Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
1
PROCESS FOR MODIFYING A POLYMERIC SURFACE
Field
The invention relates to a process for modifying a polymeric surface to
provide a
grafted polymeric coating.
Background
The modification of a substrate surface by application of a polymer coating is
a
versatile and efficient means of controlling interfacial properties such as
surface
energy (e.g. wetting behaviour), permeability, bio-activity, and chemical
reactivity.
Benefits that may be imparted to a substrate as a consequence of application
of a
polymer coating include, but are not limited to, chemical sensing ability,
wear
resistance, gas barrier enhancement, protein resistance, biocompatibility,
encouragement of cell growth and differentiation and the ability to
selectively bind
biomolecules. Methodology for forming such polymer coatings is therefore of
great
practical benefit.
For example, polymeric materials such as polystyrene have excellent
mouldability,
transparency and low cost, making them ideal for forming cell culture
substrates such
as multiwell plates, flasks and microcarrier particles. However, the
hydrophobic
surface which lacks functional groups limits the ability to control
interactions with cells
and proteins.
Surface modification may be used to enhance biocompatibility and allow
attachment
of functional groups which assist in cell binding or selection. Substrate
materials
modified by grafting of hydrophilic polymer brushes may significantly enhance
properties for cell culture applications. A number of surface grafting
techniques such
as gamma radiation, electron beam and UV-initiated grafting have been examined
but
there is a need to provide an economic treatment method which provides
excellent
control over coating properties and ultimately over the biological response.
One approach to forming polymer coatings on a polymeric surface is by using
physical or chemical adsorption techniques. Physical adsorption techniques are
most
commonly used and include dip-coating, drop casting, spin-coating, doctor
blade film
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
2
application, and roll-to roll coating. However, such coatings are prone to
delamination
upon being exposed to certain chemical and/or physical environments (e.g.
organic
solvents, temperature variations and/or mechanical abrasion).
An alternative approach to forming polymer coatings involves covalently
attaching
polymer chains to the surface of the substrate. Unlike the aforementioned
adsorption
techniques, covalently attaching the polymer chains to the substrate renders
the
coating less prone to delamination by chemical or physical means. One
particular
way of covalently attaching polymer chains to a substrate so as to form a
polymer
coating thereon utilises the so called "grafting to" technique. By this
technique, pre-
formed polymer chains are covalently attached to the substrate surface.
However,
due to diffusional and steric limitations at the substrate surface binding
sites, this
technique is prone to yielding comparatively poor grafting densities. In
addition, the
coating thickness that can be achieved by this technique is limited.
Polymer chains may also be grafted to the surface of a substrate using the so
called
"grafting from" technique. Unlike the "grafting to" technique, the "grafting
from"
technique involves polymerising monomer at the surface of the substrate so as
to
generate polymer chains "from" the surface. This technique is less prone to
the
diffusional and steric limitations of the "grafting to" technique and thereby
can more
readily afford relatively high grafting densities. However, "grafting from"
techniques
often suffer from being complex and requiring multiple steps. In particular,
the
surface of a substrate that is to be coated with the graft polymer will
generally need to
be modified or activated in some way to enable, for example, free radical
polymerisation to proceed. Thus, the substrate surface may need to undergo
glow or
corona discharge pre-treatment to promote the formation of functional groups
thereon
that can yield the required radical sites. Alternatively, free radical
initiator compounds
can be immobilised on the substrate surface that is to be grafted. Many
"grafting
from" techniques are also not capable of effectively and efficiently forming
uniform
polymer coatings on three dimensional surfaces. Furthermore, the coating
thickness
can often only be controlled within relatively narrow limits, and as the
coating
thickness typically is determined by a multitude of factors, control can be
difficult to
achieve. Accordingly, there remains scope for improving on prior art
techniques for
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
3
forming graft polymer coatings on substrates, or at the very least to provide
a useful
alternative method for preparing such graft polymer coatings.
The discussion of documents, acts, materials, devices, articles and the like
included
in this specification is solely for the purpose of providing a context for the
present
invention. It is not suggested or represented that any or all of these matters
formed
part of the prior art base or were common general knowledge in the field
relevant to
the present invention as it existed before the priority date of each claim of
this
application.
Summary
We provide a process for modifying a polymeric surface, the process
comprising;
contacting the polymeric surface with a solution comprising at least one
ethylenically unsaturated monomer; and
exposing the polymeric surface in contact with the solution to ultraviolet
light to
provide a graft-polymer of the monomer as a coating on the polymeric surface.
It is preferred that the polymeric surface and solution are free of
initiators.
In one set of embodiments the solution of ethylenically unsaturated monomer is
an
aqueous solution optionally comprising one or more water miscible solvents. It
is thus
generally preferred in this embodiment that the ethylenically unsaturated
monomer is
at least sparingly water soluble and is more preferably water soluble.
The exposure of the surface to ultraviolet light will generally be pulsed or
intermittent
exposure to UV radiation. We have found that the graft architecture from
ethylenically
unsaturated monomers and in particular water soluble ethylenically unsaturated
monomers on polymeric surfaces is significantly improved when using UV
grafting if
the polymeric surface is exposed intermittently to ultraviolet light while in
contact with
the solution of the ethylenically unsaturated monomer.
The invention may involve pulsed or intermittently exposing the polymeric
surface to
ultraviolet light. Intermittently exposing the polymeric surface to
ultraviolet light is
particularly preferred and has been found to provide significant advantages in
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
4
architecture of the graft polymer formed from the ethylenically unsaturated
monomer.
The architecture provided by intermittently exposing the polymeric surface
provides
improved swelling of the coating in aqueous environments when compared with
corresponding graft polymer coating prepared by continuous exposure.
Without wishing to be bound by theory we believe the intermittently exposing
the
polymeric surface to ultraviolet light creates differences in architecture
between
polymer layers formed during periods of exposure to ultraviolet light and
periods
without exposure to ultraviolet light.
The term intermittent exposure as used herein refers to periods of UV light
exposure
(on-periods) of duration of at least about 0.5 seconds, more preferably at
least about
1 second, more preferably at least about 2 seconds. The duration of exposure
(on-
period) may be up to about 3 minutes, more preferably up to about 60 seconds
and
still more preferably up to about 45 seconds. The period between exposures
(the off-
period) may be of duration up to about 60 minutes, more preferably up to about
30
minutes, more preferably still up to about 10 minute such as up to 5 minutes,
up to 2
minutes and up to 1 minute. The period between exposures (the off-period) may
be of
duration at least about 5 seconds, preferably at least about 10 seconds, more
preferably at least about 15 seconds, more preferably at least about 20
seconds,
more preferably still at least about 25 seconds.
In one set of embodiments the process of intermittently exposing the polymeric
surface to ultraviolet light involves periods of UV exposure in the range of
from 0.5
seconds to three minutes with the time between exposures being in the range of
from
five seconds to 60 minutes. The number of exposures to ultraviolet light is
generally at
least three exposures. In a further set of embodiments the process involves
intermittently exposing the polymeric surface comprises subjecting the surface
while
in contact with the aqueous solution to in the range of from five to one
hundred
exposures to ultraviolet light lasting in the range of from 0.5 seconds to
five minutes
such as 0.5 seconds to 3 minutes with a time gap between exposures being in
the
range of from one second to 60 minutes such as 5 seconds to 60 minutes or 10
seconds to five minutes.
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
The term pulsed exposure refers to periods of exposure (on-periods) less than
0.05 s
with intervals between exposure of less than 0.05s. Pulse widths (ON plus OFF
period) of 10 ps to 300 ps may be provided using industrial flash lamp
systems.
By a "graft" polymer is meant that the polymer chains are covalently coupled
to at
5 least the surface of the polymeric surface. The grafted polymer chains
may be
homopolymer chains or copolymer chains. By the graft polymer being a "coating"
is
meant that a plurality of polymer chains is covalently coupled to the surface
of the
polymeric surface so as to collectively form a layer of the graft polymer. The
graft
polymer chains may be crosslinked. The coating will generally modify the
surface
properties of the grafted region of the polymeric surface.
Throughout the description and the claims of this specification the word
"comprise"
and variations of the word, such as "comprising" and "comprises" is not
intended to
exclude other additives, components, integers or steps.
Detailed Description
A polymeric surface is provided upon which a polymeric coating is to be
grafted by the
method of the present invention. Examples of suitable polymeric surfaces
include,
but are not limited to surfaces comprising one or more polymers selected from
the
group consisting of, polyolefins such as polyethylene and polypropylene,
polyisobutylene and ethylene-alphaolefin copolymers, silicone polymers such as
polydimethylsiloxane; acrylic homopolymers and copolymers, such as
polyacrylate,
polymethylmethacrylate, polyethylacrylate; vinyl halide homopolymers and
copolymers, such as polyvinyl chloride; fluoropolymers such as fluorinated
ethylene-
propylene; polyvinyl ethers, such as polyvinyl methyl ether; polyvinylidene
halides,
such as polyvinylidene fluoride and polyvinylidene chloride;
polyacrylonitrile, polyvinyl
ketones; polyvinyl aromatics, such as polystyrene, polyvinyl esters, such as
polyvinyl
acetate; copolymers of vinyl monomers with each other and olefins, such as
ethylene-
methyl methacrylate copolymers, acrylonitrile-styrene copolymers, ABS resins,
and
ethylene-vinyl acetate copolymers; natural and synthetic rubbers, including
butadienestyrene copolymers, polyisoprene, polybutadiene, butadiene-
acrylonitrile
copolymers, polychloroprene rubbers, polyisobutylene
rubber,
ethylenepropylenediene rubbers, isobutylene-isoprene copolymers and
polyurethane
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
6
rubbers; polyamides such as Nylon 66 and polycaprolactam; polyesters such as
polyethylene terephthalate, alkyd resins; phenol-formaldehyde resins; urea-
formaldehyde resins, melam ine-formaldehyde resins;
polycarbonates;
polyoxyalkylenes such as polyoxyethylene, polyoxypropylene and their block
copolymers; polyimides; polyethers; epoxy resins, polyurethanes; wool; cotton;
silk;
rayon; rayon-triacetate; cellulose, cellulose acetate, cellulose butyrate;
cellulose
acetate butyrate; cellophane; cellulose nitrate; cellulose propionate;
cellulose ethers;
carboxymethyl cellulose; proteins, polypeptides; and polysaccharides.
In some embodiments, the polymeric surface is one comprised of only saturated
carbon ¨ carbon bonds and is free of double or triple carbon ¨ carbon bonds.
In such
polymeric surfaces, the minimum bond energy present is typically greater than
in
polymeric surfaces including unsaturated carbon ¨ carbon bonds. Examples of
such
polymeric surfaces may be selected from the group including polyolefins such
as
polyethylene and polypropylene, polyisobutylene and ethylene-alphaolefin
copolymers and polyvinyl aromatics, such as polystyrene, styrene copolymers,
poly-
isoprene, synthetic polyisoprene, polybutadiene, polychloroprene rubbers,
polyisobutylene rubber, ethylene-propylenediene rubbers and isobutylene-
isoprene
copolymers
In accordance with the method of the invention, the polymeric surface and
solution of
ethylenically unsaturated monomer are substantially free of radical initiator.
By being
"substantially free of radical initiator" is meant a radical initiator per se
is not included
or introduced to the polymeric surface or solution of ethylenically
unsaturated
monomer. For example, the polymerisation is to be performed in the absence of
radical initiators.
Those skilled in the art will appreciate that some monomers may, upon being
exposed
to UV radiation, decompose to afford a radical species. For avoidance of any
doubt,
monomers that are polymerised to form the graft polymer coating are not
intended to
be embraced within the definition of a radical initiator. The expression
"radical
initiator" is intended to mean compounds that are used primarily for the
purpose of
generating free radicals and includes photoinitiators such as benzophenone and
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
7
acetophenone derivatives such as diethoxy acetophenone and other radical
initiators
such as azo initiators and peroxides.
The surface may have been treated by a process such as corona discharge. Such
treatment processes are sometimes used in manufacture of polymeric articles
such
as film or cell culture plates which may be modified using the process.
Generally the
effect of corona discharge is relatively short lived so that after storage the
surface is
deactivated.
The polymeric surface to be modified may constitute all or only part of an
article to
which the method of the invention is applied. For example, the graft polymeric
coating
may be formed on at least part of a polymeric surface. In the case where the
article is
formed of polymer the bulk of the polymer may remain unmodified so that the
mechanical properties of the article are maintained. Alternatively, the
polymeric
surface to be modified may itself present as a coating on a substrate. The
substrate
may be polymeric, or alternatively may be a non-polymeric substrate such as a
glass,
ceramic or metal substrate.
In some embodiments, the polymeric surface is present on or part of an article
that is
used for the culture of cells. The cell culture device may be in a range of
structural
forms known in the art. Such structural forms include culture plates such as
microtitre
or microwell plates including comprising a multiplicity of wells such as 6,
12, 24, 48
96, 1536 or more wells and cell culture flasks. The substrate may also be in
the form
of carrier particles such as microcarrier particles. In these embodiments, it
is preferred
that the substrate surface to be modified is transparent.
Without wishing to be limited by theory, it is believed that polymerisation of
the
ethylenically unsaturated monomer in accordance with the invention occurs
predominantly at the polymeric surface such that the polymer is grafted from
that
surface. By polymer being grafted predominantly from the polymeric surface it
is in
turn believed that improved control over at least coating efficiency can be
attained.
Due to its lack of complexity, the method in accordance with the invention can
advantageously be performed with relatively low operating and capital costs.
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
8
Furthermore, the method of the invention has been found to be particularly
effective at
forming in a controlled manner graft polymer coatings with a relatively wide
range of
thicknesses on the surface of substrates having varied shapes and sizes, and
in
particular on substrates that present a three dimensional surface. Great
variability in
polymeric surface and monomer is also possible.
The method of the invention comprises contacting the polymer surface with a
solution
comprising at least one ethylenically unsaturated monomer. The solution may be
aqueous, partially aqueous, or non-aqueous. In some embodiments it is
preferable
that the solution is at least partially aqueous and further comprises at least
one water
miscible organic solvent. In other embodiments, it is preferable that the
solution is
aqueous. The choice of the most suitable solution will be dependent on the
polymeric
surface, the ethylenically unsaturated monomer, and the intended application
of the
polymeric coating.
The solution may comprise one or more solvents such as water, water miscible
solvents or mixtures of two or more thereof. Examples of water miscible
solvents
include dimethoxysulfoxide (DMSO), dimethylformamide (DMF), acetonitrile,
acetone
and alcohols such as ethanol and isopropanol. Where one or more water miscible
solvents are present in the ethylenically unsaturated monomer solution, they
will be
selected so as to not adversely affect the polymerisation reaction, the
polymeric
surface, or the resulting graft polymer coating.
The method of the invention is advantageously performed using an
environmentally
friendly aqueous solution of the ethylenically unsaturated monomer in contrast
with
conventional organic solvent based polymerisation reactions. Furthermore, the
aqueous solution is compatible with a broader range of polymeric surfaces
compared
with that available when organic solvent based reaction mediums are used. The
use
of an aqueous solution also provides a product which is readily prepared for
biological
applications such as cell culture or use as biomedical surfaces.
In a preferred embodiment, the polymeric coating is employed as cell
cultureware and
the solution is aqueous.
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
9
The concentration of monomer present in the solution will vary depending upon
the
nature of the polymer coating that is to be formed. For example, the
concentration of
the one or more ethylenically unsaturated monomers may be adjusted to tailor
the
thickness of the polymer coating. Those skilled in the art will be able to
determine the
required concentration of ethylenically unsaturated monomer for a given
polymerisation. Generally, the concentration of the one or more
ethylenically
unsaturated monomers in the solution will fall within the range of about 0.1%
(w/v) to
about 25% (w/v).
Other additives that may be present in the aqueous reaction medium include
polymerisation inhibitors. These are often present in commercially available
monomers to extend their shelf life. The fact that these inhibitors may be
present is an
advantageous feature of the invention as monomers can be used without the need
to
remove the inhibitor prior to polymerisation.
The polymerisation in accordance with the invention is preferably conducted in
a
substantially oxygen free environment. In other words, the polymerisation is
to
proceed under substantially oxygen free conditions. This may be achieved using
techniques well known to those skilled in the art. For example, the monomer
solution
and any head space above the solution may be purged with an inert gas such as
nitrogen or argon. The presence of oxygen can interfere with the efficiency of
the
polymerisation process. The fact that the reaction proceeds, albeit slowly,
despite the
presence of oxygen is an advantageous feature of the invention as it can be
used
without the need to fully remove oxygen prior to polymerisation.
