Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
~r~
HOECHST AKTI3NGESELLSCHAFT HOE 91/F 187 Dr.JA/fe
Description
Heterocyclic compounds with renin-inhibiting properties,
a process for the preparation thereof and the use thereof
EP-A 370 454, EP-A 373 497, EP-A 394 853 and EP-A 417 698
disclose amino diol derivatives which have C-terminal
heterocyclic substituents and a renin-inhibitory action.
EP-A 369 743 and J. Med. Chem. 33 (1990~, 2326-2334,
J. Med. Chem. 33 (1990), 2335-2342 and J. Med. Chem. 34
(1991), 151-157 describe alkyl alcohol and statin
derivatives which contain two heterocyclic systems linked
via an alkylamino bridge.
It has now been found, surprisingly, that compounds which
contain two heterocyclic systems linked via an alkylamino
bridge are exceptionally active and highly specific renin
inhibitors which have a substantially improved action
in vivo and a considerably enhanced absorption in the
gastrointestinal tract.
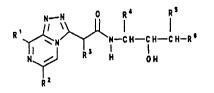
The invention therefore relates to compounds of the
formula I
R~ N--CH--CH CH--R~ t I )
in which the radicals have the following meanings:
Rl ( Cl-C8 ) -alkyl or phenyl;
R2 phenyl or pyridyl, where the pyridyl radical
optionally carries a (cl-c4)-alkyl substituent;
R3 hydrogen or a group of the formula Q-A in which Q is
pyridyl, imidazolyl, thiazolyl and pyrazolyl, and A
- 2
is methylene or ethylene,
R4 hydrogen~ (Cl-C10)-alkyl~ (C3-C3)-cycloalkyl~ (C3-C8)-
cycloalkyl-(C1-C4)-alkyl, (c6-cl2)-aryl or ~C6-C12)-
aryl-( Cl-C4 ) -alkyl;
Rs hydrogen, (C1-C10)-alkyl~ (C6 Cl2)-aryl, (C6-C12)-arYl-
(Cl-C4)-alkyl, hydroxyl or amino;
R6 a radical of the formula (II)
(CH2)~-CHR7-Z (II)
where
R7 i8 hydrogen, (Cl-C7)-alkyl, (C1-C4)-alkoxy, (Cl-C4)-
alkylthio, (Cl-C4)-alkylamino, hydroxyl, azido or
halogen, and
Z is a 5- to 7-membered heterocyclic ring which can
be benzo-fused, aromatic, partially hydrogenated or
lS completely hydrogenated and which can contain as
hetero atoms one or two identical or different
radicals from the group comprising N, O, S, NO, SO,
SO2 and which can be substituted by one or two
identical or different radicals from the series
comprising (C1-C4)-alkyl, allyl, (Cl-C4)-alkoxy,
hydroxyl, halogen, amino, mono- or di-(Cl-C4)-alkyl-
amino and CF3; and where
s is 0, 1, 2, 3 or 4; and the physiologially
tolerated salts thereof.
25 Preferred in this connection are heterocyclic compounds
in which the radicals in formula (I) have the following
meanings:
R4 isobutyl, benzyl or cyclohexylmethyl;
Rs hydrogen, (C1-C5)-alkyl, (C6-C10)-aryl, (C6-C10)-aryl-
(C1-C4)-alkyl or hydroxylr
R5 a radical of the formula (II) in which
R7is hydrogen, (C1-C4)-alkyl, (C1-C4)-alkoxy, (C1-C4)-
alkylthio, (Cl-C4)-alkylamino, hydroxyl, azido or
halogen,
35 s is 0, 1 or 2, and
Z is a heteroaryl radical from the group comprising
pyrrolyl, furyl, thienyl, imidazolyl, pyrazolyl,
~7~L7~3
-- 3 --
oxazolyl, thiazolyl, pyridyl, pyrazinyl,
pyrimidinyl, indolyl, quinolyl, isoquinolyl,
quinoxalinyl, ~-carbolinyl or a benzo-fused,
cyclopenta-, cyclohexa- or cyclohept~-fused
derivative from this group, and the other radicals
are defined as in claim 1, and the physiologically
tolerated salts thereof.
