Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02894317 2015-06-15
1 SYSTEMS AND METHODS FOR CLASSIFYING, PRIORITIZING AND INTERPRETING
2 GENETIC VARIANTS AND THERAPIES USING A DEEP NEURAL NETWORK
3 TECHNICAL FIELD
4 [0001] The following relates generally to systems and methods for
classifying, prioritizing and
interpreting genetic variants and therapies using a deep neural network.
6 BACKGROUND
7 [0002] Precision medicine, genetic testing, therapeutic
development and whole genome,
8 exome, gene panel and mini-gene reporter analysis require the ability to
accurately interpret how
9 diverse features encoded in the genome, such as protein binding sites,
RNA secondary
structures, and nucleosome positions, impact processes within cell. Most
existing approaches to
11 identifying disease variants ignore their impact on these genomic
features. Many genome studies
12 are restricted to mutations in exons that either change an amino acid in
a protein or prevent the
13 production of the protein.
14 [0003] Over the past decade, the importance of understanding
regulatory genomic
instructions and not just the protein-coding exons and genes that they control
has been
16 underscored by several observations: While evolution is estimated to
preserve at least 5.5% of
17 the human genome, only 1% accounts for exons within genes; biological
complexity often cannot
18 be accounted for by the number of genes (e.g. balsam poplar trees have
twice as many genes as
19 humans); differences between organisms cannot be accounted for by
differences between their
genes (e.g. less than 1% of human genes are distinct from those of mice and
dogs); increasingly,
21 disease-causing variants have been found outside of exons, indicating
that crucial information is
22 encoded outside of those sequences.
23 [0004] In traditional molecular diagnostics, an example workflow
may be as follows: a blood
24 or tissue sample is obtained from a patient; variants (mutations) are
identified, by either
sequencing the genome, the exome or a gene panel; the variants are
individually examined
26 manually (e.g. by a technician), using literature databases and internet
search engines; a
27 diagnostic report is prepared. Manually examining the variants is costly
and prone to human error,
28 which may lead to incorrect diagnosis and potential patient morbidity.
Automating or
29 semi-automating this step is thus beneficial. Since the number of
possible genetic variants is
large, evaluating them manually is time-consuming, highly dependent on
previous literature, and
31 involves experimental data that has poor coverage and therefore can lead
to high false negative
32 rates, or "variants of unknown significance". The same issues arise in
therapeutic design, where
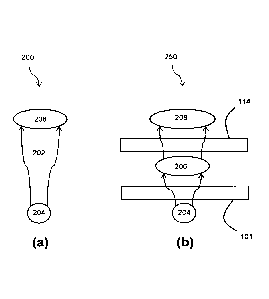
1
CA 02894317 2015-06-15
1 the number of possible therapies (molecules) to be evaluated is extremely
large.
2 [0005] Techniques have been proposed for which predicting
phenotypes (e.g., traits and
3 disease risks) from the genome can be characterized as a problem suitable
for solution by
4 machine learning, and more specifically by supervised machine learning
where inputs are
features extracted from a DNA sequence (genotype), and the outputs are the
phenotypes. Such
6 an approach is shown in Fig. 2(a). A DNA sequence 204 is fed to a
predictor 202 to generate
7 outputs 208, such as disease risks. This approach is unsatisfactory for
most complex phenotypes
8 and diseases for two reasons. First is the sheer complexity of the
relationship between genotype
9 (represented by 204) and phenotype (represented by 208). Even within a
single cell, the genome
directs the state of the cell through many layers of intricate biophysical
processes and control
11 mechanisms that have been shaped by evolution. It is extremely
challenging to infer these
12 regulatory processes by observing only the genome and phenotypes, for
example due to 'butterfly
13 effects'. For many diseases, the annoulit of data necessary would be
cost-prohibitive to acquire
14 with currently available technologies, due to the size of the genome and
the exponential number
of possible ways a disease can be traced to it. Second, even if one could
infer such models (those
16 that are predictive of disease risks), it is likely that the hidden
variables of these models would not
17 correspond to biological mechanisms that can be acted upon, unless
strong priors, such as
18 cause-effect relationships, have been built in. This is important for
the purpose of developing
19 therapies. Insisting on how a model ought to work by using these priors
can hurt model
performance if the priors are inaccurate, which they usually are.
21 [0006] Some other machine learning approaches to genetic analysis
have been proposed.
22 One such approach predicts a cell variable that combines information
across conditions, or
23 tissues. Another describes a shallow, single-layer Bayesian neural
network (BNN), which often
24 relies on methods like Markov Chain Monte Carlo (MCMC) to sample models
from a posterior
distribution, which can be difficult to speed up and scale up to a large
number of hidden variables
26 and a large volume of training data. Furthermore, computation-wise, it
is relatively expensive to
27 get predictions from a BNN, which require computing the average
predictions of many models.
28 SUMMARY
29 [0007] In one aspect, a method for computing variant-induced
changes in one or more
condition-specific cell variables for one or more variants is provided, the
method comprising:
31 computing a set of variant features from a DNA or RNA variant sequence;
applying a deep neural
32 network of at least two layers of processing units to the variant
features to compute one or more
33 condition-specific variant cell variables; computing a set of reference
features from a DNA or RNA
2
CA 02894317 2015-06-15
reference sequence; applying the deep neural network to the reference features
to compute one
2 or more condition-specific reference cell variables; computing a set of
variant-induced changes in
3 the one or more condition-specific cell variables by comparing the one or
more condition-specific
4 reference cell variables to the one or more condition-specific variant
cell variables.
[0008] In another aspect, a deep neural network for computing variant-
induced changes in
6 one or more condition-specific cell variables for one or more variants is
provided, the deep
7 neural network comprising: an input layer configured to receive as input
a set of variant features
8 from a DNA or RNA variant sequence; and at least two layers of processing
units operable to:
9 compute one or more condition-specific variant cell variables; compute a
set of reference features
from a DNA or RNA reference sequence; compute one or more condition-specific
reference cell
11 variables; compute a set of variant-induced changes in the one or more
condition-specific cell
12 variables by comparing the one or more condition-specific reference cell
variables to the one or
13 more condition-specific variant cell variables.
14 [0009] In another aspect, a method for training a deep neural
network to compute one or
more condition-specific cell variables is provided, the method comprising:
establishing a neural
16 network comprising at least two connected layers of processing units;
repeatedly updating one or
17 more parameters of the neural network so as to decrease the error for a
set of training cases
18 chosen randomly or using a predefined pattern, where each training case
comprises features
19 extracted from a DNA or RNA sequence and corresponding targets derived
from measurements
of one or more condition-specific cell variables, until a condition for
convergence is met at which
21 point the parameters are no longer updated.
22 DESCRIPTION OF THE DRAWINGS
23 [0010] The features of the invention will become more apparent in
the following detailed
24 description in which reference is made to the appended drawings wherein:
[0011] Fig. 1 is an system for cell variable prediction;
26 [0012] Fig. 2 shows a comparison of approaches to predict
phenotypes, such as disease
27 risks, from an input;
28 [0013] Fig. 3 shows a method of generating target cell variables
for training;
29 [0014] Fig. 4 shows an example deep neural network architecture
for a cell variable predictor
that predicts splicing levels;
31 [0015] Fig. 5 shows a further example deep neural network
architecture for a cell variable
3
CA 02894317 2015-06-15
1 predictor that predicts splicing levels;
2 [0016] Fig. 6 shows yet a further example deep neural network
architecture for a cell variable
3 predictor that predicts splicing levels;
4 [0017] Fig. 7 shows yet a further example deep neural network
architecture for a cell variable
predictor that predicts splicing levels;
6 [0018] Fig. 8 shows yet a further example deep neural network
architecture for a cell variable
7 predictor that predicts splicing levels;
8 [0019] Fig. 9 shows yet a further example deep neural network
architecture for a cell variable
9 predictor that predicts splicing levels;
[0020] Fig. 10 shows a method for training cell variable predictors;
11 [0021] Fi. 11 shows a system to perform non-uniform sampling of
training cases for
12 determining a mini-batch for training a deep neural network;
13 [0022] Fig. 12 shows a method for- training cell variable
predictors for ensuring a consistent
14 backpropagation signal that updates the weights connected to tissue
inputs and biases learning
towards the event with large tissue variability early on before overfilling
occurs;
16 [0023] Fig. 13 shows a method for using the outputs of the CVP for
scoring, classifying and
17 prioritizing genetic variants;
18 [0024] Fig. 14 shows a method for scoring variants by associating
cell variable changes with
19 those of other variants;
[0025] Fig. 15 shows a method for interpreting which genetic features
account for
21 variant-induced cell variable changes;
22 [0026] Fig. 16 shows a further method for interpreting which
genetic features account for
23 variant-induced cell variable changes;
24 [0027] Fig. 17 shows a further method for interpreting which
genetic features account for
variant-induced cell variable changes;
26 [0028] Fig. 18 shows a method to generate a visualization for
tissue-specific feature
27 importance; and
28 [0029] Figs. 19 shows a detailed illustration of the method to
generate a visualization for
29 tissue-specific feature importance.
