Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
METHODS AND SYSTEMS FOR WATER TREATMENT BY FLOCCULATION
FIELD OF INVENTION
The present invention relates generally to methods and systems for the
treatment of
contaminated water. More specifically, the present invention relates to
methods and systems
for water treatment which provide for removal of contaminants by flocculation.
BACKGROUND
Water treatment systems and methods are very important aspects of many
industrial operations.
Process waters are frequently contaminated with one or more components which
must be
removed prior to subsequent use of the water, or prior to disposal of the
water. Hydrocarbon
recovery operations, for example, utilize water for a number of purposes, and
efficient and
effective water treatment systems and methods are highly sought-after.
Hydrocarbon recovery
operations involving Steam-Assisted Gravity Drainage (SAGD), for example,
utilize steam to
mobilize hydrocarbons in subterranean reservoirs. Contaminants in water used
for steam
generation can cause significant issues, including scaling of boilers and
other such effects.
Methods for treating water prior to steam injection are of great interest in
the field.
Furthermore, many oilfield operations produce water from subterranean
reservoirs to the
surface. These waters, known as produced waters, often contain components
which must be
removed before the water can be recycled to generate steam, or before the
water can be
otherwise used or disposed of. Recycling of produced water typically involves
removal of
suspended solids, dissolved solids, oil, and/or scale-forming agents which
would otherwise
affect steam generation and handling equipment. Conventional produced water
treatment
equipment and processes are costly and high-maintenance. Further, produced
waters are
typically received at high temperatures and pressures, both of which typically
must be reduced
using heat exchangers before treatment using conventional processes and
systems. This also
means that post-treatment, substantial re-heating of the water is required to
produce steam for
injection downhole. Conventionally, produced water treatment involved use of
Lime Softeners
(such as Warm Lime Softener (WLS)) and related equipment.
1
CA 3027250 2018-12-12
More generally, common water treatment techniques include electrocoagulation
and
electrofloatation treatments, which are typically performed at ambient
temperatures and which
typically involve use of sacrificial iron electrodes for introducing iron
salts into the
contaminated water and triggering flocculation to allow for contaminant
separation. However,
use of such sacrificial electrodes requires frequent maintenance and upkeep in
conventional
electrocoagulation and electrofloatation water treatment systems.
Alternative, additional, and/or improved methods and/or systems for the
treatment of
contaminated waters is desirable.
SUMMARY OF INVENTION
Described herein are methods and systems for the treatment of contaminated
water. In certain
embodiments, methods and systems described herein may provide for removal of
contaminants
from, for example, a produced water from an oilfield operation. Such
contaminants may
include, for example, calcium, magnesium, silica, hardness, and/or organics,
among others.
Systems and methods provided herein may be operated at elevated temperatures
and pressures,
such as those typically encountered in a SAGD operation, which may be
particularly beneficial
when the contaminated water input is at an elevated temperature and/or when
the treated water
output is intended for use in an operation requiring the water to be heated.
Heat energy may
thus be conserved, thereby saving on heating costs. As well, methods and
systems as described
herein may allow for certain conventional water treatment apparatus to be
omitted. Further, in
certain embodiments, methods and systems described herein may allow for water
treatment
without the use of sacrificial iron electrodes to treat the contaminated
water, thereby reducing
or removing the need for electrode maintenance and/or replacement.
The present inventors have discovered that certain contaminated waters, such
as those produced
during a SAGD operation, may contain one or more flocculation inhibitors which
interfere with
contaminant removal by flocculation with flocculating ions such as iron; and
have further
discovered that such flocculation inhibitor(s) may be destroyed prior to
flocculation by an
electro- and/or chemical- reduction and/or oxidation step, thereby allowing
for contaminant
removal via flocculation. Flocculation may thus be achieved through addition
of a flocculating
2
CA 3027250 2018-12-12
ion-enriched aqueous solution into the contaminated water, which may reduce
complexity
and/or water treatment equipment demands.
Select embodiments of the present disclosure relate to, a method for treating
a contaminated
water, said method comprising:
subjecting the contaminated water to reducing or oxidizing conditions;
introducing a flocculating ion-enriched aqueous solution into the contaminated
water; and
removing at least some of at least one contaminant from the contaminated water
by ion flocculation, whereby the contaminant is captured with flocculating ion
flocks which are then separated from the contaminated water;
thereby producing a treated water.
In select embodiments of the present disclosure, the flocculating ion
comprises iron, aluminum,
or a combination thereof.
In select embodiments of the present disclosure, the step of subjecting the
contaminated water
to reducing or oxidizing conditions comprises a step of electroreduction.
In select embodiments of the present disclosure, the step of electroreduction
uses one or more
non-sacrificial electrodes.
In select embodiments of the present disclosure, the step of subjecting the
contaminated water
to reducing or oxidizing conditions comprises a step of electrooxidation.
In select embodiments of the present disclosure, the step of electrooxidation
uses one or more
non-sacrificial electrodes.
In select embodiments of the present disclosure, the step of subjecting the
contaminated water
to reducing conditions comprises a step of chemical reduction.
3
CA 3027250 2018-12-12
In select embodiments of the present disclosure, the step of chemical
reduction uses a chemical
reduction agent which is NaHS03, Na2S03, Na2S204, Na2S203, nascent hydrogen,
or a
combination thereof.
In select embodiments of the present disclosure, the step of subjecting the
contaminated water
to oxidizing conditions comprises a step of chemical oxidation.
In select embodiments of the present disclosure, the step of chemical
oxidation uses a chemical
oxidation agent which is H202, Na0C1, or a combination thereof.
In select embodiments of the present disclosure, the flocculating ion-enriched
aqueous solution
comprises an aqueous solution of Fe2+ ions.
In select embodiments of the present disclosure, the flocculating ion-enriched
aqueous solution
comprises a solution generated: by electro-flocculation using a sacrificial
iron electrode, a
sacrificial aluminum electrode, or a combination thereof; by dissolving an
iron salt, an
aluminum salt, or a combination thereof in water; from a solution of iron
vitriol; or a
combination thereof.
In select embodiments of the present disclosure, the flocculating ion-enriched
aqueous solution
is introduced into the contaminated water as a slipstream.
In select embodiments of the present disclosure, the introducing of the
flocculating ion-
enriched aqueous solution comprises: a step of pH adjustment to render silica
or other ionic
contaminants in the contaminated water reactive; a step of pH adjustment to
promote formation
of the flocculating ion flocks; or a combination thereof.
4
CA 3027250 2018-12-12
In select embodiments of the present disclosure, the step of pH adjustment to
render silica or
other ionic contaminants in the contaminated water reactive comprises
adjusting the pH to
between about 2 and about 4.
In select embodiments of the present disclosure, the pH is adjusted using HCl,
H2SO4, another
acid, or a combination thereof.
In select embodiments of the present disclosure, the step of pH adjustment to
promote
formation of the flocculating ion flocs comprises adjusting the pH to between
about 7 and about
11.
In select embodiments of the present disclosure, the pH is adjusted using
NaOH, steam
blowdown, another base, or a combination thereof.
In select embodiments of the present disclosure, the method further comprises
adding an H2S
scavenger, a chelant, a polymer, a sulphite, or a combination thereof to the
contaminated water
during treatment.
In select embodiments of the present disclosure, the flocculating ion flocs
are separated using
filtration or flotation.
In select embodiments of the present disclosure, the contaminated water is
maintained at high
temperature throughout treatment, thereby generating the treated water at high
temperature.
In select embodiments of the present disclosure, the contaminated water is
maintained at or
above about 80 C during treatment.
In select embodiments of the present disclosure, the contaminated water is
maintained at or
above about 100 C during treatment.
5
CA 3027250 2018-12-12
In select embodiments of the present disclosure, the contaminated water is a
produced water.
In select embodiments of the present disclosure, the produced water is a
produced water from
a SAGD operation.
In select embodiments of the present disclosure, the one or more contaminants
removed from
the contaminated water comprise calcium, magnesium, silica, an organic
contaminant, or a
combination thereof.
In select embodiments of the present disclosure, the method further comprising
a step of
subjecting the treated water to an electrochemical oxidation treatment or a
chemical oxidation
treatment to render organics in the treated water insoluble, and separating
the organics from
the treated water.
In select embodiments, the present disclosure relates to a method for
producing hydrocarbons
from a subterranean reservoir, said method comprising:
injecting steam, water, or a combination thereof into the subterranean
reservoir;
producing a produced water and a hydrocarbon to the surface;
treating at least a portion of the produced water using a method as defined
herein
thereby generating a treated water stream; and
using the treated water stream to provide steam, water, or a combination
thereof
for re-injection into the subterranean reservoir to produce more hydrocarbons
to the
surface.
In select embodiments, the present disclosure relates to a method for
producing hydrocarbons
from a subterranean reservoir, said method comprising:
injecting steam into the subterranean reservoir via an injection well;
6
CA 3027250 2018-12-12
producing a produced water stream and a hydrocarbon stream to the surface via
a production well, or producing a mixed produced water and hydrocarbon
emulsion
stream from the subterranean reservoir via a production well and separating
the
mixed produced water and hydrocarbon emulsion stream into a produced water
stream and a hydrocarbon stream;
treating at least a portion of the produced water stream using a method as
defined herein, thereby generating a treated water stream; and
using the treated water stream to provide steam for re-injection into the
subterranean reservoir via the same or a different injection well to produce
more
hydrocarbons to the surface.
In Select embodiments, the present disclosure relates to a system for treating
contaminated
water, the system comprising:
an input for a contaminated water;
a reducing or oxidizing unit configured to receive the contaminated water from
the input and generate reducing or oxidizing conditions in the contaminated
water;
a separation unit downstream of the reducing or oxidizing unit and in fluid
communication therewith, the separation unit configured to receive the
contaminated water from the reducing or oxidizing unit and remove flocculating
ion flocks with captured contaminant from the contaminated water;
an input for a flocculating ion-enriched aqueous solution, the input
configured
for introducing the flocculating ion-enriched aqueous solution into the
contaminated water upstream of the separation unit, at the separation unit, or
a
combination thereof; and
an output for outputting a treated water from the separation unit.
7
CA 3027250 2018-12-12
In select embodiments of the present disclosure, the flocculating ion
comprises iron, aluminum,
or a combination thereof.
In select embodiments of the present disclosure, the reducing or oxidizing
unit comprises an
electroreduction apparatus for generating reducing conditions in the
contaminated water.
In select embodiments of the present disclosure, the electroreduction
apparatus comprises one
or more non-sacrificial electrodes.
In select embodiments of the present disclosure, the electroreduction
apparatus comprises an
input for introducing a chemical reductant for generating reducing conditions
in the
contaminated water.
In select embodiments of the present disclosure, the chemical reductant is
NaHS03, Na2S03,
Na2S204, Na2S203, nascent hydrogen, or a combination thereof.
In select embodiments of the present disclosure, the reducing or oxidizing
unit comprises an
electrooxidation apparatus for generating oxidizing conditions in the
contaminated water.
In select embodiments of the present disclosure, the electrooxidation
apparatus comprises one
or more non-sacrificial electrodes.
In select embodiments of the present disclosure, the electrooxidation
apparatus comprises an
input for introducing a chemical oxidant for generating oxidizing conditions
in the
contaminated water.
In select embodiments of the present disclosure, the chemical oxidant is H202,
Na0C1, or a
combination thereof.
In select embodiments of the present disclosure, the flocculating ion-enriched
aqueous solution
comprises an aqueous solution of Fe2+ ions.
8
CA 3027250 2018-12-12
In select embodiments of the present disclosure, the flocculating ion-enriched
aqueous solution
comprises a solution generated: by electro-flocculation using a sacrificial
iron electrode, a
sacrificial aluminum electrode, or a combination thereof; by dissolving an
iron salt, an
aluminum salt, or a combination thereof in water; from a solution of iron
vitriol; or a
combination thereof.
In select embodiments of the present disclosure, the input for the
flocculating ion-enriched
aqueous solution comprises a slipstream line.
In select embodiments of the present disclosure, the system further comprises
at least one input
for a pH adjustment agent for: adjusting pH to render silica or other ionic
contaminants in the
contaminated water reactive; adjusting pH to promote formation of the
flocculating ion flocks;
or a combination thereof.
In select embodiments of the present disclosure, the adjusting the pH to
render silica or other
ionic contaminants in the contaminated water reactive comprises adjusting the
pH to between
about 2 to about 4.
In select embodiments of the present disclosure, the pH adjustment agent
comprises HC1,
H2SO4, another acid, or a combination thereof.
In select embodiments of the present disclosure, the adjusting the pH to
promote formation of
the flocculating ion flocks comprises adjusting the pH to a range between
about 7 and about
11.
In select embodiments of the present disclosure, the pH adjustment agent
comprises NaOH,
steam blowdown, another base, or a combination thereof.
9
CA 3027250 2018-12-12
In select embodiments of the present disclosure, the system further comprises
one or more
inputs configured for introducing an H2S scavenger, a chelant, a polymer, a
sulphite, or a
combination thereof to the contaminated water.
In select embodiments of the present disclosure, the separation unit comprises
a filtration
apparatus or a flotation apparatus for separating flocculating ion flocs from
the contaminated
water.
In select embodiments of the present disclosure, the system is configured to
maintain the
contaminated water at high temperature throughout treatment, thereby
generating the treated
water at high temperature.
In select embodiments of the present disclosure, the system is configured to
maintain the
contaminated water at or above about 80 C during treatment.
In select embodiments of the present disclosure, the system is configured to
maintain the
contaminated water at or above about 100 C during treatment.
In select embodiments of the present disclosure, the contaminated water
comprises a produced
water.
In select embodiments of the present disclosure, the produced water is a
produced water from
a SAGD operation.
In select embodiments of the present disclosure, the one or more contaminants
removed from
the contaminated water by the system comprise calcium, magnesium, silica, an
organic
contaminant, or a combination thereof.
In select embodiments of the present disclosure, the system further comprises:
CA 3027250 2018-12-12
a downstream electrochemical oxidation unit which is configured to receive the
treated
water output from the separation unit and to subject the treated water to an
electrochemical
oxidation treatment to render organics in the treated water insoluble; and
a downstream organics separation unit for removing insoluble organics from the
treated
water.
In select embodiments of the present disclosure, the system further comprises:
a downstream chemical oxidation unit which is configured to receive the
treated water
output from the separation unit and to subject the treated water to a chemical
oxidation
treatment to rendering organics in the treated water insoluble; and
a downstream organics separation unit for removing insoluble organics from the
treated
water.
Select embodiments of the present disclosure relate to a system for producing
hydrocarbons
from a subterranean reservoir, the system comprising:
a wellbore system comprising at least one well contacting the subterranean
reservoir, wherein the wellbore system is configured for injecting steam,
water, or a
combination thereof into the subterranean reservoir and for producing a
produced water
and a hydrocarbon to the surface; and
a system for treating contaminated water as defined herein, wherein the system
for
treating contaminated water is configured: (i) to receive at least a portion
of the
produced water from the subterranean reservoir at the input for the
contaminated water,
(ii) to treat the produced water, and (iii) to return treated water from the
output to the
wellbore system or to a different wellbore system for re-injection into the
subterranean
reservoir.
In select embodiments, the present disclosure relates to a system for
producing hydrocarbons
from a subterranean reservoir, the system comprising:
an injection well and a production well contacting the subterranean reservoir,
wherein the injection well is configured for injecting steam into the
subterranean
reservoir, and wherein the production well is configured for producing a
produced
11
CA 3027250 2018-12-12
water stream and a hydrocarbon stream, or a mixed produced water and
hydrocarbon
emulsion stream, to the surface; and
a system for treating contaminated water as defined herein, wherein the system
for
treating contaminated water is configured: (i) to receive at least a portion
of the
produced water from the subterranean reservoir at the input for the
contaminated water,
(ii) to treat the produced water, and (iii) to return treated water from the
output to the
same, or a different, injection well for re-injection into the subterranean
reservoir.
In select embodiments of the present disclosure, the injection well and the
production well are
a SAGD well pair.
In select embodiments, the present disclosure relates to a method for treating
a contaminated
water, said method comprising:
introducing a flocculating ion-enriched aqueous solution into the contaminated
water; and
removing at least some of at least one contaminant from the contaminated water
by ion flocculation, whereby the contaminant is captured with flocculating ion
flocks which are then separated from the contaminated water;
thereby producing a treated water.
In select embodiments of the present disclosure, the flocculating ion-enriched
aqueous solution
is introduced into the contaminated water as a side-stream or a slip-stream.
In select embodiments of the present disclosure, the flocculating ion-enriched
aqueous solution
is generated: by electro-flocculation of a carrier water using a sacrificial
iron electrode, a
sacrificial aluminum electrode, or a combination thereof; by dissolving an
iron salt, an
aluminum salt, or a combination thereof in a carrier water; from a solution of
iron vitriol; or a
combination thereof.
12
CA 3027250 2018-12-12
In select embodiments of the present disclosure, the ion flocculation
comprises adjusting the
pH to render silica or other ionic contaminants in the contaminated water
reactive.
In select embodiments of the present disclosure, the ion flocculation
comprises adjusting the
pH to promote formation of the flocculating ion flocks.
In select embodiments of the present disclosure, the contaminated water is
maintained at high
temperature throughout treatment, thereby generating the treated water at high
temperature.
.. Select embodiments of the present disclosure relate to a system for
treating contaminated water,
the system comprising:
an input for a contaminated water;
a separation unit configured to receive the contaminated water and remove
flocculating ion flocks with captured contaminant from the contaminated water;
an input for a flocculating ion-enriched aqueous solution, the input
configured
for introducing the flocculating ion-enriched aqueous solution into the
contaminated water upstream of the separation unit, at the separation unit, or
a
combination thereof; and
an output for outputting a treated water from the separation unit.
In select embodiments of the present disclosure, the input for the
flocculating ion-enriched
aqueous solution is a side-stream or a slip-stream.
In select embodiments of the present disclosure, the flocculating ion-enriched
aqueous solution
is generated: by electro-flocculation of a carrier water using a sacrificial
iron electrode, a
sacrificial aluminum electrode, or a combination thereof; by dissolving an
iron salt, an
aluminum salt, or a combination thereof in a carrier water; from a solution of
iron vitriol; or a
combination thereof
13
CA 3027250 2018-12-12
In select embodiments of the present disclosure, the flocculating ion
comprises iron, aluminum,
or a combination thereof.
In select embodiments of the present disclosure, the system further comprises
at least one input
for a pH adjustment agent for: adjusting pH to render silica or other ionic
contaminants in the
contaminated water reactive; adjusting pH to promote formation of the
flocculating ion flocks;
or a combination thereof.
In select embodiments of the present disclosure, the contaminated water is
maintained at high
temperature throughout treatment, thereby generating the treated water at high
temperature.
These and other features will be better understood with reference to the
following Drawings
and accompanying descriptions.
BRIEF DESCRIPTION OF DRAWINGS
FIGURE 1 shows an example of a conventional water treatment system for
treating produced
water from a SAGD operation to remove contaminants therefrom;
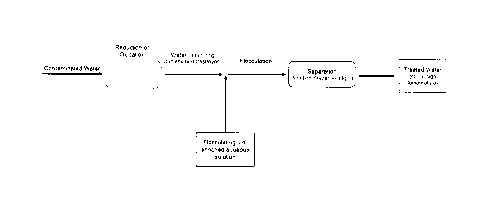
FIGURE 2 shows a flow diagram of an embodiment of a water treatment method and
system
as described herein, which avoids certain conventional apparatus and which
includes steps of
subjecting contaminated water to reducing or oxidizing conditions; introducing
a flocculating
ion-enriched aqueous solution into the contaminated water; and removing at
least some of at
least one contaminant from the contaminated water by ion flocculation, whereby
the
contaminant is captured with flocculating ion flocks which are then separated
from the
contaminated water, thereby producing a treated water;
FIGURE 3 depicts another embodiment of a water treatment method and system as
described
herein, in which produced water is subjected to electro-flocculation
treatment, followed by pH
adjustment, and then at least some of at least one contaminant is removed from
the
contaminated water by flocculation, whereby the contaminant is captured with
flocculating ion
14
CA 3027250 2018-12-12
flocks which are then separated from the contaminated water, thereby producing
a treated
water. The depicted embodiment uses an electro-flocculation unit which employs
iron-based
sacrificial electrodes to introduce iron ions, and therefore the iron
electrodes have maintenance
and upkeep considerations;
FIGURE 4 depicts an embodiment of a water treatment method and system in which
the
contaminated water is not subjected to electro-flocculation, and instead a
separate carrier water
(for example, fresh water, brackish water, or a side stream of produced water)
is subjected to
electroflocculation to generate an iron ion-enriched solution, which is then
introduced into the
contaminated water, and a pH adjustment is performed on the contaminated water
to promote
flocculation to allow for contaminant separation in a filtration or flotation
unit. However, as
described in the examples section below, when the depicted embodiment was used
to treat a
produced water sample containing flocculation inhibiting compound(s), suitable
separation
was not achieved, although in other embodiments, treatment of other
contaminated waters
containing little or no flocculation inhibiting compound(s) is expected;
FIGURE 5 depicts an embodiment of a water treatment method and system as
described herein,
which includes steps of subjecting contaminated water to reducing conditions
(via
electroreduction); introducing a flocculating ion-enriched aqueous solution
into the
contaminated water, the solution being generated by separate treatment of a
carrier water
(brackish water, fresh water, or a side stream of produced water, for example)
by
electroflocculation to introduce iron ions from a sacrificial electrode; a pH
adjustment step to
promote flocculation; and a removal step of removing at least some of at least
one contaminant
from the contaminated water by ion flocculation, whereby the contaminant is
captured with
flocculating ion flocks which are then separated from the contaminated water,
thereby
producing a treated water;
FIGURE 6 depicts another embodiment of a water treatment method and system as
described
herein, which includes steps of subjecting contaminated water to reducing
conditions (via
electroreduction); introducing an H2S scavenger to inhibit contaminants which
would
otherwise consume/block flocculating ions; introducing a flocculating ion-
enriched aqueous
solution into the contaminated water; a pH adjustment step to promote
flocculation; and a
removal step of removing at least some of at least one contaminant from the
contaminated
water by ion flocculation, whereby the contaminant is captured with
flocculating ion flocks
CA 3027250 2018-12-12
which are then separated from the contaminated water, thereby producing a
treated water;
FIGURE 7 depicts another embodiment of a water treatment method and system as
described
herein, which includes steps of subjecting contaminated water to reducing
conditions (via
electroreduction); introducing an H2S scavenger to abrogate contaminants which
would
otherwise consume/block flocculating ions; introducing a flocculating ion-
enriched aqueous
solution into the contaminated water; a pll adjustment step to promote
flocculation; a removal
step of removing at least some of at least one contaminant from the
contaminated water by ion
flocculation, whereby the contaminant is captured with flocculating ion flocks
which are then
separated from the contaminated water, thereby producing a treated water; and
steps of
introducing a chelant (to bind residual hardness to reduce scaling) and/or a
sulphite (to bind
oxygen to reduce corrosion);
FIGURE 8 depicts another embodiment of a water treatment method and system as
described
herein, which includes steps of subjecting contaminated water to reducing
conditions (via
chemical reduction); introducing a flocculating ion-enriched aqueous solution
into the
contaminated water; a pH adjustment step to promote flocculation; a removal
step of removing
at least some of at least one contaminant from the contaminated water by ion
flocculation,
whereby the contaminant is captured with flocculating ion flocks which are
then separated from
the contaminated water, thereby producing a treated water; and steps of
introducing a chelant
(to bind residual hardness to reduce scaling) and/or a sulphite (to bind
oxygen to reduce
corrosion);
FIGURE 9 depicts another embodiment of a water treatment method and system as
described
herein, which includes steps of subjecting contaminated water to oxidizing
conditions (via
electrochemical oxidation (ECO) treatment); introducing an H2S scavenger to
abrogate
contaminants which would otherwise consume/block flocculating ions;
introducing a
flocculating ion-enriched aqueous solution into the contaminated water; a pH
adjustment step
to promote flocculation; a removal step of removing at least some of at least
one contaminant
from the contaminated water by ion flocculation, whereby the contaminant is
captured with
flocculating ion flocks which are then separated from the contaminated water,
thereby
producing a treated water; and steps of introducing a chelant (to bind
residual hardness to
reduce scaling) and/or a sulphite (to bind oxygen to reduce corrosion);
FIGURE 10 depicts another embodiment of a water treatment method and system as
described
16
CA 3027250 2018-12-12
herein, which includes steps of subjecting contaminated water to electrical or
chemical
reduction conditions; introducing an H2S scavenger to abrogate contaminants
which would
otherwise consume/block flocculating ions; introducing a flocculating ion-
enriched aqueous
solution into the contaminated water; a pH adjustment step to promote
flocculation; a removal
step of removing at least some of at least one contaminant from the
contaminated water by ion
flocculation, whereby the contaminant is captured with flocculating ion flocks
which are then
separated from the contaminated water, thereby producing a treated water; a
step of subjecting
the treated water to electrochemical oxidation (ECO) treatment to render
organics in the treated
water insoluble, and separating the insoluble organics via filtration or
floatation; and steps of
introducing a chelant (to bind residual hardness to reduce scaling) and/or a
sulphite (to bind
oxygen to reduce corrosion);
Figure 11 depicts another embodiment of a water treatment method and system as
described
herein, which includes steps of subjecting contaminated water to chemical
oxidation
conditions; introducing a flocculating ion-enriched aqueous solution into the
contaminated
water; a pH adjustment step to promote flocculation; a removal step of
removing at least some
of at least one contaminant from the contaminated water by ion flocculation,
whereby the
contaminant is captured with flocculating ion flocks which are then separated
from the
contaminated water, thereby producing a treated water; and steps of
introducing a chelant (to
bind residual hardness to reduce scaling) and/or a sulphite (to bind oxygen to
reduce corrosion);
FIGURE 12 depicts another embodiment of a water treatment method and system as
described
herein, which includes steps of subjecting contaminated water to electrical or
chemical
reduction conditions; introducing a flocculating ion-enriched aqueous solution
into the
contaminated water; a pH adjustment step to promote flocculation; a removal
step of removing
at least some of at least one contaminant from the contaminated water by ion
flocculation,
.. whereby the contaminant is captured with flocculating ion flocks which are
then separated from
the contaminated water, thereby producing a treated water; a step of
subjecting the treated water
to chemical oxidation treatment to render organics in the treated water
insoluble, and separating
the insoluble organics via filtration or floatation; and steps of introducing
a chelant (to bind
residual hardness to reduce scaling) and/or a sulphite (to bind oxygen to
reduce corrosion);
FIGURE 13 shows increase in iron ion concentration in a brackish water (BW)
sample over
time during treatment in a Miniflot;
17
CA 3027250 2018-12-12
FIGURE 14 shows an efficiency curve relative to p1-1 for brackish water (BW);
FIGURE 15 shows increase in total and dissolved iron concentration as a
function of operating
time in a boiler feed water (BFW) sample;
FIGURE 16 shows increase in iron concentration in a brackish water (BW) sample
over time
during a long term treatment run with constant parameters;
FIGURE 17 shows increase in iron concentration as well as efficiency,
calculated from the data
in Figure 16 (efficiency over operating time). A flattening was observed after
the fifth hour of
operation in this experiment;
FIGURE 18 shows the titration curve obtained when checking stored Fe-ion
solutions (using
brackish water as carrier) for degradation and decomposition;
FIGURE 19 shows dependency between amperage and voltage, electrode separation,
and
conductivity for an Oil Removal Filter (ORF) outlet water sample with a
conductivity of 2.8
mS/cm;
FIGURE 20 shows the observed trend of pH over time during treatment of an ORF
outlet water
sample during testing with pH as a variable using a lab unit;
FIGURE 21 shows the observed dependency of amperage to change in voltage over
time. 10
V values were used to calculate subsequent Miniflot operation parameters. The
data suggests
that low amperage was sufficient to destroy the inhibiting compound(s);
FIGURE 22 shows the observed trend in change of pH over treatment time for
titanium
electrodes and, for comparison, the trend for graphite electrodes. Titanium
and graphite
electrodes showed different behavior when treating ORF water;
FIGURE 23 shows the amperage versus voltage curves for titanium at different
voltages (20V
and 10V) over time. The behavior is quite different for titanium, when
compared to graphite.
