Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
17a-HYDROXYLASE/Ci7,20-LYASE INHIBITORS
FIELD OF THE INVENTION
The present invention relates to cyclic urea derivatives and their use for the
treatment of various disease conditions mediated by the regulation of 17a-
hydroxylase/C17,20-lyase.
BACKGROUND
The number of people diagnosed with cancer world wide has significantly
increased and continues to rise at an alarming rate. Cancer is characterized
by an
increase in the number of abnormal cells derived from a given normal tissue,
invasion
of adjacent tissues by these abnormal cells, or lymphatic or blood-borne
spread of
malignant cells to regional lymph nodes and to distant sites (i.e.,
metastasis).
Of special interest are individuals diagnosed with androgen-dependent
disorders, such as prostate cancer, estrogen-dependent disorders (such as
breast,
uterine, and ovarian cancer), Cushing's syndrome, polycyctic ovary syndrome
(PCOS), and aldo-producing adenoma (APA).
Prostate cancer is currently the most common non-skin cancer and the second
leading cause of cancer-related death in men after lung cancer. The primary
course of
treatment for patients diagnosed with organ-confined prostate cancer is
usually
prostatectomy or radiotherapy. These treatments for prostate and breast cancer
are
highly invasive and characterized by undesirable and serious side effects.
Furthermore, a large percent of individuals who receive localized treatments
such as
surgery or radiotherapy may suffer from recurring cancer and widespread
metastases.
As with surgery and radiation therapies, there are several drawbacks to
chemotherapy,
including the fact that almost all chemotherapeutic agents are toxic, and
chemotherapy causes significant, and often dangerous, side effects, such as
severe
nausea, bone marrow depression, and immunosuppression. Additionally, many
tumor
cells are resistant or become resistant to chemotherapeutic agents through
multi-drug
resistance.
Treatments such as hormone therapy are another option for individuals
diagnosed with hormone-dependent, hormone-responsive, or hormone-sensitive
cancers, such as prostate or breast cancer. However, some individuals who have
been
1
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
administered current hormone therapy treatments may not show a significant
response
to such treatments and some may suffer from relapsing of cancer.
Currently chemo-refractory and hormone-refractory cancer patients are left
with very few treatment options and there remains an unmet need for more
effective
ways to treat cancer such as, but not limited to, prostate cancer and breast
cancer.
The demonstration by Huggins and Hodges C.V., ( Cancer Res., 1941, /, 293)
and Huggins et al in Arch.Surg.,1941, 43, 209 lead to androgen ablation being
considered as a possible approach to treatment. It has been demonstrated that
testosterone levels are reduced by orchidectomy or by administration of GnRH
analogs (gonadotropic releasing hormones). GnRH analogs can have side effects
such
as cardiovascular degeneration and osteoporosis, which are the two most
potentially
serious conditions induced by the continuous presence of GnRH . Moreover these
treatment options only eliminate testosterone production from the testes and
not that
produced by the adrenal.
In the adrenal glands, the biosynthetic cascade also leads to the formation of
gluco- and mineralcorticoids.
Since androgen and estrogen are hormones having various physiological
activities such as differentiation and proliferation of cells and the like, it
was thought
that potent and specific compounds that inhibit androgen synthesis in the
testes,
adrenals, and other tissue may be more effective for the treatment of PCa
(Njar, V. C.
0.; Brodie, A. M. H., "Inhibitors of 17a-hydroxylase-C17,20-lyase (CYP17):
Potential
agents for the treatment of prostate cancer", Current Pharm. Design, 1999, 5:
163-
180).
In order to avoid unwanted side effects, androgen biosnthesis inhibitors have
to be specific enough not to influence corticosteroid biosynthesis. A
promising novel
strategy for the treatment of prostrate cancer is the development of strong
and
selective inhibitors of CYP 17 as this would result in complete and excusive
elimination of androgen biosynthesis as suggested in Current Medicinal
Chemistry,
2005,/2, 1623-1629.
Steroid-type compounds and non-steroid-type compounds are already known
as steroid C17,20-lyase inhibitors. The steroid-type compounds are disclosed
in, for
example, WO 92/15404, WO 93/20097, EP-A 288053, EP-A 413270 and the like. As
non-steroid-type compounds, for example, in W094/27989, W096/14090 and
W097/00257 azole derivatives are described in W095/09157 1H-benzimidazole
2
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
derivatives are described in US 5,491,161, dihydronaphthalene derivatives are
described in W099/18075, and naphthalene derivatives are shown in W099/54309.
A variety of potent steroidal and non-steroidal inhibitors of CYP17 have been
reported and some have been shown to be potent inhibitors of testosterone
production
in rodent models (Njar and Brodie, above). Jarman and colleagues have
described the
hormonal impact of their most potent CYP17 inhibitor, abiraterone in patients
with
prostate cancer (O'Donnell et al., "Hormonal impact of the 17a-
hydroxylase/C17,20-
lyase inhibitors abiraterone acetate (CB7630) in patients with prostate
cancer", Br. J.
Cancer, 2004, 90: 2317-2325). Abiraterone has been discussed in patents such
as
WO 200900132, WO 2008024485, WO 2006021776, WO 09509178, WO 09320097
Non-steroidal small molecule inhibitors have been described for example in
BMC 2004,12,(4313), YM116, 2-(1H-imidazol-4-ylmethyl) -9H-carbazole, and their
effects in decreasing adrenal androgen synthesis by inhibiting C17-20 lyase
activity
in NCI-H295 human adrenocortical carcinoma cells has been described by Ideyama
Y, Kudoh M, Tanimoto K, Susaki Y, Nanya T, Nakahara T, Ishikawa H, Fujikura T,
Akaza H, Shikama H in "Jpn.J.Pharmacol.,1999,79:No. 2(213-20)". Novel non-
steroidal inhibitor of cytochrome P450 (17 alpha-hydroxylase/C17-20 lyase),
YM116,
and its role in decreased prostatic weights by reducing the serum
concentrations of
testosterone and adrenal androgens in rats has been reported by Ideyama Y,
Kudoh M,
Tanimoto K, Susaki Y, Nanya T, Nakahara T, Ishikawa H, Yoden T, Okada M,
Fujikura T, Shikama H
Proc.Am.Assoc.Cancer Res.,1998,39:89 Meet.(384)
Synthesis and biological evaluation of novel non-steroidal inhibitors of
steroid
17,20 lyase has been described by -Yoden T, Okada M, Kawaminami E, Kinoyama I,
Ideyama Y, Isomura Y in Abstr.Pap.Am.Chem.Soc.,1997 ,213 Meet.:Pt. 2(MEDI206)
Further illustrative of the background of the invention are patent
applications
such as U520080280864A1 or W028154382A1.
SUMMARY
The compounds described herein have been shown to be inhibitors of 17a-
hydroxylase/C17,20-lyase.
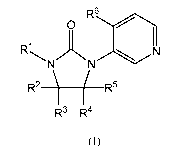
One embodiment of the present invention provides compounds of Formula (I)
3
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
R6
0
I
R1
-......... zx ........--zk...........,...... .,N
N N
R2 ( R5
R3 R4
(I)
wherein
R' is
(i) phenyl optionally substituted with 1 to 3 substituents selected from halo,
-
CN, -OH, (Ci-C6)alkyl, halo-substituted (Ci-C6)alkyl, (Ci-C6)alkoxy, -NH2, -
NH(C1-
C4)alkyl, -N((C1-C4)alky1)2, -NHC(0)-(Ci-C4)alkyl, -C(0)NH2, -C(0)-NH(C1-
C4)alkyl, -C(0)-N((Ci-C4)alky1)2, (C3-05)cycloalkyl, or a 5- to 6-membered
heterocycle,
(ii) phenyl fused to an additional phenyl, a 5- to 6-membered heteroaryl, a 5-
to 6-membered partially or fully saturated cycloalkyl, or a 5- to 6-membered
partially
or fully saturated heterocycle, where said fused phenyl is optionally
substituted with 1
to 4 substituents each independently selected from halo, -CN, (Ci-C6)alkyl,
(C1-
C6)alkoxy, hydroxy-substituted (Ci-C4)alkyl, halo-substituted (Ci-C4)alkyl,
(C3-
C5)cycloalkyl, oxo, -NH2, -NH(C1-C4)alkyl, -N((C1-C4)alky1)2, -NHC(0)-(C1-
C4)alkyl, or =N-OH,
(iii) 5- to 6-membered heteroaryl optionally substituted with 1 to 3
substituents each independently selected from halo, -CN, -OH, (Ci-C6)alkyl,
halo-
substituted (Ci-C6)alkyl, (Ci-C6)alkoxy, -NH2, -NH(C1-C4)alkyl, -N((C1-
C4)alky1)2, -
NHC(0)-(C1-C4)alkyl, -C(0)NH2, -C(0)-NH(C1-C4)alkyl, -C(0)-N((C1-C4)alky1)2,
(C3-05)cycloalkyl, or a 5- to 6-membered heterocycle, or
(iv) 5- to 6-membered heteroaryl fused to another 5- to 6-membered
heteroaryl, phenyl, 5- to 6-membered partially or fully saturated cycloalkyl,
or a 5- to
6-membered partially or fully saturated heterocycle, where said fused
heteroaryl is
optionally substituted with 1 to 4 substituents each independently selected
from halo,
-CN, (Ci-C6)alkyl, (Ci-C6)alkoxy, hydroxy-substituted (Ci-C4)alkyl, halo-
substituted
(C1-C4)alkyl, (C3-05)cycloalkyl, oxo, -NH2, -NH(C1-C4)alkyl, -N((C1-
C4)alky1)2, -
NHC(0)-(Ci-C4)alkyl, or =N-OH;
4
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
R2 and R5 are each independently CH3 or H;
R3 and R4 are each independently CH3 or H, or taken together with the carbon
atoms to which they are attached form a cyclopropyl; and
R6 is (C3-05)cycloalkyl, where the cycloalkyl is optionally substituted with
hydroxy; or a pharmaceutically acceptable salt thereof.
In another embodiment, a compound of Formula (I) is provided wherein
R' is
(i) phenyl optionally substituted with 1 to 3 substituents selected from halo,
-
CN, -OH, (Ci-C6)alkyl, halo-substituted (Ci-C6)alkyl, (Ci-C6)alkoxy, -NH2, -
NH(C1-
C4)alkyl, -N((C 1 -C4)alky1)2, -NHC(0)-(C 1-C4)alkyl, -C(0)NH2, -C(0)-NH(C1-
C4)alkyl, -C(0)-N((Ci-C4)alky1)2, or a 5- to 6-membered heterocycle,
(ii) phenyl fused to an additional phenyl, a 5- to 6-membered heteroaryl, a 5-
to 6-membered partially or fully saturated cycloalkyl, or a 5- to 6-membered
partially
or fully saturated heterocycle, where said fused phenyl is optionally
substituted with 1
to 4 substituents each independently selected from halo, -CN, (Ci-C6)alkyl,
(C1-
C6)alkoxy, hydroxy-substituted (Ci-C4)alkyl, halo-substituted (Ci-C4)alkyl,
cyclopropyl, oxo, -NH2, -NH(Ci-C4)alkyl, -N((C1-C4)alky1)2, -NHC(0)-(C1-
C4)alkyl,
or =N-OH,
(iii) 5- to 6-membered heteroaryl optionally substituted with 1 to 3
substituents each independently selected from halo, -CN, -OH, (Ci-C6)alkyl,
halo-
substituted (Ci-C6)alkyl, cyclopropyl, (Ci-C6)alkoxy, -NH2, -NH(Ci-C4)alkyl, -
N((C 1 -C4)alky1)2, -NHC(0)-(C1-C4)alkyl, -C(0)NH2, -C(0)-NH(C 1 -C4)alkyl, -
C(0)-
N((C i-C4)alky1)2, or a 5- to 6-membered heterocycle, or
(iv) 5- to 6-membered heteroaryl fused to another 5- to 6-membered
heteroaryl, phenyl, 5- to 6-membered partially or fully saturated cycloalkyl,
or a 5- to
6-membered partially or fully saturated heterocycle, where said fused
heteroaryl is
optionally substituted with 1 to 4 substituents each independently selected
from halo,
-CN, (Ci-C6)alkyl, (Ci-C6)alkoxy, hydroxy-substituted (Ci-C4)alkyl, halo-
substituted
(Ci-C4)alkyl, cyclopropyl, oxo, -NH2, -NH(C1-C4)alkyl, -N((C1-C4)alky1)2, -
NHC(0)-
(Ci-C4)alkyl, or =N-OH;
R2 and R5 are each independently CH3 or H;
R3 and R4 are each independently CH3 or H, or taken together with the carbon
atoms to which they are attached form a cyclopropyl; and
5
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
R6 is (C3-05)cycloalkyl, where the cycloalkyl is optionally substituted with
hydroxy; or a pharmaceutically acceptable salt thereof.
In one particular embodiment, R2, R3, R4 and R5 are each independently CH3
or H.
In another particular embodiment, R2, R3, R4 and R5 are each H.
In yet another particular embodiment, R2 and R5 are each H, and R3 and R4 are
taken together to form a cyclopropyl ring.
Another embodiment provides compounds of Formula (II)
R6
0
I
R1 )N N
N N
R2R5
(II)
wherein:
Rl is as defined above for the compound of Formula (I);
R2 and R5 are each independently CH3 or H (alternatively, both R2 and R5 are
H); and
R6 is (C3-05)cycloalkyl, where the cycloalkyl is optionally substituted with
hydroxy (e.g., 1-hydroxycyclopropyl, 1-hydroxycyclobutyl, or 1-
hydroxycyclopentyl);
or a pharmaceutically acceptable salt thereof.
In yet another embodiment, compounds of Formula (III) are provided
R6i
0
I
R1
,..,...._ 7N N N ........--k...........,.,.... ., N
R2
0õ. =õõ
'/R5
V
(III)
wherein Rl R2, R5 and R6 are as defined above for the compound of Formula
(II); or a
pharmaceutically acceptable salt thereof.
6
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
R6 is preferably (for compounds of Formula (I), (II) or (III)) cyclopropyl,
where the cyclopropyl is optionally substituted with hydroxy (e.g., 1-
hydroxycyclopropyl). In a particular embodiment, R6 isan unsubstituted
cyclopropyl.
Rl is preferably (for any of the compounds or embodiments described above)
is
(i) a phenyl optionally substituted with 1 or 2 substituents each
independently selected form fluoro, chloro, cyano, methyl, difluoromethyl,
trifluoromethyl, cyclopropyl, methoxy, or -C(0)NHCH3;
(ii) a fused phenyl selected from naphthalen-2-yl, naphthalen-l-yl, 1H-
indo1-5-yl, 1H-indo1-6-yl, benzothiazol-5-yl, benzothiazol-6-yl, 1,2,3,4-
tetrahydro-
quinolin-6-yl, benzo[b]thiophen-5-yl, quinolin-6-yl, quinolin-7-yl, indan-5-
yl, 1,2-
dihydroquinolin-6-yl, 1H-indazol-5-yl, 1H-indazol-6-yl, benzofuran-5-yl, 2,3-
dihydrobenzo[1,4]dioxin-6-yl, 2,3-dihydro-benzofuran-5-yl, benzo[1,3]dioxo1-5-
yl,
1,2,3,4-tetrahydro-quinolin-7-yl, quinoxalin-6-yl, benzooxazol-5-yl,
benzo[d]isoxazol-5-yl, benzo[d]isoxazol-6-yl, 1H-benzoimidazol-5-yl, 2,3-
dihydro-
1H-indazol-5-yl, 2,3-dihydro-1H-indazol-6-yl, indolin-5-yl, or 1H-benzotriazol-
5-yl,
where said fused phenyl is optionally substituted with 1 to 3 substituents
each
independently selected from fluoro, chloro, methyl, ethyl, trifluoromethyl,
methoxy,
oxo, -NH2, =N-OH or cyclopropyl;
(iii) a 5- to 6-membered heteroaryl selected from thiophen-2-yl, thiophen-
3-yl, pyridin-2-yl, pyridin-3-yl, pyridin-4-yl, pyrimidin-2-yl, pyrimidin-4-
yl,
pyrimidin-5-yl, 1H-pyrazol-4-yl, thiazol-2-yl, or isothiazol-4-yl, where said
5- to 6-
membered heteroaryl is optionally substituted with 1 to 3 substituents each
independently selected from fluoro, chloro, methyl, ethyl, isopropyl,
cyclopropyl,
hydroxy, difluoromethyl, trifluoromethyl, methoxy, -NH2, -NHC(0)CH3, -
C(0)NHCH3, or pyrrolidin-1-y1; or
(iv) a fused heteroaryl selected from benzo[b]thiophen-2-yl,
benzo[b]thiophen-3-yl, quinolin-2-yl, quinolin-3-yl, benzooxazol-2-yl,
benzothiazol-
2-yl, 4,5,6,7-tetrahydro-thieno[2,3-c]pyridin-2-yl, imidazo[1,2-a]pyridin-3-
yl,
imidazo[1,2-a]pyridin-6-yl, imidazo[1,2-a]pyridin-7-yl, 3H-imidazo[4,5-
b]pyridin-6-
yl, thieno[3,2-c]pyridin-2-yl, thieno[3,2-c]pyridin-3-yl, or 1H-indo1-3-yl,
where said
fused heteroaryl is optionally substituted with 1 to 4 substituents each
independnently
selected from fluoro, chloro, cyano, methyl, cyclopropyl, or methoxy; or a
pharmaceutically acceptable salt thereof.
7
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
In another embodiment (for any of the compounds or embodiments described
above), 1Z1 is
(i) a phenyl optionally substituted with 1 to 2 substituents each
independently
selected from fluoro, chloro, methyl, methoxy, trifluoromethyl,
difluoromethyl, or
cyano;
(ii) a fused phenyl selected from naphthalen-2-yl, quinolin-6-yl, 3,4-dihydro-
2-oxo-quinolin-6-yl, benzo[b]thiophen-5-yl, benzo[b]thiophen-6-yl,
benzo[d]isoxazol-5-yl, 1H-indazol-6-yl, 1H-indazol-5-yl, benzothiazol-6-yl,
1,2-
dihydro-3-oxo-indazol-6-yl, indan-5-yl, 1H-benzotriazol-5-yl, benzofuran-5-yl,
2,3-
dihydro-benzo[1,4]dioxin-6-yl, 2,3-dihydro-benzofuran-5-yl, or
benzo[1,3]dioxo1-5-y1
where said fused phenyl is optionally substituted with 1 to 2 substituents
each
independently selected from chloro, fluoro, methyl, ethyl, difluoromethyl,
trifluoromethyl, cyclopropyl, cyano, or amino;
(iii) a 5- to 6-membered heteroaryl selected from isothiazol-4-yl, thiophen-2-
yl, thiophen-3-yl, pyridin-2-yl, pyridin-4-yl, pyrimidin-4-yl, or pyrimidin-2-
y1 where
said isothiazol-4-yl, said thiophen-2-yl, said thiophen-3-yl, and said pyridin-
2-yl,
pyridin-4-yl, said pyrimidin-4-yl, and said pyrimidin-2-y1 are optionally
substituted
with fluoro, chloro, methyl, trifluoromethyl, difluoromethyl, cyclopropyl, or
methoxy;
Or
(iv) a fused heteroaryl selected from thieno[3,2-c]pyridin-2-yl, thieno[3,2-
c]pyridin-3-yl, thieno[3,2-c]pyridin-2-yl, imidazo[1,2-a]pyridin-7-yl, or
benzo[b]thiophen-2-yl, where said fused heteroaryl is optionally substituted
with 1 to
2 substituents each independently selected from fluoro, chloro, methyl,
difluoromethyl, trifluoromethyl, cyclopropyl, or amino; or a pharmaceutically
acceptable salt thereof.
In one particular embodiment (for any of the compounds or embodiments
described above), 1Z1 is phenyl, 4-chloro-3-fluoro-phenyl, m-tolyl, 3-methoxy-
phenyl,
3-chloro-4-fluoro-phenyl, 4-fluoro-3-methyl-phenyl, 3-trifluoromethyl-phenyl,
3-
chloro-phenyl, 4-fluoro-3-trifluoromethyl-phenyl, 3-difluoromethy1-4-fluoro-
phenyl,
3-cyano-4-fluorophenyl, 3-cyanophenyl, 3-chloro-4-cyanophenyl, 3,4-difluoro-
phenyl, 4-trifluoromethyl-phenyl; or a pharmaceutically acceptable salt
thereof.
In another particular embodiment (for any of the compounds or embodiments
described above), 1Z1 is naphthalen-2-yl, benzo[b]thiophen-5-yl, 3-
methylbenzo[b]thiophen-5-yl, 2-fluoro-3-methylbenzo[b]thiophen-5-yl, 3-
8
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
trifluoromethyl-benzo[b]thiophen-5-yl, 2-fluorobenzo[b]thiophen-5-yl, 2-
chlorobenzo[b]thiophen-5-yl, benzo[b]thiophen-6-yl, 2-fluoro-benzo[b]thiophen-
6-yl,
3-methylbenzo[b]thiophen-6-yl, 4-fluoro-benzo[b]thiophen-6-yl, 5-fluoro-3-
methylbenzo[b]thiophen-6-yl, 3-methyl-benzo[d]isoxazol-5-yl, 1H-indazol-5-yl,
1-
methyl-1H-indazol-5-yl, 3-amino-1H-indazol-5-yl, 1H-indazol-6-yl, 3-amino-1H-
indazol-6-yl, 3-methyl-1H-indazol-6-yl, 3-trifluoromethy1-1H-indazol-6-yl,
benzothiazol-6-yl, 1,2-dihydro-3-oxo-indazol-6-yl, indan-5-yl, 1H-benzotriazol-
5-yl,
3-methyl-benzofuran-5-yl, 2,3-dihydro-benzo[1,4]dioxin-6-yl, 2,3-dihydro-
benzofuran-5-yl, or 2,2-difluoro-benzo[1,3]dioxo1-5-y1; or a pharmaceutically
acceptable salt thereof.
In yet another particular embodiment (for any of the compounds or
embodiments described above), Rl is benzothiazol-6-yl, 3-methyl-benzofuran-5-
yl,
1H-indazol-6-yl, 3-methyl-1H-indazol-6-yl, or 3-trifluoromethy1-1H-indazol-6-
y1; or
a pharmaceutically acceptable salt thereof.
In yet another particular embodiment (for any of the compounds or
embodiments described above), Rl is 5-methyl-thiophen-2-yl, 5-chloro-thiophen-
2-
yl, 5-fluorobenzo[b]thiophen-2-yl, 5-trifluoromethyl-thiophen-2-yl, 5-
difluoromethyl-
thiophen-3-yl, 5-methyl-thiophen-3-yl, 2-methyl-pyridin-4-yl, 2-
trifluoromethyl-
pyridin-4-yl, 4-trifluoromethyl-pyridin-2-yl, 2-chloro-pyridin-4-yl, 2-methoxy-
pyridin-4-yl, 6-chloropyrimidin-4-yl, 6-chloro-2-methylpyrimidin-4-yl, 2-
trifluoromethyl-pyrimidin-4-yl, 4-trifluoromethyl-pyrimidin-2-yl, 2-chloro-6-
(trifluoromethyl)pyridin-4-yl, 6-cyclopropylpyrimidin-4-yl, 2-
cyclopropylpyridin-4-
yl, 5-fluoro-4-methylpyridin-2-yl, 2-cyclopropylpyrimidin-4-yl, 6-chloro-2-
(trifluoromethyl)pyrimidin-4-yl, 2,6-dichloropyridin-4-yl, 2-chloro-6-
cyclopropylpyridin-4-yl, 2-cyclopropy1-6-(trifluoromethyl)pyridin-4-yl, or 2,6-
bis(trifluoromethyl)pyridin-4-y1;
In yet another particular embodiment (for any of the compounds or
embodiments described above), Rl is 5-methyl-thiophen-2-yl, 5-chloro-thiophen-
2-
yl, 5-fluorobenzo[b]thiophen-2-yl, 5-trifluoromethyl-thiophen-2-yl, 5-
difluoromethyl-
thiophen-3-yl, 5-methyl-thiophen-3-yl, 2-methyl-pyridin-4-yl, 2-
trifluoromethyl-
pyridin-4-yl, 4-trifluoromethyl-pyridin-2-yl, 2-chloro-pyridin-4-yl, 2-methoxy-
pyridin-4-yl, 6-chloropyrimidin-4-yl, 6-chloro-2-methylpyrimidin-4-yl, 2-
trifluoromethyl-pyrimidin-4-yl, 4-trifluoromethyl-pyrimidin-2-y1; or 2-chloro-
6-
(trifluoromethyl)pyridin-4-y1; or a pharmaceutically acceptable salt thereof.
9
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
In yet another Particular embodiment, Rl is 4-chloro-thieno[3,2-c]pyridin-2-
yl,
4-chloro-thieno[3,2-c]pyridin-3-yl, thieno[3,2-c]pyridin-2-yl, 3-chloro-
imidazo[1,2-
a]pyridin-7-yl, benzo[b]thiophen-2-yl, or 4-methylthieno[3,2-c]pyridin-2-y1;
or a
pharmaceutically acceptable salt thereof.
Representative compounds of Formula (I) where R2, R3, R4 and R5 are each H
include:
1-(2-Chloropyridin-4-y1)-3-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one;
1-(4-Cyclopropylpyridin-3-y1)-3-(2-(trifluoromethyl)pyridin-4-
yl)imidazolidin-2-one;
1-(4-Cyclopropylpyridin-3-y1)-3-(naphthalen-2-yl)imidazolidin-2-one;
1-(Benzo[b]thiophen-5-y1)-3-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one;
1-(4-Cyclopropylpyridin-3-y1)-3-(3-(trifluoromethyl)phenyl)imidazolidin-2-
one;
1-(Benzo[b]thiophen-2-y1)-3-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one;
1-(6-Chloro-2-methylpyrimidin-4-y1)-3-(4-cyclopropylpyridin-3-
yl)imidazolidin-2-one;
1-(4-Cyclopropylpyridin-3-y1)-3-(4-(trifluoromethyl)pyridin-2-
yl)imidazolidin-2-one;
1-(4-Cyclopropylpyridin-3-y1)-3-(2-methoxypyridin-4-yl)imidazolidin-2-one;
1-(6-Chloropyrimidin-4-y1)-3-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one;
1-(4-Cyclopropylpyridin-3-y1)-3-(4-(trifluoromethyl)pyrimidin-2-y1)
imidazolidin-2-one;
1-(4-Cyclopropylpyridin-3-y1)-3-(2-(trifluoromethyl)pyrimidin-4-y1)
imidazolidin-2-one;
1-(4-Cyclopropylpyridin-3-y1)-3-(3-(trifluoromethyl)benzo[b]thiophen-5-y1)
imidazolidin-2-one;
1-(4-Cyclopropylpyridin-3-y1)-3-(2-fluorobenzo[b]thiophen-5-
yl)imidazolidin-2-one;
1-(4-Cyclopropylpyridin-3-y1)-3-(3-methylbenzo[b]thiophen-5-
yl)imidazolidin-2-one;
1-(Benzo[b]thiophen-6-y1)-3-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one;
1-(4-Cyclopropylpyridin-3-y1)-3-(2,2-difluorobenzo[d][1,3]dioxo1-5-
yl)imidazolidin-2-one;
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
1 -(4-Cyclopropylpyridin-3 -y1)-3 -(3 -methylb enzo [IA thiophen-6-
yl)imidazo lidin-2-one ;
1 -(4-Cyclopropylpyridin-3 -y1)-3 -(3 -methylb enz o furan-5 -yl)imidazo lidin-
2-
one ;
1 -(2-Chlorob enzo [IA thiophen-5 -y1)-3 -(4- cyclopropylpyridin-3 -
yl)imidazo lidin-2-one ;
1 -(4-Cyclopropylpyridin-3 -y1)-3 -(2,3 - dihydro-1H-inden-5 -yl)imidaz o
lidin-2-
one ;
1 -(4-Cyclopropylpyridin-3 -y1)-3 -(5 -fluorob enzo [IA thiophen-2-
yl)imidazolidin-2-one;
1 -(4-Cyclopropylpyridin-3 -y1)-3 -(3 -(trifluoromethyl)b enzo [b]thiophen-6-
yl)imidazolidin-2-one;
1 -(4-Cyclopropyl-pyridin-3 -y1)-3 -(2-fluoro-3 -methyl-b enzo [IA thiophen-5 -
y1)-
imidazo lidin-2- one ;
1 -(4-Cyclopropyl-pyridn-3 -y1)-3 -(2-fluro-b enzo (b)thiophene-6-
yl)imidazo lidin-2-one ;
1 -(4-Cyclopropyl-pyridin-3 -y1)-3 -(4-fluoro-b enzo [b]thiophen-6-
yl)imidazolidin-2-one;
1 -(4-Cyclopropyl-pyridin-3 -y1)-3 -(5 -fluoro-b enzo [b]thiophen-6-
yl)imidazolidin-2-one;
1 -(4-Cyclopropylpyridin-3 -y1)-3 -(6-fluorob enzo [IA thiophen-5 -
yl)imidazo lidin-2-one ;
1 -(4-Cyclopropylpyridin-3 -y1)-3 -(5 -fluoro-3 -methylb enzo [IA thiophen-6-
yl)imidazo lidin-2-one ;
1 -(2-Chloro-pyridin-4-y1)-3 -[4-(1 -hydroxy- cyclobutyl)pyridin-3 -yl] -
imidazo lidin-2- one ;
1 -(2-Chloropyridin-4-y1)-3 -(4-cyclop entyl-pyridin-3 -yl)imidazo lidin-2-
one ;
1 -(2-Chloropyridin-4-y1)-3 - [4-(1-hydroxycyclopenty1)-pyridin-3-
yl]imidazolidin-2-one;
1 - [4-(1 -Hydroxy-cyclopropy1)-pyridin-3 -yl] -3 -(3 -trifluoromethylpheny1)-
imidazo lidin-2- one ;
1 -(4- cyclopropylpyridin-3 -y1)-3 -(6- cyclopropylpyrimidin-4-y1) imidazo
lidin-
2-one ;
11
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
1-(4-cyclopropylpyridin-3-y1)-3-(2-cyclopropylpyridin-4-y1) imidazolidin-2-
one;
1-(4-cyclopropylpyridin-3-y1)-3-(5-fluoro-4-methylpyridin-2-y1) imidazolidin-
2-one;
1-(4-cyclopropylpyridin-3-y1)-3-(2-cyclopropylpyrimidin-4-y1) imidazolidin-
2-one;
1-(6-chloro-2-(trifluoromethyl) pyrimidin-4-y1)-3-(4-cyclopropylpyridin-3-y1)
imidazolidin-2-one;
1-(4-cyclopropylpyridin-3-y1)-3-(2, 6-dichloropyridin-4-y1) imidazolidin-2-
one;
1-(2-chloro-6-cyclopropylpyridin-4-y1)-3-(4-cyclopropylpyridin-3-y1)
imidazolidin-2-one;
1-(2-cyclopropy1-6-(trifluoromethyl) pyridin-4-y1)-3-(4-cyclopropylpyridin-3-
yl) imidazolidin-2-one; and
1-(2,6-bis(trifluoromethyppyridin-4-y1)-3-(4-cyclopropylpyridin-3-y1)
imidazolidin-2-one; or a pharmaceutically acceptable salt thereof.
