Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 03041819 2019-04-25
WO 2018/080591 PCT/US2017/029920
METHODS OF TREATMENT WITH DEUTERA TED CFTR POTENTIATORS
Reference to Related Applications
[1] This application claims the benefit of the filing date of U.S.
Provisional Application
No. 62/413,941, filed October 27, 2016, the entire content of which is
incorporated herein by
reference.
Background of the Invention
[2] Many current medicines suffer from poor absorption, distribution,
metabolism and/or
excretion (ADME) properties that prevent their wider use or limit their use in
certain
indications. Poor ADME properties are also a major reason for the failure of
drug candidates
in clinical trials. While formulation technologies and prodrug strategies can
be employed in
some cases to improve certain ADME properties, these approaches often fail to
address the
underlying ADME problems that exist for many drugs and drug candidates. One
such
problem is rapid metabolism that causes a number of drugs, which otherwise
would be highly
effective in treating a disease, to be cleared too rapidly from the body. A
possible solution to
rapid drug clearance is frequent or high dosing to attain a sufficiently high
plasma level of
drug. This, however, introduces a number of potential treatment problems such
as poor
patient compliance with the dosing regimen, side effects that become more
acute with higher
doses, and increased cost of treatment. A rapidly metabolized drug may also
expose patients
to undesirable toxic or reactive metabolites.
[3] Another ADME limitation that affects many medicines is the formation of
toxic or
biologically reactive metabolites. As a result, some patients receiving the
drug may
experience toxicities, or the safe dosing of such drugs may be limited such
that patients
receive a suboptimal amount of the active agent. In certain cases, modifying
dosing intervals
or formulation approaches can help to reduce clinical adverse effects, but
often the formation
of such undesirable metabolites is intrinsic to the metabolism of the
compound.
[4] In some select cases, a metabolic inhibitor will be co-administered
with a drug that is
cleared too rapidly. Such is the case with the protease inhibitor class of
drugs that are used to
treat HIV infection. The FDA recommends that these drugs be co-dosed with
ritonavir, an
inhibitor of cytochrome P450 enzyme 3A4 (CYP3A4), the enzyme typically
responsible for
their metabolism (see Kempf, D.J. et al., Antimicrobial agents and
chemotherapy, 1997,
41(3): 654-60). Ritonavir, however, causes adverse effects and adds to the
pill burden for
HIV patients who must already take a combination of different drugs.
Similarly, the
1
CA 03041819 2019-04-25
WO 2018/080591 PCT/US2017/029920
CYP2D6 inhibitor quinidine has been added to dextromethorphan for the purpose
of reducing
rapid CYP2D6 metabolism of dextromethorphan in a treatment of pseudobulbar
affect.
Quinidine, however, has unwanted side effects that greatly limit its use in
potential
combination therapy (see Wang, L et al., Clinical Pharmacology and
Therapeutics, 1994,
56(6 Pt 1): 659-67; and FDA label for quinidine at www.accessdata.fda.gov).
[5] In general, combining drugs with cytochrome P450 inhibitors is not a
satisfactory
strategy for decreasing drug clearance. The inhibition of a CYP enzyme's
activity can affect
the metabolism and clearance of other drugs metabolized by that same enzyme.
CYP
inhibition can cause other drugs to accumulate in the body to toxic levels.
[6] A potentially attractive strategy for improving a drug's metabolic
properties is
deuterium modification. In this approach, one attempts to slow the CYP-
mediated
metabolism of a drug or to reduce the formation of undesirable metabolites by
replacing one
or more hydrogen atoms with deuterium atoms. Deuterium is a safe, stable, non-
radioactive
isotope of hydrogen. Compared to hydrogen, deuterium forms stronger bonds with
carbon.
In select cases, the increased bond strength imparted by deuterium can
positively impact the
ADME properties of a drug, creating the potential for improved drug efficacy,
safety, and/or
tolerability. At the same time, because the size and shape of deuterium are
essentially
identical to those of hydrogen, replacement of hydrogen by deuterium would not
be expected
to affect the biochemical potency and selectivity of the drug as compared to
the original
chemical entity that contains only hydrogen.
[7] Over the past 35 years, the effects of deuterium substitution on the
rate of metabolism
have been reported for a very small percentage of approved drugs (see, e.g.,
Blake, MI et al, J
Pharm Sci, 1975, 64:367-91; Foster, AB, Adv Drug Res, 1985, 14:1-40
("Foster"); Kushner,
DJ et al, Can J Physiol Pharmacol, 1999, 79-88; Fisher, MB et al, Curr Opin
Drug Discov
Devel, 2006, 9:101-09 ("Fisher")). The results have been variable and
unpredictable. For
some compounds deuteration caused decreased metabolic clearance in vivo. For
others, there
was no change in metabolism. Still others demonstrated increased metabolic
clearance. The
variability in deuterium effects has also led experts to question or dismiss
deuterium
modification as a viable drug design strategy for inhibiting adverse
metabolism (see Foster at
p. 35 and Fisher at p. 101).
[8] The effects of deuterium modification on a drug's metabolic properties
are not
predictable even when deuterium atoms are incorporated at known sites of
metabolism. Only
by actually preparing and testing a deuterated drug can one determine if and
how the rate of
metabolism will differ from that of its non-deuterated counterpart. See, for
example, Fukuto
2
CA 03041819 2019-04-25
WO 2018/080591 PCT/US2017/029920
et al. (J. Med. Chem., 1991, 34, 2871-76). Many drugs have multiple sites
where metabolism
is possible. The site(s) where deuterium substitution is required and the
extent of deuteration
necessary to see an effect on metabolism, if any, will be different for each
drug.
[9] This disclosure relates to deuterated derivatives of ivacaftor, and
pharmaceutically
acceptable salts thereof. This disclosure also relates to compositions
comprising a deuterated
derivatives of ivacaftor and the use of such compositions in methods of
treating diseases and
conditions that are beneficially treated by administering a CFTR (cystic
fibrosis
transmembrane conductance regulator) potentiator.
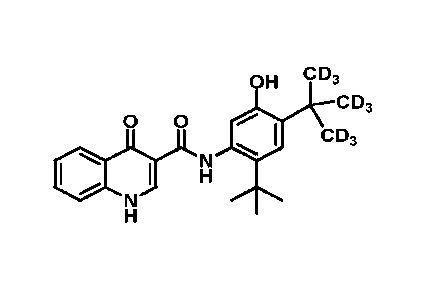
[10] Ivacaftor, also known as VX-770 and by the chemical name, N-(2,4-di-tert-
buty1-5-
hydroxypheny1)-4-oxo-1,4-dihydroquinoline-3-carboxamide, acts as a CFTR
potentiator.
Results from phase III trials of ivacaftor in patients with cystic fibrosis
carrying at least one
copy of the G551D-CFTR mutation demonstrated marked levels of improvement in
lung
function and other key indicators of the disease including sweat chloride
levels, likelihood of
pulmonary exacerbations and body weight. Ivacaftor was approved by the FDA in
2012 for
the treatment of cystic fibrosis in patients who have the G551D-CFTR mutation.
In 2014,
ivacaftor was approved for treating cystic fibrosis in patients who have one
of eight
additional mutations (G178R, S549N, S549R, G551S, G1244E, S1251N, S1255P and
G1349D) in the CFTR gene. In 2015, ivacaftor was approved for treating cystic
fibrosis in
patients who have one of 10 mutations in the CFTR gene (G551D, G1244E, G1349D,
G178R, G551S, S1251N, S1255P, S549N, S549R and R117H). Ivacaftor was granted
fast
track designation and orphan drug designation by the FDA in 2006 and 2007,
respectively,
and is marketed under the tradename Kalydeco . Ivacaftor is also approved in
combination
with VX-809 (also known as lumacaftor, a CFTR corrector) for the oral
treatment of cystic
fibrosis patients who carry the more common AF508-CFTR mutation; the
combination is
marketed under the tradename Orkambi .
[11] Despite the beneficial activities of ivacaftor, there is a continuing
need for new
compounds to treat the aforementioned diseases and conditions.
Summary of Invention
[12] It has now been found that deuterated analogs of ivacaftor (including
Compound (I),
also referred to as CTP-656, D9-ivacaftor or Compound 106) have an enhanced
metabolic
profile when administered to a subject, as compared to ivacaftor. For example,
the parent to
metabolite ratio of Compound (I) is greater than the ratio found for
ivacaftor. Compound (I)
is represented by the following structural formula:
3
CA 03041819 2019-04-25
WO 2018/080591 PCT/US2017/029920
OH CD3
CD3
0 0
110 CD3
0 I N
H
N
H (I),
or a pharmaceutically acceptable salt thereof.
[13] The enhanced pharmacokinetic profile for Compound (I) relative to
ivacaftor suggests
that Compound (I) can be efficacious at doses in the range of about 50 to
about 200 mg once
a day. Based on these discoveries, novel dosing regimens using Compound (I),
or a
pharmaceutically acceptable salt thereof, for treating a condition mediated by
CFTR in a
subject are disclosed herein.
[14] It has been further found that the exposure (blood level) of deuterated
analogs of
ivacaftor (including Compound (I), also referred to as CTP-656, D9-ivacaftor
or Compound
106) under low-fat (less than about 10 g of fat) and moderate-fat (between
about 10 g and
about 30 g of fat) conditions was similar, and was approximately 2-fold
greater than under
the fasted condition (see Example 8). In other words, the food effect results
showed that the
exposure of CTP-656 was the same regardless of the fat content of a meal. This
result is
unexpected. Because previous studies of ivacaftor had shown that food
containing high-fat
content yields better absorption of ivacaftor, the approved product labelling
for ivacaftor
requires administration with fat-containing food. However, it has now been
found that the
exposure of CTP-656 does not require consumption of a high-fat (e.g., about 60
g of fat)
meal. This could enable greater flexibility in dosing and, potentially, impact
the need for
pancreatic enzymes to aid in the digestion of fat.
[15] In a first aspect, this disclosure provides a method for treating
conditions that can be
treated by compounds that potentiate the activity of CFTR. Thus, this
disclosure provides a
method of treating a condition that is mediated by CFTR in a subject
comprising orally
administering to the subject an effective amount of Compound (I):
OH CD3
CD3
0 0
110 CD3
0 I N
H
N
H (I),
or a pharmaceutically acceptable salt thereof; wherein the Compound (I) is
administered to
the subject with food containing less than about 60 g of fat.
