Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
HYBRID MATERIALS AND NANOCOMPOSITE MATERIALS, METHODS OF
MAKING SAME, AND USES THEREOF
CROSS-REFERENCE TO RELATED APPLICATIONS
[0001] This application claims priority to U.S. provisional patent
application nos.
61/503,085, filed June 30, 2011, and 61/578,464, filed December 21, 2011, the
disclosures of
which are incorporated herein by reference.
STATEMENT REGARDING FEDERALLY SPONSORED RESEARCH
[0002] This invention was made with government support under contract
no. DE-
SC0001086 awarded by the Department of Energy. The government has certain
rights in the
invention.
FIELD OF THE INVENTION
[0003] The present invention generally relates to composite materials
and methods of
making such composite materials. More particularly, the present invention
relates to in situ
formation of nanoparticles embedded in a carbon matrix.
BACKGROUND OF THE INVENTION
[0004] Rising energy prices and unmet demand for secondary batteries
with higher
energy & power densities, higher operating voltages, improved cycling
stability, enhanced
safety, and lower initial and life cycle costs has increased interest in
lithium ion batteries
(LIB). LIBs demonstrate higher energy density, higher operating voltage and
lower self-
discharge rates compared to conventional rechargeable batteries. They have
consequently
received intense scientific and commercial interest for portable electronics
applications since
the early 1990s. In recent years, the demand for secondary (rechargeable)
batteries with better
performance, higher charge-rate capability, improved cycling stability, and
enhanced safety
has steadily increased to meet new needs for smaller, lighter, more powerful
electronic
devices, as well as to accommodate growing interests in hybrid electric and
plug-in hybrid
electric vehicles.
[0005] A crucial performance criterion is the cyclability of the
electrode materials and
a key issue in capacity retention lies in the large structural and
morphological changes many
electrode materials undergo during cyclic insertion and deinsertion of
lithium. Significantly,
these changes occur in materials following rather different lithiation
mechanisms, including
alloying, conversion, and intercalation; implying that general solutions are
required. Despite
- 1 -
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
current LIB platforms based on a graphite anode and a lithium metal oxide
(e.g., LiC002)
cathode is believed to be close to its limits due to the limited gravimetric
capacity and rate
capability of graphitic carbon as the anode material.
BRIEF SUMMARY OF THE INVENTION
[0006] The present invention provides hybrid materials, nanocomposite
materials,
methods of making such materials. Also provided are uses of such materials.
The hybrid/in
situ approach of the instant invention provides homogeneous dispersion of the
metal
precursor in the polymer matrix (e.g., a cross-linked polymer matrix).
[0007] In an aspect, the present invention provides a hybrid material. The
hybrid
material is a polymer comprising a metal precursor. The metal precursor is
chemically
bonded to the polymer. During pyrolysis of the hybrid material, nanoparticles
are formed
from the metal precursors. In an embodiment, the step of pyrolysing the hybrid
material is
carried out such that a nanocomposite material comprising a plurality of
nanoparticles, the
nanoparticles being formed from the metal component of the one or more metal
precursor
compounds, embedded in a carbon matrix is formed.
[0008] In an aspect, the present invention provides a nanocomposite
material. The
nanocomposite material has nanoparticles (e.g., metal nanoparticles, metal
oxide
nanoparticles, metal halide (e.g., metal fluoride) nanoparticles, metal boride
nanoparticles,
metal phosphate nanoparticles) embedded in a continuous phase of carbon (i.e.,
a carbon
matrix).
[0009] In an aspect, the present invention provides methods of forming
a material.
The material can be a hybrid material or nanocomposite material as described
herein.
[0010] In an embodiment, the method for forming a material comprises
the steps of:
contacting one or more monomers, one or more metal precursor compounds,
optionally, an
initiator, and, optionally, one or more solvents to form a reaction mixture,
heating the
reaction mixture such that a hybrid material comprising a plurality of metal
precursor
compounds chemically bonded to the polymer matrix is formed and, optionally,
isolating the
hybrid material. In an embodiment, the method further comprises the step of
pyrolysing the
hybrid material, such that a nanocomposite material comprising a plurality of
metal oxide
nanoparticles (or metal nanoparticles) embedded in a carbon matrix is formed.
[0011] In various embodiments, the nanocomposite material is subjected
to various ex
situ treatments such that nanoparticles of the resulting nanocomposite have
different chemical
- 2 -
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
reacted such that metal, metal halide, metal sulfide, and metal phosphate
nanoparticles are
formed or metal sulfide nanoparticles are reacted such that metal oxides,
metal halide, metal,
or metal phosphate nanoparticles are formed.
[0012] In an aspect, the present invention provides devices comprising the
hybrid
material or nanocomposite materials described herein. Examples of such devices
include
batteries (e.g., secondary batteries), on-chip inductors.
BRIEF DESCRIPTION OF THE FIGURES
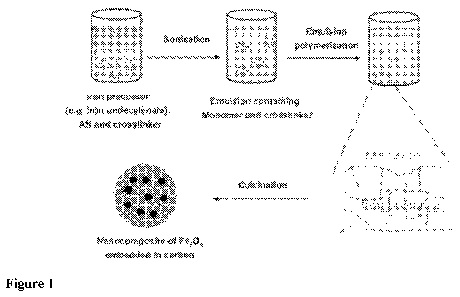
[0013] Figure 1. An example of an in situ synthesis scheme for Fe304¨C
nanocomposite.
[0014] Figure 2. Representative XRD pattern for the Fe304¨C composite
created by
pyrolysing the as-prepared PAN¨Fe(undec)3 complex.
[0015] Figure 3. Examples of (a) Morphology of polymer¨iron complex;
(b)
morphology of Fe304¨C composite; (c) EDS spectrum for the polymer¨iron
complex; and (d)
TGA data for the Fe304¨C composite.
[0016] Figure 4. Representative Raman spectrum for the Fe304¨C
composite,
deconvoluted into peaks for graphitic carbon, disordered graphite lattices and
amorphous
carbon.
[0017] Figure 5. Representative cyclic voltammograms and
voltage¨capacity profiles
for Fe304¨C nanocomposites.
[0018] Figure 6. Representative cycling performance for (a) Fe304¨C
composites run
at 1 C (924 mA h g-1); (b) composite run at 0.2 C; (c) composite run at
charging rates; (d)
bare Fe304 nanoparticles run at 1 C; and (e) bare carbon made from pyrolysis
of PAN¨DVB
run at 1 C.
[0019] Figure 7. Representative nitrogen adsorption isotherms and pore size
distribution for the Fe304¨C composite.
[0020] Figure 8. Representative (a) X-Ray diffractogram and (b) TEM
image for
MnO¨C composite.
[0021] Figure 9. Representative (a) cyclic voltammograms, (b)
voltage¨capacity
profiles of MnO¨C composite and (c) cycling performance of MnO¨C composite at
1 C (755
mA h g-1), 0.2 C and at varied charging rates, and cycling performance of pure
MnO.
- 3 -
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
nanoparticles of the following phases: Fe, Fe304, Fe203, MnO, Mn304, Sn, Co,
Co304, Cu,
Cu0; an example of a TEM image of Cu@C composite.
[0023] Figure 11. Representative XRD patterns of nanocomposites
embedding the
nanoparticles of the following phases: Ti02(anatase), V205, ZnO, Zr02; an
example of a
TEM image of Ti02@C composite.
[0024] Figure 12. Scheme for synthesizing Co@C and CoS@C
nanocomposites;
representative XRD patterns of Co@C and CoS@C nanocomposites.
[0025] Figure 13. Representative XRD pattern for Fe304@C and LiFePO4@C
nanocomposites.
[0026] Figure 14. Representative TEM image for LiFePO4@C
nanocomposites.
[0027] Figure 15. Representative XRD pattern for Mn075Fe0250@C and
LiMno 75Feo 25PO4@ C nanocomposites.
[0028] Figure 16. Representative TEM image for Mn075Fe0250@C
nanocomposite.
[0029] Figure 17. Representative TEM image for LiMn075Fe025PO4@C
nanocomposite.
[0030] Figure 18. Representative scanning electron micrographs of (a)
MS-22, (d)
MS-0; transmission electron micrographs of (b and c) MS-22, (e and MS-0;
insets of (b)
shows Mo52 nanosheet; SAED patterns of MS-22 and MS-0 in the insets of (c) and
(0
respectively.
[0031] Figure 19. An example of a schematic of the synthesis of (A)
Mo52¨carbon
nanostructure and (B) pure Mo52.
[0032] Figure 20. Representative galvanostatic charge¨discharge curves
of (a) MS-0
and (b) MS-22 at a current density of 100 mA g-1; cyclic voltammetry (CV)
curves of (c)
MS-0 and (d) MS-22 at a scan rate of 0.2 mV 5-1; (e) cycling stability of pure
Mo52 and
various Mo52¨carbon composites; (f) variation of discharge capacity as a
function of carbon
weight fraction.
[0033] Figure 21. Representative Ex situ X-ray diffraction patterns of
(a) Mo52¨
carbon (22 wt%) composite and (b) pure Mo52 after 1st discharge cycle. Peaks
marked by *
corresponds to Cu current collector; scanning electron micrographs of 1st
cycle discharged
product of (c) MS-22 and (d) MS-0.
- 4 -
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
50% carbon black in the electrode composition at various current rates in the
range of 0.4-4
A g-1.
[0035] Figure 23. Representative X-ray diffraction patterns of pure
MoS2 and MoS2-
carbon (22 wt %) composite.
[0036] Figure 24. Representative thermogravimetry analysis of pure
MoS2, MS-11,
MS-22, MS32 and MS-41.
[0037] Figure 25. Representative (a) N2 adsorption/desorption
isotherms and (b) pore
size distribution of pure MoS2, MS-22 and MS-41.
[0038] Figure 26. Representative transmission electron micrographs of (a)
MS-11 and
(b) MS-32.
[0039] Figure 27. (a) Representative cycling stability of pure MS-22
with 0%, 10%,
25% and 50% carbon black in the electrode at a current rate of (a) 100 mAg-1;
(b) at various
current rates in the range of 0.4-4 Ag-1.
[0040] Figure 28. Representative cycling stability of 550 C and 700 C
calcined
Mo52-carbon (22 wt %) composite at a current rate of (a) 100 mAg-1; (b) at
various current
rates in the range of 0.4-4 Ag-1.
