Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02875768 2016-09-20
PROCESS FOR THE PREPARATION OF PHOSPHONATE COMPOUNDS
USEFUL IN THE PREPARATION OF HIMBACINE ANALOGS
This application is a division of CA 2,594,871
Field of the Invention
This application discloses a novel process for the preparation of
himbacine analogs useful as thrombin receptor antagonists. The process
is based in part on the use of a base-promoted dynamic epimerization of a
chiral nitro center.
Background of the Invention
Thrombin is known to have a variety of activities in different cell
types and thrombin receptors are known to be present in such cell types as
human platelets, vascular smooth muscle cells, endothelial cells, and
fibroblasts. Thrombin receptor antagonists may be useful in the treatment
of thrombotic, inflammatory, atherosclerotic and fibroproliferative
disorders, as well as other disorders in which thrombin and its receptor
play a pathological role. See, for example, U.S. 6,063,847.
- 1 -
CA 02875768 2014-12-19
One thrombin receptor antagonist is a compound of the formula:
0 H H
0 _ i , \NHCOOEt
CICI
H = H
Me ¨,
N
I
SF
11
This compound is an orally bioavailable thrombin receptor
antagonist derived from himbacine. Compound 11 may be synthesized
from Compound 1:
0 H
0 õNO2
0 0
H - H
(3,NR 5R 6
1
wherein R5 and R6 are each independently selected from the group
consisting of H, alkyl, alkenyl, alkynyl, alkoxy, cycloalkyl, aryl, alkylaryl,
arylalkyl, and heteroaryl groups.
Processes for the synthesis of similar himbacine analog thrombin
receptor antagonists are disclosed in U.S. Pat. No. 6,063,847, and U.S.
publication no. 2003/0216437, methods of using thrombin receptor
antagonists are disclosed in U.S. publication no. 2004/0192753, and the
- 2 -
CA 02875768 2016-09-20
synthesis of the bisulfate salt of a particular himbacine analog is disclosed
in U.S. publication no. 2004/0176418. The present application provides a
novel process for preparing Compound 11 from Compound 1, which
process provides an improved yield and the elimination of the need for a
chiral intermediate.
Summary of the Invention
One aspect of the present invention provides a process for preparing
Compound 1:
cis
NI\ 02
0 40.0
H
0-,NR5R6
1
said process comprising the steps of:
(a) cyclizing Compound 2 in a first solvent at an elevated
temperature
0;_s).õ NO2
o NR5R6
2
wherein R5 and R6 are each independently selected from the group
consisting of H, alkyl, alkenyl, alkynyl, alkoxy, cycloalkyl, aryl, alkylaryl,
-3-
CA 02875768 2014-12-19
arylalkyl, heterocyclic and heteroaryl groups, or R5 and R6, together with
the nitrogen to which they are attached, form a 3- to 6-membered
heterocyclic compound containing 1-4 heteroatoms, to produce a first
mixture of exo isomers
trans
0
0 leip cc-43
NO2
H H
0NR5R6
said isomers having a trans-[5,6]-ring-junction and endo isomers:
cis
0 H
0 es NO2
H = H
0NR5R6
(b) epimerizing said trans-[5,6]-ring-junction in Compound 29 by
treating said first mixture with a first base to produce a second mixture
comprising cis-[5,6]-ring-junction-nitro-a isomer and cis-[5,6]-ring-
junction-nitro-13 isomer of Compound 30:
cis
0 cc+p
NO2
0 00
H H
0 NR5FR6 and
(c) treating said second mixture with a second solvent, causing said
a-isomer of Compound 30 to precipitate to produce Compound 1.
In another embodiment, the above process further comprises the
step of treating said second mixture with a second base, resulting in a
- 4 -
CA 02875768 2014-12-19
dynamic resolution of said second mixture, in which said 13-isomer of
Compound 30 is converted to a-isomer of Compound 30, and a -isomer of
Compound 30 is precipitated to produce Compound 1.
In another embodiment, said first solvent is selected from the group
consisting of xylene, N-methylpyrrolidinone, Dimethylsulfoxide, diphenyl
ether, Dimethylacetamide, and mixtures of 2 or more thereof.
In another embodiment, said temperature is between about 70 and
about 190 0C, preferably between about 80 and about 170 0C, more
preferably between about 100 and about 160 0C, still more preferably
between about 120 and about 150 0C.
In another embodiment, said first base is selected from the group
consisting of triethylamine, 1,5-diazabicyclo[4,3,0]non-5-ene 1,4-
diazabicyclo[2,2,2]octane, and 1,8-diazabicyclo[5,4,0]undec-7-ene, and
mixtures of 2 or more thereof.
In another embodiment, said second solvent is selected from the
group consisting of alcohols, ethers, ketones, esters, xylene, N-
methylpyrrolidinone, and mixtures of 2 or more thereof.
In another embodiment, the invention provides a process for preparing
Compound 2:
- 5 -
CA 02875768 2014-12-19
0
)0 ¨ *
-----) NO2
0 NR5R6
2
wherein R5 and R6 are each independently selected from the group
consisting of H, alkyl, alkenyl, alkynyl, alkoxy, cycloalkyl, aryl, alkylaryl,
arylalkyl, heterocyclic and heteroaryl groups, or R5 and R6, together with
the nitrogen to which they are attached, form a 3- to 6-membered
heterocyclic compound containing 1-4 heteroatoms, said process
comprising:
(a) converting (R)-butynol to Compound 3:
OH
)
CONR5R6
3;
(b) reducing Compound 3 to yield Compound 4:
HO .<---o
NR5Rs
4; and,
(c) reacting Compound 4 with Compound 6:
o
HO¨ . NO2
to yield Compound 2.
In yet another embodiment, the invention is directed to a process for
preparing Compound 2:
- 6 -
CA 02875768 2014-12-19
0
0 - si
)- NO2
0 NR5R6
2,
wherein R5 and R6 are each independently selected from the group
consisting of H, alkyl, alkenyl, alkynyl, alkoxy, cycloalkyl, aryl, alkylaryl,
arylalkyl, heterocyclic and heteroaryl groups, or R5 and R6, together with
the nitrogen to which they are attached, form a 3- to 6-membered
heterocyclic compound containing 1-4 heteroatoms, said process
comprising:
(a) converting (R)-butynol to Compound 3:
OH
)'\
CON R5R6
3;
(b) reacting Compound 3 with Compound 6 to yield Compound 7:
0
HO- O NO2
6;
(c) reducing Compound 7 to produce Compound 2:
o
0 ¨ ,NO2
)---1
0 NR5R6
7.
- 7 -
CA 02875768 2014-12-19
In another embodiment, the invention provides the following
compounds:
o 0
OH OS,Me3
CONPh 0 ¨ io NO2
o ¨ dighh NO2
)----__IW
CONPh2
3a 2 )--)\
, NPh2
, 7a 0 NPh2 '
2
0 H 0 H 0 H 0 H
p NO2
O isie,.NO2
O 5.5 NO2
0 oio,,NO2 o
0
H H H H H H H H
0NPh2 ,
0 NPh2 ' 0 NP÷õ
2 0 NPh2 ,
1a
0 0
O H 0 H H H H H
NO2 0 imp NO2 0 5512
0 O5,NHCO2Et
0
H H H H H H H
-,1-1 0,NPh2 ,
0 NP, õ .2 , 0 NPh2 . 0 NPh2 ,
12a 13a
0
H NH2 0 H . 00 H
0 e. 0 2Os NH2
_
H H H H H H
0/2-, NP h2,
0 NP, , .2 , 0 NPh2
NO2 NO2 0
H H H H 40 H NO2 HOOC lo NO2
O 0 , HO SO3Na HO SO3Na
8 9 ,
10 and 6 .
0
OH
io Tx
0
r
./'
N * NO2 X
4, ven HO \
N 41,
23 25 24
X=S or 0 or, NH
O 0 H
So NO2,,
0 .--- 0 NO2
0
H --A
N z X N7 X
22
---- 21
=
- 8 -
CA 02875768 2014-12-19
0
Hipip H H
\ N,
0 Oil O CO2Et
H H H H
N z X Nr X
26 27
P(0)(0E02
1,1
`= 16 HO 0
COOR7 R70
40 28 0 OR7
F 19
0
0 H a OHH NO2
NO2
0 Os ,,NO2 0 4== '1\11-12
and
H H
i=-=0 R7 H = H
oOH
0R7
18 17
5 A further understanding of the invention will be had from the
following detailed description of the invention.
Description of the Invention
10 The following definitions and terms are used herein or are otherwise
known to a skilled artisan. Except where stated otherwise, the definitions
apply throughout the specification and claims. Chemical names, common
names and chemical structures may be used interchangeably to describe
the same structure. These definitions apply regardless of whether a term is
15 used by itself or in combination with other terms, unless otherwise
indicated. Hence, the definition of "alkyl" applies to "alkyl" as well as the
"alkyl" portions of "hydroxyalkyl," "haloalkyl," "alkoxy," etc.
- 9 -
CA 02875768 2014-12-19
Unless otherwise known, stated or shown to be to the contrary, the
point of attachment for a multiple term substituent (two or more terms that
are combined to identify a single moiety) to a subject structure is through
the last named term of the multiple term substituent. For example, a
cycloalkylalkyl substituent attaches to a targeted structure through the
latter "alkyl" portion of the substituent (e.g., structure-alkyl-cycloalkyl).
The identity of each variable appearing more than once in a formula
may be independently selected from the definition for that variable, unless
otherwise indicated.
