) suspension could be used. Sonic nebulizers are
preferred
63
CA 02818071 2013-06-06
because they minimize exposing the agent to shear, which can result in
degradation
of the compound.
Ordinarily, an aqueous aerosol is made by formulating an aqueous solution
or suspension of the agent together with conventional pharmaceutically
acceptable
carriers and stabilizers. The carriers and stabilizers vary with the
requirements of
the particular compound, but typically include nonionic surfactants (Tweens,
Pluronics, or polyethylene glycol), innocuous proteins like serum albumin,
sorbitan
esters, oleic acid, lecithin, amino acids such as glycine, buffers, salts,
sugars or sugar
alcohols. Aerosols generally are prepared from isotonic solutions.
Transdermal patches have the added advantage of providing controlled
delivery of a therapeutic polymer conjugate(s) to the body. Such dosage forms
can
be made by dissolving or dispersing the agent in the proper medium. Absorption
enhancers can also be used to increase the flux of the ligand across the skin.
The
rate of such flux can be controlled by either providing a rate controlling
membrane
or dispersing the peptidomimetic in a polymer matrix or gel.
Ophthalmic formulations, eye ointments, powders, solutions and the like, are
also contemplated as being within the scope of this invention.
Pharmaceutical compositions of this invention suitable for parenteral
administration comprise one or more therapeutic polymer conjugate(s) in
combination with one or more pharmaceutically acceptable sterile isotonic
aqueous
or nonaqueous solutions, dispersions, suspensions or emulsions, or sterile
powders
which may be reconstituted into sterile injectable solutions or dispersions
just prior
to use, which may contain antioxidants, buffers, bacteriostats, solutes which
render
the formulation isotonic with the blood of the intended recipient or
suspending or
thickening agents.
Examples of suitable aqueous and nonaqueous carriers which may be
employed in the pharmaceutical compositions of the invention include water,
ethanol, polyols (such as glycerol, propylene glycol, polyethylene glycol, and
the
like), and suitable mixtures thereof, vegetable oils, such as olive oil, and
injectable
64
CA 02818071 2013-06-06
organic esters, such as ethyl oleate. Proper fluidity can be maintained, for
example,
by the use of coating materials, such as lecithin, by the maintenance of the
required
particle size in the case of dispersions, and by the use of surfactants.
These compositions may also contain adjuvants such as preservatives,
wetting agents, emulsifying agents and dispersing agents. Prevention of the
action
of microorganisms may be ensured by the inclusion of various antibacterial and
antifungal agents, for example, paraben, chlorobutanol, phenol sorbic acid,
and the
like. It may also be desirable to include isotonic agents, such as sugars,
sodium
chloride, and the like into the compositions. In addition, prolonged
absorption of the
injectable pharmaceutical form may be brought about by the inclusion of agents
which delay absorption such as aluminum monostearate and gelatin.
In some cases, in order to prolong the effect of a drug, it is desirable to
slow
the absorption of the drug from subcutaneous or intramuscular injection. This
may
be accomplished by the use of a liquid suspension of crystalline or amorphous
material having poor water solubility. The rate of absorption of the drug then
depends upon its rate of dissolution which, in turn, may depend upon crystal
size
and crystalline form. Alternatively, delayed absorption of a parenterally
administered drug form is accomplished by dissolving or suspending the drug in
an
oil vehicle.
Injectable depot forms are made by forming miCroencapsule matrices of
therapeutic polymer conjugate(s) in biodegradable polymers such as polylactide-
polyglycolide. Depending on the ratio of drug to polymer, and the nature of
the
particular polymer employed, the rate of drug release can be controlled.
Examples
of other biodegradable polymers include poly(orthoesters) and
poly(anhydrides).
Depot injectable formulations are also prepared by entrapping the drug in
liposomes
or microemulsions which are compatible with body tissue.
When the therapeutic polymer conjugate(s) of the present invention are
administered as pharmaceuticals, to humans and animals, they can be given per
se or
as a pharmaceutical composition containing, for example, 0.1 to 99.5% more
CA 02818071 2013-06-06
preferably, 0.5 to 90%) of active ingredient in combination with a
pharmaceutically
acceptable carrier.
The preparations of agents may be given orally, parenterally, topically, or
rectally. They are of course given by forms suitable for each administration
route.
For example, they are administered in tablets or capsule form, by injection,
inhalation, eye lotion, ointment, suppository, infusion; topically by lotion
or
ointment; and rectally by suppositories. Oral administration is preferred.
The phrases "parenteral administration" and "administered parenterally" as
used herein means modes of administration other than enteral and topical
administration, usually by injection, and includes, without limitation,
intravenous,
intramuscular, intraarterial, intrathecal, intracapsular, intraorbital,
intracardiac,
intradermal, intraperitoneal, transtracheal, subcutaneous,
subcuticular,.intraarticular,
subcapsular, subarachnoid, intraspinal and intrasternal injection and
infusion.
The phrases "systemic administration," "administered systemically,"
"peripheral administration" and "administered peripherally" as used herein
mean the
administration of a therapeutic polymer conjugate, drug or other material
other than
directly into the central nervous system, such that it enters the patient's
system and,
thus, is subject to metabolism and other like processes, for example,
subcutaneous
administration.
These therapeutic polymer conjugate(s) may be administered to humans and
other animals for therapy by any suitable route of administration, including
orally,
nasally, as by, for example, a spray, rectally, intravaginally, parenterally,
intracisternally and topically, as by powders, ointments or drops, including
buccally
and sublingually.
Regardless of the route of administration selected, the therapeutic polymer
conjugate(s), which may be used in a suitable hydrated form, and/or the
pharmaceutical compositions of the present invention, are formulated into
pharmaceutically acceptable dosage forms by conventional methods known to
those
of skill in the art.
66
CA 02818071 2013-06-06
Actual dosage levels of the active ingredients in the pharmaceutical
compositions of this invention may be varied so as to obtain an amount of the
active
ingredient which is effective to achieve the desired therapeutic response for
a
particular patient, composition, and mode of administration, without being
toxic to
the patient.
(c) Physical Structures Of The Subject Compositions
The subject polymers may be formed in a variety of shapes. For example, in
certain embodiments, subject polymer matrices may be presented in the form of
microparticles or nanoparticles. Microspheres typically comprise a
biodegradable
polymer matrix incorporating a drug. Microspheres can be formed by a wide
variety
of techniques known to those of skill in the art. Examples of microsphere
forming
techniques include, but are not limited to, (a) phase separation by
emulsification and
subsequent organic. solvent evaporation (including complex emulsion methods
such
as oil in water emulsions, water in oil emulsions and water-oil-water
emulsions); (b)
coacervation-phase separation; (c). melt dispersion; (d) interfacial
deposition; (e) in
situ polymerization; (f) spray drying and spray congealing; (g) air suspension
coating; and (h) pan and spray coating. These methods, as well as properties
and
characteristics of microspheres are disclosed in, for example, U.S. Pat. No.
4,652,441; U.S. Pat. No. 5,100,669; U.S. Pat. No. 4,526,938; WO 93/24150;
EPA 0258780 A2; U.S. Pat. No. 4,438,253; and U.S. Pat. 5,330,768.
To prepare microspheres of the present invention, several methods can be
employed depending upon the desired application of the delivery vehicles.
Suitable
methods include, but are not limited to, spray drying, freeze drying, air
drying,
vacuum drying, fluidized-bed drying, milling, co-precipitation and critical
fluid
extraction. In the case of spray drying, freeze drying, air drying, vacuum
drying,
fluidized-bed drying and critical fluid extraction; the components
(stabilizing polyol,
bioactive material, buffers, etc.) are first dissolved or suspended in aqueous
conditions. In the case of milling, the components are mixed in the dried form
and
milled by any method known in the art. In the case of co-precipitation, the
components are mixed in organic conditions and processed as described below.
67
CA 02818071 2013-06-06
Spray drying can be used to load the stabilizing polyol with the bioactive
material.
The components are mixed under aqueous conditions and dried using precision
nozzles to produce extremely uniform droplets in a drying chamber. Suitable
spray
drying machines include, but are not limited to; Buchi, NIRO, APV and Lab-
plant
spray driers used according to the manufacturer's instructions.
The shape of microparticles and nanoparticles may be determined by
scanning electron microscopy. Spherically shaped nanoparticles are used in
certain
embodiments, for circulation through the bloodstream. If desired, the
particles may
be fabricated using known techniques into other shapes that are more useful
for a
specific application.
In addition to intracellular delivery of a therapeutic agent, it also possible
that particles of the subject compositions, such as microparticles or
nanoparticles,
may undergo endocytosis, thereby obtaining access to the cell. The frequency
of
such an endocytosis process will likely depend on the size of any particle.
In certain embodiments, solid articles useful in defining shape and providing
rigidity and structural strength to the polymeric matrices may be used. For
example,
a polymer may be formed on a mesh or other weave for implantation. A polymer
may also be fabricated as a stent or as a shunt, adapted for holding open
areas within
body tissues or for draining fluid from one body cavity or body lumen into
another.
Further, a polymer may be fabricated as a drain or a tube suitable for
removing fluid
from a post-operative site, and in some embodiments adaptable for use with
closed
section drainage systems such as Jackson-Pratt drains and the like as are
familiar in
the art.
The mechanical properties of the polymer may be important for the
processability of making molded or pressed articles for implantation. For
example,
the glass transition temperature may vary widely but must be sufficiently
lower than
the temperature of decomposition to accommodate conventional fabrication
techniques, such, as compression molding, extrusion, or injection molding.
(d) Biodegradability And Release Characteristics
= 68
CA 02818071 2013-06-06
In certain embodiments, the polymers and blends of the present invention,
upon contact with body fluids, undergo gradual degradation. The life of a
biodegradable polymer in vivo depends upon, among other things, its molecular
weight, crystallinity, biostability, and the degree of crosslinking. In
general, the
greater the molecular weight, the higher the degree of crystallinity, and the
greater
the biostability, the slower biodegradation will be.
If a subject composition is formulated with a therapeutic agent or other
material, release of such an agent or other material for a sustained or
extended
period as compared to the release from an isotonic saline solution generally
results.
Such release profile may result in prolonged delivery (over, say 1 to about
2,000
hours, or alternatively about 2 to about SOO hours) of effective amounts
(e.g., about
0.0001 mg/kg/hour to about 1- 0 mg/kg/hour) of the agent or any other material
associated with the polymer.
A variety of factors may affect the desired rate of hydrolysis of polymers of
the subject invention, the desired softness and flexibility of the resulting
solid
matrix, rate and extent of bioactive material release. Some of such factors
include
the selection/identity of the various subunits, the enantiomeric or
diastereomeric
purity of the monomeric subunits, homogeneity of subunits found in the
polymer,
and the length of the polymer. For instance, the present invention
contemplates
heteropolymers with varying linkages, and/or the inclusion of other monomeric
elements. in the polymer, in order to control, for example, the rate of
biodegradation
of the matrix.
To illustrate further, a wide range of degradation rates may be obtained by
adjusting the hydrophobicities of the backbones or side chains of the polymers
while
still maintaining sufficient biodegradability for the use intended for any
such
polymer. Such a result may be achieved by varying the various functional
groups of
the polymer. For example, the combination of a hydrophobic backbone and a
hydrophilic linkage produces heterogeneous degradation because cleavage is
encouraged whereas water penetration is resisted.
69
CA 02818071 2013-06-06
One protocol generally accepted in the field that may be used to determine
the release rate of any therapeutic agent or other material loaded in the
polymer
matrices of the present invention involves degradation of any such matrix in a
0.1 M =
PBS solution (pH 7.4) at 37 C, an assay known in the art. For purposes of the
present invention, the term "PBS protocol" is used herein to refer to such
protocol.
In certain instances, the release rates of different polymer systems of the
present invention may be compared by subjecting them to such ea protocol. In
certain instances, it may be necessary to process polymeric systems in the
same
fashion to allow direct and relatively accurate comparisons of different
systems to be
made. For example, the present invention teaches several different means of
formulating the polymeric, matrices of the present invention. Such comparisons
may indicate that any one polymeric system releases incorporated material at a
rate
from about 2 or less to about 1000 or more times faster than another polymeric
system.
Alternatively, a comparison may reveal a rate difference of about 3, 5, 7, 10,
25, 50, 100, 250, 500 or 750 times. Even higher rate differences are
contemplated
by the present invention and release rate protocols.
In certain embodiments, when formulated in a certain manner, the release
rate for polymer systems of the present invention may present as mono- or bi-
phasic.
Release of any material incorporated into the polYmer matrix, which is often
provided as a microsphere, may be characterized in certain instances by an
initial
increased release rate, which may release from about 5 to about 50% or more of
any
incorporated material, or alternatively about 10, 15, 20, 25, 30 or 40%,
followed by
a release rate of lesser magnitude.
The release rate of any incorporated material may also be characterized by
the amount of such material released per day per mg of polymer matrix. For
example, in certain embodiments, the release rate may vary from about 1 ng or
less
of any incorporated material per day per mg of polymeric system to about 500
or
more ng/day/ing. Alternatively, the release rate may be about 0.05, 0.5, 5,
10, 25,
CA 02818071 2013-06-06
50, 75, 100, 125, 150, 175, 200, 250, 300, 350, 400, 450, or 500 ng/day/ing.
In still
other embodiments, the release rate of any incorporated material may be 10,000
ng/day/mg, or even higher_ In certain instances, materials incorporated and
characterized by such release rate .protocols may include therapeutic agents,
fillers,
and other substances.
In another aspect, the rate of release of any material from any polymer
matrix of the present invention may be presented as the half-life of such
material in
the matrix.
In addition to the embodiment involving protocols for in vitro determination
of release rates, in vivo protocols, whereby in certain instances release
rates for
polymeric systems may be determined in vivo, are also contemplated by the
present
invention. Other assays useful for determining the release of any material
from the -
polymers of the present system are known in the art.
(e) Implants and Delivery Systems
In its simplest form, a biodegradable delivery system for a therapeutic agent
consists of a dispersion of such a therapeutic agent in a polymer matrix. In
other
embodiments, an article is used for implantation, injection, or otherwise
placed
totally or partially within the body, the article comprising the subject
compositions.
It is particularly important that such an article result in minimal tissue
irritation
when implanted or injected into vasculated tissue.
Biodegradable delivery systems, and articles thereof, may be prepared in a
variety of ways known in the art. The subject polymer may be melt-processed
using
.- conventional extrusion or injection molding techniques, or these products
may be
prepared by dissolving in an appropriate solvent, followed by formation of the
=
device, and subsequent removal of the solvent by evaporation or extraction.
Once a system or implant article is in place, it should remain in at least
partial contact with a biological fluid, such as blood, internal organ
secretions,
mucus membranes, cerebrospinal fluid, and the like to allow for sustained
release of
any encapsulated therapeutic agent.
71
CA 02818071 2013-06-06
Methods of Manufacturing
Generally, compounds of the present invention can be prepared in one of two
ways: monomers bearing therapeutic agents, targeting ligands, and/or
cyclodextrin
moieties can be polymerized, or polymer backbones can be derivatized with
therapeutic agents, targeting ligands, and/or cyclodextrin moieties.
Thus, in one embodiment, the present invention contemplates the synthesis
of compounds of the invention by reacting monomers M-L-CD and M-L-D (and,
optionally, M-L-T), wherein
CD represents a cyclic moiety, such as a cyclodextrin molecule, or derivative
thereof;
L, independently for each occurrence, may be absent or represents a linker
group;
D, independently for each 'occurrence, represents the same or different
therapeutic agent or prodrugs thereof;
T, independently for each occurrence, represents the same or different
targeting ligand Or precursor thereof; and
M represents a monomer subunit bearing one or more reactive moieties
capable of undergoing a polymerization reaction with one or more other M in
the
monomers in the reaction mixture, under conditions that cause polymerization
of the
monomers to take place.
In certain embodiments, the reaction mixture may further comprise
monomers that do not bear CD, T, or D moieties, e.g., to space the derivatized
monomer units throughout the polymer.