Very particularly preferred compounds of the formula (I)
are those in which the radicals have the following
meaning:
R1 methyl, ethyl, propyl, i-propyl, butyl, i-butyl or
pentyl;
RZ phenyl or 3-pyridyl, where the pyridyl radical
optionally carries a methyl or ethyl substituent;
R4 isobutyl, benzyl or cyclohexylmethyl;
Rs hydrogen or hydroxyl;
R6 a radical of the formula (II~, in which
R7 is hydrogen or fluorine,
Z is a 2-, 3- or 4-pyridyl radical, a 2-, 4- or
5-imidazolyl radical or a 2-oxazolinyl radical,
where said heterocycles can each be substituted by
one or two identical or different radicals from the
series comprising methyl, ethyl, propyl, allyl,
fluorine, chlorine, bromine, CF3 and methoxy, and
s is 0, 1 or 2.
The chirality centers in the compounds of the formula (I)
can have the ~ or S configuration.
The term "aryl" means, for example, mono- or bicyclic
hydrocarbon ring systems with one or more aromatic rings
such as phenyl, naphthyl, tetrahydronaphthyl, indanyl and
the like. Aryl radicals can moreover be unsubstituted or
substituted by 1, 2 or 3 substituents which can be,
independently of one another, (Cl-C7)-alkyl, halogeno-
alkyl, alkoxy, alkylthio, amino, alkylamino, hydroxyl,
halogen, mercapto, nitro, formyl, carboxyl and carbamoylO
-- 4 --
Halogen is fluorine, chlorine, bromine or iodine.
(C,-C10)-Alkyl means an alkyl radical or a corresponding
unsaturated radical with 1-10 carbon atoms. The radical
can be straight-chain or branched. Corresponding state-
ments apply to radicals derived therefrom, such as, for
example, alkoxy, alkylthio, alkylamino, alkanoyl and
aralkyl.
(C3-C8)-Cycloalkyl preferably means cyclopropyl, cyclo-
butyl, cyclopentyl, cyclohexyl, cycloheptyl and cyclo-
oc~yl. If these cyclic radicals carry more than one
substituent, these can be bo~h cis and trans with respect
to one another.
The radical Z is, for example, a heteroaryl radical such
as pyrrolyl, furyl, thienyl, imidazolyl, pyrazolyl,
oxazolyl, thiazolyl, pyridyl, pyrazinyl, pyrimidinyl,
indolyl, quinolyl, isoquinolyl, quinoxalinyl,
~-carbolinyl or a benzo-fused, cyclopenta-, cyclohexa- or
cyclohepta-fused derivative of these radicals. This
heterocycle can be up to trisubstituted on a nitrogen
atom by oxide, (C1-C6)-alkyl, preferably methyl or ethyl,
phenyl or phenyl-(C1-C4)-alkyl, for example benzyl, and/or
on one or more carbon atoms by (C1-C4)-alkyl such as, for
example, methyl, phenyl, phenyl-(C1-C4)-alkyl such as, for
example, benzyl, halogen such asl for 0xample, chlorine,
hydroxyl, (C1-C4)-alkoxy such as, for example, methoxy,
phenyl-(C1-C4)-alkoxy such as, for example, benzyloxy, or
oxo, and can be partially saturated. Z preferably means
one of the following groups: 2- or 3-pyrrolyl, phenyl-
pyrrolyl, for example 4- or 5-phenyl-2-pyrrolyl, 2-furyl,
2-thienyl, 4-imidazolyl, methylimidazolyl, for example
1-methyl-2-, -4- or -5-imidazolyl, 1,3-thiazol-2-yl, 2-,
3- or 4-pyridyl, 2-, 3- or 4-pyridyl 1-oxide, 2-
pyrazinyl, 2-, 4- or 5-pyrimidyl, 2-, 3- or 5-indolyl,
substituted 2-indolyl, for example 1-methyl-, 5-methyl-,
5-methoxy-, 5-benzyloxy-, 5-chloro- or 4,5-dimethyl-2-
indolyl, 1-benzyl-2- or -3-indolyl,
-- 5 ~
4,5,6,7-tetrahydro-2-indolyl, cyclohepta[b]-5-pyrrolyl,
2-, 3- or 4-quinolyl, 4-hydroxy-2-quinolyl, 1-, 3- or
4-isoquinolyl, 1-oxo-1,2-dihydro-3-isoquinolyl,
2-quinoxalinyl, 2-benzofuranyl, 2-benzoxalolyl, 2-benzo-
thiazolyl, benzo[e]-2-indolyl or ~-carbolin-3-yl.