4
CA 02894317 2015-06-15
1 DETAILED DESCRIPTION
2 [0030] For simplicity and clarity of illustration, where
considered appropriate, reference
3 numerals may be repeated among the Figures to indicate corresponding or
analogous elements.
4 In addition, numerous specific details are set forth in order to provide
a thorough understanding of
the embodiments described herein. However, it will be understood by those of
ordinary skill in the
6 art that the embodiments described herein may be practised without these
specific details. In
7 other instances, well-known methods, procedures and components have not
been described in
8 detail so as not to obscure the embodiments described herein. Also, the
description is not to be
9 considered as limiting the scope of the embodiments described herein.
[0031] Various terms used throughout the present description may be read
and understood
11 as follows, unless the context indicates otherwise: "or" as used
throughout is inclusive, as though
12 written "and/or"; singular articles and pronouns as used throughout
include their plural forms, and
13 vice versa; similarly, gendered pronouns include their counterpart
pronouns so that pronouns
14 should not be understood as limiting anything described herein to use,
implementation,
performance, etc. by a single gender; "exemplary" should be understood as
"illustrative" or
16 "exemplifying" and not necessarily as "preferred" over other
embodiments. Further definitions for
17 terms may be set out herein; these may apply to prior and subsequent
instances of those terms,
18 as will be understood from a reading of the present description.
19 [0032] Any module, unit, component, server, computer, terminal,
engine or device
exemplified herein that executes instructions may include or otherwise have
access to computer
21 readable media such as storage media, computer storage media, or data
storage devices
22 (removable and/or non-removable) such as, for example, magnetic disks,
optical disks, or tape.
23 Computer storage media may include volatile and non-volatile, removable
and non-removable
24 media implemented in any method or technology for storage of
information, such as computer
readable instructions, data structures, program modules, or other data.
Examples of computer
26 storage media include RAM, ROM, EEPROM, flash memory or other memory
technology,
27 CD-ROM, digital versatile disks (DVD) or other optical storage, magnetic
cassettes, magnetic
28 tape, magnetic disk storage or other magnetic storage devices, or any
other medium which can
29 be used to store the desired information and which can be accessed by an
application, module, or
both. Any such computer storage media may be part of the device or accessible
or connectable
31 thereto. Further, unless the context clearly indicates otherwise, any
processor or controller set out
32 herein may be implemented as a singular processor or as a plurality of
processors. The plurality of
33 processors may be arrayed or distributed, and any processing function
referred to herein may be
5
CA 02894317 2015-06-15
1 carried out by one or by a plurality of processors, even though a single
processor may be
2 exemplified. Any method, application or module herein described may be
implemented using
3 computer readable/executable instructions that may be stored or otherwise
held by such
4 computer readable media and executed by the one or more processors.
[0033] Systems and methods described herein relate, in part, to the problem
of assessing
6 genetic variants with respect to phenotypes, such as deleteriousness for
human diseases. This
7 problem has implications in several industrial categories under the broad
umbrella of
8 'personalized medicine', including molecular diagnostics, whole genome
sequencing, and
9 pharmaceutical development.
[0034] It has been found that the effect of a variant depends on genetic
context, which
11 includes which other variants are present and, more generally, on the
genomic sequence within
12 the individual, or patient, being tested. So, whereas a particular
variant may be benign in one
13 genetic context, it may cause a disease in another genetic context. This
impacts prioritization and
14 interpretation. The following describes a process for context-dependent
genetic variant
assessment and wherein variants may be ranked and presented as a priority
list. Variant
16 prioritization can be used to increase efficiency and accuracy of manual
interpretation, since it
17 enables the technician to focus on a small subset of candidates
18 [0035] Computational procedures for prioritizing and/or
interpreting variants must generalize
19 well. Generalization refers to the ability of the computational
procedure to assess variants that
have not been seen before and that may be involved in a disease that has not
been previously
21 analyzed. A method that generalizes well should even be able to assess
variants within genes
22 that have not been previously analyzed for variants. Finally, a crucial
aspect of enabling
23 computational procedures to operate effectively is computational
efficiency since these
24 procedures may involve aggregating, organizing and sifting through large
amounts of data.
[0036] The systems and methods described herein apply deep learning to
genetic variant
26 analysis. Deep learning generally refers to methods that map data
through multiple levels of
27 abstraction, where higher levels represent more abstract entities. The
goal of deep learning is to
28 provide a fully automatic system for learning complex functions that map
inputs to outputs, without
29 using hand crafted features or rules. One implementation of deep
learning comes in the form of
feedforward neural networks, where levels of abstraction are modeled by
multiple non-linear
31 hidden layers.
32 [0037] In brief, embodiments described herein provide systems and
methods that receive as
6
CA 02894317 2015-06-15
input a DNA or RNA sequence, extracts features, and apply multiple layers of
nonlinear
2 processing units of a cell variable predictor ("CVP") to compute a cell
variable, which corresponds
3 to a measurable quantity within a cell, for different conditions, such as
tissue types. To distinguish
4 a cell variable that corresponds to a measureable quantity for a specific
condition, such as a
tissue type, from a cell variable that is a combination of measureable
quantities from multiple
6 conditions, we refer to the former as a "condition-specific cell
variable" and the latter as a
7 "non-specific cell variable". In embodiments, the CVP is applied to a DNA
or RNA sequence
8 and/or features extracted from the sequences, containing a genetic
variant, and also to a
9 corresponding reference (e.g., wild type) sequence to determine how much
the cell variable
changes because of the variant. The systems and methods can be applied to
naturally occurring
11 genomic sequences, mini-gene reporters, edited genomic sequences, such
as those edited using
12 CRISPR-Cas9, genomic sequences targeted by therapies, and other genomic
sequences. The
13 change in the cell variable in different conditions may be used to
classify disease-causing
14 variants, compute a score for how deleterious a variant is, prioritize
variants for subsequent
processing, interpret the mechanism by which a variant operates, and determine
the effect of a
16 therapy. Further, an unknown variant can be given a high score for
deleteriousness if it induces a
17 change in a particular cell variable that is similar to changes in the
same cell variable that are
18 induced by one or more variants that 0.9 known to be deleterious.
19 [0038] In embodiments, the CVP comprises a deep neural network
having multiple layers of
processing units and possibly millions of parameters. The CVP may be trained
using a dataset of
21 DNA or RNA sequences and corresponding measurements of cell variables,
using a deep
22 learning training method that adjusts the strengths of the connections
between processing units in
23 adjacent layers. Specialized training methods are described, including a
multi-task training
24 method that improves accuracy. The mechanism by which a mutation causes
a deleterious
change in a cell variable may in some instances be determined by identifying
features or groups
26 of features that are changed by the mutation and that cause the cell
variable to change, which can
27 be computed by substituting features derived from the variant sequence
one by one into the
28 reference sequence or by backpropagating the cell variable change back
to the input features.
29 [0039] If a change related to a variant of any cell variable is
large enough compared to a
reference, the variant warrants investigation for deleteriousness. The systems
described herein
31 can thus be used to prioritize genetic variants for further 'wet-lab'
investigations, significantly
32 aiding and reducing the costs of variant discovery. Furthermore, because
of the presence of cell
33 variables in the predictor, the invention can assign 'blame' to variants
that are disease causing,
7
CA 02894317 2015-06-15
1 and generate appropriate user visualizations. For example, a variant that
changes the splicing
2 'cell variable' may be targeted by a therapy that targets the splicing
pathway to remediate the
3 disease
4 [0040] As used herein, the term "reference sequence" means: in the
context of evaluating a
variant (as described below), whereupon the systems described herein compare
the variant to a
6 'reference sequence', the reference sequence is a DNA or RNA sequence
obtained using
7 genome sequencing, exome sequencing or gene sequencing of an unrelated
individual or a
8 closely related individual (e.g., parent, sibling, child). Alternatively,
the reference sequence may
9 be derived from the reference human genome, or it may be an artificially
designed sequence.