Representative compounds of Formula (II) or (III) include:
(1S,5R)-2-(4-cyclopropylpyridin-3-y1)-4-(2-(trifluoromethyl)pyridin-4-y1)-2,4-
diazabicyclo[3.1.0]hexan-3-one;
(1R,5S)-2-(4-cyclopropylpyridin-3-y1)-4-(2-(trifluoromethyl)pyridin-4-y1)-2,4-
diazabicyclo[3.1.0]hexan-3-one;
(1S ,5R)-2-(2-chloropyridin-4-y1)-4-(4-cyclopropylpyridin-3-y1)-2,4-
diazabicyclo[3.1.0]hexan-3-one;
(1R,5S)-2-(2-chloropyridin-4-y1)-4-(4-cyclopropylpyridin-3-y1)-2,4-
diazabicyclo[3.1.0]hexan-3-one;
(1S ,5R)-2-(4-cyclopropylpyridin-3-y1)-4-(2-methoxypyridin-4-y1)-2,4-
diazabicyclo[3.1 .0]hexan-3-one; and
(1R,5S)-2-(4-cyclopropylpyridin-3-y1)-4-(2-methoxypyridin-4-y1)-2,4-
diazabicyclo[3.1.0]hexan-3-one; or a pharmaceutically acceptable salt thereof.
Compounds of particular interest include: 1-(2-chloropyridin-4-y1)-3-(4-
cyclopropylpyridin-3-y1) imidazolidin-2-one;
1-(4-cyclopropylpyridin-3-y1)-3-(2-(trifluoromethyl)
pyridin-4-y1)
imidazolidin-2-one;
12
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
1-(4-cyclopropylpyridin-3-y1)-3-(3-(trifluoromethyl) phenyl) imidazolidin-2-
one;
1-(2-chloro-pyridin-4-y1)-3-[4-(1-hydroxy-cyclobuty1)-pyridin-3-y1]-
imidazolidin-2-one;
1-(6-chloro-2-methylpyrimidin-4-y1)-3-(4-cyclopropylpyridin-3-y1)
imidazolidin-2-one;
1-(4-cyclopropylpyridin-3-y1)-3-(2-methoxypyridin-4-y1) imidazolidin-2-one;
(1R,5S)-2-(4-cyclopropylpyridin-3-y1)-4-(2-(trifluoromethyl)pyridin-4-y1)-2,4-
diazabicyclo[3.1.0]hexan-3-one;
(1S ,5R)-2-(2-chloropyridin-4-y1)-4-(4-cyclopropylpyridin-3-y1)-2,4-
diazabicyclo[3.1.0]hexan-3-one;
(1R,5S)-2-(4-cyclopropylpyridin-3-y1)-4-(2-methoxypyridin-4-y1)-2,4-
diazabicyclo[3.1.0]hexan-3-one;
1-(2-chloro-6-(trifluoromethyl)pyridin-4-y1)-3-(4-cyclopropylpyridin-3-
yl)imidazolidin-2-one;
1-(6-chloro-2-(trifluoromethyl) pyrimidin-4-y1)-3-(4-cyclopropylpyridin-3-y1)
imidazolidin-2-one;
1-(4-cyclopropylpyridin-3-y1)-3-(2, 6-dichloropyridin-4-y1) imidazolidin-2-
one;
1-(2-chloro-6-cyclopropylpyridin-4-y1)-3-(4-cyclopropylpyridin-3-y1)
imidazolidin-2-one;
1-(2-cyclopropy1-6-(trifluoromethyl) pyridin-4-y1)-3-(4-cyclopropylpyridin-3-
yl) imidazolidin-2-one; and
1-(2,6-bis(trifluoromethyppyridin-4-y1)-3-(4-cyclopropylpyridin-3-y1)
imidazolidin-2-one; or a pharmaceutically acceptable salt thereof.
Other compounds include those described in the Example section below, in
particular, those comounds having an IC50 less than 1 i.IM (or 1,000 nM),
preferably,
less than 500 nM, more preferably, less than 100 nM.
In another aspect of the present invention a pharmaceutical composition is
provided which comprises a compound of Formula (I), (II), or (III), or a
pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable
carrier or
excipient. The pharmaceutical composition optionally comprises at least one
additional pharmaceutical agent (suitable pharmaceutical agents are described
herein
below).
13
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
In yet another aspect of the present invention, a method of treating a
disease,
disorder, or syndrome mediated by Cyp17 inhibition (such as those described
herein
below) is provided, where the method comprises administering a compound
according to Formula (I), (II), or (III), or a pharmaceutical composition
comprising
the compound of Formula (I), (II), or (III), and pharmaceutically acceptable
excipients, to a subject in need thereof.
Another aspect of the present invention includes a compound according to
Formula (I), (II), or (III), for use in therapy (e.g., the use of a compound
for the
treatment of a disease, disorder, or syndrome mediated by Cyp17 inhibition).
Yet another aspect of the present invention includes a method for treating a
disease, disorder or syndrome mediated by Cyp17 inhibition comprising the step
of
administering
(i) a first composition comprising a compound according to Formula (I), (II),
or (III), or a pharmaceutically acceptable salt thereof, and a
pharmaceutically
acceptable carrier or excipient; and
(ii) a second composition comprising at least one additional pharmaceutical
agent and a pharmaceutically acceptable carrier or excipient;
wherein said at least one additional pharmaceutical agent is an anticancer
agent, chemotherapy agent, or antiproliferative compound. The first and second
compositions may be administered either simultaneously or sequentially in any
order.
In one particular embodiment for each of the methods and uses described
above, the disease, disorder, or syndrome is selected from the group
consisting of
cancer (in particular, prostate cancer) and inflammation.
Definitions
As used herein, the terms "alkyl" refers to a hydrocarbon radical of the
general
formula C n1-12/1+1. The alkane radical may be straight or branched. For
example, the
term "(Ci-C6)alkyl" refers to a monovalent, straight, or branched aliphatic
group
containing 1 to 6 carbon atoms (e.g., methyl, ethyl, n-propyl, i-propyl, n-
butyl, i-
butyl, s-butyl, t-butyl, n-pentyl, 1-methylbutyl, 2-methylbutyl, 3-
methylbutyl,
neopentyl, 3,3-dimethylpropyl, hexyl, 2-methylpentyl, and the like).
Similarly, the
alkyl portion (i.e., alkyl moiety) of an alkoxy, acyl (e.g., alkanoyl),
alkylamino,
dialkylamino, and alkylthio group have the same definition as above.
14
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
"Halo-substituted alkyl" refers to an alkyl group, as defined above,
substituted
with at least one halogen atom. For example, when the halogen atom is fluoro,
common haloalkyl groups include fluoromethyl, difluoromethyl, trifluoromethyl,
2,2,2-trifluoroethyl, 2,2,2,1,1-pentafluoroethyl, and the like. Mixed halogen
substitution are also included (e.g., chlorofluoromethyl).
The term "alkenyl" refers to a monovalent group derived from a hydrocarbon
having at least one carbon-carbon double bond. The term "C2-C6-alkenyl" refers
to a
monovalent group derived from a hydrocarbon having two to six carbon atoms and
comprising at least one carbon-carbon double bond. The alkenyl group can be
unbranched or branched. Representative examples of alkenyl include vinyl, 1-
propenyl, 2-propenyl, 1-methyl-l-propenyl, 1-methy1-2-propenyl, 2-methyl-l-
propenyl, 2-methyl-2-propenyl, 1-butenyl, 2-butenyl, 3-butenyl, and so on.
The term "alkynyl" refers to a monovalent group derived from a hydrocarbon
having at least one carbon-carbon triple bond. The term "C2-C6-alkynyl" refers
to a
monovalent group derived from a hydrocarbon having two to six carbon atoms and
comprising at least one carbon-carbon triple bond. The alkynyl group can be
unbranched or branched. Representative examples include ethynyl, propynyl,
butyn-
l-yl, butyn-2-yl, and so on.
The term "hydroxy-substituted alkyl" refers to an alkyl group, as defined
above, substituted with one or more hydroxyl (-OH) groups (e.g., -CH2OH, -
CH(OH)2,
-CH(OH)-CH2OH, -CH(OH)-CH3, and so on). Preferably, the alkyl group is
substituted with 1 to 2 hydroxyl groups, more preferably one hydroxyl group.
"Halogen" or "halo" may be fluorine, chlorine, bromine or iodine (preferred
halogens as substituents are fluorine and chlorine).
The term "oxo" or ¨C(0)- refers to a carbonyl group. For example, a ketone,
aldehyde, or part of an acid, ester, amide, lactone, or lactam group.
The terms "partially or fully saturated carbocyclic ring" (also referred to as
"partially or fully saturated cycloalkyl") refers to nonaromatic rings that
are either
partially or fully hydrogenated and may exist as a single ring, bicyclic ring
or a spiral
ring. Unless specified otherwise, the carbocyclic ring is generally a 3- to 8-
membered
ring. For example, partially or fully saturated carbocyclic rings (or
cycloalkyl) include
groups such as cyclopropyl, cyclopropenyl, cyclobutyl, cyclobutenyl,
cyclopentyl,
cyclpentenyl, cyclopentadienyl, cyclohexyl, cyclohexenyl, cyclohexadienyl,
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
norbornyl (bicyclo[2.2.1]heptyl), norbornenyl, bicyclo[2.2.2]octyl, and the
like.
When optionally substituted, the group is attached via the carbocyclic ring
and not the
substituent.
The term "partially saturated or fully saturated heterocyclic ring" (also
referred
to as "partially saturated or fully saturated heterocycle") refers to
nonaromatic rings
that are either partially or fully hydrogenated and may exist as a single
ring, bicyclic
ring or a spiral ring. Unless specified otherwise, the heterocyclic ring is
generally a 3-
to 6-membered ring containing 1 to 3 heteroatoms (preferably 1 or 2
heteroatoms)
each independently selected from sulfur, oxygen and/or nitrogen. Partially
saturated
or fully saturated heterocyclic rings include groups such as epoxy,
aziridinyl,
tetrahydrofuranyl, dihydrofuranyl, dihydropyridinyl, pyrrolidinyl, N-
methylpyrrolidinyl, imidazolidinyl, imidazolinyl, piperidinyl, piperazinyl,
pyrazolidinyl, 2H-pyranyl, 4H-pyranyl, 2H-chromenyl, oxazinyl, morpholino,
thiomorpholino, tetrahydrothienyl, tetrahydrothienyl 1,1-dioxide, and the
like. When
optionally substituted, the group is attached via the heterocyclic ring and
not the
substituent. Unless specified otherwise, the heterocyclic ring may be attached
via any
valence available ring member (e.g., replacement of a H attached to the
heterocyclic
ring).
The term "fused phenyl" refers to a phenyl group fused to another ring, such
as another phenyl (i.e., naphthalene (e.g., naphthalen-2-yl, naphthalen-1-y1),
a
partially or fully saturated cycloalkyl (e.g., indan-5-yl, 2,3-dihydro-1H-
indenyl, or
tetrahydronaphthalenyl, etc.), a heteroaryl (e.g., 1H-indo1-5-yl, 1H-indo1-6-
yl,
benzothiazol-5-yl, benzothiazol-6-yl, benzo[b]thiophen-5-yl, quinolin-6-yl,
quinolin-
7-yl, isoquinolin-5-yl, isoquinolin-6-y1 isoquinolin-7-yl, isoquinolin-8-yl,
indazol-4-
yl, indazol-5-yl, indazol-6-yl, indazol-7-yl, benzofuran-4-yl, benzofuran-5-
yl,
benzofuran-6-yl, benzofuran-7-yl,benzimidazol-4-yl, or quinoxalin-6-yl,
benzooxazol-5-yl, benzo[d]isoxazol-5-yl, benzo[d]isoxazol-6-yl, 1H-
benzoimidazol-
4-yl, 1H-benzoimidazol-5-yl, 1H-benzoimidazol-6-yl, 1H-benzoimidazol-7-yl, 1H-
benzotriazol-5-yl, etc.) or a partially saturated or fully saturated
heterocycle (e.g.,
indolin-4-yl, indolin-5-yl, indolin-6-yl, indolin-7-yl, 1,2-dihydroquinolin-6-
yl,
1,2,3,4-tetrahydro-quinolin-6-yl, 1,2,3,4-tetrahydro-quinolin-7-yl, 2,3-
dihydro-1H-
benzo[d]imidazolyl, 2,3-dihydro-1H-indazolyl, 2,3-dihydrobenzo[d]oxazolyl, 2,3-
dihydrobenzo[1,4]dioxin-6-yl, 2,3-dihydro-benzofuran-5-yl, benzo[1,3]dioxo1-5-
yl,
2,3-dihydro-1H-indazol-5-yl, 2,3-dihydro-1H-indazol-6-yl, etc.), where the
group is
16
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
attached via one of the phenyl carbon atoms. When substituted, the fused
phenyl
group is attached via the phenyl and not the substituent. When substituted,
the fused
phenyl can be substituted on any valence available ring atom (e.g.,
replacement of a H
atom attached to the fused phenyl) within the fused system. For example, a
benzofuranyl group may be substituted on the phenyl or furanyl portion of the
benzofuranyl group.
The term "heteroaryl" or "heteroaromatic ring" refers to aromatic moieties
containing at least one heteratom (e.g., oxygen, sulfur, nitrogen or
combinations
thereof) within a 5- to 6-membered aromatic ring system (e.g., pyrrolyl,
pyridyl,
pyrazolyl, thienyl, furanyl, oxazolyl, imidazolyl, tetrazolyl, triazinyl,
pyrimidyl,
pyrazinyl, thiazolyl, isothiazolyl, etc.). A typical single heteroaryl ring is
generally a
5- to 6-membered ring containing one to three heteroatoms each independently
selected from oxygen, sulfur and nitrogen. When optionally substituted, the
group is
attached via the heteroaryl ring and not the substituent.
The term "fused heteroaryl" refers to a heteroaryl group fused to another
ring,
such as another heteroaryl (e.g. purinyl, thieno[3,2-c]pyridinyl (e.g.,
thieno[3,2-
c]pyridin-2-y1 and thieno[3,2-c]pyridin-3-y1), imidazo[1,2-a]pyridinyl (e.g.,
imidazo[1,2-a]pyridin-3-yl, imidazo[1,2-a]pyridin-6-yl, imidazo[1,2-a]pyridin-
7-y1
and 3H-imidazo[4,5-b]pyridin-6-y1), or benzo[b]thiophenyl, etc.), phenyl
(e.g.,
benzo[b]thiophen-2-yl, benzo[b]thiophen-3-yl, quinolin-2-yl, quinolin-3-yl,
benzooxazol-2-yl, benzothiazol-2-yl, 1H-indo1-2-yl, 1H-indo1-3-yl, isoquinolin-
l-yl,
isoquinolin-3-yl, isoquinolin-4-yl, benzofuran-2-yl, benzofuran-3-yl, indazol-
3-y1õ
benzimidazol-2-yl, etc.), a partially or fully saturated cycloalkyl (e.g.,
4,5,6,7-
tetrahydrobenzo[d]oxazolyl, 4,5,6,7-tetrahydro-1H-indolyl, 5,6,7,8-
tetrahydroquinolinyl, 5,6,7,8-tetrahydroisoquinolinyl, 4,5,6,7-
tetrahydrobenzo[b]thiophenyl, 4,5,6,7-tetrahydrobenzofuranyl, 4,5,6,7-
tetrahydro-1H-
indazolyl, 4,5,6,7-tetrahydro-1H-benzo[d]imidazolyl, or 4,5,6,7-
tetrahydrobenzo[d]oxazolyl, etc.), or a partially saturated or fully saturated
heterocycle (e.g., 8,9-dihydro-7H-purinyl, 2,3-dihydrothieno[3,2-c]pyridinyl,
4,5,6,7-
tetrahydro-thieno[2,3-c]pyridin-2-yl, 4,5,6,7-tetrahydrothieno[3,2-
c]pyridinyl, or
5,6,7,8-tetrahydroimidazo[1,2-a]pyridinyl, etc.), where the heteroaryl group
is
attached via one of the valence available heteroaryl ring atoms. When
substituted, the
heteroaryl group is attached via the fused heteroaryl and not the substituent.
When
substituted, the fused heteroaryl can be substituted on any valence available
ring atom
17
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
(e.g., replacement of a H atom attached to the fused heteroaryl) within the
fused
system. For example, an imidazo[1,2-a]pyridinyl group may be substituted on
the
imidazole or pyridine portion of the fused system.
The phrase "therapeutically effective amount" means an amount of a
compound of the present invention that (i) treats or prevents the particular
disease,
condition, or disorder, (ii) attenuates, ameliorates, or eliminates one or
more
symptoms of the particular disease, condition, or disorder, or (iii) prevents
or delays
the onset of one or more symptoms of the particular disease, condition, or
disorder
described herein. The term "animal" refers to humans (male or female),
companion
animals (e.g., dogs, cats and horses), zoo animals, marine animals, birds and
other
similar animal species. Preferred animals are mammals, in particular a human.
The phrase "pharmaceutically acceptable" indicates that the substance or
composition must be compatible chemically and/or toxicologically, with the
other
ingredients comprising a formulation, and/or the mammal being treated
therewith.
The term "compounds of the present invention" (unless specifically identified
otherwise) refer to compounds of Formula (I), (II), or (III), or
pharmaceutically
acceptable salts of the compounds, as well as, all stereoisomers (including
diastereoisomers and enantiomers), tautomers and isotopically labeled
compounds.
DETAILED DESCRIPTION
The present invention provides compounds and pharmaceutical formulations
thereof that are useful in the treatment of diseases, conditions and/or
disorders
modulated by the inhibition of 17a-hydroxylase/C17,20-lyase.
Compounds of the present invention may be synthesized by synthetic routes
that include processes analogous to those well-known in the chemical arts,
particularly in light of the description contained herein. The starting
materials are
generally available from commercial sources such as Aldrich Chemicals
(Milwaukee,
Wis.) or are readily prepared using methods well known to those skilled in the
art
(e.g., prepared by methods generally described in Louis F. Fieser and Mary
Fieser,
Reagents for Organic Synthesis, v. 1-19, Wiley, New York (1967-1999 ed.), or
Beilsteins Handbuch der organischen Chemie, 4, Aufl. ed. Springer-Verlag,
Berlin,
including supplements (also available via the Beilstein online database)).
For illustrative purposes, the reaction schemes depicted below provide
potential routes for synthesizing the compounds of the present invention as
well as
18
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
key intermediates. For a more detailed description of the individual reaction
steps, see
the Examples section below. Those skilled in the art will appreciate that
other
synthetic routes may be used to synthesize the inventive compounds. Although
specific starting materials and reagents are depicted in the schemes and
discussed
below, other starting materials and reagents can be easily substituted to
provide a
variety of derivatives and/or reaction conditions. In addition, many of the
compounds
prepared by the methods described below can be further modified in light of
this
disclosure using conventional chemistry well known to those skilled in the
art.
Scheme I below provides a potential route for synthesizing compounds of of
Formula (I), where R2, R3, R4 and R5 are each independently CH3 or H (referred
to
below as a compound of Formula (I-a).
GENERAL SCHEMES
Scheme 1
R4 R5 Rv R3H
CI
RN H2 N
(SM-2) ci y
R2
R3 0 Step 1 R4 R5
(SM-1)
NaH/DMF/THF Step 2
V
0 0
R1-X, catalyst,
R1 R Ligand, Base,
_________________________________________________ HNVNNR
Solvent, Heat
R2 ( R5 Step 3 R2 ( R5
R3 R4 R3 R4
(I-a)
In Scheme I above, R is represented by the following group (where R6 is as
defined
above).
R6
N
19
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
Step-1 & 2:
The intermediate products of Steps 1 and 2 may be synthesized using methods
analogous to those described by Kak-Shan Shia, et al., in J. Med. Chem., 2002,
45,
1644-1655 using the desired starting materials which are available
commercially or
synthesized using known procedures described in the art. For example, a
variety of 2-
chloroalkyl isocyanates can be prepared using the methods described by C K
Johnson
in J Org Chem (1967), 32(5), 1508-10. The reaction times in certain cases were
prolonged to increase the % yield as compared to reported yields in the above
mentioned J Med Chem reference.
Step-3:
The products of Step -2 obtained as described above may be converted into
the desired products by reacting with the appropriate alkyl or aryl halides
preferably
chloro / bromo alkyl or aryl derivatives using conditions well know to those
of skill in
the art, e.g., the Buchwald-Hartwig C-N coupling conditions or NaH/ DMF, and
the
like. Preferred conditions are those known as the `Buchwald-Hartwig" reaction,
e.g.,
in the presence of (a) a catalyst, such as copper iodide, (b) a base, such as
potassium
phosphate or cesium carbonate; and (c) a ligand, such as trans-1, 2-diamino
cyclohexane, in the presence of suitable solvents (e.g., 1, 4-dioxane) at
temperatures
ranging from about room temperature to the refluxing temperature of the
solvent.
When a protection group is used, then the protecting group is removed using
the
conditions appropriate for the particular protecting group used to produce
compounds
of the present invention. For a more detailed description, see the Example
section
below.
Alternatively, the substituents Rl and R may be introduced in the reverse. For
example, instead of starting with R-NH2, R1-NH2 is used as the starting
material. The
R group is then introduced in step 3 by using R-X instead of R1-X.
Scheme 2 describes how one could make the starting material (SM-1) above
where R2, R3, R4 and/or R5 are other than hydrogen.
Scheme 2
CA 02834224 2013-10-23
WO 2012/149413 PCT/US2012/035583
R4 R5R4 R5
SOCl2, DCM
CI OH DMF CI >c\yci
R2Step 1
R2
R3 0 R3 0
Sodium azide
1,4-dioxane, H20 Step 2
R4 R5 R4 R5
Toluene
85 C
>
CI
Step 3
R2
R3 0
(SM-1)
The desired chloro carboxylic acid is first converted to its corresponding
acid
chloride derivative using procedures well-known to those of skill in the art.
For
example, the carboxylic acid derivative may be treated with thionyl chloride
in the
presence of dimethylformamide (DMF) and a solvent (e.g., dichloromethane
(DCM)).
Other chlorinating agents may be used, e.g., phosphorous trichloride or
phosphorous
pentachloride. The acid chloride can then be converted to its corresponding
azide by
treatment with sodium azide. The azide is then converted to the desired
isocyanate
(SM-1) by the Curtius rearrangement, e.g., heating the azide at elevated
temperatures.
Scheme 3 describes a potential route for making SM-2, where R6 is (C3-
C5)cycloalkyl.
Scheme 3
CI R6 R6
NO2 R6-B(OH)2 )NO2 H2
I
where R6 = (C3-05)cycloalkyl (SM-2)
R6 can be introduced into SM-2 using the desired (C3-05)cycloalkyl boronic
acid in the presence of tetrakis(triphenylphosphine)palladium(0) followed by
reduction of the nitro group using standard reduction procedures well-known to
those
21
CA 02834224 2013-10-23
WO 2012/149413 PCT/US2012/035583
of skill in the art (e.g., treatment with ammonium chloride in the presence of
zinc
powder).
Scheme 4 provides an alternative synthesis for preparing compounds of
Formula (I), (II) or (III).
Scheme 4
R4 R5 R4 R5
H2N OEt R1-I O
Cul, K2CO3, DMF, R1 H?\yr
Et
H20 N
+ -IP-
R2Step 1 R2
R3 0 R3 0
DPPA, TEA,
Step 2 Toluene
R6-...
0 N
ys. --__ IR6-.._\ 0
,L, ,N
N
R1¨N>ck7z4 I )\----NH
R5 4
Cul, K3PO4, 1,4-Dioxane R1¨N R5
R2 R'7 trans-1, 2-diannino R2---- Rk7R43 -
(I) cyclohexane
Step 3
The desired Rl group may be attached to the desired amino carboxylate
compound via Buchwald-Hartwig C-N coupling conditions or NaH/ DMF, and the
like. The cyclic urea is then formed using methods analogous to those
described by
Kak-Shan Shia, et al., in J. Med. Chem., 2002, 45, 1644-1655. The pyridine
derivative may then be coupled to the imidazoline via a Buchwald-Hartwig C-N
coupling reaction described previously.
Alternatively, the unsymmetrical disubstituted-1H-imidazolin-2(31/)-ones can
be prepared by other methods discussed by T. Hafner, et al., in Synthesis
(2007) 9,
1403-1411 (e.g., Brazier, S.A, et al., J Chem Soc (1912), 101, 2352 and
Schonherr,
H.J., et al, Chem Ber (1970), 103, 1037).
Scheme 5 provides another alternative synthesis for preparing compounds of
Formula (I), where R2 (or R3) and R4 (or R5) are H (referred to below as
Formula (I-
b)).
Scheme 5
22
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
R4
R2 OH R4 R1
H 0N
r N i)r0H
0
R1¨NH2 )1' R1 _),..
HN,?---R4
Step 1 R2 0 Step 2
R2
(5a)
(5b)
Cul , K3PO4, R- I
1 ,4-Dioxane
Step 3
W
R1
)¨N
R-N ?---- R4
R2
(I-b)
In Scheme 5 above, R is represented by the following group
R6
c;zz, N
Intermediate (5a) may be formed via a Michael addition of the desired amine
(R1-NH2) to the desired acrylic acid using procedures well-known to those of
skill in
the art. For example, the amine and acrylic acid in a suitable solvent (e.g.,
toluene)
are heated at an elevated temperature (e.g., about 70 C to about 100 C) under
an
inert atmosphere. The amino acid intermediate (5a) may then be cyclized to
form the
cyclic urea intermediate (5b). For example, the cyclic urea intermediate (I-
4b) may
be formed by treating the amino acid intermediate (5a) with an activating
agent (e.g.,
diphenyl phosphoryl azide (DPPA)) in the presence of an amine (e.g.,
triethylamine)
and appropropriate solvent (e.g., toluene) at elevated temperatures. The
desired A
group may be coupled to the cyclic urea intermediate (5b) using standard
coupling
conditions described above to form the a compound of the Formula (I-b).
Scheme 6 provides a potential route for the synthesis of compounds of
Formula (II) or (III).
23
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
Scheme 6
RiFL,A,7.....-NN / \
)
11 ,
0 Pg H
Step I Step 2 0 N
V
N
R1 N
R1 Step 3 +
A01-1 6
(6a) (6b)
R1-NzN / \
0 N
(III)
The amino-protected 3-oxo-2,4-diazabicyclo[3.1.0]hexane is generally
available from commercial sources (e.g., benzyl 3-oxo-2,4-
diazabicyclo[3.1.0]hexane-
2-carboxylate which is available from Rare Chemicals Screening Compounds,
Maybridge Screening collection, Interchim Screening Library, Ambinter Stock
Screening Collection or Aurora Screening Library; or prepared using the
procedures
described by Witiak, D.T., in J. Med Chem, 1978, 21(12) 1194-1197). The Rl
group
can be introduced using the Buchwald-Hartwig reaction. For example, treating
benzyl 3-oxo-2,4-diazabicyclo[3.1.0]hexane-2-carboxylate with the desired halo-
substituted aryl or heteroaryl group (R1-X, where X is Br or I) in the
presence of
Xantphos, a Palladium catalyst (e.g.,
tris(dibenzylideneacetone(dipalladium(0)) at
elevated temperatures (e.g., about 100 C). The amino-protecting group (Pg) is
then
removed using the appropriate conditions for the particular protecting group.