The curves show a solution with similar conductivity after 30 to 40 minutes of
treatment;
FIGURE 24 shows observed increase of Fe2+ concentration in a brackish water
(BW) sample
over time during studies as described in Example 4;
18
CA 3027250 2018-12-12
FIGURE 25 shows mg/I concentrations measured for hardness during testing.
Independent of
the type of electrode used in pre-treatment, i.e. whether graphite or
titanium, target was met in
almost all cases;
FIGURE 26 shows mg/1 concentrations measured for silica during testing. Target
for active
Silica was 50 mg/l. In general, this target was met with average values for
silica of about 18 ¨
20 mg/1. Only two tests using titanium electrodes, in combination with higher
flow rate, showed
values higher than 50 mg/1;
FIGURE 27 shows silica concentrations (reactive, colloidal, and total) for
untreated process
waters from field tests that were executed at elevated temperatures.
FIGURE 28 shows silica concentrations (reactive, colloidal, and total) for
treated process
waters from field tests that were executed at elevated temperatures.
FIGURE 29 shows hardness concentrations (calculated, Ca2+, and Mg2+) for
untreated process
waters from field tests that were executed at elevated temperatures.
FIGURE 30 shows hardness concentrations (calculated, Ca2 , and Mg2+) for
untreated process
waters from field tests that were executed at elevated temperatures.
FIGURE 31 shows degradation (removal) of naphthenics at pH values of 5, 7, and
9 (A-C,
respectively) during electrolytic reduction. The slope of the degradation
curve was almost
independent of pH value;
FIGURE 32 shows degradation of naphthenic acids at pH 7 at temperatures of 20
and 50 C;
and at pH 9 and at temperatures of 20 and 50 C (A and B, respectively), during
electrolytic
reduction treatment. All other parameters were kept constant;
FIGURE 33 compares the observed degradation of naphthenic acids at room
temperature and
a pH of 7 with current, as conducted through graphite electrodes, used as a
variable. All
parameters were kept constant, with exception of current, which was changed (3
series) from
5 A, 8.3 A and finally to 10 A. The trend lines for 5 A and 8.3 A show a low
level of
degradation;
FIGURE 34 shows observed degradation of phenols during electro-chemical
reduction at pH
5, 7, and 9 (A-C, respectively). Degradation rates for phenols/phenolics were
somewhat higher
19
CA 3027250 2018-12-12
than the degradation rates observed for naphthenic acids (at least at room
temperature);
FIGURE 35 shows observed increasing degradation rates with increasing
temperature for
phenols (20 and 50 C at pH 7 in A, 20 and 50 C at pH 9 in B) during
electrolytic reduction;
FIGURE 36 shows the observed influence of current in the degradation of
phenols during
electrolytic reduction. The trend lines are according to normal standard;
FIGURE 37 shows the observed concentration of naphthenic acids and total
organic carbon
(TOC) versus treatment time using an EGO lab unit set-up. After 20 min of
treatment
naphthenic acids concentration was decreased from 18 to 7.1 mg/I, while
general TOG
concentration was reduced from 250 to 170 mg/1;
FIGURE 38 shows that removal of naphthenic acids using EGO took place faster
than the total
removal of organic carbon. At the point when approximately 50 % of naphthenic
acids are
removed, 30 % of total organics are removed (based on 18 mg/1 naphthenic acids
in feed and
250 mg/1 TOG in feed);
FIGURE 39 shows estimated design values for an EGO plant. As shown, it is
estimated that a
reduction of naphthenic acids from 18 to about 3.4 mg/1 may take a residence
time of about 2
min in such a design;
FIGURE 40 depicts a proposed design schematic of an embodiment of a design for
a direct
injection flocculation treatment pilot plant developed for the treatment of
SAGD process water
as described in Example 5;
FIGURE 41 depicts a proposed general layout of an embodiment of a design for a
direct
injection flocculation treatment pilot plant developed for the treatment of
SAGD process water
as described in Example 5; and
FIGURE 42 depicts an embodiment of a reduction vessel (A), and an embodiment
of reduction
vessel specifications (B), of a proposed design for a direct injection
flocculation treatment pilot
plant developed for the treatment of SAGD process water as described in
Example 5.
DETAILED DESCRIPTION
CA 3027250 2018-12-12
Described herein are methods and systems for the treatment of contaminated
water. It will be
appreciated that embodiments and examples are provided for illustrative
purposes intended for
those skilled in the art, and are not meant to be limiting in any way.
Conventionally, water treatment methods may have included a step of
electrocoagulation, in
which an iron-based sacrificial electrode was used to introduce Fe ions into
contaminated
water, which form iron flocks that capture contaminants and facilitate their
removal from the
water. However, such electrocoagulation treatment was typically performed at
ambient
temperature, or at temperatures between about 5 to about 90 C, and required
significant
maintenance (which may even require process streams to be taken offline for
servicing) and
operational costs to keep the sacrificial electrode(s) operational.
The present inventors sought to develop water treatment methods and systems
which are
capable of operating at high temperatures and pressures; which employ a
flocculating ion-
enriched aqueous solution, which may be separately prepared, to reduce or
eliminate use of in-
line sacrificial electrodes; and which do not require treatment of the
contaminated water with
conventional electrocoagulation. The development of such methods and systems
proved highly
challenging, due at least in part to the discovery that certain contaminated
waters such as those
produced from an oilfield operation may contain one or more flocculation
inhibiting
compounds. Indeed, experiments as described in further detail hereinbelow
revealed that
foregoing electrocoagulation treatment in favor of adding a flocculating ion-
enriched aqueous
solution to the contaminated water, while effective for certain contaminated
waters, failed to
achieve water treatment of certain contaminated water samples due to the
presence of one or
more flocculation inhibiting compounds in the contaminated water samples,
which prevented
flocculation and thus did not achieve contaminant removal.
It has now been discovered that subjecting the contaminated water to reducing
or oxidizing
conditions may be used to destroy or inactivate the flocculation inhibiting
compound(s) in the
contaminated water, thereby allowing for a flocculating ion-enriched aqueous
solution, which
may be prepared separately from the contaminated water, to be used for
contaminant removal
in a manner which does not require use of sacrificial electrodes, does not
require performing
electrocoagulation on the contaminated water, and which is fully compatible
with elevated
temperatures and pressures. In certain embodiments, the flocculating ion-
enriched aqueous
solution may be prepared separately, and may even be prepared at ambient
temperatures and
21
CA 3027250 2018-12-12
pressures, reducing complexity and operational costs. In certain embodiments,
the flocculating
ion-enriched aqueous solution may comprise a solution obtained by treating an
aqueous carrier
fluid with a conventional electrofloatation apparatus (which may be conducted
at ambient
temperature outside the main process stream, for example), a solution prepared
by dissolving
flocculating ion salts in water, or even a flocculating ion-enriched aqueous
solution obtained
or prepared from mining waste such as iron vitriol or green salt.
As described in detail herein, by combining a reducing or oxidizing pre-
treatment step with the
use of a flocculating ion-enriched aqueous solution to remove at least some of
at least one
contaminant from a contaminated water by ion flocculation, methods and systems
have now
been developed which may provide advantages and/or alternatives to
conventional approaches
as described in further detail hereinbelow.
In an embodiment, there is provided herein a method for treating a
contaminated water, said
method comprising:
subjecting the contaminated water to reducing or oxidizing conditions;
introducing a flocculating ion-enriched aqueous solution into the contaminated
water; and
removing at least some of at least one contaminant from the contaminated water
by ion flocculation, whereby the contaminant is captured with flocculating ion
flocks which are then separated from the contaminated water;
thereby producing a treated water.
The contaminated water to be treated may comprise any suitable fluid with an
aqueous
component contaminated with one or more contaminants for removal. The
contaminated water
may be fully aqueous, or may be an emulsion, for example, an aqueous emulsion
with oil. The
contaminated water may comprise a waste water, or an industrial process water,
for example.
In certain embodiments, the contaminated water may comprise, for example, a
produced water
from an oil field operation. By way of example, the waste water may comprise a
produced
water from a Steam-Assisted Gravity Drainage (SAGD) operation, or a produced
water from
another subterranean hydrocarbon mobilization process. The produced water may,
or may not,
22
CA 3027250 2018-12-12
be in an emulsion with oil(s), for example. In certain embodiments, the
contaminated water
may comprise a produced water emulsion with up to about 2,000 ppm oil, for
example.
In certain embodiments, the contaminated water may comprise one or more
contaminants to
be removed therefrom. The contaminants to be removed may include, but are not
limited to,
calcium, magnesium, silica, other water hardness, or an organic contaminant,
or any
combination thereof.
In certain embodiments, the contaminated water to be treated may be a
contaminated water
which has been subjected to one or more pre-treatment steps. By way of
example, in certain
embodiments, the contaminated water to be treated may be a contaminated water
which has
been filtered (using, for example, an ORF), or subjected to another such
treatment step, prior
to treatment with the methods described herein.
Subjecting the contaminated water to reducing or oxidizing conditions may
comprise any
suitable process step in which the contaminated water is treated under
electroreduction and/or
chemical reduction conditions, or under electrooxidation and/or chemical
oxidation conditions,
which are sufficient to destroy, degrade, remove, or inactivate at least a
portion of inhibitory
contaminants in the contaminated water (if present) which would otherwise
prevent or interfere
with subsequent flocculation of the contaminated water using a flocculating
ion-enriched
aqueous solution. In certain embodiments, it is not necessary that
contaminated water actually
comprise such inhibitory contaminant(s); rather, such a reducing or oxidizing
step may be used
to expand process versatility to both inhibitory and non-inhibitory
contaminated waters,
thereby removing the need to test for inhibitory contaminant presence in
contaminated water
to be treated.
In certain embodiments, the contaminated water may be subjected to reducing
conditions via
electroreduction. Electroreduction may comprise treating the contaminated
water by
electrolytic reduction such that electrons are introduced into the
contaminated water to destroy,
degrade, remove, or inactivate inhibitory compound(s) which would otherwise
prevent
flocculation. In certain embodiments, electroreduction treatment may employ
non-sacrificial
(i.e. non-consumptive) electrode(s), such as graphite and/or titanium
electrodes, for electrolytic
reduction. Non-sacrificial electrodes are generally lower maintenance as
compared to
consumptive electrodes. Generally, in certain embodiments, electroreduction
treatment may
comprise treatment with, for example, graphite plates installed at between
about 2cm and about
23
CA 3027250 2018-12-12
10cm separation distance, and a voltage from about 5 to about 20 volts and
between about 1
and about 16 amps of current may be applied. When using graphite plates, for
example, the
information provided in Figure 19, Figure 21, Figure 22, and Figure 23, or
other such
information, may be used to assist in selecting suitable configuration and
conditions for
.. electroreduction for a particular application, for example.
In certain embodiments, the contaminated water may be subjected to chemical
reduction-based
reducing conditions. Chemical reduction may comprise treating the contaminated
water with a
suitable chemical reducing agent such that electrons are introduced into the
contaminated water
to destroy, degrade, remove, or inactivate inhibitory compound(s) which would
otherwise
prevent flocculation. In certain embodiments, chemical reducing agents may
include one or
more of NaHS03, Na2S03, Na2S204, Na2S203, nascent hydrogen, or a combination
thereof.
In certain embodiments, the contaminated water may be subjected to a
combination of
electroreduction- and chemical reduction-based reducing conditions.
In certain embodiments, reducing conditions may include those generated by DC
electroreduction with a voltage of between 4 and 20 volts, for example.
Without wishing to be
bound by theory, in certain embodiments reducing conditions may facilitate
Kolbe electrolysis.
During such electrolysis, electrons introduced into the system may have a role
in destroying
flocculation inhibitor compound(s) in the contaminated water, due to high
energy. In certain
embodiments, reducing conditions may include those generated by a chemical
reduction agent.
In such examples, redox potential in chemical reduction may be around 0.45 v
or less.
In certain embodiments, the contaminated water may be subjected to
electrooxidation-based
oxidizing conditions. Electrooxidation may comprise treating the contaminated
water by
electrolytic oxidation such that flocculation inhibiting compound(s) in the
contaminated water
which would otherwise prevent flocculation are destroyed, degraded, removed,
or inactivated
by oxidation. In certain embodiments, electrooxidation treatment may employ
non-sacrificial
(i.e. non-consumptive) electrode(s), such as graphite and/or titanium
electrodes, for electrolytic
oxidation. Non-sacrificial electrodes are generally lower maintenance as
compared to
consumptive electrodes. Generally, in certain embodiments, electrooxidation
treatment may
comprise treatment with, for example, graphite plates installed at between
about 2cm and about
10cm separation distance, and a voltage from about 5 to about 20 volts and
between about 1
and about 16 amps may be applied.
24
CA 3027250 2018-12-12
In certain embodiments, oxidizing conditions may be those generated by
electrochemical
oxidation (ECU), for example. Where flocculation inhibiting compound(s) in the
contaminated
water are oxidized (i.e. degraded) more quickly than the average total organic
loading,
oxidizing conditions may be of particular interest. In certain embodiments,
where flocculation
inhibiting compound(s) in the contaminated water are more readily oxidized
than other bulk
organic acids or free oil (for example), oxidizing conditions may be of
particular interest.
In certain embodiments, the contaminated water may be subjected to chemical
oxidation-based
oxidizing conditions. Chemical oxidation may comprise treating the
contaminated water with
a suitable chemical oxidizing agent such that flocculation inhibiting
compound(s) in the
contaminated water which would otherwise prevent flocculation are destroyed,
degraded,
removed, or inactivated. In certain embodiments, chemical oxidizing agents may
include one
or more of H202 or Na0C1, or another suitable oxidizing agent, or a
combination thereof.
In certain embodiments, the contaminated water may be subjected to a
combination of
electrooxidation- and chemical oxidation-based oxidizing conditions.
In certain embodiments, both reducing and oxidizing conditions may be used.
For example,
low-powered ECU may be used to provide reduction/oxidation, as ECU may include
a joint
electrolytic/chemical reduction step (ECU may produce oxidizing and reducing
agents). In
certain embodiments, ECU may be employed to remove soluble organics from
contaminated
water, either as a pre-treatment step or a post-treatment step, during water
treatment.
In certain embodiments, after the contaminated water has been subjected to the
reducing or
oxidizing conditions, a flocculating ion-enriched aqueous solution may then be
introduced into
the contaminated water. In another embodiment, the flocculating ion-enriched
solution may be
introduced into the contaminated water before, during, or after the
contaminated water being
subjected to the reducing or oxidizing conditions, where conditions may be
selected such that
the flocculating ions are not appreciably deactivated (see, for example,
Example 7 below).
Flocculating ion-enriched aqueous solutions may include any suitable solution
comprising a
water-based carrier fluid and at least one flocculating ion component.
Flocculating ions may
include, for example, iron ions or aluminum ions or both. In certain
embodiments, the
flocculating ion may comprise Fe2+, and the flocculating ion-enriched aqueous
solution may
be an Fe2tenriched aqueous solution. In certain embodiments, it is
additionally contemplated
that the flocculating ion-enriched solution may be a non-aqueous, or partially
aqueous,
CA 3027250 2018-12-12
solution, slurry, or other such formulation of flocculating ions suitable for
introduction into the
contaminated water. The flocculating ion-enriched solution may comprise any
suitable solution
which provides flocculating ions to the contaminated water when introduced
thereto, thus
creating flocks which allow for separation of contaminants from the water.
Flocculating ion-
enriched solutions may be separately produced or pre-made, for example, by
dissolving
flocculating ion salts in a carrier water, or by treating a carrier water
(such as fresh water,
brackish water, a side stream of produced water, or boiler feed water, for
example) using a
consumptive electrode which introduces flocculating ions into the carrier
water (i.e. by using
an iron-based consumptive electrode of an electroflotation apparatus, for
example). In certain
.. embodiments, the flocculating ion-enriched aqueous solution may comprise a
solution
generated by electro-flocculation or electroflotation using a sacrificial iron
or aluminum
electrode, a solution generated by dissolving iron or aluminum salts in water,
a solution of iron
vitriol or green salt, or any combination thereof. In certain embodiments, the
flocculating ion
concentration of the flocculating ion-enriched aqueous solution may be
adjusted to suit the
particular application, apparatus, and contaminated water being used, so as to
provide for
contaminant removal via flocculation.
In certain embodiments, references herein to introducing a flocculating ion-
enriched aqueous
solution into the contaminated water may include one or both of introducing
flocculating ions
which are already in solution form into the contaminated water, and/or
introducing flocculating
ions which are in the form of a solid (or in another non-solution or non-
aqueous form) into the
contaminated water where the flocculating ions enter solution to produce the
flocculating ion-
enriched aqueous solution using water already in the contaminated water. By
way of example,
in certain embodiments, a step of introducing a flocculating ion-enriched
aqueous solution into
the contaminated water may comprise adding a solid or powder of a flocculating
ion salt to the
contaminated water, where the flocculating ion will dissolve into the
contaminated water to
form the flocculating ion-enriched aqueous solution therein.
As will be understood, the flocculating ion-enriched aqueous solution may be
introduced into
the contaminated water in any suitable manner. In certain embodiments, the
flocculating ion-
enriched aqueous solution may be introduced into the contaminated water as a
slip-stream, for
example. In certain embodiments, the flocculating ion-enriched aqueous
solution may be
introduced into the contaminated water via any suitable injection method known
to the person
of skill in the art having regard to the teachings herein.
26
CA 3027250 2018-12-12
Following introduction of the flocculating ion-enriched aqueous solution into
the contaminated
water, flocculation occurs, whereby flocks are formed in the contaminated
water which allow
for capture of contaminants in the contaminated water. Contaminants and flocks
may then be
separated from the contaminated water using any suitable separation technique
known to the
.. person of skill in the art. By way of example, flocks and contaminants may
be separated from
the contaminated water by filtration or floatation. In certain embodiments,
induced static
floatation (ISF), induced gas floatation (IGF), filter press, sand filter,
mixed bed filter, walnut
shell filter, cartridge filter, bag filter, stainless steel or composite
filter, and/or compact
floatation unit (CFU) equipment (with either single or multiple stage
floatation) may be used
for separation, for example. Removal of the flocks and contaminants thereby
produces a treated
water, in which a level of at least one contaminant is reduced as compared to
the contaminated
water before treatment.
In certain embodiments, the step of removing at least some of at least one
contaminant from
the contaminated water by ion flocculation may optionally include adjusting pH
of the
contaminated water to promote formation of ion flocks. In certain embodiments,
the pH may
be adjusted, for example, to a pH within a range of about 7 to about 11 in
order to promote
formation of flocks. In certain embodiments, the pH adjustment may be
performed before,
concurrently with, or after, introduction of the flocculating ion-enriched
aqueous solution into
the contaminated water, to facilitate the removal of contaminants from the
contaminated water.
In certain embodiments, the pH may be adjusted generally any suitable time
before (or during)
flocculation such that the pH of the contaminated water is suitable for
promoting formation of
ion flocks during flocculation, as reflected in the dotted line in selected
Figures.
In certain embodiments, the step of pH adjustment of the contaminated water
may be performed
in two stages. In the first stage, pH may be adjusted to render silica or
other ionic contaminants
.. in the contaminated water reactive; and in a second stage, pH may be
adjusted to promote
formation of the flocculating ion flocks. By way of example, in the first
stage, pH may be
adjusted to about 2 to about 4, and in the second stage pH may be adjusted to
within a range of
about 7 to about 11. As will be understood, pH may be adjusted in any suitable
manner known
to the skilled person. By way of example, pH may be lowered using HCI, H2SO4,
or another
.. acid, and pH may be raised using NaOH, steam blowdown, or another base, for
example.
In certain embodiments, methods as described herein may further comprise a
step of adding
one or more of an H2S scavenger, a chelant, a polymer, or a sulphite additive
to the
27
CA 3027250 2018-12-12
contaminated water during treatment. For example, in certain embodiments, an
H2S scavenger
may be added to abrogate contaminant(s) which would otherwise consume or block
flocculating ions, to promote flocculation and/or to reduce the amount of
flocculating ion to be
used. In certain embodiments, H2S scavenger may be added at any suitable time
prior to or
during flocculation, and may be added to the contaminated water, or to the
flocculating ion-
enriched solution which is then added to the contaminated water. In certain
embodiments, a
chelant may be added to the water post-treatment (i.e. post removal of one or
more
contaminants by ion flocculation), to bind with residual hardness ions to
reduce scaling. In
certain embodiments, a polymer may be added to the contaminated water before
or during
flocculation, to aid with coagulation. In certain embodiments, a sulphite may
be added to bind
with free oxygen and prevent corrosion. For example, an H2S scavenger may be
added to the
contaminated water prior to introduction of an iron ion-enriched solution, so
as to prevent the
iron ions from being blocked by (i.e. reacting with) sulfur. In certain
embodiments, a polymer
may be added to enhance flocculation and facilitate contaminant removal, for
example.
.. In certain embodiments, treated water may additionally be treated by a step
of subjecting the
treated water to electrochemical oxidation (ECO) or chemical oxidation
treatment to render
remaining organics in the treated water insoluble, and separating the organics
from the treated
water. Since residual organics may create a varnish-like scale in a boiler,
for example, such
oxidation treatment may be performed to make the organics insoluble and/or
separable (i.e. to
precipitate and/or separate organics) to avoid or reduce such scaling.
Techniques for removing
the insoluble organics from the water may include any suitable separation
methods and
apparatus known to the person of skill in the art.
In certain embodiments, temperatures and pressures under which methods and
systems as
described herein may be operated may include any of those at which fluids
produced from, for
example, a SAGD process, may typically experience:
Conventional processes for separating produced emulsion for oil sales and
further water
treatment involve a number of distinct steps, as follows. Oil-water emulsion
from the
reservoir may vary in temperature, for example from 80 C-250 C, more typically
180 C-220 C, and with a pressure of about 1,200-2,000 kPag. After emulsion is
recovered from the reservoir it may be degassed and then cooled to from 130 C-
140 C
to allow for diluent aided separation. The cooled emulsion is then treated for
coarse
28
CA 3027250 2018-12-12
oil-water separation, for example in free-water knock out (FWKO) unit(s),
and/or other
emulsion treaters, typically operating at about 800-1,500 kPag and 130 C-140 C
for
traditional gravity separation, or alternatively operating at much lower
pressures of
between 100-800 kPag and 130 C-140 C for flash treating. These conventional
emulsion treating systems typically use diluent to aid in separation of oil
and water,
where the diluent is traditionally a pentane rich natural gas liquid (NGL) or
a synthetic
crude oil, and where the diluent remains in the dewatered oil that is then
cooled and
sent to sales oil tanks.
Conventional processes for treating the produced water for re-use in steam
generation
may involve a number of distinct steps, as follows. Conventionally, produced
water
from emulsion treatment is cooled, using for example one or more heat
exchangers,
before it is subjected to de-oiling and further water treatment. This heat
exchange
process may for example decrease the produced water temperature from 130 C-140
C
to 80 C-95 C. De-oiling typically comprises several units, such as a skim tank
for bulk
oil separation, a flotation (floatation) type unit such as an induced gas or
induced static
flotation unit (IGF/ISF) for further removal of oil and suspended solids, and
a filtration
type unit such as an oil removal filter (ORF). Subsequent water treatment
typically
includes, for example, a warm lime softener (WLS), which increases the pH of
the water
and adds MagOx (Magnesium Oxide) to remove silica. The WLS is typically
followed
by an ion exchange unit where removal of scaling ions occurs. Scaling ions
typically
include dissolved calcium, magnesium, lithium and iron. A significant decrease
in
temperature of the produced water stream entering de-oiling is generally
performed for
operational reasons, particularly so that surge capacity may be carried out at
atmospheric pressure in tanks. Typical water quality from de-oiling and water
treatment may be less than 50 ppm silica, less than 0.1 ppm hardness, and less
than 1
ppm oil.
In certain embodiments, water treatment methods and systems as described
herein are
compatible with operation at elevated temperatures and pressures, such as
those typically
encountered during steam generation, produced water recovery, and treatment in
a SAGD
facilities process. Water treatment methods and systems as described herein
are also compatible
with operation at a variety of other temperatures such as ambient or room
temperatures, cooler
temperatures, warmer temperatures, and temperatures therebetween, for example.
In certain
29
CA 3027250 2018-12-12
embodiments, water treatment methods and systems as described herein may be
operable at
temperatures above about 100 C, or above about 125 C, or above about 150 C,
for example.