Partially hydrogenated or completely hydrogenated
heterocyclic rings are, for example, dihydropyridinyl,
pyrrolidinyl, for example 2-, 3- or 4-N-~ethylpyr-
rolidinyl, piperidinyl, piperazinyl, morpholino, thio-
morpholino or tetrahydrothienyl.
Salts of compounds of the formula (I) mean, inparticular, pharmaceutically utilizable and non-toxic
salts.
Salts of this type are formed~ for example, by compounds
of the formula (I) which contain acidic groups, for
example carhoxyl, with alkali metals or alkaline earth
metals such as Na, K, Mg and Ca, and with physiologically
tolerated organic amines such as, for example,
triethylamine.
Compounds of the formula (I) which contain basic groups,
for example an amino group or a guanidino group, form
salts with inorganic acids such as, for example, hydro-
chloric acid, sulfuric acid or phosphoric acid and with
organic carboxylic or sulfonic acids such as, for
example, acetic acid, citric acid, benzoic acid, maleic
acid, fumaric acid, tartaric acid or p-toluenesulfonic
acid.
The invention furthermore relates to a process for
preparing compounds of the formula tI), wherein a
fragment with a terminal carboxyl group or its reactive
derivative is coupled to a corresponding fragment with a
free amino group, where appropriate protective group( 8 )
temporarily introduced to protect other functional groups
are eliminated, and the compound obtained in this way is,
~17~ ~
-- 6 --
where appropriate, converted into a physiologically
tolerated salt.
Fragments of a compound of the formula (I) with a
terminal carboxyl group have the following formula (III)
~ N ~ 0H (~
N~J
R2
Fragments of a compound of the formula (I) with a
terminal amino group have the following formula (IV)
R~ R~
H2N-Cil--CH--CH-R~ ( I V )
OH
Methods suitable for producing an amide linkage are
described, for example, in Houben-Weyl (Methoden der
organischen Chemie [Methods of Organic Chemistry], volume
15t2), Bodanszky et al. (Peptide Synthesis, 2nd ed.
(Wiley & Sons; New York 1976)) or Gross, Meienhofer (The
Peptides-Analysis, Synthesis, ~iology (Academic Press,
New York 1979)).
The following methods are preferably used: active ester
method with N-hydroxysuccinimide or 1-hydroxybenzo-
triazole as ester component, coupling with a carbodiimide
such as dicyclohexylcarbodiimide or with propane-
phosphonic anhydride or methylethylphosphinic anhydride,and the mixed anhydride method with pivaloyl chloride
(M. Zaoral, Coll. Czech. Chem. Commun., 1962, 27, 1273).
The reaction is carried out in an inert solvent or
mixture of solvents, preferably at a temperature between
-20C and the boiling point of the reaction mixture.
~7~7
-- 7 --
Carhoxylic acids of the formula (III) were prepared by
the methods of R. H. Bradbury et al. (EP-A 369 743),
D. A. Roberts et al. (J. Med. Chem. 33 (1990), 2326-
2334), R. H. Bradbury et al. (J. Med. Chem. 33 (1990),
- 5 2335-2342) and R. H. Bradbury et al. (J. Med. Chem. 34
(1991), 151-157).
The preparation of the optically active amines of the
formula (IV) in which R4, Rs and R6 are as defined above
and which are used as starting compounds starts from
optically active ~-amino acids, whose center of asymmetry
is retained. For this purpose, an N-protected amino
aldehyde is prepared in a known manner and i8 coupled in
an aldol-analogous addition onto a corresponding hetero-
arylalkyl building block and, after elimination of the
N-protective groups, gives amino alcohols of the formula
(IV). When Rs = OH, an N-protected amino aldehyde is
likewise used as starting material and is converted into
the required intermediates for example by aldol-analogous
addition of unsaturated compounds, introduction of
suitable protective groups and subsequent epoxidation.
