Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
EXOSOMES USEFUL TO TREAT LYSOSOMAL STORAGE DISEASE
Field of the Invention
[0001] The present invention generally relates to exosomes, and more
particularly, to the
use of exosomes to treat a lysosomal storage disease.
Background of the Invention
[0002] Lysosomes are cellular organelles that function as a cellular
waste disposal
mechanism to remove unused, damaged, or excessive macro-molecules. There are
at least fifty
lysosomal enzymes that are known to function in the acidic environment of the
lysosome (pH
approximately 4.0-5.0) to break down cellular macro-molecules including;
lipids, carbohydrates,
glycolipids, proteins, nucleic acids and organelles such as mitochondria
(mitophagy). In addition
to the removal of accumulated cellular debris, the lysosomal pathway is also
involved in a
variety of cellular processes including cell secretion, signalling cascades,
energy metabolism,
plasma membrane repair, and mitochondrial homeostasis. Collectively, these
cellular processes
include autophagy/mitophagy, endocytosis and phagocytosis. The enzymes within
lysosomes are
encoded by nuclear DNA, transcribed on ribosomes in the rough endoplasmic
reticulum, and
subsequently targeted to the Golgi apparatus where they are packaged and
released in small
vesicles that ultimately fuse with endosomes to form lysosomes. A mannose-6-
phosphate
moiety is added to the lysosomal protein in the Golgi apparatus and this is
important for
lysosomal targeting through an interaction with the mannose-6-phosphate
receptors inside the
Golgi. Once the Golgi apparatus releases the vesicles, they fuse with late
endosomes, following
which the mannose-6-phosphate moieties are cleaved and the mature enzyme
remains enveloped
in the established lysosome. Extracellular proteins can enter the cell through
receptor- and non-
receptor-mediated endocytosis and form early endosomes. The early endosomes
can then fuse
together to form multi-vesicular bodies (MVBs) to become lysosomes. The
distinction between
the MVBs and some late endosomes remains unclear; however, lysosomes are
usually
characterized by the presence of a membrane protein such as lysosomal-
associated membrane
protein (LAMP), e.g. LAMP1 or 2, and intra-lysosomal enzymes such as cathepsin
D.
1
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
[0003] Lysosomal storage diseases (LSDs) are disorders resulting from
gene mutations
that lead to a defective, non-functional lysosomal protein (enzyme). There are
over two dozen
known genetic diseases that affect the lysosome. In general, the specific
genetic mutations
negatively affect the ability of the hydrolytic enzymes within the lysosome to
perform their
allotted function, thus leading to the accumulation of the precursor products
for that enzyme.
This aggregation of the intra-lysosomal precursor protein perpetuates
downstream cellular
consequences including the displacement of normal cellular contents and/or
disruption of the
lysosomes that can cause the release of hydrolytic enzymes into the cytosol
which may damage
other macromolecules crucial for cellular metabolism, redox homeostasis, and
survival.
[0004] One example of a lysosomal storage disease is Pompe disease that
results from a
genetic mutation in a protein called acid alpha-glucosidase (GAA). Mutations
in the GAA
protein lead to the progressive build-up of glycogen in skeletal and cardiac
muscle that
culminates in: (A) a severe infantile form with cardiomyopathy, respiratory
failure and
weakness, or (B) a late onset form which leads to muscle weakness and
eventually respiratory
failure.
[0005] Until recently, therapy for LSDs was designed to treat the
clinical manifestations
of the disease (i.e. anti-seizure drugs for epilepsy, analgesics for pain,
bracing for skeletal
deformities, etc.), rather than the underlying cause of the LSD. More
recently, two therapeutic
strategies have been employed to specifically address and alleviate the
biological deficiency that
leads to LSDs. The first therapy is the direct replacement of the defective
enzyme through
intravenous infusion, called enzyme replacement therapy (ERT). The second
therapy is the use
of molecular chaperones that can enhance the residual activity of a
dysfunctional enzyme and/or
stabilize ERT proteins. ERT operates on the premise that the pathology
specific defective
enzyme is replaced through direct infusion of a nascent lysosomal enzyme into
circulation. This
infused enzyme, produced via recombinant protein technology, is then taken up
by mannose-6-
phosphate receptors on the cell surface. Once inside the cell, the protein can
be internalized
through mannose-6-phosphate receptors on lysosomes, and thereby replace the
defective
enzyme.
2
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
[0006] There are a variety of issues that can arise with enzyme
replacement therapy that
limit its effectiveness. Firstly, the pH of the lysosomes is quite low whereas
blood is usually at a
pH of 7.4. Consequently, the infused enzyme is exposed to a non-native pH
environment that
causes it to denature, leading to a rapid elimination of the infused protein
from the circulation.
Therefore, the bioavailability of the infused enzyme is quite low as the total
exposure to the
affected tissues is limited. It has been estimated that only a small fraction
(1-3%) of the infused
recombinant GAA protein in Pompe disease is actually retained within skeletal
muscle 2 weeks
after the infusion. Secondly, there is a vast difference between different
tissues with respect to
their ability to respond to ERT. Skeletal muscle (especially type II fibers)
is much more resistant
to ERT therapy in comparison to cardiac muscle, when ERT is used in Pompe
disease treatment.
It is known that enzyme uptake in ERT can be enhanced using a fusion protein
containing GAA-
insulin like growth factor 2 through a glycosylation-independent lysosomal
targeting strategy
(GILT-tagged, J Blot Chem. 2013 Jan 18;288(3):1428-38). Enzyme uptake may also
be
enhanced using a carbohydrate¨remodelled approach in which additional
carbohydrate moieties
are attached to the GAA to enhance the affinity of the GAA protein for the
mannose-6-phosphate
receptor (Zhu et al., Biochem J 389, 619-628, 2005). One major issue with any
of these ERT
approaches is that present ERT can lead to the production of neutralizing
(IgG) and/or allergenic
(IgE) antibodies. Ideally, a carrier system is needed that can reduce
immunogenicity of the
infused ERT.
[0007] One of the greatest limitations of conventional intravenous
administration of ERT
is that enzymes cannot cross the blood-brain barrier. This makes conventional
ERT unusable for
lysosomal storage diseases that affect the central nervous system (e.g.
Niemann-Pick C, neuronal
ceroid lipofuscinosis, Tay-Sachs disease, Krabbe disease, etc.). Other routes
of delivery for
central nervous system diseases, i.e. intra-cerebral or intra-ventricular
delivery, pose a host of
secondary adverse side effects and therapy may not be as effective.
Consequently, it would be
desirable to have a method to deliver disease-specific ERT or mRNA to the
central nervous
system via an intravenous, sub-cutaneous, or oral route as opposed to intra-
cerebral or intra-
ventricular delivery method.
3
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
[0008] Accordingly, it would be desirable to develop a novel treatment
for pathological
conditions resulting from a deficiency in the function of a particular protein
such as a lysosomal
protein.
Summary of the Invention
[0009] It has now been determined that exosomes may be effectively used
as a vehicle to
deliver a protein and/or nucleic acid to a mammal to treat pathological
conditions resulting from
a protein deficiency such as a lysosomal storage disease.
[0010] Thus, in one aspect of the invention, exosomes that are
genetically modified to
incorporate a functional lysosomal protein and/or nucleic acid encoding a
functional lysosomal
protein.
[0011] In another aspect, a method of increasing the amount of a
lysosomal protein in
lysosomes in a mammal is provided, comprising administering to the mammal a
composition
comprising exosomes that are genetically modified to incorporate a functional
lysosomal protein
and/or nucleic acid encoding a functional lysosomal protein.
[0012] In another aspect, a method of increasing the activity of a target
protein in a
mammal is provided, comprising administering to the mammal exosomes which are
genetically
modified to incorporate a functional protein and/or nucleic acid encoding a
functional protein.
[0013] In another aspect, a method of treating a pathological condition
in a mammal
resulting from the deficiency of a protein is provided comprising
administering to the mammal
exosomes genetically engineered to incorporate the protein or nucleic acid
encoding the protein.
[0014] In a further aspect, a method of treating a lysosomal storage
disease in a mammal
is provided comprising administering to the mammal exosomes genetically
engineered to
incorporate a protein useful to treat the lysosomal storage disease or nucleic
acid encoding the
protein.
[0015] Additional aspects of the invention are include aspects and
variations set forth in
the following lettered paragraphs:
4
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
[0016] Al. An exosome produced by a process that comprises: (a)
isolating
exosomes from a biological sample from an organism or from a conditioned
medium from a
cultured cell; and (b) introducing a modification into the exosome selected
from the group
consisting of:
(i) at least one functional lysosomal protein or precursor thereof;
(ii) at least one nucleic acid comprising a nucleotide sequence that encodes
the functional
lysosomal protein or precursor thereof;
(iii) at least one fusion product comprising a lysosome targeting sequence
linked to an exosomal
membrane marker;
(iv) at least one nucleic acid comprising a nucleotide sequence that encodes
the fusion product;
and
(v) two or more of (i), (ii,) (iii), and (iv).
[0017] A2. The exosome according to paragraph Al, wherein the
isolating includes at
least one density gradient centrifugation step ideally using Percoll or other
colloidal silica
product.
[0018] A2.1 The exosome according to paragraph Al or A2, wherein the
isolating
removes vesicles that are greater than 120 nm in diameter.
[0019] A3. The exosome according to paragraph Al or A2 or A2.1,
wherein the
biological sample is from a mammal, or the cell is from a mammal or a
mammalian cell line.
[0020] A4. The exosome according to any one of paragraphs Al to A3,
wherein the
isolating removes vesicles and cellular debris less than 20 nm in diameter.
[0021] A5. An exosome that comprises a modification selected from the
group
consisting of:
(i) at least one functional lysosomal protein or precursor thereof;
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
(ii) at least one nucleic acid comprising a nucleotide sequence that encodes
the functional
lysosomal protein or precursor thereof;
(iii) at least one fusion product comprising a lysosome targeting sequence
linked to an exosomal
membrane marker;
(iv) at least one nucleic acid comprising a nucleotide sequence that encodes
the fusion product;
and
(v) two or more of (i), (ii,) (iii), and (iv).
[0022] Bl. The exosome according to any of paragraphs Al ¨ A5, having a
diameter
of 20-120 nm.
[0023] B2. The exosome according to any of paragraphs Al ¨ A5, that
comprises a
functional lysosomal protein or precursor thereof, wherein the protein is
present in a lumen of the
exosome.
[0024] B3. The exosome according to any of paragraphs Al ¨ A5, that
comprises a
nucleic acid comprising a nucleotide sequence encoding a functional lysosomal
protein or
precursor thereof, wherein the nucleic acid is present in a lumen of the
exosome.
[0025] B3.1. The exosome according to paragraph B3, wherein the nucleic
acid
comprises mRNA or modified mRNA (modRNA, e.g. 5 methyl cytosine, or N6
methyladenine)
encoding for a protein set forth in Table 1.
[0026] B4. The exosome according to paragraph B2 or B3 or B3.1, wherein
the
protein comprises one or more of the proteins set forth in Table 1.
[0027] B5. The exosome according to paragraph B4, wherein the protein
is an
enzyme.
[0028] B6. The exosome according to any one of paragraphs B2 ¨ B3.1,
wherein the
protein is selected from the group consisting of alpha-D-mannosidase, N-
aspartyl-beta-
glucosaminidase, lysosomal acid lipase, cystinosin, lysosomal associated
membrane protein-2,
6
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
alpha-galactosidase A, acid ceramidase, alpha-fucosidase, cathepsin A, acid
beta-glucosidase,
beta-galactosidase, beta-hexosaminidase A, beta-hexosaminidase B, GlcNAc-l-
phosphotransferase, beta-galactosylceramidase, lysosomal acid lipase, aryl
sulfatase A, alpha-L-
iduronidase, iduronate-2-sulphatase, heparan sulphamidase, acetyl alpha-
glucosaminidase, acetyl
CoA: alpha-glucosaminide-N-acetyltransferase, N-acetyl glucosamine-6-
sulfatase, N-acetyl
galactosamine-6-sulfatase, hyaluronidase, acetyl galactosamine-4-sulphatase,
beta-
glucuronidase, alpha-N -acetyl neuraminidase, N-actrylglucosamine-l-
phosphotransferase,
mucolipin-1, formylglycine-generating enzyme, palmitoyl-protein thioesterase-
1, tripeptidyl
peptidase I, cysteine string protein, CLN3p, CLN5p, CLN6p, CLN7p, CLN8p, acid
sphingomyelinase, NPC 1, NPC 2, acid alpha-glucosidase, cathepsin K, sialin,
alpha-N-
acetylgalactosaminidase, GM2 activator, lysosomal acid lipase.
