Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02897313 2015-07-06
WO 2014/110248
PCT/US2014/010863
CONTROLLED RELEASE FORMULATIONS OF LORAZEPAM
[1] The present application claims the benefit of priority under 35 U.S.C.
119(e) from prior U.S. provisional patent application no. 61/750,792, filed on
January 9, 2013 and from prior U.S. provisional patent application no.
61/762,836,
filed on February 8, 2013; the entire contents of each provisional application
being
incorporated herein by reference.
TECHNICAL FIELD
[2] The present invention relates to controlled release formulations of
lorazepam and to methods of treating patients with a once-a-day dose of
lorazepam.
BRIEF DESCRIPTION OF THE RELATED ART
[3] Lorazepam is the generic name for the active pharmaceutical
ingredient 7-chloro-5-(2-chloropheny1)-1,3-dihydro-3-hydroxy-2H-1,4-
benzodiazepin-2-one, which has the following structure:
0
HiyiN
= N OH
CI
CI
*
Like other benzodiazepines, lorazepam has CNS activity and has proven to be a
useful treatment for anxiety related disorders, such as: General Anxiety
Disorder or
Anxiety associated with Major Depression and others. It is almost insoluble in
water. This compound was disclosed in U.S. Patent 3,296,249.
[4] Lorazepam has been sold commercially under the brand name
ATIVANO (originally by Wyeth, now by Valeant Intl) in the form of an oral
immediate release tablet. The tablets contain 0.5 mg, 1 mg, or 2 mg of
lorazepam
and are usually administered two or three times a day (b.i.d and t.i.d,
respectively)
to achieve a total dose of 2 to 6 mg/day, though doses from 1 to 10 mg/day can
also
1
CA 02897313 2015-07-06
WO 2014/110248
PCT/US2014/010863
be used. According to the U.S. package insert material for ATIVAN : "For
anxiety, most patients require an initial dose of 2 to 3 mg/day given b.i.d.
or t.i.d."
The peak plasma concentrations (Cmax) typically occur about 2 hours (Tmax)
after
oral administration. Lorazepam has, according to the package insert, a half-
life in
human plasma of about 12 hours.
151 While the immediate release tablets of lorazepam, with a multi-
dose
per day regimen, have been available for several decades, thus far no once-a-
day
dosage form has been commercially introduced. Such a dosage form is often
desirable. Besides the benefit of convenience, a sustained release version
that
could provide 24 hour therapeutic effect, but with lower peak plasma
concentration
levels than the immediate release tablet, may reduce side effects. For this
reason,
Abrams et al. investigated a sustained release tablet containing 2 mg of
lorazepam
and compared it to a 2 mg dose of lorazepam immediate release tablets (2x1mg
tablets). S. M. L. Abrams et al., "Pharmacodynamic and Pharmacokinetic
Comparison of Two Formulations of Lorazepam and Placebo," Human
Psychopharmacology, Vol. 3, 133-138 (1988). The sustained release tablet had,
as
expected, a longer Tmax (median 8 hours) and a lower Cmax (12 ng/ml) than the
immediate release tablets (2 hours and 22 ng/ml, respectively). But the
relative
bioavailability was reduced in the sustained release tablet such that after 30
hours
the AUC was only about 85% of the AUC achieved with the immediate release
tablets. Abrams et al. also noted that "the [serum] concentrations of both
formulations were similar between 10 and 30 h[ours]." Thus, despite providing
some delay in the rise of lorazepam serum concentrations and a lower Cmax, the
sustained release tablet apparently did not serve to extend the therapeutic
duration
of lorazepam beyond that achieved with immediate release tablets.
[6] Regarding the tablet formulation, Abrams et al. does not
disclose the
design or excipients used in making the sustained release tablet. From the
plasma
concentration curve, the sustained release tablet appears to have been a first
order
release tablet, though the kinetics are not reported.
2
CA 02897313 2015-07-06
WO 2014/110248
PCT/US2014/010863
[7] The long half-life of lorazepam in blood plasma makes it a classically
disregarded candidate for the development of a once daily formulation. Also, a
drug product that provides 24 hour therapy from two doses per day, as opposed
to
three or more doses per day, is generally considered to have achieved the
majority
of patient compliance benefits. If a single daily dose formulation was
desired, an
immediate release lorazepam tablet could be used to provide a complete daily
dose
because of the long half-life of lorazepam in blood plasma. But administering
a
complete daily dose in a single immediate release dosage form would increase
the
Cmax and the peak-trough variations (concentration differences between Cmax
and
Cmin) beyond those attained in conventional b.i.d. administration (i.e., twice-
daily
dosing), and thus would likely increase the risk of drug related adverse
events, i.e.,
side effects. Using a sustained release formulation can reduce the rate of
increase
in plasma drug concentration and the value of Cmax, but runs the risk of sub-
therapeutic plasma concentration levels, especially near the end of the dosing
cycle,
and/or lower overall drug exposure than the current b.i.d. immediate release
tablet
regimen.