Unless stated, shown or otherwise known to be the contrary, all
atoms illustrated in chemical formulas for covalent compounds possess
normal valencies. Thus, hydrogen atoms, double bonds, triple bonds and
ring structures need not be expressly depicted in a general chemical
formula.
Double bonds, where appropriate, may be represented by the
presence of parentheses around an atom in a chemical formula. For
example, a carbonyl functionality, -CO-, may also be represented in a
chemical formula by -0(0)-, or -C(=0)-. One skilled in the art will be able
to determine the presence or absence of double (and triple bonds) in a
covalently-bonded molecule. For instance, it is readily recognized that a
- 10-
CA 02875768 2014-12-19
carboxyl functionality may be represented by -COOH, -C(0)0H, -C(0)OH
or -CO2H.
The term "heteroatom," as used herein, means a nitrogen, sulfur or
oxygen atom. Multiple heteroatoms in the same group may be the same or
different.
As used herein, the term "alkyl" means an aliphatic hydrocarbon
group that can be straight or branched and comprises 1 to about 24
carbon atoms in the chain. Preferred alkyl groups comprise 1 to about 15
carbon atoms in the chain. More preferred alkyl groups comprise 1 to
about 6 carbon atoms in the chain. "Lower alkyl" means alkyl groups of 1
to 6 carbon atoms in the chain. "Branched" means that one or more lower
alkyl groups such as methyl, ethyl or propyl, are attached to a linear alkyl
chain. The alkyl can be substituted by one or more substituents
independently selected from the group consisting of halo, aryl, cycloalkyl,
cyano, hydroxy, alkoxy, alkylthio, amino, -NH(alkyl), -NH(cycloalkyl), -
N(alkyl)2 (which alkyls can be the same or different), carboxy and -C(0)0-
alkyl. Non-limiting examples of suitable alkyl groups include methyl, ethyl,
n-propyl, isopropyl, n-butyl, t-butyl, n-pentyl, heptyl, nonyl, decyl,
fluoromethyl, trifluoromethyl and cyclopropylmethyl.
"Alkenyl" means an aliphatic hydrocarbon group (straight or
branched carbon chain) comprising one or more double bonds in the chain
and which can be conjugated or unconjugated. Useful alkenyl groups can
- 11 -
CA 02875768 2014-12-19
comprise 2 to about 15 carbon atoms in the chain, preferably 2 to about 12
carbon atoms in the chain, and more preferably 2 to about 6 carbon atoms
in the chain. The alkenyl group can be substituted by one or more
substituents independently selected from the group consisting of halo,
alkyl, aryl, cycloalkyl, cyano and alkoxy. Non-limiting examples of suitable
alkenyl groups include ethenyl, propenyl, n-butenyl, 3-methylbut-enyl and
n-pentenyl.
Where an alkyl or alkenyl chain joins two other variables and is
therefore bivalent, the terms alkylene and alkenylene, respectively, are
used.
"Alkoxy" means an alkyl-0- group in which the alkyl group is as
previously described. Useful alkoxy groups can comprise 1 to about 12
carbon atoms, preferably 1 to about 6 carbon atoms. Non-limiting
examples of suitable alkoxy groups include methoxy, ethoxy and
isopropoxy. The alkyl group of the alkoxy is linked to an adjacent moiety
through the ether oxygen.
The term "cycloalkyl" as used herein, means an unsubstituted or
substituted, saturated, stable, non-aromatic, chemically-feasible
carbocyclic ring having preferably from three to fifteen carbon atoms, more
preferably, from three to eight carbon atoms. The cycloalkyl carbon ring
radical is saturated and may be fused, for example, benzofused, with one
- 12 -
CA 02875768 2014-12-19
to two cycloalkyl, aromatic, heterocyclic or heteroaromatic rings. The
cycloalkyl may be attached at any endocyclic carbon atom that results in a
stable structure. Preferred carbocyclic rings have from five to six carbons.
Examples of cycloalkyl radicals include cyclopropyl, cyclobutyl, cyclopentyl,
cyclohexyl, cycloheptyl, or the like.
"Alkynyl" means an aliphatic hydrocarbon group comprising at least
one carbon-carbon triple bond and which may be straight or branched and
comprising about 2 to about 15 carbon atoms in the chain. Preferred
alkynyl groups have about 2 to about 10 carbon atoms in the chain; and
more preferably about 2 to about 6 carbon atoms in the chain. Branched
means that one or more lower alkyl groups such as methyl, ethyl or propyl,
are attached to a linear alkynyl chain. Non-limiting examples of suitable
alkynyl groups include ethynyl, propynyl, 2-butynyl, 3-methylbutynyl, n-
pentynyl, and decynyl. The alkynyl group may be substituted by one or
more substituents which may be the same or different, each substituent
being independently selected from the group consisting of alkyl, aryl and
cycloalkyl.
The term "aryl," as used herein, means a substituted or
unsubstituted, aromatic, mono- or bicyclic, chemically-feasible carbocyclic
ring system having from one to two aromatic rings. The aryl moiety will
generally have from 6 to 14 carbon atoms with all available substitutable
carbon atoms of the aryl moiety being intended as possible points of
- 13 -
CA 02875768 2014-12-19
attachment. Representative examples include phenyl, tolyl, xylyl, cumenyl,
naphthyl, tetrahydronaphthyl, indanyl, indenyl, or the like. If desired, the
carbocyclic moiety can be substituted with from one to five, preferably, one
to three, moieties, such as mono- through pentahalo, alkyl, trifluoromethyl,
phenyl, hydroxy, alkoxy, phenoxy, amino, monoalkylamino, dialkylamino,
or the like.
"Heteroaryl" means a monocyclic or multicyclic aromatic ring system
of about 5 to about 14 ring atoms, preferably about 5 to about 10 ring
atoms, in which one or more of the atoms in the ring system is/are atoms
other than carbon, for example nitrogen, oxygen or sulfur. Mono- and
polycyclic (e.g., bicyclic) heteroaryl groups can be unsubstituted or
substituted with a plurality of substituents, preferably, one to five
substituents, more preferably, one, two or three substituents (e.g., mono-
through pentahalo, alkyl, trifluoromethyl, phenyl, hydroxy, alkoxy,
phenoxy, amino, monoalkylamino, dialkylamino, or the like). Typically, a
heteroaryl group represents a chemically-feasible cyclic group of five or six
atoms, or a chemically-feasible bicyclic group of nine or ten atoms, at least
one of which is carbon, and having at least one oxygen, sulfur or nitrogen
atom interrupting a carbocyclic ring having a sufficient number of pi (TO
electrons to provide aromatic character. Representative heteroaryl
(heteroaromatic) groups are pyridinyl, pyrimidinyl, pyrazinyl, pyridazinyl,
furanyl, benzofuranyl, thienyl, benzothienyl, thiazolyl, thiadiazolyl,
- 14-
CA 02875768 2014-12-19
imidazolyl, pyrazolyl, triazolyl, isothiazolyl, benzothiazolyl, benzoxazolyl,
oxazolyl, pyrrolyl, isoxazolyl, 1,3,5-triazinyl and indolyl groups.
The term "heterocyclic ring" or "heterocycle," as used herein, means
an unsubstituted or substituted, saturated, unsaturated or aromatic,
chemically-feasible ring, comprised of carbon atoms and one or more
heteroatoms in the ring. Heterocyclic rings may be monocyclic or
polycyclic. Monocyclic rings preferably contain from three to eight atoms
in the ring structure, more preferably, five to seven atoms. Polycyclic ring
systems consisting of two rings preferably contain from six to sixteen
atoms, most preferably, ten to twelve atoms. Polycyclic ring systems
consisting of three rings contain preferably from thirteen to seventeen
atoms, more preferably, fourteen or fifteen atoms. Each heterocyclic ring
has at least one heteroatom. Unless otherwise stated, the heteroatoms
may each be independently selected from the group consisting of nitrogen,
sulfur and oxygen atoms.
The terms "Hal," "halo," "halogen" and "halide," as used herein, mean
a chloro, bromo, fluoro or iodo atom radical. Chlorides, bromides and
fluorides are preferred halides.
The term "carbonate", as used herein, is understood to include
bicarbonates.
- 15 -
CA 02875768 2014-12-19
The term "isomer", as used herein, is understood to mean one of two
or more molecules having the same number and kind of atoms and hence
the same molecular weight, but differing in respect to the arrangement or
configuration of the atoms.
The term "epimerizing", as used herein, is understood to mean
converting from one isomer to another, wherein it is the relative position of
an attached H that differs between the two isomers.
The term "precipitate", as used herein, is understood to mean to fall
out of solution as a solid.
The term "dynamic resolution", as used herein, is understood to
mean a process in which a conversion from a first isomer to a second
isomer of the same compound in a solution is thermodynamically driven by
the depletion of the second isomer from the solution by precipitation of the
second isomer.
The following abbreviations are defined: Et0H is ethanol; Me is
methyl; Et is ethyl; Bu is butyl; n-Bu is normal-butyl, t-Bu is tert-butyl,
OAc is acetate; KOt-Bu is potassium tert-butoxide; NBS is N-bromo
succinimide; NMP is 1-methyl-2-pyrrolidinone; DMAP is 4-
dimethylaminopyridine; THF is tetrahydrofuran; DBU is 1,8-
diazabicyclo[5,4,0]undec-7-ene; DMA is N,N-dimethylacetamide; n- Bu4NBr
- 16-
CA 02875768 2014-12-19
is tetrabutylammonium bromide; n-Bu4NOH is tetrabutylammonium
hydroxide, n-Bu4NH2SO4 is tetrabutylammonium hydrogen sulfate, and
"equiv." or "eq." means equivalents.