Further, the monomer solution is generally deoxygenated prior to irradiation
to reduce
scavenging of radicals. Suitable deoxygenation methods include methods known
to
those skilled in the art such as bubbling the inert gas through the monomer
solution or
freeze-thaw-pump cycles.
Examples of ethylenically unsaturated monomers that may be used in accordance
with the invention include, but are not limited to, methyl (meth)acrylate,
ethyl
(meth)acrylate, ethyl-3,3-dimethyl (meth)acrylate, butyl (meth)acrylate,
isobutyl
(meth)acrylate, isobutyl(meth)acrylate, tert-butyl (meth)acrylate, 2-
ethylhexyl
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
(meth)acrylate, isobornyl (meth)acrylate, (meth)acrylic acid, hydroxypropyl
(meth)acrylate, hydroxybutyl (meth)acrylate, (meth)acrylamide, 2-hydroxyethyl
(meth)acrylate, N-methyl (meth)acrylamide, dimethylaminoethyl (meth)acrylate,
itaconic acid, 2-carboxyethyl acrylate, styrene, p-styrene carboxylic acids, p-
styrene
5 sulfonic acids, vinyl sulfonic acid, vinyl phosphonic acid, ethacrylic
acid, alpha-
chloroacrylic acid, crotonic acid, fumaric acid, citraconic acid, mesaconic
acid, maleic
acid, glycidyl (meth)acrylate, hydroxyethyl (meth)acrylate succinate, 2-
(meth)acrylam ido-2-methy1-1-propanesulfonic acid, 2-sulfoethyl(meth)acrylate,
3-
sulfopropyl (meth)acrylate, mono-2-[(meth)acryloyloxy] ethyl succinate,
hydroxypropyl
10 (meth)acrylate, N-ethyl (meth)acrylamide, N,N-dimethyl(meth)acrylamide,
N,N-diethyl
(meth)acrylamide, N-
isopropyl(meth)acrylamide, N-(hydroxymethyl)
(meth)acrylamide, N-(2-hydroxyethyl) (meth)acrylamide, N-(2-hydroxypropyl)
(meth)acrylamide, N-methylol (meth)acrylamide, N-vinylformamide, N-
vinylacetamide,
N-vinyl-N-methylacetamide, N-(n-propyl)acrylamide, N-(n-
butyl)(meth)acrylamide, N-
tert-butyl (meth)acrylamide, cyclohexyl
(meth)acrylamide, N-(3-am inopropyl)
(meth)acrylamide, 2-am inoethyl (meth)acrylate,
N-[3-(dimethylamino)propyl]
(meth)acrylamide, N-(meth)acryloyl tris(hydroxymethyl) am
inoethane, N-
(meth)acryloyl tris(hydroxymethyl) am inoethane, diacetone (meth)acrylamide, 2-
(meth)acryloyloxy ethyl acetoacetate,
[3-(m ethacryloylam ino)propyl]
trimethylammonium chloride, [3-(methacryloyloxy)ethy1]- trimethylammonium
chloride,
[2-(m ethacryloyloxy)ethyl]dim ethyl-(3-sulfopropyl)am m onium hydroxide,
[3-
(methacryloylamino)propyl] dim ethyl(3-sulfopropyl)am m onium
hydroxide,
poly(ethylene glycol) (meth)acrylate, poly(ethylene glycol) methyl ether
(meth)acrylate, poly(propylene glycol) (meth)acrylate, poly(propylene glycol)
methyl
ether (meth)acrylate, propargyl (meth)acrylate, 4-(meth)acryloylmorpholine, N-
viny1-2-
pyrrolidone, glycerol mono(meth)acrylate, glycosyloxyethyl (meth)acrylate,
vinyl
methyl sulphone, vinyl acetate, 2-(meth)acryloxyethyl glucoside, ethylene
glycol
(meth)acrylate phosphate, ethylenically unsaturated mono-, di-, tri and
polysaccharide(s) where the saccharide moiety is net neutral, zwitterionic
monomers
such as 3-((2-(meth)acryloyloxy)ethyl)dimethylammonio) propane-l-sulfonate, 2-
((meth)acryloyloxy)ethyl 2-
(trimethylammonio)ethyl phosphate, 2-
methacryloyloxyethyl phosphorylcholine and combinations thereof.
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
11
Examples of ethylenically unsaturated monomers that may be used in accordance
with the invention also include "ligands" comprising one or more ethylenically
unsaturated groups. The term "ligand" used herein is intended to take its
common
meaning within the art being a moiety that can bind a specific biomolecule,
for
example a biomolecule expressed on the surface of a cell, in the presence of a
multitude of other biomolecules.
The polymer grafted to the polymeric surface may be a homopolymer or
copolymer,
depending on the ethylenically unsaturated monomers employed. The polymer may
further be charged or neutral, and may belong to a class of polymer selected
from the
group consisting of carboxylic acid polymers, sulfonic acid polymers, amino
polymers,
zwitterionic polymers, neutral hydrophilic polymers and hydrophobic polymers.
In some embodiments, the solution comprises a single type of ethylenically
unsaturated monomer. The ethylenically unsaturated monomer may be selected
from
any one of those described herein.
In some embodiments, the solution comprises at least 2 ethylenically
unsaturated
monomers.
Those skilled in the art will appreciate that factors such as temperature, pH
and/or the
presence or absence of water miscible co-solvent(s) may alter the solubility
of a given
monomer in a given aqueous monomer solution. Such factors can therefore be
conveniently used to promote the solubility of monomer in an aqueous solution.
In some embodiments, the ethylenically unsaturated monomer is at least
sparingly
soluble in water, and more preferably soluble in water. The term sparingly
soluble
refers to a solubility of one gram monomer to 100 mL solvent and soluble
refers to at
least 1 gram monomer to 10 m L water at 20 C.
Examples of preferred ethylenically unsaturated monomers which are water
soluble
include acrylic acid, methacrylic acid, 2-carboxyethyl acrylate, hydroxyethyl
(meth)acrylate succinate, acrylamide, methacrylamide, N-alkyl (meth)acrylam
ides
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
12
(such as N-isopropyl acrylamide), N,N-dimethyl (meth)acrylamide, N-(3-am
inopropyl)
(meth)acrylamide, 2-am inoethyl (meth)acrylate, dimethylaminoethyl
(meth)acrylate,
N-vinyl-2-pyrrolidone, 2-hydroxyethyl (meth)acrylate,
N-(2-hydroxypropyl)
(meth)acrylamide, 2-methacryloyloxyethyl phosphorylcholine,
3-sulfopropyl
(meth)acrylate, [3-(methacryloylamino)propyl] trimethylammonium chloride, [3-
(methacryloyloxy)ethy1]- trimethylammonium chloride, poly(ethylene glycol)
(meth)acrylate and methoxy poly(ethylene glycol) (meth)acrylate.
Particularly
preferred water soluble monomers include acrylic acid, 2-carboxyethyl
acrylate,
acrylamide, N-isopropyl acrylamide, N-(2-hydroxypropyl) methacrylamide, 2-
methacryloyloxyethyl phosphorylcholine, poly(ethylene glycol) (meth)acrylate
and
methoxy poly(ethylene glycol) (meth)acrylate.
In some embodiments, the solution comprises 2 monomers. A first monomer
preferably includes a carboxylic acid functional group, and the resulting
polymeric
coating immobilised on the polymeric surface then comprises carboxylic acid
groups.
Preferably, the first monomer is acrylic acid, methacrylic acid or 2-
carboxyethyl
acrylate. A second monomer is preferably providing low biofouling properties
such
that a polymeric coating formed using the method of the present invention is
non-
adhesive to mammalian cells in serum-containing culture medium. Preferably,
the
second monomer is acrylamide, poly(ethylene glycol) (meth)acrylate, methoxy
poly(ethylene glycol) (meth)acrylate, N-(2-hydroxypropyl) methacrylamide, 2-
methacryloyloxyethyl phosphorylcholine or the like as would be known in the
art. In a
particular preferred embodiment, the first monomer is acrylic acid and the
second
monomer is acrylamide. In these embodiments, the molar ratio of the first to
second
monomer in the solution can be at least 1:99 such as at least 5:95, at least
10:90 or at
least 20:80 and up to 90:10 molar such as up to 80:20 whilst retaining low
biofouling.
More preferably, the ratio is 40:60 to 80:20 such as 80:20, 70:30, 60:40,
50:50 or
40:60. In particularly preferred embodiments, the molar ratio is 40:60.
Having contacted the polymeric surface with the solution of ethylenically
unsaturated
monomer, that surface is then exposed to UV radiation so as to generate
radical
species thereon. Those skilled in the art will appreciate that UV radiation is
typically
defined as electromagnetic radiation having a wavelength shorter than visible
light,
but longer than X-rays, and therefore has a wavelength within the range of
about 10
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
13
nm to about 400 nm. There is no particular limitation concerning the
wavelength of
UV radiation that may be used in accordance with the invention provided that
it can
generate free radicals on the polymeric surface. Generally, the wavelength of
UV
radiation used will fall within the range of about 200 nm to about 400 nm. The
ultraviolet light used in the process is preferably up to 400 nm wavelength.
The
ultraviolet light used in the process is more preferably up to 300 nm
wavelength. A
range of suitable sources of ultraviolet light may be used and high intensity
microwave electrode less bulb sources are particularly preferred.
Provided that the free radicals can be generated on the polymeric surface, and
that
the surface is not adversely affected, there is also no particular limitation
as to the
intensity of the UV radiation that can be used. Generally, UV sources with an
output
of at least up to about 200 W/cm2 can be used.
In a preferred set of embodiments the aqueous solution is washed from the
surface
with water after exposure to irradiation.
The polymeric surface in contact with the solution is exposed to a period of
irradiation.
One period of irradiation may be considered an on-period, during which the
polymeric
surface is exposed to irradiation, followed by an off-period, during which the
polymeric
surface is not exposed to irradiation. For continuous exposure there is one
period,
with the on-period being of definite length and the off-period being of
indefinite length.
The present invention relates to either pulsed or intermittent irradiation,
both of which
have at least 2 periods of exposure.
For pulsed irradiation the periods of exposure (on-periods) are generally less
than
0.05 s with intervals between exposures (off periods) of less than 0.05 s.
In preferred embodiments, the polymeric surface in contact with the solution
is
exposed to intermittent irradiation. By definition, the intermittent
irradiation includes at
least 2 periods of exposure. The total number of periods of exposure may be
selected based on the desired properties of the polymeric coating. For
instance, in
applications requiring a thicker polymeric coating, or for polymer surfaces
more
susceptible to UV degradation, the number of periods of exposure can be
increased.
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
14
In preferred embodiments, a thick coating is required to render the underlying
polymeric surface 'invisible to mammalian cells cultured on the polymeric
coating. In
these embodiments, up to 100 periods of exposure may be employed.
The optimum duration of the periods of irradiation will also depend on the
nature of
the polymeric surface on to which grafting is to occur and the intensity of
irradiation. In
some instances extended periods of irradiation may lead to deterioration
and/or
deformation of the polymeric surface. For example in the case of surfaces
formed of
polystyrene exposure times not in excess of about 45 seconds are preferred,
and
more preferably less than about 30 seconds. A person skilled in the art will
be able to
determine suitable combinations of UV intensity and periods of exposure having
regard to the nature of the surface and graft monomer composition, and the
teaching
herein.
The absolute and relative durations and powers of the exposure to ultraviolet
light
(on-period) and between exposures to ultraviolet light (off-periods) should be
selected
so as to be suitable for the polymeric surface and to provide a polymeric
coating of
desired properties. Particularly important properties in some embodiments are
the
polymeric coating thickness and elastic modulus. For example, it is
hypothesised that
relatively greater on-period exposure will result in denser and thinner
polymeric
coatings, while relatively greater off-period exposure will result in softer,
thicker, more
swellable polymeric coatings.
For intermittent irradiation, the on-period should be such that the structural
integrity of
the article, polymeric surface, and/or developing polymeric coating is
maintained.
During the on-period it is hypothesised that the exposure to irradiation
causes bond
scission, relatively highly cross-linked polymer growth, and substantial heat
generation. Depending on the chemistry of the components involved in the
process,
the on-period could lead to detrimental effects. As such, the duration and
power of
the on-period should be chosen to avoid, or at least mitigate to suitable
levels, these
detrimental effects. The on-period should also be selected to be sufficient to
initiate
polymerisation.
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
For example, the on-period may be of duration of at least about 0.5 seconds,
more
preferably at least about 1 second, more preferably still at least about 2
seconds. The
on-period may be of duration up to 60 seconds, more preferably up to about 45
second, more preferably still up to about 30 seconds. In preferred
embodiments,
5 where the polymeric surface comprises polystyrene, the on-period is of
duration from
about 1 second to about 45 seconds such as about 5 seconds to about 45
seconds.
In a preferred set of embodiments the intermittent irradiation comprises on-
periods in
the range of from 1 second to 15 seconds and off-periods in the range of from
1
10 second to 60 seconds.
UV light sources with an output of up to about 200 W/cm2 have been used.
For intermittent irradiation, the off-period should be such that the final
polymeric
15 coating is of sufficient properties. During the off-period it is
hypothesised that even in
the absence of exposure to irradiation, polymer growth continues in a 'growth-
from'
non-cross-linked mode, and any heat generated during the on-period can
dissipate to
some degree. The duration of the off-period should be chosen giving
consideration to
these factors. At a certain point, polymer chain growth in this off-period
will cease,
therefore there is no anticipated benefit from an off-period longer than this,
but this
period will be dependent on the polymer surface, the monomer solvent, and the
monomer, at least.
For example, the off-period may be of duration less than about 5 minutes, more
preferably less than about 3 minutes, more preferably still less than about 2
minute.
The off-period may be of duration more than about 10 seconds, more preferably
more
than about 20 seconds, more preferably still more than about 30 seconds, more
preferably still more than about 45 seconds. In preferred embodiments, where
the
polymeric surface comprises polystyrene, the off-period is of duration from
about 20
seconds to about 60 seconds such as 20 seconds to about 50 seconds.
Without wishing to be limited by theory, it is believed that exposing the
polymeric
surface to UV radiation causes bonds that make up the molecular structure of
that
surface to undergo cleavage so as to generate radical species. The generated
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
16
radical species can then promote free radical polymerisation of the one or
more
ethylenically unsaturated monomers present within the monomer solution.
Polymerisation of the monomers in this way is believed to provide for polymer
chains
being grafted from the polymeric surface.
Without wishing to be limited by theory, it is believed that exposing the
polymeric
surface to UV radiation may also, or alternatively, cause the ethylenically
unsaturated
bonds of the monomer to undergo cleavage so as to generate radical species.
The
generated radical species can then via hydrogen abstraction from the polymer
surface
promote free radical polymerisation of the one or more ethylenically
unsaturated
monomers present within the monomer solution. Polymerisation of the monomers
in
this way is believed to provide for polymer chains being grafted from the
polymeric
surface.
Without wishing to be limited by theory, it is also believed that carbon based
radicals
are generated on the polymeric surface, and it is these radicals that are
responsible
for promoting polymerisation of the one or more ethylenically unsaturated
monomers.
Where the polymeric surface comprises a carbon based polymer, the formation of
such carbon based radicals is believed to be facilitated when the carbon based
polymer used comprises a carbon-carbon polymer backbone.
The nature of the graft polymer coating formed on the polymeric surface can
advantageously be varied to suit the intended application of the resulting
product.
One advantage of the method is that it can afford a substantially uniform and
continuous graft polymer coating on the polymeric surface, be it a two
dimensional or
a three dimensional surface. By the graft polymer coating being "substantially
uniform and continuous" is meant that it presents over the desired region of
the
polymeric surface and has an integral coating having a relatively constant
thickness.
Having said this, the graft polymer coating may of course present as a
discontinuous
coating on the polymeric surface in that it may be formed on only a part or
parts of
that surface. In that case, the graft polymer will nevertheless form a
substantially
uniform and continuous coating on those parts of the polymeric surface. For
example, it may be desirable to form a particular pattern or array of the
graft polymer
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
17
coating on the polymeric surface. This can be achieved, for example, by
passing the
UV radiation through a suitable mask that limits regions of the polymeric
surface to
UV radiation exposure.
The thickness of the graft polymer coating applied to the polymeric surface
can be
varied by adjusting parameters of the method well known to those skilled in
the art.
For example, the thickness of the graft polymer coating may be increased by
increasing the concentration of ethylenically unsaturated monomer present
within the
solution by increasing the time and/or intensity of UV radiation exposure.
