Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02823088 2013-08-08
53686-75D1
PROCESSES FOR THE PREPARATION OF
4-AMINO-2-(2,6-DIOXOPIPERIDIN-3-YL)ISOINDOLINE-1,3-DIONE
= COMPOUNDS
1. CROSS-REFERENCE TO RELATED APPLICATIONS
=
[001] This application claims priority to U.S. Provisional Application No.
=
60/696,224, filed June 30, 2005.
This application is a division of Canadian Application Serial No. 2,612,612
=
filed June 29, 2006 (parent application).
It should be understood that the expression "the present invention" or the
like
used in this specification may encompass not only the subject matter of this
divisional
application, but that of the parent application also.
2. FIELD OF THE INVENTION
[002] The present invention provides processes for the preparation of
compounds useful for
reducing levels or activity of tumor necrosis factor a in mammals. More
specifically, the
invention provides processes for the preparation of unsubstituted and
substituted 4-amino-2-
(2,6-dioxopiperidin-3-y1)isoindo1ine-1,3-dione compounds.
= 3. BACKGROUND OF THE INVENTION
[003] Excessive or unregulated production of tumor necrosis factor
a or TNF-a, has
been implicated in a number of disease conditions. These include endotoxemia
and/or Undo
shock syndrome (Tracey et al., Nature 330, 662-664 (1987) and Hinshaw et al.,
Circ. Shock
30, 279-292 (1990)), cachexia (Dezube et al., Lancet 335 (8690), 662 (1990)),
and Adult
Respiratory Distress Syndrome (Millar et al., Lancet 2 (8665), 712-714
(1989)). Certain
substituted 2-(2,6-dioxopiperidin.-3-y1)-1-oxoisoindolines have been shown to
reduce levels
of TNF-a in the literature such as International Publication No. WO 98/03502
and Muller et
al., Bioorg. Med. Chem. Lett. 9, 1625-1630 (1999).
=
=
=
1
CA 02823088 2013-08-08
53686-75D1
[0041 A substituted isoindole-1,3-dione that has demonstrated certain
therapeutic
values is 2-(2,6-dioxopiperidin-3-yl)isoindole-1,3-dione (THALOMIDTm ). This
compound
has been shown to be or is believed to be useful in treating or preventing a
wide range of
diseases and conditions including, but not limited to, inflammatory diseases,
autoimmune
diseases, cancers, heart diseases, genetic diseases, allergic diseases,
osteoporosis and lupus.
[0051 Existing methods for synthesizing unsubstituted and substituted 4-
amino-2-
(2,6-dioxopiperidin-3-yl)isoindole-1,3-dione compounds are described in U.S.
Patent Nos.
6,395,754 and 5,635,517. While these methods are enabling and useful for
preparing
unsubstituted and substituted 4-amino-2-(2,6-dioxopiperidin-3-ypisoindoline-
1,3-dione
compounds, alternative or improved methods for their preparation, particularly
in
manufacturing scale, are still needed.
la
CA 02823088 2013-08-08
WO 2007/005972
PCTMS2006/026210
[006] Citation of any reference in Section 2 of this application is not to
be construed
as an admission that such reference is prior art to the present application.
4. SUMMARY OF THE INVENTION
[007] The present invention provides efficient processes for the
preparation of
unsubstituted and substituted 4-amino-2-(2,6-dioxopiperidin-3-yl)isoindoline-
1,3-dione
compounds, particularly the unsubstituted 4-amino-2-(2,6-dioxopiperidin-3-
ypisoindoline-
1,3-dione.
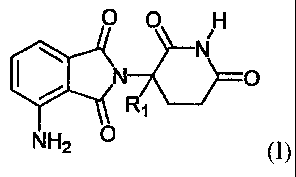
[008] In one aspect, the invention provides a process for preparing an
unsubstituted
or substituted 4-amino-2-(2,6-dioxo-3-piperidinyl)isoindole-1,3-dione compound
of Formula
0 0 H
OW II 11()
N H2
(I)
or a pharmaceutically acceptable salt, solvate including a hydrate, or
polymorph thereof,
wherein the process comprises the step of cyclizing an N-(3-aminophthaloy1)-
glutamine
compound of Formula (II) or an N-(3-aminophthaloy1)-isoglutarnine compound of
(IIA):
0 0 0 0
H 2I21H2
[101 N7tONH2 C (10 N CO2H
=
NH2NH2
OD or (11A)
or a salt thereof with a cyclizing agent of Formula (V):
= ).c
Y X=(V)
wherein R1 is H, F, benzyl, (CI-C8)alkyl, (C2-C8)alkenyl, or (C2-C8)alkynyl;
and each of X
and Y is independently an =substituted or substituted imidazolyl,
benzimidazoly1 or
benzotriazolyl. In some embodiments, RI of Formula (I) or (II) is H.
411
[009] In some embodiments, the cyclizing agent is a
carbonyldiimidazole compound
of Formula (VI):
2
CA 02823088 2013-08-08
WO 2007/005972
PCT/US2006/026210
0
R 7 jt, 1112
N N
r 5 RI4
=
R6 n3 (VI)
where each of R2, R3, R4, R5, R6 and R7 is independently H, alkyl, halo,
nitro, cyano, acyl,
alkoxy, aryloxy, alkoxycarbonyl or alkoxymethyl. In a particular embodiment,
the
carbonyldiimidazole compound is 1,1'-carbonyldiimidazole (i.e., where each of
R2, R3, R4,
R5, R6 and R7 of Formula (VI) is H). In a farther embodiment, the ratio of the
compound of
Formula (II) to 1,1'-carbonyldiimidazole is from about 1:1 to about 1:1.2.
[010] = In another embodiment, the cyclization occurs in acetonitile. In
another
embodiment, the cyclization occurs in tetrahydrofuran. In a further
embodiment, the
cyclization reaction temperature is from about 80 C to about 87 C. In another
embodiment,
the cyclization reaction time is from about 1 hour to about 5 hours.
[011] In another aspect, the invention provides a process for preparing an
imsubstituted or substituted 4-amino-2-(2,6-dioxo-3-piperidinypisoindole-1,3-
dione
compound of Formula (I) or a pharmaceutically acceptable salt or solvate or
polymorph
thereof, wherein the process comprises the step of reacting 3-aminophthalic
acid or a salt
thereof with a 3-aminoglutarimide compound of Formula (X) or a salt thereof:
R 1
H 2
0 N 0
in a solvent, wherein RI is H, F, benzyl, (CI-C8)alkyl, (C2-C8)a1keny1, or (C2-
C8)alkynyl. In
some embodiments, RI of Formula (I) or (X) is H.
[0121 In certain embodiments, the reacting step occurs in the
presence of a base, an
acid or a combination thereof. In another embodiment, the reacting step occurs
in the
presence of a base which, in some instances, can be a trialkylamine, a
substituted or
unsubstituted imidazole or a mixture thereof. In certain embodiments, the
reacting step
occurs in the presence of the base and the acid where the base may be an amine
such as
;113 triethylamine and the acid may be a carboxylic acid such as acetic
acid. In certain
embodiments, the mole ratio of triethylamine to acetic acid is from about 1:10
to about 1:1.
3
CA 02823088 2013-08-08
WO 2007/005972 PCT/US2006/026210
[013] In another embodiment, the solvent is acetonitrile. In a further
embodiment,
the reaction temperature is about 85-87 C. In a further embodiment, the
reaction time is from
about 5 to about 7 hours.
5. DETAILED DESCRIPTION OF THE INVENTION
5.1= Terminology
[014] As used herein and unless otherwise indicated, the term "halo",
"halogen" or
the like means -F, -C1, -Br or -I.
[015] As used herein and unless otherwise indicated, the term "alkyl" or
"alkyl
= group" means a saturated, monovalent, unbranched or branched hydrocarbon
chain.
Examples of alkyl groups include, but are not limited to, (C1-C8)alkyl groups,
such as methyl,
ethyl, propyl,.isopropyl, 2-methyl-1-propyl, 2-methyl-2-propyl, 2-methyl-1-
butyl, 3-methyl-
1-butyl, 2-methyl-3-butyl, 2,2-dimethyl-1-propyl, 2-methyl-1-pentyl, 3-methyl-
1-pentyl, 4-
methyl-1-pentyl, 2-methyl-2-pentyl, 3-methyl-2-pentyl, 4-methy1-2-pentyl, 2,2-
dimethyl-1-
butyl, 3,3-dinaethy1-1-butyl, 2-ethy1-1-buty1, butyl, isobutyl, t-butyl,
pentyl, isopentyl,
neopentyl, and hexyl, heptyl, and octyl. An alkyl group can be un.substituted
or substituted
with one or two suitable substituents.
[016] As used herein and unless otherwise indicated, the term "alkenyl" or
"alkenyl
group" means a monovalent, unbranched or branched. hydrocarbon chain having
one or more
double bonds therein. The double bond of an alkenyl group can be unconjugated
or
conjugated to another unsaturated group. Suitable alkenyl groups include, but
are not limited
to (C2-C8)alkenyl groups, such as vinyl, allyl, butenyl, pentenyl, hexenyl,
butadienyl,
pentadienyl, hexadienyl, 2-ethylhexenyl, 2-propy1-2-butenyl, 4-(2-methy1-3-
butene)-
pentenyl. An alkenyl group can be unsubstituted or substituted with one or two
suitable
substituents.
[017] As used herein and unless otherwise indicated, the term "alkynyl" or
"alkynyl
group" means a monovalent, unbranched or branched hydrocarbon chain having one
or more
triple bonds therein. The triple bond of an alkynyl group can be unconjugated
or conjugated
to another unsaturated group. Suitable alkynyl groups include, but are not
limited to, (C2-
C8)alkynyl groups, such as ethynyl, propynyl, butynyl, pentynyl, hexynyl,
methylpropynyl, 4-
methyl-1-butynyl, 4-propy1-2-pentynyl, and 4-butyl-2-hexynyl. An alkynyl group
can be
unsubstituted or substituted with one or two suitable substituents.