[0041] As used herein, the term "variant" means: a DNA or RNA sequence that
differs from a
11 reference sequence in one or more nucleotides, by substitutions,
insertions, deletions or any
12 other changes. The variant sequence may be obtained using genome
sequencing, exome
13 sequencing or gene sequencing of an individual. Alternatively, the
variant sequence may be
14 derived from the reference human genome, or it may be an artificially
designed sequence. For the
purpose of this invention, when a variant is being evaluated by the system,
the sequence
16 containing the variant as well as surrounding DNA or RNA sequence is
included in the 'variant'.
17 [0042] As used herein, the term "single nucleotide variant"
("SNV") means: a variant that
18 consists of a substitution to a single nt leotide.
19 [0043] As used herein, the term "variant analysis" means: the
procedure (computational or
otherwise) of processing a variant, possibly in addition to surrounding DNA or
RNA sequence that
21 establishes context, for the purpose of variant scoring, categorization,
prioritization, and
22 interpretation.
23 [0044] As used herein, the term "score" means: a numeric value
that indicates how
24 deleterious a variant is expected to be.
[0045] As used herein, the term "classification" refers to the
classification of a variant. A
26 variant may be classified in different ways, such as by applying a
threshold to the score to
27 determine if the variant is deleterious or not. The American College of
Medical Genetics
28 recommends a five-way classification: pathogenic (very likely to
contribute to the development of
29 disease); likely pathogenic (there is strong evidence that the variant
is pathogenic, but the
evidence is inconclusive); unknown significance or VUS (there is not enough
evidence to support
31 classification one way or another); likely benign (there is strong
evidence that the variant is
32 benign, but the evidence is inconclusive); benign (very likely to be
benign).
8
CA 02894317 2015-06-15
1 [0046] As used herein, the terms "rank" / "prioritization" mean:
the process of sorting the
2 scores of a set of variants to determine which variant should be further
investigated. The
3 pathogenic variants will be at the top, with the benign variants at the
bottom.
4 [0047] As used herein, the term "cell variable" means: a quantity,
level, potential, or process
outcome in the cell that is potentially relevant to the function of a living
cell, and that is computed
6 by a CVP (see below). There are two types of cell variables: a "condition-
specific cell variable" is a
7 cell variable that is measured or predicted under a specific condition,
such as a tissue type; a
8 "non-specific cell variable" is a cell variable that is derived by
combining information from across
9 multiple conditions, for example by subtracting the average cell variable
values across conditions
from the cell variable for each condition. A cell variable can often be
quantified by a vector of one
11 or more real-valued numbers, or by a probability distribution over such
a vector. Examples include
12 the strength of binding between two molecules (e.g. protein-protein or
protein-DNA binding), exon
13 splicing levels (the fraction of mRNA transcripts in a particular tissue
that contain a particular
14 exon, i.e. percent spliced in), DNA curvature, DNA methylation, RNA
folding interactions.
[0048] As used herein, the term "event" means: in the context of a splicing-
related cell
16 variable (e.g. the fraction of transcripts with an exon spliced in), an
observed (measured)
17 alternative splicing event in the cell where both the genomic features
and the corresponding
18 splicing levels are known for that particular event. Each event can be
used as either a training
19 case or a testing case for a machine learning system.
[0049] Referring now to Fig. 1, shown therein is a system 100 for cell
variable prediction,
21 comprising a machine learning unit. The machine learning unit is
preferably implemented by a
22 deep neural network, which is alternatively referred to herein as a
"cell variable predictor" ("CVP")
23 101. The CVP takes as input a set of features, including genomic
features, and produces an
24 output intended to mimic a specific cell variable. The quantification of
a cell variable can be
represented in such a system by one or more real-valued numbers on an absolute
or relative
26 scale, with or without meaningful units. In embodiments, the CVP may
provide other outputs in
27 addition to outputs intended to mimic a specific cell variable.
28 [0050] The system 100 further comprises a memory 106
communicatively linked to the CVP
29 101.
[0051] An illustrated embodiment of the CVP 101 comprising a feedforward
neural network
31 having a plurality of layers 102 (i.e. deep) is shown. Each layer
comprises one or more processing
32 units 104, each of which implements a feature detector and/or a
computation that maps an input
9
CA 02894317 2015-06-15
1 to an output. The processing units 104 n,ccept a plurality of parameter
inputs from other layers and
2 apply activation functions with associated weights for each such
parameter input to the respective
3 processing unit 104. Generally, the output of a processing unit of layer
I may be provided as input
4 to one or more processing units of layer 1 + 1.
[0052] Each processing unit may be considered as a processing "node" of the
network and
6 one or more nodes may be implemented by processing hardware, such as a
single or multi-core
7 processor and/or graphics processing unit(s) (GPU(s)). Further, it will
be understood that each
8 processing unit may be considered to be associated with a hidden unit or
an input unit of the
9 neural network for a hidden layer or an input layer, respectively. The
use of large (many hidden
variables) and deep (multiple hidden layers) neural networks may improve the
predictive
11 performances of the CVP compared to other systems.
12 [0053] In embodiments, inputs to the input layer of the CVP can
include genetic information,
13 such as sequences representing DNA, RNA, features derived from DNA and
RNA, and features
14 providing extra information (e.g. tissue type, age, sex), while outputs
at the output layer of the
CVP can include cell variables.
16 [0054] It will be appreciated that though an illustrative
feedforward network is described
17 herein, the type of neural network implemented is not limited merely to
feedforward neural
18 networks but can also be applied to any neural networks, including
convolutional neural networks,
19 recurrent neural networks, auto-encoders and Boltzmann machines.
[0055] In embodiments the system 100 comprises a secondary analysis unit
114 for receiving
21 the cell variables from the output layer and providing further analysis,
as described below.
22 [0056] The memory 106 may comprise a database for storing
activations and learned weights
23 for each feature detector, as well as for storing datasets of genetic
information and extra
24 information and optionally for storing outputs from the CVP 101. The
genetic information may
provide a training set comprising training data. The training data may, for
example, be used for
26 training the CVP 101 to predict cell variables, in which case DNA and
RNA sequences with known
27 cell variables and/or phenotypes may be provided. The memory 106 may
further store a validation
28 set comprising validation data.
29 [0057] Generally, during the training stage, the neural network
learns optimized weights for
each processing unit. After learning, the optimized weight configuration can
then be applied to
31 test data. Stochastic gradient descent can be used to train feedforward
neural networks. A
32 learning process (backpropagation), involves for the most part matrix
multiplications, which
CA 02894317 2015-06-15
1 makes them suitable for speed up using GPUs. Furthermore, the dropout
technique may be
2 utilized to prevent overfitting.
3 [0058] The system may further comprise a computing device 110
communicatively linked to
4 the CVP 101 for controlling operations carried out in the CVP. The
computing device may
comprise further input and output devices, such as input peripherals (such as
a computer mouse
6 or keyboard), and/or a display. The computing device 110 may further be
linked to a remote
7 device 112 over a wired or wireless network 108 for transmitting and
receiving data. In
8 embodiments, genetic information is received over the network 108 from
the remote device 112
9 for storage in memory 106. Cell variable predictions and lists of
variants priorities may be
displayed to a user via the display.
11 [0059] Referring now to Fig. 2, shown therein is a comparison of a
prior (Fig. 2(a)) and
12 currently described (Fig. 2(b)) machine learning process to predict
phenotypes, such as disease
13 risks or deleteriousness from a genotype. Contrary to the prior
approach, which was described
14 above, the currently described process predicts a cell variable as an
intermediate to the
phenotype. As described above, the inputs 204 to a CVP can include sequences
representing
16 DNA, RNA, features derived from DNA and RNA, and features providing
extra information (e.g.
17 tissue type, age, sex). The cell variables 206 could be, for example,
the distribution of proteins
18 along a strand of DNA containing a gene, the number of copies of a gene
(transcripts) in a cell, the
19 distribution of proteins along the transcript, and the number of
proteins. Once determined, the cell
variables can be used by the system to determine how much a variant causes the
cell variable to
21 change. By examining how much a mutation causes the cell variable to
change, the CVP can be
22 used to score, categorize, and prioritize variants. Specifically, once
determined, the cell variable
23 predictions can act as high-level features to facilitate more accurate
phenotypic predictions,
24 optionally performed at the secondary analysis unit 114. By training
predictors that predict how
genotype influences cell variables, such as concentrations of proteins, the
resultant machine
26 learning problem is modularized. Moreover, it allows variants to be
related to particular cell
27 variables, thereby providing a mechanism to explain variants.
