Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02953641 2016-12-23
WO 2016/001199 PCT/EP2015/064796
NOVEL FORMULATION AND METHOD OF SYNTHESIS
Technical Field of the Invention
The present invention relates to a drug product composition and in particular
to a
composition comprising a positron emission tomography (PET) tracer. The
composition of the present invention and its method of synthesis have certain
advantages over the prior art.
Description of Related Art
The non-natural amino acid "F-1-amino-3-fluorocyclobutane-1-carboxylic acid
(18F-
FACBC, also known as 18F-Fluciclovine) is taken up specifically by amino acid
transporters and shows promise for positron emission tomography (PET) imaging
of
prostate cancer (Nanni et at 2014 Clinical Genitourinary Cancer; 12(2): 106-
110).
Production of18F-FACBC comprises labelling of a triflate precursor compound
with
18F-fluoride:
NHBoc
Tf0 _________________________ (i) labelling <><NHBoc
vo. 18F ___________________________________________
COOEt COOEt
I II
before removal of the two protecting groups:
NHBoc (ii) deprotection <><NH2
18F _____________________________________ 3õ. 18F __
COOEt COOH
II III
Following the deprotection steps purification is carried out to remove
impurities. In the
currently-practiced methods a combination of solid phases is used: ion
retardation to
remove excess Na and excess cr left over from the deprotection steps, alumina
to
remove 18F-fluoride and a reversed phase to remove FACBC-related impurities
such as
1-amino-3-hydroxyl-cyclobutane-1-carboxylic acid (hydroxyl-ACBC) and 1-amino-3-
chloro-cyclobutane-1-carboxylic acid (chloro-ACBC).
The synthesis is currently typically carried out by means of an automated
radiosynthesis
procedure employing a so-called "cassette" or "cartridge" designed to fit
removably and
-1-
CA 02953641 2016-12-23
WO 2016/001199 PCT/EP2015/064796
interchangeably onto an automated synthesis apparatus such as those that are
commercially available from GE Healthcare, CTI Inc, Ion Beam Applications S.A.
(Chemin du Cyclotron 3, B-1348 Louvain-La-Neuve, Belgium); Raytest (Germany)
and
Bioscan (USA). The cassette comprises all the reagents, reaction vessels and
apparatus
necessary to carry out the preparation of "F-FACBC following introduction of
suitably
prepared 18F-fluoride by methods well-known in the field of PET tracer
production.
A known cassette for the synthesis of "F-FACBC is a FASTlab TM cassette from
GE
Healthcare. Each cassette is built around a one-piece-moulded manifold with 25
three-
way stopcocks, all made of polypropylene. The cassette includes a quaternary
methylammonium (QMA) solid phase extraction (SPE) cartridge, a 5 ml cyclic
olefin
copolymer reactor, one 1 ml syringe and two 5 ml syringes, spikes for
connection with
five prefilled reagent vials A-E, one water bag (100 ml), three SPE cartridges
(tC18,
HLB and alumina) and filters. The five FASTlabTm cassette reagent vials are
filled as
follows: vial A contains eluent solution comprising Kryptofix 2.2.2. and K2CO3
in
acetonitrile (MeCN), vial B contains HC1, vial C contains MeCN, vial D
contains dry
precursor compound of Formula I from the above-illustrated reaction scheme and
vial E
contains NaOH. A known method for production of "F-FACBC drug product using
this
FASTlabTm cassette is described in Example 1 of WO 2013/093025. The
radiosynthesis is started by trapping aqueous 18F-fluoride onto the QMA
followed by
elution into the reactor using eluent from vial A, and then concentrated to
dryness by
azeotropic distillation with acetonitrile from vial C. Approximately MeCN is
mixed
with precursor compound from vial D and the dissolved precursor is added to
the
reactor and heated for 3 min at 85 C. The reaction mixture is then diluted
with water
and sent through the tC18 cartridge. The reactor is washed with water and sent
through
the tC18 cartridge. The labelled intermediate, fixed on the tC18 cartridge is
washed
with water, and then incubated with NaOH for 5 min to remove the ester group.
The
deesterified intermediate is eluted off the tC18 cartridge and back into the
reactor using
water. The BOC group is hydrolysed in the reactor by adding HC1 and heating
for 5
min at 60 C. The crude 18F-FACBC is then sent through the HLB (HLB =
hydrophilic
lipophilic balanced) cartridge for removal of FACBC-related impurities, the
alumina
cartridge for removal of '8F-fluoride, and thereafter into a 30 ml product
vial containing
citrate buffer. The HLB and alumina cartridges are then washed with water,
which is
sent to the product vial. Finally, NaOH and water are added to the product
vial to
-2-
CA 02953641 2016-12-23
WO 2016/001199 PCT/EP2015/064796
provide the final purified formulation of "F-FACBC. Prior to intravenous
administration, this formulation is passed through a sterile filter.
