Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02938126 2016-07-28
WO 2015/119624
PCT/US2014/015387
COMBINATIONS OF IKKeTBK1 INHIBITORS WITH BETA ADRENERGIC
AGONISTS OR SYMPATHETIC NERVOUS SYSTEM ACTIVATORS
STATEMENT REGARDING GOVERNMENT FUNDING
This invention was made with government support under DK060591 awarded by the
National Institutes of Health. The Government has certain rights in the
invention.
FIELD
Provided herein are methods of treating obesity and obesity-related conditions
comprising the administration of combinations of IKK8/TBK1 inhibitors with
beta adrenergic
agonists or sympathetic nervous system activators, and pharmaceutical
compositions
comprising such combinations.
BACKGROUND
Obesity generates a state of chronic, low-grade inflammation in liver and
adipose
tissue accompanied by macrophage infiltration and the local secretion of
inflammatory
cytokines and chemokines that attenuate insulin action, resulting in insulin
resistance and the
subsequent development of Type 2 diabetes (Wellen and Hotamisligil, 2005;
Hotamisligil,
2006; Lumeng et al., 2007; Shoelson et al., 2007; herein incorporated by
reference in their
entireties). Numerous studies indicate a strong correlation between
inflammation and insulin
resistance across several populations (Hotamisligil, 2006; herein incorporated
by reference in
its entirety). Moreover, genetic ablation or pharmacological inhibition of
inflammatory
pathways can dissociate obesity from insulin resistance (Hotamisligil, 2006;
Shoelson et al.,
2007; herein incorporated by reference in their entireties).
The transcription factor NFKB and its inflammatory program play an important
role in
the development of insulin resistance in obese liver and adipose tissue (Yuan
et al., 2001;
Arkan et al., 2005; Wunderlich et al., 2008; Chiang et al., 2009; herein
incorporated by
reference in their entireties). NFKB is activated by the IKB kinase (IKK)
family, which has
four members: IKKa, IKKI3, IKK8, and TBK1. IKKa and IKKI3 act with the
scaffolding
partner NEMO to activate NFKB (Hacker and Karin, 2006; herein incorporated by
reference
in its entirety). Although pharmacologic inhibition or genetic ablation of
IKKI3 defined a role
for this kinase in insulin resistance (Yuan et al., 2001; Arkan et al., 2005;
herein incorporated
by reference in their entireties), the roles of the noncanonical kinases IKK8
and TBK1 are not
certain.
1
CA 02938126 2016-07-28
WO 2015/119624
PCT/US2014/015387
Obesity is a complex metabolic disorder that is caused by increased food
intake and
decreased expenditure of energy. Obesity also increases the risk of developing
type 2
diabetes, heart disease, stroke, arthritis, and certain cancers. There is
considerable evidence to
suggest that adipose tissue becomes less sensitive to catecholamines such as
adrenaline in
states of obesity, and that this reduced sensitivity in turn reduces energy
expenditure.
However, the details of this process are not fully understood.
SUMMARY
Provided herein are methods of treating obesity and obesity-related conditions
comprising the administration of combinations of IKK8/TBK1 inhibitors with
beta adrenergic
agonists or sympathetic nervous system activators, and pharmaceutical
compositions
comprising such combinations.
In some embodiments, the present invention provides methods of treating a
subject
having a condition associated with obesity, insulin resistance, or hepatic
steatosis,
comprising: administering to a subject having a condition associated with
obesity, insulin
resistance, or hepatic steatosis: (i) an IKK8 and/or TBK1 inhibitor, and (ii)
a beta adrenergic
agonist or sympathetic nervous system activator. In some embodiments,
administering causes
a reduction of body fat in the subject. In some embodiments, the subject has
or is at risk of
experiencing obesity, diabetes, or insulin resistance. In some embodiments,
diabetes is type II
diabetes. In some embodiments, the treatment results in increased glucose
metabolism,
reduction in body fat, lack of increase in body fat, increased insulin
receptor signaling,
decreased level of insulin receptor phosphorylation, reduction in or
prevention of chronic
inflammation in the liver, reduction in or prevention of chronic inflammation
in adipose
tissue, reduction in or prevention of hepatic steatosis, promotion of
metabolic energy
expenditure, reduction in circulating free fatty acids, or reduction in
cholesterol. In some
embodiments, the subject has hepatic steatosis (fatty liver disease). In some
embodiments, the
subject has steatohepatitis. In some embodiments, the subject is overweight or
obese. In some
embodiments, the subject is human.
In some embodiments, methods further comprises a step comprising testing the
subject for a disease or condition selected from the group consisting of
impaired insulin
signaling, obesity, diabetes, insulin resistance, metabolic syndrome, hepatic
steatosis, chronic
liver inflammation, and chronic inflammation in adipose tissue. In some
embodiments,
method further comprises a step of assessing the effectiveness of treatment
based upon said
testing. In some embodiments, method further comprises adjusting the treatment
based on
2
CA 02938126 2016-07-28
WO 2015/119624
PCT/US2014/015387
said assessing. In some embodiments, adjusting the treatment comprises one or
more of
altering the dose of IKK8/TBK1 inhibitor, switching to a different IKK8/TBK1
inhibitor,
altering the dose of beta adrenergic agonist or sympathetic nervous system
activator,
switching to a different beta adrenergic agonist or sympathetic nervous system
activator,
adding additional treatment.
In some embodiments, a method comprises administering a IKK8 and/or TBK1
inhibitor and a beta adrenergic agonist or sympathetic nervous system
activator that are co-
formulated in a single pharmaceutical composition. In other embodiments, the
IKK8 and/or
TBK1 inhibitor, and the beta adrenergic agonist or sympathetic nervous system
activator are
separate pharmaceutical compositions and are co-administered (e.g., within 1
hour, within 20
minutes, within 15 minutes, within 5 minutes, within 1 minute, simultaneously,
etc.).
In some embodiments, the present invention provides pharmaceutical
compositions
comprising: (i) an IKK8 and/or TBK1 inhibitor, and (ii) a beta adrenergic
agonist or
sympathetic nervous system activator. In some embodiments, the IKK8 and/or
TBK1
inhibitor comprises a small molecule. In some embodiments, the IKK8 and/or
TBK1
inhibitor comprises the structure of Formula I:
0 N R;
Ri.),õ = 1
.{
1
COOH
(I) 0
wherein R1 is a hydrogen, alkyl, phenyl, carboxyl, hydroxyl, alkoxy, or amino
group,
which may be unsubstituted or substituted by one alkyl; m is 0, 1 or 2; and R2
is alkyl,
alkoxy, halogen, nitro, hydroxy, carboxyl, butadienylene ( CH¨CH CH¨CH ),
which forms
a benzene ring with any adjacent carbon atoms or amino group, which may be
unsubstituted
or substituted by at least one alkyl, and their physiologically acceptable
salts. In some
embodiments, the IKK8 and/or TBK1 inhibitor comprises amlexanox. In some
embodiments,
the beta adrenergic agonist or sympathetic nervous system activator comprises
a small
molecule. In some embodiments, the beta adrenergic agonist or sympathetic
nervous system
activator comprises a132 adrenergic receptor agonist. In some embodiments, the
small
molecule beta adrenergic agonist or sympathetic nervous system activator is
phentermine.
