Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
WO 2022/263316
PCT/EP2022/065823
1
SYSTEM FOR ELECTROCATALYTIC CONVERSION OF CARBON OXIDES TO
MULTICARBON PRODUCTS USING A STATIONARY CATHOLYTE LAYER AND
RELATED PROCESS
TECHNICAL FIELD
The technical field generally relates to electrocatalytic conversion in
electrolyzers, and
more particularly to electroreduction process and system combining a bipolar
membrane
and a stationary catholyte layer facilitating efficient conversion of CO2
and/or CO into
multicarbon (C2 ) products.
BACKGROUND
Gigatons of CO2 need to be avoided or removed from the atmosphere each year
(see
reference 1). When powered by renewable energy sources, the fixation of
captured carbon
via electrochemical reduction of CO2 (CO2RR) offers a route to net-negative-
emission
production of multi-carbon (02+) chemicals (see reference 2). However, CO2RR
in
electrolyzers operating both with alkaline and neutral electrolytes incur
significant CO2 loss
to carbonate formation and crossover, leading to low CO2 utilization.
The industrial implementation of the CO2RR for 02+ production requires the
simultaneous
achievement of high production rates, high energy efficiencies, and high
carbon
efficiencies (see references 3 and 4). Known CO2RR electrolyzers based on
alkaline bulk
electrolytes (e.g. alkaline flow cell, Figure 1A) have achieved 02+ partial
current densities
greater than 1 A cm-2 with Faradeic Efficiency (FE) towards 02+ products
exceeding 70%
(see references 2, 4 and 5). For example, zero-gap electrolyzers based on
alkaline or
neutral bulk electrolytes (e.g. membrane electrode assembly (MEA, Figure 1B)
can deliver
about 100 mA cm-2 with total C2+ FEs of more than 70% (see references 3, 4 and
6).
For CO2RR, a catholyte having a high local pH (>12) near the cathode can be
used to
favour the CO2RR reaction with respect to the competing hydrogen evolution
reaction
(HER), to enhance the selectivity towards C2+ products(see references 2, 5 and
7 to 9).
To maintain such a high local pH, many present-day CO2RR electrolyzers use a
flowing
alkaline electrolyte/catholyte reservoir (see references 2, 4 and 5). For the
same reason,
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
2
MEAs typically use strong alkaline anion-exchange membranes (AEM) and
anolytes(see
reference 18). However, locally alkaline conditions absorb CO2 to form
carbonates:
CO2 + 20H- ¨> 0032- + H20 [1]
CO2 + OH- ¨> H003- [2]
Thus, at a steady-state, CO2 reacts with hydroxides ions to form carbonate or
bicarbonate
ions, and both reactant and electrolyte are lost, corresponding to low carbon
efficiency in
the utilization of CO2 feedstock. Recovering CO2 from carbonate/bicarbonate
can
consume as much as 60% to 70% of the energy input (see references 11 and 12).
Certain flow cells and MEAs have been designed to use neutral electrolytes
(e.g., KHCO3)
rather than strong alkaline ones in order to reduce CO2 absorption. Neutral
media flow
cells and MEAs have lower CO2 absorption than do alkaline cells, and yet,
since the
reaction drives up the local pH and creates locally alkaline conditions,
carbonate and
bicarbonate formation remain a problem (see reference 11).
Carbonate/bicarbonate ions
migrate to the anode via the AEM, to combine with protons provided from the
anode
oxygen evolution reaction, thereby releasing CO2 into the anode gas stream
(5I1). To
date, a single pass CO2 utilization (SPU, the fraction of the CO2 feed been
transformed to
products) of 02+ producing electrolyzers has remained in the range 3% to 30%
(Table Si)
(see references 6, 11, 13 to 16).
SUMMARY
The present techniques relate to carbon oxides-to-C2+ electrochemical
reduction
strategies that overcome previously-observed limits of carbon efficiency by
designing an
electroreduction system that inhibits carbon oxides crossover from cathode to
anode and
reverts formed carbonate/bicarbonate ions to carbon oxides via acidification
of the
catholyte. Carbon oxides as encompassed herein are selected from selected from
CO,
CO2 or any mixture thereof.
More particularly, in a first aspect, there is provided an electroreduction
system for
converting carbon oxides into multicarbon (02+) products; the system
comprising:
- a cathodic compartment having a reactant inlet for receiving CO, CO2 or any
mixture thereof, and comprising a cathode, the cathode comprising a catalyst
layer
that is in contact with a catholyte solution:
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
3
an anodic compartment, the anodic compartment comprising an anode and
accommodating a flowing anolyte solution;
a bipolar membrane being positioned between the cathodic compartment and the
anodic compartment, the bipolar membrane comprising:
a cation-exchange layer (CEL) in cation communication with the catholyte
solution to provide protons into the catholyte solution;
an anion-exchange layer (AEL) in anion communication with the anolyte
solution to provide hydroxide ions at a surface of the anode; and
an interfacial layer defined between the cation-exchange layer and the
anion-exchange layer for splitting water into the protons and the hydroxide
ions;
wherein the cathodic compartment and/or the anodic compartment have a product
outlet to release the C2+ products;
remarkable in that the cathodic compartment accommodates a stationary
catholyte
layer between the catalyst layer of the cathode and the CEL, the stationary
catholyte layer comprising the catholyte solution; and in that the thickness
of the
stationary catholyte layer is at most 280 pm as measured by a spiral
micrometer.
For example, the thickness of the stationary catholyte layer can be between 20
pm and
250 pm as measured by a spiral micrometer; preferably between 40 pm and 200
pm; and
more preferably between 50 pm and 150 pm; and even more preferably between 65
and
125 pm. The solid porous support can be sandwiched between the catalyst layer
of the
cathode and the CEL for direct contact therewith.
In some implementations, the cathodic compartment further comprises a solid
porous
support in between the CEL and the catalyst layer, the solid porous support
being
configured to be saturated with the catholyte solution to form the stationary
catholyte layer.
In an embodiment, the solid porous support can include polyvinylidene fluoride
(PVDF),
polytetrafluoroethylene (PTFE), polycarbonate, nylon, cellulose acetate,
cellulose nitrate,
polypropylene, alumina, or any combinations thereof.
In an embodiment, the solid porous support can have a mean pore diameter
(i.e., a mean
pore size) between 0.05 and 50 pm as determined by scanning electron
microscopy
(SEM), optionally between 0.2 and 1 pm, and further optionally of 0.45 pm.
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
4
In an embodiment, the stationary catholyte layer can have a liquid content
between 5 and
50 pLcm-2 preferably between 6 and 40 pL.cm-2; more preferably between 8 and
30
pL.cm-2; even more preferably between 8 and 30 pL.cm-2; most preferably
between 10 and
25 pLcm-2; even most preferably between 10 and 20 pL.cm-2 or between 12 and 18
pL.cm-2; when the solid porous support is saturated with the catholyte
solution; the liquid
content being determined by weighting. For example, the stationary catholyte
layer can
have a liquid content about 10 pL.cm-2.
In some implementations, the stationary catholyte layer has a thickness that
is selected to
maximize a mass transport of regenerated CO2 or CO to the catalyst layer of
the cathode
while maintaining a resistance to compression of the solid porous support.
In some implementations, the catholyte solution has a concentration of cations
between
0.25 M and 3 M; preferably between 0.4 M and 4 M; and more preferably between
0.5 M
and 2 M, and for example about 1M.
For example, the catholyte solution can be a solution of K2SO4 with a KE
concentration
equal to or greater than 0.5M.
For example, the cations in the catholyte solution can be one or more selected
from K+,
Nat, Cs, Rb+, NH4, Mg2+, Ca2+, Al3+.
For example, the catholyte solution is a solution with a cations concentration
equal to or
greater than 0.5M.
In some implementations, the catholyte solution is a buffered solution. For
example, the
buffered solution can be a solution comprising one or more selected from
KHCO3, K3PO4,
K2HPO4, KH2PO4, the buffered solution is a mixture of glycine and sodium
hydroxide, or a
mixture of H3B03 and sodium hydroxide.
In some implementations, the catholyte solution is a non-buffered solution.
For example,
the non-buffered solution can be or comprise K2SO4, KCI or any other
combinations of the
Cl- anions or S042- anions with Nat, Cs, Rb+, NH4, Mg2+, Ca2+, or Al3+
cations.
The anolyte solution can have an anolyte concentration between 2.0 M and
0.01M,
optionally about 0.1 M. In some implementations, the anolyte solution is a
neutral solution.
For example, the anolyte solution can have a pH between 7 and 10. In the
context of the
invention the anolyte solution is a neutral solution hen having a pH between 7
and 10. The
anolyte (neutral) solution can be a KHCO3, K2804, or K2HPO4 solution.
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
In some implementations, the anolyte solution is an acidic solution. For
example, the
anolyte solution has a pH between 1 and 4. For example, the anolyte solution
has a bulk
pH between 1 and 4. The acidic solution can be a H3PO4 solution, H2804
solution or a
combination thereof.
5 In some implementations, the catalyst layer of the cathode comprises
copper (Cu), silver
(Ag), platinum (Pt), carbon (C), or any combination thereof.
In some implementations, the cathode further comprises a gas diffusion layer
for
contacting the stream of CO, CO2 or any mixture thereof, and the catalyst
layer is
deposited onto the gas diffusion layer. For example, the gas diffusion layer
can be a
hydrophobic carbon paper, or a copper sputtered hydrophobic PTFE layer. In the
context
of the invention hydrophobic means a water contact angle following ISO 19403-
6:2017 of
at least 30 .
In some implementations, the anode comprises an anodic catalyst layer and an
anodic
current collector layer. For example, the anodic catalyst layer can include
one or more
selected from 1r02, Pt, Pd, Ni, Ni0x, CoOx. For example, the anodic current
collector layer
can include Ti felt, hydrophilic carbon paper, or Ni foam. In the context of
the invention
hydrophilic means a water contact angle following ISO 19403-6:2017 below 30 .
In some implementations, the interfacial layer of the bipolar membrane
comprises a water
dissociation catalyst. The water dissociation catalyst can be present as
nanoparticles. The
water dissociation catalyst can comprise one or more selected from TiO2, 1r02,
NiO, Sn02,
graphene oxide, CoOx, ZrO2, A1203, Fe(OH)3, Mn02, Ru, Rh, RuPt alloy, PtIr
alloy, Ir, Pt.
In some implementations, the AEL is a membrane comprising poly(aryl
piperidinium),
polystyrene methyl methylimidazolium, or polystyrene tetramethyl
methylimidazolium. The
GEL can comprise or consist of a sulfonated tetrafluoroethylene based
fluoropolymer-
copolymer.
In some implementations, the system can include a temperature controller
configured to
maintain an operating temperature between 20 C and 50 C, optionally about 35
C.
A single-pass utilization of the stream of CO, CO2 or any mixture thereof can
be of at least
50% for an inlet flowrate between 1 sccm and 15 sccm. The single-pass
utilization of the
stream of CO, CO2 or any mixture thereof can be of at least 60% for an inlet
flowrate
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
6
between 1 sccm and 8 sccm. The system can have a Faradeic Efficiency (FE) for
conversion into the C2, products of at least 20% during at least 20 hours of
operation and
under an applied current density between 100 and 400 mA.cm-2. For example, the
FE for
conversion into the 02+ products can be of at least 25% during at 30 hours of
operation
and the applied current density of 350 mA.cm-2.
In another aspect, there is provided a carbon oxides electroreduction process
for
converting CO, CO2 or any mixture thereof into C2, products. The process
includes:
supplying a catholyte solution and CO, CO2 or any mixture thereof to a
cathodic
compartment comprising a catalyst layer in contact with the catholyte
solution:
flowing an anolyte solution through an anodic compartment having a product
outlet
to release the C2, products, the anodic compartment comprising an anode;
providing a bipolar membrane between the cathodic compartment and the anodic
compartment, the bipolar membrane comprising:
a cation-exchange layer (CEL) in cation communication with the catholyte
solution to provide protons into the catholyte solution;
an anion-exchange layer (AEL) in anion communication with the anolyte
solution to provide hydroxide ions into the anolyte solution; and
an interfacial layer defined between the cation-exchange layer and the
anion-exchange layer for splitting water into the protons and the hydroxide
ions; and
retaining a portion of the catholyte solution as a stationary catholyte layer
between
the catalyst layer of the cathode and the CEL and in contact with the CEL
wherein
the thickness of the stationary catholyte layer is at most 280 pm as measured
by a
spiral micrometer.
In some implementations, the process can include maintaining an operating
temperature
between 20 C and 50 C, and optionally about 35 C.
In some implementations, supplying CO, CO2 or any mixture thereof to the
cathodic
compartment is performed at an inlet flowrate between 1 sccm and 15 sccm.
In some implementations, the process can include providing the cathode with an
applied
current density between 100 and 400 mA.cm-2
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
7
In some implementations, the process can include forming the stationary
catholyte layer
by providing a solid porous support between the cathode and the CEL, and
saturating the
solid porous support with the catholyte solution. For example, the saturating
can be
performed to reach a liquid content of the stationary catholyte layer between
5 and 50
pL.cm-2, optionally between 10 and 20 pL.cm-2, and further optionally about 10
pL.cm-2
when the solid porous support is saturated with the catholyte solution.
In some implementations, the catholyte solution can be supplied with a
concentration of
cations between 0.25 M and 3 M, and optionally between 0.5 M and 2 M, and
further
optionally about 1M.
In some implementations, the stationary catholyte layer can be formed with a
thickness
between 20 pm and 250 pm as measured by a spiral micrometer; preferably
between 40
pm and 200 pm, more preferably between 50 pm and 150 pm.
In some implementations, the process can include utilizing CO, CO2 or any
mixture thereof
according to a single-pass utilization of the stream of CO, CO2 or any mixture
thereof of
at least 50% for an inlet flowrate between 1 sccm and 15 sccm. For example,
the process
can include utilizing CO, CO2 or any mixture thereof according to a single-
pass utilization
of the stream of CO, CO2 or any mixture thereof of at least 60% for an inlet
flowrate
between 1 sccm and 8 sccm.
In some implementations, the process can include producing the C2, products
according
to a Faradeic Efficiency (FE) for conversion into the C2, products that is of
at least 20%
during at least 20 hours of operation and under an applied current density
between 100
and 400 mA.cm-2. For example, the process can include producing the 02+
products
according to the FE for conversion into the C2, products that is of at least
25% during at
hours of operation and the applied current density of 350 mA.cm-2.
25 It should be noted that the process can include using the system
according to all
implementations as defined herein.
The inventors have thus discovered that a cation effect can be allowed at the
cathode
surface to enable carbon oxide reduction in the acidified catholyte solution.
For example,
the electroreduction system includes a bipolar membrane and a stationary
catholyte layer
30 that maintains a catholyte solution within a cathodic compartment, to
facilitate the
participation of regenerated CO2 in CO2RR reactions. The presently designed
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
8
electroreduction system showed a single-pass CO2 utilization of more than 60%,
representing twice the previously reported state-of-art designs that produced
C2+. Owing
to its high single-pass CO2 utilization (SPU), the presently proposed
electroreduction
system minimizes the energy input associated with CO2 recovery, while enabling
comparable performance and stability to the benchmark alkaline and neutral
media AEM-
based flow cell or MEA electrolyzers.
In an embodiment 1, the invention provides an electroreduction system for
converting
carbon oxides selected from CO, CO2 or any mixture thereof into multicarbon
(C2+)
products, the system comprising:
a cathodic compartment having a reactant inlet for receiving a stream of CO,
CO2
or any mixture thereof and comprising a cathode, the cathode comprising a
catalyst
layer that is contactable with a catholyte solution:
an anodic compartment having a product outlet to release the C2, products, the
anodic compartment comprising an anode and being configured to accommodate
a flowing anolyte solution;
a bipolar membrane being positioned between the cathodic compartment and the
anodic compartment, the bipolar membrane comprising:
a cation-exchange layer (CEL) in cation communication with the catholyte
solution to provide protons into the catholyte solution;
an anion-exchange layer (AEL) in anion communication with the anolyte
solution to provide hydroxide ions at a surface of the anode; and
an interfacial layer defined between the cation-exchange layer and the
anion-exchange layer for splitting water into the protons and the hydroxide
ions;
wherein the cathodic compartment is configured to accommodate a stationary
catholyte layer between the catalyst layer of the cathode and the CEL, the
stationary catholyte layer comprising the catholyte solution.
In an embodiment 2, the system according to embodiment 1, wherein the cathodic
compartment further comprises a solid porous support in between the CEL and
the
catalyst layer, the solid porous support being configured to be saturated with
the catholyte
solution to form the stationary catholyte layer.
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
9
In an embodiment 3, the system according to embodiment 2, wherein the solid
porous
support comprises polyvinylidene fluoride (PVDF), polytetrafluoroethylene
(PTFE),
polycarbonate, nylon, cellulose acetate, cellulose nitrate, polypropylene,
alumina, or any
combinations thereof.
In an embodiment 4, the system according to embodiment 2 or 3, wherein the
solid porous
support has a mean pore size between 0.05 and 50 pm, optionally between 0.2
and 1 pm,
and further optionally of 0.45 pm.
In an embodiment 5, the system according to any one of embodiments 2 to 4,
wherein the
stationary catholyte layer has a liquid content between 5 and 50 pL.cm-2,
optionally
between 10 and 20 pLcm-2, and further optionally about 10 pL.cm-2, when the
solid porous
support is saturated with the catholyte solution.
In an embodiment 6, the system according to any one of embodiments 2 to 5,
wherein the
stationary catholyte layer has a thickness that is selected to maximize a mass
transport of
regenerated CO2 or CO to the catalyst layer of the cathode while maintaining a
resistance
to compression of the solid porous support.
In an embodiment 7, the system according to embodiment 6, wherein the
thickness of the
stationary catholyte layer is between 20 pm and 400 pm, optionally between 20
pm and
300 pm, further optionally between 20 pm and125 pm.
In an embodiment 8, the system according to any one of embodiments 2 to 7,
wherein the
solid porous support is sandwiched between the catalyst layer of the cathode
and the CEL
for direct contact therewith.
In an embodiment 9, the system according to any one of embodiments 1 to 8,
wherein the
catholyte solution has a concentration of cations between 0.25 M and 3 M, and
optionally
between 0.5 M and 2 M, and further optionally about 1M.
In an embodiment 10, the system according to embodiment 9, wherein the
catholyte
solution is a solution of K2SO4 with a K+ concentration equal to or greater
than 0.5M.
In an embodiment 11, the system according to any one of embodiments 1 to 10,
wherein
the cations in the catholyte solution are one or more selected from K+, Nat,
Cs, Rb+, NH4,
Mg2+, Ca2+, Al3+.
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
In an embodiment 12, the system according to any one of embodiments 1 to 11,
wherein
the catholyte solution is a buffered solution.
In an embodiment 13, the system according to embodiment 12, wherein the
buffered
solution is a solution comprising one or more selected from KHCO3, K3PO4,
K2HPO4,
5 KH2PO4, the buffered solution is a mixture of glycine and sodium
hydroxide, or a mixture
of H3B03 and sodium hydroxide.
In an embodiment 14, the system according to any one of embodiments 1 to 11,
wherein
the catholyte solution is a non-buffered solution.
In an embodiment 15, the system according to embodiment 14, wherein the non-
buffered
10 solution is or comprises K2SO4, KCI or any other combinations of the CI-
anions or S042
anions with Nat, Cs, RID, NH4, me, c_2-F,
a or Al3+ cations.
In an embodiment 16, the system according to any one of embodiments 1 to 15,
wherein
the anolyte solution is a neutral solution.
In an embodiment 17, the system according to embodiment 16, wherein the
anolyte
solution has a pH between 7 and 10.
In an embodiment 18, the system according to embodiment 16 or 17, wherein the
neutral
solution is a KHCO3, K2SO4, or K2HPO4 solution.