[0041] Figure 29. Representative SAED patterns of (a) pure Mo52 and
(b) Mo52-
carbon (22 wt %) composite.
[0042] Figure 30. An example of a schematic of synthesis process for
creating
organic-inorganic copolymer hybrids.
[0043] Figure 31. Representative TEM images of Fe304@C nanocomposite
(A)
before cycling and (B) after 100 charge-discharge cycles.
[0044] Figure 32. An example of an overview of the platform for
synthesizing
nanocomposites with embedded structures involving different classes of
materials.
[0045] Figure 33. Representative powder XRD patterns (A), TEM images
for Fe@C
(B) and Fe52@C (C) composites and size distribution histograms for Fe@C (D)
and Fe52@C
(E).
[0046] Figure 34. Representative (A) Cyclic voltammograms of Fe52gC;
(B) cycling
performance of Fe52@C and pristine Fe52. Red cross indicates result from
reference 28
(0.58C).
[0047] Figure 35. Representative (A) XRD pattern, (B) TEM image, (C)
STEM
image and (D) EDX spectrum for FeSn2@C nanocomposite.
- 5 -
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
cycling performance of FeSn2@C and pristine FeSn2 at 0.1C.
[0049] Figure 37. Representative (A) XRD patterns of the embedded
carbon
composites involving iron/iron oxides; (B) TEM image of 7 -Fe203@C composite;
(C) and (D)
XRD pattern and TEM image of V205gC composite; (E) and (F) XRD pattern and TEM
image of Ti02@C composite.
[0050] Figure 38. Representative (A) Cyclic voltammograms of 7 -
Fe203@C; (B)
cycling performance of 7 -Fe203@C at 0.5C, 1C and 2C and pristine Fe203 at
0.5C.
[0051] Figure 39. Representative infrared spectra of crosslinked PAN-
DVB,
Fe(C10H19C00)3 and PAN-iron composite. Inset: close-up of 1600-1700 cm-1,
normalized
using peak at 2930 cm-1.
[0052] Figure 40. Representative oxidative TGA curves for FeS2@C,
FeSn2@C and
r -Fe203@C
[0053] Figure 41. Representative voltage-capacity profiles for FeS2@C
composite run
at 0.2C and 1C.
[0054] Figure 42. Representative voltage-capacity profiles for FeSn2@C
composite
run at 0.1C.
[0055] Figure 43. Representative voltage-capacity profiles for r -
Fe203@C composite
run at 0.5C, 1C and 2C.
DETAILED DESCRIPTION OF THE INVENTION
[0056] The present invention provides hybrid materials, nanocomposite
materials,
methods of making such materials. Also provided are uses of such materials.
[0057] The hybrid/in situ approach of the instant invention provides
homogeneous
dispersion of the metal precursor in the polymer matrix (e.g., a cross-linked
polymer matrix)
and thus the pyrolysis of the hybrid is able to yield composites with
particles uniformly
dispersed in the matrix. Additionally, the synthesis of the composite via
simultaneous
creation of the active material and the carbon matrix reduces the complexity
of synthesis
procedure and lends itself to the development of low-cost/scalable production
processes.
[0058] In an aspect, the present invention provides a hybrid material.
The hybrid
material is a polymer comprising a metal precursor. The metal precursor is
chemically
bonded to the polymer. During pyrolysis of the hybrid material, nanoparticles
are formed
- 6 -
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
metal precursor compounds embedded in a polymer.
[0059] By chemically bonded it is meant that the metal precursor
(i.e., a chemical
moiety of or metal center of the metal precursor) is chemically bonded via a
chemical bond
(e.g., covalent bond, coordinate covalent bond, or ionic bond) to the polymer.
[0060] A variety of polymers can be used. Suitable polymers can be
thermally
degraded (i.e., pyrolysed) to provide a graphitic material or partially
graphitic material. The
resulting material is electrically conducting. The polymer can be a
homopolymer or a
copolymer. Examples of suitable polymers include poly(acrylonitrile),
polyvinylpyrroilidone,
polypyrrole, polyacetylene, polythiophene, polyphenylene vinylene,
polyphenylene sulfide,
polysaccharides (e.g., galactose, maltose, and glucose), acrylonitrile-
divinylbenzene
copolymer, phenol resin, and resorcinol-formaldehyde copolymer.
[0061] The metal precursor is a compound with a metal center and one
or more
ligands. The metal precursor compounds are chemically bonded to the polymer.
The metal
precursor is uniformly distributed throughout the polymer. The metal
precursors form
nanoparticles in situ during pyrolysis of the polymer. Depending on the
components of the
reaction mixture, it may be desirable the metal precursor be water soluble.
The metal
precursors are present in the hybrid material at from 10 % by weight to 90% by
weight,
including all integer % by weight values and ranges therebetween. Examples of
suitable
metal precursor compounds include metal carboxylates, metal coordination
compounds (e.g.,
metal thiolates), amino acid metal salts, and other metal-organic compounds.
[0062] By uniformly distributed it is meant there is a homogeneous
distribution of a
preponderance of the metal precursors in the polymer-based hybrid materials,
or a
homogeneous distribution of a preponderance of the nanoparticles in the
nanocomposite
materials. For the hybrid materials, there is a substantial absence of phase
separation (e.g., no
observed phase separation) between the polymer and metal precursors and/or a
substantial
absence of metal precursor aggregates (e.g., no metal precursor aggregates are
observed). For
the nanocomposite materials, there is a substantial absence of phase
separation between the
carbon matrix and nanoparticles (e.g., no observed phase separation) and/or a
substantial
absence of particle-particle aggregates (e.g., no particle-particle aggregates
are observed).
[0063] The metal precursor can be a metal carboxylate. In an
embodiment, the metal
carboxylate comprises an alkyl moiety. The alkyl moiety can be a C6 to C30
alkyl moiety,
including all integer number of carbons and ranges therebetween. The moiety
can be
- 7 -
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
substituted with a reactive chemical moiety (e.g., a carbon-carbon double
bond, and amine,
hydroxyl, carboxylate groups and combinations of such groups (which can
hydrogen bond
with moieties in the polymer/monomer)) that can be incorporated in the polymer
by a
polymerization reaction. Examples of suitable metal carboxylates include alkyl
metal
carboxylates (e.g., iron undecylenate, manganese undecylenate, tin
undecylenate, or
vanadium undecylenate), metal citrates (e.g., iron citrate, manganese citrate,
tin citrate, and
vanadium citrate), amino acid metal salts (e.g., iron aspartate), and other
metal-organic
compounds (e.g., iron gluconate).
[0064] In an embodiment, the metal precursor has a chemical moiety that
reacts with
the polymer or monomer to form a covalent bond. For example, the metal
precursor is a metal
carboxylate (e.g., iron undecylenate, manganese undecylenate, tin
undecylenate, or vanadium
undecylenate) having a carbon-carbon double bond that is copolymerized with a
monomer or
monomers.
[0065] The metal precursor can be a metal coordination compound. In an
embodiment, the metal center (e.g., Mo) of the metal precursor (e.g., ammonium
molybdenum tertrathiolate) is chemically bound to the polymer via a coordinate
covalent
bond.
[0066] In an aspect, the present invention provides a nanocomposite
material. The
nanocomposite material has nanoparticles embedded (e.g., encapsulated) in a
continuous
phase of carbon (i.e., a carbon matrix). In an embodiment, the nanocomposite
material
comprises a plurality of nanoparticles embedded in a carbon matrix.
[0067] The nanocomposite materials can include a variety of
nanoparticles. For
example, the nanoparticles can be metal nanoparticles, metal oxide
nanoparticles, metal
halide (e.g., metal fluoride) nanoparticles, metal boride nanoparticles, metal
phosphate
nanoparticles, or combinations of such nanoparticles. The nanoparticles can
include a variety
of metals. The nanoparticles can have multiple metals (e.g., metal alloys and
mixed metal
oxides). In the case of multiple metals in the nanoparticles, depending on the
composition the
individual nanoparticles can have mixed composition (alloyed nanoparticles) or
a mixture of
nanoparticles with different composition. For example, Feo 75Mno 250 can
provide alloyed
nanoparticles and Sn/FeSn2 can provide a mixture of nanoparticles with
different
compositions. The nanoparticles can be crystalline or amorphous.
[0068] Examples of suitable metal nanoparticles include Fe, Mn, and
FeSn2, FeNi3,
Al, Sn, Ge, and Si. Examples of suitable metal oxides include Fe203 (e.g., 7-
Fe203), Fe304,
- 8 -
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
Mn203, M-Mn204 (M = Li, Na, K), M003, V205, T102, M4T15012 (M = Li, Na, K,
Ag), Sn02,
SnO, Co304, and MCo02 (M = Li, Na, K). Examples of suitable metal sulfides
include MoS2,
MoS3 FeS2, FeS, Fei,S(x=0-0.2), CoS, CuS, Cu2S, TiS2, and M2S (M = Li, Na, K).
Examples
of suitable metal borides include TiB2, VB2, and LiBio. Examples of a suitable
metals
fluoride are CuF2, FeF2, and FeF3. Examples of suitable metal phosphates
include MFePat
(M=Li, Na, K) and LiMnxFei,PO4.
[0069] The nanoparticles are present at 10 % by weight to 90 % by
weight, including
all integer % by weight values and ranges therebetween. In an example, the
nanoparticles are
present at 40 % by weight to 90 % by weight.
[0070] Based on the composition of the nanoparticles and the methods
used to form
the nanoparticles, the nanoparticles can have a variety of shapes and sizes.
In various
examples, the nanoparticles have a spherical shape (e.g., Fe203 nanoparticles)
or a
rectangular shape (e.g., MoS2 nanoparticles). In the case of spherical
nanoparticles, the
diameter of the nanoparticles is from 5 nm to 500 nm, including all integer
nanometer values
and ranges therebetween, in size. In the case of rectangular nanoparticles,
the nanoparticles
have a length of 20 to 100 nm, including all integer nanometer values and
ranges
therebetween, and a thickness of 5 to 20 nm, including all integer nanometer
values and
ranges therebetween. The size can be an average size. For example, the size of
individual
nanoparticles and the average nanoparticle size can be measured by
transmission electron
microscopy.
