Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
WO 2022/129283
PCT/EP2021/086101
CHEMICAL COMPOUNDS USEFUL FOR INHIBITING NAV1.8 VOLTAGE-GATED
SODIUM CHANNELS AND TREATING NAV1.8 MEDIATED DISEASES
FIELD OF THE INVENTION
The invention relates to Nav1.8 inhibitor compounds or pharmaceutically
acceptable
salts or tautomer forms thereof, corresponding pharmaceutical compositions or
formulations,
methods or processes of compound preparation, methods, compounds for use in,
uses for
and/or combination therapies for treating pain and pain-associated diseases,
disorders and
conditions, and cardiovascular diseases, disorders, and conditions.
BACKGROUND OF THE INVENTION
Pain is a protective mechanism by which animals avoid potential tissue damage,
however there are numerous disease indications in which pain outlives its
usefulness and
becomes a disabling burden. Indications in which pain outlives its usefulness
can be broadly
categorized as those in which nerve damage or injury is the trigger
(neuropathic pain), those
in which an inflammatory response or metabolic dysregulation sensitizes the
pain response
(inflammatory pain) and those in which an injury or surgical procedure results
in a short term
elevation of pain response (post-operative/ambulatory pain).
Voltage-gated sodium channels underlie electrical signaling in all excitable
tissues by
setting the threshold and underlying the upstroke of action potentials. There
are nine distinct
isoforms of voltage-gated sodium channels. Those designated Nav1.1, Nav1.7,
Nav1.8 and
Nav1.9 are principally expressed on peripheral nerves where they control
neuronal
excitability. Na,1 .5 is the principle sodium channel isoform expressed in
cardiac myocytes,
Nav1.4 is expressed and functions in skeletal muscle, whilst Nav1.1, Nav1.2,
Nav1.3 and
Nav1_6 are widely expressed in the central nervous system (CNS) and to an
extent in the
peripheral nervous system. The principal role of these nine voltage-gated
sodium channels
is comparable in that they control sodium influx into cells but their
biophysical properties
varies which greatly influences the physiological profile of their respective
cell type (Catterall,
2012).
Currently, non-selective sodium channel inhibitors are utilized clinically as
anti-
arrhythmic and anti-seizure therapies, these include lidocaine, carbamazepine,
amitriptyline
and mexiletine. However, as these agents exhibit a lack of selectivity between
the different
sodium channel isoforms, their therapeutic utility is greatly reduced due to
adverse side
effects, largely mediated by activity in the CNS and heart. This has
stimulated efforts to
develop novel medicines which are selective for specific sodium channel
isoforms in order to
avoid side effects in the CNS and cardiovascular system.
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
The Nav1.8 channel is expressed in neurons of the dorsal root ganglia (DRG)
and
highly expressed in the small diameter neurons of this tissue which form pain
sensing C- and
A5- nerve fibers (Abrahamsen, 2008; Amaya, 2000; Novakovic, 1998). The channel
was
proposed as a therapeutic target for analgesia as soon as it was originally
cloned from rat
DRG (Akopian, 1996) due to its prominent physiological role in this tissue
type and restricted
expression profile. Nav1.8 was subsequently identified, cloned and
characterized from
human DRG tissue (Rabart 1998). The closest molecular relative of Nav1.8 is
Nav1.5 which
shares a sequence homology of - 60 %. Nav1.8 was previously known as SNS
(sensory
neuron sodium channel), PN3 (peripheral nerve sodium channel type 3), and as
it exhibits
characteristic pharmacological properties in its resistant to block by
tetrodotoxin, it is also
described as a TTX-resistant sodium channel.
Support for Na,1 .8 as a therapeutic target for pain indications comes from
several
sources. Nay1.8 has been shown to conduct the majority of current during
upstroke of the
action potential in DRG neurons (Blair & Bean, 2002) and due to its rate of re-
priming is also
critical for the ability of these neurons to fire repetitively (Blair and
Bean, 2003). Increased
expression and function of Nav1.8 has been reported in response to painful
stimuli such as
inflammatory mediators (England 1996 & Gold 1996), nerve damage (Roza 2003 &
Ruangsri
2011), and within painful neuromas (Black 2008 & Coward 2000). Knockout of the
gene
encoding Nav1.8 in mice resulted in a reduced pain phenotype in particular to
inflammatory
challenges (Akopian 1999). Knockdown of the mRNA encoding Nav1.8 also resulted
in
reduced painful phenotypes in rodent models, particularly in neuropathic
models (Lai 2002).
Pharmacological intervention via selective small molecule inhibitors has
demonstrated
efficacy in rodent models of inflammatory pain as well as neuropathic pain
(Jarvis 2007 &
Payne 2015). Supporting genetic evidence for Nav1.8 is also present in
patients with chronic
neuropathic pain where multiple gain of function mutations has been reported
to be
causative in episodic painful neuropathies and small fiber neuropathies (Faber
2012, Han
2014 & Eijkenboom 2018).
SUMMARY OF THE INVENTION
Accordingly, there is a need for the development of novel compounds,
particularly
Nav1.8 inhibitor compounds that have improved solubility and are thus more
advantageous
for alternative routes of administration, such as intravenous administration.
The invention
satisfies this need by providing prodrugs of compounds with Na,1 _8 inhibitory
activity and
uses of such prodrugs in the treatment of pain and pain associated diseases,
disorders, and
conditions, and in the treatment of cardiovascular, diseases, disorders, and
conditions. The
prodrugs of the invention have improved solubility as compared to their
respective parent
compounds, and thus can be useful for intravenous (IV) administration and
treatment of pain
2
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
and pain associated diseases, disorders, and conditions in which IV
administration may be
beneficial or preferred, such as in the treatment of acute pain.
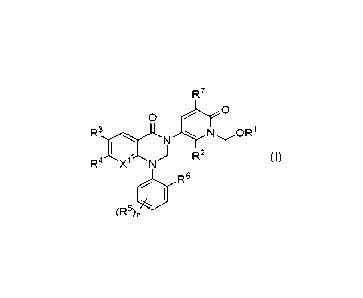
In one aspect, the invention relates to a compound of formula (I):
R7
0
R3ncitN N OR1
) R2
R4 N
R6
(R -)n (I)
or a pharmaceutically acceptable salt thereof,
wherein:
X1 is N or CH;
R1 is -P0(OH)2;
R2 is hydrogen, ¨(01_6)alkyl, -NRaRb, halo, or -(01_6)haloalkyl;
each of R3 and R4 is independently hydrogen, halo, cyano, -NRaRb, -(C1_
6)alkyl, -(Ci_6)ha10a1ky1, -0-(Ci_6)a1kyl, or -0-(Ci_6)ha10a1ky1;
each R5 is independently halo, -(01_6)alkyl, -0(01_6)alkyl, or -
0(01_6)haloalkyl;
R6 is hydrogen or ¨(C16)alkyl;
R7 is hydrogen, ¨(Ci_6)a1ky1, halo, or -(Ci_6)ha10a1ky1;
each of Ra and Rb is independently hydrogen or ¨(016)alkyl; and
n is 0, 1, or 2.
In one aspect, the invention relates to a pharmaceutical composition
comprising a
compound or a tautomer thereof, or a pharmaceutically acceptable salt thereof
as defined
herein, and a pharmaceutically acceptable excipient.
In one aspect, the invention relates to a method of inhibiting a Nav1.8
voltage-gated
sodium channel in a subject in need thereof, the method comprising
administering to the
subject a compound or a tautomer thereof, or a pharmaceutically acceptable
salt thereof as
defined herein or a pharmaceutical composition as defined herein.
In one aspect, the invention relates to a method of treatment of pain or a
pain-
associated disease, disorder, or condition in a subject in need thereof, the
method
comprising administering to the subject a therapeutically effective amount of
a compound, or
a tautomer thereof, or a pharmaceutically acceptable salt thereof as defined
herein or a
pharmaceutical composition as defined herein.
In one aspect, the invention relates to a method of treatment of atrial
fibrillation in a
subject in need thereof, the method comprising administering to the subject a
therapeutically
3
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
effective amount of a compound, or a tautomer thereof, or a pharmaceutically
acceptable
salt thereof as defined herein or a pharmaceutical composition as defined
herein.
In one aspect, the invention relates to a compound, or a tautomer thereof, or
a
pharmaceutically acceptable salt thereof as defined herein or a pharmaceutical
composition
as defined herein for use in treatment of pain or a pain-associated disease,
disorder, or
condition.
In one aspect, the invention relates to a compound, or a tautomer thereof, or
a
pharmaceutically acceptable salt thereof as defined herein or a pharmaceutical
composition
as defined herein for use in treatment of atrial fibrillation.
In one aspect, the invention relates to use of a compound, or a tautomer
thereof, or a
pharmaceutically acceptable salt thereof as defined herein or a pharmaceutical
composition
as defined herein in the manufacture of a medicament for treatment of pain or
a pain-
associated disease, disorder, or condition.
In one aspect, the invention relates to use of a compound, or a tautomer
thereof, or a
pharmaceutically acceptable salt thereof as defined herein or a pharmaceutical
composition
as defined herein in the manufacture of a medicament for treatment of atrial
fibrillation.
In one aspect, the invention relates to a compound, or a tautomer thereof, or
pharmaceutically acceptable salt thereof as defined herein, or a
pharmaceutical composition
as defined herein for use in therapy.
DETAILED DESCRIPTION OF THE INVENTION
Various publications, articles and patents are cited or described in the
background
and throughout the specification. Discussion of documents, acts, materials,
devices, articles
or the like which has been included in the present specification is for the
purpose of
providing context for the disclosure. Such discussion is not an admission that
any or all of
these matters form part of the prior art with respect to the disclosure.
Unless defined otherwise, all technical and scientific terms used herein have
the
same meaning as commonly understood to one of ordinary skill in the art to
which this
invention pertains. Otherwise, certain terms used herein have the meanings as
set forth in
the specification.
It must be noted that as used herein and in the appended claims, the singular
forms
"a," "an," and "the" include plural reference unless the context clearly
dictates otherwise.
As used herein, the conjunctive term "and/or" between multiple recited
elements is
understood as encompassing both individual and combined options. For instance,
where two
elements are conjoined by "and/or," a first option refers to the applicability
of the first element
without the second. A second option refers to the applicability of the second
element without
the first. A third option refers to the applicability of the first and second
elements together.
4
CA 03202326 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
Any one of these options is understood to fall within the meaning, and
therefore satisfy the
requirement of the term "and/or" as used herein. Concurrent applicability of
more than one of
the options is also understood to fall within the meaning, and therefore
satisfy the
requirement of the term "and/or."
Unless otherwise stated, any numerical value, such as a concentration or a
concentration range described herein, are to be understood as being modified
in all
instances by the term "about." Thus, a numerical value typically includes
10% of the
recited value. For example, the recitation of "10-fold" includes 9-fold and 11-
fold. As used
herein, the use of a numerical range expressly includes all possible
subranges, all individual
numerical values within that range, including integers within such ranges and
fractions of the
values unless the context clearly indicates otherwise.
The present invention relates to compounds of Formula (I) or pharmaceutically
acceptable salts thereof, corresponding pharmaceutical compositions, methods
or processes
of compound preparation, methods, compounds for use in, uses for and/or
combination
therapies for treating Nav1.8 mediated diseases, disorders, and conditions,
such as pain
and/or pain-associated disease(s), disorder(s) or condition(s), respectively,
and atrial
fibrillation.
The definitions for the various groups and substituent groups of any of the
Formulas
disclosed herein, or a pharmaceutically acceptable salt and/or a corresponding
tautomer
form thereof provided throughout the specification are intended to
particularly describe each
compound species disclosed herein, individually, as well as groups of one or
more
compound species.
As used herein, the term alkali metal is intended to mean the Group I
elements,
which include, but are not limited to lithium (Li), sodium (Na), or potassium
(K) and the like.
The term alkali earth metal may include, but are not limited to calcium (Ca)
or magnesium
(Mg) and the like.
As used herein, the terms "alkyl" or "straight or branched alkyl", and the
like,
represent a saturated, straight or branched hydrocarbon moiety. Exemplary
alkyls include,
but are not limited to methyl (Me), ethyl (Et), propyl (e.g., n-propyl,
isopropyl), butyl (e.g., n-
butyl, isobutyl, tert-butyl), and pentyl (e.g., n-pentyl, isopentyl,
neopentyl), etc. An alkyl
group can have a specified number of carbon atoms. When a number appears in a
subscript after the symbol "C," the subscript defines with more specificity
the number of
carbon atoms which that particular alkyl can contain. For example, the terms
"C1-C6" and
"01_6" refer to an alkyl containing 1 to 6 carbon atoms and the terms "01-04"
and "01_4" refer
to an alkyl containing 1 to 4 carbon atoms.
5
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
When the term "alkyl" is used in combination with other substituent groups,
such as
"haloalkyl" or "hydroxyalkyl", the term "alkyl" is intended to encompass a
divalent saturated,
straight or branched-chain hydrocarbon radical.
For example, the terms "haloalkyl" or "straight or branched haloalkyl" are
intended to
mean a saturated, straight or branched hydrocarbon moiety substituted with one
or more
halogens, where halogen is independently selected from: fluoro, chloro, bromo
and iodo. A
haloalkyl group can have a specified number of carbon atoms. For example, the
terms
"(Ci-06)haloalkyl" and "(C1_6)haloalkyl" refer to a saturated, straight- or
branched-chain
haloalkyl radical, having at least 1 and up to 6 carbon atoms. Likewise, the
terms "(C1_
C4)haloalkyl" and "(Ci4haloalkyl" refer to a saturated, straight- or branched-
chain haloalkyl
radical having 1 to 4 carbon atoms. "Fluorinated alkyl" or "fluoroalkyl" in
particular refers to
any alkyl group as defined above substituted with at least one fluoro atom,
e.g., one to three
fluoro atoms, such as one, two, or three fluoroatoms. Representative
haloalkyls include, but
are not limited to trifluoromethyl (-CF3), tetrafluoroethyl (-CF2CHF2),
pentafluoroethyl (-
CF2CF3) and the like.
The term "hydroxyalkyl" refers to a saturated, straight or branched
hydrocarbon
moiety substituted with one or more hydroxy groups.
As used herein, the terms "halogen" and "halo" mean fluoro (-F), chloro (-Cl),
bromo
(-Br), and iodo (-I).
"Hydroxy" or "hydroxyl" is intended to mean the radical ¨OH.
"Oxo" represents a double-bonded oxygen moiety; for example, if attached
directly to
a carbon atom forms a carbonyl moiety (C=0), or attached to an N or S forms
oxides, e.g.,
N-oxides, sulfones or sulfoxides.
The term "cyano" refers to ¨CN.
The term "amino" refers to ¨NH2. One or more hydrogen atoms of an amino group
can be replaced by a substituent such as an alkyl group, which is referred to
as an
"alkylamino." Alkylamino groups have one or both hydrogen atoms of an amino
group
replaced with an alkyl group and is attached to the parent molecule through a
bond to the
nitrogen atom of the alkylamino group. For example, alkylamino includes
methylamino (-
NHCH3), dimethylamino (-N(CH3)2), -NHCH2CH3 and the like.
"Alkoxy" refers to a group containing an alkyl radical attached through an
oxygen
linking atom, wherein alkyl is as defined above. An alkoxy group can have a
specified
number of carbon atoms. For example, the terms "(Ci-C6)alkoxy" and
"(Ci_6)alkoxy" refer to
an alkyl radical, having at least 1 and up to 6 carbon atoms attached through
an oxygen
linking atom. Likewise, the terms "(C1_C4)alkoxy" and "(C1_4)alkoxy" refer to
an alkyl radical
having at least 1 and up to 4 carbon atoms attached through an oxygen linking
atom.
6
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
Exemplary alkoxy groups include, but are not limited to, methoxy, ethoxy, n-
propoxy,
isopropoxy, n-butoxy, s-butoxy, and t-butoxy.
"Haloalkoxy" refers to an alkoxy group in which the alkyl moiety is
substituted with
one or more halogens, wherein halogen is independently selected from fluor ,
chloro,
bromo, and iodo. A haloalkoxy group can have a specified number of carbon
atoms. For
example, the term "(Ci-C6)haloalkoxy refers to a haloalkyl radical, having at
1 to 6 carbon
atoms attached through an oxygen linking atom. Representative haloalkoxy
groups include,
but are not limited to difluoromethoxy (-0CHCF2), trifluoromethoxy (-0CF3),
tetrafluoroethoxy
(-0CF2CHF2) and the like.
In accordance with convention used in the art: I¨ is used in structural
formulas
herein to depict the bond that is the point of attachment of a group, moiety
or substituent to
the core, backbone, or parent molecule structure.
When a bond to a substituent is shown to cross a bond connecting two atoms in
a
ring, then such substituent can be bonded to any atom on the ring.
