Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
WO 2022/090427 1
PCT/EP2021/080054
ANALOGUES DU 5 ALPHA-HYDROXY-6
BETA42-(1-H-IMIDAZOL-4-YL)ETHYLAMIN0]-CHOLESTAN-3 BETA-OL ET
COMPOSITIONS PHARMACEUTIQUES LE COMPRENANT POUR
UTILISATION DANS LE TRAITEMENT DU CANCER
Domaine technique
[1] L'invention se rapporte au domaine des composés stérols et plus
particulièrement à des
analogues du composé 5a-hydroxy-6612-(1H-imidazol-4-y1)-éthylamino]-cholestan-
36-ol
et compositions pharmaceutiques les comprenant pour leur utilisation dans le
traitement
du cancer.
Arrière-plan technologique
[2] Le terme cancer ou tumeur cancéreuse englobe un groupe de maladies
se
caractérisant par la multiplication et la propagation anarchiques de cellules
anormales. Si
les cellules cancéreuses ne sont pas éliminées, l'évolution de la maladie va
mener plus
ou moins rapidement au décès de la personne touchée.
[3] La prise en charge du cancer implique la chirurgie, la radiothérapie et la
chimiothérapie,
qui peuvent être utilisées seules ou en association, simultanément ou
séquentiellement.
La chimiothérapie utilise des agents antinéoplasiques qui sont des médicaments
qui
empêchent ou inhibent la maturation et la prolifération des néoplasmes. Les
agents
antinéoplasiques agissent en ciblant efficacement les cellules à division
rapide. Comme
les agents antinéoplasiques affectent la division cellulaire, les tumeurs avec
un fort taux
de croissance (telles que la leucémie myéloïde aiguë et les lymphomes
agressifs, y
compris la maladie de Hodgkin) sont plus sensibles à la chimiothérapie, car
une plus
grande proportion des cellules ciblées subit une division cellulaire à tout
moment. Les
tumeurs malignes avec des taux de croissance plus lents, comme les lymphomes
indolents, ont tendance à répondre beaucoup plus modestement à la
chimiothérapie.
Cependant, le développement de la chimiorésistance est un problème persistant
pendant
le traitement de chimiothérapie. Par exemple, le traitement conventionnel de
la leucémie
myéloïde aiguë (LMA) comprend l'administration combinée de cytarabine avec une
anthracycline, telle que la daunorubicine. Le taux de survie globale à 5 ans
est de 40 (3/0
chez les jeunes adultes et d'environ 10 % chez les patients âgés. Les taux de
réponse
varient considérablement avec le vieillissement, de 40 % à 55 % chez les
patients de plus
de 60 ans et de 24 % à 33 % chez les patients de plus de 70 ans. Ceci est
encore pire
pour les personnes âgées avec des profils cytogénétiques défavorables et le
décès dans
les 30 jours suivant le traitement varie de 10 % à 50 % avec l'âge et
l'aggravation. Par
ailleurs, la restriction de l'utilisation de ces molécules est due également à
des effets
CA 03196518 2023- 4- 24
WO 2022/090427 2
PCT/EP2021/080054
secondaires, et en particulier à l'émergence d'une toxicité cardiaque
chronique (liée aux
anthracyclines). Le taux de mortalité toxique liée à la chimiothérapie
intensive est de 10 à
20 % chez les patients de plus de 60 ans.
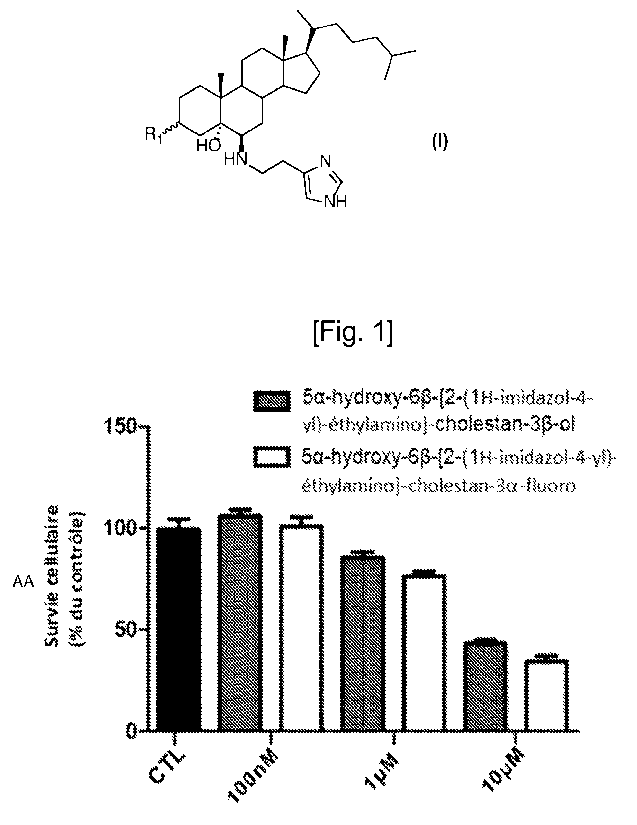
[4] Avec ce profil bénéfice-risque du régime conventionnel, seulement 30 c'/0
des personnes
âgées avec une LMA nouvellement diagnostiquée reçoivent une chimiothérapie
antinéoplasique.
[5] Au cours des dernières décennies, il n'y a eu qu'une amélioration modeste
des résultats
pour les patients plus jeunes atteints de LMA, mais aucune pour les adultes de
plus de 60
ans (la plupart des patients atteints de LMA).
[6] Il existe donc un réel besoin de développer des molécules utiles dans le
traitement de ces
tumeurs cancéreuses qui présentent des problèmes de chimiorésistance et de
toxicité
intrinsèque des médicaments antinéoplasiques. Les données précitées soulignent
la
nécessité de trouver de nouvelles approches qui combinent à la fois une
réduction des
schémas posologiques d'agents antinéoplasiques pour le traitement des tumeurs
chimiosensibles à une diminution de la résistance des tumeurs
chimiorésistantes à l'agent
antinéoplasique.
[7] On connaît du document EP3272350B1 le composé 5a-hydroxy-66-[2-(1H-
imidazol-4-y1)-
éthylamino]-cholestan-36-ol, connu sous le nom de Dendrogénine A, et nommé ci-
après
DX101, utile pour le traitement des tumeurs chimiorésistantes. La Dendrogénine
A est
capable de restaurer la sensibilité des tumeurs chimiorésistantes à un agent
antinéoplasique ou d'augmenter les effets des agents antinéoplasiques sur les
tumeurs,
ce qui permet à son tour de réduire la dose cytotoxique efficace d'agents
antinéoplasiques
contre les tumeurs chimiosensibles.
[8] Le document de De Medina et al. (Biochimie, 2021, 95(3), 482-488,
XP028982107,
Technical note: Hapten synthesis, antibody production and development of an
enzyme-
linked immunosorbent assay for detection of the natural steroidal alkaloid
Dendrogenin A)
décrit des dérivés de Dendrogénine A pour lesquels l'alcool en position 3p est
fonctionnalisé pour leur utilisation comme haptènes pour la production
d'anticorps.
[9] Le document de De Medina et al. (J.Med. Chem., 2009, 52(23), 7765-77,
XP9131948,
Synthesis of new alkylaminooxysterols with potent cell differentiating
activities:
identification of leads for the treatment of cancer and neurodegenerative
diseases) décrit
des dérivés de Dendrogénine A pour lesquels l'alcool en position 33 est
éventuellement
fonctionnalisé par un radical méthanoate ou propanoate, pour le traitement du
cancer.
CA 03196518 2023- 4- 24
WO 2022/090427 3
PCT/EP2021/080054
[10]
L'objet de la présente invention est de fournir de nouveaux composés et
analogues au
composé Dendrogénine A, utiles pour traiter les tumeurs cancéreuses, notamment
les
tumeurs chimiosensibles et/ou chimiorésistantes.
[11]
De manière surprenante, les inventeurs ont découvert que des analogues
spécifiques
du composé Dendrogénine A (nommé également DX101), présentent une activité
pharmacologique comparable à la Dendrogénine A.
Résumé
L'invention a pour premier objet un composé de formule (I) ;
R1 Hd
H
N/1-1
ou un sel pharmaceutiquement acceptable d'un tel composé,
dans lequel :
Ri est choisi parmi F, N3, OCRI-12n+1, NR23, SR2, S02R2 , avec n
8,
R2 et R3 sont indépendamment choisis parmi : H, un groupement alkyle en Cl à
08, saturé
ou insaturé, contenant éventuellement un ou plusieurs substituants choisis
parmi des
groupes allyle, carbonyle, arène et hétérocyclique,
pour son utilisation en tant que médicament, et plus particulièrement en tant
que
médicament pour faire régresser une tumeur cancéreuse de mammifère.
[12]
L'invention a pour second objet une composition pharmaceutique
comprenant dans un
véhicule pharmaceutiquement acceptable, au moins un composé de formule (I)
pour son
utilisation pour faire régresser une tumeur cancéreuse de mammifère.
Définitions
[13]
Dans cette description, à moins qu'il n'en soit spécifié autrement, il
est entendu que,
lorsqu'un intervalle est donné, il inclut les bornes supérieures et
inférieures dudit intervalle.
[14]
Dans la présente invention, tout au long de la présente description et
des
revendications annexées, les termes suivants, sauf indication contraire,
doivent être
compris comme ayant la signification suivante :
[15]
Le terme solvate est utilisé ici pour décrire un complexe
moléculaire comprenant
un composé de l'invention et contenant des quantités stoechiométriques ou sous-
CA 03196518 2023- 4- 24
WO 2022/090427 4
PCT/EP2021/080054
stoechiométriques d'une ou plusieurs molécules de solvant pharmaceutiquement
acceptable telles que l'éthanol. Le terme hydrate fait référence lorsque
ledit solvant
est de l'eau.
[16] Le terme humain fait référence à un sujet des deux sexes et à tout
stade de
développement (c'est-à-dire un nouveau-né, nourrisson, juvénile, adolescent,
adulte).
[17] Le terme patient fait référence à un animal à sang chaud, plus
préférablement un
humain, qui attend la réception ou qui reçoit des soins médicaux et/ou qui
sera l'objet d'un
acte médical.
[18] Par pharmaceutiquement acceptable il est entendu que les
ingrédients d'un produit
pharmaceutiquement acceptable sont compatibles les uns avec les autres et ne
sont pas
nuisibles pour le patient de ce produit.
[19] Le terme véhicule pharmaceutique tel qu'utilisé dans ce texte
signifie un support
ou un milieu inerte utilisé comme solvant ou diluant dans lequel l'agent
pharmaceutiquement actif est formulé et / ou administré. Des exemples non
limitatifs de
véhicules pharmaceutique comprennent les crèmes, les gels, les lotions, les
solutions et
les liposomes.
[20] Le terme administration , signifie délivrer, l'agent actif ou
l'ingrédient actif (par
exemple le composé de formule (I), dans une composition pharmaceutiquement
acceptable, au patient dans lequel une condition, un symptôme et/ou une
maladie doit être
traitée.
[21] Les termes traiter et traitement tels qu'utilisés ici incluent
atténuer, apaiser,
stopper, soigner une condition, un symptôme et/ou une maladie.
[22] Le terme analogue tels qu'utilisés ici signifie un composé ayant
une structure
chimique similaire à un autre composé de référence, mais différant de celui-ci
par un
certain composant. Il peut différer d'un ou de plusieurs atomes, groupes
fonctionnels,
sous-structures, qui sont remplacés par d'autres atomes, groupes fonctionnels
ou sous-
structures. Les analogues peuvent avoir des propriétés physiques, chimiques,
biochimiques ou pharmacologiques différentes. Dans la présente invention, les
composés
analogues sont en référence au composé Dendrogénine A. Ces analogues
présentent des
propriétés pharmacologiques identiques ou similaire par rapport au composé de
référence.
[23] Par "cancer chimiorésistant", on entend un cancer chez un patient où
la prolifération
des cellules cancéreuses ne peut être empêchée ou inhibée au moyen d'un agent
antinéoplasique ou d'une combinaison d'agents antinéoplasiques habituellement
utilisés
pour traiter ce cancer, à une dose acceptable pour le patient. Les tumeurs
peuvent être
CA 03196518 2023- 4- 24
WO 2022/090427 5 PCT/EP2021/080054
intrinsèquement résistantes avant la chimiothérapie, ou une résistance peut
être acquise
pendant le traitement par des tumeurs initialement sensibles à la
chimiothérapie.
[24] Par ''cancer chimiosensible", on entend un cancer chez un
patient qui répond aux
effets d'un agent antinéoplasique, c'est-à-dire que la prolifération des
cellules cancéreuses
peut être empêchée au moyen dudit agent antinéoplasique à une dose acceptable
pour le
patient.
[25] Le composé de formule (I) appartient au groupe des stéroïdes.
La numérotation des
atomes de carbone du composé de formule (I) suit donc la nomenclature définie
par
l'IUPAC dans Pure & Appl. Chem., Vol.61, No.10, pp.1783-1822,1989. La
numérotation
des atomes de carbone d'un composé appartenant au groupe des stéroïdes selon
l'IUPAC
est illustrée ci-dessous :
z
24
241
21 22
26
12 16 23, 24 25
11 17 27
19 D le
I g
2 e 14
10 15
A
3 7
5
4 6
[26] Dans la présente invention, les abréviations suivantes
signifient :
LMA : leucémie myéloïde aiguë;
15 Dendrogénine A : 50-hydroxy-61342-(1H-imidazol-4-y1)-éthylamino]-
cholestan-313-ol ;
MCF-7 : Michigan Cancer Foundation-7;
DMEM : Dulbecco's Modified Eagle Medium ;
SVF : sérum de veau foetal ;
ChEH : Cholestérol Epoxyde Hydrolase
20 Neuro2a : neuroblastome murin ;
CTL : Contrôle ;
MTT : bromure de 3-(4,5-dimethylthiazol-2-y1)-2,5-diphenyl tetrazolium ;
PBS : tampon phosphate salin ;
DMSO : diméthylsulfoxyde ;
DO : densité optique ou absorbance ;
CT : cholestane-313,5a,613-triol ;
OCDO : 6-oxo-cholestan-313,50-diol ;
5,6a-EC : 5,6a-epoxycholesterol ;
CA 03196518 2023- 4- 24
WO 2022/090427 6
PCT/EP2021/080054
Tam : Tamoxifène ;
CCM : chromatographie sur couche mince.
P0.: per os
LC/MS : chromatographie liquide/ spectrométrie de masse
Description des modes de réalisation
[27] L'invention concerne en premier objet un composé de formule (I)
;
HO
H
141
ou un sel pharmaceutiquement acceptable d'un tel composé, dans lequel :
Ri est choisi parmi F, N3, OCnH2n+1 NR2R3, S R2, S02R2, avec n 8,
R2 et R3 sont indépendamment choisis parmi : H, un groupement alkyle en Cl à
08, saturé
ou insaturé, contenant éventuellement un ou plusieurs substituants choisis
parmi des
groupes allyle, carbonyle, arène et hétérocyclique,
pour son utilisation en tant que médicament.
Selon un mode de réalisation, l'invention concerne un composé de formule (I) ;
HO
H
H
ou un sel pharmaceutiquement acceptable d'un tel composé, dans lequel :
Ri est choisi parmi F, N3, OCnH2n+1 NR2R3, S R2, S02R2 , avec n 8,
R2 et R3 sont indépendamment choisis parmi : H, un groupement alkyle en Cl à
08, saturé
ou insaturé, contenant éventuellement un ou plusieurs substituants choisis
parmi des
groupes allyle, carbonyle, arène et hétérocyclique,
CA 03196518 2023- 4- 24
WO 2022/090427 7
PCT/EP2021/080054
pour son utilisation en tant que médicament pour faire régresser une tumeur
cancéreuse
de mammifère.
[28] Dans la présente invention :
- le groupe carbonyle fait référence à tous les groupes fonctionnels
contenant le groupe
oxo (un atome d'oxygène doublement lié (=0) à un atonie de carbone) et peut
être choisi
parmi les aldéhydes, les cétones, l'acide carboxylique, les esters, les amides
et/ou
l'anhydride.
- le terme allyle fait référence à un groupe fonctionnel alcénique de
formule semi-
développée H2C=CH-C1-12-=
- le terme sulfonyle est un composé chimique où l'atome de soufre est combiné
à deux
atomes d'oxygène doublement liés (=0) et à son radical.
- le terme arène fait référence à tous les hydrocarbures aromatiques
monocycliques et
polycycliques
- le terme hétérocyclique fait référence à des composés aromatiques
monocycliques
et polycycliques comportant comme éléments cycliques un ou plusieurs
hétéroatomes
parmi 0, S et/ou N.
Dans la définition du composé de formule (1) selon l'invention, le radical du
carbone 3 peut
être en position a ou p. La position p étant un mode de réalisation préféré.
[29] Selon un mode de réalisation, le composé de formule (1) est un
analogue 0-aminé
dans lequel le radical Ri = NR2R3 avec R2 étant H ou COCnI-12,1-0 et R3 = H.
[30] Dans ce mode de réalisation, le composé de formule (I) est plus
particulièrement le
5a-hydroxy-6p-[2-(1H-imidazol-4-y1)-éthylamino]-3p-acétamide (nommé DX127).
[31] Dans ce mode de réalisation, le composé de formule (I) est plus
particulièrement le
5a-hydroxy-613-[2-(1H-imidazol-4-y1)-éthylamino]-3p-amino (nommé DX125).
[32] Dans ce mode de réalisation, le composé de formule (I) est plus
particulièrement le
5a-hydroxy-613-[2-(1H-imidazol-4-y1)-éthylamino]-3p-azoture (nommé DX123).
[33] Selon encore un mode de réalisation, le composé de formule (1) est le
5a-hydroxy-6p-
[2-(1H-imidazol-4-y1)-éthylamino]-cholestan-3p-fluoro (nommé DX111).
[34] Selon encore un autre mode de réalisation, le composé de formule (1)
est un analogue
0-alkylé et présente un radical Ri = OCRI-12n+i avec n 8, et est choisi parmi
:
- 5a-hydroxy-61342-(1H-imidazol-4-y1)-éthylamino]-cholestan-313-méthoxyle
(nommé
DX103)
CA 03196518 2023- 4- 24
WO 2022/090427 8
PCT/EP2021/080054
5a-hydroxy-6642-(1H-imidazol-4-y1)-éthylamino]-cholestan-36-éthoxyle (nommé
DX105)
- 5a-hydroxy-6642-(1H-1midazol-4-y1)-éthylamino]-cholestan-36-octanoxyle
(nommé
DX115)
[35] De
manière plus préférentielle encore, le composé de formule (1) est un analogue
0-
alkylé tel que
5a-hydroxy-6612-(1H-imidazol-4-y1)-éthylamino]-cholestan-36-
méthoxyle (DX103) et 5a-hydroxy-6612-(1H-imidazol-4-y1)-éthylamino]-cholestan-
36-
éthoxyle (DX105).