"n", as it is used herein, is understood to be an integer having a
value that is inclusive of the range recited thereafter. Thus "n is between 0
and 4" and "n ranges 0-4" both mean that n may have any of the values 0,
1, 2, 3 or 4.
- 17-
CA 02875768 2014-12-19
General Syntheses
The following scheme summarizes the dynamic resolution-based
approach to synthesizing Compound 11 from (R)-butynol:
0
HO - ,NO2 0
OH OH 6 Nitro-acid 0 ¨ Ai NO2
CONR5R6 t-BuCOCI NR5R6
(R)-Butynol 3 0
7
0 HO -(---\r o 4
HO - io NO2 NR5R6
6
...,
0 1. Solv/150C 0 H
2. DBU orNO2
Lindlar/H2 0 ____ ONO2 3. Dynamic crystal. , 0 lel
H - H
0NR51R6
NR5R6
2 1
0H H EtCO2C1 0 H H
Pd/C/HCOOH so __ eel
NH
2 NHCO2Et 1. Dilute NaOH
- 0 ' 2. HCI
0
H - H H ' H-
o--NR5R6 0NR5R6
13 P(0)(0E02
12
--- N
16
0 H H40 0 H H
0
,NHCO2Et OHCOEt 2
S.
e F
0
H ' I:I H - H
ol-1
00H
14 15
H H
,NHCO2Et
0 O.
H H
-
N 11
I
Si F
- 18-
CA 02875768 2014-12-19
(R)-butynol may be converted to amide 3 by either of Methods A or B:
Preparation of Amide-Method A:
0
.R OH
OH - OHX N, 5
R6
= - eC)
Li -
(R) Butyn-2-ol NR5R6
3
OP OP
xAN,R5
Li
3A 3B
P = protecting group such as THP, SiR1R2R3
Preparation of Amide-Method B:
0
R5
XAN:
OH OH
R6
PdLn/CuY/base
NR5Rs
OP
X Nµ 1.<5 PdLn/CuY/base
R6
In each of methods A and B, P is a protecting group, for example THP
or SiR1R2R3, wherein R'-R3 may be H, alkyl, alkenyl, cycloalkyl, aryl,
arylalkyl, alkylaryl, heterocyclic, and heteroaryl groups, Y is selected from
the group consisting of Cl, Br, I, and R"COO, wherein R" is selected from
the group consisting of alkyl, aryl, alkylaryl, and arylalkyl and X is a
leaving group. By way of example, X can be halogen, for example Cl, Br, or
I. As another example, X can be selected from heterocyclic rings, such as
imidazoles. L is a ligand and is selected from PR' wherein R' is selected
- 19-
CA 02875768 2014-12-19
from alkyl, aryl, alkylaryl, and NR" groups, and R" is selected from alkyl,
aryl, and alkylaryl groups. n can range from 0-8, and preferably ranges
from 0 to 4.
The butyn-2-ol is disclosed in, e.g., U.S. 6,063,847, and Methods A
and B may be performed on either the racemic or enantiopure butynol.
The butynol may be combined with a mineral acid, for example sulfuric
acid, in an organic solvent such as THF and a compound such as
hexyldimethylsilazane to provide a protecting group on the alcohol. The
protected compound may then be combined with a suitable base. A
preferred nucleophilic base is hexyllithium. The resulting metallated
compound may then be amidated by combining it with a solution
containing, e.g., diphenylcarbamylimidazole, and deprotected, to yield the
diphenylamide (Compound 3 wherein R5 and R6 are both phenyl).
The amide may then be converted to Compound 2 via either of two
routes: through vinyl alcohol 4, or through amide 7. For example, amide 3
may be combined with nitro acid 6. In one embodiment, amide 3 reacts
with a mixed anhydride of the nitro acid 6 (prepared from 6 and pivaloyl
chloride in the presence of a tert-amine base), in the presence of DMAP to
form Compound 7. The amide is subsequently subjected to hydrogenation
conditions to yield Compound 2. Preferred hydrogenation conditions
include pressurized hydrogen in the presence of a hydrogenation catalyst.
The hydrogen pressure may range from 20 to 500 psi, and a pressure of
- 20 -
CA 02875768 2014-12-19
100 psi is preferred. The hydrogenation catalyst may be a noble metal
catalyst, for example Lindlar catalyst. The hydrogenation is suitably
conducted in the presence of a solvent, preferably an aromatic solvent such
as toluene.
The yields in the above-described syntheses of Compound 3 can be
improved by suppressing side- or over- reactions that can occur as the
product (Compound 3) comes in contact with its precursors. These side- or
over- reactions can be suppressed by decreasing the residence time of the
final process step (the step resulting in Compound 3). This reduction of
residence time can be achieved by using a suitable flow operation rather
than a batch operation at this step. The reactants are introduced in
individual reactant streams which are combined and immediately mixed in
a flow-through step. This can be achieved by combining the individual flow
streams at a point near the inlet of a pump, and pumping the combined
reactant stream through a static mixer, followed by immediate quench.
Alternatively, amide 3 may be reduced to the corresponding vinyl
alcohol 4, and the alcohol is then reacted with nitro acid 6 to yield
Compound 2.
Compound 2 is subsequently cyclized to yield Compound 1. The
cyclization of 2 is conducted in a suitable solvent (e.g., hydrocarbons such
as xylene, N-methylpyrrolidinone, Dimethylsulfmdde, diphenyl ether,
-21 -
CA 02875768 2014-12-19
Dimethylacetamide and the like as well mixtures of 2 or more thereof), at
elevated temperature (e.g., between about 70 and about 190 0C, preferably
between about 80 and about 1700C, more preferably between about 100
and about 1600C, still more preferably between about 120 and about 150
oC), to produce a mixture of exo- and endo- isomers. This mixture is
treated with a suitable base to complete the epimerization at the trans
[5,61-ring-junction (29) to the cis-isomer (30). The resulting mixture
comprises the a- and [3-isomers of each Compound 29 and 30, for a total of
four isomers. The a-isomer of Compound 30 is a desirable intermediate in
the synthesis of himbacine analogs, and is herein designated Compound 1.
The resulting mixture is dynamically resolved by treatment with a
suitable base and preferential crystallization of the desired a-isomer using
a suitable solvent. The equilibrium concentrations of the a- and [3-isomers
in solution are a function of the pH of the solution, which can be modified
by addition a suitable base. Thus, the [3-isomers can be converted to the
desired a-isomers by addition of a suitable base. Simultaneously, in the
presence of a suitable solvent, the a-isomer precipitates from the solution
as a solid. In the dynamic resolution process, this precipitation tends to
deplete the a-isomer from the solution, driving the equilibrium of the 13 to a
conversion process from the 13-isomer towards the a-isomer in the solution.
Suitable bases for the steps include, for example, triethylamine, 1,5-
diazabicyclo[4,3,0]non-5-ene 1,4-diazabicyclo[2,2,2]octane, and 1,8-
- 22 -
CA 02875768 2014-12-19
diazabicyclo[5,4,0]undec-7-ene, or mixtures of 2 or more thereof. Suitable
solvent for crystallization includes hydrocarbon, alcohols, ethers, ketones,
esters, xylene, N-methylpyrrolidinone. In some embodiments, the solvent
is selected from ethanol, isopropyl alcohol, aryl alcohol alcohols, ethers,
ketones, esters, xylene, N-methylpyrrolidinone, and the mixtures of 2 or
more thereof. Advantageously, the exo-endo ratio for Compound 1 may
exceed 90:10, and may also exceed 95:5. The a:13 ratio at the nitro position
may exceed 95:5, and for example may be 98.1:1.5.
o
NO2
io NO2 Solvent . 0 Base
-
H = H
0NP5R6
(:) NR5R6
2 29
0 u+p 0a
NO2 Base and
oNO2
Precipitation
0 OS ______________________________ 0 5.
H H H
0NR5Ft6 0/2.--NR5R6
cc:13 = about 70:30 a isomer precipitates
preferentially
30 a:3 >98:2 in solid
1
The carbon-carbon double bond and the nitro group of Compound 1
may then be reduced under suitable reduction conditions to yield amine
12. Suitable reduction conditions may include contact with a
hydrogenation catalyst, such as one selected from standard noble metal
catalysts (e.g., palladium on carbon, platinum on carbon, and rhodium on
carbon, or a mixture thereof). The source of hydrogen can be hydrogen
gas, formic acid, formates, and combinations thereof. Multiple catalysts
-23-
CA 02875768 2014-12-19
may also be used. Amine 12 may then be converted to carbamate 13 by
reaction with an alkyl haloformate (e.g., ethylchloroformate,
ethylbromoformate, or ethyliodoformate). Carbamate 13 may then be
converted to the carbamate acid 14 by reaction with a base such as, for
example, a metal oxide or hydroxide, carbonate and bicarbonate, where the
metal is selected from the group consisting of lithium, sodium, potassium,
and magnesium, followed by reaction with a mineral acid. Sodium
hydroxide is a preferred base. The acid 14 is subsequently converted to
the corresponding aldehyde 15, which is reacted with phosphorus ester 16
to yield Compound 1.