As used herein, the term "compound(s) of the invention" means a compound of
any
of the Formulas disclosed herein, in any form, i.e., any salt or non-salt form
(e.g., as a free
acid or base form, or as a pharmaceutically acceptable salt thereof), any
tautomer form
thereof, and any physical form thereof (e.g., including non-solid forms (e.g.,
liquid or semi-
solid forms), and solid forms (e.g., amorphous or crystalline forms, specific
polymorphic
forms, solvates, including hydrates (e.g., mono-, di- and hemi- hydrates)),
and mixtures of
various forms.
As used herein, the term "optionally substituted" means that a group (e.g.,
alkyl, etc.),
may be unsubstituted, or the group may be substituted with one or more
specified
substituent(s) as defined herein throughout the instant specification. The
term "substituted"
as used herein with respect to a group (e.g., alkyl, etc.) means that at least
one hydrogen
atom is replaced with a non-hydrogen group, provided that all normal valencies
are
maintained and that the substitution results in a stable compound. In the case
where groups
may be selected from a number of alternative groups the selected groups may be
the same
or different. For example, various substituent groups of compound formulas as
defined in
the present invention may be optionally substituted, but are not limited to
substituents, such
as halo, cyano, amino, alkyl, haloalkyl, alkoxy, and the like.
The term "independently" when used with reference to a substituent or
heteroatom
means that where more than one substituent or heteroatom is selected from a
number of
possible substituents or heteroatoms, respectively, those substituents or
heteroatoms may
be the same or different.
7
CA 03202326 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
Compounds
Compounds of formula (I) of the present invention are prodrugs of their
respective
parent compounds, which are Nav1.8 inhibitor compounds. Upon administration of
the
prodrug, the prodrug moiety is cleaved thereby resulting in the parent
compound.
Accordingly, Nav1.8 inhibitory activity upon administration of the prodrug is
primarily due to
formation of the parent compound from cleavage of the prodrug.
The prodrugs of the present invention typically have higher aqueous solubility
than
the corresponding parent compound& This higher solubility facilitates
administration of
higher doses of the prodrug, resulting in a greater drug load per unit dosage.
Thus, the
compounds of formula (I) of the invention (La, prodrugs) may be advantageous
for
intravenous (IV) formulation and administration, and thus beneficial for use
in the treatment
of pain and pain associated diseases, disorders, and conditions in which
administration of
higher doses or administration via the IV route may be beneficial, such as
treatment of acute
pain.
The term "prodrug" refers to compounds that are drug precursors which,
following
administration and/or absorption, release the parent compound in vivo via a
metabolic
process. Typically, a prodrug has less biological activity than the parent
compound. A
prodrug may also improve the physical properties and/or efficacy of the parent
compound,
such as reduced toxicity and fewer unwanted effects through greater control of
the
absorption, blood levels, metabolic distribution and/or cellular uptake of the
parent
compound. Prodrugs may also have higher solubility than the corresponding
parent
compound.
The terms "parent compound" and "parent drug" refer to the biologically active
entity
that is released via enzymatic action of a metabolic or catabolic process, or
via a chemical
process following administration of the prodrug. The parent compound may also
be the
starting material for the preparation of the corresponding prodrug.
In one aspect, the present invention relates to a compound of Formula (I):
R7
0
R3r1LN
R41' N ) R2
R6
(R¨)n (I),
or a pharmaceutically acceptable salt thereof,
wherein:
8
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
X1 is N or CH;
R1 is -P0(OH)2;
R2 is hydrogen, ¨(01_6)alkyl, -NRaRb, halo, or -(01_6)haloalkyl;
each of R3 and R4 is independently hydrogen, halo, cyano, -NRaRb, -(C1_
6)alkyl, -(C1_6)ha10a1ky1, -0-(C1_6)a1kyl, or -0-(Ci_6)ha10a1ky1;
each of R5 is independently halo, -(Ci_6)a1ky1, -0(Ci_6)a1ky1, or -0(C1-
6)haloalkyl;
R6 is hydrogen or ¨(01_6)alkyl;
R7 is hydrogen, ¨(01_6)alkyl, halo, or -(01_6)haloalkyl;
each of R2 and Rb is independently hydrogen or ¨(C16)alkyl; and
n is 0, 1, or 2.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, X1 is N.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, X1 is CH.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, R2 is hydrogen.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, R2 is ¨(C1_6)alkyl.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, R2 is CH3.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, R2 is -NRaRb.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, R2 is -NH2.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, R3 is hydrogen, halo, or ¨(01_6)haloalkyl.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, R3 is hydrogen, -Cl, or -CF3.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, R3 is hydrogen.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, R3 is -Cl.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, R3 is -CF3.
9
CA 03202326 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, R4 is hydrogen, halo, or ¨(C1_6)ha10a1ky1.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, R4 is hydrogen, -Cl, or -CF3.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, R4 is hydrogen.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, R4 is -Cl.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, R4 is -CF3.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, one of R3 and R4 is hydrogen and the other of R3 and R4 is halo,
cyano, -NR Rb,
-(C16)alkyl, -(C1_6)haloalkyl, -0-(C1_6)alkyl, or -0-(C1_6)haloalkyl.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, one of R3 and R4 is hydrogen and the other of R3 and R4 is halo
or -(C1_
6)haloalkyl.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, one of R3 and R4 is hydrogen and the other of R3 and R4 is -Cl
or -CF3.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, each R5 is independently halo or -0(C1_6)haloalkyl.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, each R5 is independently -F or -0CF3.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, n is 1 and R5 is -F.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, n is 1 and R5 is -0CF3.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
R6
salt thereof, (R5 )n has the structure: R5a
, wherein R5a is halo, -(Ci_6)a1ky1, -
0(Ci_6)a1ky1, or -0(C1_6)ha10a1ky1
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, R52 is halo or -0(C1_6)haloalkyl.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, R5a is -F or -0CF3.
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, R5a is -F.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, R5a is -0CF3.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
R6 CH3 3-S.CH3
salt thereof, (R5 )n is: F or OCF3
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, R6 is hydrogen.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, R6is -(C1_6)alkyl.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, R6 is -CH3, -Cl2CH3, or -CH(CH3)2.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, R6 is -CH3.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, R7 is hydrogen.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, R7 is ¨(01_6)alkyl.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, R7 is halo.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof, R7 is -F or -Cl.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof,
X1 is N;
R1 is -P0(OH)2;
R2 is hydrogen or ¨(C1_6)alkyl;
one of R3 and R4 is hydrogen and the other of R3 and R4 is halo or
¨(C1_6)haloalkyl;
R5 is halo or ¨0(Ci_6)haloalkyl;
R6 is ¨(C1_6)alkyl;
R7 is hydrogen; and
n is 1.
11
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof,
X1 is N;
R1 is -P0(OH)2;
R2 is -CH3;
one of R3 and R4 is hydrogen and the other of R3 and R4 is halo or
¨(Ci_6)ha10a1ky1;
R5 is halo or ¨0(Ci_6)haloalkyl;
R6 is ¨(C16)alkyl;
R7 is hydrogen; and
n is 1 .
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof,
X1 is N;
R1 is -P0(OH)2;
R2 is -CH3;
one of R3 and R4 is hydrogen and the other of R3 and R4 is -Cl or -CF3;
R5 is -F or ¨0CF3;
R6 is -CH3;
R7 is hydrogen; and
nisi.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof,
X1 is C;
R1 is -P0(OH)2;
R2 is hydrogen or ¨(C16)alkyl;
one of R3 and R4 is hydrogen and the other of R3 and R4 is halo or
¨(01_6)haloalkyl;
R5 is halo or ¨0(C1_6)haloalkyl;
R6 is ¨(01_6)alkyl;
R7 is hydrogen; and
nisi.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof,
X1 is C;
R1 is -P0(OH)2;
R2 is -CH3;
one of R3 and R4 is hydrogen and the other of R3 and R4 is halo or
¨(C1_6)haloalkyl;
R5 is halo or ¨0(Ci_6)haloalkyl;
12
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
R6 is ¨(C1_6)alkyl;
R7 is hydrogen; and
n is 1.
In an embodiment of a compound of formula (I), or a pharmaceutically
acceptable
salt thereof,
X1 is C;
R1 is -P0(OH)2;
R2 is -CH3;
one of R3 and R4 is hydrogen and the other of R2 and R4 is -Cl or -CF3;
R6 is -F or ¨0CF3;
R6 is -CH3;
R7 is hydrogen; and
n is 1.
In another aspect, the invention relates to a compound which is selected from:
Name Structure
(5-(1-(4-Fluoro-2-methylpheny1)-4-oxo-6- o
F3cJLN -N 0, PH
3(2H)-y1)-6-methy1-2-oxopyridin-1(2H)-
Põ
HO
yl)methyl dihydrogen phosphate N
(5-(1-(4-fluoro-2-methylpheny1)-4-oxo-7- 0
(trifluoromethyl)-1,4-dihydroquinazolin- 0
3(2H)-y1)-6-methy1-2-oxopyridin-1(2H)-
N ) HO' OH
yl)methyl dihydrogen phosphate F3c
1410
(5-(6-chloro-1-(4-fluoro-2-methylphenyI)-4- 0
oxo-1,4-dihydroquinazolin-3(2H)-yI)-6- 0
NN
methy1-2-oxopyridin-1(2H)-yl)methyl LjI
N HO OH
dihydrogen phosphate
4111
(6-methy1-5-(1-(2-methyl-4- 0
(trifluoromethoxy)phenyI)-4-oxo-6- N
N
(trifluoromethyl)-1,4-dihydropyrido[2,3- I 3I HO 13E1
d]pyrimidin-3(2H)-yI)-2-oxopyridin-1(2H)- N
yl)methyl dihydrogen phosphate
ocF3
13
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
or a pharmaceutically acceptable salt thereof.
Salts
Because of their potential use in medicine, the salts of the compounds of any
of the
Formulas disclosed herein, including Formula (I) are preferably
pharmaceutically acceptable
salts. Pharmaceutically acceptable salts include, among others, those
described by Berge,
Bighley and Monkhouse J.Pharm.Sci (1977) 66, pp 1-19, or those listed in PH
Stahl and CG
Wermuth, editors, Handbook of Pharmaceutical Salts; Properties, Selection and
Use,
Second Edition Stahl/VVermuth: Wiley-VCH/VFICA, 2011. Non-pharmaceutically
acceptable
salts may be used, for example as intermediates in the preparation of a
compound of any of
the Formulas disclosed herein or a pharmaceutically acceptable salt thereof.
Suitable pharmaceutically acceptable salts can include acid or base addition
salts.
Such base additional salts can be formed by reaction of a compound of any of
the Formulas
disclosed herein, including Formula (I) of the invention with the appropriate
base, optionally
in a suitable solvent such as an organic solvent, to give the salt which can
be isolated by a
variety of methods, including crystallisation and filtration.
Such acid addition salts can be formed by reaction of a compound of any of the
Formulas disclosed herein, including Formula (I) of the invention, with the
appropriate acid,
optionally in a suitable solvent such as an organic solvent, to give the salt
which can be
isolated by a variety of methods, including crystallisation and filtration.
Salts may be prepared in situ during the final isolation and purification of a
compound
of any of the Formulas disclosed herein, including Formula (I) of the
invention. If a basic
compound of any of the Formulas disclosed herein, including Formula (I) of the
invention, is
isolated as a salt, the corresponding free base form of that compound may be
prepared by
any suitable method known to the art, including treatment of the salt with an
inorganic or
organic base. Similarly, if a compound of any of the Formulas disclosed
herein, including
Formula (I) of the invention, containing an acidic functional group such as a
phosphate group
is isolated as a salt, the corresponding free acid form of that compound may
be prepared by
any suitable method known to the art, including treatment of the salt with an
inorganic or
organic acid.
For example, when a compound of the invention is a base (contain a basic
moiety), a
desired salt form may be prepared by any suitable method known in the art,
including
treatment of the free base with an inorganic acid, such as hydrochloric acid,
hydrobromic
acid, sulfuric acid, nitric acid, phosphoric acid, and the like, or with an
organic acid, such as
acetic acid, trifluoroacetic acid, maleic acid, succinic acid, mandelic acid,
fumaric acid,
malonic acid, pyruvic acid, oxalic acid, glycolic acid, salicylic acid,
pyranosidyl acid, such as
glucuronic acid or galacturonic acid, alpha-hydroxy acid, such as citric acid
or tartaric acid,
14
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
amino acid, such as aspartic acid or glutamic acid, aromatic acid, such as
benzoic acid or
cinnamic acid, sulfonic acid, such as p-toluenesulfonic acid, methanesulfonic
acid,
ethanesulfonic acid or the like.
If an inventive basic compound is isolaied as a salt, the corresponding free
base form
of that compound may be prepared by any suitable method known to the art,
including
treatment of the salt with an inorganic or organic base, suitably an inorganic
or organic base
having a higher pKa than the free base form of the compound.
When a compound of the invention is an acid (contains an acidic moiety), a
desired
salt may be prepared by any suitable method known to the art, including
treatment of the
free acid with an inorganic or organic base, such as an amine (primary,
secondary, or
tertiary), an alkali metal or alkaline earth metal hydroxide, or the like.
Illustrative examples of
suitable salts include organic salts derived from amino acids such as glycine
and arginine,
ammonia, primary, secondary, and tertiary amines, and cyclic amines, such as
ethylene
diamine, dicyclohexylamine, ethanolamine, piperidine, morpholine, and
piperazine, as well
as inorganic salts derived from sodium, calcium, potassium, magnesium,
manganese, iron,
copper, zinc, aluminum, and lithium.
Certain of the compounds of this invention may form salts with one or more
equivalents of an acid (if the compound contains a basic moiety) or a base (if
the compound
contains an acidic moiety). The present invention includes within its scope
all possible
stoichiometric and non-stoichiometric salt forms. It will be understood that
if a compound of
any of the Formulas disclosed herein, including Formula (I) as defined herein
contains two or
more basic moieties, the stoichiometry of salt formation may include 1, 2 or
more equivalents
of acid. Such salts would contain 1, 2 or more acid counterions, for example,
a
dihydrochloride salt. Stoichiometric and non-stoichiometric forms of a
pharmaceutically
acceptable salt of a compound of any of the Formulas disclosed herein,
including Formula (I)
of the invention are included within the scope of the invention, including sub-
stoichiometric
salts, for example where a counterion contains more than one acidic proton.
Because the compounds of this invention may contain both acid and base
moieties,
pharmaceutically acceptable salts may be prepared by treating these compounds
with an
alkaline reagent or an acid reagent, respectively. Accordingly, this invention
also provides
for the conversion of one pharmaceutically acceptable salt of a compound of
this invention,
e.g., a hydrochloride salt, into another pharmaceutically acceptable salt of a
compound of
this invention, e.g., a sodium salt.
Representative pharmaceutically acceptable acid addition salts include, but
are not
limited to, 4-acetamidobenzoate, acetate, adipate, alginate, ascorbate,
aspartate,
benzenesulfonate (besylate), benzoate, bisulfate, bitartrate, butyrate,
calcium edetate,
camphorate, camphorsulfonate (camsylate), caprate (decanoate), caproate
(hexanoate),
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
caprylate (octanoate), cinnamate, citrate, cyclamate, digluconate, 2,5-
dihydroxybenzoate,
disuccinate, dodecylsulfate (estolate), edetate (ethylenediaminetetraacetate),
estolate (lauryl
sulfate), ethane-1,2-disulfonate (edisylate), ethanesulfonate (esylate),
formate, fumarate,
galactarate (mucate), gentisate (2,5-dihydroxybenzoate), glucoheptonate
(gluceptate),
gluconate, glucuronate, glutamate, glutarate, glycerophosphorate, glycolate,
hexylresorcinate, hippurate, hydrabamine (N,N'-di(dehydroabietyI)-
ethylenediamine),
hydrobromide, hydrochloride, hydroiodide, hydroxynaphthoate, isobutyrate,
lactate,
lactobionate, laurate, malate, maleate, malonate, mandelate, methanesulfonate
(mesylate),
methylsulfate, mucate, naphthalene-1,5-disulfonate (napadisylate), naphthalene-
2-sulfonate
(napsylate), nicotinate, nitrate, oleate, palmitate, p-aminobenzenesulfonate,
p-
aminosalicyclate, pamoate (embonate), pantothenate, pectinate, persulfate,
phenylacetate,
phenylethylbarbiturate, phosphate, polygalacturonate, propionate, p-
toluenesulfonate
(tosylate), pyroglutamate, pyruvate, salicylate, sebacate, stearate,
subacetate, succinate,
sulfamate, sulfate, tannate, tartrate, teoclate (8-chlorotheophyllinate),
thiocyanate,
triethiodide, undecanoate, undecylenate, and valerate.