The present inventors have found that the quality of the final "F-FACBC drug
product
obtained using the above-described known FASTlabTm cassette and process can be
somewhat variable. Residual acetonitrile levels have been found to range from
about
100 jig/ml to about 600 [tg/ml. While acceptable in terms of permitted daily
exposure
and in the context of the acceptance criteria for "F-FACBC drug product, the
amount
and observed variability is less than ideal. Furthermore, residual aluminium
have been
found to range from about 7 jig/ml to nearly 20 jig/ml, which would mean a
potential
amount of 100 [tg in a 5 ml "F-FACBC injection. Where the "F-FACBC drug
product
also comprises citrate buffer, complexes of aluminium and citrate are likely
to be
present, which is problematic as it is known that such complexes cross the
blood-brain
barrier (Rengel 2004 Biometals; 17: 669-689).
There is therefore scope to for an improved "F-FACBC drug product formulation.
Summary of the Invention
The present invention provides a drug product composition comprising "F-FACBC
that
overcomes the problems seen with known such compositions. In particular, the
composition of the present invention has an improved impurity profile, making
it safer
and more effective for imaging as compared with the prior art. Low and
predictable
levels of acetonitrile and/or aluminium in the final drug product mean that
the
composition of the invention more easily meets worldwide pharmacopeia
requirements.
In addition to a significant reduction in the concentration of aluminium in
the final drug
product, removal of the alumina cartridge has the allied advantages that a
shorter and
simplified process is permitted and that no particles arising from this
cartridge are
present, which the present inventors have noted can block the sterile filter
used prior to
injection of the drug product. Furthermore, the advantages of the present
invention are
achieved with only minor changes to the known process and without impairing
the
desirable qualities of known "F-FACBC compositions.
Detailed Description of the Preferred Embodiments
To more clearly and concisely describe and point out the subject matter of the
claimed
invention, definitions are provided in the detailed description hereinbelow
for specific
-3-
CA 02953641 2016-12-23
WO 2016/001199 PCT/EP2015/064796
terms used throughout the present specification and claims. Any
exemplification of
specific terms herein should be considered as non-limiting examples.
In one aspect the present invention relates to a positron emission tomography
(PET)
tracer composition comprising anti-l-amino -3 -18F- fluoro cyc lo butyl-1-
carboxylic acid
(18F-FACBC) characterised in that said composition comprises no more than 5.0
g/mL
dissolved aluminium (Al).
In one aspect the present invention relates to a positron emission tomography
(PET)
tracer composition comprising anti-l-amino -3 -18F- fluoro cyc lo butyl-1-
carboxylic acid
(18
F-FACBC) characterised in that said composition comprises no more than 5.0
g/mL
dissolved aluminium (Al) and no more than 50 [tg/mL acetonitrile (MeCN).
In the context of the present invention a "PET tracer composition" refers to a
composition comprising a PET tracer together with a biocompatible carrier in a
form
suitable for mammalian administration. The PET tracer composition of the
invention is
referred to hereunder also as the composition of the invention. A "PET tracer"
is
defined herein as a biologically active molecule comprising an atom which is a
positron
emitter suitable for intravenous administration to a mammalian subject
followed by
PET imaging to obtain one or more clinically-useful images of the location
and/or
distribution of the PET tracer. A "biocompatible carrier" as defined herein is
a fluid,
especially a liquid, in which a pharmaceutical is suspended or dissolved, such
that the
composition is physiologically tolerable, i.e. can be administered to the
mammalian
body without toxicity or undue discomfort. The biocompatible carrier is
suitably an
injectable carrier liquid such as sterile, pyrogen-free water for injection or
an aqueous
solution such as saline.
The compound "18F-FACBC" is represented by the following chemical structure:
H NH2
18F COOH
The term "not more than" as used herein should be understood to mean any
amount less
than and including the quoted quantity. In an idealized embodiment of the
composition
of the present invention there would be zero [tg/mL of each impurity present.
However,
-4-
CA 02953641 2016-12-23
WO 2016/001199 PCT/EP2015/064796
in reality, zero [tg/mL of an impurity is unlikely and at least a trace amount
of each
impurity remains in the composition. The term "not more than" acknowledges
that a
trace amount of one or more impurities is present in a PET tracer composition,
and
defines a concentration limit above which the composition would not be deemed
acceptable for use.
In one embodiment, the composition of the invention comprises not more than
not more
than 3.0 iug/mL dissolved Al, and in another embodiment not more than 1.5
iug/mL
dissolved Al.