3
CA 02938126 2016-07-28
WO 2015/119624
PCT/US2014/015387
BRIEF DESCRIPTION OF THE DRAWINGS
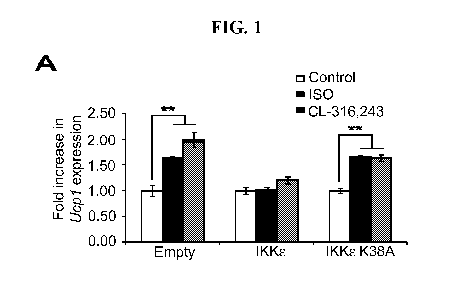
Fig. 1 shows IKK8 and TBK1 overexpression decrease sensitivity to the 13-
adrenergic/cAMP pathway in 3T3-L1 adipocytes. (A) Fold increase in Ucpl
expression in
3T3-L1 adipocytes expressing empty vector, Flag-IKK8, or Flag-IKK8 K38A
following
treatment with or without 10 [iM ISO (black bars) or 10 [iM CL-316,243 (CL,
gray bars) for
4 hr. (B) Glycerol release from 3T3-L1 adipocytes expressing empty vector
(white bars),
Flag-IKK8 (black bars), or Flag-IKK8 K38A (gray bars) treated with or without
10 [iM ISO
or 10 [LM CL. (C) Immunoblots of whole cell lysates from Figure 1B. Results
were replicated
in triplicate. D.E. stands for dark exposure and L.E. stands for light
exposure. (D)
Immunoblots of whole cell lysates from 3T3-L1 adipocytes expressing empty
vector or Flag-
IKK8 treated with or without 50 [iM FSK for 15 min. (E) cAMP levels from 3T3-
L1
adipocytes expressing empty vector, Flag-IKK8, or Flag-IKK8 K38A treated with
or without
10 1..LM ISO or 50 [iM FSK for 15 min.
Fig. 2 shows Prolonged treatment with TNFa decreases the sensitivity of
adipocytes
to 13-adrenergic stimulation in a manner dependent on the activity of IKK8 and
TBK1. (A)
Glycerol release from 3T3-L1 adipocytes treated with or without different
concentrations of
TNFa as indicated for 24 hr followed by treatment with or without 10 [iM ISO
or 50 [iM
FSK. (B) cAMP levels from 3T3-L1 adipocytes treated with or without 100 ng/ml
TNFa for
24 hr followed by treatment with or without 10 [iM ISO or 50 [iM FSK in the
presence or
absence of pretreatment of 50 [iM Amlexanox (Am). (C) cAMP levels from 3T3-L1
adipocytes treated with or without 100 ng/ml TNFa for 24 hr followed by
treatment with or
without 50 [iM FSK in the presence or absence of pretreatment of 1 [iM
CAY10576 (CAY).
(D) Immunoblots of whole cell lysates from 3T3-L1 adipocytes treated with or
without
different concentrations of TNFa as same as Figure 2A for 24 hr followed by
treatment with
or without 10 [iM ISO or 50 [iM FSK. Results were replicated in multiple
experiments. `['
indicates total HSL. `n.s.' represents non-specific band. Arrow indicates CGI-
58. (E)
Immunoblots of whole cell lysates from 3T3-L1 adipocytes treated with or
without 50 ng/ml
TNFa or 100 jig/ml poly (I:C) for 24 hr followed by treatment with or without
10 [iM ISO for
15 min in the presence or absence of pretreatment with increasing
concentrations (0, 10, 50,
and 200 [tM) of amlexanox for 30 min.
Fig. 3 shows IKK8 and TBK1 reduce cAMP levels through activation of PDE3B. (A)
cAMP levels from 3T3-L1 adipocytes expressing empty vector, Flag-IKK8, or Flag-
TBK1
treated with or without 50 [iM FSK, 250 [iM IBMX, or together for 15 min. (B)
cAMP levels
from 3T3-L1 adipocytes expressing empty vector, Flag-IKK8, or Flag-TBK1
treated with or
4
CA 02938126 2016-07-28
WO 2015/119624
PCT/US2014/015387
without 10 [iM ISO or 50 [iM FSK together with or without 10 [iM Zardaverine
(Zarda) for
15 min. (C) 32P phospho-image of in vitro kinase reaction using either
immunoprecipitated
HA-PDE3B from HEK293T cells or 1 [tg MBP (myelin basic protein) as a substrate
with
recombinant kinases as indicated. (D) Immunoblots of immunoprecipitation with
anti-HA
antibodies followed by treatment with or without CIP (top panel) and whole
cell lysates
(bottom panel) from Cos-1 cells co-expressing HA-PDE3B with Flag-IKK8/TBK1 or
Flag-
IKK8/TBK1 K38A. D.E. stands for dark exposure and L.E. stands for light
exposure. (E)
Immunoblots of GST-14-3-3 pulldown from HEK293T cells co-expressing HA-PDE3B
with
Flag-TBK1 or Flag- TBK1 K38A. Ponceau S staining shows the amount of beads
used in
GST-14-3-3 pulldown.
Fig. 4 shows IKK8 and TBK1 phosphorylate PDE3B at serine 318, resulting in the
binding of 14-3-30. (A) Summary of sites on PDE3B phosphorylated by IKK8 or
TBK1 (P-
sites) from mass spectrometry experiments. (B) Immunoblots of GST-14-3-3
pulldown from
HEK293T cells co-expressing HA-PDE3B or HA-PDE3B S318A with Flag-TBK1. Ponceau
S staining shows the amount of beads used in GST-14-3-3 pulldown. (C) GST-14-3-
3 overlay
on nitrocellulose membrane (top blot) and an immunoblot (IB) of whole cell
lysates from
HEK293T cells co-expressing HA-PDE3B or HA-PDE3B S318A with Flag-TBK1 (bottom
blot). (D) cAMP levels from 3T3-L1 adipocytes expressing empty vector, HA-
PDE3B, or
HA-PDE3B S318A treated with or without 100 ng/ml TNFa for 16 hr followed by
treatment
with or without 25 [iM FSK for 15 min.
Fig. 5 shows The IKK8/TBK1 inhibitor Amlexanox sensitizes 13-adrenergic
agonist-
stimulated lipolysis in white adipose tissue in diet-induced obese mice. (A)
Fold increase in
serum FFA (left panel) and glycerol (right panel) levels 15 min after CL-
316,243 injection in
ND- or HFD-fed mice treated with amlexanox or vehicle control for 4 days. (B)
Glycerol
release from ex vivo epididymal (left panel) and inguinal (right panel) WATs
after 1 hr
pretreatment with amlexanox or vehicle. CL-316,243 treatment was started at
time zero. (C)
Immunoblots in inguinal WAT lysates from Figure 5B after 60 min of CL-316,243
treatment.
(D) cAMP levels in epididymal WAT 20 min after CL-316,243 (CL) or saline
control
injection in HFD-fed mice treated with amlexanox or vehicle control for 4
days. (E)
Immunoblots in epididymal WAT 5 min after CL-316,243 or saline control
injection in HFD-
fed mice treated with amlexanox or vehicle control for 4 days. (F) Relative
oxygen
comsumption of mice in each treatment group.
DEFINITIONS
5
CA 02938126 2016-07-28
WO 2015/119624
PCT/US2014/015387
The terms "Inhibitor of nuclear factor kappa-B kinase subunit epsilon," "I-
kappa-B
kinase epsilon", "IKK8", and "IKKi" are used interchangeably herein to refer
to the enzyme
encoded by the IKBKE gene.
DETAILED DESCRIPTION
Provided herein are methods of treating obesity and obesity-related conditions
comprising the administration of combinations of IKK8/TBK1 inhibitors with
beta adrenergic
agonists or sympathetic nervous system activators, and pharmaceutical
compositions
comprising such combinations. In some embodiments, the present invention
provides
combinations of amlexanox or other IKKi/TBK1 inhibitors with beta-adrenergic
activators
(e.g., beta 2 or beta 3), or with agents that activate the sympathetic nervous
system (e.g.,
amphetamines, phentermine).