Representative pharmaceutically acceptable base addition salts include, but
are not
limited to, aluminium, 2-amino-2-(hydroxymethyl)-1,3-propanediol (IRIS,
tromethamine),
arginine, benethamine (N-benzylphenethylamine), benzathine (N,N'-
dibenzylethylenediamine), bis-(2-hydroxyethyl)amine, bismuth, calcium,
chloroprocaine,
choline, clemizole (1-p chlorobenzy1-2-pyrrolildine-V-ylmethylbenzimidazole),
cyclohexylamine, dibenzylethylenediamine, diethylamine, diethyltriamine,
dimethylamine,
dimethylethanolamine, dopamine, ethanolamine, ethylenediamine, L-histidine,
iron,
isoquinoline, lepidine, lithium, lysine, magnesium, meglumine (N-
methylglucamine),
piperazine, piperidine, potassium, procaine, quinine, quinoline, sodium,
strontium, t-
butylamine, and zinc.
In particular embodiments, the invention provides a pharmaceutically
acceptable salt
of of a compound of formula (I). For example, the phosphate group of R1 (-
P0(OH)2) is an
acidic moiety that is particularly likely to participate in salt formation
with cationic species
(i.e., base addition salts). Any of the base addition salts listed above can
be used to form a
pharmaceutically acceptable salt of a compound of formula (I) of the
invention. Other
functional groups of a compound of formula (I) may additionally or
alternatively participate in
salt formation with acid and/or base addition salts, such as those described
above.
In an embodiment of a pharmaceutically acceptable salt of a compound of
formula
(I), R1 is -P(0)(OH)O-M+, -P0(0-)2=21V1+, or
each M+ is independently a pharmaceutically acceptable monovalent cation; and
D2+ is a pharmaceutically acceptable divalent cation.
16
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
In some embodiments, monovalent cations (M+) suitable for use in the invention
include, but are not limited to, alkali metal ions, e.g., lithium (Lit),
sodium (Na), potassium
(K+), etc.; ammonium ions (e.g., -N(Ra)4, wherein each Ra is independently
hydrogen,
cyclohexyl, or ¨(C1_6)alkyl), the ¨(C1_6)alkyl being optionally substituted
with one or more,
suitably 1-6, -OH groups), such as NH4, ethanolamine ion (H3N
), N-methyl-
D-glucamine ion, dicyclohexylamine ion, etc. When two M+ are present, each M+
is
independently a monovalent cation, wherein each M+ is the same or different,
preferably
each M+ is the same.
In an embodiment, each M+ is independently an alkali metal ion.
In an embodiment, each M+ is independently Li, Nat, or K.
In an embodiment, each M+ is independently NH4.
In an embodiment, each M+ is independently H3N (ethanolamine
ion).
In some embodiments, divalent cations (D2+) suitable for use in the invention
include,
but are not limited to, alkaline earth metal ions, e.g., magnesium (Mg2+),
calcium (Ca2+),
strontium (Sr'), etc.; divalent aluminum ions; etc.
In an embodiment, D2+ is alkaline earth metal ion.
In an embodiment, D2+ is Mg' or Ca'.
Other monovalent and divalent cations suitable for use in the invention
include
monovalent or divalent ions of amino acid ions, such as monovalent or divalent
ions
arginine, lysine, ornithine, etc. Monovalent and divalent cations including
basic nitrogen-
containing groups can be prepared by quaternization with agents such as lower
alkyl halides
(e.g., methyl, ethyl, propyl, and butyl chlorides, bromides, and iodides),
dialkyl sulfates (e.g.,
dimethyl, diethyl, dibutyl, and diamyl sulfates), etc.
Enantiomers, Diastereomers, and Polymorphs
The compounds according to any of the Formulas disclosed herein, including
Formula (I), or a pharmaceutically acceptable salt thereof of the invention,
may contain one
or more asymmetric center(s) (i.e., also referred to as a chiral center) and
may, therefore,
exist in optically forms (e.g., as individual enantiomers, diastereomers, or
other
stereoisomeric forms, or as mixtures thereof) and racemic forms. All of these
individual
compounds, stereoisomers, and mixtures thereof are included within the scope
of the
invention.
Chiral centers, such as chiral carbon atoms, may also be present in a
substituent
such as an alkyl group. Where the stereochemistry of a chiral center present
in any of the
Formulas disclosed herein, including Formula (I), or a pharmaceutically
acceptable salt
17
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
thereof of the invention, or in any chemical structure illustrated herein, is
not specified the
structure is intended to encompass all individual stereoisomers and all
mixtures thereof.
Thus, compounds or a pharmaceutically acceptable salt thereof of the invention
containing
one or more chiral centers may be used as racemic mixtures, enantiomerically
enriched
mixtures, or as enantiomerically pure individual stereoisomers.
Individual stereoisomers of a compound according to any of the Formulas
disclosed
herein, including Formula (I), or a pharmaceutically acceptable salt thereof
of the invention,
which contain one or more asymmetric centers may be resolved by methods known
to those
skilled in the art. For example, such resolution may be carried out:
(1) by formation of diastereoisomeric salts, complexes or other derivatives;
(2) by selective reaction with a stereoisomer-specific reagent, for example by
enzymatic oxidation or reduction; or
(3) by gas-liquid or liquid chromatography in a chiral environment, for
example, on a
chiral support such as silica with a bound chiral ligand or in the presence of
a chiral solvent.
The skilled artisan will appreciate that where the desired stereoisomer is
converted into
another chemical entity by one of the separation procedures described above, a
further step
is required to liberate the desired form.
Alternatively, specific stereoisomers may be synthesized by asymmetric
synthesis
using optically active reagents, substrates, catalysts or solvents, or by
converting one
enantiomer to the other by asymmetric transformation.
When a disclosed compound or its salt is named or depicted by structure, it is
to be
understood that the compound or salt, including solvates (particularly,
hydrates) thereof, may
exist in crystalline forms, non-crystalline forms or a mixture thereof. The
compound or salt,
or solvates (particularly, hydrates) thereof, may also exhibit polymorphism
(i.e. the capacity
to occur in different crystalline forms). These different crystalline forms
are typically known
as "polymorphs."
It is to be understood that when named or depicted by structure, the disclosed
compound, or solvates (particularly, hydrates) thereof, also include all
polymorphs thereof.
Polymorphs have the same chemical composition but differ in packing,
geometrical
arrangement, and other descriptive properties of the crystalline solid state.
Polymorphs,
therefore, may have different physical properties such as shape, density,
hardness,
deformability, stability, and dissolution properties. Polymorphs typically
exhibit different
melting points, IR spectra, and X-ray powder diffraction patterns, which may
be used for
identification. One of ordinary skill in the art will appreciate that
different polymorphs may be
produced, for example, by changing or adjusting the conditions used in
crystallizing/
recrystallizing the compound.
18
CA 03202326 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
Solvates
Compounds of the invention, or pharmaceutically acceptable salts thereof may
exist
in solvated and unsolvated forms. For solvates of the compounds of the
invention, or
pharmaceutically acceptable salts thereof, that are in crystalline form, the
skilled artisan will
appreciate that pharmaceutically acceptable solvates may be formed wherein
solvent
molecules are incorporated into the crystalline lattice during
crystallization. Solvates may
involve nonaqueous solvents such as ethanol, isopropanol, DMSO, acetic acid,
ethanolamine, and ethyl acetate, or they may involve water as the solvent that
is
incorporated into the crystalline lattice. Solvates wherein water is the
solvent that is
incorporated into the crystalline lattice are typically referred to as
"hydrates." Hydrates
include stoichiometric hydrates as well as compositions containing variable
amounts of
water.
Deuterated Compounds
The invention also includes various deuterated forms of the compounds of any
of the
Formulas disclosed herein, including Formula (I) or a pharmaceutically
acceptable salt
thereof of the invention. Each available hydrogen atom attached to a carbon
atom may be
independently replaced with a deuterium atom.
A person of ordinary skill in the art will know how to synthesize deuterated
forms of
the compounds of any of the Formulas disclosed herein, including Formula (I)
or a
pharmaceutically acceptable salt thereof of the invention. For example,
deuterated
materials, such as alkyl groups may be prepared by conventional techniques
(see for
example: methyl-d3-amine available from Aldrich Chemical Co., Milwaukee, WI,
Cat.
No.489,689-2).
Isotopes
The invention also includes isotopically-labeled compounds which are identical
to
those recited in any of the Formulas disclosed herein, including Formula (I)
or a
pharmaceutically acceptable salt thereof of the invention but for the fact
that one or more
atoms are replaced by an atom having an atomic mass or mass number different
from the
atomic mass or mass number most commonly found in nature.
Examples of isotopes that can be incorporated into compounds of the invention
include isotopes of hydrogen, carbon, nitrogen, oxygen, fluorine, iodine and
chlorine such as
3H, "C, 140, 18F, 1231 or 1251.
Compounds of the invention and pharmaceutically acceptable salts of said
compounds that contain the aforementioned isotopes and/or other isotopes of
other atoms
are within the scope of the invention. Isotopically labeled compounds of the
invention, for
19
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
example those into which radioactive isotopes such as 3H or 14C have been
incorporated,
are useful in drug and/or substrate tissue distribution assays. Tritiated,
i.e. 3H, and carbon-
14, i.e. 14C, isotopes are particularly preferred for their ease of
preparation and detectability.
uC and 18F isotopes are particularly useful in PET (positron emission
tomography).
Purity
Because the compounds of the invention are intended for use in pharmaceutical
compositions it will readily be understood that they are each preferably
provided in
substantially pure form, for example at least 60% pure, more suitably at least
75% pure and
preferably at least 85%, especially at least 98% pure (% are on a weight for
weight basis).
Impure preparations of the compounds may be used for preparing more pure forms
used in
the pharmaceutical compositions.
It is recognized that the compounds of any of the Formulas disclosed herein,
including Formula (I) or a pharmaceutically acceptable salt thereof of the
invention may exist
in forms as stereoisomers, regioisomers, or diastereoisomers.
Tautomers
Moreover, compounds of the invention may exist as tautomers or in tautomeric
forms. It is conventionally understood in the chemical arts that tautomers are
structural or
constitutional isomers of chemical compounds that readily interconvert. This
reaction
commonly results in the relocation of a proton. A structural isomer, or
constitutional isomer
(per IUPAC), is a type of isomer in which molecules with the same molecular
formula have
different bonding patterns and atomic organization, as opposed to
stereoisomers, in which
molecular bonds are always in the same order and only spatial arrangement
differs. The
concept of tautomerizations is called tautomerism. The chemical reaction
interconverting the
two is called tautomerization. Care should be taken not to confuse tautomers
with depictions
of 'contributing structures' in chemical resonance. Tautomers are distinct
chemical species
and can be identified as such by their differing spectroscopic data, whereas
resonance
structures are merely convenient depictions and do not physically exist.
Synthetic Schemes and General Methods of Preparation
The present invention also relates to processes for making compounds of any of
the
Formulas disclosed herein, including Formula (I) or a pharmaceutically
acceptable salt
thereof of the invention.
The compounds of any of the Formulas disclosed herein, or a pharmaceutically
acceptable salt thereof of the invention may be made by any number of
processes using
conventional organic syntheses as described in the Schemes below and more
specifically
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
illustrated by the exemplary compounds which follow in the Examples section
herein, or by
drawing on the knowledge of a skilled organic chemist. Suitable synthetic
routes are depicted
below in the following general reaction schemes.
The synthesis provided in these Schemes are applicable for producing compounds
of
the invention as defined by any of the Formulas disclosed herein, having a
variety of
different functional groups as defined employing appropriate precursors, which
are suitably
protected if needed, to achieve compatibility with the reactions outlined
herein. Subsequent
deprotection, where needed, affords compounds of the nature generally
disclosed. While
the Schemes are shown with compounds only as defined therein, they are
illustrative of
processes that may be used to make the compounds of the invention.
Intermediates (compounds used in the preparation of the compounds of the
invention) also may be present as salts. Thus, in reference to intermediates,
the phrase
"compound(s) of formula (number)" means a compound having that structural
formula or a
pharmaceutically acceptable salt thereof.
The compounds of the invention may be obtained by using the procedures
illustrated
in the Schemes below, or by applying appropriate synthetic organic chemistry
procedures
and methodology known to those of skill in the art.
The methods provided in these Schemes can be used to prepare compounds of the
invention containing a variety of different X1, R1, R27 R37 r,47
K R5, and R6 groups (descriptions
shown above for compounds of Formula (I)) employing appropriate precursors.
Those skilled in the art will appreciate that in the preparation of compounds
of the
invention (e.g., compounds of Formula (I) or a pharmaceutically acceptable
salt thereof), it
may be necessary and/or desirable to protect one or more sensitive groups in
the molecule
or the appropriate intermediate to prevent undesirable side reactions. The
skilled artisan will
appreciate that if a substituent described herein is not compatible with the
synthetic methods
described herein, the substituent may be protected with a suitable protecting
group that is
stable to the reaction conditions. The protecting group may be removed at a
suitable point in
the reaction sequence to provide a desired intermediate or target compound.
Suitable
protecting groups for use according to the present invention are well-known to
those skilled
in the art and may be used in a conventional manner. See for example,
"Protective Groups
in Organic Synthesis" by T.W. Green and P.G.M Wets (Wiley & Sons, 1991) or
"Protecting
Groups" by P. J. Kocienski (Georg Thieme Verlag, 1994). Subsequent
deprotection, where
needed, affords compounds of the nature generally disclosed.
In some instances, a substituent may be specifically selected to be reactive
under the
reaction conditions used. Under these circumstances, the reaction conditions
convert the
selected substituent into another substituent that is either useful as an
intermediate
compound or is a desired substituent in a target compound.
21
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
While the Schemes shown below are representative of methods for preparing
compounds of Formula (I), they are only intended to be illustrative of
processes that may be
used to make the compounds of the invention.
Compound names were generated using the software naming program ChemDraw
Ultra v12.0, available from Perkin Elmer, 940 Winter Street, Waltham,
Massachusetts,
02451, USA. (http://www.perkinelmer.com/).
Scheme 1
R CO2R CO2H Esterification R5 R3'-
NH2 Hydrolysis
R4X1 NH
R5
R5
1-2 1-3
1-4
The preparation of the compounds of the present invention typically begins
with the
synthesis of N-substituted-2-aminoaromatic acid derivatives 1-4 (Scheme I).
Esterification of
a suitably substituted 2-halo aromatic acid under standard conditions provides
the
corresponding ester 1-2. Typically, esterification reactions are performed
under either acidic
conditions, in the presence of an alcohol, or under basic conditions, in the
presence of a
suitable alkyl halide. Reaction of the 2-halo aromatic ester 1-2 (X2= Cl, Br
or I) with an
appropriate aniline or amine (R5'-NH2; R5' is a substituted phenyl group)
provides the
corresponding N-substituted-2-aminoaromatic esters 1-3. Typically, this
reaction is
performed at elevated temperature, using either standard heating or microwave
irradiation,
in the presence of a catalyst, for example Pd2(dba)3 or Cu/Cu , a suitable
ligand, for
instance BINAP or Xantphos, and an inorganic base, typically Cs2CO3 or K2CO3,
in an
appropriate solvent, such as 1,4-dioxane, toluene or 2-ethoxyethanol.
Saponification of the ester 1-3 to the corresponding N-substituted-2-
aminoaromatic
acid derivatives 1-4 is typically achieved under standard basic conditions,
using bases such
as Li0H, KOH, or NaOH, in a suitable solvent or solvent system, for instance
methanol/H20,
ethanol/H20, or THF/H20. Such conditions are well-known to those of skill in
the art.
22
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
Scheme II
R7
Me
R7 R7
H2N TN OMe
OMe
R2 0 0
11-3 R3
R3
2 /1", R2
R
NH R,
x. N
R5'
1-4 R5' R5'
11-1 11-
2
The intermediate N-substituted-2-aminoaromatic acid derivatives 1-4, prepared
as
illustrated in Scheme I, can be converted to compound 11-2 as outlined in
Scheme H.
Coupling of 1-4 with a substituted methoxy pyridyl amine 11-3 under various
amide coupling
conditions known to those of skill in the art, provides the corresponding
amide 11-1. For
example, one might employ standard coupling reagents, like EDC/HOBT, HATU,
HBTU or
T3P, in the presence of an amine base, like triethylamine, or Hunig's base
(diisopropylethylamine), in a suitable solvent, typically DMF, DMA or
acetonitrile.
Alternatively, one might convert the acid to the corresponding acid chloride,
using a reagent
such as thionyl chloride or oxalyl chloride, and the like, then react the acid
chloride with a
substituted methoxy pyridyl amine 11-3, in the presence of an acid scavenger
or base, such
as pyridine, 2,6-lutidine, triethylamine or Hunig's base, in an appropriate
solvent, such as
dichloromethane or pyridine, to afford the desired coupling product 11-1.
Formation of the dihydroquinazolinone ring system, as in intermediate 11-2,
involves
reaction of intermediate 11-1 with formaldehyde or a suitable equivalent. For
instance, the
reaction may be achieved using formaldehyde, either as gaseous formaldehyde,
paraformaldehyde, or s-trioxane, in the presence of an acid, preferably PTSA
or sulfuric
acid. Alternatively, the dihydroquinazolinone mg system can be formed via
reaction of 11-1
using diiodomethane or chloroiodomethane as a formaldehyde equivalent. In this
variant of
the cyclization reaction, a base, typically Cs2CO3 or NaH, is used, in a
suitable solvent,
oftentimes acetonitrile or DMF. The choice of using formaldehyde or
diiodomethane
depends on the particular reactivity characteristics of the intermediate 11-1.