Once
the protecting group is removed, then the desired R6 substituted 3-pyridinyl
group
may then be coupled to the 3-oxo-2,4-diazabicyclo[3.1.0]hexane derivative (6b)
via a
Buchwald-Hartwig C-N coupling reaction described previously. In Scheme 6
above,
R is represented by the following group
R6
c22..N
The Example section below provides a more detailed description of the
synthetic schemes as well as other alternative processes for making compounds
of the
24
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
present invention which could be easily modified (e.g., substituting different
starting
materials) by those of skill in the art.
The compounds and intermediates described herein may be isolated and used
as the compound per se or its salt. Many of the compounds represented by
Formula
(I), (II), (III), (I-a), and (I-b) are capable of forming acid addition salts,
particularly
pharmaceutically acceptable acid addition salts. Pharmaceutically acceptable
acid
addition salts of the compound of Formula (I), (II), (III), (I-a) and (I-b)
include those
of inorganic acids, for example, hydrohalic acids such as hydrochloric acid,
hydrobromic acid or hydroiodic acid, nitric acid, sulfuric acid, phosphoric
acid; and
organic acids, for example aliphatic monocarboxylic acids such as formic acid,
acetic
acidõ propionic acid and butyric acid, aliphatic hydroxy acids such as lactic
acid,
citric acid, tartaric acid or malic acid, dicarboxylic acids such as maleic
acid or
succinic acid, aromatic carboxylic acids such as benzoic acid, p-chlorobenzoic
acid,
diphenylacetic acid or triphenylacetic acid, aromatic hydroxy acids such as o-
hydroxybenzoic acid, p-hydroxybenzoic acid, 1-hydroxynaphthalene-2-carboxylic
acid or 3-hydroxynaphthalene-2-carboxylic acid, and sulfonic acids such as
methanesulfonic acid or benzenesulfonic acid. These salts may be prepared from
compounds of Formula (I), (II), (III), (I-a) or (I-b) by known salt-forming
procedures.
Compounds of the present invention which contain acidic, e.g. carboxyl,
groups, are also capable of forming salts with bases, in particular
pharmaceutically
acceptable bases such as those well known in the art; suitable such salts
include metal
salts, particularly alkali metal or alkaline earth metal salts such as sodium,
potassium,
magnesium or calcium salts, or salts with ammonia or pharmaceutically
acceptable
organic amines or heterocyclic bases such as ethanolamines, benzylamines or
pyridine. These salts may be prepared from compounds of Formula (I), (II),
(III), (I-
a) and (I-b) by known salt-forming procedures.
In those compounds where there is an asymmetric carbon atom the compounds
exist in individual optically active isomeric forms or as mixtures thereof,
e.g. as
racemic or diastereomeric mixtures. The present invention embraces both
individual
optically active R and S isomers as well as mixtures, e.g. racemic or
diastereomeric
mixtures, thereof.
The present invention includes all pharmaceutically acceptable isotopically-
labeled compounds of the present invention wherein one or more atoms are
replaced
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
by atoms having the same atomic number, but an atomic mass or mass number
different from the atomic mass or mass number usually found in nature.
Examples of isotopes suitable for inclusion in the compounds of the invention
comprises isotopes of hydrogen, such as 2H and 3H, carbon, such as , 11C-
13C and MC,
chlorine, such as 36C1, fluorine, such as 18F, iodine, such as 1231 and 1251,
nitrogen, such
as 13N and 15N, oxygen, such as 150, 170 and 180, phosphorus, such as 32P, and
sulphur, such as 35S.
Substitution with heavier isotopes such as deuterium, i.e. 2H, may afford
certain therapeutic advantages resulting from greater metabolic stability, for
example,
increased in vivo half-life or reduced dosage requirements, and hence may be
preferred in some circumstances.
Isotopically-labeled compounds of the present invention can generally be
prepared by conventional techniques known to those skilled in the art or by
processes
analogous to those described in the accompanying Examples and Preparations
Sections using an appropriate isotopically-labeled reagent in place of the non-
labeled
reagent previously employed.
The compounds of the present invention may exist in unsolvated as well as
solvated forms with pharmaceutically acceptable solvents such as water,
ethanol, and
the like, and it is intended that the invention embrace both solvated and
unsolvated
forms. For purposes of the present invention, solvates (including hydrates)
are
considered pharmaceutical compositions, e.g., a compound of Formula (I), (II),
(III),
(I-a) or (I-b) (or pharmaceutically acceptable salt thereof) in combination
with an
excipient, wherein the excipient is a solvent.
Compounds of the present invention are useful for treating diseases,
conditions and disorders mediated by the regulation of 1 7a-hydroxylase/C17,2o-
lyase
(e.g., cancer (in particular, prostate cancer) or inflammation); consequently,
the
compounds of the present invention (including the compositions and processes
used
therein) may be used in the manufacture of a medicament for the therapeutic
applications described herein. Hence, another embodiment of the present
invention is
a pharmaceutical composition comprising a therapeutically effective amount of
a
compound of the present invention and a pharmaceutically acceptable excipient,
diluent or carrier.
A typical formulation is prepared by mixing a compound of the present
invention and a carrier, diluent or excipient. Suitable carriers, diluents and
excipients
26
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
are well known to those skilled in the art and include materials such as
carbohydrates,
waxes, water soluble and/or swellable polymers, hydrophilic or hydrophobic
materials, gelatin, oils, solvents, water, and the like. The particular
carrier, diluent or
excipient used will depend upon the means and purpose for which the compound
of
the present invention is being applied. Solvents are generally selected based
on
solvents recognized by persons skilled in the art as safe (GRAS) to be
administered to
a mammal. In general, safe solvents are non-toxic aqueous solvents such as
water and
other non-toxic solvents that are soluble or miscible in water. Suitable
aqueous
solvents include water, ethanol, propylene glycol, polyethylene glycols (e.g.,
PEG400,
PEG300), etc. and mixtures thereof. The formulations may also include one or
more
buffers, stabilizing agents, surfactants, wetting agents, lubricating agents,
emulsifiers,
suspending agents, preservatives, antioxidants, opaquing agents, glidants,
processing
aids, colorants, sweeteners, perfuming agents, flavoring agents and other
known
additives to provide an elegant presentation of the drug (i.e., a compound of
the
present invention or pharmaceutical composition thereof) or aid in the
manufacturing
of the pharmaceutical product (i.e., medicament).
The formulations may be prepared using conventional dissolution and mixing
procedures. For example, the bulk drug substance (i.e., compound of the
present
invention or stabilized form of the compound (e.g., complex with a
cyclodextrin
derivative or other known complexation agent)) is dissolved in a suitable
solvent in
the presence of one or more of the excipients. The compound of the present
invention
is typically formulated into pharmaceutical dosage forms to provide an easily
controllable dosage of the drug and to give the patient an elegant and easily
handleable product.
The pharmaceutical composition (or formulation) for application may be
packaged in a variety of ways depending upon the method used for administering
the
drug. Generally, an article for distribution includes a container having
deposited
therein the pharmaceutical formulation in an appropriate form. Suitable
containers are
well-known to those skilled in the art and include materials such as bottles
(plastic
and glass), sachets, ampoules, plastic bags, metal cylinders, and the like.
The
container may also include a tamper-proof assemblage to prevent indiscreet
access to
the contents of the package. In addition, the container has deposited thereon
a label
that describes the contents of the container. The label may also include
appropriate
warnings.
27
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
A Cyp17 inhibitor of the present invention may be usefully combined with at
least one additional pharmacologically active compound, particularly in the
treatment
of cancer. For example, a compound of the present invention, as defined above,
may
be administered simultaneously, sequentially or separately in combination with
one or
more agents selected from chemotherapy agents, e.g. mitotic inhibitors such as
a
taxane (e.g., paclitaxel or docetaxel), a vinca alkaloid (e.g., vincristine,
vinblastine,
vinorelbine or vinflunine) or other anticancer agents, e.g. cisplatin, 5-
fluorouracil or
5-fluoro-2-4(1H,3H)-pyrimidinedione (5FU), flutamide or gemcitabine. Such
combinations may offer significant advantages, including synergistic activity,
in
therapy.
A compound of the present invention may also be used in combination with
other antiproliferative compounds. Such antiproliferative compounds include,
but are
not limited to aromatase inhibitors; antiestrogens; topoisomerase I
inhibitors;
topoisomerase II inhibitors; microtubule active compounds; alkylating
compounds;
compounds which induce cell differentiation processes; cyclooxygenase
inhibitors;
MMP inhibitors; mTOR inhibitors; antineoplastic antimetabolites; platin
compounds;
compounds targeting/decreasing a protein or lipid kinase activity and further
anti-
angiogenic compounds; compounds which target, decrease or inhibit the activity
of a
protein or lipid phosphatase; gonadorelin agonists; anti-androgens; methionine
aminopeptidase inhibitors; bisphosphonates; biological response modifiers;
antiproliferative antibodies; heparanase inhibitors; inhibitors of Ras
oncogenic
isoforms; telomerase inhibitors; proteasome inhibitors; compounds used in the
treatment of hematologic malignancies; compounds which target, decrease or
inhibit
the activity of Flt-3; Hsp90 inhibitors such as 17-AAG (17-allylamino-gelda-
namycin,
NSC330507), 17-DMAG (17-dimethylaminoethylamino-17-demethoxy-
geldana-mycin, N5C707545), IPI-504, CNF1010, CNF2024, CNF1010 from
Conforma Therapeutics; temozolomide (TEMODAL); kinesin spindle protein
inhibitors, such as SB715992 or 5B743921 from GlaxoSmithKline, or
pentamidine/chlorpromazine from CombinatoRx; PI3K inhibitors; RAF inhibitors;
EDG binders, antileukemia compounds, ribonucleotide reductase inhibitors, S-
adenosylmethionine decarboxylase inhibitors, antiproliferative anti-bodies or
other
chemotherapeutic compounds. Further, alternatively or in addition they may be
used
in combination with other tumor treatment approaches, including surgery,
ionizing
radiation, photodynamic therapy, implants, e.g. with corticosteroids,
hormones, or
28
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
they may be used as radiosensitizers. Also, in anti-inflammatory and/or
antiproliferative treatment, combination with anti-inflammatory drugs is
included.
Combination is also possible with antihistamine drug substances,
bronchodilatatory
drugs, NSAID or antagonists of chemokine receptors.
The term "aromatase inhibitor" as used herein relates to a compound which
inhibits the estrogen production, i.e. the conversion of the substrates
androstenedione
and testosterone to estrone and estradiol, respectively. The term includes,
but is not
limited to steroids, especially atame-stane, exemestane and formestane and, in
part-icular, non-steroids, especially aminoglutethimide, roglethimide,
pyridoglutethimide, trilostane, testolactone, ketokonazole, vorozole,
fadrozole,
anastrozole and letrozole. Exemestane can be administered, e.g., in the form
as it is
marketed, e.g. under the trademark AROMASIN. Formestane can be administered,
e.g., in the form as it is marketed, e.g. under the trademark LENTARON.
Fadrozole
can be administered, e.g., in the form as it is marketed, e.g. un-der the
trademark
AFEMA. Anastrozole can be administered, e.g., in the form as it is marketed,
e.g.
under the trademark ARIMIDEX. Letrozole can be administered, e.g., in the form
as
it is marketed, e.g. under the trademark FEMARA or FEMAR. Amino glutethimide
can be administered, e.g., in the form as it is marketed, e.g. under the
trademark,
ORIMETEN. A combination of the invention comprising a chemo-therapeutic agent
which is an aromatase inhibitor is particularly useful for the treatment of
hormone
receptor positive tumors, e.g., breast tumors.
The term "anti-estrogen" as used herein relates to a compound which
antagonizes the effect of estrogens at the estrogen receptor level. The term
includes,
but is not limited to tamoxifen, fulvestrant, raloxifene and raloxifene
hydrochloride.
Tamoxifen can be administered, e.g., in the form as it is marketed, e.g. under
the
trademark NOLVADEX. Raloxifene hydrochloride can be administered, e.g., in the
form as it is marketed, e.g. under the trademark EVISTA. Fulvestrant can be
formulated as disclosed in US 4,659,516 or it can be administered, e.g., in
the form as
it is marketed, e.g. under the trademark FASLODEX. A combination of the
invention
comprising a chemotherapeutic agent which is an anti-estrogen is particularly
useful
for the treatment of estrogen receptor positive tumors, e.g. breast tumors.
The term "anti-androgen" as used herein relates to any substance which is
capable of in-hibiting the biological effects of androgenic hormones and
includes, but
29
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
is not limited to, bicalutamide (CASODEX), which can be formulated, e.g. as
disclosed in US 4,636,505.
The term "gonadorelin agonist" as used herein includes, but is not limited to
abarelix, goserelin and goserelin acetate. Goserelin is disclosed in US
4,100,274 and
can be administered, e.g., in the form as it is marketed, e.g. under the
trademark
ZOLADEX. Abarelix can be formulated, e.g. as disclosed in US 5,843,901.
The term "topoisomerase I inhibitor" as used herein includes, but is not
limited to topotecan, gimatecan, irinotecan, camptothecin and its analogues, 9-
nitrocamptothecin and the macromolecular camptothecin conjugate PNU-166148
(compound Al in W099/ 17804). Irinotecan can be administered, e.g. in the form
as
it is marketed, e.g. under the trademark CAMPTOSAR. Topotecan can be
administered, e.g., in the form as it is marketed, e.g. under the trademark
HYCAMTIN.
The term "topoisomerase II inhibitor" as used herein includes, but is not
limited to the anthracyclines such as doxorubicin (including liposomal
formulation,
e.g. CAELYX), daunorubicin, epirubicin, idarubicin and nemorubicin, the
anthraquinones mitoxantrone and losoxantrone, and the podophillotoxines
etoposide
and teniposide. Etoposide can be administered, e.g. in the form as it is
marketed, e.g.
under the trademark ETOPOPHOS. Teniposide can be administered, e.g. in the
form
as it is marketed, e.g. under the trademark VM 26-BRISTOL. Doxorubicin can be
administered, e.g. in the form as it is marketed, e.g. under the trademark
ADRIBLASTIN or ADRIAMYCIN. Epirubicin can be administered, e.g. in the form
as it is marketed, e.g. under the trademark FARMORUBICIN. Idarubicin can be
administered, e.g. in the form as it is marketed, e.g. under the trademark
ZAVEDOS.
Mitoxantrone can be administered, e.g. in the form as it is marketed, e.g.
under the
trademark NOVANTRON.
The term "microtubule active compound" relates to microtubule stabilizing,
microtubule destabilizing compounds and microtublin polymerization inhibitors
including, but not limited to taxanes, e.g. paclitaxel and docetaxel, vinca
alkaloids,
e.g., vinblastine, especially vinblastine sulfate, vincristine especially
vincristine
sulfate, and vinorelbine, discodermolides, cochicine and epothilones and
derivatives
thereof, e.g. epothilone B or D or derivatives thereof. Paclitaxel may be
administered
e.g. in the form as it is marketed, e.g. TAXOL. Docetaxel can be administered,
e.g., in
the form as it is marketed, e.g. under the trademark TAXOTERE. Vinblastine
sulfate
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
can be administered, e.g., in the form as it is marketed, e.g. under the
trademark
VINBLASTIN R.P. Vincristine sulfate can be administered, e.g., in the form as
it is
marketed, e.g. under the trademark FARMISTIN. Discodermolide can be obtained,
e.g., as disclosed in US 5,010,099. Also included are Epothilone derivatives
which
are disclosed in WO 98/10121, US 6,194,181, WO 98/25929, WO 98/08849, WO
99/43653, WO 98/22461 and WO 00/31247. Especially preferred are Epothilone A
and/or B.
The term "alkylating compound" as used herein includes, but is not limited to,
cyclophosphamide, ifosfamide, melphalan or nitrosourea (BCNU or Gliadel).
Cyclophosphamide can be administered, e.g., in the form as it is marketed,
e.g. under
the trademark CYCLOSTIN. Ifosfamide can be administered, e.g., in the form as
it is
marketed, e.g., under the trademark HOLOXAN.
The term "antineoplastic antimetabolite" includes, but is not limited to, 5-
Fluorouracil or 5-FU, capecitabine, gemcitabine, DNA demethylating compounds,
such as 5-azacy-ti-dine and decitabine, methotrexate and edatrexate, and folic
acid
antagonists such as pemetrexed. Capecitabine can be administe-red, e.g., in
the form
as it is marketed, e.g. under the trademark XELODA. Gemcitabine can be
administered, e.g., in the form as it is marketed, e.g. under the trademark
GEMZAR.
The term "platin compound" as used herein includes, but is not limited to,
carboplatin,
cis-platin, cisplatinum and oxaliplatin. Carboplatin can be administered,
e.g., in the
form as it is marketed, e.g. under the trademark CARBOPLAT. Oxaliplatin can be
administered, e.g., in the form as it is marketed, e.g. under the trademark
ELOXATIN.
The term "compounds targeting/decreasing a protein or lipid kinase activity";
or a "protein or lipid phosphatase activity"; or "further anti-angiogenic
compounds"
as used herein includes, but is not limited to, protein tyrosine kinase and/or
serine
and/or threonine kinase inhibitors or lipid kinase inhibitors, e.g., a)
compounds
targeting, decreasing or inhibiting the activity of the platelet-derived
growth factor-
receptors (PDGFR), such as compounds which target, decrease or inhibit the
activity
of PDGFR, especially compounds which inhibit the PDGF receptor, e.g. a N-
pheny1-
2-pyrimidine-amine derivative, e.g. imatinib, SU101, 5U6668 and GFB-111; b)
compounds targeting, decreasing or inhibiting the activity of the fibroblast
growth
factor-receptors (FGFR); c) compounds targeting, decreasing or inhibiting the
activity of the insulin-like growth factor receptor I (IGF-IR), such as
compounds
31
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
which target, decrease or inhibit the activity of IGF-IR, especially compounds
which
inhibit the kinase activity of IGF-I receptor, such as those compounds
disclosed in
WO 02/092599, or antibodies that target the extracellular domain of IGF-I
receptor or
its growth factors; d) compounds targeting, decreasing or inhibiting the
activity of the
Trk receptor tyrosine kinase family, or ephrin B4 inhibitors; e) compounds
targeting,
decreasing or inhibiting the activity of the Axl receptor tyrosine kinase
family; 0
compounds targeting, decreasing or inhibiting the activity of the Ret receptor
tyrosine
kinase; g) compounds targeting, decreasing or inhibiting the activity of the
Kit/SCFR
receptor tyrosine kinase, i.e C-kit receptor tyrosine kinases - (part of the
PDGFR
family), such as compounds which target, decrease or inhibit the activity of
the c-Kit
receptor tyrosine kinase family, especially compounds which inhibit the c-Kit
receptor, e.g. imatinib; h) compounds targeting, decreasing or inhibiting the
activity
of members of the c-Abl family, their gene-fusion products (e.g. BCR-Abl
kinase)
and mutants, such as com-pounds which target decrease or inhibit the activity
of c-
AbI family members and their gene fusion products, e.g. a N-pheny1-2-
pyrimidine-
amine derivative, e.g. imatinib or nilotinib (AM1N107); PD180970; AG957; NSC
680410; PD173955 from ParkeDavis; or dasatinib (BMS-354825); i) compounds
targeting, decreasing or inhibiting the activity of members of the protein
kinase C
(PKC) and Raf family of serine/threonine kinases, members of the MEK, SRC,
JAK,
FAK, PDK1, PKB/Akt, and Ras/MAPK family members, and/or members of the
cyclin-dependent kinase family (CDK) and are especially those staurosporine
derivatives disclosed in US 5,093,330, e.g. midostaurin; examples of further
compounds include e.g. UCN-01, safingol, BAY 43-9006, Bryostatin 1,
Perifosine;
Ilmofosine; RO 318220 and RO 320432; GO 6976; Isis 3521; LY333531/LY379196;
isochinoline compounds such as those disclosed in WO 00/09495; FTIs; BEZ235 (a
P13K inhibitor) or AT7519 (CDK inhibitor); j) compounds targeting, decreasing
or
inhibiting the activity of protein-tyrosine kinase inhibitors, such as
compounds which
target, decrease or inhibit the activity of protein-tyrosine kinase inhibitors
include
imatinib mesylate (GLEEVEC) or tyrphostin. A tyrphostin is preferably a low
molecular weight (mw<1500) compound, or a pharmaceutically acceptable salt
thereof, especially a compound selected from the benzylidenemalonitrile class
or the
S-arylbenzenemalonirile or bisubstrate quinoline class of compounds, more
especially
any compound selected from the group consisting of Tyrphostin A23/RG-50810; AG
99; Tyrphostin AG 213; Tyrphostin AG 1748; Tyrphostin AG 490; Tyrphostin B44;
32
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
Tyrphostin B44 (+) enantiomer; Tyrphostin AG 555; AG 494; Tyrphostin AG 556,
AG957 and adaphostin (4- {[(2,5-dihydroxyphenyl)methyl]amino}-benzoic acid
adamantyl ester; NSC 680410, adaphostin); k) compounds targeting, decreasing
or
inhibiting the activity of the epidermal growth factor family of receptor
tyrosine
kinases (EGFR, ErbB2, ErbB3, ErbB4 as homo- or heterodimers) and their
mutants,
such as compounds which target, decrease or inhibit the activity of the
epidermal
growth factor receptor family are especially compounds, proteins or antibodies
which
inhibit members of the EGF receptor tyrosine kinase family, e.g. EGF receptor,
ErbB2, ErbB3 and ErbB4 or bind to EGF or EGF related ligands, and are in
particular
those compounds, proteins or monoclonal antibodies generically and
specifically
disclosed in WO 97/02266, e.g. the compound of ex. 39, or in EP 0 564 409, WO
99/03854, EP 0520722, EP 0 566 226, EP 0 787 722, EP 0 837 063, US 5,747,498,
WO 98/10767, WO 97/30034, WO 97/49688, WO 97/38983 and, especially, WO
96/30347 (e.g. compound known as CP 358774), WO 96/33980 (e.g. compound ZD
1839) and WO 95/03283 (e.g. compound ZM105180); e.g. trastuzumab (Herceptin),
cetuximab (Erbitux), Iressa, Tarceva, OSI-774, CI-1033, EKB-569, GW-2016,
E1.1,
E2.4, E2.5, E6.2, E6.4, E2.11, E6.3 or E7.6.3, and 7H-pyrrolo-[2,3-
d]pyrimidine
derivatives which are disclosed in WO 03/013541; and 1) compounds targeting,
decreasing or inhibiting the activity of the c-Met receptor, such as compounds
which
target, decrease or inhibit the activity of c-Met, especially compounds which
inhibit
the kinase activity of c-Met receptor, or antibodies that target the
extracellular domain
of c-Met or bind to HGF.
Further anti-angiogenic compounds include compounds having another
mechanism for their activity, e.g. unrelated to protein or lipid kinase
inhibition e.g.
thalidomide (THALOMID) and TNP-470.
Compounds which target, decrease or inhibit the activity of a protein or lipid
phosphatase are e.g., inhibitors of phosphatase 1, phosphatase 2A, or CDC25,
e.g.
okadaic acid or a derivative thereof.
Compounds which induce cell differentiation processes are e.g. retinoic acid,
or tocopherol or tocotrienol.
The term cyclooxygenase inhibitor as used herein includes, but is not limited
to, e.g. Cox-2 inhibitors, 5-alkyl substituted 2-arylaminophenylacetic acid
and
derivatives, such as celecoxib (CELEBREX), rofecoxib (VIOXX), etoricoxib,
33
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
valdecoxib or a 5-alkyl-2-arylaminophenylacetic acid, e.g. 5-methy1-2-(2'-
chloro-6'-
fluoroanilino)phenyl acetic acid, lumiracoxib.
The term "bisphosphonates" as used herein includes, but is not limited to,
etridonic, clodronic, tiludronic, pamidronic, alendronic, ibandronic,
risedronic and
zoledronic acid. "Etridonic acid" can be administered, e.g., in the form as it
is
marketed, e.g. under the trademark DIDRONEL. "Clodronic acid" can be
administered, e.g., in the form as it is marketed, e.g. under the trademark
BONEFOS.
"Tiludronic acid" can be administered, e.g., in the form as it is marketed,
e.g. under
the trademark SKELID. "Pamidronic acid" can be administered, e.g. in the form
as it
is marketed, e.g. under the trademark AREDIA. "Alendronic acid" can be
administered, e.g., in the form as it is marketed, e.g. under the trademark
FOSAMAX.
"Ibandronic acid" can be administered, e.g., in the form as it is marketed,
e.g. under
the trademark BONDRANAT. "Risedronic acid" can be administered, e.g., in the
form as it is marketed, e.g. under the trademark ACTONEL. "Zoledronic acid"
can
be administered, e.g. in the form as it is marketed, e.g. under the trademark
ZOMETA.
The term "mTOR inhibitors" relates to compounds which inhibit the
mammalian target of rapamycin (mTOR) and which possess antiproliferative
activity
such as sirolimus (Rapamune), everolimus (CerticanO), CCI-779 and ABT578.
The term "heparanase inhibitor" as used herein refers to compounds which
target, decrease or inhibit heparin sulfate degradation. The term includes,
but is not
limited to, PI-88.
The term "biological response modifier" as used herein refers to a lymphokine
or interferons, e.g. interferon.
The term "inhibitor of Ras oncogenic isoforms", e.g. H-Ras, K-Ras, or N-Ras,
as used herein refers to compounds which target, decrease or inhibit the
oncogenic
activity of Ras e.g. a "farnesyl transferase inhibitor" e.g. L-744832, DK8G557
or
R115777 (Zarnestra).
The term "telomerase inhibitor" as used herein refers to compounds which
target, decrease or inhibit the activity of telomerase. Compounds which
target,
decrease or inhibit the activity of telomerase are especially compounds which
inhibit
the telomerase receptor, e.g. telomestatin.
The term "methionine aminopeptidase inhibitor" as used herein refers to
compounds which target, decrease or inhibit the activity of methionine
34
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
aminopeptidase. Compounds which target, decrease or inhibit the activity of
methionine aminopeptidase are e.g. bengamide or a derivative thereof.
The term "proteasome inhibitor" as used herein refers to compounds which
target, decrease or inhibit the activity of the proteasome. Compounds which
target,
decrease or inhibit the activity of the proteasome include e.g. Bortezomid
(Velcade)
and MLN 341.
The term "matrix metalloproteinase inhibitor" or ("MMP" inhibitor) as used
herein
includes, but is not limited to, collagen peptidomimetic and nonpeptidomimetic
inhibitors, tetracycline derivatives, e.g. hydroxamate peptidomimetic
inhibitor
batimastat and its orally bioavailable analogue marimastat (BB-2516),
prinomastat
(AG3340), metastat (NSC 683551) BMS-279251, BAY 12-9566, TAA211,
MMI270B or AAJ996.
The term "compounds used in the treatment of hematologic malignancies" as used
herein includes, but is not limited to, FMS-like tyrosine kinase inhibitors
e.g.
compounds targeting, decreasing or inhibiting the activity of FMS-like
tyrosine kinase
receptors (Flt-3R); interferon, 1-b-D-arabinofuransylcytosine (ara-c) and
bisulfan; and
ALK inhibitors e.g. compounds which target, decrease or inhibit anaplastic
lymphoma
kinase.
Compounds which target, decrease or inhibit the activity of FMS-like tyrosine
kinase
receptors (Flt-3R) are especially compounds, proteins or antibodies which
inhibit
members of the Flt-3R receptor kinase family, e.g. PKC412, TKI258,
midostaurin, a
staurosporine derivative, SU11248 and MLN518.
The term "HSP90 inhibitors" as used herein includes, but is not limited to,
compounds targeting, decreasing or inhibiting the intrinsic ATPase activity of
HSP90;
degrading, targeting, decreasing or inhibiting the HSP90 client proteins via
the
ubiquitin proteosome pathway. Compounds targeting, decreasing or inhibiting
the
intrinsic ATPase activity of HSP90 are especially compounds, proteins or
antibodies
which inhibit the ATPase activity of HSP90 e.g., 17-allylamino,17-
demethoxygeldanamycin (17AAG), a geldanamycin derivative; other geldanamycin
related compounds; radicicol and HDAC inhibitors. An example HSP90 inhibitor
is
AUY922.
The term "antiproliferative antibodies" as used herein includes, but is not
limited to, trastuzumab (Herceptin), Trastuzumab-DM1,erbitux, bevacizumab
(Avastin), rituximab (Rituxan), PR064553 (anti-CD40) and 2C4 Antibody. By
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
antibodies is meant e.g. intact monoclonal antibodies, polyclonal antibodies,
multispe-cific antibodies formed from at least 2 intact antibodies, and
antibodies
fragments so long as they exhibit the desired biological activity.