[36] Selon un mode de réalisation supplémentaire, le composé de formule (I)
est un
analogue soufré et présente un radical R1 = S02R2 avec R2 étant H ou OCnH2n_o,
avec n
< a
[37] Dans ce mode de réalisation, le composé de formule (1) est
préférentiellement le 5a-
hydroxy-66-[2-(1H-imidazol-4-y1)-éthylamino]-cholestan-36-méthylsulfo nyle
(nommé
DX129).
[38] Selon
un mode de réalisation, le composé de formule (I) est destiné à être utilisé
dans
le traitement du cancer du sein, de la prostate, colorectal, du poumon, de la
vessie, de la
peau, de l'utérus, du col de l'utérus, de la bouche, du cerveau, de l'estomac,
du foie, de la
gorge, du larynx, de l'oesophage, de l'os, de l'ovaire, du pancréas, du rein,
de la rétine, du
sinus, des fosses nasales, du testicule, de la tyroïde, de la vulve, le
traitement du
lymphome, lymphomes non hodgkiniens, lymphomes hodgkiniens, de la leucémie, la
leucémie myéloïde aiguë ou la leucémie lymphocytaire aiguë, le myélome
multiple,
carcinome à cellules de Merkel ou mésothéliome.
[39]
Selon un mode de réalisation, le cancer est un adénocarcinome acineux,
un carcinome
acineux, un mélanome acro-lentigineux, une kératose actinique, un
adénocarcinome, un
carcinome adénoïde kystique, un carcinome adénosquameux, un carcinome
annexiel,
une tumeur du repos surrénal, carcinome adrénocortical, carcinome sécrétant de
l'aldostérone, sarcome alvéolaire de la partie molle, carcinome améloblastique
de la
thyroïde, angiosarcome, carcinome apocrine, tumeur d'Askin, astrocytome,
carcinome
basocellulaire, carcinome basaloïde, carcinome basosquameux, cancer des voies
biliaires,
cancer de la moelle osseuse, sarcome botryoïde, carcinome bronchioalvéolaire,
adénocarcinome bronchogène, carcinome bronchogène, carcinome ex adénome
pléomorphe, chlorome, carcinome cholangiocellulaire, chondrosarcome,
choriocarcinome,
carcinome du plexus choroïde, adénocarcinome à cellules claires, cancer du
côlon,
comédocarcinome, carcinome producteur de cortisol, carcinome à cellules
cylindriques,
CA 03196518 2023- 4- 24
WO 2022/090427 9
PCT/EP2021/080054
liposarcome différencié, adénocarcinome canalaire de la prostate, carcinome
canalaire,
carcinome canalaire in situ, cancer duodénal, carcinome eccrine, carcinome
embryonnaire,
carcinome de l'endomètre, carcinome stromal de l'endomètre, sarcome
épithélioïde,
sarcome d'Ewing, carcinome exophytique, sarcome fibroblastique,
fibrocarcinome,
carcinome fibrolamellaire, fibrosarcome, carcinome folliculaire de la
thyroïde, cancer de la
vésicule biliaire, adénocarcinome gastrique, carcinome à cellules géantes,
sarcome à
cellules géantes, tumeur osseuse à cellules géantes, gliome, glioblastome
multiforme,
carcinome à cellules de granulosa, cancer de la tête et du cou, hémangiome,
hémangiosarcome, hépatoblastome, carcinome hépatocellulaire, Carcinome à
cellules de
Hürthle, cancer iléal, carcinome lobulaire infiltrant, carcinome inflammatoire
du sein,
carcinome intraductal, carcinome intra-épidermique, cancer du jéjunum, sarcome
de
Kaposi, tumeur de Krukenberg, carcinome à cellules de Kulchitsky, sarcome à
cellules de
Kupffer, carcinome à grandes cellules, cancer du larynx, mélanome de lentigo
maligna,
liposarcome, carcinome lobulaire, carcinome lobulaire in situ,
lymphoépithéliome,
lymphosarcome, mélanome malin, carcinome médullaire, carcinome médullaire de
la
thyroïde, médulloblastome, carcinome méningé, carcinome micropapillaire,
sarcome à
cellules mixtes, carcinome mucineux, carcinome muco-épidermoïde, mélanome
mucosal,
liposarcome myxoïde, myxosarcome, carcinome nasopharyngé, néphroblastome,
neuroblastome, mélanome nodulaire, cancer du rein à cellules non claires,
cancer du
poumon à cellules non petites, carcinome à cellules d'avoine, mélanome
oculaire, cancer
buccal, carcinome ostéoïde, ostéosarcome, cancer des ovaires, carcinome de
Paget,
pancréatoblastome, adénocarcinome papillaire, carcinome papillaire, carcinome
papillaire
de la thyroïde, cancer pelvien, carcinome périampullaire, tumeur phyllode,
cancer de
l'hypophyse, liposarcome pléomorphe, blastome pleuropulmonaire, carcinome
intra-
osseux primaire, cancer du rectum, carcinome des cellules rénales,
rétinoblastome,
rhabdomyosarcome, liposarcome à cellules rondes, cancer cicatriciel, cancer de
la vessie
schistosomique, carcinome schneidérien, carcinome sébacé, carcinome à cellules
en
anneau, cancer de la peau, cancer du poumon à petites cellules, ostéosarcome à
petites
cellules, sarcome des tissus mous, sarcome à cellules fusiformes, carcinome
épidermoïde,
cancer de l'estomac, mélanome à diffusion superficielle, sarcome synovial,
sarcome
télangiectasique, carcinome du canal terminal, cancer des testicules, cancer
de la thyroïde,
carcinome des cellules transitoires, carcinome tubulaire, mélanome tumorigène,
carcinome indifférencié, adénocarcinome de l'urètre, cancer de la vessie,
cancer de
l'utérus, carcinome du corps utérin, mélanome de l'utérus, cancer du vagin,
carcinome
verruqueux, carcinome villeux, liposarcome bien différencié, tumeur de Wilms
ou tumeurs
des cellules germinales.
CA 03196518 2023- 4- 24
WO 2022/090427 10
PCT/EP2021/080054
[40] Dans un mode de réalisation préféré, le composé de formule (I) est
destiné à être
utilisé dans le traitement du cancer du sein chez les mammifères.
[41] Selon un mode de réalisation, le composé est destiné à être utilisé
dans le traitement
d'un cancer ch imiosensible.
[42] Selon un mode de réalisation particulièrement préféré, le composé de
formule (I) est
destiné à être utilisé dans le traitement d'un cancer chimiorésistant.
[43] Selon un mode de réalisation, le cancer chimiorésistant est un cancer
hématologique
ou du sang, tel que la leucémie, en particulier la leucémie myéloïde aiguë ou
la leucémie
lymphocytaire aiguë, le lymphome, en particulier le lymphome non-hodgkinien et
le
myélome multiple.
[44] Selon un mode de réalisation, le cancer est chimiorésistant à la
daunorubicine, à la
cytarabine, au fluorouracile, au cisplatine, à l'acide tout-trans-rétinoïque,
au trioxyde
d'arsenic, au bortézomib ou à l'une de leurs combinaisons.
[45] Toutes les références aux composés de formules (I) comprennent les
références aux
sels, aux complexes multi-composants et leurs cristaux liquides. Toutes les
références
aux composés de formules (I) comprennent également les références aux
polymorphes
et leurs cristaux habituels.
[46] Le composé selon l'invention peut être sous forme de sels
pharmaceutiquement
acceptables. Un sel pharmaceutiquement acceptable du composé de formule (I)
comprend l'addition d'acide de celui-ci.
[47] Des sels acides appropriés sont formés à partir d'acides qui forment
des sels non
toxiques. Par exemple choisi parmi : l'acétate, l'adipate, le benzoate, le
bicarbonate, le
carbonate, le bisulfate, le sulfate, borate, camsylate, citrate, cyclamate,
édisylate, ésylate,
formate, furamate, gluceptate, gluconate, glucuronate, hexafluorophosphate,
hibenzate,
chlorhydrate de chlorure, brom hydrate, bromure, hydroiodure, iodure,
iséthionate, lactate,
malate, maléate, malonate mésylate, méthylsulfate, naphtylate, 2-napsylate,
nicotinate,
nitrate, orotate, oxalate, palmitate, pamoate, phosphate, hydrogène phosphate,
phosphate dihydrogène, pyroglutamate, saccarate, stérate, succinate, tan nate,
sels de
tartrate, tosylate, trifluoroacétate et xinofoate. De manière préféré, le sel
pharmaceutiquement acceptable du composé de formule (I) est formé à partir du
lactate.
[48] Les sels pharmaceutiquement acceptables des composés de formule (I)
peuvent être
préparés par une ou plusieurs des trois méthodes suivantes :
(i) en faisant réagir le composé de formule (I) avec l'acide désiré ;
(ii) en éliminant un groupe protecteur labile en milieu acide ou basique d'un
précurseur
CA 03196518 2023- 4- 24
WO 2022/090427 11
PCT/EP2021/080054
approprié du composé de formule (I) ou en ouvrant le cycle d'un précurseur
cyclique
approprié, par exemple une lactone ou un lactame, en utilisant l'acide ou la
base désiré ;
ou
iii) par conversion d'un sel du composé de formule (I) en un autre par
réaction avec un
acide ou une base appropriés ou au moyen d'une colonne échangeuse d'ions
appropriée.
[49]
Ces trois réactions sont généralement effectuées en solution. Le sel
obtenu peut
précipiter et être recueilli par filtration ou peut être récupéré par
évaporation du solvant.
Le degré d'ionisation du sel obtenu peut varier de complètement ionisé à
presque non
ionisé.
[50]
L'invention concerne en deuxième objet une composition pharmaceutique
comprenant
dans un véhicule pharmaceutiquement acceptable au moins un composé selon
l'invention,
comme décrit ci-dessus pour son utilisation pour faire régresser une tumeur
cancéreuse
de mammifère.
[51] Selon un mode de réalisation, la composition pharmaceutique comprend
en outre au
moins un autre agent thérapeutique.
[52] Selon un monde de réalisation préféré, cet autre agent thérapeutique
est un agent
antinéoplasique.
[53] Selon un mode de réalisation, l'agent antinéoplasique est un agent
endommageant
l'ADN tel que la camptothécine, l'irinotécan, le topotécan, l'amsacrine,
l'étoposide, le
phosphate d'étoposide, le téniposide, le cisplatine, le carboplatine,
l'oxaliplatine, le
cyclophosphamide, le chlorambucil, la chlorméthine, le busulfan, le tréosulfan
ou le
thiotépa, un antibiotique antitumoral tel que la daunorubicine, la
doxorubicine, l'épirubicine,
l'idarubicine, la mitoxantrone, la valrubicine, l'actinomycine D, la
mitomycine, la
bléomycine ou la plicamycine, un antimétabolite tel que le 5-fluorouracile, la
cytarabine, la
fludarabine ou le méthotrexate, un antimitotique tel que le paclitaxel, le
docétaxel, la
vinblastine, la vincristine, la vindésine ou la vinorelbine, ou divers agents
antinéoplasiques
tels que le bortézomib, l'acide tout- trans-rétinoïque, le trioxyde d'arsenic,
ou l'un de leurs
produits combinés.
[54] Selon un mode de réalisation, la composition pharmaceutique est
utilisée dans le
traitement du cancer chez un patient souffrant d'une tumeur qui est
chimiorésistante audit
agent antinéoplasique lorsqu'il n'est pas administré en combinaison avec un
composé
selon l'invention.
[55] Selon un mode de réalisation, la composition pharmaceutique est
utilisée dans le
traitement du cancer chez un patient souffrant d'une tumeur qui est
chimiosensible audit
CA 03196518 2023- 4- 24
WO 2022/090427 12
PCT/EP2021/080054
agent antinéoplasique, et la dose de l'agent antinéoplasique administré audit
patient en
combinaison avec un composé selon l'invention ou l'un de ses sels
pharmaceutiquement
acceptables est inférieure à la dose de l'agent antinéoplasique lorsqu'il
n'est pas
administré en combinaison avec un composé selon l'invention. En particulier,
la dose de
l'agent antinéoplasique administré audit patient en combinaison avec un
composé selon
l'invention ou l'un de ses sels pharmaceutiquement acceptables est inférieure
à la dose
de l'agent antinéoplasique administré seul, sans autre principe actif.
[56] La composition pharmaceutique selon l'invention peut également
comprendre en outre
d'autres composés thérapeutiques actifs utilisés couramment dans le traitement
de la
pathologie énoncée ci-dessus.
[57] Selon un mode de réalisation, la composition pharmaceutique de
l'invention peut être
administrée par toutes voies, notamment par voies : intradermique,
intramusculaire,
intrapéritonéale, intraveineuse ou sous-cutanée, pulmonaire, transmuqueuse
(orale,
intranasale, intravaginale, rectale), inhalation par spray nasal, utilisant
une formulation en
comprimé, capsule, solution, poudre, gel, particule ; et contenu dans une
seringue, un
dispositif implanté, une pompe osmotique, une cartouche, une micropompe ; ou
tout autre
moyen apprécié par l'artisan qualifié, bien connu dans l'art. L'administration
spécifique à
un site peut être réalisée, par exemple par voie intratumorale, infra-
articulaire,
intrabronchique, intra-abdominale, intracapsulaire, intra-cartilagineuse,
intracavitaire,
intracérébelleu se, intracérébroventriculaire, intracolique, intracervicale,
intragastrique,
intra-hépatique, intracardiaque, intraostéale,
intra-pelvien, intrapéricardique,
intrapéritonéal, intrapleural, intraprostatique, intrapulmonaire, intrarectal,
intrarénal,
intrarétinien, intraspinal, intrasynovial, intrathoracique, intra-utérin,
intravasculaire,
intravésical, intralésionnel, vaginal, rectal, buccal, sublingual, intranasal
ou transdermique
dans un dosage adapté comprenant les véhicules usuels non toxiques et
pharmaceutiquement acceptables. De manière préférée, la composition
pharmaceutique
est sous forme appropriée pour être administrée par voie intraveineuse, sous-
cutanée,
intrapéritonéale ou orale. La voie orale étant particulièrement préférée.
[58] Outre les animaux à sang chaud tels que les souris, les rats, les
chiens, les chats, les
moutons, les chevaux, les vaches et les singes, le composé de l'invention est
également
efficace sur les humains.
[59] Selon un mode de réalisation, les compositions pharmaceutiques pour
l'administration
des composés de cette invention peuvent être présentées sous forme de doses
unitaires
et peuvent être préparées par l'une des méthodes bien connues dans l'état de
l'art. Toutes
les méthodes comprennent l'étape consistant à mettre le principe actif en
association avec
CA 03196518 2023- 4- 24
WO 2022/090427 13
PCT/EP2021/080054
le support qui constitue un ou plusieurs ingrédients accessoires. En général,
les
compositions pharmaceutiques sont préparées en mettant l'ingrédient actif en
association
avec un support liquide ou un support solide finement divisé ou les deux,
puis, si
nécessaire, en façonnant le produit dans la formulation souhaitée. Dans la
composition
pharmaceutique, le composé de l'objet actif est inclus en quantité suffisante
pour produire
l'effet souhaité sur le processus ou l'état des maladies. Les compositions
pharmaceutiques
contenant le principe actif peuvent se présenter sous une forme adaptée à
l'usage oral,
par exemple sous forme de comprimés, de pastilles, de suspensions aqueuses ou
huileuses, de poudres ou de granulés dispersibles, d'émulsions, de capsules,
de sirops,
d'élixirs, de solutions, de timbres buccaux, de gel oral, de chewing-gum, de
comprimés à
mâcher, de poudre effervescente et de comprimés effervescents. Les
compositions
pharmaceutiques contenant le principe actif peuvent se présenter sous une
forme de
suspension aqueuse ou huileuse.
[60] Selon un mode de réalisation, les suspensions aqueuses contiennent les
matières
actives en mélange avec des excipients adaptés à la fabrication de suspensions
aqueuses.
Ces excipients sont des agents de suspension, par exemple la
carboxyméthylcellulose
sodique, la méthylcellulose, l'hydroxy-propylméthylcellulose, l'alginate de
sodium, la
polyvinyl-pyrrolidone, la gomme adragante et la gomme d'acacia ; les agents
dispersants
ou mouillants peuvent être un phosphatide naturel, par exemple la lécithine,
ou des
produits de condensation d'un oxyde d'alkylène avec des acides gras, par
exemple le
stéarate de polyoxyéthylène, ou des produits de condensation de l'oxyde
d'éthylène avec
des alcools aliphatiques à longue chaîne, par exemple l'heptadécaéthylène-
oxycétanol,
ou des produits de condensation de l'oxyde d'éthylène avec des esters partiels
dérivés
d'acides gras et d'un hexitol, comme le monooléate de polyoxyéthylène
sorbitol, ou des
produits de condensation de l'oxyde d'éthylène avec des esters partiels
dérivés d'acides
gras et d'anhydrides d'hexitol, par exemple le monooléate de polyéthylène
sorbitol. Les
suspensions aqueuses peuvent également contenir un ou plusieurs conservateurs,
par
exemple de l'éthyle, ou du n-propyle, du p-hydroxybenzoate, un ou plusieurs
colorants, un
ou plusieurs aromatisants, et un ou plusieurs édulcorants, comme le saccharose
ou la
saccharine.
[61] Selon un mode de réalisation, les suspensions huileuses peuvent être
formulées en
mettant en suspension le principe actif dans une huile végétale, par exemple
l'huile
d'arachide, d'olive, de sésame ou de coco, ou dans une huile minérale telle
que la paraffine
liquide. Les suspensions huileuses peuvent contenir un agent épaississant, par
exemple
de la cire d'abeille, de la paraffine dure ou de l'alcool cétylique. Des
agents édulcorants
tels que ceux mentionnés ci-dessus et des agents aromatisants peuvent être
ajoutés pour
CA 03196518 2023- 4- 24
WO 2022/090427 14
PCT/EP2021/080054
obtenir une préparation orale agréable au goût. Ces compositions peuvent être
conservées par l'ajout d'un antioxydant tel que l'acide ascorbique. Les
poudres et granules
dispersables qui conviennent à la préparation d'une suspension aqueuse par
addition
d'eau fournissent l'ingrédient actif en mélange avec un agent dispersant ou
mouillant, un
agent de suspension et un ou plusieurs conservateurs.