[0071] The nanoparticles have a narrow size distribution. For example,
the
nanoparticles have a polydispersity index of 1.001 to 1.05, including all
values to 0.001 and
ranges therebetween. In an embodiment, the nanoparticles are monodisperse
(i.e., the fraction
of nanoparticles within one standard deviation from the number average size is
greater than
or equal to 75%). In another embodiment, the fraction of nanoparticles within
one standard
deviation from the number average size is greater than or equal to 90%.
[0072] The nanoparticles are embedded in a carbon matrix. The carbon
matrix is a
partially graphitic or graphitic material. The graphitic material is a
material consisting of
graphite. The partially graphitic material is a material comprising graphite
that may also
contain disordered graphitic lattices and/or amorphous carbon. The presence of
graphite,
disordered graphitic lattices and/or amorphous carbon can be determined by
techniques such
as, for example, XRD and Raman spectroscopy. The carbon matrix is porous and
amorphous.
Aggregation of the nanoparticles in the carbon matrix is not observed (e.g.,
by TEM, SEM, or
- 9 -
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
as described herein.
[0073] The carbon matrix can have a range of porosity based the
materials and
conditions used to form the matrix. In various examples, the pores of the
carbon matrix are
less than 100nm, less than 20nm, or less than 5nm.
[0074] The nanocomposite materials exhibit desirable properties. The
nanocomposite
material is conductive. The material can have a conductivity of 10-5 to 100
S/cm. The
material can have a Vickers Hardness of the composite is 1 to 40 GPa. The
material can have
a fracture toughness of the composite is 5 to 25 MPa 111112. In various
examples, the capacity
retentions of the composites in lithium-ion batteries is greater than 90%,
greater than 95%,
greater than 98% in 100 cycles at a 1C charge/discharge rate.
[0075] In an aspect, the present invention provides methods of forming
a material.
The material can be a hybrid material or nanocomposite material as described
herein. In an
embodiment, the hybrid material is made by a method described herein. In an
embodiment,
the nanocomposite material is made by a method described herein.
[0076] In an embodiment, the method for forming a material comprises
the steps of:
contacting one or more monomers, one or more metal precursor compounds,
optionally, an
initiator, and, optionally, one or more solvents to form a reaction mixture,
heating the
reaction mixture such that a hybrid material comprising a plurality of metal
precursor
compounds chemically bonded to the polymer matrix is formed and, optionally,
isolating the
hybrid material.
[0077] In an embodiment, the reaction mixture comprises: a first
(e.g., bulk)
monomer (e.g., acrylonitrile), optionally, a second (vinyl or cross-linking)
monomer (e.g.,
divinyl benzene), a metal precursor compound (metal carboxylate) (e.g., iron
undecylenate),
an initiator (e.g., AIBN), a (anionic) surfactant (e.g., sodium dodecyl
sulfate), water, and one
or more organic solvents such that a reaction mixture that is an aqueous
emulsion is formed.
In this embodiment, the reaction mixture is, optionally, subjected to high-
shear mixing such
that a miniemulsion having oil-in-water droplets with an average size of 0.01
microns to 0.5
microns if formed. For example, high shear mixing (for bench-top scale
synthesis) can be
provided by a sonication horn operated at 500W and at 20kHz with 50%
amplitude. A larger
scale reaction may require higher power to achieve the desired shear.
[0078] In another embodiment, the reaction mixture comprises: a first
(e.g., bulk)
monomer (e.g., resorcinol), and, optionally, a second (bulk) monomer (e.g.,
formaldehyde), a
metal precursor compound (e.g., ammonium tetrathiomolybdate), and water.
- 10-
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
or four) of metal precursors, where the metal precursors each comprise a
different metal.
[0080] The hybrid material can be pyrolysed to form a nanocomposite
material.
Nanoparticles are formed in situ from the metal precursor compounds as a
result of the
pyrolysis process. The pyrolysis process can be carried out in a single step
or can have
multiple steps. For example, carbonization can comprise consecutive,
stabilization,
carbonization, and graphitization steps. The determination of pyrolysis
conditions is material
dependent and is within the purview of one having skill in the art. For
example, a single step
pyrolysis step can be from 500 to 900 C, including all values to the degree
Celsius and
ranges therebetween. For example, a multiple step pyrolysis can be 320 C for
lhour for
stabilization and 500 C for 2hrs for carbonization. Higher temperatures may be
required for
complete graphitization.
[0081] The pyrolysis step (or one of the steps of a multiple step
pyrolysis) can be
carried out in an atmosphere comprising a variety of gases. A mixture of gases
can be used.
For example, the pyrolysis step can be carried out in air (or an oxygen
containing atmosphere)
or an inert atmosphere (e.g., a nitrogen atmosphere, an argon atmosphere, or a
mixture
thereof). For example, a reactive gas such as carbon dioxide (an oxidizing
gas) can be used to
provide increased mesopore and micropore content of the carbon matrix relative
to pyrolysis
in the absence of such gas.
[0082] In an embodiment, the method further comprises the step of
pyrolysing the
hybrid material, such that a nanocomposite material comprising a plurality of
metal oxide
nanoparticles (or metal nanoparticles) embedded in a carbon matrix is formed.
[0083] In an embodiment, the resorcinol-formaldehyde hybrid polymers
are
pyrolysed in an atmosphere comprising carbon dioxide or in a carbon dioxide
gas
atmosphere. The carbon dioxide is present at atmospheric pressure or
substantially
atmospheric pressure. The use of carbon dioxide in the pyrolysis step can
provide a carbon
matrix having a desirable morphology. For example, the carbon matrix can have
an
interconnected pore structure and higher surface area than materials obtained
without using
carbon dioxide in the pyrolysis step. For example, pyrolysis of a resorcinol-
formaldehyde
hybrid polymer at 800 C in a CO2(g) atmosphere provides carbon with broad pore
size
distribution (including mesopores and micropores) with graphene-like sheet
textures. Using
carbon dioxide in the pyrolysis step can result in a loss of mass in the
resulting composite
material and increases the interconnectivity of the pores of the carbon
matrix.
-11-
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
The nanoparticles of the starting nanocomposite are subjected to reaction
conditions that
result in formation of a portion of or all nanoparticles having a different
chemical
composition than the starting nanoparticles. For example, metal oxide
nanoparticles are
reacted such that metal, metal halide, metal sulfide, and metal phosphate
nanoparticles are
formed or metal sulfide nanoparticles are reacted such that metal oxides,
metal halide, metal,
or metal phosphate nanoparticles are formed.
[0085] In an embodiment, the method further comprises reducing the
metal oxide
nanoparticles of the nanocomposite material comprising a plurality of metal
oxide
nanoparticles embedded in a carbon matrix by contacting the nanocomposite
material with a
reductant (e.g., hydrogen gas) or heating the nanocomposite material under
inert conditions
(to a temperature higher than the carbonization temperature (carbon serves as
the reductant)),
such that a nanocomposite material comprising a plurality of metal
nanoparticles embedded
in a carbon matrix is formed.
[0086] In an embodiment, the method further comprises contacting the
nanocomposite material comprising a plurality of metal oxide nanoparticles
embedded in a
carbon matrix with a sulfur compound (e.g., sulfur), halide compound (e.g.,
fluoride
compound), or phosphate compound, such that a nanocomposite material
comprising a
plurality of metal sulfide, metal halide, or metal phosphate nanoparticles
embedded in a
carbon matrix is formed.
[0087] In an embodiment, the method further comprises reducing the
metal sulfide
nanoparticles of the nanocomposite material comprising a plurality of metal
sulfide
nanoparticles embedded in a carbon matrix by contacting the nanocomposite
material with a
reductant (e.g., hydrogen gas) or heating the nanocomposite material under
inert conditions
(to a temperature higher than the carbonization temperature (carbon serves as
the reductant)),
such that a nanocomposite material comprising a plurality of metal
nanoparticles embedded
in a carbon matrix is formed.
[0088] In an embodiment, the method further comprises contacting the
nanocomposite material comprising a plurality of metal sulfide nanoparticles
embedded in a
carbon matrix with an oxygen compound, halide compound, or phosphate compound,
such
that a nanocomposite material comprising a plurality of metal oxide, metal
halide, or metal
phosphate nanoparticles embedded in a carbon matrix is formed.
- 12 -
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
hybrid material. Examples of suitable bulk monomers include acrylonitrile,
resorcinol,
formaldehyde, vinylpyrrolidone, vinyl alcohol, acrylic acid, and phenol.
[0090] The cross-linking monomer forms cross links in the polymer.
Examples of
suitable cross-linking monomers include divinylbenzene, 1,4-butadiene,
isoprene, vinylsilane,
and sulfur.
[0091] Any initiator that initiates the polymerization of the monomers
can be used.
For example radical polymerization initiators can be used. Examples of
suitable initiators
include 2,2'-azobisbutyronitrile (AIBN), benzoyl peroxide, dicumyl peroxide,
potassium
persulfate, and 4,4' -azobis(4-cyanov aleric acid).
[0092] Any surfactant that forms a suitable aqueous emulsion can be
used. For
example anionic surfactants can be used. Examples of suitable surfactants
include sodium
dodecyl sulfate, hexadecyltrimethylammonium bromide, and polysorbates.
[0093] A single solvent or mixture of solvents can be used. For
example, the solvent
can be water. In the case where water is a solvent, depending on, for example,
the
components of the reaction mixture, it may be desirable to have the pH of the
reaction
mixture be greater than 7. Examples of suitable solvents include water,
toluene, and
cyclohexane.
[0094] The selection of reaction conditions that result in formation
of the desired
nanoparticle composition is within the purview of one having skill in the art.
The
polymerization temperature for acrylonitrile is typically 60-80 C.
[0095] The steps of the method described in the various embodiments
and examples
disclosed herein are sufficient to produce hybrid materials and/or
nanocomposite materials of
the present invention. Thus, in an embodiment, the method consists essentially
of a
combination of the steps of the methods disclosed herein. In another
embodiment, the method
consists of such steps.
[0096] In an aspect, the present invention provides devices comprising
the hybrid
material or nanocomposite materials described herein. Examples of such devices
include
batteries (e.g., secondary batteries), on-chip inductors. Such device
structures and methods of
making such structures are known in the art.