For the treatment of acute myeloid leukemia (AML), compounds of formula
(I) can be used in combination with standard leukemia therapies, especially in
combination with therapies used for the treatment of AML. In particular,
compounds
of formula (I) can be administered in combination with, e.g., farnesyl
transferase
inhibitors and/or other drugs useful for the treatment of AML, such as
Daunorubicin,
Adriamycin, Ara-C, VP-16, Teniposide, Mitoxantrone, Idarubicin, Carboplatinum
and
PKC412.
The term "antileukemic compounds" includes, for example, Ara-C, a
pyrimidine analog, which is the 2-alpha-hydroxy ribose (arabinoside)
derivative of
deoxycytidine. Also included is the purine analog of hypoxanthine, 6-
mercaptopurine
(6-MP) and fludarabine phosphate.
Somatostatin receptor antagonists as used herein refers to compounds which
target, treat or inhibit the somatostatin receptor such as octreotide, and
S0M230
(pasireotide).
Tumor cell damaging approaches refer to approaches such as ionizing
radiation. The term "ionizing radiation" referred to above and hereinafter
means
ionizing radiation that occurs as either electromagnetic rays (such as X-rays
and
gamma rays) or particles (such as alpha and beta particles). Ionizing
radiation is
provided in, but not limited to, radiation therapy and is known in the art.
See
Hellman, Principles of Radiation Therapy, Cancer, in Principles and Practice
of
Oncology, Devita et al., Eds., 4th Edition, Vol. 1, pp. 248-275 (1993).
The term "EDG binders" as used herein refers a class of immunosuppressants
that modulates lymphocyte recirculation, such as FTY720.
The term "ribonucleotide reductase inhibitors" refers to pyrimidine or purine
nucleoside analogs including, but not limited to, fludarabine and/or cytosine
arabinoside (ara-C), 6-thioguanine, 5-fluorouracil, cladribine, 6-
mercaptopurine
(especially in combination with ara-C against ALL) and/or pentostatin.
Ribonucleotide reductase inhibitors are especially hydroxyurea or 2-hydroxy-1H-
isoindole-1,3-dione derivatives, such as PL-1, PL-2, PL-3, PL-4, PL-5, PL-6,
PL-7 or
PL-8 mentioned in Nandy et al., Acta Oncologica, Vol. 33, No. 8, pp. 953-961
(1994).
36
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
The term "S-adenosylmethionine decarboxylase inhibitors" as used herein
includes, but is not limited to the compounds disclosed in US 5,461,076.
Also included are in particular those compounds, proteins or monoclonal
antibodies of VEGF disclosed in WO 98/35958, e.g. 1-(4-chloroanilino)-4-(4-
pyridylmethyl)phthalazine or a pharmaceutically acceptable salt thereof, e.g.
the
succinate, or in WO 00/09495, WO 00/27820, WO 00/59509, WO 98/11223, WO
00/27819 and EP 0 769 947; those as described by Prewett et al, Cancer Res,
Vol. 59,
pp. 5209-5218 (1999); Yuan et al., Proc Natl Acad Sci USA, Vol. 93, pp. 14765-
14770 (1996); Zhu et al., Cancer Res, Vol. 58, pp. 3209-3214 (1998); and
Mordenti et
al., Toxicol Pathol, Vol. 27, No. 1, pp. 14-21 (1999); in WO 00/37502 and WO
94/10202; ANGIOSTATIN, described by O'Reilly et al., Cell, Vol. 79, pp. 315-
328
(1994); ENDOSTATIN, described by O'Reilly et al., Cell, Vol. 88, pp. 277-285
(1997); anthranilic acid amides; ZD4190; ZD6474; 5U5416; 5U6668; bevacizumab;
or anti-VEGF antibodies or anti-VEGF receptor antibodies, e.g. rhuMAb and
RHUFab, VEGF aptamer e.g. Macugon; FLT-4 inhibitors, FLT-3 inhibitors, VEGFR-
2 IgG1 antibody, Angiozyme (RPI 4610) and Bevacizumab (Avastin).
Photodynamic therapy as used herein refers to therapy which uses certain
chemicals known as photosensitizing compounds to treat or prevent cancers.
Examples of photodynamic therapy includes treatment with compounds, such as
e.g.
VISUDYNE and porfimer sodium.
Angiostatic steroids as used herein refers to compounds which block or inhibit
angiogenesis, such as, e.g., anecortave, triamcinolone, hydrocortisone, 11--
epihydrocotisol, cortexolone, 17-hydroxyprogesterone, corticosterone,
desoxycorticosterone, testosterone, estrone and dexamethasone.
Implants containing corticosteroids refers to compounds, such as e.g.
fluocinolone,
dexamethasone.
"Other chemotherapeutic compounds" include, but are not limited to, plant
alkaloids, hormonal compounds and antagonists; biological response modifiers,
preferably lymphokines or interferons; antisense oligonucleotides or
oligonucleotide
derivatives; shRNA or siRNA; or miscellaneous compounds or compounds with
other
or unknown mechanism of action.
The structure of the active compounds identified by code nos., generic or
trade
names may be taken from the actual edition of the standard compendium "The
Merck
Index" or from databases, e.g. Patents International (e.g. IMS World
Publications).
37
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
EXAMPLES
The following abbreviations used in the examples below have the
corresponding meanings:
DBU 1,8-Diazabicyclo[5.4.0]undec-7-ene
DIPA Diisopropylamine
DPPA Diphenylphosphoryl Azide
DCM Dichloromethane
DCE Dichloroethane
DMA N,N-dimethylacetamide
DMF N,N-Dimethylformamide
DMSO Dimethylsulfoxide
TEA Triethylamine
THF Tetrahydrofuran
NaBH(OAc)3 Sodium triacetoxy borohydride
Pd2(dba)3 Tris(dibenzylideneacetone)dipalladium(0)
Pd(PPh3)4 Tetrakis (triphenylphosphine)Palladium(0)
PTSA Para-toluene sulphonic acid
TES Triethyl silane
LDA Lithium Diisopropyl amide
LiHMDS Lithium bis(trimethylsilyl)amide
Xantphos 4,5-Bis(diphenylphosphino)-9,9-dimethylxanthene
TLC Thin Layer Chromatography
NMR Nuclear Magnetic Resonance
LCMS Liquid chromatography Mass spectrometry
HPLC High Performance Liquid Chromatography
Benzyl 3-oxo-2,4-diazabicyclo[3.1.0]hexane-2-carboxylate is available from
Combi-Blocks, Inc. (USA), Maybridge (United Kingdom), or Interchim (France).
38
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
Example 1
Preparation of 1-(2-chloropyridin-4-y1)-3-(4-cyclopropylpyridin-3-
yl)imidazolidin-2-
one (IA):
CI
)---aN N--..---)
0 N
(IA)
Preparation of Intermediate of 4-cyclopropy1-3-nitropyridine (I-la):
02N
/ \iN
(I-la)
4-Chloro-3-nitropyridine (100mg, 0.630mmol) and cyclopropyl boronic acid
(10.0 mg, 0.091mmol) were added to a solution of xylene (3mL) previously
purged
with argon (10 minutes). The reaction mixture was purged with argon for a
further 15
minutes, followed by the addition of potassium carbonate (174.35mg, 1.26mmol)
and
Pd(PPh3)4 (34.5mg, 0.063mmol). The resulting mixture was heated to reflux at
130 C
overnight. The reaction was monitored by TLC (30% ethylacetate in hexane). The
reaction mixture was cooled and concentrated to afford the crude product.
Purification
by column chromatography on silica gel (15% ethyl acetate in hexane) afforded
110mg of the product (100% yield). LCMS Purity: 99 %, m/z = 165 (M+1)
Preparation of Intermediate 4-cyclopropylpyridin-3-amine (I-lb):
a'A
N
NH2
(I-lb)
Zinc powder (223.2mg, 3.41mmol) and ammonium chloride solution (365mg,
6.8mmol) were added to a stirred solution of 4-cyclopropy1-3-nitropyridine (I-
la:
70mg, 0.426mmo1) in dry THF (2mL) at 0 C and the resulting mixture was stirred
at
room temperature for 1 hour. The reaction was monitored by TLC (10% methanol
in
CHC13). The reaction mixture was filtered over celite bed and washed with THF
and
39
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
filtrate was concentrated. The residue was dissolved in ethyl acetate and
washed with
water and saturated brine and dried over sodium sulphate and concentrated to
afford
450mg of the product (100% yield).
1H NMR (CDC13, 300 MHz): 6 8.10 (s, 1H), 7.95 (d, 1H), 6.90 (d, 1H), 4.1
(bs, 2H), 1.75-1.60 (m, 1H), 1.10-0.95 (q, 2H), 0.70-0.60 (q, 2H). LCMS
Purity: 85
%, m/z=135.1 (M+1)
Preparation of Intermediate 1-(2-chloroethyl)-3-(4-cyclopropylpyridin-3-y1)
urea (I-
1c):
0
)&
I H
N
(I-1c)
1-Chloro-2-isocyanatoethane (472mg, 4.477mmo1) was added drop wise to a
stirred mixture of 4-cyclopropylpyridin-3-amine (I-lb: 400mg, 2.78mmol) in
toluene
(10mL) at 0 C. The resulting mixture was stirred at room temperature for 2
hours.
The reaction was monitored by TLC (10% methanol in CHC13). The crude product
was concentrated and purified by column chromatography on silica gel (2%
methanol
in CHC13) afforded 200mg of the product (28% yield).
1H NMR (DMSO-D6, 400 MHz): 6 8.84 (s, 1H), 8.14 (s, 1H), 8.08-8.07 (d,
1H), 6.97-6.94 (t, 1H), 6.88-6.87 (d, 1H), 3.69-3.66 (t, 2H), 3.46-3.42 (dd,
2H), 1.92-
1.86 (m, 1H), 1.06-1.01 (q, 2H), 0.73-0.69 (q ,2H). LCMS Purity: 97%,
m/z=240.1
(M+1)
Preparation of Intermediate 1-(4-cyclopropylpyridin-3-y1) imidazolidin-2-one
(I-1d):
N
I ; Nr---2z N H
n
0
(I-1d)
1-(2-Chloroethyl)-3-(4-cyclopropylpyridin-3-yl)urea (I-lc: 200mg,
0.836mmo1) in dry THF was added drop wise to a stirred mixture of NaH
(40.16mg,
1.67mmol) in dry THF (5mL) at 0 C over a period of 10 minutes. The resulting
mixture was stirred at room temperature for 2 hours. The reaction was
monitored by
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
TLC (10% methanol in CHC13). The reaction mixture was quenched with methanol
and concentrated to afford the crude product. Purification by column
chromatography
on silica gel (2% methanol in CHC13) afforded 140mg of the product (94%
yield).
1H NMR (DMSO-D6, 300 MHz): 6 8.31-8.27 (m, 2H), 6.88-6.83 (m, 2H),
3.81-3.76 (t, 2H), 3.48-3.46 (t, 2H), 2.10-1.95 (m, 1H), 1.07-1.04 (q, 2H),
0.79-0.76
(q, 2H). LCMS Purity: 97 % m/z=204.1 (M+1)
Preparation of the title compound 1-(2-chloropyridin-4-yl)-3-(4-
cyclopropylpyridin-
3-yl) imidazolidin-2-one (1A):
Copper iodide (6.5mg, 0.034mmol), trans-N,N'-dimethylcyclohexane-1,2-
diamine (4.8 mg, 0.034mmol) and potassium phosphate (219.3mg, 1.034mmol) were
added to a solution of 1, 4-dioxane (5mL) previously purged with nitrogen (10
minutes). The reaction mixture was purged with argon for 10 minutes, followed
by
the addition of 1-(4-cyclopropylpyridin-3-y1) imidazolidin-2-one (I-ld: 70mg,
0.344mmo1) and 2-chloro-4-iodopyridine (99.09mg, 0.413mmol). The reaction
mixture was heated to reflux at 120 C overnight. The reaction was monitored by
TLC
(10% Me0H in chloroform). The reaction mixture was cooled and partitioned
between ethyl acetate and water. The organic layer was concentrated to yield
the
crude product. Purification by column chromatography on silica gel (2%
methanol in
CHC13) afforded 42 mg of the product (41% yield).
1H NMR (CDC13, 400 MHz): 6 8.70-8.40 (m, 2H), 8.28-8.27 (d, 1H), 7.61-
7.59 (dd, 1H), 7.53 (s, 1H), 6.90 (bs, 1H), 4.09-4.02 (m, 4H), 2.0-1.96 (m,
1H), 1.17-
1.12 (m, 2H), 0.87-0.83 (m, 2H). LCMS Purity: 99%, m/z=315 (M+1). HPLC
Purity: 95%
Example 2
Synthesis of 1-(4-cyclopropylpyridin-3-yl)-3-(2-(trifluoromethyl) pyridin-4-
yl)
imidazolidin-2-one (2A):
41
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
CF3 i---\
N / NN
/ \
0 N
(2A)
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-id: 70mg, 0.344mmo1)
was reacted with 4-bromo-2-(trifluoromethyl)pyridine (93.5mg, 0.413mmol) 1, 4-
dioxane (5mL), copper iodide (6.5mg, 0.034mmol), trans-N,N' -
dimethylcyclohexane-
1,2-diamine (4.8mg, 0.034mmol) and potassium phosphate (219mg, 1.034mmol) to
afford the crude product. Purification by preparative HPLC afforded 40mg of
the
product (33% yield).
1H NMR (CDC13, 400 MHz): 6 8.70-8.40 (m, 2H), 8.28-8.27 (d, 1H), 7.61-
7.59 (dd, 1H), 7.53 (s, 1H), 6.90 (bs, 1H), 4.16-4.04 (m, 4H), 2.2-1.95 (m,
1H), 1.17-
1.12 (m, 2H), 0.90-0.84 (m, 2H). LCMS Purity: 97%, m/z = 349.1 (M+1). HPLC
Purity: 94%
Example 3
Preparation of 1-(4-cyclopropylpyridin-3-y1)-3-(naphthalen-2-y1) imidazolidin-
2-one
(3A):
1--\
4111111* NN;
0 N
(3A)
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-id: 70mg, 0.344mmo1)
was reacted with 2-bromonaphthalene (85.68mg, 0.413mmol), 1, 4-dioxane (5mL),
copper iodide (6.5mg, 0.034mmol), trans-N,N'-dimethylcyclohexane-1,2-diamine
(4.8mg, 0.034mmol) and potassium phosphate (219mg, 1.034mmol) to afford the
crude product. Purification by column chromatography on silica gel (2%
methanol in
CHC13), followed by preparative HPLC afforded 13mg of the product (12% yield).
1H NMR (CDC13, 400 MHz): 6 8.70-8.30 (m, 2H), 8.15-8.12 (dd, 1H), 7.87 (s,
1H), 7.84-7.79 (m, 2H), 7.73 (s, 1H), 7.47-7.45 (t, 1H), 7.42-7.40 (t, 1H),
7.0-6.70 (m,
42
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
1H), 4.23-4.19 (t, 2H), 4.06-4.02 (t, 2H), 2.15-2.05 (m, 1H), 1.15-1.13 (m,
2H), 0.85-
0.84 (m, 2H). LCMS Purity: 100%, m/z= 330.2 (M+1). HPLC Purity: 98%
Example 4
Preparation of 1-(benzo[b]thiophen-5-yl)-3-(4-cyclopropylpyridin-3-yl)
imidazolidin-
2-one (4A):
1-----\
/ fit NyN-----$
S 0 ----N
(4A)
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-id: 70mg, 0.344mmo1)
was reacted with 5-bromobenzo[b]thiophene (88.1mg, 0.413mmol) 1, 4-dioxane
(5mL), copper iodide (6.5mg, 0.034mmol), trans-N,N'-dimethylcyclohexane-1,2-
diamine (4.8mg, 0.034mmol) and potassium phosphate (219mg, 1.034mmol) to
afford
the crude product. Purification by preparative HPLC afforded 15mg of the
product
(13% yield).
1H NMR (CDC13, 400 MHz): 6 8.52 (s, 1H), 8.41 (d, 1H), 7.99-7.98 (d, 1H),
7.87-7.85 (d, 1H), 7.76-7.74 (dd, 1H), 7.47-7.46 (d, 1H), 7.32-7.31 (d, 1H),
6.82-6.80
(d, 1H), 4.17-4.14 (t, 2H), 4.03-3.99 (t, 2H), 2.15-2.05 (m, 1H), 1.15-1.11
(m, 2H),
0.86-0.82 (m, 2H). LCMS Purity: 95%, m/z = 336.1 (M+1). HPLC Purity: 98%
Example 5
Preparation of 1-(4-cyclopropylpyridin-3-yl)-3-(3-(trifluoromethyl) phenyl)
imidazolidin-2-one (5A):
CF3 /-1
ONN ......--)
0 ---N
(5A)
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-id: 150mg,
0.738mmol)
was reacted with 1-bromo-3-(trifluoromethyl)benzene (831mg, 3.69mmol), 1, 4-
43
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
dioxane (10mL), copper iodide (14.0mg, 0.073mmol), trans-N,N' -
dimethylcyclohexane-1,2-diamine (8.4mg, 0.073mmo1) and potassium phosphate
(469.9mg, 2.21mmol) to afford the crude product. Purification by column
chromatography on silica gel (1.5% methanol in CHC13) afforded 205mg of the
product (80% yield).
1H NMR (CDC13, 400MHz): 6 8.70-8.40 (m, 2H), 7.90-7.84 (t, 2H), 7.51-7.47
(t, 1H), 7.35-7.27 (t, 1H), 6.85 (bs, 1H), 4.13-4.08 (t, 2H), 4.03-3.99 (t,
2H), 2.07-2.03
(m, 1H), 1.16-1.11 (m, 2H), 0.86-0.82(m, 2H). LCMS Purity: 94%, m/z = 348.1
(M+1). HPLC Purity: 96%
Example 6
Preparation of 1-(benzo[b]thiophen-2-y1)-3-(4-cyclopropylpyridin-3-y1)
imidazolidin-
2-one (6A);
1----\
=N N).(N......)
S 0 N
(6A)
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-id: 150mg,
0.738mmol)
was reacted with 2-bromobenzo[b]thiophene (188mg, 0.886mmo1), 1, 4-dioxane
(6mL), copper iodide (14.0mg, 0.0739mmol), trans-N,N' -dimethylcyclohexane-1,2-
diamine (10.5mg, 0.0739mmo1) and potassium phosphate (470mg, 2.21mmol) to
afford the crude product. Purification by column chromatography on silica gel
(0.5%
methanol in CHC13) afforded 85mg of the product (34% yield).
1H NMR (CDC13, 400MHz): 6 8.60-8.30 (m, 2H), 7.74-7.72 (d, 1H), 7.63-7.60
(d, 1H), 7.33-7.19 (m, 2H), 6.82 (s, 1H), 6.56 (s, 1H), 4.20-4.04 (m, 4H),
2.10-2.0 (m,
1H), 1.16-1.10 (m, 2H), 0.85-0.80 (m, 2H). LCMS Purity: 90%, m/z = 336.1
(M+1).
HPLC Purity: 95%
Example 7
Preparation of 1-(6-chloro-2-methylpyrimidin-4-y1)-3-(4-cyclopropylpyridin-3-
y1)
imidazolidin-2-one (7A):
44
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
CI /--\
)r--.....-NNN,...--)
N r N n
0 N
H3C
(7A)
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-id: 100mg,
0.492mmo1)
was reacted with 4,6-dichloro-2-methylpyrimidine (80.2mg, 0.492mmo1), 1, 4-
dioxane (5mL), copper iodide (9.3mg, 0.049mmol), trans-N,N' -
dimethylcyclohexane-
1,2-diamine (6.9mg, 0.049mmol) and potassium phosphate (313mg, 1.47mmol) to
afford the crude product. Purification by column chromatography on silica gel
(1%
methanol in CHC13) afforded 25mg of the product (15% yield).
1H NMR (CDC13, 400 MHz): 6 8.55-8.40 (m, 2H), 8.16 (s, 1H), 6.84 (s, 1H),
4.30-4.26 (q, 2H), 3.99-3.95 (q, 2H), 2.62 (s, 3H), 1.99-1.94 (m, 1H), 1.16-
1.11 (m,
2H), 0.86-0.81 (m, 2H). LCMS Purity: 97%, m/z = 330.1(M+1). HPLC Purity: 98%
Example 8
Preparation of 1-(4-cyclopropylpyridin-3-y1)-3-(4-(trifluoromethyl) pyridin-2-
y1)
imidazolidin-2-one (8A):
F 3C 1--\
0.--Ny N;
(8A)
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-id: 150mg,
0.738mmol)
was reacted with 2-bromo-4-(trifluoromethyl)pyridine (200.3mg, 0.88mmol), 1, 4-
dioxane (5mL), copper iodide (14.03mg, 0.073mmol), trans-N,N' -
dimethylcyclohexane-1,2-diamine (10.49mg, 0.073mmol) and potassium phosphate
(469.9mg, 2.21mmol) to afford the crude product. Purification by column
chromatography on silica gel (1% methanol in CHC13) afforded 182.5mg of the
product (71% yield).
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
1H NMR (CDC13, 300MHz): 6 8.64 (s, 1H), 8.48-8.46 (d, 2H), 7.17-7.15 (d,
2H), 6.95-6.75 (bs, 1H), 4.34-4.29 (t, 2H), 4.01-3.96 (t, 2H), 2.04-1.99 (m,
1H), 1.17-
1.10 (m, 2H), 0.86-0.81 (m, 2H). LCMS Purity: 98%, m/z = 349.0(M+1). HPLC
Purity: 98%
Example 9
Preparation of 1-(4-cyclopropylpyridin-3-y1)-3-(2-methoxypyridin-4-y1)
imidazolidin-
2-one (9A):
H3C-Cb.....,PN /
0
(9A)
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-id: 100mg,
0.492mmo1)
was reacted with 4-bromo-2-methoxypyridine (111.1mg, 0.591mmol), 1, 4-dioxane
(5mL), copper iodide (9.39mg, 0.049mmol), trans-N,N'-dimethylcyclohexane-1,2-
diamine (6.98mg, 0.049mmol) and potassium phosphate (312.9mg, 1.476mmo1) to
afford the crude product. Purification by column chromatography on silica gel
(1%
methanol in CHC13) afforded 80mg of the product (52% yield).
1H NMR (CDC13,300 MHz): 6 8.70-8.25 (m, 2H), 8.08-8.06 (d, 1H), 7.46-7.44
(dd, 1H), 6.90-6.80 (bs, 1H), 6.73 (d, 1H), 4.03-4.0 (m, 4H), 3.93 (s, 3H),
2.05-1.95
(m, 1H), 1.14-1.10 (m, 2H), 0.85-0.81 (m, 2H). LCMS Purity: 100%, m/z =
311.3(M+1). HPLC Purity: 96%
Example 10
Preparation of 1-(6-chloropyrimidin-4-y1)-3-(4-cyclopropylpyridin-3-y1)
imidazolidin-2-one (10A):
CI 1--\
)7M.--NN-.....--)
N \N N
(10A)
46
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-id: 120mg,
0.591mmol)
was reacted with 4,6-dichloropyrimidine (88.07mg, 0.591mmol), 1, 4-dioxane
(5mL),
copper iodide (11.21mg, 0.059mmol), trans-N,N' -dimethylcyclohexane-1,2-
diamine
(8.39mg, 0.059mmol) and potassium phosphate (375.96mg, 1.77mmol) to afford the
crude product. Purification by column chromatography on silica gel (0.5%
methanol
in CHC13) afforded 15.3mg of the product (9% yield).
1H NMR (CDC13, 300 MHz): 6 8.67 (s, 1H), 8.60-8.40 (m, 2H), 8.35 (s, 1H),
6.84 (s, 1H), 4.32-4.26 (t, 2H), 4.0-3.97 (t, 2H), 1.98-1.93 (m, 1H), 1.17-
1.11 (m, 2H),
0.87-0.81 (m, 2H). LCMS Purity: 100%, m/z = 316.1 (M+1). HPLC Purity: 95%
Example 11
Preparation of 1-(4-cyclopropylpyridin-3-y1)-3-(4-(trifluoromethyl) pyrimidin-
2-y1)
imidazolidin-2-one (11A):
CF3 N Ny.,/--\N
\Cr ¨ ,,.......= 3
---N 0 N
(11A)
Xantphos (23mg, 0.049mmol) and Pd2(dba)3 (22.5mg, 0.024mmol) were
added to a solution of cesium carbonate (400mg, 1.23mmol) in toluene (5mL)
previously purged with nitrogen (20 minutes). The reaction mixture was purged
with
argon for 10 minutes, followed by the addition of, 1-(4-cyclopropylpyridin-3-
yl)imidazolidin-2-one (I-id: 100mg, 0.492mmo1) and 2-chloro-4-
(trifluoromethyl)pyrimidine (0.06m1, 0.54mmol). The reaction mixture was
heated in
seal tube at 110 C for 16 hours. The reaction was monitored by TLC (10%
methanol
in chloroform). The reaction mixture was cooled, filtered and the filtrate
partitioned
between ethyl acetate and water. The organic layer was concentrated to yield
the
crude product. Purification by column chromatography on silica gel (1.5%
methanol
in chloroform) afforded 60mg of the product (35% yield).
1H NMR (CDC13, 400 MHz): 6 8.95-8.93 (d, 1H), 8.51 (s, 1H), 8.44-8.43 (d,
1H), 7.29-7.28 (d, 1H), 6.83-6.82 (d, 1H), 4.36-4.32 (t, 2H), 4.0-3.96 (t,
2H), 2.10-2.0
47
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
(m, 1H), 1.15-1.11 (m, 2H), 0.84-0.82(m, 2H). LCMS Purity: 100%, m/z = 350.1
(M+1). HPLC Purity: 97%
Example 12
Preparation of 1-(4-cyclopropylpyridin-3-y1)-3-(2-(trifluoromethyl) pyrimidin-
4-y1)
imidazolidin-2-one (12A):
//----Y-N1-1
N y N
)----=-N
1
F 3 0C N
(12A)
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-id: 100mg,
0.492mmo1)
was reacted with 4-chloro-2-(trifluoromethyl)pyrimidine (108mg, 0.591mmol), 1,
4-
dioxane (5mL), copper iodide (9.3mg, 0.049mmol), trans-N,N' -
dimethylcyclohexane-
1,2-diamine (6.9mg, 0.049mmol) and potassium phosphate (313mg, 1.47mmol) to
afford the crude product. Purification by column chromatography on silica gel
(1%
methanol in CHC13), followed by preparative HPLC afforded 20mg of the product
(12% yield).
1H NMR (CDC13, 400 MHz): 6 8.64-8.63 (d, 1H), 8.48-8.44 (m, 3H), 6.84-
6.83 (d, 1H), 4.38-4.34 (t, 2H), 4.04-4.00 (t, 2H), 2.01-1.95 (m, 1H), 1.17-
1.13 (m,
2H), 0.87-0.84 (m, 2H). LCMS Purity: 98%, m/z = 350.1 (M+1). HPLC Purity: 99%
Example 13
Preparation of 1-(4-cyclopropylpyridin-3-y1)-3-(3-(trifluoromethyl)
benzo[b] thiophen-5-y1) imidazolidin-2-one (13A):
F3C r \Nip
/
0
S
(13A)
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-id: 100mg,
0.492mmol)
was reacted with 5-bromo-3-(trifluoromethyl)benzo[b]thiophene (151mg,
0.54mmol),
1, 4-dioxane (5mL), copper iodide (9.3mg, 0.049mmol), trans-N,N'-
48
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
dimethylcyclohexane-1,2-diamine (6.9mg, 0.049mmol) and potassium phosphate
(313mg, 1.47mmol) to afford the crude product. Purification by column
chromatography on silica gel (0.5% methanol in CHC13), followed by preparative
HPLC afforded 15.4mg of the product (8% yield).
1H NMR (CDC13, 300 MHz): 6 8.52 (s, 1H), 8.43-8.41 (d, 1H), 8.15-8.12 (dd,
1H), 7.93 (s, 1H), 7.88-7.85 (d, 1H), 7.81(s, 1H), 6.81-6.79 (d, 1H), 4.18-
4.15 (m,
2H), 4.05-4.02 (m, 2H), 2.15-2.05 (m, 1H), 1.15-1.11 (m, 2H), 0.84-0.82 (m,
2H).
LCMS Purity: 98%, m/z = 404.1 (M+1). HPLC Purity: 97%
Example 14
Preparation of 1-(4-cyclopropylpyridin-3-y1)-3-(2-fluorobenzo[b]thiophen-5-y1)
imidazolidin-2-one (14A):
F
0 N
(14A)
Using analogous reagents and reaction conditions as described in step 5 of
example 1, 1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-id: 145mg,
0.714mmol) was reacted with 5-bromo-2-fluorobenzo[b]thiophene (165mg,
0.714mmol), 1, 4-dioxane (5mL), copper iodide (13.6mg, 0.071mmol), trans-N,N'-
dimethylcyclohexane-1,2-diamine (10.1mg, 0.071mmol) and potassium phosphate
(454mg, 2.14mmol) to afford the crude product. Purification by column
chromatography on silica gel (0.5-1% methanol in CHC13), followed by
preparative
HPLC afforded 53mg of the product (21% yield).
