Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 03068471 2019-12-23
WO 2019/014094
PCT/US2018/041219
METHOD FOR FORMING AN SEI LAYER ON AN ANODE
RELATED APPLICATION
This application claims the benefit of U.S. Provisional Application No.
62/530,609,
filed on July 10, 2017. The entire teachings of the above application are
incorporated
herein by reference.
BACKGROUND OF THE INVENTION
In the field of rechargeable metal-ion batteries where metal ions are shuttled
between anode and cathode, the electrolyte can react on the surface of the
electrode at
various voltages. An example of said metal ions includes lithium.
Passivation films are formed on the surface of the cathode and anode during
the
initial cycling of rechargeable metal ion batteries. The formation of the said
passivation
layers involves irreversible reactions between the metal ions, the active
coating materials,
the organic solvents and the salts dissolved in the organic solvents. The
reaction(s) can
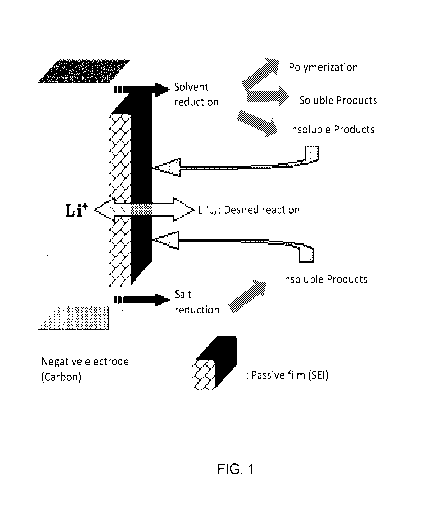
involve solvent and salt reduction forming insoluble products and polymerized
compounds,
as shown in FIG. 1. The passivation film formed at the anode is often referred
to as the
Solid Electrolyte Interface (SEI) layer and it is responsible for the majority
of the
irreversible reactions during the formation of the passivation layers.
The SEI plays a protective role that prevents and/or reduces the rate of
further
irreversible reactions of the anode with the solvent/electrolyte. An example
of the
consequences of these reactions can be seen in lithium ion batteries, which
are typically
described as having an irreversible initial loss of 5 to 40%. An ideal SEI
should be thin,
minimally porous, electrochemically inert, electrically insulating and
ionically conductive.
Formation of an SEI implies an irreversible loss that normally consumes part
of the metal
ion inventory present in the battery cathode, reducing the battery capacity.
In addition,
during this SEI formation gaseous products are formed and accumulate inside
the battery.
However, SEI formation is essential since without it the cycle life of the
battery would be
short.
The typical electrochemical formation of the SEI is often referred to as
formation
cycling. Several electrochemical formation cycling protocols may be employed
depending
on the specific chemistry. Some of the most common methods used are single-
and multi-
step current formation and pulse formation. Current can be applied while
maintaining the
cell at temperatures above room temperature but below the electrolyte boiling
point to form
1
CA 03068471 2019-12-23
WO 2019/014094
PCT/US2018/041219
specific products that are not produced during room temperature operation,
such as
inorganic metal salts. The electrochemical formation is sometimes combined
with
intermittent elevated temperature soaking steps with the elevated temperature
being kept
below the boiling point of the electrolyte. Depending on the specific
chemistry and
formation protocol applied, gas created during formation cycling may have to
be removed
to prevent pressure build up inside the cell, resulting in the introduction of
extra degassing
steps. In industry, proprietary combinations of these and other processes are
used which can
result in highly complex formation cycling protocols.
Formation protocols can have a significant economic impact on Li-ion battery
manufacturing. The formation process requires the installation of large
numbers of cycling
stations. In turn, this results in increased capital equipment cost, energy
consumption, plant
size, and temperature control requirements. Current studies have shown that
formation
cycling can account for approximately 5% of the full battery cost.
SUMMARY OF THE INVENTION
The present invention relates to the discovery of a non-electrochemical
process for
the formation of SET layers in cells in which the anodes have been pre-
alkaliated. The novel
process can eliminate the need for electrochemical formation of battery cells
by providing a
non-electrochemical formation procedure of alkaliated anodes. The non-
electrochemical
method involves soaking or maintaining the battery internal components in an
electrolyte
before conventional charging, cycling and/or operation.
For the purpose of this discussion, formation refers to the process in which a
SET is
first built in a rechargeable metal ion battery. Electrochemical formation
involves the
application of an external electrochemical driving force (voltage or
electrical current) to
form the SET on the anode surface. Non-electrochemical formation involves SET
formation
exclusively through chemical pathways, rather than electrochemical pathways.
No external
electrochemical driving force (electrical current or voltage) is utilized
during non-
electrochemical formation.
Conventional batteries (those built without pre-alkaliated anodes) are
assembled in
an inert electrochemical state, in which the anode has a near zero
potential/voltage. In this
stage there is not sufficient energy to perform the SET formation reactions
between the
anode, the organic solvents and the dissolved salts. Therefore, electrical
current or a voltage
must be applied so that the anode electrode can get to an energy state that
allows for the SET
reactions to occur.