In an alternative embodiment, the invention contemplates synthesizing a
compound of the present invention by reacting a polymer P (the polymer bearing
a
plurality of reactive groups, such as carboxylic acids, alcohols, thiols,
amines,
epoxides, etc.) with grafting agents X-L-CD and Y-L-D (and, optionally, Z-L-
T),
wherein
CD represents a cyclic moiety, such as a cyclodextrin molecule, or derivative
thereof;
72
CA 02818071 2013-06-06
L, independently for each occurrence, may be absent or represents a linker
group;
D, independently for each occurrence, represents the same or different
therapeutic agent or prodrugs thereof;
T, independently for each occurrence, represents the same or different
targeting ligand or precursor thereof;
X, independently for each occurrence, represents a reactive group, such as
carboxylic acids, alcohols, thiols, amines, epoxides, etc., capable of forming
a
covalent bond with a reactive group of the polymer; and
Y and Z, independently for each occurrence, represent inclusion hosts or
reactive groups, such as carboxylic acids, alcohols, thiols, amines, epoxides,
etc.,
capable of forming a covalent bond with a reactive group of the polymer or
inclusion complexes with CD moieties grafted to the polymer, under conditions
that
cause the grafting agents to form covalent bonds and/or inclusion complexes,
as
appropriate, with the polymer or moieties grafted to the polymer.
For example, if the polymer includes alcohols, thiols, or amines as reactive
groups, the grafting agents may include reactive groups that react with them,
such as
isocyanates, isothiocyanates, acid chlorides, acid anhydrides, epoxides,
ketenes,
sulfonyl chlorides, activated carboxylic acids (e.g., carboxylic acids treated
with an
activating agent such as PyBrOP, carbonyldihnidazole, or another reagent that
reacts
with a carboxylic acid to form a moiety susceptible to nucleophilic attack),
or other
electrophilic moieties known to those of skill in the art. In certain
embodiments, a
catalyst may be needed to cause the reaction to take place (e.g., a Lewis
acid, a
transition metal catalyst, an amine base, etc.) as will be understood by those
of skill
in the art.
In certain embodiments, the different grafting agents are reacted with the
polymer simultaneously or substantially simultaneously (e.g., in a one-pot
reaction),
or are reacted sequentially with the polymer (optionally with a purification
and/or
wash step between reactions).
Another aspect of the present invention is a method for manufacturing the
linear or branched cyclodextrin-containing polymers represented by formulae
73
CA 02818071 2013-06-06
While the discussion below focuses on the preparation of linear cyclodextrin
molecules, one skilled in the art would readily recognize that the methods
described
can be adapted for producing branched polymers by choosing an appropriate
comonomer A precursor.
Accordingly, one embodiment of the invention is a method of preparing a
linear cyclodextrin copolymer. According to the invention, a linear
cyclodextrin
copolymer of the invention may be prepared by copolymerizing a cyclodextrin
monomer precursor disubstituted with an appropriate leaving group with a
comonomer A precursor capable of displacing the leaving groups. The leaving
group, which may be the same or different, may be any leaving group known in
the
art which may be displaced upon copolymerization with a comonomer A precursor.
In a preferred embodiment, a linear cyclodextrin copolymer may be prepared by
iodinating a cyclodextrin monomer precursor to form a diiodinated cyclodextrin
monomer precursor and copolymerizing the diiodinated cyClodextrin monomer
precursor with a comonomer A precursor to form a linear cyclodextrin copolymer
having a repeating unit of formula II or III, or a combination thereof, each
as
described above. While examples presented below discuss iodinated cyclodextrin
moieties, one skilled in the art would readily recognize that the present
invention
contemplates and encompasses cyclodextrin moieties wherein other leaving
groups
such as alkyl and aryl sulfonate may be present instead of iodo groups. In a
preferred embodiment, a method of preparing a linear cyclodextrin copolymer of
the
invention by iodinating a cyclodextrin monomer precursor as described above to
form a diiodinated cyclodextrin monomer precursor of formula TVa, IVb, We or a
mixture thereof:
74
CA 02818071 2013-06-06
IMF 111,
IVa IVb
IVc
The diiodinated cyclodextrin may be prepared by any means known in the
art. (Tabushi et al. J. Am. Chem. 106, 5267-5270 (1984); Tabushi et al. J. Am.
Chem. 106, 4580-4584 (1984)). For example, P--cyclodextrin may be reacted with
biphenyl-4,4'-disulfonyl chloride in the presence of anhydrous pyridine to
form a
biphenyl-4,4'-disulfonyl chloride capped J3-cyclodextrin which may then be
reacted
with potassium iodide to produce diiodo-13-cyclodextrin. The cyclodextrin
monomer
precursor is iodinated at only two positions. By copolymerizing the
diiodinated
cyclodextrin monomer precursor with a comonomer A precursor, as described
above, a linear cyclodextrin polymer having a repeating unit of Formula la,
Ib, or a
combination thereof, also as described above, may be prepared. If appropriate,
the
iodine or iodo groups may be replaced with other known leaving groups.
Also according to the invention, the iodo groups or other appropriate leaving
group may be displaced with a group that permits reaction with a comonomer A
precursor, as described above. For example, a diiodinated cyclodextrin monomer
precursor of formula IVa, 1Vb, IVc or a mixture thereof may be aminated to
form a
diaminated cyclodextrin monomer precursor of formula Va, Vb, Vc or a mixture
thereof:
CA 02818071 2013-06-06
NH2 N H
Mir Mir
Va Vb
N H2 N H
NH2
1111,
VC
NI-I2
The diaminated cyclodextrin monomer precursor may be prepared by any
means known in the art. (Tabushi et-al. Tetrahedron Lett. 18:11527-1530
(1977);
Mungall etal., J. Org. Chem. 16591662 (1975)). For example, a diiodo-f3-
cyclodextrin may be reacted with sodium azide and then reduced to forni a
diamino-
r3-cyclodextrin). The cyclodextrin monomer precursor is aminated at only two
positions. The diaminated cyclodextrin monomer precursor may then be
copolymerized with a comonomer A precursor, as described above, to produce a
linear cyclodextrin copolymer having a repeating unit of formula 11-111 or a
combination thereof, also as described above. However, the amino functionality
of a
diaminated cyclodextrin monomer precursor need not be directly attached to the
cyclodextrin moiety. Alternatively, the amino functionality or another
nucleophilic
functionality may be introduced by displacement of the iodo or other
appropriate
leaving groups of a cyclodextrin monomer precursor with amino group containing
moieties such as, for example, HSCH2CH2NH2 (or a di-nucleophilic molecule more
generally represented by HW-(CRIR.1)n-WH wherein W, independently for each
occurrence, represents 0, S, or NR1; R1 and R2, independently for each
occurrence,
represent H, (un)substituted alkyl, (un)substituted aryl, (un)substituted
heteroalkyl,
(un)substituted heteroaryl) with an appropriate base such as a metal hydride,
alkali
or alkaline carbonate, or tertiary amine to form a diaminated cyclodextrin
monomer
precursor of formula Vd, Ve, Vf or a mixture thereof:
76
CA 02818071 2013-06-06
Vd Ve Vf
HNi NH2
NH2 2
1111,
11,
H2N
A linear oxidized cyclodextrin-containing copolymer of the invention may
also be prepared by oxidizing a reduced linear cyclodextrin-containing
copolymer of
the invention as described below. This method may be performed as long as the
comonomer A does not contain an oxidation sensitive moiety or group such as,
for
example, a thiol.
According to the invention, a linear cyclodextrin copolymer of the invention
may be oxidized so as to introduce at least one oxidized cyclodextrin monomer
into
the copolymer such that the oxidized cyclodextrin monomer is an integral part
of the
polymer backbone. A linear cyclodextrin copolymer which contains at least one
oxidized cyclodextrin monomer is defined as a linear oxidized cyclodextrin
copolymer or a linear oxidized cyclodextrin-containing polymer. The
cyclodextrin
monomer may be oxidized on either the secondary or primary hydroxyl side of
the
cyclodextrin moiety. If more than one oxidized cyclodextrin monomer is present
in
a linear oxidized cyclodextrin copolymer of the invention, the same or
different
cyclodextrin monomers oxidized on either the primary hydroxyl side, the
secondary
hydroxyl side, or both may be present. For illustration purposes, a linear
oxidized
cyclodextrin copolymer with oxidized secondary hydroxyl groups has, for
example,
at least one unit of formula VIa or VIb:
77
CA 02818071 2013-06-06
0 0
____________________________ A _____
Via
_______________________________ A __
Vlb
0 0
In formulae Vla and VIb, C is a substituted or unsubstituted oxidized
cyclodextrin monomer and A is a comonomer bound, i.e., covalently bound, to
the
oxidized cyclodextrin C. Also in formulae VIa and VIb, oxidation of the
secondary
hydroxyl groups leads to ring opening of the cyclodextrin moiety and the
formation
of aldehyde groups.
A linear oxidized cyclodextrin copolymer may be prepared by oxidation of a
linear cyclodextrin copolymer as discussed above. Oxidation of a linear
cyclodextrin copolymer of the invention may be accomplished by oxidation
techniques known in the art. (Hisamatsu et al., Starch 44:188-191(1992)).
Preferably, an oxidant such as, for example, sodium periodate is used. It
would be
understood by one of ordinary skill in the art that under standard oxidation
conditions that the degree of oxidation may vary or be varied per copolymer.
Thus
in one embodiment of the invention, a linear oxidized copolymer of the
invention
may contain one oxidized cyclodextrin monomer. In another embodiment,
substantially all cyclodextrin monomers of the copolymer would be oxidized.
Another method of preparing a linear oxidized cyclodextrin copolymer of the
invention involves the oxidation of a diiodinated or diaminated cyclodextrin
78
CA 02818071 2013-06-06
monomer precursor, as described above, to form an oxidized diiodinated or
diaminated cyclodextrin monomer precursor and copolymerization of the oxidized
diiodinated or diaminated cyclodextrin monomer precursor with a comonomer A
precursor. In a preferred embodiment, an oxidized diiodinated cyclodextrin
monomer precursor of formula Vila, VIIb, Vile, or a mixture thereof:
0 0
Vila 0 0
=
VI lb
0 0
may be prepared by oxidation of a diiodinated cyclodextrin monomer precursor
of
formulae Na, IVb, fVc, or a mixture thereof, as described above. In another
preferred embodiment, an oxidized diarninated cyclodextrin monomer precursor
of
formula VIIIa, VIIIb, VIIIc or a mixture thereof:
79
CA 02818071 2013-06-06
0 0
Villa 0 0 NH2
NH2 NH2
VIIIc
=
VilIb NH2
H2! ç NH2
may be prepared by amination of an oxidized diiodinated cyclodextrin monomer
precursor of formulae Vila, VIIb, Vile, or a mixture thereof, as described
above. In
still another preferred embodiment, an oxidized diaminated cyclodextrin
monomer
precursor of formula IXa, IXb, IX.c or a mixture thereof:
o
IXa S
IXe
H
NH2 2N
IXb NH2
H/'
NH2 2N
may be prepared by displacement of the iodo or other appropriate leaving
groups of
an oxidized cyclodextrin monomer precursor disubstituted with an iodo or other
CA 02818071 2013-06-06
appropriate leaving group with the amino or other nucleophilic group
containing
moiety such as, e.g. HSCH2CH2NH2 (or a di-nucleophilic molecule more generally
represented by HW-(CRIR2)1-WH wherein W, independently for each occurrence,
represents 0, S, or NR,; R.1 and R2, independently for each occurrence,
represent H,
(un)substituted alkyl, (un)substituted aryl, (un)substituted heteroalkyl,
(un)substituted heteroaryl) with an appropriate base such as a metal hydride,
alkali
or alkaline carbonate, or tertiary amine.
Alternatively, an oxidized diiodinated or diaminated cyclodextrin monomer
precursor, as described above, may be prepared by oxidizing a cyclodextrin
monomer precursor to form an oxidized cyclodextrin monomer precursor and then
diiodinating and/or diaminating the oxidized cyclodextrin monomer, as
described
above. As discussed above, the cyclodextrin moiety may be modified with other
leaving groups other than iodo groups and other amino group containing
functionalities. The oxidized' diiodinated or diarninated cyclodextrin monomer
precursor may then be copolymerized with a comonomer A precursor, as described
above, to form a linear oxidized cyclodextrin copolymer of the invention.
A linear oxidized cyclodextrin copolymer may also be further modified by
attachment of at least one ligand to the copolymer. The ligand is as described
above.
According to the invention, a linear cyclodextrin copolymer or linear
oxidized cyclodextrin copolymer may be attached to or grafted onto a
substrate. The
substrate may be any substrate as recognized by those of ordinary skill in the
art. In
another preferred embodiment of the invention, a linear cyclodextrin copolymer
or
linear oxidized cyclodextrin copolymer may be crosslinked to a polymer to
forrri,
respectively, a crosslinked cyclodextrin copolymer or a crosslinked oxidized
cyclodextrin copolymer. The polymer may be any polymer capable of
crosslinIcing
with a linear or linear oxidized cyclodextrin copolymer of the invention
(e.g.,
polyethylene glycol (PEG) polymer, polyethylene polymer). The polymer may also
be the same or different linear cyclodextrin copolymer or linear oxidized
cyclodextrin copolymer. Thus; for example, a linear cyclodextrin copolymer may
be
crosslinked to any polymer including, but not limited to, itself, another
linear
cyclodextrin copolymer, and a linear oxidized cyclodextrin copolymer. A
81
CA 02818071 2013-06-06
crosslinked linear cyclodextrin copolymer of the invention may be prepared by
reacting a linear cyclodextrin copolymer with a polymer in the presence of a
crosslinking agent. A crosslinked linear oxidized cyclodextrin copolymer of
the
invention may be prepared by reacting a linear oxidized cyclodextrin copolymer
with a polymer in the presence of an appropriate crosslinking agent. The
-crosslinking agent may be any crosslinking agent known in the art_ Examples
of
crosslinking agents include dihydrazides and disulfides. In a preferred
embodiment,
the crosslinking agent is a labile group such that a crosslinked copolymer may
be
uncrosslinked if desired. . .
A linear cyclodextrin copolymer and a linear oxidized cyclodextrin
copolymer of the invention may be characterized by any means known in the art.
Such characterization methods or techniques include, but are not limited to,
gel
permeation chromatography (GPC), matrix assisted laser desorption ionization-
time
of flight mass spectrometry (MALDI-TOF Mass spec), and 13C NMR., light
scattering and titration.
The invention also provides a cyclodextrin composition containing at least
one linear cyclodextrin copolymer and at least one linear oxidized
cyclodextrin
=
copolymer of the invention as described above. Accordingly, either or both of
the
linear cyclodextrin copolymer and linear oxidized cyclodextrin copolymer may
be
crosslinked to another polymer and/or bound to a ligand as described above.
Therapeutic compositions according to the invention contain a therapeutic
agent and
a linear cyclodextrin copolymer or a linear oxidized cyclodextrin copolymer,
including crosslinked copolymers, of the invention. A linear cyclodextrin
copolymer, a linear-oxidized cyclodextrin copolymer and their crosslinked
derivatives are as described above. The therapeutic agent may be any synthetic
or
naturally occurring biologically active therapeutic agent including those
known in
,the art. Examples of suitable therapeutic agents include, but are not limited
to,
antibiotics, steroids, polynucleotides (e.g., genomic DNA, cDNA, inRNA, double-
stranded RNA, and antisense oligonucleotides), plasmids, peptides, peptide
fragments, small molecules (e.g., doxorubicin) and other biologically active
macromolecules such as, for example, proteins and enzymes.
=
82
CA 02818071 2013-06-06
(g) Business Methods
Other aspects of the invention provides for certain methods of doing
business. In particular, practicing the methods of the invention may enable
novel
therapeutic compositions and improved formulations thereof. This technical
step,
when combined with one or more additional steps, provides for novel approaches
to
conduct a pharmaceutical, or preferably a life-science business. For example,
such
therapeutic prepared by the method of the invention may be tested for efficacy
as
therapeutics in a variety of disease- models, the potential therapeutic
compositions
then tested for toxicity and other safety-profiling before formulating,
packaging and
subsequently marketing the resulting formulation for the treatment of disease.
Alternatively, the rights to develop and market such formulations or to
conduct such
steps may be licensed to a third party for consideration.
Accordingly, in certain embodiments, the present invention provides a
method for conducting a pharmaceutical business, comprising:
a. manufacturing a formulation or kit including a pharmaceutical
composition of any of the compounds of claims 1-4; and
b. marketing to healthcare providers the benefits of using the
formulation or kit in the treatment of a disease or disorder.
In other embodiments, the present invention discloses a method for
conducting a pharmaceutical business, comprising:
a. providing a distribution network for selling a pharmaceutical
composition of any of the compounds of claims 1-4; and
b. providing instruction material to patients or physicians for using the
preparation in the treatment of a disease or disorder.
In certain embodiments, the present invention 'provides a method for
conducting a pharmaceutical business, comprising:
a. determining an appropriate formulation and dosage of a
pharmaceutical composition of any of the compounds of claims 1-4;
83
CA 02818071 2013-06-06
b. conducting therapeutic profiling of formulations identified in step (a),
for efficacy and toxicity in animals; and
c.
providing a distribution network for selling a preparation or .
preparations identified in step (b) as having an acceptable therapeutic
profile.