[0029] B7. The exosome according to any one of paragraphs B2 - B6,
further
comprising at least one fusion product comprising a lysosome targeting
sequence linked to an
exosomal membrane marker.
[0030] B8. The exosome according to any one of paragraphs Al - A5 or B
1, that
comprises at least one fusion product comprising a lysosome targeting sequence
linked to an
exosomal membrane marker.
[0031] B9. The exosome according to paragraph B7 or B8, wherein the
exosomal
membrane marker is selected from the group consisting of CD9, CD37, CD53,
CD63, CD81,
CD82, CD151, an integrin, ICAM-1, CDD31, an annexin, TSG101, ALIX, lysosome-
associated
membrane protein 1, lysosome-associated membrane protein 2, lysosomal integral
membrane
protein and a fragment of any exosomal membrane marker that comprises at least
one intact
transmembrane domain.
[0032] B10. The exosome according to any one of paragraphs B7 - B9,
wherein the
lysosomal targeting sequence is selected from the group consisting of lysosome-
associated
membrane protein 1, lysosome-associated membrane protein 2, lysosomal integral
membrane
protein, and a C-terminal sequence thereof comprising the sequence, G-Y-X-X-
XH, where XH is
one of glycine, valine, leucine, isoleucine, methionine, alanine, proline,
tryptophan or
phenylalanine, and X may be any amino acid.
7
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
[0033] B11. The exosome of paragraph B10, wherein the C-terminal sequence
is
selected from the group consisting of GYQSV (SEQ ID NO: 1), GYQTL (SEQ ID NO:
2),
GYQTI (SEQ ID NO: 3), GYEVM (SEQ ID NO: 4), GYEQF (SEQ ID NO: 5), AYQAL (SEQ
ID NO: 6), NYTHL (SEQ ID NO: 7), GYQRI (SEQ ID NO: 8), GYDQL (SEQ ID NO: 9),
GYKEI (SEQ ID NO: 10), GYRHV (SEQ ID NO: 11), DXXLL (SEQ ID NO: 12),
SFHDDSDEDLL (SEQ ID NO: 14), EESEERDDHLL (SEQ ID NO: 15), GYHDDSDEDLL
(SEQ ID NO: 16), ASVSLLDDELM (SEQ ID NO: 17), ASSGLDDLDLL (SEQ ID NO: 18),
VQNPSADRNLL (SEQ ID NO: 19), NALSWLDEELL (SEQ ID NO:20), TERERLL (SEQ ID
NO: 21), SETERLL (SEQ ID NO: 22), TDRTPLL (SEQ ID NO: 23), EETQPLL (SEQ ID NO:
24), ITGFSDDVPMV (SEQ ID NO: 25), DERAPLI (SEQ ID NO: 26), NEQLPML (SEQ ID
NO: 27) and DDQRDLI (SEQ ID NO: 28).
[0034] B12. The exosome according to any one of paragraphs B7 ¨ B11,
wherein the
fusion product is a fusion protein.
[0035] B13. The exosome according to paragraph B12, further wherein the
fusion
protein includes a peptide linker between the lysosome targeting sequence and
the exosomal
membrane marker.
[0036] B14. The exosome according to any one of paragraphs B7 - B13,
wherein the
lysosome targeting sequence is linked to the exosomal membrane marker with a
hydrophobic
linker comprising 4-5 hydrophobic amino acid moieties which are the same or
different and
selected from the group consisting of glycine, valine, leucine, isoleucine,
methionine, alanine,
proline, tryptophan and phenylalanine.
[0037] B15. The exosome according to any one of paragraphs B7-B13,
wherein the
fusion product includes a transmembrane domain and localizes in a membrane of
the exosome.
[0038] Cl. A composition comprising exosomes according to any one of
paragraphs
Al ¨ AS, and a pharmaceutically acceptable carrier.
[0039] C2. The composition according to paragraph Cl, wherein the
composition is
substantially free of vesicles having a diameter less than 20 nm.
8
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
[0040] C3. The composition according to paragraph Cl or C2, wherein
the
composition is substantially free of vesicles having a diameter greater than
120mm.
[0041] C4. The composition according to any one of claims Cl ¨ C3,
which exhibits a
zeta potential having a magnitude of at least 30 mV, or at least 40 mV, or at
least 50 mV, or at
least 60 mV, or at least 70 mV, or at least 80 V.
[0042] C5. The composition according ton claim C4, which exhibits a
zeta potential
of up to 200 mV, or up to 175 mV, or up to 150 mV, or up to 140 mV, or up to
130 mV, or up to
120 mV, or up to 110 mV, or up to 100 mV.
[0043] Dl. A method of increasing the amount of a lysosomal protein in
lysosomes in
a mammal, comprising administering to the mammal an exosome according to any
one of
paragraphs Al ¨ B15, or a composition according to any one of paragraphs Cl-
C5.
[0044] D2. Use of an exosome according to any one of paragraphs Al ¨
B15, or a
composition according to any one of paragraphs Cl- C5, for increasing the
amount of a
lysosomal protein in lysosomes in a mammal.
[0045] D3. A method of treating a lysosomal storage disease in a
mammal comprising
administering to the mammal an exosome according to any one of paragraphs Al ¨
B15, or a
composition according to any one of paragraphs Cl- C5.
[0046] D4. Use of an exosome according to any one of paragraphs Al ¨
B15, or a
composition according to any one of paragraphs Cl- C5, for treating a
lysosomal storage disease
in a mammal.
[0047] D5. The method or use according to any one of paragraphs D1 ¨
D4, wherein
the mammal is human.
[0048] D6. The method or use according to paragraph D5, wherein the
human has a
lysosomal storage disease selected from the group consisting of Alpha-
mannosidosis,
Aspartylglucosaminuria, Cholesteryl Ester Storage Disease, Cystinosis, Danon
Disease, Fabry
Disease, Farber Disease, Fucosidosis, Galactosialidosis, Gaucher Disease Type
I, Gaucher
9
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
Disease Type II, Gaucher Disease Type III, GM1 Gangliosidosis Type I, GM1
Gangliosidosis
Type II, GM1 Gangliosidosis Type III, GM2 - Sandhoff disease, GM2 - Tay-Sachs
disease,
GM2 - Gangliosidosis, AB variant, Mucolipidosis II, Krabbe Disease, Lysosomal
acid lipase
deficiency, Metachromatic Leukodystrophy, MPS I - Hurler Syndrome, MPS I -
Scheie
Syndrome, MPS I Hurler-Scheie Syndrome, MPS II - Hunter Syndrome, MPS IIIA -
Sanfilippo
Syndrome Type A, MPS IIIB - Sanfilippo Syndrome Type B, MPS IIIB - Sanfilippo
Syndrome
Type C, MPS IIIB - Sanfilippo Syndrome Type D, MPS IV - Morquio Type A, MPS IV
-
Morquio Type B, MPS IX - Hyaluronidase Deficiency, MPS VI - Maroteaux-Lamy,
MPS VII -
Sly Syndrome, Mucolipidosis I - Sialidosis, Mucolipidosis IIIC, Mucolipidosis
Type IV,
Multiple Sulfatase Deficiency, Neuronal Ceroid Lipofuscinosis Ti, Neuronal
Ceroid
Lipofuscinosis T2, Neuronal Ceroid Lipofuscinosis T3, Neuronal Ceroid
Lipofuscinosis T4,
Neuronal Ceroid Lipofuscinosis T5, Neuronal Ceroid Lipofuscinosis T6, Neuronal
Ceroid
Lipofuscinosis T7, Neuronal Ceroid Lipofuscinosis T8, Niemann-Pick Disease
Type A,
Niemann-Pick Disease Type B, Niemann-Pick Disease Type C, Pompe Disease,
Pycnodysostosis, Salla Disease, Schindler Disease and Wolman Disease.
[0049] D7. The method or use according to any one of paragraphs D1 ¨
D4, wherein
the mammal is human and has a disease set forth in Table 1, and the exosome
contains the
corresponding protein in Table 1, or a nucleic acid encoding said protein.
[0050] The invention further includes numerous embodiments, aspects, and
variations
that will be apparent from the drawings, details description, and claims that
follow.
[0051] These and other aspects of the invention will be described by
reference to the
following figures.
Brief Description of the Figures
[0052] Figure 1 graphically illustrates that GAA mRNA loaded unmodified
exosomes or GAA protein-loaded modified exosomes rescue GAA deficiency in
primary
fibroblasts isolated from GAA knock-out mice at higher efficiency than
conventional naked
GAA ERT. (A) Primary dermal fibroblasts isolated from GAA+/+ and GAA-/- mice
(n = 3 per
group) and were treated with naked GAA protein, empty exosomes (exosome
control), GAA
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
protein-loaded exosomes, or GAA mRNA-loaded exosomes for 48 hours. Cells were
harvested
after 48 hours and GAA activity was measured. (B) Mean SD of experiments in
(A)
independently repeated 3 times. *P < 0.05. Data were analyzed using an
unpaired t-test.
[0053] Figure 2 graphically illustrates that GAA mRNA loaded unmodified
exosomes or GAA protein-loaded modified exosomes rescue GAA deficiency in
primary
myotubes isolated from GAA knock-out mice at higher efficiency than
conventional naked
GAA ERT. (A) Primary mononuclear cells were isolated from skeletal muscle of
GAA+/+ and
GAA-/- mice (n = 3 per group). Cells were pre-plated to obtain a pure
population of myoblasts.
Cells were differentiated for 5 days into myotubes followed by treatment with
naked GAA
protein, empty exosomes (exosome control), GAA protein-loaded exosomes, or GAA
mRNA-
loaded exosomes for 48 hours. Cells were harvested after 48 hours and GAA
activity was
measured. (B) Mean SD of experiments in (A) independently repeated 3 times.
*P <0.05. Data
were analyzed using an unpaired t-test.
[0054] Figure 3 graphically illustrates that GAA mRNA loaded unmodified
exosomes or GAA protein-loaded modified exosomes rescue GAA deficiency in
primary
fibroblasts isolated from Pompe patients at higher efficiency than
conventional naked GAA
ERT. (A) Primary dermal fibroblasts isolated from three Pompe patients and
three age/gender-
matched controls, and were treated with naked GAA protein, empty exosomes
(exosome
control), GAA protein-loaded exosomes, or GAA mRNA-loaded exosomes for 48
hours. Cells
were harvested after 48 hours and GAA activity was measured. (B) Mean SD of
experiments
in (A) independently repeated 3 times. *P < 0.05. Data were analyzed using an
unpaired t-test.
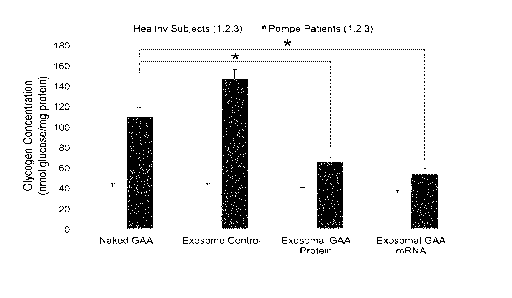
[0055] Figure 4 graphically illustrates that GAA mRNA loaded unmodified
exosomes or GAA protein-loaded modified exosomes reduced glycogen build-up in
primary
fibroblasts isolated from Pompe patients at higher efficiency than
conventional naked GAA
ERT. Primary dermal fibroblasts isolated from three Pompe patients and three
age/gender-
matched controls, and were treated with naked GAA protein, empty exosomes
(exosome
control), GAA protein-loaded exosomes, or GAA mRNA-loaded exosomes for 48
hours. Cells
were harvested after 48 hours and glycogen content was measured. Mean SD of
experiments
independently repeated 3 times. *P < 0.05. Data were analyzed using an
unpaired t-test.
11
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
[0056] Figure 5 graphically illustrates that GAA protein-loaded exosomes
therapy
reduces body weight in GAA KO mice. Body weights of GAA KO mice (n = 5-7 per
group)
treated with saline (empty exosomes), naked recombinant GAA ERT (naked GAA),
and GAA
protein-loaded exosomes ERT for 7 weeks (once a week intravenously,
corresponding to 40
mg/kg GAA). Data were analyzed using an unpaired t-test.
[0057] Figure 6 graphically illustrates that GAA protein-loaded exosome
therapy
increases strength and motor control vs. conventional naked GAA ERT in GAA KO
mice.
(A) Paw-grip endurance test, (B) grip strength test, and (C) rotarod motor
control test in GAA
KO mice (n = 5-7 per group) treated with saline (empty exosomes), naked
recombinant GAA
ERT (naked GAA), and GAA protein-loaded exosomes ERT for 7 weeks (once a week
intravenously, corresponding to 40 mg/kg GAA). *P < 0.05. Data were analyzed
using an
unpaired t-test.