2
CA 03068471 2019-12-23
WO 2019/014094
PCT/US2018/041219
When a cell is built using a pre-alkaliated anode, this anode will have an
energy
level dependent on the degree of alkaliation. The degree of pre-alkaliation
can range from
as little as 1% to as much as 99% of the total anode capacity and can depend
on the specific
anode and cathode materials and the cell negative-to-positive ratio. After pre-
alkaliation, the
anode will have a non-zero potential/voltage and therefore a non-inert energy
state. We
have discovered that when the pre-alkaliated anode has a potential voltage
higher than the
voltage at which SET can form, and if the pre-alkaliation dosage is greater
than or equal to
the 1" cycle irreversible loss of the anode, it is then possible to perform a
non-
electrochemical SET formation exclusively through chemical reactions at the
anode
electrode surface after the cell is built.
By exclusively incorporating a non-electrochemical formation for a battery
with pre-
alkaliated anodes, the SET can be formed through a preferred process that
reduces
manufacturing cost.
The invention provides a method for the formation of a cell, comprising the
steps of:
(a) Pre-alkaliating an anode to a dosage greater than or equal to the 1" cycle
irreversible
loss of the anode. The anode may be graphite, coke, other carbons, tin, tin
oxide,
silicon, silicon oxide, aluminum, lithium-active metals, alloying metal
materials, and
mixtures thereof
(b) Assembling the pre-alkaliated anode, a cathode, a separator and the
electrolyte into a
sealed cell.
(c) Soaking the cell under conditions and for a time sufficient to form an SET
layer.
(d) Optionally degassing the cell.
The invention can use commercially available pre-alkaliated anodes or include
the
pre-alkaliation step. In a preferred embodiment, the anode is pre-alkaliated
in accordance
with existing processes known in the art. The non-electrochemical soak
formation step (c)
can be implemented for periods of 1 hour to 10 days, preferably 4 hours to 5
days, or more
preferably 12 hours to 2 days. The soak temperature may be -20 C to the
boiling point of
the electrolyte, preferably 10 C to 60 C, and even more preferably 20 C to 40
C. It is
understood that a wide range of electrolytes can be used with varying boiling
points.
In one embodiment, the battery built with pre-alkaliated anodes is soaked for
a pre-
determined amount of time at ambient temperature to complete the chemical SET
formation.
3
CA 03068471 2019-12-23
WO 2019/014094
PCT/US2018/041219
In another embodiment, the battery built with pre-alkaliated anodes is soaked
for a
pre-determined amount of time at a single controlled temperature to complete
the chemical
SET formation.
In a further embodiment, the battery built with pre-alkaliated anodes is
soaked at
different ambient or controlled temperatures for a pre-determined amount of
time at each
temperature to complete the chemical SET formation.
BRIEF DESCRIPTION OF THE DRAWINGS
The foregoing and other objects, features and advantages of the invention will
be
apparent from the following more particular description of preferred
embodiments of the
invention, as illustrated in the accompanying drawings in which like reference
characters
refer to the same parts throughout the different views. The drawings are not
necessarily to
scale, emphasis instead being placed upon illustrating the principles of the
invention.
FIG. 1: SET reactions on anode.
FIG. 2: Example of anode after pre-alkaliation, but before soak.
FIG. 3: Example of anode after pre-alkaliation and soak.
FIG. 4: SET of alkaliated anodes soaked at room temperature for 24, 48 and72
hours.
FIG. 5: SET of pre-alkaliated anodes soaked for 24 hours at 25 C and 40 C.
FIG. 6: Areal Capacity of NCM vs. silicon/graphite cells (with electrochemical
vs.
non-electrochemical formation) tested over an extended range of charge and
discharge
cycles at approximately a C/2 rate.
FIG. 7: Capacity retention of full cells with electrochemical vs. non-
electrochemical
formation.
DETAILED DESCRIPTION OF THE INVENTION
Anodes comprised of graphite, coke, carbons, tin, tin oxide, silicon, silicon
oxide,
aluminum, lithium-active metals, alloying metal materials, and mixtures
thereof, such as
anodes comprised of carbon or graphite, are alkaliated during the first
charging step of the
battery operation after assembly, with the metal coming from the cathode
material. In these
cases, the cathode is the heaviest and most expensive component of the
battery. In addition,
this electrochemical formation step adds time, capital and energy resources to
the
manufacturing of the battery. Therefore, it would be desirable and of
commercial
importance to design a method for the elimination of electrochemical
formation. If a method
4
CA 03068471 2019-12-23
WO 2019/014094
PCT/US2018/041219
for non-electrochemical formation is accomplished, then significant reductions
in
manufacturing time and capital investments can be achieved.
The present invention relates to a method for the non-electrochemical
formation of
SET layers in pre-alkaliated rechargeable metal-ion batteries wherein
utilization of the said
method results in decreased manufacturing times, equipment requirements and
energy
consumption. The non-electrochemical method involves soaking or maintaining
the battery
internal components in an electrolyte before conventional charging, cycling
and/or
operation.
During conventional manufacturing, anodes comprised of carbon or graphite or
silicon or silicon/carbon blends are alkaliated during the first charging step
of the battery,
with the metal coming from the cathode material; a process referred to as
electrochemical
formation. The specialized cycling equipment used in this process is
inherently limited as to
the quantity of cells it can cycle at any one time. Each batch of cells being
cycled on the
specialized equipment typically takes 10-20 hours to complete. Therefore, one
of the
desirable goals in rechargeable metal-ion battery technology is to eliminate
the
electrochemical formation step without compromising the battery efficiency and
performance. Elimination of the electrochemical formation step will result in
lower battery
cost and elimination of a manufacturing bottleneck.