4
CA 02823088 2013-08-08
WO 2007/005972
PCT/US2006/026210
[0181 As used herein and unless otherwise indicated, the term
"substituted" as used
to describe a compound or chemical moiety means that at least one hydrogen
atom of that
compound or chemical moiety is replaced with a second chemical moiety. The
second
chemical moiety can be any suitable substituent that does not nullify the
synthetic or
pharmaceutical utility of the compoun.ds of the invention or the intermediates
useful for
preparing them. Examples of suitable substituents include, but are not limited
tó: (Cr
C8)alkyl; (C2-C8)alkenyl; (C2-C8)allcynyl; aryl; (C2-05)heteroaryl; (Cl-
C6)heterocycloalkyl;
(C3-C7)cycloalkyl; 0-(Ci-C8)alkyl; 0-(C2-COalkenyl; 0-(C2-Cs)alkynyl; 0-aryl;
CN; OH;
oxo; halo, C(0)0H; COhalo; 0(CO)halo; CF3, N3; NO2,NH2; NH((CI-Cs)alkyl);
N((CI-
C8)alky1)2; NH(ary1); N(aryl)2; (CO)NH2; (CO)NH((CI-C8)alkyl); (CO)N((q-
C8)alkyl)2;
=
(CO)NH(ary1); (CO)N(aryl)2; 0(CO)NH2; NHOH; NOH((Ci-Cs)alkY1);
NOH(ary1);0(CO)NH((C1-C8)alkyl); 0(CO)N((C1-C8)alkyl)2; 0(CO)NH(aTY1);
O(CO)N(aryl)2; CHO; CO((C1-C8)alkyl); CO(ary1); C(0)0((C1-C8)alkyl);
C(0)0(ary1);
0(C0)(( CI-C8)alkyl)- ; 0(C0)(arY1); 0(C0)0((C1-C8)alkyl); 0(C0)0(ary1); 84 Cr
C8)alkyl; S-( Ci-C8)alkenyl; S-( C1-C8)allcynyl; and S-aryl. One of skill in
art can readily
choose a suitable substituent based on the stability and pharmacological and
synthetic activity
of the compound of the invention.
[019] As used herein and unless otherwise indicated, a composition that is
"substantially free" of a compound means that the composition con ins less
than about 20%
by weight, more preferably less than about 10% by weight, even more preferably
less than
about 5% by weight, and most preferably less thRn about 3% by weight of the
compound.
[020] As used herein and unless otherwise indicated, the term
"stereochemically
pure" meons a composition that comprises one stereoisomer of a compound and is
substantially free of other stereoisomers of that compound. For example, a
stereomerically
pure composition of a compound having one chiral center will be substantially
free of the
opposite enantiomer of the compound. A stereomerically pure composition of a
compound
having two chiral centers will be substantially free of other diastereomers of
the compound.
A typical stereomerically pure compound comprises greater than about 80% by
weight of one
stereoisomer of the compound and less than about 20% by weight of other
stereoisomers of
the compound, more preferably greater than about 90% by weight of one
stereoisomer of the
411 compound and less than about 10% by weight of the other
stereoisomers of the compound,
even more preferably greater than about 95% by weight of one stereoisomer of
the compound
and less than about 5% by weight of the other stereoisomers of the compound,
and most
CA 02823088 2013-08-08
WO 2007/005972 PCT/US2006/026210
=
preferably greater than about 97% by weight of one stereoisomer of the
compound and less
than about 3% by weight of the other stereoisomers of the compound.
[0211 As used herein and unless otherwise indicated, the term
"enantiomerically
pure" means a stereomerically pure composition of a compound having one chiral
center.
[0221 As used herein and unless otherwise indicated, the terra "racemic"
or
"raceinate" means about 50% of one enantiomer and about 50% of the
corresponding
enantiomer relative to all chiral centers in the molecule. The invention
encompasses all
enanti9merica11y pure, enantiomerically enriched, diastereomerically pure,
diastereomerically
enriched, and racemic mixtures of the compounds of the invention.
[0231 As used herein and unless otherwise indicated, the term
"process(es) of the
invention" or "process(es) of preparing" or "process(es) for the preparation"
refers to the
methods disclosed herein which are useful for preparing a compound of the
invention.
Modifications to the methods disclosed herein (e.g., starting materials,
reagents, protecting
groups, solvents, temperatures, reaction times, purification) are also
encompassed by the
present invention.
[024] As used herein and unless otherwise indicated, the term "adding",
"reacting"
or the like means contacting one reactant, reagent, solvent, catalyst,
reactive group or the like
with another reactant, reagent, solvent, catalyst, reactive group or the like.
Reactants,
reagents, solvents, catalysts, reactive group or the like can be added
individually,
simultaneously or separately and can be added in any order. They can be added
in the
presence or absence of heat and can optionally be added under an inert
atmosphere.
"Reacting" can refer to in situ formation or int-molecular reaction where the
reactive groups
are in the same molecule.
[0251 As used herein and unless otherwise indicated, a reaction that is
"substantially
complete" or is driven to "substantial completion" means that the reaction
contains more than
about 80% by percent yield, more preferably more than about 90% by percent
yield, even
more preferably more than about 95% by percent yield, and most preferably more
than about
97% by percent yield of the desired product.
[026] As used herein and unless otherwise indicated, the term
"pharmaceutically
acceptable salt" includes, but is not limited to, salts of acidic or basic
groups that may be
present in the compounds of the invention. Compounds of the invention that are
basic in
nature are capable of forming a wide variety of salts with various inorganic
and organic acids.
6
CA 02823088 2013-08-08
WO 2007/005972
PCT/US2006/026210
The acids that may be used to prepare pharmaceutically acceptable salts of
such basic
compounds are those that form salts comprising pharmacologically acceptable
anions
including, but not limited to, acetate, benzenesulfonate, benzoate,
bicarbonate, bitartrate,
bromide, camsylate, carbonate, chloride, bromide, iodide, citrate,
dihydrochloride, edetate,
=
edisylate, estolate, esylate, fumarate, gluceptate, gluconate, glutamate,
glycollylarsanilate,
hexylresorcin.ate, hycirabamine, hydroxynaphthoate, isethionate, lactate,
lactobionate, malate,
maleate, mandelate, mesylate, methylsulfate, nauseate, napsylate, nitrate,
panthothenate,
phosphate/diphosphate, polygalacturonate, salicylate, stearate, succinate,
sulfate, tannate,
tartrate, teoclate, triethiodide, and pamoate. Compounds of the invention that
include an
amino group also can form phannaceutically acceptable salts with various amino
acids, in
addition'. to the acids mentioned above. Compounds of the invention that are
acidic in nature
are capable of forming base salts with various pharmacologically acceptable
cations. Non-
limiting examples of such salts include alkali metal or alkaline earth metal
salts and,
particularly, calcium, magnesium, sodium, lithium, zinc, potassium, and iron
salts.
[027] As used herein and unless otherwise indicated, the term "hydrate"
means a
compound of the present invention or a salt thereof, that further includes a
stoichiometric or
non-stoichiometeric amount of water bound by non-covalent intermolecular
forces.
[028] As used herein and unless otherwise indicated, the tenn "solvate"
means a
solvate formed from the association of one or more solvent molecules to a
compound of the
present invention. The term "solvate" includes hydrates (e.g., mono-hydrate,
dihydrate,
trihydrate, tetrahydrate, and the like).
[029] As used herein and unless otherwise indicated, the term "polymorph"
means
solid crystalline forms of a compound of the present invention or complex
thereof. Different
polymorphs of the same compound can exhibit different physical, chemical and
/or
spectroscopic properties.
[030] As used herein and unless otherwise indicated, the phrase "diseases
or
conditions related to an abnormally high level or activity of TNF-a" means
diseases or
conditions that would not arise, endure or cause symptoms if the level or
activity of TNF-a
were lower, or diseases or conditions that can be prevented or treated by a
lowering of TNF-a
4t1 level or activity.
[031] As used herein, and unless otherwise specified, the terms "treat,"
"treating"
and "treatment" contemplate an action that occurs while a patient is suffering
from the
7
CA 02823088 2013-08-08
WO 2007/005972
PCT/1JS2006/026210
specified disease or disorder, which reduces the severity or symptoms of the
disease or
disorder or retards or slows the progression or symptoms of the disease or
disorder.
[032] Acronyms or symbols for groups or reagents have the
following definition:
HPLC = high performance liquid chromatography, CH3CN = acetonitrile; DMF =
dimethyl
formamide, DMSO = dimethyl sulfindde, THF = tetrahydrofuran, CH2C12 =
methylene
chloride and CDI = 1,1 '-carbonyldiimidazole.
[0331 If there is a discrepancy between a depicted structure and
a name given that
structure, the depicted structure is to be accorded more weight. Furthermore,
if the
stereochernistry of a structure or a portion thereof is not indicated, e.g.,
with bold or dashed
lines, the structure or portion thereof is to be interpreted as encompassing
all stereoisomers of
it.
[03411 The invention can be underatood more fully by reference to
the following
detailed description and illustrative examples, which are intended to
exemplify non-limiting
embodiments of the invention.
5.2 Processes of the Invention
[0351 The present invention provides processes of preparing
unsubstituted and
substituted 4-amino-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione
compounds. In
general, the processes of the present invention may encompass improved or
efficient means
for the large scale or commercial production of unsubstituted and substituted
4-amino-2-(2,6-
dioxopiperidin-3-yl)isoindoline-1,3-dione compounds.
[0361 The unsubstituted and substituted 4-amino-2-(2,6-
dioxopiperidin-3-
yl)isoindoline-1,3-dione compounds can be used to prepare pharmaceutical
compositions
and/or dosage forms for treating a wide range of diseases and conditions
including, but not
limited to, inflammatory diseases, autoimmune diseases, cancers, heart
diseases, genetic
diseases, allergic diseases, osteoporosis and lupus. In general, the
pharmaceutical
compositions can comprise at least one of the 4-amino-2-(2,6-dioxopiperidin-3-
yl)isoindoline-1,3-dione compounds or a pharmaceutically acceptable salt,
solvate,
polymorph or stereoisomer thereof. The pharmaceutical compositions can be
administered
to patients who are treated for a wide range of diseases and conditions.