In one embodiment the composition of the present invention comprises MeCN at a
concentration not more than 20 [tg/mL.
The composition of the invention in one embodiment has an end of synthesis
(EOS)
radiochemical purity (RCP) of at least 95%, in another embodiment at least
98%, and in
yet another embodiment at least 99%.
The term "end of synthesis" refers to the point in time when the labelled
compound is
collected in the product collection vial.
EP 2119458 (Al) teaches that a more stable formulation of "F-FACBC is achieved
when the pH is maintained within the range 2.0-5.9. As discussed in WO
2013/093025,
use of citrate buffer allows the pH to be maintained within an even narrower
range,
provides resistance to degradation and enables the formulation to be
autoclaved. In one
embodiment, the composition of the present invention therefore comprises
around 50-
100 mM citrate buffer, in another embodiment around 60-90 mM citrate buffer
and in
yet another embodiment around 75-85 mM citrate buffer. The term "around" in
this
context incorporates the exact values of the ranges as well as a small
variation around
these values that would be expected by the skilled person to achieve the same
stabilisation effect.
In another aspect, the present invention provides a method to prepare a PET
tracer
composition of the invention wherein said method comprises:
(a) reacting in a reaction vessel a source of 18F-fluoride with a
precursor
compound of Formula I:
-5-
CA 02953641 2016-12-23
WO 2016/001199 PCT/EP2015/064796
LG HN¨PG2
V{N7j
H COO ¨PG1
(I);
wherein:
LG is a leaving group;
PG' is a carboxy protecting group; and,
PG2 is an amine protecting group;
to obtain a reaction mixture comprising a compound of Formula II:
H HN¨PG2
18F COO ¨PG1
(II);
wherein PG' and PG2 are as defined for Formula I;
(b) carrying out removal of PG' and PG2 to obtain a reaction mixture
comprising 1 8F-FACBC; and
(c) purifying said reaction mixture comprising 18F-FACBC by passing it
through a hydrophilic lipophilic balanced (HLB) solid phase, characterised in
that said purifying does not comprise passing the reaction mixture comprising
18F-FACBC through an alumina solid phase.
In another aspect, the present invention provides a method to prepare a PET
tracer
composition of the invention wherein said method comprises:
(a) reacting in a reaction vessel a source of 18F-fluoride with a
precursor
compound of Formula I:
LG HN¨PG2
V1171
H COO ¨PG1
(I);
wherein:
-6-
CA 02953641 2016-12-23
WO 2016/001199 PCT/EP2015/064796
LG is a leaving group;
PG' is a carboxy protecting group; and,
PG2 is an amine protecting group;
wherein said reacting step is carried out in acetonitrile;
to obtain a reaction mixture comprising a compound of Formula II:
H HN¨PG2
18F COO ¨PG1
(II);
wherein PG' and PG2 are as defined for Formula I;
(b) transferring said reaction mixture comprising said compound of
Formula
II out of said reaction vessel and carrying out removal of PG' to obtain a
reaction mixture comprising a compound of Formula III:
H HN ¨ PG2
18F COOH
(III)
wherein PG2 is as defined for Formula I;
(c) applying heat to said reaction vessel at the same time as
carrying out
removal of PG';
(d) transferring said reaction mixture comprising said compound of Formula
III back into said reaction vessel and carrying out removal of PG2 to obtain a
reaction mixture comprising 18F-FACBC;
(e) purifying said reaction mixture comprising 18F-FACBC by
passing it
through a hydrophilic lipophilic balanced (HLB) solid phase, characterised in
that said purifying does not comprise passing the reaction mixture comprising
18F-FACBC through an alumina solid phase.
The "source of 18F-fluoride" suitable for use in step (a) of the method of the
invention is
normally obtained as an aqueous solution from the nuclear reaction
180(p,n)18F. In
-7-
CA 02953641 2016-12-23
WO 2016/001199 PCT/EP2015/064796
order to increase the reactivity of fluoride and to reduce or minimise
hydroxylated by-
products resulting from the presence of water, water is typically removed from
18F-
fluoride prior to the reaction, and fluorination reactions are carried out
using anhydrous
reaction solvents (Aigbirhio et at 1995 J Fluor Chem; 70: 279-87). A further
step that
is used to improve the reactivity of 18F-fluoride for radiofluorination
reactions is to add
a cationic counterion prior to the removal of water. Suitably, the counterion
should
possess sufficient solubility within the anhydrous reaction solvent to
maintain the
solubility of the 18F-fluoride. Therefore, counterions that are typically used
include
large but soft metal ions such as rubidium or caesium, potassium complexed
with a
cryptand such as KryptofixTM, or tetraalkylammonium salts, wherein potassium
complexed with a cryptand such as KryptofixTM, or tetraalkylammonium salts are
preferred.