An additional step of the embodiment comprises providing a sales group for
.
-marketing the preparation to healthcare providers.
In still other embodiments, the present invention provides a method for .
conducting a pharmaceutical business, comprising:
a. determining an appropriate formulation and dosage of a
pharmaceutical composition of any of the compounds of claims 1-4; and
b. licensing, to a third party, the rights for further development and sale
of the formulation.
Exemplification
Materials. O-Cyclodextrin, "13-CD", (Cerestar USA, Inc. of Hammond, IN) was
dried in vacuo (<0.1 mTorr) at 120 C for 12 h before use.
OH
= 0
v.f
Ilir HO H
0 41.T.i0 01-1
O
HO
-
Olc
SOH
HO o OH 4
OH = 0
HO
HO
---\-.õ49t-___-1 Li
0
0 0 OH
HO
13-CD 13-CD
-
90 All the anhydrous solvents, HPLC grade solvents and other common organic
solvents were purchased from commercial suppliers and used without further
,
purification. Bipheny1-4,4'-disulfonyl chloride (Aldrich Chemical Company,
Inc. of
84
CA 02818071 2013-06-06
Milwaukee, WI) was recrystallized from chloroform/hexanes. Potassium iodide
was
powdered with a mortar and pestle and dried in an oven at 200 C. Polyethylene
glycol dipropanoicsuccinimide (PEG¨DiSPA, MW 3400), polyethylene glycol
dibutanoicsuccinimide (PEG¨DiSBA, MW 3400), and polyethylene glycol
dibenzotrizolecarbonate (PEG¨DiBTC, MW 3400) were purchased from Nektar
(Huntsville, AL). Polyethylene glycol di-p-nitrophenolcarbonate (PEG¨DiNPC,
MW 3400) was acquired from Sigma (St. Louis, MO). CPT was purchased from
Boehringer Ingelheim (Ingelheirn, Germany). Human plasma was purchased from
Sigma and reconstituted with DI water. Mouse plasma was prepared by centrifuge
removal of blood cells of fresh blood samples collected from BALB/C female
mice
(Charles River). 6A,6D-diiodo-6A,6D-dideoxy-16-cyclodextrin (CDDI, Scheme 2)
was
synthesized according to previous reported procedure by Hwang et. al
(Bioconjugate
Chem. 12,280-290). Deionized water (18-MQ-cm) was obtained by passing in-
house deionized water through a Barnstead E-pure purification system. NIVIR
spectra
were recorded on a Bruker AMX 500 MHzor a Varian 300 MHz spectrometer.
Mass spectral (MS) analysis was performed using either an electrospray mass
spectrometer equipped with LCQ ion trap (Thermo Finnigan) and fitted with an
electrospray ionization source or a MALDI-TOF mass spectrometer (Voyager DE-
.
PRO, Applied Biosystems). MWs=of the polymer samples were analyzed on a GPC
system equipped with a Hitachi L-6200 Intelligent Pump, an Anspec RI detector
(ERC-7512, Erma, Inc.), a Precision Detectors DLS detector (PD 2020), and
double
gel permeation columns (PL-aquagel-OH-40 8 pm 300 mm x 7.5 mm, Polymer
Laboratory) calibrated using polyethylene glycol standard and eluded using PBS
(1x) at a concentration of 20-50 mg/mL and at a 0.7 inUmin flow rate at
ambient
temperature. CD derivatives were analyzed with a C-18 reverse phase column on
a
HPLC system equipped with an UV detector (System Gold 168 Detector, Beaman
Coulter) and an evaporative light scattering EELS) detector (Sedex 75, Sedere,
France). CPT, CPT derivatives, and polymer-CPT conjugates were analyzed on
HPLC systems with a C-I8 reverse phase column (HIRPB-4438, 4.6 x 150 ram,
Richard Scientific) equipped with a fluorescence detector (FD-500, GTI/Spectro
Vision, Groton Technology, Inc.) using a gradient of potassium phosphate
buffer
CA 02818071 2013-06-06
(pH 4.1) and acetonitile. Excitation and emission wavelengths of the
fluorescence
detector were set at 370 nm and 440 nm, respectively.
Example 1: Bipheny1-4,4'-disulfonyl-A,D-Capped 13-Cyclodextrin, 1 (Tabushi et
al.
J. Am. Chem. Soc. 106, 5267-5270 (1984))
Scheme XV
oi sl=o
1
A 500 inL round bottom flask equipped with a magnetic stirbar, a Schlenlc
adapter and a septum was charged with 7.92 g (6.98 mmol) of dry P-cyclodextrin
and 250 mL of anhydrous pyridine (Aldrich Chemical Company, Inc.). The
resulting solution was stirred at 50 "C under nitrogen while 2.204 g (6.28
mmol) of
biphenyl-4,4'-disulfonyl chloride was added in four equal portions at 15 min
intervals. After stirring at 50 'C for an additional 3 h, the solvent was
removed in
vacuo and the residue was subjected to reversed-phase column chromatography
using a gradient elution of 0-40% acetonitrile in water. Fractions were
analyzed by
high performance liquid chromatography (I-IPLC) and the appropriate fractions
were
combined. After removing the bulk of the acetonitrile on a rotary evaporator,
the
resulting aqueous suspension was lyophilized to dryness. This afforded 3.39 g
(38%) of 1 as a colorless solid.
Example 2: 6A,6D-Diiodo-6A,6D-Dideoxy-p-cyclodextrin, 2 (Tabushi et al. J. Am.
Chem. 106, 4580-4584 (1984))
Scheme XVI
.11Z K1 >1
OS =41
6 0
2
86
CA 02818071 2013-06-06
A 40 mL centrifuge tube equipped with a magnetic stirbar, a Schlenk adapter
and a septum was charged with 1.02 g (7.2 mrnol) of 1, 3.54 g (21.3 minol) of
dry,
powdered potassium iodide (Aldrich) and 15 rnl, of anhydrous
N,N-dimethylformamide (DMF) (Aldrich). The resulting suspension was stirred at
80 C under nitrogen for 2 h. After cooling to room temperature, the solids
were
separated by filtration and the supernatant was collected. The solid
precipitate was
washed with a second portion of anhydrous DMF and the supernatants were
combined and concentrated in vacua. The residue was then dissolved in 14 mL of
water and cooled in an ice bath before 0.75 mL (7.3 mmol) of
tetrachloroethylene
(Aldrich) was added with rapid stirring. The precipitated product was filtered
on a
medium glass fit and washed with a small portion of acetone before it was
dried
under vacuum over P205 for 14 h. This afforded 0.90 g (92%) of 2 as a white
solid.
Example 3: 6A,6D-Bis-(2-aminoethylthio)-6A'6D-dideoxy-13-cyclodextrin, 3
(Tabushi, I: Shimokawa, K; Fugita, K. Tetrahedron Lett. 1977, 1527-1530)
Scheme XVII
HS
'SZ' NH
2
3
A 25 mL Schlenk flask equipped with a magnetic stirbar and a
septum was charged with 0_91 mL (7.37 mrnol) of a 0.81 M solution of sodium
2-aminoethylthiolate in ethanol. (Fieser, L.F.; Fieser, M. Reagents for
Organic
Synthesis; Wiley: New York, 1967; Vol. 3, pp.265-266). The solution was
evaporated to dryness and the solid was redissolved in 5mL of anhydrous DMF
(Aldrich). 6A,6D-Diiodo-6A,6D-dideoxy-P-eyelodextrin (2) (100mg, 7.38 x 10-5
mol)
was added and the resulting suspension was stirred at 60 *C under nitrogen for
2 h.
After cooling to room temperature, the solution was concentrated in vacua and
the
residue was redissolved in water. After acidifying with 0.1 N HC1, the
solution was
applied to a Toyopearl SP-650M ion-exchange column (NH4+form) and the product
was eluted with a 0 to OA M ammonium bicarbonate gradient. Appropriate
fractions
87
CA 02818071 2013-06-06
were combined and lyophilized to dryness. This afforded 80mg (79%) of 3 as a
white powder.
=
Alternative Synthesis of dicysteamine 13-CD 3.
To a solution of 4.69g (3.17 mmol) of 2 in 100 mL of degassed water was
added 0.489 g (6.34 mmol) of freshly sublimed cysteamine. The solution was
stirred under.reflux for 2 h. After cooling to room temperature and acidifying
with 1
N HC1, the solution was applied to a Toyopearl SP-650M ion-exchange column
(NH4+ form) and the product was eluted with a 0 to 0.2 M ammonium bicarbonate
gradient. Appropriate fractions were combined and lyophilized to dryness. This
procedure gave 1.87 g (39% yield) of a white solid. The solid was
characterized by
TLC (silica gel, n-PrOH-AcOEt-H20-NH3aq 5/3/3/1, detection by ninhydrin) and
exhibited a major spot corresponding to 3. Matrix-assisted laser
desorption/ionization (MALDI) time-of flight (TOF) mass spectrum was recorded
on 2 meter BUIE instrument supplied by PerSeptive Biosystems, Inc. MALDI-TOF
m/z calcd for 3: 1252, found: 1253.5 [M+H]4, 1275.5 [M+Nar, 1291.4 [1\4+Kr.
13C NIVIR (Bruker 500 MHz, D20) 5 ppm: 32.1 (S-C1-12) and 38.8 (CH2-NH2), 32.9
(C6 adjacent to S), 60.2 (C6 adjacent to OH), 70.8, 71.4, 72.5 (C2, C3, C5),
81.8
(C4), 101.7 (CI).
Example 4: 6A.,6D-Bis-(2-amino-2-carboxylethylthio)-6A'6D-dideoxy-13-
cyclodextrin, 4 (CD-BisCys)
Scheme XVIII
NH,
HS OH
0
HO SSOH
NH2 NH,
4
167 mL of 0.1 1\4 sodium carbonate buffer were degassed for 45 minutes in a
500 nil, 2-neck round bottom flask equipped with a magnetic stir bar, a
condenser
and septum. To this solution were added 1.96 g (16.2 mmol) of L-cysteine and
10.0
88
CA 02818071 2013-06-06
g (73.8 mmol) of diiodo, deoxy-P-cyclodextrin 2. The resulting suspension was
heated at a reflux temperature for 4.5 h until the solution turned clear
(colorless).
The solution was then cooled to room temperature and acidified to pH 3 using
IN
HCI. The product was precipitated by slow addition of acetone (3 times weight
ratio
of the solution). This afforded 9.0 g crude material containing CD-biscysteine
(90.0%), unreacted cyclodextrin, CD-mono-cysteine and cystine. The resulting
solid
was subjected to anionic exchange column chromatography (SuperQ650M, Tosoh
Bioscience) using a gradient elution of 0-0.4M ammonium bicarbonate. All
fractions were analyzed by HPLC. The desired fractions were combined and the
solvent was reduced to 100 rriL under vacuum. The final product was either
precipitated by adding acetone or by adding methanol (3 times weight ratio of
the
solution). 4 was obtained in 60-90% yield. 1HNMR (D20) 5 5.08 in, 7H, CD-2-
CH), 179-3.94 (m, 30H, CD-3,4-CH, CD-CH2, Cys-CH), 3.49-3.62 (m, 1411, CD-5,
6-CH), 2.92-3.30(m, 411, Cys-CH2).13C NMR (D20) 5 172.3, 101.9, 83.9, 81.6,
81.5, 73.3, 72.2, 72.0,60.7, 54.0, 34.0, 30.6. ES1/MS (m/z): 1342 [Mr, 1364
[M+
Nar. Purity of 4 was confirmed by RPLC.
=
Example 5: 6A,6D-Bis-(carboxylinethylthio)-6A.6D-dideoxy-fil-cyclodeXtrin, 5
(CDDM)
Scheme XIX
HS
HO
5
0
A 50 ml, of 0.1 M sodium carbonate solution was degassed for 2 h in a 100
mL 3-neck round bottom flask equipped with a magnetic stir bar, a condenser
and
septa. Mercaptoacetic acid (0.46 mL, 6.64 mmol) was syringed into the flask
and
pH of the solution was adjusted to 9.3 with IN sodium hydroxide. To this
resulting
solution was added 3.00 g (2.21 mmol) of di-iodo-P-cyclodextrin 2 and heated
at 80
"C for an hour. The solution temperature was increased 10 C every hour until
it
reached 100 C. After 3 h. at the reflux temperature, the clear colorless
solution was
cooled to room temperature and acidified to pH 3.5 using 1 N HC1. The crude
89
CA 02818071 2013-06-06
product was crashed out by slow addition of acetone (3 times weight ratio of
the
solution). The resulting solid was subjected to anionic exchange column
chromatography using a gradient elution of 0-0.4 M ammonium bicarbonate
solution. This afforded 1.8 g(63.4%) of 5 as a colorless solid. ESI/MS (m/z):
1281
[Mr. Purity of this compound was confirmed with 1-PLC.
Example 6: CD-Bis(Glutarnic acid-y-Benzyl ester) 6
Scheme XX
=
NH,
C COO;Lõ.....yo
-e}y'on OOR
>=11 0 0 11
0
0 6
A 50 inL round bottom flask equipped with a magnetic stirbar and a
condenser and a septum was charged with 0.101 g (0.425 mmol) of H-Glu(Obz1)-
OH and 0.15 g (0.106 mmol) of dio-ioclo l cyclodextrin 2 in 5 mL of degassed
0.1
M sodium carbonate solution. The solution mixture was heated at 100 C for 2
h.
The solution was then cooled to room temperature and acidified to pH 4 before
dialyzing in MWCO 500 membranes for 24 h. The yield of 6 was 0.142 g (83.6 N.
= Example 7 CD-BisLys(Z) 7
Scheme XXI
,CBZ
FIN
=
CBZ,NH ,CBZ
H,N
0
HO Y:11-7 OH
0 0
7
CA 02818071 2013-06-06
A 50 mL round bottom flask equipped with a magnetic stirbar and a
condenser and a septum was charged with 0.124 g (0.443 mmol) of 11-Lysine(Z)-
OH
and 0.15 g (0.111 mmol) of di-iodo-f-3-cyclodextrin 2 in 5mL of degassed 0.1M
sodium carbonate solution. The solution mixture was heated at 100 C for 4 h.
The
5 solution was then filtered and the pH of the filtrate is adjusted to 8.5
before
dialyzing in MWCO 500 membranes for 24 h. The yield of 7 was 0.124 g (68.9%).
Example 8: Synthesis of fl-cyclodextrin-Tosylate, 8 (Melton, L.D., and
Slessor,
= = K.N., Carbohydrate Research, 18, p. 29 (1971))
Scheme X.XII
Tosyl-CI
10 Tosyl
8
A 500 mL round-bottom flask equipped with a magnetic stirbar, a vacuum
adapter and a septum was charged with a solution of dry fl-cyclodextrin (8.530
g,
7.51 mmol) and 200 mL of dry pyridine. The solution was cooled to 0 C before
15 1.29 g (6.76 mmol) of tosyl chloride was added. The resulting solution
was allowed
to warm to room temperature overnight. The pyridine was removed as much as
possible in vacuo. The resulting residue was then recrystallized twice from 40
mL
of hot water to yield 7.54 (88%) of a white crystalline solid S.
20 Example 9: Synthesis of Iodo-13-cyclodextrin, 9
Scheme XXIII
KT
_______________________________________________________ )-
Tosyl
9
A round bottom flask with a magnetic stirbar and a Schlenk adapter is
25 charged with 8, 15 equivalents of Potassium iodide, and DMF. The
resulting
mixture is heated at 80 C for 3 h, after which the reaction is allowed to
cool to
room temperature. The mixture is then filtered to remove the precipitate and
the
filtrate evaporated to dryness and redissolved in water at 0 C.
Tetrachloroethylene
91
CA 02818071 2013-06-06
is added and the resulting slurry stirred vigorously at 0 C for 20 minutes.
The solid
9 is collected on a medium glass fit, triturated with acetone and stored over
P205.
Example 10: Synthesis of Cysteamine-f3-Cyclodextrin, 10
Scheme XXIV
HS
To a solution of 9 in 100 mL of degassed water is added 1 eq. of
freshly sublimed cysteamine. The solution is stirred under reflux for 2 h.
After
10 cooled to room temperature and acidified with 1 N HC1, the solution is
applied to a
Toyopearl SP-650M ion-exchange Column (NH4 form) and the product is eluted
with an ammonium bicarbonate gradient. Appropriate fractions are combined and
lyophilized to dryness to yield 10.