Compound 6 may be prepared from acrolein and nitromethane
following the scheme below. Nitromethane is treated with an inorganic
base such as metal hydroxide (e.g., Li0H, KOH, NaOH, Ca(OH)2), metal
carbonate (e.g., Li2CO3, Na2CO3, K2CO3, Cs2CO3) and acrolein in a C 1 to Cs
alcohol (e.g., methanol, ethanol, propanol, isopropanol, butanol, sec-
butanol, t-butanol, pentanols, and octanols) or a mixture of alcohols to give
crude Compound 8. To purify Compound 8, crude Compound 8 is isolated
as its metal bisulfite salt 9 by treating with a metal bisulfite reagent
selected from, NaHS03, KHS03, Na2S205, and K2S205. The bisulfite
compound 9 is converted to the purified 8 by treating with a lower alkyl
carbonyl compound (e.g., acetaldehyde, acetone, glyoxylic acid, or a salt of
glyoxylate), and a carbonate base (e.g., LiHCO3, NaHCO3, KHCO3, Na2CO3,
- 24 -
CA 02875768 2014-12-19
K2CO3) in a biphasic system containing water and a water-immiscible
solvent.
The compound 8 is cyclized by treating with a secondary amine (e.g.,
piperidine, pyrrolidine, piperazine, dialkylamines, and diarylalkylamines)
and a carboxylic acid (e.g., aliphatic and aromatic carboxylic acids) in an
organic solvent (e.g., CH2C12, chlorobenzene, t-butylmethylether, or
toluene) to produce Compound 10.
There are two methods to convert Compound 10 to Compound 6,
designated herein as Method C and Method D. In Method C, Compound
10 is first converted to Compound 6A by reacting 10 with a Wittig reagent.
The R8 in the Wittig reagent given in the scheme below is selected from Ci
to C 10 alkyl or arylalkyl groups. The Compound 6A is then converted to
Compound 6 via an inorganic base- or acid- catalyzed hydrolysis. The
applicable inorganic bases include, but are not limited to, alkaline
hydroxide, carbonate, and phosphate bases. The applicable acids include,
but are not limited to mineral and organic acids.
In method D, Compound 10 is converted directly to Compound 6 by
reacting 10 with malonic acid in a suitable solvent or solvent mixture (e.g.,
hydrocarbon solvent including halogenated solvent, aromatic solvent and
nitrogen-containing solvents). In some embodiments, the solvent is either
pyridine or toluene, or a mixture thereof. Optionally, a catalyst (e.g.,
-25-
CA 02875768 2014-12-19
piperidine, pyrrolidine, piperazine, pyridine, and triethylamine) can be
introduced to accelerate the reaction.
NO NO2
, H H
+ CH3NO _______________ I.-
0 0 0 HO SO3Na HO SO3Na
Acrolein 8 9
CO2R8
0 Ph3P=--/
NO2
NO2(Wittig reagent)
H ____________________________________
H io ___________________________________________________________ )._
Method C
0 0
8
MaIonic acid
0 J. Method D
0
Me0
io NO2 ________
3. io HO NO2
6A
6
Compound 16 can be prepared following the scheme below starting
5 from 5-bromo-2-methylpyridine N-oxide. The 5-bromo-2-methylpyridine N-
oxide is first converted to Compound 35 by treating with an anhydride
(e.g., aromatic acid anhydride, acetic anhydride, or trihalogenated acetic
anhydride) in an applicable solvent (e.g., esters, Ci to Cm hydrocarbon
solvent, or aromatic solvents, or a mixture thereof). Compound 35 is
10 converted to Compound 36 by treatment with an alcohol (e.g., methanol,
ethanol, propanol, isopropanol, butanols, and pentanols) at an elevated
temperature of about 20 to about 80 C, preferably, about 30 to about 70
0C, more preferably about 45 to about 55 C.
The synthesis of Compound 37 (with X being Cl) is disclosed in van
den Heuvel, Marco et al, J.Org. Chem., 69, 250-262 (2004). According to
the present invention, Compound 36 is converted to Compound 37
according to the scheme below, by reacting with a leaving group reagent
- 26 -
CA 02875768 2014-12-19
including a halogenating reagent (e.g., SOC12, SOBr2, PC13, PBr3, PC15, or
PBr5) or another proper leaving group reagent. In the scheme below, X is a
leaving group selected from Halogens, esters, sulfonates, and phosphates.
Compound 37 is converted to Compound 38 by treating with a
phosphite reagent. The phosphite reagent can be prepared from a
dialkylphosphite or a diarylphosphite (e.g., (R90)2P(0)H, wherein R9 is
selected from Ci-Cio alkyl, aryl, heteroaryl, and arylalkyl groups) and a
strong base (e.g., metal hydrides, RioLi, and ((Rio)3Si)2Li, wherein Rio is
selected from Ci to Cio alkyl and aryl groups).
Compound 38 is converted to Compound 16 by reacting with a
fluoroaromatic borate reagent, 3-FC6H4B(OR11)2, wherein R11 is selected
from a group consisting of Ci to Cio alkyls, aryls, heteroaryls and
hydrogen. The reaction is catalyzed using a palladium catalyst, PdLn,
wherein L is a ligand selected from PR'3 wherein R' is selected from alkyl,
aryl, alkylaryl, and NR"3 wherein R" is selected from alkyl, aryl, and
alkylaryl. Alternately, palladium on carbon ("Pd/C") may be used as the
catalyst. The preferred ligands are PPh3, P(o-To1)3, and bipyridine.
- 27 -
CA 02875768 2014-12-19
0000F3
- OH
1 N 1 1\1 ,,Ce e
Y ________________ 0
NH 00CCF3
y
Br Br
_ _
35 Br 36
_ _ P(0)(0E02
_ B(OPii)2
r P(0) (OR9)2 -
X
I 1\1
----- rjµi --0.-- y
_ Br _ PdLn
S
Br
F
37 38 16 .
The following illustrates a general scheme for the synthesis of
Compound 11 via the nitro-oxazole route:
0
HO ,- ,NO2
OH 6 0
OH
NO2
-).
t-BuC0C1
\X
N 410,
(R)-Butynol
23 N 41
X = S or 0 or, NH \ 24
X
23 HO" `
0
NO2
HO 5
6
22
- 28 -
CA 02875768 2014-12-19
0
lo
Lindlar/H2 NO2
24
N)NX
=22
0
\\NO2
1. Solv./150C
2. DBU
3. Dynamic crystal
22 H H
Nz X
=21
0 H H
os,\NH2
Pd/C/H 2 0 S.
21 H
Nz X
=26
H H
O.\\N
EtCO2C1 0 CO2Et
26 H
Nz X
=27
- 29 -
CA 02875768 2014-12-19
H H H H
1. Dilute NaOH NHCO2Et HCI esNHCO2Et
.
27 2 0 _________________ . 0
H ' 11) H H-
00H
0 H
14 15
H H
P(0)(0E02 OO ,NHCO2Et
0
...- N
-- 16 H i H
lel F
N 11
15 .
I
0 F .
- 30 -
CA 02875768 2014-12-19
0
HO - . NO2 0
OH OH 6
___________ . .
t-BuCOCI 0 ¨ IW dik NO2
)-----__
I X
(R)-Butynol
N
23 . I
N .
X=SorOor, NH \ 24
HO- 0 25
0 HO- 2---X
N' X
6 N,
0 1. Solv/150C 0 H
Lindlar/H2 0 _ NO 2. DBU _ so ,NO2
110 ` 3. Dynamic crystal.
H>.- 1=1
C(.)1=Xija
0 1 X
WV
21N22 N 411,
Pd/C/H2 0 H H. EtCO2C1 0 H H
0
,NHCO2Et 1. Dilute NaOH
Oe.,H2
___________________________ - 0 es 2. HCI ,
H - H H i H
OX OX
N it N 4s, P(0)(0E02
26 27 -- N
`- 16
H H H H
0
40HCO2 . Et so.,NHCO2Et 40
F
0 ,
H - H- H - H
(tOH
0 H
14 15
H H
0 eel ,NHCO2Et
H i H
-,.
N 11
1
---..
40 F
(R)-butynol may advantageously be protected with, for example, THP.
The THP-protected alcohol may then be permitted to react with a
substituted benzothiazole, for example 2-chlorobenzothiazole, to yield
-31 -
CA 02875768 2014-12-19
Compound 23 (where X is S). The reaction may be conducted in a solvent,
for example an organic solvent such as DMF, and in the presence of a base,
for example triethylamine. Compound 23 may then be converted to
Compound 22 by either of two routes: through vinyl alcohol 25, or
through Compound 24. The latter route may be conducted by reacting
Compound 23 with nitro acid 6 in the presence of an aromatic solvent
such as, for example, toluene, to yield Compound 24. Compound 24 is
subsequently reduced under hydrogenation conditions, for example in the
presence of hydrogen and a Lindlar catalyst, to yield Compound 22.
Compound 22 is then cyclized via a Diels-Alder reaction, followed by
treatment with a base to yield Compound 21. The cyclization of 22 is
conducted in a suitable solvent (e.g., hydrocarbons such as xylene, N-
methylpyrrolidinone, Dimethylsulfoxide, diphenyl ether,
Dimethylacetamide and the like as well mixtures of 2 or more thereof), at
elevated temperature (e.g., from about 70 to about 190 C, preferably from
about 80 to about 170 C, more preferably from about 100 to about 160 C,
still more preferably from about 120 to about 150 C), to produce a mixture
of exo- and endo- isomers. This mixture is treated with a suitable base to
complete the epimerization to produce the cis-isomer (21). Suitable bases
include, for example, triethylamine, 1,5-diazabicyclo[4,3,0]non-5-ene, 1,4-
diazabicyclo[2,2,21octane, and 1,8-diazabicyclo[5,4,0]undec-7-ene.
The following illustrates a general scheme for the synthesis of
Compound 11 via the nitro-ester route:
- 32 -
CA 02875768 2014-12-19
0
HO¨ NO2 0
6 'IP
OH OH
)----\---.