Coatings
having a gradient thickness may also be produced by having a gradient mask
between the UV source and the polymeric surface.
As used herein, the term "biomolecule" is intended to mean molecules that are
produced by an organism, tissue or cell. Biomolecules include, but are not
limited to,
peptides, oligopeptides, polypeptides, proteins, nucleic acids, nucleotides,
carbohydrates and lipids.
As an alternative to, or in combination with, forming a graft polymer coating
that
resists biomolecule adsorption and/or biofouling, it may be desirable to
include as part
of the graft polymer coating a ligand for binding with a specific biomolecule
such as a
specific biomolecule in solution or a specific biomolecule expressed on the
surface of
a cell.
By this approach, specific biomolecules per se or cells can be targeted for
attachment
to the graft polymer coating in applications such as assays and cell culture.
The graft polymer coating may be provided with such a ligand by any suitable
means.
For example, the ligand may be covalently bound to or comprise one or more of
the
ethylenically unsaturated monomers that are polymerised to form the graft
polymer
coating.
Alternatively, the graft polymer coating may be modified after it is formed so
as to
covalently couple the ligand to the surface of the graft polymer coating. In
that case,
one or more ethylenically unsaturated monomers that are polymerised to form
the
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
18
graft polymer coating may be provided with a functional group that can be used
subsequent to the formation of the graft polymer coating to facilitate the
covalent
attachment of the ligand to the graft polymer coating.
As used herein, the term "cell" refers to a live or dead cell, multicellular,
tissue or
cellular fragments, cell membrane, liposomal preparation or sub-organelle such
as
mitochondria, ribosome or nucleus. The term "cell" is also intended to include
adherent and non-adherent cell types.
The cells may be eukaryotic or prokaryotic cells.
Eukaryotic cells include those derived from animals/humans, plants, fungi, and
protists.
Prokaryotic cells include those derived unicellular microorganisms such as
bacteria
and archaea.
The term "cell" is also intended to include stem cells. In animals/humans most
adult
stem cells are lineage-restricted (multipotent) and are generally referred to
by their
tissue origin. For example, embryonic stem cells, mesenchymal stem cells,
adipose-
derived stem cells, endothelial stem cells, hematopoietc stem cells, neural
stem cells,
epithelial stem cells and skin stem cells etc.
We have found that the coatings made using continuous UV irradiation was
significantly thinner than coatings prepared using equivalent intermittent UV
irradiation conditions. Furthermore we found that the coating made using
intermittent
irradiation with delay between irradiation events was significantly thicker
than the
coating made with intermittent UV irradiation.
Analysis of the swelling ratio of the three coatings (see Table 12) indicated
that the
coatings made with intermittent UV irradiation were able to swell much more
that the
coating made using continuous UV irradiation when hydrated with PBS solution.
Without wishing to be bound by theory we believe this difference is most
likely due to
the degree of cross-linking within the coatings. Continuous UV irradiation
will lead to
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
19
four processes; (i) free radical formation, (ii) chain scission, (iii) cross-
linking
reactions, and (iv) polymer chain growth. These four processes will also occur
for
graft polymer coatings prepared using intermittent UV irradiation but the
relative
balance of the four processes will most likely be different. Assuming that the
free
radical formation is equal in both cases, intermittent UV irradiation should
lead to
more polymer growth when the sample is not being irradiated with UV than in
the
continuous case and less cross-linking within the coatings. This hypothesis is
borne
out by both the dry and hydrated thickness which were both greater in the case
of
samples prepared with intermittent UV irradiation. The reduced swelling ratio
obtained for the samples prepared using continuous UV irradiation suggest that
there
was a higher degree of cross-linking within the coatings. Thicker coatings
such as
those prepared using intermittent UV irradiation, with an equal degree of
cross-linking
would have a very similar swelling ratio. The influence of additional time
with no UV
irradiation (intermittent + delay) was to increase the polymer graft layer
thickness,
again suggesting that when the sample was not being irradiated, more polymer
growth was occurring than for shorter non-irradiation times (intermittent) and
much
more polymer chain growth was occurring than in the continuous UV irradiation
condition.
The invention will now be described with reference to the following examples.
It is to
be understood that the examples are provided by way of illustration of the
invention
and that they are in no way limiting to the scope of the invention.
EXAMPLES
Brief Description of Drawings
The Examples are described with reference to the attached drawings.
In the drawings:
Figure 1A and Figure 1B show representative high resolution XPS C is spectra
obtained on tissue culture polystyrene plates (NunclonTM 4, Nunc) before
(Figure 1A)
and after (Figure 1B) UV graft polymerisation of AAM as described in Example
1.
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
Figure 2A and Figure 2B show phase contrast images of HeLa cell attachment
after
20 hours on PS (NunclonTM 4) substrates after UV grafting of AAM (Figure 2A)
in
comparison to cell attachment on a PS (NunclonTM 4) control surface (Figure
2B) as
described in Example 3.
5 Figure 3 shows a representative high resolution XPS C is spectra obtained
on UV
graft polymers obtained from mixtures of AA and AAM monomer solutions. The
numbers (insert) refer to the percentage of AA in the monomer mixture as
described
in Example 4.
Figure 4A and Figure 4B show phase contrast images (10x objective) of L929
mouse
10 fibroblast cell attachment on NIPAM UV graft polymer coated substrates
at (Figure
4A) 37 C and (Figure 4B) after 30 min incubation at 20 C as described in
Example
6, Part B
Figure 5A and Figure 5B show phase contrast images (10 x objective) of human
MSC
attachment on NIPAM UV graft polymer coated substrates at (Figure 5A) 37 C
and
15 (Figure 5B) after 30 min incubation at 20 C as described in Example 6,
Part C.
Figure 6A and Figure 6B show representative phase contrast images (10x
objective)
of L929 cell attachment on MicroHexTM microcarrier particles coated with a
NIPAM
graft polymer coating at (Figure 6A) 37 C and (Figure 6B) after 30 min
incubation at
20 C as described in Example 7, Part B.
20 Figure 7A and Figure 7B show representative images of L929 cell
attachment on 96
well substrates coated with (Figure 7A) a 10 A AA UV graft copolymer and
(Figure
7B) the same surface after covalent immobilisation of c(RGDfK) peptide as
described
in Example 9, Part C.
Figure 8A and Figure 8B show representative images of L929 cell attachment on
MicroHexTM substrates coated with (Figure 8A) a 10 A AA UV graft copolymer
and
(Figure 8B) the same surface after covalent immobilisation of c(RGDfK) peptide
as
described in Example 10, Part C.
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
21
Figure 9 is a graph showing the cell attachment in response to coatings
prepared
using two different UV methods as a function of the composition of the
polymeric
coating in accordance with the description in Example 12, Part C.
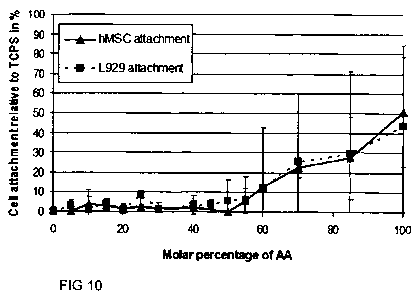
Figure 10 is a graph comparing different cell types in response to copolymer
coatings
based on acrylic acid (AA) and acrylamide (AAM) which were produced by the
initiator-free UV based coating method using intermittent UV as described in
Example
12 Part C.
Figure 11 is a graph showing the elemental ratio obtained from XPS analysis of
graft
polymer coatings with the number of UV passes as explained in Example 14 Part
B.
Figure 12 is a graph showing the change in thickness of 40 % AA-co-AAM layer
in
nanometres with the number of UV passes.
Figure 13: A, B and C are high resolution C is spectra with various
percentages of
UVA, UVB and UVC as described in Example 15, Part B.
Example 1
UV graft polymerisation from tissue culture polystyrene substrates
96 well tissue culture polystyrene (TCPS) plates (NunclonTM 4, Nunc) were used
as
received and placed into a glove box. In the glove box (under a nitrogen
atmosphere
containing < 0.2 % oxygen), 250 mg of acrylamide (AAM), poly(ethylene glycol)
methacrylate (PEGMA-OH), methoxy poly(ethylene glycol) methacrylate (PEGMA-
OMe) or N-isopropylacrylamide (NIPAM) were dissolved in 5 cm3 MilliQTM water
and
the solution purged for 10 min with nitrogen to remove residual oxygen. Each
well of
the 96 well plates was then filled with 0.15 cm3 of the monomer solution.
After
vacuum sealing of the plates into polymer bags (Sunbeam FoodSaver) in the
glove
box, the plates were placed onto a conveyor belt which ran underneath a UV
lamp
(A-200-450 nm, maximum intensity 360-390 nm, length of lamp 15 cm, output 1.8
kW, FUSION systems). The average belt speed was kept at 1.8 m=min-1 to give an
irradiation time of approximately 4 sec per pass. The vacuum sealed plates
were
exposed to UV irradiation during 30 passes under the UV lamp. In the case of
NIPAM
graft polymer coatings, plates were exposed during 20 passes under the UV
lamp.
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
22
The orientation of the plates was rotated by 90 degrees after each pass. The
wells
were then thoroughly washed with MilliQTM water using a plate washer (Thermo
Wellwash 4 MK 2) and finally air dried. For XPS analysis, the bottom of the
wells of
interest was removed using a cutting tool.
Presented in Table 1 are the elemental ratios, obtained by XPS analysis,
before and
after UV graft polymerisation on the tissue culture polystyrene plates. The
significant
changes in the 0/C ratio observed for each of the monomers and the changes
observed for the N/C ratio after graft polymerisation with the AAM and NIPAM
monomer solutions compared to that obtained for the TCPS substrate polymer
indicate successful graft polymerisation for each of the monomers. In
addition,
presented in Figure 1A and Figure 1 B are the XPS high resolution C is spectra
obtained from a tissue culture polystyrene plate before and after UV graft
polymerisation of AAM. Again the significant differences between these spectra
demonstrate the successful grafting of the AAM monomer. The high resolution
XPS
spectrum presented in Figure 1AA contains a dominant peak at 285.0 - 285.5 eV,
corresponding to the neutral carbon species C1 and C2 (C-C/C-H) and two
smaller
peaks at higher binding energy, corresponding to the C5 component (0-C=0) due
to
oxidised species originating from the surface treatment process and the C6
component corresponding to the aromatic carbon shake-up peak at approximately
292.0 eV. In comparison, the spectrum in Figure 1 B contains a peak at 285.0 -
285.5
eV, corresponding to the aliphatic carbon species C1 and C2 (C-C/C-H) and a
peak at
higher binding energy corresponding to the amide species C4 (0=C-N). The
complete attenuation of the aromatic shake-up peak in figure 1B also suggests
a
coating thickness of more than 10 nm (XPS sampling depth).
Table 1: Elemental ratios calculated from XPS survey spectra obtained on
tissue
culture polystyrene plates (NunclonTM 4, Nunc) before and after UV graft
polymerisation.
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
23
Sample O'fa N'/G
PS (NunclonTM 4) 0.079 0.000
P S-AAM 0.258 0.273
PS-PEGMA-OH 0.464 0.000
PS-PEGMA-0Me 0.416 0.000
PS-NIPAM 0.131 0.095
Example 2
UV graft polymerisation of coatings that display low protein binding
properties
Part A: Europium tagging of Human Serum Albumin (HSA)
Europium tagged human serum albumin (Eu-HSA) was prepared using the following
method. HSA (Sigma, 99%, essentially fatty acid free) was labelled using a
Delfia
Europium labelling reagent (Perkin Elmer) overnight at 4 C (pH 9.3). After
separation
of the Eu-labelled HSA from excess labelling reagent using Fast Protein Liquid
Chromatography (FPLC) (Akta Purifier, GE Healthcare) on a Superdex 75 (30/10)
size exclusion column (GE Healthcare), the Eu-HSA solution concentration was
determined via amino acid analysis (Waters Alliance HPLC). The Eu:HSA
labelling
ratio was determined in the following manner. First a number of Eu standard
solutions were prepared using various dilutions of a 100 nM Eu standard
solution and
Delfia Enhancement Solution (Perkin Elmer). The time resolved fluorescence
counts
from 100 1_ aliquots of these solutions were then obtained using a PHERAStar
multi-
well plate reader (BMG Technologies, X,ex = 337 nm, Xem = 620 nm, count delay
60 s,
count time 400 s). Comparison of the time resolved fluorescence counts
obtained
from Eu-HSA solutions of known concentration and the Eu standards was used to
calculate an Eu:HSA labelling ratio of 4.2.
Part B: Plate preparation
96 well tissue culture polystyrene plates (NunclonTM 4, Nunc) were coated with
AAM
and PEGMA-0Me graft polymers according to the experimental procedure described
in Example 1. The wells were then thoroughly washed with with MilliQTM water
using
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
24
a plate washer (Thermo Wellwash 4 MK 2) and finally air dried. Subsequently,
the
plates were analysed for the amount of protein adsorption using an assay based
on
Eu-HSA.
Part C: Protein adsorption assay
After UV graft polymerisation (Part B) each well was filled with 0.1 cm3 of a
solution
containing a solution of both Eu-HSA and HSA (1:1500 molar ratio) in phosphate
buffered saline (PBS). The total HSA concentrations used were 100, 10 and 1
g/cm3. The wells were incubated over 16 hours at room temperature, washed 6
times with PBS buffer solution and then treated with Delfia Enhancement
Solution
(Perkin Elmer) to release the Eu atoms from the adsorbed Eu-HSA. The solutions
obtained in this way were then analysed via time resolved fluorescence using a
PHERAStar multi-well plate reader (BMG Technologies, X,õ = 337 nm, Xem = 620
nm,
count delay 60 s, count time 400 s). The amount of adsorbed protein was
quantified by comparison of the counts obtained from these solutions to a
standard
curve obtained with solutions of known Eu concentration.
The results from the protein adsorption assay, carried out using three
different HSA
concentrations, are shown in Table 2. At all protein concentrations, the
adsorbed
amount detected on commercially available 96 well tissue culture polystyrene
plates
[PS (NunclonTM A)] far exceeded the amount detected on untreated polystyrene
plates (PS, Nunc) and 96 well tissue culture polystyrene plates modified by UV
graft
polymerisation with AAM [PS (NunclonTM 4) -AAM] and PEGMA-0Me [PS (NunclonTM
4)-PEGMA-0Me]. The small amounts of protein detected on the AAM and in
particular the PEGMA-0Me modified plates demonstrate that the UV graft
polymerisation method can be used to produce low protein binding surface
coatings.
CA 02877757 2014-12-23
WO 2014/000052 PCT/AU2013/000710
Table 2: Quantification of the amount of protein adsorption onto surfaces
before and
after UV graft polymerisation using Europium labelled HSA.
:
Protein concentration in solution
:
-3 \
:::(pg=cm1
:
U
-:: Adsorbed amount (ng=cm 2i
: .......:
A
PS (NunclonTM 4) 1101.0 153.5 18.0
PS 149.5 16.5 3.2
PS (NunclonTM 4) -AAM 76.0 8.8 2.5
PS (NunclonTM 4)-PEGMA-0Me 23.7 3.9 1.5
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
26
Example 3
UV graft polymerisation of coatings with low cell attachment properties
Part A: Plate preparation:
A 24 well tissue culture polystyrene plate (NunclonTM 4, Nunc) was coated with
UV
graft polymers using AAM, PEGMA-OH and PEGMA-0Me monomer solutions as per
the method described in Example 1. In the case of PEGMA-OH and PEGMA-0Me,
the inhibitor was removed from the monomer solutions using a column filled
with
inhibitor removing beads (Sigma) before transfer of the solutions to a glove
box. After
UV grafting and subsequent rinsing with MilliQTM water as per Example 1, the
plates
were extracted in a large volume of MilliQTM water for at least 72 hours
before drying
in air.