A preferred embodiment of the invention is a method for forming an SET layer
on an
anode for a rechargeable metal-ion battery, comprising the steps of:
a. Pre-alkaliating an anode to a dosage greater than or equal to the
irreversible loss of
the anode;
b. Assembling the pre-alkaliated anode, a cathode, a separator and an
electrolyte into a
sealed cell;
c. Forming an SET layer by soaking the cell without application of external
voltage or
current; and
d. Optionally degassing the cell.
When a conventional battery is built, the electrochemical formation will
result in an
irreversible loss due to the formation of the SET. This irreversible loss can
range from 5-
40% of the anode capacity depending on the kind of anode material being used.
A preferred
embodiment of the invention uses anode materials such as graphite, other
carbons, silicon,
silicon alloys, metal oxides, and combinations thereof
5
CA 03068471 2019-12-23
WO 2019/014094
PCT/US2018/041219
If an anode with an irreversible loss associated with SET formation of 15% of
the
total anode capacity is electrochemically pre-alkaliated to a dosage of 20% of
its capacity,
about 2/3 of pre-alkaliation dosage will form SET on the anode surface, and
about 1/3 of the
dosage will intercalate with the anode active materials in regions where SET
has formed, as
illustrated in FIG. 2.
As previously described, a cell is assembled using the pre-alkaliated anode, a
cathode, a separator and an electrolyte. In a preferred embodiment, the cell
is subsequently
left soaking at a controlled or ambient temperature for 1 hour to 10 days,
preferably 4 hours
to 5 days, or more preferably 12 hours to 2 days. The duration will be
affected by the pre-
alkaliation dosage via two mechanisms. First, in general, a higher level of
pre-alkaliation
causes a greater amount of SET to be formed at the anode surface during the
electrochemical
pre-alkaliation process, leaving less that must be formed during the soak-
formation step.
Therefore, cells containing anodes with higher pre-alkaliation dosages require
less soaking-
formation time than those with lower dosages. Second, higher pre-alkaliation
dosages leave
the anode at a higher energy state, as measured by cell OCV. This higher
energy state helps
with the chemical SET formation during soaking and reduces the time for the
SET formation
reactions to occur.
The non-electrochemical formation is believed to occur due to the migration of
intercalated alkali metal to areas without SET. When the cell is wetted with
electrolyte,
intercalated alkali metal migrates to areas without intercalated metal due to
concentration
gradients. As this intercalated metal migrates to areas without metal,
additional SET is
formed in those areas as illustrated in Figure 3. The electrical conductivity
of the anode
electrode distributes the potential/voltage, therefore giving areas without
metal sufficient
energy to perform SET reactions once the metal migrates into position.
A cell built with a pre-alkaliated anode has a voltage that depends on the pre-
alkaliation dosage. The voltage is higher than that of a cell built with a non-
pre-alkaliated
anode. For example, a conventional cell built with a non-alkaliated anode will
typically
have an initial open circuit voltage (OCV) of less than 0.5V, while a cell
built with a pre-
alkaliated anode will typically have an initial OCV higher than 2V. As the pre-
alkaliation
dosage is increased, the initial OCV of the resulting cell will increase, with
the upper limit
being determined by the specific anode and cathode used but being
approximately 2.9 to 3.5
V.
In a preferred embodiment, the soak temperature may be -20 C to the boiling
point
of the electrolyte, preferably 10 C to 60 C, even more preferably 20 C to 40
C. It is
6
CA 03068471 2019-12-23
WO 2019/014094
PCT/US2018/041219
understood that a wide range of electrolytes can be used with varying boiling
points.
Performing soak-formation above room temperature for a pre-determined amount
of time
can also aid the SET formation. It is well known that chemical reactions are
generally
accelerated at higher temperatures. In addition, there is a temperature
threshold below
which the reaction rates are negligible. Therefore, performing soak-formation
above room
temperature can accelerate the non-electrochemical formation and possibly form
desired
products that would otherwise not be formed at room temperature. The above
room
temperature utilized will depend on the anode level of pre-alkaliation prior
to cell assembly
and the desired SET characteristics and should not exceed the boiling point of
the specific
electrolyte. In addition, the above room temperature utilized will depend on
several factors
related to the battery components, such as solvent vapor pressure, separator
thermal stability
and anode and cathode electrode active and inactive components. However, the
temperature
should preferably be kept below the boiling point of the electrolyte to
prevent the risk of
excessive pressure build up inside the battery cell.
Depending on the specific chemistry of the cell, gaseous products may be
formed
during non-electrochemical formation, analogous to conventional
electrochemical
formation. The removal of the gaseous products through a degassing step is
easily
performed as it is part of conventional battery formation. The process of this
invention may
require a single degassing step in certain specific cases, while conventional
formation
normally requires one or more degassing steps.
When the battery built with a pre-alkaliated anode undergoes non-
electrochemical
formation to the extent of finishing SET formation, the battery can be
immediately cycled at
operational rates, eliminating the need for subsequent electrochemical
formation.
The process of the present invention will be better understood in connection
with the
following examples, which are intended as an illustration only and not
limiting of the scope
of the invention.