Optionally, the
411 pharmaceutical compositions can further comprise at least one carrier,
excipient, diluent, a
second active agent or a combination thereof. In some embodiments, the
pharmaceutical
compositions are used in the preparation of individual, single unit dosage
forms. Single unit
8
CA 02823088 2013-08-08
WO 2007/005972 PCTMS2006/026210
dosage forms are suitable for oral, mucosal (e.g., sublingual, nasal, vaginal,
cystic, rectal,
preputial, ocular, buccal or aural), parenteral (e.g., subcutaneous,
intravenous, bolus injection,
intramuscular or intraarterial), topical (e.g., eye drops or other ophthalmic
preparations),
transdermal or transcutaneous administration to a patient. Non-limiting
examples of dosage
forms include tablets, caplets, capsules (e.g., soft elastic gelatin
capsules), cachets, troches,
lozenges, dispersions, suppositories, powders, aerosols (e.g., nasal sprays or
inhalers), gels,
liquid dosage forms suitable for oral or mucosal administration to a patient,
including
suspensions (e.g., aqueous or non-aqueous liquid suspensions, oil-in-water
emulsions or a
water-in-oil liquid emulsions), solutions and elixirs, liquid dosage forms
suitable for
parenteral administration to a patient, eye drops or other ophthalmic
preparations suitable for
topical administration, and sterile solids (e.g., crystalline or amorphous
solids) that can be
reconstituted to provide liquid dosage forms suitable for parenteral
administration to a
patient.
[037] In some embodiments, the invention provides processes for preparing 4-
amino-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione compounds of Formula
(I):
0 11
(101
R 1 ____________________________________
N H 2 (1)
or a pharmaceutically acceptable salt, solvate, polymorph or stereoisomer
thereof, comprising
the step of cyclizing an N-(3-aminophthaloy1)-glutamine compound of Formula
(II), an N-(3-
aminophthaloy1)-isoglutamine compound of (IIA) or a salt thereof:
0 0 0 0
=
7tIH2
N CONH2
CO2H
NH2NH2
(II) or (ITA)
with a cyclizingagent wherein RI is H, F, benzyl, (C1-C8)alkyl, (C2-
C8)alkenyl, or (Cr
C8)alkynyl.
[038] In one embodiment, le of Fonnula (I) and/or (II) is H. In a
particular
embodiment, RI of Formula (I) and/or (II) is (CI-C8)allcyl. In a further
embodiment, of
Formula (I) and/or (II) is methyl. In another embodiment, the solvate is a
hydrate.
9
CA 02823088 2013-08-08
WO 2007/005972 = PCT/US2006/026210
[039] The cyclization of the compound of Formula (II) with the cyclizing
agent can
occur in a solvent such as acetonitrile, ethyl acetate, acetone, methyl ethyl
ketone, diethyl
ether, tetrahydrofuran,- dichloromethane, chloroform, N-methyl pyrrolidinone,
climethyl .
formamide, dimethyl sulfoxide and combinations thereof. In one embodiment, the
solvent is
acetonitrile. In another embodiment, the solvent. is boiling acetonitrile.
[040]
The reaction temperature can be any temperature useful for the
cyclization =
reaction according to a person of ordinary skill in the art. For instance, in
certain
embodiments, the cyc1i7ntion reaction temperature can vary from about 20 C to
about 100
C. In some embodiments, the cyclization reaction temperature is from about 50
C to about
90 C. In other embodiments, the cyclization reaction temperature is from
about 80 C to
about 87 C. In a particular embodiment, the cyclization reaction temperature
is the boiling
point (Le., 81-82 C at 1 atmospheric pressure) of acetonitrile.
[041] The cyclization reaction thne can be any time period useful for the
cyclization
reaction according to a person of ordinary skill in the art. For instance, in
certain
embodiments, the cyclization reaction time can vary from about 1 to about 24
hours,
depending on the reaction temperature. In general, the higher the reaction
temperature, the
shorter is the reaction time. In one embodiment, the solvent is acetonitrile,
the reaction
temperature is from about 80 C to about 87 C, and the reaction time is from
about 1 to about
hours.
[042] The cyclizing agent can be any chemical that can cause a ring
formation
reaction between the amide group and the carboxylic group of Formula (H) or
(IIA). In some
embodiments, the cyclizing agent can have the following formula:
0
Y X (v)
where each of X and Y is independently an unsubstituted or substituted
imidazolyl,
benzimidazolyl or benzotriazolyl. The cyclizing reagent of Formula (V) can be
purchased
from a commercial supplier or prepared according to any method apparent to a
person of
ordinary skill in the art. For instance, the cyclizing agent of Formula (V)
can be prepared by
reacting phosgene (C0C12) with an =substituted or substituted 1H-imidazole
compound, 1H-
benzimidazole or 1H-benzotriazole. The reaction between phosgene and a 1H-
imidazole
compound is described in Batey et al., Tetrahedron Lett., 1998, 39, 6267. The
reaction
CA 02823088 2013-08-08
WO 2007/005972 =
PCT/US2006/026210
between phosgene and a 113-benzotriazole compound is described in Katritzky et
al., J. Org.
Chem., 1997, 62, 4155.
[043] In some embodiments, the cyclizing agent is a carbonyldiimidazole
compound
having the formula:
R7 I. IR 2
N
(1-
R 5 R 4
R6 R3 (VI)
where each of R2, R3, R4, R5, R6 and R7 is independently H, alkyl, halo,
nitro, cyano, acyl,
alkoxy, aryloxy, alkoxycarbonyl or alkoxymethyl.
[044] The carbonyldiimidazole compound of Formula (VI) can be purchased
from a
commercial supplier or prepared according to any method apparent to a person
of ordinary
skill in the art. For instance, the carbonyldiimidazole compound of Formula
(VI) can be
prepared by reacting phosgene (C0C12) with an unsubstituted or substituted 1H-
imidazole
compound or a combination thereof. Some non-limiting examples of the 1H-
imidazole
compound suitable for this invention include 1H-imidazole, 2-methyl-1H-
imidazole, 1H-
imidazole-5-carbaldehyde, 2-ethyl-1H-imidazole, 2-isopropy1-1R-imidazo1e, 2-
ethy1-5-
methy1-1H-imidazole, 2-propy1-1H-imidazole, 2-nitro-1H-imidazole, 5-nitro-1H-
imidazole,
methyl 1H-imidazole-5-carboxylate, 4-(2-methoxyethyl)-1H-imidazole, 2-methy1-5-
nitro-1H-
imidazole and 5-methy1-4-nitro-1H-imidazo1e, all of which can be obtained from
a
commercial supplier such as Aldrich Chemicals, Milwaukee, WI or prepared by
methods
known to a person of ordinary skill in the art. Non-limiting examples of the
carbonyldiimidazole compound include 1,1'-carbonyldiimidazole, 2,2'-dimethy1-
1,1'-
carbonyldiimidazole, 2,2'-diethyl-1,1'-carbonyldiimidazole, 2,2'-diisopropy1-
1,1'-
carbonyldiimidazole and 2,2'-dinitro-1,1'-carbonyldiimidazole, all of which
can be obtained
commercially from a supplier such as Aldrich Chemicals, Milwaukee, WI or
prepared by the
method described above. In one embodiment, the carbonyldiimidazole compound is
1,1'-
carbonyldiimidazole.
[045] In further embodiments, the cyclizing agent is selected from Formula
(V),
411
S0C12, POC13, derivatives of SOC12, derivatives of POC13, and combinations
thereof. The
cyclization reaction can be further promoted or catalyzed by using a base in
addition to the
11
CA 02823088 2013-08-08
WO 2007/005972 PCT/US2006/026210
cyclizing agent. The base can be selected from the group consisiting of
organic amines such
as triethylamine, pyridine, derivatives of pyridine and combinations thereof.
[0461 In a
particular embodiment, the 4-amino-2-(2,6-dioxopiperidin-3-
yl)isoindoline-1,3-dione compound of Formula (I) can be prepared by cyclizing
the N-(3-
aminophthaloy1)-glutamine compound of Formula (II) or a salt thereof with 1,1'-
carbonyldiimidazole (CDI) in refluxing acetonitrile for about 3 hours as
depicted in Scheme
A below. Alternatively, the same reaction can occur in N-methyl pyrrolidinone
or
tetrahydrofuran for a time period from about 13 to about 15 hours at room
temperature. In
some embodiments, RI in Scheme A is H.
00 00 H
OH CDI
actrii 411 N NO
CONH2
Ri ___________________
NH refluxed; =NH2
2
3 hours
Formula (II) Formula (I)
SCHEME A
[047] The ratio of the compound of Formula (II) to 1,1 '-
carbonyldiimidazole can be
any ratio useful for the cyclization reaction according to a person of
ordinary skill in the art.
For instance, the ratio of the compound of Formula (II) to 1,1 '-
carbonyldiimidazole can be
from about 2:1 to about 1: 2. In some embodiments, the ratio of the compound
of Formula
(II) to 1,1 '-carbonyldiimidazole is from about 1:1 to about 1:1.5. In other
embodiments, the
ratio of the compound of Formula (II) to 1,1'-carbonyldiimidazole is from
about 1:1 to about
1 :1.2. In one embodiment, the cyclization of Formula (II) with 1,1 '-
carbonyldiimidazole
occurs in acetonitrile for 1 to 24 hours. In another embodiment, the
cyclization of Formula
(II) occurs in refluxing acetonitrile for 3 hours.
[048] In another embodiment, the 4-amino-2-(2,6-dioxopiperidin-3-
ypisoindoline-
1,3-dione compound of Formula (I) can be prepared by cyclizing the N-(3-
aminophthaloyI)-
isoglutamine compound of Formula (IIA) or a salt thereof with 1,1 '-
carbonyldiimidazole
(CDI) in a solvent, such as acetonitrile, N-methyl pyrrolidinone and
tetrahydrofuran, as
depicted in Scheme A' below. The reaction can occur at a temperature ranging
from about
room temperature to about 150 C for about 30 minutes to about 24 hours.
12
CA 02823088 2013-08-08
WO 2007/005972 PCT/US2006/026210
0 0 00
= CDI
N /\--NHc202H 1101
R1 ___________________________ solvent R1 __
NH2 NH2
Formula (IIA) =Formula (I)
SCHEME A'
[049] In one embodiment, the compound of Formula (I) can be a free amine.
Optionally, the free amine of Formula (I) can be converted into an acid salt
by reacting the
free amine of Formula (I) with the corresponding acid in a mole ratio of about
1 :1. Some
non-limiting examples of suitable acids include methanesulionic acid,
trifluoroacetic acid,
4-(trifluoromethyl)benzoic acid, p-toluenesulfonic acid, hydrochloric acid,
nitric acid,
sulfuric acid and phosphoric acid. In one embodiment, the 4-amino-2-(2,6-dioxo-
3-
piperidinyl)isoindole-1,3-dione of Formula (I) is converted into a
hydrochloride salt with
hydrochloric acid at a temperature from about 0 C to about 22 C.