23
CA 03202326 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
Scheme III
R7
R7
0
0 0
R3
N
R3,C 02 H Coupling, R3
N R5-1\11-12
R2
R2R X1 NH
R5'
111-2 11-
1
R7
0
0
"CH2=0
R2
11-2
In a variation of the methods described in Schemes I and II, the compounds of
the
present invention can be prepared as illustrated in Scheme III. Coupling of
intermediate III-1
(typically obtained from commercially available sources) with a substituted
methoxy pyridyl
amine 11-3 under various amide coupling conditions known to those of skill in
the art,
provides the corresponding amide 111-2. General conditions for forming amides
are
described in Scheme II. Subsequently, amide 111-2 can be reacted with an
appropriate
aniline or amine (R5'-NH2) under similar conditions as described for
conversion of 1-2 to 1-3 in
Scheme I to afford intermediate 11-1. Intermediate 11-1 can then be converted
to compound
11-2 according to the methods illustrated in Scheme II.
24
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
Scheme IV
R7
R7
R7
0 0 0
0 )Yo
N N NH ci N
N CI
R2
R2
R2 R4 N
R4X1N
R5'
11-2 1V-1
1V-2
R7
R7
0 0
0 ______________
OH
N _p R31 N N
R2 0P,
0
j R2 HO
R4 X1 N
R4X1N
R5' R5'
IV-3
As shown in Scheme IV, transformation of compound 11-2 to the pyridone IV-1
can be
achieved by reacting compound 11-2 with a mixture of TMS-chloride and Nal, or
a solution of
TMS-iodide, in a neutral solvent like acetonitrile, at elevated temperature.
Compound IV-1
can be reacted with chloromethyl chloroformate in the presence of an organic
base such as
DABCO in suitable solvents such as Et0Ac and DMF to provide the
chloromethylpyridone
IV-2. Reaction of compound IV-2 with potassium di-tert-butyl phosphate in the
presence of a
phase transfer catalyst such as TBAI in solvent DMF at elevated temperature
provides
compound IV-3. Removal of the tert-butyl protecting groups under acidic
conditions such as
acetic acid in acetonitrile and water provides the prodrug compounds of the
invention.
Pharmaceutical Compositions, Administration Routes, and Dosages
The compounds of the invention may be formulated into pharmaceutical
compositions prior to administration to a subject. According to one aspect,
the invention
provides a pharmaceutical composition comprising a compound of the invention
(Le. a
compound as defined by any of the Formulas disclosed herein, including Formula
(I) or a
pharmaceutically acceptable salt thereof of the invention) and one or more
pharmaceutically
acceptable excipients. According to one aspect, the invention provides a
pharmaceutical
composition comprising a compound of the invention (i.e. a compound as defined
by any of
the Formulas disclosed herein, including Formula (I) or a pharmaceutically
acceptable salt
thereof) and a pharmaceutically acceptable excipient.
In another aspect, the invention relates to a pharmaceutical composition or
formulation, which comprises: a compound of formula (I), or a pharmaceutically
acceptable
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
salt thereof of the invention; a pharmaceutically acceptable excipient(s); and
optionally one
or more other therapeutic ingredients.
The pharmaceutical compositions or formulations as defined herein typically
contain
one compound of the invention. However, in certain embodiments, the
pharmaceutical
compositions may contain more than one compound of the invention. In addition,
the
pharmaceutical compositions of the invention may optionally further comprise
one or more
additional pharmaceutically active compounds.
A pharmaceutically acceptable excipient is non-toxic and should not interfere
with the
efficacy of the active ingredient. Suitable pharmaceutically acceptable
excipients will vary
depending upon the particular dosage form chosen, route of administration,
etc. Suitable
pharmaceutically acceptable excipients include the following types of
excipients: diluents,
carriers, fillers, binders, disintegrants, lubricants, glidants, granulating
agents, coating
agents, wetting agents, solvents, co-solvents, suspending agents, emulsifiers,
sweeteners,
flavoring agents, flavor masking agents, coloring agents, anti-caking agents,
humectants,
chelating agents, plasticizers, viscosity increasing agents, antioxidants,
preservatives,
stabilizers, surfactants, and buffering agents. Examples of pharmaceutically
acceptable
excipients are described, e.g., in Remington's Pharmaceutical Sciences (Mack
Publishing
Company), The Handbook of Pharmaceutical Additives (Gower Publishing Limited),
and The
Handbook of Pharmaceutical Excigients (the American Pharmaceutical Association
and the
Pharmaceutical Press).
Pharmaceutical compositions may be adapted for administration by any
appropriate
or suitable route, for example by systemic administration (e.g., oral
administration, parenteral
administration, transdermal administration, rectal administration,
inhalation), topical
administration, etc. Parenteral administration is typically by injection or
infusion and includes
intravenous, intramuscular, and subcutaneous injection or infusion. Inhalation
refers to
administration into the patient's lungs whether inhaled through the mouth or
through the
nasal passages. Typically, administration is via the oral route or parenteral
route.
Pharmaceutical compositions adapted for oral administration may be presented
as
solid dosage forms such as tablets, capsules, caplets, troches, pills;
powders; or liquid
dosage forms such as solutions, suspensions, syrups, elixirs, or emulsion,
etc.
Pharmaceutical compositions adapted for parenteral administration (e.g.,
intravenous
administration) may be presented as solutions, suspensions, and powders for
reconstitution.
In general, pharmaceutical compositions of the invention are prepared using
conventional materials and techniques, such as mixing, blending and the like.
Some of the
methods commonly used in the art are described in Remington's Pharmaceutical
Sciences
(Mack Publishing Company).
26
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
Solid oral dosage forms, such as tablets and capsules can be prepared by
mixing a
compound of the invention with excipients such as diluents and fillers (e.g.,
starch, lactose,
sucrose, calcium carbonate, calcium phosphate and the like), binders (e.g.,
starch, acacia
gum, carboxymethyl cellulose, hydroxypropyl cellulose, crystalline cellulose,
and the like),
lubricants (e.g., magnesium stearate, talc and the like), and the like.
Pharmaceutical compositions adapted for parenteral administration can be an
injection solution prepared from powders, granules or tablets by mixing with a
carrier, such
as distilled water, saline and the like, and base and the like may be used for
pH adjustment.
In one embodiment, a pharmaceutical composition of the invention is formulated
for
oral administration.
In another embodiment, a pharmaceutical composition of the invention is
formulated
for parenteral administration, particularly intravenous administration.
The invention also provides a pharmaceutical composition comprising from 0.5
to
1,000 mg of a compound of the invention (i.e., a compound of any of the
Formulas disclosed
herein, including Formula (I) or a pharmaceutically acceptable salt thereof of
the invention)
and from 0.5 to 1,000 mg of a pharmaceutically acceptable excipient.
Compounds and pharmaceutical compositions of the invention as defined herein
may
be administered once or according to a dosing regimen, where a number of doses
are
administered at varying intervals of time for a given period of time. For
example, doses may
be administered one, two, three, or four times per day. Doses may be
administered until the
desired therapeutic effect is achieved or indefinitely to maintain the desired
therapeutic
effect. Doses of compounds of the invention may be in the range of 0.001 mg/kg
to 100
mg/kg, such as 0.001 mg/kg to 50 mg/kg. Preferably, the selected dose is
administered
orally or intravenously.
Methods, Uses, Compounds For Use in Manufacture and/or Treatment of
Diseases
In general, the invention also relates to uses of the compounds and/or
pharmaceutical compositions of the invention as defined herein for use as a
medicament or
for use in therapy.
Compounds of the invention as defined herein are inhibitors of voltage-gated
sodium
ion channels, and particularly the voltage-gated sodium ion channel Nav1.8.
The activity of
a compound utilized in this invention as an inhubitor of Nav1.8 may be assayed
according to
methods described generally in the Examples herein, or according to methods
available to
one of ordinary skill in the art.
27
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
Accordingly, in one aspect, the invention relates to uses of compounds and
pharmaceutical compositions of the invention as inhibitors of voltage-gated
sodium ion
channels, particularly Nav1.8.
In one embodiment, the invention relates to a method of inhibiting a voltage-
gated
sodium ion channel in a subject in need thereof, comprising administering to
the subject an
effective amount of a compound of the invention or a pharmaceutical
composition of the
invention as described herein. In one embodiment, the voltage-gated sodium
channel is
Nav1.8.
In embodiment, the invention relates to a compound of the invention or a
pharmaceutical composition of the invention for use in inhibiting a voltage-
gated sodium ion
channel. In one embodiment, the voltage-gated sodium channel is Nav1.8.
In one embodiment, the invention relates to use of a compound of the invention
or a
pharmaceutical composition of the invention in the manufacture of a medicament
for
inhibiting a voltage-gated sodium ion channel. In one embodiment, the voltage-
gated
sodium channel is Nav1.8.
Without wishing to be bound by any particular theory, the compounds and
compositions of the invention are particularly useful for treating a disease,
condition, or
disorder where activation or hyperactivity of Nav1.8 is implicated in the
disease, condition, or
disorder. When activation or hyperactivity of Nav1.8 is implicated in a
particular disease,
condition, or disorder, the disease, condition, or disorder may also be
referred to as a
"Nav1.8 -mediated disease, condition or disorder." Exemplary Nav1.8-mediated
diseases,
disorders, and conditions include pain and pain-associated diseases,
disorders, and
conditions, and cardiovascular diseases, disorders, and conditions such as
atrial fibrillation.
Thus, in another aspect, the invention relates to uses of compounds and
pharmaceutical compositions of the invention in methods and medicaments for
treating pain
or a pain-associated disease, disorder, or condition and/or for treating
cardiovascular
diseases, disorders, and conditions.
As used herein, "patient" or "subject" in need thereof refers to a human or
mammal.
The term "mammal" as used herein, encompasses any mammal. Examples of mammals
include, but are not limited to, cows, horses, sheep, pigs, cats, dogs, mice,
rats, rabbits,
guinea pigs, and non-human primates (NHPs), such as monkeys or apes, humans,
etc.
Suitably the subject being treated is a human.
As used herein, the terms "treat", "treating", and/or "treatment" used in
reference to a
disease, disorder, or condition mean to ameliorate or prevent the condition or
one or more
biological manifestations of the condition; to interfere with one or more
points in the
biological cascade that leads to or is responsible for the condition; to
alleviate one or more of
the symptoms or effects associated with the condition; to slow the progression
of the
28
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
condition or one or more of the biological manifestations of the condition; or
to lessen the
severity of the condition or one or more symptoms or effects associated with
the condition.
As mentioned above, "treatment" of a disease, disorder, or condition includes
prevention of
the condition. The skilled artisan will appreciate that "prevention" is not an
absolute term. In
medicine, "prevention" is understood to refer to the prophylactic
administration of a drug to
substantially diminish the likelihood or severity of a condition or biological
manifestation
thereof, or to delay the onset of such condition or biological manifestation
thereof.
As used herein, "effective amount" and "therapeutically effective amount" are
used
interchangeably. An effective amount in reference to a compound of the
invention means an
amount of the compound sufficient to treat the patient's condition, but low
enough to avoid
serious side effects (at a reasonable benefit/risk ratio) within the scope of
sound medical
judgment. An effective amount of a compound or pharmaceutically acceptable
salt thereof
and/or corresponding tautomer form thereof of the invention or corresponding
pharmaceutical composition thereof will vary according to factors, such as the
particular
compound chosen (e.g., consider the potency, efficacy, and half-life of the
compound); the
route of administration chosen; the condition being treated; the severity of
the condition
being treated; the age, size, weight, and physical condition of the patient or
subject being
treated; the medical history of the patient or subject being treated; the
duration of the
treatment; the nature of concurrent therapy; the desired therapeutic effect,
etc.
According to embodiments of the invention, a pain-associated disease, disorder
or
condition is pain caused by any one of a variety of diseases of varying
etiologies as
described throughout the present disclosure. In some embodiments, pain or a
pain-
associated disease, disorder, or condition is neuropathic pain, chronic pain,
acute pain,
nociceptive pain, inflammatory pain, musculoskeletal pain, visceral pain,
cancer pain,
idiopathic pain, multiple sclerosis, Charcot-Marie-Tooth syndrome, or
incontinence.
In some embodiments, pain or a pain-associated disease, disorder, or condition
is
acute pain.
In some embodiments, pain or a pain-associated disease, disorder, or condition
is
neuropathic pain or chronic neuropathic pain.
In some embodiments, pain or a pain-associated disease, disorder, or condition
is
neuropathic pain or chronic neuropathic pain selected from small fiber
neuropathy, small
fiber-mediated diabetic neuropathy, idiopathic small fiber neuropathy, painful
diabetic
neuropathy or polyneuropathy.
In some embodiments pain or a pain-associated disease, disorder, or condition
is
neuropathic pain selected from post-herpetic neuralgia, diabetic neuralgia,
painful HIV-
associated sensory neuropathy, trigeminal neuralgia, burning mouth syndrome,
post-
29
CA 03202326 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
amputation pain, phantom pain, painful neuroma, traumatic neuroma, Morton's
neuroma,
nerve entrapment injury, spinal stenosis, carpal tunnel syndrome, radicular
pain, sciatica
pain, nerve avulsion injury, brachial plexus avulsion, complex regional pain
syndrome, drug
therapy induced neuralgia, cancer chemotherapy induced neuralgia, anti-
retroviral therapy
induced neuralgia, post spinal cord injury pain, idiopathic small-fiber
neuropathy, idiopathic
sensory neuropathy or trigeminal autonomic cephalalgia.
In some embodiments, pain or a pain-associated disease, disorder, or condition
is
neuropathic pain or chronic neuropathic pain selected from diabetic peripheral
neuropathy,
pain caused by neuropathy, neurologic or neuronal injury, pain associated
nerve injury,
neuralgias and associated acute or chronic pain, post-herpetic neuralgia, pain
associated
root avulsions, painful traumatic mononeuropathy, painful polyneuropathy,
erythromelalgia,
paroxysmal extreme pain disorder (PEPD), burning mouth syndrome, central pain
syndromes caused by a lesion at a level of nervous system, traumatic nerve
injury, nerve
compression or entrapment, congenital insensitivity to pain (CIP),
dysmenorrheal, primary
erythromelalgia, HIV peripheral sensory neuropathy, pudendal neuralgia, spinal
nerve injury,
chronic inflammatory demyelinating polyneuropathy (CIDP), carpal tunnel
syndrome and
vasculitic neuropathy.
In some embodiments, pain or a pain-associated disease, disorder, or condition
is
visceral pain, wherein visceral pain is inflammatory bowel disease pain,
Crohn's disease
pain or interstitial cystitis pain.
In some embodiments, pain or a pain-associated disease, disorder, or condition
is
musculoskeletal pain, wherein musculoskeletal pain is osteoarthritis pain,
back pain, cold
pain, burn pain or dental pain.
In some embodiments, pain or a pain-associated disease, disorder, or condition
is
idiopathic pain, wherein idiopathic pain is fibromyalgia pain.
In some embodiments, pain or a pain-associated disease, disorder, or condition
is
chronic or acute pre-operative associated pain or chronic or acute post-
operative associated
pain. Post-operative associated pain includes ambulatory post-operative pain.
Ambulatory
surgery, also known as outpatient surgery, refers to same day surgery that
does not require
an overnight stay in a hospital or other medical facility. In some
embodiments, pre-operative
associated pain is selected from neuropathic pain or chronic neuropathic pain,
chronic
osteoarthritis pain, dental pain or inflammatory pain. In some embodiments,
post-operative
associated pain is selected from bunionectomy pain, hernia repair pair, breast
surgery pain
or cosmetic surgical pain.
In some embodiments, pain or a pain-associated disease, disorder, or condition
is
pain caused by trauma or iatrogenic medical or dental procedures. As used
herein, the term
"iatrogenic" refers to pain induced inadvertently by a medical or dental
personnel, such as
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
surgeon or dentist, during medical or dental treatment(s) or diagnostic
procedure(s), which
include, but are not limited to pain caused by pre-operative (i.e., "before"),
pen-operative
(i.e., "during" or medically induced pain during non-surgical or operative
treatment(s)) and
post-operative (Le., after, post-operative or surgical induced caused pain)
medical or dental
procedures.
In some embodiments, pain or a pain-associated disease, disorder, or condition
is
nociceptive pain, wherein nociceptive pain is post-surgical pain, cancer pain,
back and
craniofacial pain, osteoarthritis pain, dental pain or diabetic peripheral
neuropathy.