[8] A lorazepam formulation that provides a sustained release profile
with the potential for an effective and well tolerated once daily dosing
regimen
would be advantageous.
SUMMARY OF THE INVENTION
191 The present invention relates to controlled release lorazepam
compositions. A first aspect of the invention relates to a pharmaceutical
composition, comprising 0.5 to 10 mg of lorazepam in combination with
sufficient
pharmaceutically acceptable excipients to provide a solid oral dosage form
having
controlled release of said lorazepam; wherein said controlled release of
lorazepam
is:
(1) substantially zero order release; and
(2) the release of lorazepam reaches 90% within the time range of 7 to 12
hours.
The controlled release parameters are determined in a pharmaceutical
dissolution
3
CA 02897313 2015-07-06
WO 2014/110248
PCT/US2014/010863
test comprising a buffer of pH 6.8. Preferably the routine of once-daily
administration of the composition can provide the patient with 24 hours of
therapeutic effect.
[10] Another aspect of the invention relates to a controlled release
tablet, which comprises 0.5 to 10 mg of lorazepam dispersed in a controlled
release
matrix, wherein said matrix comprises polyethylene oxide. Often the
polyethylene
oxide has a molecular weight of 900,000 to 2,000,000 such as about 1,000,000.
Preferably the tablet exhibits sufficient controlled and complete release of
lorazepam to facilitate a once daily dosing regimen that provides 24 hours of
therapeutic effect.
[11] A further aspect of the invention relates to a method of treating a
lorazepam-treatable condition in a patient, which comprises administering once
a
day to a patient in need thereof a controlled release lorazepam composition
that
contains 0.5-10 mg of lorazepam that provides 24 hour therapeutic effect
during
steady state conditions. The composition exhibits the zero order controlled
release
as described above and/or is a polyethylene oxide matrix tablet as described
above.
Typical lorazepam-treatable conditions include anxiety disorders such as
Generalized Anxiety Disorder and anxiety associated with major depression, but
is
not limited thereto.
BRIEF DESCRIPTION OF THE DRAWING
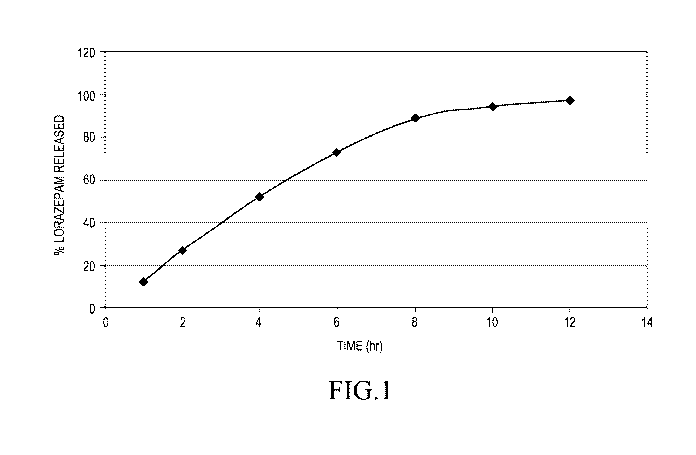
[12] Figure 1 represents the dissolution of the tablets made in the
Example using a pharmaceutical dissolution test.
DETAILED DESCRIPTION OF THE INVENTION
[13] The present invention relates to lorazepam formulations.
Providing controlled release lorazepam suitable for once daily dosing is
problematic. Slowing the rate of drug release presents the risk of a sub-
therapeutic
plasma concentration, especially in the hours before or after the daily dose
is
administered. This period of low plasma concentration can leave a patient
susceptible to break-through anxiety; e.g., the anxiety breaks through the
effect of
4
CA 02897313 2015-07-06
WO 2014/110248
PCT/US2014/010863
the drug. Giving the drug faster and/or giving more drug to compensate for the
low
plasma levels and avoiding break-through anxiety increases the risk of adverse
events (commonly referred to as side effects) such as sedation, dizziness,
memory
impairment, etc. Indeed, but for the nearly certain increase in adverse
effects as a
result of the rapid and high peak plasma concentration, a large enough
immediate
release dose of lorazepam could provide therapeutic levels for 24 hours by
simply
exploiting the inherent long plasma half-life of the drug. Thus, patient
safety issues
preclude the use of large single immediate release doses as not being
practical for
lorazepam.
[14] Commonly, an oral controlled release formulation exhibits first
order release. This means that the rate of drug release is proportional to the
amount
of drug in the formulation. As more drug is released, the rate of release
decreases.