[0097] In an embodiment, the present invention provides an electrode
comprising a
nanocomposite material. In an embodiment, a device comprises an electrode
(e.g., an anode)
comprising the nanocomposite material. In an embodiment, the present invention
provides an
on-chip inductor comprising the nanocomposite material. In an embodiment, a
device
- 13 -
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
nanocomposites containing iron or iron/nickel alloy nanoparticles.
[0098] The following examples are presented to illustrate the present
invention. They
are not intended to limiting in any manner.
EXAMPLE 1
[0099] This example describes the synthesis and characterization of
examples of
hybrid materials and nanocomposite materials of the present invention.
[0100] An in situ, scalable method for creating a variety of
transition metal oxide¨
carbon nanocomposites was developed based on free-radical polymerization and
cross-
linking of poly(acrylonitrile) in the presence of the metal oxide precursor
containing vinyl
groups. The approach yields a cross-linked polymer network, which uniformly
incorporates
nanometer-sized transition metal oxide particles. Thermal treatment of the
organic¨inorganic
hybrid material produces nearly monodisperse metal oxide nanoparticles
uniformly
embedded in a porous carbon matrix. Cyclic voltammetry and galvanostatic
cycling
electrochemical measurements in a lithium half-cell are used to evaluate the
electrochemical
properties of a Fe304¨carbon composite created using this approach. These
measurements
reveal that when used as the anode in a lithium battery, the material exhibits
stable cycling
performance at both low and high current densities. The polymer/nanoparticle
copolymerization approach can be readily adapted to synthesize metal
oxide/carbon
nanocomposites based on different particle chemistries for applications in
both the anode and
cathode of LIBs.
[0101] A facile, scalable emulsion polymerization technique for
synthesizing
transition metal oxide nanoparticles embedded in a porous carbon matrix has
been reported.
The method (illustrated in Figure 1) relies upon co-polymerization and cross-
linking of the
carbon precursor (acrylonitrile) and the nanoparticle precursor in a single-
step; it yields
polymer¨nanoparticle hybrids with uniform particle distributions at high
nanoparticle
loadings. The procedure is also applicable for large-scale production of metal
oxide¨carbon
composites required for commercial-scale LIB manufacturing processes. The
procedure was
demonstrated by using a high-capacity (924 mA h g-1) transition metal oxide
(Fe304) and
show that it is adaptable to other oxides.
[0102] A nanocomposite of metal oxide/metal and carbon has been
synthesized via a
polymerization-carbonization process. A metal precursor (a carboxylic acid
salt of the metal),
soluble in nonpolar solvents, is mixed with a monomer, a cross-linking agent
and a surfactant
- 14 -
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
yield a composite of polymer nanoparticles encapsulating the iron precursor.
The material is
then separated from the liquid phase and carbonized to give a composite
material of metal
oxide/metal-carbon nanoparticles, which may be used as the active electrode
material for
-- lithium-ion batteries. Fe304 was demonstrated as an example compound but
the method is
applicable to various metal oxides/metals. The method can also be extended to
synthesize
nanocomposites consisting of nanoparticles of other materials (e.g., other
compounds which
contain the metal such as CoS or a metal fluoride such as CuF2) embedded in a
carbon
matrix, through ex situ treatment of metal/carbon composites with sulfur,
fluorine, and other
-- materials (see, e.g., Example 2).
[0103] Reagents used in the study were purchased from Sigma-Aldrich
unless
otherwise specified and used without purification. Iron undecylenate was
synthesized by the
following procedure. 10.8 g (40 mmol) of FeC13=6H20, 4.8 g (0.12 mol) of NaOH
and 22.1 g
(0.12 mol) of undecylenic acid were added to a mixture of 80 ml of ethanol, 60
ml of water
-- and 140 ml of hexane under vigorous stifling. The mixture was heated at 70
C for 3 hours
and then the organic phase was collected using a separation funnel. After
washing with water
for 3 times, hexane was driven off from the mixture using a rotary evaporator
to obtain iron
undecylenate, a waxy solid.
[0104] In a typical reaction, 2 ml acrylonitrile (AN), 2 ml
divinylbenzene (DVB) and
-- 1.8 g of iron undecylenate were mixed to form a homogeneous solution. 3 mg
of
azobisisobutyronitrile (AIBN) and 100 mg sodium dodecyl sulfate (SDS) were
added to 25
ml of water and the former solution introduced into the aqueous phase under
sonication with
a Sonics VCX500 horn (500 W, 20 kHz, amplitude 50%). The mixture was sonicated
for 3
minutes and after a stable emulsion was formed, heated at 70 C for 12 h.
Sodium chloride
-- was added to induce aggregation of the resultant polymer¨inorganic hybrid
particles, which
were collected by centrifugation. The material obtained was heated in a
nitrogen atmosphere,
first to 320 C, held at this temperature for 1 h, then to 500 C and held for
2 h to obtain the
final metal-oxide/carbon nanocomposite product.
[0105] The crystal structures of the particles were characterized
using a Scintag
Theta¨theta PAD-X X-ray Diffractometer (Cu Ka, ) = 1.5406 A) and their
morphologies
were studied using an FEI Tecnai G2 T12 Spirit Transmission Electron
Microscope (120 kV).
Raman spectra were taken using a Renishaw In Via Confocal Raman Microscope.
Thermogravimetric analysis was performed using a TA Instruments Q5000 IR
Thermogravimetric Analyzer. Electrical conductivity measurement was made using
a Lucas
- 15 -
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
cm'. Gas adsorption analysis for porous materials was performed using a
Micromeritics
ASAP 2020 Accelerated Surface Area and Porosimetry System.
[0106] Electrochemical characterization of the composites as anode
materials in
rechargeable lithium-ion batteries was performed at room temperature in
homemade
Swagelok-type cells. The working electrode consists of 80 wt% of the active
material, 10
wt% of carbon black (Super-P Li from TIMCAL) as a conductivity aid, and 10 wt%
of
polymer binder (PVDF, polyvinylidene fluoride, Aldrich). Lithium foil was used
as the
counter and reference electrodes. A 1 M solution of LiPF6 in a 50 : 50, w/w
mixture of
ethylene carbonate and diethyl carbonate was used as the electrolyte. Celgard
2500
polypropylene membranes are used as the separator. Assembly of cell was
performed in a
glove box with moisture and oxygen concentrations below 1 ppm. The room-
temperature
electrode capacities were measured using Neware CT-3008 battery testers.
[0107] Powder X-ray diffraction was performed to determine the
crystalline phase of
the transition metal oxide. The XRD results, shown in Figure 2(a), match well
with those of
magnetite (JCPDS card no. 19-629). The broad signal in the range of 20-30 may
be due to
the presence of non-crystalline carbon in the composite, because the most
intense reflection
for graphitic carbon (002 layer) should appear at 26.8 . From the ScheiTer's
formula, the
average crystallite size of the Fe304 phase is found to be 21 nm.
[0108] Transmission Electron Micrographs (TEM) for the polymer¨particle
complex
are shown in Figure 3(a). The material generally consists of particles with
sizes in the range
200-400 nm aggregated together. Energy Dispersive X-Ray Spectroscopy (EDX) was
performed on the complex, as shown in Figure 3(c), which confirms that iron
has been
successfully incorporated in the complex. The morphology of the material after
calcination is
shown in Figure 3(b). It consists of uniformly sized Fe304 nanoparticles
embedded in a
carbon matrix and the size is consistent with the average crystallite size
calculated from the
X-Ray diffractograms (21 nm). Oxidative thermal gravimetric analysis (TGA) may
be used to
measure the weight fraction of active material Fe304 in the composite and the
data are shown
in Figure 3(d). The material is heated to 700 C under air so that Fe304 is
oxidized to Fe203
and carbon is oxidized to CO2. From the remaining weight (of Fe203), the
original weight
fraction of Fe304 is calculated to be 66%.
[0109] Other carboxylic acid salts of iron have been used as the
precursor, for
example iron oleate, but the amount of Fe304 eventually encapsulated in the
product can be
substantially lower (e.g., 33%) than for iron undecylenate. The higher
molecular weight of
- 16 -
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
efficient incorporation in the polymer complex. In addition, the fractional
weight loss of
conversion of Fe(ole)3 to Fe304 (91%) is larger than conversion of Fe(ole)3 to
Fe304 (87%),
because of the larger molecular weight of Fe(ole)3. Iron(III) acetylacetonate
(Fe(acac)3),
which has a lower molecular weight than Fe(undec)3 was also investigated. In
this case, the
higher water solubility of the particle precursor does not allow formation of
stable micelles
required for emulsion polymerization.
[0110] Polyacrylonitrile is frequently used to synthesize graphitic
materials through
calcination at high temperatures. To obtain highly graphitic carbon, PAN
should be subjected
to three consecutive processes, namely, stabilization, carbonization and
graphitization. In the
stabilization step, PAN is heated to 200-300 C in air and converted to a
cyclic or a ladder
compound, followed by treatment in nitrogen atmosphere to about 1000 C to
achieve
carbonization of the material. The third step, known as graphitization, is to
heat the material
to 1500-3000 C under argon atmosphere to improve the ordering and orientation
of the
crystallites. Because the present system contains metal oxides, if the
material is heated to
temperatures above ¨ 700 C for the sake of increasing the graphite content in
the product,
there is the possibility of carbon reducing the metal oxide to pure metal. As
a compromise, a
carbonization protocol requiring heating the material at 500 C in dry N2 was
used.
[0111] The Raman spectrum of the Fe304¨C composite is shown in Figure
4. It is
immediately noticeable that the spectrum contains two prominent peaks at
around 1350 and
1590 wavenumbers. Raman spectra for carbon materials usually contain several
peaks. In
particular, the spectrum can be deconvoluted to five bands, conesponding to
ideal graphite
(G 1580 cm-1), a disordered graphitic lattice (D1 1350 cm-1, D2 1620 cm-1 and
D4 1200
cm 1), or amorphous carbon (D3 1500 cm 1). G and D2 both come from sp2 carbon
vibrations, which can be difficult to distinguish, and in some works have been
treated as one
single component in the fitting procedure. Lorentzian functions were used in
the fitting and
the calculated positions for the peaks are: G 1596 cm-1, D1 1349 cm-1, D3 1471
cm-1, and
D4 1230 cm-1. This analysis indicates the carbon obtained in the composite is
partially
graphitic.