The "precursor compound" for step (a) of the method of the invention comprises
a non-
radioactive derivative of a radiolabelled compound, designed so that chemical
reaction
with a convenient chemical form of the detectable label occurs site-
specifically, can be
conducted in the minimum number of steps (ideally a single step), and without
the need
for significant purification (ideally no further purification), to give the
desired
radio labelled compound. Such precursor compounds are synthetic and can
conveniently be obtained in good chemical purity.
A suitable "leaving group" in the context of the compound of Formula I in step
(a) of
the method of the present invention is a chemical group that can be displaced
by
nucleophilic displacement reaction with fluoride ion. These are well-known in
the art
of synthetic chemistry. In some embodiments the leaving group of the present
invention is a linear or branched Ci_10 haloalkyl sulfonic acid substituent, a
linear or
branched C1_10 alkyl sulfonic acid substituent, a fluorosulfonic acid
substituent, or an
aromatic sulfonic acid substituent. In other embodiments of the invention the
leaving
group is selected from methanesulfonic acid, toluenesulfonic acid,
nitrobenzenesulfonic
acid, benzenesulfonic acid, trifluoromethanesulfonic acid, fluorosulfonic
acid, and
perfluoroalkylsulfonic acid. In some embodiments the leaving group is either
methanesulfonic acid, trifluoromethanesulfonic acid or toluenesulfonic acid
and in
another embodiment the leaving group is trifluoromethanesulfonic acid.
The term "protecting group" as used in connection with the substituents PG'
and PG2
-8-
CA 02953641 2016-12-23
WO 2016/001199 PCT/EP2015/064796
refers to a group which inhibits or suppresses undesirable chemical reactions,
but which
is designed to be sufficiently reactive that it may be cleaved from the
functional group
in question to obtain the desired product under mild enough conditions that do
not
modify the rest of the molecule. Protecting groups are well known to those
skilled in
the art and are described in 'Protective Groups in Organic Synthesis',
Theorodora W.
Greene and Peter G. M. Wuts, (Fourth Edition, John Wiley & Sons, 2007).
The PG' "carboxy protecting group" herein is preferably linear or branched
Ci_io alkyl
chain or an aryl substituent. The term "alkyl" used either alone or as part of
another
group is defined as any straight, branched or cyclic, saturated or unsaturated
CõH2õ+1
group. The term "aryl" refers to any C6_14 molecular fragment or group which
is
derived from a monocyclic or polycyclic aromatic hydrocarbon, or a monocyclic
or
polycyclic heteroaromatic hydrocarbon. In one embodiment of the method of the
invention PG' is selected from methyl, ethyl, t-butyl and phenyl. In another
embodiment of the invention PG' is methyl or ethyl and in yet another
embodiment PG'
is ethyl.
The PG2 "amine protecting group" herein refers to a chemical group that
suitably
prevents reaction between 18F and the amino group in the process of providing
the
compound of Formula II. Examples of suitable amine protecting groups include
various carbamate substituents, various amide substituents, various imide
substituents,
and various amine substituents. Preferably, the amine protecting group is
selected from
the group consisting of linear or branched C2_7 alkyloxycarbonyl substituents,
linear or
branched C3_7 alkenyloxycarbonyl substituents, C7_12 benzyloxycarbonyl
substituents
that may have a modifying group, C2_7 alkyldithiooxycarbonyl substituents,
linear or
branched Ci _6 alkylamide substituents, linear or branched C2_6 alkenylamide
substituents, C6_11 benzamide substituents that may have a modifying group,
C4_10 cyclic
imide substituents, C6_11 aromatic imine substituents that may have a
substituent, linear
or branched C1_6 alkylamine substituents, linear or branched C2_6 alkenylamine
substituents, and C6_11 benzylamine substituents that may have a modifying
group. In
some embodiments of the invention PG2 is selected from t-butoxycarbonyl,
allyloxycarbonyl, phthalimide, and N-benzylideneamine. In other embodiments
PG2 is
selected from t-butoxycarbonyl or phthalimide. In one embodiment of the
invention
PG2 is t-butoxycarbonyl.
-9-
CA 02953641 2016-12-23
WO 2016/001199 PCT/EP2015/064796
The term "reacting" in step (a) of the method of the invention as is well
known to those
of skill in the art refers to bringing two or more chemical substances
(typically referred
to in the art as "reactants" or "reagents") together to result in a chemical
change in one
or both/all of the chemical substances.