Applying mathematical models, it was discovered that a first order release
profile
was not well suited for developing a controlled release lorazepam tablet.
Specifically the risk of either sub-therapeutic plasma levels late in the
dosing
interval or adverse events was too great when attempting to design a first
order
release system. Instead, it was discovered that a zero order release of
lorazepam
over an extended period of time could provide safe and effective plasma
concentrations of lorazepam.
[15] Accordingly, one aspect of the present invention relates to a
pharmaceutical composition comprising 0.5 to 10 mg of lorazepam in combination
with sufficient pharmaceutically acceptable excipients to provide a solid oral
dosage form having controlled release of lorazepam. The controlled release
exhibits the following parameters: (1) substantially zero order release and
(2) the
release of lorazepam reaches 90% within the time range of 7 to 12 hours. These
controlled release parameters are determined in a pharmaceutical dissolution
test
comprising buffer of pH 6.8. For clarity, a "pharmaceutical dissolution test"
is any
in vitro dissolution test carried out under pharmaceutically reasonable
conditions.
As is well known in the art, different formulations are often tested under
different
5
CA 02897313 2015-07-06
WO 2014/110248
PCT/US2014/010863
conditions, such as basket apparatus or a paddle apparatus, varying paddle
speeds
(e.g., 50 rpm, 60 rpm, or 75 rpm), etc., in order to obtain meaningful data.
Typically dissolution testing is used to model in vivo release and/or to
differentiate
improperly made formulations from correctly manufactured ones. Developing a
suitable set of conditions for dissolution is a matter of routine practice and
skill.
For the present invention, the pharmaceutical dissolution test must include
the use
of a pH 6.8 buffer. The entire test need not be performed at pH 6.8 or in
buffer, but
at least a portion of the test must comprise the use of pH 6.8 buffer; i.e.,
two phase
testing, variable pH testing, etc., are permissible. Other than the
requirement that
an aqueous buffer solution of pH 6.8 be used at some point, the other
conditions
can be varied as appropriate. Tests that use unreasonable conditions, i.e.,
those that
would give no meaningful data and/or are designed specifically to not meet the
release parameters of the present invention are excluded. Thus, any
dissolution
test, which includes a pH 6.8 buffer, that a regulatory authority, such as the
U.S.
FDA, finds acceptable to support a filing for a controlled release lorazepam
formulation, is a dissolution test conducted under pharmaceutically reasonable
conditions and is therefore a "pharmaceutical dissolution test" for purposes
of the
present invention. In that several pharmaceutically reasonable dissolution
tests may
exist, the release parameters of the present invention need only be satisfied
in one
such test ¨ not in every possible test and regardless if not satisfied in
another test.
The pH 6.8 buffer is typically a phosphate buffered aqueous solution and may
contain additional ingredients such as in forming a complete simulated
intestinal
fluid (SIF) with (or without) enzymes as is known in the art. A typical
pharmaceutical dissolution test uses 500 ml or 900 ml of media at 37 C with
paddles at an rpm of 50 to 100 such as 60 or 75, but is not limited thereto as
explained above. When the pharmaceutical composition releases lorazepam
substantially independently of pH, then a single media is often suitable,
e.g., the
whole test can be conducted with pH 6.8 buffer. If the composition has a pH-
sensitive release (e.g., the rate of release is affected by the pH of the
media), then
6
CA 02897313 2015-07-06
WO 2014/110248
PCT/US2014/010863
different media are often desired in making a suitable, pharmaceutically
acceptable
test. For example, a two phase test may be used wherein the initial phase is
carried
out in a lower pH environment followed by the higher pH 6.8 buffer
environment.
The first phase is often 0.1 N HC1 and/or simulated gastric fluid (SGF) and
last
typically between 1 ¨ 3 hours, often 1 or 2 hours. The media is changed to
start the
second phase in pH 6.8 buffer and the test carried to completion. The initial
phase
could be a higher pH such as around pH 4 to simulate a fed stomach, etc. as is
well
known in the art. In like manner to the two phase test having low and moderate
pH, multi-phase tests could be adopted having 3, 4, or more different media
phases.
More recently, variable pH dissolution testing has become more technologically
available wherein the number of stages can be very great so as to approximate
constant pH change over all or a portion of the dissolution test.
[16] The percentage of lorazepam released during the pharmaceutical
dissolution test is stated with respect to the nominal or label amount, as is
conventional in the art. Thus, the release of 1 mg lorazepam from a 2 mg
tablet is
reported as 50% release, even if the tablet is discovered to only contain 1.94
mg of
lorazepam instead of the intended 2.0 mg. In a well-controlled process, the
actual
assayed amount is generally within +/- 5% of the label amount. The percentage
of
release at a point in time refers to the cumulative release up to that point
in time, as
per the conventional usage of these terms in the art. The amount of lorazepam
released from the beads (i.e., dissolved into the dissolution media), can be
determined by ordinary methods using routine skill.