[0058] Figure 7 graphically illustrates that GAA protein-loaded exosome
therapy
increases EDL (fast-twitch muscle) and soleus (slow-twitch muscle) mass vs.
conventional
naked GAA ERT in GAA KO mice. EDL (fast-twitch muscle; white bar) and soleus
(slow-
twitch muscle; red bar) mass in GAA KO mice (n = 5-7 per group) treated with
saline (empty
exosomes), naked recombinant GAA ERT (naked GAA), and GAA protein-loaded
exosomes
ERT for 7 weeks (once a week intravenously, corresponding to 40 mg/kg GAA). *P
< 0.05. Data
were analyzed using an unpaired t-test.
[0059] Figure 8 graphically illustrates that GAA protein-loaded exosome
therapy
increases mixed fiber-type mass vs. conventional naked GAA ERT in GAA KO mice.
(A)
Quadriceps, (B) gastrocnemius, and (C) tibialis anterior (TA) mass in GAA KO
mice (n = 5-7
per group) treated with saline (empty exosomes), naked recombinant GAA ERT
(naked GAA),
and GAA protein-loaded exosomes ERT for 7 weeks (once a week intravenously,
corresponding
to 40 mg/kg GAA). *P < 0.05. Data were analyzed using an unpaired t-test.
[0060] Figure 9 graphically illustrates that GAA protein-loaded exosome
therapy
rescues pathogenic cardiac hypertrophy and brain mass vs. conventional naked
GAA ERT
in GAA KO mice. (A) Heart and (B) brain mass in GAA KO mice (n = 5-7 per
group) treated
with saline (empty exosomes), naked recombinant GAA ERT (naked GAA), and GAA
protein-
12
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
loaded exosomes ERT for 7 weeks (once a week intravenously, corresponding to
40 mg/kg
GAA). *P < 0.05. Data were analyzed using an unpaired t-test.
[0061] Figure 10 graphically illustrates that GAA protein-loaded exosome
therapy
increases tissue GAA activity vs. conventional naked GAA ERT in GAA KO mice.
(A) GAA
activity in EDL, soleus, diaphragm, heart, and brain of GAA KO mice (n = 5-7
per group)
treated with saline (empty exosomes) or conventional naked recombinant GAA ERT
(naked
GAA). (B) GAA activity in aforementioned tissues in GAA KO mice using approach
in (A) vs.
GAA protein-loaded exosomes ERT for 7 weeks (once a week intravenously,
corresponding to
40 mg/kg GAA). *P < 0.05. Data were analyzed using an unpaired t-test.
[0062] Figure 11 graphically illustrates that GAA protein-loaded exosome
therapy
reduces tissue total glycogen content vs. conventional naked GAA ERT in GAA KO
mice.
Total glycogen content (mmol glucosyl units/kg of dry tissue weight) in
tibialis anterior (TA)
muscle, heart, and brain of GAA KO mice (n = 5-7 per group) treated with
saline (empty
exosomes), conventional naked recombinant GAA ERT (naked GAA) and exosomal
GAA. *P <
0.05. Data were analyzed using an unpaired t-test.
[0063] Figure 12 graphically illustrates that GAA mRNA-loaded exosome
therapy
increases skeletal muscle, diaphragm, and heart GAA activity in GAA KO mice.
GAA
activity is restored in fast- (EDL) and slow- (soleus) fiber-type skeletal
muscle (A), and in the
diaphragm and heart (B) in GAA KO mice (n = 5-6 per group) treated with
exosomes loaded
with GAA mRNA vs. GAA KO mice treated with empty exosomes (n = 5-6 per group).
Littermate wildtype (WT) mice were used as controls for both treatments. *P <
0.05. Data were
analyzed using an unpaired t-test.
[0064] Figure 13 graphically illustrates that GAA mRNA-loaded exosome
therapy
increases brain GAA activity in GAA KO mice. GAA activity is restored in the
brain of GAA
KO mice (n = 5-6 per group) treated with exosomes loaded with GAA mRNA vs. GAA
KO mice
treated with empty exosomes (n = 5-6 per group). Littermate wildtype (WT) mice
were used as
controls for both treatments. *P < 0.05. Data were analyzed using an unpaired
t-test.
13
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
[0065] Figure 14 graphically illustrates that GAA mRNA-loaded exosome
therapy
normalizes skeletal muscle and brain GAA gene expression in GAA KO mice. GAA
mRNA
expression is restored in the skeletal muscle (quadriceps) and brain of GAA KO
mice (n = 5-6
per group) treated with exosomes loaded with GAA mRNA vs. GAA KO mice treated
with
empty exosomes (n = 5-6 per group). Littermate wildtype (WT) mice were used as
controls for
both treatments. *P < 0.05. Data were analyzed using an unpaired t-test.
[0066] Figure 15 graphically illustrates that GAA mRNA-loaded exosome
therapy
reduces pathological glycogen content in heart and skeletal muscle of GAA KO
mice. Total
glycogen content is normalized in skeletal muscle (quadriceps and tibia/is
anterior) and heart in
GAA KO mice (n = 5-6 per group) treated with exosomes loaded with GAA mRNA vs.
GAA
KO mice treated with empty exosomes (n = 5-6 per group). Littermate wildtype
(WT) mice were
used as controls for both treatments. *P < 0.05. Data were analyzed using an
unpaired t-test.
[0067] Figure 16 graphically illustrates that GAA mRNA-loaded exosome
therapy
increases grip endurance in GAA KO mice. GAA KO mice (n = 5-6 per group)
treated with
exosomes loaded with GAA mRNA vs. GAA KO mice treated with empty exosomes (n =
5-6
per group). Littermate wildtype (WT) mice were used as controls for both
treatments. *P < 0.05.
Data were analyzed using an unpaired t-test.
[0068] Figure 17 graphically illustrates that NPC1 mRNA loaded exosome
therapy
rescues substrate accumulation in primary fibroblasts isolated from Niemann-
Pick Disease
Type C patient. Primary dermal fibroblasts isolated from three Niemann-Pick
Disease Type C
patients and three age/gender-matched controls, were treated with; media
control, empty
exosomes (exosome control), NPC1 protein-loaded exosomes, or NPC1 mRNA-loaded
exosomes for 48 hours. Cells were harvested after 48 hours and total
cholesterol and
glycosphingolipid content was measured using filipin staining. Mean SD of
experiments
independently repeated 3 times. *P < 0.05. Data were analyzed using an
unpaired t-test.
[0069] Figure 18 graphically illustrates restoration of grip strength (A)
and motor
function (B = fall time on rotarod; C = fall speed on rotarod) in NPC1¨/¨
homozygous mice
treated with NPC1 mRNA-loaded exosomes as compared to NPC1¨/¨ homozygous mice
treated
with empty exosomes.
14
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
Detailed Description of the Invention
[0070]
Exosomes are provided which have been genetically engineered to incorporate a
lysosomal protein, and/or nucleic acid encoding a lysosomal protein.
[0071]
The term "exosome" refers to cell-derived vesicles having a diameter of
between
about 40 and 120 nm, preferably a diameter of about 50-100 nm, for example, a
diameter of
about 60 nm, 70 nm, 80 nm, 90 nm, or 100 nm. Exosomes may be isolated from any
suitable
biological sample from a mammal, including but not limited to, whole blood,
serum, plasma,
urine, saliva, breast milk, cerebrospinal fluid, amniotic fluid, ascitic
fluid, bone marrow and
cultured mammalian cells (e.g. immature dendritic cells (wild-type or
immortalized), induced
and non-induced pluripotent stem cells, fibroblasts, platelets, immune cells,
reticulocytes, tumour
cells, mesenchymal stem cells, satellite cells, hematopoietic stem cells,
pancreatic stem cells,
white and beige pre-adipocytes and the like). As one of skill in the art will
appreciate, cultured
cell samples will be in the cell-appropriate culture media (using exosome-free
serum).
Exosomes include specific surface markers not present in other vesicles,
including surface
markers such as tetraspanins, e.g. CD9, CD37, CD44, CD53, CD63, CD81, CD82 and
CD151;
targeting or adhesion markers such as integrins, ICAM-1, EpCAM and CD31;
membrane fusion
markers such as annexins, TSG101, ALIX; and other exosome transmembrane
proteins such as
Rab5b,
HSP70, LAMP2 (lysosome-associated membrane protein) and LIMP (lysosomal
integral membrane protein). Exosomes may also be obtained from a non-mammalian
biological
sample, including cultured non-mammalian cells. As the molecular machinery
involved in
exosome biogenesis is believed to be evolutionarily conserved, exosomes from
non-mammalian
sources include surface markers which are isoforms of mammalian surface
markers, such as
isoforms of CD9 and CD63, which distinguish them from other cellular vesicles.
As used herein,
the term "mammal" is meant to encompass, without limitation, humans, domestic
animals such
as dogs, cats, horses, cattle, swine, sheep, goats and the like, as well as
non-domesticated animals
such as, but not limited to, mice, rats and rabbits. The term "non-mammal" is
meant to
encompass, for example, exosomes from microorganisms such as bacteria, flies,
worms, plants,
fruit/vegetables (e.g. corn, pomegranate) and yeast.
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
[0072] Exosomes may be obtained from the appropriate biological sample
using a
combination of isolation techniques, for example, centrifugation, filtration
and
ultracentrifugation methodologies. In one embodiment, the isolation protocol
includes the steps
of: i) exposing the biological sample to a first centrifugation to remove
cellular debris greater
than about 7-10 microns in size from the sample and obtaining the supernatant
following
centrifugation; ii) subjecting the supernatant from step i) to centrifugation
to remove
microvesicles therefrom; iii) microfiltering the supernatant from step ii) and
collecting the
microfiltered supernatant; iv) subjecting the microfiltered supernatant from
step iii) to at least
one round of ultracentrifugation to obtain an exosome pellet; and v) re-
suspending the exosome
pellet from step iv) in a physiological solution and conducting a second
ultracentrifugation in a
density gradient and remove the exosome pellet fraction therefrom.
[0073] Thus, the process of isolating exosomes from a biological sample
includes a first
step of removing undesired large cellular debris from the sample, i.e. cells,
cell components,
apoptotic bodies and the like greater than about 7-10 microns in size. This
step is generally
conducted by centrifugation, for example, at 1000-4000x g for 10 to 60 minutes
at 4 C,
preferably at 1500-2500x g, e.g. 2000x g, for a selected period of time such
as 10-30 minutes,
12-28 minutes, 14-24 minutes, 15-20 minutes or 16, 17, 18 or 19 minutes. As
one of skill in the
art will appreciate, a suitable commercially available laboratory centrifuge,
e.g. Thermo-
ScientificTm or Cole-ParmerTM, is employed to conduct this isolation step. To
enhance exosome
isolation, the resulting supernatant is subjected to a second optional
centrifugation step to further
remove cellular debris and apoptotic bodies, such as debris that is at least
about 7-10 microns in
size, by repeating this first step of the process, i.e. centrifugation at 1000-
4000x g for 10 to 60
minutes at 4 C, preferably at 1500-2500x g, e.g. 2000x g, for the selected
period of time.
[0074] Following removal of cell debris, the supernatant resulting from
the first
centrifugation step(s) is separated from the debris-containing pellet (by
decanting or pipetting it
off) and may then be subjected to an optional additional (second)
centrifugation step, including
spinning at 12,000-15,000x g for 30-90 minutes at 4 C to remove intermediate-
sized debris, e.g.
debris that is greater than 6 microns size. In one embodiment, this
centrifugation step is
conducted at 14,000x g for 1 hour at 4 C. The resulting supernatant is again
separated from the
debris-containing pellet.
16
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
[0075] The resulting supernatant is collected and subjected to a third
centrifugation step,
including spinning at between 40,000-60,000x g for 30-90 minutes at 4 C to
further remove
impurities such as medium to small-sized microvesicles greater than 0.3
microns in size e.g. in
the range of about 0.3-6 microns. In one embodiment, the centrifugation step
is conducted at
50,000x g for 1 hour. The resulting supernatant is separated from the pellet
for further
processing.
[0076] The supernatant is then filtered to remove debris, such as
bacteria and larger
microvesicles, having a size of about 0.22 microns or greater, e.g. using
microfiltration. The
filtration may be conducted by one or more passes through filters of the same
size, for example,
a 0.22 micron filter. Alternatively, filtration using 2 or more filters may be
conducted, using
filters of the same or of decreasing sizes, e.g. one or more passes through a
40-50 micron filter,
one or more passes through a 20-30 micron filter, one or more passes through a
10-20 micron
filter, one or more passes through a 0.22-10 micron filter, etc. Suitable
filters for use in this step
include the use of 0.45 and 0.22 micron filters.