4
CA 03041819 2019-04-25
WO 2018/080591 PCT/US2017/029920
[16] In certain embodiments, the amount of Compound (I), or a pharmaceutically
acceptable salt thereof, is in the range of about 50 mg to about 200 mg, for
example, about 50
mg, about 60 mg, about 70 mg, about 80 mg, about 90 mg, about 100 mg, about
110 mg,
about 120 mg, about 130 mg, about 140 mg, about 150 mg, about 160 mg, about
170 mg,
about 180 mg, about 190 mg, or about 200 mg. In particular, the subject is a
human. In one
embodiment, the subject is a human 6 years of age or older. Compound (I), or a
pharmaceutically acceptable salt thereof, is administered orally at any of the
foregoing
dosages. In certain embodiments, the Compound (I), or a pharmaceutically
acceptable salt
thereof, is administered orally at any of the foregoing dosages in a
pharmaceutical
formulation which is a tablet, including any tablet formulation disclosed
herein, or a
bioequivalent tablet formulation, or a granule. In an alternative first
aspect, the method
comprises administering to a subject an amount of Compound (I), or a
pharmaceutically
acceptable salt thereof, once a day, wherein the amount of Compound (I), or a
pharmaceutically acceptable salt thereof, is in the range of about 25 mg to
about 75 mg, for
example, about 25 mg, about 37.5 mg, about 50 mg, about 62.5 mg, or about 75
mg, wherein
the subject is a human 2 to less than 6 years of age and less than 14 kg; or
alternatively, is a
human 2 to less than 6 years of age and 14 kg or greater. In one aspect, the
dose for the
human 2 to less than 6 years of age and less than 14 kg is 25 mg. In one
aspect, the dose for
the human 2 to less than 6 years of age and greater than 14 kg is 37.5 mg.
Preferably,
Compound (I), or a pharmaceutically acceptable salt thereof, is administered
orally at any of
the foregoing dosages. Preferably, the Compound (I), or a pharmaceutically
acceptable salt
thereof, is administered orally at any of the foregoing dosages in a
pharmaceutical
formulation which is a granule. In certain embodiments of this aspect, the
compound is
Compound (I).
[17] A second aspect is Compound (I), or a pharmaceutically acceptable salt
thereof, for
treating conditions that can be treated by compounds that potentiate the
activity of CFTR.
The compound may be administered as disclosed herein, i.e., by administering
Compound (I)
with food containing less than 60 g of fat.
[18] A third aspect is the use of Compound (I), or a pharmaceutically
acceptable salt
thereof, for the manufacture of a medicament for treating conditions that can
be treated by
compounds that potentiate the activity of CFTR. The compound may be
administered as
disclosed herein, as disclosed herein, i.e., by administering Compound (I)
with food
containing less than 60 g of fat, e.g., in an amount in the range of 50 mg to
200 mg, once per
day.
CA 03041819 2019-04-25
WO 2018/080591 PCT/US2017/029920
[19] A fourth aspect is a pharmaceutical composition comprising a
pharmaceutically
acceptable carrier or diluent and about 50 mg to about 200 mg of Compound (I),
or a
pharmaceutically acceptable salt thereof. Specifically, the pharmaceutical
composition
comprises about 50 mg, about 60 mg, about 70 mg, about 80 mg, about 90 mg,
about 100 mg,
about 110 mg, about 120 mg, about 130 mg, about 140 mg, about 150 mg, about
160 mg,
about 170 mg, about 180 mg, about 190 mg, or about 200 mg of Compound (I), or
a
pharmaceutically acceptable salt thereof. More specifically, for example, the
pharmaceutical
composition comprises about 75, about 100, or about 150 mg of Compound Ito be
administered once per day. In a particular embodiment, the pharmaceutical
composition
comprises about 100¨ about 150 mg of Compound Ito be administered once per
day. In a
particular embodiment, the pharmaceutical composition comprises about 100 mg
of
Compound I to be administered once per day. In a particular embodiment, the
pharmaceutical
composition is a tablet. An alternative fourth aspect is a pharmaceutical
composition
comprising a pharmaceutically acceptable carrier or diluent and about 25 mg to
about 75 mg
of Compound (I), or a pharmaceutically acceptable salt thereof. Specifically,
the
pharmaceutical composition comprises about 25 mg, about 37.5 mg, about 50 mg,
about 62.5
mg, or about 75 mg of Compound (I), or a pharmaceutically acceptable salt
thereof. In a
particular embodiment, the pharmaceutical composition is a granule.
[20] In another aspect, this disclosure provides a compound as disclosed
herein (e.g.,
compound (I) or a pharmaceutically acceptable salt thereof) or a composition
comprising an
effective amount of Compound (I) (or a pharmaceutically acceptable salt
thereof) for use in
treating a condition that is mediated by CFTR in a subject, wherein the
compound is
administered with food containing less than 60 g of fat. In one embodiment,
the condition
that is mediated by CFTR is cystic fibrosis.
[21] Additional aspects and embodiments are described herein below.
Brief Description of the Drawings
[22] Figure lA depicts the mean plasma concentration (ng/mL) for CTP-656 and
ivacaftor
in the single ascending dose study.
[23] Figure 1B depicts the mean plasma concentration (ng/mL) for CTP-656 and
ivacaftor
in the single ascending dose study.
[24] Figure 2 depicts the mean plasma concentration (ng/mL) for CTP-656 and
ivacaftor
following a 150 mg oral dose.
[25] Figure 3 depicts the parent verses metabolite pharmacokinetic profile for
(a) CTP-656
6
CA 03041819 2019-04-25
WO 2018/080591 PCT/US2017/029920
and (b) Ivacaftor (Kalydeco) following a 150 mg oral dose.
[26] Figure 4A depicts the peak current potentiated by sequential additions of
test articles.
[27] Figure 4B depicts the AUC of potentiator response.
[28] Figure 4C depicts the AIsc of potentiator response for ivacaftor, CTP-
656, and D18-
ivacaftor.
[29] Figure 5 is a schematic of the single ascending dose study.
[30] Figure 6 is a scheme of the metabolites of ivacaftor and CTP-656.
[31] Figure 7A is a schematic of the crossover study for D9-ivacaftor and D18-
ivacaftor.
[32] Figure 7B depicts the mean plasma concentration (ng/mL) for D9-ivacaftor
and D18-
ivacaftor following a 25 mg oral dose.
[33] Figure 8 shows a schematic of the design of a multiple-ascending dose
trial for CTP-
656 (D9-ivacaftor). Part A: single dose pharmacokinetic comparison (with
crossover) of 150
mg CTP-656 (2x 75 mg tablets) versus 150 mg ivacaftor. Part B: assessment of
three doses of
CTP-656 (75 mg, 150 mg, and 225 mg or placebo, dosed once daily for seven
days.
[34] Figure 9 is a graph showing the plasma concentration of CTP-656 and
ivacaftor after
a single dose of CTP-656 or ivacaftor.
[35] Figure 10 is a graph showing the plasma concentration of CTP-656 and
metabolites
(left panel) and a graph showing the plasma concentration of ivacaftor and
metabolites (right
panel) after a single dose of CTP-656 or ivacaftor.
[36] Figure 11 is a graph showing the plasma concentration of CTP-656 and
metabolites
after multiple dosing (once per day for seven days) of CTP-656.
[37] Figure 12 depicts a food-effect study for CTP-656: (A) schematic of the
design of the
food-effect study; (B) a graph showing the mean plasma concentration of CTP-
656 in the
food-effect study; (C) a table showing pharmacokinetic (PK) parameters for CTP-
656 in the
food-effect study.
Detailed Description of the Invention
Definitions
[38] The term "treat" means decrease, suppress, attenuate, diminish, arrest,
or stabilize the
development or progression of a disease (e.g., a disease or disorder
delineated herein), lessen
the severity of the disease or improve the symptoms associated with the
disease.
[39] "Disease" means any condition or disorder that damages or interferes with
the normal
function of a cell, tissue, or organ.
7
CA 03041819 2019-04-25
WO 2018/080591 PCT/US2017/029920
[40] It will be recognized that some variation of natural isotopic abundance
occurs in a
synthesized compound depending upon the origin of chemical materials used in
the synthesis.
Thus, a preparation of ivacaftor will inherently contain small amounts of
deuterated
isotopologues. The concentration of naturally abundant stable hydrogen and
carbon isotopes,
notwithstanding this variation, is small and immaterial as compared to the
degree of stable
isotopic substitution of compounds of this invention. See, for instance, Wada,
E et al.,
Seikagaku, 1994, 66:15; Gannes, LZ et al., Comp Biochem Physiol Mol Integr
Physiol, 1998,
119:725.
[41] In Compound (I), any atom not specifically designated as a particular
isotope is meant
to represent any stable isotope of that atom. Unless otherwise stated, when a
position is
designated specifically as "H" or "hydrogen", the position is understood to
have hydrogen at
its natural abundance isotopic composition. Also unless otherwise stated, when
a position is
designated specifically as "D" or "deuterium", the position is understood to
have deuterium at
an abundance that is at least 3000 times greater than the natural abundance of
deuterium,
which is 0.015% (i.e., at least 45% incorporation of deuterium).
[42] The term "isotopic enrichment factor" as used herein means the ratio
between the
isotopic abundance and the natural abundance of a specified isotope.
[43] In other embodiments, a compound of this invention (i.e., Compound I has
an isotopic
enrichment factor for each designated deuterium atom of at least 3500 (52.5%
deuterium
incorporation at each designated deuterium atom), at least 4000 (60% deuterium
incorporation), at least 4500 (67.5% deuterium incorporation), at least 5000
(75% deuterium),
at least 5500 (82.5% deuterium incorporation), at least 6000 (90% deuterium
incorporation),
at least 6333.3 (95% deuterium incorporation), at least 6466.7 (97% deuterium
incorporation), at least 6600 (99% deuterium incorporation), or at least
6633.3 (99.5%
deuterium incorporation).