[17] The pharmaceutical composition of the invention provides
controlled release of lorazepam. As used herein, "controlled release" means
any
type of prolonged release of the drug beyond immediate release and is not
intended
to denote any other quality or characteristic of the release. Unless otherwise
indicated, controlled release is used synonymously with extended release and
sustained release.
7
CA 02897313 2015-07-06
WO 2014/110248
PCT/US2014/010863
[18] The controlled release for this aspect of the present
invention has
two characterizing parameters: substantially zero order release and a
specified time
to completion of release. Zero order release, as is well known in the art,
means that
the rate of release is constant over time and not proportional to the amount
of drug
in the formulation. The dissolution curve of a perfect zero order release
profile,
plotted as cumulative drug release (Y axis) vs. time (X axis), would be a
straight
line of positive slope. This is in contrast to the more common first order
release
profile, where the initial rate of release is high but slows down over time as
the
concentration of drug in the dosage form is diminished.
[19] The present invention does not require a perfect zero order
release, merely a substantially zero order release. Workers skilled in the art
are
accustomed to categorizing release as zero order or first order, etc. A
release curve
is substantially zero order when that is the best category or description;
i.e.,
declaring it first order would be less accurate or less correct. Sometimes a
worker
skilled in the art will classify the type of release for a portion of the
dissolution
release curve. For example, one portion exhibits zero order release and
another
exhibits first order. In this situation, the zero order part should comprise
at least
50%, more typically at least 60%, and often at least 70% of the amount of
lorazepam that is released, in order for the overall release to be considered
substantially zero order. For clarity, the percentage is not necessarily from
zero,
but is a continuous portion of the curve, e.g., zero order release from 10% to
60%
release of lorazepam means that 50% of the lorazepam was released under zero
order. Of course, higher portions of zero order or substantially zero order
release
are generally preferred, including at least 75%, at least 80%, at least 85%,
and at
least 90%.
[20] In more quantitative terms, a dissolution curve shows
substantially zero order release when the overall curve is fairly linear or a
significant portion is highly linear. In general the correlation coefficient
for the
zero order release linear equation Qt = Kot + Q0 is at least 0.85, more
typically at
8
CA 02897313 2015-07-06
WO 2014/110248
PCT/US2014/010863
least 0.90, and often at least 0.95 including 0.97, 0.98, and 0.99 (Qt is the
quantity
of released drug, Q0 is the initial quantity of released drug, Ko is the zero
order rate
constant, and t is time). Alternatively, a release curve may be considered to
have
zero order and first order release phases. In such an event, a curve is
substantially
zero order when at least 6 continuous hours, preferably at least 7 continuous
hours,
and more likely at least 8 continuous hours are clearly zero order release.
Such
clear phases of zero order release typically have a correlation coefficient of
at least
0.95, more preferably at least 0.99.
[21] Another
mathematical model for determining substantially zero
order release relies on the equation Qt/QT = ktn wherein Qt is the quantity of
drug
released at time t, QT is the total quantity of drug in the dosage form, k is
the
constant, and n is the release kinetics exponent. When n is about 1, then the
release
is zero order release. Substantially zero order release includes n having a
value of
0.6 to 1.2, more typically from 0.7 to 1.05, and often from 0.8 to 1.
[22] For purposes of
the present invention, a substantially zero order
release is one that meets any of the above qualitative or quantitative
criteria or
definitions of substantially zero order release, unless otherwise noted. Often
a
substantially zero order release will meet more than one criteria/definition,
but such
is not required.
[23] The
pharmaceutical composition according to this embodiment of
the invention not only has a substantially zero order release, but also
achieves 90%
release of the lorazepam within the range of 7 to 12 hours, more typically
within
7.5 to 11 hours. For clarity, the percentage is based on the stated or nominal
value
of lorazepam in the starting composition, as per the custom in the art.
Releasing
drug too rapidly can increase the risk of adverse events. Thus, the 90%
released
point should not occur too soon. But delaying release too long can increase
the risk
of incomplete absorption and/or sub-therapeutic levels. Accordingly, 90%
release
generally does not occur until 7 hours or later, more typically not earlier
than 7.5
hours, and often not earlier than 8 hours. Conversely, 90% released generally
9
CA 02897313 2015-07-06
WO 2014/110248
PCT/US2014/010863
occurs not later than 12 hours, typically before 11 hours and often by 10
hours.