[0112] Magnetite has the formula Fe2 [Fe3+2104 and adopts an inverse spinet
structure. In each unit cell (containing 8 multiples of Fe304), 8 out of 16
Fe3+ ions occupy 8
out of the 64 tetrahedral sites and all the Fe2+ ions and the remaining 8 Fe3+
ions are
distributed in 16 out of 32 octahedral sites. Lithiation of Fe304 follows the
following
pathway:
- 17 -
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
" -
(Piev JO., (Maw 13
IrePizaw 2)
_________________________________ 3t41...0 =4=3re (Maw 3)
[0113] Here parentheses denote ions in tetrahedral sites and square
brackets denote
ions in octahedral sites. During the insertion of up to 1.0 Li, the Li + ions
fill up octahedral
vacancies, with Fe3+ in the tetrahedral sites displaced to octahedral sites,
leading to the
formation of a rock-salt-like structure of Li1.0Fe304 at the end of this step.
Further insertion of
lithium involves the filling of the tetrahedral sites by Li + ions. Metallic
iron is extruded from
the rock-salt structure to accommodate the incoming Li + ions.
[0114] Cyclic voltammograms for Fe304¨C composites are shown in Figure
5(a)
(scan rate = 0.2 mV s-1). The patterns are consistent with the CV results from
other reports on
Fe304¨C composites. In the anodic process, starting from the second cycle, the
lithium
intercalation occurs mainly at around 0.7 V and in the cathodic process the
oxidation of Fe
occurs at around 1.8 V. In the first cycle, the intercalation occurred at a
lower voltage of
around 0.4 V, probably because of an overpotential arising from the crystal
structure changes
from the inverse spinet structure to the rock salt type structure. The
voltage¨capacity profiles
for the complex cycled at different charging rates (1 C or 0.5 C) are shown in
Figure 5(b).
The lithium intercalation plateaus are not as flat for the Fe304 in carbon
composites compared
to the pure oxide, probably because of reduction in crystallinity and/or
change in surface site
energetics during the process of the carbon formation for the composite.
[0115] Cycling performance data for the Fe304¨C composites are shown
in Figure 6.
The material was cycled at 1 C and 0.2 C, respectively for 100 cycles and the
performance
under different charging rates ranging from 0.5 C to 5 C was also studied.
The capacities
are calculated based on the metal oxide mass because the capacity¨voltage
profiles do not
indicate significant contribution from lithium intercalation into the carbon
host. It is apparent
from Figure 6 that the composites show very stable performance and little
fading for 100
cycles, even at 1 C charging rate. The performance is also stable for higher
charging rates (20
cycles are shown as examples). The performance of bare Fe304 nanoparticles (50
nm in size,
commercially available from Alfa Aesar) as the anode material is also shown in
Figure 6(d)
for comparison purposes; the clear improvements provided by the composite
materials are
visible from this plot. The performance of pure carbon made from pyrolysis of
PAN¨DVB at
- 18 -
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
to the lithium storage capacity.
[0116] The stable electrochemical performance of the Fe304-C
nanocomposites can
be attributed to different features of the materials. Considering the
relatively low electronic
conductivity of the carbon component, it is not a consequence of enhanced
electronic
transport afforded by the carbon. The uniformly sized Fe304 nanoparticles are
embedded in
the carbon matrix, which might serve to alleviate the volume change incurred
during the
repeated cycling. A porous, mechanically flexible reinforcement that allows
good penetration
by the electrolyte into the active material is therefore considered
advantageous. Nitrogen
adsorption analysis was performed on the composite and the surface area
measured by the
BET method is 122 m2 g-1, with the isotherms and the pore size distribution
for the composite
(calculated using BJH method) shown in Figure 7. The BET surface area of pure
carbon
obtained from pyrolysis of the PAN-DVB polymer (without Fe304 nanoparticles)
is about
three times higher, 369 m2 g-1. The pore size distribution results show that
most of the pores
are less than 10 nm in size.
[0117] The size of the Fe304 nanoparticles also seems to be an
important factor in
determining the electrochemical performance. Average diameters of Fe304
particles
synthesized using hydrothermal/solvothermal methods are usually greater than
150 nm
because the particles are typically aggregates of smaller primary
crystallites. In the current
method, the size of the Fe304 nanoparticles is relatively small and the
greater surface area and
shorter diffusion length may allow easier access of the active material by the
lithium ions.
[0118] The method developed for creating Fe304-C composites can be
applied to
synthesize nanocomposites of various other metal oxides (or other related
materials such as
pure metal) embedded in carbon matrices. Another interesting material is MnO,
which has a
theoretical lithium storage capacity of 755 mA h g-1. MnO undergoes conversion
reaction in
lithium-ion batteries: 2Li + MnO ¨> Mn + Li20 and upon lithium insertion, Mn
grains <5 nm
in size are formed. MnO-C composites can be synthesized using manganese(II)
undecylenate
as the precursor. Figure 8(a) shows the X-ray diffractogram for the MnO-C
composite, which
matches well with MnO (JCPDS card no. 07-230). Again a broad band is observed
in the
range of 20-30 , but no sharp peak could be found at 26.8 , indicating that
the carbon
component is largely amorphous. TGA is used to determine the fraction of MnO
in the
composite. Upon heating to 700 C in air, MnO is oxidized to Mn203 and the
weight fraction
of MnO in the composite is calculated to be 58% assuming all the remaining
material is
- 19 -
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
case of Fe304, MnO nanoparticles embedded in a carbon matrix are obtained.
[0119] A typical cyclic voltammogram for the MnO¨C composite is shown
in Figure
9(a) and lithium insertion/Mn2+ reduction seems to occur at around 0.5 V
against Li/Lit,
which is consistent with previous reports on MnO anode materials.
Voltage¨capacity curves
at 0.5 C and 1 C charging rates are shown in Figure 9(b) and cycling data for
the composite
run at 1 C and 0.2 C, and at varied charging rates are shown in Figure 9(c).
Similar to what
was observed for the Fe304¨C composite, little capacity fade is observed even
when the
material is subject to 1 C charging rate for over >100 cycles. Cycling at
higher charging rates
is also seen to give stable performance. Therefore a prominent feature of the
current protocol
is the ability to yield materials with stable performance at moderately high
charging rates.
[0120] In conclusion, a one-step free-radical polymerization method is
used to
synthesize cross-linked metal-oxide/poly(acrylonitrile) nanocomposites.
Pyrolysis of the
composite at moderate temperatures in an inert atmosphere yields metal-
oxide/carbon
particles comprised of uniformly distributed metal oxide nanoparticles in a
partially graphitic,
but poorly conducting carbon host. The versatility of the approach has been
demonstrated
using two different metal oxides, Fe304 and MnO. When evaluated as anode
materials in
lithium-ion batteries, composites of both materials display stable performance
at low and
high cun-ent densities.
EXAMPLE 2
[0121] This example describes the synthesis and characterization of
examples of
hybrid materials and nanocomposite materials of the present invention.
[0122] Synthesis of LiFePO4@C nanocomposite. 108mg Li0H, 221mg H3PO4
and
660mg L-ascorbic acid are dissolved in 10m1 DI water, to which 116mg Fe304@C
nanocomposite powder is added. The solution is loaded into a pressurized
container and
heated at 270 C for 12hr. The powder obtained is centrifuged and washed with
water.
[0123] Synthesis of Mn075Fe0250@C nanocomposite. Manganese (II)
undecylenate is
synthesized using the same method as iron (III) undecylenate, with MnC12 as
the Mn
precursor. 1.58g manganese (II) undecylenate and 0.75g iron (III) undecylenate
are mixed to
form a homogeneous mixture, and polymerization with acrylonitrile and
divinylbenzene is
performed using the same method as used for iron (III) undecylenate alone. The
polymerization product is collected ant heat treated in the same way to obtain
Mno 75Fe0 250@ C nanocomposite.
- 20 -
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
H3PO4 and 49mg H3P03 are dissolved in 10m1 DI water, to which 142mg
Mn075Fe0250@C
nanocomposite powder is added. The solution is loaded into a pressurized
container and
heated at 270 C for 12hr. The powder obtained is centrifuged and washed with
water.
EXAMPLE 3
[0125] The synthesis, structural characterization and electrochemical
performance of
MoS2¨carbon nanostructures is described in this example.
[0126] Composites of MoS2 and amorphous carbon were grown and self-
assembled
into hierarchical nanostructures via a hydrothermal method. Application of the
composites as
high-energy electrodes for rechargeable lithium-ion batteries was
investigated. The critical
roles of nanostructuring of MoS2 and carbon composition on lithium-ion battery
performance
are described. Pure MoS2 and 22 wt% carbon containing MoS2 materials are
designated as
MS-0 and MS-22 respectively.
[0127] Morphological investigations using SEM (Figure 18a) reveal that
the product
obtained via the hydrothermal treatment of the molybdenum sol in presence of
carbon
precursors (resorcinol and formaldehyde) takes the form of open structure of
MoS2 and
carbon. The TEM images of MS-22 (22 wt% carbon), as shown in Figures 18b and
c, indicate
that MoS2 in the composites are in the form of stacked nanosheets
homogeneously embedded
in a very thin matrix of amorphous carbon. The length and thickness of MoS2
nanosheets are
about 40 and 10 nm respectively (inset of Figure 18b). It can be observed that
the MoS2
sheets are composed of few MoS2 layers (-6 to 10 layers). A schematic of the
in situ
synthesis of MoS2¨carbon composites is shown in Figure 19A. In absence of
resorcinol and
formaldehyde, the MoS2 particles aggregate to form large MoS2 lumps (Figure
19B) as
verified by the SEM and TEM images (Figures 18d and f). On the other hand,
polycondensation of resorcinol with formaldehyde takes place during the
hydrothermal
process forming low density carbon gels. The MoS2 particles in the form of
layers
simultaneously crystallize during the hydrothermal process and are eventually
uniformly
dispersed in the carbon gel. The successive restacking of MoS2 layers is
significantly
inhibited by the carbon gel resulting in few layers of MoS2 nanosheet and
consequently self-
assembled in interconnected flakes resulting in three dimensional MoS2¨carbon
nanostructures.