In certain embodiments, water treatment methods and systems may be configured
for operation
at a temperature between about 135 C to about 250 C, for example. Accordingly,
in certain
embodiments, the contaminated water to be treated may be maintained at high
temperature
throughout treatment, thereby generating the treated water at high
temperature, saving on
energy requirements when using the treated water for injection of further
steam or water
downhole, for example. In applications where an output of treated water at
high temperature is
desired, the contaminated water may be heated during treatment by the methods
and systems
described herein; or, when the contaminated water to be treated is already
received at high
temperature, no heating, or reduced heating, during treatment may still yield
a heated treated
water. As will be understood, in certain embodiments, methods and systems
described herein
do not require a heat sink/cooling phase, and do not require heat input,
during treatment of
contaminated water. Rather, methods and systems described herein may be
configured to suit
the particular application, based on the temperature of the contaminated water
being received
and/or on the desired temperature of the treated water output. In certain
embodiments, the
contaminated water may be maintained at or above about 80 C during treatment,
or at or above
about 100 C during treatment, for example, thereby producing a heated treated
water.
In still another embodiment, there is provided herein a method for producing
hydrocarbons
from a subterranean reservoir, said method comprising:
injecting steam and/or water into the subterranean reservoir;
producing a produced water and a hydrocarbon to the surface;
treating at least a portion of the produced water using a method as defined
herein, thereby generating a treated water stream; and
using the treated water stream to provide steam and/or water for re-injection
into the subterranean reservoir to produce more hydrocarbons to the surface.
In certain embodiments, the injecting and producing may be performed as part
of a thermal in-
situ hydrocarbon recovery operation. In certain embodiments, the injecting and
producing may
be performed using an injection well and production well of, for example, a
SAGD well pair.
In certain embodiments, the injecting and producing may be performed using a
Cyclic Steam
CA 3027250 2018-12-12
Stimulation (CSS) well setup. In certain embodiments, the injecting and
producing may be
performed as a solvent-aided process (SAP) well setup, for example. In certain
embodiments,
the injecting and producing may be performed using one or more wells of
another suitable
hydrocarbon recovery operation involving steam and/or water injection for
hydrocarbon
recovery.
In yet another embodiment, there is provided herein a method for producing
hydrocarbons from
a subterranean reservoir, said method comprising:
injecting steam into the subterranean reservoir via an injection well;
producing a produced water stream and a hydrocarbon stream to the surface via
a production well, or producing a mixed produced water and hydrocarbon
emulsion
stream from the subterranean reservoir via a production well and (optionally)
separating the mixed produced water and hydrocarbon emulsion stream into a
produced water stream and a hydrocarbon stream;
treating at least a portion of the produced water stream using a method as
described herein, thereby generating a treated water stream; and
using the treated water stream to provide steam and/or water for re-injection
into the subterranean reservoir via the same or a different injection well to
produce
more hydrocarbons to the surface.
As will be understood, in certain embodiments, the produced water stream may
be produced to
the surface as an aqueous solution which may be an emulsion with hydrocarbons,
depending
on the particular operation. The produced water may, in certain embodiments,
include both an
aqueous component and an oil component, as an emulsion, for example. Where the
produced
water comprises an emulsion, the emulsion may be processed to first remove at
least a portion
of the oil-based component (either downhole or at the surface), or the
emulsion may be used
directly as contaminated water to be treated using methods and systems as
described herein. In
certain embodiments, the contaminated water may comprise a produced water
emulsion stream
with up to about 2,000 ppm oil, for example.
31
CA 3027250 2018-12-12
In certain embodiments, the injection well and production well may be a SAGD
well pair, for
example. In certain embodiments, the injection well and production well may be
a single well,
or wells, of another hydrocarbon recovery operation involving steam and/or
water injection.
In yet another embodiment, there is provided herein a system for treating
contaminated water,
the system comprising:
an input for a contaminated water;
a reducing or oxidizing unit configured to receive the contaminated water from
the
input and generate reducing or oxidizing conditions in the contaminated water;
a separation unit downstream of the reducing or oxidizing unit and in direct
or indirect
fluid communication therewith, the separation unit configured to receive the
contaminated water from the reducing or oxidizing unit and remove flocculating
ion
flocks with captured contaminant from the contaminated water;
an input for a flocculating ion-enriched aqueous solution, the input
configured for
introducing the flocculating ion-enriched aqueous solution into the
contaminated water
upstream of the separation unit, at the separation unit, or a combination
thereof; and
an output for outputting a treated water from the separation unit.
As will be understood, such systems may be for use in performing a water
treatment method
as described herein.
In certain embodiments, the reducing or oxidizing unit may be for subjecting
the contaminated
water to reducing or oxidizing conditions in which the contaminated water is
treated under
electroreduction and/or chemical reduction conditions, or under
electrooxidation and/or
chemical oxidation conditions, which are sufficient to destroy, degrade,
remove, or inactivate
at least a portion of inhibitory contaminants in the contaminated water which
would otherwise
prevent or interfere with subsequent flocculation of the contaminated water
using a flocculating
ion-enriched aqueous solution.
In certain embodiments, the reducing unit may be an electroreduction-based
reducing unit.
Electroreduction may comprise treating the contaminated water by electrolytic
reduction such
that electrons are introduced into the contaminated water to destroy, degrade,
remove, or
32
CA 3027250 2018-12-12
inactivate inhibitory compound(s) which would otherwise prevent flocculation.
In certain
embodiments, the electroreduction-based reducing unit may employ non-
sacrificial (i.e. non-
consumptive) electrode(s), such as graphite and/or titanium electrodes, for
electrolytic
reduction. By way of example, in certain embodiments a non-sacrificial
electrode may include
a doped electrode, or an Inconel, Monel, Super Duplex, or other such non-
reactive anode. Non-
sacrificial electrodes are generally lower maintenance as compared to
consumptive electrodes.
In certain embodiments, for example, electroreduction-based reducing units may
comprise a
vessel (which may be pressurized or non-pressurized), with graphite or
titanium grids operating
between about 5 and about 20 volts and about 1 to about 16 amps, for example.
In certain embodiments, the reducing unit may be a chemical reduction-based
reducing unit.
Chemical reduction may comprise treating the contaminated water with a
suitable chemical
reducing agent such that electrons are introduced into the contaminated water
to destroy,
degrade, remove, or inactivate inhibitory compound(s) which would otherwise
prevent
flocculation. In certain embodiments, chemical reducing-based reducing units
may comprise
units for introducing one or more of NaHS03, Na2S03, Na2S204, Na2S203, or
nascent hydrogen
reducing agents, or a combination thereof, into the contaminated water. In
certain
embodiments, a chemical reduction-based approach may include, for example,
direct injection
of one or more chemical reducing agents into a flowline, tank, or vessel, with
or without a static
mixer, which allows for sufficient reaction time (for example, between about
10 seconds and
about 5 minutes, or another suitable reaction time appropriate for the
particular
application/implementation) with the contaminated water.
In certain embodiments, the reducing unit may be a hybrid electroreduction-
based and chemical
reduction-based reducing unit.
In certain embodiments, the oxidizing unit may be an electrooxidation-based
oxidizing unit.
Electrooxidation may comprise treating the contaminated water by electrolytic
oxidation such
that flocculation inhibiting compound(s) in the contaminated water which would
otherwise
prevent flocculation are destroyed, degraded, removed, or inactivated by
oxidation. In certain
embodiments, electrooxidation-based oxidizing units may employ non-sacrificial
(i.e. non-
consumptive) electrode(s), such as graphite and/or titanium electrodes, for
electrolytic
oxidation. Non-sacrificial electrodes are generally lower maintenance as
compared to
consumptive electrodes. Generally, in certain embodiments, electrooxidation-
based oxidizing
units may comprise a vessel (which may be pressurized or non-pressurized),
with graphite or
33
CA 3027250 2018-12-12
titanium grids operating between about 5 and about 100 volts and about 1 to
about 50 amps,
for example.
In certain embodiments, the oxidizing unit may be a chemical oxidation-based
oxidizing unit.
Chemical oxidation may comprise treating the contaminated water with a
suitable chemical
oxidizing agent such that flocculation inhibiting compound(s) in the
contaminated water which
would otherwise prevent flocculation are destroyed, degraded, removed, or
inactivated. In
certain embodiments, chemical oxidation-based oxidizing units may comprise
units for
introducing one or more of H202 or Na0C1 oxidizing agents, or a combination
thereof, into the
contaminated water. In certain embodiments, a chemical oxidation-based
approach may
include, for example, direct injection of one or more chemical oxidation
agents into a flowline,
tank, or vessel, with or without a static mixer, which allows for sufficient
reaction time (for
example, between about 10 seconds and about 5 minutes, or another suitable
reaction time
appropriate for the particular application/implementation) with the
contaminated water.
In certain embodiments, the oxidizing unit may be a hybrid electrooxidation-
based and
chemical oxidation-based oxidizing unit.
In certain embodiments, a reducing-and-oxidizing unit may be used. For
example, low-
powered ECO may be used to provide reduction/oxidation, as ECO may include a
joint
electrolytic/chemical reduction step (ECO may produce oxidizing and reducing
agents). In
certain embodiments, ECO may be employed to remove soluble organics from
contaminated
water, either as a pre-treatment step or a post-treatment step, during water
treatment.
In another embodiment, the system may further comprise a flocculating ion-
enriched aqueous
solution generator, comprising an electro-flocculation reactor employing
sacrificial iron or
aluminum electrode(s), which is configured to generate the flocculating ion-
enriched aqueous
solution and provide the solution to the input for introducing the solution
into the contaminated
water. In certain embodiments, such a flocculating ion-enriched aqueous
solution generator
may be used for preparing the flocculating ion-enriched aqueous solution on-
site.
In certain embodiments, the input for the flocculating ion-enriched aqueous
solution may be
an input which is configured to introduce a separately prepared flocculating
ion-enriched
aqueous solution into the contaminated water. In certain embodiments, for
example, the input
for the flocculating ion-enriched aqueous solution may comprise a slipstream
line feeding into
34
CA 3027250 2018-12-12
a contaminated water line. In certain embodiments, input flocculating ion-
enriched aqueous
solution may be input at or upstream of a suitable mixing location, which may
comprise, for
example, a vessel, or a pipe, with or without a static mixer.
In still further embodiments, the system may further comprise at least one
input for a pH
adjustment agent, which is configured for:
optionally, adjusting pH to render silica or other ionic contaminants in the
contaminated
water reactive; and
adjusting pH to promote formation of the flocculating ion flocks.
By way of example, the one or more inputs for the pH adjustment agent may be
configured for:
optionally, adjusting pH of the contaminated water to a pH of about 2 to about
4 to
render silica or other ionic contaminants in the contaminated water reactive;
and
adjusting pH of the contaminated water to a range of about 7 to about 11 to
promote
formation of the flocculating ion flocks.
In certain embodiments, the one or more inputs for the pH adjustment agent may
be configured
for adjusting pH of the contaminated water at generally any suitable time
before (or during)
flocculation such that the pH of the contaminated water is suitable for
promoting formation of
ion flocks during flocculation. By way of example, where pH of the
contaminated water is
adjusted to a pH of between about 7 to about 11 to promote flocculation, the
one or more inputs
for the pH adjustment agent may be positioned for adjusting pH of the
contaminated water at
generally any suitable time prior to or during flocculation in the separation
unit (i.e. may be
upstream or at the separation unit). Where pH of the contaminated water is
first adjusted to
render silica or other ionic contaminants in the contaminated water reactive,
for example by
adjusting pH to between about 2 to about 4, the one or more inputs for the pH
adjustment agent
for such pH adjustment may be positioned for adjusting pH of the contaminated
water at
.. generally any suitable time prior to flocculation in the separation unit
(i.e. may be upstream of
the separation unit).
In certain embodiments, systems as described herein may further comprise one
or more inputs
configured for adding one or more of an H2S scavenger, a chelant, a polymer,
or a sulphite to
the contaminated water during treatment. For example, in certain embodiments,
an H2S
scavenger may be added to the contaminated water via an input, to abrogate
contaminant(s)
which would otherwise consume/block flocculating ions, to promote flocculation
and/or to
CA 3027250 2018-12-12
reduce the amount of flocculating ion to be used. In certain embodiments, H2S
scavenger may
be added at any suitable time prior to or during flocculation, and may be
added to the
contaminated water, or to the flocculating ion-enriched solution which is then
added to the
contaminated water. In certain embodiments, the H2S scavenger input may be
positioned to
input H2S scavenger prior to introduction of the flocculating ion-enriched
aqueous solution into
the contaminated water. In certain embodiments, a chelant input may be
included to add chelant
to the water post-treatment (i.e. post removal of one or more contaminants by
ion flocculation),
to bind with residual hardness ions to reduce scaling. In certain embodiments,
a polymer may
be added to the contaminated water before or during flocculation, to aid with
coagulation. In
certain embodiments, a sulphite input may be configured to add a sulphite at
any suitable time
to bind with free oxygen and prevent corrosion. For example, an H2S scavenger
input may be
included prior to the flocculating ion-enriched solution input, so as to
prevent the flocculating
ions from being consumed/blocked by sulfur. A polymer input may be included
for injecting
polymer to enhance flocculation and facilitate contaminant removal, for
example.
In another embodiment of systems as described herein, the separation unit may
comprise, for
example, a filtration or flotation apparatus for separating flocculating ion
flocs from the
contaminated water. Contaminants and flocks may be separated from the
contaminated water
using any suitable separation unit known to the person of skill in the art. In
certain
embodiments, the separation unit may comprise an induced static floatation
(ISF) unit, an
induced gas floatation (IGF) unit, filter press, sand filter, mixed bed
filter, walnut shell filter,
cartridge filter, bag filter, stainless steel or composite filter, and/or a
compact floatation unit
(CFU) (with either single or multiple stage floatation), for example.
In yet another embodiment, systems as described herein may additionally
comprise:
a downstream electrochemical oxidation (ECO) or chemical oxidation unit, which
is
configured to receive the treated water output from the separation unit and
subject the
treated water to electrochemical oxidation (ECO) or chemical oxidation
treatment,
rendering organics in the treated water insoluble; and
a downstream organics separation unit for removing insoluble organics from the
treated
water.
In still another embodiment, there is provided herein a system for producing
hydrocarbons from
a subterranean reservoir, the system comprising:
36
CA 3027250 2018-12-12
a wellbore system comprising at least one well contacting the subterranean
reservoir, the wellbore system for injecting steam and/or water into the
subterranean
reservoir and for producing a produced water and a hydrocarbon to the surface;
and
a system for treating contaminated water as defined herein, the system for
treating
contaminated water configured (i) to receive at least a portion of the
produced water
from the subterranean reservoir at the input for the contaminated water, (ii)
to treat the
produced water, and (iii) to return treated water from the output to the same,
or a
different, wellbore system for re-injection into the subterranean reservoir.
In certain embodiments, the wellbore system may be a wellbore system of a
thermal in-situ
hydrocarbon recovery operation. In certain embodiments, the wellbore system
may comprise
an injection well and production well of, for example, a SAGD well pair. In
certain
embodiments, the wellbore system may comprise a Cyclical Steam Stimulation
(CSS) well
setup. In certain embodiments, the wellbore system may comprise one or more
wells of another
suitable hydrocarbon recovery operation involving steam and/or water
injection.
In yet another embodiment, there is provided herein a system for producing
hydrocarbons from
a subterranean reservoir, the system comprising:
an injection well and a production well contacting a subterranean reservoir,
the
injection well for injecting steam into the subterranean reservoir and the
production
well for producing a produced water stream and a hydrocarbon stream, or a
mixed
produced water and hydrocarbon emulsion stream, to the surface; and
a system for treating contaminated water as described herein, the system for
treating contaminated water configured (i) to receive at least a portion of
the
produced water from the subterranean reservoir at the input for the
contaminated
water, (ii) to treat the produced water, and (iii) to return treated water
from the output
to the same, or a different, injection well for re-injection into the
subterranean
reservoir.
In certain embodiments, the produced water stream may be produced to the
surface as an
aqueous solution, or as an emulsion, depending on the particular operation.
The produced water
may thus in certain embodiments include both an aqueous component and an oil
component,
as an emulsion, for example. Where the produced water comprises an emulsion,
the emulsion
37
CA 3027250 2018-12-12
may be processed to first remove the oil component (either downhole or at the
surface) in an
emulsion separation unit of the system, or the emulsion may be used directly
as contaminated
water to be treated using methods and systems as described herein.
In certain embodiments, the injection well and production well of the system
may be a SAGD
well pair, for example. In certain embodiments, the injection well and
production well may be
wells of another hydrocarbon recovery operation involving steam injection.
Figure 1 depicts an example of a conventional water treatment system for
treating produced
water from a SAGD operation to remove contaminants therefrom. The system
includes several
treatment apparatus, including a warm lime softener (WLS) and associated
components, which
are used to treat the contaminated water. Figure 2, in contrast, depicts an
embodiment of a
water treatment method and system as described herein, which includes steps of
subjecting
contaminated water to reducing or oxidizing conditions; introducing a
flocculating ion-
enriched aqueous solution into the contaminated water; and removing at least
some of at least
one contaminant from the contaminated water by ion flocculation, whereby the
contaminant is
captured with flocculating ion flocks which are then separated from the
contaminated water,
thereby producing a treated water. The depicted method does not require WLS
apparatus, and
instead provides for water treatment via a distinct process.
Figure 3 depicts another embodiment of a water treatment method and system as
described
herein, in which produced water is subjected to electro-flocculation
treatment, followed by pH
adjustment, and then at least some of at least one contaminant is removed from
the
contaminated water by flocculation, whereby the contaminant is captured with
flocculating ion
flocks which are then separated from the contaminated water, thereby producing
a treated
water. The depicted embodiment uses an electro-flocculation unit which employs
iron-based
sacrificial electrodes to introduce iron ions, and therefore the iron
electrodes have maintenance
and upkeep considerations.
In contrast, Figure 4 depicts an embodiment of a water treatment method and
system in which
the contaminated water is not subjected to electro-flocculation, and instead a
separate carrier
water (brackish water in this example) is subjected to electroflocculation to
generate an iron
ion-enriched solution, which is then introduced into the contaminated water,
and a pH
adjustment is performed on the contaminated water to promote flocculation to
allow for
contaminant separation in a filtration or flotation unit. However, as
described in the examples
38
CA 3027250 2018-12-12
section below, when the depicted embodiment was used with a produced water
sample
containing flocculation inhibiting compound(s), suitable separation was not
achieved.
Figure 5 depicts an embodiment of a water treatment method and system as
described herein,
which includes steps of subjecting contaminated water to reducing conditions
(via
electroreduction); introducing a flocculating ion-enriched aqueous solution
into the
contaminated water, the solution being generated by separate treatment of a
carrier water
(brackish water) by electroflocculation to introduce iron ions from a
sacrificial electrode; a pH
adjustment step to promote flocculation; and a removal step of removing at
least some of at
least one contaminant from the contaminated water by ion flocculation, whereby
the
contaminant is captured with flocculating ion flocks which are then separated
from the
contaminated water, thereby producing a treated water.
Figure 6 depicts another embodiment of a water treatment method and system as
described
herein, which includes steps of subjecting contaminated water to reducing
conditions (via
electroreduction); introducing an H2S scavenger to abrogate contaminants which
would
otherwise consume/block flocculating ions; introducing a flocculating ion-
enriched aqueous
solution into the contaminated water; a pH adjustment step to promote
flocculation; and a
removal step of removing at least some of at least one contaminant from the
contaminated
water by ion flocculation, whereby the contaminant is captured with
flocculating ion flocks
which are then separated from the contaminated water, thereby producing a
treated water.
Figure 7 depicts another embodiment of a water treatment method and system as
described
herein, which includes steps of subjecting contaminated water to reducing
conditions (via
electroreduction); introducing an H2S scavenger to abrogate contaminants which
would
otherwise consume/block flocculating ions; introducing a flocculating ion-
enriched aqueous
solution into the contaminated water; a pH adjustment step to promote
flocculation; a removal
step of removing at least some of at least one contaminant from the
contaminated water by ion
flocculation, whereby the contaminant is captured with flocculating ion flocks
which are then
separated from the contaminated water, thereby producing a treated water; and
steps of
introducing a chelant and a sulphite for further reducing hardness and
preventing scaling.
Figure 8 depicts another embodiment of a water treatment method and system as
described
herein, which includes steps of subjecting contaminated water to reducing
conditions (via
chemical reduction); introducing a flocculating ion-enriched aqueous solution
into the
39
CA 3027250 2018-12-12
contaminated water; a pH adjustment step to promote flocculation; a removal
step of removing
at least some of at least one contaminant from the contaminated water by ion
flocculation,
whereby the contaminant is captured with flocculating ion flocks which are
then separated from
the contaminated water, thereby producing a treated water; and steps of
introducing a chelant
and a sulphite for further reducing hardness and preventing scaling.
Figure 9 depicts another embodiment of a water treatment method and system as
described
herein, which includes steps of subjecting contaminated water to oxidizing
conditions (via ECO
treatment); introducing an H2S scavenger to abrogate contaminants which would
otherwise
consume/block flocculating ions; introducing a flocculating ion-enriched
aqueous solution into
the contaminated water; a pH adjustment step to promote flocculation; a
removal step of
removing at least some of at least one contaminant from the contaminated water
by ion
flocculation, whereby the contaminant is captured with flocculating ion flocks
which are then
separated from the contaminated water, thereby producing a treated water; and
steps of
introducing a chelant and a sulphite for further reducing hardness and
preventing scaling.
Figure 10 depicts another embodiment of a water treatment method and system as
described
herein, which includes steps of subjecting contaminated water to electrical or
chemical
reduction conditions; introducing an H2S scavenger to abrogate contaminants
which would
otherwise consume/block flocculating ions; introducing a flocculating ion-
enriched aqueous
solution into the contaminated water; a pH adjustment step to promote
flocculation; a removal
step of removing at least some of at least one contaminant from the
contaminated water by ion
flocculation, whereby the contaminant is captured with flocculating ion flocks
which are then
separated from the contaminated water, thereby producing a treated water; a
step of subjecting
the treated water to electrochemical oxidation (ECO) treatment to render
organics in the treated
water insoluble, and separating the insoluble organics via filtration or
floatation; and steps of
introducing a chelant and a sulphite for further reducing hardness and
preventing scaling.
Figure 11 depicts another embodiment of a water treatment method and system as
described
herein, which includes steps of subjecting contaminated water to chemical
oxidation
conditions; introducing a flocculating ion-enriched aqueous solution into the
contaminated
water; a pH adjustment step to promote flocculation; a removal step of
removing at least some
of at least one contaminant from the contaminated water by ion flocculation,
whereby the
contaminant is captured with flocculating ion flocks which are then separated
from the
CA 3027250 2018-12-12
contaminated water, thereby producing a treated water; and steps of
introducing a chelant and
a sulphite for further reducing hardness and preventing scaling.
Figure 12 depicts another embodiment of a water treatment method and system as
described
herein, which includes steps of subjecting contaminated water to electrical or
chemical
.. reduction conditions; introducing a flocculating ion-enriched aqueous
solution into the
contaminated water; a pH adjustment step to promote flocculation; a removal
step of removing
at least some of at least one contaminant from the contaminated water by ion
flocculation,
whereby the contaminant is captured with flocculating ion flocks which are
then separated from
the contaminated water, thereby producing a treated water; a step of
subjecting the treated water
to chemical oxidation treatment to render organics in the treated water
insoluble, and separating
the insoluble organics via filtration or floatation; and steps of introducing
a chelant and a
sulphite for further reducing hardness and preventing scaling.
As indicated, each of the embodiments depicted in Figures 2-12 may be used to
avoid,
substitute, reduce, or replace use of a conventional treatment apparatus such
as shown in Figure
1 (such as a WLS, for example). In the embodiments depicted in Figures 3-12,
conventional
treatment apparatus which is being replaced is indicated within a shaded box.
EXAMPLE 1 ¨ Generation of Iron Ion-Enriched Aqueous Solutions
While treatment of the produced water with conventional electro-flocculation
achieved
contaminant removal, cost-benefit analysis showed that costs were similar to
current WLS-
based designs for treating produced water form an oilfield operation. As well,
the flow-through
configuration was problematic at high temperature, because high-maintenance
plates were
required in a pressure vessel for such electro-flocculation treatment,
complicating applications
where it is desirable to maintain the water at elevated
temperatures/pressures.
Accordingly, studies were performed to determine if a separately produced
flocculating ion-
enriched solution could be introduced to the contaminated produced water to
remove
contaminants via flocculation, without requiring
electroflocculation/electrocoagulation
treatment of the contaminated water. These initial experiments sought to
determine whether a
flocculating ion-enriched solution, in this example an iron ion-enriched
aqueous solution, could
41
CA 3027250 2018-12-12
be separately produced, and then slipstreamed into contaminated produced water
to achieve
contaminant removal via flocculation.
Generation of Iron Ion-Enriched Aqueous Solutions
Since treatment of industrial contaminated waters, such as produced water from
an oilfield
.. operation, is desirable, initial studies investigated options for preparing
iron ion-enriched
solutions. IGF columns are apparatus commonly employed in oilfield operations,
and these
may be operated with flow rates of several hundred m3 per hour of Skim Tank
Water, i.e. 250
m3/h. Thus, options for preparing solutions compatible with these types of
operational
parameters were investigated. It was initially hypothesized that Fe2+
concentrations of about
50 ¨ 100 mg/1 would be suitable for removing contaminants from water
efficiently
(concentrations are further studied below). Initial assumptions were that a
concentration of 75
mg/1 may be used. Based on this assumption, the total hourly amount of Fe' for
a flow rate of
250 m3/h would be about 20 kg (exactly 18.75 kg).
Accordingly, these studies, in part, sought to determine whether production of
enriched Fe2+
solution with an iron concentration of about 2 to 5 g/I using electrolytic
dissolution of iron
would be possible, and under what operating conditions. This target range
would then allow
use of only a small side stream to introduce suitable amounts of iron for
water treatment. The
volume of the side stream depends on the level of Fe2+ enrichment achieved.
This small volume
side stream would have a negligible effect on the main stream of process water
(i.e. pH,
temperature, etc., impact from the small volume of iron enriched concentrate
would be
inconsequential).
Therefore, based on the following assumptions:
STW flow rate 250m3/h;
Fe' for treatment of 75 mg/1;
18.75 kg Fe2+/h; and
Enriched Fe2+ solution of 5g/1,
the volume of the side stream would be about 3.75 m3/h.