[44] The term "isotopologue" refers to a species in which the chemical
structure differs
from Compound (I) only in the isotopic composition thereof.
[45] The term "compound," when referring to a compound of this invention,
refers to a
collection of molecules having an identical chemical structure, except that
there may be
isotopic variation among the constituent atoms of the molecules. Thus, it will
be clear to
those of skill in the art that a compound represented by a particular chemical
structure
containing indicated deuterium atoms, will also contain lesser amounts of
isotopologues
having hydrogen atoms at one or more of the designated deuterium positions in
that structure.
The relative amount of such isotopologues in a compound of this invention will
depend upon
8
CA 03041819 2019-04-25
WO 2018/080591 PCT/US2017/029920
a number of factors including the isotopic purity of deuterated reagents used
to make the
compound and the efficiency of incorporation of deuterium in the various
synthesis steps
used to prepare the compound.
[46] The invention also provides salts of Compound (I). A salt of a compound
of this
invention is formed between an acid and a basic group of the compound, such as
an amino
functional group, or a base and an acidic group of the compound, such as a
carboxyl
functional group. According to another embodiment, the compound is a
pharmaceutically
acceptable acid addition salt.
[47] The term "pharmaceutically acceptable," as used herein, refers to a
component that is,
within the scope of sound medical judgment, suitable for use in contact with
the tissues of
humans and other mammals without undue toxicity, irritation, allergic response
and the like,
and are commensurate with a reasonable benefit/risk ratio. A "pharmaceutically
acceptable
salt" means any non-toxic salt that, upon administration to a recipient, is
capable of
providing, either directly or indirectly, a compound of this invention. A
"pharmaceutically
acceptable counterion" is an ionic portion of a salt that is not toxic when
released from the
salt upon administration to a recipient.
[48] Acids commonly employed to form pharmaceutically acceptable salts include
inorganic acids such as hydrogen bisulfide, hydrochloric acid, hydrobromic
acid, hydroiodic
acid, sulfuric acid and phosphoric acid, as well as organic acids such as para-
toluenesulfonic
acid, salicylic acid, tartaric acid, bitartaric acid, ascorbic acid, maleic
acid, besylic acid,
fumaric acid, gluconic acid, glucuronic acid, formic acid, glutamic acid,
methanesulfonic
acid, ethanesulfonic acid, benzenesulfonic acid, lactic acid, oxalic acid,
para-
bromophenylsulfonic acid, carbonic acid, succinic acid, citric acid, benzoic
acid and acetic
acid, as well as related inorganic and organic acids. Such pharmaceutically
acceptable salts
thus include sulfate, pyrosulfate, bisulfate, sulfite, bisulfite, phosphate,
monohydrogenphosphate, dihydrogenphosphate, metaphosphate, pyrophosphate,
chloride,
bromide, iodide, acetate, propionate, decanoate, caprylate, acrylate, formate,
isobutyrate,
caprate, heptanoate, propiolate, oxalate, malonate, succinate, suberate,
sebacate, fumarate,
maleate, butyne-1,4-dioate, hexyne-1,6-dioate, benzoate, chlorobenzoate,
methylbenzoate,
dinitrobenzoate, hydroxybenzoate, methoxybenzoate, phthalate, terephthalate,
sulfonate,
xylene sulfonate, phenylacetate, phenylpropionate, phenylbutyrate, citrate,
lactate, f3-
hydroxybutyrate, glycolate, maleate, tartrate, methanesulfonate,
propanesulfonate,
naphthalene-1-sulfonate, naphthalene-2- sulfonate, mandelate and other salts.
In one
embodiment, pharmaceutically acceptable acid addition salts include those
formed with
9
CA 03041819 2019-04-25
WO 2018/080591 PCT/US2017/029920
mineral acids such as hydrochloric acid and hydrobromic acid, and especially
those formed
with organic acids such as maleic acid.
[49] The term "stable compounds," as used herein, refers to compounds which
possess
stability sufficient to allow for their manufacture and which maintain the
integrity of the
compound for a sufficient period of time to be useful for the purposes
detailed herein (e.g.,
formulation into therapeutic products, intermediates for use in production of
therapeutic
compounds, isolatable or storable intermediate compounds, treating a disease
or condition
responsive to therapeutic agents).
[50] The term "bioequivalent", as used herein, means a drug product showing
the absence
of a significant difference in the rate and extent to which the active
ingredient or active
moiety in a pharmaceutical equivalent to the drug product becomes available at
the site of
drug action when administered at the same molar dose under similar conditions
in an
appropriately designed study, wherein "significant difference" means that the
90%
Confidence Intervals (CI) of the test drug product must fit between 80%-125%
of the
reference drug product (see Online Training Seminar: "The FDA Process for
Approving
Generic Drugs"; www.fda.gov/Training/For HealthProfessionals/ucm090320.htm).
The Food
and Drug Administration (FDA) has issued guidelines regarding bioequivalent
drug products
including specific recommendations on the tolerable variation of inactive
ingredients in a
drug product that would likely render it a pharmaceutically equivalent form.
See, for
example, the FDA's Guidance for Industry: Submission of Summary Bioequivalence
Data for
ANDAs from May 2011, the entire contents of which are incorporated herein.
[51] "D" and "d" both refer to deuterium. "Stereoisomer" refers to both
enantiomers and
diastereomers. "Tert" and "t-" each refer to tertiary. "US" refers to the
United States of
America.
[52] "Substituted with deuterium" refers to the replacement of one or more
hydrogen
atoms with a corresponding number of deuterium atoms.
[53] In one aspect, this disclosure relates to methods of use of Compound (I),
or a
pharmaceutically acceptable salt thereof, involving certain dosing regimens
and certain
pharmaceutical compositions comprising Compound (I), or a pharmaceutically
acceptable
salt thereof. The pharmaceutical compositions and dosing regimens are useful
for treating
conditions mediated by CFTR (cystic fibrosis transmembrane conductance
regulator). In
particular, Compound (I), or a pharmaceutically acceptable salt thereof and
the
pharmaceutical compositions and methods are useful for treating conditions
that can be
treated by compounds that potentiate the activity of CFTR.
CA 03041819 2019-04-25
WO 2018/080591 PCT/US2017/029920
[54] The prescribing information for ivacaftor 150 mg tablets (marketed under
the
tradename Kalydeco ) indicates that one 150 tablet should be taken orally
every 12 hours
with fat-containing food. It has now been found that Compound (I), as
described herein, can
be administered with food having low, moderate, or high fat content, and the
patient's
exposure to CTP-656 is similar regardless of the fat content of a meal.
[55] In one aspect, the disclosure provides methods for treating conditions
that can be
treated by compounds that potentiate the activity of CFTR. The disclosure
provides a method
of treating a condition that is mediated by CFTR in a subject comprising
orally administering
to the subject an effective amount of Compound (I):
OH CD3
CD3
0 0
lel CD3
0 I N
H
N
H (I),
or a pharmaceutically acceptable salt thereof; wherein the Compound (I) is
administered to
the subject with food containing less than 60 g of fat. In certain
embodiments, Compound (I)
can be administered as a pharmaceutical composition, e.g., together with
carrier, fillers,
excipients, and the like.
[56] In certain embodiments, the condition is cystic fibrosis.
[57] In certain of the above embodiments, 50-200 mg (or 50 mg, 75 mg, 100 mg,
or 150
mg) of Compound (I), or a pharmaceutically acceptable salt thereof, is
administered to the
subject.
[58] In certain of the above embodiments, Compound (I), or a pharmaceutically
acceptable
salt thereof, is administered once per day.
[59] In certain of the above embodiments, the Compound (I) is administered in
a
pharmaceutical formulation which is a tablet.
[60] In certain of the above embodiments, the Compound (I) is administered in
a
pharmaceutical formulation which is a granule.
[61] In certain of the above embodiments, the Compound (I) is administered as
a
pharmaceutical composition, and the drug loading of Compound (I), or
pharmaceutical salt
thereof, in the composition (that is, the ratio of the weight of the Compound
(I) (or
pharmaceutical salt thereof) to the total weight of the composition (which
includes carriers,
fillers, excipients, and the like)) is not greater than 25%, or not greater
than 20%, or not
11
CA 03041819 2019-04-25
WO 2018/080591 PCT/US2017/029920
greater than 15%. In certain embodiments, the drug loading of Compound (I) (or
pharmaceutical salt thereof) in the composition is between about 10% and 15%,
or between
about 13% and 15%. In certain embodiments, the drug loading of Compound (I)
(or
pharmaceutical salt thereof) in the composition is about 13% to about 14%, or
13.5% to 14%,
or 13.6% to 13.8%.
[62] In certain of the above embodiments, any atom not designated as deuterium
in
Compound (I) is present at its natural isotopic abundance.
[63] In certain of the above embodiments, the plasma concentration of Compound
(I) in
the subject at 8 hours after administration is about 1.5 ¨ 2-fold greater than
the plasma
concentration of Compound (I) at 8 hours when the Compound (I) is administered
without
food. In certain of the above embodiments, the plasma concentration of
Compound (I) in the
subject at 16 hours after administration is about 1.5 ¨ 2-fold greater than
the plasma
concentration of Compound (I) at 16 hours when the Compound (I) is
administered without
food. In certain of the above embodiments, the plasma concentration of
Compound (I) in the
subject at 24 hours after administration is about 1.5 ¨ 2-fold greater than
the plasma
concentration of Compound (I) at 24 hours when the Compound (I) is
administered without
food.
[64] In certain of the above embodiments, the subject is a human adult or a
human
pediatric patient 6 years and older.
[65] In certain of the above embodiments, the food contains less than about 30
g of fat.
[66] In certain of the above embodiments, the food contains less than about 20
g of fat.
[67] In certain of the above embodiments, the food contains less than about 10
g of fat.
[68] In certain of the above embodiments, the food contains between about 0
and about 50
g of fat.
[69] In certain of the above embodiments, the food contains between
essentially no fat, i.e.,
is substantially fat-free.
[70] In certain of the above embodiments, the food contains between about 5
and about 20
g of fat.
[71] In certain of the above embodiments, the food contains about 7 g of fat.
In certain of
the above embodiments, the food contains about 20 g of fat.