[62]
Les sirops et élixirs peuvent être formulés avec des agents édulcorants,
par exemple
le glycérol, le propylène glycol, le sorbitol ou le saccharose. Ces
formulations peuvent
également contenir un émollient, un agent de conservation, des agents
aromatisants et
colorants.
[63] Les
compositions pharmaceutiques peuvent se présenter sous la forme d'une
suspension aqueuse ou oléagineuse injectable de manière stérile. Cette
suspension peut
être formulée selon l'art connu en utilisant les agents dispersants ou
mouillants et les
agents de suspension appropriés qui ont été mentionnés ci-dessus. La
préparation stérile
injectable peut également être une solution ou une suspension stérile
injectable dans un
diluant ou un solvant non toxique acceptable par voie parentérale, par exemple
une
solution dans du 1,3-butane diol. Les véhicules et solvants acceptables
pouvant être
utilisés comprennent ; l'eau, le liquide de Ringer et la solution isotonique
de chlorure de
sodium. En outre, des huiles fixes stériles sont traditionnellement utilisées
comme solvant
ou milieu de suspension. A cette fin, toute huile fixe peut être utilisée, y
compris les mono-
ou diglycérides synthétiques. En outre, les acides gras tels que l'acide
oléique sont utilisés
dans la préparation des produits injectables.
[64] Les compositions pharmaceutiques de la présente invention peuvent
également être
administrés sous forme de suppositoires pour l'administration rectale du
médicament. Ces
compositions peuvent être préparées en mélangeant le médicament avec un
excipient
approprié non irritant qui est solide à la température ordinaire mais liquide
à la température
rectale et qui fondra donc dans le rectum pour libérer le médicament. Ces
matières
comprennent le beurre de cacao et les polyéthylèneglycols.
[65] En outre, les compositions pharmaceutiques peuvent être administrés
par voie
oculaire au moyen de solutions ou de pommades. De plus, l'administration
transdermique
des composés en question peut être réalisée au moyen de patchs
iontophorétiques et
autres. Pour l'utilisation topique, on utilise des crèmes, des pommades, des
gelées, des
solutions ou des suspensions.
[66] Dans le traitement d'un mammifère ou patient souffrant ou risquant de
développer un
cancer, un dosage approprié de la composition pharmaceutique de cette
invention peut
généralement être d'environ 0,1 à 50 000 microgrammes (pg) par kg de poids
corporel du
CA 03196518 2023- 4- 24
WO 2022/090427 15
PCT/EP2021/080054
patient par jour, qui peut être administré en doses uniques ou multiples. Le
niveau de
dosage sera de préférence d'environ 1000 à environ 40 000 pg/kg par jour, en
fonction de
nombreux facteurs tels que la gravité du cancer à traiter, l'âge et l'état de
santé relatif du
sujet, la voie et la forme d'administration. Pour l'administration orale,
cette composition
peut être fournie sous forme de comprimés contenant 1000 à 10 0000
microgrammes de
chacun des principes actifs, en particulier 1000, 5000, 10 000, 15 000, 20
000, 25 000, 50
000, 75 000, 10 0000 microgrammes de chaque principe actif. Cette composition
peut être
administrée selon un schéma de 1 à 4 fois par jour, par exemple une ou deux
fois par jour.
Le régime posologique peut être ajusté pour fournir une réponse thérapeutique
optimale.
L'invention divulgue également un procédé de fabrication du composé de formule
(1).
[67] Selon un mode de réalisation, le procédé de fluoration en C3 comprend
une étape de
fluoration de la Dendrogénine A, réalisée à partir d'un réactif de fluoration,
par exemple le
Diéthylaminosulfur trifluoride (DAST) ou le tétrafluoroborate. La réaction de
fluoration avec
le DAST est décrite dans la littérature e Tetrahedron letters 1979, 20, 1823-
1826, A new
method for fluorination of sterols (https://doi.org/10.1016/S0040-
4039(01)86228-6). La
réaction de fluoration avec le tétrafluoroborate est décrite dans la
littérature Org. Lett.,
Vol. 11, No. 21, 2009, 5050-5053, Aminodifluorosulfinium Tetrafluoroborate
Salts as Stable
and Crystalline Deoxofluorinating Reagents
[68] Selon un mode de réalisation, le procédé de synthèse du 5a-hydroxy-
6[342-(1 I-1-
imidazol-4-y1)-éthylamino]-cholestan-313-fluoro dilactate comprend les étapes
suivantes :
- dissolution du composé 5a-hydroxy-6p-[2-(1H-imidazol-4-y1)-éthylamino]-
cholestan-3p-
fluoro dans de l'éthanol anhydre puis y ajouter de l'acide lactique ;
- agitation du mélange à température ambiante durant 3 h;
- évaporation du solvant organique.
La poudre blanche obtenue est le composé 5a-hydroxy-6[3-[2-(1H-imidazol-4-y1)-
éthylamino]-cholestan-3p-fluoro dilactate.
[69]
Selon un mode de réalisation du procédé, la température ambiante est
comprise entre
15 et 40 C, par exemple 25 ou 37, préférentiellement 20 C.
Brève description des figures
[70]
L'invention sera mieux comprise, et d'autres buts, détails, caractéristiques
et
avantages de celle-ci apparaîtront plus clairement au cours de la description
suivante de
plusieurs modes de réalisation particuliers de l'invention, donnés uniquement
à titre
illustratif et non limitatif, en référence aux dessins annexés.
CA 03196518 2023- 4- 24
WO 2022/090427 16
PCT/EP2021/080054
[71] [fig.1] La figure 1 représente les résultats d'une étude de
cytotoxicité du 5a-hydroxy-
613-[2-(1H-imidazol-4-y1)-éthylamino]-cholestan-3p-fluoro (DX111) sur les
cellules
Neuro2a via un test de bleu de trypan.
[72] [fig.2] La figure 2 illustre les résultats d'un test de viabilité
cellulaires MTT réalisé sur
des cellules tumorales mammaires MCF-7 en présence du composé 5a-hydroxy-6p-[2-
(1H-imidazol-4-y1)-éthylamino]-cholestan-3p-fluoro.
[73] [fig. 3] La figure 3 illustre les résultats de l'activité de la
Cholestérol Epoxyde Hydrolase
(ChEH) dans des cellules MCF-7 en présente du composé 5a-hydroxy-6[312-(1H-
imidazol-4-y1)-éthylamino]-cholestan-3p-fluoro.
lo [74]
[fig. 4] La figure 4 illustre le profil pharmacocinétique du composé 5a-
hydroxy-6p-[2-
(1H-imidazol-4-y1)-éthylamino]-cholestan-313-méthoxyle (DX103) en comparaison
avec le
composé Dendrogénine A (DX101).
[75] [fig. 5] La figure 5 illustre le profil pharmacocinétique du composé
5a-hydroxy-6p-[2-
(1H-imidazol-4-y1)-éthylamino]-cholestan-3p-éthoxyle (DX105) en comparaison
avec le
composé Dendrogénine A (DX101).
[76] [fig. 6] La figure 6 illustre le profil pharmacocinétique du composé
5a-hydroxy-6p-[2-
(1H-imidazo1-4-y1)-éthylamino]-cholestan-313-fluoro (DX111) en comparaison
avec le
composé Dendrogénine A (DX101).
[77] [fig. 7A] et [fig. 7B] Les figures 7A et 7B illustrent l'évolution de
la croissance tumorale
et le taux de survie chez la souris en comparant le traitement avec le DX111
et le DX101.
[78] [fig. 8] La figure 8 illustre le profil pharmacocinétique du composé
5a-hydroxy-613-[2-
(1H-imidazol-4-y1)-éthylamino]-cholestan-3[3-azoture (DX123) en comparaison
avec le
composé Dendrogénine A (DX101).
EXEMPLES
[79]
Différentes expériences ont été réalisées afin d'évaluer les caractéristiques
des
composés de formule (1).
[80]
Les composés préférés selon l'invention correspondant à la formule 1
générale dont il
est décrit ci-après la synthèse et l'activité sont les suivants :
CA 03196518 2023- 4- 24
WO 2022/090427 17
PCT/EP2021/080054
-----..
--
Et,c)
Fe,
HO H HO H HO H
DX103 DX105 DX115 DX111
N Hd fede
0
H2N Fi,. Me-ILII Hd
'H
Me-S Hv H
DX123 DX125 DX127 DX129
Les autres composés rentrant dans la portée de la formule générale, non
décrits, font partie
intégrante des composés selon l'invention.
[81] Exemple 1 : Synthèse du composé analogue 5a-hydroxy-613-[2-(1H-
imidazol-4-y1)-
éthylamino]-cholestan-313-fluoro (nommé DX111)
La première étape est une synthèse du composé 3p-fluorocholestane comprenant
les
étapes suivantes :
õ .
¨
-\---).--
r.
_________________________________________________ ,
r
,......_,... F'-' =-j-s".-
j-
fio y -"-L'' TICM
to ri. 6h
[82] 5.00 g de trifluorure de diéthylaminosulfure (d=1.22 g/ml, 31.0 mmol)
ont été dissous
on 200 ml de DCM anhydre. 6.66 grammes (g) de cholestérol (17.2 mmol) ont été
mis en
solution dans 100 millilitres (m1) de dichlorométhane anhydre et ajouté goutte
par goutte
au réactif fluoré à la température de 000. Le mélange ainsi obtenu a été
laissé sur agitation
magnétique pour 5 heures en la laissant chauffer jusqu'au la température
ambiante. A
l'issus de cette durée, la réaction a été neutralisée par addition de 100 ml
d'une solution
de NaHCO3 sat. Le mélange a été transféré dans une ampoule à décanter et la
phase
organique a été lavée par 2 fois avec NaHCO3 sat, 2 autres fois par une
solution saturée
de NaCI et une seule fois par l'eau. La phase organique a été séchée sur du
MgSO4, filtrée
et puis évaporée permettant l'obtention d'une poudre blanche. 6.61 g
correspondant au
3p-fluorocholestane a été obtenu. Le rendement final de la réaction est de 99
%.
CA 03196518 2023- 4- 24
WO 2022/090427 18
PCT/EP2021/080054
[83] 1H-NMR (500 MHz, CDCI3): O (ppm) 5.40 ¨ 5.39 (d, 1H), 4.47 ¨ 4.30 (m,
1H), 2.45 ¨
2.42 (t, 2H), 2.03 ¨ 1.95 (m, 3H), 1.90 ¨ 0.95 (m, 26H), 0.92 ¨ 0.91 (d, 3H),
0.87 - 0.85
(dd, 6H), 0.68 (s, 3H).
[84] La deuxième étape consiste à synthétiser à partir du 313-fluoro-
cholestane le composé
313-fluoro-5,6a-Epoxy-cholestane de la manière suivante :
f. J.
n)
t1 Fe
[85] 4.96 g d'acide méta-chloro-peroxybenzoïque au 77 % de pureté (22.1
mmol) ont été
dissous dans 100 ml de dichlorométhane et ajouté goutte à goutte dans un
mélange de
6.61 g de 313-fluorocholestane (17.0 mmol) dissous dans 50 ml de
dichlorométhane. Le
mélange ainsi obtenu a été mis sous agitation et maintenu à température
ambiante durant
3 heures. Le mélange obtenu a été lavé deux fois avec une solution aqueuse de
Na2S203
à 10 % en poids, deux fois une solution saturée de NaHCO3 et une solution
saturée de
NaCI. La phase organique a été séchée sur du MgSO4. anhydre. Une évaporation
sous
vide du solvant organique a été réalisée et permet l'obtention de 6.90 g d'une
poudre
blanche comprenant : 3j3-fluoro-5,6a-Epoxy-cholestane (85 (3/0 de la poudre
blanche) et
3j3-fluoro-5,613-Epoxy-cholestane (15 % de la poudre blanche). Le 3j3-fluoro-
5,6a-
epoxycholestane a été utilisé sans purification supplémentaire.
[86] 1H-NMR (500 MHz, CD0I3): O (ppm) 4.82 ¨ 4.64 (m, 1H), 2.91 ¨ 2.90 (d,
1H), 2.28 ¨
2.21 (m, 1H), 2.10 ¨ 2.06 (m, 1H), 1.97 ¨ 1.70 (m, 6H), 1.59 ¨ 0.92 (m, 23H),
0.89 ¨ 0.88
(d, 3H), 0.87 - 0.85 (dd, 6H), 0.61 (s, 3H).
[87] La troisième étape consiste à synthétiser le 5a-hydroxy-61312-(1H-
imidazol-4-y1)-
éthylaminoLcholestan-3E3-fluoro (DX111 sous forme basique) de la manière
suivante :
NH,
r
Bu( =
Fe" ¨ 130 C, 2d Fe¨ -
O
Ft%
.µ
CA 03196518 2023- 4- 24
WO 2022/090427 19
PCT/EP2021/080054
[88] 0.80 g d'histamine sous sa forme basique (7.2 mmol) a été ajouté à
une solution
butanolique de 10 ml comprenant 1.45 g du composé 3p-fluoro-5,6a-
epoxycholestane (3.6
mmol) sous agitation à 130 C. Le mélange a été maintenu sous agitation dans un
chauffage à reflux à une température de 130 C durant 48 heures.
[89]
L'avancée de la réaction peut être contrôlée par chromatographie sur couche
mince
(CCM) pour suivre la conversion du 3f3-fluoro-5,6-epoxycholestane.
[90] Après refroidissement, le mélange a été dilué dans 15 ml de méthyl
tert-butyl éther. La
phase organique a été lavée par 3 fois 15 ml d'eau.
[91] La phase organique a été séchée sur du MgSO4 anhydre, filtré et puis
évaporée
permettant l'obtention d'une huile brune. Le mélange a été purifié par colonne
chromatographique sur gel de Silice sur un automate de purification comprenant
une
colonne prépacké de 20 g éluant Acétate d'éthyle 100 %. Une poudre blanche de
0.86 g
de 5a-hydroxy-613-[2-(1H-imidazol-4-y1)-éthylamino]-cholestan-313-fluoro a été
obtenue. Le
rendement final de la réaction est de 41 (3/0 avec une pureté supérieure à 97
% mesurée
par analyse RMN (résonance magnétique nucléaire) et TLC (Chromatographie sur
couche
mince).
[92] 1H-NMR (500 MHz, CDCI3): ô (ppm) 7.54 (s, 1H), 6.80 (s, 1H), 5.05 ¨ 4.88
(m, 1H),
3.03 ¨ 2.96 (m, 1H), 2.77 ¨ 2.73 (m, 3H), 2.46 (s, 1H), 2.27 ¨ 2.20 (q, 1H),
2.00 ¨ 1.98
(d, 2H), 1.86 ¨ 0.94 (m, 31H), 0.91 ¨ 0.89 (d, 3H), 0.87 ¨ 0.85 (d, 6H), 0.67
(s, 3H).
[93]
Exemple 2: Préparation d'un sel dilactate du composé 5a-hydroxy-6512-(11-1-
imidazol-4-v1)-éthylaminol-cholestan-313-fluoro (DX111 sous forme dilactate) :
[94] Un sel dilactate du composé 5a-hydroxy-6P-[2-(1H-imidazol-4-y1)-
éthylamino]-
cholestan-313-fluoro a été préparé de la manière
suivante :
r\----)-- 0
, ..,
t i.... i
C=H /
Xy-t=-r\---)----
I '''' = - ' - -"" F : . -=
110 f t ' HOt.J. 0
1..õ N ri 311 ' =-= = .
Q,..,õ 0- 1 he -0)Lre
N
IN
il -- ..."`c [ =
Nil
[95] 267.2
mg d'acide lactique (2.97 mmol) a été ajouté à une solution de 0.76 g de 5a-
hydroxy-613-[2-(1H-imidazol-4-y1)-éthylamino]-cholestan-3p-fluoro dans 15 ml
d'éthanol
anhydre sous agitation. L'agitation a été maintenue à température ambiante 3
heures. Une
CA 03196518 2023- 4- 24
WO 2022/090427 20
PCT/EP2021/080054
évaporation sous vide du solvant organique permet l'obtention d'une poudre
blanche de
1.03 g de 5a-hydroxy-61312-(1H-imidazol-4-y1)-éthylamino]-cholestan-3p-fluoro
dilactate.
[96] 1H-NMR (500 MHz, Me0D-4d): O (ppm) 7.58 (s, 1H), 6.79 (s, 1H), 4.73 -
4.57 (m, 1 H),
3.86 - 3.82 (dd, 2H), 3.35 - 3.31 (dd, 2H), 3.18 - 3.13 (m, 1H), 3.03 ¨ 2.98
(m, 1H), 2.77 -
2.75(t, 2H), 2.70 (s, 1H), 2.12 - 2.05 (dd, 1H), 1.78- 1.76(d, 1H), 1.70 ¨
1.68 (d, 1H), 1.63
- 0.85 (m, 30H), 0.78 - 0.73 (d, 2H), 0.68 ¨ 0.66 (d, 3H), 0.61-0.60 (dd, 6H),
0.49 (s, 3H).
[97] Exemple 3 : préparation des dérivés ou analogues 3a amino et 3a
sulfure de formule
(I) :
[98] Les étapes sont les suivantes :
1) NaH, p TsCI
2) NuH
THF
H 70nC, 24h
Nu = R2S ou Ft2R3N
DOM ni-CPBA
ri, 4h
n Du0H
130 C, 48h
H H
H
Nu = R2R3KI et dans le R2S la réaction
Nu = R2R3N et R202S
donne le produit d'oxydation R202S
Mettre en agitation du cholestérol en tetrahydrofurane (THF) en présence de
NaH durant
cinq minutes à 70 C, additionner du paratoluènesulfonique chlorure (p-TsCI) et
agiter le
mélange durant 4 heures à 70 C. Additionner de l'eau, filtrer la réaction et
évaporer les
solvants organiques. Extraire le produit de la réaction avec
dichlorométhane/eau
(DCM/H20) puis sécher sur MgSO4. Éliminer les solvants organiques par
évaporation sous
vide. Le produit obtenu est utilisé tel quel pour l'étape suivante.
Solubiliser le produit
obtenu en THF sous agitation durant 12 h avec 1.1 équivalent de NuH
(Nucléophile-
hydrogène). NuH correspond à R2SH ou NHR2R3 à 70 C. La réaction est arrêtée
par
addition d'eau et les produits ont été extraits par un système AcOEt/H20. La
phase
organique a été séchée sur MgSO4 et les solvants organiques évaporés sous
vide. Les
produits cholestan-3-sulfure et cholestan-3-amino dérivés sont purifiés soit
par
chromatographie sur colonne soit par recristallisation. Le chemin réactionnel
pour obtenir
CA 03196518 2023-4-24
WO 2022/090427 21
PCT/EP2021/080054
les analogues de la Dendrogénine A sont les mêmes étapes développées pour la
synthèse
de la Dendrogénine A.