In an embodiment 19, the system according to any one of embodiments 1 to 15,
wherein
the anolyte solution is an acidic solution.
In an embodiment 20, the system according to embodiment 19, wherein the
anolyte
solution has a bulk pH between 1 and 4.
In an embodiment 21, the system according to embodiment 19 or 20, wherein the
acidic
solution is a H3PO4 solution, H2SO4 solution or a combination thereof.
In an embodiment 22, the system according to any one of embodiments 1 to 21,
wherein
the anolyte solution has an anolyte concentration between 2.0 M and 0.01M,
optionally
about 0.1 M.
In an embodiment 23, the system according to any one of embodiments 1 to 22,
wherein
the catalyst layer of the cathode comprises copper (Cu), silver (Ag), platinum
(Pt), carbon
(C), or any combination thereof.
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
11
In an embodiment 24, the system according to any one of embodiments 1 to 23,
wherein
the cathode further comprises a gas diffusion layer for contacting the stream
of CO, CO2
or any mixture thereof, and the catalyst layer is deposited onto the gas
diffusion layer.
In an embodiment 25, the system according to embodiment 24, wherein the gas
diffusion
layer is a hydrophobic carbon paper, or a copper sputtered hydrophobic PTFE
layer.
In an embodiment 26, the, the system according to any one of embodiments 1 to
25,
wherein the anode comprises an anodic catalyst layer and an anodic current
collector
layer.
In an embodiment 27, the system according to embodiment 26, wherein the anodic
catalyst layer comprises one or more selected from 1r02, Pt, Pd, Ni, Ni0x,
CoOx.
In an embodiment 28, the system according to embodiment 26 01 27, wherein the
current
collector layer comprises Ti felt, hydrophilic carbon paper, or Ni foam.
In an embodiment 29, the system according to any one of embodiments 1 to 28,
wherein
the interfacial layer of the bipolar membrane comprises a water dissociation
catalyst.
In an embodiment 30, the system according to embodiment 29, wherein the water
dissociation catalyst is present as nanoparticles.
In an embodiment 31, the system according to embodiment 29 or 30, wherein the
water
dissociation catalyst comprises one or more selected from TiO2, 1r02, NiO,
Sn02,
graphene oxide, CoOx, ZrO2, A1203, Fe(OH)3, Mn02, Ru, Rh, RuPt alloy, PtIr
alloy, Ir, Pt.
In an embodiment 32, the system according to any one of embodiments 1 to 31,
wherein
the AEL is a membrane comprising poly(aryl piperidinium), polystyrene methyl
methylimidazolium, or polystyrene tetramethyl methylimidazolium.
In an embodiment 33, the system according to any one of embodiments 1 to 32,
wherein
the CEL comprises or consists of a sulfonated tetrafluoroethylene based
fluoropolymer-
copolymer.
In an embodiment 34, the system according to any one of embodiments 1 to 33,
further
comprising a temperature controller configured to maintain an operating
temperature
between 20 C and 50 C, optionally about 35 C.
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
12
In an embodiment 35, the system according to any one of embodiments 1 to 34,
having a
single-pass utilization of the stream of CO, CO2 or any mixture thereof of at
least 50% for
an inlet flowrate between 1 sccm and 15 sccm.
In an embodiment 36, the system according to any one of embodiments 1 to 34,
having a
single-pass utilization of the stream of CO, CO2 or any mixture thereof of at
least 60% for
an inlet flowrate between 1 sccm and 8 sccm.
In an embodiment 37, the system according to any one of embodiments 1 to 36,
having a
Faradeic Efficiency (FE) for conversion into the C2+ products of at least 20%
during at least
20 hours of operation and under an applied current density between 100 and 400
mA.cm-
2.
In an embodiment 38, the system according to embodiment 37, wherein the FE for
conversion into the C2, products is of at least 25% during at 30 hours of
operation and the
applied current density of 350 mA.cm-2.
In an embodiment 39, the invention provides a carbon oxides electroreduction
process for
converting 00, 002 or any mixture thereof into 02, products, the process
comprising:
supplying a catholyte solution and a stream of CO, CO2 or any mixture thereof
to
a cathodic compartment comprising a catalyst layer in contact with the
catholyte
solution:
flowing an anolyte solution through an anodic compartment having a product
outlet
to release the C2, products, the anodic compartment comprising an anode;
providing a bipolar membrane between the cathodic compartment and the anodic
compartment, the bipolar membrane comprising:
a cation-exchange layer (CEL) in cation communication with the catholyte
solution to provide protons into the catholyte solution;
an anion-exchange layer (AEL) in anion communication with the anolyte
solution to provide hydroxide ions into the anolyte solution; and
an interfacial layer defined between the cation-exchange layer and the
anion-exchange layer for splitting water into the protons and the hydroxide
ions; and
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
13
retaining a portion of the catholyte solution as a stationary catholyte layer
between
the catalyst layer of the cathode and the CEL and in contact with the CEL.
In an embodiment 40, the process of embodiment 39, comprising maintaining an
operating
temperature between 20 C and 50 C, and optionally about 35 C.
In an embodiment 41, the process of embodiment 39 01 40, wherein supplying the
stream
of CO, CO2 or any mixture thereof to the cathodic compartment is performed at
an inlet
flowrate between 1 sccm and 15 sccm.
In an embodiment 42, the process of any one of embodiments 39 to 41,
comprising
providing the cathode with an applied current density between 100 and 400
mA.cm-2
In an embodiment 43, the process of any one of embodiments 39 to 42,
comprising
forming the stationary catholyte layer by providing a solid porous support
between the
cathode and the CEL, and saturating the solid porous support with the
catholyte solution.
In an embodiment 44, the process of embodiment 43, wherein the saturating is
performed
to reach a liquid content of the stationary catholyte layer between 5 and 50
pL.cm-2,
optionally between 10 and 20 pLcnn-2, and further optionally about 10 pLcnn-2
when the
solid porous support is saturated with the catholyte solution.
In an embodiment 45, the process of any one of embodiments 39 to 44, wherein
the
catholyte solution is supplied with a concentration of cations between 0.25 M
and 3 M, and
optionally between 0.5 M and 2 M, and further optionally about 1M.
In an embodiment 46, the process of any one of embodiments 39 to 45, wherein
stationary
catholyte layer is formed with a thickness between 20 pm and 400 pm,
optionally between
20 pm and 300 pm, further optionally between 20 pm and125 pm.
In an embodiment 47, the process of any one of embodiments 39 to 46,
comprising
utilizing the stream of CO, CO2 or any mixture thereof according to a single-
pass utilization
of at least 50% for an inlet flowrate between 1 sccm and 15 sccm.
In an embodiment 48, the process of any one of embodiments 39 to 46,
comprising
utilizing the stream of CO, CO2 or any mixture thereof according to a single-
pass utilization
of at least 60% for an inlet flowrate between 1 sccm and 8 sccm.
In an embodiment 49, the process according to any one of embodiments 39 to 48,
comprising producing the 02, products according to a Faradeic Efficiency (FE)
for
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
14
conversion into the C2+ products that is of at least 20% during at least 20
hours of operation
and under an applied current density between 100 and 400 mA.cm-2.
In an embodiment 50, the process according to embodiment 49, comprising
producing the
02+ products according to the FE for conversion into the C2, products that is
of at least
25% during at 30 hours of operation and the applied current density of 350
mA.cm-2.
In an embodiment 51, the process of any one of embodiments 39 to 50, further
comprising
using the system as defined in any one of embodiments 2 to 38.
BRIEF DESCRIPTION OF THE DRAWINGS
The attached figures illustrate various features, aspects and implementations
of the
technology described herein.
Figures 1A and 1B schematically illustrate carbonate formation and CO2
crossover mechanisms in known alkaline and neutral media CO2RR electrolyzers
(prior
art): Figure 1A: conventional AEM-based flowing-electrolyte electrolyzer,
Figure1B: zero-
gap gas-phase electrolyzer (B);
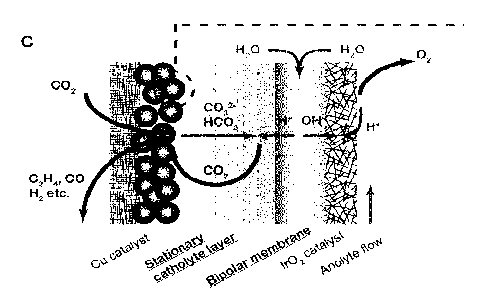
Figures 1C and 1D schematically illustrate carbonate formation and CO2
crossover mechanisms in the electroreduction system that is proposed herein:
Figure 1C:
bipolar membrane (BPM)-based stationary catholyte layer (SC)-MEA, Figure 1D:
mechanism of cation effects: potassium ions form an electrochemical double
layer on the
surface of a Cu catalyst, which modulates the local pH and prohibits the
proton adsorption,
and thereby enhances the selectivity of CO2RR and suppress that of HER (see
references
1 and 33). In SC-MEA, the carbonate/bicarbonate ions generated by the cathode
reactions
are reverted back to CO2 by the protons generated from water dissociation
inside the BPM.
The CO2 crossover in the present SC-MEA electrolyzer is thereby greatly
reduced in
comparison to known systems of Figure 1A and 1B.
Figures 1E and IF are scanning electron microscopy (SEM) images of the
cathode electrode (1E) and a cation exchange layer (CEL) / anion exchange
layer (AEL)
interface of the customized BPM used for SC-MEA in neutral anolyte (1F). The
customized
BPM consisted of a NafionTm 212 as CEL, Piperion as AEL, and a TiO2
nanoparticle layer
sandwiched in between as a water dissociation catalyst.
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
Figures 2A to 2F are graphs resulting from experimental investigations on the
CO2RR performance of the SC-MEA using neutral anolyte:
Figure 2A is a graph showing gas product FEs (`)/0) for the SC-MEA for zero
gap
(direct contact) and four catholyte solutions based on an applied current
density producing
5 maximum ethylene FE (marked on the top of each column) and at an
operating
temperature of 20 C. The dependence of cell voltage and gas FE on the current
density
can be found in Figures 7A to 70;
Figure 2B is a graph showing the dependence of cell voltage on current density
at
various operating temperatures;
10
Figure 2C is a graph showing the dependence of cathode gas products FEs on
current density at an operating temperature of 35 C;
Figure 20 is a graph showing CO2/02 ratio in an anode gas for a conventional
electrolyzer (black squares) and the present SC-MEA (grey squares) at various
current
densities. 02 and CO2 flow rates in the present SC-MEA are also indicated. The
plots
15 represent the data obtained after 1 hour of continuous electrolysis at
each current density;
Figure 2E is a graph showing the dependence of the cathode products FEs on an
inlet CO2 flow rate (sccm);
Figure 2F is a graph showing an SPU of CO2 versus the inlet CO2 flow rate. The
ideal SPU values of the conventional electrolyzers are marked for comparison.
'CO ideal'
and 'C2, (excl. acetate) ideal refer to AEM-based MEAs that produce 100% CO FE
and
100% 02, FE except for acetate (ideal SPU = 36.4%), respectively. The '
Simulated ideal'
refers to the simulated upper limit of SPU in an AEM-based MEA, assuming the
AEM-
based MEA has the same product distribution to the present BPM-based SC-MEA
operating at a CO2 flow rate of 1 sccm, and based on the assumption that one
extra CO2
is lost to carbonate per two OH- generated. The justification of the ideal SPU
simulation
can be found in SI1 of Supplemental Information.
Figures 3A to 30 are graphs resulting from experimental investigations on a
CO2RR performance of the present BPM-based SC-MEA using an acidic (pH = 2.37)
anolyte at an operating temperature of 35 C. The CO2RR performance at lower
pHs and
various temperatures are shown in Figures 14A, 14B and 15 in Supplemental
Information.
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
16
Under all the acidic conditions, the CO2 concentration in the anode gas was
below the
detection limit.
Figure 3A is a graph showing the dependence of cell voltage and cathode gas
products FEs on current density;
Figure 3B is a graph showing the dependence of the cathode products FEs on the
inlet CO2 flow rate;
Figure 3C is a graph showing the SPU of CO2 versus the inlet CO2 flow rate.
'CO
ideal' and 'C2, (excl. acetate) ideal' refer to the ideal SPU values of AEM-
based MEA
electrolyzers that produce 100% FE CO and 100% C2, except for acetate (ideal
SPU =
36.4%), respectively. The ' Simulated ideal' refers to the simulated upper
limit of SPU in
AEM-based MEA electrolyzers, assuming it has the same product distribution to
the
present BPM-based SC-MEA operating at an inlet CO2 flow rate of 1 sccm, and
based on
the assumption that one extra CO2 is lost to carbonate per two OH- generated.
The
justification of the ideal SPU simulation can be found in SI1 of Supplemental
Information;
Figure 3D is a graph showing ethylene concentration at the outlet stream for
the
electrolyzers operated at different pH and inlet CO2 flow rates.
Figures 4A to 4C are graphs showing an extended CO2RR performance of the
present BPM-based SC-MEA:
Figure 4A is a graph of ethylene FEs and cell voltages versus operating time
using
a neutral anolyte at an operating temperature of 35 C, an inlet CO2 flow rate
of 9.0 sccm,
and an applied current density of 250 mA.cm-2;
Figure 4B is a graph of ethylene FEs and cell voltages versus operating time
using
an acidic anolyte at an operating temperature of 35 C, an inlet CO2 flow rate
of 9.0 sccm
and an applied current density of 350 mA.cm-2; and
Figure 4C is a graph of the FE distribution and SPU versus operating time for
the
present BPM-based SC-MEA using an acidic anolyte (pH = 2.37) at an operating
temperature of 35 C and an inlet CO2 flow rate of 1.0 sccm.
Figures 5A to 5C are schematic representations of a hypothesized pH gradient
over layers of various cell configurations: AEM-based MEA (Figure 5A); SC-MEA
with a
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
17
pH = 10 buffer catholyte (Figure 5B); and SC-MEA with a non-buffer salt
catholyte (Figure
5C). The extent of the gradients in pH are not precisely known and are not
drawn to scale.
Figure 6 is a schematic illustration of CO2 regeneration in a stationary
catholyte
layer for a rough estimation of the impacts of a thickness of the stationary
catholyte layer
on the present BPM-based SC-MEA performance.
Figures 7A to 7C are graphs showing the FE and full cell voltage for the SC-
MEA
for various catholyte solution comprised in the stationary catholyte layer:
(Figure 7A) DI-
water; (Figure 7B) 0.25M K2SO4; (Figure 70) 2M KCI. In all cases, the anolytes
are 0.1M
KHCO3. The measurements were performed at 2000 with a CO2 inlet flow rate of
15 sccm.
Figure 8 is a graph showing ethylene FE and cell voltage with extended
operating
hours for the present BPM-based SC-MEA with a catholyte solution of 1M KHCO3
in the
stationary catholyte layer and an anolyte solution of 1M KHCO3 as the anolyte.
The
measurement was carried out at 20 'C with a CO2 flow of 9 sccm and a current
density of
100 mA cm-2.
Figures 9A to 9C relate to investigations on the CO2RR performance of an SC-
MEA based on a CEL (Nafion XL) with a catholyte solution of 0.5 M K2SO4 in the
stationary
catholyte layer and an anolyte solution of 0.5 M K2SO4 plus 0.1 (pH = 2.37) or
0.5 M (pH
= 1.84) H3PO4 as the anolyte. All the measurements were performed at an
operating
temperature of 20 'C and a CO2 flow rate of 15 sccm:
Figure 9A is a schematic representation of the present stationary catholyte
layer
in combination with an MEA;
Figure 9B is a graph showing the full cell voltage of the OEM-based SC-MEA at
various current densities;
Figure 9C is a graph showing gas products FEs of the CEM-based SC-MEA at the
same various current densities.
Figure 10 is a graph showing full cell voltage of a BPM-based SC-MEA at
various
current densities, for a water-splitting BPM including a layer of TiO2
nanoparticles as water
dissociation catalyst sandwiched by CEM and AEM (customized BPM), a
commercially
available BPM (Fumasep), and a membrane with OEM and AEM simply compressed
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
18
together. A 5 cm2 Pt/C loaded hydrophilic carbon paper and a 5 cm2 Ir02 loaded
Ti felt
were used as cathode and anode of the MEA, respectively.
Figure 11 is a graph showing a gas product FE and full cell voltage for the SC-
MEA based on commercially available BPM (Fumasep) and 0.1 M KHCO3 neutral
anolyte.
Figure 12A is a graph showing the gas products FE and full cell voltage for
the
SC-MEA based on customized BPM and 0.1 M H3PO4 + 0.5 M K2SO4 acidic anolyte,
and
Figure 12B is a graph showing the cell voltage versus operating time diagram
of the BPM-
based SC-MEA.
Figures 13A and 13B are graphs showing the dependence of gas products FE on
the current density of the BPM-based SC-MEA with 1M K of catholyte solution
in the
stationary catholyte layer and a solution of 0.1M KHCO3 (pH = 8.20) as the
anolyte for the
operating temperatures of 20 C (Figure 13A) and 50 C (Figure 13B), with the
CO2 inlet
flow rate of 15 sccm in both cases. Their cell voltages are shown in Figure
2B.
Figures 14A and 14B are graphs showing the dependence of gas products FE
(columns) and cell voltage (plot) on the current density of the BPM-based SC-
MEA with
1M K in the stationary catholyte layer and 0.1M H3PO4 + 0.5M K2SO4 (pH =
2.37) as
acidic anolyte for the operating temperatures of 20 C (Figure 14A) and 50 C
(Figure
14B), with the CO2 inlet flow rate of 15 sccm in both cases.
Figure 15 is a graph showing the dependence of gas products FE (columns) and
cell voltage (plot) on the current density of the BPM-based SC-MEA with a
catholyte
solution of 1M K+ in the stationary catholyte layer and a solution of 0.5M
H3PO4 + 0.5M
K2SO4 (pH = 1.84) as acidic anolyte for the operating temperatures of 35 C
with the CO2
inlet flow rate of 15 sccm.
Figure 16 is a graph showing a CO2: 02 ratio in an anode gas phase for the SC-
MEA using the presently customized BPM with a 0.1 M K2SO4 anolyte solution
(black), or
using a commercially available BPM (Funnasep) and a 0.1 M KHCO3 anolyte
solution (red).
The catholyte solution is 1M K+ in the stationary catholyte layer in both
cases. All the gas
samples were recorded after the cell operating for 1 hour at each current
density_ In K2SO4
anolyte, the CO2: 02 ratio is close to that in KHCO3 anolyte with the
customized BPM,
indicating that the detected anode CO2 flow in the customized BPM/KHCO3 system
(Figure
2D in the main text) was not ascribed to the acidification of bicarbonate. In
an SC-MEA
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
19
using KHCO3 anolyte, the CO2 crossover through Fumasep is similar to the case
through
customized BPM.
Figure 17: Investigations on the CO2RR performance of the SC-BPMEA using
catholyte thicknesses of 250, 125, 65 and 16 pm. All the results are collected
at 35 oC
with a CO2 flow rate of 10 sccm cm-2 (normalized by the geometric area of the
cathode),
a catholyte of 0.5 M K2SO4, and an anolyte of 0.1 M KHCO3. The dependence of
cell
voltages on current density. (b) Distribution of voltage losses measured in
the SC-BPMEA
with 65 pm thick catholyte operating at 200 mA cm-2 (cell voltage = 3.82 V).
Figure 18: Investigations on the cell voltage of the SC-BPMEA using the custom
BPM. The ohmic resistance (black) and the contribution to voltage loss (grey)
of the
stationary catholyte layer with various thicknesses. The errors in these
measurements are
below 1%.
Figure 19 Investigations on the CO2RR performance of the SC-BPMEA using
catholyte thicknesses of 250, 125, 65 and 16 pm. The dependence of the CO2RR
gas
products FE on the current density for the SC-BPMEAs with the catholyte
thickness of 250
(a), 125 (b), 65 pm (c) and 16 pm (d).