28 [0060] In one embodiment, the variant and a reference sequence are
fed into the input layer
29 of the CVP 101 and the amount of change in the cell variable is
quantified and used to score,
categorize and prioritize the variant by the secondary analysis unit 114.
31 [0061] In another embodiment, the secondary analysis unit 114
comprises a second system
32 (of similar architecture to the CVP) trained to predict a phenotype
based on the outputs of the cell
33 variable prediction systems (as illustrated in FIG. 2b). For example, in
the case of spinal muscular
11
CA 02894317 2015-06-15
1 atrophy, the cell variable could be the frequency with which the exon is
included when the gene is
2 being copied to make a protein. Other examples of cell variables include
the distribution of
3 proteins along a strand of DNA containing a gene, the number of copies of
a gene (transcripts) in
4 a cell, the distribution of proteins along the transcript, and the number
of proteins.
[0062] The CVP comprises multiple layers of nonlinear processing units to
compute the cell
6 variable using the raw DNA or RNA sequence, or features derived from the
sequence. In
7 embodiments, in order to quantify the effect of a variant, the system may
first construct a pair of
8 feature vectors corresponding to the reference sequence and the variant
sequence. Due to the
9 variant, these genomic feature vectors will be different, but without a
further cell variable predictor
it may not be possible to predict whether those differences would result in
any change in
11 phenotype. Embodiments of the predictive system may therefore infer both
the reference cell
12 variable value and the variant cell variable value using these two
distinct feature vectors. After
13 that, a distance function that combines the reference and the variant
predictions may be used to
14 produce a single score which summarizes the magnitude of predicted
effect induced by the
mutations. Example distance functions include the absolute difference in
expectation,
16 Kullback-Leibler divergence, and variation distance. Detailed
mathematical formulas of these will
17 be described in a later paragraph.
18 [0063] It will be appreciated that process 250 can rely on input
features derived from other
19 types of data besides DNA sequences (e.g. age, sex, known biomarkers) -
the above described
inputs are merely illustrative.
21 [0064] An aspect of the embodiments described herein is the use of
machine learning to infer
22 predictors that are capable of generalizing to new genetic contexts and
to new cell states. For
23 example, a predictor may be inferred using reference genome and data
profiling transcripts in
24 healthy tissues, but then applied to the genome of a cancer cell to
ascertain how the distribution of
transcripts changes in the cancer cell. This notion of generalization is a
crucial aspect of the
26 predictors that need to be inferred. If a predictor is good at
generalization, it can analyze variant
27 sequences that lead to changes in cell variables that may be indicative
of disease state, without
28 needing experimental measurements from diseased cells.
29 [0065] Process 250 may address the two problems discussed with
respect to approach 200.
Since the cell variables are more closely related to and more easily
determined from genomic
31 sequences than are phenotypes, learning predictors that map from DNA to
cell variables is
32 usually more straightforward. High-throughput sequencing technologies
are currently generating
33 massive amounts of data profiling these cell variables under diverse
conditions; these datasets
12
CA 02894317 2015-06-15
1 -- can be used to train larger and more accurate predictors. Also, since the
cell variables correspond
2 -- to intermediate biochemically active quantities, such as the
concentration of a gene transcript,
3 -- they may be good targets for therapies. If high disease risk is
associated with a change in a cell
4 -- variable compared to a healthy individyal, an effective therapy may
consist of restoring that cell
-- variable to its normal state. Embodiments may include such cell variables
as 'exon inclusion or
6 -- exclusion', 'alternative splice site selection', 'alternative
polyadenylation site selection', 'RNA- or
7 -- DNA-binding protein or microRNA specificity', and 'phosphorylation'.
8 [0066] Various aspects of the current system and method include:
the method can be applied
9 -- to raw DNA or RNA sequence or features extracted from the sequence, such
as RNA secondary
-- structures and nucleosome positions; the method can compute one or more
condition-specific cell
11 -- variables, without the need for a baseline average across conditions;
the method can detect
12 -- variants that affect all condition-specific cell variables in the same
way; the method can compare
13 -- a variant sequence to a reference sequence, enabling it to make
different predictions for the same
14 -- variant, depending on genetic context; the method can compute the
condition-specific cell
-- variables using a deep neural network, which has at least two layers of
processing units; the
16 -- method does not require disease labels (e.g., a case population and a
control population); the
17 -- method can score a variant that has never been seen before; the method
can be used to compute
18 -- a 'distance' between a variant sequence and a reference sequence, which
can be used to rank
19 -- the variant; the method can be used to compute a 'distance' between
variants, which is useful for
-- classifying unknown variants based on how similar they are to known
variants.
21 [0067] In the following sections, systems and methods for creating
a condition-specific cell
22 -- variable predictor for cassette splicing are described in further
detail. First, production of training
23 -- targets, and generation of outputs using the systems and methods will be
described.
24 -- Subsequently, the procedure for training and optimizing a deep neural
network (DNN), such as
-- the CVPs, on a sparse and unbalanced biological dataset will be described.
Subsequently,
26 -- example methods to analyze the outputs of the systems will be described.
Further, techniques to
27 -- analyze the behaviour of such a DNN in terms of its inputs and gradients
will be described.
28 [0068] Referring now to Fig. 3, shown therein is a method of
generating target cell variables
29 -- for training. During training of a neural network, a family of gradient-
following procedures are
-- performed where weights ("B") of a neural network are changed according to
the gradient of a cost
31 -- function evaluated using the prediction and the target in a training
dataset. To construct the
32 -- training procedure, the measured cell variable to be modeled is
represented in a mathematical
33 -- form, also referred to as the 'target' in a dataset. For example, in
predicting the percent-spliced-in
13
CA 02894317 2015-06-15
1 values ("PSI"), two distinct forms could be provided, the expected PSI
and a discretized version of
2 PSI.
3 [0069] To compute these targets, at block 302, the biological
measurements such as
4 RNA-Seq datasets are processed to produce a posterior probability
distribution p of PSI, using
methods such as cufflinks and the bocstrap binomial model. With posterior
probability of PSI, at
6 block 304, the expected PSI can be computed by an exact evaluation or an
approximation to the
7 following integral: E(ijJ) = fq,ijj p(*)chp. The result is a scalar value
between 0 and 1. A regression
8 model to predict the expected PSI can be trained, with the cost function
being squared loss
9 function or the cross-entropy based on a binomial distribution with
E(tli) as the probability of
success. In addition to the expected PSI, a discretized version of PSI may
also be determined at
11 block 306, which is defined by the probability mass of PSI in k
predefined bins with boundaries
12 ranging between 0 and 1. For example, using k=3 bins with a uniform bin
width, we arrive at the
13 low, mid, high' (LMH) formulation of PSI, which we also call a 'splicing
pattern'. With this
14 formulation, p(i1) is discretized to three probabilitiestpiow, pmid,
phigh} for use during training. In
particular, plow is equal to the probability that PSI is between 0 and 1/3 :
plow = fol/3 p (11i)c11,.
16 For the discretized splicing patterns, the cross entropy cost function
can be used for a
17 classification model.
18 [0070] Though the preparation of training targets according to
method 300 may be different
19 for different cell variables, the system architecture applied may be the
same or similar.
[0071] Referring now to Figs. 4 to 9, shown therein are example DNN
architectures for CVPs
21 that predicts splicing levels (IP).
22 [0072] Though the figures depict possible architecture
embodiments, the number of hidden
23 layers and the number of processing units in each layer can range widely
and may be determined
24 by hand, using data or using other information;
[0073] In an embodiment, the nodes of the DNN are fully connected, where
each connection
26 is parameterized by a real-valued weight O. The DNN has multiple layers
of non-linearity
27 consisting of hidden units. The output activation a of each hidden unit
v in layer I processes a sum
28 of weighted outputs from the previous layer, using a non-linear function
f:
29 alp = f (Ln a')
where MI represents the number of hidden units in layer I, and ao and Mo are
the input into the
14
CA 02894317 2015-06-15
1 model and its dimensionality, respectively. Different activation
functions for the hidden units can
2 be used, such as the TANH function, SIGMOID, and the rectified linear
unit (RELU).