Mixtures of diastereomers with regard to the OH-carrying
center are obtained and are separated in a manner known
per se, for example by ~ractional crystallization or by
chromatography. The diastereomeric purity is checked, for
example, by HPLC; the enantiomeric purity can be checked
in a known manner by conversion into Nosher derivatives
(H. S. Mosher et al., J. Org. Chem. 34 (1969) 2543).
N-Protected amino aldehydes are prepared by the method of
B. Castro et al. (Synthesis 1983, 676).
The aldol-analogous addition onto N-protected amino
aldehydes (preferably N-tert-butyloxycarbonyl and benzyl-
oxycarbonyl protective groups) is carried out in a
solvent which is inert to bases, such as ether, THF,
toluene, DMF, DMSO or dimethoxyethane.
Bases which can be used for the deprotonation of the
-- 8 --
heteroarylalkyl component are alkali metal alcoholates
such as potassium tert-butylate, sodium methylate, alkali
metal hydrides such as sodium or potassium hydride,
organometallic bases such as n-butyllithium, s-butyl-
lithium, methyllithium or phenyllithium, sodamide andalkali metal salts of organic ni~rogen bases such as
lithium diisopropylamide.
The necessary operations preceding and following the
preparation of compounds of the formula (I), such as
introduction and elimination of protective groups, are
known from the literature and are described~ for example,
in T. W. Greene, "Protective Groupæ Synthesis". Salts of
compounds of the formula (I~ with salt-forming groups are
prepared in a manner known per se by, for example,
reacting a compound of the formula (I) with a basic group
with a stoichiometric amount of a suitable acid.
Mixtures of stereoisomers, in particular mixtures of
diastereomers, which are produced when racemic carboxylic
acid derivatives are used, can be separated in a manner
known per se by fractional crystallization or by
chromatography.
The compounds of the formula (I) according to the
invention have enzyme-inhibiting properties, in
particular they inhibit the action of the natural enzyme
renin. Renin is a proteolytic enzyme from the class of
aspartyl proteases which is secreted as a consequence of
various stimuli (volume depletion, sodium deficiency,
~-receptor stimulation) by the juxtaglomerular cells of
the kidney into the bloodstream. There it cleaves the
decapeptide angiotensin I off the angiotensinogen which
is secreted by the liver. Angiotensin I is converted by
angiotensin converting enzyme (ACE) into angiotensin II.
Angiotensin II plays an essential part in the regulation
of blood pressure because it increases the blood pressure
directly by vasoconstriction. In addition, it stimulates
the secretion of aldosterone from the adrenal and in this
9 ~ 7~
way increases, via inhibition of sodium excretion, the
extracellular fluid volume, which in turn contributes to
increasing the blood pressure. Inhibitors of the enzyme
activity of renin bring about a reduced formation of
angiotensin I, which results in a reduced formation of
angiotensin II. The reduction in the concentration of
this active peptide hormone is the direct reason for the
blood pressure-lowering action of renin inhibitors.
The activity of renin inhibitors can be checked by
in vitro tests. This entails measuring the reduction in
the formation of angiotensin I in various systems (human
plasma, purified human renin).
1. Principle of the test
For example, human plasma which contains both renin and
angiotensinogen is incubated with the compound to be
tested at 37C. During this, angiotensin I is liberated
from angiotensinogen due to the action of renin and can
subsequently be measured in a commercially available
radioimmunoassay. This angiotensin release is inhibited
by renin inhibitors.
2. Obtaining the plasma
The blood is obtained from volunteer subjects ~about
0.5 1 per person; Bluko sampler (supplied by ASID ~onz
und Sohn, Unterschlei~heim)) and collected in partially
evacuated bottles while cooling in ice. Coagulation is
prevented by addition of EDTA (final concentration
10 mM). After centrifugation (HS 4 rotor (Sorv~ll),
3,500 rpm, 0-4C, 15 min; repeat if necessary), the
plasma is carefully removed by pipette and frozen in
suitable proportions at -30C. Only pla~mas with
sufficiently high renin activity are used for the test.