Figure 20 The exploration of the CO2RR performance and energy intensity of SC-
BPMEA with restricted reactant availability. All the measurements were
conducted at 35
C and 200 mA cm-2, and the data were collected after 2 h of continuous
operation. Carbon
balance in SC-BPMEA with 65 pm 0.5 M K2SO4 at different input CO2 flow rates.
See
Supplementary Fig. 13b for plots on a logarithmic scale.
Figure 21 The exploration of the CO2RR performance and energy intensity of SC-
BPMEA with restricted reactant availability. All the measurements were
conducted at 35
C and 200 mA cm-2, and the data were collected after 2 h of continuous
operation. The
total CO2 single-pass utilization (the 002-to-ethylene single-pass conversion
see Fig. 18)
for the SC-BPMEAs with different catholyte thickness and input CO2 flow rates.
Figure 22 The exploration of the CO2RR performance and energy intensity of SC-
BPMEA with restricted reactant availability. All the measurements were
conducted at 35
C and 200 mA cm-2, and the data were collected after 2 h of continuous
operation. (a-c)
The FE distributions and the CO2 requirements (total CO2 converted to
products) of the
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
SC-BPMEAs with different catholyte thickness and input CO2 flow rates (sccm
normalized
by electrode area). Ci refers to CO, formate and methane. C2, refers to
ethylene, ethanol,
acetate and n-propanol.
Figure 23 (a) The FE and full cell voltage for the SC-BPMEA based on 125 pm
0.5
5 M K2SO4, custom BPM and 0.1 M H3PO4 + 0.5 M K2SO4 anolyte. (b) The cell
voltage
versus operating time diagram of the cell.
Figure 24 Measurements of CO2 SPU of the SC-BPMEAs with 125 pm 0.5 M
K2SO4, operating at 300 mA cm-2 (a, b) or 200 mA cm-2 (c, d). The operating
anolytes are
(a, b) 0.1 M H3PO4 + 0.5 M K2SO4 (pH = 2.3) and (c, d) 0.1 M KOH (pH = 13.3).
All the
10 experiments were conducted at 35 C. (a, c) The FE distributions at
different input CO2
flow rates. (b, d) The total CO2 SPU and CO2-to-ethylene conversion at
different input flow
rates.
DETAILED DESCRIPTION
Techniques described herein relate to an electroreduction system that can be
used to
15 convert carbon oxides selected from CO, CO2 or any mixture thereof into
multicarbon
products with an enhanced single pass utilization (SPU) of CO2 or CO by
comparison to
know flow cells or membrane electrode assemblies (MEAs). Multiple factors can
affect the
SPU in electroreduction systems. The present electroreduction system
particularly
includes a stationary catholyte layer being configured to facilitate mass
transfer via
20 diffusion of regenerated CO2 or CO across the stationary catholyte layer
and back to an
adjacent cathode of the system.
It should be noted that the system and related process implementations that
are described
herein in relation to CO2 electroreduction can be applied to CO
electroreduction, or the
electroreduction of a mixture of CO2 and CO, without departing from the scope
of the
present techniques.
In contrast to known flow cell electrolyzers, the catholyte solution of the
present
electroreduction system is not provided flowing in and out of a cathodic
compartment but
rather remains within the cathodic compartment as a stationary catholyte layer
between a
cathode and a membrane separating the cathodic compartment from an adjacent
anodic
compartment. More particularly, referring to Figure 1C, the electroreduction
system
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
21
includes a bipolar membrane (e.g., including a cation-exchange layer (CEL), an
interfacial
layer comprising a water dissociation catalyst, and anion-exchange layer
(AEL)) that
separates an anodic compartment from a cathodic compartment. The bipolar
membrane
is used to dissociate water, thereby providing hydroxide ions to the anodic
compartment
side and protons to the cathodic compartment side. The electroreduction system
further
includes a cathode (e.g., gas diffusion layer plated with Cu catalyst) that is
positioned in
the cathodic compartment, and an anode (e.g. hydrophilic electrode such as
Ir02 catalyst
coated on a support of Ti felt) that is positioned in the anodic compartment.
As seen in
Figure 10, CO2 can be provided to the cathode via an inlet of the cathodic
compartment
so as to be converted into gas products (021-14, CO, H2) at a surface of the
cathode. A
portion of the CO2 can be lost to carbonate formation. The present
electroreduction system
further includes the stationary catholyte layer sandwiched between the bipolar
membrane
and the cathode, with the stationary catholyte layer comprising a catholyte
solution that
receives the carbonate/bicarbonate ions derived from the lost CO2 portion. As
protons are
provided from the bipolar membrane into the catholyte solution, these protons
can
combine with the carbonate/bicarbonate ions in the catholyte solution to
regenerate the
portion of CO2 that did not serve to form gas products. The regenerated CO2
can then
diffuse back to the cathode surface to form the gas products that will be
recovered in a
cathode gas mixture. In summary, the bipolar membrane can favor the conversion
of
carbonate/bicarbonate ions back to CO2 and further prevent ions crossover
between the
anodic and cathodic compartments. The stationary catholyte layer further
enables local
alkalinity to promote CO2RR in (bulk) acidified catholyte solution, and
facilitates thereby
that the regenerated CO2 participates in CO2RR reactions.
In an embodiment, the catalyst layer of the cathode comprises copper (Cu),
silver (Ag),
platinum (Pt), carbon (C), or any combination thereof. In an embodiment, the
cathode
further comprises a gas diffusion layer for contacting the CO2 stream, and the
catalyst
layer is deposited onto the gas diffusion layer. For example, the gas
diffusion layer is
hydrophobic. For example, the gas diffusion layer is a hydrophobic carbon
paper, or a
copper sputtered hydrophobic PTFE layer. In the context of the invention,
hydrophobic
means a water contact angle following ISO 19403-6:2017 of at least 30 .
In an embodiment, the anode comprises an anodic catalyst layer and an anodic
current
collector layer. For example, the anodic catalyst layer comprises one or more
selected
from Ir02, Pt, Pd, Ni, Ni0x, CoOx. For example, the current collector layer
comprises Ti
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
22
felt, hydrophilic carbon paper, or Ni foam. In the context of the invention
hydrophilicmeans
a water contact angle following ISO 19403-6:2017 below 30 .
Referring to Figure 6, the thickness of the stationary catholyte layer can be
selected to
enhance the mass transport via diffusion of the regenerated CO2 to the cathode
surface.
The stationary catholyte layer can be characterized as including a boundary
where CO2 is
regenerated and from which regenerated CO2 can diffuse towards the cathode
surface
(Cu) or the CEL, this diffusional phenomenon being driven by a concentration
gradient.
The distances from this boundary to the cathode surface and CEL are noted as
Li and L2,
respectively. The portion of the stationary catholyte layer between the
boundary and the
cathode surface can be defined as a diffusion layer having a diffusion layer
thickness Li.
The thickness of the stationary catholyte layer of the present system is
selected to
minimize the diffusion layer thickness, Li, and facilitate regenerated CO2
mass transport
while providing a mechanically robust stationary catholyte layer. The
diffusion layer
thickness L1 can be estimated based on physical properties of protons and
carbonates.
For example, one can determine the position where protons and carbonates meet
each
other by estimation of encounter problems, and considering that the speed is
proportional
to their mobility. More precise determination can be via a cross-platform
finite element
analysis, solver and multiphysics simulation software such as COMSOL .
The stationarity of the catholyte layer as defined herein thus prevents the
regenerated CO2
to be flushed away from the cathodic compartment and allows the CO2 to diffuse
back into
the cathode for conversion thereof into value-added products. In other words,
being
stationary means that the catholyte is not flowing out of the cathodic
compartment during
CO2 conversion and that the volume of the catholyte solution contained in the
stationary
catholyte layer remains between the bipolar membrane and the cathode during
operation
of the electroreduction system. One skilled in the art will understand that
the catholyte
solution can flow/move within the stationary catholyte layer according to
various mass
transport mechanisms (diffusion, migration, convection if any).
The nature of the catholyte solution of the stationary catholyte layer can be
further selected
to reduce the diffusion layer thickness. For example, it was noted from the
experimentation
discussed herein that a non-buffered catholyte solution can shorten the CO2
path length
(including the diffusion layer thickness) in comparison to a buffered
catholyte solution,
such as KHCO3.
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
23
To enhance the mechanical robustness of the stationary catholyte layer, the
stationary
catholyte layer can include a solid porous support having pores that are
saturated with the
catholyte solution. For example, the porous solid support is or comprises
polyvinylidene
fluoride (PVDF), polytetrafluoroethylene (PTFE), polycarbonate, nylon,
cellulose acetate,
cellulose nitrate, polypropylene, alumina, or any combinations thereof. The
solid porous
support has a mean pore diameter (i.e., a mean pore size) between 0.05 and 50
pm as
determined by scanning electron microscopy; for example, between 0.08 and 25
pm; for
example, between 0.10 and 10 pm; for example, between 0.15 and 5 pm; for
example,
between 0.20 and 1.0 pm; for example, between 0.30 and 0.80 pm. For example,
the solid
porous support has a mean pore size of 0.45 pm. The porous solid support can
be
provided to contact the cathode surface at one side and the cation exchange
layer of the
bipolar membrane at another side thereof. The thickness of the stationary
catholyte layer
can thus be equal to the thickness of the solid porous support.
The thickness of the stationary catholyte layer can be at most 280 pm as
measured by a
spiral micrometer.; preferably, at most 250 pm; preferably, at most 220 pm;
preferably at
most 200 pm; preferably at most 180 pm; preferably, at most 150 pm; preferably
at most
140 pm; preferably, at most 130 pm; and more preferably at most 125 pm.
The thickness of the stationary catholyte layer can be at least 20 pm as
measured by a
spiral micrometer.; preferably, at least 25 pm; preferably, at least 30 pm;
preferably at
least 40 pm; preferably at least 45 pm; preferably, at least 50 pm; preferably
at least 55
pm; preferably, at least 60 pm; and more preferably at least 65 pm.
The thickness of the stationary catholyte layer can be ranging from 20 to 280
pm as
measured by a spiral micrometer.; for example, from 20 to 250 pm; for example,
from 30
to 220 pm; for example, from 40 to 200 pm; for example, from 45 to 180 pm; for
example,
from 50 to 150 pm; for example, from 55 to 140 pm; for example, from 60 to 130
pm; for
example, from 65 to 125 pm. The thickness of the stationary catholyte layer is
measured
by a spiral micrometer. For example, the thickness of the stationary catholyte
layer can be
about 125 pm, when measured by a spiral micrometer.
However, the thickness of the stationary catholyte layer is not to be bound to
these values
and is selected to minimize the diffusion layer thickness while maintaining
the mechanical
robustness of the stationary catholyte layer. The mechanical robustness can
refer herein
to a resistance to compression that can be estimated for example via a
compression-
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
24
stress test. In other words, the stationary catholyte layer is configured to
resist
compression an maintain sufficient thickness to avoid direct contact of the
BPM with the
cathode. For example, the thickness of the stationary catholyte layer could be
inferior to
125 pm if a solid porous support having such thickness is used and yet
maintain
robustness.
When the solid porous support is saturated with the catholyte solution, the
stationary
catholyte layer has a liquid content between 5 and 50 pLcm-2; for example,
between 8
and 35 pL.cm-2; for example, between 10 and 20 pL.cm-2. For example, the
stationary
catholyte layer has a liquid content of about 10 pLcm-2
When analyzing CO2 crossover in known neutral media electrolyzers (see section
SI1 of
Supplemental Information), it was concluded that achieving high carbon
efficiency can be
achieved based on two requirements. Firstly, the carbonate/bicarbonate ions
that are
formed from CO2 absorption in locally alkaline catholyte solution should not
reach the
anode compartment side. Secondly, the carbonate/bicarbonate ions that were
formed
near the cathode should revert back to CO2, and participate in CO2RR (and not
mix with
CO2RR products).
In known neutral media flow cells, BPMs (e.g. Fumasep FBM) can be used to
convert
carbonate/bicarbonate back to CO2 and to block CO2 crossover to the anode (see
references 10 and 14). A conventional BPM can consist of a cation-exchange
layer (CEL)
laminated with an anion-exchange layer (AEL). With the CEL facing the cathode
side, the
concentration (and hence conductivity) of carbonate/bicarbonate anions in the
BPM is
substantially reduced due to the Donnan effect (see reference 17). The BPM
also
generates protons and hydroxide ions via water dissociation at the junction
between the
CEL and AEL(see references 18 and 19) under appropriate external potential.
The protons
are driven to the cathode surface, where, with judicious device engineering,
they can
intercept carbonate/bicarbonate, reverting it to 002.
However, in the BPM-based flow cells, the catholyte reacts with and absorbs
CO2. Even
at steady-state (catholyte is saturated by CO2 absorption), the CO2
regenerated at the
catholyte/BPM interface can be flushed away from the cathode surface due to
catholyte
flow, thereby releasing CO2 with gaseous CO2RR products in the cathodic
compartment,
rather than diffusing back to the cathode to participate in CO2RR (see
reference 14). As a
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
result, the CO2 loss due to carbonate/bicarbonate formation is ca. twice the
amount
transformed to products, similar to AEM-based electrolyzers.
It was thus hypothesized that using a BPM in a device without a flowing
catholyte solution,
such as a membrane electrode assembly (MEA), could prevent CO2 crossover to
the
5 anode, minimize CO2 loss via absorption to the bulk electrolyte, avoid
flushing CO2 away,
and thereby maximize the SPU. However, another issue was observed, i.e.
acidification
of the cathode (see references 1 and 20 to 22). The herein discussed
experiments (Figure
2A) showed that a Cu catalyst can produce 100% hydrogen at current densities
from 50
to 200 mA cm-2 when contacted with a cation exchange layer (CEL) of the BPM.
10 Previous reports (see references 22 and 23) have suggested inserting a
solid porous
support layer (a buffer layer) saturated with DI water or a KHCO3 solution as
the catholyte
solution between the cation-exchange membrane and an Ag catalyst layer
(cathode) to
improve the selectivity of CO2-to-CO reactions over HER. The presently
described system
was then developed to evaluate the potential of BPM-based MEA for producing
C2+, using
15 a Cu catalyst, and with the goal of a minimum of CO2 loss through
engineering the
catholyte solution. The analysis provided in section 612 of the Supplemental
Information
reveals that, in principle, the in-situ CO2 recovery in a BPM-based MEA can
potentially
become energy-efficient and may also aid on projected capital costs.
Here is demonstrated that BPM-based MEAs incorporating a stationary catholyte
layer
20 between catalyst and BPM can produce 02+ with significantly reduced CO2
loss (Figure
1C). After a series of optimizations on the stationary catholyte layer, it was
discovered that
cations (Figure 1D) enable ongoing CO2-to-02+ production on the Cu catalyst.
The BPM
was used as a proton source to regenerate CO2 in-situ and then reduce it to
02+ products
on a copper (Cu) catalyst (Figure 10). By allowing the catholyte solution to
be provided in
25 a stationary catholyte layer, the regenerated CO2 has the opportunity to
participate in
CO2RR. The presently described system can be referred to as a stationary-
catholyte MEA
(SC-MEA) or a BPM-based SC-MEA. CO2RR on Cu catalyst with an SPU of at least
60%
(02, FE of 26%) was achieved, which twice the value of the best prior known
electrolyzers
producing 02+ (see references 2,4, 15 and 24) and, as a result, the
theoretical upper limit
of SPU for neutral/alkaline C2, systems previously demonstrated was also
surpassed.
When run at a total current density of 350 rnA.cm-2, the present system
maintains an
ethylene FE at a steady rate of above 30% for more than 30 hours of continuous
operation.
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
26
Thus, the BPM of the present disclosure comprises a cation-exchange layer
(CEL) in
cation communication with the catholyte solution to provide protons into the
catholyte
solution; an anion-exchange layer (AEL) in anion communication with the
anolyte solution
to provide hydroxide ions at a surface of the anode, and an interfacial layer
defined
between the cation-exchange layer and the anion-exchange layer for splitting
water into
the protons and the hydroxide ions. The interfacial layer can comprise a water
dissociation
catalyst; with preference that the water dissociation catalyst comprises one
or more
selected from TiO2, 1r02, NiO, Sn02, graphene oxide, CoOx, ZrO2, A1203,
Fe(OH)3, Mn02,
Ru, Rh, RuPt alloy, PtIr alloy, Ir, Pt. More preferably, the water
dissociation catalyst can
be a combination of 1102 on the AEL side) and NiO on the CEL side. In an
embodiment,
the water dissociation catalyst is present as nanoparticles.
In an embodiment, the AEL is a membrane comprising poly(aryl piperidinium),
polystyrene
methyl methylimidazolium, or polystyrene tetramethyl methylimidazolium.
In an embodiment, the CEL is a NafionTm membrane (CAS number 31175-20-9)
In an embodiment, the system further comprises a temperature controller
configured to
maintain an operating temperature between 20 C and 50 C, for example, 25 C and
45 C;
for example, 30 C and 40 C optionally about 35 C.
In an embodiment, the system is having a single-pass utilization of the CO2
stream of at
least 50% for a CO2 inlet flowrate between 1 sccm and 15 sccm. In an
embodiment, the
system is having a single-pass utilization of the CO2 stream of at least 60%
for a CO2 inlet
flowrate between 1 sccm and 8 sccm. In an embodiment, the system is having a
Faradeic
Efficiency (FE) for conversion into the 02, products of at least 20% during at
least 20 hours
of operation and under an applied current density between 100 and 400 nriA.cnn-
2. In an
embodiment, the FE for conversion into the C2+ products is of at least 25%
during 30
hours of operation and the applied current density of 350 mA.cm-2.
The present disclosure also relates to a CO2 electroreduction process for
converting CO2
into C2+ products, the process comprising:
supplying a catholyte solution and CO2 to a cathodic compartment comprising a
catalyst
layer in contact with the catholyte solution:
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
27
flowing an anolyte solution through an anodic compartment having a product
outlet to
release the C2, products, the anodic compartment comprising an anode;
providing a bipolar membrane between the cathodic compartment and the anodic
compartment, the bipolar membrane comprising:
a cation-exchange layer (CEL) in cation communication with the catholyte
solution to
provide protons into the catholyte solution;
an anion-exchange layer (AEL) in anion communication with the anolyte
solution/anode?
to provide hydroxide ions into the anolyte solution; and
an interfacial layer defined between the cation-exchange layer and the anion-
exchange
layer for splitting water into the protons and the hydroxide ions;
retaining a portion of the catholyte solution as a stationary catholyte layer
between the
catalyst layer of the cathode and the CEL and in contact with the CEL
For example, the process uses the system described above.
Catholyte solution engineering for high SPU of CO2 feedstock in a CO2-to-C2.
electrolyzer
Figure 1C schematically illustrates a BPM-based SC-MEA as encompassed herein
with
related reactional and diffusional mechanisms. Figure 1E is an SEM photograph
of a
cathode that was prepared by spraying Cu nanoparticles onto a hydrophobic
carbon paper
(gas diffusion layer) for CO2RR. For example, the anode can be Ir02 supported
on titanium
felt to support the oxygen evolution reaction (0ER). Figure 1F is an SEM
photograph of a
customized BPM under reverse bias that was installed with the anion-exchange
layer
(AEL) contacting the anode, and the cation-exchange layer (CEL) contacting the
stationary catholyte layer. The cathode was then compressed onto the solid
porous
support of the stationary catholyte layer, and anodic and cathodic flow-field
plates
sandwiched the system.