[99] Le produit R202S sera obtenu par oxydation de R2S avec des
agents d'oxydation tels
que le m-CPBA ou H202.
[100] Exemple 4 : Préparation de dérivés ou analogues 313 amino et 313 sulfure
de formule
LU
Les étapes sont présentées ci-dessous :
MsCI TMS-SD2
BF3.Et.,0
Et3N
DCM
DCM
H rt, 12h Ms r1, 3h R2S
DCM TMSN,
3h BF,Et20
LIAIH4
Et20
(R2)2N d 2elf N =
H H
Dissoudre du cholestérol, ajouter du NEt3 dans du DCM et ajouter du chlorure
de mésyle
(MsCI) goutte à goutte en solution de DCM à température ambiante durant 1 h.
Agiter la
réaction durant 12 h, puis évaporer le solvant organique et cristalliser le
produit avec du
Me0H. Le produit obtenu est un solide de couleur blanche. Le produit obtenu
est utilisé
pour obtenir le 3- sulfure et 38-azoture dérivés. Le produit obtenu est
dissout en DCM
puis du TMS-SR2 pour le 38- sulfure dérivé ou du TMS-N3 pour le 313-azoture
dérivé est
ajouté à la solution. Une addition de BF3*Et20 est réalisée à température
ambiante.
Ensuite, on agite durant 3 h.
[101] Le 38-azoture est réduit en 38-amino par action de LiAIH4 en Et20 et
transformé dans
les produits de formule (I) avec réaction de R2X (X = Br, Cl ou I) en Et20 (ou
pyridine)
comme solvant. Le chemin réactionnel pour obtenir les analogues de la
Dendrogénine A
sont les mêmes étapes développées pour la synthèse de la Dendrogénine A. Le
dérivé
sulfonyle R202S sera obtenu par oxydation de R2S par des agents oxydants
usuels. Cette
méthode est détaillée dans la littérature ORGANIC LETTERS; 2009, 11, 3, 567-
570,
Practical Synthesis of 3p-Amino-5-cholestene and Related 3p-Halides Involving
i-Steroid
and Retro-i-Steroid Rearrangements (https://doi.org/10.1021/o1802343z).
CA 03196518 2023- 4- 24
WO 2022/090427 22
PCT/EP2021/080054
[102] Exemple 5 : Étude de la cytotoxicité du 5a-hydroxy-613-[2-(1H-imidazol-4-
y1)-
éthylaminecholestan-313-fluoro (nommé DX111)
[103] Pour cette expérience, un milieu de culture cellulaire a été préparé. Le
milieu de culture
est constitué d'un milieu Dulbecco's Modified Eagle Medium (DMEM,
commercialisé par
VVestburg sous la référence LO BE12-604F), comprenant 4.5 g/L glucose avec de
la L-
Glutamine, auquel y est ajouté 10 % de sérum de veau foetal (SVF). Des
cellules Neuro2a
(neuroblastome murin) sont introduites dans ce milieu de culture.
[104] II a été ensemencé des boites de 24 puits avec 10 000 cellules Neuro2a
par puits.
Après 72 heures (h) de culture en condition normale, c'est-à-dire dans un
incubateur à
une température de 37 C à 5 % de CO2, nous traitons pendant 48h les cellules
Neuro2a
avec le 5a-hydroxy-6[3-[2-(1H-imidazol-4-y1)-éthylamino]-cholestan-38-fluoro
et le 5a-
hydroxy-613-[2-(1H-imidazol-4-y1)-éthylamino]-cholestan-313-ol à 100 nM, 1 M
et 10 M.
Un contrôle (CTL) est également réalisé en utilisant le protocole décrit
précédemment
sans le traitement avec le 5a-hydroxy-61312-(1H-imidazol-4-y1)-éthylamino]-
cholestan-3a-
fluoro et le 5a-hydroxy-6812-(1H-imidazol-4-y1)-éthylaminoLcholestan-38-ol. La
survie
cellulaire est quantifiée par un test de bleu de trypan avec comptage
automatique par
l'appareil Biorad TC20 (TC20Tm Automated Cell Counter). Le test via le bleu de
trypan est
basé sur l'intégrité des membranes cellulaire, laquelle est rompue chez les
cellules mortes.
Le bleu de trypan colore les cellules mortes en bleu. L'appareil de comptage
cellulaire
Biorad TC20 compte la proportion de cellules bleues et non-bleues, et rapporte
au
pourcentage de cellules. Les résultats sont représentés figure 1. La figure 1
illustre en
ordonnée le pourcentage de survie cellulaires par rapport au groupe contrôle.
[105] II est illustré figure 1 que pour 100 nM de traitement au 5a-hydroxy-
6312-(1H-imidazol-
4-y1)-éthylamino]-cholestan-38-fluoro le pourcentage de cellule vivante reste
inchangé en
comparaison au groupe contrôle (CTL). En outre, pour des concentrations de 1
M et 10
M, le pourcentage de survie cellulaire est de 75 % et 30 /0. Il est également
observé une
activité similaire entre les deux composés testés.
En conclusion, on observe une activité cytotoxique du composé 5u-hydroxy-6312-
(1H-
imidazol-4-y1)-éthylamino]-cholestan-3j3-fluoro de formule (1) envers les
cellules tumorales
Neuro2a pour des concentrations en 5a-hydroxy-613-[2-(1H-imidazol-4-y1)-
éthylamino]-
cholestan-3a-fluoro de 1 M et 10 M.
[106] Exemple 6: Effet du 5a-hydroxy-6312-(1H-imidazol-4-y1)-éthylamino]-
cholestan-38-
fluoro sur la viabilité des cellules MCF-7
CA 03196518 2023- 4- 24
WO 2022/090427 23
PCT/EP2021/080054
[107] Un test de viabilité cellulaires a été réalisé sur des cellules
tumorales mammaires
MCF-7 (Michigan Cancer Foundation-7) surexprimant HER2 (cellules ER(-0).
Les cellules MCF-7 sont dans un milieu de culture cellulaire identique à
l'exemple 5 et sont
ensemencées dans des plaques 12 puits à 50 000 cellules par puits. 24 heures
après
ensemencement, les cellules sont traitées par le solvate véhicule comprenant
de l'eau et
de l'éthanol avec un rapport 1% d'éthanol, le 5a-hydroxy-613-[2-(1H-imidazol-4-
y1)-
éthylamino]-cholestan-3p-fluoro et le 5a-hydroxy-63-[2-(1H-imidazol-4-y1)-
éthylamino]-
cholestan-313-ol à 1, 2.5 ou 5 M. Les cellules sont observées au microscope
inversé et
photographiées via la caméra du microscope à 24 h et 48 h. Les modifications
morphologiques des cellules à 1 M sont très faibles. On observe uniquement
quelques
vésicules blanches, traduisant le début du phénomène d'autophagie, engendrant
la mort
cellulaires après 24 heures de traitement avec le 5a-hydroxy-613-[2-(1H-
imidazol-4-y1)-
éthylamino]-cholestan-3p-fluoro et le 5a-hydroxy-6p-[2-(1H-imidazol-4-y1)-
éthylamino]-
cholestan-3p-ol. Les effets sont davantage marqués à 2.5 M et 5 M avec une
augmentation du nombre de cellules mortes. En effet, il est observé après 24 h
de
traitement au 5a-hydroxy-61342-(1H-imidazol-4-y1)-éthylamino]-cholestan-33-
fluoro à 2,5
M de nombreuses vésicules blanches et des cellules qui se décrochent. Après 24
h de
traitement au 5a-hydroxy-613-[2-(1H-imidazol-4-y1)-éthylamino]-cholestan-3p-
fluoro à 5 M,
99 % des cellules observées sont surnageantes, traduisant une mort cellulaire
et 1 % des
cellules sont adhérentes et présentent des vésicules blanches. On observe
après 48 h de
traitement au 5a-hydroxy-61342-(1H-imidazol-4-y1)-éthylamino]-cholestan-33-
fluoro à 2,5
M un effet cytostatique plus important qu'à 24 h et davantage de cellules qui
s'arrondissent, traduisant la mort cellulaires. L'effet cytostatique est
illustré par une
inhibition de la prolifération cellulaire. Après 48 h de traitement au 5a-
hydroxy-6[312-(1H-
imidazol-4-y1)-éthylamino]-cholestan-3p-fluoro à 5 M, toutes les cellules
sont
surnageantes. En comparaison avec le traitement avec le 5a-hydroxy-613-[2-(1H-
imidazol-
4-y1)-éthylamino]-cholestan-313-ol, le traitement avec le 50-hydroxy-613-[2-
(1H-imidazol-4-
y1)-éthylamino]-cholestan-33-fluoro présente un effet supérieur ou égale après
24 h
d'observation et similaire après 48 h d'observation.
[108] La viabilité cellulaire est mesurée par marquage MTT à 48 heures. Ce
test est basé
sur l'utilisation du sel de tétrazolium MTT (bromure de 3-(4,5-dimethylthiazol-
2-y1)-2,5-
diphenyl tétrazolium). Le tétrazolium est réduit par la succinate
déshydrogénase
mitochondriale des cellules vivantes actives, en formazan, précipité de
couleur violette. La
quantité de précipité formé est proportionnelle à la quantité de cellules
vivantes mais
également à l'activité métabolique de chaque cellule. Ainsi, un simple dosage
de la densité
optique à 540 nm par spectroscopie permet de connaître la quantité relative de
cellules
CA 03196518 2023- 4- 24
WO 2022/090427 24
PCT/EP2021/080054
vivantes et actives métaboliquement. Après 48 heures, le milieu est aspiré,
les cellules
lavées au tampon phosphate salin (PBS) puis incubées avec du MTT (0.5 mg/mi
dans du
PBS) pendant environ 2 heures. La solution de MTT est aspirée puis les
cristaux violets
sont solubilisés dans du diméthylsulfoxyde (DMSO). La DO (densité optique) est
mesurée
à 540 nm.
[109] Les résultats de ce test sont présentés figure 2. La figure 2 illustre
en ordonnée le
pourcentage de viabilité cellulaires par rapport au groupe contrôle. Le groupe
contrôle est
réalisé de manière similaire aux groupes étudiés sans l'ajout des molécules
étudiées dans
le présent texte. En comparaison au contrôle, il est mesuré une diminution
dose
dépendante de la viabilité cellulaire en MTT pour le 5a-hydroxy-66-[2-(1H-
imidazol-4-y1)-
éthylamino]-cholestan-36-fluoro et le 5a-hydroxy-66-[2-(1H-imidazol-4-y1)-
éthylamino]-
cholestan-36-ol. Pour une concentration de 5 M, la viabilité est proche de 0
%. Cela
traduit une capacité de destruction de cellules tumorales mammaires par le
composé de
formule (I). Ces résultats sont concordant avec les observations réalisées à
24 h et 48 h
précitées.
[110] Exemple 7: Effet du 5a-hydroxy-66-[2-(1H-imidazol-4-y1)-éthylamino]-
cholestan-36-
fluor sur l'activité de la Cholestérol Epoxyde Hydrolase (ChEH) dans des
cellules MCF-
7
[111] Les composés 5,6a-epoxycholesterol (5,6a-EC) et 5,66-epoxycholesterol
(5,66-EC)
sont des oxystérols impliqués dans la pharmacologie anticancéreuse du
tamoxifène, un
médicament antitumoral largement utilisé. Ils sont tous deux métabolisés en
cholestane-
36,50,66-triol (CT) par l'enzyme cholestérol-5,6-époxyde hydrolase (ChEH), et
le CT est
métabolisé par l'enzyme HSD11B2 (116-Hydroxystéroïde déshydrogénase 2) en 6-
oxo-
cholestan-36,5a-diol (0CDO), un oncostérone promoteur de tumeur.
[112] L'expérience suivante a pour but de démontrer la capacité du 5a-hydroxy-
6612-(1H-
imidazol-4-y1)-éthylamino]-cholestan-3-fluoro à bloquer la ChEH et donc à
limiter la
métabolisation d'oncostérone, un métabolite promoteur de tumeur.
[113] Des cellules MCF-7 sont dans un milieu de culture cellulaire identique à
l'exemple 5 et
sont ensemencées dans des plaques 6 puits à 150 000 cellules par puits avec 3
puits par
condition de traitement. 24 h après ensemencement, les cellules MCF-7 sont
traitées par
du [14C]5,6a-EC (solution mère 1000X : 0.6 mM ; 20 j.ICi/j.trnol ;
concentration finale 0.6
M) seul ou en association avec du Tamoxifène (tam). Le Tamoxifène est utilisé
en tant
que témoin positif du 5a-hydroxy-6642-(1H-imidazol-4-y1)-éthylamino]-cholestan-
3a-
CA 03196518 2023- 4- 24
WO 2022/090427 25
PCT/EP2021/080054
fluoro et du 5a-hydroxy-6[342-(1H-imidazol-4-y1)-éthylamino]-cholestan-313-ol
(1 M pour
toutes les molécules).
[114] Après 24 heures de traitement, les milieux sont collectés et des
extraits lipidiques sont
préparés à partir des culots cellulaires par extraction avec 100 I_ de
chloroforme, 400 pl_
de méthanol et 300 1_ d'eau. Les extraits lipidiques sont analysés par
chromatographie
sur couche mince (CCM) en utilisant l'acétate d'éthyle (Et0Ac) en tant
qu'éluant. L'analyse
est réalisée à l'aide d'un lecteur de plaque puis par autoradiographie. Les
résultats sont
présentés figure 3. Il est observé la métabolisation quasi-totale de l'époxyde
en CT et
OCDO (puits 2 et 4) et une inhibition totale de l'activité ChEH par le
Tamoxifène et presque
totale (trace de CT) par le 5a-hydroxy-6p-[2-(1H-imidazol-4-y1)-éthylamino]-
cholestan-3a-
fluor . Des résultats similaires sont observés avec le 5a-hydroxy-63-[2-(1H-
imidazol-4-y1)-
éthylamino]-cholestan-3p-ol.
[115] En conclusion, le 5a-hydroxy-63-[2-(1H-imidazol-4-y1)-éthylamino]-
cholestan-3a-
fluor possède une activité inhibitrice de la ChEH similaire au 5a-hydroxy-
6[342-(1H-
imidazol-4-y1)-éthylamino]-cholestan-3p-ol.
[116] Exemple 8 : Synthèse du COMPOSé de formule (I) 5a-hydroxy-68-[2-(1H-
imidazol-4-y1)-
éthylamino]-cholestan-33-méthoxyle (nommé DX103)
Na' ',lel
f
60 c n.
HO
[117] Une première étape consiste à mettre en solution 4.0 grammes (g) de
cholestérol (10.3
mmol) dans 20 millilitres (m1) de tétrahydrofurane (THF). 0.80 g de NaH (60 %
dans l'huile,
20.0 mmol) a été additionné et laissé réagir pour 30 minutes à 60 C, puis 1.8
ml de
iodométhane (28.9 mmol) ont été ajoutés. Le mélange ainsi obtenu a été laissé
à 60 C
pendant la nuit, à savoir environ 10 h. Après refroidissement de la solution,
la réaction a
été neutralisée par addition de 20 ml d'eau. Le mélange a été filtré et le THF
évaporé sous
vide. Le mélange a été transféré dans une ampoule à décanter et la phase
aqueuse a été
extraite trois fois avec de l'acétate d'éthyle. Les phases organiques ainsi
obtenues ont été
combinées et séchées sur du MgSO4 puis évaporées permettant l'obtention d'une
huile.
L'huile obtenue a été dissoute dans 2 ml de Et20 et du Me0H a été ajouté
jusqu'à la
formation d'un précipité blanc. La poudre a été filtrée, lavée avec du Me0H
froid et séchée.
Une poudre blanche de 3.40 g (correspondent à 82 `Vo de rendement) de
cholestan-313-
méthoxyle a été ainsi obtenue.
CA 03196518 2023- 4- 24
WO 2022/090427 26
PCT/EP2021/080054
[118] 1H-NMR (500 MHz, CDCI3): O (ppm) 5.36 (s, 1H), 3.35 (s, 3H), 3.09 - 3.02
(q, 1H),
2.40 - 2.36 (d, 1H), 2.18 - 2.13 (t, 1H), 2.03 - 1.81 (m, 5H), 1.60 - 1.00 (m,
24H), 0.92 -
0.91 (d, 3H), 0.87 - 0.85 (dd, 6H), 0.68 (s, 3H).
[119] La deuxième étape consiste à synthétiser à partir du cholestan-33-
méthoxyle le
composé 5,6a-epoxy-cholestan-38-méthoxyle de la manière suivante :
õ,..
õ....
-...0
ed-
[120] 1.80 g d'acide méta-chloro-peroxybenzoïque (8.90 mmol) ont été dissous
dans 70 ml
de dichlorométhane et ajouté goutte à goutte dans un mélange de 2.50 g de
cholestan-
313-méthoxyle (6.24 nnmol) dissous dans 20 ml de dichlorométhane. Le mélange
ainsi
obtenu a été mis sous agitation et maintenu à température ambiante durant
trois heures.
Le mélange obtenu a été lavé avec une solution aqueuse de Na2S203 à 10 /c, en
poids,
une solution saturée de NaHCO3 et une solution saturée de NaCI. La phase
organique a
été séchée sur du MgSO4 anhydre. Une évaporation sous vide du solvant
organique a été
réalisée et permet l'obtention d'une huile transparente et visqueuse. 5 ml de
Et20 ont été
ajouté pour dissoudre l'huile, puis 25 ml d'Et0H ont été additionné et le
mélange a été
chauffé jusqu'à ébullition pour 3 fois et enfin mis à 0 C pendant la nuit pour
favoriser la
précipitation. Une poudre blanche a été filtrée, lavée avec du Me0H froid et
séché ; 1.73
g, correspondant à 67 % (avec un excès énantiomérique 90 %) de rendement de
5,6a-
epoxy-cholestan-38-méthoxyle a été ainsi obtenue.
[121] 1H-NMR (500 MHz, CDCI3): ô (ppm) 3.45 - 3.39 (m, 1H), 3.33 (s, 3H), 2.90
- 2.89 (d,
1H), 2.00 - 0.94 (m, 31H), 0.89 - 0.88 (d, 3H), 0.86 - 0.85 (dd, 6H), 0.60 (s,
3H).
[122] La troisième étape consiste à synthétiser le 5a-hydroxy-6812-(1H-
imidazol-4-y1)-
éthylamino]-cholestan-3[3.-méthoxyle (DX103 forme basique) de la manière
suivante :
i-J i õ,..
1
/-Ni-I2
$ '
_
-
P-in 1741.1
Fritf
0 H N.
1
L - 1
.