[72] In certain embodiments, the food is low-fat food or a low-fat meal. "Low-
fat food" or
a "low-fat meal" includes food or a meal having less than about 10 g of total
fat (e.g., about
7 g total fat or less). In certain embodiments, the food is moderate-fat food
or a moderate-fat
meal. "Moderate-fat food" or a "moderate-fat meal" includes food or a meal
having between
12
CA 03041819 2019-04-25
WO 2018/080591 PCT/US2017/029920
about 10 g of total fat and about 30 g total fat (e.g., about 15 g to about 25
g of total fat, or
about 20 g total fat). In certain embodiments, the food is a small meal.
[73] In certain of the above embodiments, the food comprises cheese and/or
crackers.
[74] In certain of the above embodiments, the condition is cystic fibrosis and
the subject
has one of the following mutations in the CFTR gene: G551D, G1244E, G1349D,
G178R,
G551S, S1251N, S1255P, S549N, S549R, and R117H.
[75] In certain of the above embodiments, for example, 50 mg, 60 mg, 70 mg, 80
mg,
90 mg, 100 mg, 110 mg, 120 mg, 130 mg, 140 mg, 150 mg, 160 mg, 170 mg, 180 mg,
190
mg, or 200 mg of Compound I can be administered once per day. More
specifically, for
example, 75, 100, or 150 mg of Compound I can be administered once per day. In
a particular
embodiment, 100¨ 150 mg of Compound I can be administered once per day. In a
particular
embodiment, 100 mg of Compound I can be administered once per day. In
particular
embodiments, the subject is a human. Alternatively, the method comprises
administering to a
subject an amount of Compound (I), or a pharmaceutically acceptable salt
thereof, once a
day, wherein the amount of Compound (I), or a pharmaceutically acceptable salt
thereof, is in
the range of 25 mg to 75 mg, for example, 25 mg, 37.5 mg, 50 mg, 62.5 mg, or
75 mg,
wherein the subject is a human 2 to less than 6 years of age and less than 14
kg; or
alternatively, is a human 2 to less than 6 years of age and 14 kg or greater.
In one aspect, the
dose for the human 2 to less than 6 years of age and less than 14 kg is 25 mg.
In one aspect,
the dose for the human 2 to less than 6 years of age and greater than 14 kg is
37.5 mg.
[76] In certain of the above embodiments, Compound I can be administered
immediately
before, concurrently with, or immediately after (e.g., within 30 minutes
after) the
administration or consumption of food.
[77] In certain of the above embodiments, Compound I can be administered to
the subject
with food without a pre-specified fat level or without requiring a high fat
content, e.g., a food
or meal containing less than 60 g of fat.
[78] In another aspect, the disclosure provides a kit for use in treating a
condition that is
mediated by CFTR in a subject. The kit comprises an effective amount of
Compound (I), or a
pharmaceutical composition comprising an effective amount of Compound (I):
13
CA 03041819 2019-04-25
WO 2018/080591 PCT/US2017/029920
OH CD3
CD3
0 0
lel CD3
0 I N
H
N
H (I),
or a pharmaceutically acceptable salt thereof; and instructions for
administering the
compound or the pharmaceutical composition to the subject with food without a
pre-specified
fat level or without requiring a high fat content, e.g., a food or meal
containing less than 60 g
of fat. The pharmaceutical composition can include carriers, fillers,
excipients, and the like.
[79] In certain embodiments, the condition is cystic fibrosis.
[80] In certain of the above embodiments, 50-200 mg (or 50 mg, 75 mg, 100 mg,
or 150
mg) of Compound (I), or a pharmaceutically acceptable salt thereof, is
administered to the
subject.
[81] In certain of the above embodiments, Compound (I), or a pharmaceutically
acceptable
salt thereof, is administered once per day.
[82] In certain of the above embodiments, the Compound (I) is administered in
a
pharmaceutical formulation which is a tablet.
[83] In certain of the above embodiments, the Compound (I) is administered in
a
pharmaceutical formulation which is a granule.
[84] In certain of the above embodiments, the drug loading of Compound (I) (or
pharmaceutical salt thereof) in the composition (the ratio of the weight of
the Compound (I)
(or pharmaceutical salt thereof) to the total weight of the composition (which
includes
carriers, fillers, excipients, and the like) is not greater than 25%, or not
greater than 20%, or
not greater than 15%. In certain embodiments, the drug loading of Compound (I)
(or
pharmaceutical salt thereof) in the composition is between about 10% and 15%,
or between
about 13% and 15%. In certain embodiments, the drug loading of Compound (I)
(or
pharmaceutical salt thereof) in the composition is about 13% to about 14%, or
13.5% to 14%,
or 13.6% to 13.8%.
[85] In certain of the above embodiments, any atom not designated as deuterium
in
Compound (I) is present at its natural isotopic abundance.
[86] In certain of the above embodiments, the plasma concentration of Compound
(I) in
the subject at 8 hours after administration is about 1.5 ¨ 2-fold greater than
the plasma
concentration of Compound (I) at 8 hours when the Compound (I) is administered
without
food. In certain of the above embodiments, the plasma concentration of
Compound (I) in the
14
CA 03041819 2019-04-25
WO 2018/080591 PCT/US2017/029920
subject at 16 hours after administration is about 1.5 ¨ 2-fold greater than
the plasma
concentration of Compound (I) at 16 hours when the Compound (I) is
administered without
food. In certain of the above embodiments, the plasma concentration of
Compound (I) in the
subject at 24 hours after administration is about 1.5 ¨ 2-fold greater than
the plasma
concentration of Compound (I) at 24 hours when the Compound (I) is
administered without
food.
[87] In certain of the above embodiments, the subject is a human adult or a
human
pediatric patient 6 years and older.
[88] In certain of the above embodiments, the food contains less than about 30
g of fat.
[89] In certain of the above embodiments, the food contains less than about 20
g of fat.
[90] In certain of the above embodiments, the food contains less than about 10
g of fat.
[91] In certain of the above embodiments, the food contains between about 0
and about 50
g of fat.
[92] In certain of the above embodiments, the food contains between
essentially no fat, i.e.,
is substantially fat-free.
[93] In certain of the above embodiments, the food contains between about 5
and about 20
g of fat.
[94] In certain of the above embodiments, the food contains about 7 g of fat.
In certain of
the above embodiments, the food contains about 20 g of fat.
[95] In certain embodiments, the food is low-fat food or a low-fat meal. "Low-
fat food" or
a "low-fat meal" includes food or a meal having less than about 10 g of total
fat (e.g., about
7 g total fat or less). In certain embodiments, the food is moderate-fat food
or a moderate-fat
meal. "Moderate-fat food" or a "moderate-fat meal" includes food or a meal
having between
about 10 g of total fat and about 30 g total fat (e.g., about 15 g to about 25
g of total fat, or
about 20 g total fat). In certain embodiments, the food is a small meal.
[96] In certain of the above embodiments, the food comprises cheese and/or
crackers.
[97] In certain of the above embodiments, the condition is cystic fibrosis and
the subject
has one of the following mutations in the CFTR gene: G551D, G1244E, G1349D,
G178R,
G551S, S1251N, S1255P, S549N, S549R, and R117H.
[98] In certain of the above embodiments, for example, 50 mg, 60 mg, 70 mg, 80
mg,
90 mg, 100 mg, 110 mg, 120 mg, 130 mg, 140 mg, 150 mg, 160 mg, 170 mg, 180 mg,
190
mg, or 200 mg of Compound I can be administered once per day. More
specifically, for
example, 75, 100, or 150 mg of Compound I can be administered once per day. In
a particular
embodiment, 100¨ 150 mg of Compound I can be administered once per day. In a
particular
CA 03041819 2019-04-25
WO 2018/080591 PCT/US2017/029920
embodiment, 100 mg of Compound I can be administered once per day. In
particular
embodiments, the subject is a human. Alternatively, Compound (I), or a
pharmaceutically
acceptable salt thereof, is administered once a day, wherein the amount of
Compound (I), or a
pharmaceutically acceptable salt thereof, is in the range of 25 mg to 75 mg,
for example, 25
mg, 37.5 mg, 50 mg, 62.5 mg, or 75 mg, wherein the subject is a human 2 to
less than 6
years of age and less than 14 kg; or alternatively, is a human 2 to less than
6 years of age and
14 kg or greater. In one aspect, the dose for the human 2 to less than 6 years
of age and less
than 14 kg is 25 mg. In one aspect, the dose for the human 2 to less than 6
years of age and
greater than 14 kg is 37.5 mg.
[99] In certain of the above embodiments, Compound I can be administered
immediately
before, concurrently with, or immediately after (e.g., within 30 minutes
after) the
administration or consumption of food.
[100] Conditions treatable by the methods and kits disclosed herein include
cystic fibrosis,
Hereditary emphysema, Hereditary hemochromatosis, Coagulation-Fibrinolysis
deficiencies,
such as Protein C deficiency, Type 1 hereditary angioedema, Lipid processing
deficiencies,
such as Familial hypercholesterolemia, Type 1 chylomicronemia,
Abetalipoproteinemia,
Lysosomal storage diseases, such as I-cell disease/Pseudo-Hurler,
Mucopolysaccharidoses,
Sandhof/Tay-Sachs, Crigler-Najjar type II, Polyendocrinopathy/Hyperinsulemia,
Diabetes
mellitus, Laron dwarfism, Myleoperoxidase deficiency, Primary
hypoparathyroidism,
Melanoma, Glycanosis CDG type 1, Hereditary emphysema, Congenital
hyperthyroidism,
Osteogenesis imperfecta, Hereditary hypofibrinogenemia, ACT deficiency,
Diabetes
insipidus (DI), Neurophyseal DI, Neprogenic DI, Charcot-Marie Tooth syndrome,
Perlizaeus-
Merzbacher disease, neurodegenerative diseases such as Alzheimer's disease,
Parkinson's
disease, Amyotrophic lateral sclerosis, Progressive supranuclear palsy, Pick's
disease, several
polyglutamine neurological disorders such as Huntington, Spinocerebullar
ataxia type I,
Spinal and bulbar muscular atrophy, Dentatorubal pallidoluysian, and Myotonic
dystrophy, as
well as Spongiform encephalopathies, such as Hereditary Creutzfeldt-Jakob
disease, Fabry
disease, Straussler-Scheinker syndrome, chronic obstructive pulmonary disease
(COPD), dry-
eye disease, Sjogren's disease, and a bile duct disorder or a kidney ion
channel disorder,
including, but not limited to, Bartter's syndrome and Dent's disease.