Typically at 2 hours in the pharmaceutical dissolution test, not more than 40%
of
the lorazepam has been released and preferably not more than 35%, and in some
embodiments not more than 30% has been released.
[24] The pharmaceutical composition contains 0.5 to 10 mg of
lorazepam. Lorazepam and its synthesis are well known and the drug is
generally
commercially available. Lorazepam can be amorphous or crystalline. Though it
has a diazepine ring nitrogen that could be used for forming a salt, typically
lorazepam is used as a non-salt or free base. For purposes of the present
invention,
"lorazepam" is intended to embrace all such pharmaceutical forms of lorazepam
including pharmaceutically acceptable salts, crystalline forms thereof
including
hydrates and solvates, and amorphous forms, unless noted otherwise. The amount
of lorazepam in the composition is 0.5 to 10 mg in conformance with the normal
total daily dose range. Typical amounts for commercial reasons are often from
1 to
4 mg including 1 mg, 2 mg, 2.5 mg, 3 mg, and 4 mg; though each integer from 1
to
10, inclusive, also represents suitable specific dose amounts.
[25] The pharmaceutical composition contains lorazepam in
combination with sufficient pharmaceutically acceptable excipients to provide
a
solid oral dosage form having controlled release of the lorazepam.
Pharmaceutical
excipients for making a solid oral dosage form including excipients that
alone, or in
combination, create controlled release are well known in the art. The
structure of
solid oral dosage forms that exhibit controlled release are also well known
including tablets, beads or pellets in a capsule, osmotic devices, etc.
[26] To obtain substantially zero order release from a controlled
release formulation, several design approaches are known. For example, the
osmotic device developed by ALZA Corporation is purported to provide constant
release rates over time, i.e., zero order release. These osmotic devices,
which can
have the external appearance of a tablet, generally comprise a chamber having
the
drug in the interior. The walls of the chamber take up water but do not let
the drug
CA 02897313 2015-07-06
WO 2014/110248
PCT/US2014/010863
out. The sole means of escape for the drug is through a hole (passageway) in
the
chamber, typically bored with a drill or laser. The interior often contains
additional
excipients such as to absorb water, to gel, etc. The osmotic pressure
developed
within the chamber serves to drive the drug out of the passageway in a pH
independent and relatively constant rate, which is controlled by the
excipients and
the size of the passageway. A few examples of such technology include U.S.
3,786,813; U.S. 3,845,770; and U.S. 4,624,847. A variation on this concept
where
a plug is removed in vivo to form a hole in a chamber has also been proposed
as a
suitable device for achieving zero order release in U.S. 7,195,778. A compact
that
uses hydrostatic pressure and without a chamber and hole has been proposed to
provide zero order release in U.S. 8,231,897. Another design approach uses
multi-
layer beads. Various tablets have been proposed for achieving (substantially)
zero
order release. In general the tablets rely on an erodible matrix such that
release is
(primarily) dissolution controlled instead of diffusion controlled. Erodible
matrix
tablets include biodegradable matrix types and gelling polymer matrix-types.
The
gelling polymer first swells and then undergoes dissolution/erosion at the
surface
boundary layer. Gelling polymers include hydroxypropyl methylcellulose
(HPMC), poly(ethylene oxide) (PEO) which is sometimes also referred to as
polyethylene glycol, polyvinyl alcohol (PVA), and various acrylate polymers
often
sold commercially under the brand name EUDRAGIT. Examples of the use of
gelling polymers as a matrix for controlled release tablets include U.S.
4,361,545;
U.S. 5,009,895; U.S. 5,945,125; and U.S. 6,703,045; each patent purporting to
obtain zero order release.
[27] For ease of manufacture, a tablet is the preferred solid
oral dosage
form. Typically a tablet of the invention comprises lorazepam dispersed in a
gelling polymer matrix. The matrix can comprise one or several polymers
including HPMC, PEO, PVA, etc. Blends of polymers of the same class having,
e.g., different molecular weight or cross-linking, or viscosity, etc., can
also be used.
11
CA 02897313 2015-07-06
WO 2014/110248
PCT/US2014/010863
It also possible to include non-gelling polymers in a matrix blend, so long as
the
matrix provides some degree of gelation upon sustained exposure to water.
[28] PEO is a preferred polymer matrix component in compositions of
the present invention. While HPMC is believed to be a suitable matrix polymer,
the use of PEO has been found to be especially suited for controlled release
of
lorazepam. PEO has the desired interaction with lorazepam such that the
release
can be slowed/delayed but also completed within the needed time span. This
discovery is a separate aspect of the invention; namely that PEO is generally
superior to HPMC and other matrix polymers in formulating controlled release
of
lorazepam, regardless of the release parameters. Accordingly, another
embodiment
of the invention is a controlled release tablet, which comprises 0.5 to 10 mg
of
lorazepam dispersed in a controlled release matrix, wherein the matrix
comprises
polyethylene oxide. The matrix may contain other polymers such as HPMC,
Eudragit RSPO, etc., but the amount of PEO should be the greatest of any other
gelling polymer, if present.