Example 11: Synthesis of Gly-CPT 11 (Greenwald et al., Bioorg. Med. Chem.,
1998, 6, 551-562)
Scheme XXV
0
-
N z
N
DMANDIPC 0
_____________________________________________________ 110
0 II TFA
HO 0
0
0
0 \ 0 TFA-NH!--ir
0 \ 0
11
t-Boc-glycine (0.9 g, 4.7 mmol) was dissolved in 350 mL of anhydrous
methylene chloride at room temperature, and to this solution were added D1PC
(0.75 mL, 4.7 mmol), DMAP (382 mg, 3.13 mmol) and camptothecin (0.55g,
1.57 mmol) at 0 C. The reaction mixture was allowed to warm to room
temperature
and left for 16 h. The solution was washed with 0.1 N Ha, dried and evaporated
under reduced pressure to yield a white solid, which was recrystallized from
92
CA 02818071 2013-06-06
methanol to give camptothecin-20-ester of t-Boc-glycine: 111 NMR(DMSO-d6)
7.5-8.8 (m), 7.3 (s),5.5 (s), 5.3 (s), 4 (m), 2.1 (m), 1.6 (s), 1.3 (d), 0.9
(t).
Camptothecin-20-ester of t-Boc-glycine (0.595 g, 1.06 mmol) was dissolved in a
mixture of methylene chloride (7.5 mL) and TFA (7.5 mL) and stirred at room
temperature for 1 h. Solvent was removed and the residue was recrystallized
from
methylene chloride and ether to give 0.45 g of 11.1H NMR (DMSO-d6) 87.7-8.5
(m); 7.2 (s), 5.6 (s), 5.4 (s), 4.4 (m), 2.2 (m), 1.6 (d), 1.0 (t), 13C NMR
(DMSO-d6)
M68.6, 166.6, 156.5, 152.2, 147.9,. 146.2, 144.3, 131.9, 130.6, 129.7, 128.8,
128.6,
128.0, 127.8, 119.0, 95.0, 77.6, 66.6, 50.5, 47.9, 30.2, 15.9, 7.9. ES1/MS
(m/z) =
expected 405; Found 406 (M+H).
Example 12 Synthesis of GlyGlyGly-CPT 12
Scheme XXVI
40 =
Uue.GlyGly0ly 0 N
TFA
0 = N ,
0 1 N
0 j = =
DMAPIDIPC 0
C a 11'11-Thr Cir
0
12
t-Boc-GlyGlyGly (1.359 g, 4.7 mmol) was dissolved in 350 mL of
anhydrous methylene chloride at room temperature and to this solution were
added
D1PC (0.75 mL, 4.7 mmol), DMAP (382 mg,,3.13 mmol) and camptothecin (0.55g,
1.57 mmol) at 0 C. The reaction mixture was allowed to warm to room
temperature
and left for 16 h. The solution was washed with 0.1 N HC1, dried and
evaporated
under reduced pressure to yield a white solid, which was recrystallized from
methanol to give camptothecin-20-ester of t-Boc-GlyGlyGly: 1H NMR(DMSO-d6) 5
8.40 (s), 8.25 (d), 7.91 (d), 7.78 (m), 7.65 (t), 7.26 (s), 7.05 (br, s), 5.65
(d), 5.40 (d),
5.25 (S), 5.10 (br, s), 3.75-4.42 (m), 2.15-2.35 (m), 1.45 (s),,0.95 (t)
Camptothecin-
20-ester of t-Boc-GlyGlyGly (1.5 g, 1.06 mmol) was dissolved in a mixture of
methylene chloride (10 inL) and TFA (10 mL) and stirred at room temperature
for
1 h. Solvent was removed under vacuum and the residue was re-dissolved in
methylene chloride. The solution was poured into ether to give instant
precipitate
93
=
CA 02818071 2013-06-06
(yellow). The precipitate was filtered and washed with cold ether to give 1.31
g of
12. 1H NM.R (DMSO-d6) 5 8.79 (s), 7.75-8.61 (m), 7.10 (s), 5.55 (s), 3.90-4.37
(m),
3.86 (s), 3.54 (s), 2.11-2.23 (m), 0.95 (t). ESI/MS (n/z) expected 519; Found
520
(M+H).
Stability of CPT-Peptide ester bond
11 and 12 were dissolved in PBS buffer (pH 7.4) at room temperature to
prepare a solution about 500 ug/mL. This solution was further diluted in 8.5%
H3PO4 to 10 ug/mL. Hydrolysis rate was analyzed using HPLC equipped with a Cis
RP (reverse phase) column and a fluorescence detector using a 50/50 (v/v) of
acetonitrile/potassium phosphate buffer (pH 4.1). The peaks of 11 (or 12) and
the -
released CPT (lactone form) were integrated. The stability of the ester bond
in
aqueous solution is peptide-length dependant. Thus the drug release rate
(hydrolysis
rate) can be tuned by adjusting the peptide length. See Figure 2.
Lactone Stability of CPT, 11 and 12 in Phosphate Buffered Saline (PBS)
CPT, 11 or 12 was dissolved in DMSO at 1 mg/mL and then diluted to 1
pg/mL with PBS (lx, pH 7.4). 30 pL of solution were injected into the HPLC at
room temperature at selected time intervals. The peak area from the CPT
lactone
form of CPT, 11 or 12) were integrated.
The rate of lactone ring opening for 11, 12 and CPT were studied in PBS
buffer (pH 7.4). Both 11 and 12 were very stable against ring-opening and no
carboxylate forms of 11 and 12 were detected throughout the study (7 hours).
On
the other hand, more than 60% of the CPT lactone form was transformed to its
carboxylate form in the same period of time. (See Fig. 3)
Example 13: Synthesis of Lys(BisCBZ)-CPT 13
Scheme XXVII
94
CA 02818071 2013-06-06
0 HO 0
0
N z N z
0 0
OH 0 0 0
0
CBZ CBZ
13
N, N-BisCBZ-Lysine (311 mg, 0.75 mmol) was dissolved in 56 mL of
anhydrous methylene chloride at room temperature. To this solution. were added
DIPC (0.12 mL, 0.75 mmol), DMAP (0.61 mg, 0.5 mmol) and camptothecin (0.087
g, 0.25 mmol) at 0 C. The reaction mixture was allowed to warm to room
temperature and left for 16 h. The solution was washed with 0.1 N HC1, dried
and
evaporated under reduced pressure to yield a light yellow solid, which was
recrystallized from methanol to give camptothecin-20-ester of N,N-BisCBZ-Lys
13.
Purification of 13 was satisfactory based on TLC andHPLC analysis.
Hydrolysis of 13 in aqueous solution is very slow and cannot be detected
using HPLC equipped with a UV detector. Hydrolysis rate of the ester bond of
CPT-peptide linker can be tuned not only by adjusting the length of peptide,
as
shown in Example 12, but also by using different amino acid linked directly
with
CPT's 20-0H.
The transformation of lactone form to carboxylate form of CPT and 13 were
also tested in PBS buffer. It was found that the transformation of lactone
form to
carboxylate form of compound 13 was much slower than that of free CPT,
indicating that lactone form (drug active form) can be stabilized by forming
an ester
with the ¨OH of CPT at its 20 position. (See Fig. 4)
Example 14 Synthesis Lys-Gly-CPT 14
11 is dissolved in chloroform. N, N-DiBoc-Lys-NHS (1.0 eq) is added
followed by triethylamine (1.0 eq). The mixture is stirred at rt for 16 hours
and
extracted twice with water and then dried with MgSO4. Solvent is removed under
CA 02818071 2013-06-06
high vacuum to yield N, N-DiBoc-Lys-Gly-CPT. To this compound is added a
mixture of equal volume CH2C12 and TFA and stirred at rt for 1 h. The solvent
is
then removed under vacuum. The residue is redissolved in CHCI3. Ether is added
to
the solution to crash out the product 14. The precipitate is washed several
times
=
with ether and then dried under vacuum. It is purified using a silica gel
column
chromatography to yield 14 in pure TFA salt form.
Example 15 Synthesis of Suc-Gly-CPT 15
A solution of succinic anhydride is mixed with 11(1 eq) in dry CHCI3 in the
presence of a catalytic amount of DMAP and DMA (1 eq). The mixture is stirred
at
rt for 24 hours to yield 15. 15 is purified by crystallization.
Example 16 Synthesis of Glu-Suc-Gly-CPT 16
is converted to its NHS ester using traditional DCC/NHS method. The
NHS ester of 15 is then reacted with. glutamic acid (1.0 eq) in DMSO in the
presence
of triethylamine. The solution is added to ether to precipitate 16. 16 is
purified by
15 crystallization.
Example 17 Synthesis of Glu-Bis(GlyCPT) 17
11 and Boc-Glu(NHS)-NHS (0.4 eq) are mixed in CHCI3 under argon before =
triethylamine (1 eq) is added to the mixture. The solution is stirred at it
for 16 h and
then washed with acidic water. The organic layer is dried and then solvent is
removed under vacuum. The resulting compound is purified using a silica gel
column chromatography. The purified compound is then dissolved in an equal
volume mixture of TFA and CH2Cb. The mixture is stirred at rt for 1 h and then
poured into ether. The precipitate 17 is washed with ether and dried under
vacuum.
Example 18 Synthesis of Cyclodextrin-Camptothecin 18
Scheme XXVIII
96
CA 02818071 2013-06-06
0 0 0
CO C 12 CD-NH2
N N N
0 0 0
a
CPT-CD O=<\ 0
o
Ci \ NH
CD
18
CPT (197 mg, 0.566 mmol) was vacuumed for 30 minutes. Dry chloroform
(100 mL) was added under argon. Phosgene (1.34 mL, 20% in toluene solution)
was
added at 0 C. Ice bath was removed and the solution was warmed up to room
temperature. Two hours later solvent was removed under high vacuum. Dry DMSO
(50 mL) was added to the residue, followed by 200 mg CD-NH2 (Cyclodextrin
Technology, Inc.) and triethylamine (4 mL, excess). 16 hours later, the
solution was ,
poured into 200 mL ether. Precipitate was washed with ether extensively and
then
dried. 167 mg yellow powder (13) was obtained (62% yield). TLC analysis
(silica
gel) of 18: Rf= 0 (developed with CHC13/Me0H v/v =5/1). TLC analysis of CPT:
Rf = 0.65 (developed with CHC13/Me0H v/v =5/1). Solubility: >10 mg/mL in
water. This indicates that solubility of CPT in water can be substantially
increased
when it is covalently attached to cyclodextrin molecule (free CPT solubility
in water
<0.004 mg/mL).
Example 19. Synthesis of CDDC-Dianhydride Copolymer 19,21 and its CPT
conjugate 20, 22
Scheme XXIX
97
CA 02818071 2013-06-06
=
o o
;iiiizH NH
NH2 ____________________________
0 0
19
0 OGlyCPT
TFA-GlyCPT
___________________ H
0 ty0GlyCPT
LVDGlyCPT
0 0
O 0
1+-7- 4 HOI 0
>
Licm
21
0
CPT-01y-,
TFA-GlyCPT 0 0
>
22 0Nc,o,
Gly-CPT
= A: Ethylenediamine tetraacetic dianhydride (25.6 mg, 0.1 mmol) and CDDC
5 (3, 125.3 mg, 0.1 mrnol) were dissolved in 2 mL of dry DMSO. The solution
was
heated at 50 C for 72 h. Water was added to the mixture, followed by addition
of 1
N NaOH to pH around 12. The polymer was dialyzed in 10,000 MWCO membrane
for 24 h. Precipitation was observed in the dialysis membrane. The solid was
removed and the remaining solution was dialyzed again in 10,000 MWCO
10 membrane for 24 h. A white powder 19 (75 mg) was obtained after
lyophilization.
11 is added to the polymer (19)/DMS0 solution in the presence of EDC (2
eq), NHS (1 eq), and DIEA (1.0 eq). The solution is stirred for 16 hand then
poured
into ether. The precipitate is washed with CH2C12 extensively until no free
drug is
observed in the washing solution. Compound 20 is obtained after drying under
high
15 vacuum.
B: Diethylenetriamine pentaacetic dianhydride (8.5 mg, 0.024 mmol) and
CDDC, 3 (30 mg, 0.024 mmol) were dissolved in 1-methyl-2pyridinone (2 mL).
98
CA 02818071 2013-06-06
The mixture was stirred at 64 `C for 4 days and then dialyzed in 10,000 MWCO
membrane for 2 days. A white powder 21 (3 mg) was obtained after
lyophilization.
11 is added to the polymer.(21)/DMS0 solution in the presence of EDC (2
eq), NHS (1 eq), and DIEA (1.0 eq). The solution is stirred for 16 h and then
precipitated in ether. The precipitate is washed with CH2C12 extensively until
no
free drug is observed in the washing solution. Compound 22 is obtained after
drying -
under high vacuum.
Example 20 Synthesis of CCD-Cys Copolymer 23 and its CPT conjugate 24
Scheme XXX
Mir COOH
142N.)\ Mir CH
0
OS
COOH N S
S-0 ______________________________
-
0 0 23 COON
GIY-CPT
0
TFA-GlyCPT
MEV
s
24
o
GIY-CPT
=
CCD 1 (141.3 mg, 0.1 minol) and cystitie (24 mg, 0.1 mmol) were dissolved
in dry DMSO (0.3 rnL) and pyridine (0.1 mL). The mixture was stirred under
argon
for overnight at 72 C. Water (10 mL) was added. Precipitate was filtered and
the
filtrate was dialyzed in 10,000 MWCO membrane (Spectra/Por 7) for 48 h. A
white
powder 23 (8 mg) was obtained.
11 is mixed with 23 in DMSO. EDC (2 eq), NHS (1 eq), and DIEA (1.0 eq)
are added to the solution. The solution is stirred for 16 h and then
precipitated with
ether. The precipitate is washed with CH2C12 extensively until no free drug is
observed in the washing solution. Compound 24 is obtained after drying under
high
vacuum.
99
CA 02818071 2013-06-06
Example 21 Synthesis of CD-BisGlu-Diamine Copolymer 25 and its CPT conjugate
26
Scheme 300(1OBz =
?Etz 0OBz OEM
0
HO yLNY-E7NcOH ________________
-H
0 0
OHL. OH
,GlyCPT õGlyCPT
TFA-GlyCrf
N'S:
0 0
44"
0
25 26
CD-Bis(Glutamic acid-y-Benzyl ester) 6 and ethyleneglycolbisethylamine
are dissolved in dry DMSO. EDC (3 eq) and Sulfo-NHS (2 eq) are added to the
mixture. The solution is stirred under argon for 2 days at rt. The solution is
then
transferred to a 10,000 MWCO dialysis membrane and dialyzed for 48 hours.
After
lyophilization a white powder is obtained. The solid is then dissolved in DMSO
and
methanol solvent mixture and treated with H2 in the presence of 10% Pd/C
catalyst
for 24 hours. The solution is poured into ether to crash out the product. 25
is
obtained after drying under vacuum. =
11 is mixed with 25 in DMSO solution. EDC (2 eq), NHS (1 eq), and DIEA
(1.0 eq) are added to the solution. The solution is stirred for 16 hrs and
then
precipitated with ether. The precipitate is washed with CH2C12 extensively
until no
free drug is observed in the washing solution. Compound 26 is obtained after
drying
under high vacuum.
Example 22 Synthesis of CDDM-Lys(GlyCPT) Polymer 27
Scheme XXMI
100
CA 02818071 2013-06-06
OCPT
0
C
+ HNWNU2
a o Lys-Gly-CET
0
HN-
27 0 j
OCPT
CDDM, 5, and Lys-Gly-CPT, 14, are dissolved in dry DMSO. EDC (3 eq)
and Sulfo-NHS (2 eq) are added to the mixture. The solution was stirred under
argon for 2 days at it. The solution is then poured into ether. The
precipitate 27 is
dried under vacuum.
Example 23 Synthesis of CDDC-Cys(Boc) Copolymer 28, CDDC-CysCopolymer
29 and its CPT Conjugate 30
Scheme MITI
HN000 0
H`Y`-s's-Y101-1 Hen. 0
0 NH
Bac'
S.õ
S S
28 Bee"
0 Gly-CPT
0
HQ)yGly-CPT
HN 0 0
>
0 0 NH
Gly-CPT
101
CA 02818071 2013-06-06
CDDC (3) and N,N-DiBoc-Cystine are dissolved in dry DMSO. EDC (3 eq)
and Sulfo-NHS (2 eq) were added to the mixture. The solution was stirred under
argon for 2 days at it The solution is then transferred to a 10,000 MWCO
dialysis .
membrane and dialyzed for 48 hours. After lyophilization a white powder, CDDC-
Cys(Boc) polymer 23, is obtained. To the white powder 28 is added a mixture of
HC1 and DMSO solution. The solution is stirred at rt for 1 h and then dialyzed
against water for 24 h using 10,000 MWCO membrane. 29 is obtained as a white
solid.