The "removal of PG" in step (b) of the method of the invention is suitably
carried out
by contacting the compound of Formula II, comprised within the reaction
mixture
obtained in step (a), with a carboxy deprotecting agent. A suitable carboxy
deprotecting agent may be either an acid or an alkaline solution, as is well-
known to the
skilled person (see Greene and Wuts, supra). The concentration of the carboxy
deprotecting agent is suitably just sufficient to remove the carboxy
protecting group.
Preferably the carboxy deprotecting agent is an alkaline solution. In certain
embodiments the carboxy deprotecting agent is a sodium hydroxide or a
potassium
hydroxide solution and in a preferred embodiment is a sodium hydroxide
solution, for
example of 0.5-2.0M. The temperature and the duration used for deprotection
may in
some embodiments be tailored to permit removal of PG'. For example, in certain
embodiments the reacting step is carried out at room temperature and for a
duration of
around 1-5 minutes. In one embodiment, removal of PG' is carried out by
passing the
reaction mixture comprising the compound of Formula II through a solid phase
extraction (SPE) column where the compound of Formula II binds to the solid
phase.
Once the compound of Formula II is bound, the outlet of the SPE column is
closed so
that the carboxy deprotecting agent is retained therein for a defined amount
of time. A
suitable solid phase for use in this manner is a reversed phase solid phase,
e.g. tC18.
Step (c) comprises applying heat to the reaction vessel using methods well-
known to
those of skill in the art, e.g. using a dedicated heater into which the
reaction vessel is
placed for the duration of the radiosynthesis. The application of heat must be
so that
the reaction vessel can be used for the subsequent step (d), i.e. so that the
reaction
vessel is intact and undamaged, and also so that residual solvent is
effectively removed.
This step (c) is carried out at the same time as removal of PG', i.e. after
the reaction
mixture comprising the compound of Formula II has been transferred out of said
reaction vessel. A suitable temperature for this heating step should be no
greater than
the tolerance of the reaction vessel, e.g. for a reaction vessel made from
cyclic olefin
copolymer (COC) a temperature of no greater than about 130 C and for a
reaction
-10-
CA 02953641 2016-12-23
WO 2016/001199 PCT/EP2015/064796
vessel made from polyetheretherketone (PEEK) a temperature of no greater than
about
200 C. For convenience, the temperature used to heat the reaction vessel in
step (c)
may be selected to be as close as possible to the temperature used during the
labelling
step (a). Suitable temperatures for radio labelling step (a) are in the range
of about 80-
140 C, in other embodiments 85-130 C.
The "removal of PG2" in step (d) of the method of the invention is carried out
by
contacting the compound of Formula III with an amine deprotecting agent. A
suitable
amine deprotecting agent may be either an acid or an alkaline solution, as is
well-
known to the skilled person (see Greene and Wuts, supra). The concentration of
the
amine deprotecting agent is suitably just sufficient to remove PG2. Preferably
the
amine deprotecting agent is an acid solution. A suitable acid is an acid
selected from
inorganic acids such as hydrochloric acid (HC1), sulfuric acid (H2504) and
nitric acid
(HNO3), and organic acids such as perfluoroalkyl carboxylic acids, e.g.
trifluoroacetic
acid (CF3CO2H). In certain embodiments, the amine deprotecting agent is HC1,
e.g. at a
concentration of 1.0-4.0M. Removal of PG2 is in one embodiment carried out
with heat
to allow the deprotection to proceed more rapidly. The time depends on the
reaction
temperature or other conditions. For example, in one embodiment removal of PG2
is
carried out at 60 C, with a reaction time of 5 minutes.
The aim of the "purifying" step (e) is to obtain substantially pure "F-FACBC.
The
term "substantially" refers to the complete or nearly complete extent or
degree of an
action, characteristic, property, state, structure, item, or result. The term
"substantially
pure" as used herein in the context of "F-FACBC encompasses completely pure
18F-
FACBC or "F-FACBC that is sufficiently pure to be suitable for use as a PET
tracer.
The term "suitable for use as a PET tracer" means that the purified "F-FACBC
product
is suitable for intravenous administration to a mammalian subject followed by
PET
imaging to obtain one or more clinically-useful images of the location and/or
distribution of "F-FACBC.
A "HLB solid phase" is a reversed phase solid phase having hydrophilic and
lipophilic
components suitable for a range of purposes. HLB solid phase is commercially-
available as SPE cartridges suitable for use in the method of the present
invention, e.g.
the Oasis HLB SPE cartridge.
An "alumina solid phase" is an aluminium oxide normal phase solid phase
routinely
-11-
CA 02953641 2016-12-23
WO 2016/001199 PCT/EP2015/064796
used in 18F labelling methods as a means to remove free 18F-fluoride and
optimise the
radiochemical purity of the final product. Alumina solid phase is commercially-
available as SPE cartridges suitable for use in the method of the present
invention, e.g.
the Waters Alumina N Light.