[29] The PEO used in making controlled release lorazepam tablets
typically has an average (approximate) molecular weight of at least 900,000
and
usually not greater than 5,000,000, but is not necessarily limited thereto.
Commercially available PEO, sold under the brand name POLYOX by The Dow
Chemical Corp., have molecular weights up to 7,000,000. Higher molecular
weights generally result in slower release rates. Higher concentrations of PEO
also
tend to decrease the rate of release. Thus, using more of a lower MW PEO may
provide similar release as using less of a higher MW PEO. Typically the
tablets are
formed with PEO having an average MW between 900,000 and 4,000,000, more
typically from 900,000 to about 2,000,000. If not commercially available, a
desired
MW can be achieved by blending commercially available PEO of different average
MW. In one embodiment, the PEO has a MW of about 1,000,000 (e.g., +/-
100,000).
12
CA 02897313 2015-07-06
WO 2014/110248
PCT/US2014/010863
[30] The amount of polymer matrix in the tablet is typically from 20%
to 70% of the tablet, more typically from 30% to 60% (all percentages for
tablet
ingredients refer to weight percent unless otherwise noted). As mentioned
above,
the polymer matrix is preferably composed entirely of PEO, but may be a
combination of PEO and other matrix polymers so long as PEO is the single most
prevalent type of matrix polymer. When the matrix polymer is primarily (or
exclusively) PEO having a MW from 900,000 to 1,500,000, the amount of matrix
polymer is often in the range of 35 to 55%, including 40% to 50%. Matrix
polymer
having higher MW PEO is generally used in slightly lower amounts such as 20%
to
50%, including 25% to 45%.
[31] In addition to the polymer matrix, a controlled release tablet
usually contains other excipients including diluents and lubricants. Diluents
provide bulk and can enhance tableting or tablet properties in comparison to
the use
of active and matrix polymer alone. Examples of diluents include sugars such
as
lactose or mannitol; microcrystalline cellulose; and calcium phosphates such
as
dibasic calcium phosphate dihydrate, dibasic calcium phosphate anhydrous, and
tribasic calcium phosphate. Lubricants include magnesium stearate and sodium
stearyl fumarate. Diluents often comprise from 30% to 70% of the tablet and
can be
a single diluent or a combination of diluents. Lubricants, when present,
typically
comprise 1% to 3% of the tablet weight. Because the dose of lorazepam is small
(1-10 mg), the concentration of lorazepam tends to be low and typically is
less than
10%, more typically less than 5%, and often in the 1-3% range. The combination
of polymer matrix and diluent(s) is typically 85% to 99%, more typically 90%
to
98%, and even 95% to 98%, the weight of the tablet.
[32] In one embodiment, the tablet contains lactose and calcium
phosphate as diluents. The lactose is typically lactose monohydrate and the
calcium phosphate is typically dibasic calcium phosphate. The lactose is
typically
used in an amount from 10 to 50%, such as from 20 to 40%, but is not limited
thereto. Likewise, the calcium phosphate is typically used in an amount from
10 to
13
CA 02897313 2015-07-06
WO 2014/110248
PCT/US2014/010863
50%, such as from 20 to 40%, but is not limited thereto. This dual diluent
strategy
is not limited to lactose and calcium phosphate as either one, or both, can be
replaced with another diluent. But it is preferred that at least one of
lactose and
calcium phosphate, and more preferably both, are present in the tablet within
the
above ranges.
1331 The tablets typically have a weight of 100 to 200 mg, not
including the weight of any subsequent coatings. Generally any coating applied
to
the controlled release matrix tablet is for cosmetic or stability reasons and
not for
significant or meaningful release modification.
[34] The tablets can be made by procedures known in the art. Wet
granulation and direct compression are two common techniques for making
tablets.
Direct compression is usually preferred for economic reasons and is often more
suited for gelling polymers such as PEO.
1351 The controlled release tablets containing a PEO polymer
matrix
are preferably formulated (e.g., selection of PEO MW, amount of PEO, etc.) to
achieve the above-described in vitro dissolution release parameters of
substantially
zero order release and 90% release within 7 to 12 hours, and/or the other
release
parameters as fully described above.