[0128] The X-ray diffraction patterns (XRD) of the MS-0 and MS-22
shows broad
diffraction peaks which can be indexed to 2H polytype of MoS2 crystal
structure with space
-21 -
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
composites, no peak shifts are observed suggesting that the MoS2 crystallites
are
unconstrained by chemical bonding to the carbon framework. Furthermore, no
characteristic
peak from graphitic carbon was detected in the XRD, indicating the formation
of amorphous
carbon. It can be observed that the intensity of (002) peak for MS-22 is
relatively lower
compared to pure MoS2 particles (see ESI, Figure 23). The peak at (002) is
typically observed
in bulk analogue. It suggests that the carbon inhibits the growth of (002)
planes of MoS2
crystallites, which confines its growth in the plane favoring formation of few-
layer
nanosheets in the amorphous carbon matrix. The carbon content is estimated
using
thermogravimetric analysis; the results are depicted in (see ESI, Figure 24).
It is seen that
there is successful incorporation of 11, 22, 32 and 41 wt% of carbon in MoS2-
carbon
nanostructures. The BET surface areas of MS-0, MS-22 and MS-41 are 3, 35 and
157 m2 g-1
respectively (see ESI, Figure 25). The increase in surface area with
increasing carbon content
is attributed to the porous nature of amorphous carbon.
[0129] The electrochemical properties and lithium battery performance of
all MoS2-
carbon composites using galvanostatic discharge and cyclic voltammetry
measurements was
investigated. Electrodes were prepared from the MoS2-carbon composites and a
PVDf
binder, i. e. , no carbon black or other conductivity aid was added. Figure 20
shows the
galvanostatic charge (Li removal)/discharge (Li insertion) profiles obtained
from pure and
carbon-composited MoS2 at room temperature (25 C) and at a constant current
density of
100 mA g-1 in the voltage range of 0.05-3 V. In the present study, pure MoS2
particle
exhibited a discharge capacity of 2362 mA h g-1 in the Et discharge cycle
(Figure 20a). Two
potential plateaus at 1.1 V and 0.61 V corresponding to the formation of
LixMoS2 and
conversion of MoS2 to Mo respectively are also readily apparent from Figure
20. The pure
MoS2 shows very poor Et charge and 2nd discharge capacities of 247 and 53 mA h
g-1,
respectively without any noticeable potential plateaus.
[0130] The MoS2-carbon nanocomposite structures all exhibit
significantly improved
capacity retention (Figures 20b and e). All MoS2-carbon composites manifest
the prominent
characteristic discharge potential plateaus at 1.1 V and 0.6 V in the Et
discharge cycle and
charge potential plateau at 2.3 V in all charge cycles (Figure 20b). During
subsequent
discharge cycles, the potential plateau observed at 0.6 V disappeared with
emergence of two
new inconspicuous potential plateaus at -1.9 V and 1.2 V (Figure 20b) which is
in agreement
with previous observations. The first discharge capacities of MS-11, MS-22, MS-
32 and MS-
41 are 2108, 1462, 1130, and 1078 mA h g-1 with coulombic efficiencies of 79%,
62%, 63%,
- 22 -
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
battery performance in terms of showing higher capacity and long-term
stability. MS-22
shows a discharge capacity of 755 mA h g-1 with coulombic efficiencies of 98%
after 100th
cycle at constant current density of 100 mA g-1. In contrast, MS-11, MS-32 and
MS-41 show
capacities of 10, 517 and 354 mA h g-1 after 100th cycle. Although MS-32 and
MS-41 show
good long-term capacity retention, the lower capacity possessed by both of
them is due to
higher amount of inactive carbon in the electrode compositions. It ascertains
the importance
of optimization of inactive carbon in electrode materials.
[0131] The improved cycling stability of Mo52¨carbon composites can be
attributed
to the inhibition of the side reaction of Li25 with the electrolyte that forms
a thick, gel-like
polymeric layer and manifests particle aggregation. The evidence in support of
this
explanation is provided by cyclic voltammetry (CV) (Figures 20c and d), ex
situ XRD
(Figure 21) and SEM of the 1St cycledischarged product (Figure 21). The CV
plot of MS-22
(Figures 20d) shows two distinct reduction peaks at 0.93 V and 0.37 V in the
1St cycle, which
is indicative of the respective formation of LixMoS2 and decomposition of Mo52
to Mo and
Li25. In the subsequent oxidation cycles shown until the 15th cycle, small but
sharp intensity
peaks at 1.67 V and 2.34 V can be observed. These peaks are attributed to
partial and
complete oxidation of Mo to Mo52, respectively. In the subsequent reduction
cycles, peaks at
0.93 V and 0.37 V disappear and two new small intensity peaks at 1.83 V and
1.01 V can be
observed. The peaks are in agreement with the potential plateaus observed in
charge¨
discharge curves of MS-22 (Figure 20b). It is also evident from the CV data
that even after
15th cycle, the Mo52¨carbon composite material shows excellent
electroactivity, with
negligible decrease in peak intensities. On contrary, pure Mo52 shows a
distinct reduction
peak at 0.93 V and a broad reduction peak in the voltage range of 0.05-0.5 V
in the E1
reduction cycle. Two minor intensity oxidation peaks at 1.8 V and 2.3 V can
also be observed
in the 1St oxidationcycle followed by little noticeable electrochemical
activity after the E1
cycle. The ex situ XRD of MS-0 and MS-22 both shows the signature of Mo (ICDD
no. 071-
3771) and Li25 (ICDD no. 071-4841) after 1St discharge cycle (Figure 21).
However, the peak
intensity of Li25 for MS-0 (Figure 21b) is significantly lower than MS-22
(Figure 21a). The
breadth of the CV peak at 0.05-0.5 V and decrease in Li25 XRD peak intensity
in case of
pure Mo52 particle can be explained in terms of the side reaction of Li25 with
the electrolyte.
Further evidence is provided by the ex situ SEM images of the 15t
cycledischarge products of
MS-0 (Fig. 21d) and MS-22 (Figure 21c). MS-22 maintains its original structure
whereas
pure Mo52 particles aggregated due to electrolyte degradation with Li25. As a
consequence of
-23 -
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
show higher capacity (2362 mA h g-1) but with extremely poor coulombic
efficiency of 10%
compared to all carbon¨MoS2 composites. In addition, particle agglomeration is
also a
serious cause for the poor cyclability of pure MoS2.
41, MS-11 exhibits less stable storage capacity over extended cycling.
Evidently, carbon does
have a stabilizing effect on the cycling stability of Mo52 and the composition
of the
composites can be used to influence their stability. Second, there is a
critical concentration of
carbon at which the capacity and cyclability are optimized. 22% carbon
containing Mo52¨
carbon composite exhibits the best stability. It suggests that the optimum
value of carbon in
rendering stable cycle life is around 22%. At carbon concentrations less than
22%, the Mo52
particles are not effectively coated with carbon (ESI, Figure 26a) that
promotes possible
electrochemical reaction of Li25 and electrolyte during the first discharge
process. It is
important for lithium-ion battery purposes to ensure good buffering for active
material.
Therefore, similar to MS-0, MS-11 shows higher first cycle discharge capacity
(2108 mA h
g-1) compared to other carbon¨Mo52 compositions. On the other hand, increasing
carbon
concentrations more than 22% results in thicker carbon coating (ESI, Figure
26b) and
increase in inactive mass in the electrode. Since resorcinol¨formaldehyde
synthesized carbon
is porous, the electrolyte can wet the Mo52 particle. Therefore, MS-32 and MS-
41 show
stable electrochemical activity. However, the lithium storage capacities are
lower than MS-22
due to increased proportion of inactive mass in the electrode.
[0133] The cycling stability and rate capability of MS-22 with
additional carbon
black in the electrode is shown in Figures 22 and 23 (ESI). It is observed
that incorporation of
higher amount (50%) of carbon black facilitates higher rate capability. To
further testify the
carbon quality present in the Mo52¨carbon composites, the material is calcined
at 700 C.
The lithium battery performance is shown in (see ESI, Figure 28). Heat
treatment of 700 C is
found to have deteriorating effect on the cyclability due to possible
temperature induced
crystallite growth.
[0134] A facile one-pot hydrothermal method for the synthesis of
Mo52¨carbon
nanostructures with various carbon compositions was demonstrated. The
procedure utilizes
- 24 -
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
demonstrated that incorporation of carbon provides significantly improved
cycling stability
when the material is used as a lithium battery electrode. It is also found
that an optimum level
of carbon is required to produce materials with both high lithium storage
capacity and good
electrochemical cycling stability. The improved performance is attributed to
following three
main factors. First, the porous structure of the composites allows for facile
Li + insertion¨
deinsertion into MoS2 nanosheets and for structural stresses induced by Li +
insertion¨
deinsertion to be properly accommodated since the dimension of MoS2 nanosheets
are small
(thickness ¨10 nm) and composed of few layers (-6 to 10 layers). Second,
incorporation of
Mo52 in the carbon matrix inhibits the side reaction of Li25 with electrolyte
at the interface of
Mo and carbon and finally, the carbon framework limits particle agglomeration.
The
synthesis approach reported in the present invention will be beneficial for
designing new
organic-solvent free synthesis methods for creating composite electrode
materials for lithium
batteries.
[0135] Synthesis of Mo52-carbon: The Mo52-carbon composites with varying
carbon
weight fractions were synthesized by a hydrothermal method.
Resorcinol/formaldehyde
(Sigma-Aldrich) and ammonium tetrathiomolybdate (Sigma-Aldrich) were used
respectively
as carbon and Mo52 precursors. A desired concentration (0.076 M) of aqueous
solution of
ammonium tetrathiomolybdate was added to another aqueous solution containing
resorcinol,
formaldehyde and sodium carbonate under continuous stiffing. The ratios of
resorcinol to
formaldehyde and to sodium carbonate were kept at 0.185 g m1-1 and 251
respectively
calculated on a molar basis for all Mo52-carbon composites. However, the
concentrations of
resorcinol, formaldehyde and sodium carbonate were varied to obtain various
carbon loadings
in the final product. The intense violet color sol was transferred to a Teflon-
lined stainless
steel autoclave of capacity 100 ml (70% filling) and heated at 180 C for 12
hours and then
cooled to room temperature. The resultant black product was recovered by
centrifugation and
washed with deionized water and freeze dried. The dried product was further
calcined at 550
C for 4 hours in an atmosphere of 5% H2 balanced with Ar at a heating rate of
5 Cmin-1.
Pure Mo52 was synthesized by hydrothermal treatment of ammonium
tetrathiomolybdate
(180 C for 12 h, calcination at 550 C for 4 hours under H2/Ar), but without
any addition of
resorcinol and formaldehyde. The materials were designated as MS-0, MS-11, MS-
22, MS-
32 and MS-41coffesponding to 0, 11,22, 32 and 41 wt % of carbon in the Mo52-
carbon
composites.