[050] If a racemic compound of Formula (I) is desired, a racemic N-(3-
aminophthaloy1)-glutamine compound of Formula (II) may be used in the
cyclization
reaction. Conversely, if an enantiomerically pure compound of Formula (I) is
desired, an
enantiomerically pure N-(3-aminophthaloy1)-glutamine compound of Formula (II)
may be
used. Alternatively, if an enantiomerically pure compound of Formula (I) is
desired, a
racemic mixture of Formula (I) may be prepared and then resolved into the
enantiomers by
conventional resolution techniques such as biological resolution and chemical
resolution. In
general, biological resolution uses a microbe which metabolizes one specific
enantiomer
leaving the other alone. In chemical resolution, the racemic mixture is
converted into two
diastereoisomers that may be separated by conventional techniques such as
fractional
crystallization and chromatographies. Once separated, the diasteriosomeric
forms may be
converted separately back to the enantiomers.
[051] The compound of Formula (II) can be prepared by any method known to a
person of ordinary skill in the art. For example, the compound of Formula (II)
can be
prepared by reducing the nitro group of the compound of Formula (III) to an
amine group as
depicted in Scheme B below:
13
CA 02823088 2013-08-08
WO 2007/005972 = PCT/US2006/026210
= 0 0 oNH2 0 0
OH reduction
c 1101
OH
CONH2
Ri ________________________________________________________
Ri _________________
NO2 NH2
Formula (III) Formula (II)
SCHEME B
wherein le is H, F, benzyl, (CI-C8)alkyl, (C2-C8)alkenyl, or (C2-C8)alicynyl.
In some
embodiments, RI in Scheme B is H.
[052] Similarly, the compound of Formula (IIA) can be prepared by
reducing the
nitro group of the compound of Formula (IIIA) to an amine group as depicted in
Scheme B'
below:
0 0 0 0
=
= NH2 reduction
NH2
FO2H __________________________________________ )is= CO2H
R1 ________________________________________________________
NO2 NH2
Formula (MA) Formula (IIA)
SCHEME B'
[053] In Schemes B and B' above, the compounds of Formulae (III) and
(IIIA) can
be reduced to the compounds of Formulae (II) and (IIA) respectively by any
reducing agent
known in the art that can reduce a nitro group to a primary amine. Some non-
limiting
examples of such reducing agent include hydrogen plus a catalyst (catalytic
hydrogenation),
reducing metals in an acid such as hydrochloric acid and acetic acid, sodium
sulfide in
ammonium hydroxide solution, zinc in ammonium formate solution, magnesium in
hydrazinium monoformate solution and tin dichloride in dilute hydrochloric
acid. Some non-
limiting examples of suitable hydrogenation catalyst include palladium metal
(Pd), platimun
metal (Pt), and derivatives and complexes of Pd and Pt. The hydrogenation
catalyst can be
dissolved in a solvent; or dispersed or coated on the surface of a catalyst
support such as
carbon and inorganic particles such as alumina, silica, aluminum silicates and
the like. Some
non-limiting examples of suitable reducing metals include iron, zinc amalgam,
zinc and tin.
In a particular embodiment, the reducing agent is hydrogen plus a catalyst. In
a further
embodiment, the catalyst is a Pd catalyst. In another embodiment, the catalyst
is 5% Pd/C.
In another embodiment, the catalyst is 10% Pd/C. Further, either wet or dry
hydrogenation
catalyst can be used.
14
CA 02823088 2013-08-08
WO 2007/005972 PCT/US2006/026210
[054] The catalytic hydrogenation is generally carried out at a hydrogen
pressure
that drives the reaction to substantial completion. In a particular
embodiment, the catalytic
hydrogenation is carried out at a hydrogen pressure from about 2.76 bars (L
e., 40 psi or 276
kPa) to about 4.14 bars (i.e., 60 psi or 414 kPa).
[055] In one embodiment, the catalytic hydrogenation is run at ambient
temperature.
The catalytic hydrogenation is generally perfonned until the reaction is
substantially
complete. In a particular embodiment, the catalytic hydrogenation is performed
for about 1-
24 hours at a temperature from about 15 C to about 30 C. In a further
embodiment, the
catalytic hydrogenation is performed for about 2 to 3 hours at a temperature
from about 18 C
to about 24 C.
[0561 In one embodiment, the catalytic hydrogenation occurs at a
temperature from
about 18 C to about 24 C for about 2-3 hours in methanol in the presence of
10% Pd/C.
Either wet or dry hydrogenation catalyst can be used. In a further embodiment,
the catalytic
hydrogenation occurs at a pressure from about 40 (2.76 bars or 276 kPa) to
about 50 psi (3.45
bars or 345 kPa).
[057] The catalytic hydrogenation can occur in a solvent. In one
embodiment, the
catalytic hydrogenation is conducted in a protic solvent, such as alcohols,
water, and
combinations thereof. In a further embodiment, the alcohol solvent is selected
from the
group consisting of methanol, ethanol, propanol, isopropan.ol, butanol,
isobutanol, t-butanol
and combinations thereof. In another embodiment, the catalytic hydrogenation
is conducted '
in an apolar, aprotic solvent such as 1,4-dioxane. In yet another embodiment,
the catalytic
hydrogenation is conducted in a polar, aprotic solvent such as acetone, DMSO,
DMF and
THF. In one embodiment, the solvent is a protic solvent. In a further
embodiment, the
solvent for catalytic hydrogenation is methanol. In further embodiments,
solvent mixtures
are used.
[058] If a racemic compound of Formula (II) or (IIA) is desired, a racemic
compound of Formula (III) or (IIIA) can be used. Conversely, if an
enantiomerically pure
compound of Formula (II) or (IIA) is desired, an enantiomerically pure
compound of Formula
(III) or (IIIA) can be used. Alternatively, if an enantiomerically pure
compound of Formula
(II) or (IIA) is desired, a racemic mixture of Formula (II) or (IIA) can be
prepared and then
resolved into the enantiomers by conventional resolution techniques such as
biological
resolution and chemical resolution.
CA 02823088 2013-08-08
WO 2007/005972 PCT/US2006/026210
[059] The compound of Formula (III) can be prepared by any method known to
a
person of ordinary skill in the art. For example, the compound of Formula
(III) can be
prepared by reacting 3-nitrophthalic anhydride with a glutamine of Formula
(IV) as depicted
in Scheme C below. le is as defined above. In some embodiments, le in Scheme C
is H.
0 0 0
0jH
0 + H2N CONNT-41111- * N7tRi CONH2
NO2 0 Formula (IV) NO2
Formula (III)
SCHEME C
[060] Similarly, the compound of Formula (ram can be prepared by reacting 3-
nitrophthalic anhydride with an isoglutamine of Formula (TVA) as depicted in
Scheme C'
below. RI is as defined above. In some embodiments, le in Scheme C' is H.
0 0 0 0
H2
IN 0 + HN CO2H =
N CO2H
R1
NO2 Formula (IVA) NO2
Formula (IIIA)
= SCHEME C'
[0611 The reaction between 3-nitrophthalic anhydride and the
glutamine of Formula
(IV) or the isoglutamine of Formula (NA) can occur in. a solvent such as
acetonitrile, ethyl
acetate, acetone, methyl ethyl ketone, diethyl ether, tetrahydrofuran,
dichloromethane,
chloroform, N-methyl pyrrolidinone, dimethyl formaraide, dimethyl sulfoxide
and _
combinations thereof. In one embodiment, the solvent is dimethyl formamide.
[062] The reaction temperature can be any temperature useful for
the reaction of
Scheme C or C' according to a person of ordinary skill in the art. For
instance, in certain
embodiments, the temperature of the reaction between 3-nitrophthalic anhydride
and Formula
(IV) or (IVA) can be from about 20 C to about 90 C. In some embodiments, the
reaction
temperature is from about 40 C to about 90 C. In other embodiments, the
reaction
temperature is from about 60 C to about 90 C. In further embodiments, the
reaction
fIi temperature is from about 80 C to about 90 C.
16
CA 02823088 2013-08-08
WO 2007/005972 PCT/US2006/026210
[063] The reaction time can be any time useful for the reaction of Scheme C
or C'
according to a person of ordinary skill in the art. For instance, the reaction
time can vary
from about 1 hour to about 24 hours, depending on the reaction temperature. In
general, the
higher the reaction temperature, the shorter is the reaction time. In a
particular embodiraent,
the reaction time is about 8 hours at a reaction ternperature from about 80 C
to about 90 C.
[064] If a racemic compound of Formula (III) or (IIIA) is desired, a
racemic
glutamine of Formula (IV) or (IVA) can be used. Conversely, if an
enantiomerically pure
compound of Formula (III) or (IIIA) is desired, an enantiomerically pure
glutamine of
Formula (IV) or (IVA) can be used. Non-limiting examples of glutamine of
Formula (IV)
include D-glutamine and L-glutaraine, both of which can be obtained from a
commercial
supplier such as Aldrich, Milwaukee, WI. Alternatively, if an enantiomerically
pure
compound of Formula (III) or (MA) is desired, a racemic mixture of Formula
(III) or (IIIA)
can be prepared and then resolved into the enantiomers by conventional
resolution techniques
such as biological resolution and chemical resolution.
[065] The 3-nitrophthalic anhydride can be obtained commercially from a
supplier
such as Aldrich Chemical or prepared by any known method in the art. Further,
the
compound of Formula (VII) can be prepared by reacting maleic anhydride with a
glutamine
of Formula (IV) according to the conditions described above for the reaction
between 3-
nitrophthalic anhydride with the glutamine compound of Formula (IV).
[066] Alternatively, the compound of Formula au) can be prepared according
to the
procedure depicted in Scheme D below. Referring to Scheme D below, RI is as
defined
above and R8 is alkyl such as t-butyl or aralkyl such as benzyl. In some
embodiments, RI in
Scheme D is H and R8 is t-butyl. In other embodiments, RI in Scheme D is H and
R8 is
benzyl.