42
CA 3027250 2018-12-12
The only data available for solubility of Fe2+ constants were for pure water
and at low
concentrations. However, pure water has a low conductivity which is not
preferable for
electrode configurations (only a single value is given at a pH of 7, 25 C:
Solubility 7.2 g/1).
Boiler Feed Water (BFW) and Brackish Water (BW), on the other hand, have
conductivities in
the higher range, measured respectively at 3.23 mS/cm (pH 9.16) for BFW and
18.57 mS/cm
(pH 9.772) for BW. These are more preferable, as level of conductivity is
sufficiently high to
start the process.
For these tests, Brackish Water (BW), Boiler Feed Water (BFW) were used as the
carrier water.
Characteristics of these waters were as follows:
Brackish Water was clear, contained about 6 g/I salt, mainly in the form of
NaCl, a small
amount of hardness and almost no DOC. Conductivity of BW was measured at 18.57
mS/cm,
the pH 9.77. Color was clear. Other characteristics included:
SiO2: 11 mg/1
Hardness: 0.977 mg/1
TOCtotal: 32.58 mg/1
Oil + grease: < DL
Fewt. : 0.052 mg/I; Fechss. : 0.031 mg/1
Due to the high concentration of NaCl, BW was very suitable to the production
of Fe2+ solution
in an electrolytic dissolution reactor.
Boiler Feed Water contained some organics that turn the color to dark brown.
Main
characteristics of BFW were:
Conductivity: measured at 3.23 mS/cm, the pH 9.16.
Color: clear, dark, red-brown
Other Characteristics included:
SiO2: 52 mg/I
43
CA 3027250 2018-12-12
TOC total. = 360.9 mg/1
DOC : 359.1 mg/1
Oil + grease: 23.78 mg/1
Fetot. : 0.1523 mg/1
Due to the high concentration of NaC1, the water was very suitable to the
production of Fe2+
solution in an electrolytic dissolution reactor. The organic compounds in BFW
may initially
cause foaming during EF, which disappeared mostly after some hours of
operation.
Iron ion-enriched aqueous solutions were generated using a Miniflot S-200
apparatus, which
is a compact and mobile electroflocculation plant. The plant included a feed
pump; a pH pre
adjustment with automatic HC1 dosing station; an electro flotation reactor
with up to 19 iron
electrodes, power supply with up to 10 V DC and 100 A and automatic pole
reversal capability;
a pH after-adjustment with automatic NaOH dosing station; a filter press feed
pump; and a
filter press for sludge separation and removal. After setting process
parameters, the plant
operates automatically.
For the production of enriched Fe2+ solution, the plant was operated in a
closed circuit from the
feed pump via pH pre-adjustment and reactor tanks to the pH after-adjustment
tank back to the
feed pump. The dosing pump for NaOH was shut off during this operation. Flow
rate was set
to 2001/h, and the pH value setting in the feed adjustment was varied from 9.8
(Brackish Water,
BW) down to 3. Optimal operating conditions for production of enriched Fe2+
solution with the
Miniflot were 100 A and between 4 V and 10 V. These conditions are dependent
on the
conductivity of feed water and the distance between the iron electrodes. The
distance between
the electrodes may be equal for all electrode pairs. The Miniflot plant is
equipped with 19
positions for electrodes, and the system can therefore hold between 2 and 10
electrode pairs,
depending on the conductivity of the water. For example, 10 pairs of
electrodes are used in
treating water with low conductivity. On the other hand, 2 or 3 pairs are used
for water with
relatively high conductivity. During testing, 3 to 6 electrode pairs were
used. In certain
embodiments, an industrial reactor may have one defined set of electrodes, for
example. During
testing, the number of electrode pairs was varied, as conductivity of feed
water changed with
pH value and concentration of Fe2+.
44
CA 3027250 2018-12-12
Water was pumped directly into the Miniflot. During each test phase, the water
remained in
the Miniflot in a closed circuit. In total, 3 tests were undertaken; first, a
test with BW to find
optimal process parameters. Then, a second test with BFW in the same manner as
the first test.
In the third test, optimal process parameters from the first test were taken
to produce Fe2+
solution in a long-term test run. After finishing a particular Fe2+ enrichment
run, about 50 I of
solution was removed from all the chambers (pH pre-adjustment, reactor tank
and pH after-
adjustment tank) into a smaller container and mixed. pH was controlled and the
solution was
checked for residual flocks and other solid by-products. About 3 I were then
filtrated using
standard folded fluted filters for use in subsequent lab tests.
In such manner, the following three types of Fe2+ solutions were generated:
I. Brackish Water carrier with an iron content of about 4.8 g/1 soluble iron
(first test)
2. Boiler Feed Water carrier with an iron content of about 2.65 WI
3. Brackish Water carrier from long-term run with an iron content of about
3.124 g/1
Most of the remaining water in the Miniflot, as well the water from the small
mixing tank, was
then transferred into an empty tote for disposal. Total volume of produced
enriched water was
145 1 per test.
The Miniflot plant was filled under automatic operating conditions with the
appropriate pH
setting in the pH pre-adjustment tank. The reactor plant was set to operate at
10V with
increasing amperage as the reactor basin filled up. Once the fill level
reached just below the
overflow into flock basin B 1.4, the feeding pump was stopped and a direct
connection from
tank B I .3 to the feed pump was made. This enabled the Miniflot to operate in
a closed circuit
without activating the filter press stage of the process. At this point, the
feed pump was started
again and various test runs were commenced under different conditions.
Initially, the first run
with Brackish Water was undertaken without any pH adjustment (at pH 9.8).
During testing,
pH was decreased gradually until no flocks were observed in the reactor
(acidic atmosphere).
All subsequent tests started with parameters obtained on first run with
Brackish Water.
The electrodes in the Miniflot are made of black steel without any additives
and have the
following dimensions: width 0.32 m x height 0.58 m, which corresponds to a
surface area of
0.2 m2 and a thickness of 0.01m. Before the start of each test run, the total
weight of the
CA 3027250 2018-12-12
electrodes was measured to determine and to compare iron consumption with the
analytical
data. At the start of the test program, the total weight of electrodes was
¨96.1 kg. During test
runs, the distance of separation of the electrodes had to be changed due to
changes in
conductivity. 4 electrodes were used in some runs, and as many as 7 in others.
At the end of
the runs, the final weight was measured to determine iron consumption for all
electrodes.
Brackish Water as Carrier:
Brackish Water was pumped into the Miniflot pH pre-adjustment reactor and pH
after-
adjustment tanks to overflow level. Dosing pump for NaOH was disabled during
all tests.
During the initial few hours of this run with Brackish Water, the pH dosing
pump for HCl was
also disabled. Due to this, feed water was at first treated without any pH
adjustment.
As the BW was expected to have high conductivity once dissolved iron
concentration in the
range of about 5 g/1 Fe2+ was reached, only 4 electrodes were installed in the
reactor. As a result
of this the Miniflot operated at less than full power initially. Amperage
increased gradually
with increased runtime as conductivity increased due to an increase in
dissolved Fe. Eventually
flocks appeared on top of the reactor, as solubility of formed Fe(OH)2 was
exceeded. In the
range of feed pH used, the solubility of Fe(OH)2 is only about 1 ¨2 mg/l. On
top of the reactor
some Fe(OH)2 flocks may therefore appear. In this area due to direct contact
with air, a thin
layer of flocks can oxidize into orange-brown Fe2O3 (rust). The pH value was
reduced in the
feed about every hour based on the assumption that quick test results would
confirm increasing
concentration of Fe2+ in solution. Operating parameters of the first BW test
are shown in Table
1 below:
Operat Total pH Amperage Power Number Fe- Fe Total
ing operat feed of conc.. conc. Iron
hours ing electrodes total
diss, dissol
hours mg/1 ved
A Ali
1.18 2.18 9.8 44.2 53.3 4
0.47 1.65 3.9 45.6 44.2 4
1 0 2.65 3.5 49.g 483 4
2.0 4.65 8.0 940 746 135 _
LOS. 5.73 7.5 100 128 7 2238 1001 324
2.0 7.23 7.0 100 150 7
1.0 8,73 6.5 100 150 7
2.0 10.73 6.0 100 153 7 2892 2430 419
7.5 18.23 3.0 97 436 4 5218 4990 757
46
CA 3027250 2018-12-12
(Table 1: Operating Parameters of the first BW Test)
Increase in iron ion concentration in the BW is shown in Figure 13.
Efficiency of iron production varied depending on pH. Efficiency in the
transfer of iron into
solution was calculated. A standard of 100 % efficiency is defined as the
point when all
electrons, generated by electricity, transfer into the same amount of valences
in the water or
into other substances in water. This value is measured using super clean water
and standard
platinum electrodes. Based on this standard, efficiency curve relative to pH
for BW is given in
Figure 14.
Boiler Feed Water as Carrier:
The procedure to pump Boiler Feed Water into the Miniflot was identical to the
procedure used
with Brackish Water. The plant was ran on the first day with a pH of around
7.5 in the feed
(slightly alkaline). On the second day the pH was changed to a slightly acidic
condition, i.e. a
pH of around 5. During this test run, some optimization testing was performed
by varying the
time between pole reversals. Some interesting side reactions were observed
when Brackish
Water was ran and the time of pole reversal varied, and this was followed up
during this run.
Here, pole reversal time was initially set to 15 min in the first half of the
day and 60 min in the
second half of the day.
Tests were carried out with 7 electrodes. A significant amount of foaming was
observed at the
top of the reactor during the first set of runs of day one. On the second day,
foaming was
considerably less. Organic compounds in this water were likely responsible for
the foaming
observed during start-up of the operation in the electrolytic reactor.
Overtime, foaming mostly
disappeared.
Main operating parameters are shown in Table 2:
Operati Total pH of Amperage Pole Number Fe- Fe Total
rig operat feed revers of conc. conc. Iron
hours ing al time electrodes total diss, dissol
hours min mg/I mgil ved
A
3.5 3.5 7.57.6 73.7 15 7 750 481 109
3.5 7 75-7.6 100 60 7 1247 (401.5) 180
3.5 105 5.0 100 15 7 2812 2563 407
3.5 14 5.0 100 60 7 2645 2297 383
47
CA 3027250 2018-12-12
(Table 2: Operational parameters of the second BFW as carrier medium test)
Increase in iron concentration during the test as a function of operating time
is shown in Figure
15.
A third run investigating Brackish Water as carrier over a long term run was
also performed.
During test run 1 with BW, a number of parameters were changed in order to
determine optimal
operating conditions. The assumption of better results under acidic operating
conditions was
made. Based on this, the feed water was set to a pH value of about 4. Under
these conditions
the outlet out of reactor would have a pH value of about 6. Based on
solubility data, this pH
level should be sufficiently acidic to keep dissolved iron in solution. Pole
reversal time was set
to 15 minutes and pH in feed water showed considerable fluctuation due to
overdosing with
HC1. Start-up and general operation parameters were similar to those of the
other runs.
Main operating parameters are shown in Table 3:
Operat Total pH Amperage Power Number Fe- Fe Total
ing operat feed of conc. cam, Iron
hours ing electrodes total
diss. dissol
hours mgil mgf I ved
A Ni
2.75 235 1.2 100 275 7 1588 1283
230.3
3.0 5.75 3.4 100 300 7 2357 2207
341_8
3.0 8.75 2.9 100 300 7 2565 2427
371.9
3.25 12 3.7 81.6 265.2 4 2817 2601 412.8
4.0 16 2.9 97.6 390 4 1114 3121 199.6
(Table 3: Operational Parameters during the third long term BW run)
.. Figure 16 shows increase in iron concentration in BW (long term run with
constant parameters).
Increase of iron concentration as well as efficiency was calculated from this
data, as shown in
Figure 17 (efficiency over operating time). This curve is interesting in that
a flattening was
observed after the fifth hour of operation.
Side reactions were also investigated. During operation of the Miniflot, green
flocks were
observed both in the water and on top of the reactor. This is considered
normal during most
Miniflot operations. These green flocks are the Iron Oxy-hydroxide flocks
responsible for
absorption of contaminants out of water. They will not appear, as produced
iron stays in
solution if the pH is maintained at a sufficiently low level (acidic). This
normal situation is
48
CA 3027250 2018-12-12
found, when initial feed pH is about 4 and when it is increased during
electrolysis to about 6
(see treatment of BW, optimized run). The specific formula of these green
flocks is Fe50(OH)9.
This formula shows that they are a mixture of Fe' and Fe3+ oxy-hydrates. Fe3+
is formed within
the water due to oxidation with dissolved oxygen.
These green flocks can form other oxy-hydrates with iron, and can also oxidize
in the presence
of air to an orange-brown iron oxy-hydrate Fe(OH)2Fe0(OH), which is a pre-
product of rust
(Fe2O3). This pre-product of rust does not disturb the dissolution of iron or
the water treatment
process using electro flotation.
A black coating on some of the electrodes was also observed after the process
was stopped.
This coating was relatively easy to remove (with light rinse and light
brushing). It mostly
disappeared when a pole reversal was applied at a frequency of 15 minutes.
Using this
frequency, the process was not affected by this coating. Although not directly
measured, it
appeared that there was, at least qualitatively, no impact on efficiency. This
black material
consists mostly of Magnetite, with the compositional formula of Fe2+(Fe3+)204,
i.e. a mixture
of Fe2+ and Fe3+ and oxide compound.
Storage of Enriched Fe Solutions:
After completing each of the test runs, the water was drained out of all 3
working chambers of
the Miniflot (i.e. pH pre-adjustment, reactor, after-adjustment tanks). The
water was mixed and
filtered using a fluted paper filter. The residue remaining in the fluted
filters was identified as
Fe2O3. A sample was taken for analysis to determine the amount of total and
dissolved Iron in
the filtrate. The values from these analyses were identical to those taken
directly out of the
Miniflot. Samples were stored in closed glass bottles.
After at least a week, the stored Fe solutions were checked for degradation
and decomposition.
BW Fe solution was then used to measure the titration curve and the behavior
of the solution.
The titration curve is shown in Figure 18. The behavior exhibited by the
titration curve was
absolutely normal. It is a standard "typical" curve that represents the
behavior of the Fe2+
solution across a pH range. It basically shows that Fe2+ is being dealt with
and not with Fe3+,
showing product stability. During flocking, flocks always appear green, as
expected, and flocks
become denser with time. Filtrate after filtering Fe solution at a pH of 10
confirmed that no
iron was present.
49
CA 3027250 2018-12-12
Overall, iron ion-enriched aqueous solutions were successfully prepared by
electroflocculation
both Brackish Water and Boiler Feed Water, and prepared solutions were
generally stable for
storage. Such iron ion-enriched aqueous solutions were next investigated for
water treatment
by flocculation.
Results indicate that iron-enriched solutions could be produced using
electrolysis of, for
example, brackish water (BW) or boiler feed water (BFW). By way of example,
studies indicate
that even enriched Fe2+ solutions of about 5 g Fe2 /1, or more, may be
produced in this manner.
With BW a concentration of almost 5 g/1 was obtained, and with BFW a value of
about 2.5 g/1
was reached. These values were not maximum values, but rather represent
concentrations
reached by the time the reactor was stopped. Best results during operation
were achieved when
the carrier solution had a pH of about 5 at the input stage of the reactor.
Under these operating
conditions the solution leaving the reactor had a pH of 6 ¨7. pH of the
product Fe concentrate
was not higher than 7, as solubility equilibrium of Fe2+ /Fe(OH)2 at this pH
value is given at
7.6 g Fe2+ in literature. Under these conditions the efficiency of the process
is about 60 to 70
% and there was no need to handle any high acid or caustic streams. The
efficiency is defined
as the ratio of direct energy input (in kWh/h) versus production of dissolved
Fe2+ (in g/h). The
produced Fe solution stayed relatively stable and did not change in
composition over the course
of the experiments. This was tested and confirmed with a titration curve.
Thus, enriched Fe2+ solutions may be produced using electrolysis within an
electro-flotation
.. circuit, for example. For water treatment testing, solutions having Fe2+
concentration of about
40-50mg/1, and those as low as 30mg/1, were prepared and used.
EXAMPLE 2: Introduction of Iron Ion-Enriched Aqueous Solutions for
Flocculation of
Contaminated Water
Tests were performed to investigate flocking in Skim Tank Water (STW)
contaminated water
sample following introduction of iron ion-enriched solutions such as those
generated in
Example 1 above. Studies sought to investigate the lower limits of Fe2+
concentration in the
main stream STW to demonstrate flocking, and to reduce SiO2, oil and grease
and hardness.
Tests were prepared and undertaken under normal operating conditions using
STW.
CA 3027250 2018-12-12
Skim Tank Water was used as a process water to be treated.
Pre-treatment compositional analysis showed an unusually high content of SiO2
in the STW
sample, and as a result of this, a second sample of STW, was also tested.
Characteristics of
both STW's are as follows:
Parameter Sample 1 Sample 2
co lor murky, dark grey/green Light grey-brown
pH 7.1 6.75
Conductivity in ms/cm 2.92 2.915
5102 in mg/I 4D0 300
TOC total in me 357.6 502.3
DOC in mg/I 349.7 394.9
Oil greae 191.2 141.6
Fe, in mg/ 11152 1.326*
(Table 4: STW Sample Characteristics)
While such an approach was of interest because it was hypothesized that the
contaminated
water could be maintained at high temperature and pressure, and because the
iron ion-enriched
aqueous solution was separately prepared and introduced into the contaminated
water, results
of these studies indicated that suitable water treatment and contaminant
removal was not
achieved in this contaminated water. STW produced water was not effectively
flocculated
following injection of Fe' solution separately produced by electrolytic iron
dissolution using
an EF reactor, since unforeseen difficulties were encountered. It was
hypothesized that one or
more unknown contaminants, perhaps one or more organics of unknown composition
and
origin, were interfering with flocculation-based contaminant removal from the
water sample.
During this test phase, about 3 1 of Skim Tank Water was added to each of 5
glass bottles. Into
each of these bottles was then added a certain amount of Fe" solution
(concentrate) based on
the original concentration of the Fe" solution and the desired Fe"
concentration in each of the
test bottles. Fe" concentrations of 25, 50, 75, 100 and 125 mg Fell within
each of the test
bottles were tested. It was observed that these tests could not be carried out
using the Skim
Tank Water. After pH adjustment for testing, STW turned red and no flocks were
observed.
51
CA 3027250 2018-12-12
The lab equipment included 1 gallon (4 liter) glass bottles, each with a
magnetic stirrer. The
general procedure for a set of tests was as follows:
A volume of about 20 liters of Skim Tank Water (STW) was conditioned and
prepared for
testing by adjusting the pH to 9.5. This pH adjusted STW was then used to fill
1 gallon glass
bottles to a level of 3 liters. Various amounts of enriched Fe" solution from
tests with brackish
water (BW) and boiler feed water (BFW) as carrier water were added to each of
the bottles, so
that Fe" concentration in the bottles varied from 25 mg/1, 50 mg/1, 75 mg/1 to
100 mg/I. The
exact dissolved Fe2+ concentration in BF and BFW carrier water was
respectively 4,806 mg/I
and 2,645 mg/I, measured after completing the Fe" enrichment runs. It was
assumed that the
pH stayed about the same after addition of Fe" solution since only very small
volumes were
added to each bottle. After the addition of Fe' solution, the content of each
bottle was mixed
for 20 seconds. Mixing was stopped and the formation of flocks and their
precipitation was
observed and documented.
During these tests, it was found that the color of Skim Tank Water turned a
red-black and this
made observation very difficult. Re-tests in small beakers showed absolutely
no flocking. Tests
with BFW water did show flocks after 2 days of storage, which was more in line
with initial
expectations. When these tests were repeated with BFW after 2 days of storage,
however, it
was found to be impossible to re-create flocks (in BFW).
Indeed, flocculation lab tests using enriched Fe" solution were undertaken by
adding enriched
Fe" solution to a number of water samples, including STW and BFW in its un-
adjusted pH
state and then adjusting the pH to around 9.5, where optimum flocculation was
expected. Flock
development was not observed in these tests. When Fe" solution was added to
BFW and STW
at a pH of 7, and then at a pH of 9.5, even less flocking was observed at the
higher pH. This
was consistent, since at lower pH more Fe' was in solution for flocking.
As described, the results of these studies were unexpected. None of the tested
STW samples
showed flocking when using the Fe' solutions. Instead of flocking, the color
of STW turned
from grey-brown to red-black under alkaline conditions. No flocks appeared,
but a minor
amount of very small, black, and unfilterable particles were observed.
As a comparator, STW was treated using an EF lab unit. The EF lab unit
comprised a small EF
reactor with a total volume of about 3.8 1 as well as a power supply rated at
20 V, 10 A. The
52
CA 3027250 2018-12-12
higher voltage with the power supply of the lab unit compensated for different
conductivities
in test water. It did not affect the treatment results as this is dependent on
the amperage
(constant 10 V DC). A laboratory hose pump was used to fill the reactor and to
provide a
circular flow. After treatment, the water was pumped out of the reactor for
further treatment,
i.e. pH after-adjustment and filtration. Flocculation using the EF lab unit
was confirmed.
Again, studies were undertaken to repeat flocking tests with Fe" solutions.
Five different
concentrations of Fe" solutions for adding Fe' to STW under different pH
conditions were
used. The flocking pH used in all cases was 9.5 or a bit higher. This was
tested on STW and
BFW, as well as demineralized water, drinking water and oil/water emulsion.
Results are
shown in Table 5. Normal flocking behavior was observed in some of the tested
samples, but
no flocking was observed in BFW and STW samples at pH 7. Almost no flocking
was observed
at pH 9. This indicated that with the addition of Fe" solution, better
flocking was observed at
low pH value. At lower pH there is a higher amount of dissolved iron that is
available for
flocking. No flocking at all was observed with BFW and STW.
Test no pH Amount pH re- Appearance pH after NaOH
pH atter Appearance
of re in solutio after addition addition dosing
dosing after pH
sample n adjustment
I. Tests
with de.
mineralize
tt Water
1õ1 6.49 1E0 70 Evean szakx 593 9.8 53385
2566111
11
12 6.49 150 7.7 Dnyeal, 311W.y 5.16 955
13,65,ex:en
floats
6.49 110 2.2 r3r1wn. 55818,y 553 9.53 BlaVtir:
gmen
52285. bad
2 6.49 150 95 Brown, 81,3115 5.51 ? 19.25
Brawe .7555
115282:, 52*
.L Test
with
drinking
water
21 8.1 150 70 Brewnlreen. 7.22 = 19.09 Brewn
weer,
=IT
22 8.1 150 7' 797 941 Urepsr 8ree6
2.3 41 150 412 7.81 9.7
2.2 2.1 150 9 5 G 9 13 E8836,
06:
1 Test
with
emulsion
3.1 9.62 1E0 Gieenitecit5 542 it, .. Greenwinde
Mite Walw 45,
32 9.62 150 7.7 Green fledc2, .. 8.73 .. 955
width walw 19*82, 5 why
.2w5,
3) 96* 15.0 l2 Green 41ecks 5.61 9.62 Gf-
en.,n8e
v2512, kame, 5625, 586.4,y
34 9.52 1E0 95 1192 921
le ',vale-,
water
4. Test
with BM
53
CA 3027250 2018-12-12
4,1 9.4 150 771 Srree sfro0 fI,71 5h74 &eel Neck
S1E:sok flocSss .õõ
4,2 9.271 150 7,7 flame sterSi 9,46 9..46 Sun'
L,ICe
Siam aRCS:f:
11r
4,1 9.4 150 42 Semester:I 954 54434 Stre4 btack
UTt
slaOk flocks
4,4 9.271 150 9,5 Seem solti CIA 171 16 Small temJr
time flocks
ik,:etnetts
5. Test
%vile sriv
13.1 7.47 150 74) Sone env 6.69 9.64 ::;rseell s
44d
Jr flcsks [L.. s,
': 10,1e
5.2 7,47 150 77 Scne err? mail 7.15 9 7 Small
bin*
Neck flocks
5:3 7.47 150 517 S:sne very Mel 7,9 9.8 Srr
Itons
õate
5,4 7.7 150 9,5 Ssyne g = 9,4 s
slack flusks
onflflerabk
(Table 5: Flock Tests with different waters using Fe2+ solution)
Tests were next performed using electroflocculation (via the EF test unit) in
combination with
iron ion-enriched solution. About 3.81 of STW were used to fill the reactor to
its capacity and
the unit was then operated without pH adjustment. The operating procedure was
then
undertaken with different concentrations of Fe in steps. About 80 mg/1 Fe was
used to start,
and increased up to 160 mg/1 Fe. The treated water was then pH adjusted to
about 9.5 using
NaOH and then set on the laboratory table for observation of flocking and of
sedimentation.
As the SiO2 content in STW was unusual high, a second set of test runs were
undertaken with
diluted STW (50 % STW, 50% drinking water). This took SiO2 content to about
200 mg/l. The
second set of tests were carried out in the same way as the first, but iron
concentration started
at 30 mg/1. Results are shown in Table 6:
54
CA 3027250 2018-12-12
Reaction Fe content pH after EF pH after adj.,
Visual result
time
min mel
51W pure
0 0 6,95
7 84.1 7.26 9.68 no visual flocking
8 96.1 7.33 9.91 no visual flocking
9 108.1 7.56 9.54 first sedimentation
120.1 6..95 9.94 good tlocks/sedimentation
11 132.2 7.96 9.47 good flocks/sedimentation
12 144.2 8.2 10.03 good flocks/sedimentation
13 156.2 8.35 10,12 good flocks/sedimentation
51W5050)
5 30.0 10.21 good flocks/sedimentation
6 36.0 10.26 good flocks/sedimentation
7 42.0 9.94 good flocks/sedimentation
8 48.0 9.5 good flocks/sedimentation
(Table 6: Optimization of iron content for flocking using EF lab unit)
Treatment results during addition of Fe2+ are shown in Table 7, and EF
flocking results using
a further sample of STW are shown in Table 8:
5
Conc. of iron Iron in Hardness Si01 Oil+grease -- TOC --
DOC
during product
treatment
mgil mg/I mg/P mel mg/1 m8/I meil
STW 0 0.88 7,54 400 191.2 357.6 349.7
Feed
72.1 19.69 6.14 100 22.10 227.7 232
132,2 0.702 5.59 20 20.77 176 177
STW 0 0.44 40 200 96 178 175
(50;41)
18 2.97 39.14 50 11.31 97.15 94.93
36 0.394 29.58 10 10.53 78.25 80.71
(Table 7: Treatment results for EF plus iron ion-enriched solution)
CA 3027250 2018-12-12
Reaction Fe - content pH after EF pH after-adj. Visual
result
time
min mai
STW pure
06,05.2016
0 6.98
2 24.0 7.10 9.68 no visual flocking
3 36.1 7.18 9.91 first sedimentation
4 48.1 7.3 954 good fiocksisedi mentation
60.2 7.41 9.94 good flocks/sedimentation
6 72.2 7.5 9_47 good floirksJsedimentation
(Table 8: Treatment results for EF plus iron ion-enriched solution on
additional STW sample)
Thus, STW did not flock using Fe2tenriched solutions, but did flock when EF
was applied.