Part B: Cell attachment assay using HeLa cells
Surface modified 24 well plates were sterilised by the addition of sterile PBS
(1
cm3/well, pH 7.4) which contained penicillin and streptomycin (Gibco) at
concentrations of 120 and 200 pg.cm-3, respectively, for 4 hours at room
temperature
prior to cell seeding. HeLa cells were then seeded into each test well at a
concentration of 2x105 cells/well and incubated for 24 hours at 37 C in
humidified air
containing 5 % CO2. The culture medium used was DMEM/Hams F12 (Gibco)
containing 10 % foetal bovine serum (FBS). After 20 hours incubation, four
sample
replicates were each treated with (3-(4,5-dimethylthiazol-2-y1)-2, 5-
diphenyltetrazolium
bromide) (MTT) at 500 pg/cm3 in culture medium and incubated for a further 4
hours
to obtain a quantitative value for cell attachment relative to a 24 well
tissue culture
polystyrene (NunclonTM 4, Nunc) control surface. Cell attachment on the
control
surface was set to 100 % and attachment on all other surfaces was expressed as
a
percentage of the attachment on the control surface. The MTT containing
solution
was removed from wells after the 4 hour incubation step. Wells were then
washed
three times with sterile PBS prior to the release of the MTT formazan crystals
from the
cells using 1 cm3/well of DMSO. The plates were placed on a plate shaker and
gently
agitated for 15 minutes to ensure crystals were dissolved and solutions well
mixed.
100 pl samples from each well were then transferred into wells of a 96 well
plate for
optical density measurements at X = 595 nm test wavelength and X = 655 nm
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
27
reference wavelength. At 20 hours, immediately prior to the addition of the
MTT, the
cells were also viewed by phase contrast microscopy (Olympus IMT-2, 10 x
objective
lens) and representative images of cell attachment were recorded digitally.
Presented in Table 3 are the results obtained from the MTT assay. The results
clearly show that HeLa cell attachment was reduced to very low levels on
surfaces
modified with AAM, PEGMA-OH and PEGMA-0Me UV graft polymers in comparison
to cell attachment on the PS (NunclonTM 4) control surface. This result was
further
supported by phase contrast images shown in Figure 2A and Figure 2B. Here, the
appearance of HeLa cells observed after 20 hours on AAM, PEGMA-OH and
PEGMA-0Me UV graft polymer coatings was similar, showing aggregated cells that
had not attached on the surface or loosely attached, rounded cells that were
easily
removed from the surface by shaking or rinsing. As an example, presented in
Figure
2A is a representative image of HeLa cells on an AAM modified surface. In
comparison, HeLa cells appeared firmly adherent and well spread after this
culture
period on a PS (NunclonTM 4) control surface (Figure 2B).
Table 3: HeLa cell attachment, obtained via MTT assay, after 24 hours culture
on
various surfaces. Cell attachment on the polymer grafted surfaces was
normalised to
that obtained on the PS (NunclonTM 4) control surface.
Sample 54=ti cell attachment sd
PS (NunclonTM 4)-AAM 2.30 0.30
PS (NunclonTM 4)-PEGMA-OH 0.82 0.71
PS (NunclonTM 4)-PEGMA-0Me 0.52 0.08
PS (NunclonTM 4) 100.00 3.28
Example 4
UV graft polymerisation of coatings from acrylic acid (AA) and acrylamide
(AAM) and subsequent activation and covalent binding of amines
Part A: UV grafting of coatings from AA and AAM
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
28
As described in Example 1, 5 % (w/v) aqueous solutions of acrylic acid (AA)
and
acrylamide (AAM) were degassed by purging with nitrogen for more than 15 min
in a
glove box. The solutions were then mixed to yield AAM monomer solutions
containing
%, 10 %, 20 % and 50 % (v/v) AA monomer. Solutions made in this way were then
5 transferred into the wells of 96 well plates (0.15 cm3 per well).
While still in the glove box, plates containing the solutions described above
were then
vacuum sealed into polymer bags (Sunbeam FoodSaver) and removed from the glove
box. The plates were then passed under a UV lamp (FUSION Systems) 30 times on
a
conveyor belt at a speed of approximately 1.8 m/min as described in Example 1.
After
each pass the plate was turned 90 degrees to enable more uniform UV
irradiation.
The wells of the plates were then thoroughly washed with MilliQTM water using
a
plate washer (Thermo Wellwash 4 MK 2) and air dried. For XPS analysis, the
bottom
of the wells of interest was removed using a cutting tool.
Analysis of the data presented in Figure 3 and Table 4 clearly showed that UV
graft
polymerisation of the monomer mixtures resulted in successful coatings.
Furthermore,
these results demonstrate that the composition of the UV graft polymer
coatings can
be controlled by choosing an appropriate monomer mixture. XPS spectra of the
coatings prepared with an increasing AA ratio in the monomer feed (see Figure
3)
contained an increased contribution from the C5 component, which originated
from
the carboxylic acid functionality (from the AA monomer) in the coatings. At a
monomer composition of 5 % AA, the C4 signal originating from amide bonds
(from
the AAM monomer) was dominant while at a monomer composition of 50 % AA, the
C4 and C5 signals were almost equivalent in intensity.
Table 4: The components obtained by curve fitting the XPS C is high resolution
spectra presented in Figure 3. The monomer composition is expressed as the
percentage of AA in the monomer mixture, the remaining comprising the AAM
monomer.
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
29
Monomer composition C1+C2 C3 C4 C5
5% AA 77.7 0.8 20.8 0.6
10 A AA 73.0 0.2 25.2 1.6
20% AA 71.1 1.0 17.7 7.0
50% AA 73.5 0.42 11.6 14.5
The elemental ratios presented in Table 5 confirm these results. With
increasing AA
content in the monomer feed, the amount of oxygen present in the copolymer
coating
increased, while the nitrogen content decreased.
Table 5: Elemental ratios calculated from XPS survey spectra obtained from UV
graft
polymer coatings prepared from mixtures of AA and AAM monomer solutions. The
monomer composition is expressed by the percentage of AA in the monomer
mixture.
Monomer composition 0/C WC
5% AA 0.288 0.228
10 A AA 0.317 0.233
20% AA 0.329 0.182
50% AA 0.425 0.109
Part B: NHS activation of carboxylic acid surface functional groups
A solution containing 0.125 M 1-ethy1-3-(3-dimethylaminopropyl) carbodiimide
(EDC)
and 0.125 M N-hydroxysuccinimide (NHS) in MilliQTM water was prepared and 0.05
cm3 placed into each well of a plate prepared as described in Part A. The
solution
was added to the wells immediately after preparation. After 20 minutes
incubation the
wells were washed 3 times with MilliQTM water in a plate washer (Thermo
Wellwash 4
MK 2) and dried using a stream of nitrogen. The NHS activated plate was then
immediately used for subsequent reactions.
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
Part C: Covalent immobilisation of amines on NHS activated graft polymer
coatings
A 0.1 M TFEA solution was prepared using MilliQTM water, 0.05 cm3 aliquots of
which
were then transferred into each well of a plate freshly prepared as described
in Part
B. After incubation for 24 hours, the wells were then thoroughly washed with
MilliQTM
5 water, air dried and analysed by XPS.
The increase in the F/C ratio obtained with increasing AA content (Table 6)
demonstrated that the activation of the acrylic acid with NHS was successful
and that
NHS activated surfaces prepared in this way were highly reactive towards amine
containing molecules.
10 Table 6: Elemental ratios calculated from XPS survey spectra obtained on
UV graft
polymer coatings made from mixtures of AA and AAM monomer solutions after
subsequent NHS activation and reaction with TFEA. The monomer composition is
expressed by the percentage of AA in the monomer mixture.
=Monomer composition
5`)/0 AA 0.286 0.249 0.008
10 A AA 0.260 0.221 0.035
20% AA 0.285 0.216 0.058
50% AA 0.366 0.175 0.243
15 Example 5
UV graft polymerisation of coatings from PEGMA-OH and subsequent
activation and covalent binding of amines
Si-ALAPP samples with a size of 1 cm x 1 cm were prepared as described in
20 Example 1. As per Example 1, a 5 % (w/v) solution of PEGMA-OH was
prepared in
MilliQTM and degassed by purging with nitrogen for 30 min in a glove box. In a
glove
box, to each well of a 24 well tissue culture polystyrene plate (NunclonTM 4,
Nunc),
was added a Si-ALAPP wafer as well as 0.6 cm3 of the PEGMA-OH solution.
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
31
Whilst still in the glove box, the 24 well plate containing Si-ALAPP samples
and
PEGMA-OH monomer solution was vacuum sealed into polymer bags (Sunbeam
FoodSaver) and removed from the glove box. The plates were passed 20 times
under a UV lamp (FUSION Systems) on a conveyor belt at a speed of
approximately
1.8 m/min as described in Example 1. After each pass the plate was rotated 90
degrees to enable more uniform UV irradiation. Subsequently the samples were
removed from the plate, thoroughly washed with MilliQTM water and air dried.
The
samples were then immersed for 2 hours into a 0.5 M solution of carbonyl
diimidazole
(CD!) in dry DMSO. The samples were then rinsed with MilliQTM water and
subsequently immersed into a 0.1 M solution of TFEA in PBS buffer (pH 7.4) for
7
days. Finally the samples were washed with MilliQTM water, air dried and
analysed
by XPS.
Complete attenuation of the nitrogen signal from the ALAPP coating, as may be
observed from the data presented in Table 7, after UV graft polymerisation of
PEGMA-OH indicated successful coating with a thickness of greater than 10 nm
(XPS
sampling depth). After reaction of the OH groups of the PEGMA-OH groups with
CD!,
an increase in the N/C ratio was observed (Table 7), demonstrating a
successful
surface activation reaction. Finally the presence of fluorine after the
reaction of TFEA
with the CD! activated surface (Table 7) demonstrated the reactivity of
surfaces
prepared in this way towards amine containing molecules.
Table 7: Elemental ratios calculated from XPS survey spectra obtained on PEGMA-
OH UV graft polymer coatings before and after subsequent CD! activation and
reaction with TFEA.
Sample:::0/C: NI/C:
Si-ALAPP-PEGMA-OH 0.468 0.000
0.000
Si-ALAPP-PEGMA-OH-CDI 0.453 0.032
0.000
Si-ALAPP-PEGMA-OH-CDI-TFEA 0.458 0.018 0.018
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
32
Example 6
UV graft polymerisation of coatings from NIPAM for thermo-responsive
coatings
Part A: UV grafting of coatings from NIPAM
Tissue culture polystyrene plates (4 well, NunclonTM 4, Nunc) were used as
received.
UV graft polymer coatings on these substrates were carried out using N-
isopropylacrylamide (NIPAM) monomer as per Example 1. In a glove box under a
nitrogen atmosphere, a 5 % (w/v) solution of NIPAM was prepared in MilliQTM
water
and purged for 30 min with nitrogen. Aliquots of this monomer solution (0.6
cm3) were
then transferred into each well of the 4 well plates. Whilst still in the
glove box, the
plates were vacuum sealed into polymer bags (Sunbeam FoodSaver) and removed
from the glove box. The plates were passed 20 times under a UV lamp (FUSION
Systems) on a conveyor belt at a speed of approximately 1.8 m/min as per
Example
1. After each pass the plate was rotated 90 degrees to enable more uniform UV
irradiation. Subsequently the plates were washed five times with MilliQTM
water
followed by immersion of the plates in a large volume of MilliQTM water over
72
hours. Finally the surface modified plates were air dried.
Part B: L929 cell attachment on NIPAM graft polymer modified cell culture
substrates
Mouse fibroblast cells (L929) were cultured in modified Eagles medium (MEM)
containing 10 % foetal bovine serum (FBS) and 1 % non-essential amino acids.
The
wells of the plates were sterilised by the addition of sterile PBS (0.8
cm3/well, pH 7.4)
which contained 2 % (v/v) of an antibiotic-antimycotic solution (anti-anti,
Gibco),
respectively, for 2-4 hours at room temperature prior to cell seeding. L929
cells were
seeded onto NIPAM UV graft polymer modified 4 well plates (described in Part
A) at a
seeding density of 2x105 cells/well. After an incubation period of 24 hours, a
phase
contrast image of cell attachment was taken of a representative sample while
maintaining the plate at a temperature of 37 C on a heated microscope stage.
Subsequently the heated stage was removed and the plate was allowed to cool
down
to 20 C. 30 min after removing the heated stage another image was recorded.
The
phase contrast images taken at 37 C and 20 C respectively are shown in
Figure 4A
and Figure 4B. The cells in the image taken at 37 C (Figure 4A) were adherent
and
of well spread morphology whilst the cells in the image taken at 20 C (Figure
4B)
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
33
were of rounded morphology and could be easily washed off the surface. These
cell
culture results demonstrate the thermo-responsive nature of the NIPAM UV graft
polymer coating, which leads to cell-adhesive properties at physiological
temperature
(37 C) and non-cell adhesive properties at room temperature (20 C).
Part C: MSC attachment on NIPAM graft polymer modified cell culture substrates
Mesenchymal stem cells (MSCs) were isolated from human bone marrow aspirates
using standard methodologies. That is, bone marrow was first separated over a
density gradient (1.077 gms/100 cm3) and the light density fraction collected,
washed
in PBS and then cells resuspended in a-MEM media supplemented with 20 % FBS
(pre-selected batches). Cells were then added to T-flasks at a density of
approximately 5x105 cells/cm3 and incubated at 37 C for 2-3 days. After this,
the
non-adherent fraction was removed followed by gentle washing with fresh media
and
then PBS, leaving behind adherent MSCs. Fresh alpha-MEM and FBS was added
and the cells cultured for 7 days. They were then split (-1:3, depending on
the
confluency) by using EDTA and Ca and Mg free PBS and replated with fresh media
and serum.
Human MSCs were transferred into a-MEM media supplemented with 20 % FBS and
5 ng/cm3 human recombinant FGF-2 (Prospec) and seeded onto NIPAM UV graft
polymer modified 4 well plates (described in Part A) at a seeding density of
1x105
cells/well. Prior to cell seeding, the wells of the plates were sterilised by
the addition
of sterile PBS (0.3 cm3/well, pH 7.4) which contained 2% (v/v) of an
antibiotic-
antimycotic solution (Gibco), respectively, for 2-4 hours at room temperature.
After an
incubation period of 24 hours in a humidified incubator at 37 C and 5 % CO2,
a
phase contrast image of cell attachment was taken on of representative region
of the
well while maintaining the plate at a temperature of 37 C on a temperature
controlled
microscope stage. Subsequently the temperature controlled stage was removed
and
the plate was allowed to cool down to 20 C. 30 min after removing the heated
stage
another image was recorded. The phase contrast images taken at 37 C and 20 C
respectively are shown in Figure 5A and Figure 5B. The cells in the image
taken at
37 C (5igure 5A) were adherent, well spread and almost at confluence whilst
in the
image taken at 20 C (Figure 5B) the cells were aggregated and had lifted off
the
surface in some regions. These human MSC culture results again demonstrate the
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
34
thermo-responsive nature of the NIPAM UV graft polymer coating, which leads to
cell-
adhesive properties at physiological temperature (37 C) and non-cell adhesive
properties at room temperature (20 C).
Example 7
UV graft polymerisation of coatings from NIPAM for thermo-responsive
coatings on microparticles
Part A: UV grafting of coatings from NIPAM on microcarrier particles
Tissue culture polystyrene plates (24 well, NunclonTM 4, Nunc) were used as
received. 50 mg of MicroHexTM microcarrier particles (NunclonTM 4, Nunc) were
added to each well of these plates. UV graft polymer coatings on the
microcarrier
particles were prepared out using N-isopropylacrylamide (NIPAM) monomer as per
Example 1. Briefly, in a glove box under a nitrogen atmosphere, a 5 % (w/v)
solution
of NIPAM was prepared in MilliQTM water and purged for 30 min with nitrogen.
Aliquots of this monomer solution (0.6 cm3) were transferred into each well of
the 24
well plates containing the microparticles. Whilst still in the glove box, the
plates were
then vacuum sealed into polymer bags (Sunbeam FoodSaver) and removed from the
glove box. The plates were passed 20 times under a UV lamp (FUSION Systems) on
a conveyor belt at a speed of approximately 1.8 m/min as per Example 1. After
each
pass the plate was gently agitated and rotated 90 degrees to enable more
uniform UV
irradiation. Subsequently the microparticles were washed ten times with
MilliQTM
water, centrifuging and resuspending the microparticles after each washing
step.
Finally the surface modified microparticles were incubated in a large volume
of Milli-
QTM water over 72 hours before drying under vacuum.
The MicroHexTM microcarrier particles were analysed by XPS before and NIPAM UV
graft polymerisation. Analysis of the results presented in Table 8
demonstrated that
the surface coating procedure was successful. In particular the increase in
the
calculated N/C ratio indicated the presence of the graft polymer layer on the
surface
of the microcarrier particles.
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
Table 8: Elemental ratios calculated from XPS survey spectra obtained on
MicroHex TM microcarrier particles (NunclonTM 4, Nunc) before and after
coating with a
NIPAM UV graft polymer.