[0077] The microfiltered supernatant (filtrate) may then be combined with
a suitable
physiological solution, preferably sterile, for example, an aqueous solution,
a saline solution or a
carbohydrate-containing solution in a 1:1 ratio, e.g. 10 mL of supernatant to
10mL of
physiological solution, to prevent clumping of exosomes during the subsequent
ultracentrifugation and to maintain the integrity of the exosomes. The
exosomal solution is then
subjected to ultracentrifugation to pellet exosomes and any remaining
contaminating
microvesicles (between 100-220 nm). This ultracentrifugation step is conducted
at 110,000-
170,000x g for 1-3 hours at 4 C, for example, 170,000x g for 3 hours. This
ultracentrifugation
step may optionally be repeated, e.g. 2 or more times, in order to enhance
results. Any
commercially available ultracentrifuge, e.g. Thermo-ScientificTM or BeckmanTM,
may be
employed to conduct this step. The exosome-containing pellet is removed from
the supernatant
using established techniques and re-suspended in a suitable physiological
solution.
[0078] Following ultracentrifugation, the re-suspended exosome-containing
pellet is
subjected to density gradient separation to separate contaminating
microvesicles from exosomes
based on their density. Various density gradients may be used, including, for
example, a sucrose
17
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
gradient, a colloidal silica density gradient, an iodixanol gradient, or any
other density gradient
sufficient to separate exosomes from contaminating microvesicles (e.g. a
density gradient that
functions similar to the 1.100-1.200 g/ml sucrose fraction of a sucrose
gradient). Thus, examples
of density gradients include the use of a 0.25-2.5 M continuous sucrose
density gradient
separation, e.g. sucrose cushion centrifugation, comprising 20-50% sucrose; a
colloidal silica
density gradient, e.g. PercollTM gradient separation (colloidal silica
particles of 15-30 nm
diameter, e.g. 30%/70% w/w in water (free of RNase and DNase), which have been
coated with
polyvinylpyrrolidone (PVP)); and an iodixanol gradient, e.g. 6-18% iodixanol.
The resuspended
exosome solution is added to the selected gradient and subjected to
ultracentrifugation at a speed
between 110,000-170,000x g for 1-3 hours. The resulting exosome pellet is
removed and re-
suspended in physiological solution.
[0079] Depending on the density gradient used, the re-suspended exosome
pellet
resulting from the density gradient separation may be ready for use. For
example, if the density
gradient used is a sucrose gradient, the exosome pellet is removed from the
appropriate sucrose
gradient fraction, and is ready for use, or may preferably be subjected to an
ultracentrifugation
wash step at a speed of 110,000-170,000x g for 1-3 hours at 4 C. If the
density gradient used is,
for example, a colloidal silica or a iodixanol density gradient, then the
resuspended exosome
pellet may be subjected to additional wash steps, e.g. subjected to one to
three ultracentrifugation
steps at a speed of 110,000-170,000x g for 1-3 hours each at 4 C, to yield an
essentially pure
exosome-containing pellet. As one of skill in the art will appreciate, the
exosome pellet from
any of the centrifugation or ultracentrifugation steps may be washed between
centrifugation steps
using an appropriate physiological solution, e.g. saline. The final pellet is
removed from the
supernatant and may be re-suspended in a physiologically acceptable solution
for use.
Alternatively, the exosome pellet may be stored for later use, for example, in
cold storage at 4 C,
in frozen form or in lyophilized form, prepared using well-established
protocols. The exosome
pellet may be stored in any physiological acceptable carrier, optionally
including cryogenic
stability and/or vitrification agents (e.g. DMSO, glycerol, trehalose,
polyhydroxylated alcohols
(e.g. methoxylated glycerol, propylene glycol), M22 and the like).
18
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
[0080] The described exosome isolation protocol advantageously provides a
means to
obtain mammalian exosomes which are at least about 90% pure, and preferably at
least about
95% or greater pure, i.e. referred to herein as "essentially free" from
cellular debris, apoptotic
bodies and microvesicles having a diameter less than 20 or greater than 120
nm, and preferably
free from particles having a diameter of less than 40 or greater than 120 nm,
and which are
biologically intact, e.g. not clumped or in aggregate form, and not sheared,
leaky or otherwise
damaged. Exosomes isolated according to the methods described herein exhibit a
high degree of
stability, evidenced by the zeta potential of a mixture/solution of such
exosomes, for example, a
zeta potential of at least a magnitude of 30 mV, e.g. < -30 or > +30, and
preferably, a magnitude
of at least 40 mV, 50 mV, 60 mV, 70 mV, 80 mV, or greater. The term "zeta
potential" refers to
the electrokinetic potential of a colloidal dispersion, and the magnitude of
the zeta potential
indicates the degree of electrostatic repulsion between adjacent, similarly
charged particles
(exosomes) in a dispersion. For exosomes, a zeta potential of magnitude 30 mV
or greater
indicates moderate stability, i.e. the solution or dispersion will resist
aggregation, while a zeta
potential of magnitude 40-60 mV indicates good stability, and a magnitude of
greater than 60
mV indicates excellent stability.
[0081] Moreover, high quantities of exosomes are achievable by the
present isolation
method, e.g. exosomes in an amount of about 100-2000 [tg total protein can be
obtained from 1-4
mL of mammalian serum or plasma, or from 15-20 mL of cell culture spent media
(from at least
about 2 x 106 cells). Thus, solutions comprising exosomes at a concentration
of at least about 5
[tg/[tL, and preferably at least about 10-25 [tg/[tL, may readily be prepared
due to the high
exosome yields obtained by the present method. The term "about" as used herein
with respect to
any given value refers to a deviation from that value of up to 10%, either up
to 10% greater, or
up to 10% less.
[0082] Exosomes isolated in accordance with the methods herein described,
which
beneficially retain integrity, and exhibit a high degree of purity (being
"essentially free" from
entities having a diameter less than 20 nm and greater than 120 nm), stability
and biological
activity both in vitro and in vivo, have not previously been achieved. Thus,
the present exosomes
are uniquely useful, for example, diagnostically and/or therapeutically, e.g.
for the in vivo
19
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
delivery of protein and/or nucleic acid. They have also been determined to be
non-
allergenic/non-immunogenic, and thus, safe for autologous, allogenic, and
xenogenic use.
[0083] Isolated exosomes may then be genetically engineered to
incorporate an
exogenous protein, e.g. an exogenous protein such as a lysosomal protein, or
exogenous nucleic
acid encoding a selected protein, or both. The term "exogenous" is used herein
to refer to
protein or nucleic acid originating from a source external to the exosomes.
Nucleic acid
encoding the protein may be produced using known synthetic techniques,
incorporated into a
suitable expression vector using well established methods to form a protein-
encoding expression
vector which is introduced into isolated exosomes using known techniques, e.g.
electroporation,
transfection using cationic lipid-based transfection reagents, and the like.
Similarly, the selected
protein may be produced using recombinant techniques, or may be otherwise
obtained, and then
may be introduced directly into isolated exosomes by electroporation or
transfection. More
particularly, electroporation applying voltages in the range of about 20-1000
V/cm may be used
to introduce nucleic acid or protein into exosomes. Transfection using
cationic lipid-based
transfection reagents such as, but not limited to, Lipofectamine
MessengerMAXTm
Transfection Reagent, Lipofectamine RNAiMAX Transfection Reagent,
Lipofectamine 3000
Transfection Reagent, or Lipofectamine LTX Reagent with PLUSTM Reagent, may
also be
used. The amount of transfection reagent used may vary with the reagent, the
sample and the
cargo to be introduced. For example, using Lipofectamine MessengerMAXTm
Transfection
Reagent, an amount in the range of about 0.15 uL to 10 uL may be used to load
100 ng to 2500
ng mRNA or protein into exosomes. Other methods may also be used to load
protein into
exosomes including, for example, the use of cell-penetrating peptides.
[0084] Exosomes isolated in accordance with the methods herein described,
which
beneficially retain integrity, and exhibit a high degree of purity and
stability, readily permit
loading of exogenous protein and/or nucleic acid in an amount of at least
about 1 ng nucleic acid
(e.g. mRNA) per 10 ug of exosomal protein or 30 ug protein per 10 ug of
exosomal protein.
[0085] In another embodiment, a protein-encoding expression vector as
above described,
may be introduced directly into exosome-producing cells, e.g. autologous,
allogenic, or
xenogenic cells, such as immature dendritic cells (wild-type or immortalized),
induced and non-
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
induced pluripotent stem cells, fibroblasts, platelets, immune cells,
reticulocytes, tumour cells,
mesenchymal stem cells, satellite cells, hematopoietic stem cells, pancreatic
stem cells, white
and beige pre-adipocytes and the like, by electroporation or transfection as
described above.
Following a sufficient period of time, e.g. 3-7 days to achieve stable
expression of the protein,
exosomes incorporating the expressed protein may be isolated from the exosome-
producing cells
as described herein.
[0086] Alternatively, prior to incorporation into exosomes of a selected
protein, or
nucleic acid encoding the protein, exosomes may be modified to express or
incorporate a target-
specific fusion product. For the delivery of lysosomal proteins, the target-
specific fusion product
comprises a lysosome targeting sequence, linked to an exosomal membrane
marker. The
exosomal membrane marker of the fusion product will localize the fusion
product within the
membrane of the exosome to enable the targeting sequence to direct the exosome
to the intended
target. Examples of exosome membrane markers include, but are not limited to:
tetraspanins
such as CD9, CD37, CD53, CD63, CD81, CD82 and CD151; targeting or adhesion
markers such
as integrins, ICAM-1 and CDD31; membrane fusion markers such as annexins,
TSG101, ALIX;
and other exosome transmembrane proteins such as LAMP (lysosome-associated
membrane
protein), e.g. LAMP 1 or 2, and LIMP (lysosomal integral membrane protein).
All or a fragment
of an exosome membrane marker may be utilized in the fusion product provided
that any
fragment includes a sufficient portion of the membrane marker to enable it to
localize within the
exosome membrane, i.e. the fragment comprises at least one intact
transmembrane domain to
permit localization of the portion of the membrane marker into the exosomal
membrane.
[0087] The target-specific fusion product also includes a cell or
organelle targeting
sequence such as a lysosome targeting sequence, i.e. a protein or peptide
sequence which
facilitates the targeted lysosomal uptake of the exosome. Examples of suitable
lysosomal
targeting sequences include, but are not limited to, the lysosomal targeting
sequence of LAMP
and LIMP e.g. the C-terminal sequence thereof, comprising the sequence, G-Y-X-
X-XH, where
XH is a hydrophobic residue such as glycine, valine, leucine, isoleucine,
methionine, alanine,
proline, tryptophan or phenylalanine, and X may be any amino acid. Thus,
examples of
lysosomal targeting sequences derived from the C-terminal sequence of LAMP or
LIMP include
sequences such as GYQSV (SEQ ID NO: 1), GYQTL (SEQ ID NO: 2), GYQTI (SEQ ID
NO:
21
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
3), GYEVM (SEQ ID NO: 4), GYEQF (SEQ ID NO: 5), AYQAL (SEQ ID NO: 6), NYTHL
(SEQ ID NO: 7), GYQRI (SEQ ID NO: 8), GYDQL (SEQ ID NO: 9), GYKEI (SEQ ID NO:
10), and GYRHV (SEQ ID NO: 11). Additionally, the lysosomal targeting sequence
may be a
dileucine-based motif, e.g. DXXLL (SEQ ID NO: 12), or [DE]XXXL[LI] (SEQ ID NO:
13),
such as SFHDDSDEDLL (SEQ ID NO: 14), EESEERDDHLL (SEQ ID NO: 15),
GYHDDSDEDLL (SEQ ID NO: 16), ASVSLLDDELM (SEQ ID NO: 17), ASSGLDDLDLL
(SEQ ID NO: 18), VQNPSADRNLL (SEQ ID NO: 19), NALSWLDEELL (SEQ ID NO:20),
TERERLL (SEQ ID NO: 21), SETERLL (SEQ ID NO: 22), TDRTPLL (SEQ ID NO: 23), and
EETQPLL (SEQ ID NO: 24). Other lysosomal targeting sequences include;
ITGFSDDVPMV
(SEQ ID NO: 25), DERAPLI (SEQ ID NO: 26), NEQLPML (SEQ ID NO: 27) and DDQRDLI
(SEQ ID NO: 28).