17
CA 02823088 2013-08-08
WO 2007/605972 =
PCT/US2006/026210
= 0 0 0
0
N H CIAOEt =
I N-8-0Et
.NO2 0 NO2 CI
0J¨R8
0 H2N CONH2
0 R1 0 0
= N-8-0Et Formula
(VIM 0¨R8
Triethylamine 1111 ,CON
H2
R1 _______________________________________________________________
NO2 CI Rs is alkyl or aralkyl NO2
Formula (1X)
0 = 0 0 0
0¨R8 N N¨L HC1 OH
CONH2
4110 T-c7LC ON H2 =
Ft1
NO2 NO2 CI
Formula (111)
Scheme D
[0671 Referring to Scheme D above, 3-nitzophthalimide can react
with ethyl
chlorofornaate in a solvent in the presence of a catalyst such as
triethylamine to form 3-nitro-
N-ethoxycarbonyl-phthalimide. Some non-limiting examples of suitable solvent
include
acetonitrile, ethyl acetate, acetone, methyl ethyl ketone, diethyl ether,
tetrahydrofuran,
dichloromethane, chloroform, N-methyl pyrrolidinone, dimethyl formamide,
dimethyl
sulfoxide and combinations thereof. In one embodiment, the solvent is dimethyl
sulfoxide.
The reaction temperature can be any temperature useful for the reaction of
according to a
person of ordinary skill in the art. For instance, in certain embodiments, the
reaction
temperature can be from about 0 C to about 5 C. The reaction time can be any
time useful
for the reaction according to a person of ordinary skill in the art. For
instance, the reaction
time can vary from about 1 hour to about 24 hours, depending on the reaction
temperature.
In general, the higher the reaction temperature, the shorter is the reaction
time. In a particular
embodiment, the reaction time is about 4 hours at 0-5 C.
[068] The t-butyl or benzyl N-(3-nitrophthaloy1)-glutamine of
Formula (IX) can be
purchased or prepared by reacting 3-nitro-N-ethoxycarbonyl-phthalimide with a
glutamine t-
butyl or benzyl ester of Formula (VIII) or an acid salt thereof such as a
hydrochloride salt,
where RI is H, F, benzyl, (Ci-C8)alkyl, (C2-C8)alkenyl, or (C2-C8)alkynyl; and
R8 is t-butyl or
benzyl, in a solvent in the presence of a catalyst such as triethylamine. In
some
41,1
embodiments, a racemic mixture of glutamine t-butyl ester hydrochloride is
used to prepare
of Formula (IX). In other embodiments, L-glutamine t-butyl ester hydrochloride
is used to
18
CA 02823088 2013-08-08
WO 2007/005972 = PCT/US2006/026210
prepare of Formula (IX). In further embodiments, D-glutamine t-butyl ester
hydrochloride is
used to prepare of Formula (IX). Some non-limiting examples of suitable
solvents include
acetonitrile, ethyl acetate, acetone, methyl ethyl ketone, diethyl ether,
tetrahydrofuran,
dichloromethane, chloroform, N-methyl pyrrolidinone, dimethyl formamide,
dimethyl
sulfeodde and combinations thereof. In one embodiment, the solvent is
tetrahydrofuran. The
reaction temperature can be any temperature useful for the reaction of
according-to a person
of ordinary skill in the art. For instance, in certain embodiments, the
reaction temperature
can be from about 25 C to about 100 C. The reaction time can be any time
useful for the
reaction according to a person of ordinary skill in the art. For instance, the
reaction time can
Nutty from about 1 hour to about 48 hours, depending on the reaction
temperature. In general,
the higher the reaction temperature, the shorter is the reaction time. In a
particular
embodiment, the reaction time is about 24 hours at about 65-66 C.
[069] =The reaction between hydrogen chloride and t-butyl N-(3-
nitrophthaloy1)-
glutamine of Formula (IX) in a solvent can afford the compound of Formula
(III). Some non-
limiting examples of suitable solvent include acetonitrile, ethyl acetate,µ
acetone, methyl ethyl
= ketone, diethyl ether, tetrahydrofuran, dichloromethane, chloroform, N-
methyl pyrrolidinone,
dimethyl formamide, dimethyl sulfoxide and combinations thereof. In one
embodiment, the
solvent is dichloromethane. The reaction temperature can be any temperature
useful for the
reaction of according to a person of ordinary skill in the art. For instance,
in certain
embodiments, the reaction temperature can be from about 0 C to about 100 C.
The reaction
-time can be any time useful for the reaction according to a person of
ordinary skill in the art.
For instance, the reaction time can vary from about 1 hour to about 24 hours,
depending on
the reaction temperature. In general, the higher the reaction temperature, the
shorter is the
reaction time. In a particular embodiment, the reaction time is about 16 hours
at about 20-
25 C.
[070] Referring to Scheme D, if a racemic compound of Formula (III)
is desired, a
racemic t-butyl N-(3-nitrophthaloy1)-glutamine of Formula (VIII) can be used.
Conversely, if
an enantiomerically pure compound of Formula (III) is desired, an
enantiomerically pure t-
butyl N-(3-nitrophthaloy1)-glutarnine of Formula (VIII) can be used.
Alternatively, if an
enantiomerically pure compound of Formula (III) is desired, a racemic mixture
of Formula
(III) can be prepared and then resolved into the enantiomers by conventional
resolution
techniques such as biological resolution and chemical resolution. In general,
biological
resolution uses a microbe which metabolizes one specific enantiomer leaving
the other alone.
19
CA 02823088 2013-08-08
WO 2007/005972 =
PCT/US2006/026210
In chemical resolution, the racemic mixture is converted into two
diastereoisomers that can
be separated by conventional techniques such as fractional crystallization and
chromatographies. Once separated, the diasteriosomeric forms can be converted
separately
back to the enantiomers.
=
[071] In some embodiments, the compound of Formula (MA) can be
prepared
according to the procedures depicted in Scheme D' below, which are similar to
the
procedures of Scheme D. Referring to Formulae (VILLA), (DCA) and (IIIA), RI
and R8 are as
defined above. In some embodiments, RI in Scheme D' is H and R8 is t-butyl. In
other
embodiments, RI in Scheme D' is H and R8 is benzyl.
o _ttjvH2
co-oR8
= RI o o
N-8-0Et Formula (VIIIA) =
N CO-OR8
Triethylamine
R1
NO2 I18 is alkyl or aralkyl NO2 Formula
(IXA)
0
=
0 0 _oR8 0 0
NH2 HCI NH2
co
2.--,.02H
NO2 No,
Formula (IIIA)
Scheme D'
[072] Alternatively, the 4-amino-2-(2,6-dioxopiperidin-3-ypisoindoline-1,3-
dione
compound of Formula (I), or a pharmaceutically acceptable salt, solvate,
polymorph or
stereoisomer thereof, can be prepared by reacting 3-aminophthalic acid or a
salt thereof with
a 3-aminoglutarimide compound of Formula (X) or a salt thereof:
H 2
0 N 0
(X),
in a solvent, wherein RI is H, F, benzyl, (C1-C8)alkyl, (C2-C8)alkenyl, or (C2-
C8)alkynyl. In
some embodiments, RI of Formula (X) is H.
[073] The 3-aminoglutarimide compound can be purchased commercially from a
411
supplier such as Evotec OAI, Hamburg, Germany; or prepared according to
methods
described in the literature such as Capitosti et al., Organic Letters, 2003,
Vol. 5, No. 16, pp.
CA 02823088 2013-08-08
WO 2007/005972= PCT/US2006/026210
2865-2867. In some embodiments, the 3-aminoglutarimide compound of Formula (X)
is 3-
aminoglutarimide (i.e., where RI of Formula (X) is H) or its salt. Some non-
limiting
examples of suitable salts of Formula (X) include carboxylic acid salts,
methanesulfonic acid
salt, nifluoroacetic acid salt, 4-(trifluoromethyl)benzoic acid salt, p-
toluenesulfonic acid salt,
hydrochloric acid salt, hydrobromic acid salt, nitric acid salt, sulfuric acid
salt and phosphoric
acid salt.
[0741 The above condensation or coupling reaction between the 3-
aminophthalic
acid or a salt thereof and the compound of Formula (X) or a salt thereof may
occur in the
presence of a catalyst. The catalyst may be a base, an acid such as a
carboxylic acid, or a
combination thereof. In some embodiments, the catalyst is or comprises a base.
Some non-
limiting examples of suitable bases include alkali hydroxides, alkaline
hydroxides, alkali
carboxylates (e.g., sodium acetate), alkali carbonates or hydrogen carbonates
(e.g., sodium
hydrogen carbonate), heterocyclic bases (e.g., substituted and unsubstituted
pyrrolidine,
pyrrolidinone, piperidine, piperidinone, pyrrole, pyridine, imidazole,
benzimidazole,
benzotriazole, and the like), amines and combinations thereof. In some
embodiments, the
catalyst is or comprises an amine. Some non-limiting examples of suitable
amines include
alkylamines (e.g., ethylamine), diallcylamines (e.g., diethylamine),
trialkyamines (e.g.,
triethylarnine and N,N-diisopropylethylamine), arylamines (e.g., phenylamine),
diarylamines
(e.g, diphenylamine), alkylarylamines (e.g., N-methylaniline), triarylamines
(e.g.,
triphenylamine), dialkylarylamines (e.g., N,N-dimethylaniline), and
alkydiarylamines (e.g.,
N-methyldiphenylarnine). In one embodiment, the catalyst is or comprises
triethylarnine,
unsubstituted imidazole or a combination thereof.
[0751 In certain embodiments, the catalyst is or comprises a carboxylic
acid having
Formula (XI):
R8-CO2H (XI)
=wherein R8 is alkyl, aryl, alkaryl, aralkyl, heterocyclyl or a combination
thereof. In some
embodiments, the carboxylic acid is or comprises an aliphatic carboxylic acid
such as acetic
acid. In further embodiments, the catalyst comprises at least one of the
amines and at least
one of the carboxylic acid of Formula (XI) disclosed herein. In a particular
embodiment, the
catalyst comprises triethylamine and acetic acid.