In some embodiments, pain or a pain-associated disease, disorder, or condition
is
inflammatory pain. Inflammatory pain can be pain of varied physiological
origins. In some
embodiments, inflammatory pain is selected from pain associated with
osteoarthritis,
rheumatoid arthritis, rheumatic disorder, teno-synovitis and gout, shoulder
tendonitis or
bursitis, gouty arthritis, and polymyalgia rheumatica, primary hyperalgesia,
secondary
hyperalgesia, primary allodynia, secondary allodynia, or other pain caused by
central
sensitization; complex regional pain syndrome, chronic arthritic pain and
related neuralgias
or acute pain. In some embodiments inflammatory pain is selected from pain
associated
with rheumatoid arthritis, osteoarthritis, rheumatoid spondylitis, gouty
arthritis or juvenile
arthritis. In some embodiments, inflammatory pain is selected from rheumatoid
arthritis;
rheumatoid spondylitis; gouty arthritis; juvenile arthritis; rheumatic
disorder; gout; shoulder
tendonitis or bursitis; polymyalgia rheumatica; primary hyperalgesia;
secondary
hyperalgesia; primary allodynia; secondary allodynia; or other pain caused by
central
sensitization, complex regional pain syndrome, chronic or acute arthritic pain
and related
neuralgias.
In some embodiments, inflammatory pain is selected from rheumatoid arthritis
pain or
vulvodynia.
In some embodiments, the inflammatory pain is selected from osteoarthritis,
chronic
osteoarthritis pain (e.g., hip or knee) or chronic inflammatory demyelinating
polyneuropathy.
In some embodiments pain or a pain-associated disease, disorder, or condition
is
musculoskeletal pain. In some embodiments, musculoskeletal pain is selected
from bone
and joint pain, osteoarthritis; lower back and neck pain; pain resulting from
physical trauma
or amputation. In some embodiments, musculoskeletal pain is selected from bone
and joint
pain, osteoarthritis (e.g., knee, hip), tendonitis (e.g., shoulder), bursitis
(e.g., shoulder)
tenosynovitis, lower back and neck pain, sprains, strains, or pain resulting
from physical
trauma or amputation.
In some embodiments, pain or a pain-associated disease, disorder, or condition
is
neurologic or neuronal injury associated or related pain disorders caused by
diseases
selected from neuropathy, pain associated nerve injury, pain associated root
avulsions,
31
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
painful traumatic mononeuropathy, painful polyneuropathy, erythromelalgia,
paroxysmal
extreme pain disorder (PEPD), burning mouth syndrome; central pain syndromes
caused by
a lesion at a level of nervous system); traumatic nerve injury, nerve
compression or
entrapment, congenital insensitivity to pain (CIP), dysmenorrheal, primary
erythromelalgia;
HIV peripheral sensory neuropathy; pudendal neuralgia, spinal nerve injury,
chronic
inflammatory demyelinating polyneuropathy (CIDP), carpal tunnel syndrome or
vasculitic
neuropathy.
In some embodiments, pain or a pain-associated disease, disorder, or condition
is
pain caused by trauma, or pain caused by iatrogenic, medical, or dental
procedures.
In some embodiments, pain or a pain-associated disease, disorder, or condition
is
myofascial pain; myositis or muscle inflammation; repetitive motion pain;
complex regional
pain syndrome; sympathetically maintained pain; cancer, toxins and
chemotherapy related
pain; postsurgical pain syndromes and/or associated phantom limb pain; post-
operative
medical or dental procedures or treatments pain; pain associated with HIV or
pain induced
by HIV treatment.
In some embodiments, pain or a pain-associated disease, disorder, or condition
is
neuropathic pain or other pain-associated disease, disorder, or condition
selected from
peripheral neuropathic pain, central neuropathic pain, inherited
erythromelalgia (IEM), small
fiber neuralgia (SFN), paroxysmal extreme pain disorder (PEPD), painful
diabetic
neuropathy, chronic lower back pain, neuropathic back pain, sciatica, non-
specific lower
back pain, multiple sclerosis pain, HIV-related neuropathy, post-herpetic
neuralgia,
trigeminal neuralgia, vulvodynia, pain resulting from physical trauma, post-
limb amputation
pain, neuroma pain, phantom limb pain, cancer, toxins, or chronic inflammatory
conditions.
In some embodiments, pain or a pain-associated disease, disorder, or condition
is
acute pain, chronic pain, neuropathic pain, inflammatory pain, arthritis,
migraine, cluster
headaches, trigeminal neuralgia, herpetic neuralgia, general neuralgias,
epilepsy, epilepsy
conditions, neurodegenerative disorders, psychiatric disorders, anxiety,
depression, dipolar
disorder, myotonia, arrhythmia, movement disorders, neuroendocrine disorders,
ataxia,
multiple sclerosis, irritable bowel syndrome, incontinence, visceral pain,
osteoarthritis pain,
postherpetic neuralgia, diabetic neuropathy, radicular pain, sciatica, back
pain, head pain,
neck pain, severe pain, intractable pain, nociceptive pain, breakthrough pain,
postsurgical
pain, cancer pain, stroke, cerebral ischemia, traumatic brain injury,
amyotrophic lateral
sclerosis, stress induced angina, exercise induced angina, palpitations,
hypertension, or
abnormal gastro-intestinal motility.
In some embodiments, pain or a pain-associated disease, disorder, or condition
is
femur cancer pain; non-malignant chronic bone pain; rheumatoid arthritis;
osteoarthritis;
spinal stenosis; neuropathic low back pain; myofascial pain syndrome;
fibromyalgia;
32
CA 03202326 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
temporomandibular joint pain; chronic visceral pain, abdominal pain;
pancreatic pain; IBS
pain; chronic and acute headache pain; migraine; tension headache, including,
cluster
headaches; chronic and acute neuropathic pain, post-herpetic neuralgia;
diabetic
neuropathy; HIV-associated neuropathy; trigerninal neuralgia; Charcot-Marie
Tooth
neuropathy; hereditary sensory neuropathies; 'peripheral nerve injury; painful
neuromas;
ectopic proximal and distal discharges; radiculopathy; chemotherapy induced
neuropathic
pain; radiotherapy-induced neuropathic pain; post-mastectomy pain; central
pain; spinal cord
injury pain; post-stroke pain; thalamic pain; complex regional pain syndrome;
phantom pain;
intractable pain; acute pain, acute post-operative pain; acute musculoskeletal
pain; joint
pain; mechanical low back pain; neck pain; tendonitis; injury/exercise pain;
acute visceral
pain; pyelonephritis; appendicitis; cholecystitis; intestinal obstruction;
hernias; chest pain,
cardiac pain; pelvic pain, renal colic pain, acute obstetric pain, labor pain;
cesarean section
pain; acute inflammatory, burn and trauma pain; acute intermittent pain,
endometriosis;
acute herpes zoster pain; sickle cell anemia; acute pancreatitis; breakthrough
pain; orofacial
pain including sinusitis pain, dental pain; multiple sclerosis (MS) pain; pain
in depression;
leprosy pain; Behcet's disease pain; adiposis clolorosa; phlebitic pain;
Guillain-Barre pain;
painful legs and moving toes; Haglund syndrome; erythromelalgia pain; Fabry's
disease
pain; bladder and urogenital disease, including, urinary incontinence;
hyperactivity bladder;
painful bladder syndrome; interstitial cyctitis (IC); prostatitis; complex
regional pain syndrome
(CRPS), type I and type II; widespread pain, paroxysmal extreme pain,
pruritis, tinnitis, or
angina-induced pain.
In another aspect, the invention relates to uses of compounds and
pharmaceutical
compositions of the invention in methods and medicaments for treating
cardiovascular
diseases, disorders and conditions, including atrial fibrillation and cardiac
arrhythmias.
In some embodiments, the cardiovascular disease is atrial fibrillation that is
either
idiopathic in nature or caused by a disease as defined herein. Atrial
fibrillation can be
paroxysmal atrial fibrillation, sustained atrial fibrillation, long-standing
atrial fibrillation, atrial
fibrillation with heart failure, atrial fibrillation with cardiac valve
disease, or atrial fibrillation
with chronic kidney disease. In particular embodiments, atrial fibrillation is
selected from
paroxysmal, sustained, or long-standing atrial fibrillation.
In some embodiments, the cardiovascular disease includes cardiac arrhythmias.
In one aspect, the invention relates to a method of treatment of pain or a
pain-
associated disease, disorder, or condition as defined herein in a subject in
need thereof,
comprising administering to the subject a therapeutically effective amount of
a compound of
the invention or a pharmaceutical composition of the invention as described
herein.
In an embodiment, provided is a method of treatment of acute pain or chronic
pain in
a subject in need thereof, comprising administering to the subject a
therapeutically effective
33
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
amount of a compound of the invention or a pharmaceutical composition of the
invention as
described herein.
In an embodiment, provided is a method of treatment of acute pain in a subject
in
need thereof, comprising administering to the subject a therapeutically
effective amount of a
compound of the invention or a pharmaceutical composition of the invention as
described
herein.
In an embodiment, provided is a method of treatment of pain caused by trauma;
pain
caused by iatrogenic medical or dental procedures; or pre-operative or post-
operative
associated pain in a subject in need thereof, comprising administering to the
subject a
therapeutically effective amount of a compound of the invention or a
pharmaceutical
composition of the invention as described herein.
In an embodiment, provided is a method of treatment of neuropathic pain,
nociceptive pain, inflammatory pain, musculoskeletal pain, visceral pain, or
idiopathic pain in
a subject in need thereof, comprising administering to the subject a
therapeutically effective
amount of a compound of the invention or a pharmaceutical composition of the
invention as
described herein.
In an embodiment, provided is a method of treatment of neuropathic pain or
chronic
neuropathic pain selected from small fiber neuropathy, small fiber-mediated
diabetic
neuropathy, idiopathic small fiber neuropathy, painful diabetic neuropathy or
polyneuropathy
in a subject in need thereof, comprising administering to the subject a
therapeutically
effective amount of a compound of the invention or a pharmaceutical
composition of the
invention as described herein.
In an embodiment, provided is a method of treatment of inflammatory pain
selected
from osteoarthritis, chronic osteoarthritis pain, or chronic inflammatory
demyelinating
polyneuropathy in a subject in need thereof, comprising administering to the
subject a
therapeutically effective amount of a compound of the invention or a
pharmaceutical
composition of the invention as described herein.
In one aspect, the invention relates to a method of treatment of atrial
fibrillation as
defined herein in a subject in need thereof, comprising administering to the
subject a
therapeutically effective amount of a compound of the invention or a
pharmaceutical
composition of the invention as described herein.
In one embodiment, provided is a method of treatment of atrial fibrillation,
wherein
the atrial fibrillation is paroxysmal atrial fibrillation, sustained atrial
fibrillation, long-standing
atrial fibrillation, atrial fibrillation with heart failure, atrial
fibrillation with cardiac valve disease,
or atrial fibrillation with chronic kidney disease.
34
CA 03202326 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
In another aspect, the invention provides compounds of the invention and
pharmaceutical compositions of the invention as described herein for use in
treatment of
pain or a pain-associated disease, disorder, or condition as defined herein.
In an embodiment, provided is a compound of the invention or pharmaceutical
composition of the invention for use in treatment of acute pain or chronic
pain.
In an embodiment, provided is a compound of the invention or pharmaceutical
composition of the invention for use in treatment of acute pain.
In an embodiment, provided is a compound of the invention or pharmaceutical
composition of the invention for use in treatment of pain caused by trauma;
pain caused by
iatrogenic medical or dental procedures; or pre-operative or post-operative
associated pain.
In an embodiment, provided is a compound of the invention or pharmaceutical
composition of the invention for use in treatment of neuropathic pain,
nociceptive pain,
inflammatory pain, musculoskeletal pain, visceral pain, or idiopathic pain.
In an embodiment, provided is a compound of the invention or pharmaceutical
composition of the invention for use in treatment of neuropathic pain or
chronic neuropathic
pain selected from small fiber neuropathy, small fiber-mediated diabetic
neuropathy,
idiopathic small fiber neuropathy, painful diabetic neuropathy or
polyneuropathy.
In an embodiment, provided is a compound of the invention or pharmaceutical
composition of the invention for use in treatment of inflammatory pain
selected from
osteoarthritis, chronic osteoarthritis pain, or chronic inflammatory
demyelinating
polyneuropathy.
In another aspect, the invention relates to a compound of the invention or a
pharmaceutical composition of the invention for use in treatment of atrial
fibrillation.
In one embodiment, provided is a compound of the invention or pharmaceutical
composition of the invention for use in treatment of atrial fibrillation,
wherein the atrial
fibrillation is paroxysmal atrial fibrillation, sustained atrial fibrillation,
long-standing atrial
fibrillation, atrial fibrillation with heart failure, atrial fibrillation with
cardiac valve disease, or
atrial fibrillation with chronic kidney disease.
In another aspect, the invention also provides uses of compounds of the
invention or
pharmaceutical compositions of the invention as described herein in the
manufacture of a
medicament for treatment of pain and pain associated diseases, disorders, and
conditions
as described herein.
In an embodiment, provided is use of a compound of the invention or
pharmaceutical
composition of the invention in the manufacture of a medicament for treatment
of acute pain
or chronic pain.
In an embodiment, provided is use of a compound of the invention or
pharmaceutical
composition of the invention in the manufacture of a medicament for treatment
of acute pain.
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
In an embodiment, provided is use of a compound of the invention or
pharmaceutical
composition of the invention in the manufacture of a medicament for treatment
of pain
caused by trauma; pain caused by iatrogenic medical or dental procedures; or
pre-operative
or post-operative associated pain.
In an embodiment, provided is use of a compound of the invention or
pharmaceutical
composition of the invention in the manufacture of a medicament for treatment
of
neuropathic pain, nociceptive pain, inflammatory pain, musculoskeletal pain,
visceral pain, or
idiopathic pain.
In an embodiment, provided is use of a compound of the invention or
pharmaceutical
composition of the invention in the manufacture of a medicament for treatment
of
neuropathic pain or chronic neuropathic pain selected from small fiber
neuropathy, small
fiber-mediated diabetic neuropathy, idiopathic small fiber neuropathy, painful
diabetic
neuropathy or polyneuropathy.
In an embodiment, provided is use of a compound of the invention or
pharmaceutical
composition of the invention in the manufacture of a medicament for treatment
of
inflammatory pain selected from osteoarthritis, chronic osteoarthritis pain,
or chronic
inflammatory demyelinating polyneuropathy.
In another aspect, the invention also provides uses of compounds of the
invention or
pharmaceutical compositions of the invention as described herein in the
manufacture of a
medicament for treatment of atrial fibrillation.
In an embodiment, provided is use of a compound of the invention or
pharmaceutical
composition of the invention in the manufacture of a medicament for treatment
of atrial
fibrillation, wherein the atrial fibrillation is paroxysmal atrial
fibrillation, sustained atrial
fibrillation, long-standing atrial fibrillation, atrial fibrillation with
heart failure, atrial fibrillation
with cardiac valve disease, or atrial fibrillation with chronic kidney
disease.
In another aspect, the invention relates to a compound of the invention or a
pharmaceutical composition of the invention as described herein for use in
therapy.
Combination Therapies and Uses for Therapy
Compounds and pharmaceutical compositions of the invention as described herein
can be used in combination with one or more additional therapeutic agents.
Such additional
therapeutic agents can be administered concurrently with, prior to, or
subsequent to
treatment with a compound or pharmaceutical composition of the invention as
described
herein.
In the context of this specification, the term "concurrently" when referring
to
simultaneous administration of compounds or therapeutic agents means at the
same time,
as would be the case, for example in embodiments where a compound and
additional
36
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
therapeutic agent(s) are combined in a single preparation, or when a compound
and
additional therapeutic agent(s) are administered separately but taken within a
short duration
or period of time.
In light of the foregoing, the invention also relates to a combination
therapy, which
may be a comprised of a simultaneous or co-administration, or serial
administration of a
combination of compounds or pharmaceutical compositions of the invention with
one or
more additional therapeutic agents. Such combination therapy can be used for
treatment of
pain or any pain-associated disease, disorder, or condition, or a
cardiovascular disease,
disorder, or condition as defined throughout the present specification.
Therapeutic agents suitable for use in combination with the compounds and
pharmaceutical compositions of the invention include, but are not limited to:
Acetaminophen,
Acetylsalicylic acid, Nav1.7 Inhibitors, Nav1.9 Inhibitors, anti-depressants
(i.e. such as, but
not limited to duloxetine or amitriptyline), anti-convulsants (i.e. such as,
but not limited to
pregabalin and gabapentin), opiates (i.e., such as, but not limited to
hydrocodone, codeine,
morphine, oxycodone, oxymorphone, fentanyl, and the like), etc.; and where
administration
of the above, respectively, also is determined by one of ordinary skill in the
art. In one
aspect, suitable Nav1.7 Inhibitors or Nav1.9 Inhibitors for use in the
invention, include, but
are not limited to those Nav1.7 Inhibitors or Nav1.9 Inhibitors known in the
chemical
literature.
Each component of a combination used for therapeutic purposes (e.g., compound
or
pharmaceutical composition of the invention and additional therapeutic agent)
may be
administered orally, intravenously or parenterally or in combinations thereof.