5 EXAMPLE 3 ¨ Investigating Flock-Supressing Compounds
It was hypothesized that the one or more unknown contaminants interfering with
flocculation-
based contaminant removal in Example 2 could be destroyed prior to
flocculation, thereby
allowing for contaminant removal. It was suspected that an organic substance
in STW may be
blocking the formation of flocks. It was hypothesized that some organic
complex such as
organic thiocyanate or a similar compound might be to blame. Investigations
began by treating
a small volume of STW with an ECU lab unit to effectively destroy organics in
STW. After
this treatment, it was hypothesized that flocking could again be possible.
A small ECU lab test unit was utilized. The system was basically a simple ECU
lab test unit,
designed for qualitative testing. It included a power supply (same as that
used for the EF lab
test unit), a specialized set of electrodes, a magnetic stirrer, and a
laboratory beaker. The ECO
lab test unit determined whether a wet oxidation process based on ECU can
remove soluble
organics, bacteria and other organic carbon containing compounds. Given the
unexpected
observations described above made during flocking tests with STW and BFW, the
ECU unit
was used to investigate the behavior of production water treated by hydroxyl
radicals generated
by ECU. Specifically, the aim was to destroy certain organics in Skim Tank
Water prior to re-
testing for flocking with Fe concentrate solution.
Results indicated that the ECU treated STW sample, after addition of Fe2+
solution and pH
56
CA 3027250 2018-12-12
adjustment, showed good flocks, supporting a hypothesis that an organic
compound may be to
blame for inhibiting flocculation. During ECO operation, TOC was reduced from
357.6 mg/I
to 41.09 mg/1 (DOC from 349.7 to 40.64). These results, in which ECO destroyed
of about
90% of organics, support a hypothesis that one or more organic compounds in
BFW and STW
may be preventing Fe flocking.
Experiments thus indicated that a pre-treatment step employing ECO to treat
the contaminated
produced water prior to iron flocculation via injection of a separately
produced iron ion-
enriched solution was able rescue flocculation and provide for contaminant
removal. It was
hypothesized that ECO oxidized soluble organics, perhaps including the one or
more
compounds interfering with the flocculation, using highly reactive hydroxyl
radicals generated
at the electrodes.
Dilution experiments, in which STW and BFW samples were diluted with
demineralized water,
indicated that the concentration of the inhibitory compound(s) which prevented
iron flocking
were generally present in lower concentration in BFW versus STW. Furthermore,
comparing
two different STW samples indicated that one contained about twice the
concentration relative
to the other. When STW and BFW were first diluted, and the 8mL of BW-based
Fe2+ solution
(5g/1) was added, good flocking was observed when STW and BFW was sufficiently
diluted
(i.e. between about 1:4 and about 1:10 depending on the sample.
A variety of typical oil field additives were also tested to determine whether
they inhibit Fe
flocculation. Tested additives included Petrolite RBW987; Petrolite DM08648;
Petrolite
RBW747; BPW 76325; Bulab 5901; Bulab 9773; and Bulab 9567. None were
identified as
candidates for inhibiting iron flocculation.
Further, BFW and STW samples did not contain thiocyanate (or levels were below
detection
limits), and so this was unlikely to be the inhibitor.
.. In presence of the inhibiting compound(s), results indicated that, instead
of producing the
expected Fe(OH)2 flocks, a new compound was formed, which contains the iron
produced (Fe2+
and/or Fe3+), has a red color, and is soluble in water. It is formed at pH
values that are higher
than 8 and it also remains soluble under caustic conditions. The inhibitor(s)
were not present
in brackish water that was tested.
57
CA 3027250 2018-12-12
The identity of the contaminant(s) inhibiting flocculation remains unknown.
However, it was
identified that ECO treatment was able to destroy, degrade, remove,
inactivate, or otherwise
inhibit the inhibiting contaminant(s), restoring flocculation following
addition of iron ion-
enriched aqueous solution.
EXAMPLE 4A ¨ Electroreduction, Followed by Injection of an Iron Ion-Enriched
Aqueous Solution for Flocculation of Contaminated Water, the Iron Ion-Enriched
Aqueous Solution Being Pre-Made
Experiments were performed to further investigate the nature of the inhibiting
contaminant(s),
and more importantly to identify further options for destroying or
inactivating these inhibiting
contaminant(s) which would be compatible with high temperature and high
pressure operation,
and which would facilitate the flocculating ion-enriched aqueous solution
injection approaches
detailed herein.
As discussed in Example 2, electro-chemical oxidation (ECO) rescued treatment
of STW by
iron flocculation via injection of a separately produced iron ion-enriched
solution, providing
for contaminant removal. While the identity of the inhibiting contaminant(s)
was not
determined, it was hypothesized that it might be possible to
destroy/inactivate/remove these
inhibiting contaminants using other treatments. In particular, studies were
performed to
investigate whether electrolytic reduction could abrogate the inhibiting
effects of the
contaminant(s). Further, studies were performed in particular to determine
whether electrolytic
reduction using non-consumptive electrodes could be used, since non-
consumptive electrodes
may allow for reduced maintenance and upkeep demands as compared to
consumptive
electrodes.
A test system was established using non-consumptive electrodes made of
graphite or titanium
(instead of iron), mounted in an electro-flotation reactor. Studies were
performed to determine
whether non-consumptive electrodes can be used to create an electrolytic
reduction reaction
that destroys/inactivates/removes the inhibiting compound(s), thereby
permitting flocculation
by direct injection of Fe2tenriched solution.
The lab unit included a small reactor that can hold a volume of 2.6 to 3.4 1
(depending on the
58
CA 3027250 2018-12-12
type of electrodes used). It ran on a power supply rated at 20 V, 10 A. The
higher voltage
available with the power supply of the lab unit compensated for different
conductivities found
in test waters. This did not affect the treatment results, as these depend on
the amperage
(constant 10 V DC). A laboratory hose pump is used to fill the reactor and to
provide a circular
flow. After treatment, the water was pumped out of the reactor for further
treatment, i.e. pH
after-adjustment and filtration. The apparatus used titanium electrodes to
treat ORF outlet
water. A Miniflot setup was further utilized, the Miniflot being a compact,
mobile, and fully
equipped electro-flotation plant, normally used for electro -flotation
treatment of waste water.
The equipment configuration can be altered to permit other electrolytic
processes, as used in
the production of enriched Fe' solution (see Example 1) and electro-chemical
reduction
processes using different electrodes than those normally used in electro-
flotation. Maximum
process flow rate was 2001/h. The plant included the following process steps:
pH-adjustment;
electrolytic reactor with up to 19 electrodes including power supply and pole
reversal
capability; pH after-adjustment; optional capability of polymer dosing; filter
press with pump
for flock separation; transfer pump for filtrate; and control board for manual
or for automatic
operation.
Graphite Electrodes: Graphite plates installed in the lab unit and the
Miniflot were of a
composition normally used for applications such as heating elements, although
graphite plates
typically used for water treatment are also contemplated.
Main specifications of the graphite used are:
Specific resistivity: 13 plIm
Thermal conductivity: 104 W/mK
Shore hardness: 56
Graphite electrodes used in the lab test unit had a thickness of 10 mm, and a
total area of 138
cm2. This correspond to a maximum specific amperage of 0.2 mA/cm2, which is
far below the
limits. Tests were carried out with sets of 2, 3 and 4 electrodes. The
Miniflot plant was equipped
with 10 graphite electrodes of same quality, but a thickness of 20 mm. Main
dimension were:
Width: 30 cm, height: 54 cm, area per electrode: 1,620 cm2, specific amperage:
0.2 mA/cm2.
The total reactor volume using graphite electrodes was 52.2 1, and the actual
reaction volume
59
CA 3027250 2018-12-12
for process water was up to 36.45 I once the electrodes were taken into
account. After use, no
coating, fouling or other signs of corrosion were observed on the electrode
surface. Electrodes
looked as new or unused (white spots at the bottom were caused by dried foam).
Titanium Electrodes: Titanium electrodes had the following composition: 90 %
titanium, 4%
vanadium, 6 % aluminum. This type of alloy is generally produced for the
aircraft industry.
Thickness of the titanium plates used for both the lab unit and Miniflot was 2
mm. In general,
titanium has good resistance to acids, caustics, and chlorine containing
fluids, etc. It is normally
resistant to corrosion. Dimensions of the Ti electrodes for lab test unit were
11.5 x 14 cm with
an area of 161 cm2. Specific amperage was 0.39 mA/cm2, slightly higher than
with graphite
electrodes. After use, some coating on the positive electrodes was observed.
The titanium
electrodes for the Miniflot were of the same alloy composition as for the lab
unit. The
dimensions were 58 cm x 30 cm with an area of 1,740 cm2. Specific amperage
using 10
electrodes was about 0.1mA/cm2, which was considered to be near ideal. The
rectifier power
supply for the Miniflot delivered 10 V DC with 100 A. The amperage could
decrease during
operation if the distance of the electrodes was too great for the conductivity
of the water to be
treated. The total volume of the reactor using titanium electrodes was greater
than when using
graphite electrodes because of the thinness of the titanium plates compared to
graphite. Total
reactor volume was 62.4 1, and the effective reaction volume 45.6 1.
After the tests were completed, electrodes were taken out of the reactor for
inspection. Titanium
electrodes looked worn and showed a high degree of pitting corrosion on the
surface, which
was not expected. The electrode surface not immersed in process water during
operation was
not affected by corrosion and was clean.
Runs on lab test unit with graphite and titanium electrodes
The lab unit was used to carry out initial and general tests to observe the
behavior of graphite
and titanium electrodes in the electrolytic process. Test runs with the lab
test unit took about
one hour (excluding final laboratory work). Miniflot, test runs took up to
four hours, due to the
time spent reaching operating equilibrium.
Tests were focused on investigating whether electrolysis process using
graphite or titanium
electrodes could be used to destroy the inhibiting compound(s), thereby
permitting flocking by
direct injection of Fe2+ solution. In addition to this, the following
parameters were investigated:
CA 3027250 2018-12-12
pH value for treatment;
pH trend during treatment;
voltage for treatment;
amperage for treatment; and
distance of electrode separation.
The data was used to calculate the set up for the Miniflot, as well as to
determine main process
parameters, such as pH for treatment. The data gave an initial idea on
reaction time, to permit
an adjustment to flow rate in the Miniflot, as well as on the amount of Fe'
solution for forming
flocks. During tests, no coating on the electrode surface or other unusual
behavior of electrodes
was observed. Tests performed are shown in Table 9.
Test nr Water Electrode Test purpose
treated material
LGr 1 ORF(FC) Reference test
LGr 2 ORF(FC) Graphite Basic test, settings: 20 V, 30 min
LGr 3 ORF(FC) Graphite Like basic test, but only half amperage, 20 V, 30
min
LGr 4 ORF(FC) Graphite Like basic test, but pH changed into caustic
conditions
LGr 5 ORF(FC) Graphite Like basic test, but add Fe-solution before
treatment
LGr 6 ORF(FC) Graphite Like basic test, but 10 V instead of 20 V
(like Miniflot)
LGr 7 ORF(FC) Graphite Like LGr 6, but measuring pH trend
LGr 8 STVV(FC) Graphite Like basic test, treatment of STW (FC)
LGr 9 STW(CL) Graphite Like basic test, treatment of STW (CL)
LGr 10 ORF(CL) Graphite Like basic test, treatment of ORF (CL)
LGr 11 ORF(FC) Graphite Like basic test, production of samples for
laboratory
LTi 1 ORF(FC) Titanium Basic test, settings: 20 V, 30 min
LTi 2 ORF(FC) Titanium Like basic test, but pH changed into caustic
conditions
LTi 3 ORF(FC) Titanium Like basic test, but 10 V instead of 20 V (like
Miniflot)
LTi 4 ORF(FC) Titanium Like 3, but flock test
61
CA 3027250 2018-12-12
LTi 5 ORF(FC) Titanium Like LTi 4, but measuring pH trend
LTi 6 ORF(FC) Titanium Like basic test, to measure reaction time
(Table 9: Tests performed using the Lab Unit)
Runs on Miniflot
The Miniflot plant can run automatically after setting all operating
parameters i.e. flow rate,
pH, dosing rate, etc. In this case the adaption of the system was undertaken
in two steps, due
to the presence of a relatively high amount of H2S. H2S will form FeS in the
presence of Fe2+
immediately, based on a solubility product of 10-19 mo12/12 (The solubility
product of Fe(OH)2
is with 1045 mo13/13, much higher). This FeS forms black virtually
unfilterable flocks. Only in
the presence of Fe(OH)2 can this be filtered. The consequence of this is a
higher consumption
of Fe2+ solution.
First Step: Miniflot was run with the following process steps: Transfer of
feed water using
feeding pump into reactor - pH adjustment chamber (but without pH pre-
adjustment) - gravity
drain, final drain with drain pump into waste container. In the outlet of the
reactor a small
sampling system was installed, allowing samples to be taken directly and
conveniently during
the test run without having to stop the process. The samples were then treated
with different
concentrations of Fe2+, use of polymer, etc.
Second Step: Miniflot was run with the complete process step: Transfer of feed
water using
feed pump into reactor, pH adjustment chamber, dosing of Fe2+ solution, pH
adjustment to 9.5,
drain, and finally, drain pump into the waste container. Samples for
filtration at the laboratory
were taken at the drain.
Tests performed are shown in Table 10.
Test nr Water Electrode Test purpose*
treated material
MGr 1 ORF(FC) Graphite Test full process, 92 l/h, 130 mg/I Fe,
flock tests etc,
MGr 2 ORF(FC) Graphite Reactor only, 92 l/h, tests 125 mg/ - 300 mg/I,
sampling
MGr 3 ORF(FC) Graphite Reactor only, 2311/h, tests 100 mg/ - 225 mg/I,
sampling
MGr 4 ORF(FC) Graphite Reactor only, 3081/h, tests 100 mg/ - 250 mg/I,
polym., sampling
62
CA 3027250 2018-12-12
MGr 5 ORF(FC) Graphite Reactor only, 1541/h, tests 100 mg/ - 250 mg/I,
polym., sampling
MGr 6 ORF(FC) Graphite Reactor only, determination of reaction time
MGr 7 ORF(FC) Graphite Full process, 921/h, 100 mg/I Fe, polym.,
sampling
MGr 8 ORF(FC) Graphite Full process, 3081/h, 130 mg/1 Fe, polym.,
sampling
MGr 9 ORF(FC) Graphite Full process, 921/h, scay., 130 mg/1 Fe,
polym., sampling
MGr10 ORF(FC) Graphite Full process, 92 l/h, scay., 25 - 150 mg/1 Fe,
polym., sampling
MTi 1 ORF(FC) Titanium Reactor only, 921/h, tests 25 mg/ - 175 mg/1
Fe, polym., sampling
MTi 2 ORF(FC) Titanium Reactor only, 60 l/h, tests 50 + 100 nnel
Fe, polym., sampling
MTi 3 ORF(FC) Titanium Reactor only, 60 l/h, scay., tests 50 + 100
mg/1 Fe, polym., sampling
MTi 4 ORF(FC) Titanium Full process, 60 l/h, scay., 50 mg/1 Fe,
polym., sampling
MTi 5 ORF(FC) Titanium Full process, 60 l/h, 50 mg/I Fe, polym.,
sampling
MTi 6 ORF(FC) Titanium Full process, 162 l/h, scay., 50 mg/1 Fe,
polym., sampling
MTi 7 ORF(FC) Titanium Full process,1621/h, 50 mg/1 Fe, polym.,
sampling
* Remark: All concentrations are based on calculations, analytical
data
(Table 10: Tests performed using the Miniflot Unit)
Production of Fe2+ Solution:
Fe2+ solution for step 2 of the Miniflot tests was produced by electrolysis
using the Miniflot
with iron electrodes. Final Fe2+ concentration was measured at between 4 and 5
WI. Carrier
water used to generate the =Fe2+ was BW, which is generally used as technical
water, i.e. for
dilution of chemicals, etc. The process of generating Fe2+ concentrate by this
method was tested
successfully and confirmed in Example I.
Using Miniflot and BW, about 100 1 of Fe2+ solution was generated with a
concentration of
dissolved Fe2+ of about 4 g/1. For lab tests we also used some of the
previously generated Fe2+
solution. This solution had a concentration of 4.5 g Fe2+ /I.
Table 11 shows dosing of Fe2+ solution in Miniflot as a function of flow rate
and concentration.
This data was used to set flow parameters of the Miniflot. For example, at a
flow rate of 1001/h,
and a desired Fe2+ concentration of 100 mg/I, dosing pump may be set to 2.5
1/h.
63
CA 3027250 2018-12-12
flow rate conc. Fe desired Fe amount of
solution conc. Fe sol.
l/h g/I mg/I l/h
60 3.98 100 1.5
60 3.98 125 1.9
60 3.98 150 2.3
60 3.98 175 2.6
60 3.98 200 3.0
100 3.98 100 2.5
100 3.98 125 3.1
100 3.98 150 3.8
100 3.98 175 4.4
100 3.98 200 5.0
150 3.98 100 3.8
150 3.98 125 4.7
150 3.98 150 5.7
150 3.98 175 6.6
150 3.98 200 7.5
200 3.98 100 5.0
200 3.98 125 6.3
200 3.98 150 7.5
200 3.98 175 8.8
200 3.98 200 10.1
(Table 11: Dosing of Fe2+ solution in Miniflot as a function of flow rate and
concentration)
Characteristics of treated and untreated water:
The following SAGD process waters were used:
= Brackish Water (BW): Brackish water was used as carrier for Fe2+ solution
only.
64
CA 3027250 2018-12-12
= Oil Removal Filter Outlet (ORF) water: Main tests were carried out using
ORF Outlet water.
In total, 6 totes were used to operate lab unit and Miniflot test runs.
= Skim Tank Water (STW): About 20 1 of STW were provided for bench scale
tests.
= Skim Tank water (STW) from a second source: About 20 1 of STW from a
second source
were provided for bench scale tests.
= Oil Removal Filter Outlet (ORF) from a second source: About 20 1 of ORF
from a second
source were provided for bench scale tests.
Initial analytical data on input feed waters BW, and ORF and STW from the
first source are
shown in Table 12:
Type pH Con
Ca Mg Fe Hard- Silica TOC DOC Oil+ H2S
of
water duc ness grease
tivity
mS/cm mg/I mg/I mg/I mg/I mg/I mg/I mg/I mg/I
BW 9.87 11.6 <2 <2 <0.1 <10 n.d. <0.05
n.d. <5 n.d.
(9.58) (12.42) (0.24) (0.12) (0.20) (1.11) (10) (8.371) (3.289) (b.d.) (n.d.)
ORF 7.28 2.9 4.6 0.6 <0.02 14 n.d. 500
n.d. 17 30
(7.53) (2.801) (3.0) (0.31) (0.60) (8.75) (260) (646.3) (488.8) (50.5) (n.d.)
STW 7.33 2.95 3.4 <0.4 0.02 8.4 n.d. 476
n.d. 21 n.d.
(7.12) (2.830) (2.89) (0.38) (0.20) (8.79) (265) (488.3) (432.9) (29.5) (n.d.)
Remarks: n.d.: not detected .. b.d.: below detection
limit
(Table 12: Analytical data of selected untreated waters)
Reference Testing:
As described in the Examples above, no flocking was observed in STW after
addition of Fe2+
CA 3027250 2018-12-12
solution and pH adjustment. Color of the treated water after addition of Fe"
solution turned
red-black, and no flocking was observed. On the other hand, tests using
electro-flotation with
iron electrodes achieved separation by formation of flocks. For base reference
and prior to the
scheduled tests and trials, the following two reference tests were performed:
1. After taking a small sample of ORF into a beaker, Fe" solution was added to
a concentration
of about 150 mg/1 and the solution was adjusted to a pH of 9.5 using NaOH. The
water turned
immediately black and no flocks were observed. This supports previous Example
results.
The same feed water was treated on the following day with the lab unit, but
this time the feed
water was treated with graphite electrodes first and Fe' solution was added
after treatment.
Flocking and precipitation was observed immediately. This test showed that
electroreduction
treatment with non-consumptive graphite electrodes and Fe' solution functions
to
destroy/inactivate/remove the inhibitory compound(s) which otherwise prohibit
formation of
flocks.
2. After production of Fe" solution, the Miniflot was cleaned and the plant
was operated with
Fe electrodes to treat ORF. This reference test indicated that previous
results could be
replicated with the feed water used in the subsequent testing of this Example.
Flocculation Testing using the Lab Unit and ORF Outlet Water
Tests were carried out to observe the general behavior of graphite electrodes,
and the ability to
create flocks using these electrodes after addition of Fe" solutions.
Tests with Graphite electrodes:
Basic parameters were defined as dependent on a relationship between amperage
and voltage,
electrode separation, and conductivity. These values, and the calculated
specific amperage in
mA/cm2, were used to calculate the electrode set-up. The dependency for ORF
outlet with a
conductivity of 2.8 mS/cm is given in Figure 19. Miniflot normally operates at
a maximum
voltage of 10 V. At this voltage, the separation of electrodes should be such
as to permit total
amperage of about 100 A. Using 10 V values and an active area of 414 cm2 in
the lab unit, the
separation of 10 electrodes in the Miniflot was calculated to be about 25 mm
to reach 100 A.
66
CA 3027250 2018-12-12
Later test showed operating values between 96 and 100 A, which was consistent
and supports
the calculation.
Tests with treatment time or residence time as a variable:
The lab unit was operated at 10 V and about 4.5 A for a period of
approximately 30 minutes.
Samples were taken after 15 and 30 minutes. These samples were then injected
with Fe2+
solution of 150 mg Fe2+/1 and pH adjusted. In both cases, good flocks were
observed. This
suggests that reaction time may be less than 15 min in these studies. Using
the Miniflot,
reaction time to destroy the inhibiting compound(s) was observed to be
relatively fast, i.e. less
than one minute with graphite electrodes.
Tests with pH as a variable:
Two different test runs were carried out with the lab unit to investigate the
pH parameter. In
the first run the behavior of the process with different pH-values with ORF
outlet water was
tested before treatment (pH 6.4 ¨ as delivered- and adjusted to 8.5). In the
second test the trend
of the pH-values during treatment was measured. Every minute a small sample
was taken for
measurement. The first runs with two different feed pH values showed no
observable
difference. Good flocking was observed with both runs and, as a result of
this, ORF outlet water
was treated without any pH adjustment in the feed. Trend of pH during
treatment of ORF outlet
water is shown in Figure 20 (pH of ORF changed during storage period from 6.4
to 7.3).
Values from test runs indicated three different phases during reaction:
Treatment time up to 2
mins (first degradation); Treatment time of 3-4 mins operating time (second
degradation or
result of first degradation); and Treatment time after 4 mins (typical
behavior of water
electrolysis reaction). These results suggest that an initial reaction takes
place within the first
2 minutes and another reaction after 3 ¨4 minutes.
At the end of these tests, and after Fe' addition and pH after-adjustment,
good flocks were
observed.
Tests with voltage as a variable:
Most tests were run with 20 V to apply a high amperage at this operating
voltage. In addition,
tests with 10 V were carried out to confirm treatment possibilities using
Miniflot (10 V, as a
67
CA 3027250 2018-12-12
dependency of voltage in treatment of organics could not be excluded). Figure
21 shows the
dependency of amperage to change in voltage in these studies. 10 V values were
used to
calculate Miniflot operation. The data suggests that low amperage is
sufficient to destroy the
inhibiting compound(s). All tests showed good flock development and
precipitation.
Flocculation Testing using the Miniflot and ORE Outlet Water
Lab unit tests suggested an initial Miniflot electrode separation of 25mm.
Runs were
undertaken without pH pre-adjustment, even if pH of stored waters showed
changed over time.
Miniflot tests could run at flow rates between 60 1/h up to 2001/h. Earlier
lab tests suggested a
fast reaction time, and hence a low residence time in the reactor. Initial
Miniflot tests were
conducted using only the reactor, without dosing with Fe" solution and without
pH after-
adjustment. Sampling occurred directly at the reactor outlet. Samples were
then tested for
flocking using different amounts of Fe' solution (representing different Fe'
concentrations in
the processed water).
After having identified an optimal Fe" concentrations for the Miniflot, a
complete
demonstration run of the entire process was undertaken with both graphite and
titanium
electrodes, Fe" solution dosing and pH after-adjustment. Samples were taken at
the drain of
the flock basin.
Tests with titanium and graphite electrodes:
Graphite and titanium runs were performed to compare the behavior of each of
the electrodes,
their comparative ability to destroy the inhibiting compound(s), the cost, and
overall relative
advantages and disadvantages of using graphite and titanium.
Tests with treatment time or residence time as a variable:
Reaction time using graphite electrodes was less than one minute. Taking this
into account,
when running the lab unit with titanium electrodes, operation was initially
for two minutes. At
the end of the first minute, the first sample was taken, and at the end of the
second minute the
second sample was taken. Voltage was 20 V, amperage was about 17 A in the
first minute, 10
A during the second minute (reduced amperage was caused by a reduced fluid
level in lab unit
after removal of the first sample). The first sample was then injected with
Fe" solution and
pH adjusted. The sample turned black and showed no flocks. The second sample,
after similar
68
CA 3027250 2018-12-12
treatment, showed good flocks and good precipitation. These results indicated
that titanium
electrodes had a reaction time somewhat higher than graphite electrodes.