21
CA 02823088 2013-08-08
WO 2007/005972 PCT/US2006/026210
[076] The solvent for the condensation reaction may be any solvent that can
disperse
or dissolve both the 3-aminophthalic acid or a salt thereof and the 3-
aminoglutarimide
compound of Formula (X) or a salt thereof. Non-limiting examples of suitable
solvents
include acetonitrile, ethyl acetate, acetone, methyl ethyl ketone, diethyl
ether,
tetrahydrofuran, dichloromethane, chloroform, N-methyl pyrrolidinone, dimethyl
formamide,
dimethyl sulfoxide, toluene, isopropyl acetate, isopropyl alcohol, n-propanol
and
combinations thereof. In one embodiment, the solvent is acetonitrile.
[077] The condensation reaction temperature can be any temperature useful
for the
reaction of according to a person of ordinary skill in the art. For instance,
in certain
embodiments, the condensation reaction temperature can be from about 25 C to
about 100
C.
[078] The condensation reaction time can be any time useful for the
reaction
according to a person of ordinory skill in the art. For instance, the reaction
time can vary
from about 1 to about 48 hours, depending on the reaction temperature. In
general, the higher
the reaction temperature, the shorter is the reaction time. In a particular
embodiment, the
reaction time is from about 5 hours to about 7 hours at a reaction temperature
from about 80
C to about 90 C.
[079] In one embodiment, the compound of Formula (I) is 4-amino-2-(2,6-
dioxopiperidin-3-yl)isoindoline-1,3-dione (i.e., where RI of Formula (I) is H)
which is
prepared according to Scheme E below. Referring to Scheme E, 3-aminophthalic
acid
hydrochloride [i.e., Compound (1)] reacts with 3-aminoglutarimide (i.e., where
RI of
Form.ula (X) is H) hydrochloride [i.e., Compound (2)] in a solvent such as
acetonitrile in the
presence of a catalyst Comprising triethylamite and acetic acid. In some
embodiments, the
mole ratio of triethylaraine to acetic acid is from about 1:10 to about 10:1.
In other
embodiments, the mole ratio of triethylamine to acetic acid is from about 1:10
to about 1:1.
In further embodiments, the mole ratio of triethylamine to acetic acid is
about 1:2.
0 0
triethy lam ine,
GO2H rrNH2*Hel acetic acid H
= 3b= 1.1 N
C 0 2H *(:) acetonirile
H 2.H C I N H 2
(1) (2) (3)
22
CA 02823088 2013-08-08
=
WO 2007/005972 =
PCT/US2006/026210
Scheme E
[080] The 4-amino-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione compound
of
Formula (I) can be purified by any conventional purification techniques such
as
recrystallization, extraction, chromatography and the like. In some
embodiments, the
compound of Formula (I) is purified by recrystanization. In other embodiments,
the
compound of Formula (I) is 4-amino-2-(2,6-dioxopiperidin-3-ypisoindoline-1,3-
dione (i.e.,
where R1 of Formula (I) is H) which can be purified by recrystallization with
a solvent
mixture comprising dimethyl sulfoxide and water. In further embodiments, the
ratio of
dimethyl sulfoxide to water in the solvent mixture is from about 1:10 to about
10:1 by
volume. In a further embodiment, the ratio of diinethyl sulfoxide to water in
the solvent
mixture is about 1:4 to about 1:8 by volume.
[0811 Particular embodiments of the present invention are illustrated by
the
syntheses of Examples 1-17 according to Schemes A-E and modifocations thereof.
Modifications of variables including, but not limited to, reaction solvents,
reaction times,
reaction temperatures, reagents, starting materials, and functional groups in
the particular
embodiments of the synthesis of 4-amino-2-(2,6-dioxo-3-piperidinyl)isoindole-
1,3-dione or
an acid salt thereof will be apparent to those of ordinary skill in the art.
6. EXAMPLES
Example 1 - Preparation of N-(3-nitrophthaloy1)-glutamine According to Scheme
C
[0821 A mixture Of DMF (37 L), 3-nitrophthalic anhydride (4080 g, 21.1
moles) and
L-glutamine (3020 g, 20.7 moles) was added to a round bottom flask equipped
with a
mechanical stirrer, a condenser, a thermometer, a nitrogen inlet and a heating
mantel. The
reaction mixture was stirred at 80-87 C for 8 hours. The temperature of the
reaction was
kept below 90 C at all time. The progress of the reaction was monitored by
HPLC using a
Waters Nova-Pak C18 column (3.9x150 mm, particle size = 4 micron, UV
wavelength = 240
nm, retention time = 3.64 minutes) and a 10/90 mixture of acetonitrile and
0.1% aqueous
H3PO4 by volume as an eluent at a flow rate of 1 mL/min. After the reaction
was completed,
the reaction mixture was allowed to cool to room temperature and then
concentrated to an oil
(about 90% of DMF was removed) under a reduced pressure (400 mtorr at pump) on
a
411
heating bath at 40 C. The oil was stirred with water (39.7 L) for 6 hours to
produce a slurry.
The solid in the slurry was filtered, washed with water (8.8 L), air dried and
then dried in a
vacuum oven at 60 C and <1 mm pressure. The yield of the crude product was
4915 g
23
CA 02823088 2013-08-08
=
53686-75
(92.9% purity by HPLC). The crude product was further Purified by dispersing
it in ethyl
acetate in a ratio of 10 pit, of ethyl acetate to 1 g of the crude product
After the dispersion =
was stirred overnight, it was then filtered and the solid filtered out was
dried to yield 4780 g
(70%) of the product. The product purity was found to be 99.62% by HPLC using
a Waters
Nova-Pak/C18 column (3.9x150 min, particle size = 4 micron, UV wavelength =
240 rim,
retention time = 5.0 minute-s) and an eluent inatire of Icetonitrile ind-0.1%
aqiiedtis H31504
in a ratio of 10:90 by volume at a flow rate of 1 ML/min. The produet iii DMSO-
d6-was=
characterized by a 1H NMR spectruin showing the following chemical shifts (8,
ppm):.13.32
(b, .1H), 8.33 (d, J=7.9Hz, 1H), 8.22 Hz, 1H), 8.11 (t, J=7.8 Hz, 1H), 720
(s, 1H),
6.47 (s, 111), 4.83-4.77 (dd, J=4.6 and 9.7 Hz, 1H), 2.372.12 (ni, 4H); and by
a 13C NMR
spectrum showing the following chemical shifts (8, ppm): 173.24, 170.05,
165.44, 162.77,
= 144.47, 136.71, 133.00, 128.85, 127.27, 122.55, 51.88, 31.32, 23.89. The
melting point of
the product was found to be 180-182 C. An elemental analysis yielded the
following results
in weight percent: C., 48.75; H, 3.48; N, 13.07,.whch compared With calculated
values for
C131-111N307, in weight percent: C, 48.60; H, 3.45; N, 13.08.
Example 2 - Preparation. of.N-(3-Aminophthaloy1)-glutamine According to Scheme
B
[083] A mixture of Example 1 (4780, .14.88 moles), 10% Pd/C (120
g) and methanol
(44 L) was hydrogenated at 50- psi for 2:5 hours in a-100 L hydrogenritiori
reactor. The
progress of the reaction was monitored by HPLC using a Waters Nova-Pak C18
cobitrm
(3.9x150 mm, particle size = 4 micron, UV wavelength = 240 nm, retention time
= 3.64
minutes) and an eluent mixture of acetonitrile and 0.1% aqueous H3PO4 in a
ratio of 10:90 by
volume at a flow rate of 1 ml/min. The mixture was filtered through a pad of'
CeliteTm and the
celite pad was washed with methanol (6 L). The filtrate was concentrated in
vacuo to a
gummy material. The.guriamy material was stirred with ethyl acetate (22 L)
overnight to
= fOrm a slurry. The slurry was filtered and the yellow solid filtered out
was washed with ethyl
= acetate (10 L). The yellow solid was air dried and then dried in a vacuum
oven at 60 C and
<1 mm pressure to yield 4230 g of the product. The product purity was found to
be 99.75%
by HPLC using a Waters Nova-Pak C18 column (3.9x150 mm, particle size = 4
micron, UV
-wavelength = 240 nm, retention time = 3.64 minutes) and an. eluent mixture of
acetonitrile =
and 0.1% aqueous H3PO4 in a ratio of 10:90 by volume at a flow rate of 1
mL/min. The
product in DMSO-d6 was characterized by a II-1 NMR spectruin showing the
following
= chemical shifts (8, ppm): 13.10 (b, 1H), 7.50-7.43 (dd, J=7.0 and 8.4 HZ,
1H), 7.24 (s, 1H),
7.03-6.98 (dd, J=5.0 and 8.4 Hz, 2H), 6.75 (s, 1H), 6.52 (s, 2H(, 4.70-4.64
(dd, J=4.5 and
24 =
CA 02823088 2013-08-08
=
WO 2007/005972 = PCTTUS2006/026210
10.5 Hz, 1H), 2.41-2.04 (m, 4H); and by a 13C NMR spectrum showing the
following
chemical shifts (6, ppm): 173.16, 170.81, 168.94, 167.68, 146.70, 135..41,
132.07, 121.63,
110.93, 108.68, 50.77, 31.38, 24.08. The melting point of the product was
found to be 177-
179 C. An elemental analysis yielded the following results in weight percent:
C, 53.61; H,
4.47; N, 14.31, which compared with calculated values for C13H13N305, in
weight percent: C,
53.60; H, 4.50; N, 14.43.
Example 3 - Preparation of 4-Amino-2-(2,6-dioxo-3-piperidinyl)isoindole-1,3-
dione
According to Scheme A
[084] A mixture of acetonitrile (42 L) and Example 2 (2120 g, 7.28
moles) was
added to a round bottom flask equipped with a mechanical stirrer, a condenser,
a nitrogen
inlet and a heating mantel to form a solution. When the solution was stirred
and heated to
about 40 to 45 C, 1,1'-carbonyldiimidazole (1290 g, 7.95 moles) was added.
The reaction
mixture was stirred and refluxed for 4.5 hours. The progress of the reaction
was monitored
by HPLC using a Waters Nova-Pak C18 column (3.9x150 mm, particle size = 4
micron, UV
wavelength = 240 nm, retention time = 3.64 minutes) and an eluent mixture of
acetonitrile
and 0.1% aqueous H3PO4 in a ratio of 20:80 by volume at a flow rate of 1
mL/min. After =
cooled to room temperature, the reaction mixture was filtered to yield a
yellow solid which
was subsequently washed with acetonittile (6.5L). The yellow solid was air
dried and then
dried in a vacuum oven at 60 C and <1 mm pressure to yield 1760 g (88%) of the
product.