3 [0074] Referring now to Fig. 4, shown therein is an example
architecture 400 of a deep neural
4 network that predicts alternative splicing inclusion levels in a single
tissue type i, where the
inclusion level is represented by a real-valued number Wi.
6 [0075] Inputs into the first hidden layer 406 consist of genomic
features 402 describing a
7 genomic region; these features may include binding specificities of RNA-
and DNA-binding
8 proteins, RNA secondary structures, nucleosome positions, position-
specific frequencies of short
9 nucleotide sequences, and many others. To improve learning, the features
can be normalized by
the maximum of the absolute value across all training examples. The purpose of
the first hidden
11 layer is to reduce the dimensionality of the input and learn a better
representation of the feature
12 space.
13
14 [0076] The identity of conditions (e.g., tissues) 404, which
consists of a 1-of-T binary
variables where T represent the number of conditions, are then appended to the
vector of outputs
16 of the first hidden layer, together forming the input into the second
hidden layer 408. A third
17 hidden layer 410, or additional hidden layers may be included if found
to be necessary to improve
18 generalization performance.
19 [0077] In an embodiment, the final output 412 may be a regression
model that predicts the
expected PSI.
21 [0078] Referring now to Fig. 5, in another embodiment, the
discretized PSI may be predicted
22 by a classification model 512. Fig. 5 shows an example architecture 500
of a deep neural network
23 that predicts alternative splicing inclusion levels in a single tissue
type i, where the probability
24 mass function over inclusion levels is represented by a k-valued vector,
depicted here with k = 3
values labeled (Low, Medium, High).
26 [0079] Referring now to Fig. 6, alternatively, the DNN can predict
the difference in PSI (APSI)
27 between two conditions for a particular exon. Fig. 6 shows an example
architecture 600 of a deep
28 neural network that predicts the difference between the alternative
splicing inclusion levels of two
29 tissue types (conditions) i 602 and j 604. Here, instead of one tissue
as input, two different
tissues can be supplied to the inputs.
31 [0080] Further, three classes can be generated, called decreased
inclusion 606, no change
32 608, and increased inclusion 610, which can be similarly generated, but
from the APSI
CA 02894317 2015-06-15
1 distributions. An interval can be chosen that more finely differentiates
tissue-specific alternative
2 splicing for this task, where a difference of greater than 0.15 could be
labeled as a change in PSI
3 levels. The probability mass could be summed over the intervals of -1 to -
0.15 for decreased
4 inclusion, -0.15 to 0.15 for no change, and 0.15 to 1 for increased
inclusion.
[0081] Referring now to Fig. 7, shown therein is an example architecture
700 of a deep neural
6 network that predicts the alternative splicing inclusion levels of two
tissue types i and j, where
7 the inclusion levels are represented 'by real-valued numbers IP; 702 and
P3 704 and the
8 difference in alternative splicing inclusion levels between the two
tissue types 706 is also
9 represented by a real-valued number.
[0082] In embodiments, the classification, regression, and tissue
difference codes may be
11 trained jointly. The benefit is to reuse the same hidden representations
learned by the model, and
12 for each learning task to improve the performance of another.
13 [0083] Referring now to Fig. 8, shown therein is an example
architecture 800 of a deep neural
14 network that predicts the difference between the alternative splicing
inclusion levels of two tissue
types i and j, where the probability mass function over inclusion levels is
represented by a
16 k-valued vector, depicted here with k=3 values labeled (Low, Medium,
High)- 802 and the
17 probability mass function over inclusion level differences is
represented by a d-valued vector,
18 here depicted with d=3 values labeled (Decrease, No Change, Increase)
804.
19 [0084] Referring now to Fig. 9, shown therein is an example
architecture 900 of a deep neural
network that predicts alternative splicing inclusion levels in T tissue types,
where the probability
21 mass function over inclusion levels is represented by a k-valued vector,
depicted here with k = 3
22 values labeled (Low, Medium, High). Accordingly, multiple tissues may be
trained as different
23 predictors via multitask learning. The learned representation from
features may be shared across
24 all tissues. Fig. 9 shows an example architecture of such system.
[0085] Training of the systems will now be described with reference to
Figs. 10 to 12.
26 Referring now to Fig. 10, shown therein is a method 1000 for training
the cell variable predictors of
27 the systems described above. At block 1002, the first hidden layer can
be trained using an
28 autoencoder to reduce the dimensionality of the feature space in an
unsupervised manner. An
29 autoencoder is trained by supplying the input through a non-linear
hidden layer, and
reconstructing the input, with tied weights going into and out of the hidden
layer. Alternatively, the
31 weights can be untied. This method of pretraining the network may
initialize learning near a good
32 local minimum. An autoencoder may be used instead of other
dimensionality reduction
16
CA 02894317 2015-06-15
1 techniques like principal component analysis, because it naturally fits
into the CVP's architecture,
2 and that a non-linear technique may discover a better and more compact
representation of the
3 features. At block 1004, in the second stage of training, the weights
from the input layer to the first
4 hidden layer (learned from the autoencoder) are fixed, and the inputs
corresponding to tissues are
appended. A one-hot encoding representation may be used, such that specifying
a tissue for a
6 particular training example can take the form [0 1 0 0 0] to denote the
second tissue out of 5
7 possible types. At block 1006, the reduced feature set and tissue
variables become input into the
8 second hidden layer. At block 1008, the weights connected to the second
hidden layer and the
9 final hidden layer of the CVP are then trained together in a supervised
manner, with targets being
the expected value of PSI, the discretized version of PSI, the expected value
of APSI, and/or the
11 discretized version of APSI, depending on architecture. At block 1010,
after training these final
12 two layers, weights from all layers of the CVP may be fine-tuned by
backpropagation.
13 [0086] In an alternate embodiment, the autoencoder may be omitted
altogether, and all
14 weights of neural network may be trained at once.
[0087] In one embodiment, the targets consist of (1) PSI for each of the
two tissues, and (2)
16 APSI between the two tissues. Given a particular exon and N possible
tissue types, N x N training
17 examples can be constructed. This construction has redundancy in that it
generates examples
18 where both tissues are the same in the input to teach the model that it
should predict no change
19 for APSI given identical tissue indices. Additionally, if the tissues
are swapped in the input, a
previously increased inclusion label should become decreased inclusion. The
same rationale
21 extends to the LMH classifier. Generating these additional examples is
one method to incorporate
22 this knowledge without explicitly specifying it in the model
architecture.
23 [0088] A threshold can be applied to exclude examples from
training if the total number
24 RNA-Seq junction is below a number, such as 10, to remove low signal
training examples.
[0089] In some of the embodiments, multiple tasks may be trained together.
Since each of
26 these tasks might learn at different rates, learning rates may be
allowed to differ. This is to prevent
27 one task from overfilling too soon and negatively affecting the
performance of another task before
28 the complete model is fully trained. This may be implemented by having
different learning rates for
29 the weights between the connections of the last hidden layer and the
functions used for
classification or regression for each task.
31 [0090] To train and test CVPs of the systems described herein,
data may be split into folds at
32 random for cross validation, such as five approximately equal folds.
Each fold may contain a
17
CA 02894317 2015-06-15
1 unique set of genetic information, such as exons that are not found in
any of the other folds.
2 Where five folds are provided, three of the folds could be used for
training, one used for validation,
3 and one held out for testing. Training can be performed for a fixed
number of epochs and
4 hyperparameters can be selected that give optimal area under curve
("AUC") performance or data
likelihood on the validation data. The model can then be re-trained using the
selected
6 hyperparameters with both the training and validation data. Multiple
models can be trained this
7 way from the different folds of data. Predictions from the models on
their corresponding test set
8 can then be used to evaluate the code's performance. To estimate the
confidence intervals, the
9 data can be randomly partitioned, and the above training procedure can be
repeated.
[0091] The CVP's processing unit weights may be initialized with small
random values
11 sampled from a zero-mean Gaussian distribution. Alternatively it may be
initialized with small
12 random values with a zero-mean uniform distribution. Learning may be
performed with stochastic
13 gradient descent with momentum and dropout, where mini-batches are
constructed as described
14 below. An L1 weight penalty may be included in the cost function to
improve the model
performance by disconnecting features deemed to be not useful by the
predictor. The model's
16 weights may updated after each mini-batch.
17 [0092] Referring now to Fig. 11, shown therein is a system to
perform non-uniform sampling
18 of training cases for creating a mini-batch for training a deep neural
network.