Tests with pH as a variable:
The trend of pH-values during treatment with titanium electrodes was also
measured. Every
minute a small sample was taken for pH measurement. Figure 22 shows the trend
in change of
pH over treatment time for titanium electrodes and, for comparison, the trend
for graphite
electrodes. Titanium and graphite electrodes showed completely different
behavior when
treating ORF water. There was no indication of several reactions at the
electrode surface with
titanium electrodes. On the other hand, pH trend with graphite electrodes does
show several
reactions, i.e. see minute 1-3, then minute 4-5, and finally more normal
behavior after minute
5. During the tests with titanium electrodes, the surface of the +ve electrode
became coated,
and some suspended solids in the water after treatment were also observed.
Tests with voltage as a variable:
Identical tests with titanium electrodes were also carried out using graphite.
Figure 23 shows
the curves for titanium at different voltages. The behavior is quite different
for titanium, when
compared to graphite. The curves show a solution with similar conductivity
after 30 to 40
minutes of treatment, which is unusual, as voltage and amperage were both
lower in the 10 V
test. This behavior might be caused by a coating on the surface of the
titanium electrodes, which
was observed. Some yellow to brown suspended solids were also observed in the
treated water.
This might be due to formation of TiS on the surface of the electrodes.
Based on the results of these tests as set out above, operational parameters
were selected for
subsequent water treatment runs. It was initially assumed that the Miniflot
runs would use 10
titanium electrodes, each 2mm thick. Due to the dimension of the reactor tank
of the Miniflot
the minimum electrode separation possible was 48 mm, which may result in
reduced amperage
during our tests. Tests were run without pH pre-adjustment, even in situations
where the pH of
the stored waters changed overtime. Tests could run at a flow rate of 601/h up
to 2001/h. Most
of the tests were conducted at lower flow rates. This meant that the specific
amperage of low
flow rate tests with titanium was comparable to higher flow rate tests with
graphite. Lab tests
indicated a relatively fast reaction and, hence, low residence time in the
reactor was still
sufficient.
69
CA 3027250 2018-12-12
Production of Fe2+ Solution:
A sufficient volume of Fe2+ solution was prepared for use in testing. This
solution was to be
injected into process waters pre-treated with either graphite or titanium
electrodes, to induce
flocking.
The Miniflot was used to generate a volume of Fe2+ solution in a similar setup
as was used in
previous examples, with BW used as carrier. The plant was operated in a closed
loop for about
four days using the following setup: Buffer volume ¨ feed pump pH pre-
adjustment with dosing
of HC1 - reactor ¨ overflow to drain ¨ connection drain to buffer tank.
At the end of each days' run, a sample was taken for lab analysis to test the
concentration of
dissolved iron in the solution. The pH in the pre-adjustment stage was set to
4, reactor outlet
was about 5.8 to 6. Increase of Fe2+ concentration in BW over time is shown in
Figure 24.
After completing the Fe2+ solution production run, the total volume of the
system was drained,
mixed and filled in a drum for storage. Characteristic and compositional data
of the solution in
storage is shown in Table 13.
Type of Water Diss. iron pH Conductivity
mg/I mS/cm
Fe' solution (BW 3,916 4.34 24.13
Carrier)
(Table 13: Characteristic Data of Fe2+ Solution)
Before using the concentrated Fe2+ solution, it was filtered. The quantity of
suspended solids
captured as filter sediment was not measured. Later analytics of the Fe2+
solution (carried out
7, 11 and 12 days after unfiltered storage) showed values for dissolved Fe2+
of 5,983, 6,020
and 6,100 mg/l. These may be equilibrium values based on the changing pH of
stored unfiltered
solution. The Fe' solution was used in most of the tests marked MGr and MTi
below.
Test Runs on ORF using Miniflot
Test runs with Miniflot were carried out in 3 steps. Some additional
supplementary testing was
undertaken between these three steps.
CA 3027250 2018-12-12
The first step involved the observation of flock formation with graphite
electrodes, including
the observation of flock behavior and precipitation after treatment only. In
all cases flocks were
observed with the graphite electrode tests. Concentration of Fe2+ injected
after treatment was
varied, and in addition to this, some tests were undertaken with the addition
of polymer.
In the second step, a small quantity of H2S scavenger was also added, and
results compared to
runs without the use of this scavenger. The results were equally valid for
graphite and for
titanium electrodes. Studies to investigate the reaction velocity to destroy
the organic complex
were also performed.
In the third step, performance test runs with the Miniflot were performed. In
these test runs,
parameters were changed one by one and samples were taken for internal and
external lab
analysis to determine the level of reduction of silica, hardness, TOC, DOC and
oil & grease
after treatment. Tests performed with the Miniflot are summarized in Table 8
and Table 9
above.
Floc tests with graphite electrodes:
Initial tests were carried out with Fe2+ concentrations between 125 and 300
mg/I without
addition of polymer and without H2S scavenger. At a concentration of 125 mg
Fe2-71, no flocks
were observed or precipitated, but the first precipitation was observed at 150
mg/l. Increased
concentrations provided better flocculation, and at higher concentration
flocks were seen to
float, as they were large enough to adsorb micro bubbles of gas.
The sample with the addition of 150 mg Fe2 /I with and without polymer
addition was
compared. The polymer used was Bulab 590. Polymer addition was not optimized,
but even
still polymer addition was advantageous (better flocculation and separation).
Further flock tests at lower Fe2+ concentrations showed the process water
turning black with
extremely small flocks. This suggested the formation of FeS, before Fe(OH)2
can form.
(Solubility product for FeS is much lower than for Fe(OH)2). Use of H2S
scavenger, i.e. to
remove the S2-' thereby reducing Fe2+ consumption, was therefore tested. These
tests were
mostly carried out using titanium electrodes in the Miniflot, but results were
also confirmed
using the lab unit with graphite electrodes.
Precipitation of Fe(OH)2 flocks, starting with a concentration of 25 mg/1 Fe2+
up to 175 mg/1
71
CA 3027250 2018-12-12
in incremental steps of 25 mg/1 was tested. After removal of sulfide (and
removal of some
mercaptans), flocculation and precipitation became easier and Fe2+ consumption
was
drastically reduced. Accordingly, results suggested that:
H2S scavenger combined with sulfide to form a fine yellow insoluble product,
which
may be taken out easily by Fe(OH)2 flocks.
Scavenger may be used where desirable, for example when economics of higher
iron
consumption versus amount of scavenger used is favorable.
The H2S scavenger used in these studies was called "StaSweet 6000" from CFR
Chemicals.
During tests, a concentration of 0.15 1/m3 was used.
.. Determination of reaction time:
Miniflot reactor was filled with ORF to the top. A simple system for taking
samples out of the
reactor was installed. Rectifier was turned on to 10 V and ran at 100 A and
every minute a
sample of 150 ml was taken out of the reactor for flocking tests. Fe2+
solution was added to the
samples at constant concentration of 100 mg/1 Fe2+ and pH was adjusted to 9.5.
Good flocking,
.. precipitation of flocks, and a clear supernatant was observed with all
samples including the one
taken after only one minute. Samples up to a reaction time of 5 min were
tested and showed
similar results. Results indicate that reaction time was short, i.e. less than
one minute.
Floc tests with titanium electrodes:
Initial tests were carried out with the Miniflot, using titanium electrodes
and ORF outlet water
with Pe2+ concentrations between 125 and 300 mg/1 without the addition of
either polymer or
H2S scavenger. The Miniflot was drained in the third chamber for sampling and
samples were
then taken at the drain. These samples were treated at the laboratory bench as
follows:
Using 5 to 7 single beakers, adding different amounts of Fe' solution for
different iron
concentrations, pH adjustment to 9.5 and observation for flocculation. In
other tests, some
polymer was also added, and/or some H2S scavenger.
Post treatment with titanium electrodes, iron concentrations of 75 to 200 mg/I
without the
addition of polymer or H2S scavenger (and with pH adjustment) were compared
with the same
iron concentrations and conditions, but with the addition of a few drops of
H2S scavenger.
72
CA 3027250 2018-12-12
Better flocculation and separation was observed with the scavenger, but flocs
were observed
in both conditions.
In general, it was observed that after treating ORF outlet water in the
reactor with graphite or
titanium electrodes, and with the addition of Fe" solution, good flocking was
observed in all
cases. Sedimentation of flocks was also always very good.
Flock Tests Using Standard FeSO4 Solution
Traditional electro-flotation is based on an electro-chemical reaction that
makes use of
electrons supplied by sacrificial metal electrodes. It also involves reactions
associated with the
dissolution of these electrodes. In a standard electro-flotation reactor, iron
is normally the
sacrificial or consumed electrode material used.
In the present tests, non-sacrificial (non-consumptive) graphite and titanium
electrodes and
electrolysis were used to supply the feed water with electrons, to destroy
inhibitory
compound(s) which have now been discovered to prevent the formation of Fe(OH)2
flocks.
The actual reactive material, a concentrated Fe" solution, was then produced
by electrolytic
dissolution in a side reaction using the Miniflot EF reactor. After a pre-
treatment of the feed
water with graphite or titanium electrodes to destroy the inhibitory
compound(s), the pre-
treated feed water was dosed with the Fe' concentrate and flocking was
observed. Previous
examples described above used Fe" solutions prepared by electrolytic
dissolution. The
following studies sought to determine whether commercially available Fe'
solutions could
instead be used, such as those prepared by dissolving iron salts.
Studies were performed in which the iron ion-enriched aqueous solution was
prepared by
alternative methods. Specifically, pre-made iron ion-enriched aqueous
solutions made by
dissolution of iron salts in water were tested, which did not require use of
electro-flocculation,
and instead utilize the iron ion-enriched aqueous solution as a
straightforward chemical reagent
or additive that can be slipstreamed into the produced water.
To investigate this, an FeSO4 solution was prepared with a concentration of 5g
Fell using
commercially available chemicals, and used for direct injection in place of
electrolytically
produced Fe"- (i.e. FeCl2) solution. FeSO4 was chosen, as it is used in modern
waste water
treatment systems as flocculant and to remove phosphate out of biological
waste water. It is a
73
CA 3027250 2018-12-12
by-product of titanium production, and is widely available at reasonable cost,
both as a solution
or also as solid salt.
flocculation tests with ORF outlet water pre-treated with graphite or titanium
electrodes
showed the same flocking behavior when injected with the FeSO4 solution as
when injected
with Fe2+ solution produced electrolytically in the Miniflot. The treatment
results were similar
and within the range of tolerance. No difference could be observed or measured
using Fe2+
solution produced by electrolytic dissolution of iron to form a FeCl2 solution
or by using a
FeSat solution prepared by dissolving FeSO4 salt.
Tests described above examined treatment of ORF outlet water as the
contaminated water.
Testing of three other contaminated water samples, namely two different Skim
Tank Water
(STW) samples and a second ORF outlet water, was also performed. In general,
the behavior
of these three process waters was almost identical to the behavior observed
with ORF outlet
water above. Flocking was about the same and all treatment standards were met.
Use of Polymer and H2S Scavenger
Some of the above testing included the addition of polymer and H2S scavenger
with the
injection of Fe2+ solution after pre-treatment with either graphite or
titanium electrodes. The
following observations on the effect of adding polymer and H2S scavenger were
made:
to 30 mg/I Fe2+ was sufficient to eliminate sulfide present in the water.
Black
insoluble FeS was formed in the process.
20 An
additional 25 to 50 mg/1 Fe2+ was sufficient to separate hardness and silica
by
reaction with Fe' or by absorption in the formed Fe(OH)2. Green flocks were
formed
and they exhibited good settling behavior.
With the addition of a small quantity of polymer during flock formation, it
was observed
that flocks tended to be bigger and that they tended to settle easier. Polymer
Bulab 5901
25 was used
in these studies. Although polymer use was not optimized, the amount applied
was about 0.1%. The application of H2S scavenger reduced the amount of Fe2+
solution
suitable for treatment drastically by about 25 ¨30 mg/l. H25 scavenger was
added prior
to the water treatment process directly into the feed stream. The scavenger
reacted with
the sulfide to form an insoluble organic product, which was then separated in
the
74
CA 3027250 2018-12-12
flocking step without difficulty or any additional step. Type and amount of
scavenger
was not optimized. In these tests, a product called "StaSweet 6000" supplied
by CFR
Chemicals Inc. was used. The concentration used was 0.15 1 scavenger/m3.
Analytical Results
Behavior of two ORF outlet waters and two STW waters was very similar and
independent of
the type of electrode used in pre-treatment. Final treatment results were also
very similar.
Treatment results were the same for tests run with the Miniflot and the lab
test unit.
Collected analytical data is shown in Table 14, Table 15, Table 16, and Table
17 below.
test-nr. Polymer I12S results
scaveng.
Fe conc. In lab Ca Mg hardn. Silica TOC DOC
o+g di ss. Fe
test
mg/I mg/I mg/I mg/I mg/I mg/I mg/1 ing/1 mg/I
I_Gr 8 no no 100 4.4; <2.0; 11; 9.6 8.8;
3.04 0.49 25
LGr9 no no 100 <3,0; <2.0; <5.0; 5.9;
2,43 0.22 6.99 35
LGrIO no no 100 3.9; <2.0; 9.7; 15*
*silica in feed 150 mg/i
3.27 0.33 9.54
LGr11.1 yes yes 25 2.02; 0.37; 6.59; 85;
<3.0 <2.0 40.5 82
LGr11.2 yes yes 50 7.3; 3.4; 33; 52;
6.24 2.92 27.61 SO
LGr11.3 yes yes 75 5.71; 2.83; 25.9; 31 60;
6.8 3.3 53
(Table 14: Analytical Results for Lab Unit, Graphite Electrodes)
test-nr.Polyinel 112.5 Fe conc. In results
scaveng lab test
Ca Mg hardn. Silica TOC
DOC (Hz ,diss. Fe
mg/I mg/I mg/I mg/I mg/I mg/I mg/I mg/I
LTI 1 no no 100 2.29 0.77 6.8 <1
(Table 15: Analytical Results for Lab Unit, Titanium Electrodes)
CA 3027250 2018-12-12
Test-nr.Polymel H2S Lab Results
scaveng conditions
Fe conc. Ca Mg hardn. Silica TOC
DOC ois diss. Fe
mg/I mg/I mg/I nigil rndi
MGr 1 no no - -
MGr 2 no no 250 1.01 0.13 3.06 40
MGr 3 no no 125 - -
MGr 4 no no 150 2.29 0.27 6.83 <1
MGr 5 no no 150 3 0.29 8.69 <30
MGr 6
MGr 7 yes no 100 14.5; 0.535;
38.4; <1, 30 300.4; 243.4 16_5; 1.3
10.8 0.29 28.4 280.9 12.0
MGr 8 yes no 130 5.0; 0.4; 14.1;
10,0; 294.4; 254.2; 14.4; 0.35
4.1 0.55 12 9,3 230 230 13
MGr 9 yes yes 100
MGr 10 yes yes 130 2.9; 0.14; 7.8; 45;
7.2 <2.0 19 35
(Table 116: Analytical Results for Miniflot, Graphite Electrodes)
76
CA 3027250 2018-12-12
Test-nrPolymet I-12S Lab Results
scaveng Conditions
Fe conc. Ca Mg hardn. Silica TOC DOC
ol-g
mgil rne mg/I nigji rngil mg/I
MTi 1 yes no 150 0.94; 0.27; 3.46; 12;
<3.0 <2.0 <0.5 20
MTi 2.1 yes no 50 0.22; <d-l; a54; 40;
<3.0 <2.0 <0.5 40
Mit 2.2 yes no 100 0.31; 0.17; 1.48; 25;
<3.0 <2.0 <0.50 20
MTi 3.1 yes yes 50 0.26; 0.2; 1.48; 14;
<3.0 <2.0 <0.50 20
MTi 3.2 yes yes 100 2.83; 0.37; .. 8.6; .. 5.7;
<3.0 <2.0 <0.5 0
MTi 4.1 yes yes 130 2.18; 0.22; 6.33; 15;
<3.0 <2.0 <0.50 13
MTi 4.2 yes yes 130 2.36; 0.16; 6.55; 1.9;
<3.0 <2.0 <0.50 10
Mu i 5 yes no 100 3.33; a 33; 9.69; 20;
4.1 <2.0 10 20
MTi 6.1 yes yes 50 4.22; 0.43; 12.33; 70;
4.7 <2.0 12 65
Mu i 6.2 yes yes 100 4.12; a 44; 12.11; 45;
4.9 <2.0 12 39
MTi 7.1 yes no 50 3.67; 0.46; 11.04; 80; .. 387.1
303.6 15.28
4 <2.0 10 73
Mit 7.2 yes no 100 2.91; 0.28; 8.43; 50,
3.8 <2.0 9.6 46
(Table 17: Analytical Results for Miniflot, Titanium Electrodes)
These data show, that independent of any geometric or material factors,
treatment targets were
met in nearly all cases.
Hardness:
Hardness is caused by the presence of calcium and magnesium ions, and the
calculated
combined hardness value is represented as carbonate. Target is 15 mg/l. Figure
25 shows data
measured during testing, independent of the type of electrode used in pre-
treatment, i.e.
whether graphite or titanium. In almost all cases the target was met.
77
CA 3027250 2018-12-12
Figure 25 shows consistent and near constant values for magnesium content, and
a variable and
changing concentrations of calcium. Due to these high calcium values, high
values for
hardness were calculated. It was considered that this may be caused by some
error in
measurement, or by cross contamination with drinking water. Real values for
calcium are
expected to be around 2-3 mg/I with a total hardness of 7-8 mg/l. This higher
than expected
concentration of Ca measured may be a result of cross contamination from
residual Ca(OH)2
in the Miniflot from earlier test, where we used lime slurry instead of NaOH
for pH adjustment.
Silica:
Target for active Silica is 50 mg/1. In general, this target was met with
average values for silica
of about 18 ¨ 20 mg/I. Only two tests using titanium electrodes, in
combination with higher
flow rate, showed values higher than 50 mg/1¨ see Figure 26.
Summary
Experiments herein tested whether a pre-treatment of main stream SAGD process
contaminated
water by electrolytic reduction using non-consumptive electrodes made of
graphite or titanium
could destroy inhibitory compound(s), and then permit use of a side-stream of
Fe2+ solution by
direct injection for flocking.
Non-consumable electrodes made of graphite or titanium were used. Two sources
for Fe2+ for
flocking purposes were also used, the first being a Fe2+ solution produced by
electrolytic
dissolution of iron and the second by using a Fe(H)504 solution created by
dissolving a
commercially available chemical reagent in water. In all cases, whether with
the lab test unit
or the Miniflot, whether with graphite or with titanium electrodes, good
flocking was observed
after the addition of Fe2+ solution from either source and after adjustment of
the pH to 9.5.
Precipitation time of flocks could be reduced further by adding a small amount
of polymer.
Reaction time to destroy inhibitory compound(s) which otherwise prevented the
formation of
Fe(OH)2 flocks was very short, i.e. within 1 minute with graphite,
approximately 2 minutes
with titanium electrodes. This means that an electrolytic reactor using these
non-consumable
electrodes may have a relatively small reaction volume, i.e. 5 to 10 m3 for an
industrial size of
plant, and still can pre-treat a relatively large volume, i.e. 200m3/hr, for
example. Electrical
power consumption may be less than 1/10 of the power consumption required for
standard
78
CA 3027250 2018-12-12
electro-flotation.
Both types of electrodes (graphite and titanium) performed the task of
electrolytic reduction of
the above-mentioned inhibitory compound(s). Graphite as an inert, but
conductive material,
showed absolutely no side reaction, no corrosion, no coating etc. After the
tests the electrodes
looked brand new. Electric behavior of the electrodes was constant over the
testing cycle
period. Other advantages of graphite over titanium included lower material
costs, faster
reaction time, which for graphite was less than one minute, or half of the
reaction time with
titanium. An additional advantage was the simple and easier machining and
fabrication of this
material.
Titanium, or more specifically, the alloy which was used, did not perform as
well as graphite,
but was certainly still functional. One relative disadvantage was the observed
corrosion and
pitting on the surface of titanium electrodes. In addition, a coating was
observed on some
electrodes, as well as electrical behavior that was not as constant or stable
as observed with
graphite under the conditions tested.
The physical-chemical behavior of Fe2+ ions was independent of the type of
"production" of
the iron solution. The solubility curves of Fe(OH)2 as a function of the pH
value are based on
the Fe2+ concentration, and no changes were observed whether the iron solution
was produced
electrolytically or as by-product in other metal production, for example.
Readily available
Fe(II)SO4 is used in waste water plants as a reagent to remove phosphate, as
fertilizer, and as
reduction agent in the cement industry. It is called iron vitriol or green
salt. The sulfate anion
does not change the process water composition, as sulfate is already found in
the process water.
This product is available as crystalline salt or as solution in concentrations
up to several
hundred g/1. Different Fe concentrations generally did not affect the flocking
mechanism.
The ability of a commercially available H2S scavenger to reduce Fe2+
consumption was also
tested (i.e. when dosing Fe2+ solution, initially FeS is formed, if sulfide or
mercaptans are
present in the water to be treated). H2S scavengers are organic chemicals that
can react quickly
with sulfides and mercaptans and form an insoluble compound. It was
hypothesized that this
could then reduce the amount of Fe2+ needed for flocking and for treatment.
Studies confirmed
that H25 scavenger did indeed work under these process conditions, and that
iron consumption
was reduced to about half of the amount needed without the use of a scavenger.
79
CA 3027250 2018-12-12
The above experiments successfully demonstrated that application of
electrolysis (for example,
electroreduction) using non-consumptive electrodes, followed by direct
injection of Fe"
solution to induce flocculation, treated SAGD water to suitable standards.
Appropriate
reductions in hardness and silica were obtained, and standards for reduction
of suspended
solids, oil and grease and reduction of TOC and DOC were also reached.
It was identified that an initial step of electroreduction could be performed
on the produced
water, which would destroy or inactivate the inhibiting contaminant(s) and
restore flocculation-
based contaminant removal. An initial electroreduction step may be introduced
prior to
injection of the iron ion-enriched solution (produced, in this example, from
electro-flocculation
at ambient temperature). A pH adjustment step may also be introduced following
introduction
of the iron ion-enriched aqueous solution, which further enhanced flocculation
effectiveness.
Overall, these experiments demonstrated that electrolysis could be applied
using non-
consumptive electrodes, followed by direct injection of separately prepared
Fe' solution, to
induce flocculation and thereby treat contaminated water such as, for example,
produced water
from SAGD. Both graphite and titanium were used as non-consumptive electrode
materials.
In general, both materials are chemically resistant and conductive. During
tests, graphite
performed better, as it showed no appreciable signs of corrosion or coating
during process
testing and no appreciable change in its behavior. In other tests, it was
shown that, in addition
to using Fe" solution made by electrolysis using the EF reactor (i.e. as per
Example 1), Fe"
solutions available commercially, or produced by dissolving Fe' salts, could
also be used.
EXAMPLE 4B ¨ Treating Produced Water at Elevated Temperature by
Electroreduction, Followed by Injection of an Iron Ion-Enriched Aqueous
Solution in a
Modified Miniflot
Tests were performed to further investigate the electroreduction and
flocculating ion-enriched
aqueous solution injection approaches set out in Example 4A at elevated
temperatures. In
particular, tests were performed at temperatures at or near about 80 C to
further investigate
the abrogation of contaminant inhibition by electroreduction and the treatment
of SAGD
produced waters with flocculating ion-enriched aqueous solutions.
The tests used a modified Miniflot unit based on the one described in Example
4A. The
modified Miniflot unit comprised four chambers. The first chamber (typically
configured for a
CA 3027250 2018-12-12
pH pre-adjustment) was configured for optional injection of an H2S scavenger.
The second
chamber was configured as an electrolytic reactor in a similar manner to that
described in
Example 4A with a series of ten non-consumptive graphite electrodes. The third
chamber was
configured for injection of the Fe2+ solution in a similar manner to that
described in Example
4A. The third chamber was also configured for pH adjustment and polymer
injection. The
fourth chamber was configured for sample collection. The tests involved
treating the produced
water in a process comprising the following steps: (i) optionally subjecting
the produced water
to an H2S scavenger; (ii) subjecting the produced water to reducing conditions
induced through
the graphite electrodes; (iii) introducing a flocculating ion-enriched aqueous
solution, NaOH
(for pH adjustment), and a flocculation promoting polymer into the produced
water; and (iv)
removing flocks via manual filtration using fluted filter papers. In
particular, the tests were
completed as set out in Table 18.
Test-nr. Test water Flow rate H2S Fe - concentration
scavenger
l/h mg/I
M1 ORF out 150 no 150
M2 ORF out 100 no 150
M3 ORF out 100 no 300
M4 ORF out 100 no 250, 275, 300
M5 ORF out 60 no 275
M6 ORF out 100 yes 175, 200, 300
M7 ORF out 100 yes 100, 125, 150
M8/8a ORF out 100 no 150, 175, 200, 225, 250, 275, 300
M9 ORF out 100 no 175, 200, 225, 275
M10/10a ST Inlet 100 no 200, 225, 250, 275, 300, 325, 350,
400
(Table 18: tests performed using the modified Miniflot unit ¨ "ORF out" refers
to an oil
removal filter outlet, and "ST Inlet" refers to a skim tank inlet)
In the tests of Table 18, the graphite electrodes were operated at 100 A with
voltages varying
based on conductivity. In instances where an H2S scavenger was used, the
scavenger was
injected to provide a scavenger concentration of 100 ppm. The pH was adjusted
to about 9.5
with NaOH. The polymer was injected to provide a polymer concentration of
about 100 mg/l.
Analytical data from archetypal tests of Table 18 are provided in Table 19A
and Table 19B
and relevant trends are plotted in Figures 27-30.