In some embodiments, the present invention provides a method of reducing body
fat
or preventing increase in body fat in a subject, comprising: administering to
a subject
experiencing or at risk of overweight or obese body composition a
therapeutically effective
dose of a (i) IKK8/TBK1 inhibitor and (ii)(A) a beta adrenergic agonist or (B)
sympathetic
nervous system activator. In some embodiments, the administration results in
reduction of or
prevention of increase in body fat in the subject. In some embodiments, the
subject is
experiencing or is at risk of experiencing a condition such as diabetes and
insulin resistance.
In some embodiments, administering pharmaceutical composition results in an
outcome such
as: increased glucose metabolism, increased insulin receptor signaling,
decreased level of
insulin receptor phosphorylation, reduction in or prevention of chronic
inflammation in liver,
reduction in or prevention of chronic inflammation in adipose tissue,
reduction in or
prevention of hepatic steatosis, promotion of metabolic energy expenditure,
reduction in
circulating free fatty acids, and/or reduction in cholesterol.
In some embodiments, an IKKi inhibitor is a TBK1/IKKi dual inhibitor. In some
embodiments, a TBK1/IKKi inhibitor is a small molecule. For example, in some
embodiments, a TBK1/IKKi dual inhibitor is a 2-amino-4-(3'-cyano-4'-
pyrrolidine)phenyl-
pyrimidine compound or derivatives or analogues thereof (Li et al., Int J
Cancer. 2013 Oct 6.
doi: 10.1002/ijc.28507; herein incorporated by reference in its entirety). In
other
embodiments, a TBK1/IKKi dual inhibitor is A20, TAXI BP1 (Parvatiyar et al.,
The Journal
of Biological Chemistry, 285, 14999-15009 (2010).; herein incorporated by
reference in its
entirety) or derivatives or analogues thereof In some embodiments, a TBK1/IKKi
inhibitor
is amlexanox, a derivative or analogue thereof, or a pharmaceutically
acceptable salt thereof
6
CA 02938126 2016-07-28
WO 2015/119624
PCT/US2014/015387
Amlexanox, or 2-amino-7-isopropyl-1-azaxanthone-3-carboxylic acid; 2-amino-7-
isopropy1-5-oxo-5H-chromeno[2,3-b]pyridine-3-carboxylic acid, is described in,
for example,
U.S. Pat. No. 4,143,042, herein incorporated by reference in its entirety. In
some
embodiments, the compound has the structure of Formula I:
0 y
.4,
COOfi
a
(I) 0
wherein R1 is a hydrogen, alkyl, phenyl, carboxyl, hydroxyl, alkoxy, or amino
group, which
may be unsubstituted or substituted by one alkyl; m is 0, 1 or 2; and R2 is
alkyl, alkoxy,
halogen, nitro, hydroxy, carboxyl, butadienylene ( CH¨CH CH¨CH ), which
forms a
benzene ring with any adjacent carbon atoms or amino group, which may be
unsubstituted or
substituted by at least one alkyl, and their physiologically acceptable salts.
The substituents
designated in each of the above-mentioned formulae may be substituted at
optional position
or positions of the 6-, 7-, 8-, or 9-positions of the azaxanthone ring.
In Formula (I), the alkyl group represented by R1 and R2 may be any of
straight-chain,
branched, or cyclic alkyl group having 1 to 6 carbon atoms. Typical examples
of the alkyl
group may be methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl,
tert-butyl, pentyl,
cyclopentyl, hexyl, cyclohexyl, etc.
The alkoxy group represented by R1 and R2 may, for example, be that having 1
to 4
carbon atoms in the alkyl moieties, such as methoxy, ethoxy, propoxy,
isopropoxy, butoxy,
etc.
The mono-alkyl substituted amino group represented by R1 may be that having 1
to 3
carbon atoms in the alkyl moieties, such as methylamino, ethylamino,
propylamino, or
isopropylamino. The halogen represented by R2 may be chlorine, bromine,
iodine, or
fluorine.
The alkyl substituted amino group represented by R2 includes mono- or di-alkyl
substituted ones whose alkyl moiety is that having 1 to 3 carbon atoms, e.g.,
methylamino,
ethylamino, propylamino, isopropylamino, dimethylamino, diethylamino, or
dipropylamino.
The compound of general Formula (I) can be converted to the corresponding
organic
amine salts, alkali metal salts, or ammonium salts by reacting (I) in the per
se conventional
manner with an organic amine (e.g., ethanolamine, diethanolamine, dl-
methylephedrin, 1-
(3,5-dihydroxypheny1)-L-isopropylaminoethanol, isoproterenol,
dextromethorphan, hetrazan
7
CA 02938126 2016-07-28
WO 2015/119624
PCT/US2014/015387
(diethylcarbamazine), diethylamine, triethylamine, etc.), an alkali metal
hydroxide (e.g.,
sodium hydroxide, potassium hydroxide, etc.) or ammonia, for example by mixing
them
together and heating in a suitable solvent.
In some embodiments, a sympathomimetic agent (e.g., small molecule, peptide,
RNA,
etc.) is provided that acts upon a beta adrenoceptor, having the opposite
effect of a beta
blocker. In some embodiments, a beta adrenergic receptor agonist is provided.
In some
embodiments, beta adrenergic receptor agonists mimic the action of epinephrine
and
norepinephrine signaling. In some embodiments, a beta adrenergic receptor
agonist activates
one or more of 01,132, and P3 receptors.
In some embodiments, a 01 agonist selected from Dobutamine, Isoproterenol (01
and
132), Xamoterol, epinephrine, etc. is provided. Other suitable 01 agonists are
within the scope
of the invention.
In some embodiments, a 132 agonist selected from salbutamol (albuterol in USA)
levosalbutamol (Levalbuterol in USA), fenoterol, formoterol, isoproterenol (01
and 132),
metaproterenol, salmeterol, terbutaline, clenbuterol, isoetarine, pirbuterol,
procaterol,
ritodrine, epinephrine, etc. is provided. Other suitable 132 agonists are
within the scope of the
invention.
In some embodiments, any suitable P3 agonists (e.g., Mirabegron) are within
the
scope of the invention.
In some embodiments, a beta adrenergic receptor agonist is select from the
list
including, but not limited to salbutamol (albuterol, Ventolin), levosalbutamol
(levalbuterol,
Xopenex), terbutaline (Bricanyl), pirbuterol (Maxair), procaterol,
clenbuterol, metaproterenol
(Alupent), fenoterol, bitolterol mesylate, ritodrine, isoprenaline, salmeterol
(Serevent
Diskus), formoterol (Foradil, Symbicort), bambuterol, clenbuterol,
indacaterol, arbutamine,
befunolol
bromoacetylalprenololmenthane, broxaterol, cimaterol, cirazoline, denopamine,
dopexamine,
etilefrine, hexoprenaline, higenamine, isoxsuprine, mabuterol,
methoxyphenamine, nylidrin,
oxyfedrine, prenalterol, ractopamine, reproterol, rimiterol, tretoquinol,
tulobuterol, zilpaterol,
zinterol, etc.