Each
component of a therapeutic combination may be, but is not limited to being
administered by
simultaneous administration, co-administration, or serial administration;
and/or by identical or
different routes of administration or combinations of administration routes.
In certain
embodiments, each identical or different route of administration or
combinations of
administration routes is selected from oral, intravenous or parenteral
administration.
EXAMPLES
The following examples illustrate the invention. These examples are not
intended to
limit the scope of the present invention, but rather to provide guidance to
the skilled artisan
to prepare and use the compounds, compositions, and methods of the present
invention.
While particular aspects or embodiments of the present invention are
described, the
skilled artisan will appreciate that various changes and modifications can be
made without
departing from the spirit and scope of the invention.
37
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
Synthesis Examples
It will be understood by the skilled artisan that purification methods (using
acidic or
basic modifiers) or compound workup procedures (using acidic or basic
conditions) may
result in formation of a salt of a title compound (for example, hydrobromic
acid, formic acid,
hydrochloric acid, trifluoroacetic acid, or ammonia salts of a title
compound). The invention
is intended to encompass such salts.
Final compounds were characterized with GCMS and LCMS (conditions listed
below)
and NMR. 1H NMR or 19FNMR spectra were recorded using a Bruker Avance III 500
MHz
spectrometer, Bruker Avance 400 MHz spectrometer and Varian Mercury Plus-300
MHz
spectrometer. CDCI3 is deuteriochloroform, DIVISO-d6 is
hexadeuteriodimethylsulfoxide, and
CD3OD is tetradeuteriomethanol. Chemical shifts are reported in parts per
million (ppm)
downfield from the internal standard tetramethylsilane (TMS) or the NMR
solvent.
Abbreviations for NMR data are as follows: s = singlet, d = doublet, t =
triplet, q = quartet, m
= multiplet, dd = doublet of doublets, dt = doublet of triplets, app =
apparent, br = broad. J
indicates the NMR coupling constant measured in Hertz.
Analytical methods:
1) LCMS Method: Acquity UPLC with Waters Acquity QDa mass detector using
electrospray positive [ES+ve to give M+H+] equipped with a CSH C18 column
(30mm x
2.1mm, i.d. 1.7pm packing diameter) at 45 C eluting with 0.1 % TFA in water
(solvent A)
and 0.1 % TFA in acetonitrile (solvent B), using the following elution
gradient: 1-100 %
(solvent B) over 1.85 min at a flow rate of 1.3 ml/min.
2) LCMS Method: Acquity UPLC with Waters Acquity QDa mass detector using
electrospray positive [ES+ve to give M+H+] equipped with a CSH C18 column
(30mm x
2.1mm, i.d. 1.7pm packing diameter) at 45 C eluting with formic acid in Water
(solvent A)
and formic acid in acetonitrile (solvent B), using the following elution
gradient: 1-100 %
(solvent B) over 1.85 min at a flow rate of 1.3 ml/min.
3) LCMS Method: Acquity UPLC with Waters Acquity QDa mass detector using
electrospray positive [ES+ve to give M+H ] equipped with a CSH C18 column
(30mm x
2.1mm, i.d. 1.7pm packing diameter) at 45 C eluting with 10 mM ammonium
bicarbonate in
water adjusted to pH = 10 with 25% ammonium hydroxide solution (solvent A) and
acetonitrile (solvent B), using the following elution gradient: 1-100 %
(solvent B) over 1.85
min at a flow rate of 1.3 ml/min.
38
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
4) LCMS method: Agilent 1290 Infinity II LC system with Agilent MSD 61256/6130
using multi mode (ESI and APCI +ve and ¨ve) equipped with a Sunfire 018 column
(30mm x
2.1mm, i.d. 3.5pm packing diameter) at 25 C eluting with 0.1 % formic acid in
water (solvent
A) and 0.1 % formic acid in acetonitrile (solvent B), using the following
elution gradient: 0-
100 % (solvent B) over 3.1 min and holding at 100 % for 0.8 min at a flow rate
of 1.0 ml/mm.
5) LCMS method: Agilent 1290 Infinity II LC system with Agilent MSD 6125B/6130
using multi mode (ESI and APCI +ve and ¨ve) equipped with a Atlantis dC18
column (50mm
x 4.6mm, i.d. 5.0pm packing diameter) at 25 C eluting with 0.1% TFA in water
(solvent A)
and methanol (solvent B), using the following elution gradient: 5-95 %
(solvent B) over 5.0
min and holding at 95 % for 1.5 min at a flow rate of 1.0 ml/min.
6) LCMS method: Agilent 1290 Infinity II LC system with Agilent MSD 6125B/6130
using multi mode (ESI and APCI+ve and -ve) equipped with a Zorbax XDB 018
column
(50mm x 4.6mm, i.d. 3.5pm packing diameter) at 2500 eluting with 10 mM
ammonium
acetate in water (solvent A) and acetonitrile (solvent B), using the following
elution gradient:
Solvent B: 10-95 % (solvent B) over 3.5 min and holding at 95 % for 1.0 min at
a flow rate of
1.0 ml/min.
7) LCMS method: Agilent 1290 Infinity II LC system with Agilent MSD 61256/6130
using multi mode (ESI and APCI+ve and -ve) equipped with a Xbridge 08 column
(50mm x
4.6mm, i.d. 3.5pm packing diameter) at 25 C eluting with 10 mM ammonium
bicarbonate in
water (solvent A) and acetonitrile (solvent B), using the following elution
gradient: 10-95 %
(solvent B) over 4.0 min and holding at 95 % for 1.0 min at a flow rate of 1.0
ml/min.
8) GCMS Method: Agilent 7890B GC system with Agilent MSD 5977B using El
equipped with a HP-5 column (30 m x 0.32mm, 0.25pm film thickness) at 250 C
eluting with
helium at a flow rate of 2 mL/min and 10 min run time under the following
chromatographic
run conditions: 120 C for 1 min, 40 C/min up to 300 C, hold for 4.5 min.
In the following experimental descriptions, the following abbreviations may be
used:
Abbreviation Meaning
AcOH acetic acid
aq. aqueous
BBr3 boron tribromide
39
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
BCI3 boron trichloride
BH3 borane
BINAP 2,2'-bis(diphenylphosphino)-1,1'-binaphthalene
Bn benzyl
brine saturated aqueous sodium chloride
BuLi or nBuLi butyllithium
CDI carbonyldiimidazole
0H2012 methylene chloride
CH3CN acetonitrile
COCl2 oxalyl chloride
Cs2CO3 cesium carbonate
DABCO 1,4-diazabicyclo[2.2. 2]octane
DCC dicyclohexylcarbodiimide
DCM or CH2Cl2 methylene chloride
DEAD diethyl azodicarboxylate
DEAF diethyl aminopyridine
DIAD diisopropyl azodicarboxylate
DIPEA, DIEA,
N,N-diisopropylethylamine
Hunig's base
DMA Dimethylacetamide
DMAP 4-dimethylaminopyridine
DMF N,N-dimethylformamide
DME dimethoxyethane
DMSO dimethylsulfoxide
EDC 1-[3-(dimethylamino)propyI]-3-ethylcarbodiimide
hydrochloride
Et ethyl
Et3N triethylamine
Et20 diethyl ether
Et0Ac ethyl acetate
Et0H ethanol
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
Fmoc or fmoc fluorenylmethyloxycarbonyl
g, G, gm, GM gram
GCMS gas chromatography-mass spectroscopy
h or hr hour(s)
H2 hydrogen
H202 hydrogen peroxide
H20 water
H2SO4 sulfuric acid
(0-(7-azabenzotriazol-1-y1)-N,N,N',N'-tetramethyluronium
HATU
hexafluorophosphate)
2-(1H-benzo[d][1,2,3]triazol-1-y1)-1,1,3,3-
HBTU
tetramethylisouronium hexafluorophosphate(V)
HCI hydrochloric acid
HCO2H formic acid
HOBt or HOBT 1-hydroxybenzotriazole
HPLC high performance liquid chromatography
12 Iodine
JLR jacketed lab reactor
K2003 potassium carbonate
KHSO4 potassium hydrogen sulfate
KOAc potassium acetate
L or I liter
LAH lithium aluminum hydride
LCMS liquid chromatography-mass spectroscopy
LDA lithium diisopropyl amide
LED light-emitting diode
LiOH lithium hydroxide
LHMDS lithium bis(trimethylsilyl)amide
mCPBA meta-chloroperoxybenzoic acid
MDAP mass directed autopurification
Me methyl
41
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
Me0H methanol
mg, MG milligram
MgBr2 magnesium bromide
MgSO4 magnesium sulfate
Min or mins minute(s)
ml or mL or ML milliliter
mmol millimole
Mn02 manganese dioxide
Mol, mol mole
MS mass spectrum
MTBE methyl tert-butyl ether
pw microwave
N2 nitrogen
Na(CN)BH3 sodium cyanoborohydride
NaC1 sodium chloride
Na2CO3 sodium carbonate
NaHCO3 sodium bicarbonate
NaHMDS sodium bis(trimethylsilyl)amide
NaHS03 sodium bisulfite
NaH sodium hydride
Nal sodium iodide
NaOH sodium hydroxide
Na2S03 sodium sulfite
Na2SO4 sodium sulfate
NH4C1 ammonium chloride
HCO2-NH4 ammonium formate
NH4OH ammonium hydroxide
nm nanometer
NMO 4-methylmorpholine N-oxide
NMP N-methyl-2-pyrrolidone
42
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
Pd/C palladium on carbon
PdC12(dbpf) 1 ,l'-bis(di-tert-butylphosphino)ferrocene
dichloropalladium
Pd(dppf)0I2/ [1,1'-bis(diphenylphosphino)ferrocene]
PdC12(dppf) dichloropalladium(II)
PdC12(dppf)- [1,1-bis(diphenylphosphino)ferrocene]
0H2012 adduct dichloropalladium(II), complex with
dichloromethane
Pd2 (dba) 3 tris(dibenzylideneacetone)dipalladium(0)
Pd(Ph3)4,
tetrakis(triphenylphosphine)palladium(0)
tetra kis
Pd0Ac2 or
palladium acetate
Pd(OAc)2
Pd(OH)2 palladium hydroxide
Pt FA [Bis(trifluoroacetoxy)iodo]benzene
Ph phenyl
PL HCO3 MP macroporus polystyrene supported carbonate
POCI3 phosphoryl chloride
psi Pounds per square inch
PTFE polytetrafluoroethylene
PTSOH or
PTSA or p-Toluenesulfonic acid
pTs0H
rt or RT room temperature
sat. saturated
SFC supercritical fluid chromatography
Si silica
Si SPE silica gel cartridges
5i02 silica gel
SPE solid phase extraction
T3P0 propylphosphonic anhydride
tBu or t-Bu tert-butyl group
TBAB tetrabutylammonium bromide
TBAF tetrabutylammonium fluoride
43
CA 03202326 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
TBAI tetrabutylammonium iodide
TBDMSCI tert-butyldimethylsilyl chloride
TBME tert-butylmethyl ether
2-(1H-benzotriazole-1-yI)-1,1,3,3-tetramethyluronium
TBTU
tetrafluoroborate
TEA triethylamine
TFA trifluoroacetic acid
THF tetrahydrofuran
TiCI4 titanium tetrachloride
TMS-Br or
trimethylsilyl bromide
TMSBr
TMS-CI or
trimethylsilyl chloride
TMSCI
TMSI lodotrimethylsilane or trimethylsilyl iodide
TMS-0Tf
trimethylsilyl triflate
or TMSOff
tR retention time
UPLC ultra performance liquid chromatography
Xantphos 4,5-Bis(diphenylphosphino)-9,9-dimethylxanthene
Xphos 2-Dicyclohexylphosphino-2',4',6'-
thisopropylbiphenyl
Intermediate 1
Ethyl 2-bromo-4-(trifluoromethyl)benzoate
0
CF? -Br
To a stirring solution of 2-bromo-4-(trifluoromethyl)benzoic acid (10.0 g,
37.2 mmol)
in DMF (100 mL) was added K2003 (5.65 g, 40.9 mmol) followed by ethyl iodide
(3.60 mL,
44.6 mmol) dropwise under N2 at 25 'C. The reaction mixture was stirred at the
same
temperature for 3 hours. Water (150 mL) was added and the reaction was
extracted with
Et0Ac (2 x 250 mL). The combined organic extracts were washed with brine (150
mL),
dried over Na2SO4, filtered, and concentrated. The residue was purified by
flash column
chromatography (Biotage, 100 g SNAP column, 10% Et0Ac/petroleum ether over 40
44
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
minutes) to give the title compound as a colorless oil (9.3 g, 31.3 mmol, 84 %
yield). GCMS
(m/z) 296.0 (M).
Intermediate 2
2-Ohloro-5-(trifluoromethyl)nicotinoyl chloride
F3CU0
CI
NCI
To a stirred solution of 2-chloro-5-(trifluoromethyl)nicotinic acid (50 g, 222
mmol) in
DCM (500 mL) were added oxalyl chloride (23.28 mL, 266 mmol) and DMF (1.716
mL, 22.17
mmol) at 0 C and the reaction mixture was stirred for 1 hour at RT. The
reaction mixture
was concentrated under reduced pressure to dryness under N2 to yield the title
compound
as a brown gum (52 g, 213 mmol, 96 % yield).
Intermediate 3
2-Chloro-N-(6-methoxy-2-methylpyridin-3-yI)-5-(trifluoromethyl)nicotinamide
0
I
F3C N N
I H
N Cl
A mixture of 6-methoxy-2-methylpyridin-3-amine (32.4 g, 234 mmol) and TEA (89
mL, 639 mmol) in DCM (300 mL) was added to a stirred solution of 2-chloro-5-
(trifluoromethyl)nicotinoyl chloride (52 g, 213 mmol) in DCM (300 mL) at 0 C
and the
reaction mixture was stirred for 2 hours. The reaction mixture was quenched
with ice-cold
water (500 mL) and extracted with DCM (3 x 500 mL). The combined organic
phases were
washed with water (500 mL) and brine (500 mL), dried over Na2SO4 and
concentrated in
vacuo. The crude product was purified by column chromatography (Biotage, 340 g
SiO2
column, 0-30% Et0Acipetroleum ether over 40 minutes) to give the title
compound as a
brown solid (37 g, 106 mmol, 49.9 % yield). MS (m/z) 346.0 (M+H+).
Intermediate 4
Methyl 2-((4-fluoro-2-methylphenyl)amino)-5-(trifluoromethyl)benzoate
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
0
F3C
0.--
NH
To a solution of methyl 2-bromo-5-(trifluoromethyl)benzoate (2 g, 7.1 mmol)
and 4-
fluoro-2-methylaniline (1.06 g, 8.48 mmol) in 1,4-dioxane (20 mL) under
nitrogen at room
temperature was added cesium carbonate (4.60 g, 14.13 mmol) and BINAP (0.44 g,
0.71
mmol) in a one charge. The reaction mixture was purged with nitrogen for 10
min, then
Pd2(dba)3 (0.324 g, 0.353 mmol) was added into the reaction mixture. The
reaction mixture
was stirred at 100 C for 16 h. The reaction mixture was cooled to RT and
filtered through a
Celite pad and the filtrate was concentrated onto SiO2. Purification by flash
chromatography on Si02 (25g) with 0-30% Et0Ac in petroleum ether as eluant
afforded the
title compound as a colorless solid (2.3 g, 7.0 mmol, 99 % yield). MS (m/z)
328.0 (M+H+).
Intermediates 5-6 were prepared from the indicated aryl halogen and aniline by
methods analogous to those described for Intermediate 4.
Aryl
Int. Name Structure Characterization
Aniline
halogen
ethyl 2-((4-
ethyl 2-
fluoro-2- =0 bromo-4- 4-
fluoro-2-
5 methylphenyl)a MS (m/z) 342.0
CF3 NH
(trifluoromet methylanili
mino)-4- (M+H')
hyl)benzoat
ne
(trifluoromethyl)
40
benzoate
methyl 5-chloro- methyl 2-
-f1uoro-2-
6 2-((4-fluoro-2- MS (m/z) 294.2 bromo-5-
methylanili
methylphenyl)a (M +H*) chlorobenzo
mino)benzoate
OP ate ne
46
CA 03202326 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
Intermediate 7
2-((4-Fluoro-2-methylphenyl)amino)-5-(trifluoromethyl)benzoic acid
0
F3C
OH
NH
To a solution of methyl 2-((4-fluoro-2-methylphenyl)amino)-5-
(trifluoromethyl)benzoate (2.3 g, 7.03 mmol in THF (20 mL) under N2was added
LiOH (1.68
g, 70.3 mmol). The reaction mixture was stirred at 80 C for 4 h. The reaction
mixture was
cooled to rt and filtrate was concentrated under vacuum. Crude material was
extracted with
100 mL DCM and washed with 50 mL water. Aqueous layer was acidified with 1.5 N
HCI 20
mL and extracted with DCM (100 mL) twice. Combined organic layer was dried
over sodium
sulphate, filtered and concentrated under vacuum to afford the title compound
as a yellow
solid (2.1 g, 6.7 mmol, 95 % yield). MS (m/z) 314.0 (M+H+).