[36] The pharmaceutical composition of the invention that
achieves
substantially zero order release and provides 90% release within 7 to 12
hours,
preferably provides therapeutic effect for 24 hours under steady state
conditions
with daily dosing. Quantitatively, preferred embodiments of the invention will
provide a blood plasma concentration of 10 ng/ml or more for at least 20
hours,
often at least 22 hours, and sometimes for 24 hours under steady state
conditions
over a 24 hour period (e.g., from daily-dose to daily-dose). For clarity, the
term
"steady state" is used in its ordinary sense in the pharmaceutical arts. It
does not
mean constant, but rather the dynamic equilibrium that is obtained after
consistent
successive administrations of a drug, typically several days (e.g., 5 times
the 1/2 life,
or 3-5 days in the case of lorazepam). For example, a patient already taking
14
CA 02897313 2015-07-06
WO 2014/110248
PCT/US2014/010863
lorazepam immediate release tablets on a regular schedule (two or three times
per
day) has lorazepam in his/her blood when the next dose is administered. After
ingestion, the dose is released and the amount of lorazepam in the blood
increases
to a maximum blood plasma concentration or "Cmax." The lorazepam is
concurrently being metabolized and/or removed from the blood by biological
actions of the body and so the blood plasma concentration falls. The decline
in
drug blood plasma concentration will continue until the next dose of lorazepam
is
taken. The drug blood plasma concentration will reach its lowest concentration
level, the "Cmin," just before the new dose of lorazepam is absorbed into the
blood.
The new dose causes a rise in blood plasma concentration and the cycle
repeats,
reaching the Cmax once again followed by a fall to the Cmin and a new
administration of lorazepam, etc. In contrast to the steady state, the first
dose of
lorazepam produces different blood plasma values because no lorazepam is in
the
blood at the time of the dose. The Cmin for such a single dose experiment is
zero
at the outset. The Cmax is typically noticeably lower than the steady state
Cmax.
Because the present invention is applicable for chronic administration of
lorazepam
(one or more weeks and perhaps months or years), the steady state parameters
can
be more meaningful. Indeed, in some embodiments of the present invention, a
single dose study (e.g., initial dose) will not provide a therapeutic
concentration in
the blood stream sooner than 1 hour, often not before 2 hours, and in some
embodiments not before 3 hours. In some embodiments, a minimum therapeutic
concentration can be taken as 10 ng/ml.
[37] Preferred embodiments provide a steady state Cmax from daily
dosing that is about equal to, or less than, the steady state Cmax obtained
from
dosing the same total daily amount of lorazepam via immediate release tablets
through b.i.d or t.i.d. regimens. The term "about equal" means that the steady
state
Cmax of the controlled release composition is within +/- 35%, preferably with
+/-
20% of the steady state Cmax for the corresponding immediate release tablets
given
b.i.d. or t.i.d (having the same total daily dose). In some embodiments, the
steady
CA 02897313 2015-07-06
WO 2014/110248
PCT/US2014/010863
state Cmax of the controlled release composition approximates, or is less
than, the
steady state Cmax for the corresponding immediate release tablets given b.i.d.
The
term "approximates" means +/- 15%, preferably +/- 10%. The steady state Cmin
for the controlled release composition is preferably about equal to, or
greater than,
the steady state Cmin for the corresponding immediate release tablets given
b.i.d.
or t.i.d (having the same total daily dose). In some embodiments the steady
state
Cmin approximates or is greater than the Cmin for the corresponding immediate
release tablets given b.i.d. The terms "about equal" and "approximates" have
the
same meaning as regards the Cmax.
1381 Though the steady state Cmax and Cmin values of the inventive
controlled release composition may be about equal to the Cmax and Cmin of the
corresponding dose of immediate release tablets in b.i.d. (or t.i.d.), the
controlled
release composition is believed to lower the risk of adverse events. Beyond
Cmax,
the change in the blood plasma concentration is also thought to contribute to
adverse events and/or the risk thereof The controlled release composition of
the
present has less fluctuation in blood plasma concentration; i.e., one peak and
trough
per 24 hours, versus 2 or 3 peak-trough cycles for immediate release therapy.
Moreover, the rate of increase of lorazepam blood concentration is believed to
correlate with adverse event risk: a slower rise in lorazepam blood plasma
concentration has less risk of adverse events. The composition of the present
invention in a once-daily dose form provides a slower increase in lorazepam
blood
concentration than the immediate release tablets. This difference can be
expressed
by the "Tmax," i.e., the time to regain steady state Cmax after a dose is
administered. Preferred embodiments of the pharmaceutical composition of the
invention typically provide a Tmax not sooner than 6 hours, more typically not
sooner than 8 hours, and often not sooner than 10 hours. Conversely, the Tmax
typically occurs not later than 14 hours, and usually not later than 13 hours.
A
Tmax of 12 hours +/- 1 hour (or even +/- 30 minutes) is generally preferred
for
once-daily dosing.