In some embodiments, a sympathetic nervous system activator is provided. Such
agents may activate the sympathetic nervous system by any suitable mechanism
(e.g., acting
on, increasing the release of, or inhibiting reuptake of one or more
neurotransmitters (e.g.,
norepinephrine, serotonin and dopamine epinephrine and/or adrenaline), acting
as an
adrenergic receptor agonist, etc.). Suitable sympathetic nervous system
activators may be
8
CA 02938126 2016-07-28
WO 2015/119624
PCT/US2014/015387
selected from Benzodiazepines (e.g., Diazepam (Valium), clonazepam (Klonopin),
lorazepam
(Ativan), temazepam (Restoril), flunitrazepam (Rohypnol), triazolam (Halcion),
alprazolam
(Xanax), etc.), Amphetamines (e.g., Amphetamine (Adderall), methamphetamine
(Desoxyn),
methylphenidate (Ritalin), phentermine, 4-methylaminorex, phenmetrazine
(Preludin),
methcathinone, fenfluramine (Pondimin, Fen-Phen), dexfenfluramine (Redux),
pseudoephedrine (Sudafed), ephedrine, phenylpropanolamine (old Triaminic),
phenylephrine
(Sudafed PE), etc.), phentermine, topiramate, etc. Other suitable sympathetic
nervous system
activators are within the scope of the invention.
In some embodiments, the present invention finds use in the treatment or
prevention
of overweight and obesity. The most widely accepted clinical definition of
obesity is the
World Health Organization (WHO) criteria based on BMI. Under this convention
for adults,
grade 1 overweight (commonly and simply called overweight) is a BMI of 25-29.9
kg/m2.
Grade 2 overweight (commonly called obesity) is a BMI of 30-39.9 kg/m2. Grade
3
overweight (commonly called severe or morbid obesity) is a BMI greater than or
equal to 40
kg/m2. The surgical literature often uses a different classification to
recognize particularly
severe obesity. In this setting, a BMI greater than 40 kg/m2 is described as
severe obesity, a
BMI of 40-50 kg/m2 is termed morbid obesity, and a BMI greater than 50 kg/m2
is termed
super obese. The definition of obesity in children involves BMIs greater than
the 85th
(commonly used to define overweight) or the 95th (commonly used to define
obesity)
percentile, respectively, for age-matched and sex-matched control subjects.
Secondary causes
of obesity include but are not limited to hypothyroidism, Cushing syndrome,
insulinoma,
hypothalamic obesity, polycystic ovarian syndrome, genetic syndromes (eg,
Prader-Willi
syndrome, Alstrom syndrome, Bardet-Biedl syndrome, Cohen syndrome, Borjeson-
Forssman-Lehmann syndrome, Frohlich syndrome), growth hormone deficiency, oral
contraceptive use, medication-induced obesity (e.g., phenothiazines, sodium
valproate,
carbamazepine, tricyclic antidepressants, lithium, glucocorticoids, megestrol
acetate,
thiazolidine diones, sulphonylureas, insulin, adrenergic antagonists,
serotonin antagonists
[especially cyproheptadine]), eating disorders (especially binge-eating
disorder, bulimia
nervosa, night-eating disorder), hypogonadism, pseudohypoparathyroidism, and
obesity
related to tube feeding. In some embodiments, pharmaceutical combinations and
treatments
described herein find use in the treatment of one or more of the
aforementioned secondary
causes of obesity.
In some embodiments, a subject is tested to assess the presence, the absence,
or the
level of a disease or condition (e.g., obesity and/or a related disorder,
including, but not
9
CA 02938126 2016-07-28
WO 2015/119624
PCT/US2014/015387
limited to insulin resistance, diabetes, steatosis, nonalcoholic steatotic
hepatitis, and
atherosclerosis), e.g., by assaying or measuring a biomarker, a metabolite, a
physical
symptom, an indication, etc., to determine the risk of or the presence of
obesity and/or a
related disorder, including, but not limited to insulin resistance, diabetes,
steatosis,
nonalcoholic steatotic hepatitis, and atherosclerosis, and thereafter the
subject is treated with
a pharmaceutical combination described herein based on the outcome of the
test. In some
embodiments, a patient is tested, treated, and then tested again to monitor
the response to
therapy. In some embodiments, cycles of testing and treatment may occur
without limitation
to the pattern of testing and treating (e.g., test/treat, test/treat/test,
test/treat/test/treat,
test/treat/test/treat/test, test/treat/treat/test/treat/treat, etc.), the
periodicity, or the duration of
the interval between each testing and treatment phase.
It is generally contemplated that the compositions and/or pharmaceutical
combinations according to the technology provided are formulated for
administration to a
mammal, and especially to a human with a condition that is responsive to the
administration
of such compounds. Therefore, where contemplated compounds are administered in
a
pharmacological composition or combination, it is contemplated that a
formulation may be in
the form of an admixture with a pharmaceutically acceptable carrier. For
example,
contemplated compounds and combinations can be administered orally as
pharmacologically
acceptable salts, or intravenously in a physiological saline solution (e.g.,
buffered to a pH of
about 7.2 to 7.5). Conventional buffers such as phosphates, bicarbonates, or
citrates can be
used for this purpose. Of course, one of ordinary skill in the art may modify
the formulations
within the teachings of the specification to provide numerous formulations for
a particular
route of administration. In particular, contemplated compounds may be modified
to render
them more soluble in water or other vehicle, which for example, may be easily
accomplished
with minor modifications (salt formulation, esterification, etc.) that are
well within the
ordinary skill in the art. It is also well within the ordinary skill of the
art to modify the route
of administration and dosage regimen of a particular compound in order to
manage the
pharmacokinetics of the present compounds for maximum beneficial effect in a
patient.
In certain pharmaceutical dosage forms, prodrug forms of contemplated
compounds
may be formed for various purposes, including reduction of toxicity,
increasing the organ or
target cell specificity, etc. Among various prodrug forms, acylated
(acetylated or other)
derivatives, pyridine esters, and various salt forms of the present compounds
are preferred.
One of ordinary skill in the art will recognize how to readily modify the
present compounds
to prodrug forms to facilitate delivery of active compounds to a target site
within the host
CA 02938126 2016-07-28
WO 2015/119624
PCT/US2014/015387
organism or patient. One of ordinary skill in the art will also take advantage
of favorable
pharmacokinetic parameters of the prodrug forms, where applicable, in
delivering the present
compounds to a targeted site within the host organism or patient to maximize
the intended
effect of the compound. Similarly, it should be appreciated that contemplated
compounds
may also be metabolized to their biologically active form, and all metabolites
of the
compounds herein are therefore specifically contemplated. In addition,
contemplated
compounds (and combinations thereof) may be administered in combination with
yet further
agents for treating obesity and related disorders, including, but not limited
to insulin
resistance, diabetes, steatosis, nonalcoholic steatotic hepatitis, and
atherosclerosis.
With respect to administration to a subject, it is contemplated that the
compounds
and/or combinations be administered in a pharmaceutically effective amount.
One of ordinary
skill recognizes that a pharmaceutically effective amount varies depending on
the therapeutic
agent used, the subject's age, condition, and sex, and on the extent of the
disease in the
subject. Generally, the dosage should not be so large as to cause adverse side
effects, such as
hyperviscosity syndromes, pulmonary edema, congestive heart failure, and the
like. The
dosage can also be adjusted by the individual clinician to achieve the desired
therapeutic
goal.
As used herein, the actual amount encompassed by the term "pharmaceutically
effective amount" will depend on the route of administration, the type of
subject being
treated, and the physical characteristics of the specific subject under
consideration. These
factors and their relationship to determining this amount are well known to
skilled
practitioners in the medical, veterinary, and other related arts. This amount
and the method of
administration can be tailored to achieve optimal efficacy but will depend on
such factors as
weight, diet, concurrent medication, and other factors that those skilled in
the art will
recognize.