Intermediates 8-9 were prepared from the indicated ester by methods analogous
to
those described for Intermediate 7.
Int. Name Structure Characterization Ester
2-((4-fluoro-2- OH
ethyl 2-((4-fluoro-2-
8 F3C
methylphenyl)amino)- NH MS (m/z) 311.9 (M-
methylphenyl)amino)-
4- 4- (trifluoromethyl)benzoi (trifluoromethyl)benzo
c acid ate
= '
CI
40 OH 5-chloro-2-((4-fluoro-2-
MS 011/Z) 280.0 methyl 5-
chloro-2-((4-
fluoro-2-
9 methylphenyl)amino)b
enzoic acid
411111 (M+H). methylphenyl)amino)
benzoate
Intermediate 10
2-((4-Fluoro-2-methylphenyl)amino)-N-(6-methoxy-2-methylpyridin-3-y1)-5-
(trifluoromethyl)benzamide
47
CA 03202326 2023- 6- 14
WO 2022/129283 PCT/EP2021/086101
0
F3C
Nk
NH
To a solution of 2-((4-fluoro-2-methylphenyl)amino)-5-(trifluoromethyl)benzoic
acid
(2.1 g, 6.7 mmol) , DIPEA (2.34 mL, 13.4 mmcd) and HATU (3.82 g, 10.1 mmol) in
DMF (20
mL) under nitrogen at RT was added 6-methoxy-2-methylpyridin-3-amine (1.02 g,
7.4 mmol)
dropwise over 1 min. The reaction mixture was stirred at RT for 12h. The
reaction mixture
was quenched with ice cold water (100 mL) and resulting solid was filtered and
dried under
vacuum to afford the title compound as a brown solid (2.4 g, 5.5 mmol, 82 %
yield). MS
(m/z) 434.0 (M+H+).
Intermediates 11-12 were prepared from the indicated amine and carboxylic acid
by
methods analogous to those described for Intermediate 10.
Int. Name Structure Characterization Amine
acid
2-((4-fluoro-2-
o -9
methylphenyl)amino C
2-((4-fluoro-2-
6-methoxy-
)-N-(6-methoxy-2- 0- N Ms (m/z) 433.9 2-
methylphenyl)a
11 methylpyridin-3-yI)- F3C NH
mino)-4-
(M+1-1') methylpyrid
4- (trifluoromethyl)
in-3-amine
(trifluoromethyl)benz
benzoic acid
amide
5-chloro-2-((4-fluoro- o
5-chloro-2-((4-
2- 6-methoxy-
fluoro-2-
methylphenyl)amino MS (m/z) 400.0 2-
12 NH
methylphenyl)a
)-N-(6-methoxy-2- (M +Fr) methylpyrid
4
methylpyridin-3-
11
in-3-amine mino)benzoic
acid
yl)benzamide
Intermediate 13
N-(6-methoxy-2-methylpyridin-3-y1)-2-((2-methy1-4-
(trifluoromethoxy)phenyl)amino)-5-
(trifluoromethyl)nicotinamide
48
CA 03202326 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
I
0
y F3C it, N,----,,__õN
1 H
-,
N NH
OCF3
A 1 L round bottom flask, fitted with a magnetic stir bar, was charged with 2-
chloro-N-
(6-methoxy-2-methylpyridin-3-y1)-5-(trifluoromethyl)nicotinamide (37 g, 107
mmol) and 2-
methy1-4-(trifluoromethoxy)aniline (30.7 g, 161 mmol). Toluene (500 mL) was
added
followed by cesium carbonate (69/ g, 214 mmol). The resulting reaction mixture
was
purged with nitrogen for 15 minutes before Xantphos (6.19 g, 1010 mmol) and
Pd2(dba)3
(4.90 g, 5.35 mmol) were added. The resulting dark brown reaction mixture was
stirred at
100 00 for 16 hours. The reaction mixture was allowed to cool to room
temperature and
filtered through a Celite 0 bed washing with Et0Ac (1 L). The filtrate was
concentrated
under vacuum and the crude product was purified by flash column chromatography
(Biotage,
330 g SiO2 column, 0-30% Et0Ac/petroleum ether over 90 minutes) to give the
title
compound as a brown solid (35 g, 36.5 mmol, 341 % yield). MS (m/z) 500.8 (M+H
).
Intermediate 14
1-(4-Fluoro-2-methylpheny1)-3-(6-methoxy-2-methylpyridin-3-y1)-6-
(trifluoromethyl)-
2,3-dihydroquinazolin-4(1H)-one
I
o ;7o
F3c -, N
N
N)
1.1
F
To a solution of 2-((4-fluoro-2-methylphenyl)amino)-N-(6-methoxy-2-
methylpyridin-3-
y1)-5-(trifluoromethyl)benzamide (2.4 g, 5.5 mmol) and Cs2003 (7.22 g, 22.2
mmol) in
acetonitrile (25 mL) under nitrogen at room temperature was added
diiodomethane (1.340
mL, 16.61 mmol) dropwise over 5 min. The reaction mixture was stirred at 80 00
for 16 h.
The reaction mixture was cooled to rt and filtered through Celite 8 pad. The
filtrate was
concentrated onto SiO2. Purification by flash chromatography on SiO2 (50g)
with 0-100%
Et0Ac/petroleum ether as eluant afforded the title compound as a colorless
solid (2.0 g, 4.1
mmol, 74 % yield). MS (m/z) 446.0 (M+H+).
Intermediates 15-17 were prepared from the indicated amide by methods
analogous
to those described for Intermediate 14.
49
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
Int. Name Structure Characterization Amide
oI
1-(4-fluoro-2-
o
2-((441u0r0-2-
methylpheny1)-3-(6-
methoxy-2-
N -1s.õõN
methylphenyl)amino)-
MS (m/z) 446.0
N-(6-methoxy-2-
15 methylpyridin-3-yI)-7- F3C 411 N (M+H)
methylpyridin-3-yI)-4-
(trifluoromethyl)-2,3-
(trifluoromethyl)benza
dihydroquinazolin-
mide
4(1H)-one
6-chloro-1-(4-fluoro-2-
5-chloro-2-((4-fluoro-
NN CI
2-
methoxy-2- MS (m/z) 412.0
methylphenyl)amino)-
16
methylpyridin-3-yI)- (M+Hy
N-(6-methoxy-2-
2,3-dihydroquinazolin-
methylpyridin-3-
4(1H)-one
yl)benzamide
3-(6-methoxy-2-
N-(6-methoxy-2-
methylpyridin-3-y1)-1- F3c N
methylpyridin-3-y1)-2-
(2-methyl-4- 1 MS (m/z) 512.8
((2-methyl-4-
17 (trifluoromethoxy)phen N N (M+H)
(trifluoromethoxy)phe
y1)-6-(trifluoromethyl)-
nyl)amino)-5-
2,3-dihydropyrido[2,3- 40
(trifluoromethyl)nicoti
d]pyrimidin-4(1H)-one namide
ocF3
Intermediate 18
1-(4-Fluoro-2-methylpheny1)-3-(2-methy1-6-oxo-1,6-dihydropyridin-3-y1)-6-
(trifluoromethyl)-2,3-dihydroquinazolin-4(1H)-one
0
*11-Cr\ H F3C N
N
101
To a solution of 1-(4-fluoro-2-methylpheny1)-3-(6-methoxy-2-methylpyridin-3-
y1)-6-
(trifluoromethyl)-2,3-dihydroquinazolin-4(1H)-one (0.6 g, 1.4 mmol) in
acetonitrile (10 mL)
under nitrogen at room temperature was added iodotrimethylsilane (0.54 g, 2.7
mmol)
dropwise over 5 min. The reaction mixture was stirred at 80 00 for 12 h. The
reaction
mixture was cooled to rt and concentrated under vacuum. The crude residue was
dissolved
in DCM (100 mL) and washed with sat. sodium thiosulphate (20 mL). Organic
layer was
dried over sodium sulphate and concentrated onto Celite O. Purification by
reverse phase
chromatography on C18 (40g) with 0-100% gradient with 0.1% formic acid in
acetonitrile in
0.1% formic acid in water as eluant afforded clean fractions which were
concentrated and
CA 03202326 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
the resulting precipitate with filtered, washed with water and dried to afford
the title
compound as a colorless solid (0.21 g, 0.5 mmol, 36 % yield). MS (m/z) 432.1
(M+1-1 ).
Intermediates 19-21 were prepared from the indicated intermediate by methods
analogous to those described for Intermediate 18.
Int. Name Structure Characterization
Intermediate
1-(4-fluoro-2- 1-(4-fluoro-2-
0 :cto
methylphenyI)-3-(2-
methylphenyI)-3-(6-
methyl-6-oxo-1,6-
-1-14 MS (m/z) 432.0 methoxy-2-
19 dihydropyridin-3-yI)-7- F3 N
methylpyridin-3-yI)-7-
(M +H +)
(trifluoromethyl)-2,3-
(trifluoromethyl)-2,3-
dihydroquinazolin-
dihydroquinazolin-
4(1H)-one F 4(1H)-one
6-chloro-1-(4-fluoro-2- o 6-chloro-1-
(4-fluoro-
methylphenyI)-3-(2-
rjj MS (m/z) 398.0 2-methylphenyI)-3-(6-
N- methoxy-2-
methy1-6-oxo-1,6-
20
methylpyridin-3-yI)-
dihydropyridin-3-yI)- (M +F1')
2,3-dihydroquinazolin-
2,3-
411
dihydroquinazolin-
4(1H)-one
4(1H)-one
3-(6-methoxy-2-
1-(2-methyl-4-
methylpyridin-3-yI)-1-
(trifluoromethoxy)phen F3C N .NH
(2-methy1-4-
y1)-3-(2-methy1-6-oxo- 1 j
21 1,6-dihydropyridin-3- -N -N MS (m/z) 499.2
(trifluoromethoxy)phe
(M+H') nyI)-6-
y1)-6-(trifluoromethyl)-
1110 (trifluoromethyl)-2,3-
2,3-dihydropyrido[2,3-
dihydropyrido[2,3-
d]pyrimidin-4(1H)-one OCF3
d]pyrimidin-4(1H)-one
Intermediate 22
3-(1-(Chloromethyl)-2-methy1-6-oxo-1,6-dihydropyridin-3-y1)-1-(4-fluoro-2-
methylpheny1)-6-(trifluoromethyl)-2,3-dihydroquinazolin-4(11-1)-one
0 ---7y
F3C N N Cl
N
Chloromethyl carbonochloridate (0.452 ml, 5.09 mmol) was added dropwise to a
suspension of 1-(4-fluoro-2-methylpheny1)-3-(2-methy1-6-oxo-1,6-dihydropyridin-
3-y1)-6-
(trifluoromethyl)-2,3-dihydroquinazolin-4(1H)-one (0.878g. 2.035 mmol) and
DABCO (0.183
g, 1.628 mmol) in Ethyl acetate (16.23 ml) and DMF (1.623 ml) under N2. The
reaction was
51
CA 03202326 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
stirred at 60 C for - 6 hr and then at RT for 2.5 days. The reaction was
quenched slowly
with sat. NaHCO3 (20 mL), extracted with Et0Ac and DCM (2X). The combined
organic
extracts were dried over Na2SO4 and concentrated. The residue was triturated
with a
solution of 1:2/ Et0Ac: heptane to give the title compound as an off-white
solid (0/49 g,
1.561 mmol, 77 % yield). MS (m/z) 480.3 (M+1-1 ).
Intermediates 23-25 were prepared from the indicated amide by methods
analogous
to those described for Intermediate 22.
Int. Name Structure Characterization Amide
1-(4-fluoro-2-
3-(1-(chloromethyl)-
methylphenyI)-3-(2-
2-methy1-6-oxo-1,6-
N CI methy1-6-oxo-1,6-
dihydropyridin-3-y1)- 41 NY
dihydropyridin-3-
1-(4-fluoro-2- F3 c MS (m/z) 480.0
23 YI)-7-
methylphenyI)-7-
(trifluoromethyl)-2,3-
(trifluoromethyl)-
2,3-
dihydroquinazolin-
4(1H)-one F
dihydroquinazolin-
4(1H)-one
6-chloro-1-(4-
6-chloro-3-(1- o
fluoro-2-
(chloromethyl)-2- a N õCI
methylphenyI)-3-(2-
methy1-6-oxo-1,6- I N
MS (m/z) 446.0 methy1-6-
oxo-1,6-
24 dihydropyridin-3-yI)-
(M+H).-
dihydropyridin-3-
1-(4-fluoro-2-
yI)-2,3-
methylphenyI)-2,3-
dihydroquinazolin-
dihydroquinazolin- 4(1H)-
one
4(1H)-one
1-(2-methyl-4-
3-(1-(chloromethyl)-
(trifluoromethoxy)p
2-methy1-6-oxo-1,6- henyI)-3-
(2-methyl-
dihydropyridin-3-yI)-
N CI
1-(2-methyl-4-
6-oxo-1,6-
dihydropyridin-3-
(trifluoromethoxy)Ph N MS (m/z) 546.8
enyI)-6- (M+Hr
(trifluoromethyl)-2,3-
(trifluoromethyl)-
2,3-
dihydropyrido[2,3-
d]pyrimidin-4(1H)-
OCF,
dihydropyrido[2,3-
d]pyrimidin-4(1H)-
one
one
10 Intermediate 26
Di-tert-butyl ((5-(1-(4-fluoro-2-methylpheny1)-4-oxo-6-(trifluoromethyl)-1,4-
dihydroquinazolin-3(2H)-y1)-6-methy1-2-oxopyridin-1(2H)-yl)methyl) phosphate
52
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
0
F3C
N N
NJ or '0
DMF (7.29 ml) was added to a mixture of 3-(1-(chloromethyl)-2-methy1-6-oxo-1,6-
dihydropyridin-3-y1)-1-(4-fluoro-2-methylpheny1)-6-(trifluoromethyl)-2,3-
dihydroquinazolin-
4(1H)-one (0/49 g, 1.561 mmol), potassium di-tert-butyl phosphate (0.581 g,
2.341 mmol)
and tetrabutylammonium iodide (0.029 g, 0.078 mmol) under N2 and the reaction
was stirred
at 70 C for 3 hr. The reaction was cooled to RT and quenched slowly with ice
water (40
mL). The reaction was extracted with Et0Ac, washed with water (2X), dried over
Na2SO4
and concentrated to give the title compound as a foam (0.846 g, 1.294 mmol, 83
% yield.
MS (m/z) 542.3 (M-112)1.
Intermediates 27-29 were prepared from the indicated intermediate by methods
analogous to those described for Intermediate 26.
53
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
Int. Name Structure Characterization
Intermediate
di-tert-butyl ((5-(1- 3-(1-
(4-fluoro-2-
(chloromethyl)-2-
.0 ------
methylphenyI)-4- o _.,--?- methy1-6-
oxo-1,6-
o
oxo-7- N -.... N ,o_ii,,
dihydropyridin-3-
27 (trifluoromethyl)-1,4- r c IS N,.1
(5, - MS (m/z) 542.0 (M- yI)-1-(4-fluoro-2-
dihydroquinazolin- 3 A 112)*
methylphenyI)-7-
3(2H)-y1)-6-methyl-
411
(trifluoromethyl)-
2-oxopyridin-1(2H)- 2,3-
yl)methyl) F
dihydroquinazolin-
phosphate 4(1H)-
one
di-tert-butyl ((5-(6-
6-chloro-3-(1-
chloro-1-(4-fluoro-2- o c" r -4--
(chloromethyl)-2-
methylphenyI)-4- oi 0
methyl-6-oxo-1,6-
oxo-1,4-
MS (m/z) 508.0 (M- dihydropyridin-3-
28 A dihydroquinazolin-
3(2H)-y1)-6-methyl-
112)+ y1)-1-(4-
fluoro-2-
2-oxopyridin-1(2H)- 4
methylphenyI)-2,3-
yl)methyl)
dihydroquinazolin-
F 4(1H)-
one
phosphate
3-(1-
di-tert-butyl ((6-
(chloromethyl)-2-
methy1-5-(1-(2-
methyl-4-
methyl-6-oxo-1,6-
o , ce '-----
dihydropyridin-3-
(trifluoromethoxy)ph F
enyl)-4-oxo-6- 3crxit.N ...... N 0, ,
0
--,--- p yl)-1-(2-
methyl-4-
.. 0, MS (m/z) 609.0 (M-
(trifluoromethoxy)p
29 (trifluoromethyl)-1,4- N N
A 112)* henyI)-6-
dihydropyrido[2,3-
d]pyrimidin-3(2H)- 4111
(trifluoromethyl)-
2,3-
y1)-2-oxopyridin- ocr3
dihydropyrido[2,3-
1(2H)-yl)methyl)
d]pyrimidin-4(1H)-
phosphate
one
Example 1
(5-(1-(4-Fluoro-2-methylpheny1)-4-oxo-6-(trifluoromethyl)-1,4-
dihydroquinazolin-
3(2H)-y1)-6-methy1-2-oxopyridin-1(2H)-yl)methyl dihydrogen phosphate
0 --%;=r
F3C N õ,==,. NO,p,.pH
N.J HO 0
F
Acetic acid (0.5 ml, 8.73 mmol) was added dropwise to a suspension of di-tert-
butyl
((5-(1-(4-fluoro-2-methylpheny1)-4-oxo-6-(trifluoromethyl)-1,4-
dihydroquinazolin-3(2H)-y1)-6-
54
CA 03202326 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
methyl-2-oxopyridin-1(2H)-yl)methyl) phosphate (0.3 g, 0459 mmol) in
acetonitrile (1.5 ml)
and water (1.5 ml) at RT under N2 and the reaction was stirred at 70 C for 3
hr. The
reaction was cooled and concentrated. The foam residue was rinsed with water.