Ott
MicroHex TM 0.166 0.001
MicroHexTm-NIPAM 0.157 0.123
5 Part B: L929 cell attachment on NIPAM graft polymer modified microcarrier
particles
Mouse fibroblast cells (L929) were cultured in modified Eagles medium (MEM)
containing 10 % foetal bovine serum and 1 % non-essential amino acids. L929
cells
were seeded onto NIPAM UV graft polymer modified microparticles (described in
Part
A) contained in the wells of a 4 well tissue culture polystyrene plate
(NunclonTM 4,
10
Nunc) that had been modified with a non-cell adhesive PEGMA-OH UV graft
polymer
coating as per Example 3. Each well contained 0.1 cm3 of packed surface
modified
particles. Prior to cell seeding the surface modified microcarrrier particles
were
sterilised by the addition of sterile PBS (0.6 cm3/well, pH 7.4) which
contained 2 %
(v/v) of an antibiotic-antimycotic solution (Gibco), respectively, for 2-4
hours at room
15
temperature. The cell seeding density was 2x104 cells/well. After a cell
culture period
of 24 hours, a phase contrast image of cell attachment was taken of a
representative
sample while maintaining the plate at a temperature of 37 C on a heated
microscope
stage. Subsequently the heated stage was removed and the plate was allowed to
cool down to 20 C. 30 min after removing the heated stage another image was
20
recorded. The phase contrast images taken at 37 C and 20 C respectively are
shown in Figure 6A and Figure 6B. The cells in the image taken at 37 C
(Figure 6A)
were adherent and of partially well spread morphology whilst the cells in the
image
taken at 20 C (Figure 6B) were of rounded morphology which could easily be
washed off the surface. These cell culture results demonstrate the
effectiveness of
25
the thermo-responsive NIPAM UV graft polymer coating on microparticles, which
leads to cell-adhesive properties at physiological temperature (37 C) and non-
cell
adhesive properties at room temperature (20 C).
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
36
Example 8
Evenness of coatings on three-dimensional substrates
Tissue culture polystyrene plates (96 well, NunclonTM 4, Nunc) were used as
received. UV graft polymer coatings on these plates were obtained using AAM
monomer as per Example 1. Briefly, in a glove box (under a nitrogen atmosphere
containing < 0.2 % oxygen), 250 mg of AAM were dissolved in 5 cm3 MilliQTM
water
and the solution purged for 10 min with nitrogen to remove residual oxygen.
Each well
of the 96 well plates was then filled with 0.15 cm3 of the monomer solution.
After
vacuum sealing of the plates into polymer bags (Sunbeam FoodSaver) in the
glove
box, the plates were placed onto a conveyor belt which ran underneath a UV
lamp
(A-200-450 nm, maximum intensity 360-390 nm, length of lamp 15 cm, output 1.8
kW, FUSION systems). The average belt speed was kept at 1.8 m=min-1 to give an
irradiation time of approximately 4 sec per pass. The vacuum sealed plates
were
exposed to UV irradiation during 30 passes. The orientation of the plates was
rotated
by 90 degrees after each pass. The wells were then thoroughly washed with
MilliQTM
water using a plate washer (Thermo Wellwash 4 MK 2) and finally air dried. For
XPS
analysis, the bottom as well as the wall of selected wells on the 96 well
plate was
removed using a cutting tool.
Presented in Table 9 are the elemental ratios, obtained by XPS analysis, after
UV
graft polymerisation on different regions of a representative well. Due to the
fact that
the well was filled with 0.15 cm3 of the monomer solution during UV graft
polymerisation, a coating was expected on all regions of the well except the
top of the
wall. This expectation was confirmed by the results shown in Table 9. The 0/C
and
N/C ratios obtained on top of the wall (approximately 2 mm from the top) were
similar
to those obtained on other 96 well tissue culture polystyrene samples
(NunclonTM 4,
Nunc) (see Table 1), suggesting that this part of the plate was not affected
by the UV
graft polymerisation process. However, the significant changes in the 0/C and
N/C
ratio observed for each of the other regions in comparison to the unmodified
top of the
wall suggest a successful AAM graft polymer coating in each case. In addition,
the
elemental ratios obtained from the bottom of the well, the lower part of the
wall
(approximately 2 mm from the bottom) and the middle part of the well (half way
up the
wall) show similar 0/C and N/C values close to the theoretically expected
values,
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
37
suggesting an even coating thickness of more than 10 nm (XPS sampling depth)
throughout the surface modified region of the well.
Table 9: Elemental ratios calculated from XPS survey spectra obtained on a
single
well of a partially AAM UV graft polymer modified 96 well plate. Spectra were
recorded on the different regions present in the well. These are the bottom of
the
well, the lower part of the wall (approximately 2 mm from the bottom), the
middle part
of the well (half way up the wall) and the top part of the wall (approximately
2 mm
from the top).
Reg ion OfC NIC
Bottom of well 0.259 0.259
Wall (bottom) 0.259 0.248
Wall (middle) 0.259 0.255
Wall (top) 0.085 0.010
Example 9
UV graft copolymerisation of coatings from AA and AAM on 96 well substrates,
covalent immobilisation of a peptide and effect on the cellular response
Part A: UV grafting of copolymer coatings from AA and AAM
As per Example 1, 5% (w/v) aqueous solutions of acrylic acid (AA) and
acrylamide
(AAM) were degassed by purging with nitrogen for more than 15 min in a glove
box.
A solution was made from these by mixing 10 % (v/v) AA and 90 % (v/v) AAM
monomer solution (10 % AA). The solution prepared in this way was then
transferred
into the wells of 96 well plates (0.15 cm3 per well).
While still in the glove box, plates containing the solutions described above
were then
vacuum sealed into polymer bags (Sunbeam FoodSaver) and removed from the glove
box. The plates were then passed under a UV lamp (FUSION Systems) 30 times on
a conveyor belt at a speed of approximately 1.8 m/min as described in Example
1.
After each pass the plate was rotated 90 degrees to enable more uniform UV
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
38
irradiation. The wells of the plates were then thoroughly washed with MilliQTM
water
using a plate washer (Thermo Wellwash 4 MK 2) and air dried.
Part B: NHS activation of carboxylic acid surface functional groups and
covalent
immobilisation of cRGD
A solution containing 0.125 M 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide
(EDC)
and 0.125 M N-hydroxysuccinimide (NHS) in MilliQTM water was prepared and 0.05
cm3 placed into each well of a 10% AA modified 96 well plate prepared as
described
in Part A. The solution was added to the wells immediately after preparation.
After 20
minutes incubation the wells were washed 3 times with MilliQTM water in a
plate
washer (Thermo Wellwash 4 MK 2). The NHS activated plate was then immediately
used for subsequent reactions.
An N-C terminally cyclised molecule containing a tri-amino acid motif,
arginine-
glycine-aspartic acid (c(RGDfK), Peptides International) was covalently
attached to
the polymer coating using the following method. Aliquots of a solution (0.1
cm3)
containing 200 g/mL of c(RGDfK) in PBS were added to each well of a freshly
prepared NHS activated plate described above. The solution was incubated in
the
wells overnight (15 h) at 4 C, after which the solution was removed and the
wells
washed 10 times with PBS.
Part C: L929 cell attachment on surface modified 96 well plates
Mouse fibroblast cells (L929) were cultured in modified Eagles medium (MEM)
containing 10 % foetal bovine serum (FBS) and 1 % non-essential amino acids.
The
wells of the plates were sterilised by the addition of sterile PBS (0.3
cm3/well, pH 7.4)
which contained 2 % (v/v) of an antibiotic-antimycotic solution (Gibco),
respectively,
for 2-4 hours at room temperature prior to cell seeding. L929 cells were
seeded onto
the wells of the plate with covalently immobilised c(RGDfK), freshly prepared
as
described in Part B, at a seeding density of 2x104 cells/well. In addition,
cells were
also seeded into the wells of a freshly prepared plate which had not been NHS
activated and onto which c(RGDfK) had not been covalently immobilised. After
an
incubation period of 20-22 hours, phase contrast images of cell attachment
were
taken of representative regions. The phase contrast images taken are shown in
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
39
Figure 7A and Figure 7B. The cells in the image for the sample which did not
contain
covalently immobilised c(RGDfK) (Figure 7A) were non-adherent and of rounded
morphology whilst the cells in the image taken for the sample to which
c(RGDfK) had
been covalently immobilised (Figure 7B) were adherent and of a well spread
morphology. These cell culture results demonstrate the cell adherent
properties of
the UV graft polymer coating formed from a monomer feed of 10 %AA and 90 %
AAM, to which c(RGDfK) had been covalently immobilised and the non-cell
adherent
nature of the UV graft coating to which c(RGDfK) had not been covalently
immobilised.
Example 10
UV graft copolymerisation of coatings from AA and AAM on MicroHex TM
substrates, covalent immobilisation of a peptide and effect on the cellular
response
Part A: UV grafting of copolymer coatings from AA and AAM on microcarrier
particles
Tissue culture polystyrene plates (24 well, NunclonTM 4, Nunc) were used as
received. 50 mg of MicroHexTM microcarrier particles (NunclonTM 4, Nunc) were
added to each well of these plates. UV graft polymer coatings on the
microcarrier
particles were prepared as per Example 7. Briefly, in a glove box under a
nitrogen
atmosphere, 5 % (w/v) aqueous solutions of acrylic acid (AA) and acrylamide
(AAM)
were degassed by purging with nitrogen for more than 15 min in a glove box. A
solution was made from these by mixing 10 % (v/v) AA and 90 % (v/v) AAM
monomer
solutions (10% AA). Aliquots of this 10 % AA monomer mixture (0.6 cm3) were
transferred into each well of the 24 well plates containing the
microparticles. Whilst
still in the glove box, the plates were then vacuum sealed into polymer bags
(Sunbeam FoodSaver) and removed from the glove box. The plates were passed 30
times under a UV lamp (FUSION Systems) on a conveyor belt at a speed of
approximately 1.8 m/min as per Example 1. After each pass the plates were
gently
agitated and rotated 90 degrees to enable more uniform UV irradiation.
Subsequently
the microparticles were washed ten times with MilliQTM water, centrifuging and
resuspending the microparticles after each washing step. Finally the surface
modified
microparticles were incubated in a large volume of MilliQTM water over 72
hours
before drying under vacuum prior to XPS analysis.
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
The results of XPS analysis presented in Table 10 demonstrate clearly that the
graft
polymerisation was successful on the microcarrier particles. Both the 0/C and
N/C
atomic ratios were observed to increase after the graft polymerisation
reaction. A
comparison of the relative 0/C and N/C ratio indicated a coating which
contained both
5 the AAM and AA monomers. This was confirmed by analysis of the high
resolution C
is spectra (not shown).
Table 10: Elemental ratios calculated from XPS survey spectra obtained on
MicroHex TM microcarrier particles (NunclonTM 4, Nunc) before and after
coating with a
UV graft polymer made from mixtures of 10 % (v/v) AA and 90 % (v/v) AAM
monomer
10 solutions.
ED NJ
MicroHex TM 0.166 0.001
MicroHex Tm -10 % AA 0.258 0.149
Part B: NHS activation of carboxylic acid surface functional groups and
covalent
immobilisation of c(RGDfK)
A solution containing 0.125 M 1-ethy1-3-(3-dimethylaminopropyl) carbodiimide
(EDC)
15 and 0.125 M N-hydroxysuccinimide (NHS) in MilliQTM water was prepared
and
incubated with 10% AA modified MicroHex TM microcarrier particles in such a
way that
the particles were covered by an excess of the solution. The solution was
added to
the modified microparticles immediately after preparation.
After 20 minutes
incubation with occasional shaking, the microparticles were washed 3 times
with Milli-
20 QTM water by centrifugation and resuspension in MilliQTM water. The NHS
activated
microparticles were then immediately used for subsequent reactions.
An N-C terminally cyclised molecule containing a tri-amino acid motif,
arginine-
glycine-aspartic acid (c(RGDfK), Peptides International) was covalently
attached to
the polymer coating using the following method. Aliquots of a solution (0.6
cm3)
25 containing 200 g/mL of c(RGDfK) in PBS were added to each well
containing freshly
prepared NHS activated MicroHexTM microcarrier partices prepared as described
above. Subsequently the microparticles were washed ten times with MilliQTM
water,
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
41
centrifuging and resuspending the microparticles after each washing step.
Finally the
surface modified microparticles were incubated in a large volume of MilliQTM
water
over 72 hours prior to cell culture experiments.
Part C: L929 cell attachment on surface modified microcarrier particles
Mouse fibroblast cells (L929) were cultured in modified Eagles medium (MEM)
containing 10 % foetal bovine serum and 1% non-essential amino acids. L929
cells
were seeded onto 10 % AA modified microparticles (described in Part A) and
c(RGDfK) modified microparticles (described in Part B), respectively,
contained in the
wells of a 4 well tissue culture polystyrene plate (NunclonTM 4, Nunc) that
had been
modified with a non-cell adhesive PEGMA-OH UV graft polymer coating as per
Example 3. Prior to cell seeding the micocarrier particles were sterilised by
the
addition of sterile PBS (0.3 cm3/well, pH 7.4) which contained 1 % (v/v) of an
antibiotic-antimycotic solution (Gibco), respectively, for 2-4 hours at room
temperature. Each well contained 0.1 cm3 of packed surface modified particles.
The
cell seeding density was 2x104 cells/well. After a cell culture period of 20-
22 hours, a
phase contrast image of cell attachment was taken of a representative region
for each
of the samples. The phase contrast images taken are shown in Figure 8A and
Figure
8B. The cells in the image for the sample which did not contain covalently
immobilised c(RGDfK) (Figure 8A) were non-adherent and of rounded morphology
whilst the cells in the image taken for the sample to which c(RGDfK) had been
covalently immobilised (Figure 8B) were adherent and of a well spread
morphology.
These cell culture results demonstrate the cell adherent properties of the UV
graft
polymer coating formed from a monomer feed of 10 % AA and 90 % AAM, to which
c(RGDfK) had been covalently immobilised and the non-cell adherent nature of
the
UV graft coating to which c(RGDfK) had not been covalently immobilisedthermo-
responsivethermo-responsivethermo-responsive.
Example 11
Polymer grafting: continuous versus intermittent UV irradiation
Part A: Preparation of coatings
Silicon wafers (Si) were cut into squares of approximately 7 x 7 mm dimension,
ultrasonically cleaned in a 2 % (v/v) RBS-35 surfactant solution, rinsed with
ethanol,
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
42
thoroughly rinsed with MilliQTM water, and dried under purified nitrogen. Si
wafer
pieces were further cleaned by UV/ozone treatment in a ProCleanerTM instrument
(Bioforce Nanoscience, USA) for 60 minutes immediately before use. Onto the Si
wafer pieces was deposited a cross-linked, polymeric thin film with amine
functionality
from allylamine monomer using radio-frequency glow discharge (RFGD)
techniques.
The reaction chamber was completely evacuated to a pressure of < 0.003 mbar
and
then filled with allylamine vapour to a slowly rising pressure of 0.200 mbar.
At this
time, voltage was applied across the electrodes at a frequency of 200 kHz and
load
power of 20 W for a period of 25 seconds. The resultant allylamine coatings
(Si-
ALAPP) were then rinsed in MilliQTM water before further use.
Solutions of AAM were prepared in H20 at a concentration of 10 % (w/v) in a N2
glove
box and purged, with N2, for 60 minutes. AAM solution was then applied to Si-
ALAPP
samples so that the AAM solution was 3 mm deep and sealed against oxygen
ingress
inside a polypropylene bag using a domestic vacuum food storage system
(Sunbeam). The sealed samples were then removed from the N2 glove box and
exposed to UV radiation generated by a high powered UV lamp (Fusion Systems
F5300s with 9 mm D-bulb). In normal operation, the sample is passed under the
lamp
on a conveyor (Fusion UV Systems, Inc. LC6B Benchtop Conveyor) with each pass
resulting in 2849 (UVA), 822 (UVB), 81.5 (UVC), and 2922 (UVV) mJ/cm2 (as
measured with a portable UV meter (EIT UV Power Puck II)). Knowing these
values it
was possible to equalise the UV radiation dose across different processing
protocols.
The protocols tested were 1) intermittent (22 passes on belt), 2) intermittent
+ delay
(22 passes with a 30 second delay between each), and 3) continuous (the sample
fixed in place under the lamp for a time that delivered the equivalent UV
radiation
dose as 22 passes). After the desired UV radiation exposure, the samples were
washed copiously with water and then dried under a filtered purified nitrogen
stream
prior to analysis.