1H NMR (CDC13, 300 MHz): 6 8.50 (s, 1H), 8.42-8.40 (d, 1H), 7.86 (s, 1H),
7.63 (s, 2H), 6.81-6.79 (d, 1H), 6.69-6.68 (d, 1H), 4.14-4.09 (t, 2H), 4.02-
3.96 (t, 2H),
2.11-2.03 (m, 1H), 1.16-1.09 (m, 2H), 0.88-0.82 (m, 2H). LCMS Purity: 100%,
m/z
=354.3 (M+1). HPLC Purity: 98%
Example 15
Preparation of 1-(4-cyclopropylpyridin-3-y1)-3-(3-methylbenzo[b]thiophen-5-y1)
imidazolidin-2-one (15A):
49
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
H 3 C
NN ........)
0 N
(15A)
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-id: 100mg,
0.492mmo1)
was reacted with 5-bromo-3-methylbenzo[b]thiophene (122.4mg, 0.541mmol), 1, 4-
dioxane (5mL), copper iodide (9.3mg, 0.049mmol), trans-N,N'-
dimethylcyclohexane-
1,2-diamine (5.6mg, 0.049mmol) and potassium phosphate (313mg, 1.477mmo1) to
afford the crude product. Purification by column chromatography on silica gel
(2%
methanol in CHC13) afforded 100mg of the product (58% yield).
1H NMR (CDC13, 400 MHz): 6 8.70-8.30 (m, 2H), 7.94-7.93 (d, 1H), 7.83-
7.81 (d, 1H), 7.66-7.64 (dd, 1H), 7.09(s, 1H), 6.81(s, 1H), 4.19-4.15 (m, 2H),
4.03-
3.99 (m, 2H), 2.43 (s, 3H), 2.12-2.08 (m, 1H), 1.15-1.10 (m, 2H), 0.85-0.81
(m, 2H).
LCMS Purity: 93%, m/z = 458.1 (M+1). HPLC Purity: 95%
Example 16
Preparation of 1-(benzo[b]thiophen-6-y1)-3-(4-cyclopropylpyridin-3-y1)
imidazolidin-
2-one (16A):
r \ Npj
\ /
(16A)
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-id: 100mg,
0.492mmol)
was reacted with 6-bromobenzo[b]thiophene (195mg, 0.541mmol), 1, 4-dioxane
(6mL), copper iodide (9.3mg, 0.049mmol), trans-N,N'-dimethylcyclohexane-1,2-
diamine (6.9mg, 0.049mmol) and potassium phosphate (260mg, 1.23mmol) to afford
the crude product. Purification by column chromatography on silica gel (0.5%
methanol in CHC13) afforded 120mg of the product (73% yield).
1H NMR (CDC13,400 MHz): 6 9.20-8.20 (m, 2H), 8.11 (d, 1H), 7.81-7.79 (d,
1H), 7.72-7.69 (dd, 1H), 7.35-7.34 (d, 1H), 7.29-7.27 (d, 1H), 7.15-6.55 (bs,
1H),
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
4.17-4.13 (m, 2H), 4.03-3.99 (m, 2H), 2.10-2.05 (m, 1H), 1.14-1.10 (m, 2H),
0.88-
0.81 (m, 2H). LCMS Purity: 95%, m/z = 336.1 (M+1). HPLC Purity: 97%
Example 17
Preparation of 1-(4-cyclopropylpyridin-3-y1)-3-(2,2-difluorobenzo[d]
[1,3]dioxo1-5-
vl)imidazolidin-2-one (17A):
F*00 40 Nr¨\yN
0 ----N
(17A)
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-id: 100mg,
0.492mmo1)
was reacted with 5-bromo-2,2-difluorobenzo[d][1,3]dioxole (128.4mg,
0.541mmol),
1, 4-dioxane (5mL), copper iodide (9.3mg, 0.049mmol), trans-N,N'-
dimethylcyclohexane-1,2-diamine (5.6mg, 0.049mmol) and potassium phosphate
(313.6mg, 1.447mmol) to afford the crude product. Purification by column
chromatography on silica gel (2.0% methanol in CHC13) afforded 105mg of the
product (60% yield).
1H NMR (CDC13, 400 MHz): 6 8.60-8.40 (d, 2H), 7.72 (s, 1H), 7.03 (s, 2H),
6.81-6.80 (d, 1H), 4.06-3.95 (m, 4H), 2.05-2.01 (m, 1H), 1.14-1.09 (m, 2H),
0.84-0.80
(m, 2H). LCMS Purity: 95%, m/z = 360.1 (M+1). HPLC Purity: 98%
Example 18
Preparation of 1-(4-cyclopropylpyridin-3-y1)-3-(3-methylbenzo[b]thiophen-6-y1)
imidazolidin-2-one (18A):
r\1\1.ii\)1
\ IW 0
H3C
(18A)
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-id: 100mg,
0.492mmo1)
was reacted with 6-bromo-3-methylbenzo[b]thiophene (112mg, 0.492mmo1), 1, 4-
51
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
dioxane (5mL), copper iodide (9.3mg, 0.049mmo1), trans-N,N'-
dimethylcyclohexane-
1,2-diamine (7.0mg, 0.049mmo1) and potassium phosphate (313.6mg, 1.447mmo1) to
afford the crude product. Purification by column chromatography on silica gel
(2.0%
methanol in CHC13) afforded 45mg of the product (26% yield).
1H NMR (CDC13, 400 MHz): 6 8.60-8.40 (m, 2H), 8.05 (d, 1H), 7.76-7.74 (m,
1H), 7.69-7.67 (d, 1H), 6.97 (d, 1H), 6.82 (bs, 1H), 4.20-4.10 (m, 2H), 4.05-
3.95 (m,
2H), 2.43 (s, 3H), 2.15-2.05 (m, 1H), 1.15-1.10 (m, 2H), 0.85-0.82 (m, 2H).
LCMS
Purity: 100%, m/z = 350.1 (M+1). HPLC Purity: 98%
Example 19
Preparation of 1-(4-cyclopropylpyridin-3-y1)-3-(3-methylbenzofuran-5-
vl)imidazolidin-2-one (19A):
H3C irN.i.)1\ /
/ 110
0
0
(19A)
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-id: 100mg,
0.492mmol)
was reacted with 5-bromo-3-methylbenzofuran (125mg, 0.5904mmol), 1, 4-dioxane
(4mL), copper iodide (10mg, 0.0492mmo1), trans-N,N'-dimethylcyclohexane-1,2-
diamine (7.0mg, 0.0492mmo1) and potassium phosphate (313mg, 1.476mmo1) to
afford the crude product. Purification by column chromatography on silica gel
(1.0%
methanol in CHC13) afforded 69mg of the product (42% yield).
1H NMR (CDC13, 300 MHz): 6 8.80-8.30 (m, 2H), 7.76 (s, 1H), 7.55-7.30 (m,
2H), 6.90-6.70 (bs, 1H), 4.13-4.10 (t, 2H), 4.01-3.96 (t, 2H), 2.22 (s, 3H),
2.15-2.05
(m, 1H), 1.20-1.10 (m, 2H), 0.92-0.80 (m, 2H). LCMS Purity: 91%, m/z = 334.1
(M+1). HPLC Purity: 95%
Example 20
Preparation of 1-(2-chlorobenzo[b] thiophen-5-y1)-3-(4-cyclopropylpyridin-3-
y1)
imidazolidin-2-one (20A):
52
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
I\CN---)
CI / 1 0 "0 -N
S
(20A)
Preparation of Intermediate 5-Bromo-2-chloro-benzo[b]thiophene (I-20a):
n-Butyl lithium (0.64m1, 1.2 mmol) was added drop wise to a solution of
diisopropyl amine (0.25 mL, 1.5 mmol) in dry THF at -78 C under nitrogen
atmosphere over a period of 5 minutes. The reaction mixture was stirred at -20
C for
30 minutes. To this was added 5-bromo-benzo[b]thiophene (200mg, 0.938 mmol) in
dry THF at -78 C, continued stirring for a further 30 minutes at -78 C,
followed by
the addition of N-chlorosuccinamide (225mg, 1.68mmol) in dry THF at -78 C. The
resulting mixture was stirred at room temperature for 2 hours. The reaction
was
monitored by TLC (100% hexane). The reaction mixture was partitioned between
ethyl acetate and saturated ammonium chloride. The organic layer was dried
over
sodium sulfate and concentrated to afford the crude product. Purification by
column
chromatography on silica gel (100% hexane) afforded 70mg of the product (30%
yield).
1H NMR (CDC13, 300 MHz): 67.815-7.811 (d, 1H), 7.57-7.55 (d, 1H), 7.44-
7.41 (dd, 1H), 7.12 (s, 1H).
Preparation of the title compound 1-(2-chlorobenzo[b] thiophen-5-yl)-3-(4-
cyclopropylpyridin-3-yl) imidazolidin-2-one (20A):
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(4-cyclopropylpyridin-3-y1) imidazolidin-2-one (I-id: 100mg,
0.491mmol)
was reacted with 5-bromo-2-chloro-benzo[b]thiophene-(134.6mg, 0.541mmol),
copper iodide (9.3mg, 0.049mmol), trans-N,N'-dimethylcyclohexane-1,2-diamine
(7.0mg, 0.0491mmol), potassium phosphate (260.9mg, 1.23mmol) and 1, 4-dioxane
(5mL to yield the crude product. Purification by column chromatography on
silica gel
(1.5% methanol in DCM), afforded 60.0mg of the product (33% yield)
1H NMR (CDC13, 400 MHZ): 6 8.90 (s, 1H), 8.52-8.51 (d, 1H), 7.82-7.81 (d,
1H), 7.71-7.68 (d, 1H), 7.61-7.58 (dd, 1H), 7.17-7.15 (t, 2H), 4.19-4.08 (m,
4H), 2.34-
2.29 (m, 1H), 1.50-1.45 (m, 2H), 1.11-1.06 (m, 2H). LCMS Purity: 92%, m/z =
370.0
(M+1). HPLC Purity: 94%
53
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
Example 21
Preparation of 1-(4-cyclopropylpyridin-3-y1)-3-(2,3-dihydro-1H-inden-5-
vl)imidazolidin-2-one (21A):
F-1
"
III=
NyN...;
(21A)
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-id: 100mg,
0.492mmo1)
was reacted with 5-bromo-2,3-dihydro-1H-indene (132mg, 0.5418mmo1), 1, 4-
dioxane (4mL), copper iodide (9.5mg, 0.0492mmo1), trans-N,N'-
dimethylcyclohexane-1,2-diamine (7.0mg, 0.0492mmo1) and potassium phosphate
(261mg, 1.231mmol) to afford the crude product. Purification by column
chromatography on silica gel (1.5% methanol in CHC13) afforded 12mg of the
product
(8% yield).
1H NMR (CDC13, 300 MHz): 6 8.01-7.97 (d, 1H), 7.54 (s, 1H), 7.34-6.90 (m,
3H), 6.83-6.81 (d, 1H), 4.10-3.85 (m, 4H), 3.0-2.8 (m, 4H), 2.15-1.95 (m, 3H),
1.15-
1.05 (m, 2H), 0.85-0.75 (m, 2H). LCMS Purity: 95%, m/z = 320.1 (M+1). HPLC
Purity: 97%
Example 22
Preparation of 1-(4-cyclopropylpyridin-3-y1)-3-(5-fluorobenzo[b]thiophen-2-y1)
imidazolidin-2-one (22A):
F 0
s eN
0 N I
N
(22A)
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-id: 100mg,
0.4926mmo1)
was reacted with 2-bromo-5-fluorobenzo[b]thiophene (125mg, 0.5418mmol), 1, 4-
dioxane (4mL), copper iodide (9.3mg, 0.0492mmo1), trans-N,N' -
dimethylcyclohexane-1,2-diamine (7.0mg, 0.0492mmo1) and potassium phosphate
54
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
(261mg, 1.231mmol) to afford the crude product. Purification by column
chromatography on silica gel (0.5% methanol in CHC13) afforded 85mg of the
product
(49% yield).
1H NMR (CDC13, 300 MHz): 6 8.55-8.40 (m, 2H), 7.70-7.60 (m, 1H), 7.30-
7.20 (m, 1H), 7.0-6.90 (m, 1H), 6.85-6.75 (d, 1H), 6.50 (s, 1H), 4.20-4.0 (m,
4H),
2.10-1.95 (m, 1H), 1.20-1.10 (m, 2H), 0.90-0.82 (m, 2H). LCMS Purity: 98%, m/z
=
354.1 (M+1), HPLC Purity: 93%
Example 23
Preparation of 1-(4-cyclopropylpyridin-3-y1)-3-(3-
(trifluoromethyl)benzo[b]thiophen-
6-yl)imidazolidin-2-one (23A):
CF3 4. NsirN,..,
1
0 1
S
N
(23A)
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-id: 170mg,
0.837mmol)
was reacted with 6-bromo-3-(trifluoromethyl)benzo[b]thiophene (259mg,
0.921mmol), 1, 4-dioxane (8mL), copper iodide (15.9mg, 0.083mmol), trans-N,N' -
dimethylcyclohexane-1,2-diamine (11.9mg, 0.083mmol) and potassium phosphate
(533mg, 2.51mmol) to afford the crude product. Purification by column
chromatography on silica gel (0.4% methanol in CHC13) afforded 15mg of the
product
(4% yield).
1H NMR (CDC13, 300 MHz): 6 8.70-8.40 (m, 2H), 8.29-8.28 (d, 1H), 7.9-7.89
(d, 1H), 7.79 (s, 1H), 7.70-7.66 (dd, 1H), 6.84-6.80 (bs, 1H), 4.2-4.13 (m,
2H), 4.04-
3.99 (m, 2H), 2.10-2.05 (m, 1H), 1.16-1.09 (m, 2H), 0.86-0.82 (m, 2H). LCMS
Purity: 95%, m/z = 404.1 (M+1). HPLC Purity: 95%
Example 24
Preparation of 1-(4-Cyclopropyl-pyridin-3-y1)-3-(2-flnoro-3-methyl-
benzo[b] thiophen-5-y1)-imidazolidin-2-one (24A):
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
= 1-1NI
N
0
CH3
(24A)
Preparation of Intermediate 1-(4-Bromo-phenylsulfany1)-propan-2-one (I-24a):
Br I.sr.CH3
0
(I-24a)
1-Chloro-propan-2-one (1.0 g, 11.63 mmol) and potassium carbonate (2.9 g,
21.15 mmol) were added to a solution of 4-bromo-benzenethiol (2.0 g, 10.58
mmol)
in DMF (5.0 mL) at 0 C. The resulting mixture was stirred at room temperature
overnight. The reaction was monitored by TLC (100% hexane). The reaction
mixture
quenched with ice and extracted with ethyl acetate. The organic layer was
dried over
sodium sulfate and concentrated to yield the crude product. Purification by
column
chromatography on silica gel (2% methanol in chloroform) afforded 2.0 g of the
product (80% yield).
1H NMR (CDC13, 300 MHz): 6 7.44-7.4 (d, 2H), 7.22-7.16 (d, 2H), 3.6 (s,
2H), 2.25 (s, 3H).
Preparation of Intermediate 5-Bromo-3-methyl-benzo[b] thiophene (I-24b):
CH3
Br s
(I-24b)
Polyphosphoric acid (7 g) was added to a solution of 1-(4-bromo-
phenylsulfany1)-propan-2-one (I-24a: 2.0 g, 8.163 mmol) in toluene (10 mL) and
the
resulting mixture was heated to 100 C for 5 hours. The reaction was monitored
by
TLC (100% hexane). The reaction mixture was cooled to room temperature,
quenched
with ice, basified with potassium carbonate (pH-8) and extracted with ethyl
acetate.
56
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
The organic layer was concentrated to yield the crude product. Purification by
column
chromatography on silica gel (100% hexane) afforded 1.2 g of product (66 %
yield)
1H NMR (CDC13, 300 MHz): 6 7.856-7.85 (d, 1H), 7.72-7.69 (d, 1H), 7.45-
7.41 (dd, 1H), 7.11 (s, 1H), 2.4 (s, 3H).
Preparation of Intermediate 5-Bromo-2-fluoro-3-methyl-benzo[b]thiophene (I-
24c):
CH3
Br 0\ F
S
(I-24c)
Using analogous reaction condition and workup as described in Example 20
above fro the preparation of I-20a, 5-bromo-3-methyl-benzo[b]thiophene (I-24b:
500
mg, 2.192 mmol) in dry THF was reacted with N-fluoro benzene sulfonimide (1.2
g,
3.94 mmol), diisopropyl amine (266.2 mg, 2.6315 mmol), n-butyl lithium (1.3
mL,
2.6315 mmol) to afford crude product. Purification by column chromatography on
silica gel (hexane) afforded 120 mg of the product (22 % yield).
1H NMR (CDC13, 300 MHz): 6 7.72-7.68(d, 1H), 7.54-7.5(d, 1H), 7.44-7.39
(dd, 1H), 2.11 (s, 3H).
Preparation of the title compound 1-(4-Cyclopropyl-pyridin-3-yl)-3-(2-fluoro-3-
methyl-benzo[b]thiophen-5-yl)-imidazolidin-2-one (24A):
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(4-cyclopropyl-pyridin-3-y1)-imidazolidin-2-one (I-id: 100mg, 0.4926
mmol) was reacted with 5-bromo-2-fluoro-3-methyl-benzo[b]thiophene (I-24c:
120mg, 0.4926mmo1), copper iodide (9.3mg, 0.04926mmo1), trans-N, N'-
dimethylcyclohexane-1, 2-diamine (7.0 mg, 0.04926 mmol), potassium phosphate
(313 mg, 1.4778 mmol) and 1, 4-dioxane (5 ml) to afford the crude product.
Purification by column chromatography on silica gel (2 % methanol in
chloroform)
afforded 120 mg of product (66 % yield).
1H NMR (CDC13, 400 MHz): 6 8.70-8.30 (m, 2H), 7.88-7.87 (d, 1H), 7.63-
7.61 (d,1H), 7.54-7.52 (dd, 1H), 6.81 (s, 1H), 4.16-4.12 (m, 2H), 4.02-3.98
(m, 2H),
2.25 (s, 3H), 2.12-2.05 (m, 1H), 1.13-1.10 (m, 2H), 0.85-0.81 (m, 2H). LCMS
Purity:
96%, m/z = 368.1 (M+1). HPLC Purity: 97 %
57
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
Example 25
Preparation of 1-(4-cyclopropyl-pyridin-3-yl)-3-(2-fwro-benzo(b)thiophene-6-
yl)-
imidazolidin-2-one (25A):
N
pl
F S
I I el N .1
0
(25A)
Preparation of Intermediate 6-Bromo-benzo[b]thiophene-2-carboxylic acid methyl
ester (I-25a):
01 I ...A-1
0,õ,
B r S 3
0
(I-25a)
TEA (1.29g, 12.85mmol) and mercapto-acetic acid methyl ester (1.14 g,
10.83mmol) were added to acetonitrile (50mL) previously purged with argon (10
minute). This was followed by the addition of 4-bromo-2-fluoro-benzaldehyde
(2g,
9.852mmo1) and the resulting mixture was heated to reflux at 85 C overnight.
The
reaction was monitored by TLC (20% ethyl acetate in hexane). The reaction
mixture
was cooled, concentrated, basified with 10%NaOH solution and extracted with
ethyl
acetate to afford the crude product. Purification by column chromatography on
silica
gel (5% ethyl acetate in hexane) afforded 2.4g of the product (90% yield).
1H NMR (CDC13, 300 MHz): 68.18-8.0 (dd, 2H), 7.8-7.7 (d, 1H), 7.65-7.59
(dd, 1H), 4.1-4.0 (s, 3H).
Preparation of Intermediate 6-Bromo-benzo[b]thiophene-2-carboxylic acid (I-
25b):
SI
Br sl OH
0
(I-25b)
Li0H.H20 (1.85g, 44mmol) and water (20mL) were added to a stirred
solution of 6-bromo-benzo[b]thiophene-2-carboxylic acid methyl ester (I-25a:
2.4g,
8.85mmol) in THF (25mL) at room temperature. The resulting mixture was stirred
at
room temperature for 2 hours. The reaction was monitored by TLC (100% ethyl
58
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
acetate). The reaction mixture was concentrated, acidified with 2N HC1,
filtered and
the residue was washed with n-hexane to afford 1.9g of the product (84%
yield).
1H NMR (DMSO-D6, 300 MHz): 614.0-14.03 (b, 1H), 8.4-8.39 (d, 1H), 8.28-
8.1 (d, 1H), 8.18-8.0 (d, 1H), 7.78-7.62 (dd, 1H). LCMS Purity: 99%, m/z =
255.9
(M+1)
Preparation of Intermediate 6-Bromo-benzo[b] thiophene (I-25c):
1.1 s
\
Br
(I-25c)
DBU (1.21g, 8.00mmol) was added to a stirred solution of 6-bromo-
benzo[b]thiophene-2-carboxylic acid (I-25b: 2.4g, 8.85mmol) in DMA (4mL) at
room temperature and the resulting mixture was heated in microwave at 200 C
for 1
hour. The reaction was monitored by TLC (50% ethyl acetate in hexane). The
reaction
mixture was acidified with 1N HC1 and extracted with ethyl acetate. The
organic layer
was concentrated and purified by column chromatography on silica gel (100%
hexane) to afford 320mg of the product (78% yield).
1H NMR (CDC13, 300 MHz): 6 8.18-8.0 (d, 1H), 7.7-7.6 (d, 1H), 7.5-7.59 (d,
1H), 7.59-7.42 (d, 1H), 7.3-7.29 (d, 1H).
Preparation of Intermediate 6-Bromo-2-fluoro-benzo[b] thiophene (I-25d):
101 1
Br S F
(I-25d)
Using analogous reaction condition and reagents as described Example 20 for
the preparation of I-20a, 6-bromo-benzo[b]thiophene (I-25c: 0.5g, 2.347 mmol)
in
dry THF was reacted with N-fluorobenzenesulfonimide (1.33g,4.22mmol), n-butyl
lithium (1.76 ml, 3.52 mmol), diisopropyl amine (0.57 ml, 3.99 mmol) to afford
crude
product. Purification by column chromatography on silica gel (100% hexane)
afforded 176 mg of the product (33% yield).
1H NMR (CDC13, 300 MHz): 6 7.97-7.82 (d, 1H), 7.56-7.52 (dd, 2H), 6.5-6.3
(d, 1H).
59
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
Preparation of the title compound 1-(4-cyclopropyl-pyridin-3-yl)-3-(2-fluoro-
benzo(b)thiophene-6-yl)-imidazolidin-2-one (25A):
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(4-cyclopropylpyridin-3-y1) imidazolidin-2-one (I-id:
150mg,0.738mmol)
was reacted with 6-Bromo-2-fluoro-benzo[b]thiophene (I-25d: 170.6mg,
0.738mmol), copper iodide (14.2, 0.071mmol), trans-N,N'-dimethylcyclohexane-
1,2-
diamine (10.2 mg, 0.071mmol), potassium phosphate (469.9mg, 2.21mmol) and 1, 4-
dioxane (5mL) to afford the crude product. Purification by column
chromatography
on silica gel (1% methanol in CHC13), followed by preparative HPLC afforded
11.6mg of the product (4% yield).
1H NMR (CDC13, 400 MHz): 6 8.51 (s, 1H), 8.42-8.41 (d, 1H), 8.04-8.03 (d,
1H), 7.61-7.54 (m, 2H), 6.81-6.80 (d, 1H), 6.66-6.65 (d, 1H), 4.14-4.10 (m,
2H), 4.02-
3.97 (m, 2H), 2.11-2.04 (m, 1H), 1.21-1.10 (m, 2H), 0.88-0.81 (m, 2H). LCMS
Purity: 99%, m/z = 354.1 (M+1). HPLC Purity: 91%
Example 26
Preparation of 1-(4-cyclopropyl-pyridin-3-yl)-3-(4-fluoro-benzo[b]thiophen-6-
yl)-
imidazolidin-2-one (26A):
\S 10
0
F
(26A)
Preparation of Intermediate 4-Bromo-2, 6-difluoro-benzaldehyde (I-26a):
F H
0 0
Br F
(I-26a)
Using analogous reaction condition and reagents as described in Example 20
for the preparation of I-20a above, 1-bromo-3,5-difluoro-benzene (2g, 2.10.36
mmol)
in THF was reacted with DMF (1.43g,19.68mmol), n-butyl lithium (1.56 ml, 12.4
mmol) and diisopropyl amine (0.57 mL, 15.5 mmol) to afford crude product.
Purification by column chromatography on silica gel (2% ethyl acetate in
hexane)
afforded 1.35g of the product (61% yield).
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
1H NMR (CDC13, 300 MHz): 611.5-11.0 (s, 1H), 7.4-7.2 (dd, 2H).
Preparation of intermediate 6-Bromo-4-fhwro-benzo[b]thiophene-2-carboxylic
acid
methyl ester (I-26b):
F
SI I 0
Br s 'CH3
0
(I-26b)
Using analogous reaction condition and reagents as described in Example 25
for the preparation ofI-25a, 4-bromo-2,6-difluoro-benzaldehyde (I-26a: 2.8g,
12.8mmol) was reacted with TEA (1.66g, 16.47mmol), mercapto-acetic acid methyl
ester (1.47 g, 13.83mmol) and acetonitrile (50mL) to afford crude product.
Purification by column chromatography on silica gel (2% ethyl acetate in
hexane)
afforded 2.3g of the product (64% yield).
1H NMR (CDC13, 400 MHz): 6 8.098 (d, 1H), 7.80 (s, 1H), 7.26-7.23 (m, 1H),
3.96 (s, 3H).
Preparation of intermediate 6-Bromo-4-fhwro-benzo[b]thiophene-2-carboxylic
acid
(I-26c):
F
101 I
Br S OH
0
(I-26c)
Using analogous reagents and reaction conditions as described in Example 25
for the preparation of I-25b above, 6-bromo-4-fluoro-benzo[b]thiophene-2-
carboxylic
acid methyl ester (I-26b: 2.3g, 7.85mmol) in THF (25mL) was hydrolyzed with
LiOH (1.65g, 39.38mmol) and water (20mL) to afford 1.9g of the product (83%
yield).
1H NMR (DMSO-D6, 300 MHz): 6 14.0-14.03 (b, 1H), 8.38-8.32 (d, 1H),
8.18-8.0 (s, 1H), 7.7-7.6 (dd, 1H).
Preparation of intermediate 6-Bromo-4-fhwro-benzo[b] thiophene (I-26d):
61
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
F
I I
Br S
(I-26d)
Using analogous reaction condition and reagents as described in Example 25
for the preparation of I-25c above, 6-bromo-4-fluoro-benzo[b]thiophene-2-
carboxylic
5 acid (I-26c: 1.8g, 6.47mmol) in DMA (4mL) and DBU (4.0g, 26.54mmol) were
heated in microwave to afford the crude product. Purification by column
chromatography on silica gel (100% hexane) afforded 1.45g of the product (97%
yield).
1H NMR (CDC13, 300 MHz): 6 7.9-7.8 (d, 1H), 7.5-7.48 (dd, 2H), 7.3-7.22 (d,
10 1H).
Preparation of the title comound 1-(4-cyclopropyl-pyridin-3-yl)-3-(4-fluoro-
benzo[b] thiophen-6-yl)-imidazolidin-2-one (26A):
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(4-cyclopropylpyridin-3-y1) imidazolidin-2-one (I-id: 120mg,
0.591mmol)
was reacted with 6-bromo-4-fluoro-benzo[b]thiophene (I-26d: 163.6mg,
0.738mmol),
copper iodide (11.2mg, 0.059mmol), trans-N,N' -dimethylcyclohexane-1,2-diamine
(8.9mg, 0.0591mmol), potassium phosphate (375.9mg, 1.7mmol) and 1, 4-dioxane
(5mL). Purification by column chromatography on silica gel (0.3% methanol in
CHC13) afforded 51.2mg of the product (26% yield).