81
CA 3027250 2018-12-12
Feed water
nr. Test Fe Si02 Ca Mg hardness TSS 01W Fe Fe
nr. conc.
total co II. react. ICP Hach
mg/I mg/I mg/I mg/I mg/I mg/I mg/I mg/I mg/I mg/I mg/I
1 M7 100 278 42,5 235,5 3,54 0,37 10,34 8 0,1 0,09 1,23
2 M7 125 278 42,5 235,5 3,54 0,37 10,34 8 0,1 0,09 1,23
3 M 125 291,8 40,8 251 3,55 0,36 10,34 6 9,4
0,16 1,18
4 M7 150 278 42,5 235,5 3,54 0,37 10,34 8 0,1 0,09 1,23
M 150 291,8 40,8 251 3,55 0,36 10,34 6 9,4 0,16
1,18
6 M 150 291,8 40,8 251 3,55 0,36 10,34 6 9,4 0,16 1,18
7 M 175 291,8 40,8 251 3,55 0,36 10,34 6 9,4
0,16 1,18
8 M 175 291,8 40,8 251 3,55 0,36 10,34 6 9,4
0,16 1,18
9 M9 175 344,5 72,5 272 3,78 0,39 11,1 11 - 1,77
M6 200 276 30 246 3,39 0,36 - 9,92 5 0,5 0,121
1,2
11 M8 200 276 - 283,5(?) 3,32 0,34 10 10 0,1 0,1 2
12 M9 200 344,5 72,5 272 3,78 0,39 11,1 11 - 1,77
13 M 200 323,4 74 249,4 4,14 0,53 12,53 149 98,2 0,09 2,67
14 M 225 291,8 40,8 251 3,55 0,36 10,34 6 9,4
0,16 1,18
M 225 291,8 40,8 251 3,55 0,36 10,34 6 9,4 0,16
1,18
16 M9 225 344,5 72,5 272 3,78 0,39 11,1 11 - 1,77
17 M 225 323,4 74 249,4 4,14 0,53 12,53 149 98,2 0,09 2,67
18 M4 250 288 98 190 3,48 0,38 10,3 6,5 1,3
0,097 1,59
19 M8 250 276 - 283,5(?) 3,32 0,34 10 10 0,1 0,1 2
M8 250 276 - 283,5(?) 3,32 0,34 10 10 0,1 0,1 2
21 9 250 344,5 72,5 272 3,78 0,39 11,1 11 - 1,77
22 M 250 323,4 74 249,4 4,14 0,53 12,53 149 98,2 0,09 2,67
23 M3 275 288 57 250
3,57 0,36 10,4 2068 495 0,126 0,67
24 M6 275 276 30 246 3,39 0,36 9,92 5
0,5 0,121 1,2
M8 275 276 - 283,5(?) 3,32 0,34 10 10 0,1 0,1 2
26 M9 275 344,5 72,5 272 3,78 0,39 11,1 11 - 1,77
27 M 275 324,9 255,5 4,14 0,53 12,53 149
376,5 0,089 -
28 M3 300 307,4 57,4 250 3,57 0,36 10,42 2068 495 0,16 1,18
29 M6 300 276 30 246 3,39 0,36 9,92 5
0,5 0,121 1,2
M8 300 276 - 283,5(?) 3,32 0,34 10 10 0,1 0,1 2
31 M 300 324,9 69,4 255,5 4,14 0,53 12,53 149
376,5 0,089 -
32 M 325 324,9 69,4 255,5 4,14 0,53 12,53 149
376,5 0,089 -
82
CA 3027250 2018-12-12
33 M 350 324,9 69,4 255,5 4,14 0,53 12,53 149 376,5 0,089 -
34 M 400 323,4 74 249,4 4,14 0,53 12,53 149 98,2 0,09 2,67
(Table 19A: analytical data from tests performed on untreated waters)
Treated water
nr. Test Fe S102 Ca mg Hard TSS 01W PH Fe Fe
nr. conc. -ness out
total co II. react
total solu
ble
mg/I mg/I mg/I mg/I mg/I mg/I mg/I mg/I mg/I mg/I
mg/I
1 M7 100 161 52,6 108,4 2,27 0,2 6,51 4 0 9,62 37,99 2,3
2 M7 125 139 81 58 2,33 0,22 6,71 18 0,1 9,74 36,12 4,3
3 M8a 125 140,1 29,4 110,7 2,58 0,25 7,49 16 0 9,63 27,52 0,65
4 M7 150 113 32 81 1,93 0,15 5,45 6 0,1 9,66 34 5,57
M8a 150 131,6 48,6 83,2 1,90 0,23 5,69 10 0 9,75 41,3 7,85
6 M8a 150 128,0 41,5 86,5 2,24 0,17 6,28 20 0 9,73 43,9 7,25
7 M8a 175 144,60 67,50 77,1 2,27 0,30 6,92 14 0 9,75 60,48 6,7
8 M8a 175 125,50 42,70 82,8 2,07 0,13 5,7 20 0 9,75 35,79 10,2
9 M9 175 132,30 58,50 73,8 1,75 0,22 5,26 18 0 9,78 43,54 2,84
M6 200 96,23 9,30 86,9 1,83 0,1 4,97 15 0 9,72 30,48 6,88
11 M8 200 131,2 77,0 54,2 2,71 0,23 7,72 22 0 9,12 80 0,65
12 M9 200 80,6 28,3 52,3 2,17 0,13 5,97 27 0 9,79 40,16 4,09
13 M10a 200 137,70 72,00 65,7 3,02 0,26 8,61 49 0 9,64 55,7 -
14 M8a 225 67 10,9 56,1 1,33 0,07 4,13 6 0 9,71 9,13 2,48
M8a 225 83,95 25,5 58,5 2,12 0,12 5,78 6 0 9,7 32,5 6,1
16 M9 225 63,6 13,9 50,3 1,72 0,1 4,69 15 0 9,65 12,22 6,97
17 M10a 225 120,7 63,2 57,5 2,18 0,26 6,53 46 0 9,69 67,57 -
18 M4 250 53,6
19 M8 250 72 29,5 42,5 1,97 0,12 5,41 8 0 9,66 43,3 2,1
M8 250 58 18,3 39,7 1,86 0,1 5,06 6 0 10,02 21,7 3,6
21 M9 250 71 27,9 43,1 1,19 0,15 3,6 12 0 10,08 19,08 2,81
22 M10a 250 70,9 23,6 47,3 1,95 0,11 5,33 16 0 9,77 22 1,1
23 M3 275 33,6 8,8 24,8 1,72 0,12 4,79 26 0 9,65 11,09 7,1
24 M6 275 49,1 7,6 41,5 2,36 0,01 6,26 5 0 9,84 7,9 2,62
83
CA 3027250 2018-12-12
25 M8 275 55,4 10,3 45,1 1,81 0,11 4,95 10 0 9,68 18,45 8,35
26 M9 275 48,1 13,1 35 2,21 0,14 6,07 8 0 9,73 14,85 3,19
27 M10 275 58,16 12,4 45,8 2,22 0,12 6,04 14 0 9,74 18,7 5,62
28 M3 300 12,4 -
21,4(?) 1,875 0,109 5,13 2 0 9,7 2,07 2,96
29 M6 300 14,3 - 17,7(?) 1,87 0,13 5,23 1 0 9,72 1,02 0,04
30 M8 300 28,7 - 30,8(?) 2,04 0,09 5,54 12 0 9,65 2,98 0,72
- 31 M10 300 47,8 12,9 34,9 2,32 0,16 6,46
6 0 9,88 6,08 2,95
32 M10 325 29,3
31,70 1,05 0,08 2,94 7 4,3 9,82 5,4 1,17
- 33 M10 350 31,48 5,3 26,20 1,68 0,08 4,54
4 0 10,37 3,71 2,7
34 M 10a 400 23,6 23,5 0,11 0,73 0,11 3 1 3
0 10,33 7,02 1,16
(Table 19B: analytical data from tests performed on treated waters)
Figures 27 and 28 provide information relating to silica content in the
process waters from the
tests. Figure 27 provides silica concentrations (reactive, colloidal, and
total) for untreated
process waters from the tests. As expected, total silica in feed water was
measured at between
275 mg/I and about 350 mg/l. Figure 28 provides silica concentrations
(reactive, colloidal, and
total) for treated process waters from the tests as a function of Fe2+
concentration. In Figure 28,
total silica concentration decreases with increased Fe2+ concentration, and
crosses the typical
50 mg/1 threshold SAGD water processing at an Fe2+concentration of between
about 280mg/1
and about 300 mg/1.
Figures 29 and 30 provide information relating to hardness content in the
process waters from
the tests. Figure 29 provides hardness concentrations (calculated, Ca2', and
Mg2+) for untreated
process waters from the tests. As expected, calculated hardness in feed water
was measured at
between 10 mg/1 and about 12 mg/l. Figure 30 provides hardness concentrations
(calculated,
Ca2 , and Mg2+) for treated process waters from the tests as a function of
Fe2+ concentration.
In Figure 30, calculated hardness concentration decreases with increased Fe2
concentration
and is reduced by about 50 % at an Fe2+ concentration of between about 280
mg/1 and about
320 mg,/1 (the concentration range correlated with silica concentrations
approaching the typical
50 mg/1 threshold SAGD water processing in Figure 28).
In addition to the foregoing tests, a series of control tests were also
completed. In particular,
about 2.5 1 of untreated process water (i.e. process water not subjected
reducing conditions)
was removed from the Miniflot unit and divided into twelve aliquots. The
various aliquots were
84
CA 3027250 2018-12-12
treated with Fe2 solutions to provide concentrations ranging from about 150
mg/1 to about
1000 mg/1 (in increments of about 50 mg/1 and 100 mg/1) and then pH-adjusted
to 9.5.
Qualitative observations from the control tests are set out in Table 20.
Fe conc. Observation
mg/I
150 ¨ 400 complete black
450 very fine flocks
500 very fine flocks
550 some very small flocks
600 some very small flocks
650 flock and murky
700 flock and murky
800 almost clear phase with flocks
900 almost clear phase with flocks
1000 clear phase, flocks, filterable
(Table 20: observations from control tests)
The observations of Table 20 indicate that, in the absence of an electrolytic
reduction step, it
was not possible to treat process water to the required specifications without
requiring
uneconomical concentrations of Fe2+ concentrations.
In summary, the forgoing tests indicate that electrolysis (for example,
electroreduction using
non-consumptive electrodes), followed by direct injection of Fe2 , a polymer
and a pH
adjusting agent, provided treated SAGD water to suitable standards.
Appropriate reductions in
hardness and silica were obtained. Presently described water treatment
processes may provide
advantages over previous practices for SAGD process water treatment. By way of
example,
water treatment methods and systems as described herein may be compatible with
high
temperature operation which may provide for better energy efficiency and
energy conservation.
In turn this may provide for lower total operating costs, may reduce amounts
of sludge
generated, may provide for overall process simplification, and/or may be
compatible with
existing process equipment for implementation.
EXAMPLE 5 ¨ Direct Injection Flocculation Treatment Pilot Plant Designs for
Treatment of SAGD Process Water
Design considerations for an embodiment of a direct injection flocculation
treatment pilot plant
for treatment of SAGD process water are described. The pilot plant embodiment
is designed to
treat up to 6m3/h SAGD process water under SAGD operating conditions, i.e.
design
CA 3027250 2018-12-12
temperature of up to 170 C and design pressure of up to 1.450 kPa. It
represents a scale-up
factor of approximately 1:29 to 1:33 to a full commercial sized treatment
train of between 175
to 200m3/h.
In this non-limiting system embodiment, the system provides for electrolytic
reduction using
non-consumable graphite electrodes; injection of concentrated Fe2+ solution
(optionally also
including the preparation or production of Fe2+ solution); polymer and H2S
scavenger injection;
and pH adjustment to permit the formation of Fe(OH)2 flocks. Initial
electrolytic reduction is
performed to destroy some inhibitory compound(s) which otherwise would
interfere with the
formation of Fe(OH)2 flocks. Flock containing contaminants are then separated
from treated
process water by a two stage IGF system.
In this embodiment, direct injection flocculation is used in the treatment of
SAGD process
water, and may operate under normal conditions found in SAGD, i.e. elevated
temperature and
pressure. With the ability to operate under these conditions, the creation of
a heat sink is
avoided during process water treatment. The maintenance of conditions of
elevated
temperature across the treatment platform may significantly improve the energy
efficiency and
overall economics of water treatment in SAGD.
Experiments have demonstrated that such methods and systems may be used to
remove various
contaminants in the process water to a high degree (i.e. Silica, hardness,
suspended solids, oil
and grease, etc.). Initial tests were carried out at room temperature and
atmospheric pressure
and at somewhat higher temperature, i.e. 60 C. In this example, a pilot plant
is described which
is designed to operate at both standard ambient operating conditions and at
operating conditions
that are normally found in SAGD.
The main process steps in this example are as follows:
Electrochemical reduction,
Dosing of chemicals including flocculation, and
separation of contaminants together with flocks in an IGF column.
Figure 40 depicts a general process description, and Figure 41 depicts a
layout-general
arrangement drawing of the designed pilot plant of this example. The pilot
plant may be
86
CA 3027250 2018-12-12
operated in parallel to current SAGD process water treatment. It may be
operated at ambient
conditions (i.e. atmospheric pressure and room temperature - approx. 20C), and
up to more
typical operating pressures and temperatures, as found in SAGD, i.e. 850 kPa
and 140 C. Inlet
and process outlet connections will operate under similar conditions. Process
water will be
pumped into the system using turbine pump P 01. Incoming process conditions
will be
measured by PIR 03, FIRCAL 01 and TIR 01. FIRC 01 will trigger the by-pass
valve FCV 01,
which allows for different flow rates through the plant.
In the first dosing step I-12S scavenger will be added according to a dosing
rate established from
previous tests. Dosing pump P 02 (flow rate adjustable via process control
system (PCS))
pumps the chemical from the delivery and storage tote PT 02. PT 02 is
installed in the spill
containment pan PP 02 for safety reasons and will be equipped with the level
switch LSAL 02,
which triggers a low-level alarm and shuts down the dosing pump P 02 in case
of empty
container.
H2S scavenger will be dosed into the main process stream via injection valve
CV 01 and mixed
using the static mixer M 01. Process water with H2S scavenger will then enter
the reduction
stage with reaction tank T 01.
The reduction stage consists of the tank T 01 holding a set of internally
installed graphite
electrodes. The power supply for electro chemical reduction is applied in the
process water
between the electrodes. Temperature measurements will be taken at the inlet
and outlet of tank
T 01. The graphite electrodes will be powered via isolated copper bars inside
the tank, which
are also used as a support rack to keep the horizontally arranged electrodes
in place. Positive
and negative copper bars are fixed in the top of the rectangular flange via
isolated feedthroughs.
The power supply is in the electrical room. DC current (10 V, 500 A) will be
transferred to
tank T 01 via insulated copper bus bars.
In total, 17 electrodes will be installed in the reduction tank T 01(9
cathodes, 8 anodes). Process
water will flow inside the reaction tank T 01 and through the electrodes only.
Temperatures at
in and outlet points will be measured using TIR 02 and TIR 03. Within PCS the
temperature
difference between these points will be measured and monitored at TDIAH 04.
For example,
increasing temperature sets of an alarm indicating possible coating of the
electrodes or other
causes of decreased efficiency at the electrochemical reaction, resulting in
increasing
87
CA 3027250 2018-12-12
temperature due to heat input. To avoid critical buildup of heat in the
system, DC supply R 01
will only deliver DC, when pump P 01 is working and flow rate within the
process is higher
than at a minimum value, measured with FIRCAL -01.
Within the power supply R 01 voltage and amperage will be monitored. Low
values at EIAL
(V) and EIAL (A) will trigger an alarm, as no electrochemical reaction happens
(caused by
isolating problems or other problems).
Process water flows into next dosing station, where FeCl2 solution will be
added using P 03.
Dosing pump P 03 (flow rate adjustable via PCS) pumps the chemical from the
delivery and
storage tote PT 03. PT 03 sits on a spill containment pan PP 03 for safety
reasons and will be
equipped with the level switch LSAL 03. This will trigger a low-level alarm
and will shut down
the dosing pump P 03 if the container is empty.
FeCl2 solution will be dosed into the main process stream via injection valve
CV 02 and will
be mixed with the static mixer M 02. Process water with FeCl2 solution will
enter the third
dosing station, where the pH in the process water is increased.
NaOH will be added using P 04. Dosing rate at dosing pump P 04 will be
controlled at
measurement point QICAL 01, which measures the pH value. Dosing pump P 04
pumps NaOH
from the delivery and storage tote PT 04. PT 04 sits on the spill containment
pan PP 04 for
safety reasons and will be equipped with the level switch LSAL 04. This switch
will give a
low-level alarm and will shut down dosing pump P 04 if the container is empty.
NaOH will be dosed into the main process stream via injection valve CV 03 and
mixed with
the static mixer M 03. PH adjusted process water flows through about 10 m
piping for sufficient
retention time to form Fe(OH)2 flocks, before it enters the last dosing
station.
In the last dosing station polymer will be added to improve flocculation of
the formed Fe(OH)2
flocks. Polymer solution will be added using P 04. Dosing pump P 04 (flow rate
adjustable via
PCS) pumps the chemical from the delivery and storage tote PT 04. PT 04 sits
on the spill
containment pan PP 05 for safety reasons and will be equipped with the level
switch LSAL 04,
which will trigger the low-level alarm LSAL 05 and will shut down dosing pump
P 04 if the
container is empty.
Polymer solution will be dosed into the main process stream via injection
valve CV 04 and will
88
CA 3027250 2018-12-12
be mixed with the static mixer M 04. Static mixer M 04 will be specially
designed for polymer
mixing. However, a rinsing device will be included in the design in case of
plugging. When
the pilot plant operation is stopped, the ball valves V 16 and V17 will be
closed and the mixer
M 04 can be rinsed with water via V 18 and 19.
.. For optimal flocculation, a retention time of 30 seconds may be provided by
increasing the
piping to a diameter of 200 mm for a length of about 1.6 m, prior to the
process water entering
the IGF column T 02 for flock and sludge separation.
The flock and sludge separation is based on the known IGF system including
recycle pump and
gas injection. Natural gas will be used for flotation.
Process water exits column T 02 via V 27 and measuring station QIR 02 (o+g)
and flows back
into main process stream. Flocks and sludge will exit the column T02 via V 28
and level control
valve LCV 01 and will be added to process water stream to exit the pilot
plant. Flow rate of the
side stream is measured with FIR 02.
Basic layout and rough dimensions: Initial dimensions and the basic layout of
the pilot plant
are shown in Figure 41.
IGF Columns: The IGF set-up as shown in Figure 40 and Figure 41 is a
representation of an
IGF column for illustrative purposes only. By way of example, a 2 Stage IGF
may be used as
an integrated component within the pilot plant and not as an add-on.
Figure 42 (A) depicts an embodiment of a reduction vessel, and Figure 42 (B)
provides an
embodiment of a reduction vessel specification example.
In certain embodiments, produced water may contain an oil component, and floc-
rich
coagulated oil may be separated from the water using, for example, a filter
press. In other
embodiments, particularly where scale-up is of interest, filter press-based
floc removal may be
substituted for, for example, floatation-based equipment such as induced
static floatation (ISF),
induced gas floatation (IGF), and compact floatation units (CFU) equipment
(with either single
or multiple stage floatation), for example. These systems may effectively
deoil, and remove
silica and hardness, from contaminated produced water, in a manner compatible
with high
temperature (up to 220 C, for example) and pressure. In certain embodiments,
produced treated
water stream may thus be directly used for steam generation, for example
OTSGs, with
89
CA 3027250 2018-12-12
minimal additional treatment and without excessive scaling.
EXAMPLE 6¨ Reduction and Oxidation of Naphthenic Acids/Naphthenates and
Phenols
in SAGD Process Water
Methods and systems described herein include a pre-treatment step to
destroy/inactivate/remove inhibitory compound(s) in contaminated water which
would
otherwise interfere with flocculation following addition of a flocculating ion-
enriched aqueous
solution. In this example, additional studies were performed to determine
whether naphthenic
acids and/or phenols could also be destroyed in the same pre-treatment step.
Electroreduction
and Electro-chemical Oxidation (ECO) pre-treatment steps were both studied for
ability to
destroy naphthenic acids.
As described hereinbelow, while electroreduction pre-treatment was able to
degrade
naphthenic acids and phenols, the energy input required was not favourable. In
contrast, ECO
showed effective removal of naphthenic acids. Of interest was the observed
oxidation of
naphthenic acids quickly, and at the early stage of oxidation of organics
relative to the oxidation
of total organics (TOC's). This supports an effective and efficient method to
remove such
fouling components from contaminated water such as, for example, SAGD process
water prior
to steam generation.
The problem of fouling, often seen in SAGD and other oil processing equipment
that involves
water and steam, has been linked to the formation of naphthenic salts (mainly
with Ca2+) etc.,
and related to naphthenic acids and phenol(s) found in petroleum and in
process water(s).
Naphthenic acids are believed to be more or less soluble in water, depending
on their molecular
weight and the pH of the water. It seems, that when pH is less than the 5-6
range, naphthenic
acids are either soluble in water or form small droplets as emulsion in water.
In this range Ca2+
and Mg2+ are also soluble. In the range between pH 6-7, salt formation starts
and emulsified
drops go into solution as surface tension changes drastically. Naphthenic
acids soluble in water
are then able to form Canaphthenates. With pH >8, the Ca-naphthenates become
insoluble and
CA 3027250 2018-12-12
can cause problems, i.e. fouling. If the removal of naphthenic acids can be
demonstrated using
a pre-treatment step, this may offer yet another advantage to the presently
described water
treatment methods and systems.
Test procedure for reduction and oxidation tests
ORF outlet water was used in these treatment studies. Table 21 shows physical
and analytics
data of the ORF outlet feed water, obtained for the water at time of
collection and prior to use
in experiments (columns 3 versus 4).
Fluid type ORF Outlet ORF Outlet
Collection Point FC F701 MIT F701 Tote
Sample ID 103350 Ce12027
Date Collected 201&04-18 2027-0/-24
Tate 1 pH was measured
An fytics Data pH 7.3-7.4 during testing
Conductivity mSicm 2.92
COD mg/I 2,350
DOC 240
TOC mg/I 740 250
04-0 mg/I 9.9
TSS mg/I 22
Difference attributed to lab
Naphth.Acids mg/I 33 18 procedure
Phenols 45 51
Hardness mg/I 8.1
Ca mg/I 3.2
Mg mg/I
Silica mg/I 130
Fe mg/I
H25 mei 9.7 2.4
Total
OrgSulphur mg/I 8.7 2.2
(Table 21: ORF outlet water physical and analytical data)
Reduction tests:
Approximately 201 of ORF outlet process water was prepared in a pail. A hose
pump was used
to pump the prepared process water into the bottom of the reactor. Overflow
was collected for
sampling. The lab reactor was equipped with 4 graphite electrodes (2 positive,
2 negative). To
undertake tests at higher temperature, a small heat exchanger was assembled
using copper
tubing, installed in a bath of heated water. In this way, temperature could be
increased in-line
to about 50 C.
91
CA 3027250 2018-12-12
The effect of the various treatment parameters on the following constituents
was measured:
Naphthenic acids and phenols; hardness; Ca; Mg; total organic Sulphur; TOC;
DOC; TSS;
TDS; 0+G. Parameters that were varied during tests included: pH at treatment;
amperage;
voltage; temperature; retention time (flow rate, specific amperage, specific
power).
Tests were performed at room temperature and at 50 C. About 200 1 process ORF
outlet water
was transferred into an open barrel and pH-adjusted using HC1 for the tests at
pH 5 and 7, and
using NaOH for tests at pH 9. Mixing was done using a turbine pump installed
in the barrel.
One barrel with pH-adjusted water provided enough water for about 9 runs.
After having used
up the pH-adjusted water, a new batch was prepared in the same manner. pH
adjusted water
was pumped using a hose pump with adjustable speed, so that the tests could be
run at different
flow rates. Test water entered the test reactor at the bottom and exited the
reactor via overflow
into a pail. All samples were taken out of this pail without further
treatment.
The reactor was equipped with four graphite electrodes (dimensions: 11.5 x 12
cm, area 138
cm2). Distance between the electrodes was 2 cm. pH outlet and temperature
differences
between in and outlet of reactor were measured directly at time of testing.
Changes at the outlet
pH during the test runs with process water at pH 7 and 9 were observed, so
repeats of these
runs later at higher temperatures were performed, but otherwise under the same
conditions.
Oxidation tests:
Oxidation tests were undertaken by electro-chemical oxidation using a small
lab ECU "beaker"
set-up. The ECO set-up utilized proprietary, specially coated ultra-high
potential Boron Doped
Diamond (UHP-BDD) electrodes that use Niobium or Tantalum as carrier material.
These
electrodes permit the highest currently available electro-chemical potential
to be applied to
completely oxidize organics. Tests showed that this ECU can completely oxidize
soluble
organics to CO2 and water. The lowest TOC after ECU that we observed during
qualitative
testing was 2 mg/l. Initial ECU studies on TOC reduction included the
following time study
shown in Table 22:
92
CA 3027250 2018-12-12
time voltage amperage remarks
min V A
0 9.10 10.2 foe m, chlo rine oda r
9.40 10.2 color turns dark
9.30 10.2 color turns lighter
9.10 10.2 color turns lighter
10.10 10.2 color turns lighter
9.50 10.2 color turns lighter
9.00 10.2 calor almost yellow
12.50 10.2 yellow
SO 11.10 10.2 yellow, no foam
90 10.30 10.2 Color
lighter
100 10.00 10.2 color
hghter
110 9.60 10.2 color
lighter
/20 9.40 10.2 almost no color, Anne chbrine ado]
130 10.90 10.2 no chlorine, but some ozone odor
140 10.30 10.2 ozone odor
150 9.70 10.2 stop, take sample
(Table 22: Extract of ECU testing protocol)
A noteworthy observation was the presence of reaction stages based on the
following
characteristics:
5 foam production, chlorine odor;
no foam production, but change of color from dark-brown to yellow; and
almost no color, no chlorine odor, but presence of ozone odor.
These reaction stages suggested that organics were not uniformly oxidized by
ECU, but that
different organics may be oxidized at different rates and at distinct stages.
Studies were thus
10 performed to determine whether naphthenic acids could be
destroyed/oxidized at a faster rate
than the overall reduction in organics by ECU, and at the initial stages of
oxidation. Studies
focused on measuring the concentration of naphthenic acids after different
treatment times to
see if a complete or partial oxidation of these compounds would be observed,
and whether
changes occur quickly or in a slow manner. Reaction temperature in the process
water was
15 maintained to below 50 C. The first two runs were used to estimate the
efficiency of the
oxidation process relative to total energy input.
93
CA 3027250 2018-12-12
Tests were carried out using a beaker which held the ECO electrode set. A
magnetic stirrer was
used to mix the feed water and to push some of the feed water through the
narrow space
between the electrodes. During ECO tests, about 1 I of water was used for
sampling purposes.