In some embodiments, a single pharmaceutical composition comprising both a (i)
IKK8/TBK1 inhibitor and (ii) (A) a beta adrenergic agonist or (B) sympathetic
nervous
system activator is provided. In other embodiments, separate pharmaceutical
compositions
are administered, one comprising an IKK8/TBK1 inhibitor and another comprising
a beta
adrenergic agonist and/or sympathetic nervous system activator. Dosing and
scheduling of
administration of separate pharmaceutical compositions may be determined
and/or adjusted
jointly or separately.
The dosage amount and frequency are selected to create an effective level of
the
compound without substantially harmful effects. When administered orally or
intravenously,
11
CA 02938126 2016-07-28
WO 2015/119624
PCT/US2014/015387
the dosage will generally range from 0.001 to 10,000 mg/kg/day or dose (e.g.,
0.01 to 1000
mg/kg/day or dose; 0.1 to 100 mg/kg/day or dose, 1 to 100 mg/kg/day or dose,
or amounts
therein).
In some embodiments, a single dose is administered to a subject. In other
embodiments, multiple doses are administered over two or more time points,
separated by
hours, days, weeks, etc. In some embodiments, pharmaceutical compositions are
administered over a long period of time (e.g., chronically), for example, for
a period of
months or years (e.g., 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, or more months
or years). In such
embodiments, pharmaceutical compositions may be taken on a regular scheduled
basis (e.g.,
daily, weekly, etc.) for the duration of the extended period.
Methods of administering a pharmaceutically effective amount include, without
limitation, administration in parenteral, oral, intraperitoneal, intranasal,
topical, sublingual,
rectal, and vaginal forms. Parenteral routes of administration include, for
example,
subcutaneous, intravenous, intramuscular, intrastemal injection, and infusion
routes. In some
embodiments, amlexanox, a derivative thereof, or a pharmaceutically acceptable
salt thereof,
is administered orally.
Pharmaceutical compositions preferably comprise one or more compounds of the
present technology associated with one or more pharmaceutically acceptable
carriers,
diluents, or excipients. Pharmaceutically acceptable carriers are known in the
art such as
those described in, for example, Remingtons Pharmaceutical Sciences, Mack
Publishing Co.
(A. R. Gennaro edit. 1985), explicitly incorporated herein by reference for
all purposes.
Accordingly, in some embodiments, the composition (comprising one agent or a
pharmaceutical composition) is formulated as a tablet, a capsule, a time
release tablet, a time
release capsule; a time release pellet; a slow release tablet, a slow release
capsule; a slow
release pellet; a fast release tablet, a fast release capsule; a fast release
pellet; a sublingual
tablet; a gel capsule; a microencapsulation; a transdermal delivery
formulation; a transdermal
gel; a transdermal patch; a sterile solution; a sterile solution prepared for
use as an
intramuscular or subcutaneous injection, for use as a direct injection into a
targeted site, or
for intravenous administration; a solution prepared for rectal administration;
a solution
prepared for administration through a gastric feeding tube or duodenal feeding
tube; a
suppository for rectal administration; a liquid for oral consumption prepared
as a solution or
an elixir; a topical cream; a gel; a lotion; a tincture; a syrup; an emulsion;
or a suspension.
In some embodiments, the time release formulation is a sustained-release,
sustained-
action, extended-release, controlled-release, modified release, or continuous-
release
12
CA 02938126 2016-07-28
WO 2015/119624
PCT/US2014/015387
mechanism, e.g., the composition is formulated to dissolve quickly, slowly, or
at any
appropriate rate of release of therapeutic agents over time.
In some embodiments, the pharmaceutical preparations and/or formulations of
the
technology are provided in particles. Particles as used herein means nano or
microparticles
(or in some instances larger) that can consist in whole or in part of the
therapeutic agents as
described herein. The particles may contain the preparations and/or
formulations in a core
surrounded by a coating, including, but not limited to, an enteric coating.
The preparations
and/or formulations also may be dispersed throughout the particles. The
preparations and/or
formulations also may be adsorbed into the particles. The particles may be of
any order
release kinetics, including zero order release, first order release, second
order release, delayed
release, sustained release, immediate release, and any combination thereof,
etc. The particle
may include, in addition to the preparations and/or formulations, any of those
materials
routinely used in the art of pharmacy and medicine, including, but not limited
to, erodible,
nonerodible, biodegradable, or nonbiodegradable material or combinations
thereof. The
particles may be microcapsules which contain the formulation in a solution or
in a semi-solid
state. The particles may be of virtually any shape.
Both non-biodegradable and biodegradable polymeric materials can be used in
the
manufacture of particles for delivering the preparations and/or formulations.
Such polymers
may be natural or synthetic polymers. The polymer is selected based on the
period of time
over which release is desired. Bioadhesive polymers of particular interest
include bioerodible
hydrogels described by H. S. Sawhney, C. P. Pathak and J. A. Hubell in
Macromolecules,
(1993) 26: 581-587, the teachings of which are incorporated herein by
reference. These
include polyhyaluronic acids, casein, gelatin, glutin, polyanhydrides,
polyacrylic acid,
alginate, chitosan, poly(methyl methacrylates), poly(ethyl methacrylates),
poly(butylmethacrylate), poly (isobutyl methacrylate),
poly(hexylmethacrylate),
poly(isodecyl methacrylate), poly(lauryl methacrylate),
poly(phenylmethacrylate),
poly(methyl acrylate), poly(isopropyl acrylate), poly(isobutyl acrylate), and
poly(octadecyl
acrylate).
In some embodiments, the pharmaceutical compositions are formulated with a
buffering agent. The buffering agent may be any pharmaceutically acceptable
buffering
agent. Buffer systems include citrate buffers, acetate buffers, borate
buffers, and phosphate
buffers. Examples of buffers include citric acid, sodium citrate, sodium
acetate, acetic acid,
sodium phosphate and phosphoric acid, sodium ascorbate, tartartic acid, maleic
acid, glycine,
13
CA 02938126 2016-07-28
WO 2015/119624
PCT/US2014/015387
sodium lactate, lactic acid, ascorbic acid, imidazole, sodium bicarbonate and
carbonic acid,
sodium succinate and succinic acid, histidine, and sodium benzoate and benzoic
acid.
EXPERIMENTAL
Obesity produces a chronic inflammatory state involving the NFKB pathway,
resulting
in persistent elevation of the noncanonical IKB kinases IKKi and TBK1.
Experiments
conducted during development of embodiments of the present invention
demonstrate that
these kinases attenuate P-adrenergic signaling in white adipose tissue.
Treatment of 3T3-L1
adipocytes with specific inhibitors of these kinases restored P-adrenergic
signaling and
lipolysis attenuated by TNFa and Poly (I:C). Conversely, overexpression of the
kinases
reduced induction of Ucpl, lipolysis, cAMP levels, and phosphorylation of
hormone sensitive
lipase in response to isoproterenol or forskolin. Noncanonical IKKs reduce
catecholamine
sensitivity by phosphorylating and activating the major adipocyte
phosphodiesterase PDE3B.
In vivo inhibition of these kinases by treatment of obese mice with the drug
amlexanox
reversed obesity-induced catecholamine resistance, and restored PKA signaling
in response
to injection of a P-3 adrenergic agonist. These studies indicate that by
reducing production of
cAMP in adipocytes, IKK8 and TBK1 contribute to the repression of energy
expenditure
during obesity.