1H NMR (CDC13, 400 MHz): 6 9.20-8.20 (m, 2H), 7.80 (s, 1H), 7.61-7.57 (dd,
1H), 7.40-7.37 (d, 1H), 7.35-7.32 (d, 1H), 6.83-6.81 (d, 1H), 4.15-4.10 (m,
2H), 4.03-
4.00 (m, 2H), 2.08-2.04 (m, 1H), 1.15-1.10 (m, 2H), 0.88-0.82 (m, 2H). LCMS
Purity: 93%, m/z = 354.1 (M+1). HPLC Purity: 94%
Example 27
Preparation of 1-(4-cyclopropyl-pyridin-3-yl)-3-(5-fluoro-benzo[b]thiophen-6-
yl)-
imidazolidin-2-one (27A):
62
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
r\NI.i)
\ I 0
F
(27A)
Preparation of Intermediate 4-Bromo-2, 5-difhwro-benzaldehyde (I-27a):
F
0
Brs F
(I-27a)
n-Butyl lithium (3.6 m1,7.7 mmol) was added drop wise to a solution of 1,4-
dibromo-2,5-difluoro-benzene (2g, 7.35mmol) in dry ether at -78 C under
nitrogen
atmosphere and the resulting mixture was stirred at -78 C for 30 minutes. This
was
followed by the addition of DMF (0.85m1, 11.03mmol) in dry THF. The resultant
was stirred at room temperature for 1 hour. The reaction was monitored by TLC
(5%
ethyl acetate in hexane). The reaction mixture was partitioned between ethyl
acetate
and saturated ammonium chloride. The organic layer was concentrated and
purified
by column chromatography on silica gel (2% ethyl acetate in hexane) to afford
600mg
of the product (37% yield).
1H NMR (CDC13, 300 MHz): 6 10.27-10.26 (d, 1H), 7.61-7.57 (t, 1H), 7.49-
7.44 (q, 1H)
Preparation of Intermediate 6-Bromo-5-fhwro-benzo[b]thiophene-2-carboxylic
acid
methyl ester (I-27b):
F
0 \ 0¨CH3
Br S 0
(I-27b)
Using analogous reaction condition and reagents as described in Example 25
for the preparation of I-25a above, 4-bromo-2,5-difluoro-benzaldehyde (I-27a:
1.71g,
7.73mmol) was reacted with TEA (1.4mL, 10.05mmol), mercapto-acetic acid methyl
ester (920mg, 8.51mmol) and acetonitrile (50mL) to afford crude product.
63
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
Purification by column chromatography on silica gel (5% ethyl acetate in
hexane)
afforded 2.3g of the product (Yield 64%).
1H NMR (CDC13, 300 MHz): 6 8.1-7.95 (m, 2H), 7.65-7.55 (d, 1H), 3.95 (s,
3H).
Preparation of Intermediate 6-Bromo-5-fluoro-benzo[b]thiophene-2-carboxylic
acid
(I-27c):
F 0
\ OH
Br S 0
(I-27c)
Using analogous reagents and reaction conditions as described in Example 25
for the preparation of I-25b above, 6-bromo-5-fluoro-benzo[b]thiophene-2-
carboxylic
acid methyl ester (I-27b: 0.3g, 1.0mmol) in THF (10mL) was hydrolyzed with
LiOH
(87mg, 2.04mmol) and water (2mL) to afford 250mg of the product (yield 88%)
1H NMR (DMSO-D6, 300 MHz): 6 8.54-8.48 (d, 1H), 8.06-7.94 (m, 2H).
Preparation of Intermediate 6-Bromo-5-fluoro-benzo[b]thiophene (I-27d):
F
g I I
Br S
(I-27d)
Using analogous reaction condition and reagents as described in Example 25
for the preparation of I-25c above, 6-bromo-5-fluoro-benzo[b]thiophene-2-
carboxylic
acid (I-27c: 250mg 0.89mmol) in DMA (4mL) and DBU (550mL, 3.6mmol) were
heated in microwave to afford the crude product. Purification by column
chromatography on silica gel (100% hexane) afforded 180mg of the product (86%
yield).
1H NMR (CDC13, 300 MHz): 6 8.06-8.0 (d, 1H), 7.58-7.48 (t, 2H), 7.28-7.24
(d, 1H)
Preparation of the title compound 1-(4-cyclopropyl-pyridin-3-yl)-3-(5-fluoro-
benzo[b]thiophen-6-yl)-imidazolidin-2-one (27A):
64
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(4-cyclopropylpyridin-3-y1) imidazolidin-2-one (I-id:
100mg,0.491mmol)
was reacted with 6-bromo-5-fluoro-benzo[b]thiophene (I-27d: 136mg,
0.5901mmol),
copper iodide (28mg, 0.147mmol), trans-N,N'-dimethylcyclohexane-1,2-diamine
(21mg, 0.147mmol), potassium phosphate (313.9mg, 1.7mmol) and 1, 4-dioxane
(5mL) to afford the crude product. Purification by column chromatography on
silica
gel (1% methanol in CHC13) afforded 80mg of the product (46% yield).
1H NMR (CDC13, 300 MHz): 6 8.85-8.20 (m, 2H), 8.10-8.0 (d, 1H), 7.60-7.48
(m, 2H), 7.32-7.20 (s, 1H), 7.0-6.8 (bs, 1H), 4.20-4.10 (m, 4H), 2.20-2.05 (m,
1H),
1.20-1.10 (m, 2H), 0.88-0.82 (m, 2H). LCMS Purity: 99%, miz = 354.0 (M+1).
HPLC Purity: 96%
Example 28
Preparation of 1-(4-cyclopropylpyridin-3-y1)-3-(6-fluorobenzo[b]thiophen-5-y1)
imidazolidin-2-one (28A):
r \ Npj
0 \ /
S F
(28A)
Preparation of Intermediate 5-bromo-2, 4-difluorobenzaldehyde (I-28a):
0
B r 0H
F F
(I-28a)
Using analogous reagents and reaction conditions as described in Example 27
for the preparation of I-27a above, 1,5-dibromo-2,4-difluorobenzene (3g,
11.07mmol)
in dry ether was reacted with DMF (1.05mL, 14.39mmol) and n-butyl lithium
(6.08
ml, 11.56 mmol to afford the crude product. Purification by column
chromatography
on silica gel (2% ethyl acetate in hexane) afforded 1.5g of the product (61%
yield).
1H NMR (CDC13, 300 MHz): 6 10.2 (s, 1H), 8.14-8.06 (t, 1H), 7.06-6.98 (t,
1H).
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
Preparation of intermediate methyl 5-bromo-6-fluorobenzo[b]thiophene-2-
carboxylate (I-28b):
Br
=i I S 0,CH3
F
0
(I-28b)
Using analogous reaction condition and reagents as described in Example 25
for the preparation ofI-25a above, 5-bromo-2,4-difluorobenzaldehyde (I-28a:
475mg,
2.149mmol) was reacted with TEA (0.748mL, 5.372mmo1), mercapto-acetic acid
methyl ester (0.211mL, 2.36mmol) and DMSO (4 mL) to afford crude product.
Purification by column chromatography on silica gel (1.5% ethyl acetate in
hexane)
afforded 30mg of the product (5% yield).
1H NMR (DMSO-D6, 300 MHz): 6 8.08-8.06 (d, 1H), 7.95 (s, 1H), 7.62-7.59
(d, 1H), 3.95 (s, 1H).
Preparation of Intermediate 5-bromo-6-fluorobenzo[b]thiophene-2-carboxylic
acid
(I-28c):
Br
=i I S OH
F
0
(I-28c)
Using analogous reagents and reaction conditions as described in Example 25
for the preparation of I-25b above, methyl 5-bromo-6-fluorobenzo[b]thiophene-2-
carboxylate (I-28b: 0.3g, 1.038mmol) in THF (10mL) was hydrolyzed with LiOH
(300mg, 1.038mmol) and water (2mL) to afford 240mg of the product (85% yield)
1H NMR (DMSO D6, 300 MHz): 6 8.40-8.38 (d, 1H), 8.19-8.16 (m, 1H), 8.09-
8.05 (m, 1H).
Preparation of Intermediate 5-bromo-6-fluorobenzo[b]thiophene (I-28d):
66
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
Br
g I I
F S
(I-28d)
Using analogous reaction condition and reagents as described in Example 25
for the preparation of I-25c above, 5-bromo-6-fluorobenzo[b]thiophene-2-
carboxylic
acid (I-28c: 240mg 0.875mmo1) in DMA (4mL) and DBU (0.523mL, 3.503mmo1)
were heated in microwave to afford the crude product. Purification by column
chromatography on silica gel (100% hexane) afforded 110mg of the product (55%
yield).
1H NMR (CDC13, 400 MHz): 6 8.00-7.98 (d, 1H), 7.63-7.61 (d, 1H), 7.44-7.43
(d, 1H), 7.26-7.24 (t, 1H).
Preparation of title compound 1-(4-cyclopropylpyridin-3-y1)-3-(6-fluorobenzo-
[b] thiophen-5-y1) imidazolidin-2-one (28A):
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-id: 90g, 0.443mmo1)
was
reacted with 5-bromo-6-fluorobenzo[b]thiophene (I-28d: 102mg, 0.443mmo1), 1, 4-
dioxane (4mL), copper iodide (9mg, 0.044mmol), trans-N,N'-dimethylcyclohexane-
1,2-diamine (19mg, 0.132mmol) and potassium phosphate (282mg, 1.329mmo1) to
afford the crude product. Purification by preparative HPLC afforded 9mg of the
product (6% yield).
1H NMR (CDC13, 300 MHz): 6 8.55 (s, 1H), 8.43-8.41 (d, 1H), 8.0-7.97 (d,
1H), 7.66-7.62 (d, 1H), 7.43-7.41 (d, 1H), 7.29-7.26 (d, 1H), 6.83-6.81 (d,
1H), 4.10-
4.01 (m, 4H), 2.13-2.10 (m, 1H), 1.18-1.15 (m, 2H), 0.84-0.82 (m, 2H).. LCMS
Purity: 98%, m/z = 354.1 (M+1). HPLC Purity: 96%
Example 29
Preparation of 1-(4-cyclopropylpyridin-3-y1)-3-(5-fluoro-3-
methylbenzo[b]thiophen-
6-yl)imidazolidin-2-one (29A):
67
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
S N \ /
r\N.il)
\ IW 0
F
H3C
(29A)
Preparation of Intermediate 1-(3-bromo-4-fluorophenylthio) propan-2-one (I-
29a):
F 0
_CH3
Br S" y
0
(I-29a)
Using analogous reagents and reaction conditions as described in Example 24
for the preparation of I-24a above, 3-bromo-4-fluoro-benzenethiol (2.0g, 9.756
mmol)
was reacted with 1-chloro-propan-2-one (0.86 mL, 10.73 mmol), DMF (6.0 mL),
and
potassium carbonate(2.69g, 19.51 mmol) to afford crude product which was
purified
by column on silica gel (5% ethyl acetate in hexane) to afford 2.1 g of the
product (82
% yield).
1H NMR (CDC13, 300 MHz): 6 7.6-7.5 (dd, 1H), 7.3-7.2 (m, 1H), 7.1-7.0 (t,
1H), 3.6 (s, 2H), 2.25 (s, 3H). LCMS Purity: 94%, m/z = 320.0 (M+1)
Preparation of Intermediate 6-bromo-5-fluoro-3-methylbenzo[b]thiophene (I-
29b):
CH3
F
\
Br's
(I-29b)
Using analogous reagents and reaction conditions as described in Example 24
for the preparation of I-24b above, 1-(3-bromo-4-fluoro-phenylsulfany1)-propan-
2-
one (I-29a: 2.1g, 8.015mmol) in toluene (10 mL) was cyclized with
polyphosphoric
acid (8 g) to yield crude product which was purified by column chromatography
on
silica gel (100% hexane) to afford 1.4 g of product (72% yield).
1H NMR (CDC13, 300 MHz): 6 8.02-7.96 (d, 1H), 7.72-7.64 (q, 1H), 7.44-7.38
(d, 1H), 2.4 (s, 3H).
68
CA 02834224 2013-10-23
WO 2012/149413 PCT/US2012/035583
Preparation of the title compound 1-(4-cyclopropylpyridin-3-yl)-3-(5-fluoro-3-
methylbenzo[b]thiophen-6-yl)imidazolidin-2-one (29A):
Using analogous reagents and reaction conditions as described in Example 1
above, 4-cyclopropyl-pyridin-3-y1)-imidazolidin-2-one (I-id: 150mg,0.7389
mmol)
was reacted with 6-bromo-5-fluoro-3-methyl-benzo[b]thiophene (I-29b:
216mg,0.8866 mmol), 1, 4-dioxane (5mL), copper iodide (14.07mg, 0.0738mmo1),
trans-N,N'-dimethylcyclohexane-1,2-diamine (10.4mg, 0Ø0738 mmol) and
potassium phosphate (469.9mg, 2.2167mmo1) to afford the crude product.
Purification
by column chromatography on silica gel (1% methanol in chloroform), followed
by
preparative HPLC afforded 62 mg of the product (22% yield).
1H NMR (CDC13, 400 MHz): 6 8.52 (s, 1H), 8.41-8.40 (d, 1H), 8.03-8.01 (d,
1H), 7.46-7.43 (d, 1H), 7.14 (s, 1H), 6.81-6.80 (d, 1H), 4.20-4.10 (m, 2H),
4.05-3.99
(m, 2H), 2.40 (s, 3H) 2.2-2.1 (m, 1H), 1.17-1.14 (m, 2H), 0.83-0.82 (m, 2H).
LCMS
Purity: 100%, m/z = 368.1 (M+1). HPLC Purity: 97%
Example 30
Preparation of (1R,5S)-2-(2-chloropyridin-4-yl)-4-(4-cyclopropylpyridin-3-yl)-
2,4-
diazabicyclo[3.1.0]hexan-3-one (Enantiomer I. 30A-I) and (1 S,5R)-2-(2-
chloropyridin-4-yl)-4-(4-cyclopropylpyridin-3-yl)-2,4-diazabicyclo[3.1.0]hexan-
3-
one (Enantiomer II: 30A-II):
CI El..7Arl-
CI H,,,A,1-
N N
0 N 0 N
(30A-I) (30A-II)
Preparation of Intermediate 4-cyclopropyl-3-iodopyridine (I-30a):
C
NOA
I
(I-30a)
Isoamyl nitrite (2.62gm, 22.38mmol) was added to the stirred solution of 4-
cyclopropylpyridin-3-amine (1.0g, 7.46mmol) in dry THF (15mL) under argon
atmosphere. This was followed by the addition of diiodomethane (3.0 mL,
69
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
22.38mmol) and copper iodide (1.42g, 7.46mmol). The resulting mixture was
refluxed at 80 C for 1 hour. The reaction mixture was cooled, filtered and the
filtrate
was partitioned between ethyl acetate and water. The organic layer was
concentrated
to yield the crude product. Purification by column chromatography on silica
gel (15%
ethyl acetate in hexane) afforded 600mg of the product (34% yield).
1H NMR (CDC13, 400MHz): 6 8.84 (s, 1H), 8.35-8.34 (d, 1H), 6.73-6.71 (d,
1H) 2.10-2.04 (m, 1H), 1.17-1.12 (m, 2H), 0.78-0.74 (m, 2H).
Preparation of Intermediate benzyl 4-(2-chloropyridin-4-y1)-3-oxo-2,4-
diazabicyclo[3.1.0]hexane-2-carboxylate (I-30b)
16\,, 0 =
CI
N I 0
(I-30b)
Using analogous reagents and reaction conditions as described in Example 1
above, benzyl 3-oxo-2,4-diazabicyclo[3.1.0]hexane-2-carboxylate (200mg,
0.861mmol) was reacted with 2-chloro-4-iodopyridine (227mg, 0.947mmo1),
xantphos (45mg, 0.077mmol), Pd2(dba)3 (24mg, 0.025mmol), cesium carbonate
(421mg, 1.29mmol) and 1, 4-dioxane (12mL) at 100 C for 3 hours. Purification
by
column chromatography on silica gel (50% ethyl acetate in hexane) afforded 220
mg
of the product (75% yield).
LCMS Purity: 96%, m/z = 344.0 (M+1)
Preparation of Intermediate 2-(2-chloropyridin-4-y1)-2,4-
diazabicyclo[3.1.0]hexan-3-
one (I-30c):
C, A
--- N ./1\1H
N N 1 11
0
(I-30c)
A solution of benzyl 4-(2-chloropyridin-4-y1)-3-oxo-2,4-diazabicyclo-
[3.1.0]hexane-2-carboxylate (I-30b: 20.0mg, 0.058mmol) in 3.0mL of 6N HC1 was
heated to 100 C for 1 hour. The resulting mixture was basified with 1N NaOH.
The
reaction mixture was cooled and partitioned between ethyl acetate and water.
The
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
organic layer was concentrated and washed with hexane to afford 10mg of the
product
(83% yield).
1H NMR (CDC13, 300 MHz): 6 8.28-8.26 (d, 1H), 7.61-7.60 (t, 2H), 5.7 (s,
1H), 3.6-3.5 (m, 1H), 3.35-3.25 (m, 1H), 1.05-1.02 (q, 1H), 0.62-0.59 (q,
1H)..
LCMS Purity: 90%, m/z = 210.0 (M+1)
Preparation of the title compound 2-(2-chloropyridin-4-yl)-4-(4-
cyclopropylpyridin-
3-yl)-2,4-diazabicyclo[3.1.0]hexan-3-one (racemic mixture) (30A):
Using analogous reagents and reaction conditions as described in Example 1
above, 2-(2-chloropyridin-4-y1)-2,4-diazabicyclo[3.1.0]hexan-3-one (I-30c:
90mg,
0.43mmol) was reacted with 4-cyclopropy1-3-iodopyridine (I-30a: 116mg,
0.47mmol), copper iodide (8mg, 0.043mmol), trans-N,N'-dimethylcyclohexane-1,2-
diamine (6.1mg, 0.043 mmol), potassium phosphate (228mg, 1.07mmol) and 1, 4-
dioxane (5mL) to afford the crude product. Purification by column
chromatography
on silica gel (1.5% methanol in CHC13) afforded 15mg of the product (11%
yield).
1H NMR (CDC13, 400 MHz): 6 8.51 (s, 1H), 8.45-8.44 (d, 1H), 8.32-8.31 (d,
1H), 7.71-7.67 (m, 2H), 6.85-6.84 (d, 1H), 3.72-3.61 (m, 2H), 2.07-2.03 (m,
1H),
1.28-1.10 (m, 3H), 0.93-0.78 (m, 3H).. LCMS Purity: 97%, m/z = 327.1 (M+1).
HPLC Purity: 96%
The racemic mixture of 2-(2-chloropyridin-4-y1)-4-(4-cyclopropylpyridin-3-
y1)-2,4-diazabicyclo[3.1.0]hexan-3-one (30A) was further separated by chiral
preparative HPLC (mobile phases: A: n-hexane; B: IPA, column: CHIRALPAK
AD-H (250 X lOmm, 5p), flow: 7 mL/minute, elution: isocratic (80% of A : 20%
of
B), diluent: IPA, approximate retention times of peaks: 15.0 minutes (peak I)
and 24.0
minutes (peak II)) to obtain two pure isomers: Enantiomer I (30A-I) and
Enantiomer
II (30A-II).
Enantiomer I (30A-I):
1H NMR (CDC13, 400 MHz): 6 8.60-8.38 (m, 2H), 8.31-8.29 (d, 1H), 7.69-
7.66 (m, 2H), 6.86 (s, 1H), 3.71-3.66 (m, 1H), 3.64-3.60 (m, 1H), 2.09-2.01
(m, 1H),
1.35-1.10 (m, 3H), 0.95-0.75 (m, 3H). LCMS Purity: 100%, m/z = 327.1 (M+1).
HPLC Purity: 99%
Enantiomer II (30A-II):
71
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
1H NMR (CDC13, 400 MHz): 6 8.60-8.40 (m, 2H), 8.31-8.29 (d, 1H), 7.69-
7.66 (m, 2H), 6.85 (s, 1H), 3.71-3.59 (m, 2H), 2.08-2.01 (m, 1H), 1.24-1.09
(m, 3H),
0.92-0.77 (m, 3H). LCMS Purity: 100%, m/z = 327.1 (M+1). HPLC Purity: 100%
Example 31
Preparation of (1S,5R)-2-(4-cyclopropylpyridin-3-y1)-4-(2-methoxypyridin-4-y1)-
2,4-
diazabicyclo[3.1.0]hexan-3-one (Enantiomer I. 31A-I), and (1R,5S)-2-(4-
cyclopropylpyridin-3-y1)-4-(2-methoxypyridin-4-y1)-2,4-
diazabicyclo[3.1.0]hexan-3-
one (Enantiomer II: 31A-II):
0
Li 3,, ..3...., r rs-0 H,,A,µH
. .--
N 11 N
0 N 0 N
(31A)
Preparation of Intermediate benzyl 4-(2-methoxypyridin-4-y1)-3-oxo-2,4-
diazabicyclo[3.1.0]hexane-2-carboxylate (I-31a):
16\ 0 *
H3C,o....NraN
0
N N I 0
(I-31a)
Using analogous reagents and reaction conditions as described in Example 1
above, benzyl 3-oxo-2,4-diazabicyclo[3.1.0]hexane-2-carboxylate (50mg,
0.215mmol) was reacted with 4-bromo-2-methoxypyridine (44.5mg, 0.237mmol),
xantphos (11mg, 0.019mmol), Pd2(dba)3 (6.0mg, 0.006mmol), cesium carbonate
(105mg, 0.32mmol) and 1, 4-dioxane (3.0 mL) in seal tube at 100 C for 2.5
hours.
Purification by column chromatography on silica gel (50% ethyl acetate in
hexane)
afforded (35% ethyl acetate in hexane) afforded 30 mg of the product (41%
yield).
1H NMR (CDC13, 300 MHz): 6 8.12-8.10 (d, 1H), 7.47-7.34 (m, 6H), 6.95-
6.94 (d, 1H), 5.4-5.3 (q, 2H), 3.95 (s, 3H), 3.94-3.82 (m, 1H), 3.48-3.43 (m,
1H), 1.2-
1.14 (q, 1H), 0.75-0.70 (m, 1H).
72
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
Preparation of Intermediate 2-(2-methoxypyridin-4-yl)-2,4-diazabicyclo[3.1.0]
hexan-
3-one (I-31b):
..3.....
---- NyNH
N /
0
(I-3 lb)
Using analogous reagents and reaction conditions as described in Example 30
for the preparation of I-3 0c above, benzyl 4-(2-methoxypyridin-4-y1)-3-oxo-
2,4-
diazabicyclo[3.1.0]hexane-2-carboxylate (150.0mg, 0.442 mmol) was treated with
10.0mL of 6N HC1 to yield the crude product. Purification by column
chromatography on silica gel (1.5% methanol in chloroform) afforded 60mg of
the
product (67% yield).
1I-1 NMR (DMSO-D6, 300 MHz): 6 8.18 (s, 1H), 8.02-8.00 (d, 1H), 7.38-7.32
(dd, 1H), 6.957-6.951 (d, 1H).
Preparation of the title compound 2-(4-cyclopropylpyridin-3-yl)-4-(2-
methoxypyridin-
4-yl)-2,4-diazabicyclo[3.1.0]hexan-3-one (31A):
Using analogous reagents and reaction conditions as described in Example 1
above, 2-(2-methoxypyridin-4-y1)-2,4-diazabicyclo[3.1.0]hexan-3-one (1-3 lb:
140mg, 0.682mmo1) was reacted with 4-cyclopropy1-3-iodopyridine (I-30a: 200mg,
0.819mmol), copper iodide (13mg, 0.0682mmo1), trans-N,N'-dimethylcyclohexane-
1,2-diamine (10 mg, 0.0682mmo1), potassium phosphate (435mg, 2.05mmol)and 1, 4-
dioxane (10mL) to yield the crude product. Purification by column
chromatography
on silica gel (1.5% methanol in chloroform) afforded 120mg of the product (57%
yield).
1f1 NMR (CDC13, 400 MHz): 6 8.51 (s, 1H), 8.42-8.41 (d, 1H), 8.11-8.09 (d,
1H), 7.47-7.46 (dd, 1H), 6.96 (d, 1H), 6.83-6.82 (d, 1H), 3.95 (s, 3H), 3.68-
3.55 (m,
2H), 2.09-2.04 (m, 1H), 1.20-0.9 (m, 3H), 0.91-0.79 (m, 3H). LCMS Purity: 98%,
m/z = 323.1 (M+1). HPLC Purity: 99%
The racemic mixture of 2-(4-cyclopropylpyridin-3-y1)-4-(2-methoxypyridin-4-
y1)-2,4-diazabicyclo[3.1.0]hexan-3-one (31A) was further separated by chiral
preparative HPLC (mobile phases: A: n-hexane, B: IPA, column: CHIRALPAK
73
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
AD-H (250 X lOmm, 5p), flow: 5 mL/minute, elution: isocratic (75% of A: 25% of
B), diluent: ethanol, approximate retention times of peaks: 18.8 minutes (peak
I) &
26.5 minutes (peak II)) to obtain two pure isomers: Enantiomer I (3 1A-I) and
Enantiomer II (31A-II).
Enantiomer I (3 1A-I)
1H NMR (CDC13, 400 MHz): 6 8.51 (s, 1H), 8.42-8.41 (d, 1H), 8.11-8.10 (d, 1H),
7.48-7.46 (dd, 1H), 6.96 (d, 1H), 6.83-6.82 (d, 1H), 3.95 (s, 3H), 3.68-3.58
(m, 2H),
2.10-2.04 (m, 1H), 1.25-1.07 (m, 3H), 0.91-0.79 (m, 3H). LCMS Purity: 100%,
m/z =
323.2(M+1)
HPLC Purity: 97%
Enantiomer II (31A-II).
1H NMR (CDC13, 400 MHz): 6 8.16-8.06 (bs, 1H), 7.48-7.46 (dd, 1H), 7.0-6.4
(bs,
1H), 3.96 (s, 3H), 3.68-3.57 (m, 2H), 2.06-2.04 (m, 1H), 1.25-1.10 (m, 3H),
0.91-0.77
(m, 3H). LCMS Purity: 100%, m/z = 323.3 (M+1) HPLC Purity: 98%
Example 32
Preparation of (1S,5R)-2-(4-cyclopropylpyridin-3-y1)-4-(2-
(trifluoromethyl)pyridin-4-
v1)-2,4-diazabicyclo[3.1.0]hexan-3-one (Enantiomer I. 32A-I), and (1R,5S)-2-(4-
cyclopropylpyridin-3-y1)-4-(2-(trifluoromethyl)pyridin-4-y1)-2,4-
diazabicyclo[3.1.0]hexan-3-one (Enantiomer II: 32A-II):
H,
H
CF3 .r1-
CF3 ,A01-.:__)
0..-NN / \ 0.-N N / \
N 11 N [I ,
0 N 0 N
(32A-I) (32A-II)
Preparation of Intermediate benzyl 3-oxo-4-(2-(trifluoromethyl)pyridin-4-y1)-
2,4-
diazabicyclo[3.1.0]hexane-2-carboxylate (I-32a):
16\ 0 =
F3C)N.(I\I--(
N' j 8 0
(I-32a)
74
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
Using analogous reagents and reaction conditions as described in Example 1
above, benzyl 3-oxo-2,4-diazabicyclo[3.1.0]hexane-2-carboxylate (100mg,
0.43mmol) was heated with 4-bromo-2-(trifluoromethyl)pyridine (107mg,
0.474mmo1), xantphos (22.36mg, 0.038mmol), Pd2(dba)3 (12mg, 0.012mmol), cesium
carbonate (210mg, 0.64mmol) and 1, 4-dioxane (6.0mL) at 100 C for 1 hour to
yield
the crude product. Purification by column chromatography on silica gel (35%
ethyl
acetate in hexane) afforded 120 mg of the product (74% yield).
1H NMR (CDC13, 300 MHz): 6 8.67-8.65 (d, 1H), 8.01 (s, 1H), 7.85-7.83 (dd,
1H), 7.5-7.34 (m, 5H), 5.42-5.3 (m, 2H), 3.94-3.90 (m, 1H), 3.56-3.51 (m, 1H),
0.81-
0.76 (m, 2H). LCMS Purity: 82%, m/z = 378.1 (M+1)
Preparation of Intermediate 2-(2-(trifluoromethyl) pyridin-4-yl)-2,4-
diazabicyclo-
[3.1.0]hexan-3-one (I-32b):
F3C
)--NA\
NJ0\ / y NH
0
(I-32b)
Using analogous reagents and reaction conditions as described in Example 30
for the preparation of I-3 0c, benzyl 3-oxo-4-(2-(trifluoromethyl)pyridin-4-
y1)-2,4-
diazabicyclo[3.1.0]hexane-2-carboxylate (I-32a: 55 mg, 0.1458 mmol) was
treated
with 4.0mL of 6N HC1 to yield the 40mg of the product (85% yield).
1H NMR (CDC13, 300 MHz): 6 8.61-8.59 (d, 1H), 7.97-7.96 (d, 1H), 7.79-7.77
(dd, 1H), 5.74 (s, 1H), 3.61-3.57 (m, 1H), 3.35-3.28 (m, 1H), 1.1-1.05 (q,
1H), 0.64-
0.61 (q, 1H).