In the method of the invention, steps (a)-(c) or (a)-(e) are carried out in
sequence.
In one embodiment of the method of the present invention, the substituent LG
in the
compound of Formula I is a linear or branched C1_10 haloalkyl sulfonic acid
substituent,
a linear or branched Ci_10 alkyl sulfonic acid substituent, a fluorosulfonic
acid
substituent, or an aromatic sulfonic acid substituent. Examples of LG include
methanesulfonic acid, toluenesulfonic acid, nitrobenzenesulfonic acid,
benzenesulfonic
acid, trifluoromethanesulfonic acid, fluorosulfonic acid, and
perfluoroalkylsulfonic
acid. In one embodiment LG is trifluoromethanesulfonic acid.
In one embodiment of the method of the present invention the substituent PG'
in the
compounds of Formula I and II is a linear or branched C1_10 alkyl chain or an
aryl
substituent. For example, PG' can be methyl, ethyl, t-butyl or phenyl. In one
embodiment PG' is methyl or ethyl. In another embodiment, PG' is ethyl.
In one embodiment of the method of the present invention the substituent PG2
in the
compounds of Formulas I-III is a carbamate substituent, an amide substituent,
an imide
substituent or an amine substituent.
Examples include t-butoxycarbonyl,
allyloxycarbonyl, phthalimide, and N-benzylideneamine. In one embodiment, PG2
is t-
butoxycarbonyl.
The method of the present invention may further comprise the step of
formulating the
purified reaction mixture obtained in step (e) with citrate buffer. In one
embodiment,
this formulating step results in a concentration of 50-100 mM citrate buffer,
in another
embodiment 60-90 mM citrate buffer and in yet another embodiment 75-85 mM
citrate
buffer.
In one embodiment, the method of the invention is automated, e.g. carried out
on an
automated synthesis apparatus. 18F-labelled PET tracers are often conveniently
prepared on automated radiosynthesis apparatus. By
the term "automated
radiosynthesis apparatus" is meant an automated module based on the principle
of unit
operations as described by Satyamurthy et at (1999 Clin Positr Imag; 2(5): 233-
253).
-12-
CA 02953641 2016-12-23
WO 2016/001199 PCT/EP2015/064796
The term "unit operations" means that complex processes are reduced to a
series of
simple operations or reactions, which can be applied to a range of materials.
Suitable
automated synthesiser apparatus are commercially available from a range of
suppliers
including: GE Healthcare Ltd (Chalfont St Giles, UK); CTI Inc. (Knoxville,
USA); Ion
Beam Applications S.A. (Chemin du Cyclotron 3, B-1348 Louvain-La-Neuve,
Belgium); Raytest (Straubenhardt, Germany) and Bioscan (Washington DC, USA).
Commercial automated radiosynthesis apparatus also provide suitable containers
for the
liquid radioactive waste generated as a result of the radiopharmaceutical
preparation.
Automated radiosynthesis apparatus are not typically provided with radiation
shielding,
since they are designed to be employed in a suitably configured radioactive
work cell.
The radioactive work cell, also termed a hot cell, provides suitable radiation
shielding
to protect the operator from potential radiation dose, as well as ventilation
to remove
chemical and/or radioactive vapours.
Preferred automated radiosynthesis apparatus of the present invention are
those which
interact with a disposable or single use "cassette" (also commonly referred to
as a
"cartridge") which comprises all the reagents, reaction vessels and apparatus
necessary
to carry out the preparation of a given batch of radiopharmaceutical. By use
of such
cassettes the automated radiosynthesis apparatus has the flexibility to be
capable of
making a variety of different radiopharmaceuticals with minimal risk of cross-
contamination, by simply changing the cassette. The cassette approach also has
the
advantages of: simplified set-up and hence reduced risk of operator error;
improved
GMP (Good Manufacturing Practice) compliance; multi-tracer capability; rapid
change
between production runs; pre-run automated diagnostic checking of the cassette
and
reagents; automated barcode cross-check of chemical reagents vs the synthesis
to be
carried out; reagent traceability; single-use and hence no risk of cross-
contamination,
tamper and abuse resistance.
The cassette has been simplified by removal of the alumina cartridge. The
alumina
cartridge was present in prior cassette configurations to remove residues of
free '8F.
fluoride from insufficient purification and/or from radio lysis. However, the
present
inventors have found that the rest activity on the alumina cartridge is very
low (0.1-
0.3%) indicating both a robust purification process and a low degree of
radiolysis.
These data suggest that the alumina cartridge is superfluous and can be
removed. This
-13-
CA 02953641 2016-12-23
WO 2016/001199
PCT/EP2015/064796
has the additional benefit of there being no risk of any particles from the
alumina
cartridge being present in drug product, which pose a risk of blocking the
sterile filter.