Example #1
The following is a detailed example of non-electrochemical formation of cells
with
pre-alkaliated anodes, tested in half cells. The anode used has an
irreversible capacity loss
of approximately 10.5% and it was pre-alkaliated to approximately 15% of its
total
capacity. Alkaliated anodes composed of a silicon-graphite mix are punched to
the desired
size of approximately 1.5 by 1.5 cm. The anode electrodes are then assembled
against
lithium metal of approximately the same size in a pouch cell assembly. The
separator used
7
CA 03068471 2019-12-23
WO 2019/014094
PCT/US2018/041219
was Celgard 2320. The electrolyte used was 1M LiPF6 in 3:7 (EC:EMC) with 2% VC
and
10% FEC with moisture levels below lOppm. A vacuum was applied to the cell
during
sealing to remove the gas present and improve electrode and separator wetting.
Each cell
was soaked, without externally applied voltage or current, for 24, 48 or 72
hours at room
temperature or 40 C. All the battery tests were carried out in a custom-made
environmental
chamber controlled at 26 C. A Maccor model 4300 battery tester was used to
test the pouch
cells. Each cell was operated at slow cycling rates analogous to formation
cycles. For
comparison, analogous cells with non-alkaliated anodes were constructed. For
those skilled
in the art, it is well known that the amount of SET formed can be estimated
from the initial
irreversibility of the cell. By subtracting the irreversible loss of a pre-
alkaliated anode from
that of a non-alkaliated anode, we can infer the SET formed via non-
electrochemical
formation in the cells with pre-alkaliated anodes.
FIG. 4 shows the inferred SET of pre-alkaliated anodes soaked at room
temperature
for 24, 48 and 72 hours. The result shows that there is an increase of the
inferred SET when
the cell is soaked for longer than 24 hours, therefore non-electrochemical
formation of the
alkaliated anode is occurring. FIG. 5 shows the inferred SET of pre-alkaliated
anodes soaked
for 24 hours at 25 C and 40 C. There is an increase of the inferred SET at the
higher
temperature which shows that SET formation reactions in alkaliated anodes can
be
accelerated with elevated temperatures. The soaking time and temperature
parameters need
be optimized for the specific anode and cell chemistry. However, FIG. 2 and
FIG. 3 show
the feasibility of non-electrochemical cell formation when using pre-
alkaliated anodes.
Example #2
The following is a detailed example of full cell preparation and processing.
Pre-
alkaliated anodes composed of a silicon-graphite mix are punched to the
desired size of
approximately 3 by 5 cm. The anode used has an irreversible capacity loss of
approximately
10.5% and it was pre-alkaliated to approximately 15% of its total capacity.
The anode
electrodes are then assembled against NCM cathodes of approximately the same
size in a
pouch cell assembly. The separator used was Celgard 2320. The electrolyte used
was 1M
LiPF6 in 3:7 (EC:EMC) with 2% VC and 10% FEC with moisture levels below lOppm.
A
vacuum was applied to the cell during sealing to remove the gas present and
aid in electrode
and separator wetting. The cell was soaked for 24 hrs at room temperature.
After this, a
small incision was made on the corner of the pouch, a vacuum was applied and
the cell was
given a final seal while under vacuum. All the battery tests were carried out
in a custom-
8
CA 03068471 2019-12-23
WO 2019/014094
PCT/US2018/041219
made environmental chamber controlled at 26 C. A Maccor model 4300 battery
tester was
used to test the pouch cells. One cell was operated at the normal cycling
rates without the
use of the conventional electrochemical formation cycles at the start. For
comparison,
another cell was operated at normal cycling rates after two slow
electrochemical formation
.. cycles were used at the start.
FIG. 6 and FIG. 7 show the areal capacity and capacity retention of the cells
with
non-electrochemical and electrochemical formation, tested over an extended
range of
charge and discharge cycles at approximately a C/2 rate. The results show the
effectiveness
of the invention and of eliminating the electrochemical formation by comparing
the cells
through an extended number of cycles.
In a preferred embodiment of the invention, lithiation is used in the pre-
alkaliating
step. There exist processes known in the art such as that found in US
20130327648 Al
(Grant et al.), incorporated herein for reference, which have yielded
excellent results with
the current invention.
A method for fabricating a lithiated anode which provides increased amounts of
lithium available for cycling, improved reversible capacity during charge and
discharge of a
rechargeable battery and a consequent lighter battery is preferred.
Electrolytic field plates
are held at a voltage necessary to establish a field between the anode and the
field plate, and
to lithiate the anode, such as to plate or intercalate lithium onto a foil, or
into an anode
substrate or sheet, or to form an SEI layer upon the anode. A typical
operating voltage for
this is 4.1V. An appropriate reference electrode, such as Ag/AgNO3 non-aqueous
reference
from Bioanalytical Systems, Inc., located close to the targeted negative
electrode may be
preferred to monitor the anode conditions. It is possible to operate the field
plates in either
voltage or current control mode. With current control, the full operating
potential may not
be immediately obtained. This operation under current control may result in
lower initial
operating voltages. This lower voltage may prefer secondary side reactions
instead of the
dissociation of the lithium halide salt (e.g. LiC1) and the resulting
intercalation of the anode
material. Operating under voltage control can ensure that the field plate
potential is
immediately set to a sufficient potential to favor the dissociation of the
lithium halide salt
(e.g. 4.1 Volt for LiC1) and to minimize secondary side reactions. Current
control can
alternatively be used if the subsequent operating voltage remains above the
lithium halide
salt dissociation threshold. This can be done by setting a sufficiently high
initial current
density (e.g. between about 0.5 and 2 mA/cm2, preferably about 1 mA/cm2) that
will favor
the dissociation rather than secondary side reactions. An oxidizing current is
applied at the
9
CA 03068471 2019-12-23
WO 2019/014094
PCT/US2018/041219
field plate, so there is a need to use an inert material or a conductive
oxide. In one
embodiment, the inert material comprising the field plate is selected from
glassy carbon,
tantalum, gold, platinum, silver, and rhodium. The inert material comprising
the field plate
is selected from platinum, gold or carbon. Preferably, the inert material
comprising the field
plate is carbon or glassy carbon. The field plates may also be comprised of a
base material
such as stainless steel that is plated with an inert conductive material such
as gold, platinum,
or glassy carbon. The field plates are immersed within the bath, with the
anode passing
between the field plates. The field plates can be operated as a single entity
at a single
controlled voltage or current density, or multiple plates can be implemented
that allow for
.. independent control of voltage or current density over multiple zones.