In contrast with previous works that used 420 to 800 pm glass microfiber
filters (see
references 22 and 23), the present system can include a porous stationary
catholyte layer
having a thickness that is below 400 pm, e.g. about 125 pm and including a
solid porous
support that is configured to be saturated with the catholyte solution. The
solid porous
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
28
support can be, for example, a PVDF filter membrane having a mean pore size
(i.e.
diameter) of 0.45 pm and configured to receive a liquid content of about 10
pLcm-2. In
addition to apparent benefits such as lower CO2 absorption capacity (ca. 0.1
mmol cm-2)
and lower ohmic resistance, the lower thickness of this porous stationary
layer allows for
improving the mass transport efficiency of the in-situ recovered CO2 to
catalyst
(quantitatively simulated in section SI3 of Supplemental Information provided
further
below) in comparison to known systems.
It was further discovered that the composition of the stationary catholyte
layer greatly
impacts CO2RR performance in SC-MEA (analyzed and rationalized in sections SI2
and
SI3 of Supplemental Information provided further below). Rather than DI-water
or KHCO3
as used in previous studies (see references 22 and 23), the catholyte solution
can be
designed as a non-buffered catholyte solution, e.g. K2SO4, in order to
introduce cation
effects as a means to promote selectivity to CO2RR over HER (see references 25
and 26).
Under external potential, cations such as K can form an electrochemical
double layer on
the catalyst surface (see Figure 1D), introducing changes in polarity,
absorption
preference, local pH, and local CO2 concentration, as observed and modeled
before (see
references 25 and 26). Increasing K+ concentration from 0 to 2 M in the
stationary catholyte
layer was seen to enhance CO2RR selectivity. At optimized current densities
(referring to
Figures 7A to 7C further detailed in Supplemental Information), the ethylene
FE was
observed to increase from 0.5% (0 M) and 2.5% (0.5 M) to 25% (1 M) and 27% (2
M). The
enhancement of CO2RR selectivity (Figures 2A and 7A to 7C) as K concentration
increases, implies a predominant role of cation effects in promoting CO2RR in
the present
BPM-based SC-MEA. Despite higher ethylene FE and lower hydrogen FE, 2 M lc in
the
stationary catholyte also resulted in carbonate salt precipitation at the back
of the carbon
paper of the cathode, which caused the loss of K+ and obstructed the mass
transport of
CO2 over time (see reference 27).
Therefore, the cation concentration of the catholyte solution is ranging from
0.25 M to 6.00
M; for example, from 0.50 M to 3 M; for example, from 0.50 M to 2.80 M, for
example from
0.75 M to 2.50 M, for example from 1.00 M to 2 M; for example, the cation
concentration
of the catholyte solution is about 1 M. In an embodiment, the cations in the
catholyte
solution are one or more selected from K+, Na, Cs, Rb+, NH4, Mg2+, Ca2+, Al3+.
For
example, the catholyte solution can be a solution of K2SO4 having a K+
concentration
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
29
equal to or greater than 0.5M; preferably equal to or greater than 1.0 M, more
preferably
equal to or greater than 1.5 M. For example, the catholyte solution can be a
solution of
K2804 having a K+ concentration of at most 3.00 M.
In an embodiment, the catholyte solution is buffered. For example, the
buffered solution is
a solution comprising one or more selected from deionized (DI) water, KHCO3,
K3PO4,
K2HPO4, KH2PO4 or the buffered solution is a mixture of glycine- and sodium
hydroxide,
or a mixture of H3B03 and sodium hydroxide.
In another embodiment, the catholyte solution is non-buffered. For example,
the non-
buffered solution is or comprises K2SO4, KCI or any other combinations of the
CI- anions
or S042- anions with Nat, Cs, Rb+, NH4, me, c_2 ,
a or Al3+ cations.
The anolyte solution has an anolyte concentration between 2.0 M and 0.01M; for
example,
between 1.5 M and 0.05M; for example, between 1.0 M and 0.08 M; for example,
between
0.5 M and 0.1 M. For example, the anolyte solution has an anolyte
concentration of about
0.1 M.
In an embodiment, the anolyte solution is neutral. For example, the anolyte
solution has a
pH between 7.0 and 10.0; preferably a pH between 7.5 and 9.5. For example, the
anolyte
(neutral) solution is a KHCO3, K2SO4, or K2HPO4 solution.
In another embodiment, the anolyte solution is acidic. For example, the
anolyte solution
has a pH between 1.0 and 4.0; for example, between 1.5 and 3.5; for example,
between
2.0 and 3Ø For example, the acidic solution is an H3PO4 solution, H2SO4
solution or a
combination thereof.
The mechanism of preventing CO2 crossover in the SC-MEA is depicted in Figure
1C.
Under applied potential, the protons generated at the CEL/AEL interface of the
BPM
migrate to the cathode, while the carbonate/bicarbonate ions generated at the
cathode
migrate to the GEL side of the BPM. The carbonate/bicarbonate ions are
reverted to CO2
when being intercepted by the protons near the interface between the
stationary catholyte
layer and CEL (at the boundary of the diffusion layer) and subsequently
diffuse back to
the cathode to participate in CO2RR.
Proton-induced CO2 regeneration could also be accomplished by coupling a
cation-
exchange membrane and acidic anolyte since the anodic OER can supply protons.
It was
observed that the CO2 crossover in this system is below the detection limits.
However, the
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
experimental and theoretical analyzes showed that this system does not operate
continuously because of co-ion transport and water balance issues (discussed
in SI3 of
Supplemental Information).
BPM prevents CO2 crossover in SC-MEA using neutral anolyte
5 To compare the CO2 crossover in the present BPM-based SC-MEA with the
known AEM-
based neutral media electrolyzers, the CO2RR performance of the present BPM-
based
SC-MEA was firstly measured, using a flowing neutral anolyte solution (0.1 M
KHCO3, pH
-8.2), and the results are summarized in Figures 2A to 2F.
The operating temperature on CO2RR performance was then optimized and it was
10 discovered that 35'C can be the optimal temperature in the presently
designed BPM-
based SC-MEA (see Figures 2B, 20, 13A and section SI5 of Supplemental
Information).
A CO2/02 ratio of an anodic gas mixture (also referred to as an anode gas) was
further
studied to evaluate the capability to prohibit CO2 crossover in the BPM-based
SC-MEA,
the key to achieving a high SPU (SI1) (see references 13 and 14). In agreement
with
15 previous studies (see references 13 and 14), the AEM-based MEA showed
002/02 ratios
very close to 2 for current densities from 100 to 300 mA.cm-2 (Figure 2D),
indicating that
the majority of the anionic charge carrier in AEM-based MEA is 0032-, which
causes ca.
one molecule of CO2 loss per two electrons transferred.
Conversely, the CO2/02 ratio in the anode gas produced in the present BPM-
based SC-
20 MEA is one order of magnitude lower than that in AEM-based MEA,
evidencing the
prevention of CO2 crossover. The detected anode CO2 flow is not ascribed to
the
acidification of KHCO3, as supported by the performed control experiments
(Figure 16 in
SI6 of Supplemental Information). The CO2/02 ratio of the anode gas decreased
as the
operating current density increased, which was assigned to the fact that
higher current
25 density (also higher cell voltage) decreases the pH at the CEL surface
and lowers the
effective diffusion coefficient of CO2 and HCO3-/003- in the CEL where they
must move
against the outward flow of hydrated H+ (see references 19 and 28). Given that
in the SC-
MEA, CO2 crossover is greatly reduced, and the catholyte solution is provided
in the
stationary catholyte layer, the CO2 that was regenerated from
carbonate/bicarbonate ions
30 can diffuse back to the cathode for participating in CO2RR. Therefore,
the present BPM-
based SC-MEA is shown to present high CO2 SPU at a low inlet CO2 flow rate.
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
31
By decreasing the inlet CO2 flow rate, it was further demonstrated that a 6PM-
based SC-
MEA operating in neutral (pH = 8.20) anolyte solution and at 35 C can achieve
an SPU of
about 61%, which is a significant improvement in carbon efficiency compared to
the known
neutral-media AEM-based CO2RR electrolyzers. Figure 2E shows FEs of gas
products at
the cathode within a given range of CO2 feed rates (Figure 2C). Lowering the
CO2 flow
rate from 15 (Figure 3a) to 8 sccm (Figure 3b) was shown not to cause a
significant change
in the gas product distribution. However, a further decrease in CO2 flow rate
led to the
domination of HER over the CO2RR, likely due to the limited CO2 mass transport
(see
reference 15). At the CO2 flow rates investigated, it was observed that 10% of
the CO2RR
FE was 'missing', which can be attributable to the liquid products being
oxidized at the
anode and/or being trapped in the stationary catholyte layer and thus not
found in the
analysis. The SPU was calculated by substituting the products FE values into
Equation
(3) in section 511 of Supplemental Information and is reported in Figure 2F.
As the flow
rate decreases, the SPU of SC-MEA increases from 23% (8 sccm) to 61% (1 sccm),
exceeding the upper limit of the SPU for the ordinary electrolyzers producing
fully CO
(50%) or fully C2, (excluding acetate) products (25%), and is also higher than
the state-of-
art reported SPU (30%) for producing 02+ products (see reference 15). The
upper limit of
SPU for an AEM-based electrolyzer was also simulated. Assuming that the AEM-
based
electrolyzer had a similar product distribution and one extra CO2 is lost to
carbonate per
two OH- generated, the upper limit of the resulting SPU should be in the range
of 13% to
15% (red zone in Figure 2F, simulation details in section 511 of Supplemental
Information).
Acidic anolyte further suppresses CO2 crossover
When using a neutral anolyte solution in the present BPM-based SC-MEA, the CO2
crossover ¨ despite being significantly reduced ¨ was not down to zero. In the
neutral
anolyte solution, some of the CO2 generated in the stationary catholyte layer
may diffuse
through the BPM's CEL, combining with hydroxide ions in the AEL and migrate to
the
anode. When using an acidic anolyte solution, it was hypothesized that the
hydroxide ions
in the AEL could be partially replaced by the anionic species in the anolyte
solution, such
as H2PO4-, S042-, which might inhibit carbonate/bicarbonate formation and
crossover. It
was indeed observed that when operating in an acidic anolyte solution with a
bulk pH of
2.37 (0.1 M H3PO4+ 0.5 M K2SO4), the CO2 content at the anodic gas stream was
below
the detection limit in the operating current density range from 100 to 400 mA
cm-2.
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
32
The impacts of operating temperature and anolyte acidity on the performance of
the
present BPM-based SC-MEA (Figure 3A, 14A, 14B, 15 and SI5) was studied. Under
optimized operating conditions (e.g., 35 C, anolyte pH = 2.37), the BPM-based
SC-MEA
exhibited an ethylene FE of 38% at an applied current density of 350 mA cm-2.
The
dependence of cathode FE on the inlet CO2 flow rate is shown in Figure 3B. In
an acidic
anolyte solution, the CO2 mass transport limitation occurs when the CO2 inlet
flow rate is
reduced from 4 sccm to 2 sccm, which is lower than that in a neutral anolyte
solution. This
effect also leads to a higher maximum outlet ethylene concentration in the
acidic anolyte
solution (8.6%) than that in the neutral anolyte solution (6.2%), both of
which were
achieved at an inlet CO2 flow rate of 4 sccm (Figure 3D). These results are
ascribed to the
better efficiency (as seen from the anode gas analysis) of CO2 recycling in
the acidic
anolyte solution ¨ this compensates for the low CO2 feeding when mass
transport limits
set in. At an inlet CO2 flow rate of 1 sccm, the BPM-based SC-MEA achieved an
SPU of
60% (Figure 3C): which is twice the highest experimental SPU reported for
known AEM-
based MEA that produced C2, (see reference 15). As a reference, a simulated
AEM-based
MEA showed an SPU of 15 1%.
Table 1: Summary of energy penalty associated with CO2 recovery energy
consumption
for SC-MEA, simulated AEM-based MEA (Figure 30), and benchmark neutral media
AEM-
based MEA electrolyzer from literature.
SC-MEA (acidic, SC-MEA (acidic, Reference 15
1sccm) 4 sccm) MEA
CO2 SPU (%) 60 3 39 3 30
Ethylene FE ( /0) 14 2 37 0.5 25
Inlet CO2 flow rate
1 4 2
(sccm)
CO2 recovery (GJ per
2.8 0.3 6.6 0.7 10
Ton utilized CO2) a
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
33
CO2 recovery (GJ per
Ton produced ethylene) 20 3 46 3 60
a
a All the evaluated devices are flowing-catholyte-free MEA cells operating in
neutral
or acidic anolytes, of which the CO2 absorption is negligible. Therefore, a
4.3
GJ/Ton CO2 separation energy consumption (see reference 29) was implemented
based on the amine capture process from mixed gas for all the unutilized CO2
from
both cathode and anode.
Table 1 summarizes and compares the CO2 recovery energy consumptions of the SC-
MEA (acidic anolyte), and the literature benchmark neutral media AEM-based MEA
electrolyzer (see reference 15). Coupling the advantages of minimized CO2
crossover
(enabled by BPM) and acidic bulk anolyte solution, the present BPM-based SC-
MEA can
allow a 86% and 72% reduction in energy penalty associated with CO2 recovery
compared
to the simulated MEA electrolyzer and literature benchmark neutral media
e1ectr01yzer15,
respectively. These results highlight the need for high-SPU CO2RR devices,
i.e., lower
energy consumption.
CO2RR stability of the present BPM-based SC-MEA
The stability of the proposed BPM-based SC-MEA operating under optimized
conditions
(in terms of ethylene FE) (Figures 4A and 4B) was then investigated. In both
neutral
(Figure 4A) and acidic (Figure 4B) anolyte solutions, the SC-MEA was fed with
9 sccm
CO2 and exhibited stable cell voltages at around 5 V and ca. 30% to 40%
ethylene FE
throughout 25 hours (neutral) or 30 hours (acidic) of continuous operation.
The operating
time of the SC-MEA under the conditions that enable high SPU (i.e., anolyte
bulk pH of
2.37, the operating temperature of 35 C, and inlet CO2 flow rate of 1 sccm)
was also
extended. Under this condition, the SC-MEA was less stable than the ones fed
by 9 sccm
CO2 ¨ the C2, FE dropped by ca. 37% after 5 hours accompanied by decreased
SPU. In
the SC-MEA fed by 9 sccm CO2 flow, the products could be carried out by
unreacted CO2.
In the case of 1 sccm CO2 inlet flow rate, the mass exchange efficiency can be
lower than
the 9 sccm cases, which can lead to the over-accumulation of CO2RR products
and
consequently lower the activity of Cu catalyst (see references 30 to 32). This
over-
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
34
accumulation issue is a newly discovered challenge in high-SPU electrolyzers,
calling for
innovations of system design and catalyst in the future. Nevertheless,
throughout the 5
hours of continuous operation, SC-MEA maintained SPUs greater than that of the
neutral
and alkaline media electrolyzers.
Several alternative implementations and examples have been described and
illustrated
herein. The implementations of the BPM-based SC-MEA described above are
intended
to be exemplary only. A person of ordinary skill in the art would appreciate
the features of
the individual implementations and the possible combinations and variations of
the
components. A person of ordinary skill in the art would further appreciate
that any of the
implementations could be provided in any combination with the other
implementations
disclosed herein. It is understood that the developed design may be embodied
in other
specific forms without departing from the central characteristics thereof
(e.g., CO2
crossover limitation, regeneration of absorbed CO2, and control of the
diffusion layer in the
stationary catholyte layer). The present implementations and examples,
therefore, are to
be considered in all respects as illustrative and not restrictive, and the
system and process
proposed herein are not to be limited to the details given herein.
Accordingly, while the
specific implementations have been illustrated and described, numerous
modifications
can come to mind.
For example, the cathode of the system could be further designed to include a
macroporous gas diffusion layer, microporous gas diffusion layer, a metallic
layer
containing Cu with/without Al and/or Zn and/or Mg in any form (ionic or
reduced or
nanoparticles), short-side-chain iononners (e.g., Nafion or similar ionic
polymers), an
organic molecular compound (e.g., pyridine) either in free form or grafted
into any of the
above layer.
EXPERIMENTAL RESULTS
A single-pass CO2 utilization of at least 60% at a production rate of 91
nnA.cnn-2 toward C2+
products was achieved. When run at a total current density of 350 mA.cm-2, the
present
BPM-based SC-MEA electrolyzer delivered an average ethylene Faradaic
Efficiency (FE)
of 35% for over 30 hours. CO2 loss due to crossover was inferiorto 0.1%.
Materials
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
Phosphoric acid (H3PO4, 85%), potassium sulfate (K2SO4, 99%), potassium
bicarbonate
(KHCO3, 99.7%), potassium chloride (KCI, 99%), potassium hydroxide (KOH,
99.95%),
copper nanoparticles (25 nnn), NafionTM 1100W (5 wt.% in a mixture of lower
aliphatic
alcohols and water) and isopropanol (IPA, 99%) were purchased from Sigma
Aldrich and
5 used as received. Titanium oxide nanoparticles (T102, Aeroxide P25) and
PVDF
membrane filter (0.45 pm pore size, 125 pm thickness) were purchased from
Fisher
Scientific and used as received. NafionTM 212, NafionTm XL, Fumasep (FAS-PET-
130) and
titanium (Ti) felt were purchased from Fuel Cell Store. Iridium(IV) chloride
hydrate
(Premion , 99.99%, metals basis, Ir 73% min) was purchased from Alfa Aesar.
The water
10 used in this study was 18 MO Milli-Q deionized- (DI-) water. Nafion
membranes were
activated through the following procedure: 1 hour in 80 C 1M H2SO4 ¨ 1 hour
in 80 C
H202 ¨ 1 hour in 1M H2SO4 ¨ stored in DI-water. Fumasep was used as received
and
stored in 1M KCI. Piperion (40 pm) was purchased from VV7Energy and stored in
0.5M
KO H.
15 Fabrication of a water dissociation catalyst layer of the customized
bipolar membrane
(BPM)
The water dissociation catalyst layer was fabricated following a similar
procedure in a
previous report (see reference 18). TiO2 nanoparticles ink were prepared by
sonicating
the mixture of TiO2, DI-water, and IPA with the weight ratio of 1: 833: 2833
for 30 minutes.
20 TiO2 nanoparticle ink was spray-coated onto a Nafion 212 membrane, of
which the edges
were sealed by Kapton tapes. The exposed membrane dimension was 2.2 cm x 2.2
cm.
The nominal loading of TiO2 is 0.2 mg cm-2. The TiO2-coated Nafion was
immediately used
for assembling electrolyzers once prepared.
Anode and cathode preparation
25 For the CO2RR, cathode gas diffusion electrodes (GDEs) were prepared by
spray-
depositing a catalyst ink dispersing 1 mg mL-1 of Cu nanoparticles and 0.25 mg
mL-1 of
NafionTM 1100W in methanol onto a hydrophobic carbon paper. The mass loading
of Cu
NPs in the GDE was kept at 1.5 mg/cm2. The GDEs were dried in the air
overnight before
experiments.
30 For the OER, the anode electrodes were prepared following a recipe
described in Ozden,
A., Li, F., Garcia De Arquer, F.P., Rosas-Hernandez, A., Thevenon, A., Wang,
Y., Hung,
S.F., Wang, X., Chen, B., Li, J., et al. (2020). High-rate and efficient
ethylene
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
36
electrosynthesis using a catalyst/promoter/transport layer. ACS Energy Lett.
5, 2811-
2818; and Gabardo, C.M., O'Brien, CF., Edwards, J.P., McCallum, C., Xu, Y.,
Dinh, CT.,
Li, J., Sargent, E.H., and Sinton, D. (2019). Continuous Carbon Dioxide
Electroreduction
to Concentrated Multi-carbon Products Using a Membrane Electrode Assembly.
Joule 3,
2777-2791 (see references 6 and 15). The electrode preparation procedure
involves:
etching the Ti felt in hydrochloric acid at 70 C for 40 min; rinsing the
etched Ti felt with DI
water; immersing the Ti felt into an Ir(IV) chloride hydrate solution; drying
and sintering the
Ir(IV) loaded Ti felt. The loading, drying, and sintering steps were repeated
until a final Ir
loading of 1.5 mg cm-2 was achieved.