Plasmas with low renin activity are activated
(prorenin ~ renin) by a cold treatment (-4C, 3 days).
- 10 - 2~
3. Test procedure
Angiotensin I is determined using the Renin-Maia- kit
(Serono Diagnostics S.A., Coinsins, ~witzerland~. The
plasma is incubated in accordance with the instructions
given there with:
Incubation mixture: 1000 ~1 of plasma (thawed at 0-4C)
100 ~1 of phosphate buffer
(pH 7.4)
(addition of 10-~ M
ramiprilat)
10 ~1 of PMSF solution
10 ~1 of 0.1% Genapol PFIC
12 ~1 of DMSO or test product
The test products are generally dissolved Io-2 M in 100~
dimethyl sulfoxide (DMSO) and appropriately diluted with
DMSO; the maximum content of DMSO in the incubation
mixture is 1~.
The mixtures are mixed in ice and, for the incubation,
placed in a waterbath (37C) for 1 hour. A total of 6
samples (each 100 ~1) are taken from an additional
mixture without inhibitor and without further incubation
for determination of the initial angiotensin I content of
the plasma used.
The concentrations of the test products are chosen so
that approximately the range of 10-90~ enzyme inhibition
is covered (at least five concentrations~. At the end of
the incubation time, three 100 ~1 samples from each
mixture are frozen in precooled Eppendorf tubes on dry
ice and stored at about -25C for the angiotensin I
determination (mean of three single samples).
Angiotensin I radioimmunoassay (RIA)
The instructions for use of the RIA kit (Renin-Maia kit,
-- 1 1 --
Serono Diagnostics S.A., Coinsins, Switzerland) are
followed exactly.
The calibration plot covers the range from 0.2 to 25.0 ng
of angiotensin I per ml. The baseline angiotensin I
content of the plasma is subtracted from all the measure-
ments. The plasma renin activity (PRA) is reported as ng
of ang I/ml x hour. PRA values in the presence of the
test substances are related to a mixture without
inhibitor (= 100~) and the ICso is read off as % remaining
activity against the concentration (M) of the test
product (logarithmic scale).
The compounds of the formula (I) described in the present
invention show inhibitory actions in the in vitro test at
concentrations of about 10-5 to 10-1 mol/l.
Renin inhibitors bring about a lowering of blood pressure
in animals deprived of salt. Since human renin differs
from the renin of other species, primates (marmosets,
rhesus monkeys) are used for the in vivo test of renin
inhibitors. Primate renin and human renin are substan-
tially homologous in their sequence. Endogenous reninsecretion is stimulated by i.v. injection of furosemide.
The test compounds are subsequently administered and
their action on blood pressure and heart rate is
measured. ~he compounds of the present invention are
active in this test in an i.v. dose range of about
0.1-5 mg/kg and on intraduodenal administration by
gastroscope in the dose range of about 1-50 mg/kg. The
compounds of the formula (I) described in the present
invention can be used as antih~pertensives and for the
treatment of heart failure.
HIV protease is cut autocatalytically out of the GAG-POL
polypeptide and subsequently cleaves the precursor
peptide p5S into the core antigens pl7, p24 and pl4. It
is thus an essential enzyme whose inhibition interrupts
the life cycle of the virus and suppresses its growth.
12 ~ ~ i d
Biological tests showed that the compounds according to
the invention have an enzyme inhibitory action and
inhibit viral enzymes such as HIV protease too. The HIV
protease inhibiting action has particular importance and
S qualifies the compounds according to the invention in
particular for the therapy and prophylaxis of diseases
caused by infection with HIV. The compounds of the
formula (I) according to the invention show inhibitory
actions in the in vitro tests used at concentrations of
about 10-4 to 10-~ mol/l.
The present invention also relates to the use of com-
pounds of the formula (I) for producing pharmaceuticals
for the therapy of high blood pressure and the treatment
of congestive heart failure, and for the therapy and
prophylaxis of viral diseases, in particular of diseases
caused by HIV, and to said pharmaceuticals.