19 [0093] To promote neural networks to better discover patterns in
the inputs that help to
distinguish tissue types or genomic features, a system is provided for biasing
the distribution of
21 training events in the mini-batches. The system comprises training cases
separated into
22 "high-variance" cases and "low-variance" cases. The set of high-variance
training cases is thus
23 selected by thresholding each case's variance across tissue types or
genomic features. In the
24 illustrated embodiment the "high-variance" cases are provided in a
database 1106, and the
"low-variance" cases are provided in a database 1108. The system further
comprises switches
26 1104 and multiplexers 1102. In use, each row of a mini-batch 1110 is
sampled either from a list of
27 high- or low-variance training cases, depending on a probabilistic {0,1}
switch value. The resulting
28 mini-batch of genomic features and corresponding cell variable targets
can be used for training,
29 such as for training the architectures in Figs. 6 and 7.
[0094] Referring now to Fig. 12, shown therein is a method for training
cell variable predictors
31 for ensuring a consistent backpropagation signal that updates the
weights connected to tissue
32 inputs and biases learning towards the event with large tissue
variability early on before overfitting
33 occurs. According to a method 1200, at block 1202, all training cases
are separated into a
18
CA 02894317 2015-06-15
1 database of "high-variance" cases and a database of "low-variance" cases,
where the variance of
2 each training case is measured as "variance of the 4) training targets
across tissue types" and the
3 threshold for separating high/low is any pre-determined constant. At
block 1204, all events that
4 exhibit large tissue variability are selected, and mini-batches are
constructed based only on these
events. At each training epoch, training cases can be further sampled (with or
without
6 replacement) from the larger pool of events with low tissue variability,
of some pre-determined or
7 randomized size typically smaller than equal to one fifth of the mini-
batch size. A purpose of
8 method 1200 is to have a consistent backpropagation signal that updates
the weights connected
9 to the tissue inputs and bias learning towards the event with large
tissue variability early on before
over-fitting occurs. As training progresses, the splicing pattern of the
events with low tissue
11 variability is also learned. This arrangement effectively gives the
events with large tissue
12 variability greater importance (i.e. more weight) during optimization.
This may be beneficial to
13 improve the models' tissue specificity.
14 [0095] With the above methods for training, techniques to reduce
overfitting can be applied to
the system to provide an embodiment of a CVP with dropout. Along with the use
of GPUs, CVPs
16 comprising of deep neural networks may be a competitive technique for
conducting learning and
17 prediction on biological datasets, with the advantage that they can be
trained quickly, have
18 enough capacity to model complex relationships, and scale well with the
number of hidden
19 variables and volume of data, making them potentially highly suitable
for 'omic' datasets.
[0096] Additionally, the performance of a CVP depends on a good set of
hyperparameters.
21 Instead of conducting a grid search over the hyperparameter space,
Bayesian frameworks can be
22 used to automatically select a model's hyperparameters. These methods
use a Gaussian Process
23 to search for a joint setting of hyperparameters that optimize a
process's performance on
24 validation data. It uses the performance measures from previous
experiments to decide which
hyperparameters to try next, taking into account the trade-off between
exploration and
26 exploitation. This method eliminates many of the human judgments
involved with hyperparameter
27 optimization and reduces the time required to find such hyperparameters.
Alternatively,
28 randomized hyperparameter search can be performed, where the
hyperparameters to be
29 optimized is sampled from a uniform distribution. These methods require
only the search range of
hyperparameter values to be specified, as well as how long to run the
optimization for.
31 [0097] In the following paragraphs, methods for using the outputs
of the CVP for scoring,
32 classifying and prioritizing genetic variants (with reference to Fig.
13); for scoring variants by
33 associating cell variable changes with those of other variants (with
reference to Fig. 14); and for
19
CA 02894317 2015-06-15
1 interpreting which genetic features account for variant-induced cell
variable changes (with
2 reference to Figs. 15 to 18), will be described.
3 [0098] The systems described above can be used to compute a set of
condition-specific
4 scores for how deleterious a variant is. For instance, a variant may be
found to have a high
deleteriousness score in brain tissue, but not in liver tissue. In this way
the condition-specific cell
6 variables computed as described above can be used to compute condition-
specific
7 deleteriousness scores. To classify variants as pathogenic, likely
pathogenic, unknown
8 significance (VUS), likely benign or benign, and to prioritize or rank a
set of variants, these sets of
9 scores can be combined.
[0099] According to a method 1300, to quantify the effect of a SNV (single
nucleotide
11 variation) or a combination of mutations (called in general a variant)
using a CVP, at block 1302, a
12 pair of feature vectors are constructed corresponding to the reference
sequence and the variant
13 sequence. Due to the mutation, these genomic feature vectors will be
different, but without a
14 further CVP it may not be possible to predict whether those differences
will result in any change in
phenotype. At block 1304, the predictive system is therefore used to compute
both the reference
16 cell variable value and the mutant cell variable value for each
condition, using these two distinct
17 feature vectors. After that, at block 1306, a distance function that
combines the reference and the
18 mutant predictions can be used to produce a single score for each
condition, which summarizes
19 the magnitude of predicted effect induced by the mutations. Because
large change of cell
variables is likely to cause diseases, without further information about a
particular diseases and a
21 particular cell variable, high scoring mutations are assumed to cause
diseases.
22 [0100] Examples of distance functions are the expected difference,
Kullback-Leibler
23 divergence, and variation distance. In the following, we describe each
of these distance functions
24 in detail using a LMH splicing predictor as an example.
[0101] The expected difference represents the absolute value of the
difference induced by
26 the mutation in the expected value of a cell variable. For an LMH PSI
predictor, the predicted
27 reference splicing patterns fplowwt,Pmiciwt,Phighwt} and the predicted
mutant splicing patterns
28 tpiowmut, prnidmut, phighmut) are computed using the reference and
mutant feature vectors as
29 inputs. Then, the expected value of the predicted cell variable with and
without the mutation is
computed, denoted as iliwtand illmut. The expected value is a weighted average
of the PSI
31 values corresponding to the center of the bins used to define the
splicing pattern. As described
32 above, if three bins are used with uniform spacing, reference PSI is
computed by iliwt =
CA 02894317 2015-06-15
1 1/6 Pluwwt + 1/2 Pmidwt + 5/6 phighwt. In the same way, mutant PSI is
computed by tit,õõt =
2 1/6 Thowmut + 1/2 pmidmut + 5/6 phighmut. The final score is the absolute
difference between
3 the expected PSI: s = I dr
This can be combined across conditions, by computing
4 the maximum absolute difference across conditions.
[0102] Kullback-Leibler (KL) divergence is an information theoretic measure
of difference
P(i)
6 between probability distributions P and Q: DKL(P, Q) = iP (i)log-o. Due
to the asymmetric
7 nature of the KL divergence, either s = D (13 P
- KL \- wt, -mut, Or s
DKL(Pmut, Pwt) can be used as the
8 distance measure. The KL divergence can be computed for each condition
and the sum (or
9 average) KL divergence can be computed across conditions, or the maximum
KL divergence can
be computed across tissues.
11
[0103] The variation distance is another measure of difference between
probability
12 distributions. It is the sum of absolute value of the predicted
probabilities. In the LMH splicing
13 predictor example, s = EsEflow,mid,high) pmut_pwtI. Again, this can be
computed for each
14 condition and then the sum or maximum can be taken across conditions.
[0104] Once the score of a variant has been computed at block 1306, at
block 1308 the score
16 can be thresholded and/or combined with other information to classify
the variant as pathogenic,
17 likely pathogenic, unknown significance (VUS), likely benign or benign.
18
[0105] Further, at block 1310, given a set of variants, the score of every
variant can be
19 computed and the set of variants can be reordered so that the highest-
scoring (most deleterious)
variants are at the top of the list and the lowest-scoring variants are at the
bottom of the list.
21
[0106] Referring now to Fig. 14, a method 1400 is shown for scoring,
classifying and
22 prioritizing variants. The method 1400 comprises by, at block 1402,
associating the cell variable
23 changes of variants with those of other variants with known function.
For instance, suppose the
24 system 100 determines that a variant that has never been seen before
causes a change in a
particular cell variable, say the cassette splicing level of a specific exon.
Suppose a nearby variant
26 whose disease function is well-characterized causes a similar change in
the exact same cell
27 variable, e.g., the splicing level of the same exon. Since mutations act
by changing cellular
28 chemistry, such as the splicing level of the exon, it can be inferred
that the unknown variant likely
29 has the same functional impact as the known variant. The system can
ascertain the 'distance'
between two variants in this fashion using a variety of different measures.