[101] In a specific embodiment, the condition is cystic fibrosis in a subject
such as a human
patient in need thereof. In another specific embodiment, the condition is
chronic obstructive
pulmonary disease in a subject such as a human patient in need thereof. In
certain
embodiments, the subject is a human patient having the G551D-CFTR mutation. In
certain
16
CA 03041819 2019-04-25
WO 2018/080591 PCT/US2017/029920
embodiments, the subject is a human patient having one of the following
mutations in the
CFTR gene: G178R, S549N, S549R, G551S, G1244E, S1251N, S1255P or G1349D. In
certain embodiments, the subject is a human patient having one of the
following mutations in
the CFTR gene: G551D, G1244E, G1349D, G178R, G551S, S1251N, S1255P, S549N,
S549R and R117H. In another example of the foregoing embodiments, the compound
is
administered orally once a day.
[102] In certain of the foregoing embodiments, the compound is administered
optionally in
combination with a second agent. In certain embodiments, the subject is a
human patient
having the AF508-CFTR mutation. Examples of second agents include CFTR
correctors,
such as lumacaftor (VX-809) or tezacaftor (VX-661). In some embodiments
wherein
Compound (I), or a pharmaceutically acceptable salt thereof, is administered
optionally in
combination with a second agent, the amount of Compound (I), or a
pharmaceutically
acceptable salt thereof, is administered once daily at 50 mg to 200 mg each
time, for
example, about 50 mg, 60 mg, 70 mg, 80 mg, 90 mg, 100 mg, 110 mg, 120 mg, 130
mg,
140 mg, 150 mg, 160 mg, 170 mg, 180 mg, 190 mg, or about 200 mg.
Alternatively, wherein
Compound (I), or a pharmaceutically acceptable salt thereof, is administered
optionally in
combination with a second agent, the amount of Compound (I), or a
pharmaceutically
acceptable salt thereof, is administered once daily at 25 mg to 75 mg each
time, for example,
about 25 mg, 37.5 mg, 50 mg, 62.5 mg, or about 75 mg.
[103] Effective doses will also vary, as recognized by those skilled in the
art, depending on
the diseases treated, the severity of the disease, the route of
administration, the sex, age and
general health condition of the subject, excipient usage, the possibility of
co-usage with other
therapeutic treatments such as use of other agents and the judgment of the
treating physician.
[104] For pharmaceutical compositions that comprise a second therapeutic
agent, an
effective amount of the second therapeutic agent is between about 20% and 100%
of the
dosage normally utilized in a monotherapy regime using just that agent.
Preferably, an
effective amount is between about 70% and 100% of the normal monotherapeutic
dose. The
normal monotherapeutic dosages of these second therapeutic agents are well
known in the art.
See, e.g., Wells et al., eds., Pharmacotherapy Handbook, 2nd Edition, Appleton
and Lange,
Stamford, Conn. (2000); PDR Pharmacopoeia, Tarascon Pocket Pharmacopoeia 2000,
Deluxe Edition, Tarascon Publishing, Loma Linda, Calif. (2000), each of which
references
are incorporated herein by reference in their entirety.
[105] It is expected that some of the second therapeutic agents referenced
above will act
synergistically with the compounds of this invention. When this occurs, it
will allow the
17
CA 03041819 2019-04-25
WO 2018/080591 PCT/US2017/029920
effective dosage of the second therapeutic agent and/or Compound (I), or a
pharmaceutically
acceptable salt thereof, to be reduced from that required in a monotherapy.
This has the
advantage of minimizing toxic side effects of either the second therapeutic
agent or
Compound (I), or a pharmaceutically acceptable salt thereof, synergistic
improvements in
efficacy, improved ease of administration or use and/or reduced overall
expense of compound
preparation or formulation.
[106] In another embodiment, any of the above methods of treatment comprises
the further
step of co-administering to the subject in need thereof one or more second
therapeutic agents.
The choice of second therapeutic agent may be made from any second therapeutic
agent
known to be useful for co-administration with ivacaftor. The choice of second
therapeutic
agent is also dependent upon the particular disease or condition to be
treated. Examples of
second therapeutic agents that may be employed in the methods of this
invention are those set
forth above for use in combination compositions comprising Compound (I), or a
pharmaceutically acceptable salt thereof, and a second therapeutic agent.
[107] In particular, the combination therapies of this invention include co-
administering a
Compound (I), or a pharmaceutically acceptable salt thereof, or a
pharmaceutically
acceptable salt thereof and a second therapeutic agent such as VX-809
(lumacaftor) or VX-
661 (tezacaftor), to a subject in need thereof for treatment. In certain
embodiments, the
subject is a human patient having the AF508-CFTR mutation (in particular, a
human patient
homozygous for the F508del mutation).
[108] The term "co-administered" as used herein means that the second
therapeutic agent
may be administered together with Compound (I), or a pharmaceutically
acceptable salt
thereof, as part of a single dosage form (such as a composition of this
invention comprising a
compound of the invention and an second therapeutic agent as described above)
or as
separate, multiple dosage forms. Alternatively, the additional agent may be
administered
prior to, consecutively with, or following the administration of Compound (I),
or a
pharmaceutically acceptable salt thereof. In such combination therapy
treatment, both
Compound (I), or a pharmaceutically acceptable salt thereof, and the second
therapeutic
agent(s) are administered by conventional methods. The administration of a
composition of
this invention, comprising both Compound (I), or a pharmaceutically acceptable
salt thereof,
and a second therapeutic agent, to a subject does not preclude the separate
administration of
that same therapeutic agent, any other second therapeutic agent or Compound
(I), or a
pharmaceutically acceptable salt thereof, to said subject at another time
during a course of
treatment.
18
CA 03041819 2019-04-25
WO 2018/080591 PCT/US2017/029920
[109] Effective amounts of these second therapeutic agents are well known to
those skilled
in the art and guidance for dosing may be found in patents and published
patent applications
referenced herein, as well as in Wells et al., eds., Pharmacotherapy Handbook,
2nd Edition,
Appleton and Lange, Stamford, Conn. (2000); PDR Pharmacopoeia, Tarascon Pocket
Pharmacopoeia 2000, Deluxe Edition, Tarascon Publishing, Loma Linda, Calif.
(2000), and
other medical texts. However, it is well within the skilled artisan's purview
to determine the
second therapeutic agent's optimal effective-amount range.
[110] In one embodiment of the invention, the effective amount of the second
therapeutic
agent is less than its effective amount would be where Compound (I), or a
pharmaceutically
acceptable salt thereof, is not administered. In this way, undesired side
effects associated
with high doses of either agent may be minimized. Other potential advantages
(including
without limitation improved dosing regimens and/or reduced drug cost) will be
apparent to
those of skill in the art.
[111] In yet another aspect, the invention provides the use of Compound (I),
or a
pharmaceutically acceptable salt thereof, alone or together with one or more
of the above-
described second therapeutic agents in the manufacture of a medicament, either
as a single
composition or as separate dosage forms, for treatment or prevention in a
subject of a disease,
disorder or symptom set forth above. Another aspect of the invention is
Compound (I), or a
pharmaceutically acceptable salt thereof, for use in the treatment or
prevention in a subject of
a disease, disorder or symptom thereof delineated herein.
[112] In one embodiment, any atom not designated as deuterium is present at
its natural
isotopic abundance in Compound (I), or a pharmaceutically acceptable salt
thereof.
[113] The synthesis of Compound (I), or a pharmaceutically acceptable salt
thereof, may be
readily achieved by the methods described U.S. Patent No. 8,865,902, the
teachings of which
are incorporated herein by reference. In particular, Example 3 of U.S. Patent
No. 8,865,902
describes a synthesis of Compound (I).
[114] Such methods can be carried out utilizing corresponding deuterated and
optionally,
other isotope-containing reagents and/or intermediates to synthesize the
compounds
delineated herein, or invoking standard synthetic protocols known in the art
for introducing
isotopic atoms to a chemical structure.
[115] The invention also provides pharmaceutical compositions comprising an
effective
amount of Compound (I), or a pharmaceutically acceptable salt thereof; and a
pharmaceutically acceptable carrier. The carrier(s) are "acceptable" in the
sense of being
compatible with the other ingredients of the formulation and, in the case of a
19
CA 03041819 2019-04-25
WO 2018/080591 PCT/US2017/029920
pharmaceutically acceptable carrier, not deleterious to the recipient thereof
in an amount used
in the medicament.
[116] Pharmaceutically acceptable carriers, adjuvants and vehicles that may be
used in the
pharmaceutical compositions of this invention include, but are not limited to,
ion exchangers,
alumina, aluminum stearate, lecithin, serum proteins, such as human serum
albumin, buffer
substances such as phosphates, glycine, sorbic acid, potassium sorbate,
partial glyceride
mixtures of saturated vegetable fatty acids, water, salts or electrolytes,
such as protamine
sulfate, disodium hydrogen phosphate, potassium hydrogen phosphate, sodium
chloride, zinc
salts, colloidal silica, magnesium trisilicate, polyvinyl pyrrolidone,
cellulose-based
substances, polyethylene glycol, sodium carboxymethylcellulo se,
polyacrylates, waxes,
polyethylene-polyoxypropylene-block polymers, polyethylene glycol and wool
fat.
[117] If required, the solubility and bioavailability of the compounds of the
present
invention in pharmaceutical compositions may be enhanced by methods well-known
in the
art. One method includes the use of lipid excipients in the formulation. See
"Oral Lipid-
Based Formulations: Enhancing the Bioavailability of Poorly Water-Soluble
Drugs (Drugs
and the Pharmaceutical Sciences)," David J. Hauss, ed. Informa Healthcare,
2007; and "Role
of Lipid Excipients in Modifying Oral and Parenteral Drug Delivery: Basic
Principles and
Biological Examples," Kishor M. Wasan, ed. Wiley-Interscience, 2006.