Example 1
IKKt and TBK1 overexpression decrease sensitivity to the 13-adrenergic/cAMP
pathway
in 3T3-L1 adipocytes
Sympathetic activation of adipose tissue is involved in maintaining energy
balance by
stimulating lipolysis and fat oxidation (Coppack et al., 1994; Langin, 2006;
Festuccia et al.,
2011; herein incorporated by reference in their entireties). Activation of P-
adrenergic
signaling by either P-adrenergic agonists or cold exposure in white and brown
adipose tissue
initiates a cascade of events through cyclic AMP (cAMP), culminating in the
transcriptional
upregulation of Ucpl, which results in increased proton leak and energy
expenditure
(Himms-Hagen et al., 2000; Cao et al., 2004; Yehuda-Shnaidman et al., 2010;
herein
incorporated by reference in their entireties). Compared to wild-type (WT)
controls, IKK8-
deficient mice exhibit increased energy expenditure while on a high fat diet
(HFD),
accompanied by increased expression of Ucpl in white adipose depots (Chiang et
al., 2009;
herein incorporated by reference in its entirety). Increased energy
expenditure in IKK8-
deficient mice was only seen in HFD-fed mice (Chiang et al., 2009; herein
incorporated by
14
CA 02938126 2016-07-28
WO 2015/119624
PCT/US2014/015387
reference in its entirety), indicating that upon induction of IKK8 during
obesity, the kinase
represses an increased adaptive thermogenic response to overnutrition. This
effect was further
analyzed by overexpressing IKK8 in 3T3-L1 adipocytes and examining Ucpl gene
expression after treatment with the non-selective 13-adrenergic agonist,
isoproterenol (ISO), or
the 133-adrenergic agonist, CL-316,243. Fold difference in Ucpl gene
expression was
calculated by normalization of relative Ucpl mRNA levels in treated relative
to control
samples. Treatment of empty vector-expressing cells with ISO or CL-316,243
resulted in a
1.6-fold or twofold increase in Ucpl mRNA levels, respectively (Figure 1A).
The induction
of Ucpl gene expression in response to ISO or CL-316,243 was blunted when WT
IKK8was
overexpressed in these cells. However, expression of the kinase-inactive
mutant of IKK8
K3 8A (Fitzgerald et al., 2003; herein incorporated by reference in its
entirety) was less
effective, but still modestly repressed Ucpl expression.
In addition to increased Ucpl expression, IKK8 knockout mice also exhibited
increased lipolysis and fat oxidation (Chiang et al., 2009; herein
incorporated by reference in
its entirety), suggesting that decreased lipolysis in adipose tissue from
obese mice might
result in part from increased expression of IKK8 and TBK1 (Chiang et al.,
2009; herein
incorporated by reference in its entirety). The obesity-dependent increase was
modeled in the
noncanonical IKKs by overexpressing IKK8 in 3T3-L1 adipocytes, followed by
assay of
glycerol release in response to ISO or CL-316,243. Although both isoproterenol
and CL-
316,243 increased lipolysis in empty vector-expressing cells, overexpression
of WT
IKK8reduced the lipolytic effects of isoproterenol and CL-316,243 by greater
than 40%, and
also reduced basal glycerol release (Figure 1B). The reduction in lipolysis by
IKK8
overexpression was accompanied by dramatically reduced phosphorylation of HSL
and
perilipin in response to ISO or CL-316,243 (Figure 1C). Expression of the
catalytically
inactive kinase was less effective in blocking lipolytic signaling, although
the levels of
protein achieved by overexpression were lower compared to the WT kinase
(Figure 1B,C,
Figure 1). Overexpression of TBK1 reduced phosphorylation of HSL in response
to
isoproterenol or the adenylyl cyclase activator, forskolin (Figure 1).
Identical results were
obtained when IKK8 was overexpressed in 3T3-L1 adipocytes stimulated with
forskolin
(Figure 1D), as detected by western blotting with an anti-phospho-PKA
substrate motif
antibody.
Overexpression of IKK8 also repressed the phosphorylation of p38 (p-p38) in
response to forskolin (Figure 1D) or isoproterenol (Figure 1), whereas
overexpression of
IKK8K38A was without effect (Figure 1). While glycerol release is likely the
result of
CA 02938126 2016-07-28
WO 2015/119624
PCT/US2014/015387
changes in HSL and perilipin phosphorylation, it is important to note that we
have not
directly assayed whether re-esterification of glycerol intermediates are also
affected. Taken
together, these data indicate that similar to what is observed in obesity,
overexpression of
IKK8 or TBK1 can repress lipolytic signaling. It is contemplated that the
partial effectiveness
of the kinase-inactive mutants reflects their activation of endogenous IKK8 or
TBK1 kinases
due to dimerization (Larabi et al., 2013; Tu et al., 2013; herein incorporated
by reference in
their entireties).
Since PKA signaling is responsible for Ucpl induction in response to
catecholamines
(Klein et al., 2000; Cao et al., 2001; herein incorporated by reference in
their entireties);
experiments were conducted during development of embodiments of the present
invention to
investigate IKK8 and TBK1 induced reduction of13-adrenergic sensitivity of
adipocytes by
decreasing cAMP levels. IKK8 overexpression in 3T3-L1 adipocytes reduced by
greater than
80% the increase in cAMP levels produced by both isoproterenol and forskolin,
whereas
overexpression of IKK8 K38A did not (Figure 1E). Previous studies have shown
that
decreased sensitivity to adrenergic stimuli in adipose tissue can result from
reduced 0-
adrenergic receptors (Reynisdottir et al., 1994; herein incorporated by
reference in its
entirety) or increased expression of a2-adrenergic receptors (Stich et al.,
2002; herein
incorporated by reference in its entirety).
Example 2
Prolonged treatment with TNFa decreases the sensitivity of adipocytes to 13-
adrenergic
stimulation in a manner dependent on the activity of IKKt and TBK1
Obesity is accompanied by infiltration of proinflammatory macrophages into
adipose
tissue; these cells secrete inflammatory cytokines, such as TNFa, which
generate insulin
resistance by stimulating catabolic pathways (Hotamisligil, 2006; Lumeng et
al., 2007; Ye
and Keller, 2010; Ouchi et al., 2011; herein incorporated by reference in
their entireties).
Although TNFa is known to increase lipolysis in adipocytes (Zhang et al.,
2002; Souza et al.,
2003; Green et al., 2004; Plomgaard et al., 2008; herein incorporated by
reference in their
entireties), there is also evidence of a counterinflammatory response in
obesity that may serve
to repress energy expenditure (Gregor and Hotamisligil, 2011; Saltiel, 2012;
Calay and
Hotamisligil, 2013; Reilly et al., 2013; herein incorporated by reference in
their entireties).
Experiments were conducted during development of embodiments of the present
invention
using TNFa to model the inflammatory milieu of obese adipose tissue in cell
culture to
determine whether the cytokine might also regulate 13-adrenergic signaling in
this context.
16
CA 02938126 2016-07-28
WO 2015/119624
PCT/US2014/015387
While short-term treatment with TNFa augmented the increase in cAMP produced
by
forskolin treatment, this effect declined after 12 hr. After 24 hr of
exposure, TNFa inhibited
the production of the second messenger produced by forskolin (Figure 2). Thus,
the catabolic
effects of the proinflammatory cytokine TNFa in adipocytes are transient and
followed by an
inhibitory phase.
Treatment of 3T3-L1 adipocytes with TNFa for 24 hr induced the expression of
IKK8
and increased TBK1 phosphorylation at the active site in a manner that was
dependent on the
activity of IKKI3 and the NFKB pathway (Reilly et al., 2013; herein
incorporated by reference
in its entirety). Experiments were conducted during development of embodiments
of the
present invention to determine whether the repression of13-adrenergic
sensitivity produced by
longer-term treatment with TNFa is due to increased activity of the
noncanonical IKKs.