'2 ri'
CA 03196518 2023- 4- 24
WO 2022/090427 27
PCT/EP2021/080054
0.81 g d'histamine (7.30 mmol) sous sa forme basique a été ajoutée à une
solution
butanolique de 10 ml comprenant 1.50 g du composé 5,6a-epoxy-cholestan-3[3-
méthoxyle
(3.62 mmol) sous agitation. Le mélange a été maintenu sous agitation dans un
chauffage
à reflux à une température de 130 oc durant 48 heures.
L'avancée de la réaction peut être contrôlée par chromatographie sur couche
mince
(CCM) pour suivre la conversion du 5,6a-epoxy-cholestan-3[3-méthoxyle.
Après refroidissement, le mélangea été dilué dans 10 ml de méthyl tert-butyle
éther. La
phase organique a été lavée par 2 fois avec 10 ml d'eau puis une fois avec 10
ml d'une
solution saturée de NaCI.
La phase organique a été séchée sur du MgSO4 anhydre. Le mélange a été purifié
par
colonne chromatographique sur un automate de purification. L'éluant utilisé
est un
mélange Acétate d'éthyle-Méthanol en rapport 90-10 %. Une poudre blanche de
1.32 g
de 5a-hydroxy-6[312-(1H-imidazol-4-y1)-éthylaminoLcholestan-3[3-méthoxy a été
obtenue.
Le rendement final de la réaction est de 69 % avec une pureté supérieure à 95
"Yo mesurée
par analyse RMN (résonance magnétique nucléaire) et TLC (Chromatographie sur
couche
mince).
[123] 1H-NMR (500 MHz, Me0D-4d): ô (ppm) 7.62 (s, 1H), 6.88(s, 1H), 3.71 ¨3.65
(m, 1H),
3.34 (s, 3H), 2.98 ¨ 2.97 (d, 1H), 2.78 ¨ 2.77 (m, 3H), 2.45 (s, 1H), 2.03 ¨
2.00 (m, 1H),
1.94 ¨ 1.83 (m, 3H), 1.65 ¨ 1.01 (m, 27H), 0.95 ¨ 0.94 (d, 3H), 0.91 - 0.89
(d, 6H), 0.71
(s, 3H).
[124] Exemple 9 : Préparation d'un sel dilactate du composé 5a-hydroxy-6312-
(1H-
imidazol-4-y1)-éthylaminol-cholestan-35-méthoxyle (DX103 forme dilactate)
[125] Un sel dilactate du composé 5a-hydroxy-6[3-[2-(1H-imidazol-4-y1)-
éthylamino]-
cholestan-313-méthoxyle a été préparé de la manière suivante :
H OH
0
0
3i1 '0
I N INe
= OH
`--NH
[126] 21.0 mg d'acide lactique (1.89 mmol) a été ajouté à une solution de 0.50
g de 5a-
hydroxy-6[342-(1H-imidazol-4-y1)-éthylamino]-cholestan-313-méthoxyle (0.95
mmol) dans
15 ml d'éthanol anhydre sous agitation. L'agitation a été maintenue à
température
CA 03196518 2023- 4- 24
WO 2022/090427 28
PCT/EP2021/080054
ambiante 3 heures. Une évaporation sous vide du solvant organique permet
l'obtention
d'une poudre blanche de 0.52 g de 5a-hydroxy-61342-(1H-imidazol-4-y1)-
éthylamino]-
cholestan-313-méthoxyle dilactate.
[127] 1H-NMR (500 MHz, Me0D-4d): O (ppm) 7.61 (s, 1H), 6.84 (s, 1H), 3.93 -
3.89 (q, 2H),
3.62 - 3.57 (m, 1H), 3.39 - 3.09 (m, 8H), 3.21 - 3.16 (m, 1H), 2.84 - 2.74 (m,
3H), 1.91 -
1.81 (m, 2H), 1.70 - 0.79 (m, 31H), 0.73 - 0.72 (d, 3H), 0.68 - 0.66 (d, 6H),
0.56 (s, 3H).
[128] Exemple 10 : Synthèse du composé de formule (I) 5a-hydroxy-61312-(1H-
imidazol-4-
v1)-éthvlaminol-cholestan-3(3-éthoxyle (nommé DX105)
La première étape est une synthèse du composé cholestan-313-éthoxy comprenant
les
lo étapes suivantes :
Kiil Et,
,
III
4.00 g de cholestérol (10.3 mmol) a été mis en solution dans 20 ml de THF.
0.82 g de
NaH (60 (3/0 dans l'huile, 20.0 mmol) a été additionné et laissé réagir pour
30 minutes à
60 C, puis 1.9 ml de iodoéthane (28.9 mmol) a été ajouté. Le mélange ainsi
obtenu a été
laissé à 60 C pendant la nuit. Après refroidissement de la solution, la
réaction a été
neutralisée par addition de 20 ml d'eau. Le mélange a été filtré et le THF
évaporé sous
vide. Le mélange a été transféré dans une ampoule à décanter et la phase
aqueuse a été
extraite trois fois avec d'acétate d'éthyle. Les phases organiques ainsi
obtenues ont été
combinées et séchées sur du MgSO4 puis évaporées permettant l'obtention d'une
huile.
L'huile obtenue a été dissoute dans 2 ml de Et20 et du Me0H a été ajouté
jusqu'à la
formation d'un précipité blanc. La poudre a été filtrée, lavée avec du Me0H
froid et séché.
Une poudre blanche de 2.12 g (correspondent à 49 43/0 de rendement) de
cholestan-313-
éthoxyle a été ainsi obtenue.
[129] 11-1-NMR (500 MHz, CD0I3): O (ppm) 5.35 (s, 1H), 3.53-3.51 (q, 2H), 3.17
- 3.14 (m,
1H), 2.38 -2.35 (d, 1H), 2.22 -2.17 (t, 3H), 2.02 - 1.79 (m, 5H), 1.60- 0.94
(m, 27H),
0.92 - 0.91 (d, 3H), 0.87 - 0.85 (dd, 6H), 0.67 (s, 3H).
CA 03196518 2023- 4- 24
WO 2022/090427 29
PCT/EP2021/080054
[130] La deuxième étape consiste à synthétiser à partir du cholestan-3[3-
éthoxyle le
composé 5,6a-epoxy-cholestan-313-éthoxyle de la manière suivante :
-1"---n. I \,- rn (21'13/Ç
...... _ _....... . fr--\ -
---\\/--
E.f::.,1
R -ih ....,,...-.....0
u.
[131] 1.44 g d'acide méta-chloro-peroxybenzoïque (correspondant à 6.43 mmol)
ont été
dissous dans 50 ml de dichlorométhane et ajouté goutte à goutte dans un
mélange de 2.0
g de cholestan-3[3-éthoxyle (4.82 mmol) dissous dans 10 ml de dichlorométhane.
Le
mélange ainsi obtenu a été mis sous agitation et maintenu à température
ambiante durant
3 heures. Le mélange obtenu a été lavé avec une solution aqueuse de Na2S203 à
10 %
en poids, une solution saturée de NaHCO3 et une solution saturée de NaCI. La
phase
organique a été séchée sur du MgSO4 anhydre. Une évaporation sous vide du
solvant
organique a été réalisée et permet l'obtention d'une huile transparente et
visqueuse. 5 ml
de Et20 ont été ajouté pour dissoudre l'huile, puis 25 ml d'Et0H ont été
additionné et le
mélange a été chauffé jusqu'à ébullition pour 3 fois et puis mis à 0 C pendant
la nuit pour
favoriser la précipitation. Une poudre blanche a été filtrée, lavée avec du
Me0H froid et
séchée : 0.72 g, correspondant à 35 (3/0 (avec un excès énantiomérique k90 %)
de
rendement de 5,6a-epoxy-cholestan-313-éthoxy a été ainsi obtenu.
[132] 1H-NMR (500 MHz, CDCI3): (5 (ppm) 3.55 ¨ 3.46 (m, 3H), 2.89 ¨ 2.88 (d,
1H), 2.04 ¨
0.93 (m, 34H), 0.89 ¨ 0.88 (d, 3H), 0.86 - 0.85 (dd, 6H), 0.60 (s, 3H).
[133] La troisième étape consiste à synthétiser le 5a-hydroxy-6f3-[2-(1H-
imidazol-4-y1)-
éthylamino]-cholestan-313-éthoxy (DX105 sous forme basique) de la manière
suivante :
. H
¨NFI2
='-ç I
.. ,.......e, .. ,
,_, i
r=iii
13`'.'C, . -:.Ei h
L1.1 1
[134] 0.31 g d'histamine sous sa forme basique (correspondent à 2.74 mmol) a
été ajouté à
une solution butanolique de 5 ml comprenant 0.51 g du composé 5,6a-epoxy-
cholestan-
313-éthoxyle à 1.18 mmol sous agitation. Le mélangea été maintenu sous
agitation dans
un chauffage à reflux à une température de 130 C durant 48 heures.
CA 03196518 2023- 4- 24
WO 2022/090427 30
PCT/EP2021/080054
L'avancée de la réaction peut être contrôlée par chromatographie sur couche
mince
(CCM) pour suivre la conversion du 5,6a-epoxy-cholestan-3f3-éthoxyle.
Après refroidissement, le mélange a été dilué dans 5 ml de méthyl tert-butyle
éther. La
phase organique a été lavée par 2 fois avec 5 ml d'eau puis une fois avec 5 ml
d'une
solution saturée de NaCI.
La phase organique a été séchée sur du MgSO4 anhydre. Le mélange a été purifié
par
colonne chromatographique sur un automate de purification. L'éluant utilisé
est un
mélange Acétate d'éthyle-Méthanol en rapport 90-10. Une poudre blanche de 0.28
g de
5a-hydroxy-6[3-[2-(1H-imidazol-4-y1)-éthylamino]-cholestan-3[3-éthoxyle a été
obtenue. Le
rendement final de la réaction est de 44 % avec une pureté supérieure à 97 %
mesurée
par analyse RMN (résonance magnétique nucléaire) et TLC (Chromatographie sur
couche
mince).
[135] 1H-NMR (500 MHz, Me0D-4d): ô (ppm) 7.62 (s, 1H), 6.89 (s, 1H), 3.82
¨3.76 (m, 1H),
3.57 ¨ 3.52 (q, 2H), 3.05 ¨ 3.00 (m, 1H), 2.85 ¨ 2.80 (m, 3H), 2.50 (s, 1H),
2.03 ¨ 1.83 (m,
5H), 1.65 ¨ 1.51 (m, 7H), 1.42 ¨ 1.01 (m, 22H), 0.96 ¨ 0.94 (d, 3H), 0.91 -
0.89 (d, 6H),
0.72 (s, 3H).
[136] Exemple 11 : Préparation d'un sel dilactate du composé 5a-hydroxy-6542-
(1H-
imidazol-4-y1)-éthylamino]-cholestan-3f3-éthoxyle (DX105 sous forme dilactate)
[137] Un sel dilactate du composé 5a-hydroxy-613-[2-(1H-imidazol-4-y1)-
éthylamino]-
cholestan-313-éthoxyle a été préparé de la manière suivante :
0
OH
1 ..õ.====-=..r,
1
HiTS Hl"
1;1,1 -F1,N1
OH
0 =i
NH '2-- Nil
C H
[138] 166.2 mg d'acide lactique (1.85 mmol) a été ajouté à une solution de
0.50 g de 5a-
hydroxy-613-[2-(1H-imidazo1-4-y1)-éthylamino]-cholestan-3f3-éthoxyle (0.92
mmol) dans 5
ml d'éthanol anhydre sous agitation. L'agitation a été maintenue à température
ambiante
pour 3 heures. Une évaporation sous vide du solvant organique permet
l'obtention d'une
poudre blanche de 0.20 g de 5a-hydroxy-61312-(1H-imidazol-4-y1)-
éthylaminoLcholestan-
3[3-éthoxyle dilactate.
CA 03196518 2023- 4- 24
WO 2022/090427 31_
PCT/EP2021/080054
[139] 1H-NMR (500 MHz, Me0D-4d): O (ppm) 7.61 (s, 1H), 6.84 (s, 1H), 3.92
¨3.89 (q, 2H),
3.60 - 3.57 (m, 1H), 3.39 - 3.09 (m, 7H), 2.84 ¨ 2.74 (m, 3H), 1.91 ¨ 1.81 (m,
2H), 1.70 -
0.79 (m, 34H), 0.73 ¨ 0.72 (d, 3H), 0.67 - 0.65 (d, 6H), 0.54 (s, 3H).
[140] Exemple 12: Synthèse du composé de formule (I) 5a-hydroxy-6(342-(1H-
imidazol-4-
y1)-éthylaminoi-cholestan-313-octanoxyle (nommé DX115)
La première étape est une synthèse du composé cholestan-313-octanoxyle
comprenant
les étapes suivantes4xf :
f Nal' 0:0
- =
1,F
ti0er
4.00 g de cholestérol a été mis en solution dans 20 ml de tétrahydrofurane.
0.84 g de NaH
10 a été additionné et laissé réagir pour 30 minutes à 60 C, puis 3.0 g
d'isooctane ont été
ajoutés. Le mélange ainsi obtenu a été laissé à 60 C pendant la nuit Après
refroidissement
de la solution, la réaction a été neutralisée par addition de 20 ml d'eau. Le
mélange a été
filtré et le THF évaporé sous vide. Le mélange a été transféré dans une
ampoule à
décanter et la phase aqueuse a été extraite trois fois avec de l'acétate
d'éthyle. Les phases
15 organiques ainsi obtenues ont été combinées et séchées sur du MgSO4 puis
évaporées
permettant l'obtention d'une huile.
L'huile obtenue a été dissoute dans 2 ml de Et20 et du Me0H a été ajouté
jusqu'à
formation d'un précipité blanc. La poudre a été filtrée, lavée avec du Me0H
froid et séchée.
Une poudre blanche de 2.5 g (correspondent à 48 %) de cholestan-33-octanoxyle
a été
20 ainsi obtenue.
[141] 1H-NMR (500 MHz, CDCI3): O (ppm) 5.35 (s, 1H), 3.45 - 3.43 (q, 2H), 3.15
¨ 3.10 (q,
1H), 2.37 ¨ 2.35 (d, 1H), 2.21 ¨ 2.16 (t, 1H), 2.02 ¨ 1.95 (m, 2H), 1.90 ¨
1.84 (m, 3H), 1.58
¨ 0.97 (m, 39H), 0.92 ¨ 0.91 (d, 3H), 0.87 - 0.86 (dd, 6H), 0.67 (s, 3H).
[142] La deuxième étape consiste à synthétiser à partir du cholestan-3p-
octanoxyle le
25 composé 5,6a-époxycholestan-33-octanoxyle de la manière suivante :
_ .
Lf,f3A
=
C411 . .
81-117,0 CH
0.90 g d'acide méta-chloro-peroxybenzoïque (correspondent à 4.0 mmol) ont été
dissous
CA 03196518 2023- 4- 24
WO 2022/090427 32
PCT/EP2021/080054
dans 40 ml de dichlorométhane et ajouté goutte à goutte dans un mélange de
1.50 g de
cholestan-33-octanoxyle à 3.0 mmol dissous dans 10 ml de dichlorométhane. Le
mélange
ainsi obtenu a été mis sous agitation et maintenu à température ambiante
durant 3 heures.
Le mélange obtenu a été lavé avec une solution aqueuse de Na2S203 à 10 % en
poids,
une solution saturée de NaHCO3 et une solution saturée de NaCI. La phase
organique a
été séchée sur du MgSO4 anhydre. Une évaporation sous vide du solvant
organique a été
réalisée et permet l'obtention une huile transparente et visqueuse. 5 ml de
Et20 ont été
ajouté pour dissoudre l'huile, puis 25 ml de Me0H ont été additionné et le
mélange a été
chauffé jusqu'à ébullition pour 3 fois et enfin mis à 0 C pendant la nuit pour
favoriser la
précipitation. Une poudre blanche a été filtrée, lavée avec du Me0H froid et
séchée : 1.19
g, correspondent à 77 % de rendement (avec un excès énantiomérique 90 %) de
5,6a-
epoxy-cholestan-3p-octanoxyle a été ainsi obtenue.
[143] 1H-NMR (500 MHz, CDCI3): ô (ppm) 3.51 ¨ 3.37 (m, 3H), 2.88 ¨ 2.87 (d,
1H), 2.02 ¨
1.87 (m, 4H), 1.84 ¨ 1.76 (m, 1H), 1.69 ¨ 1.67(m, 1H), 1.58¨ 1.45(m, 7H), 0.89
¨ 0.88
(m, 42H), 0.60 (s, 3H).
[144] La troisième étape consiste à synthétiser le 5a-hydroxy-6[3-[2-(1H-
imidazol-4-y1)-
éthylamino]-cholestan-313-octanoxyle (DX115 sous forme basique) de la manière
suivante :
Il-- il H
= F. Fi1-1 C7E-1e---
'13 d Fi
I- No
N
en,111510
11-1µd
[145] 0.48 g d'histamine sous sa forme basique (correspondant à 4.31 mmol) a
été ajoutée
à une solution butanolique de 10 ml comprenant 1.1 g du composé 5,6a-epoxy-
cholestan-
3p-octanoxyle à 2.14 mmol sous agitation. Le mélange a été maintenu sous
agitation dans
un chauffage à reflux à une température de 130 C durant 48 heures.
L'avancée de la réaction peut être contrôlée par chromatographie sur couche
mince
(CCM) pour suivre la conversion du 5,6a-epoxy-cholestan-3p-octanoxyle.
Après refroidissement, le mélange a été dilué dans 10 ml de méthyl tert-butyl
éther. La
phase organique a été lavée par 2 fois avec 10 ml d'eau puis une fois avec 10
ml d'une
solution saturée de NaCI.
La phase organique a été séchée sur du MgSO4 anhydre. Le mélange a été purifié
par
colonne chromatographique sur un automate de purification. L'éluant utilisé
est un
CA 03196518 2023- 4- 24
WO 2022/090427 33
PCT/EP2021/080054
mélange Acétate d'éthyle/Méthanol en rapport 95/5. Une poudre blanche de 0.74
g de 5a-
hydroxy-61342-(1H-imidazol-4-y1)-éthylamino]-cholestan-313-octanoxyle a été
obtenue. Le
rendement final de la réaction est de 55 % avec une pureté supérieure à 95 %
mesurée
par analyse RMN (résonance magnétique nucléaire) et TLC (Chromatographie sur
couche
mince).
[146] 1H-NMR (500 MHz, Me0D-4d): O (ppm) 7.59 (s, 1H), 6.86 (s, 1H), 3.79 -
3.74 (q, 1H),
3.49 - 3.47 (q, 2H), 2.95 - 2.90 (m, 1H), 2.78 - 2.70 (m, 3H), 2.40 (s, 1H),
2.01 - 1.84 (m,
5H), 1.62 - 1.54 (m, 9H), 1.39 - 1.02 (m, 30H), 0.95 - 0.89 (d, 12H), 0.70 (s,
3H).