The process has been improved by the addition of a concurrent step of removal
of
residual acetonitrile from the reactor while the deesterification step
proceeds on the
tC18 cartridge. This results in a final drug product having a lower and more
predictable
concentration of residual acetonitrile than that obtained using prior art
methods.
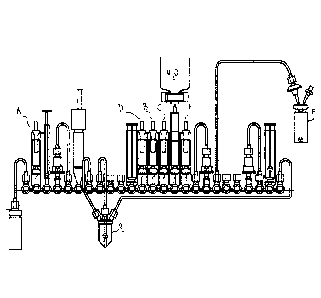
Brief Description of the Figures
Figure 1 illustrates an exemplary cassette to carry out the method of the
invention.
Brief Description of the Examples
The following non-limiting examples serve to illustrate particular embodiments
of the
subject matter of the present invention.
List of Abbreviations used in the Examples
BOC tert-Butyloxycarbonyl
DP drug product
HLB hydrophobic-lipophilic balance
K222 Kryptofix 222
MeCN acetonitrile
QMA quaternary methyl ammonium
RAC radioactive concentration
RCP: radiochemical purity
Examples
Comparative Example 1: Prior Art Synthesis of 18 F-FACBC
1(i) FASTlab CassetteAll radiochemistry was performed on a commercially-
available GE FASTlabTm with single-use cassettes. Each cassette is built
around a one-
piece-moulded manifold with 25 three-way stopcocks, all made of polypropylene.
Briefly, the cassette includes a 5 ml reactor (cyclic olefin copolymer), one 1
ml syringe
and two 5 ml syringes, spikes for connection with five prefilled vials, one
water bag
(100 ml) as well as various SPE cartridges and filters. Fluid paths are
controlled with
-14-
CA 02953641 2016-12-23
WO 2016/001199 PCT/EP2015/064796
nitrogen purging, vacuum and the three syringes. The fully automated system is
designed for single-step fluorinations with cyclotron-produced 18F-fluoride.
The
FASTlab was programmed by the software package in a step-by-step time-
dependent
sequence of events such as moving the syringes, nitrogen purging, vacuum, and
temperature regulation. Vial A contained K222 (58.8 mg, 156 mop, K2CO3 (8.1
mg,
60.8 Rmol) in 79.5% (v/v) MeCN(aq) (1105 1). Vial B contained 4M HC1 (2.0
m1). Vial
C contained MeCN (4.1m1). Vial D contained the precursor (48.4 mg, 123.5 mop
in
its dry form (stored at ¨20 C until cassette assembly). Vial E contained 2 M
NaOH (4.1
m1). The 30 ml product collection glass vial was filled with 200 mM trisodium
citrate
(10 m1).
1(ii) Production of18F-Fluoride
No-carrier-added 18F-fluoride was produced via the 180(p,n)18F nuclear
reaction on a
GE PETtrace 6 cyclotron (Norwegian Cyclotron Centre, Oslo). Irradiations were
performed using a dual-beam, 30 A current on two equal Ag targets with HAVAR
foils
using 16.5 MeV protons. Each target contained 1.6 ml of? 96% [180]water
(Marshall
Isotopes). Subsequent to irradiation and delivery to a hotcell, each target
was washed
with [160]water (Merck, water for GR analysis). Aqueous 18F-fluoride was
passed
through the QMA and into the 180-H20 recovery vial. The QMA was then flushed
with
MeCN and sent to waste.
J(iii) '8F-Fluoride Labelling
The trapped 18F-fluoride was eluted into the reactor using eluent from vial A
and then
concentrated to dryness by azeotropic distillation with acetonitrile (vial C).
MeCN was
mixed with precursor in vial D from which the dissolved precursor was added to
the
reactor and heated to 85 C.
1(iv) Removal of Ester Protecting Group
The reaction mixture was diluted with water and sent through the tC18
cartridge.
Reactor was washed with water and sent through the tC18 cartridge. The
labelled
intermediate, fixed on the tC18 cartridge was washed with water, and then
incubated
with 2M NaOH after which the 2M NaOH was sent to waste.
1(v) Removal of BOC Protecting Group
The labelled intermediate (without the ester group) was then eluted off the
tC18
-15-
CA 02953641 2016-12-23
WO 2016/001199 PCT/EP2015/064796
cartridge into the reactor using water. The BOC group was hydrolysed by adding
4M
HC1 and heating the reactor.
1(0 Purification
The reactor content with the crude "F-FACBC was sent through the HLB and
Alumina
cartridges and into the 30 ml product vial. The HLB and Alumina cartridges
were
washed with water and collected in the product vial.