Long-term treatment with TNFa repressed the induction of Ucpl gene expression
in response
to 13-adrenergic stimuli (Figure 2), whereas the expression of IKK8 mRNA
(Ikbke) was
upregulated. Treatment of 3T3-L1 adipocytes with TNFa for 24 hr decreased
glycerol release
in response to both isoproterenol and forskolin in a dose-dependent manner
(Figure 2A).
TNFa treatment also decreased isoproterenol- and forskolin-stimulated cAMP
production; an
effect that was largely rescued by preincubation of cells with the selective,
but structurally
unrelated inhibitors of IKK8 and TBK1, amlexanox (Figure 2B) (Reilly et al.,
2013; herein
incorporated by reference in its entirety) or CAY10576 (Figure 2C) (Bamborough
et al.,
2006; herein incorporated by reference in its entirety). Isoproterenol-
stimulated13-adrenergic
signaling was also decreased by treatment of cells with TNFa (Figure 2D), as
manifested by
decreased phosphorylation of HSL, perilipin, and other proteins recognized by
the PKA
substrate motif antibody, whereas IKK8 expression was concurrently upregulated
and TBK1
phosphorylation was increased by the treatment with TNFa. Pretreatment of 3T3-
L1
adipocytes with amlexanox also blocked the inhibitory effect of TNFa on
isoproterenol-
stimulated 13-adrenergic signaling, as determined by western blotting with an
anti-phospho-
PKA substrate motif antibody, antiphospho-HSL, and anti-phospho-perilipin
antibodies
(Figure 2E). Phosphorylation of p38 in response to isoproterenol was also
dramatically
augmented by amlexanox in a dose-dependent manner. Previous studies showed
that the toll-
like receptor 3 (TLR3) agonist, Poly (I:C), results in the direct activation
of IKK8 and TBK1
(Hemmi et al., 2004; Clark et al., 2009; Clark et al., 2011; herein
incorporated by reference in
their entireties). Similar to TNFa, treatment of 3T3-L1 adipocytes with Poly
(I:C)
simultaneously reduced stimulation of cAMP production, lipolysis and
phosphorylation in
response to 13-adrenergic stimulation (Figure 2), and the inhibitory effects
of Poly (I:C) on the
17
CA 02938126 2016-07-28
WO 2015/119624
PCT/US2014/015387
sensitivity to isoproterenol stimulation were partially restored by amlexanox
pretreatment,
but not to the extent that was observed with TNFa treatment (Figure 2E). These
results
indicate that obesity-associated inflammation leads to the activation of IKK8
and TBK1,
which produces reduced sensitivity of adipocytes to 13-adrenergic stimulation.
Example 3
IKKt and TBK1 reduce cAMP levels through activation of PDE3B
cAMP levels can also be regulated by phosphodiesterases, which cleave the
second
messenger and in the process dampen cAMP-dependent signals. Phosphodiesterase
3B
(PDE3B) is the major PDE isoform expressed in adipocytes (Zmuda-Trzebiatowska
et al.,
2006; herein incorporated by reference in its entirety). Genetic ablation or
pharmacological
inhibition of PDE3B in cells and in vivo revealed an important role for the
enzyme in lipid
and glucose metabolism (Choi et al., 2006; Berger et al., 2009; Degerman et
al., 2011; herein
incorporated by reference in their entireties). Phosphorylation and activation
of PDE3B by
insulin in adipocytes is thought to be mediated by Akt, and cAMP itself acts
as a negative
feedback regulator of its own levels by promoting PKA-dependent
phosphorylation and
activation of PDE3B (Degerman et al., 2011; herein incorporated by reference
in its entirety).
Experiments conducted during development of embodiments of the present
invention
demonstrated that cAMP production was impaired in forskolin or isoproterenol-
stimulated
3T3-L1 adipocytes overexpressing IKK8 (Figure 1E), therefore additional
experiments were
conducted to determine whether noncanonical IKKs might desensitize adrenergic
stimulation
through increased activity of PDE3B in adipocytes. Pretreatment with a
nonspecific
phosphodiesterase inhibitor, IBMX, in 3T3-L1 adipocytes expressing IKK8 or
TBK1 rescued
the full stimulation of cAMP production in response to forskolin (Figure 3A).
Interestingly,
the selective PDE3B and PDE4 inhibitor, Zardaverine (Schudt et al., 1991;
herein
incorporated by reference in its entirety), also blocked the inhibitory
effects of IKK8 and
TBK1 overexpression on cAMP levels in response to isoproterenol and forskolin
in 3T3-L1
adipocytes (Figure 3B), suggesting an important role for PDE3B as a target of
the
noncanonical IKKs.
It was next examined whether IKK8 and TBK1 directly phosphorylate PDE3B to
regulate cAMP levels. Recombinant TBK1, Akt and PKA were incubated in vitro
with [y-
3211ATP and purified PDE3B as a substrate. Phosphorylation was assessed by SDS-
PAGE
followed by autoradiography. TBK1 directly catalyzed the phosphorylation of
PDE3B;
phosphorylation was also produced by incubation with Akt and PKA, as
previously reported
18
CA 02938126 2016-07-28
WO 2015/119624
PCT/US2014/015387
(Kitamura et al., 1999; Palmer et al., 2007; herein incorporated by reference
in their
entireties) (Figure 3C). IKK8 also catalyzed this phosphorylation in vitro.
This increase in
phosphorylation produced by in vitro incubation with TBK1, IKK8 and PKA was
also
detected when PDE3B was blotted with antibodies that recognize the 14-3-3
binding motif
(Figure 3). When purified PDE3B
was incubated with the same amount of recombinant TBK1 and canonical IKKI3
kinases in
vitro, phosphorylation of PDE3B by IKKI3 was barely detectable, indicating a
level of
specificity in which PDE3B is a better target of the noncanonical IKKs (Figure
3). This
phosphorylation was dose-dependent with respect to ATP (Figure 3).
To determine whether IKK8 can phosphorylate PDE3B in cells, IKK8 and its
inactive
mutant K38A were co-expressed with HA-tagged PDE3B in HEK293T cells, followed
by
immunoprecipitation (IP) with anti-HA antibodies. Expression of IKK8 in cells
caused a shift
in electrophoretic mobility of PDE3B, and this shift was not detected when
IKK8 K38A was
expressed (Figure 3). Phosphorylation of PDE3B was also detected after
expression of IKK8
but not its kinase-inactive mutant K38A in cells, as detected by blotting with
antibodies that
recognize the 14-3-3 binding motif. To determine whether this molecular shift
was dependent
on phosphorylation of PDE3B, HA-PDE3B was co-expressed in Cos-1 cells along
with
IKK8, TBK1 or their kinase inactive mutants, and HA immunoprecipitates were
treated with
or without calf intestinal phosphatase (CIP). Expression of both of the wild-
type kinases
reduced the electrophoretic mobility of PDE3B, which could be reversed by
treatment with
the phosphatase (Figure 3D, compare lane 3, 7 to lane 4, 8). Neither of the
kinase-inactive
mutants had an effect (Figure 3D, compare lane 5, 9 to lane 6, 10).