Part B: Characterisation of coatings
XPS analysis of the coatings prepared was carried out and the results obtained
are
presented in Table 11. After RFGD thin film deposition (Si-ALAPP) the
composition of
the sample was as expected for coatings of this sort, being rich in C and N.
After
continuous irradiation of the sample in AAM monomer solution, both the 0 and N
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
43
atomic percentages increased, consistent with a graft polymer coating
comprising
mainly polyacrylamide (compare to PAAM theoretical composition). Irradiation
of the
samples in the presence of AAM monomer solution in an intermittent fashion and
an
intermittent + delay fashion also produced coatings which conformed to the
theoretical expectations for a graft polyacrylamide coating. High resolution C
is
spectra (not shown) confirmed the presence of polyacrylamide coatings in all
three
cases. It is generally not possible to estimate the thickness of coatings with
the XPS
technique unless the thickness is less than the analysis depth of the XPS
technique
(5- 10 nm).
Table 11: Atomic percentages and elemental ratios obtained from XPS analysis
of
graft polymer coatings formed using continuous, intermittent or intermittent +
delay
conditions. Also included for comparison are the analysis results obtained for
the Si-
ALAPP sample and the theoretical composition of polyacrylamide (PAAM).
Sample 0/C
N/C
(at. %) (at. %) (at. %)
PAAM (theoretical) 60.7 19.7 19.7 0.33 0.33
Si-ALAPP 76.8
12.6 10.4 0.16 0.14
Continuous 64.6
18.3 17.1 0.28 0.27
Intermittent 62.3 18.8 16.8 0.30 0.27
Intermittent + delay 60.4 19.6 16.9 0.32 0.28
In order to estimate the thickness of the graft polymer coatings formed using
the three
conditions under investigation, profilometry was used. In this case, coated
samples
were scratched with the tip of a syringe needle to expose the Si wafer
beneath. The
depth of the scratch was then measured with a profilometer (Dektak by Veeco)
for
various scratches / locations and referenced to the Si-ALAPP substrate
surface. The
results of replicate profilometry experiments are presented in Table 12. The
data for
the thickness of the Si-ALAPP sample is not included (typically 25-30 nm
thick).
Thickness data for graft polymer coatings are referenced to the surface of the
Si-
ALAPP substrate.
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
44
Clearly indicated by the data presented in Table 12 was that the dry thickness
of the
coatings prepared increased in the order: continuous < intermittent <
intermittent +
delay. Clearly the dry thickness coating produced using continuous UV
irradiation
was significantly lower than that of the two coatings produced under
intermittent UV
irradiation conditions. Furthermore, the coating made using intermittent +
delay UV
irradiation was significantly thicker than the coating made with intermittent
UV
irradiation.
For cell culture applications, the hydrated thickness of the coatings produced
is of
more relevance than the dry thickness. In order to estimate the hydrated
thickness of
the coatings, an Atomic Force Microscope (AFM) technique was implemented. Here
a silica colloid particle (diameter - 4 m) was glued (Epon 1004, Shell) to
the
cantilever spring to provide a probe of known geometry (i.e. spherical).
Interactions
between the silica colloid and the graft polymeric coatings in phosphate
buffered
saline (PBS) (pH 7.4) solution were then measured as a function of the
separation
distance. As the silica colloid makes contact with the coating, a repulsive
force is
generated, the range of which provides an estimate of the hydrated thickness
of the
coating. Interaction forces were measured using an MFP-3D AFM (Asylum
Research, Santa Barbara, CA) at several locations on the samples which were
mounted inside a fluid cell. The spring constant of the cantilever was
determined
using the resonance method of Cleveland et al. (Cleveland, J. et al., (1993),
Rev. Sci.
Instrum., 64, 403-5) and the radius of the silica particle was determined
using optical
microscopy. The deflection of the cantilever as a function of piezo distance
travelled
was scaled to the force as a function of separation distance using the MFP-3D
software. Reference measurements were made using a non-compressible surface
such as a Si wafer piece and the inverse slope of the deflection in hard
contact was
used to calibrate the photodetector.
Table 12: Results obtained for graft polymeric coatings prepared by
continuous,
intermittent or intermittent + delay conditions. Data were obtained for the
dry
thickness using profilometry. The data for the hydrated thickness was obtained
using
AFM direct force measurements in PBS solution. The swelling ratio was
calculated
from the dry and hydrated thickness values. The modulus of the samples was
obtained from analysis of the interaction force data and comparison to
theoretical
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
predictions calculated using Hertz theory (reference for Hertz theory and the
approach used is: Dimitriadis, E.K., et al. (2002), Biophysical J., 82, 2798-
2810).
Reported results are averages obtained from analysis of three locations on
replicate
samples.
Swelling Modulus
Sample Thickness Thickness
Ratio (Pa)
:==
Continuous 136 38 708 20 5.2 200
Intermittent 231 11 4056 155 17.5
90
Intermittent + delay 302 10 5549 288 18.3
90
5
Analysis of the hydrated thickness data for graft polymeric coatings prepared
using
either continuous, intermittent and intermittent + delay UV irradiation
conditions
presented in Table 12 show clearly that the coating made using continuous UV
irradiation was significantly thinner than the two coatings prepared using
intermittent
10 UV irradiation conditions. Furthermore the coating made using
intermittent + delay UV
irradiation was significantly thicker than the coating made with intermittent
UV
irradiation.
Analysis of the swelling ratio of the three coatings (see Table 13) indicated
that the
coatings made with intermittent UV irradiation were able to swell much more
that the
15 coating made using continuous UV irradiation when hydrated with PBS
solution. This
is most likely due to the degree of cross-linking within the coatings.
Continuous UV
irradiation will lead to four processes; (i) free radical formation, (ii)
chain scission, (iii)
cross-linking reactions and (iv) polymer chain growth. These four processes
will also
occur for graft polymeric coatings prepared using intermittent UV irradiation
but the
20 relative balance of the four processes will most likely be different.
Assuming that the
free radical formation is equal in both cases, intermittent UV irradiation
should lead to
more polymer growth when the sample is not being irradiated with UV than in
the
continuous case and less cross-linking within the coatings. This hypothesis is
borne
out by both the dry and hydrated thickness which were both greater in the case
of
25 samples prepared with intermittent UV irradiation. The reduced swelling
ratio obtained
for the samples prepared using continuous UV irradiation suggest that there
was a
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
46
higher degree of cross-linking within the coatings. Thicker coatings such as
those
prepared using intermittent UV irradiation, with an equal degree of cross-
linking would
have a very similar swelling ratio. The influence of additional time with no
UV
irradiation (intermittent + delay) was to increase the polymeric coating graft
layer
thickness, again suggesting that when the sample was not being irradiated,
more
polymer growth was occurring than for shorter non-irradiation times
(intermittent) and
much more polymer chain growth was occurring than in the continuous UV
irradiation
condition.
Also reported in Table 12 are the modulus values for the three coating
conditions
obtained from analysis of the AFM direct interaction force data and by fitting
of the
force curves with model data generated using Hertz theory. Here it is clear
that the
coatings produced using the two intermittent conditions are slightly softer
than those
prepared using continuous irradiation. This data supports the hypothesis that
there
was less cross-linking within the coatings made using intermittent UV
irradiation.
Example 12
Cellular response to different coating architectures
Part A: Formation of initiator free UV graft copolymer coatings using
intermittent
exposure of UV radiation
As per Example 1, 10 % (w/v) aqueous solutions of different molar ratios (0-
100%) of
the monomers acrylic acid (AA) and acrylamide (AAM) were degassed by purging
with nitrogen for more than 15 min in a glove box (oxygen concentration
<0.1%). The
solutions prepared in this way were then transferred into the wells of 96 well
tissue
culture polystyrene (TCPS) plates (NunclonTM 4, Nunc). The volume of monomer
solution added to each well was 0.07 cm3. While still in the glove box, plates
containing the monomer solutions described above were then vacuum sealed into
polymer bags (Sunbeam FoodSaver) and removed from the glove box. The plates
were then passed under a UV lamp (Fusion Systems F5300s, 9 mm D-bulb) 35 times
on a conveyor belt (Fusion Systems LC6B Benchtop Conveyor) at a speed of
approximately 1.8 m/min. After each pass the plate was rotated 180 degrees to
enable more uniform UV irradiation. The plates were then thoroughly washed
with
running MilliQTM water followed by incubation in a large volume of MilliQTM
water
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
47
over 72 hours, with daily water changes, at room temperature to remove any
remaining monomer or non-covalently bound polymer. Finally the multiwell plate
samples were air dried.
Part B: Formation of UV graft copolymer coatings based on macro-initiators
96 well tissue culture polystyrene plates (NunclonTM 4, Nunc) were introduced
into a
radio frequency glow discharge plasma reactor described elsewhere [Griesser
HJ.,
Vacuum 39 (1989) 485]. Plates were placed onto a rectangular copper electrode
having the same dimensions as the base of the multiwell plate. Deposition of
an
allylamine plasma polymer (ALAPP) thin film was then carried out for 25 s at a
power
of 20 W, a frequency of 200 kHz and an initial monomer pressure of 0.33 mbar.
Subsequently, the in-house synthesised macro-initiator poly(acrylic acid-co-
diethyl-
dithiocarbamic acid 4-vinyl-benzyl ester) (PI) described elsewhere [L.
Meagher, H.
Thissen, P. Pasic, R.A. Evans, G. Johnson, W02008019450-A1] was covalently
immobilised on the amine functionalised multiwell plate surface by incubation
with a
mixture of 90 % (v/v) DMF (Merck) and 10 % (v/v) MilliQTM water containing 3.8
mg/m L N-(3-dimethylaminopropyI)-N'-ethylcarbodiimide hydrochloride (EDC)
(Sigma)
at room temperature over 2 hours at a PI concentration of 1 % (w/v). Plates
were then
washed 3 times with a mixture of 90 % (v/v) DMF (Merck) and 10 % (v/v)
MilliQTM
water and 3 times with MilliQTM before drying in air.
10 % (w/v) aqueous solutions containing different molar ratios (0-100 %) of
acrylic
acid (AA) and acrylamide (AAM) monomers were degassed by purging with nitrogen
for more than 15 min in a glove box (oxygen concentration <0.1 %). The
solutions
prepared in this way were then transferred into the wells of PI modified ALAPP
treated 96 well tissue culture polystyrene plates. The volume of monomer
solution
added to each well was 0.20 cm3. While still in the glove box, plates
containing the
monomer solutions described above were then vacuum sealed into polymer bags
(Sunbeam FoodSaver) and removed from the glove box. The plates were then
placed
under a UV lamp (Spectroline, model XX-15A) and irradiated continuously at an
intensity of 10 mW/cm2 in a custom-built box for 6 hours to achieve
polymerisation.
The plates were then thoroughly washed at least 3 times with MilliQTM water
followed
by incubation in a large volume of MilliQTM water over 72 hours at room
temperature
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
48
to remove any remaining monomer or non-covalently bound polymer. Finally the
multiwell plate samples were air dried.
Part C: Characterisation of coating composition and the cellular response
X-ray photoelectron spectroscopy (XPS) analysis was carried out on homo- and
copolymer coatings prepared using AA and AAM solutions on TCPS (Part A) or
TCPS-ALAPP-PI (Part B) substrates, respectively using the two different UV
based
coating methods. The results obtained are presented in Table 13 and 14.
Analysis of
the results demonstrated that a coating was successfully grown from the
substrates in
all cases. The 0/C and N/C elemental ratios observed on coatings produced by
the
initiator-free intermittent UV coating method (Part A) were close to the
expected
theoretical values for homopolymeric or copolymeric coatings derived from the
specified monomer solutions, providing evidence that the molar ratio of AA and
AAM
in the coatings were similar to the molar ratio in the monomer feed solutions.
A clear
trend was also observed for both the 0/C and N/C ratios. The same trend was
observed on coatings prepared by the macro-initiator-based UV method (Part B).
However, in the latter case the observed elemental ratios were somewhat
different
from the expected theoretical values due to the reduced coating thickness
achieved
with this method, which resulted in coatings that had a dry thickness less
than the
probe depth of the XPS technique (i.e. some of the data contained a
contribution from
both the coating and from the substrate).
Table 13: Average elemental ratios obtained from XPS analysis of graft homo-
and
copolymer coatings prepared on TCPS substrates using AA and AAM solutions of
varying composition. Coatings were formed using initiator-free intermittent UV
graft
polymerisation (Part A).
Mo I /0 AA 0/C N/C
0 0.299 0.281
5 0.301 0.256
10 0.339 0.230
15 0.341 0.224
20 0.366 0.212
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
49
25 0.396 0.201
30 0.399 0.187
40 0.414 0.170
50 0.505 0.131
55 0.455 0.134
60 0.453 0.140
70 0.510 0.076
85 0.562 0.045
100 0.566 0.033
Table 14: Average elemental ratios obtained from XPS analysis of graft homo-
and
copolymer coatings prepared on TCPS-ALAPP-PI substrates using AA and AAM feed
solutions of varying composition. Coatings were formed using macro-initiator-
based
UV graft polymerisation (Part B).
Sample 0/C N/C
ALAPP 0.152 0.112
ALAPP-PI 0.157 0.099
0 mol % AA 0.240 0.241
5 mol % AA 0.278 0.241
10 mol % AA 0.291 0.224
20 mol % AA 0.333 0.203
50 mol % AA 0.302 0.092
100 mol % AA 0.332 0.058
The attachment of cells to coatings was evaluated using either HeLa cells,
human
mesenchymal stem cells (hMSC) or L929 mouse fibroblasts. Cell culture
experiments
were carried out using surface modified 96 well tissue culture polystyrene
plates as
well as 96 well tissue culture polystyrene control plates (NunclonTM 4, Nunc).
Samples prepared using the initiator-free intermittent UV coating method were
sterilised by gamma irradiation using a dose of 15 kGy (Steritech). Samples
prepared
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
using the macro-initiator-based UV coating method were sterilised immediately
before
cell culture by incubation with a solution of phosphate buffered saline (PBS)
containing penicillin and streptomycin at concentrations of 120 and 200
pg/cm3,
respectively over 4 hours at room temperature.
5 HeLa cell attachment was assessed at a seeding density of 2x104
cells/well in fresh
Dulbecco's modified Eagle's medium (DMEM)/Hams F12 medium supplemented with
10% foetal bovine serum (FBS), penicillin, streptomycin and glutamine.
Human mesenchymal stem cell (hMSC) attachment was assessed at a seeding
density of 7875 cells/well in Mesenculte-XF medium (StemcellTM Technologies).
10 L929 cell attachment was assessed at a seeding density of 7875
cells/well in MEM +
GlutaMAXTm-I medium (Gibco) supplemented with 10 % FBS, 1 % v/v non-essential
amino acids, and 1 % v/v Anti-Anti.
For each cell type, plates were incubated for 24 hours at 37 C in humidified
air
containing 5 % CO2
15 In the case of HeLa cells, the quantification of cell attachment was
carried out by
washing of the wells with 200 pL of culture medium to remove suspended and
loosely
bound cells after 24 hours incubation. (3-(4,5-dimethylthiazol-2-y1)-2, 5-
diphenyltetrazolium bromide) (MTT) in DMEM/Hams F12 solution was then added to
each well and plates incubated for 4 hours at 37 C. The medium was removed
from
20 each well and replaced with DMSO (100 pL/well). Plates were agitated
gently to
dissolve the stain for 15 minutes on a plate shaker prior to colorimetric
measurement
of cell viability at a wavelength of 595 nm. The absorbance values measured
from the
test samples were expressed as a percentage of those measured in tissue
culture
polystyrene (TCPS) control well.
25 In the case of hMSC cells, the quantification of cell attachment was
carried out by
washing of the wells with 200 pL of culture medium to remove suspended and
loosely
bound cells after 24 hours incubation. 100 pL [3-(4,5-dimethylthiazol-2-y1)-5-
(3-
carboxymethoxypheny1)-2-(4-sulfopheny1)-2H-tetrazolium (MTS) in culture medium
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
51
was then added to each well and plates incubated for 3 hours at 37 C and 5 %
CO2.
Results were read with a microplate reader (BioTek) at 490 and 655 nm. The
difference between the readings from both wavelengths were obtained and
averaged.
The data was then normalised by comparison to the reading obtained from the
TCPS
surface.