The product purity was found to be 99.57% by HPLC using a Waters Nova-Pak C18
column
(3.9x150 mm, particle size = 4 micron, UV wavelength = 240 nm, retention time
= 3.64
minutes) and an eluent mixture of acetonitile and 0.1% aqueous H3PO4 in a
ratio of 20:80 by
volume as at a flow rate of 1 mL/min. The product in DMSO-d6 was characterized
by a 11-1
NMR spectrum showing the following chemical shifts (6, ppm): 11.10 (s, 1H),
7.47(t, 3=7.9
Hz, 1H), 7.03-6.99 (dd, J=4.8 and 8.4 Hz, 2H), 6.52 (s, 2H), 5.09-5.02 (dd,
J=5.3 and 12.4
Hz, 1H), 2.96-2.82 (m, 1H), 2.62-2.46 (m, 2H), 2.07-2.00 (m, 1H); and by a 13C
NMR
spectrum showing the following chemical shifts (8, ppm): 172.82, 170.11,
168.57, 167-37,
146.71, 135.46, 131.99, 121.70, 110.97, 108.52, 48.47, 30.97, 22.14. The
melting point of
the product was found to be 315.5-317.5 C. An elemental analysis yielded the
following
results in weight percent: C, 56.98; H, 3.86; N, 15.35, which compared with
calculated values
for Ci3H1 IN304, in weight percent: 57.14; H, 4.06; N, 15.38.
CA 02823088 2013-08-08
WO 2007/005972 = - PCT/US2006/026210
Example 4 - Preparation of 3-Nitro-N-ethoxycarbonyl-phthalimide According to
Scheme D
=
[0851 Ethyl chloroforrnate (1.89 g, 19.7 mmol) was added dropwise over
10 minutes
to a stirred solution of 3-nitrophthalimide (3.0 g, 15.6 mmol) and
triethylamine (1.78 g, 17.6
mmol) in DMF (20 mL) at about 0-5 C under nitrogen. The reaction was allowed
to warm
to room temperature and stirred for 4 hours. The reaction mixture was slowly
added to an
agitated mixture of ice and water (60 mL). The slurry was filtered and the
solid was
crystallized from CHC13 (15 mL) and petroleum ether (15 mL) to yield 3.1 g
(75%) of the
product as an off-white solid: mp 100.0-100.5 C;IHNMR. (CDC13) 8 8.25(d, 1=7.5
Hz, 1H),
8.20(d, J=8.0 Hz, 1H),. 8.03(t, J=7.9 Hz, 1H), 4.49(q, J=7.1 Hz, 2H), 1.44(t,
J=7.2 Hz, 3H);
13C NMR. (CDC13) 8 161.45, 158.40, 147.52, 145.65, 136.60, 132.93, 129.65,
128.01, 122.54,
64.64, 13.92; HPLC, Waters Nova-Pak/C18, 3.9x150 mm, 4 micron, 1 mL/min, 240
nm,
30/70 CH3CN/0.1% H3PO4(aq), 5.17 min (98.11%); Anal. calculated for C1al8N206
: C,
50.00; H, 3.05; N, 10.60. Found: C, 50.13; H, 2.96; N, 10.54.
Example 5 - Preparation oft-Butyl N-(3-nitrophthaloy1)-L-glutamine
[086] A mixture of Example 4 (1.0 g, 3.8 mmol), L-glutamine t-butyl ester
hydrochloride (0.9 g, 3.8 mmol) and triethylamine (0.54 g, 5.3 mmol) in THF
(30 mL) was
refluxed for 24 hours. The THF solvent was removed in vacuo and the residue
was dissolved
in CH2C12 (50 mL). The CH2Cl2 solution was washed with water (2x15 mL) and
brine (15
mL) and then dried. The solvent was removed and the residue was purified by
flash
chromatograph (CH2C12:Et0Ac/7:3) to give 0.9 g (63%) of a glassy material: NMR
(CDC13) 6 8.15(d, J=7.9 Hz, 2H), 7.94(t, J=7.8 Hz, 1H), 5.57(b, 2H), 4.84(dd,
J=5.1 and 9.7
Hz, 1H), 2.53-2.30(m, 4H), 1.43(s, 9H); HPLC, Waters Nova-Pak/C18, 3.9x150 mm,
4
micron, 1 mL/min, 240 mn, 30/70 CH3CN/0.1% H3PO4(aq), 6.48 min (99.68%);
Chiral
Analysis, Daicel Chiral Pak AD, 0.4x25 Cm, 1 mL/min, 240 nm, 5.32 min.
(99.39%); Anal.
calculated for C171-119N307 : C, 54.11; H, 5.08; N, 11.14. Found: C, 54.21; H,
5.08; N, 10.85.
Example 6 - Preparation of N-(3-Nitrophthaloy1)-L-glutamine
[087] Hydrogen chloride gas was bubbled into a stirred cold (5 C) solution
of
Example 5 (5.7 g, 15.1 mmol) in CH2C12 (100 mL) for 25 minutes. The mixture
was then
stirred at room temperature for 16 hours. Ether (50 mL) was added and the
resulting mixture
was stirred for 30 minutes. The slurry was filtered to yield 4.5 g of solid,
which was used in
the next reaction: NMR (DMSO-d6) 8 8.36(dd, J=0.8 and 8.0 Hz, 1H), 8.24(dd,
J=0.8 and
26
CA 02823088 2015-02-06
53686-75D1
7.5 Hz, 114), 8.11(t, J=7.9 Hz, 1H), 7.19(b, 1H), 6.72(b, 1H), 4.80(dd, J=3.5
and 8.8 Hz, 1H),
2.30-2.10(m, 4H).
Example 7 - Preparation of (S)-3-(3'-Nitrophthalimido)-piperidine-2,6-dione
[088] A suspension mixture of Example 6 (4.3 g, 13.4 mmol) in
anhydrous CH2C12
(170 mL) was cooled to -40 C with an isopropyl alcohol (IPA)/dry ice bath.
Thionyl
chloride (1.03 mL, 14.5 mmol) was added dropwise followed by pyridine (1.17
mL, 14.5
mmol). After 30 minutes, triethylanaine (2.06 mL, 14.8 mmol) was added and the
mixture
was stirred at about -30 to -40 C for 3 hours. The mixture was filtered and
washed with
CH2C12to yield 2.3 g (57%) of the crude product. The crude product was
recrystallized from
acetone (300 mL) to yield 2 g of the product as a White solid: mp 259.0-284.0
C (dec.);111
NMR (DMSO-d6) 8 11.19(s, 1H), 8.34(d, J=7.8 Hz, 1H), 8.23(d, J=7.1 Hz, 1H),
8.12(t, J=7.8
Hz, 1H), 5.25-5.17(dd, J=5.2 and 12.7 Hz, 1H), 2.97-2.82(m, 1H), 2.64-2.44(m,
2H), 2.08-
2.05(m, 1H); 13C NMR (DMSO-d6) 8 172.67, 169.46, 165.15, 162.50, 144.42,
136.78,
132.99, 128.84, 127.27, 122.53, 49.41, 30.84, 21.71; HPLC, Waters Nova-
Pak/C18, 3.9x150
mm, 4 micron, 1 mL/min, 240 mn, 10/90 CH3CN/0.1 %H3PO4(aq), 4.27 min.(99.63%);
Anal.
calculated for Ci3H9N306 : C, 51.49; H, 2.99;N, 13.86. Found: C, 51.67;
H,.2.93; N, 13.57.
Example 8 - Preparation of (S)-3-(3'-Aminophthalimido)-piperidine-2,6-dione
[089] A mixture of (S)-3-(3'-nitrophthalimido)-piperidine-2,6-dione (0.76
g, 2.5
mraol) and 10% Pd/C (0.3 g) in acetone (200 mL) was hydrogenated in a Parr-
Shaker
TM
apparatus at 50 psi of hydrogen for 24 hours. The mixture was filtered through
celite and the
filtrate was concentrated in vacuo. The solid was stirred with hot ethyl
acetate for 30 minutes
to give 0.47 g (69%) of the product as a yellow solid: mp 309-310 C; 1H NMR
(DMSO-d6)
11.10 (s, 1H), 7.47(dd, J=7.2 and 8.3 Hz, 1H), 7.04-6.99(dd, J=6.9 and 8.3 Hz,
2H), 6.53(s,
214), 5.09-5.02(dd, J=5.3 and 12.4 Hz, 1H), 2.96-2.82(m, 1H), 2.62-2.46(m,
2H), 2.09-
1.99(m, 1H); 13C NMR (DMSO-d6) 8 172.80, 170.10, 168.57, 167.36, 146.71,
135.44,
131.98, 121.69, 110.98, 108.54, 48.48, 30.97, 22.15; HPLC, Waters Nova-
Pak/C18, 3.9x150
mm, 4 micron, 1 mL/min, 240 nm, 15/85 CH3CN/0.1 % H3PO4(aq), 4.99 min.
(98.77%);
Chiral analysis, Daicel Chiral Pak AD, 0.46x25 cm, 1 mL/min, 240 tun, 30/70
Hexane/IPA
9.55 min.(1.32%), 12.55 min(97.66%); Anal. calculated for C13H1tN304: C,
57.14; H, 4.06;
N, 15.38. Found: C, 57.15; H, 4.15; N, 14.99.
Example 9 - Preparation of t-Butyl N-(3-nitrophthaloy1)-D-glutamine
[090] A mixture of Example 4 (5.9 g, 22.3 mmol), D-glutamine t-butyl ester
(4.5 g,
= 22.3 mmol) and triethylamine (0.9 g, 8.9 mmol) in THF (100 mL) was
refluxed for 24 hours.