The anode typically comprises a compatible anodic material which is any
material
which functions as an anode in an electrolytic cell. The term anode is
equivalent to the
terms negative electrode, conductive foil, anode sheet, anode substrate, or
non-reactive
plating-capable foil. In one embodiment, anodes are lithium-intercalating
anodes. Examples
of materials that comprise lithium-intercalating anodes include but are not
limited to carbon,
graphite, tin oxide, silicon, silicon alloys, silicon oxide, binders such as
polyvinylidene
difluoride (PVDF), lithium polyacrylate (LiPAA), polyacrylic acid (PAA),
carboxymethyl
cellulose (CMC), styrene-butadiene rubber (SBR) or polyimide (PI), and
mixtures thereof
In a further embodiment, lithium-intercalating anode materials are selected
from graphite,
cokes, mesocarbons, carbon nanowires, carbon fibers, silicon nanoparticles or
other metal
nanomaterials and mixtures thereof In another embodiment, alloying metals such
as tin or
aluminum may be used to host the lithium metal as a result of the lithiation.
A reducing
current is applied to the anode in such a way as to intercalate the lithium.
The anode is
bathed in a solution comprising a non-aqueous solvent and at least one
dissolved lithium
salt. The term non-aqueous solvent is a low molecular weight organic solvent
added to an
electrolyte which serves the purpose of solvating the inorganic ion salt.
Typical examples of
a non-aqueous solvents are butylene carbonate, propylene carbonate, ethylene
carbonate,
vinylene carbonate, vinyl ethylene carbonate, dimethyl carbonate, diethyl
carbonate,
dipropyl carbonate, methyl ethyl carbonate, acetonitrile, gamma-butyrolactone,
triglyme,
.. tetraglyme, dimethylsulfoxide, dioxolane, sulfolane, room temperature ionic
liquids (RTIL)
and mixtures thereof In one embodiment, a non-aqueous solvent is selected from
ethylene
carbonate, vinylene carbonate, vinyl ethylene carbonate, gamma-butyrolactone,
and
mixtures thereof In a second embodiment, a non-aqueous solvent is gamma-
butyrolactone.
In a third embodiment, an additive can be introduced to support high quality
SET formation.
CA 03068471 2019-12-23
WO 2019/014094
PCT/US2018/041219
The additive could be vinylene carbonate, ethylene carbonate or maleic
anhydride. In a
fourth embodiment, a gas such as CO2 or SO2 is sparged into the non-aqueous
solution in
order to: increase salt solubility; increase the ionic conductivity; support
the formation of an
Li2CO3 or Li2S03 SET layer; and increase the lithiation efficiency.
The term alkali metal salt refers to an inorganic salt which is suitable for
use in a
non-aqueous solvent. Examples of suitable alkali metal cations comprising an
alkali metal
salt are those selected from Lit, Nat, K+, Rb+, Cs, Fr, and mixtures thereof
Examples of
suitable halogen anions comprising an alkali metal salt are those selected
from F-, Cl-, Br-,
I-, and mixtures thereof In one embodiment, the alkali metal salt is selected
from LiF,
LiCl, LiBr, NaF, NaCl, NaBr, KF, KC1, KBr, and mixtures thereof Other salts
such as
LiNO3 may be used, but in the preferred embodiment, the alkali metal salt is
the halide
LiCl.
Inexpensive salts with gaseous decomposition products can be halides such as
LiCl,
LiBr, and LiF. LiCl and other simple salts can be difficult to dissolve or
ionize in non-
aqueous solvents. Solvents such as propylene carbonate (PC), dimethyl
carbonate (DMC),
and acetonitrile support only trace amounts of LiCl in solution without the
use of a
complexing agent such as A1C13. A1C13 and other complexing agents can be
difficult to
handle in regard to moisture management and high corrosivity. In addition,
some solvents
that can dissolve halide salts, such DMSO or tetrahydrofuran (THF), do not
allow complete
ionization of the salt, and/or attack the binding polymers in the anode
composites. Gamma-
butyrolactone has been found to facilitate the dissolution and ionization of
the desirable
alkali metal halide salts. It combines good solubility of the alkali metal
halide salts with
compatibility with TFE Teflon, PVDF, butadiene rubber and other binders. The
use of
halide salts with gaseous decomposition products such as LiCl prevents the
production of
solid precipitates during the lithiation process. Since the lithiation process
products are
primarily lithium ions and gas, there are few solid precipitates or
intermediate compounds
that can accumulate in the non-aqueous solvent solution. Removal of dissolved
gas from
the non-aqueous solvent solution is preferred over solid precipitates during
long term
continuous operation of a production system.