[118] Another known method of enhancing bioavailability is the use of an
amorphous form
of a compound of this invention optionally formulated with a poloxamer, such
as LUTROLTm
and PLURONICTM (BASF Corporation), or block copolymers of ethylene oxide and
propylene oxide. See United States patent 7,014,866; and United States patent
publications
20060094744 and 20060079502.
[119] The pharmaceutical compositions of the invention include those suitable
for oral
administration. Other formulations may conveniently be presented in unit
dosage form, e.g.,
tablets, sustained release capsules, granules, and in liposomes, and may be
prepared by any
methods well known in the art of pharmacy. See, for example, Remington: The
Science and
Practice of Pharmacy, Lippincott Williams & Wilkins, Baltimore, MD (20th ed.
2000).
[120] Such preparative methods include the step of bringing into association
with the
molecule to be administered ingredients such as the carrier that constitutes
one or more
accessory ingredients. In general, the compositions are prepared by uniformly
and intimately
bringing into association the active ingredients with liquid carriers,
liposomes or finely
divided solid carriers, or both, and then, if necessary, shaping the product.
CA 03041819 2019-04-25
WO 2018/080591 PCT/US2017/029920
[121] In certain embodiments, the compound is administered orally.
Compositions of the
present invention suitable for oral administration may be presented as
discrete units such as
capsules, sachets, or tablets each containing a predetermined amount of the
active ingredient;
a powder or granules; a solution or a suspension in an aqueous liquid or a non-
aqueous liquid;
an oil-in-water liquid emulsion; a water-in-oil liquid emulsion; packed in
liposomes; or as a
bolus, etc. Soft gelatin capsules can be useful for containing such
suspensions, which may
beneficially increase the rate of compound absorption. In a specific
embodiment, the
compound is administered orally as a tablet. Alternatively, the compound is
administered
orally as a granule.
[122] In the case of tablets for oral use, carriers that are commonly used
include lactose and
corn starch. Lubricating agents, such as magnesium stearate, are also
typically added. For
oral administration in a capsule form, useful diluents include lactose and
dried cornstarch.
When aqueous suspensions are administered orally, the active ingredient is
combined with
emulsifying and suspending agents. If desired, certain sweetening and/or
flavoring and/or
coloring agents may be added. In another embodiment, the composition is in the
form of a
tablet. In certain embodiments, exemplary formulations for the tablet are
disclosed in US.
Patent No. 8,754,224, the teachings of which are herein incorporated by
reference.
[123] In a particular embodiment, the tablet contains 150 mg of Compound (I),
or a
pharmaceutically acceptable salt thereof, and the following inactive
ingredients: colloidal
silicon dioxide, croscarmellose sodium, hypromellose acetate succinate,
lactose monohydrate,
magnesium stearate, microcrystalline cellulose, and sodium lauryl sulfate. In
certain
embodiments, the tablet film coat contains carnauba wax, FD&C Blue #2, PEG
3350,
polyvinyl alcohol, talc, and titanium dioxide. In certain embodiments, the
tablet is printed
with a printing ink; the printing ink may contain ammonium hydroxide, iron
oxide black,
propylene glycol, and shellac. In another particular embodiment, the tablet
contains 75 mg of
Compound I (CTP-656, D9-ivacaftor), together with the following inactive
ingredients:
microcrystalline cellulose, lactose monohydrate, colloidal silicon dioxide,
croscarmellose
sodium, magnesium stearate, and sodium lauryl sulfate. Multiple tablets may be
administered
to provide a suitable once-daily dose (e.g., two 75 mg tablets administered
together for a
150 mg once-daily dose). In another particular embodiment, the tablet
comprises granules
compressed with extra-granular material; the granules comprise about 17.1
percent (by
weight of the tablet) of an amorphous dispersion of Compound I (wherein the
amorphous
dispersion comprises about 80% substantially amorphous Compound I by weight of
the
dispersion, hypromellose acetate succinate (HPMCAS) (about 19.5 percent by
weight of the
21
CA 03041819 2019-04-25
WO 2018/080591 PCT/US2017/029920
dispersion) and sodium laurel sulfate (about 0.5 percent by weight of the
dispersion)), a
compression aid such as microcrystalline cellulose (e.g., Avicel PH101) (about
39.0 percent
by weight of the tablet), a diluent/filler such as lactose monohydrate 316
(about 38.9 percent
by weight of the tablet) a surfactant such as sodium lauryl sulfate (SLS)
(about 0.50 percent
by weight of the tablet), a disintegrant such as croscarmellose sodium (e.g.,
Ac-di-sol) (about
1.50 percent by weight of the tablet) and a lubricant such as Hyqual magnesium
stearate
(about 0.20 percent by weight of the tablet), and the extracellular matrix
comprises a
disintegrant such as croscarmellose sodium (e.g., Ac-di-sol) (about 1.50
percent by weight of
the tablet), a glidant such as colloidal silicon dioxide (about 0.50 percent
by weight of the
tablet) and additional lubricant such as magnesium stearate (e.g., Hyqual
(about 0.80 percent
by weight of the tablet). Thus, in one embodiment, the invention provides a
pharmaceutical
composition comprising about 17.1 wt % of a solid dispersion by weight of the
composition,
wherein the dispersion comprises about 80 wt % of substantially amorphous
Compound I
(CTP-656) by weight of the dispersion, about 19.5 wt % of hypromellose acetate
succinate
(HPMCAS) by weight of the dispersion, and about 0.5 wt % SLS by weight of the
dispersion;
about 39.0 wt % of microcrystalline cellulose by weight of the composition;
about 38.9 wt %
of lactose monohydrate by weight of the composition; about 3 wt % of sodium
croscarmellose by weight of the composition; about 0.5 wt % of SLS by weight
of the
composition; about 0.5 wt % of colloidal silicon dioxide by weight of the
composition; and
about 0.8 wt % of magnesium stearate by weight of the composition. In certain
embodiments,
the tablet comprises 75 mg of Compound I (CTP-656). In other embodiments, the
tablet
comprises 100 mg of Compound I (CTP-656). In still other embodiments, the
tablet
comprises 150 mg of Compound I (CTP-656).
[124] In another particular embodiment, the granule is enclosed in a unit-dose
packet
containing 25 mg, 50 mg or 75 mg of Compound (I), or a pharmaceutically
acceptable salt
thereof. Each unit-dose packet of Compound (I) oral granules contains 25 mg of
Compound
(I), 50 mg of Compound (I) or 75 mg of Compound (I) and the following inactive
ingredients: colloidal silicon dioxide, croscarmellose sodium, hypromellose
acetate succinate,
lactose monohydrate, magnesium stearate, mannitol, sucralose, and sodium
lauryl sulfate. In
another embodiment, the composition is in the form of a granule. In certain
embodiments,
exemplary formulations for the granule are disclosed in US. Patent No.
8,883,206, the
teachings of which are herein incorporated by reference.
[125] In another embodiment, a composition of this invention further comprises
a second
therapeutic agent. The second therapeutic agent may be selected from any
compound or
22
CA 03041819 2019-04-25
WO 2018/080591 PCT/US2017/029920
therapeutic agent known to have or that demonstrates advantageous properties
when
administered with a compound having the same mechanism of action as ivacaftor.
[126] Preferably, the second therapeutic agent is an agent useful in the
treatment of a variety
of conditions, including cystic fibrosis, Hereditary emphysema, Hereditary
hemochromatosis,
Coagulation-Fibrinolysis deficiencies, such as Protein C deficiency, Type 1
hereditary
angioedema, Lipid processing deficiencies, such as Familial
hypercholesterolemia, Type 1
chylomicronemia, Abetalipoproteinemia, Lysosomal storage diseases, such as I-
cell
disease/Pseudo-Hurler, Mucopolysaccharidoses, Sandhof/Tay-Sachs, Crigler-
Najjar type II,
Polyendocrinopathy/Hyperinsulemia, Diabetes mellitus, Laron dwarfism,
Myleoperoxidase
deficiency, Primary hypoparathyroidism, Melanoma, Glycanosis CDG type 1,
Hereditary
emphysema, Congenital hyperthyroidism, Osteogenesis imperfecta, Hereditary
hypofibrinogenemia, ACT deficiency, Diabetes insipidus (DI), Neurophyseal DI,
Neprogenic
DI, Charcot-Marie Tooth syndrome, Perlizaeus-Merzbacher disease,
neurodegenerative
diseases such as Alzheimer's disease, Parkinson's disease, Amyotrophic lateral
sclerosis,
Progressive supranuclear palsy, Pick's disease, several polyglutamine
neurological disorders
asuch as Huntington, Spinocerebullar ataxia type I, Spinal and bulbar muscular
atrophy,
Dentatorubal pallidoluysian, and Myotonic dystrophy, as well as Spongiform
encephalopathies, such as Hereditary Creutzfeldt-Jakob disease, Fabry disease,
Straussler-
Scheinker syndrome, COPD, dry-eye disease, Sjogren's disease, and a bile duct
disorder or a
kidney ion channel disorder, including, but not limited to, Bartter's syndrome
and Dent's
disease.
[127] In a specific embodiment, the second therapeutic agent is an agent
useful in the
treatment of cystic fibrosis. In certain embodiments, the second therapeutic
agent is an agent
useful in the treatment of cystic fibrosis in a human patient having the G551D-
CFTR
mutation. In certain embodiments, the second therapeutic agent is an agent
useful in the
treatment of cystic fibrosis in a human patient having any of the following
mutations in the
CFTR gene: G178R, 5549N, 5549R, G5515, G1244E, 51251N, 51255P or G1349D. In
certain embodiments, the second therapeutic agent is an agent useful in the
treatment of
cystic fibrosis in a human patient having any of the following mutations in
the CFTR gene:
G551D, G1244E, G1349D, G178R, G5515, S125 1N, 51255P, 5549N, 5549R and R117H.
[128] In one embodiment, the second therapeutic agent is VX-809 (lumacaftor)
or VX-661
(tezacaftor). In certain embodiments, the subject is a human patient having
the AF508-CFTR
mutation (in particular, a human patient homozygous for the F508del mutation).