27
CA 02823088 2015-02-06
53686-75D1
The mixture was diluted with CH2C12 (100 mL) and washed with water (2x50 mL),
brine (50
mL) and dried. The solvent was removed in vacuo and the residue was purified
by flash
chromatography (2% CH3OH in CH2C12) to afford 6.26 g (75%) of the product as a
glassy
material: 1H NMR (CDC13) 8 8.12(d, J=7.5 Hz, 211), 7.94(dd, J=7.9 and 9.1 Hz,
1H), 5.50(b,
11.1), 5.41(b, 1H), 4.85(dd, J=5.1 and 9.8 Hz, 1H), 2.61-2.50(m, 2H), 2.35-
2.27(m,2H), 1.44(s,
9H); 13C NMR (CDC13) 5 173.77, 167.06, 165.25, 162.51, 145.07, 135.56, 133.78,
128.72,
127.27, 123.45, 83.23, 53.18, 32.27, 27.79, 24.42; HPLC, Waters Nova-Pak/C18,
3.9x150
mm, 4 micron, 1 mL/min, 240 rim, 25/75 CH3CN/0.1 % H3PO4(aq) 4.32
min.(99.74%);
TM
Chiral analysis, Daicel Chiral Pak AD, 0.46x25 cm, 1 mL/min, 240 nm, 55/45
Hexane/TPA
5.88 min.(99.68%); Anal. calculated for C171119N307: C, 54.11; H, 5.08;N,
11.14. Found: C,
54.25; H, 5.12; N, 10.85.
Example 10 - Preparation of N-(3-Nitrophthaloy1)-D-glutamine
[091] Hydrogen chloride gas was bubbled into a stirred cold (5 C) solution
of
Example 9 (5.9 g, 15.6 mmol) in CH2C12 (100 mL) for 1 hour then stirred at
room
temperature for another hour. Ether (100 mL) was added and stirred for another
30 min. The
mixture was filtered, washed with ether (60 mL) and dried (40 C, <1 nun Hg) to
afford 4.7 g
(94%) of the product: 1H NIVIR (DMSO-d6) 8 8.33(d, J=7.8 Hz, 1H), 8.22(d,
J=7.2 Hz, 1H),
8.11(t, J=7.8 Hz, 1H), 7.19(b, 1H), 6.72(b, 111), 4.81(dd, J=4.6 and 9.7 Hz,
111), 2.39-2.12(m,
4H); 13C NMR (DMSO-d6) 8 173.21, 169.99, 165.41, 162.73, 144.45, 136.68,
132.98,
128.80, 127.23, 122.52, 51.87, 31.31, 23.87.
Example 11 - Preparation of (R)-3-(3'-Nitrophthalimido)-piperidine-2,6-dione
[092] A suspension mixture of Example 10 (4.3 g, 13.4 mmol) in anhydrous
CH2Cl2
(170 mL) was cooled to -40 C with IPA/dry ice bath. Thionyl chloride (1.7 g,
14.5 mmol)
was added dropwise followed by pyridine (1.2 g, 14.5 mmol). After 30 minutes,
triethylamine (1.5 g, 14.8 mmol) was added and the mixture was stirred at -30
to -40 C for 3
hours. The mixture was filtered, washed with CH2C12 (50 mL) and dried (60 C,
<1 mm Hg)
to give 2.93 g of the product. Another 0.6 g of the product was obtained from
the methylene
chloride filtrate. Both fractions were combined (3.53 g) and recrystallized
from acetone (450
mL) to afford 2.89 g (71%) of the product as a white solid: mp 256.5-257.5
C;11-1NMR.
(DMSO-d6) 5 11.18(s, 1H), 8.34(dd, J=0.8 and 7.9 Hz, 1H), 8.23(dd, J=0.8 and
7.5 Hz, 1H),
8.12(t, 3=7.8 Hz, 1H), 5.22(dd, J=5.3 and 12.8 Hz, 1H), 2.97-2.82(m, 1H), 2.64-
2.47(m, 2H),
2.13-2.04(m, 1H); 13C NMR. (DMSO-d6) 5 172.66, 169.44, 165.14, 162.48, 144.41,
136.76,
132.98, 128.83, 127.25, 122.52, 49.41, 30.83, 21.70; HPLC, Waters Nova-
Pak/C18, 3.9x150
28
CA 02823088 2013-08-08
WO 2007/005972
PCIMS2006/026210
MM, 4 micron, 1 mL/min, 240 nm, 10/90 CH3CN/0.1 % H3PO4(aq) 3.35 min.(100%);
Anal.
calculated for C0119N306: C, 51.49; H, 2.99; N,13.86. Found: C, 51.55; H,
2.82; N, 13.48.
Example 12 - Preparation of (R)-3-(3'-Aminophthalimido)-piperidine-2,6-dione
[093] A mixture of Example 11 (1.0 g, 3.3 mmol) and 10% Pd/C
(0.2 g) in acetone
(250 mL) was hydrogenated in a Parr-Shaker apparatus at 50 psi of hydrogen for
4 hours.
The mixture was filtered through celite and the fitrate was concentrated in
vacuo. The yellow
solid was slurried in hot Et0Ac (20 niL) for 30 minutes to give 0.53 g (59%)
of the product
as a yellow solid: nip 307.5-309.5 C; 1HNMR (DMSO-d6) 8 11.06(s, 1H), 7.47(d ,
1=7.0 and
8.4 Hz, 1H), 7.02(dd, J=4.6 and 8.4 Hz, 2H), 6.53(s, 2H), 5.07(dd, 1=5.4 and
12.5 Hz,
1H),2.952.84(m, 1H), 2.62-2.46(m, 2H), 2.09-1.99(m, 1H); 13C NMR (DMSO-d6) 8
172.78,
170.08, 168.56, 167.35, 146.70, 135.43, 131.98, 121.68, 110.95, 108.53, 48.47,
30.96, 22.14;
HPLC, Waters Nove-pak/CI8, 3.9x150 mm, 4 micron, 1 mL/min, 240 mn, 10/90
CH3CN/0.1
% H3P64(a4), 3.67 min.(99.68%); Chiral analysis, Daicel Chiral Pak AD, 0.46x25
cm, 1
mL/min, 240 nm, 30/70 Hexane-DA 7.88 min. (97.48%); Anal. calculated for
CoHnNiat:
C, 57.14; H, 4.06; N, 15.38. Found: C, 57.34; H, 3.91; N, 15.14.
Example 13 - Preparation of 4-Amino-2-(2,6-dioxo-3-piperidiny1)isoindo1e-113-
dione
According to Scheme E
10941 A mixture of 3-aminophthalic acid hydrochloride (200 g,
0.92 mol, from
Prosynth Ltd., Suffolk, UK), 3-aminoglutarimide hydrochloride (159 g, 0.96
mol, from
Evotec OM, Hamburg, (iermany), acetonitrile (2.0 L), and acetic acid (577 g,
9.6 mol, from
Fisher Scientifc) was charged into a reaction vessel. After the mixture was
stirred for 15
minutes, triethylamine- (465.0 g, 4.6 mol, from Aldrich, Milwaukee, WI) was
added dropwise
over 30-35 minutes while the reaction temperature was maintained at 20-25 C.
Next, the
reaction mixture was stirred further for 10-15 minutes and then refluxed at
about 85 to 87 C
for about 5 to 7 hours or until the in-process control, i.e., IIPLC AP at 240
nm, indicates that
<2% of the 3-aminophthalic acid remained in the reaction mixture. After the
reaction
mixture was cooled to about 20 to 25 C over 1-2 hours, 1.0 L of water was
charged over 15-
30 minutes at about 20 to 25 C. The resulting mixture was stirred at about 15
to 20 C for
about 20 to 30 minutes to provide a yellow solid precipitate, which was
filtered, washed with
DI water (3 x 1.0 L) and acetonitrile (2 x 500 mL), and then dried at about 35
to 40 C in
.?11 vacuo to a constant weight at 210.0 g (84 %).
29
CA 02823088 2013-08-08
WO 2007/005972 =
PCT/US2006/026210
=
Example 14 - Preparation of 4-Amino-2-(2,6-dioxo-3-piperidinyl)isoindole-1,3-
dione
[095] Example 14 was prepared similarly according to the procedure for
Example 13
except that there was no acetic acid; the amount of triethylamine was reduced
from 4.6 mol to
3.2 mol; and the refluxing time was increased from about 5 to 7 hours to about
47 hours. The
amount of 4-Amino-2-(2,6-dioxo-3-piperidinyl)isoindole-1,3-dione in the
reaction mixture
was found to be 94%.
Example 15 - Preparation of 4-Amino-2-(2,6-dioxo-3-piperiditayl)isoindole-1,3-
dione
[096] Example 15 was prepared similarly according to the procedure for
Example 13
except that there was no acetic acid and the 4.6 mol of triethylamine was
replaced with 9.2
mole of irnidazole. The amount of 4-Amino-2-(2,6-dioxo-3-piperidinypisoindole-
1,3-dione
in the reaction mixture was found to be 92%.
Example 16 - Preparation of 4-Amino-2-(2,6-dioxo-3-piperidinyl)isoindole-1,3-
dione
[097] Example 16 was prepared similarly according to the procedure for
Example 13
except that the 4.6 mol of triethylamine was replaced with 9.2 mole of
imidazole. The
amount of 4-Amino-2-(2,6-dioxo-3-piperidinyl)isoindole-1,3-dione in the
reaction mixture
was found to be 85%.
Example 17 - Recrystallization of 4-Amino-2-(2,6-dioxo-3-piperidinyl)isoindole-
1,3-
dione
[098] The 4-amino:2-(2,6-dioxo-3-piperidinyl)isoindole-1,3-dione racemates
and
stereomers such as Examples 3, 8, and 12-16 can be purified by
recrystallization as described
below. A mixture of crude Example 13 (200 g) and DMSO (800 mL) was charged
into a
reaction vessel. The resulting slurry was heated to about 45 to 50 C and then
stirred until
full dissolution of the solid was achieved (about 10 to 15 minutes). The
resulting solution
was clarified at about 45 to 50 C followed by a DMSO (400 mL) line rinse at
about 45 to 50
C. The solution was added to purified water (7.2 L) at about 75 to 80 C over
at least 60
minutes. The resulting suspension was cooled to about 15 to 20 C over at
least 1.5 hours and
stirred at the same temperature for about 1.5 to 2 hours. The suspension was
filtered and the
solid was washed with purified water (2 x 2 L). The purified product was dried
under
vacuum at about 35 to 40 C until constant weight is attained. The yield of
the purified
product was 196.8 g (98% recovery). The melting point of the purified product
was found to
be 321-323 C.
CA 02823088 2015-02-06
53686-75D1
[099] The present invention is not to be limited in scope by the
specific
embodiments disclosed in the examples that are intended as illustrations of a
few aspects of
the invention.
31