Because the system
31 computes variant-induced changes in a cell variable for different
conditions, this information can
21
CA 02894317 2015-06-15
1 be used to more accurately associate variants with one another. For
example, two variants that
2 induce a similar cell variable change in brain tissue would be associated
more strongly than two
3 variants that induce similar cell variable changes, but in different
tissues.
4 [0107] Unlike many existing systems, the methods and systems
described here can be used
to score, classify, prioritize and interpret a variant in the context of
different reference sequences.
6 For instance, when a child's variant is compared to a reference sequence
obtained from the
7 reference human genome, the variant may have a high score, but when the
same variant is
8 compared to the reference sequences obtained from his or her unaffected
parents, the variant
9 may have a low score, indicating that the variant is likely not the cause
of the disease. In contrast,
if the child's variant is found to have a high score when it is compared to
the reference sequences
11 obtained from his or her parents, then it is more likely to be the cause
of the disease. Another
12 circumstance in which different reference sequences arise is when the
variant may be present in
13 more than one transcript, which can occur because transcription occurs
bidirectionally in the
14 genome, there may be alternative transcription start sites, there may be
alternative splicing, and
for other reasons.
16 [0108] Referring now to Figs. 15 to 19, methods will now be
described to identify the impact of
17 features (which may include nucleotides) on a cell variable CVP
prediction.
18 [0109] It can be useful to determine why a variant changes a cell
variable and leads to
19 disease. A variant leads to a change in DNA/RNA sequence and/or a change
in the DNA/RNA
features extracted from the sequence. However, which particular changes in the
sequence or
21 features are important. An SNV may change more than one feature (e.g., a
protein binding site
22 and RNA secondary structure), but because of contextual dependence only
some of the affected
23 features play an important role.
24 [0110] To ascertain this, the system 100 can determine which
inputs (nucleotides or
DNA/RNA features) are responsible for changes in cell variables. In other
words, it is useful to
26 know how important a feature is overall for making a specific
prediction, and it is also useful to
27 know in what way the feature contributes to the prediction (positively
or negatively).
28 [0111] Referring now to Fig. 15, a first method 1500 to identify
the impact of features on a cell
29 variable CVP prediction works by computing, at block 1502, the features
for the sequence
containing the variant and the features for the sequence that does not have
the variant. At block
31 1504, both feature vectors are fed into the cell variable predictor to
obtain the two sets of
32 condition-specific cell variables. At block 1506, a single feature from
the variant sequence is
22
CA 02894317 2015-06-15
1 copied into the corresponding feature in the non-variant sequence and the
system is used to
2 compute the set of condition-specific cell variables. At block 1508, this
is repeated for all features
3 and the feature that produces the set of condition-specific cell
variables that is most similar to the
4 set of condition-specific cell variables for the variant sequence is
identified. This approach can be
extended to test a set of pairs of features or a set of arbitrary combinations
of features.
6 [0112] Referring now to Fig. 16, a second method 1600 to identify
the impact of features on a
7 cell variable CVP prediction evaluates the impact of a subset S g [1,
...,n} of input features
8 x = (x1, xn) on the corresponding cell variable prediction z = f (x). The
method consists of, at
9 block 1602, constructing a new set of input features 2 = (21, kn) where
for each feature index
i E S in the subset the value gi has been replaced with the median value of xi
across the
11 training dataset. At block 1604, this new feature vector is then sent
through the cell variable
12 prediction system in question, resulting in a new prediction'2 f (5e) .
For a splicing cell variable
13 predictor, this entails replacing genomic feature xi with its median
value across all events (all
14 exons) in the training set. The impact of feature subsets of the same
size are comparable,
including all cases when ISI = 1. Among comparable feature subsets, those that
correspond to
16 the largest decrease in performance may be deemed to have high impact.
At block 1606, the
17 overall importance of a feature (as opposed to its importance for a
specific training or test case)
18 with regard to a particular dataset (e.g. a training or test set) can be
determined as the average or
19 median of all its impact scores across all cases in that dataset.
[0113] Referring now to Fig. 17, a third method 1700 is described to
identify the impact of
21 features on a cell variable CVP prediction. At block 1702, an example
from the dataset is given as
22 input to the trained model and forward propagated through a CVP
comprising of a neural network
23 to generate an output. At block 1704, the target is modified to a
different value compared to the
24 predicted output; for example, in classification, the class label would
be modified so that it differs
from the prediction. At block 1706, the error signal is backpropagated to the
inputs. The resulting
26 signal describes how much each input feature needs to change in order to
make the modified
27 prediction, as well as the direction. The computation is extremely
quick, as it only requires a single
28 forward and backward pass through the CVP, and all examples can be
calculated in parallel.
29 Features that need to be changed the most are deemed to be important. At
block 1708, the overall
importance of a feature (as opposed to its importance for a specific training
or test case) with
31 regards to a particular dataset (e.g. a training or test set) can be
determined as the average or
32 median of amount of change across all cases in that dataset. The benefit
of this approach
33 compared to the first is it can model hew multiple features operate
simultaneously.
23
CA 02894317 2015-06-15
1 [0114] Referring now to Fig. 18, a complementary method 1800 based
on the method of 1700
2 to analyze a CVP is to see how features are used in a tissue-specific
manner. At block 1802, this
3 extension simply receives examples from the dataset corresponding to
particular tissues, and, at
4 block 1804, performs the procedure as described above [110]. In cases
where the cell variable
predictor is tissue-specific (e.g. Figs. 4-9) this procedure yields tissue-
specific feature importance
6 information.
7 [0115] Referring now to Fig. 19, shown therein is a detailed
illustration of a method 1900 to
8 generate a visualization for tissue-specific feature importance based on
the method described in
9 1700 and 1800. At block 1902, input comprising examples from a dataset
corresponding to a
particular tissue is provided to the CVP. At block 1904, tissue-specific cell
variable predictions are
11 provided by the CVP. At block 1906, targets are constructed based on the
cell value predictions,
12 such that there is a mismatch between the prediction and the target. At
block 1908, an update
13 signal is computed which describes how the weights of the connection
need to change to make
14 the prediction match the target. At block 1910, an update signal
backpropagated to the input,
Afeature, is further computed. At blo,k 1912, examples from the dataset are
sorted by tissue
16 types. At block 1914, the overall importance of features for each tissue
is computed by taking the
17 mean of the magnitude of the update signal over the entire dataset. At
block 1916, a
18 visualization is generated, where the importance of each feature is
colored accordingly for each
19 tissue.
[0116] The systems and methods described here can also be used to determine
whether a
21 therapy reverses the effect of a variant on a pertinent cell variable.
For example, an SNV within an
22 intron may cause a decrease in the cell variable that corresponds to the
inclusion level of a nearby
23 exon, but an oligonucleotide therapy that targets the same region as the
SNV or a different one
24 may cause the cell variable (inclusion level) to rise to its original
level. Or, a DNA editing system
such as CRISPR-Cas9 may be used to edit the DNA, adding, remove or changing a
sequence
26 such that the cell variable (inclusion level) of the exon rises to its
original level. If the method
27 described here is applied to a variant and a reference sequence obtained
from the reference
28 genome or an unaffected family member, and the cell variable is found to
change by a certain
29 amount, or if the cell variable has been measured to change by a certain
amount, the following
technique can be used to evaluate putative therapies to see if they correct
the change. In the case
31 of therapies that target the variant sequence, such as by protein-DNA or
protein-RNA binding or
32 by oligonucleotide hybridization, the effect of the therapy on the
variant can be computed using
33 the CVP, where the reference is taken to be the variant sequence and the
"variant sequence" is
24
CA 02894317 2015-06-15
1 now taken to be the variant sequence modified to account for the effect
of the therapy. If the
2 therapy targets a subsequence of the variant, that subsequence may be, in
silico, modified by
3 randomly changing the nucleotides, setting them all to a particular
value, or some other method.
4 Alternatively or additionally, when features are extracted from the
modified sequence, features
that overlap, fully or partially, with the targeted subsequence may be set to
values that reflect
6 absence of the feature. The reference (the original variant) and the
modified variant are then fed
7 into the CVP and the change in the cell variable is computed. This is
repeated with a wide range of
8 therapies, and the efficacy of each therapy can be determined by how much
the therapy-induced
9 change in the cell variable corrects for the original variant-induced
change. In the case of a DNA
editing system, such as CRISPR-Cas9, the procedure is even more
straightforward. The
11 reference is taken to be the original variant, and the variant is taken
to be the edited version of the
12 variant. The output of the CVP then indicates by how much the cell
variable will change because
13 of the editing.