Suc-Gly-CPT 15 is mixed with 29 in DMSO solution. EDC (2 eq), NHS (1
eq), and DEEA (1.0 eq) are added to the solution. The solution is stirred for
16 h
under argon and then precipitated with ether. The precipitate is washed with
ether
until no free drug is observed in the washing solution. Compound 30 is
obtained
after drying under high vacuum.
. .
Example 24 Synthesis of biodegradable CD-Polyphosphoester Polymer 31 and its
CPT conjugates 32
Scheme XXXII/
0 cu., 0 cn,
) \mcisc,43.), ( II 1 a II 1
ON 0 > -- " ¨
2>EP O¨C¨C 0 1,
..,'
e:7...,..
.
0 H H CI
NaOH Tr7 0
1H3 0 CH, 0 CH,
¨(11-0¨C¨C 0 ) ( W . c 0 ) ( I! .
____c____0
I
NI-I H H, I
OCPT H H2 n 1
CI
,/-- H H2 -1-
1TA-GlyCPT
32
Synthesis of the biodegradable polyphosphoester can be found in Wang J et
al, JAC'S, 2001, 123, 9480-9481.
. .
102
CA 02818071 2013-06-06
Polyphosphoester is mixed with 10 (0.5 eq of repeat unit) in DMSO. EDC (2
eq), NHS (1 eq), and DIEA (1.0 eq) are added to the solution. The solution is
stirred
= for 16 hrs and then precipitated with ether. The obtained CD-
polyphosphoester 31 is
dissolved in DMSO. To the solution is added 11 (0.5 eq of repeat unit), EDC (2
eq),
NHS (1 eq), and DIEA (1.0 eq). The solution is stirred for 16 h and then
precipitated with ether. The precipitate is washed with ether extensively
until no free
drug is observed in the washing solution. Compound 32 is obtained after drying
under high vacuum.
Example 25 Synthesis of CD Copolymer¨CPT conjugate 33 with polyethylene
backbone via radical polymerization
Scheme XXXV
<
-o ( ___ 0
AIBN
_________________________________________________ 0
0
0
07 0/ HN 0 0 HN
\11 or ATRP
CPT CPT
0 0
0 0
33
Acrylate monomers of CPT, triethyleneglycol trionomethylether, and CD-
monocystamine can be synthesized from N-Acryloxysuccinimide (Polysciences,
Inc.). These monomers are mixed in 1:1:1 ratio in dry DMSO. AIBN is added to
the mixture under argon. The solution is stirred at rt for 24-48 hrs until the
solution
becomes viscous. Polymer-CPT conjugate 33 is precipitated with ether and dried
under vacuum.
Example 26 Synthesis of CD-graft-poly(ethylene-alt-maleic anhydride)-
GlyGlyGlyCPT 34
Scheme XXXVI
103
CA 02818071 2013-06-06
s
0
n
H,N'P' , I
\ In ,
t n
0 0 0 0 0
0 0Ifa - - 0 FM 0- HN
0
NIN 0 0
Thi)
)1 c)../1
iLNII .
õLorni s
tint
IIN
\ N
----- ---- N ' \
, 0 0
\ 0
0 0
0
34
Poly(ethylene-alt-maleic anhydride) (Aldrich) is dissolved in DMSO. 10 (0.4
eq of repeat unit) and 12 (0.4 eq of repeat) are added. The solution is heated
at 70
C for 16 hrs and then precipitated with ether. The obtained CD-graft-
poly(ethylene-alt-maleic anhydride)-GlyGlyGlyCPT 34 is dried under high
vacuum.
Example 27 Synthesis of Polyglutamate-CD-CPT Conjugate 35 .
Scheme =WTI
I]
).õ...N.
/
,.. .
Hoo
104
CA 02818071 2013-06-06
Polyglutamate (from Sigma-Aldrich) is mixed with 10 (0.5 eq of repeat unit)
and 11 (0.5 eq of repeat unit) in DMSO. EDC (3 eq), NHS (2 eq), and DLEA (1.0
eq) are added to the solution. The solution is stirred for 16 hr and then
precipitated
with ether. After drying under high vacuum, polyglutamate-CD-CPT conjugate 35
is obtained.
Example 28: Synthesis and Characterization of CD-BisCys-Peg3400 Copolymers
36 and their CPT Conjugates 37.
A. Synthesis and Characterization of CD-BisCys-Peg3400 Copolymers 36 .
Scheme )00CVIIIa .
,, . o , ,, ..,i.`=- = 10,
,....4.5x ,,õ on W-01.
As n A....%.-01
It-oN
11.1.4 110 . PEG -DiSPA (MW =i4i 00 IDa) y
c,Ir.sti
V
S- I
Q
0.1\17011 rs ...---r 1 ' __ = f-trA,... s2 ( II -
' .P. 0
. Con DMSO/D1EA, TEA or DP.I4P
.... .
s
(.1.;:f...7nles,. - -.- = ,
4 .
36a-f
Poly(CDECys,rA-PEG)
Abbr.: PA = propanolcrtninebond between PEG and CO
Scheme XXXVIllb =
-
on .
r a;tz.. a .,õ'..r`-'-µ,'"
,r,41
1.7./ olt n,,-Ott Xi- I fin' int 0.kon
Ito 0 0 HO 110 n
110 0
TIo 00 00
0.
s 0 t". 5..arComonomer
õ,,,
. , .
1314SO(DIEA
0 0 00
0 nn
-
4
36g-i
PEG-DiSBA (MW =3400 f...) 0 Poly(CDDCys.BA-PEG)
,....--'
o
Q-1?'.11-".-'-{"."--)z-- =====,;",1%._;,-Z3 3611 12= f"H-"-`'.-"--17-'.,----
-''.. )
PEG-D.11TC (MW =3440 if3a) . Poly(CDDCTs-CR-PEG)
3G E.
Opr =
PEG-DiNPC (MW = 3400 i.D.2) Poly(CDISICys-C13-PEG)
=
.... Abbr: BA = butanoiannide bond; CB =
carbanrne bond
,
= ,
105
CA 02818071 2013-06-06
Synthesis of Poly(CDDCys-PA-PEG), 36a 4 (after
precipitation with
acetone, 63 mg, 0.047 mmol) and PEG-DiSPA (MW 3400, 160 mg, 0.047 mmol)
were dried under vacuum for 8 hours. Anhydrous DMSO (1.26 mL) was added to
the mixture under argon. After 10
minutes of stirring, anhydrous
diisopropylethylamine (DIEA, 19 pL, 2.3 eq.) was added under argon. The
reaction
mixture was stirred under argon for 120 h. The polymer containing solution was
dialyzed using a 10,000 MWCO. membrane (Spectra/Por 7) against water for 48 h
and lyophilized to yield 196 mg 36a (90%, Table 1). M. = 57.4 lcDa, Mõ = 41.7
kDa, /1/1õ/Adõ = 1.38. 'H NMR (D20) 8 5.08 (m, CD-2-H), 4.27 (m, Cys-CH), 2.72-
3.76 (m, CD-3,4,5,6-CH, CD-CH2, PEG-CH2), 2.44 (m, Cys-CH2).
Synthesis of other poly(CDDCys-PA-PEG) (36b-1), Poly(CDDCys-BA-PEG)
(36g) Poly(CDDCys-CB-PEG) (3611-0 were achieved under polymerization
condition similar to that of 36a. Details for the polymerization conditions,
monomer
selection, polymer molecular weight, polydispersity and yields are listed in
Table 1.
36g: 'H NMR (D20) 8 5.10 (m, CD-2-H), 4.25-4.37 (m, Cys-CH), 2.72-3.86 (m,
CD-3,4,5,6-CH, CD-CH,, PEG:CH2), 2.21 (m, Cys-CH7). 36h-i: 'H NMR (D20) 5
5.05 (m, CD-2-11.), 4.56 (m, Cys-CH), 2.70-3.93 (rn, CD-3,4,5,6-CH, CD-CH2,
PEG-
CH2), 2.38 (m, -OCH2CH2CH2C(0)-NH-), 2.34 (m, Cys-CH,), 1.90 (in, -
OCH2CH2CH2C(0)-NH-).
Addition of a non-nucleophilic organic base (such as DMA) was essential for
this polymerization as no viscosity changes of the polymerization solutions
were
observed after 48 hours if no base was added. When 2.3 eq. of DEEA were added,
the viscosity of the polymerization solution increased dramatically after 4-6
hours of
reaction. D1EA deprotonates the amino groups of 4 to render them more
nucleophilic for coupling with PEG-DiSPA. There were essentially no
differences in
the polymerizations if other bases, such as TEA or DMAP, were used (36b-e,
Table
1). Polymerization using 4 recovered by the two different precipitation
methods
(acetone and methanol) produced polymers with different MWs. 4 that was
purified
by the methanol-precipitation method (contains no free cystine) gave higher MW
polymer (36d-e as compared to the less pure 4 that was obtained from the
acetone-
106
CA 02818071 2013-06-06
precipitation method (36a). Polymerization of 4 with PEG-DiSPA typically
produced polymer yields greater than 90%.
4 was polymerized with other activated monomers such as PEG-DiSBA, PEG-
S DiBTC, and PEG-
DiNPC. Reaction of 4 with PEG-DiSBA gave polymer 36g with
- similar linkages as 36a-f (amide bond, but one more ¨Cli2 group than 36a-f
at the
linker) with M, over 100 kDa, while reaction of 4 with PEG-DiBTC and PEG-
DiNPC generated polymers 36h and 361, respectively, with connecting carbamate
moiety and M,õ's over 50 kDa (Table 1).
Table 1. Polymerization of 4 with difunctionalized PEG
PEG Polymerization M,,, M, Ma Yield
CDP Base
Comonomer time (h) (kDa) (kDa) Mõ
(%)
36aa PEG-DiSPA DIEA 120 57.4 41.7 1.38 90
36ba PEG-DiSPA DMAP 120 54.2 38.1 1.42 91
36ea PEG-DiSPA TEA 120 57.4 42.6 135 91
36db . PEG-DiSPA DIEA 120 93.6 58.0 1.48 96
36eb PEG-DiSPA DIEA 144 97.3 58.0 1.67 94
36fb PEG-DiSPA DIEA 2 35.3 25.6 1.38 95
36g PEG-DiSBA DIEA 120 114.7 77.9 1.47 96
3611 PEG-DiBTC DIEA 120 67.6 39.4 1.47 95
.36i PEG-DiNPC DIEA 120 86.5 57.2 1.51 96
a 4 was washed with acetone before polymerization.
4 was washed with methanol before polymerization.
Polymers 36a-i are highly soluble in aqueous solution. They can be easily
dissolved in water or phosphate buffered saline (PBS) solution at
concentrations of
at least 200 mg/mL. Solubility of these polymers in aqueous solution at
concentrations higher than 200 mg/mL was not attempted due to the high
viscosity.
These polymers were also soluble in DMF, DMSO and methanol, slightly soluble
in
CH3CN and CHC13, but insoluble in Tiff and ethyl ether.
107
CA 02818071 2013-06-06
Molecular Weight Control of CD Polymers 4 (after precipitation with
methanol) (56.2 mg, 0.0419 mmol) and PEG-DiSPA (147 mg, 0.0419 mmol) were
,
dried under vacuum for 4-8 hours. To the mixture was added dry DMSO (1.1 mL)
under argon. After 10 minutes stirring, DMA (16 ,uL, 2.2 eq) was added under
=
argon. A portion of polymerization solution (150 pL) was removed and
precipitated
with ether at selected times (2 h, 18 h, 43 h, 70 h, 168 h and 288 11). MWs of
the
precipitated polymers were determined as described above.
As shown in Figures 5a and 5b, molecular weights of 36 can be controlled by.
adjusting polymerization time.
B. Synthesis of Poly(CDDCys-PA-PEG)-CPTConjugates (FIGGG6, LGGG1O,
11G6, HGGG10). .
Scheme XXXIX
_6- =
: - -o
- - . i , (ft..- = -. 0 . Z.---
= . .I: ' -.1 0
..,...
C:42:
N 641',0.'111
0
Ili HO = ION '
0
.-...' N " 36e m
HO -
if =
0
N \ ./ ______ >- - E tfs--,4.3.
C
1 _sn RE/NHS 0H tiot0 _-
lifyo
o NH
n,'
'---0 6
0
.".. N =
SFA..NH 0
I.0 - 0
01
i 11 .
1 =
0
x i õ = 0 ..... .
...:5 .
_ .
A.
.,...), Lu.
10. = w a =
. \/ ;',.:.;./ "' scr',,,,,D . .
yr' 0 I
0 rk
=
--..\ = 36e or 36f . , ,.,,0 -10 N
>,-
4.. 10
0
EDONIIS --(--- ii:t . oil o.
q
1111 110 tS
,_.µ Cl 01 03' 0
M? . n!
TYA.NbR 0 0yy-4.
/-1
12 . Ho 00
0
-AL
n 1 ' ¨ . II-
W- .
N
-
108
CA 02818071 2013-06-06
Synthesis of Poly(CDDCys-PA-PEG)-GlyGlyGly-CPT (HGGG6) 36e (1.37
g, 0.30 mmol of repeat unit) was dissolved in dry DMSO (136 mL). The mixture
was stirred for 10 minutes. 12 (419 mg, 0.712 mmol, 2.36 eq), DIEA (0.092 mL,
0.712 mmol, 2.36 eq), EDC (172 mg, 0.903 mmol, 3 eq), and NHS (76 mg, 0.662
mmol, 2.2 eq) were added to the polymer solution and stirred for ca. 15 hours.
The
polymer was precipitated with ethyl ether (1 L). The ether was poured out and
the
precipitate was washed with CH3CN (3 x 100 mL). The precipitate was dissolved
in
water 600 mL. Some insoluble solid was filtered through 0.2 jum filters. The
solution was dialyzed using 25,000 MWCO membrane (Spectra/Por 7) for 10 h at
10-15 C in DI water. Dialysis water was changed every 60 minutes. The polymer-
drug conjugate solution was sterilized by passing it through 0.2 uM filters.
The
solution was lyophilized to yield ayellow solid HGGG6 (1.42 g, 85% yield).
Synthesis of Poly(CDDCys-PA-PEG)-GlyGlyGly-CPT (LGGG10)
Conjugation of 12 to 36f was performed in a manner similar to that used to
produce
HGGG6 except that this conjugate Was dialyzed with 10,000 MWCO membrane
(Spectra/Por 7) instead of with 25,000 MWCO membrane. The yield of LGGG10
was 83%.
Synthesis of Poly(CDDCys-PA-PEG)-Gly-CPT (HG6)
Conjugation of 11
to 36e was performed in a manner similar to that used to produce HGGG6. The
. 20 yield of HG6 was 83%.
Synthesis of Poly(CDDCys-PA-PEG)-GlyGlyGly-CPT (HGGGIO) 36e (1.5
g, 0.33 mmol of repeat unit) was dissolved in dry DMSO (150 mL). The mixture
was stirred for 10 minutes. 12 (941 mg, 1.49 mmol, 4.5 eq), D1EA (0.258 mL,
1.49
mmol, 4.5 eq), EDC (283 mg, 1.49 mmol, 4.5 eq), and NHS (113 mg, 0.99 mmol, 3
eq) was added to the polymer solution and stirred for ca. 24 hours. Another
portion
of EDC (142 mg, 0.75 mmol, 2.3 eq) and NHS (56 mg, 0.5 mmol, 1.5 eq) were
added to the conjugation solution. The polymer was stirred for an additional
22
hours. The workup procedure was the same as that for the synthesis of HGGG6.
The yield of HGGG10 was 77%.
Determination of wt% CPT on the Conjugates Stock solutions
of
HGGG6, LGGG10, 11G6 and HGGG10 were prepared at a concentration of 10
mg/mL in DMSO. An aliquot of corresponding stock solution was diluted to 100
109
CA 02818071 2013-06-06
,ug/mL using 1 N NaOH. CPT was completely hydrolyzed in this basic solution
and
transformed to its carboxylate form within 2 h at room temperature. An aliquot
of
this solution was diluted to 10 pg/mL using 8.5% H3PO4, and the CPT
carboxylate
form was transformed to its lactone form. 30 pL of this solution was injected
into
the HPLC. The peak area from the CPT lactone form was integrated and compared
to a standard curve.