[147] Exemple 13 : Préparation d'un sel dilactate du composé 5a-hydroxy-6842-
(1H-
imidazo1-4-y1)-éthylamino]-cholestan-313- octanoxyle (DX115 sous forme
dilactate)
[148] Un sel dilactate du composé 5a-hydroxy-68-[2-(1H-imidazo1-4-y1)-
éthylamino]-
cholestan-313-octanoxyle a été préparé de la manière suivante :
Pic
0
C,11, C,11
FiC: F crlye
F,1
1-tOhl
rt ' I N
OH
iH.0-
OH
[149] 166.2 mg d'acide lactique (1.85 mmol) a été ajouté à une solution de
0.57 g de 5a-
hydroxy-613-[2-(1H-imidazol-4-y1)-éthylamino]-cholestan-313-octanoxyle (0.92
mmol) dans
5 ml d'éthanol anhydre sous agitation. L'agitation a été maintenue à
température ambiante
pendant trois heures. Une évaporation sous vide du solvant organique permet
l'obtention
d'une poudre blanche de 0.59 g de 5a-hydroxy-6842-(1H-imidazol-4-y1)-
éthylamino]-
cholestan-313-octanoxyle dilactate.
[150] 1H-NMR (500 MHz, Me0D-4d): O (ppm) 7.71 (s, 1H), 6.94 (s, 1H), 4.02 -
3.98 (q, 2H),
3.72 - 3.65 (q, 1H), 3.41 - 3.31 (m, 3H), 3.21 - 3.16 (m, 1H), 2.95 - 2.92 (t,
2H), 2.86 - 2.85
(d, 1H), 2.04 - 1.99 (t, 1H), 1.96 - 1.93 (d, 1H), 1.83 - 1.59 (m, 7H), 1.49 -
1.04 (m, 38H),
0.98 - 0.89 (m, 2H), 0.85 - 0.84 (d, 3H), 0.81 - 0.77 (m, 9H), 0.66 (s, 3H).
[151] Exemple 14: Synthèse du composé 5a-hydroxy-6842-(1H-imidazol-4-y1)-
éthylam ino]-
cholestan-3f3-azoture (nommé DX123)
[152] La première étape est la synthèse du composé 3-mesylcholestane
comprenant les
étapes suivantes :
CA 03196518 2023- 4- 24
WO 2022/090427 34
PCT/EP2021/080054
_
j12-'s--\\--')'-
HO' .."'-''''' 09C rt on. ..._,S.Ø,,
_...,...-,....:1,....õ,./
[153] Dans un ballon de 1L à 0 C ont été solubilisés 40 g de cholestérol (0.1
mol) et 22 ml
de NEt3 (d=0.88 g/ml, 0.19 mol) en 340 ml de dichlorométhane anhydre. 10 ml de
méthanesulfonyl chloride (1.48 g/ml, 0.13 mol) ont été solubilisé dans 40 ml
de
dichlorométhane anhydre et ajouté goutte par goutte à la solution contenant le
cholestérol.
Le mélange ainsi obtenu a été laissé sur agitation magnétique pour toute la
nuit en la
laissant chauffer jusqu'à température ambiante.
A l'issus de cette durée, la réaction a été monitorée par TLC et concentré sur
vide jusqu'au
2/3 du volume initiale. L'ajout de 500 ml de Me0H nous permet d'obtenir 46.4 g
d'un
précipité blanc correspondant au produit souhaité (97 % de rendement).
[154] 1H-NMR (500 MHz, CDCI3): O (ppm) 5.42 - 5.41 (d, 1H), 4.55 - 4.49 (q,
1H), 3.00 (s,
3H), 2.56 - 2.45 (m, 2H), 2.05 - 1.96 (m, 3H), 1.92 - 1.75 (m, 3H), 1.60 -
0.93 (m, 23H),
0.92 - 0.90 (d, 3H), 0.87 - 0.85 (dd, 6H), 0.67 (s, 3H).
[155] La deuxième étape consiste à synthétiser à partir du 313-mesyl
cholestérol le composé
3p-azoture-cholestane de la manière suivante :
Ir I
0 r., fr - -' -
#1_,./e----\
--t :: [-, Nee0
:.,r
,
[156] Dans un ballon de 500 ml nous avons ajouté en séquence à température
ambiante :
23.27 g de 3p-mésylcholesterol (50.1 mmol), 100 ml de dichlorométhane anhydre,
7.5 ml
d'azoture de triméthylsilyle (d=0.868 g/ml, 56.5 mmol) et enfin 12.5 ml de
diéthyléthérate
de trifluorure de bore (d=1.15 g/ml, 101.3 mmol). Le mélange ainsi obtenu a
été laissé sur
agitation magnétique pendant 3 heures.
A l'issus de cette durée, la réaction a été neutralisée par addition de 100 ml
d'une solution
de NaOH 2M. Les produits organiques ont été extrait deux fois avec
dichlorométhane. Les
phases organiques ont été combinées et rincées deux fois avec une solution
saturée de
NaCI. La phase organique a été séchée sur du MgSO4, filtrée et puis évaporée
permettant
l'obtention d'une solide. Le brut de la réaction a été purifié par colonne
chromatograph igue
CA 03196518 2023- 4- 24
WO 2022/090427 35
PCT/EP2021/080054
éluant hexane 100 %. Une poudre blanche-jaunâtre de 13.33 g correspondant au
313-
azoture-cholestane a été ainsi obtenue. Le rendement final de la réaction est
de 65%.
[157] 1H-NMR (500 MHz, CDCI3): O (ppm) 5.39 - 5.38 (d, 1H), 3.23 - 3.17 (q,
1H), 2.30 -
2.28 (d, 2H), 2.03 - 1.97 (m, 2H), 1.91 - 1.81 (m, 3H), 1.60 - 0.94 (m, 24H),
0.92 - 0.91
(d, 3H), 0.87 - 0.86 (dd, 6H), 0.68 (s, 3H).
[158] La troisième étape de synthèse consiste à synthétiser à partir du 313-
azoturecholestane
le composé 313-azoture-5,6a-epoxycholestane de la manière suivante :
I--- --\1----
I 117 1 l'HA
-
I - -p. 111_ 7
N3 '" -'' rt 't=
[159] 950 mg d'acide méta-chloro-peroxybenzoïque au 77% de pureté (4.24 mmol)
ont été
dissous dans 15 ml de dichlorométhane et ajouté goutte à goutte dans un
solution de 1.3
g de 3f3-azoturecholestane (3.16 mmol) dissous dans 15 ml de dichlorométhane.
Le
mélange ainsi obtenu a été mis sous agitation et maintenu à température
ambiante durant
3 heures. Le mélange obtenu a été lavé deux fois avec une solution aqueuse de
Na2S203
à 10 % en poids, deux fois avec une solution saturée de NaHCO3 et une fois
avec une
solution saturée de NaCI. La phase organique a été séchée sur du MgSO4anhydre.
Une
évaporation sous vide du solvant organique a été réalisée et permet
l'obtention de 1.35 g
d'une poudre blanche correspondant au mélange de : 3-azoture-5,6-
epoxycholestane
(83 (3/0 du total) et 33-azoture-5,63-epoxycholestane (17 (3/0 de la poudre
blanche). Le
produit final a été utilisé sans purification supplémentaire.
[160] 11-1-NMR (500 MHz, CD0I3): O (ppm) 3.63 - 3.56 (q, 1H), 2.94 - 2.93 (d,
1H), 2.13 -
2.08 (t, 1H), 1.97 - 0.94 (m, 30H), 0.89 - 0.88 (d, 3H), 0.86 - 0.85 (dd, 6H),
0.61 (s, 3H).
[161] La quatrième étape est la synthétise du 5a-hydroxy-613-[2-(1H-imidazol-4-
y1)-
éthylamino]-cholestan-313-azoture (DX123 sous forme neutre) de la manière
suivante :
H2N.õ...., ..., ,.. N..
---) --\---\\_._
,. .
1-- Nil
- -
FIN,,.......,...yr N
N.:-.)- , ---eej .1 ._, i:r C h
CA 03196518 2023- 4- 24
WO 2022/090427 36 PCT/EP2021/080054
[162] 864 mg d'histamine sous sa forme basique (7.77 mmol) a été ajouté à une
solution
butanolique de 20 ml comprenant 2.02 g du composé 313-azoture-5,6-
epoxycholestane
à 83 % (3.9 mmol) sous agitation à 130 C. Le mélangea été maintenu sous
agitation dans
un chauffage à reflux à une température de 130 C durant 48 heures.
L'avancée de la réaction peut être contrôlée par chromatographie sur couche
mince
(CCM) pour suivre la conversion du 33-azoture-5,6a-epoxycholestane.
Après refroidissement, le mélange a été dilué dans 15 ml de méthyl tert-butyl
éther. La
phase organique a été lavée par 3 fois avec 15 ml d'eau.
La phase organique a été séchée sur du MgSO4anhydre, filtré et puis évaporée
permettant
1.0 l'obtention d'une huile brune. Le mélange a été purifié par colonne
chromatographique sur
gel de Silice sur un automate de purification comprenant une colonne prépacké
de 40 g
éluant dichlorom éthane-acétate d'éthyle 75-25 % jusqu'au 0-100 /0. Une
poudre blanche
de 890 mg de 5a-hydroxy-613-[2-(1H-imidazol-4-y1)-éthylamino]-cholestan-3p-
azoture a
été obtenue. Le rendement final de la réaction est de 42 % avec une pureté
supérieure à
97 % mesurée par analyse RMN (résonance magnétique nucléaire) et TLC
(Chromatographie sur couche mince).
[163] 11-1-NMR (500 MHz, Me0D-4d): ô (ppm) 7.55 (s, 1H), 6.81 (s, 1H), 3.73 -
3.67 (q, 1H),
2.90 - 2.85 (m, 1H), 2.72 - 2.62 (m, 3H), 2.33 (s, 1H), 2.05 - 2.00 (t, 1H),
1.96 - 1.94 (m,
1H), 1.84 - 1.77 (m, 1H), 1.74 - 1.72 (m, 1H), 1.62 - 0.97 (m, 27H), 0.89 -
0.88 (d, 3H),
0.85 - 0.84 (d, 6H), 0.64 (s, 3H).
[164] Exemple 15: Préparation d'un sel dilactate du composé 5a-hydroxy-61312-
(1H-
imidazol-4-y1)-éthylamino]-cholestan-3(3-azoture (DX123 forme dilactate)
.,;---,
0
- )= - HIC; -11.I'e'
N3 ç c "rf
otetd-'
H Nerfr-) I
-9 Fie; .: i
`1,-,N,_,,,...,_
e,
-
-0- FIN N EiCy-- - ,..,
ri. :i h .."(..)-
NH
0
[165] 63.5 mg d'acide lactique (0.77 mmol) a été ajouté à une solution de 210
mg de 5a-
hydroxy-613-[2-(1H-imidazol-4-y1)-éthylamino]-cholestan-3f3-azoture dans 4 ml
d'éthanol
anhydre sous agitation. L'agitation a été maintenue à température ambiante
pour 3 heures.
Une évaporation sous vide du solvant organique permet l'obtention d'une poudre
blanche
CA 03196518 2023- 4- 24
WO 2022/090427 37
PCT/EP2021/080054
de 263.5 mg de 5a-hydroxy-6612-(1H-imidazol-4-y1)-éthylamino]-cholestan-313-
azoture
dilactate.
[166] 1H-NMR (500 MHz, Me0D-4d): ô (ppm) 7.67 (s, 1H), 6.92 (s, 1H), 4.01 -
3.97 (m, 2H),
3.73 - 3.67 (q, 1H), 3.34 - 3.29 (m, 1H), 3.19 - 3.13 (m, 1H), 2.91 - 2.88 (t,
2H), 2.81 (s,
1H), 2.20 - 2.15 (t, 1H), 1.94 - 1.92 (d, 1H), 1.77 - 1.75 (m, 3H), 1.66 -
1.58 (m, 4H), 1.47
- 0.98 (m, 26H), 0.95 - 0.87 (m, 2H), 0.83 - 0.82 (d, 3H), 0.77 - 0.76 (dd,
6H), 0.65 (s, 3H).
[167] Exemple 16: Synthèse d'un sel trichlorure du composé 5a-hydroxy-6642-(1H-
imidazo1-4-y1)-éthylamino]-cholestan-313-amino (nommé DX125, sous forme
trichloride)
[168] La réaction qui va permettre de synthétiser un sel trichlorure du 5a-
hydroxy-6[342-(1H-
imidazol-4-y1)-éthylamino]-cholestan-313-amino à partir du 5a-hydroxy-613-[2-
(1H-imidazol-
4-y1)-éthylamino]-cholestan-313-azoture est la suivante :
- .....,'"..,.,___
' ''"ji)F12 eeS )= .>
.7r.i
2, ' = .n. ._ .,_=
1 "ICI
MN .., Ati 717) - C . 2h .i.,1=::N
,,,õ....... !,.
LNI-1
ILlIA1-1
[169] 730 mg de triphénylphosphine (2.8 mmol) a été ajouté à une solution de
8.0 ml THF
de 300 mg de 5a-hydroxy-6p12-(1H-imidazol-4-y1)-éthylamino]-cholestan-313-
azoture
(0.56 mmol) sous agitation à 70 C. Le mélange a été maintenu sous agitation
dans un
chauffage à reflux à une température de 70 C durant 2 heures. Puis, 0.5 ml
d'eau
(correspondent à 27.8 mmol) a été additionné et maintenu l'agitation pour deux
autres
heures à 70 C. L'avancée de la réaction est contrôlée par chromatographie sur
couche
mince (CCM), puis le mélange des solvants a été évaporé. La poudre blanche
obtenue a
été dissoute avec 20 ml de dichlorométhane et transféré dans une ampoule à
décanter
contenant 20 ml d'une solution aqueuse de HCI (1 ml de HCI 37% dans 19 ml
d'eau), la
phase aqueuse a été lavée trois fois avec du dichlorométhane. La phase aqueuse
a été
séchée sous vide permettant l'obtention d'une poudre blanche. La poudre a été
reprise
avec du dichlorométhane et filtré une dernière fois pour éliminer les
dernières traces de
triphénylphosphine. La procédure nous permet d'obtenir 350 mg d'un sel
trichlorure de 5a-
hydroxy-613-[2-(1H-imidazo1-4-y1)-éthylamino]-cholestan-313-amino avec un
rendement
quantitatif et une pureté supérieure à 95 %.
CA 03196518 2023- 4- 24
WO 2022/090427 38
PCT/EP2021/080054
[170] 1H-NMR (500 MHz, Me0D-4d): $5 (ppm) 8.90 (s, 1H), 7.56 (s, 1H), 3.67 -
3.60 (q, 1 H),
3.56 - 3.41 (m, 4H), 3.28 - 3.27 (d, 1H), 2.61 - 2.56 (t, 1H), 2.08 - 2.05 (d,
1H), 1.99 - 1.11
(m, 28H), 1.06 - 1.00 (dd, 1H) 0.96 - 0.94 (d, 3H), 0.89 - 0.88 (dd, 6H), 0.78
(s, 3H).
[171] Exemple 17: Synthèse du composé 5a-hydroxy-6(3-[2-(1H-imidazol-4-y1)-
éthylamino]-
cholestan-313-acétamide (nommé DX127)
[172] La première étape de synthèse est la réduction du groupement azoture en
amine en
position 3 du dérivé cholestan-313-azoture.
I ieM
_______________________________________________ -3>
>
N3 OCnr h H2N
[173] 5.21 g de cholestan-313-azoture (12.7 mmol) ont été dissous dans 60 ml
de
tétrahydrofurane (THF) puis 5 portions d'environ 480 mg de LiALH4 ont été
additionnés
toutes les 15 minutes pour un total de 2.32 g (61.1 mmol). Le mélange ainsi
obtenu est
laissé sur agitation magnétique pendant 3 heures. A l'issus de cette durée, la
réaction a
été neutralisée par addition de quelques gouttes de Na2CO3 5 % aq (ajouté
délicatement).
La phase organique a été extrait trois fois avec de l'AcOEt et les phases
organiques ont
été combinés. La solution résultante a été séchée sur du MgSO4, filtrée et
puis évaporée
permettant l'obtention d'un solide. Une poudre blanche suffisamment propre de
3.78 g
correspondant au 313-amino-cholestane a été ainsi obtenue. Le rendement final
de la
réaction est de 77 %.
[174] 1H-NMR (500 MHz, 0DCI3): O (ppm) 5.32 - 5.31 (d, 1H), 2.63 - 2.57 (q,
1H), 2.17 -
2.13 (m, 1H), 2.08 - 1.93 (m, 4H), 1.85 - 1.81 (m, 2H), 1.72 - 1.68 (m, 1H),
1.43 - 0.84
(m, 33H), 0.68 (s, 3H).
[175] La deuxième étape consiste à synthétiser à partir du cholestan-3-amine
le composé
cholestan-313-acétamide de la manière suivante :
A r
pyricline r
0:
DC
rté c rmµ
H
[176] 3.78 g de cholestan-313-amine (9.8 mmol) ont été dissous dans 20 ml de
dichlorométhane anhydre, puis 16 mi de pyridine anhydre (198 mmol) et 5.0 g
d'anhydride
CA 03196518 2023- 4- 24
WO 2022/090427 39
PCT/EP2021/080054
acétique (49.0 mmol) ont été ajoutés à la réaction. Le mélange ainsi obtenu a
été mis sous
agitation et maintenu à température ambiante pour toute la nuit. Le mélange
obtenu a été
lavé trois fois avec une solution aqueuse de HCI 0.1 M et la phase organique a
été séchée
sur du MgSO4 anhydre, filtrée et séchée sous vide. L'huile obtenu a été
dissous avec 30
ml de chloroforme, 90 ml de Me0H a été additionné et le mélange a été chauffé
jusqu'à
ébullition pour 3 fois et jusqu'à la réduction du volume des solvants de 2/3
et enfin mis à
0 C pour favoriser la précipitation. Une poudre blanche a été obtenue,
filtrée, lavée avec
du Me0H froid et séchée : 2.41 g, correspondent à 58 % de rendement de
cholestan-36-
acétamide a été ainsi obtenue.
lo
[177] 11-1-NMR (500 MHz, 0D013): O (ppm) 5.36 - 5.35 (d, 1H), 5.32 - 5.30 (d,
1H), 3.73 -
3.65 (q, 1H), 2.32 - 2.29 (d, 1H), 2.09 - 1.79 (m, 9H), 1.60 - 0.95 (m, 22H),
0.92 - 0.90
(d, 3H), 0.87 - 0.85 (dd, 6H), 0.67 (s, 3H).