In the case of L929 cells, the quantification of cell attachment was carried
out by
washing of the wells with 200 pL of culture medium to remove suspended and
loosely
bound cells after 24 hours incubation. 100 pL [3-(4,5-dimethylthiazol-2-y1)-5-
(3-
carboxymethoxypheny1)-2-(4-sulfopheny1)-2H-tetrazolium (MTS) in MEM medium
(Gibco) containing 10 % FBS and minimal essential amino acids was then added
to
each well and plates incubated for 3 hours at 37 C and 5 % CO2. Results were
read
with a microplate reader (BioTek) at wavelengths of 490 and 655 nm. The
differences
between the readings at both wavelengths were obtained and averaged. The data
was then normalised by comparison to the reading obtained from the TCPS
surface.
Figure 9 shows cell attachment results in response to coatings prepared using
two
different UV methods as a function of the composition of the polymeric
coating. The
coatings were either homopolymers or copolymers formed from defined molar
ratios
of acrylic acid (AA) and acrylamide (AAM). For initiator-free UV based
coatings
produced by intermittent UV exposure (Part A), hMSC attachment was effectively
reduced to levels below 10 % of the cell attachment obtained on TCPS for molar
percentages of AA up to 55 %. In comparison, for coatings prepared using the
macro-
initiator based UV approach (Part B), HeLa cell attachment was only
effectively
reduced to levels below 10% of the value obtained on TCPS for molar
percentages of
AA which were below 10%. Lines are drawn to guide the eye (-13).
Analysis of the data presented in Figure 9 clearly suggests that, for the same
molar
ratio of AA and AAM in copolymer coatings present on a polystyrene substrate
surface, different cell attachment responses were observed depending on the
coating
method. For the initiator-free UV based coatings produced by intermittent UV
irradiation (described in Part A), cell attachment can be effectively reduced
to levels
below 10% of the TCPS value for molar percentages of AA up to 55%. In
comparison,
for coatings prepared using the macro-initiator based UV approach, cell
attachment
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
52
was onlyeffectively reduced to levels below 10% of the TCPS value for molar
percentages of AA below 10%. These results were obtained using human
mesenchymal stem cells (hMSC) and HeLa cells, respectively. Furthermore,
analysis
of the data presented in Figure 10 clearly shows that similar cell attachment
results
were obtained with different cell types (with hMSCs and L929 cells) on the
same
coating. This result was obtained over the whole range of AA/AAM molar
compositions for coatings, produced by the initiator-free UV based coating
method
using intermittent UV. The results clearly demonstrate that similar cell
attachment
results were achieved. For both cell types, cell attachment was effectively
reduced to
levels below 10% of TCPS for molar percentages of AA of up to 55%. Lines are
drawn to guide the eye.
For comparison, analysis of the data presented in Example 13 shows clearly
that for a
40 mol% AA-co-AAM surface prepared using continuous irradiation, the same low
L929 adhesion was observed.
Figure 10 shows the response of different cell types to copolymer coatings
based on
acrylic acid (AA) and acrylamide (AAM). Coatings were produced by the
initiator-free
UV based coating method using intermittent UV. Similar cell attachment results
were
obtained using hMSCs and L929 cells. For both cell types, cell attachment was
effectively reduced to levels below 10% of TCPS for molar percentages of AA of
up to
55%. Lines are drawn to guide the eye (-13).
Overall the data in Figure 9 and 10 clearly support the hypothesis that
different
coating architectures were produced by the two very different UV based
polymerisation methods, and that this difference was responsible for the
observed
differences in the cellular response (i.e. cell attachment). Furthermore, the
data
clearly demonstrate that coatings produced by the initiator-free UV based
coating
method were more effective at preventing cell attachment of different cell
types over a
much wider range of molar ratios of AA and AAM compositions. Due to the fact
that
cell attachment was enabled by the non-specific adsorption of serum proteins,
it can
be concluded that the initiator-free UV based coating method is more effective
at
preventing non-specific serum protein adsorption over a much wider range of AA
and
AAM molar ratios.
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
53
Example 13
Polymer grafting: continuous versus intermittent UV irradiation
Part A: Preparation of coatings
Silicon wafers (Si) were cut into squares of 7 x 7 mm dimension,
ultrasonically
cleaned in a 2 % (v/v) RBS-35 surfactant, 2 % (v/v) ethanol, solution,
thoroughly
rinsed with MilliQTM water, and dried with a high velocity, filtered stream of
purified
nitrogen gas. Si wafer pieces were further cleaned by UV/ozone treatment in a
ProCleanerTM instrument (Bioforce Nanoscience, USA) for 60 minutes immediately
before use. Silicon Wafer samples were then were then introduced into a radio
frequency glow discharge plasma reactor described elsewhere [Griesser HJ.,
Vacuum
39 (1989) 485]. Samples were placed onto a round lower copper electrode having
the
same dimensions as the top electrode. Deposition of an allylamine plasma
polymer
(ALAPP) thin film was then carried out for 25 s at a power of 20 W, a
frequency of 200
kHz and an initial monomer pressure of 0.20 mbar. The resulting allylamine
coatings
(Si-ALAPP) were left in air until further use.
A 7.5% (w/v) aqueous solution containing 40 mol% acrylic acid (AA) and 60 mol%
acrylamide (AAM) was prepared in a nitrogen glove box where it was also
transferred
into PTFE vessels containing Si-ALAPP samples. The volume of monomer solution
added to each vessel was 4 mL. While still in the glove box, vessels
containing the
monomer solutions were then vacuum sealed into polymer bags (Sunbeam
FoodSaver) and removed from the glove box. The sealed samples were then
exposed to UV radiation generated by a high powered UV lamp (Fusion UV Systems
LH6 with 9 mm D-bulb). In normal operation, the sample was positioned under
the
lamp on a fixed stage and irradiated. A pneumatic shutter was programmed to
open
and close, such that defined "on" periods of exposure and defined "off"
periods of
non-exposure could be set. A portable UV meter (E IT UV Power Puck II)) was
used to
determine the total energy and irradiance reaching the samples under any given
settings. Knowing these values, it was then possible to determine equal UV
radiation
doses across different processing protocols.
The different processing protocols tested are shown in Table 15.
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
54
Table 15
Sample
OW (s) OFF6 (s) cycles UVC (J/cm2)
Continuous 1 15 n/a 1
0.58
Continuous 2 30 n/a 1
0.99
Continuous 3 45 n/a 1
1.41
Continuous 4 60 n/a 1
1.83
Continuous 5 75 n/a 1
2.25
Continuous 6 90 n/a 1
2.67
Continuous 7 120 n/a 1
3.51
Intermittent (Continuous 6) - 20 OFF - 1 1 20 53 2.67*
Intermittent (Continuous 6) - 20 OFF ¨ 2 1.9 20 35 2.67*
Intermittent (Continuous 2) - 20 OFF ¨ 1 1 20 19 0.99*
Intermittent (Continuous 2) - 20 OFF ¨ 2 2.9 20 9 0.99*
Intermittent (Continuous 2) - 20 OFF ¨ 3 4.7 20 6 0.99*
Intermittent (Continuous 2) - 20 OFF ¨ 4 10 20 3 0.99*
Intermittent (Continuous 2) - 20 OFF ¨ 5 15 20 2 0.99*
Intermittent (Continuous 2) - 1 OFF - 1 1 1 19 0.99*
Intermittent (Continuous 2) - 10 OFF¨ 1 1 10 19 0.99*
Intermittent (Continuous 2) - 30 OFF ¨ 1 1 30 19 0.99*
Intermittent (Continuous 2) - 60 OFF ¨ 1 1 60 19 0.99*
Intermittent (Continuous 2) - 1 OFF - 3 4.7 1 6 0.99*
Intermittent (Continuous 2) - 40 OFF ¨ 3 4.7 40 6 0.99*
Intermittent (Continuous 2) - 60 OFF ¨ 3 4.7 60 6 0.99*
= *Target values.
= &Values represent settings used on the equipment.
After the desired UV radiation exposure, the samples were washed copiously
with
water and then dried under a filtered purified nitrogen stream prior to
analysis.
CA 02877757 2014-12-23
WO 2014/000052 PCT/AU2013/000710
Part B: Characterisation of coatings
XPS analysis of the coatings prepared was carried out and the results obtained
are
presented in Table 16.
Table 16: Atomic percentages and elemental ratios obtained from XPS analysis
of
5 graft polymer coatings formed using continuous and intermittent processing
conditions.
0 N C
Sample
0/C N/C
(at. %) (at. %) (at. %)
Si-ALAPP (series 1) 17.5 8.5 73.4 0.24
0.12
Continuous 1 26.2 9.1 63.5 0.41
0.14
Continuous 2 26.4 9.2 63.5 0.42
0.14
Continuous 3 27.4 9.1 62.4 0.44
0.15
Continuous 4 27.3 9.1 62.2 0.44
0.15
Continuous 5 27.4 9.0 62.3 0.44
0.14
Continuous 6 27.2 9.0 62.7 0.43
0.14
Continuous 7 25.0 10.9 63.1 0.40
0.17
Si-ALAPP (series 2) 17.6 8.5 73.0 0.24
0.12
Intermittent (Continuous 6) - 20 OFF - 1 27.2 9.3 62.3 0.44
0.15
Intermittent (Continuous 6) - 20 OFF - 2 27.6 9.3 61.8 0.45
0.15
Intermittent (Continuous 2) - 20 OFF - 1 27.6 9.2 61.9 0.45
0.15
Intermittent (Continuous 2) - 20 OFF - 2 27.7 9.1 61.9 0.45
0.15
Intermittent (Continuous 2) - 20 OFF - 3 27.5 9.2 61.9 0.44
0.15
Intermittent (Continuous 2) - 20 OFF - 4 27.8 9.2 61.7 0.45
0.15
Intermittent (Continuous 2) - 20 OFF - 5 27.3 9.1 62.3 0.44
0.15
Si-ALAPP (series 3) 16.0 12.4 71.3 0.22
0.17
Intermittent (Continuous 2) - 1 OFF - 1 27.6 9.9 62.2 0.44
0.16
Intermittent (Continuous 2) - 10 OFF - 1 27.3 9.8 62.7 0.43
0.16
Intermittent (Continuous 2) - 30 OFF - 1 27.3 9.5 62.9 0.43
0.15
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
56
Intermittent (Continuous 2) - 60 OFF - 1 27.5 9.6 62.6 0.44
0.15
Intermittent (Continuous 2) - 1 OFF - 3 27.6 9.8 62.4 0.44
0.16
Intermittent (Continuous 2) - 40 OFF - 3 26.6 9.9 63.3 0.42
0.16
Intermittent (Continuous 2) - 60 OFF - 3 27.1 9.7 62.9 0.43
0.15
After allylamine plasma polymer thin film deposition (Si-ALAPP), the
composition was
as expected. After continuous irradiation of the sample in monomer solution,
both the
0 and N atomic percentages increased, consistent with a graft copolymer
coating
consisting of acrylamide and acrylic acid.
Ellipsometry was used to estimate the thickness of the graft polymer coatings
formed
(JA Woolam Co, M2000). Phase data were collected at 4 angles (60, 65, 70, and
75
degrees) for 20 seconds at each angle. The data was fitted using a Tauc-
Lorentz
general oscillator model. The results of ellipsometry experiments are
presented in
Table 17.
Table 17: Thickness data derived from ellipsometric analysis of graft polymer
coatings formed using continuous and intermittent conditions.
Sample Thickness* (nm)
Si-ALAPP (series 1) 29.04
Continuous 1 15.2
Continuous 2 33.81
Continuous 3 63.21
Continuous 4 95.64
Continuous 5 115.93
Continuous 6 151.98
Continuous 7 NM
Si-ALAPP (series 2) 29.52
Intermittent (Continuous 6) - 20 OFF - 1 212.8
Intermittent (Continuous 6) - 20 OFF - 2 196.44
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
57
Intermittent (Continuous 2) - 20 OFF - 1 28.68
Intermittent (Continuous 2) - 20 OFF - 2 22.61
Intermittent (Continuous 2) - 20 OFF - 3 37.17
Intermittent (Continuous 2) - 20 OFF - 4 38.33
Intermittent (Continuous 2) - 20 OFF - 5 37.59
Si-ALAPP (series 3) 32.9
Intermittent (Continuous 2) - 1 OFF - 1 122.09
Intermittent (Continuous 2) - 10 OFF - 1 66.66
Intermittent (Continuous 2) - 30 OFF - 1 96.97
Intermittent (Continuous 2) - 60 OFF - 1 115.53
Intermittent (Continuous 2) - 1 OFF - 3 108.95
Intermittent (Continuous 2) - 40 OFF - 3 134.79
Intermittent (Continuous 2) - 60 OFF - 3 127.83
= NM = not measured because of insufficient coating quality
= *thickness of layer (i.e. not including underlying ALAPP layer)
Sample 'Continuous 7' could not be measured. On some replicate samples, the
ALAPP had delaminated from the Si substrate owing to elevated temperature and
degradation from the long continuous exposure times. On other replicate
samples, the
coating that had formed was very non-homogenous.
Sample 'Continuous 6' appeared to be on the verge of delamination, and in a
few
areas of some replicate samples delamination occurred. However, the coating
thickness was able to be measured. Comparing sample 'Continuous 6' and samples
'Intermittent (Continuous 6) -20 OFF ¨ 1' and 'Intermittent (Continuous 6) -20
OFF ¨
2', where all three samples received the same UVC radiation dose of 0.99
J/cm2, it is
evident that moving from a continuous to an intermitted exposure regime has
lead to
both a) an ability to prepare coatings without delamination of the ALAPP from
the Si,
and b) thicker coatings. The increase in thickness obtained in this series of
samples
with varying intermittent exposure compared to continuous exposure is
consistent
with the dataset presented in Example 11. In Example 11, intermittent UV
exposure
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
58
resulted in thicker coatings which swelled in PBS to a higher degree and had a
lower
modulus. It is reasonable to assume that the same trends would be present in
the
dataset presented here.
One of the clear trends apparent in the dataset is that when the "on" time is
varied but
the UVC is kept constant, the coating thickness obtained is similar (and
generally
thicker than the thickness obtained with continuous irradiation). Analysis of
this data
allows the conclusion that the total UVC dose is very important in determining
the
thickness of the coating.
There are also some clear trends which may be observed when considering the
impact of the "off" time during sample preparation. Example 11 demonstrated
that a
delay after irradiation resulted in thicker coatings. We concluded that this
was due to
continued polymerisation after the samples were removed from the UV lamp and
that
delays before being placed under the UV lamp again allowed for additional
polymerisation and a thicker coating. Here we can see that when the "off" time
was
increased (equivalent to an increased delay time), a greater coating thickness
was
obtained. For example an "off" time increase of 10 to 60 s (for samples:
Intermittent
(Continuous 2) ¨ 10 OFF ¨ 1, Intermittent (Continuous 2 ¨ 30 OFF ¨ 1, and
Intermittent (Continuous 2) - 60 OFF ¨ 1), the thickness increased from 67 to
116 nm.
Thus where the "on" time is considered, it is the total UVC exposure which
contributes
to increased thickness. In the case of the "off" time, the length of the "off"
time which
contributes to increased thickness.
The culture of L929 fibroblasts with a covalently immobilised, cell adherent
cyclic
RGDfK peptide were also carried out using a protocol similar to that in
previous
Examples and compared to controls (no attached peptide). In all cases, the
cells
attached and spread well on the coatings. No significant differences were
noted in the
cell number, cell circularity or the area occupied by the cells. Control
surfaces
prepared by continuous irradiation also resisted cellular adhesion.
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
59
Example 14
Polymer grafting: intermittent UV irradiation of increasing cycles
Part A: Preparation of coatings
A 7.5% (w/v) aqueous solution containing 40 mol% acrylic acid (AA) and 60 mol%
acrylamide (AAM) was prepared in a nitrogen glove box where it was also
transferred
into the wells of 48- well tissue culture polystyrene plates containing Si-
ALAPP
samples as per Example 13. The volume of monomer solution added to each well
was 227 uL. While still in the glove box, plates containing the monomer
solutions
were then vacuum sealed into polymer bags (Sunbeam FoodSaver) and removed
from the glove box. The plates were then passed under a UV lamp (Fusion UV
Systems LH6, 9 mm D-bulb) for 5, 15, 25, 35, or 45 times on a conveyor belt
(Fusion
UV Systems DRS 10/12 Conveyor). The wafers were then removed from the
monomer solution and thoroughly washed at least 3 times with MilliQTM water
followed by incubation in a large volume of MilliQTM water over 72 hours at
room
temperature to remove any remaining monomer or non-covalently bound polymer.
Finally the samples were air dried.