Gamma-butyrolactone also has a capable electrochemical window, including the
lithium potential near -3 volts vs. a standard hydrogen electrode (SHE). It is
a capable
electrolyte with high permittivity and low freezing point, and can dissolve
and ionize up to a
1 M concentration of LiCl. A modest amount of heat can be used to reach this
value. In one
embodiment, the heat to dissolve and ionize up to a 1 M concentration of LiCl
is between
11
CA 03068471 2019-12-23
WO 2019/014094
PCT/US2018/041219
about 30 C and 65 C, such as between about 38 C and 55 C. However, it has been
discovered that solubility of LiC1 decreases with an increase in temperature.
Thus, a
preferred temperature for the pre-alkaliation step is about room temperature,
or between
about 20 C and 30 C. The lithiation tank can also have an internal circulating
pump and
distribution manifold to prevent localized salt concentration deprivation.
Dissolved gas such as CO2 or SO2 can enhance the lithiation process. It
increases the
solubility of the salt, the ionic conductivity of the non-aqueous solvent, and
doubles the
efficiency of lithiation. Since CO2 is inexpensive, easily dried, chemically
safe, and a
potential building block gas for a high quality SET layer, it has been
selected as the
preferred dissolved gas. CO2 preferentially reacts with trace H20 and Li +
during the
lithiation process to form a stable, insoluble SET material (Li2O, Li2CO3
etc.). The moisture
level in the lithiation tank is driven down by the consumption of CO2 and H20
according to
this process, and care is given to control the moisture level in the tank to
between about 5 to
ppm. In this way, anode lithiation with a quality SET material is produced
continuously.
15 The intercalation or plating process for lithium ions (or generally
lithiation) from 1
M LiC1 salt in gamma-butyrolactone solvent will occur at about 4.1 volts
measured between
the anode sheet and the reference electrode up to a reducing current density
of 2mA/cm2 or
more. As intercalation rates are increased too far beyond this current
density, dendrites or
lithium plating may begin to take place which harm the final battery or
electrochemical cell
20 performance. This will vary depending on the graphite porosity etc. In
order to control both
the currents and dependent voltages accurately, it may be necessary to divide
the field plate
into zones. Other metals can also be plated or intercalated with this method
including
sodium as an example. As mentioned above, the byproduct of the intercalation
process
when using a halide alkali metal salt is an evolving gas at the counter
electrode (field plate).
In a preferred embodiment, the evolving gas is selected from F2, C12, Br2, and
mixtures
thereof In a more preferred embodiment, the evolving gas is C12.
Prior to entering the lithiation bath, the anode material can be pre-soaked in
an
electrolyte solution. The pre-soaking of the anode material will ensure full
wetting of the
material prior to the start of the lithiation process. This pre-soak bath can
contain a non-
aqueous solvent with or without a lithium salt, with or without a sparge gas,
and with or
without an SET promoting additive. Preferably, the pre-soak step is without
lithium salt.
The evolution of gas at the field plate or counter electrode can result in
evolving gas
entering into, and/or being released from, the bath solution. As a result,
controlling the
12
CA 03068471 2019-12-23
WO 2019/014094
PCT/US2018/041219
build-up of dissolved and released gas is desired to avoid corrosion, as for
example, in the
hypothetical case of trace water contamination reacting with chlorine gas, to
form HC1
during chlorine gas evolution. The tank assembly can be configured to control
the
introduction of moisture into the system by using a dry gas blanket on top of
the liquid. In
one embodiment, the dry gas (1-10ppm moisture) is selected from helium (He),
neon (Ne),
argon (Ar), krypton (Kr), xenon (Xe), sulfur hexafluoride (SF6), nitrogen
(N2), dry air,
carbon dioxide (CO2) and mixtures thereof In a preferred embodiment, the dry
gas is
selected from nitrogen, argon, carbon dioxide, dry air and mixtures thereof
Moisture
ingress can also be controlled by having a long narrow gap entry and exit
tunnel for the
anode film where a counter flowing dry gas is used to mitigate air entry into
the system.
A process and apparatus that continuously controls moisture, gas, and small
quantities of lithiated organic compounds during a continuous lithiation
process is preferred.
Liquid is drawn from a bath through a series of valves. The liquid can be
delivered in a
batch mode to a refluxing unit, or it can be continuously circulated through a
conditioning
loop including distillation or reverse osmosis. The reflux unit can take
batches of material
through a vacuum refluxing process that will remove both accumulated gas as
well as
moisture from the liquid. In one embodiment, the accumulated gas is selected
from F2, C12,
Br2, and mixtures thereof In a more preferred embodiment, the accumulated gas
is C12. The
use of reflux conditioning instead of a distillation process can prevent a
change in the salt
concentration of the working fluid which would result in a loss of salt
content through
precipitation. Once the batch liquid has been refluxed for a designated period
of time, the
liquid can be returned to the bath with a lower moisture and gas content. The
size and rate
of the reflux unit can be matched to the moisture ingress rate and to the gas
production rate
in order keep the bath liquid at optimum conditions. The reflux rate can be
increased
through use of multiple simultaneous batches and through the use of high rate
reflux
equipment such as a rotary evaporator and high vacuum conditions. The reflux
batch
moisture content typically decays in an exponential fashion and the turnover
rate can be
tuned for optimal moisture control with minimal energy input and equipment
cost.