1(vii) Formulation
2M NaOH and water was added to the product vial, giving a purified drug
product (DP)
with a total volume of 26 ml.
(lviii) Acetonitrile Concentration
Acetonitrile (MeCN) concentration was determined using a gas chromatographic
system with FID, an automated liquid injector, a fused silica capillary column
with USP
stationary phase G43 (6% cyanopropylpheny1-94% dimethyl polysiloxane) and a
reporting integrator or data system with reintegration capacity. 1000 [tg/ml
of MeCN
was used as a standard. Blank was prepared by transferring 1 ml of purified
water to a
2 ml GC crimp cap vial, which was capped immediately. 1 ml of the standard was
transferred to a 2 ml GC crimp cap vials and capped immediately. 0.20 ml of
the
sample was transferred to a 2 ml GC crimp cap vial with low volume insert
(0.25 ml)
and capped immediately. The experimental conditions of the GC instrument were
as
follows:
Carrier gas flow, Helium: 2.0 ml/min
Oven temperature program: 40 C for 6 minutes then 20 C/min to 240 C for 4
minutes
Injector temperature : 225 C
Split ratio : 10:1
Detector : FID
Detector temperature : 250 C
Hydrogen flow rate: 30 ml/min
Air flow rate : 400 ml/min
-16-
CA 02953641 2016-12-23
WO 2016/001199
PCT/EP2015/064796
Make up gas flow rate (He) : 25 ml/min
The experimental conditions of the automatic liquid injector were as follows:
Solvent pre washes : 3
Sample pumps : 3
Solvent post washes : 3
Injection volume: 1 ml
The column was conditioned at 250 C for at least one hour prior to use.
One injection of each standard and two replicate injections of the sample
solution were
performed in addition to blank injections in the following order:
1. Blank
2. Calibration standard
3. Calibration standard
4. Blank
5. Sample, replicate 1
6. Sample, replicate 2
7. Blank
The concentration of each analyte, Csample was calculated in jig/ml using the
following
formula:
A
C = =
where:
Asample: Peak area of the analyte in sample
Cstd: Concentration of the analyte in calibration standard (pg/m1)
Astd: Peak area of the analyte in calibration standard, average of 2
injections
(/ ix) Aluminium Concentration
-17-
CA 02953641 2016-12-23
WO 2016/001199 PCT/EP2015/064796
Aluminium concentration was determined by inductively coupled plasma atomic
emission spectroscopy (ICP-AES).
(1x) Radiochemical Parameters
Radiochemical purity (RCP) and radioactive concentration (RAC) of "F-FACBC
were
measured.
RCP was determined by thin layer chromatography (TLC). The TLC strip was
eluted
using a mobile phase consisting of acetonitrile:methanol:water:acetic acid,
20:5:5:1 v/v.
The RCP and any radiochemical impurities including 18F-fluoride were reported
as
percentages of the net sum of all peaks.
(lxi) Results
The following results were obtained:
Production # RAC (MBq/m1) RCP(/0)TO MeCN ( g/m1) Al ( g/m1)
1 1915 >99 506 14
2 1804 >99 324 14
3 1950 >99 302 13
4 1698 >99 89 15
5 1570 >99 596 17
6 1815 >99 218 15
Example 2: Synthesis of18F-FACBC using Inventive Method
2(i) Modified Sequence
A modified FASTlabTm cassette was used, as illustrated in Figure 1. The
sequence
described in Example 1 was used except that the sequence included the extra
heating/purging of the reaction vessel. The hydrolysis step was replaced with
two
steps, the first step of which included hydrolysis and in parallel heating of
the reactor at
85 C, nitrogen purging (600 mbar HF) of the reaction vessel and vacuum (-600
mbar).
The second step also included hydrolysis but heating of the reaction vessel
was stopped.
Nitrogen purging (600 mbar HF) and vacuum (-600 mbar) were used for cooling of
the
reaction vessel. Furthermore, the alumina SPE was removed and the sequence was
changed to transfer the product directly to the formulation buffer vial after
the HLB
cartridge step.
-18-
CA 02953641 2016-12-23
WO 2016/001199
PCT/EP2015/064796
2(h) Analysis
The analysis methods as described in Example 1 were used.
Production # RAC (MBq/m1) RCP(/0)TO MeCN ( g/m1) Al ( g/m1)
7 3112 99.1 20 0.7
8 3900 99.1 20 0.8
9 1631 99.1 21 1.3
731 99.9 22 0.8
11 1831 99.8 25 0.8
12 1654 99.9 24 1.3
13 1573 99.1 21 1.1
14 1750 99.4 23 1.1
788 99.0 19 1.1
16 1023 99.2 17 1.1
-19-