Preparation of the title compound 2-(4-cyclopropylpyridin-3-yl)-4-(2-
ftrifluoromethyl)pyridin-4-yl)-2,4-diazabicyclo[3.1.0]hexan-3-one as a racemic
mixture (32A):
Using analogous reagents and reaction conditions as described in Example 1
above, 2-(2-(trifluoromethyl)pyridin-4-y1)-2,4-diazabicyclo[3.1.0]hexan-3-one
(I-32b:
60mg, 0.2469mmo1) was treated with 4-cyclopropy1-3-iodopyridine (I-30a:
66.5mg,
0.2716mmol), copper iodide (4.7mg, 0.02469mmo1), trans-N,N' -
dimethylcyclohexane-1,2-diamine (3.5 mg, 0.02469mmo1), potassium phosphate
(157.02mg, 0.7407mmo1), 1-4-dioxane (2mL) was heated to reflux at 115 C for 4
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
hours to yield the crude product. Purification by column chromatography on
silica gel
(1.5% methanol in chloroform), followed by preparative HPLC afforded 45mg of
the
product (51% yield)
1H NMR (CDC13, 400 MHz): 6 8.66-8.64 (d, 1H), 8.52-8.46 (d, 2H), 8.07-8.06
(d, 1H), 7.89-7.87 (dd, 1H), 6.86-6.85 (d, 1H), 3.77-3.73 (m, 1H), 3.68-3.64
(m, 1H),
2.09-2.02 (m, 1H), 1.29-1.23 (q, 1H), 1.22-1.10 (m, 2H), 0.95-0.89 (m, 2H),
0.87-0.78
(m, 1H). LCMS Purity: 100%, m/z = 361.2 (M+1). HPLC Purity: 99%
The racemic mixture of 2-(4-cyclopropylpyridin-3-y1)-4-(2-(trifluoromethyl)
pyridin-4-y1)-2,4-diazabicyclo[3.1.0]hexan-3-one (32A) was further separated
by
chiral preparative HPLC (mobile phases: A: n-hexane: B: IPA, column:
CHIRALPAK AD-H (250 X 10 mm) 5i.tm, flow: 5.0 mL/minute, elution: isocratic
(80% of A: 20% of B), diluent : IPA, approximate retention times of peaks :
13.0
minutes (peak I) & 21.0 minutes (peak II)) to obtain two pure isomers:
Enantiomer I
(32A-I) and Enantiomer II (32A-II).
Enantiomer I (32A-I):
1H NMR (CDC13, 400 MHz): 6 8.65-8.64 (d, 1H), 8.51 (s, 1H), 8.45-8.44 (d,
1H), 8.06 (s, 1H), 7.89-7.87 (d, 1H), 6.85-6.84 (d, 1H), 3.76-3.64 (m, 2H),
2.06-2.03
(m, 1H), 1.35-1.10 (m, 3H), 0.95-0.75 (m,3H). LCMS Purity: 100%, m/z = 361.1
(M+1). HPLC Purity: 100 %
Enantiomer II (32A-II):
1H NMR (CDC13, 400 MHz): 6 8.65-8.64 (d, 1H), 8.51 (s, 1H), 8.45-8.44 (d,
1H), 8.06 (s, 1H), 7.89-7.87 (d, 1H), 6.85-6.84 (d,1H), 3.76-3.64 (m, 2H),
2.06-2.03
(m, 1H), 1.35-1.10 (m, 3H), 0.95-0.75 (m, 3H). LCMS Purity: 100%, m/z = 361.2
(M+1). HPLC Purity: 99%
Example 33
Preparation of 1-(2-chloro-pyridin-4-y1)-3-14-(1-hydroxy-cyclobuty1)-pyridin-3-
ylr
imidazolidin-2-one (33A):
76
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
CI
HO r\N--6
I 0
(33A)
Preparation of Intermediate 1-(2-chloroethyl)-3-(2-chloro-pyridin-4-y1) urea
(I-34a):
0
HNANCI
H
NCI
(I-33a)
Using analogous reagents and reaction conditions as described in Example 1
above, 4-amino-2-chloro pyridine (3.0g, 23.33mmol) in toluene (30mL) was
reacted
with 1-chloro-2-isocyanatoethane (3.69g, 35.0mmol) and purified by column
chromatography on silica gel (1.5% methanol in DCM) to afford 3.3g of the
product
(61% yield).
114 NMR (CDC13, 300 MHz): 6 8.50 (s, 1H), 8.10 (d, 1H), 7.50 (d, 1H), 7.30-
7.20 (m, 1H), 6.1 (bs, 1H), 3.70-3.60 (m, 4H).
Preparation of Intermediate 1-(2-chloro-pyridin-4-y1) imidazolidin-2-one (I-
33b):
CI
H N
II N
0
(I-33b)
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(2-chloroethyl)-3-(2-chloro-pyridin-4-y1) urea (I-33a: 7.0g,
30.042mmol) in
dry THF (40mL) was reacted with 60%NaH (1.08g, 45.06mmol) in dry THF (30mL)
to afford the crude product. Purification by column chromatography on silica
gel (1%
methanol in DCM) afforded 5.40g of the product (92% yield).
114 NMR (DMSO-D6, 300 MHz): 6 8.20-8.18 (d, 1H), 7.65 (d, 1H), 7.53-7.49
(m, 2H), 3.90-3.84 (m, 2H), 3.47-3.41 (m, 2H). LCMS Purity: 98%, m/z = 198.1
(M+1). HPLC Purity: 98%
Preparation of Intermediate 1-(3-Bromo-pyridin-4-y1)-cyclobutanol (I-33c):
77
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
Br H
1
N
(I-33c)
3-bromopyridine (1g, 6.329mmo1) was reacted with cylcobutanone (0.93 mL,
6.369 mmol), n-butyl lithium (4.02 mL, 7.64 mmol) and diisopropyl amine (0.99
mL,
7.005 mmol) in dry THF to afford the crude product. Purification by column
chromatography on silica gel (40% ethyl acetate in hexane) afforded 802 mg of
the
product (55% yield). LCMS Purity: 79%, m/z=227.9 (M+1)
Preparation of the title compound 1-(2-chloro-pyridin-4-yl)-314-(1-hydroxy-
cyclobutyl)-pyridin-3-ylrimidazolidin-2-one (33A):
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(2-chloro-pyridin-4-y1)-imidazolidin-2-one (I-33b: 100mg, 0.5076
mmol)
was reacted with 1-(3-bromo-pyridin-4-y1)-cyclobutanol (1-33c: 127mg, 0.5583
mmol) copper iodide (9.6mg, 0.0507mmol), trans-N,N'-dimethylcyclohexane-1,2-
diamine (21 mg, 0.1522 mmol), potassium phosphate (269 mg, 1.269 mmol) and 1,
4-
dioxane (3 mL) to afford crude product. Purification by column chromatography
on
silica gel (2% methanol in CHC13), followed by preparative HPLC afforded 25 mg
of
pure product (14 % yield)
114 NMR (CDC13, 400 MHz): 6 8.59-8.53 (d, 2H), 8.31-8.29 (d, 1H), 7.57-7.55
(dd, 1H), 7.49 (d, 1H), 7.33-7.32 (d, 1H), 4.76 (s, 1H), 4.10-4.09 (m, 4H),
2.45-2.34
(m, 6H). LCMS Purity: 96%, m/z = 345.1 (M+1). HPLC Purity: 99%
Example 34
Preparation of 1-(2-chloropyridin-4-yl)-3-(4-cyclopentyl-pyridin-3-yl)-
imidazolidin-
2-one (34A):
CI
--- --N/
N
(34A)
78
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
Preparation of Intermediate 1-(3-Bromo¨pyridin-4-y1)¨cyclopentanol (I-35a):
NocBr 0
I
HO
(I-34a)
Using analogous reaction condition and reagents as described in Example 20
yield).
1H NMR (CDC13, 300 MHz): 6 8.68 (s, 1H), 8.48-8.46 (d, 1H), 7.60-7.58 (d,
1H), 2.41-2.34 (m, 3H), 2.05-1.90 (m, 7H), 1.25-1.21 (m, 1H)
LCMS Purity: 97%, m/z = 242.1 (M+1)
N Br
1
/
111
(I-34b)
PTSA (819mg, 4.315mmol) was added portionwise to a stirred mixture of 1-
(3-bromo¨pyridin-4-y1)¨cyclopentanol (I-34a: 520mg, 2.15mmol) in 25mL of
LCMS Purity: 100%, m/z = 224.1 (M+1)
Preparation of Intermediate 3-bromo-4-cyclopentyl-pyridine (I-34c):
79
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
NUS Br
(I-34c)
Platinum oxide (20mg) was added to a mixture of 3-bromo-4-cyclopent-1-
enyl-pyridine (I-34b: 140mg, 0.622mmo1) in toluene (5mL) and stirred overnight
under hydrogen atmosphere. The reaction was monitored by TLC (20% ethyl
acetate
in hexane). The reaction mixture was filtered through celite pad and washed
with
toluene and concentrated to afford crude product. Purification by column
chromatography (8% ethyl acetate in hexane) afforded 98mg of the product (71%
yield).
1H NMR (CDC13, 300 MHz): 6 8.64 (s, 1H), 8.42-8.40 (d, 1H), 7.21-7.19 (d,
1H), 3.37-3.34 (m, 1H), 2.18-2.12 (m, 2H), 1.87-1.73 (m, 6H).
Preparation of the title compound 1-(2-chloropyridin-4-yl)-3-(4-cyclopentyl-
pyridin-
3-yl)-imidazolidin-2-one (34A):
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(2-chloro-pyridin-4-y1) imidazolidin-2-one (I-33b: 75mg, 0.38mmol)
was
reacted with and 3-bromo-4-cyclopentyl-pyridine (I-34c: 95mg, 0.418mmol)
copper
iodide (7.22mg, 0.38mmol), trans-N,N'-dimethylcyclohexane-1,2-diamine
(16.21mg,
0.114mmol), potassium phosphate (241.9mg, 1.15mmol) and 1, 4-dioxane (5mL) to
yield the crude product. Purified by preparative HPLC afforded 2.50mg of the
product
(2% yield).
1H NMR (CDC13, 300 MHz): 6 8.70-8.40 (m, 2H), 8.30-8.28 (d, 1H), 7.70-
7.45 (m, 2H), 7.36 (s, 1H), 4.1-3.90 (m, 4H), 3.30-3.0 (m, 1H), 2.30-2.0 (m,
3H),
1.95-1.70 (m, 5H). LCMS Purity: 100%, m/z = 343.3 (M+1). HPLC Purity: 96%
Example 35
Preparation of 1-(2-chloropyridin-4-yl)-314-(1-hydroxy cyclopentyl)-pyridin-3-
yli-
imidazolidin-2-one (35A):
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
%)H
CI /--\
)0,N yN / \
Nk i
0 N
(35A)
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(2-chloro-pyridin-4-y1) imidazolidin-2-one (I-33b: 100mg, 0.5mmol)
was
reacted with 1-(3-bromo¨pyridin-4-y1)¨cyclopentanol (I-34a: 135mg, 0.558mmol),
copper iodide (9.6mg, 0.05mmol), trans-N,N'-dimethylcyclohexane-1,2-diamine
(21.62mg, 0.15mmol), potassium phosphate (322.87mg, 1.52mmol) and 1, 4-dioxane
(5mL) to yield the crude product. Purification by preparative HPLC afforded
20mg of
the product (11% yield).
ifl NMR (DMSO-D6, 400 MHz): 6 8.52-8.51 (m, 2H), 8.26-8.24 (d,1H), 7.69
(d, 1H),7.61-7.59 (dd, 1H), 7.56-7.55 (d,1H), 5.09 (s, 1H), 4.1-3.99 (m, 3H),
3.90-
3.75 (bs, 1H), 2.10-1.9, (m, 3H), 1.78 -1.63 (m, 5H). LCMS Purity: 98%, m/z =
359.1 (M+1). HPLC Purity: 96%
Example 36
Preparation of 114-(1-hydroxy-cyclopropy1)-pyridin-3-y1]-3-(3-trifluoromethyl-
phenyl)-imidazolidin-2-one (36A):
CF3. N1r
r-----\ H
N _
1
0 Nr
(36A)
Preparation of Intermediate 1-(2-Chloro-ethyl)-3-(3-trifluoromethyl-phenyl)-
urea (I-
36a):
H H
F3C 00
(I-36a)
Using analogous reagents and reaction conditions as described in Example 1
above, 3-trifluoromethyl-phenylamine (2.0g, 12.412mmol) in toluene (20mL) was
81
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
reacted with 1-chloro-2-isocyanatoethane (1.96 g, 18.618 mmol to give 3.2g of
pure
product(96 %)
1H NMR (CDC13, 300 MHz): 6 7.60-7.20 (m, 5H), 5.75 (bs, 1H), 3.80-3.45
(m, 4H). LCMS Purity: 100%, m/z = 266.9 (M+1)
Preparation of Intermediate 1-(3-trifluoromethyl-phenyl)-imidazolidin-2-one (I-
36b):
-NH
F3C 0
(1-36b)
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(2-chloro-ethyl)-3-(3-trifluoromethyl-pheny1)-urea (1-36a: 3.2g,12.02
mmol) was reacted with sodium hydride (870 mg, 18.03 mmol) in dry THF (35mL to
give 2.9g of product(95%)
1H NMR (CDC13, 300 MHz): 6 7.86-7.70 (m, 2H), 7.50-7.40 (t, 1H), 7.35-7.24
(m, 1H), 5.20 (bs, 1H), 4.0-3.90 (t, 2H), 3.70-3.60 (t, 2H). LCMS Purity:
100%, m/z
= 231 (M+1)
Preparation of Intermediate 312-0xo-3-(3-trifluoromethyl-phenyl)-imidazolidin-
l-
vlrisonicotinic acid ethyl ester (I-36c):
CF3 0.,...õ.0CH2CH3
. NIMN
1i i=
0
N
(I-36c)
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(3-trifluoromethyl-pheny1)-imidazolidin-2-one (1-36b: 200mg, 0.869
mmol)
was reacted with 3-bromo-isonicotinic acid ethyl ester (239 mg, 1.042 mmol),
copper
iodide (16.5mg, 0.869mmo1), trans-N, N'-dimethylcyclohexane-1,2-diamine (37.01
mg, 0.26 mmol), potassium phosphate (460.5 mg, 2.172 mmol) and 1, 4-dioxane (5
mL) to afford crude product. Purification by column chromatography on silica
gel (60
% ethyl acetate in hexane) afforded 280 mg of pure product (85 % yield).
82
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
1H NMR (CDC13, 300 MHz): 6 8.64-8.61 (m, 1H), 7.84-7.82 (d, 2H), 7.73-
7.72 (d, 1H), 7.47 (t, 1H), 7.40-7.32 (m, 1H), 4.38-4.31 (q, 2H), 4.13-4.09
(m, 4H),
1.34-1.30 (t, 3H).
Preparation of the title compound 114-(1-hydroxy-cyclopropyl)-pyridin-3-yl]-3-
(3-
trifluoromethyl-phenyl)-imidazolidin-2-one (36A):
2.5 M Solution of EtMgBr (0.8mL, 1.974 mmol) was added drop wise to a
solution of titanium(IV)isopropoxide (187.3mg, 0.659 mmol) in diethyl ether
(10 mL)
for a period of 15 minutes at -78 C under nitrogen atmosphere. The resulting
mixture
was stirred for 90 minutes. This was followed by the addition of 342-oxo-3-(3-
trifluoromethyl-pheny1)-imidazolidin-l-y1]-isonicotinic acid ethyl ester (I-
36c: 125
mg, 0.329 mmol) in diethyl ether (3 mL) at -78 C and continued stirring at
room
temperature overnight. The reaction was monitored by TLC (80 % ethyl acetate
in
hexane). The reaction mixture was cooled and partitioned between ethyl acetate
and
1N HC1. The organic layer was concentrated to yield crude product which was
purified by preparative HPLC to afford 8 mg of the product (6 % yield).
1H NMR (CDC13, MHz): 6 8.52 (s, 1H), 8.41 (d, 1H), 7.99-7.98 (d, 1H), 7.87-
7.85 (d, 1H), 7.76-7.74 (dd, 1H), 7.47-7.46 (d, 1H), 7.32-7.31 (d, 1H), 6.82-
6.80 (d,
1H), 4.17-4.14 (m, 2H), 4.03-3.99 (m, 2H), 2.15-2.05 (m, 1H), 1.15-1.11 (m,
2H),
0.86-0.82 (m, 2H). LCMS Purity: 95%, m/z = 336.1(M+1). HPLC Purity: 98%
Example 37
Preparation of 1-(2-chloro-6-(trifluoromethyl)pyridin-4-yl)-3-(4-
cyclopropylpyridin-
3-yl)imidazolidin-2-one (37A):
n\l.ill)
CIN..1 \ /
N 0
CF3
(37A)
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-id: 110mg,
0.5412mmol)
was reacted with 2-chloro-4-iodo-6-(trifluoromethyl)pyridine (200mg,
0.6494mmo1),
1, 4-dioxane (5mL), copper iodide (10mg, 0.0541mmol), trans-N1,N2-
dimethylcyclohexane-1,2-diamine (23mg, 0.1623mmol) and potassium phosphate
(345mg, 1.6236mmol) to afford the crude product. Purification by column
83
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
chromatography on silica gel (1.5% methanol in CHC13), followed by prep HPLC
afforded 22mg of the product (10% yield).
1H NMR (CDC13, 300 MHz): 6 8.65-8.35 (bs, 2H), 8.0 (s, 1H), 7.7 (s, 1H),
6.90-6.80 (bs, 1H), 4.15-4.05 (m, 4H), 2.0-1.9 (m, 1H), 1.20-1.10 (m, 2H),
0.92-0.80
(m, 2H). LCMS Purity: 98%, m/z = 383.1 (M+1). HPLC Purity: 99%
Example 38
Preparation of 1-(4-cyclopropylpyridin-3-y1)-3-(6-cyclopropylpyrimidin-4-y1)
imidazolidin-2-one (38A):
1\11\1 Nr¨\YN
0 N
(38A)
Preparation of Intermediate 1-(6-chloropyrimidin-4-y1)-3-(4-cyclopropylpyridin-
3-y1)
imidazolidin-2-one (I-38a):
iYN Nr¨NYN / \
0 N
CI
(1-38a)
Using analogous reagents and reaction conditions as described in Example 11
above, 1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-id: 500mg, 2.46mmol)
was reacted with 4,6-dichloro-pyrimidine (403mg, 2.70mmol), 2-
dicyclohexylphosphino-2',4',6'-triisopropylbiphenyl (117mg, 0.246mmo1),
Pd2(dba)3
(112mg, 0.123mmol), cesium carbonate (2.009g, 6.15mmol) and toluene (10 mL) in
seal tube at 110 C for 14 hours. Purification by column chromatography on
silica gel
(1.5% methanol in chloroform) afforded 140 mg of the product (18.18% yield).
1HNMR (CDC13, 300MHz): 6 8.688-8.684 (d, 1H), 8.48-8.46 (m, 2H), 8.375-
8.371 (d, 1H), 6.85-6.83 (d, 1H), 4.33-4.27 (t, 2H), 4.03-3.98 (t, 2H), 2.05-
1.95 (m,
1H), 1.21-1.12 (m, 2H), 0.88-0.82 (m, 2H). LCMS: 99.53%, m/z = 315.9 (M+1).
84
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
Preparation of 1-(4-cyclopropylpyridin-3-y1)-3-(6-cyclopropylpyrimidin-4-y1)
imidazolidin-2-one (38A):
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(6-chloropyrimidin-4-y1)-3-(4-cyclopropylpyridin-3-yl)imidazolidin-2-
one
(I-38a: 140mg, 0.44mmol) was reacted with cyclopropyl boronic acid (46mg,
0.533mmo1), Pd(PPh3)4 (26mg, 0.02mmol), potassium carbonate (123mg,
0.888mmo1) and xylene (10 mL) in seal tube at 125 C for 14 hours. Purification
by
column chromatography on silica gel (1% methanol in chloroform) afforded 55 mg
of
the product (38.73% yield).
11-INMR (CDC13, 300MHz): 6 8.70 (s, 1H), 8.48 (s, 1H), 8.45-8.43 (d, 1H),
8.15 (s, 1H), 6.83-6.81 (d, 1H), 4.30-4.25 (t, 2H), 4.00-3.94 (t, 2H), 2.00-
1.96 (m,
2H), 1.16-1.04 (m, 6H), 0.86-0.83 (m, 2H). LCMS: 98.39%, m/z = 322.2 (M+1).
HPLC: 90.41%.
Example 39
Preparation of 1-(4-cyclopropylpyridin-3-y1)-3-(2-cyclopropylpyridin-4-y1)
imidazolidin-2-one (39A):
1----\
?y
N
0 ---N
(39A)
Preparation of Intermediate 1-(2-chloropyridin-4-y1)-3-(4-cyclopropylpyridin-3-
y1)
imidazolidin-2-one (I-39a):
CI
0 ---C4
\ / N
Na,13.
(I-39a)
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-id: 200mg,
0.985mmo1)
was reacted with 2-chloro-4-iodo pyridine (283mg, 0.118mmol) 1, 4-dioxane
(10mL),
copper iodide (19mg, 0.0985mmo1), trans-N,N'-dimethylcyclohexane-1,2-diamine
(14mg, 0.0985mmo1) and potassium phosphate (626mg, 2.95mmol) to afford the
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
crude product. Purification by column chromatography on silica gel (2%
methanol in
chloroform) afforded 220 mg of the product (71.10% yield).
LCMS: 100%, m/z = 315.1 (M+1).
Preparation of 1-(4-cyclopropylpyridin-3-y1)-3-(2-cyclopropylpyridin-4-y1)
imidazolidin-2-one (39A):
1-(2-chloropyridin-4-y1)-3-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-
39a: 220mg, 0.7mmol) was reacted with cyclopropyl boronic acid (72mg,
0.84mmol),
Pd(PPh3)4 (41mg, 0.035mmol), potassium carbonate (194g, 1.4mmol) and xylene
(10
mL) in seal tube at 125 C for 14 hours. Purification by column chromatography
on
silica gel (1% methanol in chloroform) afforded 75 mg of the product (33.48%
yield).
ifINMR (CDC13, 400MHz): 6 8.48 (s, 1H), 8.43-8.42 (d, 1H), 8.34-8.33 (d,
1H), 7.50-7.49 (d, 1H), 7.24-7.22 (dd, 1H), 6.81-6.80 (d, 1H), 4.09-3.97 (m,
4H),
2.05-1.98 (m, 2H), 1.15-1.10 (m, 2H), 1.05-0.94 (m, 4H), 0.85-0.81 (m, 2H).
LCMS:
100%, m/z = 321.2 (M+1). HPLC: 98.90%.
Example 40
Preparation of 1-(4-cyclopropylpyridin-3-y1)-3-(6-methoxypyrimidin-4-y1)
imidazolidin-2-one (40A):
1----\
0 N
0
µCH3
(40A)
Using analogous reagents and reaction conditions as described in Example 11
above, 1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-id: 100mg,
0.492mmo1)
was reacted with 4-chloro-6-methoxypyrimidine (78mg, 0.541mmol), 2-
dicyclohexyl-
phosphino-2',4',6'-triisopropylbiphenyl (23mg, 0.0492mmo1), Pd2(dba)3 (23mg,
0.024mmol), cesium carbonate (400mg, 1.23mmol) and toluene (10 mL) in seal
tube
at 115 C for 14 hours. Purification by column chromatography on silica gel (2%
methanol in chloroform) afforded 50 mg of the product (34.6% yield).
ifINMR (CDC13, 400MHz): 6 8.53 (s, 1H), 8.47 (s, 1H), 8.43-8.42 (d, 1H),
7.66 (s, 1H), 6.82-6.80 (d, 1H), 4.30-4.26 (t, 2H), 3.97-3.93 (m, 5H), 2.02-
1.98 (m,
86
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
1H), 1.15-1.10 (m, 2H), 0.85-0.80 (m, 2H). LCMS: 98.15%, m/z = 313.1 (M+1).
HPLC: 99.04%.
Example 41
Preparation of 1-(4-cyclopropylpyridin-3-y1)-3-(6-methylpyrimidin-4-y1)
imidazolidin-2-one (41A):
1-----\
N2N NYN \
0 N
H3C
(41A)
Using analogous reagents and reaction conditions as described in Example 11
above, 1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-id: 100mg,
0.492mmo1)
was reacted with 4-chloro-6-methylpyrimidine (73mg, 0.541mmol), 2-dicyclohexyl-
phosphino-2',4',6'-triisopropylbiphenyl (23mg, 0.0492mmo1), Pd2(dba)3 (23mg,
0.024mmol), cesium carbonate (400mg, 1.23mmol) and toluene (10 mL) in seal
tube
at 115 C for 14 hours. Purification by column chromatography on silica gel (2%
methanol in chloroform) afforded 30 mg of the product (21.1% yield).
11-11\IMR (CDC13, 300MHz): 6 8.78 (s, 1H), 8.48 (s, 1H), 8.45-8.43 (d, 1H),
8.16 (s, 1H), 6.83-6.81 (d, 1H), 4.31-4.28 (t, 2H), 4.01-3.95 (t, 2H), 2.49
(s, 3H), 2.02-
1.98 (m, 1H), 1.18-1.11 (m, 2H), 0.88-0.82 (m, 2H). LCMS: 100%, m/z = 296.1
(M+1). HPLC: 92.00%.
Example 42
Preparation of 1-(4-cyclopropylpyridin-3-y1)-3-(5-fluoro-4-methylpyridin-2-y1)
imidazolidin-2-one (42A):
1----\
N
F X 0 N
H3C
87
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
(42A)
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-id: 100mg,
0.492mmo1)
was reacted with 2-bromo-5-fluoro-4-methylpyridine (102mg, 0.541mmol) 1, 4-
dioxane (5mL), copper iodide (9mg, 0.0492mmo1), trans-N,N'-dimethylcyclohexane-
1,2-diamine (21mg, 0.147mmol) and potassium phosphate (313mg, 1.47mmol) to
afford the crude product. Purification by column chromatography on silica gel
(80%
ethyl acetate in hexane) afforded 60 mg of the product (39.21% yield).
11-11\IMR (CDC13, 300MHz): 6 8.19-8.17 (d, 1H), 8.05 (s, 1H), 7.27 (s, 1H),
4.27-4.22 (t, 2H), 3.96-3.91 (t, 2H), 2.30 (s, 3H), 2.08-2.00 (m, 1H), 1.13-
1.10 (m,
2H), 0.83-0.81 (m, 2H). LCMS: 100%, m/z = 314.1 (M+1). HPLC: 92.09%.
Example 43
Preparation of 1-(4-cyclopropylpyridin-3-y1)-3-(2-cyclopropylpyrimidin-4-y1)
imidazolidin-2-one (43A):
ki i-----\
--
0 N
(43A)
Preparation of Intermediate 4-chloro-2-cyclopropylpyrimidine (I-43a):
CI N
YA
N
(I-43a)
POC13 (5mL) was added to 2-cyclopropylpyrimidin-4-ol (100mg, 0.735mmo1)
and refluxed at 150 C for 2.5 hours. The resulting mixture was concentrated,
the
concentrate was basified with dilute sodium bicarbonate solution and diluted
with
DCM. The organic layer was washed with brine, dried over sodium sulphate and
concentrated to afford 125mg of the crude product as a pale brown liquid.
11-11\IMR (CDC13, 300MHz): 6 8.42-8.40 (d, 1H), 7.09-7.07 (d, 1H), 2.25-2.20
(m, 1H), 1.24-1.14 (m, 4H).
88
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
Preparation of 1-(4-cyclopropylpyridin-3-y1)-3-(2-cyclopropylpyrimidin-4-y1)
imidazolidin-2-one (43A):
Using analogous reagents and reaction conditions as described in Example 11
above, 1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-id: 100mg,
0.492mmo1)
was reacted with 4-chloro-2-cyclopropylpyrimidine (I-43a: 92mg, 0.591mmol), 2-
dicyclohexylphosphino-2',4',6'-triisopropylbiphenyl (24mg, 0.0492mmo1),
Pd2(dba)3
(23mg, 0.0246mmo1), cesium carbonate (400mg, 1.23mmol) and toluene (5 mL) in
seal tube at 110 C for 14 hours. Purification by column chromatography on
silica gel
(2% methanol in chloroform), followed by preparative HPLC afforded 25 mg of
the
product (% yield).
11-11NMR (CDC13, 300MHz): 6 8.46 (s, 1H), 8.43-8.42 (d, 1H), 8.36-8.34 (d,
1H), 8.00-7.97 (dd, 1H), 6.81-6.80 (d, 1H), 4.28-4.23 (t, 2H), 3.97-3.92 (t,
2H), 2.19-
2.14 (m, 1H), 2.01-1.95 (m, 1H), 1.15-0.99 (m, 6H), 0.87-0.79 (m, 2H). LCMS:
96.64%, m/z = 322.1 (M+1). HPLC: 98.82%.
Example 44
Preparation of 1-(4-cyclopropylpyridin-3-y1)-3-(2-methylpyrimidin-4-y1)
imidazolidin-2-one (44A):
H3C N r---\
N Y N / \
0 N
(44A)
Using analogous reagents and reaction conditions as described in Example 11
above, 1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-id: 100mg,
0.492mmo1)
was reacted with 4-chloro-2-methylpyrimidine (68mg, 0.541mmol), 2-dicyclohexyl-
phosphino-2',4',6'-triisopropylbiphenyl (23mg, 0.0492mmo1), Pd2(dba)3 (23mg,
0.024mmol), cesium carbonate (400mg, 1.23mmol) and toluene (5 mL) in seal tube
at
110 C for 14 hours. Purification by column chromatography on silica gel (1.5%
methanol in chloroform) afforded 76 mg of the product (52% yield).