[0088] To increase the efficiency of lysosome targeting, the targeting
sequence may be
linked to the exosomal membrane marker with a hydrophobic linker comprising 4-
5 hydrophobic
amino acid moieties, including one or more of glycine, valine, leucine,
isoleucine, methionine,
alanine, proline, tryptophan or phenylalanine, which are the same or
different. Thus, the
hydrophobic linker may include 4-5 glycine residues, or 4-5 of any one of the
other hydrophobic
amino acids, or may include a mixture of 2 or more hydrophobic amino acids.
[0089] Exosomes incorporating the lysosome-specific fusion product may be
produced
using recombinant technology. In this regard, an expression vector encoding
the target-specific
fusion product is introduced by electroporation or transfection into exosome-
producing cells
isolated from an appropriate biological sample. As one of skill in the art
will appreciate, it is
also possible to produce the fusion product using recombinant techniques, and
then introduce the
fusion product directly into exosome-producing cells using similar techniques,
e.g.
electroporation, transfection using cationic lipid-based transfection
reagents, and the like.
Following a sufficient period of time, exosomes generated by the exosome-
producing cells, and
including the fusion product, may be isolated as described.
[0090] The desired lysosomal protein, nucleic acid encoding the protein
or both may be
introduced into isolated exosomes incorporating a lysosome-targeting fusion
product (modified
lysosome-targeting exosomes) as previously described, using electroporation or
transfection
22
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
methods. Addition to the exosome of both the desired lysosomal protein and
nucleic acid
encoding the same lysosomal protein may increase delivery efficiency of the
protein.
[0091] Exosomes genetically engineered to incorporate a protein, and/or
nucleic acid
encoding the protein, may be used to deliver the protein and/or nucleic acid
to mammal in vivo in
the treatment of a pathological condition or disease in which the protein is
defective or absent to
upregulate the activity of the protein and thereby treat the disease. In one
embodiment,
exosomes incorporating a lysosomal protein, including exosomes modified to
include a
lysosome-targeting fusion product and unmodified exosomes (i.e. exosomes not
including a
lysosome-targeting fusion product) may be used to deliver a protein (or
nucleic acid encoding the
protein) to lysosomes in vivo in the treatment of a pathological condition or
disease such as a
lysosomal storage disease (LSD), a disease in which a protein is defective or
missing in
lysosomes. Examples of lysosomal storage diseases that may be treated using
the present
engineered exosomes are set out in Table 1 below, and identify the protein
required to treat the
disease and the NCBI (National Centre for Biotechnology Information) reference
which provides
mRNA transcript sequence information, as well as protein sequence information.
Table 1.
Alpha-mannosidosis alpha-D-mannosidase NM 000528.3
Aspartylglucosaminuria N-aspartyl-beta-glucosaminidase NM 000027.3
Cholesteryl Ester Storage Disease lysosomal acid
lipase NM 000235.3
Cystinosis Cystinosin NM 001031681.2
lysosomal associated membrane
Danon Disease protein-2 NM 002294.2
Fabry Disease alpha-galactosidase A NM 000169.2
Farber Disease acid ceramidase NM 177924.3
Fucosidosis alpha-fucosidase NM 000147.4
Galactosialidosis cathepsin A NM 000308.2
Gaucher Disease - Type I acid beta-glucosidase NM 000157.3
Gaucher Disease - Type II acid beta-glucosidase NM 000157.3
Gaucher Disease - Type III acid beta-glucosidase NM 000157.3
GM1 - Gangliosidosis (types I, II, III) beta-
galactosidase NM 000404.2
GM2 - Sandhoff disease (I/J/A) beta-hexosaminidase A NM 001292004.1
23
CA 02962081 2017-03-22
WO 2016/044947
PCT/CA2015/050959
GM2 - Tay-Sachs disease beta-hexosaminidase A + B NM 000520.4
GM2 - Gangliosidosis, AB variant GM2 activator
(GM2A) NM 000405.4
I-Cell Disease/Mucolipidosis II GlcNAc-l-phosphotransferase NM 024312.4
Krabbe Disease beta-galactosylceramidase NM 000153.3
Lysosomal acid lipase deficiency lysosomal acid
lipase NM 000235
Metachromatic Leukodystrophy arylsulfatase A NM 000487.5
MPS I - Hurler Syndrome alpha-L-iduronidase NM 000203.4
MPS I - Scheie Syndrome alpha-L-iduronidase NM 000203.4
MPS I Hurler-Scheie Syndrome alpha-L-iduronidase NM 000203.4
MPS II - Hunter Syndrome iduronate-2-sulphatase NM 000202.6
MPS IIIA - Sanfilippo Syndrome Type A heparan sulphamidase NM 000199.3
MPS IIIB - Sanfilippo Syndrome Type B acetyl alpha-glucosaminidase NM
000263.3
acetyl CoA: alpha-glucosaminide-N-
MPS IIIC - Sanfilippo Syndrome Type C acetyltransferase NM 152419.2
MPS IIID - Sanfilippo Syndrome Type D N-acetyl glucosamine-6-sulfatase NM
002076.3
MPS IV - Morquio Type A N-acetyl galactosamine-6-sulfatase
NM_000512.4
MPS IV - Morquio Type B beta-galactosidase NM 000404.2
MPS IX - Hyaluronidase Deficiency Hyaluronidase
NM 033159.3
acetyl galactosamine-4-sulphatase
MPS VI - Maroteaux-Lamy (arylsulphatase B) NM 000046.3
MPS VII - Sly Syndrome beta-glucuronidase NM 000181.3
Mucolipidosis I - Sialidosis alpha-N -acetyl neuraminidase NM
001304450.1
N-actrylglucosamine-l-
Mucolipidosis IIIC phosphotransferase NM 032520.4
Mucolipidosis Type IV mucolipin-1 NM 020533.2
Multiple Sulfatase Deficiency formylglycine-generating enzyme NM
001042469.1
Neuronal Ceroid Lipofuscinosis Ti palmitoyl-
protein thioe ste rase -1 NM 000310.3
Neuronal Ceroid Lipofuscinosis T2 tripeptidyl
peptidase I NM 001020382.2
Neuronal Ceroid Lipofuscinosis T3 CLN3p NM
001283889.1
Neuronal Ceroid Lipofuscinosis T4 cysteine string
protein NM 025219.2
Neuronal Ceroid Lipofuscinosis T5 CLN5p NM
006493.2
Neuronal Ceroid Lipofuscinosis T6 CLN6p NM
017882.2
Neuronal Ceroid Lipofuscinosis T7 CLN7p NM
152778.2
Neuronal Ceroid Lipofuscinosis T8 CLN8p
NMO18941.3
Niemann-Pick Disease Type A acid sphingomyelinase NM 000543.4
Niemann-Pick Disease Type B acid sphingomyelinase NM 000543.4
24
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
Niemann-Pick Disease Type C NPC 1/ NPC 2 NM
000271.4
Pompe Disease acid alpha-glucosidase NM
000152.3
Pycnodysostosis cathepsin K NM
000396.3
Salla Disease sialin (sialic acid transporter)
NMO12434.4
Schindler Disease alpha-N-acetylgalactosaminidase NM
000262.2
Wolman Disease lysosomal acid lipase NM
000235.3
[0092] As one of skill in the art will appreciate, the lysosomal protein
for incorporation
into exosomes according to the invention may be a functional native mammalian
lysosomal
protein, including for example, a protein from human and non-human mammals, or
a
functionally equivalent protein. The term "functionally equivalent" is used
herein to refer to a
protein which exhibits the same or similar function (retains at least about
30% of the activity of
native lysosomal protein) to the native protein, and includes all isoforms,
variants, recombinant
produced forms, and naturally-occurring or artificially modified forms, i.e.
including
modifications that do not adversely affect activity and which may increase
cell uptake, stability,
activity and/or therapeutic efficacy. The term "functionally equivalent" also
refers to nucleic
acid, e.g. mRNA, DNA or cDNA, encoding a lysosomal protein, and is meant to
include any
nucleic acid sequence which encodes a functional lysosomal protein, including
all transcript
variants, variants that encode protein isoforms, variants due to degeneracy of
the genetic code,
artificially modified variants, and the like. Protein modifications may
include, but are not limited
to, one or more amino acid substitutions (for example, with a similarly
charged amino acid, e.g.
substitution of one amino acid with another each having non-polar side chains
such as valine,
leucine, alanine, isoleucine, glycine, methionine, phenylalanine, tryptophan,
proline; substitution
of one amino acid with another each having basic side chains such as
histidine, lysine, arginine;
substitution of one amino acid with another each having acidic side chains
such as aspartic acid
and glutamic acid; and substitution of one amino acid with another each having
polar side chains
such as cysteine, serine, threonine, tyrosine, asparagine, glutamine),
additions or deletions;
modifications to amino acid side chains, addition of a protecting group at the
N- or C- terminal
ends of the protein, addition of oligosaccharides such as phosphorylated
mannopyranosyl
oligosaccharides including at least one mannose-6-phosphate (e.g. mannose-6-
phospate (M6P),
phosphopentamannose, bi- and tri- antennary mannopyranosyl oligosaccharides
(bis-M6P and
tri-M6P)), galactose, mannose, N-acetylglucosamine, and fucose, fusion
products (e.g. with Fc
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
peptide), and the like. Suitable modifications will generally maintain at
least about 70%
sequence similarity with the active site and other conserved domains of native
lysosomal protein,
and preferably at least about 80%, 90%, 95% or greater sequence similarity.
Nucleic acid
modifications may include one or more base substitutions or alterations,
addition of 5' or 3'
protecting groups, and the like, preferably maintaining significant sequence
similarity, e.g. at
least about 70%, and preferably, 80%, 90%, 95% or greater.
[0093] Modified or unmodified exosomes including a selected protein, such
as a
lysosomal protein, or nucleic acid encoding the protein in accordance with the
invention, may be
formulated for therapeutic use by combination with a pharmaceutically or
physiologically
acceptable carrier. The expressions "pharmaceutically acceptable" or
"physiologically
acceptable" means acceptable for use in the pharmaceutical and veterinary
arts, i.e. not being
unacceptably toxic or otherwise unsuitable for physiological use. As one of
skill in the art will
appreciate, the selected carrier will vary with intended utility of the
exosome formulation. In one
embodiment, exosomes are formulated for administration by infusion or
injection, e.g.
subcutaneously, intraperitoneally, intramuscularly or intravenously, and thus,
are formulated as a
suspension in a medical-grade, physiologically acceptable carrier, such as an
aqueous solution in
sterile and pyrogen-free form, optionally, buffered or made isotonic. The
carrier may be distilled
water (DNase- and RNase-free), a sterile carbohydrate-containing solution
(e.g. sucrose or
dextrose) or a sterile saline solution comprising sodium chloride and
optionally buffered.
Suitable sterile saline solutions may include varying concentrations of sodium
chloride, for
example, normal saline (0.9%), half-normal saline (0.45%), quarter-normal
saline (0.22%), and
solutions comprising greater amounts of sodium chloride (e.g. 3%-7%, or
greater). Saline
solutions may optionally include additional components, e.g. carbohydrates
such as dextrose and
the like. Examples of saline solutions including additional components,
include Ringer's
solution, e.g. lactated or acetated Ringer's solution, phosphate buffered
saline (PBS), TRIS
(hydroxymethyl) aminomethane hydroxymethyl) aminomethane)-buffered saline (TB
5), Hank's
balanced salt solution (HBSS), Earle's balanced solution (EBSS), standard
saline citrate (SSC),
HEPES-buffered saline (FIBS) and Gey's balanced salt solution (GBSS).
26
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
[0094] In other embodiments, the present exosomes are formulated for
administration by
routes including, but not limited to, oral, intranasal, enteral, topical,
sublingual, intra-arterial,
intramedullary, intrauterine, intrathecal, inhalation, ocular, transdermal,
vaginal or rectal routes,
and will include appropriate carriers in each case. For oral administration,
exosomes may be
formulated in normal saline, complexed with food, in a capsule or in a liquid
formulation with an
emulsifying agent (honey, egg yolk, soy lecithin, and the like). Exosome
compositions for
topical application may be prepared including appropriate carriers. Creams,
lotions and
ointments may be prepared for topical application using an appropriate base
such as a
triglyceride base. Such creams, lotions and ointments may also contain a
surface active agent.
Aerosol formulations may also be prepared in which suitable propellant
adjuvants are used.
Other adjuvants may also be added to the composition regardless of how it is
to be administered,
for example, anti-microbial agents, anti-oxidants and other preservatives may
be added to the
composition to prevent microbial growth and/or degradation over prolonged
storage periods.
[0095] Modified and unmodified exosomes according to the present
invention are useful
in a method to treat a pathological condition involving a defective/missing
protein, or a condition
involving lack of expression of a protein, e.g. a lysosomal storage disease.