Assembly of the stationary catholyte membrane electrode assembly (SC-MEA)
The MEA set (5 cm2) was purchased from Dioxide Materials. A cathode was cut
into a 2.1
cm x 2.1 cm piece and placed onto the MEA cathode plate with a flow window
with a
dimension of 2.23 cm x 2.23 cm. The four edges of the cathode were sealed by
Kapton
tapes, which also made the flow window fully covered. The exposed cathode area
was
measured every time before the electrochemical tests, which was in the range
of 3.1 to
4.2 cnn2. Onto the cathode, a PVFD filter membrane (2 cm x 2 cm) saturated
with desirable
electrolyte (sonicate in electrolyte for 15 minutes to degas) was carefully
placed. This
PVDF layer serves as the 'stationary catholyte layer.' Note that the BPMs used
in neutral
and acidic conditions were a customized one and a commercially available one
(Fumasep), respectively, to achieve a better compromise of CO2RR performance
and
stability. The considerations of membrane selection can be found in SI4 of
Supplemental
Information. When using customized BPM, a TiO2 coated Nafion was placed onto
the
stationary catholyte layer with the TiO2 layer facing up, then covered by a
Piperion (5 cm
x 5 cm) membrane. When using Fumasep BPM, the membrane was placed with its
cation-
exchange layer (CEL) facing the cathode side. An Ir02 loaded Ti felt (2.5 cm x
2.5 cm)
was placed onto the anion-exchange layer (AEL) of the BPM.
Scanning electron microscopy (SEM)
Images of cathode and customized BPM were captured by an FEI Quanta FEG 250
environmental SEM (see Figures lE and 1F).
Electrochemical measurements
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
37
Throughout all experiments, the cathode side was flowed by CO2 with a flow
rate of 15
sccm unless otherwise specified, while the anode side was fed with neutral or
acidic
electrolyte at 10 mL/min by a peristaltic pump. The electrochemical
measurements were
performed with a potentiostat (Autolab PGSTAT204 with 10A booster). The cell
voltages
reported in this work are not iR corrected. The exemplified flow rate of
anolyte should not
be taken as a limitation and different values (other than 10 mUmin) would
provide an SPU
of at least 60% as encompassed herein.
Product analysis
The CO2RR gas products, oxygen, and CO2 were analyzed by injecting the gas
samples
into a gas chromatograph (Perkin Elmer Clarus 590) coupled with a thermal
conductivity
detector (TCD) and a flame ionization detector (FID). The gas chromatograph
was
equipped with a Molecular Sieve 5A Capillary Column and a packed Carboxen-1000
Column with argon as the carrier gas. The volumetric gas flow rates in and out
of the cell
were measured with a bubble column. The FE of a gas product is calculated as
follows:
FEi = xi x rev.; x njiF
Where x, is the volume fraction of the gas product i, V is the outlet gas flow
rate in L s', P
is atmosphere pressure 101.325 kPa, R is the ideal gas constant 8.314 J mol-1
K-1, T is
the room temperature in K, n, is the number of electrons required to produce
one molecule
of product F is the Faraday Constant 96485 C m01-1, and J is the total current
in A.
The liquid products from the cathode side of the SC-MEA were collected using a
cold trap
cooled to 0 C. The collected liquid was combined with anolyte (some crossover
liquid
product) for quantifying by the proton nuclear magnetic resonance spectroscopy
CH NMR)
on an Agilent DD2 500 spectrometer in D20 using water suppression mode and
dimethyl
sulfoxide (DMS0) as the internal standard. For each plot of liquid product
quantification,
fresh anolyte was used, and the duration of the collection is 30 minutes. The
FE of a liquid
product is calculated as follows:
FE i = rn x
ft
Where m, is the quantity of the liquid product i in mole, t is the duration of
product collection
(1800 seconds).
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
38
The CO2 SPU calculation is detailed in section 511 of Supplemental Information
provided
below.
Sll Analysis and comparison of CO2 single-pass utilization among state-of-art
CO2RR electrolyzers
The ordinary CO2RR electrolyzers suffer from an upper limit of CO2
utilization, depending
on their product distributions, as analyzed below:
In the presence of hydroxide ions, CO2 molecules react with OH- (reactions [1]
and [2])
faster than being electrochemically reduced because the reaction kinetics are
more
favorable (see references 11, 13 and 14).
CO2 + 20H- ¨> C032- + H20 [1]
CO2 + OH- ¨> HCO3- [2]
Meanwhile, the major cathode reactions in neutral or alkaline media include:
CO2 + H20 + 2e- ¨> CO + 20H- [3]
CO2 + H2O + 2e- ¨> HC00- + OH- [4]
2CO2 + 5H20 + 8e- ¨> CH3C00- + 70H- [5]
2CO2 + 8H20 + 12e- ¨> C21-14+ 120H- [6]
2CO2 + 9H20 + 12e- ¨> C2H5OH + 120H- [7]
CO2 + 6H20 + 8e- ¨> CH4+ 80H- [8]
3CO2 + 13H20 + 18e- ¨> C3H7OH + 180H- [9]
2H20 + 2e- ¨> H2+ 20H- [10]
All these reactions generate hydroxide, of which the rate (in mole per second,
MOH) is:
J x FE[i] x k[i]
MOH (1)
Where J is the current in amps, FEN is the faradaic efficiency of the specific
reaction [3-
10], F is the Faraday constant, and kfil is the number of OH- generated per
electron
transferred in the specific reaction [3-10]. For the cathode reactions [4] and
[5], the kid
values are 0.5 and 0.875, respectively; for the other cathode reactions the
kfij values are
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
39
I. Figures 1A and 1B show that, in neutral media, the in-situ generated
hydroxide reacts
with CO2 to form carbonate and/or bicarbonate, which migrate to anode, combine
with
protons (generated by the oxygen evolution reaction), and release CO2 into
anode gas
stream. This phenomenon is known as CO2 crossover (see references 11 to 13).
Therefore, the 002/02 ratio in anode gas provides insight into the identity of
the anionic
charge carrier(s) that combine with the I-I+ generated on the anode (see
reference
14),Ideally, if the charge carrier is H003- or 0032-, the 002/02 ratio in the
anode gas stream
is 4 0r2, respectively (see reference 14). While the other charge carriers
like OH-, HC00-
or CH3000- do not release CO2 by acidification at the anode.
Based on the analysis above, the inlet CO2 (Cm) is balanced by four parts: the
CO2 in
outstreann (CO, the electrochemically reduced CO2 (C2), the absorbed CO2 (C3),
and the
crossover CO2 (Ca). In other words, the mass balance of CO2 (in mole per
second) is:
= + C2 C3 C4 (2)
The carbon utilization efficiency is evaluated by single-pass utilization
(SPU):
C2
SPU = ¨ (3)
Cin
In conventional flow cells and MEAs, some studies have demonstrated that C/
can be
negligible compared to C2 and C4 by carefully tuning Cm. When the CO2
absorption in the
system reaches a steady-state, C3 is almost zero. Therefore, the upper limit
of SPU is:
C2
SPUurnit = _______________________________________________________ (4)
C2 C4
C2 = >jX FE[i]
(5)
n[i] F
Where J is the current in amp, FEN is the faradaic efficiency of the specific
reaction [3-9],
n111 is the number of electrons transferred per consumed CO2 in the specific
reaction [3-9],
and F is the Faraday constant. In neutral media, C4 ranges from 0.5 to 1 times
MoH,
depending on the species of charge carrier cross the AEM. To evaluate the
upper limit of
the SPU, C4 = 0.5 MOH. Substituting (1) and (5) into (4) gives:
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
FE
Etl
n[i]
(6)
SP Ulimtt ___________________ 1
Eli' FEN X (- 0.5k[i])
n[i]
Therefore, in the conventional flow cells and MEAs operating in neutral media,
the upper
limits of SPU depend on their product distributions. For example, the SPU
upper limits of
the systems that produce 100% FE of CO (n = 2; kfil = 1) or 100% FE of
ethylene ON =
6; kg = 1) are 50% or 25%, respectively.' Notably, HER does not contribute to
C2 but still
5 generates hydroxide that can drive CO2 crossover. Accordingly, the CO2RR
performances
of the electrolyzers that show state-of-art SPU in the references were
identified and
summarized in Table Si. None of the reported electrolyzers can achieve an SPU
exceeding 30% for 02, production, and 44% for CO production.
The SPU measurement missed 10-13% of the product FE likely because some liquid
10 product was trapped in the stationary catholyte layer or migrated and
got oxidized on the
anode. Therefore, the SPU reported in this work are the minimum values. The
upper limit
of CO2 SPU was also simulated for the SC-MEA under various conditions and are
listed
in Table S1. The upper limit SPU values without considering the missing FE are
indicated
in the brackets.
15 Nevertheless, the missing FE is taken into account for calculating the
upper limit SPU of
the electrolyzer to make a conservative comparison of SPU. The missing FE can
be
ascribed to three groups of liquid products, i.e., formate, acetate, and
ethanol/propanol.
However, formate FE is only 5% of the liquid product in the SC-MEA operating
under all
the circumstances, and ascribing the missing FE to formate will result in a
total CO2
20 consumption exceeding the inlet CO2 amount; the missing FE to acetate,
ethanol, and
propanol is ascribed here. This simulation gives the ranges of upper limit SPU
for the SC-
MEA under different conditions (Table S1). In the main text, these ranges are
depicted in
Figure 2F and 30.
25 Table S1 Summary of the CO2 single-pass utilization (SPU) of the flow
cells (FC) and
membrane electrode assembly (MEA) in literature operating at the current
densities over
100 mA cm-2. The stationary catholyte MEA (SC-MEA) operating under different
conditions
are listed for comparison. The upper limit SPU is simulated by substituting
the reported
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
41
FE distribution into Equation (6). The experimental SPU is calculated from
Equation (3).
The electrolyzers producing C2, are indicated by references are bold.
FE ____________________________ distribution
Experimental
CO2 flow SPU SPU
Cell type/
: Upper Limit
(sccm) H2 CO C2H4 CH4 Et0H Acetate Formate Propanol
Upper Limit Experimental
SPU
references
Alkaline
7 0.07 0.2 0.45 0 0.175 0 0 0 -
24% -
FC9
Alkaline
50 0.07 0.06 0.7 0.02 0.1 0.05 0 0 -
0.5% - FC9
Alkaline
17 0.36 0.64 0 0 0 0 0 0 - 21% -
FC7
Neutral
100 0.02 0.95 o o o o o o - 7% -
MEM
Neutral
2 0.59 0.00 0.24 0.08 0.09 0.025 0.015 0.01
- 30% -
MEA9
Neutral
15 0.23 0.77 0 0 0 0 0 0 - 44% -
MEA1
Alkaline
100 0.28 0.72 0 0 0 0 0 0 - 40% -
MEAll
Neutral
45 0.05 0.15 0.45 0.02 0.2 0.04 0.02
0.04 4% FC3
Neutral
45 0.08 0.18 042 0.02 0.15 0.03 0.03 0.05 - 3% -
FC
(BPM)2
13.7
Neutral
1 0.62 0.007 0.12 0.001 0.052 0.046 0.008 0.012 1.0% 61%
4.5 SC-MEA
(9.1%) a
(this work)
15.5
Acidic
1 0.56 0.002 0.14 0.007 0.03 0.09 0.004 0.002 1.1% 60%
3.9 SC-MEA
(10.0%) a
(this work)
25.0
Acidic
4 0.21 0.042 0.37 0.002 0.11 0.085 0.012 0.031 1.0% 39%
1.6 SC-MEA
(this work)
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
42
SI2 Analysis of intrinsic energy consumption associated with in-situ CO2
regeneration in SC-MEA and the comparison with AEM-based MEAs
Bipolar membranes losses
With the application of an appropriate external potential, water dissociation:
H20 ¨> H+ +
OH-, occurs at the interface of the CEL/AEL, and the protons and hydroxides
serve as
charge carriers in CEL and AEL, respectively. Under standard conditions (25
C, 1 atm,
with activities of H+ and OH- at 1 M in the CEL and AEL, respectively), the
electric potential
across the BPM is -0.83 V at equilibrium. The electric potential energy
difference for H+
and OH- across the BPM exactly compensates for the difference in activity such
that the
electrochemical potential is the same everywhere at equilibrium (see reference
18). For
net current to flow, an additional electric potential must be applied across
the membrane
causing a deviation from the open-circuit value of -0.83 V. This deviation is
typically called
the water dissociation overpotential and represents the losses associated with
generating
H+ and OH- and transporting it out of the interfacial layer between CEL and
AEL and out
of the BPM. Often, it is stated that a BPM induces a "thermodynamic" voltage
loss of 0.83
V - however as discussed above, this is incorrect - the losses can be quite
small. For
example, with appropriate materials and operating conditions, the cell voltage
of a BPM-
based water electrolyzer can be lower than that of an AEM-based electrolyzer
at the
current density up to 500 mA cm-2 (see reference 18). BPM electrolyzers can
begin to split
water with a total voltage of <2 V, which would be impossible if there were an
intrinsic
830 mV penalty for using the BPM.
With a pH = 8.20 anolyte, the SC-MEA has a cell voltage of 3.7 V at 100 mA cm-
2 and 50
C (Figure 2B in the main text), very close to the AEM-based MEA (3.4 to 3.5 V)
operating
under similar conditions and the same anolyte (see reference 16). The 0.2 to
0.3 V cell
voltage gap is likely ascribed to two factors: the ohmic loss due to thicker
BPM (ca. 50 pm
CEL + 40 pm AEL) than AEM (ca. 40 pm); the cathode pH gradient (see Figures 5A
to
5C).
Energy loss in stationary catholyte layer
In the present SC-MEA design, the stationary catholyte layer is a 125 pm-thick
0.5 M
K2SO4 solution (conductivity 0.15 S cm-1). Although the total ionic
conductivity of this
catholyte is large, the H+ / OH- / HCO3 / 0032- conductivity is very small.
Because these
are the relevant ionic charge carriers in carbon dioxide electrolysis at
steady state, a large
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
43
pH gradient between Cu and the bulk catholyte is induced. Establishing this pH
gradient
is a source of an additional concentration overpotential. As shown in Figure
5A, there is
no cathode pH gradient in AEM-based MEA as long as fresh base (e.g. alkaline
KOH) is
fed to the cathode to react with and capture CO2 (of course this induces a
different
efficiency loss, namely the need to generate base externally to the system).
In the present SC-MEA (Figures 5B and 5C), the local pH of Cu is considered to
be high
(see reference 38) (>13) due to the continuous generation of OH- from cathode
reactions.
As shown in Figure 5B, if the catholyte is a pH = - 10 buffer solution (e.g.,
glycine-sodium
hydroxide or potassium carbonate/bicarbonate), ApH is likely - 3, which would
increase
the cell voltage by ca. 0.18 V. Accordingly, the extra energy (Gex) consumed
in SC-MEA
with pH = 10 buffer catholyte from carbonate or bicarbonate is calculated as
follows:
Gõ = 0.059 x ApH xF x J
In other words, SC-MEA is likely to save energy compared to ex-situ CO2
capture (3.5 to
4.7 GJ per ton -see reference 29), especially as the costs of renewable
electricity
decrease, and if cross-over of the buffer ions can be minimized or eliminated.
Previous techno-economic analysis has concluded that (see reference 39) for an
AEM-
based MEA, even under the optimistic evaluations (zero-ohmic loss, cell
voltage 2 V, SPU
40%, 90% 02+ FE, half the CO2 loss and current density 1 A cm-2), the total
cost for
producing 1 ton of ethylene from CO2RR is $1,300, higher than the ethylene
market price
($1,000), of which $750 is spent for electricity and $520 is spent for ex-situ
002 recovery.
Using the same optimistic metrics (except for SPU, which can be optimized to
100% in
SC-MEA), the in-situ 002 recovery in SC-MEA (if using pH = 10 buffer
catholyte) can cut
the CO2 capture cost ($520 ¨> $0), with a 9% (0.18 V to 2 V) increase to
electricity cost
($750 ¨> $817), making the production cost for 1 ton of ethylene to $847.
Therefore, SC-
MEA has the potential to make the 002-to-ethylene electrolysis economically
feasible.
Nevertheless, the buffer catholyte is not used in the present SC-MEA. In the
buffer
catholyte SC-MEA, CO2 is generated at the boundary of CEL and catholyte and
diffuses
across the catholyte layer (125 pm) to Cu. Previous simulation works have
suggested that
such a long diffusion path cannot support a high rate (>100 mA cm-2) CO2RR
(see
reference 38). This effect also explains the experimental observation when
using KHCO3
(with some buffer capability) as the stationary catholyte, as shown in Figure
8 discussed
in section SI3. In this work, the primary purpose is to demonstrate that the
SC-MEA can
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
44
effectively prohibit CO2 crossover and promote high SPU in conjunction with a
BPM. A
non-buffered catholyte like K2SO4 can shorten the regenerated CO2 diffusion
path. As
shown in Scheme 1C, the migration of protons from the CEL to Cu acidifies the
stationary
catholyte, making the diffusion path shorter than that in a buffer catholyte.
However, this
phenomenon also creates a larger pH gradient than that in buffer catholyte SC-
MEA. The
pH gradient is greater at higher current densities, as needed to drive the
larger
proton/hydroxide fluxes.
In the future, by adopting strategies from membrane science and materials
engineering,
the catholyte thickness can be reduced to below 10 pm and a buffer catholyte
can be used
with minimum cross-over that will minimize the pH gradients developed within
the system
and thus the concentration overpotential losses associated with them. Coupling
with the
efforts of BPM materials and catalysts development, it is expected that the SC-
MEA may
be a high-total-energy-efficient system for CO2-to-C2, production.
SI3 Additional information for the catholyte engineering towards high SPU
CO2RR
With the present BPM-based SC-MEA, the CO2 consumed for CO2RR is provided by
two
sources: the inlet CO2 flow (gas), and the regenerated CO2 (dissolved form) in
the
stationary catholyte layer. As the upper limit of the CO2 SPU for most of C2,
is 25%, the
CO2 regeneration procedure in the SC-MEA will supply 75% of the CO2
consumption if the
target is to achieve 100% SPU. Thus, the mass transport effectiveness of the
regenerated
CO2 is an important consideration in the SC-MEA, which is determined by the
thickness
of the stationary catholyte layer because a too thick catholyte layer cannot
effectively
deliver the regenerated CO2 to Cu catalyst, as analyzed below.
At steady-state, the net current in the stationary catholyte layer of the SC-
MEA should be
primarily driven by the electromig ration of protons/hydroxide and
carbonate/bicarbonate
ions simultaneously generated from water dissociation of BPM and reactions
[1]/[2],
respectively. Referring to Figure 6, the protons and carbonate/bicarbonate
ions combine
somewhere in the stationary catholyte layer, forming a virtual boundary where
CO2 is
regenerated and diffuses towards Cu and CEL driven by the concentration
gradient. The
distances from this boundary to the Cu and CEL are noted as Li and L2,
respectively. The
zones between the boundary and Cu/CEL are noted as Zone 1 and Zone 2 for
convenient
discussion. Before the electrolysis, the CO2 concentration is zero everywhere
in the
stationary catholyte layer.
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
After the electrolysis starts and proceeds, the CO2 concentration at the
boundary gradually
arises, driving the generated CO2 (dissolved form) to diffuse towards Cu and
CEL. The
CO2 (dissolved form) that diffuses deeper into the stationary catholyte layer
towards the
CEL is not consumed by CO2RR but accumulates in Zone 2 until reaching a
concentration
5 close to that at the boundary (then no driving force). In the other
direction, the CO2 diffuses
towards Cu is consumed for CO2RR, forming a concentration gradient in Zone 1,
which
creates a continuous CO2 diffusion flux from boundary to Cu. Note that the
real CO2
(dissolved form) concentration distribution in Zone 1 deviates from linear due
to the local
pH gradient. Zone 1 can be described as a diffusion layer (see reference 40).