Pharmaceutical products contain an effective amount of
the active substance of the formula (I), preferably
together with an inorganic or organic pharmaceutically
utilizable excipient. Administration can be intranasally,
intravenously, subcutaneously or orally. The dosage of
the active substance depends on the warm blooded species,
the body weight, age and on the mode of administration.
The pharmaceutical products of the present invention are
produced in dissolving, mixing, granulating or coating
processes known per se.
For a form for oral administration, the active compounds
are mixed with the additives customary for this purpose,
such as excipient, stabilizers or inert diluents, and
converted by conventional methods into suitable dosage
forms such as tablets, coated tablets, hard gelatin
capsules, aqueous, alcoholic or oily solutions. Examples
of inert vehicles which can be used are gum arabic,
magnesia, magnesium carbonate, potassium phosphate,
lactose, glucose, magnesium stearyl fumarate or starch,
- 13 -
in particular corn starch. This preparation can be
carried out both as dry and wet granules. Examples of
suitable oily excipients or solvents are vegetable or
animal oils, such as sunflower oil and fish liver oil.
For subcutaneous or intravenous administration, the
active compounds or the physiologically tolerated salts
thereof are converted, if required with the substances
customary for this purpose, such as solubilizers, emulsi-
fiers or other auxiliaries, into solutions, suspensions
or emulsions. Examples of suitable solvents are: water,
physiological saline solutions or alcohols, for example
ethanol, propanediol or glycerol, as well as sugar
solutions such as glucose or mannitol solutions, or else
a mixture of the various solvents mentioned.
15 List of abbreviations used:
Boc tert.-Butoxycarbonyl
BuLi n-Butyllithium
DCC Dicyclohexylcarbodiimide
DCI Desorption Chemical Ionisation
DNP 2,4-Dinitrophenyl
DME Dimethoxyethane
DMF Dimethylformamide
EA Ethyl acetate
FAB Fast atom bombardment
h Hour
HOBt l-Hydroxybenzotriazole
M Molecular peak
MS Mass spectrum
min Minutes
NEM N-Ethylmorpholine
RT Room temperature
THF Tetrahydrofuran
Trt Triphenylmethyl
The following examples illustrate the preparation of the
compounds according to the invention.
14
Example 1
N-[2-(8-Isobutyl-6-phenyl-1,2,4-triazolo[4,3-a]pyrazin-3-
yl)ethanoyl]-(2S,3R,4S)-l-cyclohexyl-3,4-dihydroxy-~-
(2-pyridyl)-2-hexylamine
la) (2S,3R,4S)-2-tert.-Butyloxycarbonylamino-l-cyclo-
hexyl-3,4-dihydroxy-6-(2-pyridyl)-hexane
2.8 ml of n-butyllithium are added to 186 mg of
2-picoline in 20 ml of THF at -78C. After the mixture
has warmed up to room temperature it is stirred for 30
min and then cooled to -40C. 2 mmol of (2RS,3R,4S)-3-
tert . -butyldimethylsilyloxy-4-tert . -
butyloxycarbonylamino-5-cyclohexyl-1,2-epoxypentane
(disclosed in EP-A 189 203, Example 6) are added
(dissolved in 10 ml of THF). The mixture is stirred at
room temperature for 10 h and then diluted with water and
extracted with methyl tert.-butyl ether. The crude
product after concentration is dissolved in THF and
stirred with 10 ml of a 1 M solution of
tetrabutylammonium fluoride in THF at 0C for 1 h.
Dilution with water, extraction with EA and concentration
result in 300 mg of the (2S,3R,4S) isomer (MS (FAB): 393
(M+H)) and 240 mg of the (2S,3S,4S) isomer (MS (FAB): 393
(M~H)) after separation by chromatography on silica gel.
lb) N-[2-(8-Isobutyl-6-phenyl-1,2,4-triazolo[4,3-a]-
pyrazin-3-yl)-ethanoyl]-(2S,3R,4S)-l-cyclohexyl-3,4-
dihydroxy-6-(2-pyridyl)-2~hexylamine
400 mg of (2S,3R,4S)-2-tert.-butyloxycarbonylamino-1-
cyclohexyl-3,4-dihydroxy-6-(2-pyridyl)hexane (compound
from Example la) are dissolved in 5 ml of abs. CH2Cl2,
5 ml of trifluoroacetic acid are added and the resulting
solution is stirred at room temperature for 3 h. The
residue obtained after concentration is taken up in 1 M
NaHCO3 solution, the mixture is extracted three times
with EA, and the combined EA phases are dried over Na2SO~.