The refluxing unit can be placed after a salt dosing unit. The salt dosing
unit can be
used to add and mix the desired salt into the non-aqueous solvent solution.
The temperature
of the dosing unit can be held to maximize the solubility of the salt in the
electrolyte and the
elevated temperature can also be used as a pre-heating step for the refluxing
unit. In one
embodiment, the dosing unit maintains an elevated process temperature of
between about
30 C and 65 C, such as between about 38 C and 55 C. However, it has now been
13
CA 03068471 2019-12-23
WO 2019/014094
PCT/US2018/041219
discovered that a preferred temperature is about room temperature, or between
about 20 C
and 30 C. The benefit of dosing in the salt in a dosing unit before the
refluxing unit is that
the salt does not have to be in a completely dry state. Removing the moisture
from a solid
phase salt can be very difficult. Once a salt is dissolved into solution,
however, the water
content of the salt can be removed through the refluxing process. Maintaining
the dosing
unit at an elevated temperature increases the solubility of the lithium salt
in the non-aqueous
solvent and ensures full dissolution of the salt prior to the refluxing unit.
The conditioning/replenishment loop operates in a continuous mode and can also
be
used to remove dissolved gases from the bath liquid through use of a membrane
contactor.
The gas output from the membrane contactor and the reflux unit can be passed
through a
scrubber to capture any effluent, such as chlorine gas, produced by the
process. In one
embodiment, the dissolved gases are selected from F2, C12, Br2, and mixtures
thereof In a
more preferred embodiment, the dissolved gas is C12. The bath liquid can also
be paired
against either vacuum or a dry gas within the membrane contactor in order to
remove
unwanted gases. In one embodiment, the dry gas is selected from helium (He),
neon (Ne),
argon (Ar), krypton (Kr), xenon (Xe), sulfur hexafluoride (SF6) nitrogen (N2),
carbon
dioxide (CO2), dry air and mixtures thereof In a preferred embodiment, the dry
gas is
selected from nitrogen, argon, carbon dioxide, dry air and mixtures thereof
An inline heater can be used to establish or maintain an elevated tank
temperature to
maintain consistent bath operating conditions, even with variations in
facility temperature,
as discussed above. As the lithiation reaction is exothermic, it can be
desirable to cool the
bath.
A filter unit can be used to remove any accumulated particulate contamination.
The
filter unit can be located at various points in the loop including prior to
the pump and after
the salt dosing unit. The filter unit can be used to remove particulates from
the non-aqueous
solvent in cases where a non-halide lithium salt such as LiNO3 is used such
that a
precipitate is formed at the field plates.
Lithium halide salt can be added to the non-aqueous solvent using the salt
dosing
unit. An excess of solid lithium salt can be maintained within the dosing unit
to keep the
lithium salt concentration within the loop and within the bath at the desired
level (i.e., a
saturated solution of about 0.5 M to 1.0 M) over long periods of time. The
dosing unit can
be configured to keep the solid salt from entering the bath or refluxing unit.
By dosing salt
prior to the refluxing unit, there is no need to separately dry the salt with
its high water
14
CA 03068471 2019-12-23
WO 2019/014094
PCT/US2018/041219
binding energy in its granular state. In one embodiment, the lithium salt
within the salt
dosing unit is selected from LiF, LiC1, LiBr, and mixtures thereof In a
preferred
embodiment, the lithium halide salt within the salt dosing unit is LiCl.
Dissolved lithium
salts can be carried through the rest of the loop. The fluid circulation loop
pump rate can be
matched to maintain a constant lithium salt concentration in the tank. For a
given anode
substrate process rate, a matching loop circulation rate will dose the same
amount of lithium
salt as the lithiation process consumes. As the anode process rate is
increased or decreased,
the loop circulation rate can be modified to maintain an equilibrium state
within the bath.
Depending on the specific tank conditions, the bath fluid can be treated using
a
circulating loop, a refluxing unit or a distillation unit as shown in Figures
2 and 4. A
circulating loop can dose in salt, remove dissolved gases, control the bath
temperature and
removed particulate contaminants. A refluxing unit is effective at removing
dissolved gases
and for removing moisture content without reducing the salt content of the
solution. A
distillation unit is effective at removing dissolved gases, removing moisture
content,
removing all salt content and removing lithiated organic compounds. The output
from the
distillation unit can be fed back into a dosing and refluxing unit to
reestablish the salt
content if required. The effluent from the distillation unit can be collected
and treated to
recover used salt for reuse in the lithiation process. For example, DMC
solvent will rinse
away all but the insoluble salt so that the salt may be re-introduced into the
dosing unit.
Recirculating loops, refluxing unit and distillation units can be shared
across multiple tanks
that have different input and output requirements as a means of minimizing
equipment size
and cost.
When the anode is lithiated to the extent of the irreversible and extended
cyclic loss
amount, it can be assembled into a rechargeable battery or electrochemical
cell with a
smaller amount of lithium-bearing cathode material than would otherwise be
required,
thereby improving the specific capacity, specific energy, volumetric capacity
density and
volumetric energy density of the cell while reducing its cost.
When the anode is lithiated to the extent of the irreversible and extended
cyclic loss
amount, as well as the intended cycling amount, it can be assembled into a
battery or
electrochemical cell with a cathode material that does not initially contain
lithium. This type
of cathode material can be much less expensive than lithium containing cathode
materials,
and examples include, but are not limited to, Mn02, V205 and polyaniline.