The diffusion
10 layer thickness, Li, has great impacts on the CO2 mass transport (see
reference 40). Since
the electric field of the stationary catholyte layer can be considered
homogeneous (see
reference 41), Li and L2 are known to be proportional to the mobility of the
corresponding
ions. Given that L1+L2 is the total thickness of the stationary catholyte
layer, a thinner
stationary catholyte layer has a thinner CO2 diffusion layer (Li), which is
beneficial for CO2
15 mass transport from bulk to catalyst (see reference 40). In the future,
thinner, yet
mechanically robust porous layers, can be employed to improve the mass
transport of the
regenerated CO2, as discussed in SI2.
A previous work (see reference 22) showed that in an MEA cell, inserting a
solid porous
supporting layer saturated with DI water in between Ag catalyst and the cation-
exchange
20 layer of the BPM can improve the FE for converting CO2 to CO. In the
system, this strategy
was also attempted, and the electrolyzer shows a >94% FE towards hydrogen, and
a high
full-cell voltage of 6-9 V, even under optimized conditions, as shown in
Figure 7A. On the
other hand, the cation effect plays an important role in the SC-MEA, which can
suppress
the HER and promote CO2RR under acidified environment near the Cu catalyst, as
shown
25 in Figure 7B, 7C, and 12A.
The K2SO4 catholyte in the stationary catholyte layer will gradually be
partially transformed
to KHCO3 over time owing to reactions [1] and [2] as well as the slow leakage
of S042- to
anolyte. To study the impact of such transformation, the CO2RR performance of
SC-MEA
was measured when a 1 M KHCO3 buffer electrolyte is used as catholyte at the
beginning.
30 It was found that using 1 M KHCO3, the SC-MEA shows an expectable C2I-14
FE of ca. 23%
at the current density of 100 mA cm-2. However, its stability is poor in that
the FE gradually
decreases by > 50% after the initial 5 hours. This experiment has been
repeated three
times, and a typical FEs and cell voltage vs. time curve is displayed in
Figure 8. HCO3-
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
46
has a higher buffer capacity than S042-, and the pH gradient built up in KHCO3
is thus
expected to be smaller than that in K2SO4. The CO2 diffusion layer thickness
in KHCO3 is
thus likely greater than that in 0.5 M K2804 catholyte (analyzed above). With
the thicker
CO2 diffusion layer, the regenerated CO2 may gradually accumulate at the
boundary
(Figure 6) because the concentration gradient-driven CO2 diffusion flux is
lower than the
CO2 generation rate. When the accumulated aq. CO2 reaches the saturated
concentration,
it could bubble out periodically and physically damage the catalyst layer
and/or stationary
catholyte layer. This phenomenon may also be the reason for the periodical
voltage
fluctuation in Figure 8.
To this end, the stationary catholyte layer was engineered in SC-MEA to a 125
pm thick,
0.5 M non-buffer K2504 solution.
Since it was discovered that the cation effect enables the CO2RR even in an
acidified
environment, it was attempted to extend the stationary catholyte layer
strategy in an MEA
cell using CEM and an acidic anolyte pH <2.37, expecting a lower cell voltage
than the
BPM-based cells while maintaining high SPU. The configuration of this system
is shown
in Figure 9A. Some previous studies have confirmed that, in neutral
electrolytes, using a
cation exchange membrane (CEM) instead of AEM even increases the amount of CO2
crossover to the anode gas (see reference 13). In contrast, it was found that
in the acidic
MEA cell, the CO2 crossover was eliminated. The anode gas CO2 contents were
below
the detection limit of the GC for the current density in the range between 100
to 300 mA
cm-2. This observation should be ascribed to the lower pH near the stationary
catholyte
layer/CEM interface, as shown in Figure 9A.
This configuration shows lower full cell voltage (Figure 9B) in comparison to
the BPM-
based SC-MEA presented in the main text in part due to the low resistance of
the CEM.
Meanwhile, it has a reasonable CO2RR selectivity over HER because the Win the
anolyte
can migrate to the Cu surface to induce the cation effect. However, this
design was not
amenable to steady state operation without continuous addition of acid and
salt to the
anolyte and catholyte as the initial pH gradient will be eliminated due to co-
ion transport
and neutralization. In fact, the CO2RR selectivity of this system gradually
drops over time,
and after ca. 3 hours, this system produces 100% hydrogen at the cathode. In
addition, it
was observed that this design periodically eject electrolyte from the cathode
flow channel,
probably due to a water balance issue:
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
47
On the anode, the OER generates one proton per one electron transfer:
2H20 ¨ 4e- ¨> 4H+ + 02
Since a OEM is used in this system, the charge carriers across the membrane
are K+ and
H. K+ is adsorbed at the Cu surface to form an electrochemical double layer,
and the
migration of K+ terminates until reaching the equilibrium between electric
potential and
chemical potential, which usually takes tens of seconds (see reference 19) H+
migrates
to the cathode side combines with OH- (or 0032-/HCO3-), which produces water
at the
cathode side. The water balance for different cathode reactions ([3-9] in
section SI1) was
accordingly calculated:
Table S2. The water balance in a CEM-based SC-MEA.
Water balance in the cathode Water balance in the anode
Product
(Mole) (Mole)
(1 Mole)
Consumed Generated Net Consumed
CO 1 2 +1 1
HC00- 1 1 0 1
CH3000- 5 7 +2 4
02H4 8 12 +4 6
C2H5OH 9 12 +3 6
CH4 6 8 +2 4
H2 2 2 0 1
As seen, most of the cathode reactions generate water on the cathode side. The
generated water keeps diluting and pushing out the electrolyte in the
stationary catholyte
layer, of which the volume is small (ca. 10 pL per cm2 electrode area). This
phenomenon
results in the flooding (also confirmed by experimental observation) of the
cathode and
continues loss of K+ in the system, degrading CO2RR performance because the
concentration of K is critical to CO2RR performance for the catalyst (Figures
7A to 70).
SI4 Additional considerations to BPM in SC-MEA
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
48
In a neutral anolyte solution, a customized BPM was adopted, which consisted
of a layer
of TiO2 nanoparticles as a water dissociation catalyst sandwiched by a CEM and
an AEM.
In an acidic anolyte solution, a commercially available BPM (Fumasep) was used
to
achieve the best compromise among CO2RR performance, cell voltage, and
stability.
Water splitting measurement was firstly conducted to compare the resistance of
customized BPM and Fumasep. Figure 10 shows that the BPM with TiO2 water
dissociation catalyst (black plots) has lower resistance than the one without
water
dissociation catalyst (blue plots) and commercially available Fumasep BPM.
When been used in the SC-MEA with neutral anolyte (Figure 11), Fumasep shows
similar
product distributions and a slightly better CO2 crossover inhibiting
capability (Figures 2D,
and 16) but higher cell voltage comparing with the one based on customized
BPM, which
is expectable. Therefore, the customized BPM was adopted in the neutral
anolyte studies.
For the SC-MEA using acidic anolyte, the customized BPM also promotes lower
cell
voltage than Fumasep (Figure 12A). However, this system always fails within 4-
6 hours
of continuous operating due to the short circuit issue, and a typical voltage
versus
operating time diagram is shown in Figure 12B. It is suspected that this is
caused by the
growth of Cu dendrites that physically penetrated through BPM and contact with
the
anode. Cu could be partially dissolved by acid and electrochemically re-
deposited onto
the catalyst layer, forming sharp dendrites (see reference 42). Differently,
it was found that
the Fumasep-based SC-MEA is more stable (Figure 4B in the main text) probably
because
Fumasep is mechanically rigid, so Cu dendrites cannot penetrate. To this end,
Fumasep
was adopted in the acidic anolyte studies.
S15 Temperature and pH effects on CO2RR performance in SC-MEA
Elevating the operating temperature reduced the full cell voltage at given
current densities
(Figure 2B in the main text), which is attributed to accelerated water
dissociation, reduced
cross-membrane ohmic loss, and improved electrode kinetics (see reference 18).
As
displayed in Figure 2C, 13A and 13B, with increasing operating temperature,
the optimal
ethylene FE first increased from 25% (20 C, 200 mA cm-2) to 35% (35 C, 250 mA
cm-2)
and then decreased to 30% (50 C, 150 mA cm-2). This trend is attributed to the
trade-off
between CO2RR and HER kinetics as well as the CO2 solubility (detrimental to
FE) (see
references 1, 43 and 44). It is thus concluded that 35 C can be an optimal
temperature in
the present device design.
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
49
SI6 Control experiment of CO2 crossover
Referring to Figure 16, all the gas samples were recorded after the cell
operating for 1
hour at each current density. In K2SO4, the CO2: 02 ratio is close to that in
KHCO3 for
customized BPM, indicating that the detected anode CO2 flow in the customized
BPM/KHCO3 system (Figure 2D in the main text) was not ascribed to the
acidification of
bicarbonate. In an SC-MEA using KHCO3 anolyte, the CO2 crossover through
Fumasep
is similar to the case through customized BPM.
Further results regarding the thickness and the composition of the stationary
catholyte ¨(SC) layer
Finite-element numerical simulations of the stationary catholyte (SC)-layer
The present invention founds that the composition and thickness of the
catholyte layer
influence the local pH, the efficiency of CO2 regeneration and, thereby, the
overall cell
performance. A one-dimensional multiphysics model in COMSOLO was applied to
investigate the catholyte layer in BPM-based CO2RR electrolyzers.
The CO2 reactant is provided by two sources: the inlet CO2 flow (gas) and the
regenerated
CO2 (dissolved form, aq.) in the catholyte. To achieve high SPU, it is
necessary to restrict
the gaseous CO2 feed. Under a restricted gaseous CO2 availability, the cathode
CO2
supply relies more on regeneration: in an ideal case with 100% SPU and 100%
C2,
selectivity, regeneration contributes 75% of the consumed CO2. Thus, the mass
transport
of regenerated CO2 is most critical, and that transport is governed by
catholyte
composition and thickness.
At steady-state, electrolysis creates a pH gradient through the catholyte
layer: the pH is
high near the cathode and low near the CEL. The protons and (bi)carbonate ions
recombine in the catholyte, forming CO2 (aq.) that diffuses, in response to a
concentration
gradient, to the Cu catalyst.
Simulations resolve the local cathode environment as a function of dimensions,
electrolyte
and running conditions. The modeled thicknesses of 250, 125, 65 and 16 pm were
selected to correspond to commercially available materials.
Use of a buffering catholyte (e.g. KHCO3) leads to a thick CO2 (aq.) diffusion
layer¨close
to the catholyte thickness, since the CO2 (aq.) is generated near the CEL
surface. This
effect reduces the CO2 (aq) mass-transfer efficiency.
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
In contrast, with a non-buffering catholyte layer (e.g. 0.5 M K2504) with
thicknesses of
250, 125, and 65 pm, the local pH values near the cathode are greater than 11,
which is
sufficient to promote selectivity towards CO2RR over HER. Reducing the SC-
layer
thickness to 16 pm results in a cathode pH of 8.7, implying a lower
selectivity toward
5 CO2RR.
Fig. 1c and ld show the simulated concentration profiles of CO2 (aq.) in the
non-buffering
SC-layer. At steady-state, the CO2 (aq.) is continuously supplied to the
cathode to
participate in CO2RR, forming a concentration gradient (the boundary was
defined here
as the position where CO2 concentration is 1% lower than the saturated
concentration) to
10 the cathode surface. Prior studies have termed the zone between
the cathode and this
boundary the diffusion layer (see reference 40). The thickness of the
diffusion layer
controls the efficiency of CO2 (aq.) mass transport (see reference 40).
According to the
simulations, the thicknesses of the diffusion layers are 75, 35, 12 and 5 pm
for the
catholyte layers with the thicknesses of 250, 125, 65 and 16 pm, respectively.
For
15 reference, the CO2 (aq.) diffusion layer thickness in H-cells (all
CO2 supplied in dissolved
form) is typically 40-100 pm, and this does not support current densities
exceeding 100
mA cm-2. It was expected that diffusion layers <40 pm, and a corresponding
catholyte
thickness <150 pm, are required for sufficient mass transport in a non-
buffering catholyte.
To achieve similar mass transport in a buffering catholyte, the total
thickness could not
20 exceed 12 pm, and the cathodic pH would not be sufficiently
alkaline for selective CO2RR.
The simulation results suggest the following design principles for the
catholyte layer in a
BPM-based electrolyzer: the local cathode pH and the diffusion layer thickness
of the
regenerated CO2 increase as the catholyte thickness increases; the buffering
capacity of
the catholyte increases the diffusion layer thickness and reduces transport.
Precise control
25 of the thickness of a non-buffering catholyte should thus offer a
route to high SPU, CO2RR
selectivity and reaction rate.
System design for high SPU of CO2 feedstock
Guided by the above analysis, the inventors focused on a stationary catholyte
bipolar
30 membrane electrode assembly (SC-BPMEA) electrolyzer and
incorporated a judiciously-
designed catholyte layer and BPM.
The cathode was prepared by spraying Cu nanoparticles onto a hydrophobic
carbon gas-
diffusion layer for CO2RR. The anode was 1102 supported on Ti felt for the
oxygen evolution
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
51
reaction (OER). A BPM under reverse bias was employed with the AEL contacting
the
anode and the CEL contacting the SC-layer (porous support saturated with
electrolyte).
The cathode was compressed onto the porous layer, and the anode and cathode
flow-
field plates sandwiched the system.
The BPM employed in this work sandwiched TiO2 nanoparticles as the water
dissociation
catalyst (see reference 18). This custom BPM can lower the cell voltage by -1
V compared
with commercial BPMs (e.g. Fumasep). The full cell voltage of such custom BPM-
based
electrolyzers is close to that of AEM systems.
Measurements of the 002/02 ratio in the anode gas stream show that the SC-
BPMEA
effectively prevents CO2 crossover, as required for high SPU (see references
13 and 14).
In agreement with the previous studies (see references 13 and 14), the AEM-
based MEA
(AEMEA) showed an anode CO2/02 ratio of -2 for current densities ranging from
100 to
300 mA cm-2. In conventional AEMEAs, the anionic charge carriers are C032-,
and thus
suffer the loss of one molecule of CO2 for every two electrons transferred.
The anode
CO2/02 ratio in the SC-BPMEA (0.06 at 200 mA cm-2) is one order of magnitude
lower.
Control experiments confirm that the CO2 detected in the anode is not due to
acidification
of anolyte (using 0.1 M K2SO4 instead of 0.1 M KHCO3 resulted in a similar CO2
/02 ratio).
The anode 002/02 ratio decreases as the operating current density increases,
an effect
that was ascribed to an increased flux of protons toward the cathode. This
flux decreases
the pH at the CEL surface and reduces the diffusion of CO2 and H003-/003- in
the CEL
(see references 19 and 28).
Impact of the thickness of the SC-layer on CO2RR.
The thickness of the stationary catholyte was found to have a major impact on
cell voltage.
The cell voltage of the SC-BPMEA decreases as the thickness of the SC-layer
decreases
(Fig. 17) from 250 pm (5.1 V, 200 mA cm-2) to a minimum at 65 pm (3.8 V, 200
mA cm-2).
Further thinning the catholyte to 16 pm resulted in higher voltage (4.4 V, 200
mA cm-2) -
an effect of the lower-porosity support layer used in the 16 pm case (< 20%
vs. > 70% for
the thicker layers). A longer ion migration path and higher ohmic resistance
partially
explain the 0.67 V cell voltage increase as the stationary catholyte thickness
increases
from 65 to 125 pm. Based on the independently measured ohmic resistance
(Supplementary Fig. 18), increasing the SC-layer thickness from 65 to 125 pm
imposes
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
52
an ohmic voltage increase of merely 0.07 V at 200 mA cm-2. Similarly, compared
to 65
pm, the 250 pm SC-layer increases the ohmic voltage loss by 0.24 V at 200 mA
cm-2,
while the cell voltage increases by 1.3 V.
The simulations indicate that the thicker SC-layer results in longer transport
distances for
dissolved 002. The CO2 regeneration rate inside the SC-layer also depends on
the current
density, and for thicker SC-layers (e.g. > 125 pm), CO2 bubbles are more prone
to form
near the CEL. These bubbles obstruct ion migration, increasing the ohmic
resistance of
the SC-BPMEA. Electrochemical impedance spectroscopy measurements also support
this finding. An applied current of 200 mA cm-2 resulted in an insignificant
change to the
high-frequency resistance (HFR) of the SC-BPMEA with a 65 pm-thick SC-layer;
while, in
contrast, the HFR of the SC-BPMEA with a 125 pm-thick SC-layer increased by
120%
after applying 200 mA cm-2 for 20 min, leading to a cell voltage 0.6 V higher
than for the
65-pm SC-layer.
The cell voltage of the SC-BPMEA with a 65 pm SC-layer operating at 200 mA cm-
2 is 3.8
V, comparable to the AEM-based neutral-media MEAs operating at similar
conditions
(difference < 0.05 V) (see references 5, 6 and 15). This result demonstrates
that the cell
voltage of a BPM-based CO2RR electrolyzer can be as low as that of an AEM-
based
electrolyzer with a current density of up to 200 mA cm-2, while suppressing
unwanted
crossover and providing high SPU.
The thickness of the SC-layer also affects selectivity towards CO2RR. With
thicknesses of
65, 125 and 250 pm, the H2 Faraday efficiencies (FEs) are consistent (- 20% at
200 mA
cm-2, Fig. 19a-19c), confirming that high local pH conditions are maintained
the cathode
in these cases. However, reducing the thickness to 16 pm increases the H2 FE
to 88% at
200 mA cm-2 (Fig. 19d), consistent with a cathodic pH that is reduced due to
fast proton
transport through a thin SC-layer. Without restricting CO2 availability (the
performance in
Fig. 19a-19d was recorded at a CO2 flow rate of 10 sccm cm-2), the SC-BPMEAs
with the
SC-layer thickness of 65, 125 and 250 pm show similar ethylene FE of 35-43%.
Assessment of SPU in SC-BPMEA.
By suppressing the crossover of CO2 (e.g. <0.5% of total CO2 input at 200 mA
cm-2, Fig.
2d and 20), the SC-BPMEA surpasses the SPU of conventional CO2-to-C2,
electrolyzers,
in which carbonate is the dominant charge carrier. Measuring the CO2 SPUs with
a
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
53
restricted CO2 flow rate is a direct approach to determining the upper bound
of SPU in the
CO2RR electrolyzers.
As the inlet CO2 flow rate decreased, the C2+ FE of the SC-BPMEA at 200 mA cm-
2
decreased, accompanied by an increase in the H2 FE (Fig. 4a, 4b and 4c). With
SC-layer
thicknesses of 65 pm (Fig. 22c), as the input 002 flow rate decreases from
1.17 to 0.58
and 0.29 sccm cm-2, the 02+ FE decreases from 49% to 48% and 34%, while the H2
FE
increases from 23% to 31% and 64%. This shift is consistent with a 002 mass
transport
limitation (see references 6 and 15).
The stationary catholyte thickness affects the SPU of the SC-BPMEA. The SPU
gradually
increases up to 21, 61 and 78% for the SC-BPMEAs with SC-layer thicknesses of
250,
125 and 65 pm, respectively (Fig. 21). These results demonstrate that high CO2
conversion efficiencies are possible using SC-BPMEAs with SC-layer thicknesses
of 125
and 65 pm.
For a given CO2 flow rate, a thicker SC-layer produces a lower SPU (Fig. 21).