Previous studies suggested that IKK8 and TBK1 bind to their respective
substrates
through a sequence that includes a ubiquitin-like domain (ULD) proximal to
their kinase
domain. This domain is highly conserved among the IKK family members, and is
49%
identical between IKK8 and TBK1 (Ikeda et al., 2007; May et al., 2004; herein
incorporated
by reference in their entireties). To confirm that PDE3B is a bona fide
substrate of IKK8 and
TBK1, a GST-ULD domain fusion protein was prepared from TBK1 and incubated
this
fusion protein with 3T3-L1 adipocyte lysates. The fusion protein specifically
precipitated
endogenous PDE3B from these lysates (Figure 3). To explore further the
interaction of these
two proteins, WT TBK1 and its K38A mutant were co-expressed with HA-tagged
PDE3B in
HEK293T cells, and immunoprecipitated the protein with anti-HA antibodies.
Kinase-
inactive TBK1 was preferentially co-immunoprecipitated with PDE3B, whereas the
interaction of PDE3B with WT TBK1 was barely detectable (Figure 3). These data
indicate
19
CA 02938126 2016-07-28
WO 2015/119624
PCT/US2014/015387
that TBK1 and IKK8 associate with substrates such as PDE3B, and subsequently
dissociate
upon phosphorylation.
Too test further the role of PDE3B phosphorylation by IKK8 and TBK1 in
initiating
its interaction with 14-3-313, a GST-14-3-30 fusion protein was prepared which
was incubated
with lysates from HEK293T cells co-expressing TBK1 with PDE3B. PDE3B was
preferentially pulled down by GST-14-3-313 after phosphorylation by TBK1 but
not by its
inactive K38A mutant, whereas GST beads alone enriched neither PDE3B nor its
phosphorylated form (Figure 3E).
Example 4
IKKt and TBK1 phosphorylate PDE3B at serine 318, resulting in the binding of
14-3-313
To evaluate the regulatory role of PDE3B phosphorylation by IKK8 and TBK1, we
determined which sites are phosphorylated. HA-PDE3B was co-expressed in Cos-1
cells with
IKK8 and TBK1, and phosphorylated PDE3B was enriched by IP with anti-HA
antibodies.
Phosphorylation sites on human PDE3B were then determined by LC-MS/MS mass
spectrometry. This analysis revealed that serines 22, 299, 318, 381, 463, 467,
and 503 were
phosphorylated by both kinases; there were no differences between the kinases
(Figure 4A).
Interestingly, the phosphorylation profile of PDE3B matched neither known Akt
or PKA
profiles (Lindh et al., 2007). However, phosphorylation on serine 299 and
serine 318 had
previously been identified on mouse PDE3B (residues equivalent to Serine 277
and 296 in
mouse PDE3B) in adipocytes and hepatocytes in response to both insulin and
forskolin
(Lindh et al., 2007). While several serine residues are known to be
phosphorylated on PDE3B
in response to stimuli, serine 318 (human) is the best characterized. This
residue resides in a
consensus phosphorylation sequence for both Akt and PKA, and also serves as a
consensus
14-3-3 binding motif once phosphorylated (Lindh et al., 2007; Palmer et al.,
2007). We thus
created a Ser318Ala (S318A) mutant of PDE3B, and examined its interaction with
a GST-14-
3-313 fusion protein or by GST-14-3-3 overlay assay. Interestingly, despite
incubation with
TBK1, the phospho-defective, S318A mutant of PDE3B, did not specifically
interact with
GST-14-3-3f3, whereas the wild-type protein did (Figure 4B,C). In a GST pull-
down assay,
the molecular shift of PDE3B S318A was still detected by western blot (Figure
4B),
indicating that phosphorylation of PDE3B by TBK1 on other sites still
occurred, but were not
crucial for 14-3-30 binding.
To examine the functional importance of the phosphorylation of PDE3B at Serine
318, we overexpressed WT PDE3B and its S318A mutant in 3T3-L1 adipocytes, and
tested
CA 02938126 2016-07-28
WO 2015/119624
PCT/US2014/015387
the response of the cells to TNFa. Overexpression of WT PDE3B in cells reduced
the
attenuation of forskolin-stimulated cAMP production and phosphorylation of HSL
produced
by TNFa, whereas PDE3B S318A was ineffective (Figure 4D, Figure 4¨figure
supplement
1A,B). These data suggest that although IKK8 and TBK1 can phosphorylate PDE3B
on
several sites, serine 318 may be particularly important in the regulation of
phosphodiesterase
function by promoting the interaction between PDE3B and 14-3-3 ft More
importantly, this
residue is the major site mediating the negative effects of IKK8 and TBK1 on
sensitivity of
adipocytes to 13-adrenergic stimulation.
Example 5
The IKKeTBK1 inhibitor Amlexanox sensitizes 13-adrenergic agonist stimulated
lipolysis in white adipose tissue in diet-induced obese mice
To test the functional importance of the noncanonical IKKs in maintaining
energy
balance in vivo, experiments were conducted during development of embodiments
of the
present invention to investigate whether the administration of a selective
inhibitor of IKK8
and TBK1, amlexanox, can reverse diet-induced catecholamine resistance in
rodentsMice
were fed a high fat or normal diet, treated them with amlexanox by oral gavage
for 4 days
(prior to the point when weight loss is seen), and then gave a single
intraperitoneal (IP)
injection of the 133-adrenergic agonist CL-316,243. Injection of CL-316,243
stimulated a
threefold increase in serum FFA and glycerol levels in both vehicle and
amlexanox-treated
mice on normal diet (ND). The effect of CL-316,243 to increase serum FFAs was
significantly attenuated in HFD-fed, vehicle-treated mice. However, HFD-fed
mice treated
with amlexanox responded like normal diet mice, despite the fact that they
were weight
matched with control HFD-fed mice (Figure 5A). The fold increase in serum
glycerol levels
was also significantly higher in amlexanox-treated HFD mice, as compared to
vehicle-treated
HFD-fed mice. In addition, ex vivo pretreatment of white adipose tissues from
mice on a
HFD with amlexanox enhanced glycerol release (Figure 5B). This effect was more
pronounced in the inguinal fat depot, where amlexanox pretreatment increased
phosphorylation of HSL, perilipin, and other proteins recognized by the PKA
substrate motif
antibody in response to CL-316,243 treatment compared to vehicle-pretreated
tissues (Figure
5C). Amlexanox also concurrently increased the phosphorylation of TBK1 at
Ser172 due to
the relief of feedback inhibition, as previously reported with other
inhibitors (Clark et al.,
2009; Reilly et al., 2013).
21
CA 02938126 2016-07-28
WO 2015/119624
PCT/US2014/015387
To examine whether inhibition of TBK1 and IKK8 with amlexanox reverses
resistance to catecholamineinduced lipolysis in vivo by increasing stimulation
of cAMP
production, cAMP levels we measured in epididymal adipose tissue from mice on
HFD after
CL-316,243 IP injection. Levels of cAMP were increased after CL-316,243 IP
injection in
mice on HFD pretreated with amlexanox (Figure 5D). Consistent with this, HSL
phosphorylation was also increased after CL-316,243 IP injection of HFD-fed
mice
pretreated with amlexanox (Figure 5E).
To examine whether inhibition of catecholamine resistance in obese adipose
tissue by
targeting noncanonical IKKs with amlexanox can lead to increase energy
expenditure in diet-
induced obese mice, oxygen consumption rates were measured of vehicle or
amlexanox-
treated HFD-fed mice after a single injection of CL-316,243 in metabolic
cages. The effect of
CL-316,243 to increase energy expenditure was more pronounced in
amlexanoxtreated HFD-
fed mice, as compared to vehicle-treated HFD-fed mice (Figure 5F). These data
indicate that
targeting the noncanonical IKKs with the selective inhibitor amlexanox
ameliorated
catecholamine resistance in obese adipose tissue.
22