11-11NMR (CDC13,400MHz): 6 8.48-8.41 (m, 3H), 8.08-8.06 (d, 1H), 6.83-6.81
(d, 1H), 4.31-4.27 (t, 2H), 3.99-3.95 (t, 2H), 2.64 (s, 3H), 2.03-1.97 (m,
1H), 1.15-
89
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
1.10 (m, 2H), 0.88-0.81 (m, 2H). LCMS: 98.50%, m/z = 296.1 (M+1). HPLC:
96.16%.
Example 45
Preparation of 1-(6-chloro-2-(trifluoromethyl) pyrimidin-4-y1)-3-(4-
cyclopropylpyridin-3-y1) imidazolidin-2-one (45A):
CI [----\
---)--*--¨N N....;
i y \
0 N
F3C
(45A)
Using analogous reagents and reaction conditions as described in Example 11
above, 1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-id: 100mg,
0.492mmo1)
was reacted with 4,6-dichloro-2-trifluoromethylpyrimidine (120mg, 0.541mmol),
xantphos (25mg, 0.04428mmo1), Pd2(dba)3 (15mg, 0.01476mmo1), cesium carbonate
(225mg, 0.6888mmo1) and 1,4-dioxane (5 mL) in a sealed tube for 5 hours.
Purification by column chromatography on silica gel (1% methanol in
chloroform)
followed by preparative HPLC afforded 25 mg of the product (13.24% yield).
11-11\IMR (CDC13, 300MHz): 6 8.51-8.46 (m, 2H), 7.26 (m, 1H), 6.85-6.83 (d,
1H), 4.38-4.32 (t, 2H), 4.05-4.00 (t, 2H), 2.00-1.90 (m, 1H), 1.16-1.11 (m,
2H), 0.88-
0.84 (m, 2H). LCMS: 96.71%, m/z = 383.6 (M+1). HPLC: 98.68%.
Example 46
Preparation of 1-(4-cyclopropylpyridin-3-y1)-3-(2, 6-dichloropyridin-4-y1)
imidazolidin-2-one (46A):
CI 1----\
N.----3
N 1
N 0 N
CI
(46A)
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-id: 500mg,
2.4630mmol)
was reacted with 2,6-dichloro-4-iodopyridine (810 mg, 2.9556 mmol), copper
iodide
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
(47mg, 0.2463mmo1), trans-cyclohexane-1,2-diamine (0.089 mL, 0.7389 mmol),
potassium phosphate (1.56 g, 7.3891 mmol) and 1, 4-dioxane (10 mL) to afford
crude
product. Purification by column chromatography on silica gel (2% methanol in
chloroform) afforded 800 mg of pure product (93.02 % yield).
11-11\IMR (CDC13, 300MHz): 6 8.48-8.43 (m, 2H), 7.57 (s, 2H), 6.85-6.81 (d,
1H), 4.04 (s, 4H), 2.00-1.90 (m, 1H), 1.20-1.10 (m, 2H), 0.9-0.8 (m, 2H).
LCMS:
97.01%, m/z = 348.8 (M+1). HPLC: 99.40%.
Example 47
Preparation of 1-(2-chloro-6-cyclopropylpyridin-4-y1)-3-(4-cyclopropylpyridin-
3-y1)
imidazolidin-2-one (47A):
p---:-:\ Nr-1
%----)Or IN
CI
(47A)
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(4-cyclopropyl pyridin-3-y1)-3-(2,6-dichloropyridin-4-y1)
imidazolidin-2-one
(I-46a: 100mg, 0.2865mmo1) was reacted with cyclopropyl boronic acid (74mg,
0.8593mmo1), Pd(PPh3)4 (7mg, 0.0057mmol), sodium carbonate (69g, 0.6590mmo1)
and toluene (5 mL) in seal tube at 120 C for 12 hours. Purification by column
chromatography on silica gel (1% methanol in chloroform), followed by
preparative
HPLC afforded 20 mg of the product (20% yield).
11-INMR (CDC13, 400MHz): 6 8.49-8.45 (m, 2H), 7.524-7.520 (d, 1H), 7.21-
7.20 (d, 1H), 6.84-6.82 (d, 1H), 4.08-3.99 (m, 4H), 2.03-1.96 (m, 2H),1.17-
1.12 (m,
2H), 1.07-0.96 (m, 4H), 0.87-0.83 (m, 2H). LCMS: 100%, m/z = 355.1 (M+1).
HPLC: 98.74%.
Example 48
Preparation of 1-(2-cyclopropy1-6-(trifluoromethyl) pyridin-4-y1)-3-(4-
cyclopropylpyridin-3-y1) imidazolidin-2-one (48A):
91
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
A r--\N--)
N(
\ -N
AN ,) I
CF3
(48A)
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-id: 260mg,
1.2807mmol)
was reacted with 2-chloro-4-iodo-6-(trifluoromethyl)pyridine (471 mg, 1.5369
mmol),
copper iodide (24mg, 0.1280mmol), trans-cyclohexane-1,2-diamine (15 mg, 0.1280
mmol), potassium phosphate (816mg, 3.8421 mmol) and 1, 4-dioxane (5 mL) to
afford crude product. Purification by column chromatography on silica gel (1%
methanol in chloroform) afforded 250 mg of pure product (51.1 % yield).
11-INMR (CDC13, 400MHz): 6 8.55-8.42 (bs, 2H), 7.64-7.62 (d, 2H), 6.85 (s,
1H), 4.13-4.02 (m, 4H), 2.10-1.98 (m, 2H), 1.18-0.99 (m, 6H), 0.87-0.83 (m,
2H).
LCMS: 98.89%, m/z = 388.9 (M+1). HPLC: 99.67%.
Example 49
Preparation of 1-(2,6-bis(triflnoromethyl)pyridin-4-y1)-3-(4-
cyclopropylpyridin-3-y1)
imidazolidin-2-one (49A):
F3CNõ,i
1 \I:\D -N
NI
CF3
(49A)
Using analogous reagents and reaction conditions as described in Example 11,
1-(4-cyclopropylpyridin-3-yl)imidazolidin-2-one (I-1c1: 90mg, 0.4408mmo1) was
reacted with 4-chloro-2,6-trifluoromethylpyridine (100mg, 0.4007mmol), 2-
dicyclohexyl-phosphino-2',4',6'-triisopropylbiphenyl (10mg, 0.02mmol),
Pd2(dba)3
(19mg, 0.02mmol), cesium carbonate (183mg, 0.561 Ommol) and toluene (5 mL) in
seal tube at 110 C for 14 hours. Purification by column chromatography on
silica gel
(2% methanol in chloroform) followed by preparative HPLC afforded 81 mg of the
product (48.79% yield).
92
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
11-INMR (CDC13, 300MHz): 6 8.48-8.46 (m, 2H), 8.12 (s, 2H), 6.85-6.84 (d,
1H), 4.20-4.07 (m, 4H), 1.98-1.93 (m, 1H), 1.18-1.11 (m, 2H), 0.88-0.83 (m,
2H).
LCMS: 100%, m/z = 417.4 (M+1). HPLC: 98.38%.
Example 50
Preparation of 1-(4-cyclopropy1-2-hydroxypyridin-3-y1)-3-(2-(trifluoromethyl)
pyridin-4-y1) imidazolidin-2-one (50A):
F3C
N2----N N
Y 1
0
HO N
(50A)
Preparation of Intermediate 4-cyclopropy1-3-iodopyridine 1-oxide (I-50a):
I
(I-50a)
m-Chloroperbenzoic acid (408mg, 2.36mmol) was added to the solution of 4-
cyclopropy1-3-iodopyridine (I-30a: 200mg. 0.816mmol) in DCM (10mL) and the
resulting mixture was stirred at room temperature for 3 hours. The reaction
was
monitored by TLC. The reaction mixture was quenched with ice and diluted with
DCM. The organic layer was washed with dilute NaOH solution, water and brine
solution, dried over sodium sulphate and concentrated to afford 200 mg of the
product
(94.7% yield).
11-INMR (DMSO-D6, 300MHz): 6 8.59-8.58 (d, 1H), 8.09-8.06 (dd, 1H), 6.91-
6.89 (d, 1H), 2.05-1.90 (m, 1H), 1.08-1.04 (m, 2H), 0.74-0.72 (m, 2H). LCMS:
99.45%, m/z = 261.4 (M+1).
Preparation of Intermediate 4-cyclopropy1-3-(2-oxo-3-(2-(trifluoromethyl)
pyridin-4-
v1) imidazolidin-1-y1) pyridine 1-oxide (I-50b):
F3C [¨A
Nr I NYN / \
0 N+
6-
93
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
(I-50b)
Using analogous reagents and reaction conditions as described in Example 1
above, 1-(3-trifluoromethyl-pheny1)-imidazolidin-2-one (I-36b: 60mg, 0.259
mmol)
was reacted with 4-cyclopropy1-3-iodopyridine 1-oxide (I-50a: 81 mg, 0.31
mmol),
copper iodide (5mg, 0.025mmol), trans-N, N'-dimethylcyclohexane-1,2-diamine (4
mg, 0.025 mmol), potassium phosphate (165 mg, 0.77 mmol) and 1, 4-dioxane (5
mL)
to afford crude product. Purification by column chromatography on silica gel
(1.5%
methanol in chloroform) afforded 65 mg of pure product (71.4 % yield).
11-1NMR (CDC13, 400MHz): 6 8.63-8.62 (d, 1H), 8.19-8.18 (d, 1H), 8.10-8.08
(dd, 1H), 7.92-7.91 (d, 1H), 7.73-7.71 (dd, 1H), 6.84-6.83 (d, 1H), 4.15-4.11
(m, 2H),
4.05-4.01 (m, 2H), 1.96-1.92 (m, 1H), 1.16-1.11 (m, 2H), 0.81-0.78 (m, 2H).
LCMS:
100%, m/z = 365.1 (M+1). HPLC: 99.70%.
Preparation of 1-(4-cyclopropy1-2-hydroxypyridin-3-y1)-3-(2-(trifluoromethyl)
pyridin-4-y1) imidazolidin-2-one (50A):
4-Cyclopropy1-3-(2-oxo-3-(2-(trifluoromethyl) pyridin-4-y1) imidazolidin-l-
yl)pyridine 1-oxide (I-50b: 100mg, 0.273mmo1) was heated with acetic anhydride
(5mL) at 120 C for 3 hours. The resulting mixture was concentrated, basified
with
dilute sodium bicarbonate solution, and diluted with DCM. The organic layer
was
dried over sodium sulphate and concentrated to afford the crude product.
Purification
by column chromatography on silica gel (3% methanol in chloroform), followed
by
preparative HPLC afforded 8 mg of the product (8% yield).
11-1NMR (CD30D, 300MHz): 6 8.53-8.51 (d, 1H), 8.30-8.29 (d, 1H), 7.68-7.65
(dd, 1H), 7.36-7.33 (d, 1H), 5.90-5.88 (d, 1H), 4.18-3.93 (m, 4H), 3.81-3.75
(m, 1H),
2.18-2.13 (m, 1H), 1.15-1.11 (m, 2H), 0.95-0.93 (m, 2H). LCMS: 98.62%, m/z =
364.9 (M+1). HPLC: 96.87%.
PHARMACOLOGICAL TESTING
The abbreviations listed below and used in the preparations below have the
corresponding meanings.
CYP Cytochrome P450
CPM Counts per minute
Cyt b5 Cytochrome b5
DMSO Dimethyl sulfoxide
94
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
DHEA Dehydroepiandrosterone
NADPH Nicotinamide adenine dinucleotide phosphate
Human and rat -Cytochrome P450, 17-20 lyase
1) Cytochrome P450, 17-20 lyase (CYP17-lyase) assay development using
recombinant human CYP17 enzyme and 17-a-hydroxy pregnenolone [21-3H] as the
substrate.
Cytochrome P450, 17-a-Hydroxylase, 17-20 lyase (CYP17) is a multi
functional enzyme that plays a key role in the biosynthesis of steroid
hormones. It
catalyses both conventional hydroxylation and also the carbon-carbon bond
cleavage
reactions (Peter Lee-Robichaud et al, Biochem.J, (1997) 321, 857-63). In the
hydroxylation reaction, it converts progesterone and pregnenolone to the
corresponding hydroxylated products 17-a-hydroxy progesterone and 17-a-hydroxy
pregnenolone. In the lyase reaction, it catalyzes the conversion of these
hydroxylated
substrates to Androstenedione and Dehydroepiandrosterone (DHEA) respectively.
In
the Cyp17 lyase assay described here, the conversion of 17-a- hydroxy
pregnenolone
to Dehydroepiandrosterone and acetic acid is being monitored..
The hydroxylation and cleavage activities are catalyzed sequentially at the
common active site of Cyp17 and proceed through transfer of two electrons from
NADPH via its redox partner, cytochrome P450 reductase (CPR). The reaction
mechanism for each activity is thought to involve formation of distinct iron-
oxygen
complexes. Cytochromeb5 selectively stimulates the lyase activity and has no
significant effect on its hydroxylase activity. Lyase activity is stimulated
by
cytochrome b5 up to 10-fold in reconstituted assays with insignificant
stimulation of
the hydroxylase activity (MK Akthar et al, Journal of Endocrinology (2005)
187, 267-
274 and Katagiri M et al, Biophysical Research Communications (1982) 108, 379-
384).
Assay method was adopted from a published protocol with some
modifications to suit our requirements (Dmitry N Grigoryev et al, Analytical
Biochemistry, (1999) 267, 319-330). The conversion of 17-a-hydroxy
pregnenolone
to Dehydroepiandrosterone is accompanied by the release of acetic acid. In the
Cyp17
lyase assay, 17-a-hydroxy pregnenolone labeled with tritium (3H) at position
21 is
used as the substrate. Chloroform extraction removes the radioactive steroids
and
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
acetic acid is taken into aqueous layer. The tritiated acetic acid released in
the assay
thus extracted is quantified to determine the enzyme activity.
Initial buffer conditions were, 50 mM Phosphate buffer, pH 7.5 was used as
the starting buffer for Cyp17 lyase activity based on the data published in US
patent
publication No. US2004/0198773 Al. This buffer was found to be suitable for
regular Cyp17 lyase assay. Human Cyp 17 gene was cloned and expressed in
Adenoviral expression system in A549 cell lines. The purified cell membrane
preparations were used as the source for Human CYP17 enzyme. Total protein
concentration: 8 mg/mt.
To identify the appropriate concentration of the enzyme required for the
assay,
concentration dependent enzyme activity was determined at a substrate (17-a-
hydroxypregnenolone [21-3H]) concentration of 0.5 M (Vincent C.O. Nijar, et
al., J
Med Chem, (1998) 41, 902-912). The protein activity was found to be in the
linear
range up to 20 g, the highest concentration tested. Based on the enzyme
concentration curve and stock concentration, 15 g was selected for the assay.
At this
protein concentration, the S/N ratio was 30, with a good signal window M
(CP
- ¨Pos Ctrl ¨
CPMBlank 1650)
Km (Michaelis Menton constant) is a measure of the binding affinity of
substrate to the enzyme. 17-a-hydroxy pregnenolone [21-3H] is a substrate for
17, 20
lyase enzyme. Km for this substrate was determined by monitoring the tritiated
acetic
acid release as a function of substrate concentration. Concentration of 17-a-
hydroxy-
pregnenolone [21-3H] was varied from 0.03125 M to 1 M. For the Km
determination, the data was fit to a hyperbolic equation (Graphpad Prism
software
IV). The Km was estimated as 0.25 M, close to the reported value. (Dmitry N.
Grigoryev et al, Analytical Biochemistry (1999) 267, 319-330)
For routine screening, the assay was set up with 16 ps of enzyme in 50 pL
reaction volume. 17a- hydroxy pregnenolone [21-3H] was added to a final
concentration of 0.25 tM. NADPH is used at a final concentration of 4.2 mM.
Total
reaction volume was made up to 50 pL with 50 mM Phosphate buffer pH 7.5. The
reaction mixture was incubated at room temperature for 90 minutes with gentle
shaking. The reaction was stopped by the addition of 100 pL of buffer. 500 pL
of 5%
freshly prepared activated charcoal was added to the solution and mixed well
by
vortexing. The samples were centrifuged at 17568 x g for 5 minutes. (14000
rpm).
96
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
The supernatant was carefully transferred to fresh tube and 1.3 mL of
scintillation
fluid was added, mixed by vortexing.
The radioactivity was measured in a 1450 MicroBeta TriLuxTm scintillation
counter from Wallac-Perkin Elmer , USA. The measurements were carried out in
2.0
mL eppendorfrm tubes. Each tube was counted for 1 minute. The amount of
tritiated
acetic acid released is proportional to the lyase activity. Percent lyase
activity in
presence of inhibitor was calculated using the formula given below.
CPM sample ¨ CPM blank
% Lyase activity ¨ --------------------------------- x 100
CPM Pos. Ctrl ¨ CPM Blank
Sample: Enzyme reaction in presence of inhibitor.
Positive control: Enzyme reaction without inhibitor but containing DMSO at 1%
final
concentration.
Blank- Contains all reagents except enzyme.
% Inhibition = 100% ¨ % Lyase activity
For IC50 determination, the % inhibition was plotted as a function of
inhibitor
concentration. The data was fitted to sigmoidal equation using Graphpad Prism
software IV to generate IC50 values.
Dose-response studies by standard compounds Abiraterone and Ketoconazole
were carried out as part of assay optimization.
For the rat CYP 17 lyase model:
The same procedure described above was used but using rat testes microsomes
as the source and with a substrate concentration of 0.5 M.
The results for the compounds tested from the Examples above using the assay
above are listed in Table 1 below.
Table 1
Example Lyase IC 50 nM
Compounds
No. Human /Rat
1-(2-Chloropyridin-4-y1)-3-(4-
1A cyclopropylpyridin-3- 8/2.7
yl)imidazolidin-2-one
1-(4-Cyclopropylpyridin-3-y1)-3-
2A (2-(trifluoromethyl) pyridin-4-y1) 9/1.1
imidazolidin-2-one
97
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
Example Lyase IC 50 nM
Compounds
No. Human /Rat
1-(4-Cyclopropylpyridin-3-y1)-3-
3A (naphthalen-2-y1) imidazolidin-2- 26/NA
one
1-(Benzo[b]thiophen-5-y1)-3-(4-
4A cyclopropylpyridin-3-y1) 13/NA
imidazolidin-2-one
1-(4-Cyclopropylpyridin-3-y1)-3-
SA (3-(trifluoromethyl) phenyl) 6/NA
imidazolidin-2-one
1-(Benzo[b]thiophen-2-y1)-3-(4-
6A cyclopropylpyridin-3-y1) 13/NA
imidazolidin-2-one
1-(6-Chloro-2-methylpyrimidin-4-
7A y1)-3 -(4-cyclopropylpyridin-3 -y1) 41/NA
imidazolidin-2-one
1-(4-Cyclopropylpyridin-3-y1)-3-
8A (4-(trifluoromethyl) pyridin-2-y1) 22/NA
imidazolidin-2-one
1-(4-Cyclopropylpyridin-3-y1)-3-
9A (2-methoxypyridin-4-y1) 42/4.6
imidazolidin-2-one
1-(6-Chloropyrimidin-4-y1)-3-(4-
10A cyclopropylpyridin-3 -y1) 121/NA
imidazolidin-2-one
1-(4-Cyclopropylpyridin-3-y1)-3-
11A (4-(trifluoromethyl) pyrimidin-2- 58%@10 mM/NA
yl) imidazolidin-2-one
1-(4-Cyclopropylpyridin-3-y1)-3-
12A (2-(trifluoromethyl) pyrimidin-4- 99/NA
yl) imidazolidin-2-one
1-(4-Cyclopropylpyridin-3-y1)-3-
(3-(trifluoromethyl) 11/NA
13A
benzo[b]thiophen-5-y1)
imidazolidin-2-one
1-(4-Cyclopropylpyridin-3-y1)-3-
14A (2-fluorobenzo[b]thiophen-5-y1) 26/NA
imidazolidin-2-one
1-(4-Cyclopropylpyridin-3-y1)-3-
15A (3-methylbenzo[b]thiophen-5-y1) 6.7/NA
imidazolidin-2-one
1-(Benzo[b]thiophen-6-y1)-3-(4-
16A cyclopropylpyridin-3-y1) 7/NA
imidazolidin-2-one
1-(4-Cyclopropylpyridin-3-y1)-3-
17A (2,2-difluorobenzo[d][1,3]dioxo1-5- 58/NA
yl)imidazolidin-2-one
98
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
Example Lyase IC 50 nM
Compounds
No. Human /Rat
1-(4-Cyclopropylpyridin-3-y1)-3-
18A (3-methylbenzo[b]thiophen-6-y1) 76/NA
imidazolidin-2-one
1-(4-Cyclopropylpyridin-3-y1)-3-
19A (3-methylbenzofuran-5- 2.7/NA
yl)imidazolidin-2-one
1-(2-Chlorobenzo[b]thiophen-5-
20A y1)-3-(4-cyclopropylpyridin-3-y1) 106/NA
imidazolidin-2-one
1-(4-Cyclopropylpyridin-3-y1)-3-
21A (2,3-dihydro-1H-inden-5- 17/NA
yl)imidazolidin-2-one
1-(4-Cyclopropylpyridin-3-y1)-3-
22A (5-fluorobenzo[b]thiophen-2-y1) 7/NA
imidazolidin-2-one
1-(4-Cyclopropylpyridin-3-y1)-3-
(3-(trifluoromethyl) 1213/NA
23A
benzo[b]thiophen-6-
yl)imidazolidin-2-one
1-(4-Cyclopropyl-pyridin-3-y1)-3-
(2-fluoro-3-methyl- 19/NA
24A
benzo[b]thiophen-5-y1)-
imidazolidin-2-one
1-(4-Cyclopropyl-pyridin-3-y1)-3-
25A (2-fluoro-benzo(b)thiophene-6-y1)- 30/NA
imidazolidin-2-one
1-(4-Cyclopropyl-pyridin-3-y1)-3-
26A (4-fluoro-benzo[b]thiophen-6-y1)- 6/NA
imidazolidin-2-one
1-(4-Cyclopropyl-pyridin-3-y1)-3-
27A (5-fluoro-benzo[b]thiophen-6-y1)- 461/NA
imidazolidin-2-one
1-(4-Cyclopropylpyridin-3-y1)-3-
28A (6-fluorobenzo[b]thiophen-5-y1) 548/NA
imidazolidin-2-one
1-(4-Cyclopropylpyridin-3-y1)-3-
(5-fluoro-3-
29A 65%@10 mM/NA
methylbenzo[b]thiophen-6-
yl)imidazolidin-2-one
(1R,5S)-2-(2-Chloropyridin-4-y1)-
4-(4-cyclopropylpyridin-3-y1)-2,4- 161/NA
30A-I
thazabicyclo[3.1.0]hexan-3-one
(Enantiomer I)
(1S,5R)-2-(2-Chloropyridin-4-y1)-
4-(4-cyclopropylpyridin-3-y1)-2,4- 6/NA
30A-II
thazabicyclo[3.1.0]hexan-3-one
(Enantiomer II)
99
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
Example Lyase IC 50 nM
Compounds
No. Human /Rat
(1S,5R)-2-(4-Cyclopropylpyridin-
3-y1)-4-(2-methoxypyridin-4-y1)- 169/NA
31A-1
2,4-diazabicyclo[3.1.0]hexan-3-one
(Enantiomer I)
(1R,5S)-2-(4-Cyclopropylpyridin-
3-y1)-4-(2-methoxypyridin-4-y1)- 12/NA
31A-II
2,4-diazabicyclo[3.1.0]hexan-3-one
(Enantiomer II)
(1S,5R)-2-(4-Cyclopropylpyridin-
3-y1)-4-(2-(trifluoromethyl)pyridin- 184/NA
32A-I
4-y1)-2,4-diazabicyclo[3.1.0]hexan-
3-one (Enantiomer I)
(1R,5S)-2-(4-Cyclopropylpyridin-
3-y1)-4-(2-(trifluoromethyl)pyridin- 6.9/3
32A-II
4-y1)-2,4-diazabicyclo[3.1.0]hexan-
3-one (Enantiomer II)
1-(2-Chloro-pyridin-4-y1)-3-[4-(1-
33A hydroxy-cyclobuty1)-pyridin-3-y1]-
45/NA
imidazolidin-2-one
1-(2-Chloropyridin-4-y1)-3-(4-
34A cyclop entyl-pyridin-3 13/NA
-y1)-
imidazolidin-2-one
1-(2-Chloropyridin-4-y1)-3-[4-(1-
35A hydroxy cyclopenty1)-pyridin-3-
341/NA
yl]-imidazolidin-2-one
1-[4-(1-Hydroxy-cyclopropy1)-
36A pyridin-3-y1]-3-(3-trifluoromethyl- 27/NA
phenyl)-imidazolidin-2-one
1-(2-Chloro-6-(trifluoromethyl)-
37A pyridin-4-y1)-3-(4-cyclopropyl-
5/NA
pyridin-3-yl)imidazolidin-2-one
1-(4-Cyclopropylpyridin-3-y1)-3-
38A (6-cyclopropylpyrimidin-4-y1) 48/NA
imidazolidin-2-one
1-(4-Cyclopropylpyridin-3-y1)-3-
39A (2-cyclopropylpyridin-4-y1) 12/NA
imidazolidin-2-one
1-(4-Cyclopropylpyridin-3-y1)-3-
40A (6-methoxypyrimidin-4-y1) 84/NA
imidazolidin-2-one
1-(4-Cyclopropylpyridin-3-y1)-3-
41A (6-methylpyrimidin-4-y1) 70/NA
imidazolidin-2-one
1-(4-Cyclopropylpyridin-3-y1)-3-
42A (5-fluoro-4-methylpyridin-2-y1) 22/NA
imidazolidin-2-one
100
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
Example Lyase IC 50 nM
Compounds
No. Human /Rat
1-(4-Cyclopropylpyridin-3-y1)-3-
43A (2-cyclopropylpyrimidin-4-y1) 36/NA
imidazolidin-2-one
1-(4-Cyclopropylpyridin-3-y1)-3-
44A (2-methylpyrimidin-4-y1) 397/NA
imidazolidin-2-one
1-(6-Chloro-2-(trifluoromethyl)
45A pyrimidin-4-y1)-3-(4- 10/NA
cyclopropylpyridin-3-y1)
imidazolidin-2-one
1-(4-Cyclopropylpyridin-3-y1)-3-
46A (2, 6-dichloropyridin-4-y1) 1.9/NA
imidazolidin-2-one
1-(2-Chloro-6-cyclopropylpyridin-
47A 4-y1)-3-(4-cyclopropylpyridin-3-y1) 5.2/NA
imidazolidin-2-one
1-(2-Cyclopropy1-6-
48A (trifluoromethyl) pyridin-4-y1)-3- 4.75/NA
(4-cyclopropylpyridin-3-y1)
imidazolidin-2-one
1-(2,6-Bis(trifluoromethyl)pyridin-
49A 4-y1)-3-(4-cyclopropylpyridin-3-y1) 6/NA
imidazolidin-2-one
1-(4-Cyclopropy1-2-
hydroxypyridin-3-y1)-3-(2-
50A 51%@10 M/NA
(trifluoromethyl) pyridin-4-y1)
imidazolidin-2-one
Comparison of the methyl pyridine compounds (i.e., compounds of Formula I
where, R6 is methyl) described in PCT Publication No. WO 2010/149755 with the
corresponding cyclopropyl derivatives (compounds of Formula I where, R6 is
cyclopropyl) described herein showed that replacement of methyl by cyclopropyl
typically resulted in improvement in potency of inhibition of human CYP17 in
biochemical and cell based assays as well as improved selectivity over CYP1A2.
In
addition, the cylcopropyl pyridine derivatives typically exhibited improved
metabolic
stability in activated microsomes that resulted in longer half lives and
lowering of
clearance of the compound in vivo.
The compounds of present invention in free form or in salt form, exhibit
valuable pharmacological properties, e.g. inhibition of CYP17 lyase, e.g. as
indicated
in the in vitro tests provided above and are therefore useful for therapy
mediated by
101
CA 02834224 2013-10-23
WO 2012/149413
PCT/US2012/035583
such inhibition. For example, the compounds of the present invention are
useful in
the treatment of inflammation and cancer (in particular, prostate cancer) in a
mammal
(preferably, a human).
Thus, as a further embodiment, the present invention provides the use of a
compound of the present invention in therapy. In a further embodiment, the
therapy is
selected from a disease mediated by the regulation of 17a-hydroxylase/C17,20-
lyase.
In another embodiment, the invention provides a method of treating a disease
which is treated by the regulation of 17a-hydroxylase/C17,20-lyase comprising
administration of a therapeutically acceptable amount of a compound of the
present
invention. In a further embodiment, the disease is prostate cancer.
102