14 [0117] Thus, what has been provided is, essentially, a system and
method for computing
variant-induced changes in one more condition-specific cell variables. An
exemplary method
16 comprises computing a set of features from the DNA or RNA sequence
containing the variant,
17 applying a network of at least two layers of processing units (the deep
neural network) to the
18 variant features to compute the one or more condition-specific variant
cell variables, computing a
19 set of features from a reference DNA or RNA sequence, applying the deep
network to the
reference features to compute the one or more condition-specific reference
cell variables, and
21 computing the variant-induced changes in the one or more condition-
specific cell variables by
22 comparing the one or more condition-specific reference cell variables to
the one or more
23 condition-specific variant cell variables. In embodiments, the number of
condition-specific cell
24 variables is at least two.
[0118] The deep neural network may be trained using a dataset of examples,
where each
26 example is a measured DNA or RNA sequence and a corresponding set of
measured values of
27 the condition-specific cell variables, one for each condition, and where
the condition-specific cell
28 variables are not normalized using a baseline that is determined by
combining the
29 condition-specific cell variables across two or more conditions.
[0119] The set of features may include a binary matrix with 4 rows and a
number of columns
31 equal to the length of the DNA or RNA sequence and where each column
contains a single '1' and
32 three 'O's and where the row in which each '1' occurs indicates the
nucleotide at the
33 corresponding position in the DNA or RNA sequence. The set of features
includes a set of
CA 02894317 2015-06-15
1 features may be computed using the recognition path of an autoencoder
that is applied to the
2 binary matrix. The autoencoder may be trained using a dataset of binary
matrices computed using
3 a set of DNA or RNA sequences of fixed length. The set of features may
also include real and
4 binary features derived from the DNA or RNA sequence.
[0120] At least part of the deep network may be configured to form a
convolutional network
6 and/or recurrent network. Part of the deep network that is a recurrent
network may be configured
7 to use of long-term short-term memory.
8 [0121] The deep neural network may be trained using a dataset of
feature vectors extracted
9 from DNA or RNA and a corresponding set of measured values of cell
variables. The training
method may adjust the parameters of the deep neural network so as to minimize
the sum of the
11 error between the measured cell variables and the output of the deep
neural network. The error
12 may be the squared difference between the measured cell variable and the
corresponding output
13 of the neural network. The error may be the absolute difference between
the measured cell
14 variable and the corresponding output of the neural network. The error
may be the
Kullback-Leibler divergence between the measured cell variable and the
corresponding output of
16 the neural network. Stochastic gradient descent may be used to train the
deep neural network.
17 [0122] Dropout may be used to train the deep neural network.
18 [0123] The hyperparameters of the deep neural network may be
adjusted so as to minimize
19 the error on a separate validation set.
[0124] The deep neural network may be trained using multitask learning,
where the outputs of
21 the deep neural network are comprised at least two of the following: a
real-valued cell variable, a
22 probability distribution over a discretized cell variable, a probability
distribution over a real-valued
23 cell variable, a difference between two real-valued cell variables, a
probability distribution over a
24 discretized difference between two real-valued cell variables, a
probability distribution over the
difference between two real-valued cell variables.
26 [0125] An input to the deep neural network may indicate the
condition for which the cell
27 variable is computed and the deep neural network is applied repeatedly
to compute each
28 condition-specific cell variable.
29 [0126] The output of the deep neural network may comprise one real
value for each condition
and the variant-induced change for each condition may be computed by
subtracting the computed
31 reference cell variable from the computed variant cell variable.
26
CA 02894317 2015-06-15
1 [0127] The output of the deep neural network may comprise a
probability distribution over a
2 discrete variable for each condition and the variant-induced change for
each condition may be
3 computed by summing the absolute difference between the computed
probabilities for the
4 reference cell variable and the variant cell variable.
[0128] The output of the deep neural network may comprise a probability
distribution over a
6 discrete variable for each condition and the variant-induced change for
each condition may be
7 computed using the Kullback-Leibler divergence between the computed
probabilities for the
8 reference cell variable and the variant cell variable.
9 [0129] The output of the deep neural network may comprise a
probability distribution over a
discrete variable for each condition and the variant-induced change for each
condition may be
11 computed by first computing the expected value of the reference cell
variable and the variant cell
12 variable, and then subtracting the expected value of the reference cell
variable from the expected
13 value of the variant cell variable.
14 [0130] The variant-induced changes in the one or more condition-
specific cell variables may
be combined to output a single numerical variant score. The variant score may
be computed by
16 summing the variant-induced changes across conditions. The variant score
may be computed by
17 summing the squares of the variant-induced changes across conditions.
The variant score may
18 be computed by summing the outputs of a nonlinear function that are
computed by applying the
19 nonlinear function to the variant-induced changes across conditions.
[0131] At least two variants and corresponding reference sequences may be
independently
21 processed to compute the variant-induced changes in one or more
condition-specific cell
22 variables for each variant and corresponding reference sequence. At
least two variants and
23 corresponding reference sequences may be independently processed to
compute the variant
24 score for each variant and corresponding reference sequence. The variant
scores may be used to
prioritize the variants by sorting them according to their scores. Thresholds
may be applied to the
26 score to classify the variant as deleterious or non-deleterious, or to
classify the variant as
27 pathogenic, likely pathogenic, unknown significance, likely benign or
benign, or to classify the
28 variant using any other discrete set of labels. A validation data
consisting of variants, reference
29 sequences, and labels may be used to compute the thresholds that
minimize classification error.
The scores may be combined with additional numerical information before the
variants are sorted.
31 The scores may be combined with additional numerical information before
the thresholds are
32 applied. The scores may be combined with additional numerical
information before the thresholds
33 are applied.
27
=
CA 02894317 2015-06-15
1 [0132] For one or more pairs of variants, the distance between the
two variants in each pair
2 may be computed by summing the output of a nonlinear function applied to
the difference
3 between the change in the condition-specific cell variable for the first
variant and the change in the
4 condition-specific cell variable for the second variant. The nonlinear
function may be the square
operation. The nonlinear function may be the absolute operation.
6 [0133] The deleteriousness label of an unknown variant may be
determined by computing the
7 distance of the variant to one or more variants of known deleteriousness
and outputting the label
8 or the score of the closest known variant. The deleteriousness value of
an unknown variant may
9 be determined by computing the distance of the variant to one or more
variants of known
deleteriousness and then computing the weighted average of their labels or
scores, where the
11 weights are nonlinear functions of the distances. Two or more unknown
variants may be
12 prioritized, by sorting them according to their deleteriousness values.
13 [0134] The mini-batches used during multitask training may be
balanced so that the number
14 of cases that exhibit a large difference is similar to the number of
cases that exhibit a small
difference.
16 [0135] The genetic variant may be a single nucleotide variant. The
genetic variant may
17 contain two or more distinct single nucleotide variants. The genetic
variant may be a combination
18 of substitutions, insertions and deletions and not be a single
nucleotide variant. The genetic
19 variant may be obtained by sequencing the DNA from a patient sample.
[0136] The reference sequence may be obtained by sequencing the DNA from a
close
21 relative of the patient. The reference sequence may be any DNA or RNA
sequence and the
22 variant sequence may be any DNA or RNA sequence, but where the reference
sequence and the
23 variant sequence are not identical.
24 [0137] The features may include position-dependent genetic
features such as conservation.
[0138] The most explanatory feature may be determined by examining each
feature in turn,
26 and computing a feature-specific variant feature vector by copying the
feature derived from the
27 variant sequence onto the features derived from the reference sequence;
using the deep neural
28 network to compute the variant-induced changes in the one or more
condition-specific cell
29 variables for that feature-specific variant identifying the feature whose
corresponding
feature-specific variant-induced changes in the one or more condition-specific
cell variables are
31 most similar to the variant-induced changes in the one or more condition-
specific cell variables.
32 [0139] The similarity between the feature-specific variant-induced
changes in the one or more
28
CA 02894317 2015-06-15
1 condition-specific cell variables and the variant-induced changes in the
one or more
2 condition-specific cell variables may be computed by summing the squares
of their differences.
3 [0140] Although the invention has been described with reference to
certain specific
4 embodiments, various modifications thereof will be apparent to those
skilled in the art without
departing from the spirit and scope of the invention as outlined in the claims
appended hereto.
29