0 ¨ NO2
COOR2 t-BuCOCI OR2
(R)-Butynol 28 0
19
0 1. SoIv/150C 0 H a
2. DBU
Lindlar/H2 0 _ O NO2 3, Dynamic crystal 0 es -No2
_
H - H
00 R2
00 R7
18 17
0 HH H EtCO2C1 H
Pd/C/H 2 a
_____________ ' 0 Ole ''NI-12 _____ ' 0 41140'NHCO2Et .
H - 111 H - Ill
---.
0 OH 0 OH
20 P(0)(0E02 14
H
16
,NHCO2Et
0 H H 0 Oe '
=
0 4010.''NHCO2Et F H H
H - Ill
oi')H N 11
I
SF
(R)-butynol is converted to a benzyl ester 28 (where R7 is benzyl).
Compound 28 reacts with the mixed anhydride of the nitro acid 6
(prepared from 6 and pivaloyl chloride in the presence of a tert-amine
5 base), in the presence of DMAP, to form Compound 19. Compound 19 is
reduced under hydrogenation conditions, e.g., in the presence of hydrogen
and a Lindlar catalyst, to yield ester 18. Ester 18 is subsequently cyclized
to yield Compound 17 as follows. The cyclization of 18 is conducted in a
suitable solvent (e.g., hydrocarbons such as xylene, N-methylpyrrolidinone,
- 33 -
CA 02875768 2014-12-19
Dimethylsulfoxide, diphenyl ether, Dimethylacetamide and the like as well
mixtures of 2 or more thereof), at elevated temperature (e.g., from about 70
to about 230 C, preferably, from about 80 to about 170 C, more preferably,
from about 130 to about 160 C, still more preferably, from about 140 to
about 150 C), to produce a mixture of exo- and endo- isomers. This
mixture is treated with a suitable base to complete the epimerization to the
cis-isomer 17 as described previously. The carbon-carbon double bond
and the nitro group of Compound 17 may then be reduced under suitable
reduction conditions to yield amine 20. Suitable reduction conditions may
include contact with a hydrogenation catalyst, such as one selected from
standard noble metal catalysts. Multiple catalysts may also be used. A
preferred reduction catalyst is palladium on carbon. The source of
hydrogen can be hydrogen gas, formic acid, formates, and combinations
thereof.
The experimental conditions disclosed herein are preferred
conditions, and one of ordinary skill in the art can modify them as
necessary to achieve the same products.
EXAMPLES
Example 1 - Preparation of 3-(5-Nitro-cyclohex-1-eny1)-acrylic acid
(Compound 6) and its Salt:
A. Preparation of Compound 9 from Acrolein
- 34 -
CA 02875768 2014-12-19
CH3NO2 NO2 NO2
1.4 KOH/Me0H-H H Na2S205 H
-20 C
0 0 0 or NaHS03
HO SO3Na HO SO3Na
Acrolein 8 9
To a solution of potassium hydroxide (3.1 g, 0.05 mol) in methanol
(450m1) were added nitromethane (39 ml, 0.69 mol) and isopropanol
(450m1) under nitrogen atmosphere. The resulting mixture was cooled to a
temperature between -200C and -250C. Acrolein (120 ml, 1.74 mol) was
then added slowly in about 3 to 3.5 hours while maintaining the
temperature between -200C and -250C. After stirring at the same
temperature for 1 hour, the reaction was quenched with acetic acid (4 m1).
The reaction mixture was warmed up to room temperature and a solution
of sodium metasulfite (135 g, 0.67 mol) in water (700 ml) was slowly added
at about 250C. After stirring the resulting suspension for 1 hour, the
mixture was cooled to 100C and stirred for another hour. White solid was
obtained after filtration and drying under vacuum. The product was carried
to the next step without further purification. Yield: 219g, 83%. 1H NMR
(400 MHz, DMSO-d6) 8 1.41-1.64 (m, 2H), 1.76-1.99 (m, 6H), 3.79-3.85 (m,
2H), 4.63 (m, 1H), 5.44 (t, J =6.2Hz, 2H).
B. Preparation of 4-Nitro-heptanedial
-35 -
CA 02875768 2014-12-19
0
NO2NO2
F.Ity0H
HH
/NaHCO3
0
HO SO3Na HO SO3Na 0 0
9 8
To a suspension of sodium 1,7-dihydroxy-4-nitro-heptane-1,7-
disulfonate, 9, (219 g, 0.57 mol) in methylene chloride (1.6L) was added a
solution of glyoxylic acid (160 g, 1.7 mol) and sodium bicarbonate (150 g,
1.78 mol) in water (2L). The resulting mixture was stirred at room
temperature for 30 to 60 minutes until all solids were dissolved. The
organic layer was split and the aqueous layer was extracted with methylene
chloride twice (2 x 400 ml). Combined extracts were then concentrated to
give a colorless oil. The product was carried to next step without further
purification. Yield: 85g, 86%. 1H NMR (400 MHz, CDC13) 6 2.09-2.24 (m,
4H), 2.58 (m, 4H), 4.61 (m, 1H), 9.77 (s, 2H). 13C NMR 6 26.2, 39.9, 86.9,
200Ø
C. Preparation of 5-Nitro-cyclohex-1-enecarbaldehyde:
NO2 0
Pyrrolidine
/Benzoic Acid NO2
0 8 0
20 To a solution of 4-Nitro-heptanedial (35.2 g, 0.2 mol) in methylene
chloride (0.7L) were added pyrrolidine (2 ml, 0.024 mol) and benzoic acid
(1.46 g, 0.012 mol), and the resulting mixture was refluxed for 10 to 15
- 36 -
CA 02875768 2014-12-19
hours. The reaction mixture was cooled to room temperature, washed with
1N HC1 (170 ml), saturated with NaHCO3 (170 ml) and water (170 ml) and
concentrated to give brownish oil with a purity of about 80%. The product
was carried to the next step without further purification. Yield: 32.2g,
75%. 1H NMR (400 MHz, CDC13) d 2.29-2.34 (m, 2H), 2.46-2.64 (m, 2H),
2.85-2.88 (m, 2H), 4.74 (m, 1H), 6.86 (m, 1H), 9.50 (s, 1H).
D. Preparation of 3-(5-Nitro-cyclohex-1-eny1)-acrylic acid
0
0 /-0H
NO2 0 HO2C lo NO
Pyridine
10 6
To a solution of 5-nitro-cyclohex-1-enecarbaldehyde (18g, 0.116 mol)
in pyridine (36 ml) was added malonic acid (41 g, 0.394 mol). The resulting
suspension was heated to 60 00 for about 7 hours. After cooling to a
temperature between 150C and 200C, 6N HC1 (72 ml) was slowly added into
the reaction mixture to adjust the pH to between 1.5 and 2 while
maintaining the temperature between 200C and 2500. The mixture was
then extracted with methylene chloride three times (1X180 ml, 2X90 ml).
The combined extracts were washed with 1N HC1 (48 ml), water (48 ml) and
concentrated to a volume of 36 ml. The concentrate suspension was cooled
to 00C and 50C for 1 hour. Light yellow solid was obtained after filtration
and drying under vacuum. Yield: 10g, 60%. Mp 158-160 C. 1HNMR (400
-37 -
CA 02875768 2014-12-19
MHz, DMSO-d6): 6 2.10 - 2.33 (m, 4H), 2.73 (m, 2H), 4.96 (m, 1H), 5.83 (d,
J = 20 Hz, 1H), 6.28 (s, 1H), 7.27 (d, J = 20 Hz, 1H), 12.3 (s, 1H).
Example 2 - Alternative Method for Preparing 3-(5-Nitro-cyclohex-1-eny1)-
acrylic acid (Compound 5) via Wittig Reagent:
0
0 CO2Me
H
I
NO2 Me0 _ lo NO2
(Wittig reagent)
___________________________________________ in
6A
To a solution of 10 (67 g, 432 mmol) in 1 L of methanol at 0 0C was
added 144.4 g (432 mmol) of the Wittig reagent. The resulting mixture was
agitated at 0 0C for 3 hrs. The solvent was removed under reduced
10 pressure. The residue was extracted with Me0Bu-t twice. The extract was
filtered to remove any solid, washed with brine, and concentrated. The
residue was chromatographed on a silica gel column, eluting with
hexane/ethyl acetate (10/1) to give 9.2 g cis and 55.1 g (60.4%) trans
product. 1H NMR (CDC13) d 7.31 (d, J = 11.3 Hz, 1H), 6.18 (m, 1H), 5.84 (d,
J = 15.9 Hz, 1H), 4.74-4.68 (m, 1H), 3.76 (s, 3H), 2.81-2.74 (m, 2H), 2.50-
2.04 (m, 4H).
0 0
Me0 ____ NO2 NaOH HO _
lop NO2
_____________________________________________ i
6
Next, to a flask were added 2.1 g of the methyl ester, 9.6 ml of Me0H
20 and 2.4 ml of water. To the mixture at about 5 0C was added dropwise
-38 -
CA 02875768 2014-12-19
0.96 ml of 50% NaOH. The mixture was allowed to warm to room
temperature and stirred at this temperature for about 24 hrs. The reaction
mixture was neutralized with HOAc to pH between 4 and 5 and the
methanol was removed under reduced pressure. The residue was extracted
with 3 X 50 ml Et0Ac. The Et0Ac layer was concentrated to give 1.5 g of
nitroacid 6 (76.5%).