The
residue was purified by reverse phase (EZ Prep Isco, C18 Aq 15.5g Gold column,
30-80%
gradient, acetonitrile with 0.1% formic acid/water with 0.1% formic acid, 30
mUmin flow rate,
12.3 min overall run time) to give the title compound as a white solid (64.7
mg, 0.120 mmol,
26.0% yield). 1H NMR (400 MHz, DMSO-d6) 6:11.60 (br s, 2H), 8.09 (d, J=2.4 Hz,
1H), 7.66
(br d, J=8.3 Hz, 1H), 7.52-7.38 (m, 2H), 7.37-7.28 (m, 1H), 7.26-7.15 (m, 1H),
6.43-6.30 (m,
2H), 5.80-5.60 (m, 2H), 5.55 (d, J=9.3 Hz, 0.6H), 5.1-5.2 (m, 0.8H), 4.75 (br
d, J=9.3 Hz,
0.6H), 2.3-2.4 (m, 3H), 2.25 (br s, 3H). MS (m/z) 542.0 (M+H ).
Examples 2-4 were prepared from the indicated Intermediate by methods
analogous
to those described for Example 1.
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
Ex. Name Structure Characterization
Intermediate
(5-(1-(4-fluoro-2-
di-tert-butyl ((5-(1-
methylpheny1)-4- 1H NMR (400 MHz,
(4-fluoro-2-
oxo-7- o -;-----r DMSO-d6) 6: 8.07 (d, J =
methylphenyI)-4-
(trifluoromethyl)- ._, p 8.0 Hz, 1H), 7.46-
7.37
oxo-7-
1,4- N -1-NI-).---,POH (m, 2H), 7.35 - 7.13 (m,
dihydroquinazolin- F3c N) H 5H), 6.47 - 6.25 (m,
2H), (trifluoromethyly
2
1,4-
3(2H)-y1)-6- 5.57 (br s, 2H), 5.51
(br s,
methyl-2-
I. 0.5H), 5.13 (br s, 1H),4.74 dihydroquinazolin-
3(2H)-y1)-6-methyl-
oxopyridin-1(2H)- (br s, 0.5H), 2.44 -
2.31
2-oxopyridin-1(2H)-
yl)methyl F (m, 3H), 2.24 (s, 3H)
yl)methyl)
dihydrogen MS (m/z) 542.0 (M+H)
phosphate
phosphate
1H NMR (400 MHz,
DMSO-d6) 6: 7.80 (d, J =
(5-(6-chloro-1-(4-
2.6 Hz, 1H), 7.46 (d, J =
fluoro-2-
di-tert-butyl ((5-(6-
methylpheny1)-4- o ----..."-e) 9.8 Hz, 1H), 7.40
(dd, J =
chloro-1-(4-fluoro-
r, o 8.8, 2.6 Hz, 1H), 7.36 -
oxo-1,4- 7.27 (m, 2H), 7.22 - 7.12
2-methylphenyI)-4-
dihydroquinazolin-
N) HO OH
(TI, 1H), 6.39 (d, J = 9.6
3 3(2H)-yI)-6-
oxo-1,4-
dihydroquinazolin-
Hz, 1H), 6.35 - 6.17 (m,
methy1-2-
3(2H)-yI)-6-methyl-
oxopyridin-1(2H)- 0 1H), 5.83 - 5.69 (m, 2H),
5.46 (brs, 0.6H), 5.21 -
2-oxopyridin-1(2H)-
yl)methyl
yl)methyl)
F 4.97 (m, 0.8H), 4.69 (br
s,
dihydrogen
phosphate
0.6H), 2.43 - 2.28 (m, 3H),
phosphate
2.23 (s, 3H)
MS (m/z) 508.0 (M+H)
(6-methy1-5-(1-(2- 1H NMR (400 MHz,
di-tert-butyl ((6-
methyl-4- DMSO-d6) 6: 8.60 (s, 1H),
methy1-5-(1-(2-
(trifluoromethoxy)p o 8.32 (d, J = 2.40 Hz,
1H), methy1-4-
o
henyI)-4-oxo-6- H N D 0 7.55-7.15 (m, 6H), 6.30 (t,
(trifluoromethoxy)p
F3c..,.., ....,..-.,..õ.--,N
(trifluoromethyl)- I ) '---HO,ID''OH J = 10.00 Hz, 1H),
5.70- henyI)-4-oxo-6-
1,4- '-N N 5.50 (m, 2.5H), 5.35 (d, J
(trifluoromethyl)-
4
dihydropyrido[2,3- = 10 Hz, 0.5H), 5.22 (d,
J 1,4-
d]pyrimidin-3(2H)-
4111 = 10 Hz, 0.5H) 4.92 (d, J dihydropyrido[2,3-
y1)-2-oxopyridin- = 9.6 Hz, 0.5H), 2.42 (s,
d]pyrimidin-3(2H)-
1(2H)-yl)methyl ocF3 3H), 2.24 (d, J = 6.8 Hz,
yI)-2-oxopyridin-
dihydrogen 3H)
1(2H)-yl)methyl)
phosphate MS (m/z) 609.0 (M+H)-
phosphate
Biological Assays
The Na,1.8 Inhibitor compounds or pharmaceutically acceptable salts thereof of
the
invention are useful for treatment of pain, pain disorders or conditions, pain-
related disorders or
conditions or pain caused by diseases, respectively, such as those defined
throughout the
instant application.
The biological activity of the compounds of the invention can be determined
using
suitable assays, such as those measuring such inhibition and those evaluating
the ability of the
56
CA 03202326 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
compounds to inhibit voltage gated sodium channel Na, 1.8 in vitro or in
animal models of
infection.
Biological Assay Example 1:
Human embryonic kidney 293 cells (HEK293) expressing human Nav1.8, human
Na,[31 and human TREK1 (HEK293-Na,1 .8) were grown at 37 C, 5% CO2 in 150cm2
flasks.
HEK293-Nav1.8 were passaged every 2-3 days into T175 cell culture flasks when
confluency
reached 80 ¨ 90 %.
Pharmacological assessment of the compounds of the invention was performed
using HEK293-Nav1.8 in combination with an assay developed on the QPatch 48
HTX
electrophysiological system. HEK293-Nav1.8 were prepared on the day of use by
removing
culture media, washing in DPBS, adding Acculase (2m1 to cover the surface,
aspirate lml
then 1.5 min at 37 C) followed by addition of CHO-SFM II to stop the enzyme
digestion and
in order to obtain a suspension of 3 x 106 cell/rnL.
Compound was prepared in an extracellular solution of the following
composition:
NaCI (145 mM), KCI (4 mM), CaCl2 (2 mM), MgCl2 (2 mM), HEPES (1 mM), Glucose
(10
mM), pH 7.4 with NaOH Osmolality 300 mOsM/L. The intracellular solution was
used of the
following composition: CsF (115 mM), CsCI (20 mM), NaCI (5 mM), EGTA (10 mM),
HEPES
(10 mM), Sucrose (20 mM), pH 7.2 with CsOH Osmolality 310 mOsm/L.
Utilizing the voltage-clamp mode in the QPatch 48 HTX system a half
inactivation
state voltage protocol (V1/2) was used to determine pharmacological activity
of compounds of
the invention at Nay1.8 ion channels. A V112 protocol was utilized with the
following voltage
steps: a holding voltage of -100 mV was established followed by a 20 ms
voltage step to 0
mV (P1), followed by an inactivating voltage step at -46 mV for 8 seconds,
followed by a step
to -100 mV for 20 ms, before a 20 ms step to CImV (P2) before returning to the
holding
voltage of -100 mV. This voltage protocol was repeated at a frequency of
0.07Hz., current
magnitude was quantified at the P2 step throughout the recording. Inhibition
of the measured
current amplitude with the compounds of the invention was analyzed by fitting
a 6 - 8 point
dose-response curve allowing determination of the fifty percent inhibition
concentration
(IC50). Within the QPatch HTX software, P2 current was normalized according to
measurements made at baseline after compound and after positive reference
compound and
fit to the following equation:
(Input ¨ Baseline)
n. Icpp = Normalized Current = ______________________________________
(FullResponse ¨ Baseline)
To assess current run-down over the course of the experiment vehicle-only
wells
were utilized and the normalized current with vehicle-only (n. Ivõ) was
determined. To
57
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
correct the compound response for run-down, the currents were corrected
according the
following formula:
(n. icpp n. IvEH)
n. IRD_Correct = ____________________________________________
(1 ¨ n. IvEH)
Compounds of the invention and the corresponding parent compounds (see Table
1A
for structures) were tested for activity against Nav1. 8 sodium channels in
the above assay
in one or more experimental runs and the results are shown in Table 1 below.
Potency of
the compounds of the invention is reported as a pIC50 value. The pIC50 value
is the
negative log of the 1050 value, wherein the IC50 value is the half maximal
inhibitory
concentration measured in molar (M). Potency of the compounds of the invention
is
compared to the potency of the parent compound. For compounds tested in more
than one
experimental run, the 131050 value is reported as an average.
Table 1
Compounds of the Invention Parent Compounds
Parent [Nav1.8]
Compound [Nav1.8]
Compound pIC50
Example No. pIC50
No.
1 6.8 1A 8
2 6.6 2A 7.7
3 6.9 3A 8
4 6.1 4A 7.5
Table 1A
Compound Structure Parent Structure
Example Compound
No. No.
1 1A
o o
F3C N Q,OH F3C
N
NH
NJ HO oN
58
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
2 o 2A oo
F3C N
N) H
F3C N
3 o %--e) 3A
0
CI CI
.NH
N) H
1411
4 0 ce 4A o
NH
HC H
NN
OCF3
OCF3
Biological Assay Example 2:
Kinetic solubility measurement using Charged Aerosol Detector (CAD). The
aqueous
kinetic solubility at pH 7.4 was determined by measuring the concentration of
solute in
solution after precipitation from DMSO stock solution. The DMSO stock solution
was diluted
20-fold with phosphate buffered saline (PBS) pH 7.4 and the solubility of the
compound was
measured after 1 hour equilibration at room temperature by HPLC-CAD.
Calibration
standards of Ketoconazole and Primidone were prepared by serial dilutions in
DMSO at
concentrations ranging from 0.016 to 4.5 mg/ml to produce the calibration
curve used to
determine the solubility of the compounds as previously described in Max W.
Robinson et al,
Use of Calculated Physicochemical Properties to Enhance Quantitative Response
When
Using Charged Aerosol Detection, Anal. Chem., 2017, 89 (3), pp 1772-1777,
which is herein
incorporated by reference.
CAD solubility of the compounds of the invention and the corresponding parent
compounds was measured as described above and the results are shown in Table 2
below.
59
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
Table 2
Prodrug Compounds of the Parent Compounds
Invention
CAD Parent CAD
Compound solubilitya Compound solubilityb
Example No.
(pg/mL) No. (pg/mL)
1 >260 1A 48
2 >193 2A 51
3 102 3A 54
4 181 4A 87
a CAD solubility was measured in multiple experimental runs and the data
reported as an
average for Compound Example Nos. 1 and 3. CAD solubility was measured in one
experimental run for Compound Example Nos. 2 and 4.
b CAD solubility is reported as the average of multiple experimental runs.
Biological Assay Example 3: Rat IV/PO Study
An in vivo rat pharmacokinetic study was conducted to determine whether the
prodrug compounds of the invention are converted to the respective parent
compound upon
administration. The rat pharmacokinetic study was conducted with a crossover
design on
two study days with a one-day recovery period between each study day. Three
male, dual
catheterized (femoral vein and carotid artery) Han Wistar rats were used for
the study. Each
rat was also implanted with a gastric catheter for oral dose administration.
Rats were dosed
at 1 mg/kg by a 60 minute intravenous (IV) infusion (femoral vein cannula),
then
subsequently oral dosed at 2 mg/kg via the gastric cannula, with 48 hours
between dosing
sessions. Dose solutions of the compound of Example 3 were prepared in 20%
Cavitron/5%
DMSO/75% water (IV) and in 6% Cavitron/5% DMS0/89%water (PO) without pH
adjustment. The dose solutions were filtered using a 0.22 p filter. The pH of
the final dosing
solutions was 6Ø
During the intravenous study leg, blood samples were collected from the
carotid
artery catheter at target times of 15, 30, 60 (end of infusion), 65, 75, 90,
120, 240, 360, 480,
720, and 1440 minutes following the initiation of the intravenous infusion of
the compound of
Example 3. During the oral study leg, blood samples were collected prior to
dosing and at
target times of 15, 30, 60, 90, 120, 180, 240, 360, 480, 720, and 1440 minutes
following oral
administration. Blood samples, 100 pL, were mixed with 100 pL phosphatase
inhibitor, a 50
pL aliquot of the blood and inhibitor mixture was transferred to a non-
heparinized tube and
CA 03202328 2023- 6- 14
WO 2022/129283 PCT/EP2021/086101
stored at approximately -80 C until analyzed. The concentrations in the
filtered dose
solutions were confirmed by preparing stepwise dilutions first into 50%
aqueous acetonitrile
with 0.1% formic acid then into heparinized male Wistar Han blood:inhibitor to
achieve
determined nominal concentrations. Triplicate 50 pL aliquots were removed and
were
frozen and stored at approximately 80 C until analyzed by LC-MS/MS as
described below.
LC-MS/MS was used to quantify the compound of Example 3 and the corresponding
parent compound of Example 3A in the biological samples generated in the above
described
in vivo study. Samples were prepared by protein precipitation followed by LC-
MS/MS
analysis employing positive-mode ionization against a set of calibration
standards for the
compounds prepared in the same matrix. Pharmacokinetic parameters for the
study was
derived from the concentration versus time profiles. Key pharmacokinetic
parameters such
as AUCo¨ (extrapolated area under the blood concentration-time curve), AUCo_t
(area under
the blood concentration-time curve to the last time point with quantifiable
drug), Cmax
(maximum concentration), Tmax (time Cmax is achieved), CL (systemic blood
clearance),
Vdss (steady-state volume of distribution), MRT (mean residence time), and
t112 (half-life)
were determined for the compound of Example 3. The key pharmacokinetic
parameters
such as AUCo¨, AUCo_t, Cmax, Tmax, MRT, and t112 (half-life) were determined
for the
parent compound Example 3A. Descriptive statistical data of pharmacokinetic
parameters
were calculated, including the mean and standard deviation (SD) using
Microsoft Excel.
The data are shown below in Tables 3A and 3B. Data are reported as mean SD
(N=3).
Table 3A
Route
Parameter
Intravenous Oral
Dose (mg/kg) 0.87 0.01 1.7 0.0
Cmax (ng/mL) 260 24
Tmax (h)b 1.0, 0.25, 1.0
Half-life (h)b 0.42, 3.8, 4.8
Compound of
Example 3 MRT (h)b 0.14, 0.87, 1.2
__________________________________________________________________ There were
no
(Prodrug) CL 58 9 quantifiable
(mL/min/kg)
concentrations
Vdss (L/kg)b 0.44, 3.6 3.8 following
oral
AUCo-t ____________________________________________________________
administration
0.24 0.04
(pg.h/mL)
AUCo-
0.25 0.03
(pg.h/mL)
Bioavailability
(%)
61
CA 03202328 2023- 6- 14
WO 2022/129283
PCT/EP2021/086101
b Values listed individually due to variability
Table 3B
Route
Parameter
Intravenous Oral
Dose (mg/kg)
Cmax (ng/mL) 170 26 170 41
Trinax (h) 1.1 0.05 1.2 0.3
Compound of Half-life (h) 3.7 0.4 3.4 0.5
Example 3A
(Parent Compound) MRT (h) 5.0 0.3 TO 0.2
CL (nnUmin/kg)
Vdss (L/kg)
AUCo_t(pg.h/mL) 0.58 0.06
1.3 0.1
AUCo¨ (pg.h/mL) 0.63 0.06
1.4 0.1
Bioavailability (%)
It is to be understood that the invention is not limited to the embodiments
illustrated
hereinabove and the right is reserved to the illustrated embodiments and all
modifications
coming within the scope of the following claims.
62
CA 03202328 2023- 6- 14