[178] La troisième étape consiste à synthétiser le 5,6-epoxy-cholestan-3[3-
acétamide de la
manière suivante :
cr PA
0
N - M
)4.1
H
[179] 1.19 g d'acide méta-chloro-peroxybenzoïque au 77% de pureté (5.3 mmol)
ont été
dissous dans 10 ml de dichlorométhane et ajouté goutte à goutte dans un
mélange de
1.61 g de cholest-3[3-acétamide (3.8 mmol) dissous dans 25 ml de
dichlorométhane. Le
mélange ainsi obtenu a été mis sous agitation et maintenue à température
ambiante
durant 3 heures. Le mélange obtenu a été lavé deux fois avec une solution
aqueuse de
Na2S203 à 10 % en poids, deux fois une solution saturée de NaHCO3 et une
solution
saturée de NaCI. La phase organique a été séchée sur du MgSO4 anhydre. Une
évaporation sous vide du solvant organique a été réalisée et permet
l'obtention de 1.65 g
d'une poudre blanche comprenant : 5,6a-Epoxy-cholest-313-acétamide (60 % de la
poudre
blanche) et 5,60-Epoxy-cholest-33-acétamide (40 % de la poudre blanche). Le
5,6a-
Epoxy-cholest-3[3-acétamide a été utilisé sans purification supplémentaire.
[180] 11-1-NMR (500 MHz, CD0I3): O (ppm) 5.29 - 5.28 (d, 1H), 4.05 - 3.99 (q,
1H), 2.89 -
2.88 (d, 1H), 2.08 - 0.84 (m, 43H), 0.60 (s, 3H).
[181] La quatrième étape consiste à synthétiser le 5a-hydroxy-6[3-[2-(1H-
imidazol-4-y1)-
éthylamino]-cholestan-3[3-acetamide (DX127 sous forme neutre) de la manière
suivante :
CA 03196518 2023- 4- 24
WO 2022/090427 40
PCT/EP2021/080054
1-12N r47
p,( I
13c2deL, 1
H H
H N
[182] 0.47 g d'histamine sous sa forme basique (correspondent à 4.26 mmol) a
été ajouté à
une solution butanolique de 20 ml comprenant 1.65 g du composé 5,6u-epoxy-
cholestan-
3p-acétamide à 60 A correspondent à 0.99 mmol sous agitation. Le mélange a
été
maintenu sous agitation dans un chauffage à reflux à une température de 130 C
durant
48 heures. L'avancée de la réaction peut être contrôlée par chromatographie
sur couche
mince (CCM) pour suivre la conversion du 5,6a-epoxy-cholestan-3p-acétamide.
Après
refroidissement, le mélange a été dilué dans 20 ml de méthyl tert-butyle
éther. La phase
organique a été lavée par 2 fois avec 20 ml d'eau et trois fois avec 20 ml
d'une solution
saturée de NaCI. La phase organique a été séchée sur du MgSO4 anhydre. Le
mélange a
été purifié par colonne chromatographique sur un automate de purification.
L'éluant utilisé
est un mélange dichlorométhane-méthanol-ammoniac en rapport 75-20-5 %. Une
poudre
blanche de 0.37 g de 5a-hydroxy-6[312-(1H-imidazol-4-y1)-éthylamino]-cholestan-
313-
acetamide a été obtenue. Le rendement final de la réaction est de 30 % avec
une pureté
supérieure à 97 % mesurée par analyse RMN (résonance magnétique nucléaire) et
TLC
(Chromatographie sur couche mince).
[183] 1H-NMR (500 MHz, Me0D-4d): ô (ppm) 7.56 (s, 1H), 6.81 (s, 1H), 4.15 ¨
4.08 (q, 1H),
2.91 ¨ 2.88 (m, 1H), 2.74 ¨ 2.68 (m, 3H), 2.35 (s, 1H), 1.99 ¨ 1.94 (m, 2H),
1.87 ¨ 1.77 (m,
5H), 1.66 ¨ 0.97 (m, 28H), 0.90 ¨ 0.88 (d, 3H), 0.85 - 0.84 (d, 6H), 0.65 (s,
3H).
[184] Exemple 18 : Préparation d'un sel dilactate du composé 5a-hydroxy-61312-
(1H-
imidazol-4-y1)-éthylamino]-cholestan-3P-acétamide (DX127 sous forme dilactate)
[185] Un sel dilactate du composé 5a-hydroxy-61342-(1H-imidazol-4-y1)-
éthylamino]-
cholestan-313-acétamide a été préparé de la manière suivante:
CA 03196518 2023- 4- 24
WO 2022/090427 41_ PCT/EP2021/080054
0
fe
N 0
0 el. HO)jï1-1 0
0
-)( /11', N elolle
H 1-1_91 H
N r\
+ -
H HO Et0H
H rt, 3h
N
---=01-1
[186] 120.6 mg d'acide lactique (1.34 mmol) ont été ajoutés à une solution de
370 mg de
5a-hydroxy-6[3-[2-(1H-imidazol-4-y1)-éthylamino]-cholestan-3[3-acetamide dans
5 ml
d'éthanol anhydre sous agitation. L'agitation a été maintenue à température
ambiante pour
3 heures. Une évaporation sous vide du solvant organique permet l'obtention
d'une poudre
blanche de 490 mg de 5a-hydroxy-61342-(1H-imidazol-4-y1)-éthylamino]-cholestan-
313-
acétamide dilactate.
[187] 1H-NMR (500 MHz, Me0D-4d): O (ppm) 7.69 (s, 1H), 6.91 (s, 1H), 4.08 -
4.03 (m, 1H),
3.37 - 3.27 (m, 1H), 3.18 - 3.12 (m, 2H), 2.91 - 2.88 (t, 2H), 2.77 (s, 1H),
2.07 - 2.02 (t, 1H),
1.93 - 1.90 (d, 1H), 1.79 (s, 3H), 1.76 - 0.88 (m, 36H), 0.82 - 0.81 (d, 3H),
0.75 - 074 (dd,
6H), 0.63 (s, 3H).
[188] Exemple 19 : Synthèse du composé 5a-hydroxy-65-[2-(1H-imidazol-4-y1)-
éthylamino]-
cholestan-313-méthylsulfonyle (nommé DX129)
[189] La première étape consiste à synthétiser à partir du 313-
mésylcholestérol le composé
3[3-méthylthio-cholestane de la manière suivante :
_ --- . f.,="'S t':::'...lp
1.),,,..:2 j
eicfj .-.
[190] Dans un ballon de 500 ml, nous avons ajouté en séquence à température
ambiante :
10.62 g de 3-mésylcholestérol (22.9 mmol), 50 ml de dichlorométhane, 5.0 g de
triméthyl(méthylthio)silane (41.6 mmol) et 8.0 ml de diéthyléthérate de
trifluorure de bore
(d=1.15 g/ml, 64.8 mmol). Le mélange ainsi obtenu a été laissé sur agitation
magnétique
pour 3 heures.
A l'issus de cette durée, la réaction a été neutralisée par addition de 100 ml
d'une solution
de NaOH 2M. La phase organique a été extraite deux fois avec du dichlométhane.
Les
phases organiques ont été combinées et rincées deux fois avec une solution
saturée de
NaCI. La phase organique a été séchée sur du MgSO4, filtrée et puis évaporée
permettant
CA 03196518 2023- 4- 24
WO 2022/090427 42
PCT/EP2021/080054
l'obtention d'un solide. Le brut réactionnel a été purifié par colonne
chromatographique
sur gel de silice éluant hexane 100 /.3. Une poudre blanche de 6.04 g
correspondant au
313-méthylthio-cholestane a été ainsi obtenue. Le rendement final de la
réaction est de
63%.
[191] 1H-NMR (500 MHz, CDCI3): O (ppm) 5.33 (s, 1H), 2.71 - 2.65 (m, 1H), 2.30
- 2.26 (m,
2H), 2.11 (s, 3H), 2.02 - 0.94 (m, 29H), 0.92 - 0.91 (d, 3H), 0.87 - 0.85 (dd,
6H), 0.67 (s,
3H).
[192] La deuxième étape de synthèse consiste à synthétiser à partir du 313-
méthylthio-
cholestane le composé 3[3-méthylsulfony1-5,6-epoxy-cholestane de la manière
suivante :
- . ).--- \--y-
41.
Ci L.1
[193] 6.30 g d'acide méta-chloro-peroxybenzoïque au 77 % de pureté (28.1 mmol)
a été
dissout dans 40 ml de dichlorométhane et une solution de 2.9 g de 3-méthylthio-
cholestane (6.8 mmol) s dans 20 ml de dichlorométhane a été ajoutée goutte à
goutte. Le
mélange ainsi obtenu a été mis sous agitation et maintenue à température
ambiante
durant 3 heures. Le mélange obtenu a été lavé deux fois avec une solution
aqueuse de
Na2S203 à 10 % en poids, trois fois avec une solution saturée de NaHCO3 et une
fois avec
une solution saturée de NaCI. La phase organique a été séchée sur du MgSO4
anhydre.
Une évaporation sous vide du solvant organique a été réalisée et permet
l'obtention de
2.10 g d'une poudre blanche. Le brut de la réaction a été purifié par colonne
chromatographique éluant initial hexane 100% et puis des mélanges d'hexane et
AcOEt.
Le produit souhaité a été purifié par colonne chromatographique sur gel de
silice avec
l'éluent hexanes-AcOEt 55-45%v. Une poudre blanche de 380 mg correspondant au
3-
méthylthio-5,6epoxy-cholestane a été ainsi obtenue. Le rendement final de la
réaction est
de 12%.
[194] 1H-NMR (500 MHz, CDCI3): O (ppm) 3.25 - 3.20 (m, 1H), 3.02-3.01 (d, 1H),
2.83 (s,
3H), 2.11 - 2.09 (m, 1H), 1.98 - 1.93 (m, 3H), 1.87 - 1.79 (m, 3H), 1.57 -
0.93 (m, 24H),
0.89 - 0.88 (d, 3H), 0.86 - 0.85 (dd, 6H), 0.61 (s, 3H).
[195] La troisième étape est la synthèse du 5a-hydroxy-6[312-(1H-imidazol-4-
y1)-
éthylamino]-cholestan-3[3-méthylsulfonyl (DX129 sous forme basique) de la
manière
CA 03196518 2023- 4- 24
WO 2022/090427 43
PCT/EP2021/080054
suivante :
V.'
N F-1
1:30-c 111 d' 'u H i
[196] 338 mg d'histamine sous sa forme basique (3.04 mmol) a été ajoutée à une
solution
butanolique de 5 ml comprenant 350 mg du composé 3-méthylsulfony1-5,6-epoxy-
cholestane (0.75 mmol) sous agitation à 130 C. Le mélangea été maintenu sous
agitation
dans un chauffage à reflux à une température de 130 OC durant 48 heures.
L'avancée de
la réaction peut être contrôlée par chromatographie sur couche mince (CCM)
pour suivre
la conversion du 3-méthylsulfony1-5,6-epoxy-cholestane.
Après refroidissement, le mélange a été dilué dans 5 ml de méthyl tert-butyl
éther. La
phase organique a été lavée par 3 fois avec 15 ml de chlorure de sodium
saturée.
La phase organique a été séchée sur du MgSO4 anhydre, filtrée et puis évaporée
permettant l'obtention d'une huile brune. Le brut de la réaction a été purifié
par colonne
chromatographique éluant initiale AcOEt 100 %, puis des mélanges AcOEt-Me0H.
Le
produit souhaité était purifié avec le mélange AcOEt-Me0H 75-25 %. Une poudre
jaune
de 190 mg correspondant au 5a-hydroxy-61312-(1H-imidazol-4-y1)-éthylamino]-
cholestan-
3-méthylsulfonyl est obtenue. Le produit est purifié une deuxième fois par
colonne
chromatographique pour avoir une pureté supérieure à 97
[197] % mesurée par analyse RMN (résonance magnétique nucléaire) et TLC
(Chromatographie sur couche mince). Une poudre blanche de 167.4 mg a été
obtenue.
Le rendement final de la réaction est de 39 %.
[198] 11-1-NMR (500 MHz, Me0D-4d): ô (ppm) 7.63 (s, 1H), 6.89 (s, 1H), 3.49 -
3.40 (m, 1H),
3.04 - 3.02 (m, 1H), 2.89 (s, 3H), 2.81 - 2.78 (m, 3H), 2.50 (s, 1H), 2.44 -
2.40 (t, 1H),
2.02 - 2.01 (m, 1H), 1.94 - 1.92 (m, 1H), 1.88 - 1.83 (m, 1H), 1.76 - 1.00 (m,
30H), 0.90
- 0.89 (d, 3H), 0.85 - 0.84 (d, 6H), 0.66 (s, 3H).
[199] Exemple 20: Préparation d'un sel dilactate du composé 5a-hydroxy-61312-
(1H-
imidazol-4-y1)-éthylamino]-cholestan-313-méthylsulfonyl (DX129 sous forme
dilactate)
CA 03196518 2023- 4- 24
WO 2022/090427 44
PCT/EP2021/080054
1.111
N Se HO)L-i:
N
0
d's\b H Et0H H
rt, 3h +
1-1(5F12_
H
00
141
H
[200] 51.6 mg d'acide lactique (1.34 mmol) a été ajouté à une solution de
165.0 mg de 5a-
hydroxy-66-[2-(1H-imidazol-4-y1)-éthylamino]-cholestan-36-méthylsulfo nyl dans
5 ml
d'éthanol anhydre sous agitation. L'agitation a été maintenue à température
ambiante pour
3 heures. Une évaporation sous vide du solvant organique permet l'obtention
d'une poudre
blanche de 216.6 mg de 5a-hydroxy-6612-(1H-imidazol-4-y1)-éthylamino]-
cholestan-3[3-
méthylsulfonyl dilactate.
[201] 1H-NMR (500 MHz, Me0D-4d): O (ppm) 7.77 (s, 1H), 6.98(s, 1H), 3.52 -3.47
(m, 1H),
3.45 - 3.41 (m, 1H), 3.18 - 3.14 (m, 1H), 2.96 - 2.94 (t, 2H), 2.89 - 2.87 (m,
4H), 2.59 -
2.55 (t, 1H), 2.02 - 2.00 (m, 1H), 1.97 - 1.95 (m, 1H), 1.86 - 1.80 (m, 2H),
1.75 - 1.65 (m,
5H) 1.57 - 0.95 (m, 29H), 0.90 - 0.89 (d, 3H), 0.84 - 0.82 (dd, 6H), 0.72 (s,
3H).
[202] Exemple 21 : Étude pharmacocinétique du DX103
[203] L'étude suivante est un dosage par LC/MS dans le plasma des différentes
molécules
sur 3 jours (11 points de mesures au final). Les graphiques sont toujours
présentés en
comparaison avec le DX101 qui est la référence.
[204] Protocole
Groupe 1 Groupe 2
Administration du DX101 DX103
composé
Dose 50 mg/kg 50 mg/kg
Voie d'application orale orale
Animaux rat rat
Taille du groupe 3 3
Échantillons plasma plasma
Dosage DX101 DX103 et DX101
CA 03196518 2023- 4- 24
WO 2022/090427 45
PCT/EP2021/080054
Échantillonnage plasmatique à 0 (sans injection), 5, 10, 15, 30 min, 1h 4h 8h
24h 48h 72h (11
points)
[205] Le profil pharmacocinétique du DX103 en comparaison avec le DX101 est
donné à la
figure 4. Les résultats sont les suivants :
DX101 DX103 5
Aire sous la courbe 143297 105351
Concentration max (nM) 140 135
Temps pour Cmax (min) 480 240
[206] Conclusion : Le profil du DX103 montre une absorption plus rapide dans
l'organisme
et une biodisponibilité légèrement inférieure au DX101.
[207] Exemple 22 : Étude pharmacocinétique du DX105
[208] L'étude suivante est un dosage par LC/MS dans le plasma des différentes
molécules
sur 3 jours (11 points de mesures au final). Les graphiques sont toujours
présentés en
comparaison avec le DX101 qui est la référence.
Protocole
Groupe 1 Groupe 2
Administration du DX101 DX105
composé
Dose 50 mg/kg 50 mg/kg
Voie d'application orale orale
Animaux rat rat
Taille du groupe 3 3
Échantillons plasma plasma
Dosage DX101 DX105 et DX101
Échantillonnage plasmatique à 0 (sans injection), 5, 10, 15, 30 min, 1h 4h 8h
24h 48h
72h (11 points)
[209] Le profil pharmacocinétique du DX105 en comparaison avec le DX101 est
donné à la
figure 5. Les résultats sont les suivants :
CA 03196518 2023- 4- 24
WO 2022/090427 46
PCT/EP2021/080054
DX101 DX105
Aire sous la courbe 143297 145590
Concentration max (nM) 140 197
Temps pour Cmax (min) 480 240
[210] Le DX105 montre une biodisponibilité équivalente (voir très légèrement
supérieure) au
DX101. En revanche, il présente une absorption beaucoup plus rapide et une
concentration maximale beaucoup plus importante, ce qui permet de prétendre à
de bonne
perspective in vivo.
[211] Exemple 23 : Étude pharmacocinétique du DX111
[212] L'étude suivante est un dosage par LC/MS dans le plasma des différentes
molécules
sur 3 jours (11 points de mesures au final). Les graphiques sont toujours
présentés en
comparaison avec le DX101 qui est la référence.
Groupe 1 Groupe 2
Administration du DX101 DX111
composé
Dose 50 mg/kg 50 mg/kg
Voie d'application orale orale
Animaux rat rat
Taille du groupe 3 3
Échantillons plasma plasma
Dosage DX101 DX111 et DX101
Échantillonnage plasmatique à 0 (sans injection), 5, 10, 15, 30 min, 1h 4h 8h
24h 48h 72h
(11 points)
[213] Le profil pharmacocinétique du DX111 en comparaison avec le DX101 est
donné à la
figure 6. Les résultats sont les suivants :
DX101 DX111
Aire sous la courbe 143297 368921
Concentration max (nM) 140 515
Temps pour Cmax (min) 480 240
CA 03196518 2023- 4- 24
WO 2022/090427 47
PCT/EP2021/080054
Cette étude de pharmacocinétique par voie orale montre que le 0X1 11 démontre
une
absorption de 3 fois supérieure au DX101. De plus, le DX111 possède une
concentration
maximale plus importante ainsi qu'une absorption plus rapide.
[214] Exemple 24 : Étude pharmacocinétique du DX123
[215] L'étude suivante est un dosage par LC/MS dans le plasma des différentes
molécules
sur 3 jours (11 points de mesures au final). Les graphiques sont toujours
présentés en
comparaison avec le DX101 qui est la référence.