The terms "treat",
"treating" or "treatment" are used herein to refer to methods that favorably
alter a pathological
condition such as a lysosomal storage disease or other disease in which there
is a protein
deficiency, including those that moderate, reverse, reduce the severity of, or
protect against, the
progression of the target disease. Thus, for use to treat such a pathological
condition, a
therapeutically effective amount of modified or unmodified exosomes, for
example, carrying a
selected lysosomal protein, or nucleic acid encoding a selected lysosomal
protein, are
administered to a mammal. The term "therapeutically effective amount" is an
amount of
exosome required to treat the condition, while not exceeding an amount which
may cause
significant adverse effects. Exosome dosages that are therapeutically
effective will vary on
many factors including the nature of the condition to be treated as well as
the particular
individual being treated. Appropriate exosome dosages for use include dosages
sufficient to
result in an increase in activity of the target protein in the patient by at
least about 10%, and
preferably an increase in activity of the target protein of greater than 10%,
for example, at least
20%, 30%, 40%, 50% or greater. In one embodiment, the dosage may be a dosage
of exosome
27
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
that delivers from about 0.1 mg/kg to about 100 mg/kg, such as 0.1-50 mg/kg,
or 0.1-10 mg/kg,
of the desired protein, or an exosome dosage that delivers a sufficient amount
of nucleic acid to
yield about 0.1 mg/kg to about 100 mg/kg, such as 0.1-50 mg/kg, or 0.1-10
mg/kg, of the desired
protein. For example, the dosage of mRNA encoding a particular protein may be
in the range of
about 1 ug/kg to 1 mg/kg to treat a given disease such as an LSD. The term
"about" is used
herein to mean an amount that may differ somewhat from the given value, by an
amount that
would not be expected to significantly affect activity or outcome as
appreciated by one of skill in
the art, for example, a variance of from 1-10% from the given value.
[0096] As will be appreciated by one of skill in the art, exosomes
comprising a lysosomal
protein, and/or nucleic acid encoding the protein, for example, to treat a
lysosomal storage
disease, may be used in conjunction with (at different times or
simultaneously, either in
combination or separately) one or more additional therapies to facilitate
treatment, including but
not limited to; protein-specific modifications (i.e., GILT-tagged or
carbohydrate re-modelled
(mannose-6-phosphate enriched)), exercise, molecular chaperone compounds, or
substrate
reduction therapies (e.g. Miglustat). With respect to molecular chaperones,
the addition of a
molecular chaperone may be included within the exosome or administered
separately up to 2
hours prior to exosome infusion to increase stability of the lysosomal protein
and further enhance
ERT stability when exosomes are delivering protein. Examples of such
chaperones include, but
are not limited to, endogenous proteins such as Hsc70, Hsp40, Hsp70, Hsp90,
Hip, and BAG-1;
chemical compounds such as the imino sugars, N-butyl-deoxynojirimycin, 1-
deoxygalactonojirimycin; and like compounds.
[0097] The modified and unmodified exosomes of the present invention
provide many
advantages over current treatment methods such as enzyme replacement therapy.
At the outset,
exosomes exhibit greater uptake than other delivery means into tissues and
into organelles such
as lysosomes given that exosomes are naturally part of the lysosomal/endosomal
recycling
pathway, and thus, represent a physiological treatment method. In addition,
the present
exosomes provide improved stability and protection from degradation and
denaturation to
proteins/enzymes or nucleic acid that they deliver. This in turn results in
higher protein or
nucleic acid delivery rates. Exosomes can also cross the blood-brain barrier,
allowing for the
delivery of protein or nucleic acid to the central nervous system and, thereby
being useful to treat
28
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
conditions that involve the brain, e.g. LSDs such as neuronal ceroid-
Lipofuscinosis (Batten
disease), Tay-Sachs disease, metachromatic leukodystrophy and Niemann-Pick C.
Further, the
use of exosomes to deliver protein/nucleic acid results in a minimal immune
reaction to the
protein or nucleic acid being delivered because exosomes may be obtained from
cells that do not
induce any significant immunogenic response or which are not toxic (e.g.
exosomes from
immature dendritic cells). Moreover, the present exosomes may be tailored to
incorporate a
targeting sequence that results in enhanced recognition and fusion with the
target organelle, e.g.
lysosomes, to result in increased specificity with respect to targeted
protein/nucleic acid delivery.
[0098] Embodiments of the invention are described by reference to the
following specific
examples which are not to be construed as limiting.
Example 1 ¨ Treatment of acid a-glucosidase deficiency
Production of Human and Mouse GAA mRNA and Protein
[0099] Human acid a-glucosidase (GAA - NCBI Reference Sequence: NM
000152.3)
and mouse GAA (NCBI Reference Sequence: NM 008064.3) cDNA from skeletal muscle
were
sub-cloned into mammalian vector (pGEX GST-fusion vector; GE Healthcare Life
Sciences).
The vector was maintained using competent E. coil DH5alpha line (Life
Technologies). The
pGEX mammalian vector was then transfected into Chinese Hamster Ovary Cells
(CHO; ATCC
Cat. CCL-661) for mass production of active GAA enzyme. To isolate recombinant
GAA in its
processed active form, CHO cells transfected with GAA-pGEX vector were lysed
and CHO cell
lysate was cleared using ultra-performance resins for GST-tagged fusion
protein purification (GE
Healthcare Life Sciences). Over 80% of the recombinant protein was eluted
after 3 washes.
Elution #1 and Elution #2 were combined to obtain a high yield of protein. GST
tag was
removed from active GAA using PreScission Protease (GE Healthcare Life
Sciences).
[00100] To synthesize GAA mRNA, GAA cDNA was sub-cloned and amplified from
skeletal muscle (from mouse and human). Using conventional PCR, start codon
(ATG) and
Kozak sequence (GCCACC) were introduced. This cDNA was then cloned into the
pMRNA" plasmid using EcoRI and BamHI restriction enzyme sites followed by
transformation
of competent E. coil DH5alpha line (Life Technologies). The colony containing
the vector with
positive insert was amplified. The vector was isolated from these colonies
(Qiagen) and T7 RNA
29
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
polymerase-based in vitro transcription reaction was carried out. An anti-
reverse cap analog
(ARCA), modified nucleotides (5-Methylcytidine-5'-Triphosphate and
Pseudouridine-5'-
Triphosphate) and poly-A tail were incorporated into the mRNAs to enhance the
stability and to
reduce the immune response of host cells. DNase I digest and phosphatase
treatment was carried
out to remove any DNA contamination and to remove the 5' triphosphates at the
end of the RNA
to further reduce innate immune responses in mammalian cells, respectively.
The clean-up spin
columns were used to recover GAA mRNA for downstream encapsulation in
engineered
exosomes.
Production of Non-immunogenic Exosomes Expressing CD9-LamplTS Fusion Plasmid
[00101] Exosomes expressing CD9-Lamp 1 TS fusion plasmid were then
prepared for
introduction of GAA (protein) into lysosomes. Dendritic cells (DC) were
isolated from mouse
bone marrow progenitor cells and from human peripheral blood mononuclear cells
(collected
using Ficoll gradient separation of human blood). Briefly, femur and tibia
were carefully
harvested from mice and were flushed with MSS media to collect bone marrow
progenitor
cells. The bone marrow progenitor cells were cultured in GlutaMAX-DMEM media
(Life
Technologies) containing 10% FBS, 1mM sodium pyruvate, 0.5% penicillin-
streptomycin, and
mouse recombinant granulocyte/macrophage colony-stimulating factor (R&D
Systems). For
human dendritic cell isolation, blood was collected in EDTA-lavender tubes
followed by dilution
of blood with 4x PBS buffer (pH 7.2 and 2 mM EDTA). 40 mL of diluted cell
suspension was
carefully layered over 20 mL of Ficoll gradient. The gradient was centrifuged
at 400x g for 60
minutes followed by collection of the interphase layer containing the
mononuclear cells. The
mononuclear cells were cultured in IMDM media (BD Biosciences) containing 10%
FBS, 1%
glutamine, 0.5% penicillin-streptomycin, and human recombinant
granulocyte/macrophage
colony-stimulating factor (R&D Systems). Both human and mouse dendritic cells
were further
purified using EasySepTM Mouse and Human Pan-DC Enrichment Kit (Stem Cell
Technologies).
Dendritic cells were then cultured with the aforementioned media (GlutaMAX-
DMEM media for
mouse DC and IMDB media for human DC). Media was pre-spun at 170,000x g for 2
hours at
37 C for 4 days to ensure that the subsequent exosome pellet would not be
contaminated with
bovine microvesicles and/or exogenous exosomes.
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
[00102] To prepare unmodified exosomes (not tagged with a membrane fusion
product),
cells were grown to about 80% confluency before exosome collection as
described below. To
prepare modified exosomes (tagged with a membrane fusion product), on the
third day, DC were
transfected with mammalian expression CD9-Lamp 1 TS fusion plasmid 0.1-1 ug
(depending on
cell density) using Lipofectamine 3000 reagent (Life Technologies). The CD9-
Lamp 1 TS fusion
plasmid was made using Gateway technology and vectors (Life Technologies)
with amplified
skeletal muscle (mouse and human) cDNA that corresponds to LAMP1 protein
lysosomal
targeting sequence (for human: MAAPGSARRPLLLLLLLLLLGLMHCASA (SEQ ID NO: 29);
for mouse: MAAPGARRPL LLLLLAGLAHGASALFEVKN (SEQ ID NO: 30)) + first 10
amino acids of mature LAMP1 protein (for human: AMFMVKNGNG (SEQ ID NO: 31);
for
mouse: LFEVKNNGTT (SEQ ID NO: 32)). The CD9 cDNA exosome marker (for mouse:
NM 007657; for human: NM 001769.3) was amplified from mouse and human
dendritic cell
cDNA). The lysosomal targeting sequence and the exosome marker were linked via
PCR, and
then were incorporated into a mammalian expression vector. On the fifth day,
the dendritic cells
were washed and combined with fresh growth media (GlutaMAX-DMEM media for
mouse DC
and IMDB media for human DC, both media containing pre-spun exosome depleted
FBS).
[00103] The dendritic cells were then grown to about 80% confluency in
alpha minimum
essential medium supplemented with ribonucleosides, deoxyribonucleosides, 4 mM
L-glutamine,
1 mM sodium pyruvate, 5 ng/mL murine GM-CSF, and 20% fetal bovine serum. For
conditioned
media collection, cells were washed twice with sterile PBS (pH 7.4, Life
Technologies) and the
aforementioned media (with exosome-depleted fetal bovine serum) was added.
Conditioned
media from human and mouse immature dendritic cell culture was collected after
48 hours. The
media (10 mL) was spun at 2,000x g for 15 min at 4 C to remove any cellular
debris. This is
followed by an optional 2000x g spin for 60 min at 4 C to further remove any
contaminating
non-adherent cells. The supernatant was then spun at 14,000x g for 60 min at 4
C. The resulting
supernatant was spun at 50,000x g for 60 min at 4 C. The supernatant was then
filtered through a
40 pm filter, followed by filtration through a 0.22 pm syringe filter (twice).
The supernatant was
then carefully transferred into ultracentrifuge tubes and diluted with an
equal amount of sterile
PBS (pH 7.4, Life Technologies). This mixture was then subjected to
ultracentrifugation at
100,000x-170,000x g for 2 hours at 4 C using a fixed-angle rotor. The
resulting pellet was re-
31
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
suspended in PBS and re-centrifuged at 100,000x-170,000x g for 2 hours at 4 C.
The pellet was
resuspended carefully with 25 mL of sterile PBS (pH 7.4, Life Technologies)
and then added
gently on top of 4 mL of 30%/70% PercollTM gradient cushion (made with 0.22
[tm filter
sterilized water) in an ultracentrifuge tube. This mixture was spun at
100,000x-170,000x g for 90
minutes at 4 C. With a syringe, the exosomal pellet-containing fraction at the
gradient interface
was isolated carefully, diluted in 50 mL of sterile PBS (pH 7.4, Life
Technologies), followed by
a final spin for 90 minutes at 100,000x-170,000x g at 4 C to obtain purified
exosomes. The
resulting exosomal pellet was resuspended in sterile PBS or sterile 0.9%
saline for downstream
use. Exosomal fraction purity was confirmed by sizing using a Beckman DelsaMax
dynamic
light scattering analyzer showing minimal contamination outside of the 40-120
nm size range,
and by immuno-gold labelling/Western blotting using the exosome membrane
markers, CD9,
CD63, TSG101 and ALIX.