CA 03068471 2019-12-23
WO 2019/014094
PCT/US2018/041219
EXAMPLES
The following is a detailed example of an anode preparation and processing. 25
micron
thick copper foil was cleaned with isopropyl alcohol and Kimberly-Clark
Kimwipes to
remove oil and debris and then dried in air. A solution was prepared by adding
2.1 grams of
1,000,000 weight PVDF powder from Arkema Fluoropolymers Div. to 95 ml of dry
NMP
solvent from Aldrich Chemical. The solution was mixed with a stir bar
overnight to fully
dissolve the PVDF material. The solution was kept in the dark to prevent the
light sensitive
solvent from reacting. 33.9 ml of this PVDF solution was then added to 15
grams of Conoco
Philips CPreme G5 graphite and 0.33 grams of acetylene black and stirred for 2
hours in a
.. ball mill at 600 RPM without any mixing balls. The resulting slurry was
cast onto the
copper foil using a vacuum hold down plate with heating capability. The
finished graphite
thickness after casting and drying at 120 C was about 100 microns or 14
mg/cm2. The
anode sheet was then die punched into 15 mm diameter discs and then pressed at
about
3000 psi and 120 C for use in a 2032 button cell assembly. The copper/graphite
anode discs
were then vacuum baked at 125 C and about 1 mTorr in a National Appliance
Company
model 5851 vacuum oven for at least 12 hours.
The anode discs were then transferred into a Terra Universal dry air glove box
with -
65 C dew point air supplied by compressed dry air passed through a Kaeser two
stage
regenerative drier. The anode discs were then vacuum infiltrated with a GBL
solvent with a
0.5 M concentration of LiC1 salt solution. This electrolyte solution had been
prepared by
heating to 90 C and then vacuum refluxing at about 1 mTorr for 6 hours to
remove moisture
down to about 10 ppm. The anode discs were allowed to soak for a half hour at
vacuum
conditions, a half hour in atmospheric pressure conditions and a half hour in
the lithiation
vessel itself prior to any currents being passed. The lithiation vessel
included a constant
bubbling of CO2 gas to achieve a saturation level and a temperature of 30 C.
Test leads
from a Maccor 4300 battery tester were connected to the anode sample (red
working) and
glassy carbon (black counter) electrode. Voltage at the working electrode is
monitored via a
Ag/AgNO3 non-aqueous electrode. A reducing current of 2 mA/cm2 was applied to
the
graphite anode until a total of 1 mAhr/cm2 was achieved. The pre-lithiated
anode disc was
.. then rinsed in pure distilled GBL and vacuum dried. The anode discs were
then assembled
against either LiFePO4 or LiCo02 12mm diameter cathode discs. The separator
used was
Celguard 2400, and about 0.2 ml of electrolyte was used in the assembly. The
electrolyte
was 1:1:1 EC:DMC:DEC with 1M LiPF6 salt and 1% VC with moisture levels at
about 10
ppm. A vacuum was applied to the assembled cell to remove bubbles before
crimping in an
16
CA 03068471 2019-12-23
WO 2019/014094
PCT/US2018/041219
MTI model MT-160D crimping tool. Subsequent electrical tests were then
performed on the
battery tester unit using a 12 hour delay, two about C/12 formation cycles to
at least 3.7
volts, about C/3 charge/discharge cycles, and 20 minute rest steps between
them. All the
battery tests were carried out in a home-made environmental chamber controlled
to 26 C.
A Maccor model 4300 battery tester was used to test the CR2032 size button
cells
assembled with a CPreme graphite anodes, LiFePO4 or LiCo02 cathodes, and
Celguard
2400 separators. Electrolyte solutions containing a 1:1:1 mixture of
EC:DMC:DEC with 1
molar concentration of LiPF6 salt and 1% VC were used. Both anodes and
cathodes were
cast with PVDF binders. First charge and discharge cycles, often called the
formation
.. cycles, were performed at a current rate of approximately C/12. The first
cycle irreversible
loss using pre-lithiated and non-pre-lithiated graphite anodes mounted against
LiFePO4
cathodes can be compared. The initial absolute charge capacity of the two
samples is
different due to extraneous packaging variation. The irreversible losses are
representative of
the methods described, however. The reversible capacity of the button cell can
be 56%. The
reversible capacity of the button cell when matched to a pre-lithiated anode
can be 98%. A
typical LiCo02/graphite vs. a LiCo02/pre-lithiated graphite, but otherwise
identical sample
can be tested over an extended range of charges and discharge cycles at
approximately a
C/3 rate. The results indicate that there is a long lasting benefit to the
battery cell due to
pre-lithiation using the methods described.
An example of a salt other than LiC1 that has been used by the inventor to
achieve
lithiation is LiNO3. Reasonable rates of lithiation have been achieved with
LiNO3. When
the anodes pre-lithiated using LiNO3were paired with LiFePO4 cathodes,
however, poor
cycling resulted, possibly due to an unidentified byproduct. This problem can
be solved by a
slightly more complicated removal process such as an additional anode rinse.
While there has been illustrated and described what is at present considered
to be the
preferred embodiment of the present invention, it will be understood by those
skilled in the
art that various changes and modifications may be made and equivalents may be
substituted
for elements thereof without departing from the true scope of the invention.
Therefore, it is
intended that this invention not be limited to the particular embodiment
disclosed, but that
the invention will include all embodiments falling within the scope of the
appended claims.
17