23
CA 03041819 2019-04-25
WO 2018/080591 PCT/US2017/029920
[129] In another embodiment, the invention provides separate dosage forms of
Compound
(I), or a pharmaceutically acceptable salt thereof, and one or more of any of
the above-
described second therapeutic agents, wherein Compound (I), or a
pharmaceutically
acceptable salt thereof, and second therapeutic agent are associated with one
another. The
term "associated with one another" as used herein means that the separate
dosage forms are
packaged together or otherwise attached to one another such that it is readily
apparent that the
separate dosage forms are intended to be sold and administered together
(within less than
24 hours of one another, consecutively or simultaneously).
[130] In the methods and pharmaceutical compositions of the invention,
Compound (I), or a
pharmaceutically acceptable salt thereof, is present in an effective amount.
As used herein,
the term "effective amount" refers to an amount which, when administered in a
proper dosing
regimen, is sufficient to treat the target disorder.
[131] This disclosure also provides a pharmaceutical product. In one aspect,
the product
comprises: a) an oral dosage form comprising an effective amount of Compound
(I):
OH CD3
CD3
0 0
lel CD3
0 I N
H
N
H (I),
or a pharmaceutically acceptable salt thereof; and b) prescribing information
for
administering the oral dosage form, wherein the prescribing information
includes: i) dosage
and administration information for adults and pediatric patients 6 years and
older instructing
the administration of 50-200 mg of Compound (I) taken orally with food
containing less than
60 g of fat. In certain embodiments, the oral dosage form comprises about 75,
about 100, or
about 150 mg of Compound I. In certain of the above embodiments, the
prescribing
information instructs the oral administration of Compound (I) once per day. In
certain of the
above embodiments, the prescribing information instructs the oral
administration of
Compound (I) with food containing less than 30 g of fat, or less than 20 g of
fat, or less than
g of fat. In certain of the above embodiments, the prescribing information
instructs the
oral administration of Compound (I) with food containing between 0 and 50 g of
fat, or
between 5 and 20 g of fat. In certain embodiments, the prescribing information
instructs the
oral administration of Compound (I) with food containing about 7 g of fat. In
certain
embodiments, the prescribing information instructs the oral administration of
Compound (I)
with food containing about 20 g of fat. In certain embodiments, the
prescribing information
24
CA 03041819 2019-04-25
WO 2018/080591 PCT/US2017/029920
instructs the oral administration of Compound (I) with food containing about
low-fat food or
a low-fat meal. In certain embodiments, the prescribing information instructs
the oral
administration of Compound (I) with moderate-fat food or a moderate-fat meal.
In certain
embodiments, the prescribing information instructs the oral administration of
Compound (I)
with a small meal. In certain embodiments, the pharmaceutical product is used
in the claimed
methods and the claimed kit as described above.
[132] In certain of the above embodiments, the food comprises cheese.
[133] In any of the aspects and embodiments herein, the oral dosage form
comprises a
pharmaceutical composition of Compound (I) with a pharmaceutically acceptable
carrier.
[134] The interrelationship of dosages for animals and humans (based on
milligrams per
meter squared of body surface) is described in Freireich et al., Cancer
Chemother. Rep, 1966,
50: 219. Body surface area may be approximately determined from height and
weight of the
subject. See, e.g., Scientific Tables, Geigy Pharmaceuticals, Ardsley, N.Y.,
1970, 537.
Examples
[135] Example 1. Phase 1 Single Ascending Dose (SAD) clinical trial
[136] Ten healthy male and female volunteers were enrolled in a single
ascending dose
study of 3 doses of CTP-656 (75, 150 and 300 mg), with a cross-over comparison
of 150 mg
CTP-656 and 150 mg Kalydeco (the trade name of ivacaftor) (Figure 5). Doses
of CTP-
656 were administered as an aqueous suspension, and Kalydeco was administered
as a tablet.
All doses of CTP-656 and Kalydeco were administered within 30 minutes after
the start of a
high-fat containing breakfast. There was a 7 day washout between doses. The
objective of the
study was to compare the pharmacokinetics of single ascending doses (75, 150,
and 300 mg)
of CTP-656, to compare the pharmacokinetics of a single dose of 150 mg CTP-656
and 150
mg Kalydeco and to assess the safety and tolerability of CTP-656.
[137] Overall, CTP-656 administered as single doses at doses 75 mg, 150 mg and
300 mg
(following a high-fat meal) was generally well tolerated in healthy male and
female subjects.
Kalydeco tablets, administered as a single oral dose of 150 mg (following a
high-fat meal)
were also generally well tolerated in healthy male and female subjects.
[138] There were no clinically significant differences in safety assessments
between CTP-
656 (75 mg, 150 mg or 300 mg) and Kalydeco (150 mg) and no apparent dose-
related trends
in subjects following CTP-656.
[139] The Tma, for CTP-656 was similar between each of the treatments, with
the apparent
terminal half-life of 14-17 hours. The apparent terminal half-life of 150 mg
of Kalydeco
CA 03041819 2019-04-25
WO 2018/080591 PCT/US2017/029920
(11.18 hours) was shorter than for CTP-656.
[140] For CTP-656, the relationship between dose and exposure was found to be
approximately linear using log-log regression analysis, with the increase in
exposure being
greater than dose proportional across dose levels (75 mg, 150 mg and 300 mg)
(Figures la
and lb).
[141] A summary of the pharmacokinetic properties of CTP-656 and Kalydeco is
presented
in the chart below:
CTP-656 CTP-656 Kalydeco CTP-656
Dosed Drug
75 mg 150 mg 150 mg 300 mg
PK Parameter Mean (CV%)
Tmax 5.00 5.00 5.00 5.00
()a (5.00-12.00) (5.00-10.00) (3.00-12.00) (5.00-10.00)
Cmax 838 2,212 1,101 4,968
(ng/mL) (22) (26) (46) (23)
C24h, 270 712 169 1,540
(ng/mL) (36) (40) (38) (39)
AUC0( 16,581 44,916 12,925 105,179
(ng*hr/mL) (31) (36) (32) (34)
T1/2 14.1 15.0 11.2 17.3
(hr) (17) (21) (16) (14)
CL/F 4.9 3.8 13.3 3.2
(L/hr) (29) (44) (49) (36)
a Median (Range) For 150 mg of CTP-656, Cmax was approximately 2-fold, AUC0f
was
approximately 3.5-fold, and C241r was 4.2-fold that of Kalydeco tablets. The
CL/F
(clearance over bioavailability, a measure of the clearance of an orally-dosed
drug) for CTP-
656 was approximately 30% that of Kalydeco (Figure 2).
[142] A comparison of the pharmacokinetic properties of CTP-656 and Kalydeco
for the
150 mg dose is presented in the chart below:
CTP-656 Kalydeco Ratio of CTP-656 to
Dosed Drug
(n=9) (n=9) Kalydeco
PK Parameter Mean (CV%)
26
CA 03041819 2019-04-25
WO 2018/080591 PCT/US2017/029920
T a
max
5.0-10.0 a 3.0-10.0 ---
(hr)
C12 1306.7 412.8
3.2
(ng/mL) (27) (33)
C24hr 712.2 168.9
4.2
(ng/mL) (40) (38)
AUC0-24hr 27289.0 9876.4
(29) 2.8
(ng*hr/mL) (33)
C max 2212.2 1101.0
( 26) 2.0
(ng/mL) (46)
T1/2 15.00 11.18
(21) 1.3
(hr) (16)
a
range
[143] Therefore, based on the enhanced PK profile for CTP-656 relative to
Kalydeco ,
CTP-656 has potential to show efficacy at doses in the range of 50-200 mg QD
(once daily).
[144] Example 2. Parent vs. Metabolite Pharmacokinetic Profile
[145] As shown in Figure 3, deuteration dramatically impacts the metabolism of
the
deuterated ivacaftor analog CTP-656 compared to ivacaftor after a single dose.
Ivacaftor,
CTP-656, and their metabolites are shown in Figure 6. There is a significant
reduction in the
production of the metabolites D8-M1 and D6-M6 from CTP-656 relative to the
production of
the metabolites M1 and M6 from ivacaftor. Thus, the parent-to-MI ratio of AUC0-
241r for
CTP-656/D8-M1 is 2.0, compared to 0.58 for the ratio of ivacaftor to Ml. The
parent-to-M1
ratio of C. and C24hr for CTP-656/D8-M1 are 2.1 and 2.2, respectively,
compared to 0.54
and 0.55, respectively, for ivacaftor/M1. Further, the parent-to-M6 ratio of
AUC0_241r for
CTP-656/D6-M6 is 4.0, compared to 1.5 for the ratio of ivacaftor to M6. The
parent-to-M6
ratio of C. and C24hr for CTP-656/D6-M6 are 4.3 and 2.5 respectively, compared
to 1.4 and
0.97, respectively, for ivacaftor/M6. As seen in Figure 3 (a), CTP-656 is the
most abundant
species in plasma at all times measured, in contrast to ivacaftor (b), where
the M1 metabolite
predominates after 2 hours, and the level of the M6 metabolite reaches the
level of ivacaftor
after about 8 hours. As a result, the most pharmacologically-active species
(i.e., the parent) is
the most abundant species for CTP-656, but not for ivacaftor.
CTP-656 PK Parameters: Parent/Metabolite Ratios
27
CA 03041819 2019-04-25
WO 2018/080591 PCT/US2017/029920
AUC 0-24hr Cmax C24hr
CTP-656/D8-M1 2.0 2.1 2.2
CTP-656/D6-M6 4.0 4.3 2.5
Ivacaftor PK Parameters: Parent/Metabolite Ratios
AUC 0-24hr Cmax C24hr
Ivacaftor/M1 0.58 0.54 0.55
Ivacaftor/M6 1.5 1.4 0.97
[146] In summary, CTP-656 demonstrated a superior pharmacokinetic profile
compared to
Kalydeco, the current standard of care for treatment of cystic fibrosis
patients. Results of the
Phase 1 trial also showed that CTP-656 was well-tolerated and its safety
profile was
comparable to Kalydeco. In the Phase 1 cross-over comparison of CTP-656 and
Kalydeco,
CTP-656 demonstrated a superior pharmacokinetic profile compared to Kalydeco
including a
reduced rate of clearance, longer half-life, substantially increased exposure
and greater
plasma levels at 24 hours. An analysis of metabolites in plasma also showed
that the overall
exposure profile of CTP-656 differed from that of Kalydeco in that the
majority of plasma
exposure in the case of CTP-656 was due to parent drug, whereas with Kalydeco
the majority
of plasma exposure was due to a less-active metabolite and an approximately
equivalent
amount of an inactive metabolite was also observed. It is believed that the
longer half-life
and lower metabolite levels measured for CTP-656 offer a dosing advantage to
the patient.