In the SC-
BPMEA, reactant CO2 is available from the inlet gas stream and regeneration in
the SC-
layer. With unrestricted CO2 supply (Fig. 19a-Fig 19c), the H2 FEs are similar
for different
stationary cathode layer thicknesses, indicating that both the CO2
availability and local pH
are unaffected by catholyte thickness under excess supply conditions. The
simulations
suggest that the thicker SC-layer results in a lower dissolved CO2 flux to the
cathode due
to the smaller concentration gradient. Compared to the SC-BPMEAs with thinner
SC-
layers, CO2 availability with thicker SC-layers decreases more significantly
with reducing
CO2 flow rate, leading to a more dramatic increase in H2 FE (Fig. 22a-22c).
The experimental trends are generally consistent with those of the
simulations. The SC-
BPMEA with a dissolved CO2 diffusion layer thicker than 75 pm (representing a
250-pm
SC-layer) fails to surpass the SPU limit because of insufficient mass
transfer. In contrast,
a 65-pm SC-layer facilitates efficient mass transport of the regenerated CO2
(diffusion
layer thickness of 12 pm) and simultaneously promotes high local cathode pH.
It was found (see Fig. 23a-b and Fig. 24a-d) that SC-BPMEAs using acidic and
alkaline
electrolytes achieve carbon efficiencies comparable to those using neutral
electrolytes.
The compatibility of SC-BPMEAs with a range of electrolytes offers flexibility
in the
selection of cathode and anode catalysts. In contrast, in prior art, acidic
002-to-C2+
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
54
electrolyzers have only been demonstrated with precious metal anodes. Indeed
For the
SC-BPMEA using acidic anolyte, the custom BPM enables a lower cell voltage
than
Fumasep. However, this system always fails within -4-6 h of continuous
operation due to
an apparent short-circuit issue, and the typical voltage versus operation
duration is shown
in Fig. 23b. We suspect this is caused by the growth of Cu dendrites that
physically
penetrated through BPM and contact with the anode. Cu could be partially
dissolved by
acid and electrochemically re-deposited onto the catalyst layer, forming sharp
dendrites.
Differently, itw as found that the Fumasep-based SC-BPMEA is more stable,
probably
because Fumasep is mechanically reinforced and thus more rigid, so Cu
dendrites cannot
easily penetrate.
The SC-BPMEA shows > 50-h stability operating at 200 mA cm-2 with limited CO2
availability (CO2 input flow rate of 1.42 sccm cm-2). This operating stability
is competitive
with that of the neutral-electrolyte-based CO2-to-C2, electrolyzers.
Can a cation-exchange membrane replace the BPM in SC-BPMEA?
The inventors attempted to extend the SC-layer strategy in a CEM-based MEA
cell (i.e.
SC-CEMEA, Fig 9a) using an acidic anolyte with pH <2.4, expecting a lower cell
voltage
than the SC-BPMEA while maintaining high SPU. It was found that in the SC-
CEMEA, the
CO2 crossover was essentially eliminated. This observation is ascribed to the
lower pH
near the stationary catholyte layer/CEM interface, as shown in Fig. 9a.
SC-CEMEA shows a lower full cell voltage (Fig. 9b) compared to the SC-BPMEA
presented, partly due to the lower resistance of the CEM and the absence of
water
dissociation overpotential. Meanwhile, it has a reasonable CO2RR selectivity
over HER
(Fig. 9c) due to the cation effect and high local pH induced by the presence
1K+ in the SC-
layer (Fig. 9a). However, this design is not amenable to steady-state
operation without
continuous addition of acid and salt to the anolyte, as the initial pH
gradient will be
eliminated due to co-ion transport and neutralization. We found the CO2RR
selectivity
decreases over time and approaches 100% H2 after -3 h.
It was also observed that the SC-CEMEA design periodically ejects electrolyte
from the
cathode flow channel, likely due to poor water balance. On the anode, the OER
generates
one proton per one electron transfer. The charge carriers across the CEM are
primarily
H+, although neutral ion pairs will diffuse as well. At the cathode K+ makes
up the
electrochemical double layer at the Cu surface, and the steady-state 1K+
profiles are
governed by the electric and chemical-potential gradients that develop under
operation,
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
which usually takes tens of seconds (see reference 19). H+ migrates to the
cathode and
combines with OH- (or C0321HCO3-), producing water at the cathode. The protons
also
drag water molecules (-1 per proton) by electro-osmosis. It was accordingly
calculated
the water balance for different cathode products as listed in Table 2. The
water generated
5 and transported to the cathode appears to dilute and push out the
electrolyte in the
stationary catholyte layer, of which the volume is small (ca. 10 pL per cm2
electrode area).
This phenomenon results in flooding of the cathode (as confirmed
experimentally) and
loss of supporting electrolyte, thus degrading performance. In the BPMEA
design, it is
likely that the BPM slows co-ion transit across the membrane, compared to the
OEM, by
10 the large outward flux of OH- and H+ from the water dissociating
junction.
Table 2. The cathode-anode water balance in an SC-CEMEA.
Product Water balance in the cathode (mol)
Water balance in the anode (mol)
(1 mol) Consumed Generated Dragged in
Net Consumed and dragged out
CO 1 2 2 +3 3
HC00- 1 1 1 +1 2
CH3C00- 5 7 7 +9 11
C2H4 8 12 12 +16 18
C2H5OH 9 12 12 +15 18
CH4 6 8 8 +10 12
H2 2 2 2 +2
15 Energy assessment of the SC-BPMEA with optimal SC-layer
The energy costs (measured in gigajoules per tonne of the target product,
GJ/t) for a 002-
to-02+ electrolyzer include the electrolysis electrical energy, cathodic
stream separation,
and anodic stream separation. CO2RR performance metrics of importance include
cell
voltage, target product FE, SPU and CO2 crossover (see reference 12). High SPU
and
20 high energy efficiency have not been accomplished simultaneously in 02+
electroproduction. In SC-BPMEAs, a higher SPU reduces the energy required for
cathode
separation, but the accompanying decrease in the ethylene selectivity (Fig.
22c) elevates
the specific energy requirement. Atotal energy assessment of the SC-BPM EA was
carried
out and other state-of-art CO2-to-ethylene electrolyzers and summarized the
results in
25 Table 3.
Table 3 Comparison of the energy intensity between various 002-to-ethylene
electrolyzers and this work. All the energy costs are normalized per ton of
ethylene
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
56
produced. The energy intensities of the SC-BPMEA operating at other CO2 input
flow
rates are listed in Table S3 of Supplementary Information.
This work
This work
Neutral- Acidic Acidic MEAa
Metrics (10 sccm
(1.17 sccm
MEA a 15 flow cell a (1) 1(2)
CM -2)
cm-
2)
Cell type MEA Flow cell MEA Sc-
SC-BPMEA
Electrolyte Neutral Acidic Acidic Neutral
Neutral
Full cell voltage (V) 3.75 4.20 3.80 3.82
3.82
Ethylene FE (`)/0) 45 28 36 42
40
Current density (mA cm-2) 150 1200 100 200
200
Input CO2 flow rate 8 3 0.8 10
1.17
(sccm cm -2)
Total CO2 SPU (%) 3 78 34 4.1
35
CO2-to-ethylene (%) 12 28 7.6 2.0
17
Demonstrated stability 100 14 12 52
Energy intensity (GJ per ton ethylene)
Electrolyzer electricity 345 620 436 385
395
Cathode separation 38 17 28 85
15
Anode separation 116 0 b 0 b 0 b
0 b
Overall energy 499 637 465 470
410
' The energy intensities of reference CO2-to-ethylene devices operating under
the reported conditions are
calculated, and those that provide the lowest energy intensity are presented
in this table.
b Crossover of CO2 in the acidic flow cell, acidic MEA and SC-BPMEA are each
lower than 0.5% of input
CO2. Therefore, we assume the anodic separation energy to be 0.
0) Huang, J. E. et al. CO 2 electrolysis to multi-carbon products in strong
acid.
Science 372, 1074-1078 (2021)
(2) O'Brien, C. P. et al. Single Pass CO2Conversion Exceeding
85% in the
Electrosynthesis of Multicarbon Products via Local CO2Regenerati on. ACS
Energy
Lett. 6, 2952-2959 (2021)
In such systems, CO2 and OH- react to form carbonate continuously. This
carbonate has
to be recovered to maintain the CO2RR performance of such a system, consuming
5.5 GJ
per tonne CO2. In the alkaline CO2RR electrolyzers, ca. 63 tonne of CO2
transforms to
carbonate to produce 1 tonne of ethylene, representing an energy penalty of
350 GJ. This
costs at least $1,900 per tonne of ethylene, while its market price is $800-
1000 per tonne.
The alkaline electrolyzers thus do not allow for ethylene electrochemical
production to be
yet profitable.
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
57
In neutral-media CO2RR electrolyzers, recovering the CO2 from the anodic gas
stream
results in significant energy costs. In the context of highly selective
conversion (i.e., CO2-
to-ethylene with unity selectivity), the recovery process requires an energy
input of 52 GJ
to produce every tonne of product. In practice, due to non-unity product
selectivity, the
process is even more prohibitive, i.e., requiring an energy penalty of 80 -
130 GJ for
producing one tonne of ethylene.
As the SPU increases from 4 to 35%, we found a dramatic decrease in energy
associated
with cathode separation - from 85 to 15 GJ/t ethylene (Table 3), with the
ethylene FE
reduced by only 2%. Further increasing the SPU beyond 35% does not
substantially
reduce the energy cost associated with cathodic separation. This finding
agrees with a
recent energy analysis that in a (bi)carbonate-free CO2-to-C2, electrolyzer,
improving SPU
over 40% offers an insignificant benefit to the downstream separation cost.
Pursuing an
SPU > 35% decreases ethylene FE by more than 4% when using the SC-BPMEA, and
thus the increased input electricity cost exceeds the savings in the cathodic
separation
(Table 3). Therefore, 35% SPU is the most favourable condition for the present
SC-
BPMEA.
The energy intensity of producing ethylene in SC-BPMEA is -30% lower than that
in
conventional neutral-electrolyte-based CO2 electrolyzers (Table 3). In
conventional
neutral-electrolyte 002-to-ethylene electrolyzers, the CO2 crossover (at least
70%) costs
60-90 GJ per ton of ethylene to recover CO2 from the anodic 02 stream'.
Notably, this
energy penalty cannot readily be reduced, independent of optimizing catalysts
and
operating conditions (e.g. input CO2 flow rates, reaction rates, operating
temperature and
pressure). In contrast, crossover CO2 in SC-BPMEA is < 0.5% of the total CO2
input,
minimizing the energy cost of anodic separation.
Recently, CO2-to-ethylene conversion has been achieved in acidic electrolytes
in both flow
cell and MEA configurations. These systems enabled CO2 SPUs exceeding 75% and
also
mitigated the energy cost associated with anodic separation (Table 3). Owing
to the
strongly acidic environment, the flow cell enables an ethylene FE of 28% at a
full-cell
potential of 4.2 V. The acidic MEA used an anion-exchange ionomer coating on
the
catalyst layer to promote CO2RR over HER. The modification of the surface with
the anion
exchange ionomer resulted in a higher ohmic loss, and thus the cell required
potentials of
3.8 V and 4.4 V at 100 mA cm-2 and 200 mA cm-2, respectively. These devices
thus
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
58
eliminated the anodic CO2/02 separation energy but at the penalty of larger
cell voltages
and/or lower ethylene FEs. In contrast, SC-BPMEA shows a cell voltage of 3.8 V
at 200
mA cm-2 with an ethylene FE of 42% - voltages and selectivities comparable to
the best
conventional neutral-electrolyte CO2-to-ethylene MEAs (see reference 15).
Compared to
acidic systems, the energy intensity of the SC-BPMEA is 36% and 12% lower than
acidic
flow cell and acidic MEA, respectively (Table 3).
The inventors have demonstrated a BPM-based CO2-to-02, MEA, with a judiciously-
designed SC-layer between catalyst and BPM, that overcomes the (bi)carbonate-
formation reactant loss issue without compromising performance. The
composition and
thickness of the SC-layer determine the CO2RR performance and SPU via a strong
influence on the local pH and the chemistry and transport of CO2 The buffering
capacity
and the thickness of the SC-layer determine the efficiency of the
regeneration, the
transport, and the availability of reactant CO2. These effects were predicted
in simulations
and supported by experiments. The SC-BPMEA design largely eliminates the
energy
penalty associated with the CO2 loss in electrochemical CO2 reduction.
The performance of the SC-BPMEA might be further improved using, for example,
ionic
liquid or other organic salts as the catholyte, and by optimizing the
porosity, structure and
hydrophobicity of the porous support layers. The CO2RR performance of the SC-
BPMEA
might be improved with new cathodic catalysts, optimizing the loading and
processing of
the catalyst layer, and by implementing BPMs with further-lowered water-
dissociation
voltage loss. Broadly, the SC-BPMEA is a useful platform for evaluating CO2RR
catalysts
operating with high CO2 utilization. The strategy and findings presented here
are also
relevant to the electrochemical systems such as nitrate reduction and
(bi)carbonate
reduction, where controlling dissimilar microenvironments near each electrode
is useful,
and the exchange/transport of species (other than OH- or H+) between cathode
and anode
is problematic.
Methods
Materials
Phosphoric acid (H3PO4, 85%), potassium sulfate (K2SO4, 99%), potassium
bicarbonate
(KHCO3, 99.7%), potassium chloride (KCI, 99%), potassium hydroxide (KOH,
99.95%),
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
59
copper nanoparticles (25 nm), NafionTm 1100W (5 wt. % in a mixture of lower
aliphatic
alcohols and water) and isopropanol (IPA, 99%) were purchased from Sigma
Aldrich and
used as received. Titanium oxide nanoparticles (TiO2, Aeroxide P25) were
purchased from
Fisher Scientific and used as received. The porous supports were also
purchased from
Fisher Scientific: 125 pm PVDF (0.45 pm pore size), 65 pm PTFE (0.44 pm pore
size) and
16 pm PC (0.4 pm pore size). NafionTM 212, NafionTM XL, Fumasep (FAS-PET-130)
and
titanium (Ti) felt were purchased from Fuel Cell Store. Iridium(IV) chloride
hydrate
(Premion , 99.99%, metals basis, Ir 73% min) was purchased from Alfa Aesar.
The water
used in this study was 18 MO Milli-Q deionized- (DI-) water. Nafion membranes
were
activated through the following procedure: 1 h in 80 C 1M H2SO4¨ 1 h in 80 C
H202¨ 1
h in 1 M H2SO4 ¨ stored in DI-water. Fumasep was used as received and stored
in 1 M
KCI. Piperion (40 pm) was purchased from W7Energy and stored in 0.5 M KOH.
Fabrication of water dissociation catalyst layer of the custom bipolar
membrane
(BPM)
The water dissociation catalyst layer was fabricated following a similar
procedure in a
previous report17. TiO2 nanoparticles inks were prepared by sonicating the
mixture of TiO2,
DI-water, and IPA with the weight ratio of 1: 833: 2833 for 30 min. TiO2
nanoparticle ink
was spray-coated onto a Nafion 212 membrane, of which the edges were sealed by
Kapton tape. The exposed membrane dimension was 2.2 cm x 2.2 cm. The nominal
loading of TiO2 is 0.2 mg cm-2. The TiO2-coated NafionTM was immediately used
for
assembling electrolyzers once prepared.
Electrode preparation
For the CO2RR, we prepared the gas diffusion electrodes (GDEs) by spray-
depositing a
catalyst ink dispersing 1 mg mL-1 of Cu nanoparticles and 0.25 mg mL-1 of
NafionTM 1100W
in methanol onto a hydrophobic carbon paper. The mass loading of Cu NPs in the
GDE
was kept at 1.5 mg/cm2. The GDEs were dried in the air overnight prior to
experiments.
The OER electrode preparation procedure involves: etching the Ti felt in
hydrochloric acid
at 70 C for 40 min; rinsing the etched Ti felt with DI water; immersing the
Ti felt into an
Ir(IV) chloride hydrate solution; drying and sintering the ft-loaded Ti felt.
The loading,
drying, and sintering steps were repeated until a final Ir loading of 1.5 mg
cm-2 was
achieved.
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
Assembly of the stationary catholyte membrane electrode assembly (SC-BPMEA)
The MEA set (5 cm2) was purchased from Dioxide Materials. A cathode was cut
into a 2.1
cm x 2.1 cm piece and placed onto the MEA cathode plate with a flow window
with a
5 dimension of 2.2 cm x 2.2 cm. The four edges of the cathode were sealed
by Kapton tape,
which also made the flow window fully covered. The exposed cathode area was
measured
every time before the electrochemical tests, in the range of 3.1 to 4.2 cm2.
Onto the
cathode, a porous support layer (2 cm x 2 cm with various thicknesses, 250 pm
was
stacking two 125 pm-thick PVDF) saturated with desirable electrolyte
(sonicated in
10 electrolyte for 15 min to degas) was carefully placed. This porous
support layer serves as
the 'stationary catholyte layer (SC-layer).' The considerations of membrane
selection can
be found in SI2 and SI4 of the Supplementary Information. When using the
custom BPM,
a TiO2-coated Nafion membrane was placed onto the SC-layer with the TiO2 layer
facing
up, then covered by a Piperion (5 cm x 5 cm) membrane. When using Fumasep BPM,
the
15 membrane was placed with its cation-exchange layer (CEL) facing the
cathode side. An
Ir02 loaded Ti felt (2 cm x 2 cm) was placed onto the anion-exchange layer
(AEL) of the
BPM.
Scanning electron microscopy (SEM)
20 Images of cathode and custom BPM were captured by an FEI Quanta FEG 250
environmental SEM.
Electrochemical measurements
Throughout all experiments, CO2 flowed to the cathode side at 10 sccm cm-2
unless
25 otherwise specified, while the anode side was fed with neutral 0.1 M
KHCO3 at 10 mUrnin
by a peristaltic pump unless otherwise specified. The electrochemical
measurements were
performed with a potentiostat (Autolab PGSTAT204 with 10A booster). The cell
voltages
reported in this work are not iR corrected. The system was allowed to
stabilize at the
specific conditions for > 1000 seconds before recording the results. All the
error bars
30 represent standard deviations based on three measurements.
Product analysis
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
61
The CO2RR gas products, oxygen, and CO2 were analyzed by injecting the gas
samples
into a gas chromatograph (Perkin Elmer Clarus 590) coupled with a thermal
conductivity
detector (TCD) and a flame ionization detector (FID). The gas chromatograph
was
equipped with a Molecular Sieve 5A Capillary Column and a packed Carboxen-1000
Column with argon as the carrier gas. The volumetric gas flow rates in and out
of the cell
were measured with a bubble column. The FE of a gas product is calculated as
follows:
VP niF
FEi = xi x x (1)
Where x, is the volume fraction of the gas product i, V is the outlet gas flow
rate in L s', P
is atmosphere pressure 101.325 kPa, R is the ideal gas constant 8.314 J mo1-1
K-1, T is
the room temperature in K, n, is the number of electrons required to produce
one molecule
of product F is the Faraday Constant 96485 C mo1-1, and J is the total current
in A.
The liquid products from the cathode side of the SC-BPMEA were collected using
a cold
trap cooled to 0 C. The collected liquid was combined with anolyte (some
crossover liquid
product) for quantifying by the proton nuclear magnetic resonance spectroscopy
CH NMR)
on an Agilent DD2 500 spectrometer in D20 using water suppression mode and
dimethyl
sulfoxide (DMSO) as the internal standard. For each plot of liquid product
quantification,
fresh anolyte was used, and the duration of the collection was 30 min. The FE
of a liquid
product is calculated as follows:
FEi = mi xn.; (2)
Where m, is the quantity of the liquid product i in mole, t is the duration of
product collection
(1800 seconds).
COMSOL one-dimensional modeling
The electrochemical reaction model was performed by COMSOL Multiphysics
version 5.5.