11 and 12 were conjugated to 36e or 36f (Table 2) using conventional
coupling methods. Due to the instability of the ester linker of 11 and 12 in
aqueous
solution, the conjugation was conducted in anhydrous DMSO under argon. An
organic base was required to deprotonate the TFA salts of 11 and 12 to
facilitate the
coupling. For polymer conjugation with 12, the weight percent (wt%) drug
loading
was around 6-10%. The theoretical maximum drug loading is around 13% using
PEG with MW of 3400 Da; maximum values can be increased by decreasing the
MW of the PEG segments. Solubilities of all conjugates in water or PBS were
more
than 200 mg/mL (equivalent to a 12-20 mg CPT/mL for 6-10 wt% drug loading,
respectively). Details for the TIGGG6, LGGG10, HG6, and HGGG10 are
summarized in Table 2.
Table 2. Properties of polymer-CPT conjugates.
Mu, of parent polymer
Conjugate My/gib Linker CPT (wt%)
(x 10-3)
F1GGG6
97 1.7 triglycine 6.1
LGGG10 35 1.6 triglycine 10.2
HG6 97 1.7 glycine 6.8
HGGG10 97 1.7 triglycine 9.6
'Abbreviations: H = High Mu, polymer (97 IcDa), L = Low M1 polymer (35
kDa), GGG = triglycine linker, G = glycine linker, 6 = drug loading around
6 wt%, 10 = drug loading around 10 wt%.
b Polymer polydispersity as measured by light scattering techniques(26)
110
CA 02818071 2013-06-06
C. Release of CPT from HGGG6 and 1-1G6
Release of CPT in PBS HGGG6 and HG6 were prepared at I mg/mL in PBS (lx,
pH 7.4). A 100 p.L aliquot of the solution was transferred to a 1.5 mL
Eppendorf
tube and incubated at 37 'C. The incubated samples were quenched at selected
time
intervals and stored at -80 C until the analysis. Each solution was diluted
with
8.5% H3PO4 to a 5 mL total volume in a volumetric flask. 30 fiL of such
solution
was injected into the HPLC. The peak area from the CPT laetone form was
integrated and compared to a standard curve.
Analysis for the release of CPT from HGGG6 and HG6 in PBS containing
acetyl cholinesterase (an esterase, 100 units/mL), in KH2PO4 buffer (pH 6.1,
0.1 .1\4)
and in the ,K1-171304 buffer (pH 6.1, 0.1 M) containing cathepsin B (a
cysteine
proteinase, 200 pM, preaetivated on ice for 30 minutes in this buffer
containing 2
mM DTT and 1 mM EDTA) were performed in a manner similar to that described
above for PBS alone.
Release of CPT in Human Plasma An aliquot of HGGG6 and HG6 stock
solution were diluted to give final concentration of 0.5 mg/mL in PBS (lx, pH
7.4).
This solution was added to a lyophilized powder of human plasma to
reconstitute
100% human plasma by the recommended amount. The solution was divided into
equal volume (250 pL) to 1.5 mL Eppendorf tubes, incubated at 37 C, and
stopped
at selected time point. Samples were stored at ¨80 C until the analysis.
Samples
were separated from plasma by solid phase extraction columns. The solid phase
extraction cartridge (Oasis HLB lcc cartridge from Waters) was pre-conditioned
with 1 mL of acetonitrile and then with 1 mL of 8.5% H3PO4 before loading.
Samples were acidified with equal volume of 8_5% H3PO4 prior to loading. After
the acidified solution was loaded on the cartridge, the bed was washed with 3
x lmL
of water. Released CPT and polymer conjugate were eluted with 3 x 1 mL of a
solution mixture of acetonitrile and potassium phosphate buffer (pH 4.1)
(60/40 v/v).
The eluted solution was diluted to 5 naL total volume in a 5 mL volumetric
flask. 30
pL of such solution was injected into the HPLC. The peak area from the CPT
lactone form was integrated and compared to a standard curve.
111
=
CA 02818071 2013-06-06
Release of CPT from HGGG6 and HG6 in PBS containing 4% human
plasma (PBS/reconstituted human plasma solution = 96/4 (v/v)), in mouse plasma
and in reconstituted human albumin (PBS solution) were performed in a manner
=
similar to that described above for pure human plasma.
In PBS (lx, pH 7.4), the half-lives (An) for releasing CPT from HG6 and
HGGG6 were 59h and 321i, respectively. The half-lives decreased to 25h and
22h,
respectively, in the presence of 4% human plasma, and to 1.7h and 1.6h,
respectively, in 100% human plasma ("HP") and 2.6h and 2.2h, respectively, in
100% mouse plasma ("MT"). CPT release rates for both 1HG6 and HGGG6 in the
presence of albumin ("Alb") or acetyl cholinesterase ("Ac Cho") were on the
same
order of magnitude as in PBS. In a buffer solution at a pH lower than PBS (pH
6.1)
with or without the enzyme cathepsin B (active at pH 6.1), less than 50% of
total
conjugated CPT was released from both 11G6 and HGGG6 for times up to 144 h
(Table 3).
Table 3. Half-life (tin, in hour) of the release of CPT from EIG6 and
IIGGG6a
4% pH 6.1 Cath B
Conjugate PBS", liPd MP' Albf Ac Chog
HpC bufferh (pH 6.1)1
11G6 59 25 62 33
1.7 2.6 >144 >144
1IGGG6 32 22 1.6 2.2 73 43 >144 >144
a tip is defined as time (hours) for the release of half of the total
conjugated CPT.
Abbreviations: HP means human plasma, MP means mouse plasma.
b pH 7.4 PBS lx. buffer.
c Reconstituted human plasma mixed with PBS (v/v = 4/96). =
d Reconstituted human plasma
e Fresh mouse plasma
112
CA 02818071 2013-06-06
fin reconstituted human albumin PBS buffer
g In the presence of acetyl cholinesterase PBS solution (100 units/mL).
Ii pH 6.1 phosphate buffer (0.11\4)
pH 6.1 phosphate buffer in the presence of Cathepsin B
Release of CPT in Solution at Different pH. HGGG6 and HG6 were
prepared at 1 ing/mL in buffer solution with pHs ranging from acidic (pH =
1.2) to
basic (pH = 13.1) and incubated at 37 C for 24h. An aliquot of each solution
was
diluted with 8.5% H3PO4 to about 100 ,ug/mL. 30 fiL of such solution was
injected
into HPLC. The peak area from the CPT lactone form was integrated and compared
to a standard curve.
The pH of aqueous solution has a significant effect on the CPT release rates
from both 11G6 and HGGG6. The amounts of CPT released from 11G6 and
HGGG6 at 37 C after 24 h in buffer solutions with pHs ranging from 1.1 to
13.1
are illustrated in Figure 6. The glycinyl-CPT ester bonds of both 11G6 and
HGGG6
were very stable in acidic pH (1.1 to 6.4) as less than 7% of CPT were
released in 24
h.
/C50 via MTT Assay The human ovarian carcinoma A2780 cell line was
obtained from the European Collection of Cell Cultures (Salisbury, Wiltshire,
UK.).
The human colorectal adenocarcinoma HT29, human prostate carcinoma PC-3, and
human colonic adeoncarcinoma LS174T cell lines were obtained from the American
Type Culture Collection (Rockville, MD). Cells were seeded in 96-well plates
at
5000 cells/well and grown in medium containing 10% fetal bovine serum at 37 C
for 24 h in a humidified 5% CO2 atmosphere. The medium was replaced with fresh
medium containing CPT, 36e, HGGG6 or 11G6 in concentrations ranging from 1
nM to 10 ,uM of CPT and 36e (CPT equivalent for HGGG6 and HG6). At each
concentration three wells per plate were treated. The effect of the compounds
on
cell growth was measured by the MTT assay after 72 h. The medium was removed,
the cells were rinsed with PBS, MTT solution was added at a concentration of
0.5
mg/mL, and the plates were incubated for 4 h at 37 C. The medium was removed
and the formazan crystals were solubilized in DMSO. Absorbance was measured at
113
CA 02818071 2013-06-06
560 nin using a SPECTRAFluor Plus plate reader (Tecan, Durham, NC). The
percentage of cell survival was calculated relative to untreated cells, and
/C50's were
determined from plots of dose versus cell survival. IC50 data of CPT, 36e,
HGGG6 =
or 11G6 are listed in Table 4.
Table 4. IC50 of CPT, unconjugated polymer 36e and CPT conjugates HG6 and _
HGGG6 in various cell lines
Cell Line 36e (uM) CPT (pM) 11G6 (uM) HGGG6 (,uM)
LS174T >300 0.005 0.050 0.010
HT29 300 0.020 0.050 0.030
A2780 100 0.007 0.025 0.020
PC3 >300 0.075 0.25 0.15
Example 29: Poly-CD-BisCys-Peg3400-Ala-CPT 37
36e (54 mg, 0.012 mmol of repeat unit) was dissolved in dry DMSO (226 mL)
and stirred for 10 minutes. TFA-Ala-CPT which is prepared similar to 11 (15
mg,
0.028 mmol, 2.36 eq), DIEA (4.88 mL, 0.028 mmol, 2.36 eq), DCC (24.52 mg, 0.12
mmol, 10 eq), and NHS (13.6 mg, 0.12 mmol, 10 eq) were added to the polymer
solution. The mixture was stirred for about 16 hours. The polymer was
precipitated
with ether (40 inL) and washed with ether (2 x 30 mL) and with CH3CN (2 x 10
mL). It was then redissolved in pH 4 aqueous solution (10 mL) and dialyzed at
room
temperature for 48h using 25,000 MWCO membrane. The solution was then passed
through a sterilized 0.2 um filter and then lyophilized.to yield 37(46 mg,
85%).
Weight percent of drug loading was calculated to be 5.5% using HPLC equipped
with a fluorescence detector after releasing CPT from 37 using base. Free CPT
in
37 is <1%.
Example 30: Poly-CD-BisCys-Peg3400-Leu-CPT 38
114
CA 02818071 2013-06-06
36e (54 mg, 0.012 mmol of repeat unit) was dissolved in dry DMSO (226 mL)
and stirred for 10 minutes. 11,A.-Leu-CPT which is prepared similar to 11 (16
mg,
0.028 mmol, 2.36 eq), DIEA (4.88 mL, 0.028 mmol, 2.36 eq), DCC (24.52 mg, 0.12
mmol, 10 eq), and NHS (13.6 mg, 0.12 mmol, 10 eq) were added to the polymer
solution. The mixture was stirred for about 16 hours. The polymer was
precipitated
with ether (40 mL) and washed with ether (2 x 30 mL) and with CH3CN (2 x 10
mL). It was then redissolved in pH 4 aqueous solution (10 mL) and dialyzed at
room
temperature for 48 h using 25,000 MWCO membrane. The solution was then passed
through a sterilized 0.2 1.im filter and then lyophilized to yield 38 (42 mg,
78%).
Weight percent of drug loading was calculated to be 5.0% using HPLC equipped
with a fluorescence detector after releasing CPT from 38 using base. Free CPT
in
38 is < 1%.
Example 31: Synthesis of CD-BisCys-BisPeg-FITC 39
Scheme )0CXX
.74- FluorNe1Ssccn-Pcg5k
-NHSHprl)0O DlEA
Olt
COOH HOOC
0 0 011
01 40 dr-
OH HO .11611. 0 0
39
=
=.
4 (25 mg, 0.0186 mmol) and FITC-Peg5000-NHS (Shearwater, 186 mg,
0.0373 mmol) were dissolved in dry DMSO (2 mL). DIEA (0.0094 mL, 0.056
mmol, 3 eq) was added to the mixture. The mixture was kept in dark and stirred
for
24 hours. Water (10 mL) was then added and the solution was dialyzed in dark
using 10,000 MWCO for about 48 hours. After lyophilization a yellow polymer 39
was obtained. Polymer was characterized by MS and Ili NMR.
Example 32: Synthesis of Bis-suecinimidyl succinatePeg3400 (Bis-SS-PEG) (40a)
and biodegradable CD-BisCys-SS-Peg3400 (40b) and its CPT conjugate 41
Scheme =XI
115
CA 02818071 2013-06-06
=
0 0
0
OH 0
DMAP 0 0
=
0 0 0 0
-S S
DCC HO 0 0
OH )m
0 40a 0
0
rr
0 0
Gly-CPT
_________________________________________________________________________ )1.
0 0
HO 0 0 014 40b
o
===,õ o
0
CPT-01y0
0 Gly-CPT
Degradabk.ler bond linkage
41
A 100 mL round bottom flask equipped with a magnetic stirbar and a septum
- 5 was charged with 10 g (2.99 mmol) of polyethylene glycol Mw 3350,
2.0 g (20
mmol) of succinic anhydride and 50 mL of anhydrous pyridine. After stirring
the
solution at 50 C for 16 h, the solvent was removed by rotary evaporator. The
residue was redissolved in 30 mL of water and extracted with 10 mL of
chloroform
three times. The organic layer was dried over MgSO4 and filtered. The solvent
was
then concentrated and precipitated out in diethyl ether. This resulted in 9.6
g of bis-
succinimidyl Peg3400 at a yield of 90.6%. The product was analyzed by reverse-
phase columned High Performance Liquid Chromatography.
A 100 mL round bottom flask equipped with a magnetic stirbar and a septum
was charged with 2 g (0.56 mmol) of bis-suecinimidyl Peg3400 and 10 mL of
anhydrous dichloromethane. To this solution was added 0.324 g (2.82 mmol) of N-
hydroxyl succinimide. The solution mixture was then cooled in an ice bath and
added 0.58 g (2.82 mmol) of 1,3-diuclohexylcarbodiimide. After leaving at room
temperature for 24 h, the solution was filtered and precipitated out in 150 mL
of
diethyl ether. Dissolution in 10 mL dichloromethane and precipitation in
diethyl
ether was repeated two times. This afforded 1.74 g (82_9 %) of Bis-SS-PEG 40a.
It
1 I 6
CA 02818071 2013-06-06
was analyzed by reverse-phase columned High Performance Liquid
Chromatography.
CD-BisCys-SS-Peg3400 Polymer 40b
A 50-mL pearl shaped flask was charged with 100 mg (0.0746 mmol) of 4 and
254 mg (0.0746 mmol) of 40a. The combined solids were dried under vacuum for
24 hours before the addition of lmL of anhydrous DMSO and 2.2 equivalents
(0.164
mmol) of DIEA. The solution mixture was stirred at room temperature for 3 days
and then precipitated out in diethyl ether. This yielded 100% of 40b.
Molecular
weight was analyzed on a Hitachi HPLC system equipped with an Anspec RI
detector, a Precision Detectors DLS detector, and a ProgeI-TSK G3000pwxL
column
using 0.1 M PBS as eluant at a 0.7 mLmin-1 flow rate. M = 93,000, Mn = 61,000
and M/Mr, = 1.5.
CD-BisCys-SS-Peg3400-G1yG1yGly-CPT conjugate 41
40b (201.8 mg, 0.044 mmol of repeat unit), TFA-GlyGlyGly-CPT 12 (66 mg,
0.105 mmol, 2.36 eq), EDC (25.5 mg, 0.133 mmol, 3 eq), and NHS (11 mg, 0.0977
mmol, 2.2 eq) were dissolved in dry DMSO (6 mL) and stirred for 30 minutes.
DIEA (19 !IL, 0.105 mmol, 2.36 eq), added to the polymer solution. The mixture
was stirred for about 41 hours. The polymer was crashed out with diethyl ether
(250
mL) and washed with acetonitrile (3x25 mL). It was then re-dissolved in pH 4
water (10mg/mL) and dialyzed at room temperature for 24 hours using 10,000
MWCO membrane. The solution was then passed through a sterilized 0.2 p.m
filter
and then lyophilized to yield 41 (128 mg, 52%). Weight percent of drug loading
was calculated to be 6.95% using HPLC equipped with a fluorescence detector
after
releasing CPT from 41 using base..
Hydrolysis of 41 was set up in human plasma (100% solution) at lmg/mL.
Aliquot solutions (100 pL) were placed in 1.5mL eppendorf tubes and incubated
in
37 C water bath. Then, the samples were acidified with 100 pL of 8.5% H3PO4
and
loaded on pre-conditioned solid phase extraction cartridge. It was eluted with
60:40
117
CA 02818071 2013-06-06
(v/v) acetonitrile: K112PO4 buffer. Free CPT (lactone form) was analyzed on
HPLC/Fluorescence detector using acetonitrile/ KH2PO4 buffer. Half-life was
determined to be 3 h.