Part B: Characterisation of coatings
XPS analysis of the coatings prepared was carried out and the results obtained
are
presented in Figure 11. As the number of passes increases, the composition
determined changed from that of ALAPP towards the theoretical composition of a
40
mol% poly(AA-co-AAM) copolymer, indicating an increasing thickness of the
coating
to one greater than the XPS sampling depth of approximately 10 nm. The
observed
deviation from the theoretical composition at higher UV passes may be due to
the
kinetics of the co-polymerisation.
Ellipsometry was used to estimate the thickness of the graft polymer coatings
formed
(JA Woolam Co, M2000). Phase data were collected at 4 angles (60, 65, 70, and
75
degrees) for 20 seconds at each angle. The data was fitted using a Tauc-
Lorentz
general oscillator model. The results of ellipsometry experiments are
presented in
Figure 12.
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
The culture of L929 fibroblasts with a covalently immobilised, cell adherent
cyclic
RGDfK peptide were also carried out using a protocol similar to that in
previous
Examples and compared to controls (no attached peptide). In all cases, the
cells
attached and spread well on the coatings. No significant differences were
noted in the
5 cell number, cell circularity or the area occupied by the cells. The one
exception was
for the sample prepared at 5 UV passes, where cell adhesion was observed
presumably because the cell was sensing the underlying ALAPP.
Example 15
Polymer grafting: effect of blocking UVB and/or UVC
10 Part A: Preparation of coatings
Silicon wafers (Si) were cut into squares of 7 x 7 mm dimension,
ultrasonically
cleaned in a 2 % (v/v) RBS-35 surfactant, 2 % (v/v) ethanol, solution,
thoroughly
rinsed with MilliQTM water, and dried under purified nitrogen. Si wafer pieces
were
further cleaned by UV/ozone treatment in a ProCleanerTM instrument (Bioforce
15 Nanoscience, USA) for 60 minutes immediately before use. Silicon Wafer
samples
were then were then introduced into a radio frequency glow discharge plasma
reactor
described elsewhere [Griesser HJ., Vacuum 39 (1989) 485]. Samples were placed
onto a round lower copper electrode having the same dimensions as the top
electrode. Deposition of an allylamine plasma polymer (ALAPP) thin film was
then
20 carried out for 25 s at a power of 20 W, a frequency of 200 kHz and an
initial
monomer pressure of 0.20 mbar. The resulting allylamine coatings (Si-ALAPP)
were
then rinsed in MilliQTM water before further use.
A 7.5% (w/v) aqueous solution containing 40 mol% acrylic acid (AA) and 60 mol%
acrylamide (AAM) was prepared in a nitrogen glove box where it was also
transferred
25 into PTFE vessels containing Si-ALAPP samples. The volume of monomer
solution
added to each vessel was 4 mL. While still in the glove box, vessels
containing the
monomer solutions were then vacuum sealed into polymer bags (Sunbeam
FoodSaver) and removed from the glove box. In addition, filters were placed
inside
the polymer bags in some cases in order to attenuate the intensity of UVA, UVB
and
30 UVC on the sample surface. The conditions used specifically were: 100 %
UVA, 100
% UVB and 100 % UVC; 100 % UVA, 30 % UVB and 0 % UVC; and finally 90 %
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
61
UVA, 0 % UVB and 0 % UVC. These values were determined using the Power Puck
intensity measuring device and various filters. The sealed samples were then
removed from the N2 glove box and exposed to UV radiation generated by a high
powered UV lamp (Fusion UV Systems LH6 with 9 mm D-bulb). In normal operation,
the sample was positioned under the lamp on a fixed stage. A pneumatic shutter
was
programmed to open and close, such that defined "on" periods of exposure and
defined "off" periods of non-exposure were achieved. A portable UV meter (EIT
UV
Power Puck II)) was used to determine the total energy and type of radiation
reaching
the samples under any given setting. In each case, the samples were subjected
to 20
cycles of UV exposure where the UV was "on" for 2 s and "off" for 10 s in each
cycle.
Part B: Characterisation of coatings
XPS results obtained on the coatings are presented in Table 18, where it may
be
observed that the compositions of all three coatings obtained were very
similar, both
in terms of atomic percentages, but also in terms of the elemental ratios 0/C
and N/C.
Shown in Figure xxx are representative high resolution C is spectra. The shape
for
the three spectra obtained for each of the samples analysed were also very
similar,
suggesting that not only the compositions were similar but also the relative
proportions of carbon based functional groups. Analysis of the XPS data
suggested
that grafting occurred in the presence of all three types of UV light
investigated, i.e.
UVA, UVB and UVC and that all coatings produced had a thickness that was
greater
than the XPS analysis depth of approximately 10 nm.
Table 18: Atomic percentages and elemental ratios obtained from XPS analysis
of
graft polymer coatings formed using intermittent conditions with varying
percentages
of UVA, UVB and UVC.
0
SAMPLE
0/C N/C
(at. %) (at. %) (at. %)
Sample 2 (100 % UVA, 100% UVB, 100% UVC) 27.7 9.0 60.9
0.45 0.15
Sample 3 (90 % UVA, 0% UVB, 0% UVC) 26.8 9.2 63.4
0.42 0.14
Sample 4 (100 % UVA, 30% UVB, 0% UVC) 27.5 9.4 62.2
0.44 0.15
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
62
Whilst the coatings made with the varying percentages of UVA, UVB and UVC were
compositionally similar, the thickness varied in the order 100 % UVA, 100 %
UVB and
100 % UVC was thicker than 100 % UVA, 30 % UVB and 0 % UVC was thicker than
90 % UVA, 0 % UVB and 0 % UVC.
Example 16
Polymer grafting: argon atmosphere
Part A: Preparation of coatings
7.5 % (w/v) aqueous solutions of different molar ratios (0-100%) of the
monomers
acrylic acid (AA) and acrylamide (AAM) were degassed by three cycles of freeze-
pump-thaw in a air-tight vessel and then transferred into an argon-filled
glove box
(oxygen concentration <0.03%). The solutions prepared in this way were then
transferred into the wells of 48 well tissue culture polystyrene (TCPS) plates
(NunclonTM 4, Nunc), with some wells containing Si-ALAPP samples as per
Example
13. The volume of monomer solution added to each well was 172 pL (without
wafer)
and 227 uL (with wafer). While still in the glove box, plates containing the
monomer
solutions described above were then vacuum sealed into polymer bags (Sunbeam
FoodSaver) and removed from the glove box. The plates were then passed under a
UV lamp (Fusion UV Systems LH6, 9 mm D-bulb) 40 times on a conveyor belt
(Fusion UV Systems DRS 10/12 Conveyor). The plates and wafers were then
thoroughly washed with MilliQTM water followed by incubation in a large volume
of
MilliQTM water over 72 hours, with daily water changes, at room temperature to
remove any remaining monomer or non-covalently bound polymer. Finally the
multiwell plate samples and wafers were air dried.
Part B: Characterisation of coatings
XPS analysis of the coatings prepared was carried out and the results obtained
are
presented in Table 19. Analysis of the data obtained from the XPS analysis
suggested that the coating compositions were as expected. For example, the
atomic
percentage of nitrogen in the coating decreased as the mole percentage of AAM
monomer in the feed solution was decreased. In parallel with a decrease in the
nitrogen content, an increase in the oxygen content was observed as the mole
percentage of AA in the monomer feed was increased.
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
63
Table 19: Atomic percentages and elemental ratios obtained from XPS analysis
of
graft polymer coatings formed using intermittent conditions in an Argon
atmosphere.
SAMPLE 0
(at. %) (at. %) (at. %)
0/C N/C
Si-ALAPP
15.6 12.6 71.8 0.22 0.18
0 mol% AA-co-AAM (ie AAM) 16.6 19.3 64.1
0.26 0.30
mol% AA-co-AAM 18.8 17.0 64.1
0.29 0.27
mol% AA-co-AAM 21.2 14.7 64.1
0.33 0.23
mol% AA-co-AAM 23.3 12.4 64.3
0.36 0.19
mol% AA-co-AAM 25.9 10.2 63.8
0.41 0.16
mol% AA-co-AAM 27.5 8.8 63.8
0.43 0.14
mol% AA-co-AAM 27.8 8.2 63.9
0.44 0.13
mol% AA-co-AAM 31.1 5.2 63.8
0.49 0.08
mol% AA-co-AAM 32.4 3.7 63.8
0.51 0.06
mol% AA-co-AAM 34.1 2.0 63.5
0.54 0.03
100 mol% AA-co-AAM (ie AA) 35.7 0.5 63.8
0.56 0.01
In order to estimate the thickness of the graft polymer coatings formed
ellipsometry
5 was used. Phase data were collected at 4 angles (60, 65, 70, and 75
degrees) for 20
seconds each angle. The data was fitted using a Tauc-Lorentz general
oscillator
model. The results of ellipsometry experiments are presented in Table 20.
Analysis of
the data obtained indicated that small differences in thickness were apparent
which
depended on the composition of the monomer feed (i.e. the mole percentage of
AA
10 and AAM monomers) and that combinations of AA and AAM above 20 mol % AA
gave the thickest coatings while the homopolymer coatings appeared to be
slightly
thinner.
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
64
Table 20. Thickness data derived from ellipsometric analysis of graft polymer
coatings formed using intermittent conditions in an Argon atmosphere
SAMPLE Thickness* (nm)
Si-ALAPP 27.5
0 mol% AA-co-AAM (i.e. AAM) 125.2
mol% AA-co-AAM 178.9
mol% AA-co-AAM 200.0
mol% AA-co-AAM 231.4
mol% AA-co-AAM 248.0
mol% AA-co-AAM 221.3
mol% AA-co-AAM 228.4
mol% AA-co-AAM 228.2
mol% AA-co-AAM 243.5
mol% AA-co-AAM 215.2
100 mol% AA-co-AAM (ie AA) 206.6
= *thickness of layer in question (i.e. not including underlying ALAPP
layer)
5 This data set clearly demonstrates that the coatings can be prepared in
the presence
of argon as well as nitrogen gas.
Example 17
Polymer grafting using monomers from a wide range of chemical classes
Part A: Preparation of coatings
10 Aqueous solutions or aqueous solutions containing up to 50 % DMSO (v/v)
were
prepared using the monomers listed in Table 21 at concentrations ranging from
0.4 to
1.0 M were prepared in a glove box under a nitrogen atmosphere. Small volumes
(40
¨ 300 uL) of the solutions were added to the wells of multiwall plates (96
well). At
least one row of empty wells was left between rows of wells containing monomer
CA 02877757 2014-12-23
WO 2014/000052
PCT/AU2013/000710
solution to avoid cross-contamination of the polymer coatings. While still in
the glove
box, plates containing the monomer solutions were then vacuum sealed into
polymer
bags (Sunbeam FoodSaver) and removed from the glove box. The plates were then
passed under a UV lamp (Fusion UV Systems LH6, 9 mm D-bulb) for up to 60 times
5 on a conveyor belt (Fusion UV Systems DRS 10/12 Conveyor). The plates
were then
thoroughly washed at least 3 times using the solution in which the polymer
coating
was formed, followed by at least 3 washes with MilliQTM water followed by
incubation
in a large volume of MilliQTM water over 72 hours at room temperature to
remove any
remaining monomer or non-covalently bound polymer. After a further and final
10 washing, the samples were air dried, double bagged and sterilised using
gamma
irradiation at a dose of 15 KGy prior to characterisation using X-ray
photoelectron
spectroscopy (XPS).
Part B: Characterisation of coatings
XPS analysis of the coatings prepared was carried out and the results obtained
are
15 presented in Table 21. Also included for comparison is a typical surface
composition
of the base substrate, tissue culture treated polystyrene (TCPS). As may be
readily
observed, the surface composition of coatings formed from the monomers listed
in
Table 21 were all distinctly different from that of the TCPS substrate,
suggesting that
a coating was formed in each case. The surface of the TCPS substrate contains
only
20 carbon and oxygen. In some cases, the composition was intermediate
between the
TCPS composition and that of the theoretical composition for the polymer
coating of
interest. In this case the thickness of the coating formed was less than the
XPS
sampling depth and the surface composition contained a contribution from the
underlying substrate. In many cases the surface composition of the coatings
formed
25 on top of the TCPS substrate was very similar to the theoretical
composition,
indicating that the coating was at least as thick as the XPS sampling depth
(10 nm).
Table 21: Elemental composition (atomic %) obtained from XPS analysis of UV
graft
polymer coatings prepared using monomer solutions prepared with the monomers
listed in the left hand column.
CA 02877757 2014-12-23
WO 2014/000052 PCT/AU2013/000710
66
Monomer/Sample Name Composition (atomic %)
C 0 N S Ca Na Cl K Si
TCPS 82.0 18.0
N-isopropylacrylamide 75.6 12.2 12.2
Methyl methacrylate 71.6 27.6
0.8
2-Carboxyethyl acrylate 63.0 37.0
3-Sulfopropyl acrylate, Potassium
49.2 38.5 0.6 7.4 0.9 3.5
salt
Mono-2-(methacryloyloxy)ethyl
64.3 35.7
succinate
N-(3-
Am inopropyl)methacrylam ide 71.2 14.2 13.5 0.6 0.6
hydrochloride
2-Aminoethyl methacrylate
62.3 27.7 8.7 0.3
0.4
hydrochloride
2-Hydroxyethylmethacrylate 67.6 32.4
Poly(ethylene glycol) methyl ether
68.8 31.2
methacrylate [MW 475]
Methyl acrylate 74.8 25.1 0.1
Poly(propylene glycol) methyl
72.2 27.8
ether acrylate [MW202]
4-Hydroxybutyl acrylate 71.8 26.2 2.0
2-Methacryloyloxy ethyl
67.1 32.9
acetoacetate
Acrylic Acid 62.8 37.2
Methacrylic acid 69.6 30.4
Hydroxyethylacrylate succinate 60.7 35.4 3.9
2-Acrylamido-2-methy1-1-
55.7 27.8 6.9 6.1 3.6
propanesulfonic acid
[3-(Methacryloylamino)propyl]
73.3 13.5 10.1 0.7 2.4
trimethylammonium chloride
[3-(Methacryloyloxy)ethy1]-
72.0 19.2 5.8 0.5 2.6
trimethylammonium chloride
2-Hydroxyethylacrylate 64.6 35.4
Poly(ethylene glycol)
67.5 32.5
methacrylate [MW 360]
Poly(ethylene glycol) methyl ether 70.7 29.3
methacrylate [MW 1100]
Poly(ethylene glycol) methyl ether
70.6 29.4
methacrylate [MW 2080]
Poly(ethylene glycol)
68.3 31.8
methacrylate [MW 526]
Acrylamide 64.0 18.6 17.5
1-Viny1-2-pyrrolidone 76.9 12.9 10.2
N,N,-Dimethylacrylamide 72.7 14.1 13.2
CA 02877757 2014-12-23
WO 2014/000052 PCT/AU2013/000710
67
N-[3-(Dimethylamino)propyl]
72.4 16.3 11.0 0.4
methacrylamide
Methacrylamide 70.5 16.7 12.8
(2-Dimethylaminoethyl)
73.2 19.2 7.3
methacrylate
N-(2-Hydroxypropyl)
70.3 19.9 9.8
methacrylamide
[2-
(Methacryloyloxy)ethyl]dimethyl-
63.9 26.8 4.7 4.6
(3-sulfopropyl)ammonium
hydroxide
[3-(Methacryloylamino)propyl]
dimethyl(3-sulfopropyl)ammonium 65.1 21.9 8.7 4.3
hydroxide
4-Acryloylmorpholine 70.2 21.0 8.5
0.5
N-(hydroxymethyl)acrylamide 62.6 24.4 12.8
0.2
N-2-hydroxyethyl acrylamide 64.5 23.6 11.3
0.5
N-Methacryloyl
tris(hydroxymethyl) 64.7 27.1 7.4
0.7
aminomethane
Sulfopropylmethacrylate
54.9 33.4 1.1 6.2 0.9 3.5
Potassium Salt
Diacetone acrylamide 75.3 16.5 8.2
N,N-Diethyl acrylamide 78.2 11.1 10.7
N-Ethyl acrylamide 70.2 16.6 13.2
N-(n-propyl)acrylamide 74.8 13.7 11.5
Hydroxypropyl acrylate 67.4 31.7 0.9
N-tert-Butyl methacrylamide 75.0 18.8 6.2
N-tert-Butyl acrylamide 75.8 15.9 8.3
N-(n-Butyl)methacrylamide 77.9 13.3 8.7