Example 3 - Preparation of Compound 3a:
0 OH
OH 05 eq HMDS
0-Si\¨ Hex-Li Ph2NAWN
NPh2
Li
H 2S 04
0
(R)-3-Butyn-2-ol 3a
The following procedures can be operated on either the racemic or
the enantiopure starting butyn-2-ol. To a stirred solution of sulfuric acid
(conc., 40 L) in THF (240 mL) were sequentially added (R)-3 butyn-2-ol (40
g, 0.57 mol) and then hexylmethyldisilazane (49.6g, 0.31 mol) at room
temperature. The solution was refluxed for 3-4 hours and then slowly
cooled to -40 C. The resulting mixture was slowly charged in hexyllithium
(2.5M in hexane, 249 mL, 0.62 mol) while maintaining the temperature at -
40 0C. This solution and a solution of diphenylcarbamylimidazole (180 g,
0,68 mol) in a mixed solvent of THF (1088 mL) and toluene (435 mL) were
mixed using pumps through a chilled static mixer and directly quenched
into 5N sulfuric acid (560 mL, -50C). The quenched solution was warmed
to 250C and stirred for 1 hour. The organic layer was separated, washed
with 5N sulfuric acid (80 mL) and then twice with 10% brine (200 mL each
- 39 -
CA 02875768 2014-12-19
time). The pH of the final brine wash was adjusted to 5-7 with a 5%
NaHCO3 solution. The organic layer was then distilled and replaced with
toluene (440 mL). The toluene solution was added to heptane (400mL) at
850C, cooled slowly to 20 C and filtered. The filtered cake was washed with
a mixed solution of toluene (80 mL) and heptane (80 mL). The cake was
then dried in vacuum oven at 50 C to afford the title compound in 84%
molar yield (120.6 g, purity 99%). Mp 1050C. 11-1 NMR (400MHz, DMSO-d6)
5 1.04 (d, J=6.4Hz, 3H), 5 4.27 (dq, J=5.6 Hz, 6.4 Hz, 1H), 6 5.49 (d, J = 5.6
Hz, 1H), 6 7.2-7.5 (m, 10H); 13C NMR (DMSO-d6) 6 23.7, 56.3, 76.9, 96.4,
126.8, 127.0, 128.5, 129.2, 129.4, 129.6, 141.5, 142.2, 152.9.
Example 4 - Preparation of Compound 7a:
02N 40 COOH
0
HO 6 k NO2
O
,
C0NPh2 C0NPh2
3a la
To a flask were charged sequentially Compound 6 (90 g, 0.46 mole)
and toluene (500 mL). The suspension was cooled to about 0 0C, and N-
methylmorpholine (91 mL, 0.83 mole) and trimethylacetyl chloride (56 mL,
0.46 mole) were slowly added while keeping the reaction temperature below
5 C. The reaction mixture was agitated for 1 hour at 0 0C and assayed for
completion of formation of mixed anhydride (< 10% of UB remains). A
solution of 3a (100 g, 0.38 mole) in toluene (400 mL) and THF (220 mL)
- 40 -
CA 02875768 2016-09-20
was added while keeping the reaction temperature below 5 C. This was
followed by addition of a solution of 4-dimethylaminopyridine (5.5 g, 0.046
mole) in THF (45 mL). The mixture was agitated at about 0 C for 8-12
hours until reaction completion (<0.2% EB remains). Reaction was
quenched by adding a solution of 2.0 N H2SO4 (400 mL), warmed up to 25
C and filtered through a pad of CeliteTM. The layers were separated and
the organic layer was washed with 5% K2CO3 solution (3x300 mL) to
remove excess 6 (<1% of 6 remains). The mixture was washed with 5%
NaC1 solution (300 mL), filtered through a pad of celite, and concentrated
to about 500 mL final volume. Solution yield 90-95%. 1H NMR (CDC13,
400 MHz) 6 7.05-7.35 (m, 11H), 6.13 (br, 1H), 5.62 (dd, J = 16, 4 Hz, 1H),
5.31 (q, J = 7 Hz, 1H), 4.67 (m, 1H), 2.62-2.78 (m, 2H), 2.58 (br, 2H), 2.05
(m, 2H), 1.22 (d, J = 7 Hz, 3H).
Example 5 - Preparation of Compound 2a:
o o
o id& ¨ NO2 Lindlar/H2 0 io
NO2
- >---)
Nph,
0 0 NPh2
7a 2a
To a solution of 7a in toluene (200 mL, 50.0 g active, 112.5 mmol)
were charged Lindlar catalyst (2.5 g of 5% Pd / CaCO3 with 5% Pb
poisoned, 1.2 mmol) and quinoline (1.5 mL, 11.6 mmol). The mixture was
hydrogenated using 100 psi hydrogen at 25-30 C until reaction completion
as judged by HPLC. After removal of catalyst by filtration, toluene was
-41 -
CA 02875768 2014-12-19
replaced with ethyl alcohol by regulated vacuum distillation of about 40 C.
The product was dynamically crystallized from ethyl alcohol (180 mL) at
40 C in the presence of triethyl amine (8.5 mL). The reaction mixture was
slowly cooled to 5 C over a period of 4 hours. After stirring at 5 C for 3
hours, the product was filtered and washed with cold ethyl alcohol. The
product was dried at 60 C in a vacuum oven with nitrogen purge overnight
to give 2a as a yellow crystalline solid. Yield: 73.7 %. Mp 113-115 0C. 1H
NMR (400 MHz, CDC13) 8 1.48 (d, J = 6.4 Hz, 3H), 2.21-2.46 (m, 4H), 2.80
(m, 2H), 4.71 (m, 1H), 5.81-5.91 (m, 3H), 6.19 (m, 1H), 6.29 (q, J = 6.4 Hz,
1H), 7.28-7.37 (m, 11H).
Example 6 - Preparation of Compound la:
0
0
NO2
0 ,1102
110 0 00's
H =
Ph2N(0)C 2a la C(0)NPh2
Into a 2 L 3-neck round bottom flask was placed 2a (25 g, 0.056 mol)
and ethyl acetate (210 mL). The contents were stirred until Compound 2a
completely dissolved. The solution was washed with 0.25 M H2SO4 (75 mL)
and with water (3 x 75 mL). The organic phase was concentrated under
reduced pressure to about 200 mL, and 1-methyl-2-pyrrolidinone (50 mL)
was added. The solution was heated under distillation mode until a
- 42 -
CA 02875768 2014-12-19
temperature of 145 C was attained. The solution was held at this
temperature for 3.5 h. The solution was cooled to room temperature, and
DBU (0.57 mL, 6.8 mol%) was added. The solution was stirred for 1 h and
was quenched with 0.1 M H2SO4 (125 mL) and the product was extracted
into ethyl acetate (125 mL). The organic phase was washed with water
(125 mL) and was treated with DARCO-G60 (2.5 g) at 65 C for 1 h. The
suspension was filtered through a pad of Celite while the solution remained
hot. The solution was concentrated by atmospheric distillation to 38 mL.
The remaining ethyl acetate was replaced with isopropyl alcohol by
azeotropic distillation. The volume of the solution was adjusted to 225 mL.
The solution was diluted with ethyl alcohol (denatured with 0.5% toluene,
100 mL). The solution was slowly cooled to about 65 C, and DBU (0.29
mL, 3.4 mol%) was added. The suspension was slowly cooled to 15 C and
held at this temperature for 5 h. The product was filtered and washed with
a 2:1 mixture of isopropyl alcohol and ethyl alcohol (50 mL). 19.3 g was
obtained upon drying for 24 h at 50 C (90.2 wt % purity, 17.4 g active,
72.5% yield). Mp 151.8 C. 1H NMR (400 MHz, CDC13): 5 0.99 (m, 1H),
1.56 (d, J=6.0 Hz, 3H), 2.03 (m, 1H), 2.25-2.31 (m, 1H), 2.42-2.53 (m, 2H),
2.62-2.76 (m, 3H), 2.86-2.91 (m, 1H), 2.96-3.00 (m, 1H), 4.28-4.36 (m, 1H),
4.67-474 (m, 1H), 5.42 (hr s, 1H), 7.22-7.53 (m, 10H).
Example 7 - Preparation of Compound 13a:
-43-
CA 02875768 2014-12-19
0 H H H0 H H
0 0_0',NO2
0 d&..NH2
esNHCOOEt
WgIPI
_
H H H H H
CONPh2
roONPh2 -o0NPh2
la 12a 13a
To a three-neck flask equipped with an agitator, thermometer and
nitrogen inlet were sequentially added la (100g), THF (600 ml), 10%
palladium on carbon (50% wet, 35g) and water (400 ml). The mixture was
agitated for about 10 minutes at room temperature and then heated to
about 50 C. Formic acid (70 ml) was added slowly while the temperature
was maintained between 45 and 55 C. The reaction mixture was agitated
for 4 hours at 45-55 C. After the reaction was judged complete by HPLC,
the reaction mixture was cooled to 20 C and the pH was adjusted to 1 - 2
with 25% H2SO4 (60mL). THF (200mL) was added to the reaction mixture,
which was then filtered through a pad of Celite to remove the catalyst. A
mixed solution of THF (300 mL), water (300 ml) and 25% H2SO4 (5 mL) was
used to rinse the flask and catalyst, and filtered through the Celite. The
combined solution containing compound 12a was charged back into a
clean flask and the mixture was cooled to below 10 C. The pH was
adjusted to about 9 with 25% NaOH (30 mL) at below 10 C and NaC1 (150
g) was then added. The mixture was warmed to 20 C and two phases were
separated. The aqueous phase was extracted with THF (400 mL) and the
combined organic phases were washed with a brine solution (40 g of NaC1
in 200 mL of water). The organic layer was cooled to 5 C and triethyl
amine (56 mL) was added. Then ethyl chloroformate (23.6 mLml) was
added slowly. The mixture was warmed to 20 C and stirred for 30
- 44 -
CA 02875768 2014-12-19
minutes. After the reaction was judged complete, 200 ml of MTBE and 100
mL of water were added to the reaction mixture, followed by the slow
addition of 100 mL of 25% H2SO4. The two phases were separated and the
organic layer was washed with 200 ml of 12% H2SO4. The organic layer
was then concentrated and azeotropically distilled with 2B ethanol and 250
ml water was added at 70-80 C. The compound 13a was precipitated out
from ethanol-water with seeding at 55-65 C. After agitating for 1 hour at
55-65 C, 150 ml water was added at this temperature and held for 1 hour.