Degradation of CD-BisCys-SS-Peg3400 Polymer 40b
50mg/mL of 40b solution was prepared in human plasma reconstituted in
PBS (pH 7.4) solution. 100 pL aliquots were incubated at 37 C. Each sample
tube _
was taken out at a specific time point and crashed out in 9004 cold methanol.
The
solution was centrifuged and the supernatant was analyzed on a 1-1PLC/ELS
detector.
The resulting spectrum is shown in Figure 7.
Methods for increasing drug weight percent loading
Method I. Synthesis of CD-BisC'ys-Peg Copolymer with a short Peg linkage and
its
GlyCPT conjugate
Example 33: Synthesis of CD-BisCys-Peg (short PEG, e.g., Peg200-Peg2000) and
its CPT Conjugate 42
Scheme XXXXII
= - H
n'
0 0 0
-0 Yj 0
=
0- 0
O/
=
0 0
N
. 110
42
Synthesis of polymer and drug conjugate 42 are same as 36, 37, and 38 in
Example 28. =
1 1 8
CA 02818071 2013-06-06
_._ .
Method II. Synthesis of CD-Biscps-Peg Copolymer with multiple drug molecules
on each loading site.
Example 34: Synthesis of CD-BisCys-Peg and its GluBis(GlyCPT) Conjugate 43.
= 5 Scheme XXXXIII
eHs W H
=s'''/\,o
/
m n
/
./.o 0/õ,-,. 0
HN HN 0 -
0 0
0
Oil. r Oil. r 23 PEG Mw
=3400
=--.. N ---, N
2 CPT per CD =
Maximum drug loading 13.4 %
N/ \ N/ \
4110' 0
0 o
o
o o o o
=-=., --,,, ------ PEG Mw
=3400
4 CPT per CD
Maximum drug loading 25 %
UN MN HN UN
0
--, N -.-._ N --=-- N --... N
N'" N/ \ N'" U"" =
-
0 * * *
43
36 and Glu-Bis(Gly-CPT) 17 are dissolved in DMSO. EDC (3 eq), NHS (22
eq), and DIEA (2.2 eq) are added to the solution. CD-BisCys-Peg-GluBis(GlyCPT)
43 is precipitated with CH3CN and washed with the same solvent until no free
drug
is detected using UV or TLC. 43 is dried under high vacuum.
Example 35: Synthesis of PEI-CD-CPT conjugate 44 (Branched CD-Polymer with
CPT conjugates)
= 119
CA 02818071 2013-06-06
A: Synthesis of branched PEI-cyclodextrin polymer
Scheme =OCR/
NH
NH,
H\
=
NH NR, FIN
NH HIV
Tosyl
NH is1112
FIN NH NI-I, NH
NH
NH
PEI (29 mg, Aldrich Mw 25,000) was dissolved in dry DMSO (2 In.L).
Cyclodextrin monotosylate (448 mg, Cyclodextrin Technologies Development,
Inc.)
was added to the solution under N2. The cloudy solution turned clear after the
mixture was stirred at 70 C for about 1 hour. The solution turned slightly
yellow
after 48 hours at such temperature under N2.
Thesolution was transferred to a Spectra/Por MWCO 10,000 membrane and
dialyzed against water for 4 days. Water was then removed by lyophilization. A
white powder was obtained (120-140 mg) after the solution was lyophilized.
Cyclodextrin/PEI ratio was calculated based on the proton integration of III
NMR.
B: Synthesis of branched PEI-CD-CPT conjugate
Scheme XXXXV
120
CA 02818071 2013-06-06
\17
UN IN
V17 .
NilUN NT!
7
- (
EDC/1411S --1
_____________________________________ >¨ ._1(-)L
Nil
LIIII.kµrj'P. " ZUII 1'11111, ?NH .
= 0 01
LII
111i Nil
11,14 o
4 4
PEI-CD and Suc-Gly-CPT 15 (1.0 eq) are dissolved in DMSO. EDC (3 eq),
NHS (2.2 eq), and DIEA (1 eq) are added to the solution. PEI-CD-Gly-CPT 44 is
precipitated with ether, washed extensively with this solvent, and dried under
high
vacuum.
Example 36: Synthesis of Ad-PE03400-Ad 45
Scheme XXXXVI
0 0 0 0
NI-12 + N-0-C-CH,-CH2---0-PEG3406-0-CH2-CH2-C-0 --N - -
0 = 0
0 0
H 11 11
N¨C¨CH2-CH2-0¨PEG3400-0¨CH2-CH2¨C¨N
240 mg of 1-aminoadamantane (1.60 rnmol, Aldrich) and 272 mg of
15 PEG3400(SPA)2 (0.080 mmol, Shearwater Polymers) was added to a glass
vial
equipped with a stirbar. To this was added 5 mL of dichloromethane, and the
solution was stirred overnight. The next day, the solution was filtered to
remove the
n-hydroxysuccinimide byproduct and the dichloromethane was removed in vacuo.
The residue was dissolved in water and centrifuged to remove excess 1-
121
CA 02818071 2013-06-06
aminoadamantane. The supernatant was then dialyzed overnight in Pierce's Slide-
-
A-Lyzer with MWC0-3500: The solution was then lyophilized to afford 248 mg of
a white fluffy solid of Ad-PEG3400-Ad 45.
Example 37: Synthesis of DiCyclodextrin PEG 46
362 mg of CD-NH2 (0_32 mmol, Cyclodextrin Technology, Inc.) and 436 mg
of PEG3400(SPA)2 (0.128 mmol, Shearwater Polymers) were added to a glass vial
equipped with a stirbar. To this vial was added 4.36 mL of DMSO, and the
solution
was stirred for 72 hrs. The solution was dialyzed using 2000 MWCO membrane for
4 days in water. 46 (603 mg, 86%) was obtained as a white powder after
lyophilization.
Example 38: Synthesis of Inclusion polymer 47 using DiAD-Peg 45 and DiCD-
PEG 46
46 (54.6 mg, 0.01 mmol) and 45 (34 mg, 0.01 mmol) were mixed in water
(0.27 mL) and stirred for overnight. The solution is very viscous. Polymer 47
was
crashed out with ether and dried under vacuum
Example 39: Synthesis of Inclusion Polymer CPT-Conjugate 48 between DiCD-
PEG 46 and a CPT-Di-AD compound
Synthesis of diadamantane crosslinker: Bis-(2(1-adamantypethyl)phosphate
(Zhang et al., J. Am. Chem. Soc1997, 119, 1676-1681)
Scheme )oomill
10 0 4- Pyridine
0 0 le 0
OCI-13 OH
Anhydrous pyridine (10 mL, Aldrich, Milwaukee, WI) was cooled in an ice
bath and methyl dichlorophosphate (1.488 g, 10 mmol, Aldrich, Milwaukee, WI)
was added dropwise. The mixture was kept cold for a further 15 mm. During this
122
CA 02818071 2013-06-06
period a precipitate of N-methylpyridinium dichlorophosphate formed. 1-
. Adamantane ethanol (4.758 g, 26.4 mmol, Aldrich, Milwaukee, WI) was
added, and
the sealed mixture was stirred overnight at room temperature. It was then
poured
into 10% NaHCO3 solution (50 "IQ and the pyridine was evaporated under vacuum.
The slightly yellow solid was dissolved in 1 L of water and extracted with
ether
(three 150 la portions). The aqueous phase was acidified with 2 N HCI to pH 1,
and then extracted with three 150 mL portions of CHC13:n-BuOH (7:3). The
combined organic layer (ether and CHC13:n-BuOH) was washed with water and a
slightly yellow precipitate was formed in the mixed solvents, at which point
the
solvents were evaporated under vacuum. A slightly yellow solid was formed and
was recrystallized from acetone/hexane. The solid was dried under vacuum,
yield 60
%.
Scheme =Mil
0
o'"No
) ________________________________________ 10
OH
= pi
\ 0
0 0
Bis-(2(1-adamantyl)ethyl)phosphate and 11 are mixed in DMSO. EDC (3
eq), NHS (2.2 eq), and DIEA (1 eq) are added to the solution. Solution is
stirred
under argon for 16 hours. Bis-(2(1-adamantypethyl)phosphate-Gly-CPT is
precipitated with ether, washed extensively with this solvent, and dried under
high
vacuum. This compound and Di-CD-PEG 46 are mixed in DMSO to form inclusion
polymer-CPT conjugate 48.
Example 40: Synthesis of AD-Peg-Folate 49
123
CA 02818071 2013-06-06
The following procedure is the synthesis of AD-Peg5000-Folate. This method
can be adapted for the synthesis of any Guest molecule-Linker-Ligand tri-
component molecules.
I. Synthesis of AD-Peg-NH,
266 mg of FM0C-PEG5000-NHS (78.2 Imo!, Shearwater Polymers,
Huntsville AL) were added to a glass vial equipped with a magnetic stirbar. 10
eq.
of 1-adamantane-methylamine (1.5 mmol, Aldrich) dissolved in 3 mL of
dichloromethane were then added and the solution stirred overnight at room
temperature. The solvent was removed in vacuo and water was added to the
remaining solution to dissolve the PEG product. The solution was centrifuged
at
20K rcf for 10 minutes, whereupon the adamantane-methylamine phase-separated
as
a denser liquid. The aqueous portion was collected and water removed in vacuo.
The remaining viscous liquid was redissolved in 20% piperidine in DMF for FMOC
deprotection and stirred for 30 minutes at room temperature. The solvent was
removed in vacuo, washed several times with MAP, redissolved in water, and run
on
an anionic exchange column to remove unreacted PEG. The first fractions were
collected and lyophilized to yield 222 mg of a white, fluffy powder (76%
yield) of
the desired product which was confirmed by MALDI-TOF analysis.
2. Synthesis of N-HYdravsuccinimide-folate (1VHS-Jblate)
Scheme )00=
0
011 11,N N
13
DCC/NHS N
14;:rIN.,)= 0
0
OH 25
- 011 0 it
0
0 OH
NHS-folate is synthesized according to the method of Lee and Low (Lee, R.
.1; Low, P. S. J Biol. Chem. 1994, 269, 3198-3204). Folic acid (5 g, 11.3
mmol;
124 =
CA 02818071 2013-06-06
Sigma) is dissolved in DMSO (100 ml) and triethylamine (2.5 ml) and reacted
with
N-hydroxysuccinimide (2.6 g, 22.6 mrnol) and dicyclohexylcarbodiimide (4.7 g,
22.7 minol) over-night at room temperature. The solution is filtered and
concentrated under reduced pressure. NHS-folate is precipitated using diethyl
ether
(yellow-orange precipitate), and washed 2-3 times in anhydrous ether, dried
under
vacuum, and stored at ¨20 C.
3. AD-Feg5000-Folate
Scheme L
N
IN 111_2-o
-AD-UII,
-r 0
0 0 on
49
=
AD-Peg5000-NH2 and NHS-Folate are mixed at 1:1 eq. in DMSO solution.
DIEA (2 eq) is added. The mixture is allowed to stir at room temperature for
overnight. The DMSO solution is then dialyzed against 3500 MWCO (Spectra/For
7) membrane for 48 hours. AD-Peg5000-Folate 49 is obtained after
lyophilization.
Example 41: Formulation of the AD-Peg-Folate and a CD-Polymer-CPT conjugate
A typical procedure: A CD-polymer CPT conjugate is dissolved in a D5W buffer.
A D5W solution containing 0.1-I eq (mols of repeat unit) of AD-Peg-Folate
solution -
is added to polymer solution. Particle sizes of polymer are measured before
and
after adding of AD-Peg-Folate using light scattering. This solution is used
for either
in vitro or in vivo test for the analysis of folate targeting effects.
Example 42: Covalent linking a targeting ligand (e.g., folate) to a CD-CPT
polymer
conjugate (eg PEI-CD-GlyCPT) 50
Scheme LI
125
CA 02818071 2013-06-06
Fr: "ti
c" c'
7
_
¨71
MI HI, NU
-C"
IMI.
LI
:1?-1
PEI-CD-GlyCPT 44 and Folate-NHS (0.1-0.5 eq) are mixed in DMSO and stirred
5 for 24 hours. The polymer is crashed out with ether and washed
extensively with
this solvent until no free folate molecule can be detected. The resulting CD-
polymer-CPT-Folate conjugate 50 is dried under vacuum.
Example 43: Crosslinking of CD-CPT polymer using Ad-Peg-Ad. 51
10 A typical procedure: AD-Peg-AD 45 (0.01-0.1 eq of CD-polymer repeat
unit) and
CD-polymer CPT conjugate are mixed in a minimum amount of DMSO. The
resulting viscous solution is precipitated with ether and dried under vacuum
to yield
lightly crosslinked polymer 51. 51 should still maintain small particle size
in
solution, have good water solubility, and have higher molecular weight than
parent
15 CD-polymer CPT conjugate.
Example 44: In vivo tests of carnptothecin polymer conjugates HGGG6, LGGG10,
HG6, and HGGG10 (Synthesized according to Example 28).
20 A. In Vivo Toxicity and Blood Chemistry Analysis from Dosing With Parent
Polymer 36e
The toxicity of 36e and its effects on blood chemistry were evaluated in
female Charles River nude mice (13-14 weeks old). Four
treatment groups of six
mice each were treated with a D5W solution of 36e at a dose of 240 mg/kg, 160.
126
CA 02818071 2013-06-06
=
mg/kg, 80 mg/kg or D5W alone by i.v. tail vein injection on Days 1, 5, and 9,
respectively. Dosing volume was determined based upon a ratio of 200 pl for a
20 g
mouse, and was scaled appropriately according to the actual 13W of the mice.
The
BWs of the mice were followed daily for the first 5 days and then twice a
week,
thereafter. Blood samples (150-200 pl) were collected from each mouse by retro-
orbital bleeding under isoflourane on Day 12. Samples from three mice in each
group were used for complete blood count (CBC) analyses, while blood samples
from the remaining three mice in each group were processed for blood chemistry
analyses. The study was stopped at Day 23. All mice were euthanized by cardiac
puncture under CO2, and blood from each mouse was collected for CBC and blood
chemistry analysis in the same manner as on Day 12.
There was no significant difference in BW loss, CBC or blood chemistry data
between any of the 36e treated groups and D5W control group throughout the
study,
and no time dependent effects were observed over 23 days for all the treated
groups.
36e was well tolerated by mice at the Maximum dose treated (240 mg/kg).
B. Determination of Maximum Tolerable Dose (MTD) for CDP-CPT
Conjugates.
The MTD was determined using female Charles River nude mice (15-16 weeks old)
for 11G6, LGGG10, HGGG10. A 5% (w/v) of dextrose solution (D5W) of the
polymer-CPT conjugates was freshly prepared before each injection. Doses for
the
treatment groups ranged from 2.25 mg CPT/kg to 54 mg CPT/kg. Dosing was
administered intravenously (i.v.) by tail vein injection on Days 1, 5, and 9.
The
dosing volume was determined based upon a ratio of 200 /al for a 20 g mouse,
and
. was scaled appropriately according to actual body weight (BW) of the mice.
Three
to five mice were used in each treatment group. The BWs of the mice were
followed
daily for the first 5 days and then twice a week, thereafter. The MTD was
defined as
the highest administered dose that resulted in a decrease of mean group BW of
less
than 20% or the highest administered dose that did not result in death of any
animal
in that group. The maximum mean body weight loss and treatment related deaths
for all treated groups are listed in Table 5.
127
CA 02818071 2013-06-06
Table 5. Treatment response for the MTD study. Nude mice (n= 3-6) were treated
i.v. by tail vein injection.
Agent mg/kga Max %BW loss; Dayb NTR/Nc
D5W - -2.5%; Day 3 0/6
36e 240 -2M%; Day 3 0/6
36e 160 -3.5%; Day 13 0/6
36e 80 -2.3%; Day 3 0/6
LGGG10 54 -20.6%; Day 3 3/3
LGGG10 36 -9.3%; Day 13 0/3
LGGG10 18 0 0/3
LGGG10 9 0 0/5
LGGG10 415 -0.8%; Day 13 0/5
EIG6 54. -28.5%; Day 3 ` 3/3
HG6 36 -23.9%; Day 3 3/3
11G6 18 -22.1%; Day 3 3/3
11G6 9 -6.1%; Day 9 0/5 .