[00104] Purified exosomes were suspended in a 100-140 [EL of pre-chilled
electroporation
buffer (1.5 mM potassium phosphate pH 7.2, 25 mM KC1, and 21% (vol/vol)
OptiPrep for GAA
mRNA electroporation of pre-engineered exosomes OR 1.5 mM potassium phosphate
pH 7.2, 25
mM KC1, 250 mM trehalose, 1 mM inositol, and 21% (vol/vol) OptiPrep for GAA
protein
electroporation of pre-engineered exosomes). Exosomes were then counted and
sized by
NanoSight nanoparticle tracking analysis (NanoSight, Ltd.). Yield was about 1
x 109 particles
around ¨100 nm in size. Using the PierceTM BCA protein quantification assay
(Thermo
Scientific), the yield of exosomes was estimated and found to be between 10 ¨
15 ug of
exosomes.
Introduction of GAA mRNA or protein into exosomes
[00105] Electroporation mixture was prepared by carefully mixing CD9-LAMP1-
tagged
(modified) exosomes and GAA protein in 1:1 ratio in electroporation buffer.
Similarly,
electroporation mixture was prepared by mixing non-tagged (unmodified)
exosomes and GAA
mRNA in 1:1 ratio in electroporation buffer. Electroporation was carried out
in 0.4 mm
electroporation cuvettes at 400 mV and 125 pF capacitance (pulse time 14
milliseconds (ms) for
mRNA and 24 ms for protein) using Gene Pulse XCell electroporation system
(BioRad). After
electroporation, exosomes were resuspended in 20 mL of 0.9% saline solution
followed by
ultracentrifugation for 2 hours at 170,000x g at 4 C. For in vitro and in vivo
exosome
32
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
administration, GAA (mRNA or protein)-loaded exosomes (CD9-LAMP1 tagged
exosomes
carrying GAA protein or non-tagged exosomes carrying GAA mRNA) were re-
suspended in 5%
(wt/vol) glucose in 0.9% saline solution. Alternatively, exosomes were loaded
with GAA mRNA
or protein using cationic lipid-based transfection reagents (Lipofectamine
MessengerMAXTm
Transfection Reagent, Life Technologies). After transfection, exosomes were
spun for 2 hours at
170,000x g at 4 C followed by re-suspension in 5% (wt/vol) glucose in 0.9%
sterile saline
solution.
GAA mRNA or GAA protein-loaded exosomes rescue GAA deficiency in primary
fibroblasts
and myotubes
[00106] Four GAA heterozygous breeding mouse pairs (GAA-/ ) were obtained
from
Jackson Laboratories (Maine, USA) to generate homozygous GAA knock-outs (GAA-/-
) and
wild-type littermates (GAA'). Mice were genotyped at 1 month of age using a
standard
genotyping kit (REDExtract-N-Amp Tissue PCR Kit; Sigma Aldrich). Homozygous
GAA
knock-out mutants exhibited significant deficits in front-limb muscle strength
as early as
1.5 months of age. During breeding, all animals were housed three to five per
cage in a 12-h
light/dark cycle and were fed ad libitum (Harlan-Teklad 8640 22/5 rodent diet)
after weaning.
The study was approved by the McMaster University Animal Research and Ethics
Board under
the global Animal Utilization Protocol # 12-03-09, and the experimental
protocol strictly
followed guidelines put forth by Canadian Council of Animal Care.
[00107] Primary fibroblasts and myotubes from GAA+/+ (wild-type mice with
normal
GAA activity) and GAA-/- (GAA knock-out mice, model of Pompe Disease) were
harvested
using standard isolation techniques. Myoblasts were differentiated into
myotubes for exosome-
treatment experiments. Fibroblasts and myotubes were treated with recombinant
murine GAA
protein (40 mg/kg) or 10 ug (total exosomal protein) of empty murine exosomes
(not loaded with
GAA mRNA or protein), exosomal GAA protein (modified exosomes loaded with
murine GAA
protein, 40 mg/kg of recombinant GAA in 10 ug of total exosomal protein), or
exosomal GAA
mRNA (mRNA dose equivalent to delivery of 40 mg/kg GAA, ¨100-150 ng mRNA, in
10 ug of
total exosomal proteins in which exosomes were unmodified) for 48 hours in pre-
spun growth
media devoid of bovine microvesicles and exosomes.
33
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
[00108] A standard fluorometric enzyme assay, originally described by
Reuser et al.
(Biochem Biophys Res Commui'L 1978 Jun 29;82(4):1176-82), was used to
determine acid a-
glucosidase (GAA; EC 3.2.1.20) activity of treated and untreated fibroblasts
and myotubes. In
brief, cellular lysates were prepared by homogenizing cells (fibroblasts and
myotubes) in 200 [EL
mannitol buffer (70 mM sucrose, 220 mM mannitol, 10 mM HEPES, 1 mM EGTA,
protease
inhibitor mixture (Complete Tablets, Roche), pH 7.4). Following BCA assay
(Pierce) for
colorimetric determination of protein concentration, 10 [IL of each sample (in
triplicate) was
mixed with 20 [IL of the artificial acid a-glucosidase substrate, 4-methyl-
umbelliferyl a-d-gluco-
pyranoside, in 0.2 M sodium acetate [NaAc] buffer, pH 3.9, heated to 65 C in
a 96 well black
plate. Standards were prepared from a 5 mM 4-methylumbelliferone/50% ethanol
stock by serial
dilution in 0.2 M NaAc buffer (pH 3.9), loaded in 10 [IL triplicates, and
mixed with 20 [EL 0.2 M
NaAc buffer (pH 3.9). The samples were then incubated in the dark for 1 h at
37.5 C and the
reaction was terminated by adding 200 [EL of 0.5 M sodium carbonate (pH 10.7).
The release of
the product, 4-methylumbelliferone, from the substrate is proportional to acid
a-glucosidase
activity (nmol/mg protein/hr), and the resulting fluorescence was read at 360
nm
excitation/460 nm emission with a monochromator-based microplate detection
system (Tecan).
[00109] Primary fibroblasts (Figure 1) and primary myotubes (Figure 2)
show partial to
complete rescue of GAA activity when treated with exosomal GAA protein
(modified exosomes)
or mRNA (unmodified exosomes), respectively, which is 2 to 8-fold higher than
GAA rescue
achieved by treatment with naked GAA (representative of conventional enzyme
replacement
therapy (ERT)).
GAA mRNA or GAA protein-loaded exosomes prevent GAA deficiency in primary
dermal
fibroblasts isolated from Pompe patients
[00110] Primary dermal fibroblasts (grown from skin biopsies used to
diagnose Pompe
disease) from three Pompe patients and three healthy age/gender-matched
controls were treated
with naked human GAA recombinant protein or empty exosomes (not loaded with
GAA mRNA
or protein), exosomal GAA protein (modified exosomes loaded with human GAA
protein), or
exosomal GAA mRNA (unmodified exosomes loaded with human GAA mRNA) for 48
hours in
pre-spun growth media devoid of bovine microvesicles and exosomes.
34
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
[00111] GAA activity of treated and untreated fibroblasts was determined
as described
above. Primary fibroblasts from Pompe patients show partial to complete rescue
of GAA activity
when treated with exosomal GAA protein or mRNA, respectively (Figure 3). The
rescue in GAA
activity using GAA-exosome therapy was about 2.5 to 6-fold higher than that
achieved by
treatment with naked GAA (conventional ERT).
[00112] This was further confirmed by a significant decrease in fibroblast
total glycogen
content (Figure 4). To determine glycogen content, cell lysate was treated
with 30% KOH,
boiled for 15 min, and stored on ice for 30 min. Glycogen was precipitated
using 66% ethanol
followed by centrifugation (5000x g for 10 min at 4 C). The resulting pellet
was resuspended in
300 [EL of 0.1 M acetic acid. Glycogen concentration was determined by
examining release of
glucose after digestion of 100 pL of glycogen-acetic acid mix with 0.1 U
Aspergillus niger
amyloglucosidase (Sigma) at 55 C for 30 min. The glucose concentration after
hydrolysis is
proportional to glucose reaction with ATP to form glucose-6-phosphate which
converts NADP
into NADPH, spectrophotometrically determined at 340 nm.
[00113] Fibroblasts treated with naked GAA protein and exosomal GAA
protein were
labeled with DAPI (nuclear marker), phalloidin (cellular cytoskeletal marker),
and
carboxyfluorescein succinimidyl diacetate ester (non-specific labeling of
protein GAA) and
subsequently imaged. GAA rescue using exosomal delivery of GAA mRNA and
protein was
shown to be due to more efficient uptake thereof as compared to the uptake of
naked GAA
protein.
Example 2 ¨ Treatment of GAA deficiency in vivo with Protein-loaded Exosomes
Methods
[00114] Breeding of GAA mice. Four GAA heterozygous breeding pairs (HET;
GAA-1+;
6ne /6 +) were obtained from Jackson Laboratories (Maine, USA) to generate
homozygous GAA
knock-outs (MUT; GAA; 6neo/6neo). Mice were genotyped at 1 month of age using
a standard
genotyping kit as per vendor's instructions (REDExtract-N-Amp Tissue PCR Kit;
Sigma
Aldrich). During breeding and throughout the experimental period, animals were
housed in
standard cages with 12-h light/dark cycles and free access to water/rodent
chow (Harlan Teklad
8640 22/5) at McMaster University's Central Animal Facility. The study was
approved by
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
McMaster University's Animal Research and Ethics Board, and the experimental
procedures
strictly followed guidelines published by the Canadian Council of Animal Care.
[00115] Production of Non-immunugenic GAA protein-loaded Exosomes for
Treatment
of GAA mice. Mouse GAA protein was produced as described previously in Example
1. To
produce exosomes from mouse dendritic cells (DC), DC were harvested also as
described in
Example 1. GAA protein was loaded into the exosomes (unmodified) using the
protein loading
method as in Example 1. GAA protein-loaded exosomes were resuspended in 0.9%
saline
solution for treatment. GAA mice were divided into three treatment groups (n =
7 per group):
saline, naked GAA protein, and GAA protein-loaded exosome (EXO GAA). Mice were
treated
once a week for 7 weeks. Dosage of naked GAA protein and GAA protein-loaded
exosome was
40 mg of GAA protein/kg, given intravenously in 0.9% sterile saline. The
amount of GAA
loaded in exosomes was measured against a standard curve of GAA activity using
purified
recombinant GAA. The mice were tested for paw-grip endurance test, grip
strength, and motor
function at the beginning and end of the study (24 hours prior to harvesting
tissue). Skeletal
muscle (quadriceps, tibialis anterior, EDL, soleus, and diaphragm), heart, and
brain were
harvested from mice in all treatment groups.
[00116] GAA enzyme activity. A standard fluorometric enzyme assay as
described above
was used to determine acid a-glucosidase activity in cellular lysates prepared
from fibroblasts
and myotubes.
[00117] Extraction of total glycogen. Total glycogen content in tissues
(tibialis anterior,
heart, and brain) was determined as previously described (Devries et al.,
2005, Adamo et al.,
1998). Briefly, snap-frozen tissue samples were freeze dried for 48 h,
powdered, and dissected
free of any blood and connective tissue and weighed. Ice-cooled 1 M HC1 (100
[IL) was added to
2-5 mg of tissue powder in 5 mL glass tubes, followed by vortexing and
pressing the tissue
powder against the tube walls with a glass rod. Prior to hydrolysis, tubes
were weighed and
sealed with glass stoppers, and thereafter placed in a water bath (100 C) for
2 h, after which the
tubes were reweighed and any change in weight was rectified with the addition
of deionized
water. Lastly, hydrolyzed samples were neutralized with 2 M Trizma base,
vortexed, centrifuged
at 3000 rpm for 5 min, transferred to 1.5 mL polyethylene tubes, and stored at
¨ 80 C.
36
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
[00118] Fluorometric analysis of total glycogen. Total glycogen content in
the tissue
samples (tibialis anterior, heart, and brain) was determined using a
fluorometric assay modified
for a monochromator-based microplate detection system (Tecan Safire 2, Tecan
Group Ltd,
Mannedorf, Switzerland). In brief, 10 pi, of glucose standards (2.5 04-600 uM)
and neutralized
samples was loaded in triplicate onto a 96 well black plate. Next, 190 uL of
assay buffer (50 mM
Tris, 1 mM MgC12, 0.5 mM DTT, 300 uM ATP, 50 tM NADP, 0.02 U/mL glucose-6-P
dehydrogenase in DW, pH 8.1) was added to each well, which was followed by a 5-
min
incubation step in the dark and a baseline reading at 340 nm excitation/460 nm
emission.