Protocole
Groupe 1 Groupe 2
Administration du DX101 DX123
composé
Dose 50 mg/kg 50 mg/kg
Voie d'application orale orale
Animaux rat rat
Taille du groupe 3 3
Échantillons plasma plasma
Dosage DX101 DX123 et DX101
Echantillonnage plasmatique à 0 (sans injection), 5, 10, 15, 30min, 1h 4h 8h
24h 48h 72h
(11 points)
[216] Le profil pharmacocinétique du DX123 en comparaison avec le DX101 est
donné à la
figure 8. Les résultats sont les suivants :
DX101 DX123
Aire sous la courbe 3019 6711
Concentration max (nM) 150 721
Temps pour Cmax (min) 4 4
[217] Cette première analyse de pharmacocinétique par voie orale montre que le
DX123
présente une biodisponibilité deux fois plus élevée en comparaison au DX101.
Ces
résultats permettent de prétendre à de bonne perspective in vivo du DX123.
[218] Exemple 25: Étude de la cytotoxicité des analogues du DX101 selon
l'invention sur
les cellules 4T1
CA 03196518 2023- 4- 24
WO 2022/090427 48
PCT/EP2021/080054
[219] Les tests de viabilité cellulaires ont été réalisés sur des cellules
tumorales mammaires
murine 4T1 caractérisées comme triples négatives (HER2-, ER-, PR-).
[220] Pour cette expérience, un milieu de culture cellulaire a été préparé. Le
milieu de culture
est constitué d'un milieu Dulbecco's Modified Eagle Medium (DMEM,
commercialisé par
Westburg sous la référence LO BE12-604F), comprenant 4.5 g/L glucose avec de
la L-
Glutamine, auquel y est ajouté 10 % de sérum de veau foetal (SVF) et 50 U/ml
de
pénicilline /streptomycine. Les cellules 4T1 sont introduites dans ce milieu
de culture.
[221] II a été ensemencé des boites de 96 puits avec 2000 cellules 4T1 par
puits. Après 72
heures (h) de culture en condition normale, c'est-à-dire dans un incubateur à
une
température de 37 C à 5 % d'02, nous traitons pendant 48h les cellules 4T1
avec le
DX101, DX103, DX111, DX123, DX125, DX127 et DX129 à 100 nM, 1 M, 2.5 M et 10
M. Une condition contrôle (CTL) est également réalisée en parallèle en
utilisant le
protocole décrit précédemment sans traitement avec les molécules DX101, DX103,
DX111, DX123, DX125, DX127 ou DX129.
[222] La viabilité cellulaire est mesurée par trois différentes méthodes. Pour
la première
méthode, un marquage MTT est réalisé à 48 heures. Ce test est basé sur
l'utilisation du
sel de tétrazolium MTT (bromure de 3-(4,5-dimethylthiazol-2-y1)-2,5-diphenyl
tetrazolium).
Le tétrazolium est réduit par la succinate déshydrogénase mitochondriale des
cellules
vivantes actives, en formazan, précipité de couleur violette. La quantité de
précipité
formée est proportionnelle à la quantité de cellules vivantes mais également à
l'activité
métabolique de chaque cellule. Ainsi, un simple dosage de la densité optique à
550 nm
par spectroscopie permet de connaître la quantité relative de cellules
vivantes et actives
métaboliquement. Après 48 heures, le milieu est aspiré, et les cellules sont
incubées avec
du MTT (0.5 mg/mi dans du milieu de culture) pendant environ 3 heures. La
solution de
MTT est aspirée et les cristaux violets sont solubilisés dans du
diméthylsulfoxyde (DMSO).
La DO (densité optique) est mesurée à 550 nm. Le pourcentage de viabilité est
alors
déterminé dans chaque puits par rapport au CTL et l'IC50 (concentration pour
laquelle il
reste 50 % de cellules vivantes) est déterminée pour chaque molécule avec le
logiciel
Prism à l'aide d'une droite de régression non linéaire (log(inhibitor) vs.
Response).
[223] Pour la deuxième méthode, le pourcentage de viabilité est déterminé à
l'aide du
dosage de l'activité de l'enzyme LDH (lactate déshydrogénase) dans les
surnageants
cellulaires à l'aide du kit non radioactive cytotoxicity assay kit (Promega).
La LDH est une
enzyme libérée dans le surnageant des cellules mortes. Plus l'activité LDH
dans le
surnageant est élevée, plus la mort cellulaire est importante. Dans cet essai
enzymatique,
la LDH libérée convertit un sel de ttrazolium violet en formazan de couleur
rouge,
CA 03196518 2023- 4- 24
WO 2022/090427 49
PCT/EP2021/080054
absorbant à 490 nm. L'intensité de la couleur rouge est proportionnelle au
nombre de
cellules morte. Après 48h de traitement, les surnageants sont transférés dans
une
nouvelle plaque 96 puits et sont incubés pendant 30 minutes en présence du
substrate
mix à température ambiante. La réaction est arrêtée à l'aide du réactif stop
stolution et
l'absorbance est déterminée à 490 nM. Le pourcentage de mort cellulaire est
déterminé
ici à l'aide d'un contrôle 100 % d'activité maximale de la LDH (réalisé à
partir de cellules
non traitées incubées en présence du lysis solution 45 minutes à 37 C juste
avant l'ajout
du substrate mix), et la viabilité cellulaire dans chaque puits est alors
déduite de ce
pourcentage. L'IC50 est alors déterminée comme expliqué dans le paragraphe
précédent.
[224] Pour la troisième méthode, le pourcentage de viabilité est déterminé à
l'aide du kit
CellTox Green Cytotoxicity Assay (Promega). Ce test mesure la mort cellulaire
via un
changement de l'intégrité membranaire. Ce test utilise une sonde de type
cyanine qui ne
pénètre pas dans les cellules quand elles sont vivantes, mais, qui se lie à
l'ADN des
cellules mortes, elles perméables à la sonde, la rendant alors fluorescente.
En
conséquence, plus la fluorescence dans les puits est élevée, plus la mort
cellulaire est
importante. Après 48h de traitement, les cellules sont incubées pendant 15
minutes
minimum en présence du Celltox green reagent à température ambiante et la
fluorescence
est lue
¨emission 485 nm/A
¨excitation 590 nm. Le pourcentage de mort cellulaire est déterminé
à l'aide du contrôle 100 % de mort cellulaire (réalisé à partir de cellules
non traitées
incubées en présence du lysis solution 30 minutes à 37 C avant l'ajout du
Celltox green
reagent), et la viabilité cellulaire dans chaque puits est alors déduite de ce
pourcentage.
L'IC50 est alors déterminée comme expliqué précédemment.
[225] Les résultats des IC50 de ces tests sont présentés dans les Tableaux la,
lb et 1 c.
Dans ces tableaux :
- a Le seuil de significativité a été calculé par comparaison du LogIC50 du
composé au
LogIC50 du DX101 avec min n=3 et un test de One-way ANOVA suivi d'un post-test
de
Dunn's
_ nb représente le nombre de test indépendant avec 4 à 10 réplicats pour
chaque condition.
[226] Tableau la:
IC50 (IIM) MIT
Composé nb Moyenne IC50 p valuea
LogIC50 (IIM)
CA 03196518 2023- 4- 24
WO 2022/090427 50
PCT/EP2021/080054
(iSEM)
DX101 10 0.64 0.05 4.40
DX103 2 0.50 0.02 3.16
DX111 5 0.23 0.03 1.69 <0.05
DX115 3 1.57 0.12 >10 <0.001
DX123 6 0.77 0.04 5.94 >0.05
DX125 4 2.16 0.23 >10 <0.0001
DX127 2 1.07 0.04 11.67
DX129 1 1.18 0.17 15.10
[227] Tableau lb:
Icso (p M LDH
Composé nb Moyenne IC50 p valuea
Log1C50 (11M)
( SEM)
DX101 10 0.51 0.07 3.24
DX103 2 0.64 0.09 4.40 >0.05
DX111 5 0.27 0.13 1.88 >0.05
DX115 3 1.24 0.07 >10 >0.05
DX123 5 0.16 0.19 1.45 >0.05
DX125 3 1.56 0.63 >10 <0.01
DX127 2 0.85 0.44 7.09
DX129 1 0.59 0.18 3.87
[228] Tableau lc :
IC50 (n) Green Cytotoxicity Assy
CA 03196518 2023- 4- 24
WO 2022/090427 51_
PCT/EP2021/080054
Composé nb Moyenne IC50 p valuea
Log1C50 (11M)
( SEM)
DX101 8 0.43 0.08 2.68
DX103 1 0.42 0.13 2.63 >0.05
DX111 9 0.34 0.11 2.18 >0.05
DX115 3 1.40 0.05 > 10 <0.001
DX123 6 0.32 0.13 > 10 >0.05
DX125 4 1.35 0.25 >10 <0.001
DX127 2 1.04 0.13 10.89
DX129 1 1.21 0.10 16.18
[229] II est illustré dans les Tableaux la, lb et 1 c que pour le DX111, 11050
est
significativement inférieure (jusqu'à un facteur de 2,5) à celle du DX101,
indiquant une
activité cytotoxique supérieure à celle du DX101. En outre, l'activité du
DX123 a une
tendance à être supérieure à celle du DX101, et les activités du DX125, DX127
et du
DX129 sont inférieures à celle du DX101.
[230] Exemple 26 : Étude de la cytotoxicité des analogues du DX101 selon
l'invention sur
les cellules BT-474
[231] Les tests de viabilité cellulaires ont été réalisés également sur des
cellules tumorales
mammaires humaines BT-474 (caractérisée comme étant triples positives HER2+,
ER+,
PR+). Les cellules BT-474 sont dans un milieu de culture cellulaire identique
à l'exemple
précédent et sont ensemencées dans des plaques 24 puits à 70 000 cellules par
puits,
pour la détermination de la viabilité cellulaire à l'aide du bleu Trypan, ou
dans des plaques
96 puits à 13 000 cellules par puits pour la détermination de la viabilité
cellulaire à l'aide
du test MTT ou LDH. Après 96 heures (h) de culture en condition normale, c'est-
à-dire
dans un incubateur à une température de 37 C à 5 0/0 d02, nous traitons
pendant 48h les
cellules BT-474 avec le DX101, DX103, DX105, DX111, DX123 et DX127 à 100 nM, 1
M,
2.5 M et 10 M. Un contrôle est également réalisé en utilisant le protocole
décrit
précédemment sans traitement avec les molécules DX101, DX103, DX105, DX111,
DX123 et DX127.
CA 03196518 2023- 4- 24
WO 2022/090427 52
PCT/EP2021/080054
[232] Après une digestion à la trypsine de 10 minutes à 37 C, la survie
cellulaire a été
quantifiée également par un test de bleu de trypan avec comptage automatique
par
l'appareil Biorad TC20 (TC20Tm Automated Cell Counter). Le test via le bleu de
trypan est
basé sur l'intégrité des membranes cellulaire, laquelle est rompue chez les
cellules mortes.
Le bleu de trypan colore les cellules mortes en bleu. L'appareil de comptage
cellulaire
Biorad TC20 compte la proportion de cellules bleues et non-bleues, et rapporte
au
pourcentage de cellules. Le pourcentage de viabilité est alors déterminé dans
chaque
puits par rapport au cellules non traitées et l'IC50 est déterminée comme
expliqué dans
l'exemple précédent. Les résultats sont représentés sur le Tableau 2.
Également, le
pourcentage de viabilité des cellules BT-474 a été déterminé à l'aide du test
MIT et LDH,
réalisé comme décrit dans l'exemple précédent.
[233] Les résultats sont représentés dans les Tableaux 2a, 2b et 2c. Dans ces
tableaux :
- a Le seuil de significativité a été calculé par comparaison du LogIC50 du
composé au
LogIC50 du DX101 avec min n=3 et un test de One-way ANOVA suivi d'un post-test
de
Dunn's (excepté pour le test LDH où la p-value a été calculée par un t-test)
_ nb représente le nombre de test indépendant avec 3 à 10 réplicas pour chaque
condition.
[234] Tableau 2a:
IC50 (p.M) MTT
Composé nb Moyenne
IC50 p yaluea
LogIC50 (IIM)
( SEM)
6 /
DX101 0.88 0.03 7.53
4
DX103 0.85+0.05 7.08
>0.05
5
DX105 0.86 0.04 4.08 >0.05
5
DX111 0.95+0.10 8.94 >0.05
[235] Tableau 2b:
IC50 (IIM) LDH
Composé nb Moyenne
IC50 p values
LogIC50 (IIM)
CA 03196518 2023- 4- 24
WO 2022/090427 53
PCT/EP2021/080054
( SEM)
6
DX101 1.03 0.06 10.64
0
DX103
0
DX105
3
DX111 0.89 0.04 7.82 >0.05
[236] Tableau 2c:
IC50 (IIM) Trypan Blue
Composé nb Moyenne ICso p valuea
LogIC50 (IIM)
( 5E M)
6
0X101 0.61 0.20 4.03
4
DX103 0.42 0.25 2.62 >0.05
DX105 0.55 0.14 3.57 >0.05
DX111 60.78 0.15 5.96 >0.05
[237] II est illustré dans les Tableaux 2a, 2b et 2c que l'activité des
molécules DX103, DX105,
5
DX111 et DX127 est similaire à celle du DX101, et que celle du DX123 a la
tendance à
être supérieure à celle du DX101 dans cette lignée. Toutes ces molécules sont
donc
pressenties comme de bons candidats pour un développement industriel car elles
ont des
données de biologique similaires ou supérieures au DX101.
[238] Exemple 27 : Effet du composé analogue DX111 sur la croissance tumorale
in vivo
[239] Toutes les procédures sur les animaux ont été conduites selon les
directives de notre
institution après avoir été approuvée par le comité d'éthique. Les cellules
4T1 ont, été
cultivées comme précédemment, été dissociées dans la trypsine, lavées deux
fois dans
du PBS froid et re-suspendues dans du PBS à 1.5 million/ml. Les tumeurs 4T1
ont été
obtenues par transplantation sous-cutanée de 0.150 million de cellules dans
100 [IL dans
le flanc de souris Balb/c femelle (9 semaines, Janvier). Quand les tumeurs ont
atteint un
volume de 50-100 mm3, les souris ont été gavées de 40 mg/kg de DX101 ou de 40
mg/kg
CA 03196518 2023- 4- 24
WO 2022/090427 54
PCT/EP2021/080054
de DX111 ou du véhicule contrôle (eau). Le traitement a été réalisé tous les
jours jusqu'à
la fin de l'expérience (volume tumoral> 1000 mm3). Le volume tumoral a été
déterminé
tous les jours à l'aide d'un pied à coulisse et calculé à l'aide de la formule
:1/2 X (Longueur
Largeur2). Le pourcentage d'inhibition de la croissance tumorale a été
déterminé selon
la formule suivante : 100 X (1- (Volume tumoral, jour 7/ volume tumoral jour
0) mol 1) / (1-
(volume tumoral, jour 7/ volume tumoral jour 0) véhicule).
[240] La méthode de Kaplan-Meier a été utilisée pour comparer la survie des
animaux.
[241] II est illustré sur la Figure 7A que le DX111 présente un effet
supérieur au DX101 sur
la réduction de la croissance tumorale ("p<0.01, test one-way ANOVA et post-
test de
Tukey). L'inhibition de la croissance tumorale à 7 jours a en outre été
déterminée à 67 %
pour les animaux traités au DX111 et à 48 % pour les animaux traités au DX101.
[242] En outre, l'analyse de la survie des animaux, illustrée également sur la
Figure 7B,
indique une meilleure médiane de survie des animaux traités au DX111 (Log-rank
Mantel-
Cox test, *p<0.05 et ns, non significatif ; Logrank test for trend, **p<0.01).
Également, on
y observe que après 15 jours de traitements, la survie est de 25 % pour les
animaux traités
au DX111 alors qu'elle est de 0 % pour les animaux traités au DX101. La
médiane de
survie après traitement du DX101 (40 mg/kg) se situe à 9 jours alors que celle
du DX111
(40 mg/kg) se situe à 10 jours.
[243] En conclusion, l'effet du DX111 est beaucoup plus important in vivo sur
l'inhibition de
la croissance tumorale et influe fortement sur la survie des animaux.
[244] Exemple 28 : Étude pour déterminer la pharmacocinétique et la
biodisponibilité du
DX111 par voie orale chez le rat.
[245] Protocole : L'étude a été réalisée sur quatre groupes décrits ci-après.
Groupe 1 Groupe 2 Groupe 3 Groupe 4
Administration du DX101 DX101 DX111 DX111
composé
Dose 50 mg/kg 5 mg/kg 50 mg/kg 5 mg/kg
Voie d'application orale I.V. orale I.V.
Animaux rat rat rat rat
Taille du groupe 3 3 3 3
Échantillons plasma plasma plasma plasma
CA 03196518 2023- 4- 24
WO 2022/090427 55
PCT/EP2021/080054
Dosage DX101 DX101 DX111 DX111
[246] Prélèvement plasmatique à 0 (sans injection), 15, 30min, lh, 4h, 8h,
24h, 48h, 72h
[247] Les résultats sont donnés dans le Tableau 3
[248] Tableau 3
Composé DX101 DX111
DX101 DX111
Dose po (mg/kg) 50 50 Dose iv (mg/kg) 5
5
Cmax [ng/ml] 77 309 Cmax [ng/ml] 3308
2970
Tmax [h] 4 4
ASC(t0-tlast) 1546 3228 ASC(t0-tlast)
6205 3516
Vd [L/kg] 29
43
Vss [L/kg] 19
12
T1/2 2nd phase [h] 39 24 T1/2 2nd phase [h] 28
21
Clairance[ml/min/kg] 412 240 Clairance[ml/min/kg] 11
25
F 3 0/0 10 `)/0
[249] De façon surprenante, les résultats montrent que le composé analogue
DX111
présente une biodisponibilité 3 fois supérieure au composé de référence DX101
en
diminuant le temps de demi-vie d'élimination. La concentration maximale
plasmatique
obtenue avec le DX111 est 4 fois supérieure à celle du DX101 et la clairance
est divisée
par 2.
[250] Bien que l'invention ait été décrite en liaison avec plusieurs modes de
réalisation
particuliers, il est bien évident qu'elle n'y est nullement limitée et qu'elle
comprend tous les
équivalents techniques des moyens décrits ainsi que leurs combinaisons si
celles-ci
entrent dans le cadre de l'invention.
L'usage du verbe comporter , comprendre ou inclure et de ses formes
conjuguées n'exclut pas la présence d'autres éléments ou d'autres étapes que
ceux
énoncés dans une revendication.
CA 03196518 2023- 4- 24