After cooling to 15-25 C, the mixture was agitated for an additional 3 hours
at 15-25 C and then the product was filtered and washed with ethanol-
water. The product was dried at 50-60 C to provide an off-white solid (86g,
Yield: 85%). Mp 188.2 C 1FINMR (CDC13) 8 7.25 - 7.55 (m, 10 H), 4.89(m,
1H), 4.51 (bs, 1H), 4.09 (d, J = 6.98 Hz, 2H), 3.49 (brs, 1H), 2.41 (m, 2H),
2.25 ( m, 1H), 2.06 (d, J = 10.8 Hz, 2H), 1.96 (d, J = 10.9 Hz, 1H), 1.83
(ddd, J = 13.5, 6.09, 2.51 Hz, 1H), 1.63(m, 1H), 1.52 (d, J = 5.8 Hz, 3H),
1.23 (m, 5H), 1.17 (q, J = 11.5 Hz, 2H), 0.92 (q, J = 11.5 Hz, 1H). MS (ESI)
for M+H calcd. 491 Found: 491.
Example 8 - Preparation of Compound 14a:
0
H H H H
NHCO2Et NHCO2Et
0 0
5%aq Nayi
THF
H H 11 )
tONPh2 L-02H
13a 14a
-45-
CA 02875768 2014-12-19
To a 250-mL 3-neck flask equipped with an agitator, thermometer,
and a reflux condenser, were added 10 g of 13a (20.4 mmol) and THF (50
mL). To this solution was added an aqueous solution of 5% (w/w) sodium
hydroxide (50 mL). The reaction mixture was then heated to and agitated
at 40 0C for about 4 hours. When the hydrolysis reaction was judged
complete, toluene (50 mL) was added and the mixture was agitated at a
rather fast rate for about 10 minutes. The organic phase containing the
by-product was separated from the aqueous phase containing product.
The organic phase was back-extracted with 5% aqueous NaOH solution (50
mL). The combined aqueous solutions were extracted twice with toluene (2
x 50 mL) and the organic extracts were discarded. To the aqueous solution
were added a solvent mixture of toluene (25 mL) and THF (50 mL). The
resulting mixture was cooled to between 0 to 5 C. A 2 N hydrochloric acid
aqueous solution (about 59 mL) was added to adjust the pH of the mixture
from - 13 to 2.5 at 0 to 50C. The aqueous phase was then separated from
the organic phase and extracted with a solvent mixture of toluene (25 mL)
and THF (50 mL). The organic phase and organic wash were combined and
diluted with THF (50 mL). The mixture was then concentrated
atmospherically to a final moisture content of < 0.05% by repeated
distillations, if necessary. The crude product was used in the next step
without further isolation and purification (containing 6.80 g, 99% yield).
11-I-NMR (CD3CN) 8 9.72 (bs, 1H), 7.17 - 7.41 (Ph in toluene), 5.45 (bs, 1H),
4.68 (dt, J = 5.90, 16.0, 1H), 4.03 (q, J = 7.10, 2H), 3.45 - 3.50 (m, 1H),
2.50 - 2.65 (m, 2H), 2.45 (dd, J = 5.64, 11.5, 1H), 2.36 (methyl in toluene),
- 46 -
CA 02875768 2014-12-19
1.83 (m, 4 protons), 1.34- 1.50 (qt, J = 2.91, 11.0, 1H), 1.32 (d, J = 5.91,
3H), 1.15- 1.25 (m, 6H), 0.95- 1.05 (m, 2H).
Example 9 - Preparation of Compound 36:
rOOCCF3 (OH
e
TF -
AA Me0H ,,.,(131 0
I N
NH 00CCF3 +
I r I
y _____________________________ y ________________
¨ _ Br Me0OCCF3
y
Br Br
35 36
To a solution of 5-bromo-2-methylpyridine N-oxide (10.0 g, 5.32
mmol) in Et0Ac (50.0 ml) at 0 C was added dropwise trifluoroacetic
anhydride (9.8 ml, 6.92 mmol.) while keeping the temperature below 50 C.
After the completion of the addition, the mixture was heated to between 75
and 80 C and stirred for at least 1 h. HPLC assay of the mixture indicated
reaction completion when 5-bromo-2-methylpyridine N-oxide is < 5%.
Upon completion, the mixture was cooled below 50 C and Me0H
(10.0 ml) was added. The mixture was heated for at least 1 h at 50 C.
The solution was concentrated under vacuum and Me0H was removed by
displacement with Et0Ac (40.0 ml) and concentrated to a volume of 30 ml.
To the concentrate was added toluene (20.0 ml) and the solution cooled to -
10 C over 2 h. The crystalline solid was filtered and washed with cold
toluene and dried overnight under vacuum at 35 C to provide 10.1 g (63%)
of 36. Mp 89 - 92 C. 11-1 NMR (DMSO-d6) 64.56 (s, 2 H), 7.49 (d, 1 H), 8.1
(dd, J = 2.3, 2.3 Hz, 1 H), 8.64 (d, J = 2.1 Hz, 1 H).
-47 -
CA 02875768 2014-12-19
Example 10 - Preparation of Compound 16:
F(0) (0E02
r OHr P(0) (0E02 F B(OH)2
N
N = F3CO2H
N
yBr Br
Br
36
37 38
16
A. Preparation of 37:
A slurry of 36 (10.0 g, 33.1mmol) in TBME (100 ml) was treated with
20% potassium carbonate (20 ml) solution and stirred at room temperature
for 1 h. The layers were separated and the organic layer was washed with
water. The TBME and water were removed by atmospheric distillation and
azeotropic distillation with acetonitrile (100 ml) and further concentrated
under vacuum to a volume of 40 ml. A Karl Fischer was performed to
confirm the removal of water (KF < 0.2). To the acetonitrile concentrate
was added dropwise thionyl chloride (3.2 ml, 43.7 mmol) while keeping the
temperature below 45 C. The reaction mixture was then heated at 45 C
for 2 h at which time an HPLC assay indicated complete reaction. The
reaction mixture was cooled to 25 C and quenched with water (20 ml)
while keeping the temperature below 40 C. The reaction mixture was
slowly poured into a mixture of 20% sodium carbonate (40 ml) and toluene
(100 ml), stirred for 10 min and the layers were partitioned. The toluene
extract was concentrated under reduced pressure to a volume of about 20
ml. A KF was performed to confirm the removal of water (KF < 0.2).
B. Preparation of 38
-48-
CA 02875768 2014-12-19
To a dry reaction vessel was charged a solution of lithium
bis(trimethylsily1) amide 1.3M in THF (51 ml, 66.2 mmol) and diethyl
phosphite (13 ml, 101.6 mmol) while keeping the temperature under 25 C.
The solution was stirred at 25 C for at least 1 h. The toluene solution
containing 37 from above was added over 1 h and the resulting mixture
was stirred at 25 C for at least 2 h at which time an HPLC assay indicated
complete reaction. Upon completion, the solution was quenched into 5%
sodium chloride (50 ml). The aqueous layer was extracted with toluene (50
ml). The combined organic layer was concentrated under reduced pressure
to a volume of about 20 ml. Toluene (80 ml) was then added and the
solution was washed with a 20% solution of potassium carbonate to
remove diethyl phosphate, confirmed by 1H NMR (< 20 mol%). The toluene
solution was then washed with water and concentrated under reduced
pressure to a volume of about 40 ml of Compound 38 solution.
C. Preparation of 16:
[001] To a reaction vessel was charged sodium carbonate (8 g; 75.5
mmol), 30 ml of water and stirred until dissolved. To this solution were
added 3-fluorophenylboronic acid (6 g; 42.9 mmol) and 5% Pd/C 50% wet
(0.5 g). The toluene solution of Compound 38 from above was then added
and the mixture was heated to 75 C for at least 5 h at which time an
HPLC assay indicated complete reaction. Upon completion, the reaction
mixture was cooled to 25 C and filtered to remove the Pd/C catalyst. The
layers were separated and the organic layer was washed and concentrated
under reduced pressure to about 20m1. Heptane (20 ml) was slowly added,
-49 -
CA 02875768 2014-12-19
seed crystals were added, and the mixture was cooled to -10 C over 2 h.
The crystalline solid was filtered, washed with heptane and dried overnight
under vacuum at 30 C to provide 8 g (75%). Mp 61 - 63 C. 6 1.3 (t, J =
7.08 Hz, 3H), 3.42 (s, 1H), 3.49 (s, 1H), 4.1 (q, J = 7.08 Hz, 2H), 7.04 -
7.11
(m, 1H), 7.23 - 7.3 (m, 1H), 7.32 - 7.3 (m, 1H), 7.32 -7.36 (m, 1H), 7.39 -
7.48 (m, 1H), 7.81 (ddd, J = 8.08, 2.3, 0.41 Hz, 1H), 8.74 (d, J = 2.36, 1H).