This simulation was built upon previous modeling work. The local pH and
different species
concentrations were simulated for different catholyte thicknesses (16 pm, 65
pm, 125 pm,
and 250 pm). Two different catholytes (K2SO4 and KHCO3) were used in the
simulation.
All the chemical reactions between species were considered in this one-
dimensional
modeling. The simulation included a 50 pm thick gas diffusion layer (GDL), a
0.1 pm thick
Cu cathode catalyst (CL), a catholyte region with various thicknesses
indicated above,
and a cation exchange layer (CEL) boundary.
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
62
Constant concentration (Dirichlet) boundary conditions were used.
Specifically, a constant
concentration 37.8 mM of CO2 was assumed within the GDL layer, as this region
is in
direct contact with the input CO2 flow and thus assumed to be at equilibrium
with gas
phase CO2 over this region for the purposes of the simulation. The BPM was
interpreted
as a boundary with a constant species concentration (1 M H30 at the CEL
surface),
because it was assumed to generate protons as the dominant ionic charge
carrier at a
constant rate under constant current density (200 mA cm-2).
A user-controlled mesh is employed in the COMSOL simulation. Edge type of mesh
is
used for GDL, CL, catholytes, respectively. Specifically, the mesh
distribution is predefined
with an interval of 500 nm for GDL and catholytes, and an interval of 5nm for
CL.
Five different electrode reactions were considered at the cathode catalyst
layer in this
simulation. Specifically, the hydrogen evolution reaction and CO2 reduction
reactions to
CO, CH4, 02H4, and C2H5OH occurred at the cathode catalyst layer. In SC-BPMEA,
the
catalyst layer is immersed in a catholyte. Thus the simulation considers no
gas-phase
transport in the catalyst layer. The carbonate equilibrium reactions,
corresponding
catholyte buffer reactions, and a water dissociation reaction were considered
in the
catholyte region. The electrochemical reaction rates of the specific products
were
determined from experimental results. They are calculated based in the same
manner as
previous work17.
The electrochemical reactions at cathode catalyst layer:
2H20 + 2e- 112 20H-
CO2+ H20 +2e- ¨> CO +20H-
CO2 H2 0 8e- ¨> CH4 + 80 H-
2CO2+ 8H20 +12e- C2H4 + 120H-
2CO2+ 9H20 +12e- C2H5OH + 12011-
The heterogenous electrochemical reaction rates are determined by the
following
equations:
¨ * _______________________________ (3)
niF Lcarcayst
'total (-FEco FE'cH4 FEc2H4 FEczlison * _____________
rCO2 =
(4)
F \ 2 8 12 12 Lcata/yst
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
63
total
r0H¨ = (5)
F Lcatalyst
Where /i represents the partial current density for CO, CH4, C2H4, and C2H5OH
occurred
at the cathode catalyst layer, respectively. ni represents the number of
electrons
transferred per mole reactant. F represents faraday's constant. /roral
represents the total
current density. The FEs for the specific product is determined by the
experimental results.
E represents the catalyst porosity value. Lcataiyõ represents the cathode
catalyst length.
The chemical reactions at the catholyte region and the corresponding forward
kr rate
constants and reverse kr rate constants taken from the literature:
Reaction kf kr
CO2 +H20 H + HCO 0.036 [s-1] 7.83x104
HCO3 44 11+ + co 2.5 [s-1] 5x1019 [M-
1s-1]
CO2 + OH- HCO 2.23x103[M-1s-1] 4.85x10-
8
HCO + 0H CO + H20 6x109[M-1s-1] 1.2 [s-1]
K2504 21C+ + S0,21- 1x107 [s-1] 7.96x107 [M-
1-120 44 H + OH- 108 [s-1] 1019 [M-1s-1]
The Transport of Diluted Species physics model was used. The Nernst-Planck set
of
equations governed the species diffusion, and they were calculated in the same
manner
as previous work.13,14 Migration was ignored for simplicity as the experiments
were
performed in the concentrated electrolyte. The ion species transport is thus
calculated by
solving the two equations below.
8ci aft
+ = Ri (6)
ji = Daiaci
(7)
Di = * (8)
TF,i
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
64
= ET," (9)
Where Lis the molar flux, and ri represents the heterogeneous electrode
reactions for CO2
reduction that were modelled at the cathode catalyst layer. RI represents the
rates of the
homogeneous reactions indicated above. The Millington and Quirk model is used
to
determine the effective diffusivity, D1. Ep represents porosity coefficient.
TF,, represents
tortuosity coefficient.
The porosity value of 0.6 was used for the cathode catalyst and the porosity
value of 1 for
the catholyte region. The species diffusion coefficients are listed below.
Species Diffusion coefficients Di(10-9 m2s-1)
CO2 1.91
H20 2.57
K2SO4 1.39
KHCO3 1.20
K+ 1.98
H+ 9.31
OH- 5.26
HCO3- 1.185
C032- 0.923
S042- 1.07
Henry's law and sets of Sechenov equation are applied to calculate the CO2
concentration.
The concentration of CO2 in electrolytes depends on temperature and pressure.
It is
estimated in the same manner as previous work. The Sechenov coefficients are
listed
below.37
Species Sechenov coefficients
hc,o,c02 -0.0172
h-r,c02 -0.000338
hK 0.0922
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
hoH 0.0839
hiHco3 0.0967
hc03 0.1423
Energy assessment
We evaluated the energy consumptions for electrolyzer electricity, cathodic
separation,
and anodic separation in the context of ethylene. We consider the state-of-the-
art CO2RR
systems from the literature, including alkaline flow-cell electrolyzers,
neutral MEA
5 electrolyzers, acidic flow-cells and MEAs. This consideration is based on
the performance
metrics, including selectivity, productivity, and full-cell voltage ¨ the
combination reflects
as energy intensity of producing multi-carbon products (i.e. ethylene). The
proximity of
these performance metrics will help refine the effect of anodic and cathodic
separation on
the energy requirement for producing ethylene. We summarize the input
parameters to
10 the model for all the systems. The energy assessment model, as well as
the assumptions,
are based on the previous work. Ideally, it will be interesting to use
experimental/modelling
data corresponding to the exact gas composition from the CO2-to-C2, device.
However, at
present, there is a gap in published literature. We therefore employed one of
the most
widely used models (i.e. biogas upgrading) as the best approximation for
evaluating the
15 energy cost associated with cathode gas separation. The details of
calculations for the
carbon regeneration (for alkaline flow cell) and cathodic separation (for all
the
electrolyzers), can be found in previous work. The anodic separation (for
neutral MEA
electrolyzer) is modelled based on an alkaline capture solvent. The amount of
CO2
crossover to the anode is calculated for one tonne of ethylene produced. The
energy
20 required to separate the CO2/02 mixture is calculated based on a recent
report by Carbon
Engineering, in which 5.25 GJ/tonne CO2 thermal energy and 77 kWh/tonne CO2
are
reported to be required to capture CO2 and release at 1 bar. This energy
consumption is
a typical value for the alkaline capture process. For acidic flow-cell and MEA
electrolyzers,
we assume no energy cost associated with the anodic separation considering no
CO2
25 availability at the anodic gas stream.
REFERENCES - The following references are incorporated herein by reference:
1. Kibria, M.G., Edwards, J.P., Gabardo, C.M., Dinh, C.T.,
Seifitokaldani, A., Sinton,
D., and Sargent, E.H. (2019). Electrochemical CO2 Reduction into Chemical
30 Feedstocks: From Mechanistic Electrocatalysis Models to System Design.
Adv.
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
66
Mater. 31, 1-24.
2. Garcia de Arquer, F.P., Dinh, C.T., Ozden, A., Wicks, J., McCallum, C.,
Kirmani,
A.R., Nam, D.H., Gabardo, C., Seifitokaldani, A., Wang, X., et al. (2020). CO2
electrolysis to multicarbon products at activities greater than 1 A cm-2.
Science
367 (6478), 661-666 (doi: 10.1126/science.aay4217)
3. De Luna, P., Hahn, C., Higgins, D., Jaffer, S.A., Jaramillo, T.F., and
Sargent, E.H.
(2019). What would it take for renewably powered electrosynthesis to displace
petrochemical processes? Science 364 (6438) (doi: 10.1126/science.aav3506)
4. Dinh, C.T., Burdyny, T., Kibria, G., Seifitokaldani, A., Gabardo, C.M.,
Pelayo
Garcia De Arguer, F., Kiani, A., Edwards, J.P., De Luna, P., Bushuyev, 0.S.,
et al.
(2018). CO2 electroreduction to ethylene via hydroxide-mediated copper
catalysis
at an abrupt interface. Science. 360, 783-787.
5. Li, F., Thevenon, A., Rosas-Hernandez, A., Wang, Z., Li, Y., Gabardo,
C.M.,
Ozden, A., Dinh, C.T., Li, J., Wang, Y., et al. (2020). Molecular tuning of
002-to-
ethylene conversion. Nature 577, 509-513.
6. Ozden, A., Li, F., Garcia De Arguer, F.P., Rosas-Hernandez, A.,
Thevenon, A.,
Wang, Y., Hung, S.F., Wang, X., Chen, B., Li, J., et al. (2020). High-rate and
efficient ethylene electrosynthesis using a catalyst/promoter/transport layer.
ACS
Energy Lett. 5,2811-2818.
7. Ross, M.B., De Luna, P., Li, Y., Dinh, C.T., Kim, D., Yang, P., and
Sargent, E.H.
(2019). Designing materials for electrochemical carbon dioxide recycling. Nat.
Catal. 2, 648-658.
8. Li, F., Li, Y.C., Wang, Z., Li, J., Nam, D.H., Lum, Y., Luo, M., Wang,
X., Ozden,
A., Hung, S.F., et al. (2020). Cooperative 002-to-ethanol conversion via
enriched
intermediates at molecule-metal catalyst interfaces. Nat. Catal. 3, 75-82.
9. Wang, Y., Wang, Z., Dinh, C.T., Li, J., Ozden, A., Golam Kibria, M.,
Seifitokaldani,
A., Tan, CS., Gabardo, C.M., Luo, M., et al. (2019). Catalyst synthesis under
CO2 electroreduction favours faceting and promotes renewable fuels
electrosynthesis. Nat. Catal.
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
67
10. Vermaas, D.A., and Smith, W.A. (2016). Synergistic Electrochemical CO2
Reduction and Water Oxidation with a Bipolar Membrane. ACS Energy Lett. 1,
1143-1148.
11. Rabinowitz, J.A., and Kanan, M.W. (2020). The future of low-temperature
carbon
dioxide electrolysis depends on solving one basic problem. Nat. Commun. 11, 10-
12.
12. Ozden, A., Wang, Y., Li, F., Luo, M., Sisler, J., Thevenon,
A., Rosas-Hernandez,
A., Burdyny, T., Lum, Y., Yadegari, H., et al. (2021). Cascade CO2
electroreduction enables efficient carbonate-free production of ethylene.
Joule, 1-
14.
13. Ma, M., Clark, E.L., Chorkendorff, K.T.T.S.D., and Seger, B. (2020).
Insights into
the carbon balance for CO2 electroreduction on Cu using gas diffusion
electrode
reactor designs. Energy Environ. Sci. 13, 977-985.
14. Ma, M., Kim, S., Chorkendorff, I., and Seger, B. (2020). Role of ion-
selective
membranes in the carbon balance for CO2 electroreduction: Via gas diffusion
electrode reactor designs. Chem. Sci. 11,8854-8861.
15. Gabardo, CM., O'Brien, C.P., Edwards, J.P., McCallum, C., Xu, Y., Dinh,
C.T., Li,
J., Sargent, E.H., and Sinton, D. (2019). Continuous Carbon Dioxide
Electroreduction to Concentrated Multi-carbon Products Using a Membrane
Electrode Assembly. Joule 3, 2777-2791.
16. Larrazabal, G.O., Strom-Hansen, P., Heli, J.P., Zeiter, K.,
Therkildsen, K.T.,
Chorkendorff, I., and Seger, B. (2019). Analysis of Mass Flows and Membrane
Cross-over in CO2 Reduction at High Current Densities in an MEA-Type
Electrolyzer. ACS Appl. Mater. Interfaces 11,41281-41288.
17. Mohammad, A.W., Teow, Y.H., Ang, W.L., Chung, Y.T., Oatley-Radcliffe,
D.L.,
and Hilal, N. (2015). Nanofiltration membranes review: Recent advances and
future prospects. Desalination 356, 226-254.
18. Oener, S.Z., Foster, M.J., and Boettcher, S.W. (2020).
Accelerating water
dissociation in bipolar membranes and for electrocatalysis. Science 369, 1099¨
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
68
1103 (2020).
19. Oener, S.Z., Ardo, S., and Boettcher, S.W. (2017). Ionic
Processes in Water
Electrolysis: The Role of Ion-Selective Membranes. ACS Energy Lett. 2, 2625-
2634.
20. Yan, Z., Hitt, J.L., Zeng, Z., Hickner, M.A., and Mallouk, T.E.
Improving the
efficiency of CO2 electrolysis by using a bipolar membrane with a weak-acid
cation exchange layer. Nat. Chem.
21. Li, Y.C., Zhou, D., Yan, Z., Goncalves, R.H., Salvatore, D.A.,
Berlinguette, OP.,
and Mallouk, T.E. (2016). Electrolysis of CO2 to Syngas in Bipolar Membrane-
Based Electrochemical Cells. ACS Energy Lett. 1, 1149-1153.
22. Salvatore, D.A., Weekes, D.M., He, J., Dettelbach, K.E., Li, Y.C.,
Mallouk, T.E.,
and Berlinguette, C.P. (2018). Electrolysis of Gaseous CO2 to CO in a Flow
Cell
with a Bipolar Membrane. ACS Energy Lett. 3, 149-154.
23. Delacourt, C., Ridgway, P.L., Kerr, J.B., and Newman, J. (2008). Design
of an
Electrochemical Cell Making Syngas (CO+H[sub 2]) from CO[sub 2] and H[sub
2]0 Reduction at Room Temperature. J. Electrochem. Soc. 155, B42.
24. Ma, W., Xie, S., Liu, T., Fan, Q., Ye, J., Sun, F., Jiang, Z., Zhang,
Q., Cheng, J.,
and Wang, Y. (2020). Electrocatalytic reduction of CO2 to ethylene and ethanol
through hydrogen-assisted C¨C coupling over fluorine-modified copper. Nat.
Catal. 3, 478-487.
25. Resasco, J., Chen, L.D., Clark, E., Tsai, C., Hahn, C., Jaramillo,
T.F., Chan, K.,
and Bell, A.T. (2017). Promoter Effects of Alkali Metal Cations on the
Electrochemical Reduction of Carbon Dioxide. J. Am. Chem. Soc. 139, 11277-
11287.
26. Singh, M.R., Kwon, Y., Lum, Y., Ager, J.W., and Bell, A.T. (2016).
Hydrolysis of
Electrolyte Cations Enhances the Electrochemical Reduction of CO2 over Ag and
Cu. J. Am. Chem. Soc. 138, 13006-13012.
27. Xu, Y., Edwards, J.P., Liu, S., Miao, R.K., Huang, J.E.,
Gabardo, CM., O'Brien,
OP., Li, J., Sargent, E.H., and Sinton, D. (2021). Self-Cleaning CO 2
Reduction
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
69
Systems: Unsteady Electrochemical Forcing Enables Stability. ACS Energy Lett.
6, 809-815.
28. Bui, J.C., Digdaya, I., Xiang, C., Bell, A.T., and Weber, A.Z. (2020).
Understanding Multi-Ion Transport Mechanisms in Bipolar Membranes. ACS Appl.
Mater. Interfaces 12, 52509-52526.
29. Zhang, Y., Ji, X., and Lu, X. (2014). Energy consumption analysis for
CO 2
separation from gas mixtures. Appl. Energy 130, 237-243.
30. Xie, J.F., Huang, Y.X., Li, W.W., Song, X.N., Xiong, L., and Yu, H.Q.
(2014).
Efficient electrochemical CO2 reduction on a unique chrysanthemum-like Cu
nanoflower electrode and direct observation of carbon deposite. Electrochim.
Acta
139, 137-144.
31. Qiu, Y.L., Zhong, H.X., Zhang, T.T., Xu, W. Bin, Li, X.F., and Zhang,
H.M. (2017).
Copper Electrode Fabricated via Pulse Electrodeposition: Toward High Methane
Selectivity and Activity for CO2 Electroreduction. ACS Catal. 7, 6302-6310.
32. Jermann, B., and Augustynski, J. (1994). Long-term activation of the
copper
cathode in the course of CO2 reduction. Electrochim. Acta 39, 1891-1896.
33. Lee, G., Li, Y.C., Kim, J.Y., Peng, T., Nam, D.H., Sedighian
Rasouli, A., Li, F.,
Luo, M., 1p, A.H., Joo, Y.C., et al. (2021). Electrochemical upgrade of CO2
from
amine capture solution. Nat. Energy 6, 46-53.
34 Ma, S., Sadakiyo, M., Luo, R., Heima, M., Yamauchi, M., and Kenis,
P.J.A.
(2016). One-step electrosynthesis of ethylene and ethanol from CO 2 in an
alkaline electrolyzer. J. Power Sources 301, 219-228.
35 Verma, S., Hamasaki, Y., Kim, C., Huang, W., Lu, S., Jhong,
H.R.M., Gewirth,
A.A., Fujigaya, T., Nakashima, N., and Kenis, P.J.A. (2018). Insights into the
Low
Overpotential Electroreduction of CO2 to CO on a Supported Gold Catalyst in an
Alkaline Flow Electrolyzer. ACS Energy Lett. 3, 193-198
36 Jeng, E., and Jiao, F. (2020). Investigation of CO2 single-
pass conversion in a
flow electrolyzer. React. Chem. Eng. 5, 1768-1775
CA 03219519 2023- 11- 17
WO 2022/263316
PCT/EP2022/065823
37 Jana, C. (2020). High carbonate ion conductance of a robust
PiperION membrane
allows industrial current density and conversion in a zero-gap carbon. Energy
Environ. Sci. 13, 4098-4105
38 Burdyny, T., and Smith, W.A. (2019). CO2 reduction on gas-
diffusion electrodes
5 and why catalytic performance must be assessed at commercially-
relevant
conditions. Energy Environ. Sci. 12, 1442-1453
39 Sisler, J., Shaihroz Kha, 1p, A.H., Jaffer, M.W.S.S.A., and
Sargent, E.H. (2021).
Ethylene Electrosynthesis : A Comparative Techno-economic Analysis of Alkaline
vs Membrane Electrode Assembly vs CO2-00- C2H4 Tandems. ACS Energy
10 Lett. 6,997-1002.
40 Xing, Z., Hu, L., Ripatti, D.S., Hu, X., and Feng, X. (2021).
Enhancing carbon
dioxide gas-diffusion electrolysis by creating a hydrophobic catalyst
microenvironment. Nat. Commun. 12, 1-11.
41 Singh, MR., Clark, E.L., and Bell, A.T. (2015). Effects of
electrolyte, catalyst, and
15 membrane composition and operating conditions on the performance of
solar-
driven electrochemical reduction of carbon dioxide. Phys. Chem. Chem. Phys.
17,
18924-18936.
42 Rashid, N., Ahmad, M., and Ingole, P.P. (2020). Dendritic
copper microstructured
electrodeposits for e ffi cient and selective electrochemical reduction of
carbon
20 dioxide into Cl and C2 hydrocarbons. J. CO2 Util. 38, 385-397.
43 Dufek, E.J., Lister, T.E., and Mcllwain, M.E. (2011). Bench-
scale electrochemical
system for generation of CO and syn-gas. J. Appl. Electrochem. 41, 623-631.
44 Lee, W., Kim, YE., Youn, M.H., Jeong, S.K., and Park, K.T.
(2018). Catholyte-
Free Electrocatalytic CO2 Reduction to Formate. Angew. Chennie - Int. Ed. 57,
25 6883-6887
CA 03219519 2023- 11- 17