Atotal of five tests were undertaken, with reaction times of 20, 40, 60, 80
and 120 minutes. As
reaction time increased, the formation of a precipitate was observed, which
was believed to be
,
a "new' organic compound. At this point, the early formation of foam had
stopped. Precipitate
after 80 min of treatment formed a flock that could not be oxidized in further
treatment, as only
soluble organics are typically treated with this process. The formation of
this "new" organic
compound took place at the later stage of ECO treatment.
Analytical Results of Reduction Tests
Table 23 shows analytical data for reduction test runs performed.
test data ' -- - analytical dat a
test on. set voltage det. flow-
mate. centric pat 00d. temp. COD '00C TOC dsg 050 fuel, plisen. fwd. Maid Cs
Mg fe, re, 02133 HIS 1001
1,71 -wave-sag - tete ARV fla Off. filtered
120141 filleted attititit wok 5
S = 0 eted spec.
V A 7:0708 qn no ns so, -'3' 91,11
403/32 170 31372 313/1 993.11 mgAS 78811 762/1 17311 8980 177370
813,72 7881i 593,0 0147!
1.1<4 37<133)1 1300 740 43 45
.3.1 3-.7
!,?1,23 .. = = .. . = Me< sin 2130 1.31 = 240 '
244 932 11 Ili 51 300 <60 130 1A 2.2
1.1.1 ' 0.0 . 015 5.02 = 200 7..20.3 30.00 5.06 1 130
240 8.8 20 15 49 8.1 cos 13.3 '2 906 140 005 001
1.0/ ; 0.0 : 9.35 sno 1.04 i bo.o 43.noo 5_02 04 210 HO 730
27 17 47 8.1 <0.4 32 .322 .05 140 1.2 1.1
12.3 :5,0 9..30 . 5.03 0.50 20.0,04700 5.02 2 210 240 81 14
16 48 11.0 <794 .3.1. '2 10.47 140 1.1 1.2
1.1.4 ' 5.0 20 300 0.2.9 : 130 1L562.13 4.28 23. .220
140 1.4 24 16 40 /5 .115 .11 . 2 10.6 130 1.2
12
1.1.1 I SA 13.50 830 . 2420 11.20.0 39340 ' 522. 90 1.421 177
52 24 17 45 6.9 521 3.0 .10 , <0.3 0.08 150 , 02
172
1.1.2 SA 130 8.0 LW i 60.03.9280 0,10 IS 130 240 5.1
0.4 17 42 S. 7 0200 15. 300 , Ø3 1.1 140 2.5
2.3
1.2<1.21323,0 9000.13) 40.02301240 5.532 LS 200 280 154
0 8 132 44 11 160 4.1 31.0 0.44 1.3 140 2.0
15
1.24 1 "3 1430 . 6200 029 i 15.0 7.024,30 .6.14, 46 140 250
01 34 14 18 0.32 9.22 14 .1.0 0.51 1.3 13203
2.2 21
10.1 4.0 id 20 1020 2.10 120.0 57320 .5.01 10
140 260 3.2 12 13, 41 80 5.64 16 710 <0.179 0.40 160 , 1.1
1<3
1.3.2 5.0 13 60 . 1320 1.00 . 6.3.0 97930 0.1.6 2.4 240 150
5.1 11 16 _ 42 8.5 11 3.5 31.0 0.30 0.91 140
151 1.3
1.3.3 = 5.0 13.60 10.20 00.50 = 30.05340.40 5.10 21 240
240 94 24 17 45 8.4 0.74 34 .10 03.8 1.3 140
2.4 2.2
1.1.4 '0.0 13.00 10.20 0.25 . 15.02.91140 520 55 , 2710
270 0.1 372 16 39 8.1 851 3.3 11.0 0..4 2,9
150 1.9 10
21.1 = 1.0 9.80 5.00 0.00 1120.0 110.00 244 0 230
234 III 2.5 18 718 5.3 8.58 3.1 '10 10.4 7251
120 2.4 2.2
11.1 7.0 9.00 5.00 1.00 . 00.0304500 6.69 '
00 * 17.11 2221 ' 33 4 3.12 4 10 "48 " 0.4 ' 8 3.3 ' 3.4 '10 7331
"0048 "140 ' 2.0 ' 1:3
2.1.1 7.0 9.60 500 0.50 30.0 33371 5.27 1.5
- 2.,:., ' 2013 ' 0.0 r 6.5 - 10 - 41 ' 8.77 881 ' 3.4 .00 303 ..
0112 .. 1337 .= 11 ' 2.2
1.1.4 7.0 9.50 43301 0.25 15.0 1320130 7.04 0 2
' 230 " 26,11 ' 9.2 ' 4.7 .. 17 ' 45 ' 22 4 9.02 ' 1.3
31.0 ... 059 ' 1.5 P. 110 ' 1.9 * 111
2.2.1 '7.0 12.00: 720' 200 120.0 22320 137 0 ,,,
' 240 '224 '8.0 ' s.s r lb r 45 " 8.1 "9.21 ' 13
.71.0 37130 .. 0678 1o77 p2.) . 20
22.? 10 12.20 3.23 1.00 60.0 646.110 733.
'31', ' 250 ' 270 ' 7.2 P32,3 "113 P41 PO4 ' 3 18 P34 .10 Ø1 ' 0.15
P1<0 ... 1.4 1.3
32/9 2.0 .02.00 7.33 0.50 30.0(122.50
4.313 0 1' 2131 " 1311 '29 "3.1 4 17 ''11 ' 4.3 '108 '3.4 .1 0 <0.3
"01.92 '1.8301 r 1,3.' 1.4
22.4 .100 12.30 ' 4.30 0.25 . 15.0 2.324.40 8.02 '
24 '220 140 "1.9 74,) ' 1.5 ' 37) " 0.1 013 r 3.3 </A * 010 ' 1,3
"1.30 "22 ' 2.1
3 3,1 7.0 1020 1002 2.01 120.0 44090) 507 r 115
230 "200 " 70 4.5 . Ib r 41 905 <0.5 .15 1172 13) 131 '130
' 1Ø ' 11
, 2 70 /550 10 A 1.09 501, 9314.0 7.08 ' ' 3 4
"220< "2011 '4 7877 P3.4 v r 45 30,5. Arab 735 310 ':302
'3, ' 130 "10 006
. /.1.1 7Ø y 60 10001 004 140 1.124.00 e.32 ' 1 '
200 ' 234 ' 1.'1 4 i'f S ' 14. "44 105 901 614 310 '.40 7 6
'030 . 3.3' 17
' 5 ' . 160 200 r 12 ' 10 ' IS 40 309 Ø5 315 110
3,30 7 6 . 130 ' 1.5 1 12
4.1.1 9,0 1000 5.00 2.00 120.0 220.00 9,084 0 0
02.0 9Ø 9.10 900 2.00 690 327.09 3333 ' 0 0.438310.0
at 503 3150.484
1.171 0.0 9.50 5.03 0.50 0.0 00.00 862 '"010
35.4 4.0 9.40 5.00 43.25 10.0 1.:50.39 = 0.5.5
3,-.4 : Iiii., I..i i . bur rm Ensidinsdalts 6 set
t<464parti 373P.33 ' 2:0 P230 "3.1 ' 3 0 ' 4/ P320 Ø5 ". 1.10
"7Ø ' 23
3.01 9.0 4.90 ' 5.00 = 2.00 420.0 24300 0.08 r A ' 140 "230
"83. "1.0 - 10 "44 <0.9 .$0.5 731. 310 59 <3. "130 "1.9 '
1.8
3.1.1 909.90 500 1.00 : 60.0 390.02 211 "0.0 ' 240 4 240
' 8.7 r 4.4 ..= II ' 48 -- .05 -- .05 -- 114 -- .10 -- 33.0 -- . 8 .. 130 ..
2.0 ' 2.6
41.3 0.0 9.210 5.02 0.50 30.040300 9.03 ' 00 ' 2.30
210 ' 8,2 37.3 Id ... 18 <0.3 ,4215 <1.3 <10 44.0 e.6
'1.30 ..= 3,7 ... 2.5
41.4 30 9.90 5.03 an 1,5.01,47114115 3243 ' 2. 0
...2i0 .22.2.5 * FA "073 "II. r 46 '.046 305 115 .10 <1.0
'3' 120 1 0 ' 18
32.1 ' 2.0 WO 730 ' 2.00 :120.0 135.80 AM "0 140 r 214 4
9.0 4 2.6 ' 17 46 .04 = 839 <10 7:170 e3.0 " 0.68 =- 130
1.1" 1:5
32.2 9.0 12.50 7.10 1.93 50.0 92600 0,02 "04
"230) "377 "9.8 <1.0 ' lb 45 30.5 "3,3.0 316 110 1/0 * 1.1 r
130 ' 2.0 . 24
22..3 9.0 12.19 840' 0.50 10.0 Lceo.ea '93 ' 1 5 .' 200
100 '15.6 ' S8 = 18 ' 71.5 <0.5 006 716 .30 '.320 d 6 P.
130 ' 2.2 ' 2.1
r
3.2.4 30 1250 9.10 i 0.15 . 10 0 5476.10 703 4 ' 240 *180
" 074 '4 1 12 " 41 d2.5 .00 7:14 110 7:1 0 , 4 ' 120 r 280
222
53.0 9.0 1400 = 10.00 2.00 .1,203 11080 924 1 P. 240
160 ":31, ' 1 1 ' 12 "13 <03 "'.23 735 310 ':30 .. 0.65 "100 "16
2<
11.2 . 4.0 13.00 tom 1.00 60.0 64000 ts T1 '
LS '140 p100 5.2 "51 "17 P40 3115 p8,15 310 .19 <3.0 p0221
'130 ' 07 ' 1.6
11.3 9.0 15.00 10.00 0.50 30.0 1.20000 8.03 '-
2 ' "230 '270 "0.3 3.2 lb P43 <05 "3.24 315 1172 730 "0.42
"3220 r 1.5 . 14
1.3.4 . 9.0 . 14 40 IOC* 0.25 18.0 2.920/10
7.41 ' 032, '230 '270 ' 142 ' 3.7 IS '31 <0.3 "3.8) <Is 410
140 ' :384 ' 1.30 "1.4 13
21.1 .. 2.00õ. 1140 10.02 0.50 . 30.0 50000 .. Li. 12 "
.31 ' 310 P23.3 "1.4 ' 4.4 P1.3 P42 30.70 ' 3 27 310 .10 3.3.0
r. 0777 '130 ' 26 l'. 13
4 1.2 . 2.481 12100 1000 .00 13.0 33000 7.17 "
40 ' 1.00 Ir. Pk1 ' 7,1 . 1.1. ' lb 4 33 313. ' nos <ts , in
<4.0 1.1 p131' ' 1.2 ' 1.1
1 23 200 WO 10.20 . 0.28 13.0 3,2000 246 P3
"29,11 "390 I'. 7.9 * IA ' lb 11 .05 ' 819 319 d10 .3.0 21 '130
21 10
4.1.1 9.3(1) 11160 i 10.05 = 17.50 10.0 5120/20 317 ' 30
"2.30 "ZOO "1.), <1 ' 17 - 46 305 = 7.84 7.10 110 <30 . 0.114 r
130 - 1.1 .. 10
9.1/ 100. 1002 , 10.310 0.25 15.0 1,134.313 . 2.44 '
40 "2.20 250 '113 14 ' 116 ' 39 .08 "3,63 A:15 310 '.30
1,0 ' 130 ' 1.6 r 10
0.1.1 900- 11 40 Valk 0.15 15 0 7.121.23 757
"7 =9 ' 110 10.0 ' 06 ' 3 4 16 0 41 Ø5 "12 715.
182 ':10 r 0 91 110 11 ..= 7
94
CA 3027250 2018-12-12
(Table 23: Analytical Data for Reduction Test Runs)
Analytical Results of ECO Tests
Table 21 shows analytical data for reduction test runs performed.
AnAytical results from (CO tests lelectro chemical oxidation)
heed at Data lime lume matto temp. ratter istripera, TOC DOC
011ograme 04006004 46e na6 vedwaiee reductlee cement
swank a time lama= add TOC 6149100evit
I WA C V A met pet 416/4 VaAfl.
41411. tO
Off rata 24.04.2017 vote 250 242 9,9 111 51
WM,
C0.1 24042017 OM 1 TO 25 11.4 _10 170 ..31. ad.
7,1 n.d, 1200 60,56 foam:, dJ= =
GM 2 2404.2012 latIS 1 :40 57 14-12,3 10 110 .4..
ad 34 4.4 56/40 111,11 , eta
KO. 3 20,31.7017 09135 1 fie 20 14,2.15,e 10 SS
11-Ø4 25212017 10A0 1 40 26 14,5=16A 10 47
FC0.5 75 64.2017 13,15 I 120 70 14.6.17.11 10 22 a
4
*marks 1 'Aetna defecten Omit ..der tedetattper Afait, ed. 0011 409111104
(Table 21: Analytical Data for Oxidation Test Runs; commas are decimal points)
Discussions
In total, 46 single reduction tests were performed. At the completion of each
test, water samples
were taken for analysis. Specific attention was paid to the concentrations of
naphthenic acids
and phenols. In addition, the change in concentration of various other
components was also
tracked.
Components like TOC, DOC, oil and grease, silica, H2S and total organic S
showed no change
when parameters during reduction tests were changed to optimize for
degradation of
naphthenic acids and/or phenols. These data support these components typically
tending to
react chemically with Fe' or to get absorbed by Fe(OH)2. They were not
affected by electro-
chemical reduction.
Analytical values observed related to naphthenic acids, phenols and total
suspended solids
(TSS) were assessed.
Removal of Naphthenic Acids - Reduction:
Electro-chemical reduction tests were conducted to measure the effect of
parameters like pH,
flow rate, temperature, voltage, amperage. This was done by taking one of
these parameters as
a constant, and testing the effect of the other parameters against this
constant. The specific
energy, measured in kWh/m3, was chosen as a measure of degradation of
naphthenic acids or
phenols. This main parameter involved the following single variables: Flow
rate, retention
CA 3027250 2018-12-12
time, power input (voltage and amperage). Furthermore, it implied an operating
cost and it can
be used to compare operating costs of electro-chemical reduction against other
operating costs
like electro-flotation, etc.
In general, a low rate of removal of naphthenic acids was observed during
electro-chemical
reduction tests. Figure 31 shows degradation (removal) of naphthenics at pH
values of 5, 7,
and 9 (A-C, respectively). pH variation and treatment at room temperature were
not material
parameters for optimization of degradation of naphthenic acids. Basically, the
slope of the
degradation curve was almost independent of pH value. Degradation levels
observed were
generally poor under the conditions tested.
Another important operating parameter is temperature. As previously mentioned,
most of the
reduction tests were conducted at room temperature to establish general
trends. Two runs were
also conducted at higher temperature, i.e. 50 C, using a "heat exchanger" set-
up. Figure 32
shows degradation of naphthenic acids at pH 7 at temperatures of 20 and 50 C;
and at pH 9
and at temperatures of 20 and 50 C (A and B, respectively). All other
parameters were kept
constant.
No clear differences in degradation of naphthenic acids by electro-chemical
reduction were
observed at either 20 or 50 C, nor at pH 7 or pH 9.
Figure 33 compares the degradation of naphthenic acids at room temperature and
a pH of 7
with current, as conducted through graphite electrodes, used as a variable.
This figure shows
an unusual behavior. All parameters were kept constant, with exception of
current, which was
changed (3 series) from 5 A, 8.3 A and finally to 10 A. The trend lines for 5
A and 8.3 A show
a low level of degradation. At 10 A, no slope was seen for the trend line. The
line is flat,
indicating no degradation. Under these conditions the specific surface
amperage is 24 mA/cm2,
which may be too high for the requirement of the reduction reaction using this
type of graphite
electrode.
Overall, degradation of naphthenic acids by electro-chemical reduction was
poor, and
apparently independent of temperature, and of changes in pH under the
conditions tested.
Removal of Phenols ¨ Reduction:
In addition to the tests for degradation of naphthenic acids by electro-
chemical reduction, the
96
CA 3027250 2018-12-12
behavior of phenols to electro-chemical reduction was also tested. Degradation
of phenols is
shown in Figure 34 at pH 5, 7, and 9 (A-C, respectively). Degradation rates
for phenolics were
somewhat higher than the degradation rates observed for naphthenic acids (at
least at room
temperature). However, operating costs were still high. The behavior at higher
temperature
(20 C instead of 50 C) was different from the behavior observed for naphthenic
acids
degradation. Figure 35 shows increasing degradation rates with increasing
temperature for
phenols (20 and 50 C at pH 7 in A, 20 and 50 C at pH 9 in B).
Electro-chemical reduction treatment for phenols show two different trend
lines based on
temperature for both pH 7 and for pH 9. This is quite different from the
behavior observed for
naphthenic acids. A basic projection of behavior to 80 C and 140 C would
estimate a 50 %
degradation of phenols at 80 C with operating costs based on about 10 kWh/m3
and a further
estimated 50 % degradation of phenols at 140 C with operating costs based on
about 6
kWh/m3.
Figure 36 shows the influence of current in the degradation of phenols. The
trend lines are
according to normal standard and show behavior as expected.
Although the observed degradation of phenols was somewhat faster than the
degradation of
naphthenic acids, based on the amount of power required to achieve meaningful
levels of
reduction in naphthenic acid/phenol levels under the conditions tested, this
approach was not
favoured. As will be understood, an economical decision may influence
applications where
such treatment may be desired. In certain embodiments, such as where higher
energy inputs
are not cost prohibitive or otherwise undesirable, such reduction treatment
may be employed.
Removal of Naphthenic Acids ¨ Oxidation (ECO):
Specially coated Niobium or Tantalum BDD electrodes may be used to oxidize all
organic
compounds to carbon dioxide and water and oxides of sulfur and phosphorus
through a process
called electro-chemical oxidation (ECO). In general, the speed and efficiency
of this "ECO"-
process is high, i.e. approaching 100% efficiency. Oxidation of organic matter
occurs in steps,
with some organics oxidized or destroyed immediately, and others in a second
or third step or
stage of oxidation. The electrodes are non-consumptive and permanent.
97
CA 3027250 2018-12-12
Tests were carried out using an ECO lab unit set-up. Figure 37 shows the
concentration of
naphthenic acids and total organic carbon (TOC) versus treatment time. After
20 min of
treatment naphthenic acids concentration was decreased from 18 to 7.1 mg/I,
while general
TOC concentration was reduced from 250 to 170 mg/l. This shows that naphthenic
acids were
oxidized faster than the overall mixture of organics in the feedwater.
Figure 38 shows that removal of naphthenic acids took place much faster than
the total removal
of organic carbon. At the point when approximately 50 % of naphthenic acids
are removed,
only 30 % of total organics are removed (based on 18 mg/1 naphthenic acids in
feed and 250
mg/I TOC in feed). These values may be optimized using different geometries
for electrodes.
Figure 37 shows the reduction in naphthenic acids and TOC over time using a
small electrode
package with a separation of 2 mm between electrodes. During the first run the
temperature
increase was measured in the beaker containing the process water and the
electrodes. The
beaker was not cooled. In subsequent tests, a water bath was used to cool down
the beaker
during treatment. Energy input is defined by voltage and amperage. Energy
output can be
measured by the temperature increase of the test water. Temperature increase
can be caused
by:
oxidation heat of organic matter through use of these electrodes, and
heat from unused energy, used to generate radicals that are not used to
oxidize organics.
It is estimated that the degradation of naphthenic acids in a properly
designed ECO plant may
even reach about 100% efficiency, or about 20 times faster than what was
observed in these
initial studies. Figure 39 shows estimated design values for an ECO plant. As
shown, it is
estimated that a reduction of naphthenic acids from 18 to about 3.4 mg/1 may
take a residence
time of about 2 min. The final content of TOC may be around 100 mg/1 at this
point.
Discussions:
Lab tests with ECO have shown that electro-chemical oxidation was effective in
removing
naphthenic acids. It is also known that phenols are effectively and
efficiently removed by ECO,
and that the reaction time is as fast for phenols as for naphthenic acids,
possibly even faster.
Of interest was an observed destruction of naphthenic acids at the initial
stage of the oxidation
98
CA 3027250 2018-12-12
process, especially when compared to the overall oxidation of TOC's. After 20
minutes of
treatment, concentration of naphthenic acids was decreased from 18 to 7.1
mg/1, a reduction of
61%, while general TOC concentration was only reduced from 250 to 170 mg/1, a
reduction of
32%. This supports that the destruction of naphthenic acids took place quickly
and early in the
oxidation reaction process.
Based on this, a rough calculation on the efficiency of ECO for the
destruction of naphthenic
acids in an industrial sized set-up was made. In certain embodiments, it may
be possible to
reduce naphthenic acid levels from about 18mg/1 untreated to about 3mg/1 with
a plant
residence time of about 2 minutes.
In certain embodiments, ECO may offer a beneficial additional finishing
treatment step, after
water treatment and before water is used in steam generation of SAGD, for
example.
Results obtained further indicate that in certain embodiments, using ECO to
provide oxidizing
conditions to destroy flocculation inhibiting compound(s) in the contaminated
water may
additionally provide for removal of naphthenic acids/naphthenates and/or
phenols, in addition
to certain other organics when present in the contaminated water.
EXAMPLE 7 ¨Additional Water Treatment System Design Configurations
Particular embodiments of methods and systems for treating contaminated water
are described
in this Example. The following exemplified water treatment system and method
embodiments
each include a reducing (chemical or electrical) or oxidizing (chemical or
electrical) unit/step
which subjects the contaminated water to reducing or oxidizing conditions; a
separation
unit/step for removing flocs/contaminant(s) from the contaminated water (in
these examples, a
filtration or floatation-based separation unit/step); and an input/step of
introduction for a
flocculating ion (in these examples, iron ions) into the contaminated water to
cause
flocculation. The exemplified system and method embodiments may optionally
further include
one or more inputs/addition steps for optionally inputting one or more of the
following
additional agents: an H2S scavenger; a pH adjustment agent; a chelant; a
sulphite; and/or a
polymer. These examples are intended for non-limiting purposes to illustrate
that a wide variety
of configurations and sequences of operations of systems and methods as
described herein are
99
CA 3027250 2018-12-12
contemplated, which may be employed depending on the particular application.
By way of example, a preferred exemplary process sequence for treating a
contaminated water
may include the following:
Reduction (Chemical or Electrical) ¨> H2S Scavenger (optional) ¨> Iron Rich
Water ¨>
pH Adjustment (optional) --> Separation (optional) ¨> Chelant (optional) -->
Sulphite
(optional)
The present inventors have identified that flocks form particularly well when
iron-rich water is
added to the contaminated water at a suitable pH, such as a pH between about 7-
11. It has
further been identified that for treating contaminated water which contains,
or which may
contain, one or more flocculation inhibiting compound(s), a reduction or
oxidation step may
be performed prior to flocculation, so as to destroy/degrade/remove the
flocculation inhibiting
compound(s) (if present). As well, it has been observed that adding an H2S
scavenger prior to
flocculation may be used to suppress the negative impact of sulfur on
flocculation. A chelant
may be used after flocculation and separation, so as to reduce scaling
properties of the treated
water without interfering with the flocculating ions used for
flocculation/separation. Sulphite
may be used to attack oxygen, and may be introduced at generally any suitable
process stage.
The inventors have additionally identified that a one- or two-stage pH
adjustment may be
performed to assist with flocculation and contaminant removal. By way of
example, a pH
adjustment to a pH of about 7 to about 11 may be performed at generally any
suitable stage
prior to or during flocculation to assist with flocculation. Where a two-stage
pH adjustment is
used, the pH may be adjusted to a pH of about 2-4 to make silica in the
contaminated water
reactive at generally any suitable stage prior to flocculation, and the pH may
then be adjusted
to a pH of about 7 to about 10 prior to or during the flocculation step to
promote flocculation
and contaminant removal.
The inventors have further identified that where an oxidation step is used to
destroy/degrade/remove flocculation inhibiting compound(s) from the
contaminated water,
such oxidation may additionally remove certain organics from the contaminated
water.
Furthermore, where output treated water still contains at least some organics,
a downstream
oxidation step may be performed on the treated water to remove organics
therefrom. In certain
embodiments, an oxidation step may be used two or more times during water
treatment, for
100
CA 3027250 2018-12-12
example.
Based on these observations, and the studies described in detail throughout
the present
specification, additional examples of exemplary process sequences of methods
and systems for
treating a contaminated water as described herein may include one or more of
the following:
= Reduction -->
Scavenger ¨> Iron Rich Water ---> pH Adjustment ¨> Separation ¨>
Chelant
= Oxidation ¨> Scavenger ¨> Iron Rich Water ¨> pH Adjustment ¨> Separation
¨>
Chelant
= Reduction pH Adjustment ¨> Scavenger ¨> Iron Rich Water ¨> Separation ¨>
Chelant
= pH Adjustment --> Reduction ¨> Scavenger ¨> Iron Rich Water Separation
Chelant
= pH Adjustment --> Scavenger ¨> Reduction ¨> Iron Rich Water -->
Separation ¨>
Chelant
= Reduction ¨> Scavenger ¨> Iron Rich Water ¨> pH Adjustment ¨> Separation ¨>
Oxidation ¨> Separation ¨> Chelant
= Reduction ¨> Scavenger ¨> Iron Rich Water ¨> pH Adjustment ¨* Separation
¨>
Oxidation ¨> Chelant ¨> Separation
= Oxidation ¨> Scavenger --> Iron Rich Water ¨> pH Adjustment ¨* Separation
--->
Oxidation ¨> Chelant ¨> Separation
= Oxidation ¨> pH Adjustment ¨> Scavenger ¨> Iron Rich Water ¨* Separation
¨>
Oxidation ¨> Chelant ¨> Separation
= Iron Rich Water ¨* Scavenger ¨> Reduction ¨> pH Adjustment ¨> Separation
¨>
Chelant
The person of skill in the art having regard to the teachings herein will
understand that various
other configurations may be possible, and may be employed to suit the
particular application.
These examples are provided for illustrative purposes to demonstrate that a
variety of
configurations/sequences are contemplated herein.
101
CA 3027250 2018-12-12
One or more illustrative embodiments have been described by way of example. It
will be
understood to persons skilled in the art that a number of variations and
modifications can be
made without departing from the scope of the invention as defined in the
claims.
102
CA 3027250 2018-12-12