- 15 _ 2~r'~
The residue after concentration is dissolved in 5 ml of
absolute DMF, 3l6.6 mg of 2-(8-isobutyl-6-phenyl-l,2,4-
triazolo[413-a]pyrazin-3-yl)acetic acid (disclosed in
EP-A 369,743, Example l), 208.4 mg of N,N-dicyclohexyl-
carbodiimide and 206.8 mg of l-hydroxybenzotriazole are
addedl the pH of the solution is adjusted to 9 with
N-ethylmorpholinel and it is ~tirred at room temperature
for 50 h. Filtration is followed by dilution with EA and
washing once each with saturated NaHCO3 solutionl water
and saturated brinel and chromatography with a
CH2Cl2/CH3OH mixture as mobile phase yields 207.4 mg of
the title compound.
MS(FAB):585 (M+H)
Example 2
N-[2-(8-Isobutyl-6-phenyl-ll2,4-triazolo[4,3-a]pyrazin-3-
yl)-3-(3-pyridyl)propionyl]-(2S,3R,4S)-l-cyclohexyl-3,4-
dihydroxy-6-(2-pyridyl)-2-hexylamine
The title compound results from reaction of 2-[8-iso-
butyl-6-phenyl-1,2,4-triazolo[4,3-a]pyrazin-3-yl]-3-(3-
pyridyl)propionic acid (disclosed in EP-A 369 743,
Example 3) with the compound from Example la by the
process of Example lb.
MS (FAB):676 (M+H)
Example 3
N-[2-(8-Propyl-6-(3-pyridyl)-l,2,4-triazolo[4,3-a]-
pyrazin-3-yl)ethanoyl]-(2S,3R,4S)-l-cyclohexyl-3,4-
dihydroxy-6-(2-pyridyl)-2-hexylamine
The title compound is prepared from 8-propyl-6-(3-
pyridyl)-ll214-triazolo[4l3-a]pyrazin-3-yl-acetic acid
(disclosed in EP-A 369 7431 Example 4) and the compound
from Example la by the process detailed in Example lb.
MS (FAB): 572 (M+H)
16 2~ 3
Example 4
N-[2-(8-Propyl-6-(3-pyridyl)-1,2,4-triazolo[4,3-a]-
pyrazin-3-yl)-3-(3-pyridyl)-propionyl]-t2S,3R,4S)-l-
cyclohexyl-3,4-dihydroxy-6-(2-pyridyl)-2-hexylamine
The title compound is prepared from 2-[8-propyl-6-(3-
pyridyl)-1,2,4-triazolo[4,3-a]pyrazin-3-yl]-3-(3-
pyridyl)propionic acid (disclosed in EP-A 369 743,
Example 6) and the compound from Example la by the
process of Example lb.
10 MS tFAB): 663 (M+H)
Example 5
N-[3-Imidazol-4-yl)-(2RS)-2-(8-propyl-6-(3-pyridyl)-
1,2,4-triazolo[4,3-a]pyrazin-3-yl)-propionyl]-(2S,3R,4S)-
l-cyclohexyl-3,4-dihydroxy-6-(2-pyridyl)-2-hexylamine
The title compound is prepared from 3-(1-triphenylmethyl-
imidazol-4-yl)-2-[8-propyl-6-(3-pyridyl)-1,2,4-triazolo-
[4,3-a]pyrazin-3-yl]-propionic acid (disclosed in EP-A
369 743, Example 27) and the compound from Example la by
the process of Example lb and subsequent elimination of
the Ni~-Trt protective group with 90~ strength trifluoro-
acetic acid and working up by extraction.
MS (FAB): 652 (M+H)