- 25 -
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
diffraction (Scintag theta¨theta PAD-X-ray Diffractometer; Cu-Ka radiation,
ii, = 1.5406 A).
The morphology was observed by scanning electron microscopy (SEM, LEO 1550
FESEM)
and transmission electron microscopy (TEM, FEI Tecnai G2 T12). Specific
surface area
(BET) was obtained from nitrogen adsorption-desorption isotherms
(Micromeritics ASAP
2020). Estimation of carbon content in Mo52-carbon composites was done using
thermogravimetric analysis (TGA, TA Instruments Q5000). TGA experiments were
performed by heating the sample in air from room temperature to 700 C at a
heating rate of
Cmin-1. For the working electrode, slurry of the active material and carbon
black (Super P
10 Timacal) was prepared with PVdF (Sigma) in a weight ratio of
Mo52:CB:PVDf = 90:0:10,
80:10:10, 65:10:10, 40:50:10 in N-methyl-pyrrolidone (NMP). The slurry was
cast on a
copper foil and dried in vacuum at 120 C for 12 h. Room temperature cyclic
voltammetry
(CV, CH608 CH Instruments) and galvanostatic charge/discharge cycling (Maccor)
were
done in 2032 coin-type cells with pure metal Li (Aldrich) as anode, Whatman
glass fibre as
separator and 1M LiPF6 in ethylene carbonate (EC, Aldrich) and dimethyl
carbonate (DMC,
Aldrich) (1:1 w/w) as an electrolyte.
EXAMPLE 4
[0137] This example describes the synthesis and characterization of
examples of
hybrid materials and nanocomposite materials of the present invention.
materials, nanoscale organic-inorganic hybrid materials created using the
approach are
attractive as anodes and cathodes for next-generation lithium and other
rechargeable battery
systems. Additionally, the platform is very versatile and through ex situ
conversion or
utilization of multiple precursors, can be applied to various classes of
materials including
metal oxides, metals, metal sulfides and alloys. The approach also lends
itself to the
development of scalable processes for production of nanostructured battery
materials.
[0139] This general approach for synthesizing metal oxide-, metal
sulfide-, and metal
alloy-carbon nanocomposites ameliorates the physical and chemical stresses
associated with
repeated insertion and de-insertion of lithium present a fundamental challenge
to further
- 26 -
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
case investigated the nanocomposites manifest improved electrochemical
stability whether
they are applied as anodes or cathodes in a secondary battery.
[0140] An approach for the in situ synthesis of nanoparticles embedded
in a carbon
matrix through a miniemulsion polymerization technique was shown. The as
prepared
carbon-nanoparticle hybrid materials can be facilely modified ex situ to
significantly increase
the range of materials chemistries that can be achieved by the method. The
approach is based
on the in situ synthesis of inorganic nanoparticles and organic polymers from
precursors
capable of forming chemical cross-links with each other. After pyrolysis of
the organic phase,
the process yields a well-defined nanostructured material comprised of
discrete inorganic
nanoparticles embedded in a porous carbon matrix. Post treatment of the
embedded particles
creates carbon-nanoparticle hybrids based on metals, metal alloys, and a
variety of other
particles attractive for lithium battery applications.
[0141] Emulsion polymerization is a widely used method for
synthesizing polymer
latexes for applications such as adhesives and coatings. The method typically
uses monomers
with low water solubility, stabilized by surfactant in an aqueous media. The
polymerization
rate is limited by the diffusion of reactive monomer, through the aqueous
phase, from
monomer droplets to monomer-swollen polymer particles where polymerization
takes place.
Application of high shear force to the emulsion yields a so-called
miniemulsion, comprised of
droplets with small sizes, usually 0.01-0.5 um, compared to 1-10 um in
conventional
emulsion polymerization. Because of the high surface area of monomer droplets
in a
miniemulsion, nucleation takes place mainly via radical entry into the
emulsified monomer
droplets and reaction proceeds through polymerization of the monomers in these
small
droplets. If more than one monomer chemistry is employed simultaneously or
sequentially,
the approach can be used to create copolymers with different architectures. If
the
polymerization reaction is performed in the presence of guest species miscible
with the
monomer (e.g. dyes, metal complexes, etc.), the guest species can be embedded
in the
polymer particle host. However, a common drawback is that only relatively low
loadings (a
few percent by weight) of the guest can be achieved. A new method to overcome
this
drawback and demonstrate the applicability of the method to synthesize
composites involving
various types of LIB electrode materials was demonstrated.
[0142] Chemicals and materials synthesis. Chemical reagents were
purchased from
Sigma-Aldrich and used without purification. Fe304@C nanocomposite was
synthesized
according to a previously reported procedure. 2m1 acrylonitrile (AN), 2m1
divinylbenzene
-27 -
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
azobisisobutyronitrile (AIBN) and 100mg sodium dodecyl sulfate (SDS) were
added to 25m1
of water and the former solution added dropwise to the aqueous phase under
sonication with
a Sonics VCX500 horn (500W, 20kHz, amplitude 50%). The mixture was sonicated
for 3
minutes and after a stable emulsion was formed, heated at 70 C for 12 hrs.
Sodium chloride
was added to induce aggregation of the resultant polymer-inorganic hybrid
particles, which
were collected by centrifugation. The material obtained was heated in an argon
atmosphere,
first to 320 C, held at this temperature for lhr, then to 500 C and held for
2hrs to obtain the
Fe304@C nanocomposite product. Fe304@C was then ground into powder and heated
at
650 C in a tube furnace under a 7% H2 (balance Ar) gas environment for 2hrs to
obtain
Fe@C powders. The latter is mixed with 2x mass of sulfur, loaded into a Pyrex
tube, sealed
and heated at 500 C for 4hrs to obtain Fe52@C nanocomposite. The product is
washed with
CS2 to remove any residual elemental sulfur. To synthesize y-Fe203@C and a-
Fe203@C
composites, Fe304@C powder is heated in air at 350 C for 5hrs and 390 C for
lhr,
respectively, to obtain the products. To synthesize V205@C, VC13 is used as
the starting
material to synthesize V(C10f119C00)3. After polymerizing with acrylonitrile,
the material is
pyrolysed at 500 C in argon for 2hrs and then heated in air at 390 C for
lhourto obtain
V205@C. To synthesize FeSn2@C nanocomposite, tin undecylenate
(Sn(C10f119C00)2) was
synthesized in a similar fashion as iron undecylenate, except with SnC12 as
the starting
material. 2.2g Fe(C10f119C00)3 and 1.8g Sn(C10f119C00)2 were mixed first, 2m1
AN and 2m1
DVB were added, and then the rest of the procedure was carried out as above.
[0143] The crystal structures of the particles were characterized
using Scintag Theta-
theta PAD-X X-Ray Diffractometer (Cu Ka, 2,= 1.5406 A) and their morphologies
were
studied using ELI Tecnai G2 T12 Spirit Transmission Electron Microscope
(120kV).
Thermogravimetric analysis was performed using TA Instruments Q5000 IR
Thermogravimetric Analyzer.
[0144] Cell assembly and testing. Electrochemical characterization of
the composites
as anode materials in rechargeable lithium-ion batteries was performed at room
temperature
in 2032 coin-type cells. The working electrode consisted of 80 wt% of the
active material, 10
wt% of carbon black (Super-P Li from TIMCAL) as a conductivity aid, and 10 wt%
of
polymer binder (PVDF, polyvinylidene fluoride, Aldrich). Copper foil was used
as the
current collector for nanocomposites targeted for application as the LIB anode
and aluminum
for those targeted as cathodes. Lithium foil was used as the counter and
reference electrode
for evaluating both that anode and cathode materials. A 1 M solution of LiPF6
in a 50:50 w/w
- 28 -
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
2500 polypropylene membranes are used as the separator. Assembly of cell was
performed in
a glove box with moisture and oxygen concentrations below 1 ppm. The room-
temperature
electrode capacities were measured using Neware CT-3008 battery testers.
Cyclic
voltammetry was performed with a CHI600D potentiostat.
[0145] Figure 30 illustrates the chemistry of a miniemulsion
polymerization
methodology that could be used to create organic-inorganic hybrid copolymers
with high
inorganic loadings. Using an organic monomer (e.g. acrylonitrile, or AN) and
the metal salt
of an unsaturated carboxylic acid (e.g. iron (III) undecylenate) and
divinylbenzene as
crosslinker the method yields well-defined iron oxide nanoparticles uniformly
embedded in a
polyacrylonitrile host (Figure 31A). FTIR spectra of iron (III) undecylenate
and the AN-iron
copolymer composite are compared in Figure 39. Significant decrease in the
intensity of the
C=C stretch peak at around 1640 cm-1 (normalized with respect to C-H stretch
at around 2910
-1 i
cm ) s observed, showing that as expected many of the double bonds in iron
(III)
undecylenate have been eliminated during polymerization.
[0146] Upon thermal treatment, the as prepared polyacrylonitrile (PAN)
¨
nanoparticle hybrids are transformed into carbon-Fe304 nanocomposites
characterized by the
uniform distribution of Fe304 in a partially graphitic carbon host was
demonstrated. When
evaluated as the anode in a lithium ion battery, the material showed
significantly improved
cycling stability and capacity retention relative to anodes based on pristine
Fe304
nanoparticles. The performance enhancement brought about by the in situ
synthesis approach
was argued to largely originate from the uniform separation of the embedded
nanoparticles
achieved in the composites, which simultaneously minimizes aggregation of the
active
nanostructures, facilitates electron transport, and maximizes the degree to
which the carbon
framework is able to absorb and isolate mechanical stresses produced by
structural changes.
Figure 31B shows the TEM image of the nanocomposite after 100 charge-discharge
cycles. It
indicates that with the mechanical support provided by the carbon matrix, the
pulverization of
the active material nanoparticles is mitigated, which is the source of the
observed
improvement in cyclability.