11G6 4.5 -4.4%; Day 5 0/5
11G6 2.25 -2.9%; Day 9 0/5
HGGG10 54 --- 3/3 ,
HGGG10 36 -34%; Day 5 3/3
IIGGG10 18 -16%; Day 3 1/3
HGGG10 9 -3.3%; Day 9 0/5
HG,GG10 4_5 -2_5%; Day 9 0/5
a Mg CDP /kg for the CDP polymer and mg CPT/kg for the three
conjugates tested. .
b Maximum body weight (BW) loss observed post injection
C Number of treatment-related deaths (NTR) to the number of mice treated
('1)
The MTD of LGGG10, 111G6, and HGGG10 were determined to be 36 mg
CPT/kg, 9 mg CPT/kg, and 9 mg CPT/kg, respectively. Based on the structural
128
CA 02818071 2013-06-06
=
similarities between HGGG6 and HGGG10, it is expected that the MTD for these
two groups are similar. Therefore, the MTD of HGGG6 (not tested) was assumed
to be 9 mg CPT/kg.
C. Antitumor Efficacy Study.
The antitumor efficacy study was performed using female Charles River nude
mice (15-16 weeks old). A fragment (1 mm3) of human LS174T colon carcinoma
tissue was implanted subcutaneously (s.c.) into the right flank of each test
mouse
approximately 14-18 days before dosing. The tumor volume was determined by
measuring the tumor in two dimensions with calipers and calculated using the
formula: tumor volume = (length x width2)/2. Tumor volume was converted to
tumor weight assuming 1 mm3 is equal to 1 mg tumor in weight. Treatment was
initialized when mean tumor size reached approximately 60-100 mg (Day 1). The
animals were sorted into twelve groups. Each group consisted of seven mice
with
tumor sizes ranging from 62.5-144.0 mg with group mean tumor sizes of 88.6-
90.7
mg_ Mice in each group were treated according to the protocol listed in Table
6. All
conjugate treatments were administered intravenously by tail vein injection.
Tumor
sizes were measured twice a week for the duration of the experiment. At the
end of
study, tumors from each euthanized mouse were harvested and frozen at ¨80 C.
Table 6. Dosing protocol for efficacy study.'
Dose (nig
Gr'oup Agent Route Scheduled
CPT/kg)b
D5 W i.v. Q4D x 3
9 CPT 9 i .p. Q4D x
3 Irinotecau 100f i.p. Qwk x 3
4 HGGG6 9 i.v. Q4D x 3
5 HGGG6 4.5 i.v. Q4D x 3
129
CA 02818071 2013-06-06
6 LGGG10 36 i _v. Q4D x 3
7 LGGG10 18 i.v. Q4D x 3
8 LGGG10 9 i.v. Q4D x 3
9 11G6 9 i.v. Q4D x 3
11G6 4.5 i.v. Q4D x 3
11 HGGGIO 9 i.v. Q4D x 3
12 HGGGIO 4.5 i.v. Q4D x.3
a Seven mice were used in each group
b Doses are equivalent of CPT except for group 3
i.p. = intraperitoneal, i.v. = intravenous
d Administration schedules were abbreviated as: Q4D x 3 = three injection with
5 four-day
intervals, Qwk x 3 = three injection with one-week interval, first dose was
initialized
on day 1 for all groups.
The scheduled third dose was not given due to the emerging toxicity
100 mg irinotecan/kg
Each animal was euthanized .when tumor weight reached the predetermined
endpoint size (1,500 mg). The time-to-endpoint (TTE) for each mouse was
calculated from the equation: ITE, = (log(endpoint-b))/m, where b and m are
the
intercept and the slope, respectively, of the line obtained by linear
regression of a
log-transformed tumor growth data set comprised of the first observation that
exceeded the study endpoint volume and the three consecutive observations that
immediately preceded the attainment of the endpoint volume. Animals that do
not
reach the endpoint were assigned a 11'E, value equal to the last day of the
study (114
days). Animals classified as treatment-related deaths (Ttt.) were assigned a
1TE.
value equal to the day of death. Animals classified as non-treatment-related
death
(NTR) are excluded from TIE calculations. Tumor growth delay (TGD), defined as
the increase in the median time to endpoint (TTE) in a treatment group
compared to
the control group, was one parameter investigated to evaluate treatment
efficacy.
130
CA 02818071 2013-06-06
TGD is calculated as the difference between the median TTE for a treatment
group
and the median TIE of the control group (TGD = T - C) and is expressed in
days,
and as a percentage of the median FIE of the control group; %TGD = (T-C)/C
where T is equal to median TTE for a treatment groups and C is equal to median
1TE for the control, Group 1.
Toxicity. Animals were weighed daily on Days 1-5, then twice weekly
thereafter. Mice were examined frequently for overt signs of any adverse, drug-
related side effects. Acceptable toxicity for cancer drugs in mice is defined
by the
NCI as a group mean body-weight loss of less than 20% during the study, and
not.
more than one toxic death among seven treated animals.
Results for this efficacy study that include median ap, values, median tumor
burden on day 114, treatment response and deaths are summarized in Table 7.
Table 7.
Median
NThei
Median Tumor P vs P vs
Group ______________ T-Cb c/oTGDc NNTRI
1-lha Burden in D5W1' CPT'
NEug
mg (Nsci)
1 34.9 -(0) 0/1/6
2 5L4 16.5 47% -(0) 2/0/5 0.2128 -
3 68.7 33.8 97% 1152(3) 0/0/4 0.0002 0.0253
4 114.0 79.1 227% 256 (5) 1/0/1 0.0040 0.0115
5 65.6 30.7 88% 566 (2) 0/1/4 0.0046 0.1369
6 100.0 65.1 187% 666 (3) 4/0/0 0.0272 0.0289
7 75.6 40.7 117% 221 (3) 0/0/4 0.0018 0.0601
8 63.2 28.3 81% 700(1) 1/0/5 0.0006 0.1064
9 114.0 79.1 227% 394 (4) 0/0/3 0.0002 0.0028
10 74.2 39.3 113% 668(2) 1/1/3 0.0016 0.0673
11 114.0 79.1 227% 500 (5) 1/0/1 0.0040 0.0050
12 78.0 43.1 123% 1010 (2) 0/0/6 0.0006 0.0392
a 1-11, = Time (Days) to endpoint (1500 mg)
131
CA 02818071 2013-06-06
b T-C =-- Difference between TTE (Days) of treated versus control group
%TGD = [(T-C)/C]
d
Mice surviving
e NTR = Number of treatment-related death
fNNTR = Number of non-treatment-related death
g NEu = Number of mice euthanized after reaching endpoint
h P value versus the D5V%i treatment group (Group 1)
P value versus the CPT treatment group (Group 2)
One NTR death on day 72 was observed in the control animals freated with
D5W. Tumors in the other six control mice grew to the 1500 mg endpoint size,
yielding a median 1-11, of 34.9 days (Table 7).
Two treatment-related deaths were reported on Day 9 for CPT at 9 mg/kg. Thus,
CPT must be considered to be toxic at this dose in this experiment. The median
TTE
for this group was 51.4 days, corresponding to a 16.5 day T-C and a 47% TGD,
relative to untreated control. mice (not significant). No animal in Group 2
survived to
Day 114.
Group 3 received irinotecan i.p. at 100 mg/kg (Qwk x 3). The median TTE for
Group 3 was 68.7 days, corresponding to a significant 33.8 day T¨C and a 97%
TGD, relative to control mice (P <0.01). Three animals survived to Day 114
with a
median tumor burden of 1,152 mg. No regressions were recorded.
Groups 4 and 5 received HGGG6 i.v. Q4D x 3 at 9 and 4.5 mg CPT/kg,
respectively. One treatment-related death was observed on Day 16 in Group 4,
and
one NTR death was recorded on Day 37 in Group 5. The median TTE for Group 4
was 114 days, the maximum possible value in this study. This TTE value
corresponds to a significant 79.1 day T¨C and a 227% TGD, relative to control
(P <
0.01). Tumors in five mice of Group 4 did not reach the 1,500 mg endpoint.
These
five mice had a median tumor burden of 256 mg on Day 114. The median TTE for
Group 5 was 65.6 days, and corresponds to a significant 30.7 day T¨C and an
88%
TGD, relative to control (P < 0.01).
Groups 6-8 were treated with LGGG10 i.v. Q4D x 3 at 36, 18, and 9 mg
CPT/kg, respectively. Although no death was observed in MTD study using this
132
CA 02818071 2013-06-06
conjugate in non-tumor bearing mice at 36 mg CPT/kg (Table 5), four treatment-
related deaths were recorded in Group 6 when tumor-bearing mice were given at
this
dose, two on Day 16 and one each on Days 75 and 100. These results indicate
that
36 mg CPT/kg is probably over the MTD of LGGG10. As shown in Table 5, no
significant body weight loss was recorded in the MTD study when the mice were
dosed at 18 mg CPT/Icg, indicating that this dose is below the MTD. Therefore,
the
MTD of LGGG10 lies somewhere between 18 to 36 mg CPT/kg. The median TTE
for Group 7 (18 mg CPT/kg) was 75.6 days. This 1.-1E value corresponds to a
significant 40.7 day T¨C and a 117% TGD, relative to control mice (P < 0.01).
Three mice in this group had a median tumor burden of 221 mg on Day 114. One
late TR death was recorded on Day 103 in Group 8 (9 mg CPT/kg). The median
'FIFE for Group 8 was 63.2 days. This TTE value corresponds to a significant
28.3
day T¨C and an 81% TGD, relative to untreated control mice (P < 0.01). The
remaining mouse in this group had a tumor burden of 700 mg on Day 114.
Groups 9 and 10 were dosed with 11G6 i.v. Q4D x 3 at 9 and 4.5 mg CPT/kg,
respectively. One FR and one NTR death were recorded in Group 10 on Days 47
and 84, respectively. The median TTE for Group 9 was the maximum, 114 days.
This TTE value corresponds to a significant 79.1 day T¨C and a 227% TGD,
relative to untreated control mice p < 0.01). Four mice in Group 9 had a
median
tumor burden of 394 mg on Day 114. The median TUE for Group 10 was 74.2 days.
This F1L, value corresponds to a significant 39.3 day T¨C and a 113% TGD,
relative to control mice (P < 0.01). The remaining two mice in Group 10 had a
median tumor burden of 668 mg on Day 114.
Groups 11 and 12 were dosed with HGGG10 i.v. Q4D x 3 at 9 and 4.5 mg
CPT/kg, respectively. One treatment-related death was recorded on Day 16 in
Group
11. The median TTE for Groups 11 and 12 were 114 days and 78 days,
respectively.
The TTE value for Group 11 corresponds to a significant 79.1 day T¨C and a
227%
TGD, relative to control mice (F < 0.01). Tumors in five mice in Group 11 did
not
reach the endpoint; these five mice had a median tumor burden of 500 mg on Day
114. The TTE value of Group 12 corresponds to a significant 43.1 day T¨C and a
123% TGD, relative to control mice (P < 0.01). The remaining two mice in this
group had a median tumor burden of 1,010 mg on Day 114.
133
CA 02818071 2013-06-06
The tumor growth curve as a function of time for the D5W, CPT, irinotecan,
LGGG10 at its highest non-toxic dose tested (18 mg CPT/kg), and the other
three
conjugates with high MW polymer (HGGG6, 11G6, HGGG10) at their MTDs are
shown in Figure 8. The three high MW conjugates administered at their MTDs
=
displayed more prolonged tumor growth inhibition compared to D5W, CPT and
irinotecan. The median tumor growth curves for HGGG6, HG6 and HGGGIO that
are illustrated in Figure 9 show that there is a distinct dose response for
all three of
these polymers when dosed as their MTD and at half of their MTD. The medium
tumor growth curves for ILGGG10 and EIGGG10 each dosed at 9 mg CPT/kg as =
illustrated in Figure 10 demonstrate that high MW polymer-drug conjugate has
greater antitumor effect when compared to the low MW conjugates presumably due
to the enhanced accumulation (EPR effect) and reduced renal clearance.
Mean Body Weight Loss of Mice. Mean body weight (MBW) losses as a
function of time are plotted for D5W, CPT, irinotecan and the three conjugates
containing high MW polymer at their MTDs (Fig. 11). Maximum MBW losses
observed in Group 2 (CPT) and the two conjugates with the triglycine linker
dosed
at their MTDs (Groups 4 and 11) were 13.1%, 18.3%, and 12.6%, respectively.
Maximum MBW loss of IIG6 (3.4%), the only conjugate with a glycine linker, was
similar to the maximum MBW loss recorded for irinotecan (5.0%). Negligible
(<5%) maximum group mean body-weight losses were recorded in all the other
treatment groups and in the D5W. group. Mean body weight returned to baseline
levels for all treatment groups following cessation of therapy.
D Correlation of Tumor size of enthanized mouse and the CPT concentration in
corresponding tumor.
Each tumor harvested from mice at the completion of the LS1 74t xenog,raft
mouse study was thawed and placed in a 2 ml lysis tube (Lysing Matrix D,
Qbiogen). 300 1L of lysis reagent (Cellytic ¨MT Mommalian Tissue
Lysis/Extraction reagent) was added to each tube. The tissue was homogenized
on a
FastPrep FP12 homogenizer (Qbiogen) at 5 m/s for 40 sec. Homogenization was
repeated six times with a 10 min interval between successive homogenization.
The
134
CA 02818071 2013-06-06
homogenized solution was centrifuged at 14000g forl 5 mm at 10 C. 90 p.L of
the
solution was syringed out to which 10 1.t.L IN NaOH was added. An aliquot of
400
iL Me014 was added to this solution after allowing the homogenized solution to
stand for 2h at room temperature. The solution was centrifuged for 15 mm at
14000
g. The supernatant (270 u.L) was mixed with 30 uL 1N HC1 and injected into an
HPLC for analysis. The correlation of CPT concentration (ng/mg tissue) to
tumor
size (in mg) is illustrated in Figure 12. CPT concentration was inversely
correlated =
to tumor size.
Example 45: Synthesis of Poly(CDDC-PEG)-Amphotericin B 52 via amide linker
Scheme LII
AmE1 funt3
0 0
n 2. AmB
0
52
Poly(CDDC-PEG) (788 mg) and 1,1'-carbonyl diimidazole (CDI, 1.45 g, 50
eq.) were stirred in anhydrous DMSO (10 mL) in the presence of DMAP (429 mg,
eq) for 16 h. Ether (200 mL) was added to the mixture to precipitate
poly(CDDC-PEG)-carbonyl-imidazole. The resulting yellow solid was washed with
ether 2 x 200 mL and dried under vacuum. The solid was dissolved in anhydrous
DMSO (15 mL), followed by adding AmB (332 mg, 2 eq) and DMAP (43.0 mg,
20 2eq). The solution was stirred in dark for 48 h and dialyzed in water
using 25000
MWCO membrane for 3 days. The solution was then filtered using 0.2 gm filter
and lyophilized. A yellow solid (920 mg) 52 was obtained. The wt% of AmB is
around 13%.
Example 46: Synthesis of Poly(CDDC-PEG)-Anphotericin B 53 via imine linker
135
CA 02818071 2013-06-06
0
11a104
, 11
DMSO k
sasdlo-34..
es "
_fte r
co
A¨C;)
( I
53
3 and PEG-DiSPA (1:1 ratio) were dried under vacuum at room temperature.
DMSO (10 mg of 3/mL DMSO) was added to the solid and followed by adding of
D1EA (2 eq) to the mixture. Polymer was crashed out with excess ether 5 days
later
and dialyzed using 25000 MWCO membrane for 48 It. The yield of poly(CDDC-
PEG) is 80-95%. The Mw of polymer was determined using GPC to be 70-100
lcDa.
Poly(CDDC-PEG) (1.124g, 0.25 mmol) was dissolved in water (55 naL).
Na104 (0.264g, 5 eq.) was added. The solution was stirred in dark at room
temperature for 20 mm and stored at 4 C for 24h in dark. BaCh solution was
added
(5.05 eq) to the solution to give immediate precipitation of Ba(104)2. The
precipitate
was filtered. Saturated Na2CO3 solution was added to adjust pH to 11.
Amphotericin B (343mg, 1.5 eq) was then added to solution and stirred at rt in
dark
for 48h. The pH of solution was maintained to be 11 by adding NaOH (0.1N)
throughout the reaction. The solution was dialyzed at 4 C for 48 h using
25000
MWCO and lyophilized to give 1.03 g polymer-AmB conjugate 53 as a yellow
= powder. The wt% of AmB is determined to be 18 using UV spectrometer at
405
urn.
D. References
Additional cyclodextrin-containing polymers that can be modified according
to the teachings of the present invention, as well as methods of preparing.
such
polymers, are disclosed in U.S. Patents 6,509,323; 7,091,192;
7,375,096; 7,166,302 and 7,018,609.
136