[147] Example 3- Measurement of CTP-656 and Ivacaftor activity
[148] Chloride transport of G551D-CFTR overexpressed in Fischer Rat Thyroid
cells was
measured in an Ussing chamber apparatus. The short circuit current (TO was
monitored after
basolateral permeabilization with 10011M amphotericin and activation of CFTR
by 1011M
forskolin. Testing was performed in the presence of a chloride gradient at 35
C. Test articles
were applied in an additive and sequential manner to epithelia at 0.0008 p,M,
0.004 p,M,
0.02 p,M, and 0.111M along with 0.5 [IL, 2.0 [IL, 0.5 [IL, and 2.5 [IL
additions of the DMSO
vehicle. Values are the means of responses from each test concentration
applied to six
epithelia. (Figure 4A)
[149] Chloride transport of homozygous F508del-CFTR human bronchial epithelial
cell
monolayers (patient code CFFT027G) was measured in an Ussing chamber
apparatus. The
short circuit current (TO was monitored after blockade of sodium current
through the
epithelial sodium channel (ENaC) with 3011M amiloride and activation of CFTR
by 1011M
28
CA 03041819 2019-04-25
WO 2018/080591 PCT/US2017/029920
forskolin. Symmetric physiologic saline solutions at 27 C were used for
temperature
correction of F508del-CFTR. Test articles were applied in an additive and
sequential manner
to epithelia at 0.0008 pM, 0.004 pM, 0.02 pM, and 0.111M along with 0.5 [IL,
2.0 [IL, 0.5 [IL,
and 2.5 [IL additions of the DMSO vehicle. Values are the means of responses
from each test
concentration applied to six epithelia. (Figure 4B and Figure 4C).
[150] Therefore, D9-, D18-ivacaftor and ivacaftor provided equivalent in vitro
CFTR
potentiation.
OH CD3
CD3
0 0 #CD3
0 I N
H
N D3C CD3
H CD3
D18-ivacaftor
[151] Example 4- Human Crossover Study for D9-ivacaftor and D18-ivacaftor
[152] Six healthy volunteers were enrolled in a cross-over comparison of 25 mg
D9-
ivacaftor (CTP-656) and 25 mg D18-ivacaftor (Figure 7A). The objective of the
study was to
compare the pharmacokinetics of a single dose of 25 mg D9-ivacaftor and 25 mg
D18-
ivacaftor. Doses of D9-ivacaftor (CTP-656) and D18-ivacaftor were administered
as an
aqueous suspension. All doses of D9-ivacaftor (CTP-656) and D18-ivacaftor were
administered to fasted subjects (three subjects per group). There was a 7 day
washout
between doses.
[153] A comparison of the pharmacokinetic properties of D9-ivacaftor and D18-
ivacaftor is
presented in the chart below:
Dosed Drug D9-ivacaftor D18-ivacaftor
Mean (CV%)
PK Parameter
T max 3.0 2.5
(ho a (2.0-4.0) (2.0-5.0)
Cmax 270 233
(ng/mL) (24) (18)
C24hr 52.6 42.3
(ng/mL) (28) (16)
29
CA 03041819 2019-04-25
WO 2018/080591 PCT/US2017/029920
AUCO-inf 3,812 3,196
(ng*hr/mL) (26) (15)
a Median (Range)
[154] It was found that D9-ivacaftor showed a superior PK profile compared to
D18-
ivacaftor (Figure 7B).
[155] Example 5- Preparation of tablet form
[156] CTP-656 (D9-ivacaftor) (17.1% by weight of an 80% amorphous dispersion
(the
dispersion contains 80% of Compound I by weight of the dispersion,
hypromellose acetate
succinate (HPMCAS) (about 19.5 percent by weight of the dispersion) and sodium
laurel
sulfate (about 0.5 percent by weight of the dispersion)) was blended with
microcrystalline
cellulose (Avicel PH101) (39.00 percent by weight), lactose monohydrate 316
(38.9 percent
by weight) sodium lauryl sulfate (0.50 percent by weight), croscarmellose
sodium (Ac-di-sol)
(1.50 percent by weight) and Hyqual magnesium stearate (0.20 percent by
weight) in a bottle
blender. The blend was compacted and milled to form granules, which are sieved
through
#20 and #80 mesh sieves. The granules and remaining fines are blended with
additional
croscarmellose sodium (Ac-di-sol) (1.50 percent by weight), colloidal silicon
dioxide (0.50
percent by weight) and additional Hyqual magnesium stearate (0.80 percent by
weight), and
the final blend compressed into tablets using a rotary press. Each tablet
contains 75 mg CTP-
656 (D9-ivacaftor).
[157] Example 6- Human Crossover Study for D9-ivacaftor and ivacaftor
[158] Healthy volunteers were enrolled in a cross-over comparison of 150 mg D9-
ivacaftor
(CTP-656) and 150 mg ivacaftor (Kalydeco) (Figure 8, left panel). The
objective of the study
was to compare the safety, tolerability, and pharmacokinetics of a single dose
of 150 mg D9-
ivacaftor and 150 mg ivacaftor. Doses of D9-ivacaftor (CTP-656) and ivacaftor
were
administered as tablets (the D9-ivacaftor was administered as two 75 mg
tablets). All doses
of D9-ivacaftor (CTP-656) and ivacaftor (Kalydeco) were administered to fed
subjects (high
fat breakfast) (four subjects per sequence). There was a 7 day washout between
doses. Blood
samples were taken at intervals.
[159] The results are shown in Figures 9 and 10. It was found that CTP-656 had
approximately 3-fold enhanced C241r and AUC0_241r compared to ivacaftor
(Figure 9). Oral
clearance of CTP-656 was about one-third that of ivacaftor. The half-life of
CTP-656 was
about 15 hours, which is about 40% greater than the half-life of ivacaftor
(about 11 hours).
Further, the ratio of CTP-656 to metabolites D-M1 and D-M6 (see Figure 6) was
higher than
CA 03041819 2019-04-25
WO 2018/080591 PCT/US2017/029920
the ratio of ivacaftor to the metabolites M1 and M6 (Figure 10).
[160] Example 7- Human Multiple Ascending Dose Study for D9-ivacaftor
[161] Healthy volunteers were enrolled in a multiple-ascending dose study of
D9-ivacaftor
(CTP-656) (Figure 8, right panel). The objective of the study was to compare
the safety,
tolerability, and pharmacokinetics of D9-ivacaftor at three doses for seven
days, compared to
placebo. Doses of D9-ivacaftor (CTP-656) and placebo were administered as
tablets (the D9-
ivacaftor was administered as one, two or three 75 mg tablets, for doses of 75
mg, 150 mg, or
225 mg). All doses of D9-ivacaftor (CTP-656) were administered to fed subjects
(high fat
breakfast; about 60 g fat)) (8 subjects per sequence received CTP-656, two
subjects per
sequence received placebo). There was a 7 day washout between doses. Blood
samples were
taken at intervals and the plasma concentration of CTP-656.
[162] The results are shown in Figure 11. It was found that steady state
plasma levels of
CTP-656 were reached after about three days of dosing. CTP-656 showed a dose-
proportional increase in exposure with repeated dosing for the 150 mg dose
relative to the
75 mg dose. The 225 mg dose group showed higher than dose-proportional
exposure. The
ratio of CTP-656 to metabolites D-M1 and D-M6 in plasma was greater than one.
The CTP-
656 and D-M1 accumulation ratio was about 1-6 to 1.8 for key exposure
parameters C241r and
AUC0-241r. No serious adverse events were reported; the majority of adverse
events reported
were mild in severity.
[163] Example 8 ¨ Food-Effect Study for D9-ivacaftor
[164] The relative bioavailability of CTP-656 under fasted and fed conditions,
with low and
moderate amounts of fat, was assessed in 15 healthy volunteers.
[165] The food-effect study was an open-label, single-dose, crossover study.
Subjects
fasted overnight prior to randomization in a 1:1:1 ratio to one of three
sequences (Fig. 12A).
All subjects received a single dose of 150 mg of CTP-656 (two 75 mg tablets,
prepared as in
Example 5) either under fasting conditions or 30 minutes following the start
of a low-fat
(about 7 g of fat) or moderate-fat (about 20 g of fat) meal. The plasma
concentrations of
CTP-656 and certain metabolites were measured as applicable.
[166] It was found that exposure to CTP-656 under low-fat and moderate-fat
conditions was
similar, and was approximately 2-fold greater that under the fasted condition
(Figures 12B
and 12C). In particular, the mean Cmax, AUC and C24hr for CTP-656 under both
fed
conditions was approximately 2-fold greater than under fasted conditions. The
mean Cmax,
AUC and C241r for the major metabolite (D-M1) was approximately 1.7-fold
higher in the fed
versus fasted conditions. CTP-656 was well-tolerated under all conditions. In
addition, the
31
CA 03041819 2019-04-25
WO 2018/080591
PCT/US2017/029920
exposure to CTP-656 under low-fat and moderate-fat conditions was similar to
the exposure
to CTP-656 under high-fat conditions seen in Example 7.
[167] The food effect results show that the exposure of CTP-656 was similar
regardless of
the fat content of a meal. In addition, the safety, tolerability and
pharmacokinetic profile of
CTP-656 observed to date supports its development as a once-daily CFTR
potentiator. This
profile may result in a number of potential advantages over current
treatments, including
simplified dosing, enhanced efficacy and reduced drug-drug interaction.
[168] Without further description, it is believed that one of ordinary skill
in the art can,
using the preceding description and the illustrative examples, make and
utilize the
compounds of the present invention and practice the claimed methods. It should
be
understood that the foregoing discussion and examples merely present a
detailed description
of certain preferred embodiments. It will be apparent to those of ordinary
skill in the art that
various modifications and equivalents can be made without departing from the
spirit and
scope of the invention.
32