Example 11 - Preparation of Compound 23a:
OH
OTHP S
+ ioN
¨CI - 21,i-S
THP-(R)-Butynol 23a N it
To a flask were added 21g (124mmol) of 2-chlorobenzothiazole
chloride, 30g of KI, 150m1 of DMF, 2.7g of CuI, 8.4g of Pd(PPh3)4, 50m1 of
Et3N, and 118 ml of THP protected (R)-butynol. The mixture was stirred at
room temperature for 18 hrs. Most of the solvent was removed under
reduced pressure. Water was added and the product was extracted with a
mixture of t-BuOMe and hexane. The organic layer was washed with brine
and concentrated to give an oil. The oil was dissolved in 250m1 of Me0H
and treated with Ts0H for the deprotection. The mixture was heated at 50
0C for a few hrs. The pH was adjusted to between 7 and 8 with NaOH.
Most of the solvent was removed. The residue was chromatographed on a
silica gel column, eluting with Et0Ac/hexane to give 19.7g of 23a (78%). 1H
NMR (CDC13) 6 7.98-7.96 (m, 1H), 7.76-7.74 (M, 1H), 7.45-7.33 (m, 1H),
4.82-4.76 (m, 1H), 3.43 (d, J = 5.4 Hz, 1H), 1.55 (d, J = 6.7 Hz, 3H).
- 50 -
CA 02875768 2014-12-19
Example 12 - Preparation of Compound 24a:
0
OH 0 0 ¨ NO2
+ HO ¨ NO2 __________
23a N 6 N
24a
The same procedure for the preparation of 7a described above in
Example 4 was followed starting with 15 g of 23a to give, after column
purification, 17g of 24a. 1H NMR (CDC13) 67.99-7.97 (m, 1H), 7.79-7.77
(m, 1H), 7.48-7.35 (m, 2H), 7.31 (d, J = 15.9 Hz, 1H), 6.15 (bs, 1H), 5.80-
5.74 (m, 2H), 4.72-4.58 (m, 1H), 2.82-2.65 (m, 2H), 2.50-2.05 (m, 4H), 1.61
(d, J = 6.7 Hz, 3H).
Example 13 - Preparation of Compound 22a:
0
NO2
0 Lindlar/H2
NO2
N S
N
24a 41, 22a
The same procedure for the conversion of 7 to 2 (Example 5) was
followed starting from 15g of 24a to give, after column purification, 17g
product. 1H NMR (CDC13) 6 8.18 (d, J = 8.4 Hz, 1H), 7.98 (d, J = 8.4 Hz,
1H), 7.45-7.37 (m, 1H), 7.32-7.28 (m, 1H), 7.24 (d, 15.8 Hz, 1H), 6.59 (dd,
J = 11.8, 1.3 Hz, 1H), 6.55-6.40 (m, 1H), 6.15-6.08 (m, 1H), 6.00 (dd, J =
-5'-
CA 02875768 2014-12-19
11.8, 8.2 Hz, 1H), 5.76 (d, J = 15.8 Hz, 1H), (m, 1H), 2.80-2.65 (m, 2H),
2.46-2.05 (m, 4H), 1.50 (m, 3H).
Example 14 - Preparation of Compound 21a via DieIs-Alder Reaction:
0 0
ip NO2
1. Solv./150C 0 ONO
2. DBU
N S N S
= 22a
111. 21a
The same procedure for the conversion of 2a to la (Example 6) was
followed starting from 0.34g of 22a. The ratio of exo:endo was determined
by HPLC and NMR and found to be 60:40.
Example 15 - Preparation of Compound 19a:
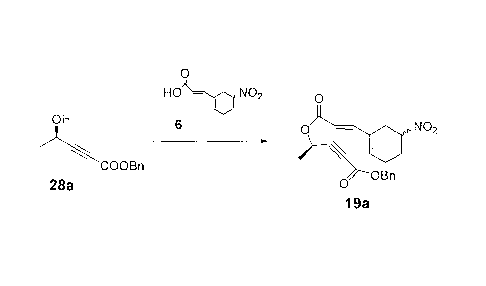
0
HO ¨ NO2 0
OH 6
0 ¨ Ahh NO2
COOBn t-BuCOCI
OBn
28a 0
19a
To a flask under nitrogen were added 1.48g of nitro acid 6 and 9 ml
of toluene. To this mixture was added dropwise 2.4 ml of Et3N to dissolve
all solid. To the cooled mixture at between 0 and 5 C were added 0.9 ml of
pivaloyl chloride and 30mg of 4-dimethylaminopyridine (DMAP). The
resulting mixture was stirred at between 0 and 5 C for 18 hrs. The
reaction mixture was poured into 10 ml of water. The layers were
separated and the organic layer was washed with NaHCO3 and water and
concentrated under reduced pressure. The residue was chromatographed
- 52 -
CA 02875768 2014-12-19
on a silica gel column, eluting with Hexane/Et0Ac to give 1.56g of 19a
(81%). 1H NMR (CDC13) 67.45-7.31 (m, 6H), 6.28-6.18 (m, 1H), 5.81 (d, J =
15.9 Hz, 1H), 5.62 (q, J = 6.8 Hz, 1H), 5.20 (s, 2H), 4.78-4.68 (m, 1H), 3.88-
3.70 (m, 2H), 2.52-2.15 (m, 4H), 1.57 (d, J = 6.8 Hz, 3H).
Example 15 - Preparation of Compound 18a:
o 0
Lindlar/H2io NO2
)----0 ¨ iIIIPt
___ NO2
0 0-0Bn - )-----
0 OBn
19a 18a
To a 100 ml Parr flask were added 1.4g of 19a, 25 ml of toluene, 0.14
g of Lindlar catalyst (Alfa Chem), and 0.1 ml of quinoline. The flask was
evacuated 3 times with nitrogen and vacuum and filled with hydrogen to
psi. The flask was shaken at room temperature for 3.5 hrs. The
mixture was filtered through a pad of celite and washed with toluene. The
filtrate was washed with 3X30 ml 1N HC1 solution and 30 ml brine. The
layers were separated and the organic layer was dried over MgSO4 and
15 concentrated to give 1.36 g (97%) of yellow oil. 1H NMR (CDC13) 6 7.45-
7.10
(m, 7H), 6.41-6.31 (m, 1H), 6.28-6.15 (m, 2H), 5.90-5.78 (m, 2H), 5.18 (s,
2H), (m, 1H), 2.88-2.70 (m, 2H), 2.50-2.15 (m, 6H), 1.41 (d, J = 6.5 Hz, 3H).
Example 16 - Preparation of Compound 17a:
- 53 -
CA 02875768 2014-12-19
0 0 a
1. Solv/1 50 C
110 NO2
2. DBU 0 'NO2
H H
0^0Bn
OJ''OBn
18a 17a
To a flask were added 0.17 g of 18a and 3 ml of xylene. The mixture
was heated to 150 0C for about 6 hrs and cooled to between 30 and 35 0C.
To the cooled mixture was added 1.5 ml of DBU. The resulting solution
was heated at between 30 and 35 0C for lh to complete the epimerization of
the initial trans product at the [6,5] junction to the cis product. There were
a total of four isomers generated. The exo:endo ratio of the Diels-Alder
reaction was about 78:22 and the a:13 ratio was about 80:20. The solvent
was removed under reduced pressure and the residue was purified on a
silica gel column to give 0.082g (48%) of the desired exo product and
0.025g (15%) of the endo product. Exo-isomer (a:I3 mixture): 1 NMR
(CDC13) 67.45-7.32 (m, 5H), 5.51 (bs, 1H), 5.22-5.10(m, 2H), 4.65 (bs, 1H,
a-isomer), 4.46-4.30 (m, 2H), 3.37-3.30 (m, 1H), 3.14-3.09 (m, 1H, 13-
isomer), 2.94-2.89 (m, 1H), 2.75-1.75 (m, 7H), 1.12 (d, J = 6.1 Hz, 3H, a-
isomer), 1.11 (d, J = 5.0 Hz, 3H, [3-isomer). Endo-isomer 1H NMR (CDC13) 6
7.40-7.30 (m, 5H), 5.80 (bs, 1H), 5.25 (d, j = 11.9 Hz, 1H), 4.60 (d, J = 11.9
Hz, 1H), 4.58-4.48 (m, 1H), 4.15-4.05 (m, 1H), 3.35-3.25 (m, 1H), 3.06 (t, J
= 5.7 Hz, 1H), 2.95-2.88 (m, 1H), 2.65-2.50 (m, 2H), 2.40-2.30 (m, 1H),
2.28-2.20 (m, 1H), (m, 2H), 1.42 (d, J = 6.5Hz, 3H), 1.05-0.95 (m, 1H).
Example 17 - Preparation of Compound 20:
- 54 -
CA 02875768 2016-09-20
0 et 0 H H
0 40.9NO2 Pd/C/H2 0 OWNH2
H = H H H
0 OH
0 OH
17a 20
To a Parr flask were added 0.47 g of 17a, 35m1 of Et0Ac, and 0.51 g
of Pt/C. The flask was evacuated with nitrogen and vacuum 3 times, filled
with hydrogen to 100 psi and was shaken for about 24 hrs as monitored by
NMR. The mixture was filtered and washed with Me0H. The filtrate was
concentrate to give 0.29 g of a gray solid. 1H NMR (Acetic acid-d4) 6 (a:13 =
78:22) 4.80-4.68 (m, 1H), 3.78 (bs, 1H, (3-isomer), 3.41-3.28 (m, 1H, a-
isomer), (m, 3H), 2.20-1.00 (m, 10H), 1.33 (d, J = 5.8Hz, 3H).
While the present invention has been described in conjunction with
the specific embodiments set forth above, many alternatives, modifications
and variations thereof will be apparent to those of ordinary skill in the art.
-55 -