[0147] A goal of the present work is to illustrate the versatility of the
synthesis
method and to evaluate the generality of the hybrids produced. Figure 32 shows
an
abbreviated list that identifies the variety of hybrid materials relevant for
application in
lithium battery electrodes that can be synthesized using the approach. Because
of the large
number of Fe-based compounds and alloys that are of interest for LIB
applications,
- 29 -
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
compounds; the enhancements were subsequently evaluated in their properties by
using the
composites as anodes or cathodes for LIBs. Results on other examples (e.g.
materials based
on vanadium and titanium) will also be discussed.
[0148] As illustrated in Figure 32, carbon-Fe304 nanocomposites (Fe304@C)
synthesized using the procedures outlined earlier can be reduced to Fe@C
composites either
by heating the material in an H2 environment or simply by heating the
composite in an inert
gas to a temperature somewhat higher than the carbonization temperature
(whereby carbon
serves as the reducing agent). The XRD pattern of the material obtained after
heating
Fe304@C to 650-700 C under H2 is shown in Figure 33A, which is unambiguously
assigned
to the a-Fe (JCPDS card #06-0696). Figure 33B is a transmission electron
micrograph of the
material showing that the materials are comprised of well-dispersed ca. 30 nm
Fe
nanoparticles, which is consistent with the average crystallite size of 29nm
deduced from
XRD.
[0149] Because the carbon matrix is porous, it allows the infusion of other
chemical
agents, which can react with the embedded Fe nanoparticles. FeS2 is a
promising cathode
material for lithium batteries because of its high reversible capacity (625
mAh/g), low cost
and low toxicity. It is well-known in primary lithium battery applications and
high
temperature thermal batteries, but its use in room-temperature rechargeable
cells has been
hindered by the material's limited cyclability. A vapor infusion procedure was
used to react
the Fe@C composites with sulfur at 500 C. Figures 33A and C are the
conesponding XRD
and TEM patterns for this material. The XRD pattern is unambiguously assigned
to FeS2
(JCPDS card # 42-1340) and reveals that reaction with sulfur has nearly
doubled the
crystallite size to 54nm; again consistent with results from TEM, which show
uniformly
distributed ca. 55nm FeS2 particles in the carbon host. Particle size
histograms obtained from
TEM images for Fe@C and FeS2@C composites are shown in Figures 33(D) and (E),
with
average sizes of 29.7 3.8nm and 53.8 9.9nm, respectively. Through oxidative
TGA (Figure
40) the weight fraction of FeS2 in the product is found to be 75%.
[0150] Figure 34 report results from cyclic voltammetry and
galvanostatic cycling
measurements performed using the as prepared FeS2@C composites. In the
cathodic scan of
the first cycle, FeS2 follows a two-step lithiation: FeS2+ 2Li+ + 2e
Li2FeS2 (-2V) and
Li2FeS2 + 2Li+ + 2e Fe + 2Li2S (-1.4V). In the anodic scans, the material
is converted to
Li2FeS2 at around 1.8V and then to Li2FeS2 (0<x<0.8) at around 2.5V. At room
temperature
if the material is driven to high potentials (above 2.45V), instead of
regenerating the FeS2
- 30 -
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
transformation from Li2õFeS2(hexagonal) to FeSx (hexagonal). Subsequent
cycling occurs
between Li2õFeS2 and Fe/Li25. Voltage-capacity profiles at different charging
rates are
shown in Figure 41. Plateaus at ¨2V and 1.4-1.5V in the discharge curve
correspond to
lithiation peaks in the anodic scan. The gravimetric capacity is calculated
with respect to
active material mass (the same for the materials to follow in this work). The
cyclability is
compared with commercially available pristine Fe52, showing the enhancement of
cycling
performance brought about by forming nanocomposites with carbon.
[0151] When more than one metal precursor is used, the approach
yielded
nanocomposites with alloy nanoparticles embedded in the carbon matrix. An
example of this
is the iron-tin alloy, which is being actively investigated as an anode
material in LIBs. Alloys
of tin with another metal (e.g. Sb, Co, Fe, Ni) are able to provide some
alleviation effect for
the pulverization of tin through the mechanical protection offered by the
other metal which
gets extruded during lithiation. The incorporation of such alloy nanoparticles
in a carbon
matrix provides a means of additional mechanical support so that the cycling
stability of the
material may be further enhanced.
[0152] FeSn2@C nanocomposites can be synthesized using a combination
of iron and
tin precursors, as confirmed by XRD (Figure 35A). EDX indicates the presence
of iron and
tin in the composite (Figures 35C and 35D) and yields an atomic ratio of
Fe/Sn=0.59. The
weight fraction of FeSn2in the composite is determined using oxidative TGA to
be 68%
(Figure 40). The TEM images are shown in Figure 35B. There appears to be a
broader
distribution of nanoparticle size compared to other materials (e.g. metal
oxides, sulfides)
synthesized using the same approach. The reason may be that tin has a
relatively low melting
point and liquid tin likely formed droplets with broad size distribution
before reacting with
iron. Cyclic voltammograms of the material are shown in Figure 36A. The
electrochemical
reaction of FeSn2 in LIB can be expressed as follows: FeSn2+ 8.8Li+ + 8.8e
2Li445n + Fe
and Li445n Sn + 4.4 Li + + 4.4e. The reversible capacity of the material
results from the
repeated alloying and dealloying of lithium with tin. Multiple lithiation
peaks occur in the CV
indicating the multi-step reaction associated with Li-Sn alloying. Some of the
important
intermediate phases include Li75n3 (formed at ¨0.45V vs. Li/Lit) and Li75n2(-
0.28V).
Overlapping with the SEI formation peak may have caused some broadening of
these
lithiation peaks. The cycling performance of the composite at different rates
is compared with
-31 -
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
enhancement over the bare material is clearly seen.
[0153] Ex situ treatment may also be performed on metal oxides
themselves to yield
the oxides with different valences of the metal. This brings about a method to
overcome the
limitations on the types of metal oxides that can be synthesized using the
current approach.
There are two main reasons for this limitation. One is that in general metal
salts with higher
valences has a higher tendency to hydrolyze and the corresponding carboxylic
acid salt may
be more difficult to synthesize. For example, only Mn(C10H19C00)2 can be
synthesized using
the current approach and not Mn(C10fl19C00)3. The other is that with a given
precursor,
usually only one type of metal oxide can be obtained from the direct pyrolysis
of the
precursor. For example, the pyrolysis of the Fe(C10H19C00)3precursor only
yields magnetite
and does not directly give maghemite or hematite. With ex situ oxidation, the
composites
involving lower-valence metal oxides may be transformed into composites
containing metal
oxides with higher valences, which cannot be directly made. For example, Fe304
(magnetite)
may be oxidized to maghemite or hematite and MnO may be oxidized to form
Mn304. The
XRD patterns of a-Fe@C (JCPDS card #06-0696), Fe304@C (#19-0629), a-Fe203@C
(#33-
0664) and y-Fe203@C (#25-1402) are shown in Figure 37A and TEM image for y-
Fe203@C
in 6B.
[0154] Another example is vanadium. V(V) salt is not stable in water
and V205
cannot be directly synthesized using this approach. However, V(III) salt may
be used to
synthesize V(C10H19C00)3 precursor which can be pyrolysed to form V02@C which
is then
oxidized in air to give V205@C composite. The XRD patterns and TEM images of
V205@C
(JCPDS card #41-1426) and Ti02@C (anatase, JCPDS card #21-1272) are shown in
Figures
37C-F.
[0155] The electrochemical performance of y-Fe203@C was tested. a-Fe203 has
been
extensively investigated as LIB electrode materials undergoing either
intercalation
mechanism at low levels of lithiation or conversion reaction at high levels of
lithiation and
there have also been some reports on y-Fe203. Cyclic voltammograms of they-
Fe203@C
composite synthesized using the current method are shown in Figure 38A. Fe203
nanoparticles follow the reversible conversion reaction Fe203+ 6Li+ +6e 2Fe
+3Li20
when fully lithiated. The large peak in the first cathodic scan at ¨0.5V vs.
Li/Lit is usually
attributed to SEI formation and in the subsequent cycles lithiation of Fe203
takes place at
¨0.8V corresponding to the reduction of Fe3+ to Fe . The broad peak centered
at ¨1.7V in the
-32-
CA 02840747 2013-12-30
WO 2013/003836
PCT/US2012/045188
cyclic voltammograms indicates stable cycling performance, which is shown in
Figure 38B at
1C, 0.5C and 2C charging rates.
[0156] Since the active material is incorporated in an amorphous
carbon matrix,
which does not make a significant contribution to the lithiation capacity, it
is useful to
determine the effect of the carbon. Using as an example the Fe304-carbon
nanocomposite
containing 66% by weight Fe304 (924mAh/g) and the balance carbon (40mAh/g),
the
gravimetric theoretical capacity of the composite is 620 mAh/g. From mercury
porosimetry,
the pore volume of carbon is found to be 0.5516 ml/g and assuming the bulk
densities of
magnetite and amorphous carbon to be 5.2 and 2.1 g/cm3, respectively, the
volumetric
theoretical capacities of magnetite and the composite are 4.81 and 1.30
Ah/cm3. Therefore the
employment of the porous carbon matrix comes at the cost of a reduced
volumetric capacity,
which can be limited in an actual battery design by engineering the porosity
and weight
fraction of the carbon matrix to achieve desired gravimetric and volumetric
capacity goals
while preserving the improving cyclability imparted by the porous carbon
support.
[0157] In conclusion, a platform has been developed whereby through
the
copolymerization of organic and inorganic starting materials and formation of
a hybrid
followed by calcination, embedded nanostructures consisting of uniformly sized
nanoparticles incorporated in a porous carbon matrix may be synthesized in
situ. Either by
mere in situ reaction, or combined with ex situ engineering of the embedded
material, a wide
variety of embedded nanostructures may be synthesized which show enhanced
lithium
storage performance over the bare material. The method obviates the relatively
stringent
experimental control required in many other methods of creating carbon
composites and
provides a convenient way to prevent the aggregation of particles. Therefore
the process
lends itself to cheap and facile scale-up. Besides the materials, which have
been demonstrated,
additional categories of materials can be made using the current approach
(e.g. silicon and
phosphates), which is part of the ongoing work.
[0158] While the invention has been particularly shown and described
with reference
to specific embodiments (some of which are preferred embodiments), it should
be understood
by those having skill in the art that various changes in form and detail may
be made therein
without departing from the spirit and scope of the present invention as
disclosed herein.
-33 -