16
CA 02897313 2015-07-06
WO 2014/110248
PCT/US2014/010863
[39] Specific embodiments of the present invention relate to a
pharmaceutical composition that contains 2 mg of lorazepam and provides for
once-daily dosing. Such a composition preferably provides a steady state Cmax
of
26 ng/ml or less, usually 23 ng/ml or less when administered once daily. The
Cmin, however, does not fall below therapeutic levels. Preferably the Cmin is
at
least 10 ng/ml, sometimes at least 11 ng/ml, and can be at least 12 ng/ml,
when
administered once daily. The Tmax is typically within the range of 10 to 14
hours
after once-daily administration.
[40] Typically the composition of the present invention exhibits
dose proportionality within the range of 1-6 mg of lorazepam. The
proportionality
is typically with respect to the AUC (total exposure) but is also preferably
found
with the steady state Cmax. The following approximation can apply to preferred
embodiments regarding the steady state Cmax. Each 1 mg of lorazepam provides a
steady state Cmax of not greater than 10 ng/ml + 20%. Thus under this
embodiment, a 2 mg dose preferably provides a steady state Cmax within the
range
of 20 to 24 ng/ml or less; a 3 mg dose preferably provides a steady state Cmax
within the range of 30 to 36 ng/ml or less, etc.
[41] For clarity, all of the values for steady state Cmax, Cmin, and
Tmax can be for a single subject but more commonly are an average of multiple
subjects, e.g., multiple patients, multiple participants in a bioavailability
study, etc.
Also, the steady state values can be calculated from a single dose study by
methods
known in the art. Such calculated values are suitable for determining the
steady
state values for purposes of the present invention.
[42] The pharmaceutical compositions of the invention can be used to
treat any lorazepam-treatable condition. These conditions are most often
related to
the treatment or management of anxiety including anxiety related disorders.
Examples include, but are not limited to: Generalized Anxiety Disorder and
anxiety associated with major depression. But other uses for lorazepam can
also
apply to this invention; e.g., PTSD, insomnia and/or sleep disorders, bipolar
17
CA 02897313 2015-07-06
WO 2014/110248
PCT/US2014/010863
disorder, obsessive-compulsive disorder (OCD), social anxiety disorder,
convulsions, etc. The pharmaceutical compositions of the present invention are
generally administered once per day. Though the dose is usually administered
once
daily, some clinicians may elect to divide the total daily dose amount for
some
patients into one or more administrations per day.
[43] The invention will be further described with respect to the
following non-limiting example.
Example
[44] Pharmaceutical tablets having a PEO matrix were made having
the following nominal composition.
Reference Unit Composition
to Quality
Component Standards Function mg/Tablet % w/w
Lorazepam USP Active ingredient 2.0 1.35
Calcium hydrogen phosphate USP Filler/Compression 38.5 25.65
dihydrate (EMCOMPRESS ) aid
Polyethylene oxide NF Release control 67.5 45.00
(Polyox WSR N-12K) agent
Lactose monohydrate USP/NF Filler 40.5 27.00
Magnesium stearate USP/NF Lubricant 1.5 1
Total 1150.0 100.0
...............................................................................
.
...............................................................................
..
..............
USP = United States Pharmacopeia.
NF = National Formulary.
[45] The tablets are typically made by screening lorazepam (20 mesh
sieve) and the excipients (30 mesh sieve), except magnesium stearate, and
mixing
in a V-blender for approximately 20 minutes in total at 25 RPM. To the powder
mixture is added the magnesium stearate and mixed for approximately 2 minutes.
The resulting blend is tableted using 8 mm round standard concave punches/dies
to
form a batch of 6500 lorazepam tablets.
[46] The dissolution of the tablets is measured by subjecting a sample
tablets to an in vitro dissolution test. The conditions and typical results
are shown
below.
18
CA 02897313 2015-07-06
WO 2014/110248
PCT/US2014/010863
Apparatus USP Apparatus 2 Volume 500 mL
Rotation Speed 50 RPM Temperature 37 C 0.5 C
Number of
Medium pH 6.8 Phosphate Buffer Units 3
Collection Time (h)
1 2 4 6 8 10 12
Percent Dissolved 12 27 52 73 90 94 97
Mean
11 25 48 68 85 92 96
Min
14 29 55 76 93 97 98
Max
RSD 12.4 7.4 6.9 6.0 4.5 2.7 1.0
RSD = Relative Standard Deviation.
[47] The above pharmaceutical dissolution test shows that the tablets
have a substantially zero order release and reach 90% release at 8 hours. The
mean
dissolution values are plotted in figure 1.
[48] Each of the patents and articles mentioned above are incorporated
herein by reference. The invention having been described it will be obvious
that
the same may be varied in many ways and all such modifications are
contemplated
as being within the scope of the invention as defined by the following claims
19