Thereafter, the final reaction was commenced by adding 5 pL hexokinase to each
sample and the
plate was incubated for 30 min at RT as described previously. Net fluorescence
values were
obtained by subtracting the baseline read (and blanks) from the final read and
unadjusted
glycogen concentrations were calculated using the regression formula from the
standard curve.
Total glycogen concentrations were adjusted by an extraction dilution factor
(dry tissue weight
and volume acid) and reported as mmol/mg dry tissue weight.
Results
[00119] GAA protein-loaded exosome therapy enhances strength and motor
control,
increases muscle mass, and reduces pathogenic cardiac hypertrophy vs.
conventional naked
GAA ERT in GAA KO mice. 7-week treatment of GAA KO mice with GAA protein-
loaded
exosomes resulted in reduced body weight (Figure 5), and increased grip
endurance time (A),
grip strength (B), and rotarod fall time (C)(Figure 6) in GAA KO mice when
compared to GAA
KO mice treated with naked GAA protein (conventional enzyme replacement
therapy (ERT)).
Additionally, GAA KO mice treated with GAA protein-loaded exosomes showed
improvements
in predominantly slow- or fast-twitch fiber skeletal muscle mass (soleus and
EDL, respectively)
along with an increase in mass of mixed fiber-type muscles (quadriceps,
gastrocnemius and
tibialis anterior (TA)) in comparison to GAA KO mice treated with conventional
naked GAA
protein ERT (Figures 7 and 8). A significant reduction in heart mass and brain
mass (to normal
levels) of GAA KO mice treated with GAA protein-loaded exosomes was also
observed when
compared to GAA KO mice treated with conventional naked GAA protein ERT
(Figure 9).
37
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
[00120] GAA protein-loaded exosome therapy restores GAA activity in all
tissue tested
vs. conventional naked GAA ERT in GAA KO mice. 7-week treatment in GAA KO mice
with
GAA protein-loaded exosomes restored GAA activity in slow-, fast-, and mixed
fiber type
skeletal muscle, diaphragm, and heart to a greater degree (Figure 10B) than
conventional naked
GAA ERT (Figure 10A). Importantly, GAA activity was restored in brain
confirming that GAA
protein-loaded exosomes cross the blood-brain barrier and restores GAA enzyme
activity in
neuronal tissue (brain, Figure 10B).
[00121] GAA protein-loaded exosome therapy clears pathological
accumulation of
glycogen. Additionally, 7-week treatment of GAA protein-loaded exosomes in GAA
KO mice
reduced pathological build-up of glycogen in skeletal muscle and heart to a
greater degree than
that conventional naked GAA ERT in GAA KO mice (Figure 11). Similarly,
clearance of total
glycogen in the brain following treatment with GAA protein-loaded exosomes,
but not following
treatment with GAA ERT, was observed, depicting that GAA-packaged exosomes do
cross the
blood-brain barrier and have functional consequences (Figure 11). Inability to
cross the blood-
brain barrier is a major limitation of conventional intravenous administration
of ERT.
Example 3 ¨ Treatment of GAA deficiency in vivo with mRNA-loaded Exosomes
GAA mRNA-loaded exosomes therapy restores GAA activity and GAA mRNA in all
tissue
tested to wild-type mice levels
[00122] A 7-week intravenous treatment of GAA knockout (KO) mice (as
above) with
GAA mRNA-loaded unmodified exosomes (no including a lysosomal targeting fusion
product).
Exosomes were loaded with an mRNA dose equivalent to delivery of 40 mg/kg GAA,
¨100-150
ng mRNA, in 10 ug of total exosomal proteins, isolated and prepared as
described in Example 1,
was conducted.
[00123] Treatment of GAA KO mice with GAA mRNA-loaded exosomes restored
GAA
activity in fast- (EDL) and slow- (soleus) fiber-type skeletal muscle,
diaphragm, and heart, as
compared to that in GAA KO mice treated with empty exosomes (Figures 12A and
12B). Empty
exosomes and GAA mRNA-loaded exosomes were given to wild-type mice (WT) as
controls.
GAA activity was restored in GAA KO brain depicting that GAA mRNA-loaded
exosomes
38
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
could not only cross the blood-brain barrier, but GAA mRNA delivered via
exosomes did induce
functional changes in neuronal tissue (Figure 13). It should be noted that the
mice were harvested
four days after the last intravenous bolus of GAA mRNA-loaded exosomes and the
GAA activity
was maintained in all tissues tested. GAA mRNA expression was maintained in
skeletal muscle
(quadriceps femoris) and brain in GAA KO mice treated with GAA mRNA-loaded
exosomes
four days after the last injection (Figure 14). This result shows that there
are physiological
alterations in mRNA stabilizing proteins and/or the miRNA network in GAA KO
mice only
(where GAA protein activity is negligible) that prevents degradation of the
GAA mRNA
delivered, compared to WT mice where mRNA levels do not increase significantly
(since they
have optimal GAA activity). It was concluded that the effect of the mRNA
(packaged in
exosomes) therapy persists for much longer periods of time as compared to
conventional enzyme
replacement therapy. Additionally, GAA mRNA-loaded exosomes resulted in
substrate reduction
(total glycogen content) in skeletal muscle (quadriceps and tibialis anterior)
and heart in GAA
KO mice to normal physiological wild-type levels (Figure 15).
[00124] Interestingly, tissues from WT mice treated with GAA mRNA-loaded
exosomes
did not show an up-regulation/overexpression of GAA activity indicating that
there are inherent
pathways that protect against an abnormal increase in GAA activity above
physiological levels.
From the perspective of treatment safety, this indicates the potential to
avoid non-specific effects
of very high levels of GAA activity (such as levels achieved by strategies
like AAV-mediated
GAA induction) using exosomal protein or nucleic acid delivery.
GAA mRNA-loaded exosomes therapy normalized neuromuscular function in GAA KO
mice
to wildtype mice levels
[00125] The 7-week treatment of GAA mRNA-loaded exosomes in GAA KO mice
normalized grip endurance in the wire hang test (marker of neuromuscular
function) to wild-type
mice levels, while grip endurance in GAA KO mice treated with empty exosomes
was not
restored (control group; Figure 16). Wild-type mice treated with GAA mRNA-
loaded exosomes
maintained normal grip endurance (Figure 16).
39
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
Example 4 ¨ Treatment of Niemann-Pick disease, type Cl deficiency
[00126]
To determine the utility of the present exosomes to treat lysosomal storage
disease
that primarily affects the CNS (i.e., Niemann-Pick type C, neural ceroid
lipofuscinosis, Krabbe
disease, metachromaticleukodystrophy, etc.), the following studies were
conducted.
[00127]
An in vitro study was conducted in which primary dermal fibroblasts from three
patients with genetically confirmed Niemann-Pick disease, type Cl (NPC1)
mutations were
treated for 48 h as follows: media control (no therapy), "empty" exosomes,
exosomes + human
rNPC1 mRNA (NPC1 mRNA was cloned from the human genome (NCBI Accession #:
NM 000271.4), and exosomes + human rNPC1 protein (human rNPC1 mRNA was cloned
from
human genome and expressed in CHO cells). Exosome treatment was with 10 ug of
exosomes
(unmodified with a lysosomal tag) in 0.9% sterile saline loaded with either
NPC1 mRNA (100
ng) or rNPC1 protein (100 ug). Isolation of exosomes and loading with NPC1
mRNA and
protein was conducted using methods similar to those described for GAA in
Example 1.
[00128]
Outcome measured was filipin staining (showing the accumulation of cholesterol
and glycosphingolipids). A complete restoration of normal filipin staining in
patient cells treated
with NPC1 mRNA-loaded exosomes, and near complete restoration of filipin
staining in patient
cells treated with NPC1 protein-loaded exosomes resulted indicating NPC1
restoration (Figure
17).
Example 5 ¨ Treatment of Niemann-Pick disease, type Cl in vivo
[00129]
Breeding of NPC1 mice. Four NPC1 heterozygous breeding pairs (HET;
NPC1; 6./6
+) were obtained from Jackson Laboratories (Maine, USA) to generate
homozygous NPC1 knock-outs (MUT; NPC1-
/-;
)
Mice were genotyped at 1 month of
age using a standard genotyping kit as per vendor's instructions (REDExtract-N-
Amp Tissue
PCR Kit; Sigma Aldrich). During breeding and throughout the experimental
period, animals
were housed in standard cages with 12-h light/dark cycles and free access to
water/rodent chow
(Harlan Teklad 8640 22/5) at McMaster University's Central Animal Facility.
The study was
approved by McMaster University's Animal Research and Ethics Board, and the
experimental
procedures strictly followed guidelines published by the Canadian Council of
Animal Care.
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
[00130] The NPC1-/- homozygous mice are known to have reduced levels of
myelin in the
cerebellum, and the astrocytes and microglia in these mice proliferate and
occupy areas of
neuronal loss or degeneration. Weight loss is accompanied by a progressive
motor coordination
deficit, or ataxia, at least as early as postnatal day 45 (P45). The lifespan
of homozygous animals
is reduced to a mean of 76 days.
[00131] At 30 days of age, NPC1-/- homozygous mice and littermate wildtype
mice
(NPC1+1+) were treated (intravenously) with either empty exosomes (10 ug of
total exosomal
protein) or exosomes loaded with mouse npcl mRNA (3.33 ng of mRNA per g of
mouse in 10
ug of total exosomal protein) in 0.9% saline solution per week for 5 weeks.
[00132] The mice were tested for paw-grip endurance, grip strength, and
motor function at
the beginning and end of the study (24 hours prior to harvesting tissue). The
NPC1-/-
homozygous mouse treated with NPC1 mRNA-loaded exosomes exhibited many fold
higher
values for paw-grip endurance, grip strength and motor function as compared to
an NPC1-/-
homozygous mice treated with empty exosomes (Figure 18). The untreated NPC4-
mouse was so
ataxic that the animal facility staff had to provide gel and food on the floor
of the cage due to
complete inability to reach water and food, whilst the treated mouse was
indistinguishable from
wild type mice.
Example 6 ¨ Treatment of 13-g1ucocerebrosidase deficiency
[00133] Gaucher disease is a genetic disorder in which glucocerebroside (a
sphingolipid)
accumulates in cells and certain organs. It is characterized by bruising,
fatigue, anemia, low
blood platelet count and enlargement of the liver and spleen, and is caused by
hereditary
deficiency of the enzyme, 13-glucocerebrosidase or glucosylceramidase (GBA).
The following
experiment is conducted to determine if this enzyme could be delivered to
cells using exosomes.
[00134] Primary dermal fibroblasts grown from skin biopsies obtained from
three Gaucher
patients and three healthy age/gender-matched controls are treated with Gba
mRNA (cloned
from the human genome (NCBI Accession #: NM 000157.3) empty exosomes (not
loaded with
Gba mRNA), or exosomal Gba mRNA (bioengineered exosomes loaded with human Gba
mRNA cloned from human genome) for 48 hours in pre-spun growth media devoid of
bovine
41
CA 02962081 2017-03-22
WO 2016/044947 PCT/CA2015/050959
microvesicles and exosomes. Treatment is with exosomes (10 ug of total
exosomal protein in
0.9% sterile saline solution) loaded with Gba mRNA (100 ng) (exosome isolation
and loading is
conducted as described in Example 1 for exosome isolation and GAA exosome
loading).
Exosomes may be modified with a fusion lysosomal targeting product.
[00135] Gba activity of treated and untreated dermal fibroblasts may be
determined as
previously described (Pasmanik-Chor, M, et al, Biochem 1, 317:81-88, 1996).
Primary
fibroblasts from Gaucher patients are expected to show partial to complete
rescue of GBA
activity when treated with exosomal Gba mRNA.
[00136] Remarkably, the foregoing demonstrates for the first time that
mRNA and protein
can be delivered using the present bioengineered exosomes to completely
correct a genetic defect
in vivo in which there is a protein/enzyme deficiency. Thus, the present
method results in
upregulation of a target protein in vivo by at least about 10%, and preferably
by at least about
50% or more, including upregulation of the protein to normal, wild-type
levels, and further to
effect recovery of normal function, activity and anatomy in a mammal.
[00137] Given the demonstration that the present exosome-mediated therapy
can
effectively treat three different lysosomal storage diseases that collectively
represent the main
tissues that are variably affected in all lysosomal storage diseases (e.g.
brain, muscle, heart, liver,
spleen, bone marrow and bone) and that the cellular pathophysiology of each
lysosomal storage
disease is the same, as one skilled in the art will appreciate, the present
exosome-mediated
therapy is applicable to all genetic disorders that affect the lysosome.
[00138] Relevant portions of references referred to herein are
incorporated by reference.
42